Note: Descriptions are shown in the official language in which they were submitted.
CA 02331602 2001-10-25
METHOD OF MANUFACTURING A POSITIVE ELECTRODE ACTIVE
MATERIAL OF A SECONDARY BATTERY
This application is a divisional application of
Canadian Patent Application Serial No. 2,110,097, filed
November 26, 1993, and entitled "Secondary Battery"
BACKGROUND OF THE INVENTION
Field of the Invention
The present invention relates to a secondary
battery which can repeatedly be used, and more
particularly to a reliable secondary battery capable of
preventing short circuit occurring due to dendrite even
if the battery is repeatedly charged and discharged.
Related Background Art
Since global warming is expected to take place due
to the greenhouse effect caused from an increase in CO~
and so forth, construction of thermal power plants
encounters problems. Accordingly, it has been considered
feasible to perform so-called load levelling for the
purpose of effectively using generators by accumulating
electric power at night in secondary batteries at homes
to level the load.
There arises another desire for a development of
secondary battery which exhibits a high energy density
for use in an electric car that does not exhaust air
contamination substances. Further, development of a high
performance secondary battery has been needed for use as
a power source for portable equipment, such as a book-
type personal computer, a word processor, a video camera
and a portable telephone.
A locking chair type lithium ion battery capable
CA 02331602 2001-10-25
-2-
of serving as the foregoing high performance secondary
battery and comprising a positive electrode active
material comprising lithium ions introduced into an
interlayer compound thereof and a negative electrode
active material comprising carbon has been developed and
partially put into practical use.
However the lithium ion battery has not achieved
the high energy density that is the original
characteristics of the lithium battery which uses the
metal lithium as the positive electrode active material.
The reason why a large capacity lithium accumulator of
the type that uses the lithium metal as the positive
electrode has not been put into practical use is that
generation of dendrite of the lithium (tree branch like
crystal) which is the main cause of a short circuit
cannot be prevented yet.
The lithium battery, nickel-zinc battery and the
air-zinc battery encounter the difficulty that lithium
or zinc is, as described above, deposited on the surface
of the negative electrode at the time of charge. At this
time, the current density is locally raised on the
negative electrode surface depending upon the surface
condition, causing lithium or zinc to be selectively
deposited in the foregoing place. The deposited metal
grows (dendrite) in the form of tree branch with the
progress of the charge and discharge cycles while
penetrating a separator until it reaches the positive
electrode, causing a short circuit to occur.
CA 02331602 2001-10-25
-3-
The dendrite reaction mechanism is considered as
follows. Since lithium or zinc that deposits at the time
of charge has a considerable reactivity, it reacts with
electrolyte solution or water or the like in the
electrolyte solution, causing an insulating film to be
formed which has a large resistance. Therefore, the
current density in the foregoing portion is raised at
the time of the next charge, resulting in that the
dendrite can further easily glow. It leads to a fact
that a short circuit takes place between the negative
electrode and the positive electrode, resulting in that
the charge cannot be performed.
If the short circuit has taken place considerably,
the energy of the battery will be consumed in a very
short time, causing heat to be generated. As a result,
the solvent of the electrolyte solution can be
decomposed, resulting in generation of gas. Therefore,
gas is generated, causing the internal pressure to be
raised. In this case, an accident of an exposure or fire
can be generated for the worst. Therefore, there has
been a desire for a long life deposition of lithium that
does not easily cause the internal short circuit even if
the charge and the discharge are repeated.
Also nickel-zinc batteries and air-zinc batteries
generate dendrite of zinc due to repetition of charging
and discharging, the dendrite penetrating the separator.
As a result the zinc negative electrode and the positive
electrode encounter a short circuit. Therefore, the
CA 02331602 2001-10-25
-4-
foregoing conventional technology suffers from an
excessively short cycle life.
SUMMARY OF THE INVENTION
An object of the present invention is to provide a
lithium, lithium alloy, zinc or zinc alloy secondary
battery capable of overcoming the foregoing problems
experienced with the conventional secondary batteries
and exhibiting a long cycle life.
In order to overcome the foregoing problems
experienced with the conventional technology, the
inventors of the present invention have made energetic
studies. As a result, a fact was found that generation
of dendrite of lithium or zinc can be prevented by
forming a film permitting ions relating to battery
reactions to pass through on the surface of the negative
electrode.
The present invention is characterized in that a
secondary battery comprises a negative electrode made of
a negative electrode active material, a separator, a
positive electrode made of a positive electrode active
material, an electrolyte solution, a collecting
electrode and a battery case, wherein the surface of the
negative electrode is covered with a film permitting
ions relating to battery reactions to pass through.
The material of the film has a molecular structure
or small apertures which do not permit the negative
electrode active material which precipitates on the
CA 02331602 2001-10-25
-5-
negative electrode but which permit ions relating to the
battery reactions to pass through.
The present invention is characterized in that the
foregoing material of the film has been electron
donative elements or groups for enabling the ions
relating to the battery reactions to be easily conducted
in the film.
The electron donative element is exemplified by
oxygen atoms, nitrogen atoms, sulfur atoms and
transition metal atoms respectively having a paired
electron, a non-paired electron or electron d. The
electron donative group is exemplified by a ring
compound and a compound having a carbon double bond
having electron n or an aromatic ring.
The film formed on the surface of the negative
electrode according to the present invention is
characterized in that it cannot be dissolved by the
electrolyte.
The inventor of the present invention found a fact
that treatment of the surface of the negative electrode
with a nitrogen compound or a halogen compound, which is
active in a gas phase, will prevent the generation of
lithium dendrite.
According to the present invention, there is
provided a secondary battery having a negative electrode
active material composed of lithium, a separator, a
positive electrode active material, an electrolyte, a
collector and a battery case, wherein at least the
CA 02331602 2001-10-25
-6-
surface of the lithium negative electrode opposing the
positive electrode is treated with reactive and gaseous
material containing nitrogen or a halogen element.
According to the present invention, there is
S provided a battery comprising a negative electrode, a
separator, a positive electrode and an electrolyte,
wherein one or more layers selected from a group
consisting of a conductor layer, a semiconductor layer
and an insulating layer are formed between the negative
electrode and the separator.
If the negative electrode active material is
lithium or lithium alloy, the foregoing layer is formed
into a micropore structure having small apertures
permitting at least lithium ions to pass through. If
zinc or zinc alloy is used, the small apertures permit
hydride ions to pass through.
The small apertures permitting ions to pass through
may be realized by the molecular structure of the
material or by a manufacturing method. The small
apertures can be easily formed by, for example,
injecting an electrolyte into the foregoing layer at the
time of forming the layer to manufacture the battery,
the electrolyte being eluting to form the micropores.
Another method may be employed in which a foaming
material is added at the time of forming the foregoing
layer and then the micropores are formed by heat
treatment or the like.
The structure of the stacked layers may be a single
CA 02331602 2001-10-25
7
layer or a mufti-layer composed of two or more layers or
composed of a conductor layer, a semiconductor layer, an
insulating layer and a composite layer containing two or
more types of elements or compounds.
Further, a fact was found that the separator
partially including a film-shape member (hereinafter
sometimes called a "metal oxide film") of a metal oxide
formed by a mold made of a bimolecular film forming
compound is able to prevent short circuit in the battery
occurring between the negative electrode and the
positive electrode even if dendrite is generated in the
negative electrode.
According to the present invention, there is
provided a secondary battery comprising a negative
electrode made of a negative electrode active material,
a positive electrode made of a positive electrode active
material and a separator which separating the positive
electrode active material and the negative electrode
active material from each other, wherein at least a
mufti-layer metal oxide is present between the positive
electrode and the negative electrode.
Another fact was found that an arrangement that
surface of the positive electrode is covered with a thin
film made of an insulating material or a semiconductor
which is free from electron conduction and which permits
ions relating to battery reactions will prevent short
circuit in the battery between the negative electrode
and the positive electrode even if dendrite is generated
CA 02331602 2001-10-25
g _
in the negative electrode.
According to the present invention, there is
provided a secondary battery at least comprising a
negative electrode, a separator, a positive electrode,
an electrolyte, a collector and a battery case, wherein
at least the surface of the positive electrode opposing
the negative electrode is covered with one or more thin
film layers selected from a group consisting of an
insulating layer, a semiconductor layer, a layer
composed of an insulating material and a semiconductor
which permit ions relating to the battery reactions to
pass through.
Another fact was found that the utility of a
positive electrode made of a compound of one or more
types of transition metals, whose crystal grain size of
500A or less, provides a secondary lithium battery
having high capacity, large energy and long cycle life.
According to the present invention, there is
provided a lithium secondary battery at least comprising
a negative electrode active material, a separator, a
positive electrode active material through which ions
can be introduced/discharged due to charge/discharge, an
electrolyte which is an ion conductor, a collecting
electrode and a battery case, wherein the main component
of the positive electrode active material 13104 is a
compound of one or more type of transition metal and a
group 6A element and having a crystal grain size of 500A
or less.
CA 02331602 2001-10-25
-9-
The main component material of the positive
electrode active material has a structure of an
aggregate selected from a group consisting of amorphous,
microcrystal, a mixture of amorphous, microcrystal and a
mixture of amorphous, microcrystal and multi-crystal.
The arrangement that the positive electrode active
material of the lithium secondary battery is made of
compound of the transition metal having a structure of
the aggregate selected from a group consisting of
amorphous, microcrystal, a mixture of amorphous and a
microcrystal and a mixture of an amorphous, a
microcrystal and a multi-crystal, and the group 6A
element and having a crystal grain size of 500 A or
less, more~preferably 200 A enables the following
effects to be obtained:
(1) Since the reactive area of the positive
electrode active material can be enlarged, the
electrochemical reactions at the time of charge and
discharge can be made smooth, and therefore the
chargeable capacity can be enlarged.
(2) The introduction and the discharge of lithium
ions at the time of the charge and the discharge prevent
the distortion of the positive electrode active
material, causing the cycle life to be lengthened.
It is preferable that the specific area of the
positive electrode active material mainly composed of
the compound of the transition metal and the group 6A
element be 50 mz/g or more in a state before the material
CA 02331602 2001-10-25
-I~-
is formed into the positive electrode, more preferably
100 mz/g or more .
The employment of the compound of the transition
metal and the group 6A element containing hydrogen will
improve in the charge and discharge cycle
characteristics.
By subjecting the positive electrode active
material to a lipophilic treatment using an organic
metal compound, the solid-liquid reactions between the
electrolyte and the positive electrode active material
can be made further smooth at the time of charge and the
discharge.
The compound of the transition metal and the group
6A element is exemplified by a metal oxide such as a
nickel oxide, a cobalt oxide, a titanium oxide, an iron
oxide, a vanadium oxide, a manganese oxide, a molybdenum
oxide, a chrome oxide or a tungsten oxide, a metal
sulfide such as a molybdenum sulfide, an iron sulfide or
a titanium sulfide, a hydride such as an oxy iron
hydride or their mixtures.
By employment of metal lithium having a film
through which lithium ions are able to pass to form the
negative electrode active material of the secondary
battery, a lithium secondary battery exhibiting a long
life and a high energy density can be obtained.
According to the present invention, there is
provided a method of manufacturing a positive electrode
active material of a lithium secondary battery at least
CA 02331602 2001-10-25
comprising the step of forming a compound of a
transition metal and a group 6A element, the raw
material of which is one or more types of materials
selected from a group consisting of the transition
metal, the salt of the transition metal, an organic
metal compound of the transition metal, a hydride of the
transition metal hydroxide, a hydroxide transition
metal, a carbonyl compound of a transition metal and a
transition metal oxide and which has a structure of an
aggregate having a crystal grain size of 500 A or less,
more preferably 200 A or less and selected from a group
consisting of amorphous, microcrystal, a mixture of
amorphous and microcrystal and a mixture of amorphous,
microcrystal and mufti-crystal.
According to the present invention, there is
provided a method of manufacturing a positive electrode
active material which is a compound of a transition
metal and a group 6A element, the method comprising the
steps of
employing one or more types of reactions
selected from a group consisting of a reaction between
a salt of the transition metal and alkali, a hydrolysis
decomposition reaction of an organic transition metal
compound and a reaction between the transition metal and
alkali to prepare a hydride of the transition metal;
employing a dehydrating reaction or decomposition
of the salt of the transition metal or the organic
transition metal compound in a gas phase or a reaction
CA 02331602 2001-10-25
-12-
between the salt of the transition metal or the
decomposed material of the organic transition metal
compound or vapor of the transition metal and the group
6A element or the group 6A compound;
melting one or more types of materials selected
from a group consisting of the transition metal and the
transition metal compound to be allowed to react with
one or more types of materials selected from a group
consisting of the group 6A element and the compound of
the group 6A element; and
rapidly cooling the materials to form an aggregate
having a crystal grain size of 500 A or less and formed
into a structure selected from a group consisting of
amorphous, microcrystal, a mixture of amorphous and
microcrystal and a mixture of amorphous, microcrystal
and mufti-crystal.
In another aspect, the present invention provides a
secondary battery comprising: a negative electrode
having a negative electrode active material, a positive
electrode substantially made of a positive electrode
active material; a separator arranged between the
negative electrode and the positive electrode; an
electrolyte provided between said positive and negative
electrodes; and a film with a thickness of 50A and 100
~,m and of a molecular structure of large voids and fine
holes covering at least separator side of said negative
electrode for operating to preclude a contact between
said negative electrode and said electrolyte
CA 02331602 2001-10-25
-13-
substantially through which passes ion which contributes
to the battery operation.
BRIEF DESCRIPTION OF THE DRAWINGS
Fig. 1 is a basic structural view which illustrates
a secondary battery according to the present invention;
Fig. 2 is a schematic cross sectional view which
illustrates a flat battery to which the present
invention is applied;
Fig. 3 is a schematic cross sectional view which
illustrates an example of a cylindrical battery to which
the present invention is applied;
Fig. 4 is a schematic view which illustrates an
example of a partial structure of a negative electrode
covered with a film through which ions relating to
battery reactions are able to pass;
Fig. 5 is a basic structural view which illustrates
another embodiment of the secondary battery according to
the present invention;
Fig. 6 is a schematic structural view which
illustrates an apparatus for subjecting lithium for use
in the secondary battery according to the present
invention to surface treatment;
Fig. 7 is a schematic structural view which
illustrates an apparatus for subjecting lithium for use
in the secondary battery according to the present
invention to surface treatment;
Fig. 8 is a schematic view which illustrates an
CA 02331602 2001-10-25
- 14-
effect of the present invention;
Figs. 9A to 9H are views which illustrate examples
of layer stacking patterns according to the present
invention including a conductor layer, a semiconductor
layer and an insulating layer between the negative
electrode and the separator thereof;
Fig. 10 is another basic structural view which
illustrates the secondary battery according to the
present invention;
Fig. 11 is a schematic cross sectional view which
illustrates another cylindrical battery to which the
present invention is applied;
Fig. 12 is another basic structural view which
illustrates the secondary battery according to the
present invention; and
Fig. 13 is another basic structural view which
illustrates the secondary battery according to the
present invention.
DETAILED DESCRIPTION OF THE PREFERRED EMBODIMENTS
Structure of Batterv
A secondary battery according to the present
invention comprises a negative electrode, a separator, a
positive electrode, an electrolyte and a collector. Fig.
1 is a basic structural view which illustrates the
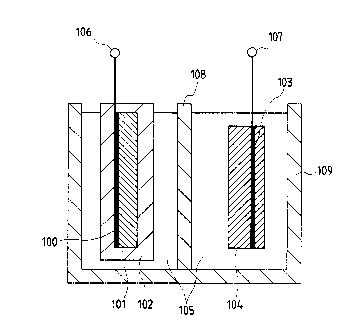
secondary battery. Referring to Fig. l, reference
numeral 100 represents a negative collector, 101
represents a negative electrode substantially made of
CA 02331602 2001-10-25
-15-
negative electrode active material, 102 represents a
shell, 103 represents a positive collector, 104
represents a positive electrode substantially made of
positive electrode active material, 105 represents an
electrolyte solution (electrolyte), 106 represents a
negative terminal, 107 represents a positive terminal,
109 represents a case for the secondary battery, and 108
represents a separator. Figs. 5, 10 and 12 illustrate
another basic structure in which a multi-layer oxide
film 10102, an ion permeable film 10102 and a layer 5102
applied with lithium surface treatment in place of the
film 102. If the lithium battery comprises the negative
electrode 101 made of the negative electrode active
material which is lithium or lithium alloy, lithium ions
in the electrolytic solution 105 are introduced into the
space between the layers of the positive electrode
active material of the positive electrode 104 through
the permeable film due to the discharge reaction in a
case of the structure shown in Fig. 12. Simultaneously,
lithium ions are dissolved and discharged from the
negative electrode active material 101 into the
electrolyte 105 through the film 102 and the multi-layer
metal oxide 10102. In the charging reaction, lithium
ions in the electrolyte solution 105 are, in the form of
lithium metal, precipitated into the negative electrode
active material through the film 102 (dendrite can
easily grow if the film 102 is not present).
Simultaneously, lithium between the layers of the
CA 02331602 2001-10-25
- 16-
positive electrode active material 104 is dissolved and
discharged into the electrolyte solution 105. Although
lithium ions precipitated during the charging reaction
are in a very active state to react with small-quantity
water, oxygen, impurities or solvent in the electrolyte
contained, the structure in which the surface of the
negative electrode 101 is covered with the film 102
prevents direct contact between the precipitated lithium
and the electrolyte solution. Therefore, the generation
of dendrite, which causes the battery short circuit to
occur, can be prevented.
In a case of an alkali battery comprising the
negative electrode active material 101 made of zinc or
zinc alloy, the discharge reaction takes place in such a
manner that hydroxyl ions in the electrolyte solution
105, similarly to the above, react with the negative
electrode active material 101 of the negative electrode
101 through the film 102. Simultaneously hydroxyl ions
are discharged into the electrolyte solution 105 from
the positive electrode active material of the positive
electrode 104. The charging reaction is performed in
such a manner that hydroxyl ions are discharged from the
negative electrode 101 into the electrolyte solution 105
through the film 102 (if the film 102 is not present at
this time, zinc ions in the electrolyte solution 105
easily cause dendrite to glow on the negative
electrode). Simultaneously, hydroxyl ions in the
electrolyte solution 105 react with the positive
CA 02331602 2001-10-25
- 17-
electrode 104. Similarly to the case where the negative
electrode active material is lithium, the presence of
the film 102 prevents generation of zinc dendrite at the
time of charging.
Therefore, the present invention is able to prevent
short circuits in the battery, lengthen the life of the
secondary battery and improve the safety.
If the foregoing negative electrode active material
is lithium or lithium alloy, lithium ions are used in
the reactions in the battery. In the case of the alkali
battery containing zinc as the negative electrode active
material, hydroxyl ions are used in the reactions. The
batteries comprising zinc as the negative electrode
active material is typified by nickel-zinc battery, air-
zinc battery and bromine-zinc battery (however, the
bromine-zinc battery comprises zinc ion as the negative
side ion that are used in the inside reactions).
Method of Forming Nectative Electrode
The negative electrode can be formed by any one of the
following methods as well as the method for forming the
same by directly covering the surface of the negative
electrode active material with a coating material by a
dipping method, a spraying method or a CVD method.
It is effective to employ a method in which a
conductive and porous matrix (the base), for example,
sponge-like or fiber metal or carbon, having
communication holes is covered with a coating material
through which ions for use in the reactions in the
CA 02331602 2001-10-25
-18-
battery can be passed through, and then lithium or zinc,
which is the negative electrode active material, is
caused to electrochemical precipitate and adhere so that
the negative electrode is formed. If a porous and
conductive matrix has a wide specific area, the current
density per unit area at the time of the charge and the
discharge can be lowered. Therefore, the growth of the
dendrite can be prevented and accordingly the charging/
discharging efficiency can be improved.
It is preferable that the film made of a coating
material, through which ions can be passed through, the
small apertures serving as the space, in which the
lithium can be precipitated, be formed into a 3D net
shape.
Another effective method may be employed which has
steps of directly immersing a porous and conductive
matrix in the melted negative electrode active material,
taking out the matrix, covering the surface of the
conductive matrix with the negative electrode active
material or electrochemically covering the same with the
negative electrode active material, and applying a
coating material through which ions for use in the
reactions in the battery can be passed to the surface.
Fig. 4 is a schematic view which illustrates a
partial structure of a negative electrode formed by
covering a conductive matrix 400 with a negative
electrode active material 401 and a coating material 402
through which ions for use in the reactions in the
CA 02331602 2001-10-25
-19-
battery can be passed.
Material for Coverina the Surface of Negative Electrode
The coating material for covering the surface of
the negative electrode will now be described (1 to 7).
1. Film Having an Inorganic Glass Structure
By covering the surface of the negative electrode
active material with the film having an inorganic glass
structure, through which ions for use in the reactions
in the battery can be passed, the reactions between
lithium and water or oxygen can be prevented,, causing
easiness in handling if the negative electrode active
material is lithium. Further, the direct contact between
the lithium and the electrolyte solution does not take
place. Therefore, the growth of the polymer film formed
from the solvent of the electrolyte solution on the
surface of lithium can be prevented. If the negative
electrode active material is zinc, the elution of zinc
into the electrolyte solution can be prevented. As a
result, formation of dendrite can be prevented and
therefore the cycle life against charge and discharge
can be lengthened. Since the film having the inorganic
glass structure is flame retardant or incombustible,
safety in an emergency breakage or the like can be
improved. The foregoing inorganic glass may be made any
one of a metal oxide selected from a group consisting of
silica, titanium oxide, alumina, zirconia oxide,
magnesium oxide, tantalum oxide, molybdenum oxide,
tungsten molybdenum, tin oxide, indium oxide, iron
CA 02331602 2001-10-25
-20-
oxide, chrome oxide, aluminum phosphate, iron phosphate,
silicon phosphate and their mixtures. In particular, it
is preferable to employ silica, titanium oxide, alumina,
zirconia oxide or aluminum phosphate.
A sol-gel method is one of adequate methods for
forming the inorganic glass. however, the fact that
lithium has a low melting point of 181°C causes a
necessity to arise for the operation of directly
applying the foregoing material to the surface of
lithium to be performed at low temperature. Since
lithium reacts with water and alcohol, the operation
must be performed in a circumstance in which no water
and alcohol is present (the case in which the inorganic
glass is previously applied to the conductive matrix or
the case where the negative electrode active material is
zinc are excluded).
Therefore, the sol-gel method, which is the typical
inorganic coating method at low temperature, must be
performed with any particular means. The raw material
for the material having the inorganic glass structure is
obtained in such a manner that an acid or a base and
water are added to a solution of alcohol of an organic
metal compound such as a metal alkoxide to hydrolyze the
raw material so as to form colloid particles having
metal atom-oxygen atom bonds, and then the solvent is
substituted by a non-hydric solvent except alcohol.
The surface coating of the negative electrode
active material is formed in a manner comprising steps
CA 02331602 2001-10-25
-21 -
of directly applying the foregoing colloid solution, or
applying a solution, in which a monomer or an organic
polymer or both organic polymer and a crosslinking
material are dissolved in the colloid solution, and
polymerizing it or drying and hardening it. By combining
the organic polymers, strength against cracks and
separations can be improved. If the electrolyte for
forming the battery is dissolved in the colloid solution
to form the film, wettability with the electrolytic
solution can be improved, causing ions to be moved
easily.
As an alternative to alkoxide, any one of the
following organic metal compound may be employed: acetyl
acetone salt, an alkyl metal compound, acetyl acetone
metal salt, naphthene acid metal salt, and octyl acid
metal salt.
The organic polymer for combining the organic
polymers is exemplified by epoxy resin, polyester,
polyimide, polyethylene, polypropylene, polyurethane,
polystyrene, polyethylene glycol, nylon, fluorine resin
and silicon resin.
The polymer crosslinking material is exemplified by
diisocyanate, polyisocyanate prepolymer, block
isocyanate, organic peroxide, polyamine, oxims, nitroso
compound, sulfur or sulfur compound, selenium, magnesium
oxide, lead oxide and zinc oxide. As an alternative to
using the crosslinking material, a method may be
employed in which radial rays or electron rays or
CA 02331602 2001-10-25
-22-
ultraviolet rays are applied to polymerize or crosslink
the polymer.
As an application method, a dipping method, screen
printing, spraying or a roll coating method may be
employed. The viscosity of the liquid to be applied must
adequately be adjusted to be adaptable to the
application method.
In order to facilitate the movement of the charge
at the time of the charging operation, powder or fiber
or whisker of conductive material such as carbon or
titanium may be mixed with the foregoing film forming
solution.
It is preferable that the thickness of the film to
be formed on the surface of the negative electrode
IS active material ranges from 50 A to 100 ~, further
preferably ranges from 100 A to 10 ~.. The optimum
thickness of the film differs depending upon the density
or the void ratio of the film and considerably differs
depending upon the type of the electrolytic solution.
The thickness of the film can be adjusted by
changing the concentration of the main material in the
coating liquid for forming the film.
2. Polymer Film of Derivative of Aromatic Hydrocarbon
Compound
By covering the surface of the negative electrode
active material with a polymer film of a derivative of
an aromatic hydrocarbon compound, the reaction between
lithium and water or oxygen can be prevented in the case
CA 02331602 2001-10-25
-23-
where the negative electrode active material is lithium.
It leads to a fact that handling can be facilitated.
Further, the contact between lithium and electrolyte
solution can be prevented. Therefore, the growth of a
polymer film, which is formed from the solvent of the
electrolytic solution, on the surface of lithium can be
prevented. If the negative electrode active material is
zinc, elution of zinc into the electrolyte solution is
prevented by the film.
As a result, formation of dendrite can be prevented
and therefore the life against the charge and discharge
cycle can be lengthened. The derivative of the aromatic
hydrocarbon for forming the charge moving complex with
lithium is one or more derivatives selected from a group
consisting of naphthalene, anthracene, phenanthlene,
naphthacene, pyrene, triphenylene, perillene, picene,
benzopyrene, coronene and ovalene. The polymer for use
to form the coating material can be prepared by
polymerization or copolymerization of vinyl monomer,
monomer of acetylene derivative or dicarboxylic acid and
a monomer such as glycol. The polymerization of the
vinyl monomer can be performed by radical or ion
polymerization. The monomer of the acetylene derivative
can be polymerized while using a chloride of tungsten as
a catalyzer. The dicarboxylic acid and diamine can be
polycondensed and the dicarboxylic acid and glycol can
as well as be polycondensed. The monomer of the aromatic
derivative for forming the polymer is exemplified by 2 -
CA 02331602 2001-10-25
-24-
vinyl naphthalene, 2 - vinyl pyridine, 9 - vinyl
anthracene, 9, 10 - anthracene dipropionic acid, 9, 10 -
bis (phenyl ethyl) anthracene and 5, 12 - bis (phenyl
ethynyl) naphthalene. It is preferable to use 2 - vinyl
S naphthalene or 9 - vinyl anthracene.
A starting material for the radical polymerization
is exemplified by azobisisobutylonitryl (AIBN),
benzoylperoxide (BPO) and t-butylhydroperoxide. A
starting material for the cation polymerization is
exemplified by an acid such as H2S04, H3P04, HC104, CC13 or
CO,H and Friedel-Craft catalyzes such as BF3, A1C13, TiCl9
or SnCl9. A large ring compound having an aromatic ring
can be polymerized by dehydrogenation in which the
Friedel-Craft catalyzer and an oxidizer are combined to
each other. A starting material for the anion
polymerization may be an alkaline metal compound or an
organic metal compound.
As an alternative to the foregoing method, a
polymer into which an aromatic group can be obtained by
subjecting the side chain of each polymer to a
substitution reaction with a derivative of an aromatic
compound. Another method may be employed in which an
electrolytic polymerization reaction is caused to take
place in an electrolytic solution containing a monomer
mixed therein to form directly a polymer of an aromatic
compound on the surface of lithium.
When the surface of lithium is applied with a
coating by using the foregoing polymer solution, it is
CA 02331602 2001-10-25
- 25 -
preferable to use a polymer solution dehydrated and
deoxidized sufficiently in inactive gas dehydrated
sufficiently. It is preferable to use a solvent in the
foregoing solution which has been dehydrated with active
alumina, molecular sheave, phosphorus pentaoxide or
calcium chloride. As an alternative to this, it is
preferable depending upon the type of the solvent that
the solvent be distilled under presence of alkaline
metal in inactive gas to remove impurities and to be
dehydrated (however, the necessity of strictly
controlling water can be eliminated when the polymer is
previously applied to the conductive matrix or when the
negative electrode active material is zinc).
An electrolyte may previously be mixed when the
foregoing film is formed. It leads to a fact that
wettability between the electrolytic solution and the
film can be improved, causing ions to easily pass
through the film. In order to facilitate the movement of
the charge at the time of charging, conductive powder,
such as carbon or titanium, fiber or whisker may be
mixed at the time of forming the film.
Since the performance of the battery deteriorates
if the polymer coating film is dissolved in an organic
solvent of the electrolyte, it is preferable to be
crosslinked in such a manner, for example, ultraviolet
rays, electron rays or radial rays are applied or a
crosslinking material, such as a radical generating
agent, is used.
CA 02331602 2001-10-25
-26-
It is preferable that the thickness of the film to
be formed on the surface of the negative electrode
active material ranges from 50 A to 100 ~, more
preferably ranges from 100 A to 10
The optimum thickness of the film differs depending
upon the density or the void ratio of the film and
considerably differs depending upon the type of the
electrolyte solution. The thickness of the film can be
adjusted by changing the concentration of the main
material in the coating liquid for forming the film.
3. Organic Metal Compound
When the surface of lithium and the organic metal
compound react with each other, bonding with lithium
atoms takes place so that a film having a surface which
is organic-bonded is formed. As a result, the
wettability (the lipophilic property) is improved,
causing lithium ions to be easily introduced/discharged
at the time of the charge and discharge. Further, the
surface coating film prevents the direct contact between
lithium and the organic solvent, causing the formation,
on the surface of lithium, of a polymerized film of the
organic solvent, which increases the resistance in the
battery, can be prevented. As a result, formation of
dendrite can be prevented, and therefore life against
the charge and discharge cycle can be lengthened.
Further, reactions between lithium and water during the
manufacturing process can be prevented and accordingly
handling can be made easier. If the content of lithium
CA 02331602 2001-10-25
-27-
in the film is high in the lithium battery according to
the present invention, rapid reactions of lithium at an
emergency breakage can be prevented.
The foregoing organic metal compound may be a
material selected from a group consisting of: metal
alkoxide, alkaline metal compound, acetyl acetone metal
salt, naphthene acid metal salt, or oxtyl acid metal
salt of metal, such as titanium, aluminum, silicon,
zirconium, tantalum, magnesium, indium, tin, molybdenum,
tungsten or germanium. Among the foregoing organic metal
compounds, it is preferable to use a metal compound of
silicon or titanium or aluminum because of easy forming
of the film and excellent stability of the formed film.
The organic silicon compound may be alkoxysilane,
alkylsilane, halogenated silane, siloxane, silane
containing vinyl group, amino group, epoxy group,
methacrylic group or mercaptal group introduced thereto,
hydrogen - denatured, vinyl - denatured, hydroxyl group
denatured, amino - denatured, carboxylic group
denatured, chloro - denatured, epoxy denatured,
methachryloxy - denatured, mercapto - denatured,
fluorine - denatured, long - chain - alkyl denatured or
phenyl - denatured polysiloxane, alkylene oxide
denatured siloxane copolymer, silicon - denatured
copolymer, alkoxysilane - denatured polymer, silicon
denatured urethane or silicon - denatured nylon.
The organic titanium compound may be
alkoxytitanium, titanium chelate, titanium acylate or
CA 02331602 2001-10-25
-28-
titanium polymer.
The organic aluminum compound may be
alkoxyaluminum, alkylaluminum or a halogenated aluminum.
The organic silicon-titanium compound may be a
tyranopolymer of a silicon polymer crosslinked to the
main chain of a polycarbosilane skeleton with a titanium
compound.
Also a material prepared by introducing a
derivative of an organic metal compound into a polymer
by substitution may be used as the coating material.
The film may be formed by directly applying the
organic compound or by applying it after diluted with a
solvent if the organic compound is in the form of
liquid. If the organic compound is in the form of a
solid, a solution dissolved in a solvent can be applied.
The organic compound may be applied by dipping, screen
printing, spraying or roll coating. The viscosity of the
foregoing coating liquid must adequately be adjusted to
be suitable for the employed coating method.
By mixing the organic metal compound at the time of
forming the film, the wettability of the electrolyte
solution can be improved and accordingly ions can easily
be introduced/discharged at the time of the charge and
the discharge. Further, the movement of the charge can
be made easier at the time of the battery charge by
mixing powder or fiber or whisker of conductive material
such as carbon or titanium at the time of applying the
organic metal compound.
CA 02331602 2001-10-25
-29-
It is preferable that the thickness of the film to
be formed on lithium ranges from 50 A to 100 ~, more
preferably 100 A to 10 ~. The optimum thickness of the
film differs depending upon the density or the void
ratio of the film and considerably differs depending
upon the type of the electrolyte solution. The thickness
of the film can be adjusted by changing the
concentration of the main material in the coating liquid
for forming the film.
4. Fluororesin Coating Material
By using fluorine resin of a type having an ether
bond to cover the surface of lithium, the surface
coating process can be completed easily and the contact
between lithium and the film can be improved.
The presence of oxygen atoms of the fluorine resin
having the ether bond makes easier the coordination of
lithium ions, enabling lithium ions to be moved easily
in the fluorine resin.
Further, the surface coating film prevents the
direct contact between the lithium metal and the organic
solvent, and therefore the formation of a polymerized
film of the organic solvent, which increases the
internal resistance of the battery, on the surface of
lithium can be prevented.
As a result, the formation of dendrite can be
prevented and therefore the life against the charge and
discharge cycle can be lengthened.
By covering the surface of lithium with the
CA 02331602 2001-10-25
-30-
fluorine resin, reactions of lithium with water or
oxygen can be prevented, causing handling to be
facilitated.
Since the fluorine resin is a flame retardant
resin, it exhibits safety at the time of an emergency
fire accident.
The fluorine resin for covering the surface of
lithium is exemplified by: a copolymer with a vinyl
monomer, such as vinyl ether, dioxysol, dioxyne or
dioxycene having an ether bond with fluoroethylene or
dime monomer derivative or a copolymer with a vinyl
monomer, such as vinyl ether, dioxysol or dioxyne,
dioxycene having a fluorized ether bond with a dime
compound, such as ethylene. In particular, it is
preferable to use a copolymer with vinyl ethex having an
ether bond with fluoroethylene. The fluoroethylene may
be a fluoroethylene derivative such as
tetrafluoroethylene, chlorotrifluoroethylene, vinylidene
fluoride or vinyl fluoride. The fluoroethylene copolymer
containing the ether bond can be polymerized by a
solution, suspension, block or emulsion polymerization.
As a starting material, a peroxide, alkyl boron, light
or radial rays may be employed.
The fluororesin can be coated on lithium metal by
any one of the following methods.
a. A solution of the fluororesin is applied by
spraying, screen printing, by using a coater or by
dipping.
CA 02331602 2001-10-25
-31 -
b. The fluororesin is directly coated to the
surface of lithium by a vacuum evaporation method such
as sputtering.
c. A polymer film is directly formed by plasma
polymerization under an atmosphere of monomer which is
the raw material for the fluororesin.
If the lithium surface is coated by using the
fluororesin solution, it is preferable to use, in an
inactive gas dehydrated sufficiently, a fluororesin
solution dehydrated and deoxidized sufficiently. It is
preferable to use a solvent in the foregoing solution
which has been dehydrated with active alumina, molecular
sheave, phosphorus pentaoxide or calcium chloride. As an
alternative to this, it is preferable depending upon the
type of the solvent that the solvent be distilled under
presence of alkaline metal in inactive gas to remove
impurities and to be dehydrated. However, the necessity
of strictly controlling water can be eliminated when
lithium is electrochemically inserted and allowed to
adhere between the fluororesin and the conductive
matrix.
An electrolyte may previously be mixed when the
foregoing film is formed. It leads to a fact that
wettability between the electrolyte solution and the
film can be improved, causing ions to easily pass
through the film. In order to facilitate the movement of
the charge at the time of charging, conductive powder,
such as carbon or titanium, fiber or whisker may be
CA 02331602 2001-10-25
-32-
mixed at the time of forming the film.
Since the performance of the battery deteriorates
if the fluororesin film is dissolved in an organic
solvent of the electrolyte solution, it is preferable
that the film is crosslinked.
As an alternative to the foregoing method of
coating the lithium surface with the fluororesin
solution, another method may be employed in which a
fluorocompound, such as tetrafluoroethylene is
polymerized with the main raw material with plasma to be
applied to the surface. It is preferable to employ
another method for improving the contact and strength of
the film in which oxygen, hydrogen, helium, argon,
nitrogen, silane, hydrocarbon or the like is mixed with
the fluorocompound which is the main material. The
plasma can effectively be generated by a DC or RF glow
discharge method, a microwave discharge method or a
laser beam irradiation method.
It is preferable that the thickness of the film
to be formed on the surface of lithium ranges from 50 A
to 100 ~, more preferably ranges from 100 A to 10 ~. The
optimum thickness of the film differs depending upon the
density or the void ratio of the film and considerably
differs depending upon the type of the electrolyte
solution. The thickness of the film can be adjusted by
changing the concentration of the main material in the
coating liquid for forming the film.
5. Large Ring Compound
CA 02331602 2001-10-25
-33-
By coating the surface of the negative electrode
active material with a large ring compound through which
ions for use in the reactions in the battery can be
passed, the reactions between lithium and water or
oxygen can be prevented if the negative electrode active
material is lithium. As a result, handling can easily be
performed. Further, the direct contact between lithium
and the electrolyte solution can be prevented, and
therefore the growth of a polymer film on the surface of
lithium to be formed from the solvent of the electrolyte
solution can be prevented. If the negative electrode
active material is zinc, the dissolution of zinc into
the electrolyte solution can be prevented. As a result,
the formation of dendrite can be prevented, causing the
life against the charge and discharge cycle can be
lengthened.
The large ring compound is a large ring compound
having heteroatoms composed of one or more types of
atoms selected from a group consisting of oxygen,
nitrogen and sulfur. In order to cause ions for use in
the reactions in the battery to be passed
satisfactorily, a compound having one or more structures
selected from a group consisting of ring polyether, ring
polyamide, ring polythioether, azacrown ether, ring
thioether, thiocrown ether, cryptand, cycrum, nonactyne
and bariomicine each having a hole having a radius
larger than the radius of ions to be used in the
reactions in the battery, thyracrown, cyclodextrin,
CA 02331602 2001-10-25
-34-
cyclophane, phtharocyanine and porphyrin each of which
is crown ether having silicon atoms. It is preferable to
use crown ether polymer, bariomicine, phthalocyanine or
porphylin.
By covering the surface of the negative electrode
active material with the large ring compound, the
movement of ions for use in the battery reactions
between the electrolyte solution and the negative
electrode active material can be made easier. Therefore,
local generation of zinc or lithium dendrite on the
negative electrode can be prevented. Further, reactions
between fresh lithium or zinc generated at the time of
the charge with the solvent of the electrolyte solution
can be prevented.
IS The surface coating of the large ring compound to
be applied to the surface of the negative electrode
active material can be performed in any of the following
methods.
a. A polymer solution obtained, by
polymerization, from the derivative of the large ring
compound is applied by dipping, spraying, screen
printing and coater coating.
b. A mixture of the binder polymer and the
derivative of the large ring compound is applied, and
then it is crosslinked so that the film is formed.
c. The derivative of the large ring compound is,
as a monomer, dissolved in an electrolyte solution, an
electric field is applied to the solution, and then the
CA 02331602 2001-10-25
-35-
film is formed on the surface of the negative electrode
active material or the conductive matrix by electrolytic
polymerization.
d. Lithium is immersed in a solution of the
derivative of the large ring compound, which can be
anion-polymerized by lithium, so that the polymer film
is formed.
e. A polymer is applied, the polymer being
obtained by heating and condensing a large ring compound
having an aromatic ring and formaldehyde in a formic
acid.
f. The film is formed by sputtering the large ring
compound or the polymer of the same or by plasma-
polymerizing the same.
An electrolyte may be mixed at the time of forming
the foregoing film. As a result, the wettability between
the electrolyte solution and the film can be improved,
causing ions to be easily passed through the film. In
order to facilitate the movement of the charge at the
time of the charging operation, powder or fiber or
whisker of conductive material such as carbon or
titanium may be mixed at the time of forming the film.
As the polymer for use in the coating solution,
poly [(dibenzo - 18 - crown - 6) - coformaldehyde] or
the like can be used. A polymer for coating can newly be
prepared by the following polymerization reactions. By
condensation polymerization of a large ring compound
having, at the terminative group thereof, carboxylic
CA 02331602 2001-10-25
-36-
group or amino group or a hydroxyl group, polyamide can
be obtained if the carboxylic group and the amino group
react with each other. Polyester can be obtained if the
carboxylic group and the hydroxyl group react with each
other. The vinyl compound of the large ring compound or
dime of the large ring compound enables an additive
polymer to be obtained by radical polymerization, cation
polymerization or anion polymerization. The starting
material of the radical polymerization may be
azobisisobutylnitryl (AIBN), benzoylperoxide (BPO) or t-
butylhydroperoxide. A starting material for the cation
polymerization is exemplified by an acid such as H2S09,
H3P09, HClOq, CC13 or COZH and Friedel-Craft catalyzer
such as BF3, A1C13, TiClq or SnClq . A large ring compound
having an aromatic ring can be polymerized by
dehydrogenation in which the Friedel-Craft catalyzer and
an oxidizer are combined to each other. A starting
material for the anion polymerization may be an alkaline
metal compound or an organic metal compound.
As the monomer of the large ring compound for use
in the polymerization may be crown ether/(+) - 18 -
crown - 6 - tetracarboxylic acid, 1, 5, 9, 13, 17, 21 -
hexathiacyclotetrakosan - 3, 11 - 19 - triol, l, 5, 9,
13 - tetrathiacyclohexadecan - 3, 11 - diol, 1 - aza -
12 - crown - 4, 1 - aza - 15 - crown - 5, 1 - aza - 18 -
crown - 6, 1, 4, 10, 13 - tetraxyso - 7, 16 -
diazocyclooctadecan, 1, 4, 10 - trioxa - 7, 13 -
diazacyclopentadecan, or 6, 8 - dioxabicyclo [3. 2. 1] -
CA 02331602 2001-10-25
-37-
oxtane - 7 - on. As an alternative to this, dibenzocrown
ether can be used.
In the foregoing polymerization, a copolymer of two
more types of derivatives of the large ring compounds or
a copolymer of the large ring compound and another
monomer may be used as well as the polymer of the
derivatives of the large ring compounds. A polymer
obtainable by introducing the derivative of the large
ring compound into a polymer by substitution may be
employed.
When a battery is manufactured, a polymer must be
selected so as not to be dissolved in the solvent of the
electrolyte solution or the polymer crosslinking
reactions are caused to proceed so as not be dissolved
in the electrolyte solution.
The derivative of the large ring compound having,
at the terminative group thereof, carboxylic group or
amino group or a hydroxyl group or having a vinyl bond
or a dime bond and a crosslinking material are mixed in
the polymer serving as the binder, and then the mixed
material is hardened. The crosslinking material is
selected from a group consisting of disocyanate, a
polyisocyanate prepolymer, block isocyanate, an organic
peroxide, polyamine, oxims, a nitroso compound, sulfur,
a sulfur compound, selene, a magnesium oxide, a lead
oxide and a zinc oxide. The organic peroxide is
exemplified by dicumyl - peroxide, 2, 5 - dimethyl - 2,
5 - di - (t - butyl - peroxy) hexane, 1, 3 - bis - (t-
CA 02331602 2001-10-25
-38-
butyl - peroxy isopropyl) benzene, 1, 1 - bis - (t-butyl
- peroxy) - 3, 3, 5 - trimethyl - cyclohexane, n-butyl -
4, 4 - bis - (t-butylperoxy) valelate, 2, 2 - bis - (t-
butyl - peroxide) butane, t-butyl - peroxy - benzene,
and vinyl - tris - (t-butyl - peroxy) silane. As an
accelerating agent, a guanidine, aldehyde - amine,
aldehyde - ammonia, thiazol, sulfonamide, thiourea,
thiuram, dithiocarbamate, xanthate accelerating agent is
used.
Another coating method using the binder polymer is
exemplified by a method in which a mixture of the large
ring compound and the binder polymer is applied, and
then radial rays, electron rays or ultraviolet rays are
applied to cause the applied material to be crosslinked.
As a method for covering the negative electrode
active material by the electrolytic polymerization, a
monomer, such as dibenzocrown ether, is mixed in the
electrolyte solution, and then the electrolytic
polymerization is performed while using the negative
electrode active material or the conductive matrix as an
anode. The solvent of the electrolyte solution is
exemplified by acetonitryl (CH3CN) , benzonitryl (C6HSCN) ,
propylene carbonate (PC), dimethylformamide (DMF),
tetrahydrofuran (THF) , nitrobenzene (C6HSN0z) ,
dichloroethane, diethoxyethane, chlorobenzene, Y -
butyrolactone and dioxolan and their mixture. It is
preferable that the solvent be dehydrated with active
alumina, molecular sheave, phosphorus pentaoxide or
CA 02331602 2001-10-25
-39-
calcium chloride. As an alternative to this, it is
preferable depending upon the type of the solvent that
the solvent be distilled under presence of alkaline
metal in inactive gas to remove impurities and to be
dehydrated. The supporting electrolyte is an acid, such
as H2S0q, HC1 or HN03 or salt composed of monovalent
metal ion (Li+, K+, Na+, Rb+ or Ag+) or tetraammonia ion
(tetrabutyl ammonia ion (TBA+) and tetraethyl ammonia ion
(TEA+) ) and Lewis acid ion (BF9-, PF6-, AsF6- or C104-) . It
is preferable that the foregoing salt is refined by re-
crystallization or it is heated under lowered pressure
to sufficiently dehydrate and deoxidize the salt.
As the monomer, crown ether/benzo - 15 - crown -
5, crown ether/benzo - 18 - crown - 6, crown ether/N -
phenylaza - 15 - crown - 5, crown ether/dibenzo - 18 -
crown - 6, crown ether/dibenzopyridino - 18 - crown - 6,
crown ether/dibenzo - 24 - crown - 8, 1, 13 - bis (8 -
quinolyl) - 1, 4, 7, 10, 13 - pentaoxatridecan, 5, 6 -
benzo - 4, 7, 13, 16, 21, 24 - hexaoxa - 1, 10 -
diazabicyclo [8. 8. 8] - hexakosan, 5, 6 - 14, 15 -
dibenzo - 4, 7, 13, 16, 21, 24 - hexaoxa - 1, 10 -
diazabicyclo [8. 8. 81 - hexakosan, bis [(benzo - 15 -
crown - 5 -) - 15 - ilmethyl] pimelate, crown
ether/dibenzo - 30 - crown - 10, N, N' - dibenzyl - 1,
4, 10, 13 - tetraoxa - 7, 16 - diazacyclooxtadecan,
dilithiumphthalocyanin, 4' - nitrobenzo - 15 - crown -
5, 3, 6, 9, 14 - tetrathiabicyclo [9. 2. 1] tetradeca -
11, 13 - dime and their mixture.
CA 02331602 2001-10-25
-40-
Since the performance of the battery deteriorates
if the polymer film for covering the surface of the
negative electrode active material is dissolved in the
electrolyte solution, it is preferable to be
crosslinked.
It is preferable that the thickness of the film to
be formed on the surface of lithium ranges from 50 A
to 100 ~, further preferably ranges from 100 A to 10
The optimum thickness of the film differs depending upon
the density or the void ratio of the film and
considerably differs depending upon the type of the
electrolyte solution. The thickness of the film can be
adjusted by changing the concentration of the main
material in the coating liquid for forming the film.
6. Polymer (polyphosphazene) Film in Which Phosphor
Atoms and Nitrogen Atoms are Alternately Bonded in a
Phosphor-Nitrogen Double Bond Manner
The surface of the negative electrode active
material is covered with a polymer (polyphosphazene)
film through which ions for use in the reactions in the
battery can be passed and in which phosphor atoms and
nitrogen atoms are alternately phosphor-nitrogen double
bond. As a result, lithium and the electrolyte solution
do not come in contact with each other if the negative
electrode active material is lithium. Therefore,
formation of the polymer film from the solvent of the
electrolyte solution on the surface of lithium can be
prevented. If the negative electrode active material is
CA 02331602 2001-10-25
-41 -
zinc, elution of zinc into the electrolyte solution can
be prevented. As a result, formation of dendrite can be
prevented, and therefore the life against charge and
discharge cycle can be lengthened.
Since the lithium battery according to the present
invention comprises the polyphosphazene film covering
lithium is a flame retardant film, safety against
emergency breakage can be improved.
The polymer for use as the covering material can be
obtained by heating a dichloropolyphosphazene trimer to
200 to 300°C and by ring-opening polymerization. The
dichloropolyphosphazene trimer can be synthesized from
phosphorus pentachloride and ammonia chloride or
ammonia. Any one of the following catalyzer is used at
the time of the polymerization: benzoic acid, sodium
benzonate, 2, 6 - di - p - cresol, water, methanol,
ethanol, nitromethane, ether, heteropoly acid, sulfur,
zinc, tin and sodium. Further, various type of
polyorganophosphazene can be obtained by substituting
chloride atoms of polydichlorophosphazene by an organic
reagent or an organic metal reagent.
If the surface of lithium is coated with the
foregoing polymer solution, it is preferable that the
polymer solution dehydrated and deoxidized sufficiently
be used in inactive gas dehydrated sufficiently
(however, the necessity of strictly controlling water
can be eliminated when the polymer is previously applied
to the conductive matrix or when the negative electrode
CA 02331602 2001-10-25
-42-
active material is zinc).
It is preferable to use a solvent in the foregoing
solution which has been dehydrated with active alumina,
molecular sheave, phosphorus pentaoxide calcium
chloride. As an alternative to this, it is preferable
depending upon the type of the solvent that the solvent
be distilled under presence of alkaline metal in
inactive gas to remove impurities and to be dehydrated
(however, the necessity of strictly controlling water
can be eliminated when the polymer is previously applied
to the conductive matrix or when the negative electrode
active material is zinc).
An electrolyte may previously be mixed when the
foregoing film is formed. It leads to a fact that
wettability between the electrolyte solution and the
film can be improved, causing ions to easily pass
through the film. In order to facilitate the movement of
the charge at the time of charging, conductive powder,
such as carbon or titanium, fiber or whisker may be
mixed at the time of forming the film.
Since the performance of the battery deteriorates
if the polymer coating film is dissolved in an organic
solvent of the electrolyte, it is preferable to be
crosslinked in such a manner, for example, utraviolet
rays, electron rays or radial rays are applied or a
crosslinking material, such as a radical generating
agent, is used.
It is preferable that the thickness of the film to
CA 02331602 2001-10-25
- 43 -
be formed on the surface of the negative electrode
active material ranges from 50 A to 100 ~, more
preferably ranges from 100 A to 10
The optimum thickness of the film differs depending
upon the density or the void ratio of the film and
considerably differs depending upon the type of the
electrolyte solution. The thickness of the film can be
adjusted by changing the concentration of the main
material in the coating liquid for forming the film.
7. Other Organic Polymer Film
The surface of the negative electrode is covered
with an organic polymer containing one or more types of
elements selected from a group consisting of oxygen,
nitrogen and sulfur and permitting ions relating to the
battery reactions to pass through. The direct contact of
fresh negative electrode active materials precipitated
during the charging reactions can be prevented due to
the foregoing cover film. Therefore, the negative
electrode is not covered with a substance prepared due
to the reactions with the electrolyte solution and
having low conductivity. As a result, the growth of
dendrite can be prevented.
The organic polymer containing oxygen is
exemplified by cellulose, alkyl cellulose,
nitrocellulose, acetyl cellulose, chitin, chitosan,
polyethylene glycol, polyethylene oxide, polypropylene
oxide, polyvinyl alcohol, polyvinyl acetate,
polylactide, polylactone, poly - 3 - hydroxyalcanoate,
CA 02331602 2001-10-25
-44-
polyglycol acid, polyacetic acid, polydioxanon, glycol
acid - lactone copolymer, polyethylene terephthalate,
polyphenylene oxide, polyether etherketone and the like.
It is preferable to use acetyl cellulose, chitin or
polyvinyl alcohol.
The organic polymer containing nitrogen is
exemplified by collagen, chitin, chitosan, polyurethane,
polyimide, polyether imide and the like.
The organic polymer containing sulfur is
exemplified by polyphenylene sulfide, polysulfon and
polyethersulfon.
The negative electrode is covered with the
foregoing organic polymer film formed in such a manner
that the solution of the organic polymer is applied and
dried and then a crosslinking reactions are caused to
occur. Another method of forming the film may be
employed in such a manner that the organic polymer is
used as a target in a sputtering method. Another method
may be employed in which a monomer serving as the
organic polymer is plasma-polymerized to form the film.
The application may be completed by dipping,
spraying or screen printing.
The crosslinking reactions can be performed by any
one of the following methods: irradiation of ultraviolet
rays, electron rays or radial rays; or decomposition of
a radical generating material such as azobisbutylonitryl
or peroxy benzoil. The reason why the organic polymer of
the film is crosslinked is that elution of the film into
CA 02331602 2001-10-25
-45-
the electrolyte solution must be prevented.
The thickness of the film must range so far as ions
in the electrolyte solution relating to the battery
reactions are able to pass through the same. It differs
depending upon the material and the void ratio of the
film and the type of the ion. It is preferable that the
0
mean thickness ranges from 10 A to 100 ~, more
preferably 100 A to 10 ~. If the ion permeability
through the film is unsatisfactory, an electrolyte may
be mixed at the time of forming the film.
If the electrolytic solution of the battery is a
water soluble solution and it is not hydrophilic, it
is preferable to perform a treatment using a silane
coupling material or a titanate coupling material to
attain hydrophilic characteristics.
Although the various covering materials and coating
methods have been described while employing the negative
electrode active material is directly covered with the
film, another method may be employed in which the
conductive matrix is previously covered with the coating
material and then the negative electrode active material
is introduced.
Negative Electrode Active Material
As the negative electrode active material 101, lithium,
lithium alloy, zinc or zinc alloy is used. The lithium
alloy may contain one or more types of elements selected
from a group consisting of magnesium, aluminum,
titanium, tin, indium, boron, gallium, potassium,
CA 02331602 2001-10-25
-46-
sodium, calcium, zinc and lead and the like. The zinc
alloy may contain one or more types of elements selected
from a group consisting of aluminum, indium, magnesium,
tin, titanium, copper, lead, tin, lithium and mercury
and the like.
The negative electrode active material for the
alkali-zinc battery may be zinc, zinc alloy, zinc oxide
or zinc hydroxide, the negative electrode active
material being uniformly kneaded with a bonding material
or a kneading solution to obtain paste. The paste is
applied to a collector followed by drying them so that a
negative plate is obtained.
The bonding material is exemplified by polyvinyl
alcohol, a cellulose material such as methyl cellulose
or carboxymethyl cellulose, a polyolefin material such
as polyethylene, a fluororesin such as
polytetrafluoroethylene and a polyamide resin such as
nylon.
The kneading solution may be an organic solvent
such as ethylene glycol or water containing oxo acid
salt such as sodium phosphate or the like.
The collector may be an iron plate applied with
nickel plating and having apertures, a foam metal or
nickel mesh or the like.
Positive Electrode
The positive electrode is formed by mixing a
positive electrode active material, conductive powder
and a bonding material on the collector. In order to
CA 02331602 2001-10-25
-47-
easily form the positive electrode active material, a
solvent-resisting resin, such as polypropylene or
polyethylene or fluororesin is used as the bonding
material if necessary. In order to further easily
collect electric currents, the conductive powder is
mixed at the time of the formation. The material of the
conductive powder is exemplified by various carbon,
copper, nickel and titanium and the like.
Positive Electrode for Lithium Secondar Batter
The positive electrode active material 104 of the
lithium secondary battery is made of a compound having
layers through which lithium ions can be passed, the
compound being a transition metal such as a metal oxide
exemplified by manganese oxide, vanadium oxide,
molybdenum oxide, chrome oxide, cobalt oxide, nickel
oxide, titanium oxide, iron oxide, and tungsten oxide or
a metal sulfide exemplified by titanium sulfide,
molybdenum sulfide, iron sulfide and shebrell phase
sulfide (MyMo6Se_Z (M: metal such as copper, cobalt or
nickel)). The transition metal element may be an element
having partially shell d or shell f. The metal selenide
is exemplified by niobium selenide. The metal hydroxide
is exemplified by oxyhydroxide. The conductive polymer
is exemplified by polyacetylene, polyparaphenyline,
polyaniline, polythiophene, polypyrol and
polytriphenylamine. The composite oxide is exemplified
by LiMri2_xMr.Oq and LiCO"Nil_x02.
The positive electrode is manufactured in such a
CA 02331602 2001-10-25
-48-
manner that paste obtained by adding a bonding material,
such as polyethylene, polypropylene or a fluororesin, to
the positive electrode active material 104 is pressed
against the positive electrode collector 103. In order
S to improve the collecting performance of the positive
electrode, it is preferable to add conductive powder to
the paste.
The conductive powder may be a carbon material
such as acetylene black or metal such as copper, nickel
or titanium. The collector 103 may be fiber-like, porous
or mesh-shape carbon material, stainless steel,
titanium, nickel, copper, platinum or gold.
Positive Electrode for Nickel-Zinc Battery
The positive electrode of the nickel-zinc battery
is categorized to a paste type electrode formed by
directly charging nickel hydroxide powder into the
collector and a sintering type formed by immersing
nickel hydroxide into a small apertures of the nickel
sintered plate.
The paste type positive electrode is formed in such
a manner that paste obtained by uniformly kneading an
additive, such as nickel or cobalt, with a bonding
material or a kneading solution is applied to the
collector followed by drying them.
The bonding material and the collector are made of
materials of the same type as those of the zinc negative
electrode.
The sintered type positive electrode is
CA 02331602 2001-10-25
-49-
manufactured in such a manner that a sintered plate
obtained by sintering nickel powder on the nickel-plated
iron plate having apertures is immersed in a mixed
solution of nickel salt serving as a main active
material and cobalt salt serving as an additive and then
caused to react with an alkali solution of, for example,
sodium hydride so that nickel hydroxide is impregnated
into the sintered plate.
Positive Electrode for Air-Zinc Secondary Battery
The positive electrode of the air-zinc secondary
battery is made of a material composed of an air
electrode, a water-repellent film and a diffuser paper
sheet.
The catalyzer of the air electrode is prepared in
such a manner that silver, manganese dioxide, nickel-
cobalt composite oxide or platinum is added to a carbon
material, such as porous carbon (active carbon) or
carbon black, porous nickel, copper oxide or the like
having a specific area of 200 to 1000 m2/g.
The water-repellent film is provided to prevent
leakage of the electrolyte solution passed through the
air electrode to the outside of the battery. The water-
repellent film is made of a fluororesin such as
polytetrafluoroethylene. The diffuser paper sheet is
provided for the purpose of uniformly supplying oxygen
to the overall surface of the air electrode, the
diffuser paper sheet being made of cellophane or the
like.
CA 02331602 2001-10-25
-50-
A zinc-bromine battery comprising the negative
electrode active material which is zinc includes bromine
to serve as the positive electrode active material.
Coating of Positive Electrode
In order to prevent generation of dendrite causing
the short circuit at the time of the charge, the surface
of the positive electrode is covered with a film through
which ions for use in the reactions in the battery can
be passed so that the life of the battery against the
cyclic usage can be lengthened.
The coating material may be a polymer of the
derivative of a large ring compound, a polymer of the
derivative of an aromatic hydrocarbon, fluororesin,
silicon resin, titanium resin, polyolefin, inorganic
oxide, nitride, carbide or halide. It is effective to
improve the safety of the lithium secondary battery to
cover the positive electrode with a flame retardant or
noncombustible material, such as fluororesin,
polyphosphazene, an inorganic oxide, nitride, carbide or
halide .
If the film through which ions for use in the
reactions in the battery can be passed is a multi-layer
metal oxide film formed by using a bimolecular film as a
mold, an effect of a separator can be obtained and an
effect of preventing the short circuit between the
negative electrode and the positive electrode can
further be improved.
Electrolyte
CA 02331602 2001-10-25
-51-
The electrolyte is used as it is or in the form of
a solution in which it is dissolved in a solvent or
after it has been solidified by adding a gelatinizing
material, such as a polymer, to the solution. Generally
an electrolyte solution, in which the electrolyte is
dissolved in a solvent, is held in a porous separator.
The conductance of the electrolyte must be raised
as much as possible because it relates to the internal
resistance of the battery and considerably affects the
current density at the time of the charge and the
discharge. It is preferable that the conductance at
25°C be 1 x 10-3 S/cm or higher, more preferably 5 x
10-3 S/cm or higher.
[In a Case Where Negative Electrode Active Material is
Lithium or Lithium Alloy]
The electrolyte is made of an acid, such as H2S09,
HC1 or HN03, a salt composed of Lewis acid ion (BF9-,
PF6-, AsF6 or C109 ) or their mixture. Further, positive
ions, such as sodium ions, potassium ions,
tetraalkylammonium ions, and the Lewis acid ions may be
used together. It is preferable that the foregoing salt
is heated under lowered pressure to sufficiently
dehydrate and deoxidize the salt.
The solvent of the electrolyte is exemplified by
acetonitryl (CH3CN) , benzonitryl (C6H5CN) , propylene
carbonate (PC), ethylene carbonate (EC), dimethyl
formamide (DMF), tetrahydrofuran (THF), nitrobenzene
(C6HSN02), dichloroethane, diethoxyethane, chlorobenzene,
CA 02331602 2001-10-25
-52-
- butylolactone, dioxolan, sulforan, nitromethane,
dimethylsulfide, dimethylsuloxide, dimethoxyethane,
methyl formate, 3 - methyl - 2 - oxydazolidinone, 2 -
methyltetrahydrofuran, sulfur dioxide, phosphoryl
chloride, thionyl chloride, sulfulyl chloride, dimethyl
formaldehyde, Y - buthlolactone and tetrahydrofuran and
their mixture.
It is preferable that the foregoing solvent be
dehydrated with active alumina, molecular sheave,
phosphorus pentaoxide or calcium chloride. As an
alternative to this, it is preferable depending upon the
type of the solvent that the solvent be distilled under
presence of alkaline metal in inactive gas to remove
impurities and to be dehydrated.
It is preferable to gel the electrolyte solution to
prevent leakage of the electrolyte solution. As the
gelling material, it is preferable to use a polymer of a
type which swells when it absorbs the solvent of the
electrolyte solution, the gelling material being a
polymer exemplified by a polyethylene oxide, polyvinyl
alcohol and an polyacryl amide.
[In a Case Where the Negative Electrode Active Material
is Zinc or Zinc Alloy]
As the electrolyte solution, salt of alkali or zinc
borate is used which is a sole or a mixed solution of
potassium hydroxide, sodium hydroxide, lithium hydroxide
or ammonium hydroxide.
It is preferable to gel the electrolyte solution to
CA 02331602 2001-10-25
-53-
prevent leakage of the electrolyte solution. As the
gelling material, it is preferable to use a polymer of a
type which swells when it absorbs the solvent of the
electrolyte solution, the gelling material being a
polymer exemplified by a polyethylene oxide, polyvinyl
alcohol and an polyacryl amide.
In a case of a battery such as a bromine - zinc
battery except for the alkali battery, a salt such as
zinc borate is used.
The solid electrolyte is manufactured in such a
manner that a polymer compound of a polyethylene oxide
(PEO) type and salt of the electrolyte are dissolved in
the foregoing non-water-soluble solvent to be gelled and
then they are developed on, for example, a flat board to
evaporate the non-water-soluble solvent. The PEO polymer
compound is exemplified by polyethylene oxide or a poly
(methoxyethoxyphosphazene) crosslinked by polyethylene
oxide or isocyanate.
The electrolyte solution for the alkali-zinc
secondary battery may be a solution of sodium hydroxide,
potassium hydroxide or lithium hydroxide or their
mixture.
Separator
The separators (108, 208 and 308) are provided to
prevent short circuits between the positive electrode
and the negative electrode. They also act to hold the
electrolyte solution.
The separator must meet the following conditions.
CA 02331602 2001-10-25
-54-
(1) The separator cannot be dissolved in the
electrolyte solution and must be stable with respect to
the same.
(2) The separator is able to absorb a large
quantity of the electrolyte solution and must exhibit
satisfactory holding force.
(3) The separator must have small apertures
through which lithium ions and hydroxide ions can be
passed.
(4) The separator must have small apertures each
having a size which is able to prevent the penetration
of dendrite.
(5) The separator must be mechanically strong so
as not to be broken or deformed excessively when it is
wound .
The material that is able to meet the foregoing
conditions is exemplified by a unwoven fabric or a
micropore structure of glass, polypropylene,
polyethylene, or fluororesin.
Also a metal oxide film having small apertures or a
resin film formed by combining metal oxides may be used.
If a metal oxide film in the form of a multilayer
structure is used, dendrite cannot easily pass through
it and therefore an effect of preventing the short
circuits can be obtained. If a fluororesin film which is
a flame retardant or a glass or a metal oxide film which
is a noncombustible material is used, the safety can
further be improved.
CA 02331602 2001-10-25
-SS-
Since the electrolyte solution of the alkali-zinc
secondary battery is a water-type solvent, a hydrophilic
separator must be used which is exemplified by a non-
woven fabric or a micropore structure of nylon,
polypropylene or hydrophilic polypropylene.
Collector
Collectors 100, 103, 200 and 300 are made of fiber,
porous or mesh-like carbon, stainless steel, titanium,
nickel, copper, platinum or gold.
Shape and Structure of Batted
The battery is formed into a flat, cylindrical,
square (rectangular) or a sheet shape battery. The
spiral and cylindrical structure is enabled to have a
large electrode area by winding while interposing the
separator between the negative electrode and the
positive electrode so that a large electric current can
be caused to flow at the time of the charge and the
discharge. The rectangular type battery enables an
accommodation space for accommodating the secondary
battery to be used effectively. The structure may be a
single-layer structure and a multi-layer structure.
Figs. 2 and 3 illustrate the schematic cross
sectional views which respectively illustrate an example
of a single-layer flat battery and a spiral and
cylindrical battery. Referring to Figs. 2 and 3,
reference numeral 201 and 301 represent negative
electrodes covered with films, and 200 and 300 represent
CA 02331602 2001-10-25
-56-
collectors for the negative electrodes 201 and 301.
Reference numeral 203 and 303 represent positive
electrode made of the positive electrode active
material. Reference numeral 304 represents a collector
for the positive electrode, and 206 and 306 represent
negative terminals (caps for the negative electrodes 201
and 301). Reference numeral 207 and 307 represent outer
cases (cases serving as positive electrode cases and
battery cases), 208 and 308 represent separators which
hold the electrolyte solution, 210 and 310 represent
insulating packings, and 311 represents an insulating
plate.
Referring to Fig. 5, reference numeral 5201 and
5301 represent lithium members subjected to surface
treatment. In the structure shown in Fig. 5, reference
numeral 201 and 310 represent lithium members subjected
to surface treatment.
The batteries shown in Figs. 2 and 3 are
manufactured in such a manner that the separators 208
and 308 are held between the negative electrodes 201,
301 subjected to the surface treatment and the positive
electrodes 203 and 303 to be placed in the positive
electrode cases 207 and 307 followed by injecting the
electrolyte solution. Then, the negative electrode caps
206, 306, the insulating packing 210 and 310 are
assembled so that the batteries are manufactured.
In the case of the lithium battery, it is
preferable that the preparation of the material and
CA 02331602 2001-10-25
- 57 -
assembling of the battery are performed in dry air from
which water has sufficiently be removed or in a dry
inactive gas.
Battery Case (Outer Case)
S The battery case may be a metal outer case also
serving as an output terminal or a plastic resin case.
The positive electrode cases 207, 307, the negative
electrode caps 206 and 306 are made of stainless steel,
and in particular, titanium clad stainless steel or
copper clad stainless steel or a steel plate applied
with nickel-plating.
Although the structures shown in Figs. 2 and 3
comprises the positive electrode cases 207 and 307 also
serving as the battery cases and the output terminals.
The battery case may be made of metal such as aluminum
or zinc, plastic such as polypropylene or a composite
material of metal, glass fiber and plastic as well as
the stainless steel.
Insulating Packing
The insulating packings 210 and 310 may be made of
fluororesin, polyamide resin, polysulfon resin or
rubber.
Cap
The capping method may be a bonding method, welding
method, soldering method or a glass sealing method as
well as a caulking method using a gasket such as an
insulating packing.
Insulating Plate
CA 02331602 2001-10-25
- 5g -
The insulating plate 311 for insulating the inside
of the battery may be made of an organic resin or
ceramics.
Safety Valve
A safety valve (not shown in Figs. 2 and 3) using
rubber, a spring or a metal ball may be used to serve as
a safety means to act if the internal pressure in the
battery has been raised.
The basic structure of another embodiment of the
lithium secondary battery according to the present
invention comprises a side opposing at least the
positive electrode, the side being composed of a lithium
negative electrode subjected to treatment using a
reactive gas containing nitrogen or halogen, a
separator, a positive electrode active material,
electrolyte and a collector. Fig. 5 is a basic
structural view which illustrates the lithium secondary
battery according to the present invention. Referring to
Fig. 5, reference numeral 100 represents a collector for
a negative electrode, 101 represents a negative
electrode active material (lithium or lithium alloy),
102 represents a surface treatment layer for the lithium
portion, 103 represents a collector for the positive
electrode, 104 represents a positive electrode active
material, 105 represents an electrolyte solution, 106
represents a negative terminal, 107 represents a
positive terminal, 108 represents a separator, and 109
represents a battery case. In the discharge reactions,
CA 02331602 2001-10-25
-59-
lithium ions in the electrolyte solution 105 are
introduced into the interlayer of the positive electrode
active material 104.
Simultaneously, lithium ions are eluted from the
lithium negative electrode 101 into the electrolyte
solution 105 by way of a treatment-applied surface 5102.
In the charging reactions, lithium ions in the
electrolyte solution 105 are, lithium metal ions are
precipitated to the lithium negative electrode 101 by
way of the treatment-applied surface 5102.
Simultaneously, lithium in the interlayer of the
positive electrode active material 104 is eluted into
the electrolyte solution 105.
Surface Treatment for Lithium Necrative Electrode
The nitrogen compound for treating the surface of
lithium or the lithium alloy is exemplified by nitrogen,
ammonia and nitrogen trifluoride. If nitrogen is used as
the nitride, it must be activated as to be formed into a
plasma form by DC or high frequency discharge or by
laser beam application. Also other nitrides are enabled
to have improved reactivity when formed into plasma.
The halogen compound is exemplified by fluorine,
chlorine, bromine, iodine, hydrogen fluoride, hydrogen
chloride, hydrogen bromide, chlorine trifluoride,
methane tetrafluoride, methane tetrachloride, methane
trifluoride, methane fluoride trichloride, sulfur
hexafluoride and boron trichloride. The inactive gas
such as carbon halide must be formed into plasma as to
CA 02331602 2001-10-25
-60-
have improved reactivity. It is preferable that the very
active gas such as fluorine, chlorine, hydrogen
fluoride, hydrogen chloride or chloride trifluoride be
diluted with inactive gas such as argon gas or helium
gas.
The nitrogen compound and the halogen compound may
be mixed with each other or contain oxygen gas, hydrogen
gas, argon gas, helium gas, xenon gas or the like added
thereto to improve the activity of the reactive gas or
to control the activity. The negative electrode may be
treated with plasma of hydrogen gas or argon gas prior
to treating the surface of the negative electrode so
that a fresh negative electrode surface is caused to
appear.
More specifically, according to the present
invention, there is provided a secondary battery
manufactured in such a manner that active nitrogen
plasma or fluorine plasma or hydrogen fluoride is
brought into contact with the surface of lithium to
cause reactions to take place, an inactive thin film
made of lithium nitride or lithium fluoride and allowing
lithium ions to pass through is formed on the surface of
lithium, and the lithium portion is used as the negative
electrode. As a result, the direct contact of lithium
precipitated at the time of the charge with the
electrolyte solution is prevented so that the generation
of dendrite at the time of the charge is prevented.
Therefore, a lithium battery exhibiting a long life
CA 02331602 2001-10-25
-61 -
against the charge and discharge cycle can be obtained.
Further, the application of the foregoing treatment to
the surface of lithium prevents the reactions between
lithium and water. Further, handling can be made easier.
It is preferable that the thickness of the
treatment layer to be formed on the surface of lithium
ranges from 10 A to 1 ~., more preferably 50 A to 1000 A.
The thickness is adjusted by the reaction time or the
concentration of the reaction gas.
It is preferable that the concentration of nitrogen
atoms or halogen atoms in the treatment layer formed on
the surface of lithium be gradually lowered from the
surface to the inside portion of lithium.
Positive Electrode Active Material
The positive electrode active material 5104 may be
a material into which lithium can be introduced into the
interlayer thereof, the material being exemplified by a
metal oxide such as a nickel oxide, cobalt oxide,
titanium oxide, iron oxide, vanadium oxide, manganese
oxide, molybdenum oxide, chrome oxide or tungsten oxide,
or a metal sulfide such as molybdenum sulfide, iron
sulfide or titanium sulfide, a hydroxide such as oxy
iron hydroxide, or a conductive polymer such as
polyacetylene, polyaniline, polypyrol or polythiophene.
In order to make easier the formation of the
positive electrode active material, solvent-resisting
resin, such as polypropylene, polyethylene or
fluororesin is used as a bonding material if necessary.
CA 02331602 2001-10-25
-62-
In order to further facilitate collection of electric
current, it is preferable to mix conductive powder at
the time of molding the positive electrode active
material. The conductive powder may be carbon black,
copper, nickel or titanium.
Fig. 8 illustrates an example a pattern of stacked
layers according to the present invention in which a
conductive layer, a semiconductor layer and an
insulating layer are formed between the negative
electrode and the separator. Referring to Fig. 8,
reference numeral 001 represents a negative electrode,
002 represents a dendrite, 003 represents the conductive
layer or the semiconductor layer, 004 represents a
separator, 005 represents an insulating layer, 006
represents an electrolyte solution, and 007 represents a
positive electrode.
Fig. 8 is a schematic view which illustrates an
effect of the present invention in an example in which
the insulating layer through which lithium ions can be
passed and a conductive layer are stacked on the surface
of the lithium negative electrode.
When the charging mode, lithium (or zinc) is
precipitated on to the negative electrode 001. At this
time, a portion in which the current density is high is
locally generated on the negative electrode 001
depending upon the projections and pits of the surface
and upon the thickness of the insulating film. Lithium
(or zinc) is selectively precipitated, causing the
CA 02331602 2001-10-25
-63-
dendrite 002 to grow. With proceeding of the charging or
discharging cycle, the dendrite 002 reach the conductive
layer 003, When a short circuit state between the
dendrite 002 and the conductive layer 003 is realized,
the current density at the negative electrode is lowered
at the time of the charge. Therefore, the further growth
of the dendrite 002 is prevented, and therefore
penetration of the dendrite 002 through the separator
004 to reach the positive electrode 007 can be
prevented.
By covering the negative electrode with the ion
permeable insulating layer 005, active lithium (or zinc)
precipitated at the time of the charge cannot easily
react with the electrolyte solution. Therefore, the
generation of the growth of the insulating film can be
prevented.
Figs. 9A to 9H illustrate various examples in which
one or more types of layers selected from the ion
permeable conductor, semiconductor and insulating
material are formed between the negative electrode and
the separator. Reference numerals shown in Figs. 9A to
9H are the same as those shown in Fig. 8. The present
invention is not limited to the structure shown in Figs.
9A to 9H.
It is preferable that the conductive layer to be
formed between the negative electrode and the separator
be made of carbon, Ni, Ti, Pt, A1, Pb, Cr, Cu, V, Mo, W,
Fe, Co, Zn or Mg, more preferably made of carbon, Ni or
CA 02331602 2001-10-25
-64-
Ti.
If the semiconductor layer is formed in place of
the conductive layer, a similar effect obtainable from
the conductive layer can be obtained. If the
semiconductor layer is employed, the current density at
the time of the growth of dendrite is higher than that
in the case where the conductive layer is formed.
However, the conductivity is lowered as compared with
the case where the conductive layer is formed.
Therefore, an advantage can be realized that the easy
conduction with the positive electrode is prevented.
The semiconductor layer may be made of diamond, Si,
nickel oxide, copper oxide, manganese oxide, titanium
oxide, zinc oxide, zirconium oxide, tungsten oxide,
molybdenum oxide or vanadium oxide.
The insulating layer may be made of a halide such
as lithium fluoride or magnesium fluoride, a nitride
such as silicon nitride, a carbon such as silicon
carbide, or a polymer such as polyethylene,
polypropylene or fluororesin.
The forming method and materials of the conductive
layer, the semiconductor layer, the insulating layer and
the composite layer will now be described. When lithium
is used, the following two desires must be met.
If water is left in the raw material, water and
lithium react with each other. Therefore, water must be
previously removed by a method by dehydration using
active alumina, molecular sheave, phosphorus pentaoxide
CA 02331602 2001-10-25
- 65 -
or calcium chloride. If a solvent is used, it is
sometime preferable that the solvent be distilled under
presence of alkaline metal in inactive gas to remove
impurities and to be dehydrated.
The temperature at which the foregoing layer is
applied to the surface of the negative electrode must be
lower than a level at which the negative electrode
active material is melted.
Conductive Layer
As a typical example of the conductive layer,
manufacturing method of carbon conductive layer or a
metal conductive layer made of, for example, Ni or Ti,
will now be described.
Carbon
The form of the carbon crystal is categorized to
crystal, amorphous, and a mixture of them. The carbon to
be applied may be carbon powder or carbon fiber or a
carbon paper sheet or the like obtainable by paper
machining.
The surface of the negative electrode can be
covered with carbon by any one of the following methods.
(1) A solution obtained by uniformly dispersing
carbon powder or carbon fiber in an organic solvent such
as toluene or xylene is, under inactive atmosphere of
Ar, is applied to the surface of the negative electrode
by a spraying method, screen printing method, a coater
method or a dipping method followed by drying the
solution, and then carbon are pressed against the
CA 02331602 2001-10-25
-66-
surface .
(2) The carbon paper is stacked on the surface of
the negative electrode, and then the carbon is pressed
against the surface.
(3) A vacuum evaporation method such as sputtering
using carbon as the target is performed so that the
surface of the negative electrode is covered with
carbon.
(4) A CVD (Chemical Vapor Deposition) method is
performed under the presence of an organic compound
which is the raw material of carbon so that the negative
electrode is covered with carbon.
In the foregoing coating method, the carbon powder
or the carbon fiber must be pressed against the surface
so that the contact is improved. The pressing work is
performed by using a pressing machine or a roller press.
The temperature at the time of the pressing work must be
lower than a level at which the negative electrode
active material is melted. It is preferable that the
thickness of the coating range from 10 to 100 ~,m, more
preferably from 50 mm or thinner in order to prevent
reduction of the quantity of the contained negative
electrode active material.
The pressing method to be performed after the
carbon paper has been stacked may be a method using a
pressing machine or a roller press, a method in which
the positive electrode plate and the negative electrode
plate are wound while interposing the separator, and a
CA 02331602 2001-10-25
-67-
method in which winding pressure or stacking pressure is
applied while interposing the carbon paper at the time
of stacking the carbon paper. The latter method is able
to eliminate a process of previously pressing the carbon
paper against the negative electrode and the carbon
paper can be pressed in the winding or the stacking
process. It is preferable that the carbon paper having a
thickness ranging from 150 to 300 ~m be pressed to have
a thickness ranging from 75 to 150 ~.m.
The sputtering coating method is performed in such
a manner that carbon is used as the target in an
inactive atmosphere of argon and DC or RF discharge is
performed so that the surface of the negative electrode
is covered with carbon.
The raw material of the CVD carbon coating method
may be saturated hydrocarbon such as methane,
unsaturated hydrocarbon such as acetylene, ethylene,
propylene or benzene, carbon monoxide, alcohol or
acetone. The exciting method is exemplified by a method
using plasma, laser or heating filament. It is
preferable that the thickness of the carbon applied be 1
~,m or thinner, more preferably 1000 A.
In the case of the plasma CVD method, the power
source for the glow discharge may be a high frequency or
a DC power source. The high frequency source may be a
usual band source such as radio frequency (RF), VHF or
microwave source. The wavelength of the radio waves is
typified by 13.56 MHz, while that of the microwave is
CA 02331602 2001-10-25
-68-
typified by 2.45 GHz.
If the laser CVD is employed, an ultraviolet ArF
eximer laser or infrared ray CO2 is used as the laser
beam source.
Metal Such as Ni or Ti
The metal such as Ni or Ti is applied to the
surface of the negative electrode by a sputtering method
as is employed to apply carbon, a CVD method, an
electron beam evaporation method or a cluster ion beam
evaporation method.
The target for use in the sputtering method may be
Ni or Ti or the like, when a composite film is formed,
two or more kinds of targets are used to perform either
or both of the targets are subjected to the sputtering
process. The sputtering method must be performed under
an inactive atmosphere of argon similarly to carbon.
The CVD coating method employs the following
materials as the raw materials.
As the raw material for Ni and Ti, a solution in
which organic metal such as acetylacetone complex of
nickel (or titanium) is dissolved in non-water-soluble
solvent such as hexane, acetone or toluene or a solution
in which a halide such as nickel chloride (or titanium)
is dissolved in a non-water-soluble solvent such as
ethanol is subjected to bubbling in a carrier gas
(hydrogen or the like) and then the solution is
introduced into the CVD reaction chamber to cause the
CVD reactions to take place so that coating is
CA 02331602 2001-10-25
-69-
performed.
When a composite film of Ni and carbon is formed, a
hydrocarbon such as methane is used together with a
metal compound.
Semiconductor Layer
The semiconductor layer is exemplified by diamond
Si, nickel oxide, copper oxide, cobalt oxide, manganese
oxide, titanium oxide, zinc oxide, zirconium oxide,
tungsten oxide, molybdenum oxide or vanadium oxide. The
coating method may be a sputtering method, an electron
beam evaporation method, a plasma CVD method, a light
CVD method, a laser CVD method or a heat CVD method.
Si
As the target for use in the sputtering method, Si
or the like is used.
In the CVD method, the raw material for Si may be
hydroxide gas such as SiH4 or Si2H6, fluorine gas such as
SiHF3, SiH2F2 or SiH3F, or a chlorine gas such as SiHCl3,
SiH2C12 or SiHCl3. If the foregoing raw material is in
the form of liquid, it is heated as to be in the form of
vapor or subjected to bubbling by a carrier gas before
it is introduced. If a composite film of Si and carbon
material is formed, hydrocarbon such as methane is used
together with the foregoing gas. A compound comprising
carbon, P or B may be adequately mixed.
Oxide such as Nickel Oxide and Titanium Oxide
The nickel oxide and titanium oxide can be prepared
by a sol-gel method in which alkoxide such as nickel or
CA 02331602 2001-10-25
-70-
titanium or organic metal is dissolved in alcohol and
then it is hydrolyzed, an anodic oxidation method in
which a solution in which nickel salt or titanium salt
is dissolved is electrolyzed, or a CVD method or an
electron beam evaporation method to introduce oxygen gas
into the reaction chamber.
Insulating Layer
As the insulating layer, a halide, a nitride, a
carbide or resin such as polyethylene (PE),
polypropylene (PP) or fluororesin is used. As the
coating method, a sputtering method, a plasma CVD method
or a coating method is used.
Other resins may be used such that a resin obtained
by gasifying a monomer of an organic polymer and by
plasma polymerizing it or a resin obtained by sputtering
an organic polymer or a film of an organic polymer may
be used. In this case, the resin member must have small
aperture through which ions can be passed and must not
react with the electrolytic solution.
Nitride
In the sputtering method, a target comprising
nitride exemplified by silicon nitride or lithium
nitride is used. As an alternative to this, Si or Li is
used as the target, nitrogen gas or ammonia is used as
the reaction gas and sputtering is performed in the
foregoing state.
PE and PP
CA 02331602 2001-10-25
-71-
In the plasma CVD method, SiH9 or Si2H6 and ammonia,
nitrogen gas or NF3 are used together as the raw
materials.
In the sputtering method, a PE or PP target is used
to perform sputtering to cover the insulating layer.
In the plasma polymerization method, ethylene is
used as the raw material in the case of PE, while
propylene is used in the case of PP.
Fluororesin
In the sputtering method, the target comprises a
polymer or a copolymer such as polytetrafluoroethylene,
polytrifluoroethylene, vinyl fluoride, vinylidene
fluoride or dichlorodifluoroethylene.
As the raw material gas for use in the plasma
polymerization method, tetrafluoroethylene,
trifluoroethylene, vinyl fluoride, vinylidene fluoride
or dichlorofluoroethylene is used.
A fluororesin film having micropores can be used.
Composite Layer
The composite layer is selected from a group
consisting of the foregoing conductor, semiconductor and
insulating material. By using two or more types or raw
materials, the composite layer is formed by a sputtering
method of a CVD method. As an alternative to this, two
or more types of powder selected from a group consisting
of conductor powder, semiconductor powder and insulating
material powder are melted in a melted or dissolved
resin so that the film for use as the composite film is
CA 02331602 2001-10-25
-72-
manufactured.
Another structure may be employed as a preferred
structure in which the concentrations (content) of the
conductor, semiconductor and the insulating material in
the composite layer are change continuously or
discontinuously in the direction of the thickness of the
layer.
The apertures in the layer can be formed in such a
manner that: an electrolyte is mixed with the raw
material at the time of applying the conductor,
semiconductor and the insulating material by the
sputtering method or the CVD method to add the
electrolyte into the conductor, semiconductor and the
insulating material. The foregoing electrolyte is eluted
into the electrolyte solution of the battery so that a
micropore structures are formed in the conductor,
semiconductor and the insulating material. Since lithium
ions and hydroxide ions can easily be introduced and
discharged in the micropores, the charging and
discharging efficiencies can be improved. Since the pore
has a small size, the growth of the dendrite can be
prevented, and therefore the life against the charging
and discharging cycle can be lengthened.
Stacked Structure
The conductor, semiconductor and the insulating
material layers may be formed into a single layer or a
multi-layer composed of two or more layers.
The stacking method comprises a step of stacking a
CA 02331602 2001-10-25
-73-
film selected from the group consisting of the
conductor, semiconductor and the insulating material
between the negative electrode and the separator. The
film is stacked on the surface of the negative
electrode, or stacked between the negative electrode and
the separator in a non-contact manner or stacked on the
surface of the separator. As an alternative to this, the
negative electrode or the separator or a substance
through which ions can be passed may be used as the base
on which the conductor, semiconductor and the insulating
material are stacked by a sputtering method or a CVD
method.
Thickness
The optimum thickness of the single, multi-layer or
the composite layer composed of the one or more types of
layers selected from the group consisting of the
conductor, semiconductor and the insulating material
differs depending upon the void ratio, the aperture
distribution, the material of the layers and the number
of layers. If the material includes a large volume gaps
as the polymer material, it is preferable the thickness
be 10 ~m or thinner, more preferably 1 ~m or thinner. If
a precise material such as the inorganic compound is
used, it is preferable to make the thickness to be 1 ~m
or thinner, more preferably 1000 A or thinner.
Multi-Layer Metal Oxide
The multi-layer metal oxide 10102 must contain one
or more types of materials selected from a group
CA 02331602 2001-10-25
-74-
consisting of alumina, titanium oxide, silica, selium
oxide, zirconia oxide, magnesium oxide, chrome oxide,
calcium oxide, tin oxide, indium oxide and germanium
oxide.
The multi-layer metal oxide film is formed by
molding a bimolecular film in a mold. By forming the
metal oxide film by using the bimolecular film, a metal
oxide film having small apertures and a large specific
area and formed into a multi-layer film structure can be
obtained.
Method of Preparing Multi-Layer Metal Oxide
The preparation is usually in such a manner that: a
sol in which very fine particles of the metal oxide is,
in a collide manner, dispersed in a solvent, such as
water, is added to a supersonic dispersion solution of
water or buffer solution of a film forming compound for
forming a bimolecular film selected depending upon the
type of the sol and upon the surface charge so that a
uniformly-dispersed solution is prepared; then it is
developed on a fluororesin film or a glass plate
followed by developing the solution; and a cast film is
manufactured. If the uniformly-dispersed film cannot be
obtained, the any one of the following methods is
employed:
(1) A cast film is formed by adding a low-melting point
organic solvent, such as alcohol, chloroform, acetone or
tetrahydrofuran, is added to the film forming compound
and dispersing the materials, and then water or a buffer
CA 02331602 2001-10-25
-75-
solution is added to the solution from which the solvent
has been gradually evaporated, and then the mixture is
dispersed with ultrasonic waves.
(2) Water or a buffer solution is added to the cast film
described in (1), and the material is heated to a level
higher than the phase transition temperature of the
film.
(3) The film forming compound is dissolved in an organic
solvent, such as diethylether or ethylalcohol, and the
solution is injected into water and a buffer solution.
As a method of obtaining the bimolecular film which
serves as the mold of the film shape of the metal oxide,
the mechanical strength can be improved by any one of
the following methods:
(a) The film forming compound is impregnated into a
porous polymer film to form the bimolecular solid film.
(b) A hydrophobic polymer or a hydrophilic polymer and
the film forming compound are dissolved in a solvent,
and the solution is developed so that a cast film is
obtained.
(c) A solution of a polymer electrolyte having a charge
opposing that of the ionic film forming compound is
mixed with a solution in which the ionic film forming
compound is dispersed so that sedimentation of polyion
complex is obtained, and then the polyion complex is
dissolved in an organic solvent followed by developing
it so that the cast film is obtained.
Alternatively, any one of the following methods may
CA 02331602 2001-10-25
-76-
be employed: a chloric acid (surface active agent)
method which is used at the time of preparing ribosome;
a freezing-melting method; an inverse-phase evaporation
method; and a macro-ribosome preparation method.
The film forming compound may be a compound having
both hydrophobic group and a hydrophilic group
(substance having amphipathic property) is used. The
film forming compound may be an ammonia compound, an
anion compound, a nonion compound and a polymerable
compound depending upon the molecular structure. The
hydrophobic group of the film forming compound is
categorized to a hydrocarbon, fluorine carbide,
unsaturated hydrocarbon, and unsaturated fluorine
carbide. A material containing a chromophore structure
introduced thereto in order to improve the molecular
orientation in the film forming compound may be used.
The film forming compound is exemplified by
p - octyloxyaniline hydrochloride,
p - (octyloxy) - p' - hydroxyazobenzene,
p - (10 - octyloxy) - p~ - octyloxyazobenzene,
dodecyl - N - [p - {p - ( 10 -
bromodecyloxy)phenylazo~benzoil]L - alaninate, L -
glutamic acid didodecylester hydrochloride, N - [11 -
bromoundecanoyl] - L glutamic acid didodecylester,
dimethyldihexadecylammonium bromide, N - [(3 -
(trimethylammonio)etyloxybenzoil] didodecyl - L -
glutamic acid bromide,
dioctadecylmethyldimethylammomonium bromide, N - [11 -
CA 02331602 2001-10-25
77
hydroxyundecanoyl] - L - glutamic acid
ditetradecylester, N - [11 - phospholoundecanoyl] - L -
acid ditetradecylester, 1, 2 - bis
(hexadecyloxycarbonyl) ethane - 1 - sulfonic acid
sodium, N - [(2 - oxo - 1, 3 , 2 - oxazaphosphoryl) - 11
- oxadodecanoyl] - L - glutamic acid didoecylester, N -
[(2 - trimethylammonio - ethylphosphonate)undecanoyl] -
L - glutamic acid didodecylester, N - [(2 - ammomonio -
ethylphosphonate)undecanoyl] - L - glutamic acid
didodecyl ester and 1, 3 - hexadecyl - 2 - polyethylene
glycolylglyceline.
It is preferable that the surface of the multi-
layer metal oxide film of a non-water-soluble battery of
a type comprising the negative electrode active material
which is lithium is subjected to lipophillic treatment
using an organic metal compound such as a silane
coupling material or titanate coupling agent.
In order to lower the current density at the
leading portion of the dendrite at the time of the
charge and to prevent the growth of dendrite, the
surface of the multi-layer metal oxide film on the
negative electrode side may be applied with a conductive
material by evaporation or plating.
Very Fine Particle Sol of Metal Oxide
The dispersed sol of very fine particles of the
metal oxide is usually obtained by an acid, a base and
water are added to an alcohol solution of an organic
metal compound such as metal alkoxide as to be
CA 02331602 2001-10-25
_ 7g
hydrolyzed to form colloid of the very fine particles of
the metal oxide. The dispersion medium of the very fine
particle sol is obtained by substituting to an organic
solvent if necessary.
The alkoxide is typified by tetramethoxysilane,
tetraethoxysilane, aluminum isopropoxide, and titanium
isopropoxide. Another metal organic compound such as
acetyl acetone complex salt, alkyl metal compound,
acetylacetone metal salt, naphthenate metal salt or
octylate metal salt may be used.
The very fine particles of the metal oxide can be
obtained by another method for obtaining the same from
the gas phase reaction of the vapor of the organic metal
compound or the metal. The obtained very fine particles
of the oxide are dispersed in a solvent so that a
dispersed sol is prepared.
If the negative electrode active material is
lithium or lithium alloy, lithium ions relate to the
battery reactions. In the case of an alkali battery
comprising the negative electrode active material which
is zinc, hydroxyl ions relate to the same. The typical
battery of a type comprising the negative electrode
active material which is zinc is exemplified by a
nickel-zinc battery and an air-zinc battery.
By covering the surface of the positive electrode
104 of the battery with a film 12102 which is made of an
insulating material or a semiconductor through which
ions relating to the battery reactions can be passed,
CA 02331602 2001-10-25
-79-
dendrite of lithium or zinc grown from the negative
electrode 101 through the separator 108 during the
repetition of the charge and the discharge are not
substantially brought into contact with the conductor or
a collector in the positive electrode 104. As a result,
short circuits in the battery can be prevented, and
therefore the life of the secondary battery can be
lengthened. Further, the safety can be improved.
Covering of Positive Electrode of Secondary Battery
As the insulating material of the coating material
of the positive electrode of the secondary battery, any
one of the following materials may be used which is
selected from a group consisting of a polymer of the
derivative of a large ring compound, a polymer of the
derivative of an aromatic hydrocarbon, fluororesin,
silicon resin, titanium resin, polyolefin, inorganic
oxide, nitride, carbide and halide. It is effective for
the positive electrode of the lithium secondary battery
to be covered with the polymer of the derivative of the
large ring compound, the polymer of the derivative of
the aromatic hydrocarbon or the fluororesin.
Polymer of Derivative of Large Rina Compound
The large ring compound is a ring compound of a
type comprising hetero-atoms of one or more types
selected from a group consisting of oxygen, nitrogen,
sulfur and phosphorus. The large ring compound is a ring
polyether having hole each having a radius larger than
the radius of the lithium ion. The large ring compound
CA 02331602 2001-10-25
-80-
has one or more structures selected from a group
consisting of ring polyamine, ring polythioether,
azacrown ether, ring thioether, thiocrown ether,
cryptand, cyclam, nonactine, variomicine, thyracrown
which is a crown ether containing silicon atoms,
cyclodextrin, cyclofan, phthalocyanin and porphyrin
compound.
The surface of the positive electrode is covered
with the large ring compound by any one of the following
methods.
a. A polymer solution obtainable from
polymerization of the derivative of the foregoing large
ring compound is applied by a coating method such as
dipping, spraying, screen-printing or coating
application method.
b. A mixture in which the derivative of the large
ring compound is mixed with the polymer serving as the
binder is applied and then crosslinked to form the film.
c. The derivative of the large ring compound is
used as a monomer to be dissolved in an electrolyte
solution, and then an electric field is applied to
electrolyze and polymerize the material so that the film
is formed on the surface of the positive electrode.
d. The molded positive electrode is immersed in a
solution of the derivative of the large ring compound,
which anion-polymerizes, so that the polymer film is
formed.
e. A polymer obtainable by heating and condensing
CA 02331602 2001-10-25
- ~1 -
the large ring compound having an aromatic ring and
formaldehyde in a formic acid is applied.
f. The film is formed by sputtering of a polymer
of the large ring compound or the derivative of the
large ring compound or by plasma-polymerizing the large
ring compound.
An electrolyte may be mixed at the time of forming
the film. As a result, the wettability between the
electrolyte solution and the film can be improved,
causing ions to easily pass through the film.
The polymer for uses as the coating solution may be
poly [(dibenzo - 18 - crown - 6) - co - formaldehyde]
or the like. The coating polymer can be newly formed by
the following polymerizing reactions: a large ring
compound having a carboxylic group or an amino group or
a hydroxyl group at the terminal group thereof is
condensed and polymerized so that polyamide is obtained
if the carboxylic group and the amino group react with
each other. If the carboxylic group and the hydroxyl
group react with each other, polyester can be obtained.
The vinyl compound of the large ring compound or dime
of the large ring compound can be obtained in the form
of an addition polymer by radical polymerization, ration
polymerization or anion polymerization. The starting
material in the radical polymerization may be
azobisisobutylonitryl (AIBN), benzoylperoxide (BPO) or
t - butylhydroperoxide. A starting material for the
ration polymerization is exemplified by an acid such as
CA 02331602 2001-10-25
-82-
HZSOq, H3P0q, HClOq, CC13COZH and Friedel-Craft catalyzer
such as BF3, A1C13, TiClq or SnCl4. A large ring compound
having an aromatic ring can be polymerized by
dehydrogenation in which the Friedel-Craft catalyzer and
S an oxidizer are combined to each other. A starting
material for the anion polymerization may be an alkaline
metal compound or an organic metal compound.
As the monomer of the large ring compound for use
in the polymerization may be crown ether/(+) - 18 -
crown - 6 - tetracarboxylic acid, 1, 5, 9, 13, 17,
21 - hexathiacyclotetrakosan - 3, 11 - 19 - triol, 1, 5,
9, 13 - tetrathiacyclohexadecan - 3, 11 - diol, 1 - aza
- 12 - crown - 4, 1 - aza - 15 - crown - 5, 1 - aza - 18
- crown - 6, 1, 4, 10, 13 - tetraxyso - 7, 16 -
diazocyclooctadecan, 1, 4, 10 - trioxa - 7, 13 -
diazacyclopentadecan, or 6, 8 - dioxabicyclo [3. 2. 1] -
oxtane - 7 - on. As an alternative to this, dibenzocrown
ether can be used.
In the foregoing polymerization, a copolymer of two
more types of derivatives of the large ring compounds or
a copolymer of the large ring compound and another
monomer may be used as well as the polymer of the
derivatives of the large ring compounds. A polymer
obtainable by introducing the derivative of the large
ring compound into a polymer by substitution may be
employed.
CA 02331602 2001-10-25
-83-
When a battery is manufactured, a polymer must be
selected so as not to be dissolved in the solvent of the
electrolyte solution or the polymer crosslinking
reactions are caused to proceed so as not be dissolved
in the electrolyte solution.
The derivative of the large ring compound having,
at the terminative group thereof, carboxylic group or
amino group or a hydroxyl group or having a vinyl bond
or a dime bond and a crosslinking material are mixed in
the polymer serving as the binder, and then the mixed
material is hardened. An accelerating material may be
mixed at this time. The crosslinking material is
selected from a group consisting of disocyanate, a
polyisocyanate prepolymer, block isocyanate, an organic
peroxide, polyamine, oxims, a nitroso compound, sulfur,
a sulfur compound, selene, a magnesium oxide, a lead
oxide and a zinc oxide. The organic peroxide is
exemplified by di - cumyl - peroxide, 2, 5 - dimethyl -
2, 5 - di - (t - butyl - peroxy) hexane, 1, 3 - bis -
(t-butyl - peroxy isopropyl) benzene, 1, 1 - bis - (t-
butyl - peroxy) - 3, 3, 5 - trimethyl - cyclohexane, n -
butyl - 4, 4 - bis - (t - butylperoxy) valelate, 2, 2 -
bis - (t - butyl - peroxide) butane, t - butyl - peroxy
- benzene, and vinyl - tris - (t - butyl - peroxy)
silane. As an accelerating agent, a guanidine, aldehyde
- amine, aldhyde-ammonia, thiazol, sulfonamide,
thiourea, thiuram, dithiocarbamate, xanthate
accelerating agent is used.
CA 02331602 2001-10-25
-84-
Another coating method using the binder polymer is
exemplified by a method in which a mixture of the large
ring compound and the binder polymer is applied, and
then radial rays, electron rays or ultraviolet rays are
applied to cause the applied material to be crosslinked.
As a method for covering the positive electrode by
the electrolytic polymerization, a monomer, such as
dibenzocrown ether, is mixed in the electrolyte
solution, and then the electrolytic polymerization is
performed while using the negative electrode active
material or the conductive matrix as an anode. The
solvent of the electrolyte solution is exemplified by
acetonitryl (CH3CN) , benzonitryl (C6HSCN) , propylene
carbonate (PC), dimethylformamide (DMF), tetrahydrofuran
(THF) , nitrobenzene (C6HSN02) , dichloroethane,
diethoxyethane, chlorobenzene, Y - butyrolactone and
dioxolan and their mixture.
It is preferable that the solvent be dehydrated
with active alumina, molecular sheave, phosphorus
pentaoxide or calcium chloride.
The supporting electrolyte is an acid, such as
H~SO4, HCl or HN03 or salt composed of monovalent metal
ion (Li+, K+, Na+, Rb+ or Ag+) or tetraammonia ion
(tetrabutyl ammonia ion (TBA+) and tetraethyl ammonia ion
(TEA+) ) and Lewis acid ion (BFq-, PF6-, AsF6- or ClOq-) . It
is preferable that the foregoing salt is refined by re-
crystallization or it is heated under lowered pressure
to sufficiently dehydrate and deoxidize the salt.
CA 02331602 2001-10-25
- 8s -
As the monomer, crown ether/benzo - 15 - crown - 5,
crown ether/benzo - 18 - crown - 6, crown ether/N -
phenylaza - 15 - crown - 5, crown ether/dibenzo - 18 -
crown - 6, crown ether/dibenzopyridino - 18 - crown - 6,
crown ether/dibenzo - 24 - crown - 8, 1, 13 - bis (8 -
quinolyl) - 1, 4, 7, 10, 13 - pentaoxatridecan, 5, 6 -
benzo - 4, 7, 13, 16, 21, 24 - hexaoxa - 1, 10 -
diazabicyclo [8. 8. 8] - hexakosan, 5, 6 - 14, 15 -
dibenzo - 4, 7, 13, 16, 21, 24 - hexaoxa - 1, 10 -
diazabicyclo [8. 8. 8] - hexakosan, bis [(benzo - 15 -
crown - 5 -) - 15 - ilmethyl] pimelate, crown
ether/dibenzo - 30 - crown - 10, N, N' - dibenzyl - 1,
4, 10, 13 - teraoxa - 7, 16 - diazacyclooxtadecan,
dilithiumphthalocyanin, 4' - nitrobenzo - 15 - crown -
5, 3, 6, 9, 14 - tetrathiabicyclo [9. 2. 1] tetradeca -
11, 13 - dime and their mixture.
Since the performance of the battery deteriorates
if the polymer film for covering the surface of the
negative electrode active material is dissolved in the
electrolyte solution, it is preferable to be
crosslinked.
It is preferable that the thickness of the film
to be formed on the surface of lithium ranges from 10 A
to 100 ~, further preferably ranges from 50 A to 10
The optimum thickness of the film differs depending upon
the density or the void ratio of the film and
considerably differs depending upon the type of the
electrolyte solution. The thickness of the film can be
CA 02331602 2001-10-25
-86-
adjusted by changing the concentration of the main
material in the coating liquid for forming the film.
Polymer of Derivative of Aromatic Hydrocarbon
As the derivative of the aromatic hydrocarbon for
forming the charge moving complex is one or more types
of derivatives selected from a group consisting of
naphthalene, anthracene, phenanthlene, naphthacene,
pyrene, triphenylene, perillene, picene, benzopyrene,
coronene and ovalene.
The polymer for use to form the coating material
can be prepared by polymerization or copolymerization of
vinyl monomer, monomer of acetylene derivative or
dicarboxylic acid and diamine, and dicarboxylic acid and
glycol. The polymerization of the vinyl monomer can be
IS performed by radical or ion polymerization. The monomer
of the acetylene derivative can be polymerized while
using a chloride of tungsten as a catalyzer. The
dicarboxylic acid and diamine can be polycondensed and
the dicarboxylic acid and glycol can as well as be
polycondensed.
The monomer of the aromatic derivative for forming
the polymer is exemplified by 2 - vinyl naphthalene, 2 -
vinyl pyridine, 9 - vinyl anthracene, 9, 10 - anthracene
dipropionic acid, 9, 10 - bis (phenyl ethyl) anthracene
and 5, 12 - bis (phenyl ethynyl) naphthalene.
A starting material for the radical polymerization
is exemplified by azobisisobutylonitryl (AIBN),
benzoylperoxide (BPO) and t - butylhydroperoxide. A
CA 02331602 2001-10-25
starting material for the cation polymerization is
exemplified by an acid such as HzS04, H3P0q, HClOq, CC13 or
C02H and Friedel-Craft catalyzer such as BF3, A1C13, TiCl4
or SnCl9. A large ring compound having an aromatic ring
S can be polymerized by.dehydrogenation in which the
Friedel-Craft catalyzer and an oxidizer are combined to
each other. A starting material for the anion
polymerization may be an alkaline metal compound or an
organic metal compound.
As an alternative to the foregoing method, a
polymer into which an aromatic group can be obtained by
subjecting the side chain of each polymer to a
substitution reaction with a derivative of an aromatic
compound. Another method may be employed in which an
electrolytic polymerization reaction is caused to take
place in an electrolyte solution containing a monomer
mixed therein to form directly a polymer of an aromatic
compound on the surface of positive electrode.
When the surface of the positive electrode is
applied with a coating by using the foregoing polymer
solution, it is preferable to use a polymer solution
dehydrated and deoxidized sufficiently in inactive gas
dehydrated sufficiently. It is preferable to use a
solvent in the foregoing solution which has been
dehydrated with active alumina, molecular sheave,
phosphorus pentaoxide or calcium chloride. As an
alternative to this, it is preferable depending upon the
type of the solvent that the solvent be distilled under
CA 02331602 2001-10-25
_ $g
presence of alkaline metal in inactive gas to remove
impurities and to be dehydrated.
An electrolyte may previously be mixed when the
foregoing film is formed. It leads to a fact that
wettability between the electrolyte solution and the
film can be improved, causing ions to easily pass
through the film.
Since the performance of the battery deteriorates
if the polymer coating film is dissolved in an organic
solvent of the electrolyte, it is preferable to be
crosslinked in such a manner, for example, ultraviolet
rays, electron rays or radial rays are applied or a
crosslinking material, such as a radical generating
agent, is used.
Fluororesin
The fluororesin for covering the surface of the
positive electrode is exemplified by ethylene
tetrafluoride-ethylene copolymer, ethylene tetrafluoride
chloride, ethylene tetrafluoride - per -
fluoroalkylvinyl ether copolymer, ethylene tetrafluoride
- propylene hexafluoride copolymer, vinylidene fluoride
resin, vinyl fluoride resin, and ethylene tetrafluoride
resin. Since the foregoing materials are not dissolved
in a solvent, it is preferable to employ sputtering or
plasma polymerization to cover the surface of the
positive electrode with the fluororesin.
Among the fluororesins, those having an ether bond
is able to facilitate the surface covering because they
CA 02331602 2001-10-25
-89-
can easily be dissolved in a solvent and to improve
the affinity with lithium ions. The fluororesin having
the ether bond is exemplified by: a copolymer of
ethylene fluoride and vinyl monomer such as vinyl ether,
dioxysol, dioxyne or dioxycene having an ether bond or
dienemonomer derivative or a copolymer with a vinyl
monomer, such as vinyl ether, dioxysol or dioxyne,
dioxycene having a fluorized ether bond with a dime
compound, such as ethylene. The fluoroethylene may be a
fluoroethylene derivative such as tetrafluoroethylene,
chlorotrifluoroethylene, vinylidenefluoride or vinyl
fluoride. The fluoroethylene copolymer containing the
ether bond can be polymerized by a solution, suspension,
block or emulsion polymerization. As a starting
material, a peroxide, alkyl boron, light or radial rays
may be employed.
The fluororesin can be coated on lithium metal by
any one of the following methods.
a. A solution of the fluororesin is applied by
spraying, screen printing, by using a coater or by
dipping.
b. The fluororesin is directly coated to the
surface of lithium by a vacuum evaporation method such
as sputtering.
c. A polymer film is directly formed by plasma
polymerization under an atmosphere of monomer which is
the raw material for the fluororesin.
If the lithium surface is coated by using the
CA 02331602 2001-10-25
-90-
fluororesin solution, it is preferable to use, in an
inactive gas dehydrated sufficiently, a fluororesin
solution dehydrated and deoxidized sufficiently. It is
preferable to use a solvent in the foregoing solution
which has been dehydrated with active alumina, molecular
sheave, phosphorus pentaoxide or calcium chloride. As an
alternative to this, it is preferable depending upon the
type of the solvent that the solvent be distilled under
presence of alkaline metal in inactive gas to remove
impurities and to be dehydrated.
An electrolyte may previously be mixed when the
foregoing film is formed. It leads to a fact that
wettability between the electrolyte solution and the
film can be improved, causing ions to easily pass
through the film.
Since the performance of the battery deteriorates
if the fluororesin film is dissolved in an organic
solvent of the electrolyte solution, it is preferable
that the film is crosslinked.
As an alternative to the foregoing method of
coating the surface with the fluororesin solution having
the ether bond, another method may be employed in which
ethylene fluoride and vinyl monomer are used as the main
raw material as to be plasma-polymerized so that the
surface is covered. In order to easily cause the plasma
polymerization to take place easily or to improve the
contact of the film and the strength of the same, it is
preferable to add, to the fluorine compound serving as
CA 02331602 2001-10-25
-91-
the raw material, oxygen, hydrogen, helium, argon,
nitrogen, silane, hydrocarbon or the like. The plasma
can effectively be generated by a DC or RF glow
discharge method, a microwave discharge method or a
laser beam irradiation method. The fluororesin having
the ether bond may be sputtered to cover the surface of
the positive electrode.
It is preferable that the thickness of the film
to be formed on the surface of lithium ranges from 10 A
to 100 ~, more preferably ranges from 50 A to 10 ~.. The
optimum thickness of the film differs depending upon the
density or the void ratio of the film and considerably
differs depending upon the type of the electrolyte
solution. The thickness of the film can be adjusted by
changing the concentration of the main material in the
coating liquid for forming the film. If the plasma
polymerization or sputtering is performed, the thickness
can be adjusted by controlling the deposition time
period.
Silicon Resin
The organic silicon compound may be any one of a
material selected from a group consisting of
alkoxysilane, alkylsilane, halogenated silane, siloxane,
silane containing vinyl group, amino group, epoxy group,
methacrylic group or mercaptal group introduced thereto,
hydrogen - denatured, vinyl - denatured, hydroxyl group
denatured, amino - denatured, carboxylic group
denatured, chloro - denatured, epoxy denatured,
CA 02331602 2001-10-25
-92-
methachryloxy - denatured, mercapto - denatured,
fluorine - denatured, long-chain-alkyl denatured or
phenyl - denatured polysiloxane, alkylene oxide
denatured siloxane copolymer, silicon - denatured
copolymer, alkoxysilane - denatured polymer, silicon -
denatured urethane or silicon - denatured nylon.
If the organic compound is liquid, the film can be
formed by a direct coating method or the organic
compound is diluted in a solvent and then applied. If
the organic compound is solid, a solution dissolved in a
solvent may be applied. The application method may be a
dipping method, a screen printing method, a spraying
method, a roll coating method or the like. The viscosity
of the coating solution must adequately be adjusted to
be adaptable to the coating method.
Titanium Polymer
A titanium polymer obtained by causing an organic
titanium compound to act on an organic polymer may be
used. For example, a thiranopolymer of silicon polymer
formed by cross linking the main chain of a
polycarbosilane skeleton with the titanium organic
compound may be used.
As an alternative to the titanium polymer, a
material obtained by introducing the derivative of an
organic metal compound, such as an organic aluminum
compound, into a polymer by substitution reactions may
be used as the coating material.
Polyphosphazene
CA 02331602 2001-10-25
-93-
Polyphosphazene which is a polymer in which
phosphor atoms and nitrogen atoms alternately form
phosphor-nitrogen double bonds can be obtained by
heating a dichloropolyphosphazene trimer to 200 to 300°C
and by ring-opening polymerization. The
dichloropolyphosphazene trimer can be synthesized from
phosphorus pentachloride and ammonia chloride or
ammonia. Any one of the following catalyzer is used at
the time of the polymerization: benzoic acid, sodium
benzonate, 2, 6 - di - p - cresol, water, methanol,
ethanol, nitromethane, ether, heteropoly acid, sulfur,
zinc, tin and sodium.
Further, various type of polyorganophosphazene can
be obtained by substituting chloride atoms of
polydichlorophosphazene by an organic reagent or an
organic metal reagent.
If the surface of positive electrode is coated with
the foregoing polyphosphazene, it is preferable that the
polymer solution dehydrated and deoxidized sufficiently
be used in inactive gas dehydrated sufficiently. It is
preferable to use a solvent in the foregoing solution
which has been dehydrated with active alumina, molecular
sheave, phosphorus pentaoxide or calcium chloride. As an
alternative to this, it is preferable depending upon the
type of the solvent that the solvent be distilled under
presence of alkaline metal in inactive gas to remove
impurities and to be dehydrated.
An electrolyte may previously be mixed when the
CA 02331602 2001-10-25
-94-
foregoing film is formed. It leads to a fact that
wettability between the electrolyte solution and the
film can be improved, causing ions to easily pass
through the film.
Since the performance of the battery deteriorates
if the polymer coating film is dissolved in an organic
solvent of the electrolyte, it is preferable to be
crosslinked in such a manner, for example, ultraviolet
rays, electron rays or radial rays are applied or a
crosslinking material, such as a radical generating
agent, is used.
It is preferable that the thickness of the film to
be formed on the surface of lithium ranges from 10 A
to 100 ~C, more preferably ranges from 50 A to 10 ~. The
optimum thickness of the film differs depending upon
the density or the void ratio of the film and
considerably differs depending upon the type of the
electrolyte solution. The thickness of the film can be
adjusted by changing the concentration of the main
material in the coating liquid for forming the film.
Polyolefin
As polyolefin, polyethylene or polypropylene may be
used. Polyolefin is used in such a manner that the
positive electrode is dipped in a solution dissolved in
a solvent such as tetrahydrofuran or o-dichlorobenzene,
the positive electrode is then dried and crosslinked by
ultraviolet, electrons, radial rays or the like so that
the film is formed on the surface of the positive
CA 02331602 2001-10-25
-95-
electrode.
The film can be formed by sputtering or plasma CVD
method. In the material in the plasma CVD method,
ethylene gas or propylene gas may be used.
Inoraanic Oxide
The inorganic glass is made of a material, such as
silica, titanium oxide, alumina, zirconia oxide,
magnesium oxide, tantalum oxide, molybdenum oxide,
tungsten molybdenum, tin oxide, indium oxide, iron
oxide, chrome oxide, aluminum phosphate, iron phosphate,
silicon phosphate and their mixtures. A sol-gel method
is one of adequate methods for forming the inorganic
glass. The raw material for the material having the
inorganic glass structure is obtained in such a manner
that an acid or a base and water are added to a solution
of alcohol of an organic metal compound such as a metal
alkoxide to hydrolyze the raw material so as to form
colloid particles having metal atom-oxygen atom bonds,
and then the colloid solution is directly applied to the
surface of the positive electrode. As an alternative to
this, a solution in which a monomer or an organic
polymer or the organic polymer and a crosslinking
material are dissolved in the collide solution is
applied, and then the solution is polymerized or dried
and polymerized so that the film is formed. By forming
the composite organic polymer, strength against cracks
and separation can be improved. If the electrolyte
forming the lithium battery is dissolved in the collide
CA 02331602 2001-10-25
-96-
solution to form the film, the wettability with the
electrolyte solution can be improved and ions are
enabled to move easily.
As an alternative to alkoxide, any one of the
following organic metal compound may be employed: acetyl
acetone complex salt, an alkyl metal compound, acetyl
acetone metal salt, naphthene acid metal salt, and octyl
acid metal salt.
The organic polymer for combining the organic
polymers is exemplified by epoxy resin, polyester,
polyimide, polyethylene, polypropylene, polyurethane,
polystyrene, polyethylene glycol, nylon, fluorine resin
and silicon resin.
The polymer crosslinking material is exemplified by
diisocyanate, polyisocyanate prepolymer, block
isocyanate, organic peroxide, polyamine, oxims, nitroso
compound, sulfur or sulfur compound, selenium, magnesium
oxide, lead oxide and zinc oxide.
As an alternative to using the crosslinking
material, a method may be employed in which radial rays
or electron rays or ultraviolet rays are applied to
polymerize or crosslink the polymer.
As an application method, a dipping method, screen
printing, spraying or a roll coating method may be
employed. The viscosity of the liquid to be applied must
adequately be adjusted to be adaptable to the
application method.
Another method of forming the film by the glass
CA 02331602 2001-10-25
-97-
inorganic oxide may be employed, for example, any one of
the following evaporation method or a CVD method may be
employed: a sputtering method, an electron beam
evaporation method, a plasma CVD (Chemical Vapor
Deposition) method, and a laser CVD method.
The sputtering process and the electron beam
evaporation can be performed by a method in which the
oxide material is directly evaporated or by a method in
which silicon or metal vapor and oxygen gas are caused
to react with each other to form the film.
The plasma CVD method and the laser CVD method is
performed in such a manner that oxygen gas and any one
of a hydroxide or halide of silicon or metal and an
organic metal compound are used as the raw materials to
be decomposed by discharge or laser so that the film is
formed.
The inorganic oxide can be used to form the film by
another method, that is, an electrochemical method
comprising the steps of forming a metal film made of
aluminum titanium, tantalum, niobium by sputtering or
electron beam, anode-oxidizing the film by using an
oxalic acid, a phosphoric acid or ammonia borate as an
electrolyte solution to form the oxide film.
A film forming method may be employed which
utilizes the equilibrium reaction in the water solution
to precipitate and cause the oxide film to grow on the
surface of the dipped substrate. In this case, silica (a
silicon oxide), titanium oxide or vanadium oxide film
CA 02331602 2001-10-25
-98-
can be formed. A specific method of forming the silica
film comprises the step of dipping the positive
electrode in a solution in which silica is saturated in
a water solution of hydrofluosilisic acid to form the
film.
Nitride
The nitride can be obtained from silicon nitride,
titanium nitride, aluminum nitride or boron nitride or
the like. The nitride film can be formed on the positive
electrode by sputtering, an electron beam evaporation
method, a plasma CVD method or a laser CVD method.
The sputtering and the electron beam evaporation
methods are categorized to a method in which the nitride
material is directly evaporated and a method in which
vapor of silicon, titanium or aluminum and nitrogen
plasma generated from nitrogen gas or ammonia gas are
caused to react with each other to form the film.
The plasma CVD method and the laser CVD method
respectively comprise the step of decomposing the raw
material gas, such as nitrogen gas, ammonia gas or
nitrogen trifluoride and a hydroxide such as silicon,
titanium or aluminum, halide or an organic metal
compound by discharge or laser to form the film.
Carbide
The carbide can be obtained from a material
exemplified by amorphous carbon, silicon carbide,
titanium carbide, vanadium carbide and tungsten carbide.
The carbide film can be formed by a similar method
CA 02331602 2001-10-25
-99-
employed to form the nitride.
The sputtering and the electron beam evaporation
methods are categorized to a method in which the
material from which the carbide is obtained is directly
evaporated and a method in which silicon, titanium,
vanadium or tungsten and carbon are used as the raw
materials to form the film.
The plasma CVD method and the laser CVD method
respectively comprise the step of, by laser, decomposing
the material, such as hydrocarbon, from which carbon is
obtained and a hydroxide such as silicon, titanium,
vanadium or tungsten or a halide or an organic metal
compound to form the film.
Halide
The halide can be obtained from a material selected
from a group consisting of lithium fluoride, magnesium
fluoride, sodium fluoride, potassium fluoride, barium
fluoride or lithium chloride.
The halide film can be formed by a sputtering
method or an electron beam evaporation method. A CVD
method such as the plasma CVD method or the laser CVD
method may be employed.
Semiconductor
The semiconductor for covering the positive
electrode is made of a material exemplified by diamond
(carbon), silicon, nickel oxide, copper oxide, vanadium
oxide, tin oxide and zinc oxide.
Alkali metal, phosphorus or boron may be added as
CA 02331602 2001-10-25
- 100 -
impurities.
The semiconductor film may be formed by sputtering,
electron beam evaporation, plasma CVD or laser CVD as
well as the foregoing method of forming the inorganic
acid film.
Coverina of Positive Electrode of Nickel-Zinc Secondarv
Battery or Air-Zinc Secondary Battery
The material and the method of covering the
positive electrode of a nickel-zinc secondary battery or
air-zinc secondary battery may be the same as those
employed to cover the positive electrode of the lithium
secondary battery. Also a polymer of a type which is
dissolved in an organic solvent but which is not
dissolved in water may be used. If the covering material
has water repellant characteristics, a hydrophilic group
must be introduced or must be subjected to hydrophilic
treatment using a silane coupling material.
Thickness of Film Coverina the Surface of Positive
Electrode
The thickness of the film for covering the surface
of the positive electrode can be controlled by adjusting
the concentration of the solution if the surface is
covered by utilizing the liquid layer reactions
performed by the polymer solution or the sol-gel method.
The same can be controlled by adjusting the deposition
time if sputtering, the electron beam evaporation, the
CVD method or the plasma polymerization is employed.
Aperture in Coating Film on Positive Electrode Surface
CA 02331602 2001-10-25
- 100 -
impurities.
The semiconductor film may be formed by sputtering,
electron beam evaporation, plasma CVD or laser CVD as
well as the foregoing method of forming the inorganic
acid film.
Covering of Positive Electrode of Nickel-Zinc Secondary
Battery or Air-Zinc Secondary Battery
The material and the method of covering the
positive electrode of a nickel-zinc secondary battery or
air-zinc secondary battery may be the same as those
employed to cover the positive electrode of the lithium
secondary battery. Also a polymer of a type which is
dissolved in an organic solvent but which is not
dissolved in water may be used. If the covering material
has water repellant characteristics, a hydrophilic group
must be introduced or must be subjected to hydrophilic
treatment using a silane coupling material.
Thickness of Film Covering the Surface of Positive
Electrode
The thickness of the film for covering the surface
of the positive electrode can be controlled by adjusting
the concentration of the solution if the surface is
covered by utilizing the liquid layer reactions
performed by the polymer solution or the sol-gel method.
The same can be controlled by adjusting the deposition
time if sputtering, the electron beam evaporation, the
CVD method or the plasma polymerization is employed.
Aperture in Coating Film on Positive Electrode Surface
CA 02331602 2001-10-25
- 101 -
The apertures and the void ratio of the coating
film for covering the surface of the positive electrode
can be controlled by adjusting the concentration of the
solution or the drying condition if covering is
S performed by using the polymer solution or by utilizing
the sol-gel method. Further, any one of the following
methods may be employed: a method for forming the film
while causing foam to be generated by adjusting the
concentration of the foaming material; a method having
the steps of forming a film in which the content of the
electrolyte is adjusted, and dissolving the electrolyte;
a method for controlling the evaporation rate; and a
method for adjusting the mixture ratio of the reactive
materials.
If the negative electrode is covered with the
coating material of a type employed to cover the
positive electrode, growth of dendrite from the negative
electrode can be prevented. Therefore, short circuits
in the battery can be prevented in effect obtainable
from covering the positive electrode. As a result, the
battery cycle can further be lengthened.
Then, a positive electrode according to the present
invention composed of transition metal and group 6A
element will now be described.
Transition Metal Element
The transition metal element in the positive
electrode active material according to the present
invention may be an element having partially shell d or
CA 02331602 2001-10-25
- 102 -
shell f and selected from a group consisting of T, Zr,
Hf, V, Nb, Ta, Cr, Mo, W, Wn, Tc, Re, Fe, Ru, Os, Co,
Rh, Ir, Ni, Pd, Pt, Cu, Ag and Au. Any one of the first
transition system metal is mainly used which is
exemplified by Ti, V, Cr, Mn, Fe, Co, Ni or Cu. A
compound of the transition metal and the group 6A
element is manufactured by a material exemplified by the
transition metal, the salt of the transition metal, an
oxide of the transition metal and a hydroxide of the
transition metal.
Group 6A Element
The positive electrode active material according to
the present invention comprises the group 6A element
exemplified by 0, S, Se, Te and Po. Among the foregoing
elements, 0 or S is mainly employed. A compound of the
transition metal and the group 6A element can be
manufactured by a material exemplified by a compound of
a hydroxide, halide, halide oxide or oxide of sulfur,
selene, tellurium, polonium.
Additive Element
By adding elements except the transition metal, the
distortion of the positive electrode active material
occurring due to the introduction/discharge of lithium
ions can be relaxed. The element can be effectively
added by a method comprising the step of adding the
salt, the halide or an organic metal compound of one or
more types of elements selected from a group consisting
of lithium, magnesium, sodium, potassium, aluminum,
CA 02331602 2001-10-25
-103-
zinc, calcium, barium, lead, indium, boron, silicon,
tin, phosphor, arsenic, antimony, bismuth, chlorine and
fluorine. It is preferable that the atom ratio of the
additive element with respect to the transition metal
element be 1 or less.
Method of Manufacturing Compound of Transition Metal and
Group 6A Element
The compound of the transition metal and the group
6A element according to the present invention is
manufactured by any one of methods categorized to a
method in which the compound is prepared from the
solution of the salt of the transition metal or the
organic transition metal compound by way of a hydroxide,
a method comprising the steps of melting the transition
metal or the transition metal compound and rapidly
cooling it, and a method in which the transition metal
compound is caused to react in a gas phase.
The method of preparing the transition metal oxide
from the hydroxide is exemplified by a method comprising
the step of baking or drying hydroxide in air or in an
oxygen atmosphere to prepare powder. A specific example
of preparing the transition sulfide from the hydroxide
is exemplified by a method comprising the step of baking
a hydroxide in a hydrogen reduction atmosphere in which
a hydrogen sulfide is mixed to prepare powder.
The rapid cooling method for preparing the
transition metal oxide comprises the steps of melting
the transition metal compound, such as the transition
CA 02331602 2001-10-25
- 104 -
metal or the transition metal oxide, and spraying oxygen
gas containing inactive gas mixed thereto to a rotating
disc to prepare the powder.
The method of preparing the transition metal oxide
by the gas phase reactions comprises the step of
oxidizing or hydrolyzing the salt of the transition
metal or causing vapor of the transition metal to react
with the group 6A element or a compound of the group 6A
element or decomposing the organic transition metal
compound to prepare the powder.
It is preferable that each process be performed at
400°C or lower, more preferable 300°C or lower.
The liquid phase reaction method, the gas phase
reaction method and the melting and rapid cooling method
will now be described.
Liquid Phase Reaction
A method of preparing the hydroxide for use in the
main reaction in the method of manufacturing the
material from the solution by way of the hydroxide is
exemplified by:
Method of Preparincx Hydroxide
It is preferable that the transition metal
hydroxide be prepared by reactions between the salt of
the transition metal and alkali or by hydrolysis of the
organic transition metal compound or by reactions
between the transition metal and alkali. It is
preferable that the preparation temperature be 150°C or
lower, more preferably 100°C or lower.
CA 02331602 2001-10-25
- 105 -
Reactions between Salt of Transition Metal and Alkali
By causing alkali to react with the salt of the
transition metal, a hydroxide of the transition metal is
sedimentated to be prepared. By mixing salts of two or
more types of the transition metals, a hydroxide of a
composite transition metal can be obtained.
The salt of the transition metal is typified by a
carbonate, a nitrate, a halide, a sulfate, a sulfamate,
acetate, oxalate, critate, tertrate, formate or
ammonate.
The alkali may be lithium hydroxide, sodium
hydroxide, potassium hydroxide or ammonia hydroxide. As
an alternative to this, urea or thiourea may be used
which raises the pH by generating hydroxide ions when
heated.
It is preferable that an organic acid or inorganic
acid or an amine is, in a small quantity, added or an
organic solvent such as alcohol is added at the time of
causing alkali to react with the water solution of the
transition metal salt in order to fine the sediment
particles of the hydroxide.
By vibrating the sediment with ultrasonic waves,
the sediment particles can be fined. Therefore, the
specific area can be enlarged.
Hydrolysis Reaction of Organic Transition Metal Compound
The hydroxide of the transition metal can be
prepared by hydrolyzing an organic metal compound of the
transition metal, such as alkoxide, acetyl acetenate,
CA 02331602 2001-10-25
- 106 -
octylate or naphthalate.
The hydrolysis of alkoxide is specifically
performed in such a manner that alkoxide of the
transition metal is dissolved by water, alcohol or
ethanol amine or the like, and an inorganic acid such as
hydrochloric acid or an organic acid such as acetic acid
or ammonia hydroxide or amine is added.
The alkoxide of the transition metal may be a
material selected from a group consisting of
Mn ( OC2H5 ) z , Mn ( OC3H~ ) 2 , Mn ( OCqH9 ) 2 , Ni ( OC2H5 ) , Ni ( OC3H~ )
2 ,
Ni (OC9H9) 2, Co (OC2H5) 2, Co (OC3H,) 2, Co (OC9H9) 2, Ti (OC2H5) 2,
Ti (OC3H7) 2, Ti (OCqH9) 2, Fe (OCzHS) 2, Fe (OC3H~) z, Fe (OCQH9) 2,
Cu ( OC2H5 ) 2 , Cu ( OC3H, ) 2 , Cu ( OCqH9 ) 2 , VO ( OCH3 ) 3 , VO ( OC2H5
) 3 ,
VO (OC3H~) 3, VO (OCqH9) 3 and Y (OCqH9) 3.
The acetylacetonate of the transition metal is
exemplified by Cu (CSH~02) 2, Co (C5H,02) 2, (H20) 2, Co (CSH~02) 3,
Ni ( CSH~OZ ) 2 ( H20 ) 2 , Mn ( CSH~OZ ) z ( H20 ) 2 , Cr ( CSH-,OZ ) 3 , VO
( CSH702 ) z ,
Fe (CSH,02) 3, Ti (OCqH9) 2 (CSH~O) z, La (CSH,02) 3, Y (CSH~02) 3, and
Zr ( CSH~OZ ) q .
The octylate of the transition metal is exemplified
by Cu ( C,H15C00 ) Z , Ni ( C~H15C00 ) z , Fe ( C7H15C00 ) 2 , Mn ( C~H15C00 )
2 ,
Co (C~H15C00) 2, Zr (C,H15C00) 2, Y (C~H15C00) 2, and La (C,H15C00) 2.
The naphtate of the transition metal is exemplified
by the salt of naphthate expressed by a general formula
C~Hz~ ~Oz, that is, cobalt naphthenate, copper naphthenate,
manganese naphthenate, iron naphthenate, nickel
naphthenate, vanadium naphthenate, yttrim naphthenate
and lanthanum naphthenate.
CA 02331602 2001-10-25
- 107 -
The alcohol is exemplified by methyl alcohol, ethyl
alcohol, isopropyl alcohol, ethylene glycol and
propylene glycol.
Dehydrating Reaction
In order to obtain the oxide of the transition
metal by dehydration from the transition metal hydroxide
prepared by the foregoing solution reactions, it is
preferable that the transition metal hydroxide is
immersed in an organic solvent such as alcohol or
acetone which is mixed with eater to sufficiently
substitute water, and then it is dried in a vacuum state
at 100°C or higher. As an alternative to this, heating
and dehydration is performed by using microwaves. If the
drying temperature is too high, crystallization is
enhanced and therefore the hydroxide groups are
decreased. Therefore, it is preferable that the
temperature be 400°C or lower. It is preferable that the
frequency of the microwaves be a frequency which can
easily be absorbed by water.
In order to enlarge the specific area, another
method may be employed in which the dehydration is
performed by freezing and drying.
~droaen Treatment
Hydrogen is mixed into the dried atmosphere at the
time of drying the transition metal oxide followed by
performing heat treatment. As an alternative to this, a
method is employed in which the transition metal
hydroxide or the transition metal oxide is subjected to
hydrogen plasma treatment. The hydrogen plasma can be
CA 02331602 2001-10-25
- 108 -
generated by a hydrogen gas discharge method or by a
method of exciting and decomposing the hydrogen gas by
laser beams.
Introduction of Group 6A Element Except Oxygen
A method is employed in which the transition metal
hydroxide or the transition metal oxide is treated with
hydrogen sulfide or hydrogen selenide or the group 6A
element is mixed at the time of preparing the transition
metal hydroxide.
Gas Phase Reaction Method
As a method of preparing a compound of the
transition metal and the group 6A element, a gasified
transition metal salt or an organic transition metal
compound or vapor of the transition metal and the group
6A element or the group 6A element compound are caused
to react with each other in the gas phase to prepare
powder. Another method may be employed in which the
gasified transition metal salt or the organic transition
metal compound containing the group 6A element is
decomposed in the gas phase to prepare the compound of
the transition metal and the group 6A element.
If the transition metal salt or the organic
transition metal compound is in the form of solid, it is
heated as to be formed into vapor or it is heated as to
be formed into liquid. Then, carrier gas is bubbled as
to be introduced into the reaction chamber. If the
transition metal salt or the organic transition metal
compound is in the form of liquid, it may be heated as
to be formed into vapor or carrier gas is bubbled as to
CA 02331602 2001-10-25
- 109 -
be introduced into the reaction chamber.
The salt of the transition metal may mainly be a
halide such as a chloride. As an alternative to this, a
carbonate, a nitrate, a sulfate, sulfominate, acetate,
citrate, tertrate, formate or ammonate. The chloride is
exemplified by VOC13, MnCl2, MoClS, TiCl4, NiCl2, CoCl2,
FeCl3, WC16, YC13 and ZrCl4.
The raw material from which the group 6A element
can be obtained is exemplified by the group 6A element,
the hydroxide of the group 6A element and the halide of
the group 6A element.
By mixing hydrogen gas in the foregoing gas phase
reaction, a compound of the transition metal and the
group 6A element containing hydrogen can be prepared.
In the foregoing gas phase reaction, it is
preferable to employ any one of the following methods: a
heat CVD (Chemical Vapor Deposition) method, a plasma
CVD method, a laser CVD method, a filament method, a
reactive sputtering method and the electron beam method.
Sputtering is performed by heat in the heat CVD
method, by charge in the plasma CVD method, by heat
energy or light energy of laser beams in the laser CVD
method, by heat of a filament made of tungsten or the
like in the filament method, and in the reactive
sputtering method in the reactive gas atmosphere, and by
electron beam heating in the electron beam method. As a
result, gas phase reactions are respectively performed
so that the material is prepared. It is preferable that
the raw material be in the form of solid in the reactive
CA 02331602 2001-10-25
- 110-
sputtering method or the electron beam method.
Rapid Coolincr Method
In this case, any one of the following methods may
be employed: a gun method comprising the steps of
generating, while breaking a Mylar film, shock waves by
helium gas, in which oxygen or hydrogen sulfide is
mixed, as to be sprayed to the transition metal or the
transition metal compound melted with high frequency
waves, and causing blown compound powder to impact
against a cooling steel plate in the form of a slide
disposed below so that the temperature is rapidly
cooled; a method in which a molten bath is dispersed by
spraying with inactive gas jet in which oxygen or
hydrogen sulfide is mixed; and an atomization method in
which molten bath of the transition metal or the
transition metal compound is sprayed in an atmosphere
containing the group 6A element such as oxygen or group
6A element compound such as hydrogen sulfide as to be
formed into powder. By mixing hydrogen gas into the
inactive gas, hydrogen can be introduced into the
product.
It is preferable that the rapid cooling rate be 101
to 108 K, more preferable 10z to 108 K.
The melting and heating furnace may be a crucible
furnace, an induction furnace, an arc furnace or an
electron beam furnace.
The method of causing the alkali to directly react
with the transition metal is exemplified by a method in
which metal such as vanadium is caused to react with
CA 02331602 2001-10-25
- 111 -
molten alkali to prepare an oxide.
Conductor Core
When conductor powder is mixed at the time of
preparing the compound of the transition metal and the
group 6A element to cause the compound of the transition
metal and the group 6A element to grow while using the
conductor powder as the core, the collecting efficiency
can be raised. Therefore, the introduction and discharge
of lithium ions can be made easier, and therefore the
battery capacity can be enlarged.
The conductor powder may be made of one or more
types of materials selected from a group consisting of
carbon, titanium, nickel, cobalt, iron, chrome,
manganese, vanadium, platinum, paradium, copper, silver,
gold, zinc, tin, indium, lead, tungsten and molybdenum.
It is preferable to use one or more types of elements
selected from a group consisting of carbon, titanium,
nickel, cobalt, iron, chrome, manganese, vanadium and
platinum.
The shape of the conductor powder is formed into
one or more types of shapes selected from a group
consisting of a spherical shape, a flake shape, a needle
shape and a fiber shape. As a result, also the compound
powder of the transition metal and the group 6A element
can be formed into the spherical, flake, needle or fiber
shape. Therefore, the efficiency of the electron
movement between the positive electrode active material
can be raised, and therefore the charging and
discharging efficiency can be improved.
CA 02331602 2001-10-25
- 112-
It is preferable that the diameter of the conductor
powder be 10 A to 100 ,u when measured by an electron
microscope, more preferably 10 A to 10 ,u.
Crushina of Positive Electrode Active Material
The prepared positive electrode active material
must be crushed as to have an adequate grain size if
obtained positive electrode active material is in the
form of a block.
It is preferable to crush the positive electrode
active material by using, while combining, means
selected from a group consisting of a compression
crushing machine, a shearing crushing machine, an impact
crushing machine, a roll mill, a roller mill, a high-
speed rotational mill, a ball mill, a medium stirring
mill, a jet mill, a mortar and a stamping mill.
Coatina Film Made of Conductor
The prepared compound of the transition metal and
the group 6A element is covered with a conductor thin
film by chemical plating (non-electrolytic plating) or
by evaporation. As a result of the foregoing process,
the current collecting efficiency can be raised, the
introduction/discharge of lithium ion can be facilitated
and the battery capacity can be enlarged.
The conductor thin film may be made of one or more
types of materials selected from a group consisting of
carbon, titanium, nickel, cobalt, iron, chrome,
manganese, vanadium, platinum, paradium, copper, silver,
gold, zinc, tin, indium, lead, tungsten and molybdenum.
It is preferable to use one or more types of elements
CA 02331602 2001-10-25
- 113 -
selected from a group consisting of carbon, titanium,
nickel, cobalt, iron, chrome, manganese, vanadium and
platinum.
The chemical plating method is a method in which
metal ions are deoxidized by a deoxidizer such as
formaldehyde to precipitate the metal film.
The evaporation is performed by any one of the
following methods: a method in which vapor of metal is
generated by electron beams or laser beams as to be
applied to the subject; a method in which a carbon or
metal target is sputtered as to be applied to the
subject; a method in which hydrocarbon or organic
solvent or an organic metal compound is decomposed by
discharge or laser or heat as to be applied to the
subject. The decomposition of the hydrocarbon and the
organic solvent enables a carbon film to be formed,
while the decomposition of the organic metal compound
enables a metal film to be formed.
It is preferable that the thickness of the
conductor film ranges from 50 A to 1 ,u.
Lipophilic Treatment
The positive electrode active material is subjected
to the lipophilic treatment in such a manner that the
organic metal compound is immersed in a solution, in
which an organic solvent is dissolved, and then it is
dried.
It is preferable that the organic metal compound
for use in the lipophilic treatment be an organic metal
compound such as silane coupling material or organic
CA 02331602 2001-10-25
- 114-
titanate. If dilution is performed, the dilution
concentration with respect to the solvent ranges from
0.05 to 2 wt%.
The silane coupling material is exemplified by
vinyltrimethoxysilane, vinylethoxysilane, N - (2 -
aminoethyl) 3 - aminopropylmethyl dimethoxysilane, N -(2
- aminoethyl) 3 - aminopropyltrimethoxysilane, 3 -
aminopropyltriethoxysilane, 3 -
glycidexypropyltrimethoxysilane, 3 -
glycidexypropylmethyldimethoxysilane, 2 - (3,4 -
epoxycychlohexyl) ethyltrimethoxysilane, 3 -
chloropropylmethyldimethoxysilane, 3 -
chloropropyltrimethoxysilane, 3 -
methacryloxytrimethoxysilane, 3 -
mercaptotrimethoxysilane, and N - [2 -
(vinylpenzilamino) ethyl] - 3 -
aminopropyltrimethoxysilane.
The organic titanate is exemplified by tetra - i -
propoxytitanium, tetra - n - butoxytitanium, tetrakis (2
- ethylhexyloxy) titanium, tetrastearyloxytitanium,
di - i - proxy~bis (acetylacetate) titanium,
dihydroxy~bis (lactato)titanium, titanium - i -
propoxyoctylene glycol, titanium stearate, propane
dioxytitanbis (ethylacetoacetate), propanedioxytitanium
(acetylacetonate) (ethylacetoacetate), oxotitanbis
(monoammonium oxalate), tri - n -
butoxytitanmonostearate, and titan polymer.
Analysis of Transition Metal and Group 6A Element
Measurement of Size of Crystal Grain
CA 02331602 2001-10-25
115 -
The size of crystal grains was evaluated from the
peak half width and the angle of diffraction of X-ray
diffraction curve in accordance with the following
Scherrer's Formula:
t= 0.9?~/Bcos6B (Scherrer's Formula)
where t: size of crystal grain
?~: wave length of X-ray beam
B: peak half width
6B: angle of diffraction
It is preferable that the average size of the
crystal grain of the positive electrode active material
which is the compound of the transition metal and the
group 6A element for use in the secondary battery
according to the present invention be 500 A or less when
calculated by using the foregoing Scherrer's Formula,
more preferably 200 A or less.
Observation of Crystal Structure
The crystal structure of the positive electrode
active material and the group 6A element prepared by the
method according to the present invention can be
observed in such a manner that the waveform of the
radial distribution function can be observed by X-ray
diffraction, the diffraction pattern can be observed by
reflecting high speed electron beam diffraction (RHEED)
and the waveform of the X-ray diffraction curve can be
observed.
The radial distribution function can be obtained by
Fourier transforming the dispersion intensity of
measured X-ray or neutron beam. The radial distribution
CA 02331602 2001-10-25
- 116 -
function is expressed by the presence possibility of
atoms with respect to an arbitrary atom or a function of
the deviation distance from an average numerical
density. If the specimen is made of amorphous material,
a moderate peak curve can be obtained. If the specimen
is crystal material, a discontinuous a sharp peak can be
obtained.
The RHEED enables a halo pattern to be observed if
the specimen is made of amorphous material, enables a
ring pattern to be observed if the specimen is made of
microcrystal material and enables a spot pattern to be
observed if the specimen is made of a multi-crystal
material.
The dispersion angle and the dispersion intensity
obtainable in the X-ray small angle dispersion method
also enables of fluctuation of ununiform density
peculiar to the amorphous material.
Further, the differential thermal analysis enables
heat absorption or heat generation due to the structural
relaxation or the crystallization structural change
occurring to the temperature rise to be observed if the
specimen is amorphous material. If a hydroxide group is
present, heat absorption due to dehydration can be
observed.
By using the foregoing means, the structure of the
compound of the transition metal and the group 6A
element prepared by the manufacturing method according
to the present invention can be analyzed so that the
amorphous structures the microcrystal structure and the
CA 02331602 2001-10-25
- 117-
multi-crystal structure are confirmed.
Analysis of Hydrogen
The compound of the transition metal containing
hydrogen and the group 6A element is subjected to
qualitatively analyzed by SIMS (Secondary Ion Mass
Spectrometry) analysis.
Manufacturing of Positive Electrode
The positive electrode is manufactured in such a
manner that the bonding material, and conductive powder
if necessary, is mixed with the powder of the compound
of the transition metal and the group 6A element
manufactured by the foregoing method, and then formed
into the positive electrode together with the collector.
It is preferable that the forgoing forming process be
performed in dry air from which water has sufficiently
be removed, more particularly under inactive gas
atmosphere.
The conductive powder acts to enhance the electron
conduction and facilitate the current collection because
the active material, which is the compound of the
transition metal and the group 6A element, does not
substantially have the electron conductivity.
The conductive powder may be carbon material, such
as acetylene black, ketchen black or graphite powder, or
metal, such as nickel, titanium, copper or stainless
steel. It is preferable that the mixture ratio of the
conductive powder with respect to the positive electrode
active material be 1 or less.
CA 02331602 2001-10-25
- 118 -
The bonding material bonds the positive electrode
active material powder to one another and prevents
generation of cracks in the charge and discharge cycle
to prevent the separation from the collector. The
bonding material may be one or more types of resin which
are stable against the organic solvent and which are
selected from a group consisting of fluororesin,
polyethylene, polypropylene and silicon resin. It is
preferable that the foregoing resin be in the form of
liquid or solution or having a low melting point.
Further, it is preferable that the solvent be removed
and the resin is crosslinked during the process of
manufacturing the positive electrode. As a result, the
content of the bonding material in the positive
electrode can be lowered and the capacity of the battery
can be improved. The liquid resin or the resin which can
be dissolved in the solvent is exemplified by
fluororesin having an ether bond and silicon resin. If
the fluororesin having the ether bond is used, the
concentration can be lowered when it is dissolved in a
solvent. Therefore, the content in the positive
electrode can be lowered as much as possible and the
void ratio can be raised. Further, the state is very
stable after crosslinking has been performed so that a
satisfactory effect can be obtained upon the charge and
discharge cycle.
It is also preferable that dehydration is performed
with heat generated by microwaves and a vacuum drier is
used to dehydrate the positive electrode after the
CA 02331602 2001-10-25
- 119-
positive electrode has been formed.
It is preferable that the mixture ratio of the
bonding material with respect to the positive electrode
active material be 0.1 or less.
Examples
Examples of the present invention will now be
described. It should be noted that the present invention
is not limited to the examples below.
Example 1-1
A flat type battery having a simple structure,
exhibiting assembling easiness and having a cross
sectional structure schematically shown in Fig. 2 was
assembled to evaluate the characteristics of the
secondary battery while mainly evaluating the life
against cycle operation.
First, a solution in which colloidal silica
manufactured by Shokubai Kasei and subjected to
hydrophobic treatment in an atmosphere of dry argon gas
was dispersed in N, N - dimethylformamide was dehydrated
by active alumina. Then, lithium metal foil to which
titanium mesh collector 200 was pressed against the
reverse side was immersed in the colloidal silica
solution prepared by the foregoing method, and then
dried at 120°C so that a lithium negative electrode 201
covered with silica was manufactured.
A positive electrode active material 203 was
manufactured in such a manner that a mixture of
dehydrated and electrolyzed manganese dioxide and
lithium carbonate was heated so that a lithium-manganese
CA 02331602 2001-10-25
- 120 -
composite oxide was prepared. Then, tetrafluoroethylene
polymer powder was mixed, and then pressed against the
titanium mesh as to be formed into a designed shape.
The electrolyte solution was obtained by
dissolving, by 1 M (mol/1), lithium arsenate
hexafluoride salt in an equally mixed solvent of
propylene carbonate (PC) and dimethoxyethane (DME).
A separator 208 was formed in such a manner that
a separator made of polypropylene and having small
apertures was sandwiched by unwoven polypropylene
sheets.
The assembling process was performed in such a
manner that the separator 208 was place between the
negative electrode 201 and the positive electrode 203
followed by inserting them into a positive electrode
case 207 made of titanium clad stainless steel. Then, an
electrolyte solution was injected followed by sealing
the positive electrode case 207 with a negative cap 206
made of titanium clad stainless steel and an insulating
packing 210 made of fluororubber so that a secondary
battery was manufactured.
Comparative Example 1-1
A lithium secondary battery was manufactured by a
method similar to Example 1-1 except for excluding the
surface covering treatment using the metal lithium foil
performed in Example 1-1.
Comparative Example 1-2
A battery similar to that according to Example 1-1
was manufactured.
CA 02331602 2001-10-25
- 121
The preparation of the materials and assembly were
performed in an atmosphere of dry argon similarly to
Example 1-1.
First, aluminum trichloride and phosphoric acid
S were allowed to reach with each other in ethyl alcohol,
and gradually heated to 100°C so that glass form
aluminum phosphate was obtained. Then, it was dispersed
in n-hexane so that a film forming solution was
prepared. The film forming solution was applied to a
lithium metal foil to which the titanium mesh collector
was pressed. Then, a film was formed at 120°C. Then,
similar processes to those according to Example 1-1 were
performed so that a lithium battery was manufactured.
Example 1-3
A battery similar to that according to Example
1-1 was manufactured.
The preparation of the materials and assembly were
performed in an atmosphere of dry argon similarly to
Example 1-1.
First, acetic acid and water were added to an ethyl
alcohol solution of tetrabutoxytitanium to be
hydrolyzed. Then, diethylamine was added so that
colloidal titanium oxide was formed. Then, colloidal
titanium oxide was dissolved in a xylene solution of
polyethylene so that a film forming solution was
prepared. The film forming solution was applied to the
lithium metal foil to which the stainless mesh collector
was pressed followed by drying it at 100°C. Then,
electron beams were applied to crosslink the
CA 02331602 2001-10-25
- 122 -
polyethylene so that a film was formed. Then, similar
processes to those according to Example 1-1 were
performed so that a lithium battery was manufactured.
Example 1-4
A battery similar to that according to Example 1-1
was manufactured.
The preparation of the materials and assembly were
performed in an atmosphere of dry argon similarly to
Example 1-1.
A toluene solution of triethylaluminum was mixed
with an isopropyl alcohol solution of
tetrabutoxytitanium, followed by adding acetic acid and
water as to be hydrolyzed. Then, diethylamine was added
so that colloidal titanium oxide-alumina was formed.
Then, the colloidal titanium oxide-alumina was dispersed
in a toluene solution which acrylic resin and epoxy
resin were dissolved so that the film forming solution
was prepared. The film forming solution was applied to a
lithium metal foil to which the stainless mesh collector
was pressed, and then a film was formed at 80°C . Then,
similar processes to those according to Example 1-1 were
performed so that a lithium battery was manufactured.
Example 1-5
A battery similar to that according to Example 1-1
was manufactured.
The preparation of the materials and assembly were
performed in an atmosphere of dry argon similarly to
Example 1-1.
First, acetic acid and water were added to an
CA 02331602 2001-10-25
- 123 -
ethyl alcohol solution of tetraethoxysilane as to be
hydrolyzed. Then, diethyl amine was added so that
colloidal silica oxide was formed. Then, the colloidal
silica was dispersed in an acetonitryl solution of
polyethylene glycol, followed by adding
azobisisobutylonitryl and arsenic hexafluoride lithium
salt so that a film forming solution was prepared. The
film forming solution was applied to the lithium metal
foil to which the stainless mesh collector was pressed,
followed by drying it at 100°C as to crosslink
polyethylene glycol so that a film was formed. Then,
similar processes to those according to Example 1-1 were
performed so that a lithium battery was manufactured.
Example 1-6
A battery similar to that according to Example 1-1
was manufactured.
First, felt-shape carbon was immersed in silica
coating solution NT-6326, which was manufactured by
Nissan Kagaku and to which boron tetrafluoride lithium
salt was added, and taken out from it. Then, it was
hardened at 110°C for 20 minutes and 300°C for 30 minutes
so that silica coating was performed. The obtained felt-
shape carbon covered with the silica was used as the
cathode and the lithium metal was used as the anode in
an electrolyte solution in which arsenic hexafluoride
lithium salt was, by 1 M (mol/1), dissolved in an equal-
quantity mixture solvent of propylene carbonate and
dimethoxyethane dehydrated sufficiently. As a result,
lithium was introduced and allowed to adhere to the
CA 02331602 2001-10-25
- 124 -
felt-shape carbon covered with silica so that the
negative electrode 201 was manufactured.
Then, similar processes to those according to
Example 1-1 were performed so that a battery was
manufactured.
Example 1-7
A battery similar to that according to Example 1-1
was manufactured.
First, paste obtained by mixing zinc oxide
powder, metal zinc powder and water glass was injected
into foamed nickel (Celmet) manufactured by Sumitomo
Denko. Then, the material was hardened at 80°C for 20
minutes and 200°C for 30 minutes so that the negative
electrode 201 was formed.
Then, paste obtained by mixing, with nickel
hydroxide, nickel powder, cobalt powder, carboxylic
methyl cellulose serving as the bonding material,
ethylene glycol and water was injected into the Celmet
manufactured by Sumitomo Denko. Then, it was dried and
pressed so that the positive electrode 203 was formed.
The separator was formed in such a manner that
polypropylene film subjected to hydrophilic treatment
and having small apertures was sandwiched by unwoven
polyamide sheets. The electrolyte solution was a 30 wt%
potassium hydroxide solution. The assembly was performed
similarly to Example 1-1 so that a nickel-zinc secondary
battery was manufactured.
Comparative Example 1-2
A nickel-zinc secondary battery was manufactured by
CA 02331602 2001-10-25
-125-
a method similar to Example 1-7 except that the negative
electrode was manufactured by mixing polyethylene
serving as the bonding material and ethylene glycol in
place of the water glass.
Example 1-8
A battery having a structure similar to that
according to Example 1-1 was manufactured. However, a
positive electrode case having small apertures for
receiving oxygen in air was employed.
The negative electrode 201 was formed similarly to
that according to Example 1-7.
The positive electrode 203 was formed in such a
manner that the positive catalyzer layer was formed by
adding manganese dioxide to active carbon, and a water
repellant film made of polytetrafluoroethylene and
cellulose-type diffusion paper were stacked on the
positive catalyzer layer.
The separator 208 comprises a cellophane film,
while the electrolyte solution comprises a 30 wt%
potassium hydroxide solution.
The battery was assembled in such a manner that the
separator 208 was held between the negative electrode
201 and the positive electrode 203, followed by
inserting them into the positive electrode case 207
having small apertures for receiving oxygen in air and
made of titanium clad stainless steel. Then, the
electrolyte solution was injected, and sealing was
performed by the negative cap 206 made of the titanium
clad stainless steel and the insulating packing 210 made
CA 02331602 2001-10-25
- 126
of fluorine rubber so that the air-zinc secondary
battery was manufactured.
Comparative Example 1-3
An air-zinc secondary battery was manufactured by a
method similar to that according to Example 1-8 except
that the negative electrode according to Comparative
Example 1-2 was used.
Evaluation of Performance of Secondar Batter
The performance of lithium secondary batteries
according to Examples 1-1 to 1-6 and Comparative Example
1-1, the nickel-zinc secondary battery according to
Example 1-7 and Comparative Example 1-2, and the airzinc
secondary battery according to Example 1-8 and
Comparative Example 1-3 was evaluated. The evaluation
was performed by a charge and discharge cycle test under
the following conditions with respect to the cycle life
of the batteries according to Comparative Examples 1-1,
1-2 and 1-3. The conditions for the cycle test were made
as follows: the charge and discharge was performed by
0.2C (electric current which was 0.2 times
capacity/time), pause for 30 minutes and a cut-off
voltage of 1.0V was applied. A charging/discharging
apparatus HJ-1O1M6 manufactured by Hokuto Electric was
used. The charge/discharge test was commenced at
discharge, the battery capacity was evaluated by the
quantity of the third discharge and the cycle life was
evaluated by the number of cycles when the battery
capacity had deteriorated to 60% or less.
The cycle life of each battery with respect to the
CA 02331602 2001-10-25
- 127
cycle life of the battery according to Comparative
Example 1-1 which was made to be 1 was as shown in Table
1.
As can be understood from the results of
comparisons made between Examples 1-1 to 1-6 and
Comparative Example 1-1, between Example 1-7 and
Comparative Example 1-2 and between Example 1-8 and
Comparative Example 1-3, the cycle life can
considerably be lengthened due to use of the foil made
of the negative electrode active material according to
the present invention and arranged in such a manner that
the surface is covered with the film having an inorganic
glass structure through which lithium ions can be
passed.
CA 02331602 2001-10-25
- 128 -
Table 1
Cycle Life according
to the present
Secondary Battery Manufactured invention
Cycle Life
Exam 1e accordin to
P Comparative g
Example Comparative Example
Example 1-1 Comparative
Example 1-1 2.5
Example 1-2 Comparative
Example 1-1 2,0
Example 1-3 Comparative
Example 1-1 1.5
Example 1-4 Comparative
Example 1-1 1.5
Example 1-5 Comparative
Example 1-1 3.0
Example 1-6 Comparative
Example 1-1 3.4
Example 1-7 ~ Comparative
Example 1-2 1.5
Example 1-8 Comparative
Example 1-3 2.0
CA 02331602 2001-10-25
- 129 -
Example 2-1
A flat battery which had a simple structure, which
could be assembled simply and which had a cross
sectional shape schematically shown in Fig. 2 was
assembled to evaluate the characteristics of the
secondary battery while evaluating the cycle life.
First, a peroxide benzoyl was, in an atmosphere of
dry argon gas, dissolved in a toluene solution of poly
(2-vinyl napthalene) manufactured by Aldrich Chemical
Company, Inc. Then, a lithium metal foil to which the
stainless mesh collector 200 was pressed from the
reverse side was immersed in it. Then, heat treatment at
100°C was performed so that a lithium electrode 201
covered with the poly (2-vinyl napthalene) was
manufactured.
The positive electrode active material 203
comprised a lithium-manganese composite oxide prepared
by heat treatment subjected to a mixture of dehydrated
electrolytic manganese dioxide and lithium carbonate and
a graphite mixture. Then, tetrafluoroethylene polymer
powder was mixed and then pressed against a titanium
mesh as to be formed.
The electrolyte solution was prepared by
dissolving, by 1 M (mol/1), arsenate hexafluoride
lithium salt in an equal-quantity mixture solvent of
propylene carbonate (PC) and dimethoxyethane (DME).
The separator 208 was formed by sandwiching
a separator made of propylene and having small apertures
by unwoven polypropylene sheets.
CA 02331602 2001-10-25
130
Comparative Example 2 1
A lithium secondary battery was manufactured by a
method similar to Example 2-1 except for that the metal
lithium foil surface covering treatment according to
Example 2-1 was omitted.
Example 2-2
A battery similar to that according to Example 2-1
was manufactured.
The preparation of materials and assembling of the
battery were performed in an atmosphere of dry argon
similarly to Example 2-1.
First, 0.03 M azobisisobutylnitryl was added to a
5M tetrahydrofuran solution of 9-vinyl anthracene,
following by performing polymerization at 45°C so that a
polymer solution was obtained. Then, the polymer
solution was applied to lithium metal foil to which a
titanium mesh collector was pressed followed by drying
the polymer solution. Then, ultraviolet rays were
applied so that a lithium electrode covered with the
polyvinyl antracene film was manufactured. Then, similar
processes to those according to Example 2-1 were
performed so that the lithium secondary battery was
manufactured.
Example 2-3
A battery similar to that according to Example
2-1 was manufactured.
The preparation of materials and assembling of the
battery were performed in an atmosphere of dry argon
similarly to Example 2-1.
CA 02331602 2001-10-25
- 131 -
First, 9, 10-antracene dipropionic acid and
ethylene glycol of the same mol and a small quantity of
zinc acetate were injected into a reaction chamber
followed by dehydrating and condensing them at 200°C in
S an argon gas flow. Then, azobisisobutlonitryl was added,
and the lithium metal foil to which a stainless steel
mesh collector was pressed was immersed followed by
taking out it. Then, it was heated to 100°C so that a
film was formed on the surface of lithium. Then, similar
processes to those according to Example 2-1 were
performed so that the lithium secondary battery was
manufactured.
Example 2-4
A battery similar to that according to Example 2-1
was manufactured.
The preparation of materials and assembling of the
battery were performed in an atmosphere of dry argon
similarly to Example 2-1.
Tantalum pentachloride was dissolved in toluene at
80°C, and a toluene solution of 5, 12-bis(phenylethynyl)
naphthalene was added followed by polymerizing them at
80°C. An obtained polymer was cleaned with methyl
alcohol, dried and again dissolved
in toluene so that a film forming solution was prepared.
Then, a lithium metal foil to which a stainless steel
mesh collector was pressed was immersed, followed by
raising it and drying the same. Then, electron beams
were applied to crosslink the material so that a film
was formed on the surface of lithium. Then, similar
CA 02331602 2001-10-25
- 132 -
processes to those according to Example 2-1 were
performed so that the lithium secondary battery was
manufactured.
Example 2-5
A lithium secondary battery similar to that
according to Example 2-1 was manufactured except that
peroxide benzoyl was dissolved in a toluene solution of
poly (2-vinyl naphthalene), and then arsenate
hexafluoride lithium salt was added so that the film
forming solution was prepared.
20
CA 02331602 2001-10-25
-133-
Example 2-6
A battery similar to that according to Example 2-1
was manufactured.
The preparation of materials and assembling of the
S battery were performed in an atmosphere of dry argon
similarly to Example 2-1.
Felt-form carbon, which had been dehydrated and
dried, was immersed in lithium dissolved in a stainless
container, followed by cooling the same so that felt-
form carbon was manufactured. Then, it was immersed in a
toluene solution of poly (2-vinyl naphthalene) to which
azobisisobutylonitryl was added, followed by raising the
felt carbon. It was then dried at 80°C, irradiated with
ultraviolet rays to be crosslinked so that the negative
electrode 201 was manufactured. Then, similar processes
to those according to Example 2-1 were performed so that
the lithium secondary battery was manufactured.
Evaluation of Performance of Lithium Secondar Batter
The performance of lithium secondary batteries
according to Examples 2-1 to 2-5 and Comparative Example
2-1 was evaluated. The evaluation was performed by a
charge and discharge cycle test under the following
conditions with respect to the cycle life of the
batteries according to Comparative Example 2-1. The
conditions for the cycle test were made as follows: the
charge and discharge was performed by 0.2C (electric
current which was 0.2 times capacity/time), pause for 30
minutes and a cut-off voltage of 1.0V was applied. A
charging/discharging apparatus HJ-101M6 manufactured by
CA 02331602 2001-10-25
134 -
Hokuto Electric was used. The charge/discharge test was
commenced at discharge, the battery capacity was
evaluated by the quantity of the third discharge and the
cycle life was evaluated by the number of cycles when
the battery capacity had deteriorated to 600 or less.
The cycle life of each battery with respect to the
cycle life of the battery according to Comparative
Example 1-1 which was made to be 1 was as shown in Table
2.
As can be understood from the results of
comparisons made between Examples 2-1 to 2-5 and
Comparative Example 2-1, the cycle life can considerably
be lengthened due to use of the negative electrode
comprising the lithium foil covered with the polymer of
the derivative of the aromatic hydrocarbon compound
according to the present invention.
Table 2
Cycle Life Cycle Life of
Manufactured Lithium Battery of Comparative
Example Example
Comparative Example 2-1 1.0
Example 2-1 3.0
Example 2-2 1,5
Example 2-3 2.0
Example 2-4 3.0
Example 2-5 3.5
Example 2-6 3,7
CA 02331602 2001-10-25
- 135 -
Example 3-1
A flat battery which had a simple structure, which
could be assembled simply and which had a cross
sectional shape schematically shown in Fig. 2 was
assembled to evaluate the characteristics of the
secondary battery while evaluating the cycle life.
First, a titanium mesh collector 200 was pressed
from the reverse side of the lithium metal foil in an
atmosphere of dry argon gas. Then, the lithium metal
foil was immersed in a dioxane solution of
tetrabutoxytitanium, followed by drying it so that the
lithium electrode 201 having a film formed as described
above was manufactured.
The positive electrode active material 203 was
prepared in such a manner that a mixture of a dehydrated
and electrolyzed manganese deoxide and a lithium
carbonate were heated so that a lithium-manganese
composite oxide was prepared. Then, tetrafluoroethylene
polymer powder was mixed, and then it was pressed
against a titanium mesh as to be formed as designed.
The electrolyte solution was prepared in such a
manner that arsenate hexafluoride lithium salt was, by 1
M (mol/1) was dissolved in an equal quantity mixture
solvent of propylene carbonate (PC) and dimethoxyethane
( DME ) .
The separator 208 was manufactured in such a manner
that a propylene separator having small apertures was
sandwiched by unwoven polypropylene sheets.
CA 02331602 2001-10-25
- 136 -
Comparative Example 3-1
A battery similar to that according to Example 3-1
was manufactured except that the metal lithium foil
surface treatment was omitted.
Comparative Example 3-2
A battery similar to that according to Example 3-1
was manufactured.
The preparation of materials and assembling of the
battery were performed in an atmosphere of dry argon
similarly to Example 3-1. A solution in which peroxide
benzoyl was added to a hexane solution of Tirano Coat
(polytitanocarbosilane) manufactured by Ube Kosan was
applied to a lithium metal foil to which a titan mesh
collector was pressed, followed by drying it. Then, it
was heated to 80°C, and then heated to 150°C so that a
lithium electrode having a Tirano Coat film formed
thereon was manufactured.
Then, similar processes to those according to
Example 3-1 were performed so that the lithium secondary
battery was manufactured.
Example 3-3
A battery similar to that according to Example 3-1
was manufactured.
The preparation of materials and assembling of the
battery were performed in an atmosphere of dry argon
similarly to Example 3-1.
First, 2, 4, 6-trimethyl-tris (3, 3, 3-
trifluoropropyl)cyclotrisiloxane was heated to 76°C in
an argon gas. Then, calcium silate catalyzer was added
CA 02331602 2001-10-25
- 137 -
and polymerization was performed so that a polymer was
obtained. Then, peroxide benzoyl was added to the
polymer, and then a lithium metal foil to which the
stainless steel mesh collector was pressed was immersed
S in the solution, and rayed from the solution. Then, heat
treatment at 100°C was performed so that a lithium
electrode having a polysiloxane film formed thereon was
manufactured.
Then, similar processes to those according to
Example 3-1 were performed so that the lithium secondary
battery was manufactured.
Example 3-4
A battery similar to that according to Example 3-1
was manufactured.
The preparation of materials and assembling of the
battery were performed in an atmosphere of dry argon
similarly to Example 3-1.
First, azobisiosbutylonitryl was added to
tetramethyldivinyldisiloxane, and then a lithium metal
foil to which a stainless steel mesh collector was
pressed was immersed, followed by raising it. Then,
ultraviolet rays were applied so that a lithium
electrode covered with the polysiloxane film was
manufactured.
Then, similar processes to those according to
Example 3-1 were performed so that the lithium secondary
battery was manufactured.
Example 3-5
A battery similar to that according to Example 3-1
CA 02331602 2001-10-25
138 -
was. manufactured.
The preparation of materials and assembling of the
battery were performed in an atmosphere of dry argon
similarly to Example 3-1.
A lithium secondary battery was manufactured
similarly to Example 3-2 except that lithium was
immersed in a solution in which arsenate hexafluoride
lithium salt was added to a solution in which peroxide
benzoyl was added to a hexane solution of Tirano Coat
(polytitanocarbosilane) manufactured by Ube Kosan.
Evaluation of Performance of Lithium Secondar Batter
The performance of lithium secondary batteries
according to Examples 3-1 to 3-S and Comparative Example
3-1 was evaluated. The evaluation was performed by a
charge and discharge cycle test under the following
conditions with respect to the cycle life of the
batteries according to Comparative Example 2-1. The
conditions for the cycle test were made as follows: the
charge and discharge was performed by 0.2C (electric
current which was 0.2 times capacity/time), pause for 30
minutes and a cut-off voltage of 1.0V was applied. A
charging/discharging apparatus HJ-1O1M6 manufactured by
Hokuto Electric was used. The charge/discharge test was
commenced at discharge, the battery capacity was
evaluated by the quantity of the third discharge and the
cycle life was evaluated by the number of cycles when
the battery capacity had deteriorated to 600 or less.
The cycle life of each battery with respect to the
cycle life of the battery according to Comparative
CA 02331602 2001-10-25
- 139 -
Example 1-1 which was made to be 1 was as shown in Table
3.
As can be understood from the results of
comparisons made between Examples 3-1 to 3-5 and
Comparative Example 3-1, the cycle life can considerably
be lengthened due to use of the negative electrode
comprising the lithium foil covered with the organic
metal compound according to the present invention.
Table 3
Cycle Life Cycle Life of
Manufactured Lithium Battery of Comparative
Example Example
Comparative Example 3-1 1.0
Example 3-1 3.0
Example 3-2 2.5
Example 3-3 1.5
Example 3-4 2.0
Example 3-5 3.0
Example 4-1
A flat battery which had a simple structure, which
could be assembled simply and which had a cross
sectional shape schematically shown in Fig. 2 was
assembled to evaluate the characteristics of the
secondary battery while evaluating the cycle life.
First, a titanium mesh collector 200 was pressed
against the lithium metal foil from the reverse side in
an atmosphere of dry argon. Then, the lithium metal foil
CA 02331602 2001-10-25
- 140 -
was immersed in a teflon AF solution which was a
copolymer of tetrafluoroethylene and 2, 2-
bistrifluoromethyl - 4, 5 - difluoro-1, 3 - dioxysol and
which was manufactured by Dupont, followed by bring the
lithium metal foil. As a result, the lithium negative
electrode 201 covered with amorphous fluororesin was
manufactured. The positive electrode active material 203
comprised a lithium-manganese composite oxide prepared
by heating a mixture of dehydrated and electrolyzed
manganese dioxide and lithium carbonate and a mixture of
graphite. Then, tetrafluoroethylene polymer powder was
mixed, and pressed against the titanium mesh as to be
formed as designed. The electrolyte solution was
prepared by dissolving, by 1 M (mol/1), arsenate
hexafluoride lithium salt in an equal quantity mixture
solvent of propylene carbonate (PC) and dimethoxyethane
(DME). The separator 208 was manufactured in such a
manner that a propylene separator having small apertures
was sandwiched by unwoven polypropylene sheets. The
battery was assembled in such a manner that the
separator 208 was held between the negative electrode
201 and the positive electrode 203, followed by
inserting them into a positive electrode case 207 made
of titanium clad stainless steel, followed by injecting
the electrolyte solution. Then, the negative electrode
cap 206 made of the titanium clad stainless steel and
the insulating packing 210 made of fluorine rubber were
used for sealing so that the lithium secondary battery
was manufactured.
CA 02331602 2001-10-25
141 -
Comparative Example 4-1
A lithium secondary battery was manufactured under
the same conditions as those according to Example
4-1 except that the metal lithium foil surface covering
was omitted.
Example 4-2
A battery similar to that according to Example 4-1
was manufactured.
The preparation of materials and assembling of the
battery were performed in an atmosphere of dry argon
similarly to Example 4-1. The lithium metal foil to
which a titanium mesh collector was pressed was inserted
into a chamber of a sputtering apparatus, and then
retained gas was exhausted to a vacuum level of 5 x 10-6
Torr. Then, argon gas was allowed to flow to control the
internal pressure to 3 x 10-3 Torn followed by performing
RF discharge to generate plasma for use in sputtering
using Neoflon PFA which was a copolymer of
tetrafluoroethylene and perfluoroalkylvinyl ether and
manufactured by Daikin as a target. As a result, a
lithium electrode having a film formed thereon was
manufactured. Then, similar processes to those according
to Example 4-1 were performed so that the lithium
secondary battery was manufactured.
Example 4-3
A battery similar to that according to Example 2-1
was manufactured.
The preparation of materials and assembling of the
battery were performed in an atmosphere of dry argon
CA 02331602 2001-10-25
- 142 -
similarly to Example 4-1. The lithium metal foil to
which a stainless steel mesh collector was pressed was
inserted into a chamber of an RF (Radio Frequency)
plasma CVD apparatus, and then the retained gas was
exhausted to realize a vacuum level of 2 x 10-6 Torr.
Then, vinylidene fluoride was introduced into the
chamber, and isobutyl vinyl ether was introduced into
the chamber while using hydrogen gas as the carrier gas.
While controlling the internal pressure to 1 Torr, RF
discharge was caused to take place so that a plasma
polymer film was formed on the surface of lithium. Then,
similar processes to those according to Example 4-1 were
performed so that the lithium secondary battery was
manufactured.
Example 4-4
A battery similar to that according to Example 4-1
was manufactured.
The preparation of materials and assembling of the
battery were performed in an atmosphere of dry argon
similarly to Example 4-1. A lithium secondary battery
was manufactured similarly to Example 4-1 except that
the titanium mesh collector was pressed from the reverse
side, and then it was immersed in a xylene solution of
Lumiflon which was a copolymer of tetrafluoroethylene
and non-fluorine-type vinyl ether, which was
manufactured by Asahi Glass and to which isocyanate and
graphite powder were added. Then, it was dried at 140°C
for 10 minutes so that the lithium electrode covered
with the amorphous fluororesin was manufactured.
CA 02331602 2001-10-25
- 143 -
Example 4-5
A battery similar to that according to Example 4-1
was manufactured.
The preparation of materials and assembling of the
battery were performed in an atmosphere of dry argon
similarly to Example 4-1. First, coating fluororesin was
prepared. An automatic crepe having a stirring machine
received 200 parts of pure water, 40 parts of vinyl
fluoride, 60 parts of ethylvinyl ether, 0.6 parts of
perofluorocarboxylic acid, 0.2 parts of persulfuric
ammonium and 3 parts of water. Then, the solution was
heated to 46°C while maintaining the pressure at 42.5
atmosphere to perform polymerization for 8 hours. An
obtained polymer was cleaned with hot methanol, followed
by drying it. Then, it was dissolved in tetrahydrofuran
dehydrated sufficiently, followed by adding peroxide
benzoyl. Then, the lithium metal foil,to which a
stainless steel mesh collector was pressed was immersed
followed by raising it. Then, it was heated to 100°C so
that a lithium electrode covered with fluororesin was
manufactured.
Then, similar processes to those according to
Example 4-1 were performed so that the lithium secondary
battery was manufactured.
Example 4-6
A battery similar to that according to Example 2-1
was manufactured.
First, a lithium electrode was manufactured. The
lithium metal foil to which a stainless steel mesh
CA 02331602 2001-10-25
- I 44 -
collector was pressed was injected into a vacuum chamber
of a parallel and flat type plasma CVD apparatus to
which a 13.56 MHz high frequency power source was
connected in such a manner that it was placed perpen-
dicular to the parallel and flat electrode. Then,
retained gas was exhausted to realize a vacuum level of
10-5 Torr. Then, 10 sccm of tetrafluoroethylene, 2 sccm
of ethylene, 2 sccm of hydrogen, 1 sccm of helium and 1
sccm of oxygen were introduced into the vacuum chamber,
which was a reaction chamber, and then the internal
pressure was maintained at 0.8 Torr. Then, 200 watts of
high frequency power was supplied to the parallel and
flat electrode so that a plasma polymer film of the
fluororesin was formed on the surface of lithium. By
using the lithium electrode covered with the fluororesin
prepared by the foregoing method so that a lithium
secondary battery was manufactured by the similar
processes to those according to Example 4-1.
Evaluation of Performance of Lithium Secondar Batter
The performance of lithium secondary batteries
according to Examples 4-1 to 4-6 and Comparative Example
4-1 was evaluated. The evaluation was performed by a
charge and discharge cycle test under the following
conditions with respect to the cycle life of the
batteries according to Comparative Example 4-1. The
conditions for the cycle test were made as follows: the
charge and discharge was performed by 0.2C (electric
current which was 0.2 times capacity/time), pause for 30
minutes and a cut-off voltage of 1.0V was applied. A
CA 02331602 2001-10-25
145 -
charging/discharging apparatus HJ-1O1M6 manufactured by
Hokuto Electric was used. The charge/discharge test was
commenced at discharge, the battery capacity was
evaluate the quantity of the third discharge and the
S cycle life was evaluate by the number of cycles when the
battery capacity had deteriorated to 60% or less.
The cycle life of each battery with respect to the
cycle life of the battery according to Comparative
Example 4-1 which was made to be 1 was as shown in Table
4 .
As can be understood from the results of
comparisons made between Examples 4-1 to 4-6 and
Comparative Example 4-1, the cycle life can considerably
be lengthened due to use.of the negative electrode
comprising the lithium foil covered with the fluororesin
having the ether bond according to the present
invention.
CA 02331602 2001-10-25
- 146 -
Table 4
Cycle Life Cycle Life of
Manufactured Lithium Batter
y of Comparative
Example Example 4-1
Comparative Example 4-1 1.0
Example 4-1 3.5
Example 4-2 1.5
Example 4-3 2.0
Example 4-4 3.0
Example 4-5 2.5
Example 4-6 1.5
Example 5-1
A flat battery which had a simple structure, which
could be assembled simply and which had a cross
sectional shape schematically shown in Fig. 2 was
assembled to evaluate the characteristics of the
secondary battery while evaluating the cycle life.
In an atmosphere of dried argon gas, the titanium
mesh collecting electrode 200 was pressed to the reverse
side of the lithium metal foil. Then, the lithium metal
foil was immersed in a tetrahydrofuran solution of
Cryptofix 222 B Polymer which was a large ring compound
polymer and which was manufactured by E. Merch. Then, it
was dried, and then heated to 150°C so that a lithium
electrode 201 covered with the large ring compound
polymer was manufactured.
The positive electrode active material comprised a
CA 02331602 2001-10-25
- 147
lithium-manganese composite oxide prepared by heat
treatment applied to a mixture of dehydrated and
electrolyzed manganese dioxide and lithium carbonate and
a mixture of graphite. Then, tetrafluoroethylene polymer
was mixed, and then pressed against the stainless steel
mesh as to be formed as designed.
The separator 208 was manufactured in such a manner
that a propylene separator having small apertures was
sandwiched by unwoven polypropylene sheets.
The electrolyte solution was prepared by
dissolving, by 1 M (mol/1), arsenate hexafluoride
lithium salt in an equal quantity mixture solvent of
propylene carbonate (PC) and dimethoxyethane (DME).
Assembling of the battery were performed similarly to
Example 1-1.
Comparative Example 5-1
A lithium secondary battery was manufactured
similarly to Example 5-1 except that the metal lithium
foil surface covering was omitted.
Example 5-2
A battery similar to that according to Example 5-1
was manufactured.
The preparation of materials and assembling of the
battery were performed in an atmosphere of dry argon
similarly to Example 2-1. A lithium metal foil, to which
a titanium mesh collector was pressed, was placed in
acetonyl solution in which 0.1 M of a monomer of benzo-
15-crown-5 and 0.2 M of boron tetrafluoride
tetrabutylammonium salt as to be electrolyzed and
CA 02331602 2001-10-25
- 148 -
polymerized with a voltage level of 3 V while using a
platinum electrode as the cathode electrode. As a
result, a lithium electrode having a large ring compound
polymer film formed thereon was manufactured.
Then, similar processes to those according to
Example 5-1 were performed so that the lithium secondary
battery was manufactured.
Example 5-3
A battery similar to that according to Example 5-1
was manufactured.
The preparation of materials and assembling of the
battery were performed in an atmosphere of dry argon
similarly to Example 5-1. First, (+) - 18 - crown - 6 -
tetracarboxylic acid (0.4 mol), 1, 4 - butanediol (0.8
mol), tetratitanate - n - butylester (0.08 g) and
butylhydroxytinoxide (0.03 g) were mixed. Then, the
temperature was maintained at 220°C, and distillation
was performed for 60 minutes to remove products such as
water. Then, the product and titanate tetra-n-butylester
(0.02 g) were injected into an automatic crepe, and the
pressure was lowered and heating to 250°C was performed
for 22 hours so that a polymer was obtained.
Trylenediisocyanate was added to the thus obtained
polymer, and then the lithium metal foil, to which a
titanium mesh collector was pressed, was immersed. Then,
the lithium metal foil was raised, and then it was
heated, and dried at 80°C so that the surface of the
lithium foil was covered with a large ring compound
polymer.
CA 02331602 2001-10-25
- 149 -
Then, similar processes to those according to
Example 5-1 were performed so that the lithium secondary
battery was manufactured.
Example 5-4
A battery similar to that according to Example 2-1
was manufactured.
The preparation of materials and assembling of the
battery were performed in an atmosphere of dry argon
similarly to Example 5-1. A lithium metal foil, to which
a titanium steel mesh collector was pressed, was
immersed in a toluene solution of 3, 3'- dibenzyl - 1,
4, 8, 11 - tetraoxacyclotetradecan to which
azobisisobutylnitryl and arsenate hexafluoride lithium
salt were added. Then, the lithium metal was raised from
the solution, and then ultraviolet rays were applied to
crosslinking so that the surface of the lithium metal
foil was covered.
Then, similar processes to those according
to Example 5-1 were performed so that the lithium
secondary battery was manufactured.
Example 5-5
A battery similar to that according to Example 2-1
was manufactured.
The preparation of materials and assembling of the
battery were performed in an atmosphere of dry argon
similarly to Example 5-1. Styrene and 4, 7, 13, 16, 21,
24 - hexaoxa - 1, 10-diazobicyclo [8.8.8] hexakosan were
dissolved in a tetrahydrofuran solution of naphthalene
degasified and dehydrated sufficiently. Then, graphite
CA 02331602 2001-10-25
- 150 -
powder was mixed. The lithium metal foil, to which a
titanium mesh collector was pressed, was immersed to
perform polymerization reaction, followed by drying as
to be hardened. As a result, the surface of lithium was
covered with a large ring compound polymer.
Then, similar processes to those according to
Example 5-1 were performed so that the lithium secondary
battery was manufactured.
Example 5-6
A battery similar to that according to Example 5-1
was manufactured.
The preparation of materials and assembling of the
battery were performed in an atmosphere of dry air.
A porous nickel sheet manufactured by Osaka Gas was
dehydrated and dried as to be used as an anode,
while a platinum electrode was used as a cathode so that
electrolytic polymerization was performed in an
acetonitryl solution in which 0.1 M of benzo-18-crown-6
and 0.2 M or boron tetrafluoride tetrabutylammonia salt
were dissolved. As a result, a porous nickel sheet
covered with a crown ether polymer was manufactured.
Then, an electrolyte solution was prepared in such a
manner that 1M of boron tetrafluoride lithium salt was
dissolved in an equal-quantity mixture solvent of
propylene carbonate and dimethoxyethane. An obtained
porous nickel sheet thus covered was used as a cathode
and lithium metal was used as an anode. Then, lithium
was inserted between the porous nickel sheet cover and
the nickel as to be allowed to adhere so that the
CA 02331602 2001-10-25
-151-
negative electrode 203 was formed.
Then, similar processes to those according to
Example 5-1 were performed so that the lithium secondary
battery was manufactured.
Example 5-7
A nickel-zinc battery having a similar structure as
that of the battery according to Example 5-1 was
manufactured.
First, paste was prepared by mixing polyvinyl
alcohol, phthalocyanine zinc, zinc oxide powder, zinc
powder, formaldehyde, formic acid and water. The paste
was applied to a nickel mesh and heated so that the
negative electrode 201 was manufactured.
Then, paste obtained by mixing nickel powder,
cobalt powder, carboxylic cellulose serving as a bonding
material, ethylene glycol and water with nickel
hydroxide was injected into foamed nickel (Celmet)
manufactured by Sumitomo Denko. Then, it was dried and
pressed so that a positive electrode 203 was formed.
The separator 208 was formed in such a manner that
a polyamide film subjected to hydrophilic treatment and
having small apertures was sandwiched by unwoven
polyamide sheets. The electrolyte solution was a 30 wto
potassium hydroxide solution. The assembly was performed
similarly to Example 5-1 so that a nickel-zinc secondary
battery was manufactured.
Comparative Example 5-2
A nickel-zinc secondary battery was manufactured
except that the negative electrode 201 was formed by
CA 02331602 2001-10-25
- 152
applying paste obtained by mixing zinc oxide powder,
zinc powder, polyvinyl alcohol and ethylene glycol to
the nickel mesh followed by drying.
Example 5-8
A battery having a structure similar to that
according to Example 5-1 was manufactured. However, a
positive electrode case having small apertures for
receiving oxygen in air was used.
The negative electrode 201 was manufactured in such
a manner that paste was applied to a nickel mesh
followed by heating to dry the negative electrode 201,
the paste being obtained by mixing zinc oxide powder,
zinc powder, polyacrylamide, water, formaldehyde,
lithium hydroxide and phthalocyanine zinc.
The positive electrode 203 was formed in such a
manner that a positive catalyzer layer was formed by
adding manganese dioxide to active carbon, and a water
repellant film made of polytetrafluoroethylene and
cellulose dispersed paper were stacked on the positive
catalyzer layer.
The separator 209 comprised a cellophane film and
the electrolyte solution comprised 30 wto solution of
potassium hydroxide.
Assembling was performed in such a manner that the
separator 208 was held between the negative electrode
201 and the positive electrode 203, and they were
inserted into the positive electrode case 207 having
small apertures for receiving oxygen in air and made of
titanium clad stainless steel. Then, the electrolyte
CA 02331602 2001-10-25
- 153 -
solution was injected and sealing was performed by using
the negative electrode cap 206 made of titanium clad
stainless steel and the insulating packing made of
fluorine rubber. As a result, an air-zinc secondary
battery was manufactured.
Comparative Example 5-3
An air-zinc secondary battery was manufactured
similarly to Example 5-8 except that the negative
electrode according to Comparative Example 5-2 was used.
Evaluation of Performance of Lithium Secondary Battery
The performance of lithium secondary batteries
according to Examples 5-1 to 5-5 and Comparative Example
5-1 was evaluated. The evaluation was performed by a
charge and discharge cycle test under the following
conditions with respect to the cycle life of the
batteries according to Comparative Example 1. The
conditions for the cycle test were made as follows: the
charge and discharge was performed by 0.2C (electric
current which was 0.2 times capacity/time), pause for 30
minutes and a cut-off voltage of 1.0V was applied. A
charging/discharging apparatus HJ-1O1M6 manufactured by
Hokuto Electric was used. The charge/discharge test was
commenced at discharge, the battery capacity was
evaluate the quantity of the third discharge and the
cycle life was evaluate by the number of cycles when the
battery capacity had deteriorated to 60% or less.
The cycle life of each battery with respect to the
cycle life of the battery according to Comparative
Example 5-1 which was made to be 1 was as shown in Table
CA 02331602 2001-10-25
- 154
2.
As can be understood from the results of
comparisons made between Examples 5-1 to 5-6 and
Comparative Example 2-1 and those between Example 5-7
and Comparative Example 5-3, the cycle life can
considerably be lengthened due to use of the negative
electrode comprising the lithium or zinc covered with
the large ring compound polymer according to the present
invention.
Table 5
Cycle Life Cycle Life .
Lithium Battery of of
Manufactured
Example Comparative
Comparative Example
Example
Exam 1e
Example 5-1 Comparative
3,0
Example 5-1
Example 5-2 Comparative
2.5
Example 5-1
Example 5-3 Comparative
3.5
Example 5-1
Example 5-4 Comparative
4.0
Example 5-1
Example 5-5 Comparative
3.0
Example 5-1
Example 5-6 Comparative
4.2
Example 5-1
Example 5-7 Comparative
2.8
Example 5-2
Example 5-8 Comparative
2,~
Example 5-2
CA 02331602 2001-10-25
- 155 -
Example 6-1
A flat battery which had a simple structure, which
could be assembled simply and which had a cross
sectional shape schematically shown in Fig. 2 was
assembled to evaluate the characteristics of the
secondary battery while evaluating the cycle life.
First, the titanium collector 200 was pressed against
the reverse side of the lithium metal foil in an
atmosphere of dry argon gas, and then the lithium metal
foil was immersed in a toluene solution PPZ-U1001
manufactured by Idemitsu. Then, it was previously dried,
and ultraviolet rays were applied so that the lithium
electrode 201 covered with phosphazene was manufactured.
The positive electrode active material 204 was
prepared in such a manner that a lithium-manganese
composite oxide was prepared by heating a mixture of
dehydrated and electrolyzed manganese dioxide and
lithium carbonate. Then, tetrafluoroethylene polymer
powder was mixed followed by pressing to a titanium mesh
as to be formed as designed.
The electrolyte solution was prepared by
dissolving, by 1 M (mol/1), arsenate hexafluoride
lithium salt in an equal quantity mixture solvent of
propylene carbonate (PC) and dimethoxyethane (DME). The
separator 208 was manufactured in such a manner that a
propylene separator having small apertures was
sandwiched by unwoven polypropylene sheets.
Comparative Example 6-1
A lithium secondary battery was manufactured
CA 02331602 2001-10-25
- 156 -
similarly to Example 6-1 except that the surface
covering of the metal lithium foil was omitted.
Comparative Example 6-2
A battery similar to that according to Example 6-1
was manufactured. The preparation of materials and
assembling of the battery were performed in an
atmosphere of dry argon similarly to Example 6-1. First,
hexachlorotriphosphazene was repeatedly degasified,
melted and solidified, and then it was heated to 250°C
as to be polymerized so that polydichlorophosphazene was
obtained. Then, a tetrahydrofuran solution of aniline
was added to a benzene solution of
polychlorophosphazene, followed by heating and refluxed.
Then, the solution was allowed to stand to remove
aniline hydrochloride by filtering followed by again
sedimented it and drying it. Then, it was again
dissolved in tetrahydrofuran so that a solution of poly
[bis (phenylamino)phosphazene] was prepared. Then, the
foregoing solution was applied to the lithium metal
foil, to which a titanium mesh collector was pressed,
followed by drying the solution. Then, ultraviolet rays
were applied so that a lithium electrode covered with
polyphosphazene was manufactured. Then, similar
processes to those according to Example 6-1 were
performed so that the lithium secondary battery was
manufactured.
Example 6-3
A battery similar to that according to Example 5-1
was manufactured. The preparation of materials
CA 02331602 2001-10-25
- 157 -
and assembling of the battery were performed in an
atmosphere of dry argon similarly to Example 6-1. First,
a benzene solution of polydichlorophosphazene was, while
being stirred, slowly dropped in a tetrahydrofuran
solution of sodium alkoxide of trifluoroethanol,
followed by heating and refluxing it. Then, it was
neutralized, filtered, cleaned with water and ethyl
alcohol, and again sedimented with acetone and water,
followed by drying. As a result, polybis
(trifluoroethoxy) phosphazene was obtained. The acetone
solution of the polyfluorokoxyphosphazene was applied to
the lithium metal foil, to which a stainless steel mesh
collector was pressed. Then, it was dried, and then
irradiated with electron beams so that a lithium
electrode covered with polyphosphazene was manufactured.
Then, similar processes to those according to Example 6-
1 were performed so that the lithium secondary battery
was manufactured.
Example 6-4
A battery similar to that according to Example 5-1
was manufactured. The preparation of materials and
assembling of the battery were performed in an
atmosphere of dry argon similarly to Example 6-1. First,
sodium alkoxide of naphthalene ethanol was, in dioxane,
prepared from naphthalene ethanol and sodium hydroxide.
Then, tetra-n-butyl ammonia bromide was added to the
sodium alkoxide, and then a dioxane solution of
polydichlorophosphazene was added while being dripped.
The thus-obtained mixed was subjected to a heating
CA 02331602 2001-10-25
- 158
reactions at 80°C, and the solvent was somewhat removed.
Then, it was again sedimented with tetrahydrofuran and
water, and then it was refined, followed by Soxhlet-
extracting it. As a result, polydiphenoxyphosphazene was
obtained. Then, azoisobutylnitryl was added to a
tetrahydrofuran solution of the obtained polymer. Then,
the lithium metal foil, to which a stainless steel mesh
collector was pressed, was immersed and raised from it
followed by drying it and subjecting to a heat treatment
set to 80°C. As a result, a lithium electrode covered
with polyphosphazene was manufactured. Then, similar
processes to those according to Example 6-1 were
performed so that the lithium secondary battery was
manufactured.
Example 6-5
A battery similar to that according to Example 5-1
was manufactured. The preparation of materials and
assembling of the battery were performed in an
atmosphere of dry argon similarly to Example 6-1. A
lithium secondary battery was manufactured similarly to
Example 6-1 except that arsenate hexafluoride lithium
salt was added to a toluene solution of PPZ-U1001
manufactured by Idemitsu, and then lithium was immersed
in the thus-prepared solution.
Evaluation of Performance of Lithium Secondary Battery
The performance of lithium secondary batteries
according to Examples 6-1 to 6-5 and Comparative Example
6-1 was evaluated. The evaluation was performed by a
charge and discharge cycle test under the following
CA 02331602 2001-10-25
- 159 -
conditions with respect to the cycle life of the
batteries according to Comparative Example 6-1. The
conditions for the cycle test were made as follows: the
charge and discharge was performed by 0.2C (electric
current which was 0.2 times capacity/time), pause for 30
minutes and a cut-off voltage of 1.0V was applied. A
charging/discharging apparatus HJ-lOlM6 manufactured by
Hokuto Electric was used. The charge/discharge test was
commenced at discharge, the battery capacity was
evaluate the quantity of the third discharge and the
cycle life was evaluate by the number of cycles when the
battery capacity had deteriorated to 60% or less. The
cycle life of each battery with respect to the cycle
life of the battery according to Comparative Example 6-1
which was made to be 1 was as shown in Table 6. As can
be understood from the results of comparisons made
between Examples 6-1 to 6-5 and Comparative Example 6-1,
the cycle life can considerably be lengthened due to use
of the negative electrode comprising the lithium or zinc
covered with the large ring compound polymer according
to the present invention.
CA 02331602 2001-10-25
- 160
. Table 6
Cycle Life Cycle Life:of
Manufactured Lithium Battery of Comparative
Example Example 2-1
Comparative Example 6-1 1.0
Example 6-1 2.0
Example 6-2 1.5
Example 6-3 2.0
Example 6-4 3.0
Example 6-5 2,5
Example 7-1
A flat battery which had a simple structure, which
could be assembled simply and which had a cross
sectional shape schematically shown in Fig. 2 was
assembled. The preparation of materials and assembling
of the battery were performed in an atmosphere of dry
argon.
First, the titanium mesh collector 200 having a
lead was pressed against the reverse side of the lithium
metal foil in an atmosphere of dry argon. Then, it was
immersed in a nitromethane solution of acetyl cellulose
to which azobisisobutylotryl and boron tetrafluoride
lithium salt. Then, it was dried, and irradiated with
ultraviolet rays so that a film was formed. As a result,
the lithium negative electrode 201 was manufactured
The positive electrode active material was prepared
in such a manner that a lithium-manganese oxide was
CA 02331602 2001-10-25
-161-
prepared by mixing electrolyzed manganese dioxide and
lithium carbonate at a ratio of 1:0.4 and by heating the
mixture at 800°C. Then, ketchen black and Super Konak,
which is a fluororesin paint manufactured by Nihon Yushi
were mixed to the prepared lithium-manganese oxide.
Then, it was pressed against a nickel mesh as to be
formed as designed followed by performing heat treatment
set to 170°C. As a result, the positive electrode 203
was manufactured.
The separator 208 was manufactured in such a manner
that a porous layer-shape alumina film, an unwoven
polypropylene sheet and polypropylene separator having
small apertures were sandwiched.
The electrolyte solution was prepared by
dissolving, by 1 M (mol/1), tetrafluoride borate lithium
salt in an equal quantity mixture solvent of propylene
carbonate (PC) and dimethoxyethane (DME).
The assembly was performed in such a manner that
the negative electrode 201, the positive electrode 203
and the separator 208 were sandwiched as to be inserted
into the positive electrode case 207 made of titanium
clad stainless steel, and then the electrolyte solution
was injected. Then, sealing was performed by the
negative cap 206 made of the titanium clad stainless
steel and the insulating packing 210 made of fluorine
rubber so that a lithium secondary battery was
manufactured.
Example 7-2
A battery similar to that according to Example 7-1
CA 02331602 2001-10-25
- 162 -
and shown in Fig. 2 was manufactured.
The preparation of materials and assembling of the
battery were performed in an atmosphere of dry argon
similarly to Example 7-1.
S A lithium secondary battery was manufactured by a
method similar to that according to Example 7-1.
First, the titanium mesh collector 200 having a
lead was pressed against the reverse side of the lithium
metal foil in an atmosphere of dry argon gas. Then, it
was immersed in a tetrahydrofuran solution of a
polyethylene oxide to which peroxide benzoyl was added,
and then it was dried at 110°C, and ultraviolet rays
were applied so that a film was formed. As a result, the
lithium negative electrode 201 was manufactured.
The positive electrode active material was prepared
in such a manner that a lithium-manganese oxide was
prepared by mixing electrolyzed manganese dioxide and
lithium carbonate at a ratio of 1:0.4 and by heating the
mixture at 800°C. Then, ketchen black and
tetrafluoroethylene polymer powder were mixed with the
prepared lithium-manganese oxide. Then, it was pressed
against a nickel mesh as to be formed as designed
followed by performing heat treatment set to 250°C. As a
result, the positive electrode 203 was manufactured.
The separator 208 was manufactured by sandwiching a
unwoven polypropylene sheet and a polypropylene
separator having small apertures.
The electrolyte solution was prepared by
dissolving, by 1 M (mol/1), tetrafloride borate lithium
CA 02331602 2001-10-25
- 163 -
salt in a solvent of propylene carbonate (PC).
Then, similar processes to those according to
Example 7-1 were performed so that the lithium secondary
battery was manufactured.
Example 7-3
A battery similar to that according to Example 7-1
and shown in Fig. 2 was manufactured.
The preparation of materials and assembling of the
battery were performed in an atmosphere of dry argon
similarly to Example 7-1.
First, the lithium surface film was manufactured in
such a manner that lithium foil to which the nickel mesh
collector 200 was pressed was placed in a sputtering
apparatus. Then, a degree of vacuum of 1.5 x 10-4 pascal
by lowering the pressure and by degasifying the inside
of the sputtering apparatus. Then, argon gas was
introduced by 5 sccm, and the internal pressure in the
film forming chamber was maintained at 6 x 10-1 pascal to
perform sputtering while using polyether sulfon as a
target so that the lithium negative electrode 201
covered with chitosan having a thickness of 500 A was
obtained.
Then, similar processes to those according to
Example 7-2 were performed so that the lithium secondary
battery was manufactured.
Example 7-4
Under the same conditions as those according to
Example 7-3 except that lithium foil to which the nickel
mesh collector 200 was pressed was placed in a
CA 02331602 2001-10-25
- 164 -
sputtering apparatus. Then, a degree of vacuum of 1.5 x
10-q pascal by lowering the pressure and by degasifying
the inside of the sputtering apparatus. Then, argon gas
was introduced by 5 sccm, and the internal pressure in
the film forming chamber was maintained at 6 x 10-1
pascal to perform sputtering while using chitosan as a
target so that the lithium negative electrode 201
covered with chitosan having a thickness of 1000 A was
obtained.
Then, similar processes to those according to
Example 7-2 were performed so that the lithium secondary
battery was manufactured.
Example 7-5
A flat nickel-zinc secondary battery which had a
simple structure, which could be assembled simply and
which had a cross sectional shape schematically shown in
Fig. 2 was manufactured.
The negative electrode was manufactured in such a
manner that ethylene tetrafluoride polymer powder
serving as a bonding material was added to a mixture of
zinc powder and zinc oxide powder as to be pressed
against the two sides of a copper punching metal plate
so that the negative electrode was formed. The negative
electrode was immersed in an acetone-ethylalcohol
solution of acetyl cellulose to which
azobisisobutylonitryl was added, and then it was dried
and heated to 110°C. Then, ultraviolet rays were applied
so that a film was formed. As a result, the zinc
negative electrode 201 was manufactured.
CA 02331602 2001-10-25
- 165 -
The positive electrode 203 was manufactured in such
a manner that nickel hydroxide was impregnated in a
sintered nickel electrode plate and it was covered with
an acetylcellulose film similarly to the negative
electrode.
The separator 208 was manufactured in such a manner
that a hydrophilic unwoven nylon sheet and a nylon film
having small apertures were sandwiched.
The electrolyte solution comprised 30 wt% potassium
hydroxide water solution to which lithium hydroxide was
added.
The battery was assembled similarly to Example 7-1.
Example 7-6
A flat nickel-zinc secondary battery which
had a simple structure, which could be assembled simply
and which had a cross sectional shape schematically
shown in Fig. 2 was manufactured.
Under the same conditions as those according to
Example 7-5 except that the formed zinc negative
electrode was placed in a sputtering apparatus, and the
inside pressure was lowered to a degree of vacuum
of 1.5 x 10-9 Pascal by lowering and degasifying the
inside. Then, argon gas was introduced by 5 sccm, and
the internal pressure in the film forming chamber was
maintained at 6 x 10-1 Pascal. Then, sputtering was
performed while using collagen as a target so that a
zinc negative electrode covered with collagen having
a thickness of 1000 A was obtained.
The positive electrode 203 was manufactured by
CA 02331602 2001-10-25
- 166 -
impregnating nickel hydroxide into a sintered nickel
electrode plate.
Then, similar processes to those according to
Example 2-1 were performed so that the nickel-zinc
secondary battery was manufactured.
In order to evaluate the performance of the
batteries according to the foregoing Examples, batteries
according to Comparative Examples were manufactured.
Comparative Example 7-1
A lithium secondary battery was manufactured by a
method similar to Example 7-2 except that the surface of
the metal lithium foil was not covered.
Comparative Example 7-2
A nickel-zinc secondary battery was manufactured by
a method similar to Example 7-6 except that the surface
of the zinc negative electrode was not covered.
Evaluation of Performance of Lithium Secondary Batter~r
The performance of lithium secondary batteries and
nickel-zinc secondary batteries according to Examples
and Comparative Examples was evaluated. The evaluation
was performed by a charge and discharge cycle test under
the following conditions with respect to the cycle life
of the batteries according to Comparative Examples.
The conditions for the cycle test were made as
follows: the charge and discharge was performed by 0.2C
(electric current which was 0.2 times capacity/time),
pause for 30 minutes and a cut-off voltage of 1.0V was
applied. A charging/discharging apparatus HJ-1O1M6
manufactured by Hokuto Electric was used: The
CA 02331602 2001-10-25
- 167 -
charge/discharge test was commenced at discharge, the
battery capacity was evaluate the quantity of the third
discharge and the cycle life was evaluated by the number
of cycles when the battery capacity had deteriorated to
600 or less.
The cycle life of batteries according to Examples
of the present invention with respect to the cycle life
of the batteries according to Comparative Examples which
was made to be 1.0 was as shown in Table 7.
Table 7
Cycle Life Cycle Life
Lithium Battery of of
Manufactured
Example Comparative
Example Comparative
Example
Exam 1e
Example 7-1 Comparative
3.6
Example 7-1
Example 7-2 Comparative
1.7
Example 7-1
Example 7-3 Comparative
1.3
Example 7-1
Example 7-4 Comparative 1.6
Example 7-1
Example 7-5 Comparative 1.8
Example 7-2
Example 7-6 Comparative 1.4
Example 7-2
As can be understood from the results of
comparisons made between Examples 7-1 to 7-4 and
Comparative Example 7-1 and those between Example 7-5
and Comparative Example 7-2, the cycle life can
CA 02331602 2001-10-25
- 168 -
considerably be lengthened due to use of the secondary
battery according to the present invention.
Example 8
A battery which had a simple structure, which could
be assembled simply and which had a cross sectional
shape schematically shown in Fig. 2 was manufactured.
First, an RF (Radio Wave Frequency) discharge
plasma processing apparatus having a structure
schematically shown in Fig. 6 was used to apply surface
treatment to a lithium negative electrode. In an
atmosphere of dry argon gas, a nickel mesh collector was
pressed against the reverse side of the lithium metal
foil. In order to protect the surface of lithium, a
polyester film was interposed at the time of coiling
them. The coil-shape lithium foil 400 was mounted on a
conveyance roll 407 of the plasma processing apparatus,
and then the retained gas was exhausted to realize a
degree of vacuum of 2 x 10-6 Torr. Then, 20 sccm of
nitrogen gas and 10 sccm of argon gas were introduced to
a plasma processing chamber 405 through a gas
introduction pipe 406. While controlling the internal
pressure in the plasma processing chamber to 0.5 Torr,
waves having a high frequency of 13.56 MHz were supplied
by 200 watts to cause discharge to take place. Then, a
winding roll 408 was used to hold and take up the
protection film comprising the polyester film so that
the surface of lithium was plasma-treated. Then, the
lithium foil subjected to the plasma treatment was cut
so that a negative electrode was manufactured. The
CA 02331602 2001-10-25
- 169 -
protection film to be interposed at the time of the
winding operation in the plasma process, a separator for
a battery may be used.
The positive electrode active material 203 was
prepared in such a manner that a lithium-manganese oxide
was prepared by mixing electrolyzed manganese dioxide
and lithium carbonate at a ratio of 1:0.4 and by heating
the mixture at 850°C. Then, graphite and
tetrafluoroethylene polymer powder were mixed to the
prepared lithium-manganese oxide. Then, it was pressed
against a nickel mesh as to be formed into a desired
positive electrode.
The electrolyte solution was prepared by
dissolving, by 1 M (mol/1), tetrafloride borate lithium
salt in an equal quantity mixture solvent of propylene
carbonate (PC) and dimethoxyethane (DME).
The separator 208 was manufactured in such a manner
that a polypropylene separator having small apertures
was sandwiched by unwoven polypropylene sheets.
Example 9
A battery similar to that according to Example 8
and shown in Fig. 2 was manufactured.
The preparation of materials and assembling of the
battery were performed in an atmosphere of dry argon
similarly to Example 8.
A lithium secondary battery was manufactured by a
method similar to that according to Example 8.
First, a microwave discharge plasma processing
apparatus having a structure schematically shown in Fig.
CA 02331602 2001-10-25
- 170 -
7 was used to apply surface treatment to a lithium
negative electrode. In an atmosphere of dry argon gas, a
nickel mesh collector was pressed against the reverse
side of the lithium metal foil, and then disposed on a
sheet processing substrate holder 501 followed by
injecting them in a load chamber 508 of the microwave
discharge plasma processing apparatus. Then, the
retained gas in the load chamber 508 was exhausted to
realize a degree of vacuum of 1 x 10-6 Torr. Then, a gate
valve 509 was opened, and the substrate holder was
conveyed to the plasma processing chamber 502 and the
gate valve 509 was closed. Then, 10 sccm of carbon
tetrafluoride gas and 5 sccm of hydrogen gas were
introduced into the plasma processing chamber 502
through the gas introduction pipe 504. While controlling
the internal pressure in the plasma processing chamber
to 0.01 Torr, microwaves having a frequency of 2.45 GHz
were supplied by 100 watts through a wave guide pipe 506
and a microwave introduction window 505 to cause
discharge to take place. As a result, the surface of
lithium was plasma-processed. The lithium foil plasma-
processed was used as the negative electrode.
Then, similar processes to those according to
Example 8 were performed so that the lithium secondary
battery was manufactured.
Example 10
Under the same conditions as those according to
Example 9 except that 10 sccm of gas of nitrogen
trifluoride was, as the lithium surface treatment
CA 02331602 2001-10-25
-171-
gas, introduced through the gas introduction pipe 504 in
place of the 10 sccm of the carbon tetrafluoride gas and
the 5 sccm of the hydrogen gas. As a result, the surface
of lithium was processed similarly to Example 9. Then,
similar processes to those according to Example 8 were
performed so that the lithium secondary battery was
manufactured.
Example 11
Under the same conditions as those according to
Example 8 except that 5 sccm of chlorine trifluoride
gas, 2 sccm of oxygen gas and 100 sccm of helium gas
were, as the lithium surface treatment gas, introduced
through the gas introduction pipe 504 in place of the 5
sccm of chlorine trifluoride gas and 2 sccm of oxygen
gas. Then, the surface of lithium was processed while
omitting discharge.
Then, similar processes to those according to
Example 8 were performed so that the lithium secondary
battery was manufactured.
In order to compare and evaluate the performance of
the batteries according to the foregoing Examples, the
following comparative battery was manufactured.
Comparative Example 8
A lithium secondary battery was manufactured
similarly to Example 8 except that the metal lithium
foil, to which the nickel mesh was pressed, was, as it
is, used as the negative electrode.
Evaluation of Performance of Secondary Batterv
The performance of lithium secondary batteries
CA 02331602 2001-10-25
- 172 -
according to Examples and Comparative Example was
evaluated. The evaluation was performed by a charge and
discharge cycle test under the following conditions with
respect to the cycle life of the batteries according to
Comparative Example.
The conditions for the cycle test were made as
follows: the charge and discharge was performed by 0.2C
(electric current which was 0.2 times capacity/time),
pause for 30 minutes and a cut-off voltage of 1.0V was
applied. A charging/discharging apparatus HJ-1O1M6
manufactured by Hokuto Electric was used. The
charge/discharge test was commenced at discharge, the
battery capacity was evaluated the quantity of the third
discharge and the cycle life was evaluated by the number
of cycles when the battery capacity had deteriorated to
60% or less.
The cycle life of batteries according to Examples
of the present invention with respect to the cycle life
of the battery according to Comparative Example which
was made to be 1.0 was as shown in Table 8.
As can be understood from the results of
comparisons made between Examples 8 to 11 and
Comparative Example 8, the cycle life can considerably
be lengthened due to use of the secondary battery
according to the present invention.
CA 02331602 2001-10-25
-173-
Table 8
Cycle Life Cycle Life
of of
Lithium Battery Manufactured Example Comparative
Example 8
Comparative 1.0
Example
8
Example 8 2.5
Example 9 2.0
Example 10 3.0
Example 11 l . '7
Example 12
A liquid-rich test cell similar to the apparatus
shown in Fig. 1 was used to conduct tests.
The preparation of materials and assembly of the
battery were performed in an atmosphere of dry Ar.
Lithium metal foil, to which a titanium collector was
pressed, was inserted into the chamber of an RF plasma
CVD apparatus. Then, the retained gas was exhausted to
realize a degree of vacuum of 2 x 10-6 Torr. Then,
tetrafluoroethylene, ethylene, hydrogen, helium and
oxygen were introduced into the chamber, and the
internal pressure was maintained at 0.8 Torr. Then, high
frequency power of 200 watts was supplied to a parallel
and flat electrode so that a plasma polymer film of
fluororesin was formed on the foregoing sample to have a
thickness of 100 A. Then, acetylene gas was, as raw
material gas, introduced into the chamber, and the
pressure in the chamber was controlled to 0.1 Torr, and
CA 02331602 2001-10-25
- 174 -
then RF discharge was performed so that a carbon film
was formed on the surface of lithium to have a thickness
of 200 A so that a sample negative electrode was
manufactured (see Fig. 9A) .
The positive electrode active material was prepared
by heating a mixture of dehydrated and electrolyzed
manganese dioxide, lithium carbonate and black lead. The
lithium-manganese composite oxide was mixed with
tetrafluoroethylene polymer powder, and then it was
pressed against titanium mesh to be formed into a
desired positive electrode.
The separator was manufactured by sandwiching a
polypropylene separator having small apertures and
unwoven polypropylene sheets.
The electrolyte solution was prepared by dissolving
1 M of arsenate hexafluoride lithium salt in an equal-
quantity mixture solvent of propylene carbonate and
dimethoxyethane.
A lithium secondary battery was manufactured as
shown in Fig. 1.
Example 13
Under the same conditions as those according to
Example 12 except that lithium metal foil covered with a
plasma polymer film of fluorine resin was inserted
into the chamber of the RF plasma CVD apparatus. Then,
the retained gas was exhausted to realize a degree of
vacuum of 2 x 10-6 Torr. Monosilane gas was, as the raw
material gas, introduced into the chamber, and then the
pressure in the chamber was controlled to 0.1 Torr.
CA 02331602 2001-10-25
- 175 -
Then, RF discharge was performed so that an amorphous
silicon film was formed on the surface of the lithium
metal foil to have a thickness of 100 A as to be used as
a sample electrode. A battery similar to that according
to Example 12 was manufactured except that the foregoing
sample electrode was used as the negative electrode.
Example 14
Petroleum type pitch was spinned by a flow method,
and subjected to heat treatment in an atmosphere of
inactive gas so that black lead fiber having a specific
area of 10 m2/g was obtained. By heating the fiber to
completely remove water, and dispersed in toluene
dehydrated by molecular sieves as to be applied to the
lithium metal foil. Then, the material was dried and
pressed by a pressing machine so that a sample was
obtained (comprised a fiber layer having a thickness of
50 Vim). Then, a titanium mesh collector was pressed
against the reverse side of the sample so that a
negative electrode was obtained. A battery similar to
that according to Example 12 was manufactured except
that the foregoing negative electrode was used as the
negative electrode.
Exa~le 15
A solution in which Lumifron, which was fluororesin
paint manufactured by Asahi Glass, and lithium
hexafluoride salt were dissolved was applied to the
surface of the separator, and then it was pressed. A
battery similar to that according to Example 12 was
manufactured except that the separator was disposed as
CA 02331602 2001-10-25
- 176 -
shown in Fig. 9F.
Example 16
A battery was manufactured under the same
conditions as those according to Example 12 except that
black lead fiber having a specific area of 10 m2/g or
more was paper-made and thus-obtained black lead paper
(having a thickness of 200 ,um) was disposed between the
negative electrode and the separator as shown in Fig.
9B.
Example 17
The preparation of materials and assembling of the
battery were performed in an atmosphere of dry argon.
Lithium metal foil, to which a titanium mesh collector
was pressed, was inserted into the chamber of an RF
plasma CVD apparatus. Then, the retained gas was
exhausted to realize a degree of vacuum of 2 x 10-6 Torr.
A hexane solution of acetylacetone complex of nickel was
used as the material, and it was bubbled with hydrogen
gas as to be introduced into the chamber. The pressure
in the chamber was controlled to 1 Torr, and RF
discharge was performed so that a nickel film was formed
on the surface of the metal foil to have a thickness of
200 A as to be served as a sample electrode. A battery
was manufactured under the same conditions as those
according to Example 12 except that the foregoing sample
electrode was used as the negative electrode.
Example 18
RF discharge was performed under the same
conditions as those according to Example 17 except that
CA 02331602 2001-10-25
- 177 -
oxygen gas was used as the material together with the
hexane solution of acetylacetone complex of nickel. As a
result, a nickel oxide film was formed on the surface of
the lithium metal foil to have a thickness of 50 F. to
serve as a sample electrode. A battery was manufactured
under the same conditions as those according to example
12 except that the foregoing sample electrode was used
as the negative electrode.
CA 02331602 2001-10-25
178
Example 19
The preparation of materials and assembling of the
battery were performed in an atmosphere of dry argon.
Lithium metal foil, to which a titanium mesh collector
was pressed, was inserted into the chamber of the RF
plasma CVD apparatus. Then, the retained gas was
exhausted to realize a degree of vacuum of 2 x 10-6
Trorr. Monosilane gas and ammonia gas were, as raw
material gas, introduced into the chamber, and then the
pressure in the chamber was controlled to 0.1 Torr to
perform RF discharge. As a result, a silicon nitride
film was formed on the lithium metal foil to have a
thickness of 200 A as to be serve as a sample electrode.
A battery was manufactured under the same conditions as
those according to Example 12 except that the foregoing
sample electrode was used as a negative electrode.
Exam_pl a 2 0
The RF discharge was performed under the same
conditions as those according to Example 17 except that
methane gas was used as the raw material together with
the hexane solution of acetylacetone complex of
titanium. As a result, a composite film of titanium and
carbon was formed on the surface of the lithium metal
foil to have a thickness of 250 A to serve as a sample
electrode. A battery was manufactured under the same
conditions as those according to example 12 except that
the foregoing sample electrode was used as the negative
electrode.
CA 02331602 2001-10-25
- 179 -
Example 21
A polypropylene separator was inserted into the
chamber of the RF plasma CVD apparatus. Then, the
retained gas was exhausted to realize a degree of vacuum
of 2 x 10-6 Torr. Acetylene gas was introduced into the
chamber, and then the pressure in the chamber was
controlled to 0.1 Torr and RF discharge was performed.
As a result, a carbon film was formed on the surface of
the separator to have a thickness of 200 P.. A battery
was manufactured under the same conditions as those
according to example 12 except that the foregoing
separator was used and disposed as shown in Fig. 9F.
Example 22
Under the same conditions as those according to
Example 21 except that monosilane gas was used as the
raw material gas to cover the separator with an
atmosphere silicon film. A battery was manufactured
under the same conditions as those according to example
12 except that the separator was used and disposed as
shown in Fig. 9F.
Example 23
The preparation of materials and assembling of the
battery were performed in an atmosphere of dry argon. A
polypropylene separator was inserted into the chamber of
the RF plasma CVD apparatus. Then, the retained gas was
exhausted to realize a degree of vacuum of 10-5 Torr.
Then, tetrafluoroethylene ethylene, Hz, helium and oxygen
were introduced into the chamber, and the internal
pressure was maintained at 0.8 Torr. High frequency
CA 02331602 2001-10-25
- 180 -
power was, by 200 watts, supplied to a parallel and flat
electrode so that a plasma polymer film of fluororesin
was formed on the separator. Then, acetylene gas was, as
the raw material gas, introduced into the chamber, and
the internal pressure in the chamber was controlled to
0.1 Torr to perform RF discharge. As a result, a carbon
film was formed to have a thickness of 200 A. A battery
was manufactured under the same conditions as those
according to example 12 except that the foregoing
separator was used and disposed as shown in Fig. 9G.
Example 24
The preparation of materials and assembling of the
battery were performed in an atmosphere of dry argon. A
fluororesin film having micropores was inserted into the
chamber of the RF plasma CVD apparatus. Then, the
retained gas was exhausted to realize a degree of vacuum
of 2 x 10-6 Torr. A hexane solution of acetylacetone
complex of nickel was, as the raw material, used as to
be bubbled with hydrogen gas as to be introduced into
the chamber. The pressure in the chamber was controlled
to 1 Torr to perform RF discharge so that a nickel film
was formed on the surface of the separator to have a
thickness of 300 A. A battery was manufactured under the
same conditions as those according to example 12 except
that the foregoing film was used and disposed as shown
in Fig. 19C.
Example 25
Under the same conditions as those according to
Example 24 except that monosilane gas was used as the
CA 02331602 2001-10-25
- 181 -
raw material gas to form an amorphous silicon film on
the fluororesin film having micropores. A battery was
manufactured under the same conditions as those
according to example 12 except that the foregoing film
was used and disposed as shown in Fig. 9C.
Example 26
The preparation of materials and assembling of the
battery were performed in an atmosphere of dry argon.
Lithium metal foil, to which a titanium mesh collector
was pressed, was inserted into the chamber of the RF
plasma CVD apparatus. Then, the retained gas was
exhausted to realize a degree of vacuum of 2 x 10-6 Torr.
Monosilane gas and acetylene gas were, as the raw
material gas, introduced into the chamber, and then the
pressure in the chamber was controlled to 0.1 Torr to
perform RF discharge. As a result, the surface of the
lithium metal foil was covered with silicon carbide
film having a thickness of 300 A to serve as a sample
electrode. A battery was manufactured under the same
conditions as those according to example 12 except
that the foregoing sample electrode was used as the
negative electrode.
Example 27
The preparation of materials and assembling of the
battery were performed in an atmosphere of dry argon.
Lithium metal foil, to which a titanium mesh collector
was pressed, was inserted into the chamber of the RF
plasma CVD apparatus. Then, the retained gas was
exhausted to realize a degree of vacuum of 5 x l0-6 Torr.
CA 02331602 2001-10-25
- 182 -
Then, Ar gas was allowed to flow to make the inside of
the chamber to be an inactive atmosphere. Then, the
pressure in the chamber was lowered to 3 x 10-3 Torr, and
black lead and Si were used as the target for use in the
S RF discharge. Sputtering of Si was commenced at the
initial stage of the discharge and the proportion of
black lead sputtering was gradually raised so that a
composite layer of carbon and Si was formed on the
surface of the lithium metal foil to have a thickness of
300 A to serve as a sample electrode. A battery was
manufactured under the same conditions as those
according to example 12 except that the foregoing sample
electrode was used as the negative electrode.
Example 28
A battery was manufactured under the same
conditions as those according to example 27 except that
black lead, Si and polytetrafluoroethylene were used as
target and the thus-formed composite film was stacked on
the surface of the lithium metal foil.
Example 29
The preparation of materials and assembling of the
battery were performed in an atmosphere of dry argon.
The sample according to Example 12 was inserted into the
chamber of the RF plasma CVD apparatus. Then, the
retained gas was exhausted to realize a degree of vacuum
of 10-5 Torr. Then, tetrafluoroethylene ethylene,
hydrogen, helium and oxygen were introduced into the
chamber, and the internal pressure was maintained at 0.8
Torr. Then, high frequency power was, by 200 watts,
CA 02331602 2001-10-25
-183-
supplied to a parallel and flat electrode to form a
plasma polymer film of fluororesin on the foregoing
sample so that a sample electrode was obtained. A
battery was manufactured under the same conditions as
those according to example 12 except that the foregoing
sample electrode was used as the negative electrode.
Example 30
The preparation of materials and assembling of the
battery were performed in an atmosphere of dry argon.
Lithium metal foil, to which a titanium mesh collector
was pressed, was inserted into the chamber of sputtering
apparatus. Then, the retained gas was exhausted to
realize a degree of vacuum of 5 x 10-6 Torr. Ar gas was
allowed to flow to make the inside of the chamber to be
an inactive atmosphere. Then, pressure in the chamber
was lowered to 3 x 10-3 Torr, and black lead and LiAsF6
were used as the targets to perform RF discharge. As a
result, carbon and LiAsF6 were formed on the surface of
the lithium metal foil to have a thickness of 300 A so
that a sample electrode was obtained. A battery was
manufactured under the same conditions as those
according to example 12 except that the foregoing sample
electrode was used as the negative electrode.
Example 31
The separator according to Example 21 and covered
with carbon was used, and a fluororesin film having
micropores was stacked on the coated carbon as shown in
Fig. 9H. A battery was manufactured under the same
conditions as those according to example 12 except that
CA 02331602 2001-10-25
- 184 -
the foregoing film was used.
Examgl a 3 2
The spiral and cylindrical battery shown in Fig. 3
was used in the test, the battery being KR-A type
battery having a contour of 17.0 mm and 50.5 mm high.
Paste obtained by, together with ethylene glycol,
kneading zinc oxide and metal zinc serving as the main
active material and polyvinyl alcohol serving as the
bonding material was applied to an iron plate applied
with nickel plating having apertures. Then, drying and
pressing were performed so that a zinc electrode plate
was obtained.
The zinc electrode plate was inserted into the
chamber of the RF plasma CVD apparatus to perform RF
discharge in such a manner that the proportion of
tetrafluoroethylene was high in the initial stage of the
discharge process and the proportions of acetylene and
oxygen were raised in the following stage of the
discharge process. As a result, a fluororesin film
having a thickness of 200 A was formed on the surface of
the zinc electrode plate so that a sample electrode was
obtained.
The positive electrode plate was manufactured in
such a manner that paste was obtained by kneading a
solution in which nickel and cobalt were added to nickel
hydroxide and carboxydimethyl cellulose serving as a
bonding material, and water was added to the kneaded
solution. The paste was injected into foamed metal
(Celmet manufactured by Sumitomo Denko), and it was
CA 02331602 2001-10-25
-185-
dried and pressed.
A separator (Cell Guard manufactured by Cellanese)
was used which was manufactured by integrating a film
having small apertures and unwoven polypropylene sheet.
The electrolyte solution comprised 30 wt% potassium
solution.
Assembly was performed in such a manner that a
wound group in which a separator was interposed between
the negative electrode and the positive electrode was
inserted into a battery case made of titanium clad
stainless steel. Then, the electrolyte solution was
injected, and the negative electrode cap made of
titanium stainless steel and an insulating packing made
of fluorine rubber were inserted and they were caulked
so that a nickel-zinc secondary battery was
manufactured.
Exam~ale 33
The flat type battery shown in Fig. 2 was used to
conduct the test.
Tetrafluoroethylene polymer powder was mixed to
zinc oxide and metal zinc, and then was pressed against
a nickel mesh as to be formed into a zinc electrode
plate. The zinc negative electrode was inserted into
chamber of the RF plasma CVD apparatus, and then
fluororesin having a thickness of 50 A and carbon having
a thickness of 200 A were formed on the surface of the
zinc electrode plate so that a negative electrode was
formed.
A positive electrode catalyzer layer was formed by
CA 02331602 2001-10-25
- 186 -
adding manganese dioxide to active carbon. Then, a water
repellant film made of polytetrafluoroethylene and
dispersed paper made of cellulose were stacked on the
positive electrode catalyzer layer so that a positive
electrode was formed. A separator made of cellophane and
30 wt% potassium hydroxide serving as the electrolyte
solution were employed.
Assembly was performed in such a manner that a
separator was interposed between the negative electrode
and the positive electrode and they were inserted into a
battery case made of stainless steel and having small
apertures. Then, the electrolyte solution was injected,
and then a negative electrode cap made of titanium clad
stainless steel and an insulating packing made of
fluorine rubber were used to perform sealing. As a
result, an air-zinc secondary battery was manufactured.
Comparative Example 12
A battery was manufactured under the same
conditions as those according to example 12 except that
a negative electrode, which comprised lithium metal foil
having no carbon film, was used.
Comparative Example 13
A battery was manufactured under the same
conditions as those according to example 31 except that
a negative electrode, which comprised a zinc electrode
plate which was not covered with the composite film of
the fluororesin and carbon, was used.
Comparative Example 14
A battery was manufactured under the same
CA 02331602 2001-10-25
- 1g7 -
conditions as those according to example 33 except that
a negative electrode, which comprised a zinc electrode
plate which was not covered with the composite film of
the fluororesin and carbon, was used.
The batteries respectively according to Examples 12
to 31 and Comparative Example 12 were charged with a
current of 0.2 C to a level of 4.0 V, then paused for 30
minutes, and then discharged with a current of 0.2 C to
a level of 2.8 V. The foregoing test was repeated,
resulting in as shown in Table 9.
The batteries respectively according to Examples 32
and 33 and Comparative Examples 13 and 14 were charged
with a current of 0.2 C to 150 %, then paused for 30
minutes, and then discharged with a current of 0.2 C to
1. o V .
The results of the cycle life tests of the
respective Examples with respect to Comparative Examples
are shown in Table 9 while making the cycle life of the
batteries according to Comparative Examples 12 to 14 to
be 1. As can be understood from the results shown in
Table 9, the charge/discharge cycle life can signifi-
cantly be lengthened as compared with Comparative
Examples when a single layer, a multi-layer or a
composite layer is disposed between the negative
electrode and the separator, the single layer, the
multi-layer or the composite layer being the conductor
layer made of carbon or nickel or titanium, the
semiconductor layer made of silicon or the metal oxide,
and the insulating layer made of the halide, nitride,
CA 02331602 2001-10-25
- Igg -
carbide or the organic polyer (according to respective
Examples).
Table 9
Manufactured Cycle Life of
Secondary Example
Battery
Cycle Life of
Examples Comparative Examples Comparative Example
Example 12 Comparative Example 12 2.5
Example 13 Comparative Example 12 2.1
Exam 14 Com arative Exam 12 1.5
1e 1e
Exam 15 Com arative Exam 12 1.3
1e 1e
Example 16 Com arative Exam 12 1.3
1e
Example 17 Comparative Example 12 2.2
Example l8 Comparative Example 12 2.3
Exam 19 Com arative Exam 12 2.0
1e 1e
Example 20 Comparative Exam 12 2.1
1e
Example 21 Comparative Exam 12 1.6
1e
Example 22 Comparative Example 12 1.5
Example 23 Comparative Exam 12 1.7
1e
Example 24 Comparative Exam 12 1.5
1e
Exam 25 Com arative Example 12 1.5
1e
Exam 26 Com arative Exam 12 2.2
1e 1e
Example 27 Comparative Example 12 2.1
Example 28 Comparative Example 12 2.2
Exa~le 29 Comparative Example 12 2.0
Example 30 Comparative Example 12 2.6
Example 31 Comparative Example 12 1.6
Exam 32 Comparative Example 13 2.2
1e
Example 33 Comparative Example 14 2.2
~
CA 02331602 2001-10-25
- 189 -
Example 34
A polysiloxane film was formed by the following
method.
As the film forming compound, 0.05 M (mol/Q) an
amphipathic compound N-[b-(trimethylammonio)
ethyloxybenzoyl]-didodecyl-L-glutamic acid bromide and
0.15 M of trimethoxymethyl silane were processed with
supersonic waves for 3 minutes as to be dispersed in
water. The dispersion solution was developed on a
tetrafluoroethylene polymer sheet (Goatex manufactured
by Japan Goa Tex) and allowed to stand at 25°C and 60%
relative humidity for 3 days so that a multi-layer
bimolecular film was obtained. The film was treated with
ammonia gas in a sealed glass container to hydrolyze and
condense the methoxysilane group. Ethyl alcohol was used
to extract and remove the amphipathic compound so that a
polysiloxane film was obtained on the Goa Tex sheet.
Manufacturing of Secondary Battery
A lithium secondary battery which had a simple
structure, which could be assembled simply and which had
a cross sectional shape schematically shown in Fig. 2
was manufactured.
The positive electrode active material 203 was
prepared in such a manner that electrolyzed manganese
dioxide and lithium carbonate were mixed at a ratio of
1:0.4, and then heated at 800°C so that a lithium-
manganese oxide was prepared. Then, graphite and
tetrafluoroethylene polymer powder were mixed to the
prepared lithium-manganese oxide, and then pressed
CA 02331602 2001-10-25
- 190 -
against a nickel mesh as to be formed into the positive
electrode.
Then, silica sol dispersion ethyl alcohol
(manufactured by Shokubai Kasei) was mixed in water in
which N-[b-(trimethylammonio)ethyloxybenzoyl]-didodecyl-
L-glutamic acid bromide, which was the film forming
compound, was dispersed. The positive electrode was
dipped in the foregoing dispersion solution, dried at
room temperature, further dried at 80°C, and cleaned
with hexane to remove the film forming compound. Then,
it was dried at 250°C in a vacuum atmosphere so that the
positive electrode 203 covered with the multi-layer
silica film was manufactured.
In an atmosphere of dry argon gas, titanium mesh
collector 200 was pressed against the reverse side of
the lithium metal foil, and then the lithium metal foil
was immersed in a solution of Lumiflon, which was a
copolymer of ethylene tetrafluoride and vinyl ether and
which was manufactured by Asahi Glass, and then dried as
to be hardened. As a result, the lithium negative
electrocde 201 covered with fluororesin was
manufactured.
The electrolyte solution was prepared by
dissolving, by 1 M (mol/Q) , boron tetrafluoride lithium
salt in an equal quantity mixture solvent of propylene
carbonate (PC) and dimethoxyethane (DME).
The separator 208 comprised a composite member
of the polysiloxane film prepared as described above and
the Goa Tex sheet.
CA 02331602 2001-10-25
-191-
Assembly was performed in such a manner that the
separator 208 was interposed between the negative
electrode 201 and the positive electrode 203 as to be
inserted into the positive electrode case 207 made of
titanium clad stainless steel. Then, the electrolyte
solution was injected, and then the negative electrode
cap 206 made of titanium clad stainless steel and the
insulating packing 210 made of fluorine rubber were used
to seal so that a lithium secondary battery was
manufactured.
Example 35
Preparation of Multi-Layer Metal Oxide
A titanium oxide was prepared by the following
method.
A film forming compound, that is,
dihexadecylphosphate was mixed to a sol dispersed
solution of very small particles of titanium oxide
(manufactured by Idemitsu), and dispersed with
ultrasonic waves similarly to Example 34. Then, it was
developed on a tetrafluoroethylene polymer membrane
filter. Then, it was dried at room temperature,
resulting in a cast film to be obtained which was then
cleaned with ethyl alcohol. Then, it was baked at 300°C
so that a titanium oxide film was formed.
The separator 208 was manufactured in such a manner
that the thus-formed titanium oxide, unwoven
polypropylene sheets and a polypropylene separator
having small apertures were sandwiched.
A battery was manufactured under the same
CA 02331602 2001-10-25
- 192 -
conditions as those according to example 34 except the
foregoing arrangements.
Example 36
A lithium secondary battery was manufactured under
the same conditions as those according to example 35
except that the titanium oxide film was immersed in 0.1%
ethylalcohol solution of silane coupling SH6020
(manufactured by Toray Dowconing).
Example 37
Preparation of Multi-Layer Metal Oxide
A cast film obtained similarly to Example 35 was
based at 800°C so that an alumina film was manufactured,
the cast film being obtained from a film forming
compound brought into class four from N-(11-
bromoundecanoyl)-L-glutamic acid didodecylester with
triethylamine and amorphous alumina super fine particle
sol.
The N-X11-bromoundecanoyl)-L-glutamic acid
didodecylester was prepared in such a manner that L
glutamic acid didodecylester hydrochloride salt was
synthesized from L-glutamic acid and dodecylalcohol, and
then the L-glutamic acid didodecylester hydrochloride
salt, triethylamine, cyanophsophoric acid diethyl and
11-bromoundecan acid were used.
Assembly of Secondary Battery
A nickel-zinc secondary battery which had a simple
structure, which could be assembled simply and which had
a cross sectional shape schematically shown in Fig. 2
was manufactured.
CA 02331602 2001-10-25
-193-
The positive electrode was manufactured by
impregnating nickel hydroxide into a sintered nickel
electrode plate.
The negative electrode was formed in such a manner
S that tetrafluoride ethylene polymer powder serving as
the bonding material was added to a mixture of zinc
powder and zinc oxide powder and they were pressed
against the two sides of a copper punching metal to be
formed into the desired shape. Then, alumina trichloride
and phosphoric acid were, in ethyl alcohol, caused to
react with each other at 0°C as to dip the negative
electrode. Then, it was gradually heated up to 100°C so
that glass-type aluminum phosphate film was formed on
the negative electrode.
The electrolyte solution comprised 30 wto potassium
hydroxide containing lithium hydroxide added thereto.
The battery was assembled similarly to Example 34.
Example 38
A nickel-zinc secondary battery was manufactured
under the same conditions as those according to Example
37 except that the surface of the negative electrode was
not covered with the aluminum phosphate.
Example 39
A nickel-zinc secondary battery was manufactured
under the same conditions as those according to Example
37 except that a positive electrode manufactured as
follows was used.
A dispersion solution in which was mixed a film
forming compound obtained by bringing N-(11-
CA 02331602 2001-10-25
- 194 -
bromoundecanoyl)-L-glutamic acid didodecylester into
class four with triethylamine, amorphous alumina very
small particles sol and polyvinyl alcohol water solution
containing crosslinking material added thereto was
developed on the surface of the positive electrode
formed by causing a sintered nickel plate to impregnate
nickel hydroxide, followed by drying the developed
solution. Then, crosslinking reactions were caused to
take place at 120°C, and cleared with ethyl alcohol.
Then, the material was dried in a vacuum atmosphere so
that a positive electrode covered with a composite film
of alumina and polyvinyl alcohol was manufactured.
In order to compare and evaluate the foregoing
batteries, the following comparative batteries were
manufactured.
Comparative Example 34
A lithium secondary battery was manufactured under
the same conditions as those according to example 35
except that no titanium oxide was used and the lithium
of the negative electrode was not covered.
Comparative Example 35
A nickel-zinc secondary battery was manufactured
under the same conditions as those according to example
37 except that no alumina was used and zinc of the
negative electrode was not covered.
Evaluation of Performance of Secondary Battery
The performance of lithium secondary batteries
according to Examples and Comparative Examples was
CA 02331602 2001-10-25
- 195 -
evaluated. The evaluation was performed by a charge and
discharge cycle test under the following conditions with
respect to the cycle life of the batteries according to
Comparative Examples.
The conditions for the cycle test were made as
follows: the charge and discharge was performed by 0.2C
(electric current which was 0.2 times capacity/time),
pause for 30 minutes and a cut-off voltage of 1..0V was
applied. A charging/discharging apparatus HJ-101M6
manufactured by Hokuto Electric was used. The
charge/discharge test was commenced at discharge, the
battery capacity was evaluated by the quantity of the
third discharge and the cycle life was evaluated by the
number of cycles when the battery capacity had
deteriorated to 60% or less.
The cycle life of each battery with respect to the
cycle life of the battery according to Comparative
Examples which was made to be 1 was as shown in Table 2.
As can be understood from the results of
comparisons made between Examples 34 to 36 and
Comparative Example 34 and those between Examples 37 to
39 and Comparative Example 35, the cycle life can be
lengthened due to use of the secondary battery having
the structure according to the present invention.
CA 02331602 2001-10-25
- 196 -
Table 10
Secondary Battery Manufactured Cycle Life of
Example
Cycle Life
of Comparative
Example
Example Comparative Example
Example 34 Comparative Example 34 5.4
Example 35 Comparative Example 34 2.1
Example 36 Comparative Example 34 2.3
Example 37 Comparative Example 35 2.6
Example 38 Comparative Example 35 1,9
Example 39 Comparative Example 35 I 2.2
~
Example 40
A lithium secondary battery which had a simple
structure, which could be assembled simply and which had
a cross sectional shape schematically shown in Fig. 2
S was manufactured.
The positive electrode active material 203 was
manufactured in such a manner that electrolyzed
manganese dioxide and lithium carbonate were mixed at a
ratio of 1:0.4. Then, the mixture was heated to 800°C so
that a lithium-manganese oxide was prepared. Graphite
and tetrafluoroethylene polymer powder were added to the
prepared lithium-manganese oxide, and pressed against a
nickel mesh as to be formed into the positive electrode.
Then, the positive electrode was immersed in a
solution of Teflon AF which was a copolymer of
CA 02331602 2001-10-25
- 197 -
tetrafluoroethylene and 2, 2-bistrifluoromethyl-4,5-
difluoro-1, 3-dioxol and which was manufactured by
Dupont, and then it was dried. As a result, the positive
electrode 203 covered with the fluororesin was
manufactured.
In an atmosphere of dry argon gas, the titanium
mesh collector 200 was pressed against the reverse side
of the lithium metal foil, and then the lithium metal
foil was immersed in a solution which was a copolymer of
ethylene tetrafluoride and vinyl ether and which was
manufactured by Asahi Glass. Then, it was dried as to be
hardened so that the lithium negative electrode 201
covered with the fluororesin was manufactured.
The electrolyte solution was prepared by
dissolving, by 1 M (mol/Q), boric acid tetrafluoride
lithium salt in an equal quantity mixture solvent of
propylene carbonate (PC) and dimethoxyethane (DME).
The separator 208 was manufactured in such a manner
that a polypropylene separator having small apertures
was sandwiched by unwoven polypropylene sheets.
The assembly was performed in such a manner that
the separator 208 was held between the negative
electrode 201 and the positive electrode 203 as to be
inserted into the positive case 207 made of titanium
clad stainless steel. Then, the electrolyte solution was
injected, and then the negative electrode cap made of
titanium clad stainless steel and the insulating packing
210 made of fluorine rubber were used for sealing so
that a lithium secondary battery was manufactured.
CA 02331602 2001-10-25
- 198 -
Example 41
A lithium battery was manufactured under the same
conditions as those according to example 40 except the
process for covering the positive electrode.
The positive electrode was manufactured similarly
to Example 40. Then, the positive electrode was immersed
in an acetonitryl solution, in which were dissolved 0.1
M of monomer of benzo-15-crown-5 and 2.0 M of
electrolytic boronic acid tetrafluoride tetrabutyl
ammonium salt. Then, the platinum electrode was used as
the cathode electrode, and voltage of 3 V was applied to
perform electrolysis and polymerization so that a large
ring compound polymer covering film was formed on the
surface of the positive electrode.
In an atmosphere of dry argon gas, the titanium
mesh collector 200 was pressed against the lithium metal
foil so that the negative electrode was manufactured.
Then, similar processes to those according to
Example 40 were performed so that the lithium secondary
battery was manufactured.
Examgle 42
A lithium secondary battery which had a simple
structure, which could be assembled simply and which had
a cross sectional shape schematically shown in Fig. 2
was manufactured.
The positive electrode was manufactured by a
similar process according to Example 40. Then, peroxide
benzoyl and boronic acid tetrafluoride lithium were
dissolved in a tetrahydrofuran solution of poly (2-vinyl
CA 02331602 2001-10-25
199 -
naphthalene) manufactured by Aldrich Chemical Company,
Inc. Then, the positive electrode was immersed in it,
and then heated to 100°C so that the positive electrode
203 covered with poly (2-vinyl naphthalene) was
manufactured.
Then, similar processes to those according to
Example 41 were performed so that the lithium secondary
battery was manufactured.
Example 43
The positive electrode was manufactured by a
process similar to that according to Example 40. Then,
boronic acid tetrafluoride lithium salt was added and
dissolved in a toluene solution PPZ-U1001 manufactured
by Idemitsu. Then, the positive electrode was immersed,
and previously dried, and then ultraviolet rays were
applied so that the positive electrode 203 covered with
polyphosphazene was manufactured.
Then, similar processes to those according to
Example 41 were performed so that the lithium secondary
battery was manufactured.
Example 44
The positive electrode was manufactured by a
process similar to that according to Example 40. The
thus-manufactured positive electrode was injected into a
sputtering apparatus. Then, the retained gas was
exhausted to realize a degree of vacuum of 2 x 10-6 Torr.
Then, argon gas, which was a mixture of 10% nitrogen gas
and 5o acetylene gas, was allowed to flow. The internal
pressure was controlled to 3 x 10-3 Torr, and lithium
CA 02331602 2001-10-25
200 -
fluoride was made to be a target of sputtering. As a
result, the positive electrode covered with carbon and a
film made of lithium nitride cor~taining fluorine were
formed.
Then, similar processes to those according to
Example 41 were performed so that the lithium secondary
battery was manufactured.
Example 45
A nickel-zinc secondary battery which had a simple
structure, which could be assembled simply and which had
a cross sectional shape schematically shown in Fig. 2
was manufactured.
The positive electrode was manufactured in such a
manner that zinc hydroxide was impregnated in a sintered
nickel electrode plate. Then, acetic acid and water were
added to an ethyl alcohol solution of tetraethoxy silane
to be dehydrated. Then, diethylamine was added so that
colloidal silica was formed. Then, the positive
electrode was immersed in the colloidal solution of the
silica, and dried at 100°C. As a result, a silica film
was formed on the surface of the positive electrode.
The negative electrode was manufactured in such a
manner that ethylene tetrafluoride polymer powder
serving as a bonding material was added to a mixture of
zinc powder and zinc oxide powder, then they were
pressed to the two sides of a copper punching metal
as to be formed into the negative electrode.
The electrolyte solution comprised 30 wt% potassium
hydroxide water solution to which lithium hydroxide was
CA 02331602 2001-10-25
- 201 -
added.
The battery was assembled similarly to Example 40.
In order to compare and evaluate the performance of
the batteries according to Examples, the following
comparative batteries were manufactured.
Comparative Example 40
A lithium secondary battery was manufactured under
the same conditions as those according to example 40
except that the positive electrode and the negative
electrode were not covered.
Comparative Example 41
A nickel-zinc secondary battery was manufactured
under the same conditions as those according to example
45 except that the surface coating was omitted.
Evaluation of Performance of Secondar Batter
The performance of the secondary batteries
according to Examples and Comparative Examples was
evaluated. The evaluation was performed by a charge and
discharge cycle test under the following conditions with
respect to the cycle life of the batteries according to
Comparative Examples. The conditions for the cycle test
were made as follows: the charge and discharge was
performed by 0.2C (electric current which was 0.2 times
capacity/time), pause for 30 minutes and a cut-off
voltage of 1.0V was applied. A charging/discharging
apparatus HJ-1O1M6 manufactured by Hokuto Electric was
used.
The charge/discharge test was commenced at
discharge, the battery capacity was evaluate the
CA 02331602 2001-10-25
-202-
quantity of the third discharge and the cycle life was
evaluate by the number of cycles when the battery
capacity had deteriorated to 60% or less.
The cycle life of each battery with respect to the
cycle life of the battery according to Comparative
Examples which was made to be 1 was as shown in Table
11.
As can be understood from the results of
comparisons made between Examples 40 to 44 and
Comparative Example 11 and those between Example 45 and
Comparative Example 41, the cycle life can be lengthened
due to use of the secondary battery having the structure
according to the present invention.
Table 11
Secondary Battery Manufactured Cycle Life of
Example
Cycle Life
of Compatarive
Example
Example Comparative
Example
Example 40 Comparative Examp7.e40 5.1
Example 41 Comparative Example 40 2,0
Example 42 Comparative Example 40 1.4
Example 43 Comparative Example 40 1.6
Example 44 Comparative Example 40 1,g
Example 45 Comparative Example 41 1,~
CA 02331602 2001-10-25
- 203 -
Pre aration of Positive Electrode Active Material
Methods of preparing the positive electrode active
material according to the present invention are
exemplified in Preparation Methods 46 to 55 and the
conventional preparation methods are exemplified by
Comparative Preparation Methods.
Preparation Method 46
A lithium-manganese oxide was prepared as follows.
Manganese acetate was dissolved in water, and then
super fine nickel powder ENP-005 manufactured by
Sumitomo Denko was suspended in a water solution of
manganese nitrate. Then, a water solution of lithium
hydroxide was dripped until the pH was 8 or higher while
vibrating the suspension solution with supersonic
vibrations of 20 kHz so that sedimentation was
generated. Then, ethyl alcohol was added, and
supernatant liquid of the solution including the
sediment was removed by decantation. The ethylalcohol
cleaning and the decantation were repeated. Then, it was
dissolved in O.lo methyl alcohol solution of Sila Ace
S210 (vinylmethoxysilane) which was a silane coupling
material manufactured by Chisso. Then, the solvent was
removed by a centrifugal separator. The obtained
sedimentation was dried at 120°C, and dried at 200°C in a
vacuum drier so that grains of manganese oxide were
prepared.
The size of crystal grains was measured from the
half value width of the X-ray analysis curve with
respect to a manganese oxide and the angle of
CA 02331602 2001-10-25
-204-
diffraction in accordance with the Scherrer's Equation.
The size of the crystal grain was 60 A or larger.
The RHEED pattern resulted in a ring pattern like a
halo pattern.
The X-ray radial distribution function resulted in
a continuous and moderate peak curve.
The scattering angle and the scattering intensity
obtainable from the X-ray small angle scattering method
resulted in non-uniform density fluctuation to be
observed.
The specific area was measured by the BET method,
resulting in that the specific area was 123 m2/g.
Preparation Method 47
Vanadium oxide was prepared as follows.
Vanadium pentaoxide was gradually added to a water
solution of. lithium hydroxide as to be dissolved. While
applying supersonic vibrations, the water solution was
sprayed into liquid nitrogen as to be frozen. Then, the
temperature was raised to -20°C, and the pressure was
lowered so that freezing and drying were performed to
dehydrate and dry the material. Obtained grains were
dried at 150°C, and further dried in a vacuum drier at
250°C so that grains of vanadium oxide were obtained.
Then, the sample was immersed in 0.1% isopropylalcohol
solution of tetra-iso-propoxytitanium, and then the
solvent was removed by a centrifugal separator. The
obtained sedimentation was dried at 120°C, and then
dried at 200°C in a vacuum state so that grains of
manganese oxide were prepared.
CA 02331602 2001-10-25
- 205 -
Although the size of crystal grains was intended to
measure from the half value width of the X-ray analysis
curve and the angle of diffraction in accordance with
the Scherrer's Equation, it could not be measured
because the diffraction curve resulted in a broad form.
The RHEED pattern resulted in a halo pattern.
The X-ray radial distribution function resulted in
a continuous and moderate peak curve.
The scattering angle and the scattering intensity
obtainable from the X-ray small angle scattering method
resulted in non-uniform density fluctuation to be
observed.
The specific area was measured by the BET method,
resulting in that the specific area was 105 m2/g.
Preparation Method 48
A lithium-nickel oxide was prepared as follows.
Nickel acetate was dissolved in a mixture solvent
of acetic acid, ethyl alcohol and water. While vibrating
the solution with supersonic vibrations of 20 kHz, an
ethyl alcohol solution of ethoxylithium, which was
alkoxide, was dripped as to be mixed. Then, the solution
was heated to 80°C to enhance the hydrolysis
decomposition reactions so that sol was generated. The
supernatant liquid of the solution including the sol-
shape sedimentation was removed by decantation, cleaned
with ethyl alcohol, the decantation was repeated and the
solvent was removed by a centrifugal separator. The
obtained sedimentation was dried at 150°C, and then
suspended in a non-electrolyzed nickel plating solution
CA 02331602 2001-10-25
-206-
Ni-701 manufactured by Kojundo Kagaku. Then, it was
heated to 70°C, nickel coating was performed, and water
cleaning and decantation were repeated. Then, ethyl
alcohol cleaning was performed, and decantation was
repeated, and then the solvent was removed by a
centrifugal separator. The sample was dried at 230°C in
a vacuum state so that grains of nickel oxide were
obtained.
Although the size of crystal grains was intended to
measure from the half value width of the X-ray analysis
curve and the angle of diffraction in accordance with
the Scherrer~s Equation, it could not be measured
because the diffraction curve with respect to an oxide
resulted in a broad form.
The RHEED pattern resulted in a ring pattern
considered due to nickel plating. The pattern before the
nickel plating process resulted in a halo pattern.
The X-ray radial distribution function resulted in
a continuous and moderate peak curve. The scattering
angle and the scattering intensity obtainable from the
X-ray small angle scattering method resulted in non-
uniform density fluctuation to be observed.
The specific area was measured by the BET method,
resulting in that the specific area was 210 m'/g.
Preparation Method 49
A lithium-nickel-cobalt oxide was prepared by the
following method.
Nickel nitrate and cobalt nitrate were dissolved in
water, and then a water solution of lithium hydroxide
CA 02331602 2001-10-25
- 207 -
was dripped to a solution of the nickel nitrate and
cobalt nitrate while vibrating with supersonic
vibrations of 20 kHz until the pH was 8 or higher so
that sedimentation was generated. Then, ethyl alcohol
was added so that the supernatant liquid of the solution
including the sedimentation was removed by decantation.
the ethyl alcohol cleaning and the decantation were
repeated and the solvent was removed by a centrifugal
separator. The obtained sedimentation was dried at
120°C, dried at 200°C in a vacuum state so that grains of
a nickel cobalt oxide were prepared.
The size of crystal grains was measured from the
half value width of the X-ray analysis curve and the
angle of diffraction in accordance with the Scherrer's
Equation. The size of the crystal grain was 140 A.
The RHEED pattern resulted in a ring pattern like a
halo pattern.
The X-ray radial distribution function resulted in
a continuous and moderate peak curve.
The scattering angle and the scattering intensity
obtainable from the X-ray small angle scattering method
resulted in non-uniform density fluctuation to be
observed.
The specific area was measured by the BET method,
resulting in that the specific area was 160 m'/g.
Preparation Method 50
Vanadium-molybdenum oxide was prepared as follows.
Vandyl sulfate and molybdenum sulfate were added to
water as to be suspended, and acetic acid was gradually
CA 02331602 2001-10-25
- 208 -
added as to be dissolved. Then, a water solution of
lithium hydroxide was dripped to a solution of the
vanadium oxide and acetic acid while vibrating with
supersonic vibrations of 20 kHz until the pH was 8 or
S higher so that sedimentation was generated. Then, ethyl
alcohol was added so that the supernatant liquid of the
solution including the sedimentation was removed by
decantation. The ethyl alcohol cleaning and the
decantation were repeated and the solvent was removed by
a centrifugal separator. The obtained sedimentation was
dried at 120°C, dried at 200°C in a vacuum state so that
grains of a vanadium oxide were prepared.
The size of crystal grains was measured from the
half value width of the X-ray analysis curve and the
angle of diffraction in accordance with the Scherrer's
Equation. The size of the crystal grain was 80 A.
The RHEED pattern resulted in a ring pattern like a
halo pattern.
The X-ray radial distribution function resulted in
a continuous and moderate peak curve.
The scattering angle and the scattering intensity
obtainable from the X-ray small angle scattering method
resulted in non-uniform density fluctuation to be
observed.
The specific area was measured by the BET method,
resulting in that the specific area was 100 m2/g.
Preparation Method 51
Vanadium-molybdenum oxide was prepared by the
following method.
CA 02331602 2001-10-25
- 209 -
The vanadium oxide and the molybdenum oxide were
mixed at a ratio of 7:3, and heated up to 800°C as to be
melted and mixed so that a molten bath was made. Then,
the molten bath was dispersed by jet gas which was a
mixture of 200 oxygen and 2o hydrogen with argon gas. It
was sprayed at high speed to a cooled and retaining
metal disc so that grains of vanadium oxide and
molybdenum oxide were prepared.
The size of crystal grains was measured from the
half value width of the X-ray analysis curve and the
angle of diffraction in accordance with the Scherrer's
Equation. The size of the crystal grain was 110 A.
The RHEED pattern resulted in a ring pattern having
weak intensity.
The X-ray radial distribution function resulted in
a continuous and moderate peak curve.
The scattering angle and the scattering intensity
obtainable from the X-ray small angle scattering method
resulted in non-uniform density fluctuation to be
observed.
The specific area was measured by the BET method,
resulting in that the specific area was 60 m2/g.
Preparation Method 52
Titanium sulfide was prepared by the following
method .
Hydrogen gas was allowed to flow by 500 sccm into a
reaction chamber of a plasma CVD apparatus degasified as
to be vacuum state. The pressure was maintained at 10
Torr and discharge was caused to take place at a high
CA 02331602 2001-10-25
- 210 -
frequency wave of 13.56 MHz. Then, 200 sccm of helium
gas was, as a carrier gas,' bubbled in a hexane solution
of tetrabutoxytitanium, and injected by 200 sccm through
a nozzle into the reaction chamber of the plasma CVD
apparatus. Simultaneously, 250 sccm of hydrogen sulfide
was introduced as to be reacted in a gas phase to
capture grains of titanium sulfide by a capturing
machine.
The size of crystal grains was measured from the
half value width of the X-ray analysis curve and the
angle of diffraction in accordance with the Scherrer's
Equation. The size of the crystal grain was 200 A.
The RHEED pattern resulted in a ring pattern having
weak intensity.
The X-ray radial distribution function resulted in
a continuous and moderate peak curve.
The scattering angle and the scattering intensity
obtainable from the X-ray small angle scattering method
resulted in non-uniform density fluctuation to be
observed.
The specific area was measured by the BET method,
resulting in that the specific area was 175 m2/g.
Preparation Method 53
Lithium-iron-cobalt oxide was prepared by the
following method.
A mixture solution of 0.5 mol/1 cobalt chloride and
1 mol ferric chloride mixed at a ratio of 1:l was
gradually added to a 5 mol/1 water solution of lithium
hydroxide while stirring and bubbling argon gas. Then,
CA 02331602 2001-10-25
-211 -
reaction chamber was set to 100°C to be matured. After
the maturation, it was injected into cooled water, and
cleaning was performed with water cooled by water by
means of decantation until the pH of the solution was 8.
It was dried at 200°C in a vacuum state, and crushed by
a ball mill in an atmosphere of argon gas.
The size of crystal grains was measured from the
half value width of the X-ray analysis curve and the
angle of diffraction in accordance with the Scherrer's
Equation. The size of the crystal grain was 150 A.
The RHEED pattern resulted in a ring pattern having
weak intensity to be observed.
The X-ray radial distribution function resulted in
a continuous and moderate peak curve.
The scattering angle and the scattering intensity
obtainable from the X-ray small angle scattering method
resulted in non-uniform density fluctuation to be
observed.
The specific area was measured by the BET method,
resulting in that the specific area was 2100 m2/g.
Example 54
Manganese Acetate, magnesium chloride and urea were
added to a solution in which 300 g of vanadium pentoxide
was dissolved in 2 litters of hydrochloric acid,
followed by heating the solution to 95 to 95°C for 10
minutes to generate ammonia. Lithium hydride solution
was dripped to make the pH to generate sediment. Then,
decantation and water cleaning were repeated, and then
cleaning with ethyl alcohol was performed, and the
CA 02331602 2001-10-25
- 212 -
material was dried by a spray drier. Then, the sample
was dried at 200°C in a vacuum state.
The size of crystal grains was measured from the
half value width of the X-ray analysis curve and the
angle of diffraction in accordance with the Scherrer's
Equation. The size of the crystal grain was 90 A.
The RHEED pattern resulted in a ring pattern like a
halo pattern.
The X-ray radial distribution function resulted in
a continuous and moderate peak curve.
The scattering angle and the scattering intensity
obtainable from the X-ray small angle scattering method
resulted in non-uniform density fluctuation to be
observed.
The specific area was measured by the BET method,
resulting in that the specific area was 80 m2/g.
Preparation Method 55
A lithium-copper-cobalt oxide was prepared by the
following method.
An oxalic acid was added to a water solution in
which a copper sulfate and a cobalt nitrate were
dissolved. Then, supersonic vibrations were applied, and
lithium hydroxide was dripped until the pH was 7 so that
sedimentation was generated. Water cleaning and
decantation were repeated, and then, a water solution of
lithium hydroxide was added, supersonic vibrations were
applied, and an ethyl alcohol was added. Decantation and
cleaning with ethyl alcohol were repeated, and the
material was dried by using a spray drier. Further, the
CA 02331602 2001-10-25
- 213 -
material was dried at 200°C in a vacuum state.
The size of crystal grains was measured from the
half value width of the X-ray analysis curve and the
angle of diffraction in accordance with the Scherrer's
Equation. The size of the crystal grain was 160 A.
The RHEED pattern resulted in a ring pattern having
weak intensity.
The X-ray radial distribution function resulted a
continuous and moderate peak curve.
The scattering angle and the scattering intensity
obtainable from the X-ray small angle scattering method
resulted in non-uniform density fluctuation to be
observed.
The specific area was measured by the BET method,
resulting in that the specific area was 50 m'/g.
Other Analyses
The SIMS analysis resulted that the positive
electrode active materials according to Preparation
Methods 46 to 55 contained hydrogen and lithium. Also
the dehydration peak of each TG (Thermogravimetric
analysis), DTA (Differential Thermal Analysis) and DSC
(Differential Scan Thermal Heating Value Measurement)
and the absorption spectrum of FTIR (Fourier Transform
Infrared) resulted that a hydroxyl group was present.
Comparative Preparation Method 46
A lithium-manganese oxide was prepared by the
following method.
Powder of electrolyzed manganese dioxide
manufactured by Mitsui Kinzoku and lithium carbonate
CA 02331602 2001-10-25
- 214 -
were mixed with each other at a ratio of 1:0.4 and they
were heated at 800°C so that lithium manganese oxide was
prepared.
The size of crystal grains was measured from the
half value width of the X-ray analysis curve and the
angle of diffraction in accordance with the Scherrer's
Equation. The size of the crystal grain was 600 A or
larger.
The RHEED pattern resulted in a ring pattern in
which a spot pattern could be confirmed.
The X-ray radial distribution function resulted in
discontinuous peak curve.
The specific area was measured by the BET method,
resulting in that the specific area was 40 m2/g.
Comparative Preparation Method 47
A reagent manufactured by Wako was dried at 400°C
in a vacuum state.
The size of crystal grains was measured from the
half value width of the X-ray analysis curve and the
angle of diffraction in accordance with the Schemer's
Equation. The size of the crystal grain was 800 f~.
The RHEED pattern resulted in a ring pattern in
which a spot pattern could be confirmed.
The X-ray radial distribution function resulted in
discontinuous peak curve.
The specific area was measured by the BET method,
resulting in that the specific area was 4 m2/g.
Comparative Preparation Method 48
A lithium-nickel oxide was prepared by the
CA 02331602 2001-10-25
- 215 -
following method.
A lithium carbonate and nickel nitrate were mixed
at an equal mol ratio of 1:1 and the mixture was heated
to 800°C so that a lithium-nickel oxide was prepared.
The size of crystal grains was measured from the
half value width of the X-ray analysis curve and the
angle of diffraction in accordance with the Scherrer's
Equation. The size of the crystal grain was 2000 A or
larger.
The RHEED pattern resulted in a ring pattern in
which a spot pattern could be confirmed.
The X-ray radial distribution function resulted in
discontinuous peak curve.
The specific area was measured by the BET method,
resulting in that the specific area was 50 m2/g.
Comparative Preparation Method 49
A lithium-nickel-cobalt oxide was prepared by the
following method.
A lithium carbonate, nickel carbonate and cobalt
carbonate were mixed at a mol ratio of 10:3:7 and the
mixture was heated to 900°C for 20 hours as to be
decomposed so that a nickel-cobalt oxide was prepared.
The size of crystal grains was measured from the
half value width of the X-ray analysis curve and the
angle of diffraction in accordance with the Scherrer's
Equation. The size of the crystal grain was 1100 A or
larger.
The RHEED pattern resulted in a ring pattern in
which a spot pattern could be confirmed.
CA 02331602 2001-10-25
- 216 -
The X-ray radial distribution function resulted in
discontinuous peak curve.
The specific area was measured by the BET method,
resulting in that the specific area was 40 m2/g.
Comparative Preparation Method 50
A vanadium-molybdenum oxide was prepared by the
following method.
A vanadium oxide and a molybdenum oxide were mixed
at a ratio of 7:3, and the mixture was heated to 800°C
in a platinum crucible as to be melted and mixed. Then,
the mixture was cooled gradually so that the block-shape
vanadium oxide and molybdenum oxide were prepared. They
were crushed by a roller mill so that grains of vanadium
oxide-molybdenum oxide were prepared.
The size of crystal grains was measured from the
half value width of the X-ray analysis curve and the
angle of diffraction in accordance with the Scherrer's
Equation. The size of the crystal grain was 700 A or
larger.
The RHEED pattern resulted in a ring pattern in
which a spot pattern could be confirmed.
The X-ray radial distribution function resulted in
discontinuous peak curve.
The specific area was measured by the BET method,
resulting in that the specific area was 10 m2/g.
Comparative Preparation Method 51
A titanium sulfide was prepared by the following
method.
A titanium disulfide powder manufactured by Kojundo
CA 02331602 2001-10-25
-217-
Kagaku was dried at 400°C in a vacuum state.
The size of crystal grains was measured from the
half value width of the X-ray analysis curve and the
angle of diffraction in accordance with the Scherrer's
Equation. The size of the crystal grain was 900 A or
larger.
The RHEED pattern resulted in a ring pattern in
which a spot pattern could be confirmed.
The X-ray radial distribution function resulted in
discontinuous peak curve.
The specific area was measured by the BET method,
resulting in that the specific area was 50 m2/g.
Comparative Preparation Method 52
A lithium-iron-cobalt oxide was prepared by the
following method.
Lithium carbonate, iron acetate and cobalt
carbonate were mixed at the same mole ratio, and
decomposed at 600°C in air so that an iron cobalt oxide
was prepared. Then, it was crushed by a ball mill so
that grains were obtained.
The size of crystal grains was measured from the
half value width of the X-ray analysis curve and the
angle of diffraction in accordance with the Scherrer's
Equation. The size of the crystal grain was 1000 A or
larger .
The RHEED pattern resulted in a ring pattern in
which a spot pattern could be confirmed.
The X-ray radial distribution function resulted in
discontinuous peak curve.
CA 02331602 2001-10-25
-218-
The specific area was measured by the BET method,
resulting in that the specific area was 40 m2/g.
Comparative Preparation Method 53
A manganese containing magnesium added thereto-
vanadium oxide was prepared by the following method.
A manganese dioxide, vanadium pentaoxide and
magnesium hydroxide were mixed at a mol ratio of
10:10:1, and then they were decomposed at 700°C in air.
As a result, the manganese containing magnesium added
thereto-vanadium oxide was prepared. Then, they were
crushed by a ball mill so that grains were obtained.
The size of crystal grains was measured from the
half value width of the X-ray analysis curve and the
angle of diffraction in accordance with the Scherrer's
Equation. The size of the crystal grain was 1300 A or
larger.
The RHEED pattern resulted in a ring pattern in
which a spot pattern could be confirmed.
The X-ray radial distribution function resulted in
discontinuous peak curve.
The specific area was measured by the BET method,
resulting in that the specific area was 27 mz/g.
Comparative Preparation Method 54
A lithium-copper-cobalt oxide was prepared by the
following method.
A lithium carbonate, cobalt carbonate and copper
carbonate were mixed at the same mol ratio, and then
they were decomposed at 600°C in air. As a result, the
manganese containing magnesium added thereto-vanadium
CA 02331602 2001-10-25
-219-
oxide was prepared. Then, they were crushed by a ball
mill so that grains were obtained.
The size of crystal grains was measured from the
half value width of the X-ray analysis curve and the
angle of diffraction in accordance with the Scherrer's
Equation. The size of the crystal grain was 1100 A or
larger.
The RHEED pattern resulted in a ring pattern in
which a pot pattern could be confirmed.
The X-ray radial distribution function resulted in
discontinuous peak curve.
The specific area was measured by the BET method,
resulting in that the specific area was 10 m2/g.
Analyzing Apparatus
The positive electrode active material prepared by
Preparation Methods 46 to 55 and Comparative Preparation
Methods 46 to 54 were analyzed by using the following
apparatuses. The X-ray diffraction measurement was
performed by using MXP3VA manufactured by MacScience.
The RHEED measurement was performed by using JEM-
100SX manufactured by Nihon Denshi.
The specific area measurement by the BET method was
performed by using GEMIN12300 manufactured by
Micromeritex.
As a result of the comparisons between the
transition metal and the group 6A element according to
Preparation Methods according to the present invention
and those according to Comparative Preparation Methods,
the compounds according to the present invention
CA 02331602 2001-10-25
-220-
exhibited smaller particle grain size as compared with
the results of Comparative Preparation Method while
having an amorphous or microcrystal structure.
Manufacturing of Lithium Secondary Batte
Lithium secondary batteries were manufactured by
using the positive electrode active materials prepared
by the foregoing preparation methods.
Example 46
The positive electrode active material prepared by
the foregoing Preparation Method 46 was used to
manufacture a battery which had a simple structure,
which could be assembed simply and which had a cross
sectional shape schematically shown in Fig. 2.
In an atmosphere of dry argon gas, the negative
electrode active material 201 was used in such a manner
that the titanium mesh collector 200 was pressed against
the reverse side of the lithium metal foil. Then, the
surface of lithium was covered with a fluororesin thin
film by using thin solution of Lumiflon, which was a
fluororesin paint manufactured by Asahi Glass, so that
the negative electrode was manufactured.
Acetylene black powder and xylene solution of the
Lumiflon, which was a fluororesin paint manufactured by
Asahi Glass, were mixed with the positive electrode
active material which was prepared by Preparation Method
46 and which was lithium-manganese oxide. The mixture
was applied to the titanium mesh, and it was hardened at
80°C, and then heated with microwaves. As a result, the
positive electrode 203 was formed.
CA 02331602 2001-10-25
- 221 -
The electrolyte solution was prepared by
dissolving, by 1 M (mol/1), boronic acid tetrafluoride
lithium salt in an equal quantity mixture solvent of
propylene carbonate (PC) and dimethoxyethane (DME).
The separator 208 was manufactured in such a manner
that a polypropylene separator having small apertures
was sandwiched by unwoven polypropylene sheets.
The assembly was performed in such a manner that
the separator 208 was sandwiched between the negative
electrode 201 and the positive electrode 203, followed
by inserting them into a positive electrode case 207
made of titanium clad stainless steel. Then, the
electrolyte solution was injected. Then, the negative
electrode cap 206 made of the titanium clad stainless
steel and the insulating packing 210 made of fluorine
rubber were used for sealing so that the lithium
secondary battery was manufactured.
Example 47
The vanadium oxide prepared by Preparation Method
47 was used as the positive electrode active material, a
battery having a cross sectional shape schematically
shown in Fig. 2 was manufactured.
First, the nickel mesh collector was pressed
against the reverse side of the lithium metal foil in an
atmosphere of dry argon gas so that the negative
electrode was manufactured.
Acetylene black powder and Super Konak F which was
a fluororesin paint manufactured by Nihon Yushi were
mixed with the vanadium oxide serving as the positive
CA 02331602 2001-10-25
- 222 -
electrode active material and prepared by Preparation
Method 2, followed by adding xylene by a small quantity.
The mixture was applied to the nickel mesh, and it was
hardened at 150°C. As a result, the positive electrode
was manufactured.
Then, similar processes to those according to
Example 46 were performed so that the lithium secondary
battery shown in Fig. 2 was assembled.
Example 48
By using the positive electrode active material
prepared by Preparation Method 48, a battery which had a
simple structure, which could be assembled simply and
which had a cross sectional shape schematically shown in
Fig. 2 was assembled.
First, the nickel mesh collector was pressed
against the reverse side of the lithium metal foil in an
atmosphere of dry argon gas so that the negative
electrode was manufactured.
Acethylene black powder and tetrafluoroethylene
polymer powder were mixed with the lithium-nickel oxide
prepared by Preparation Method 48 and serving as the
positive electrode active material. The mixture was
pressed against the nickel mesh with heat as to be
formed into the positive electrode 203.
Then, similar processes to those according to
Example 46 were performed so that the lithium secondary
battery shown in Fig. 2 was assembled.
Example 49
The lithium-nickel-cobalt oxide prepared by
CA 02331602 2001-10-25
- 223 -
Preparation Method 49 was used as the positive electrode
active material so that the battery shown in Fig. 2 was
manufactured by the process similar to that according to
Example 48.
Example 50
The vanadium-molybdenum oxide prepared by
Preparation Method 50 was used as the positive electrode
active material so that the battery shown in Fig. 2 was
manufactured by the process similar to that according to
Example 48.
Example 51
The vanadium-molybdenum oxide prepared by
Preparation Method 51 was used as the positive electrode
active material so that the battery shown in Fig. 2 was
manufactured by the process similar to that according to
Example 48.
Example 52
The titanium sulfide prepared by Preparation Method
52 was used as the positive electrode active material so
that the battery shown in Fig. 2 was manufactured by the
process similar to that according to Example 48.
Example 53
The lithium-iron-cobalt oxide prepared by
Preparation Method 53 was used as the positive electrode
active material so that the battery shown in Fig. 2 was
manufactured by the process similar to that according to
Example 48.
Examgle 54
The manganese containing magnesium added thereto-
CA 02331602 2001-10-25
- 224 -
vanadium oxide prepared by Preparation Method 54 was
used as the positive electrode active material so that
the battery shown in Fig. 2 was manufactured by the
process similar to that according to Example 48.
Example 55
The lithium-copper-cobalt oxide prepared by
Preparation Method 55 was used as the positive electrode
active material so that the battery shown in Fig. 2 was
manufactured by the process similar to that according to
Example 48.
Comparative Example 46
By using the positive electrode active material
prepared by Comparative Preparation Method 46, the
battery having a cross sectional shape schematically
shown in Fig. 2 was manufactured.
First, the titanium mesh collector 200 was pressed
against the reverse side of the lithium metal foil in an
atmosphere of dry argon so that the negative electrode
was manufactured.
Acetylene black powder and tetrafluoroethylene
polymer powder were mixed to the lithium-manganese oxide
prepared by Comparative Preparation Method 46 and
serving as the positive electrode active material as to
be, with heat, pressed and formed into the positive
electrode 203.
Then, similar processes to those according to
Example 46 were performed so that the lithium secondary
battery was assembled.
Comparative Example 47
CA 02331602 2001-10-25
-225-
The vanadium oxide prepared by Comparative
Preparation Method 47 was used as the positive electrode
active material so that the battery shown in Fig. 2 was
manufactured by the process similar to that according to
Example 48.
Comparative Example 48
The lithium-nickel oxide prepared by Comparative
Preparation Method 48 was used as the positive electrode
active material so that the battery shown in Fig. 2 was
manufactured by the process similar to that according to
Example 48.
Comparative Example 49
The lithium-nickel-cobalt oxide prepared by
Comparative Preparation Method 49 was used as the
positive electrode active material so that the battery
shown in Fig. 2 was manufactured by the process similar
to that according to Example 48.
Comparative Example 50
The vanadium-molybdenum oxide prepared by
Comparative Preparation Method 50 was used as the
positive electrode active material so that the battery
shown in Fig. 2 was manufactured by the process similar
to that according to Example 48.
Comparative Exa~le 51
The titanium sulfide prepared by Comparative
Preparation Method 51 was used as the positive electrode
active material so that the battery shown in Fig. 2 was
manufactured by the process similar to that according to
Example 48.
CA 02331602 2001-10-25
- 226 -
Comparative Example 52
The lithium-iron-cobalt oxide prepared by
Comparative Preparation Method 52 was used as the
positive electrode active material so that the battery
shown in Fig. 2 was manufactured by the process similar
to that according to Example 48.
Comparative Example 53
The manganese containing magnesium added thereto-
vanadium oxide prepared by Comparative Preparation
Method 53 was used as the positive electrode active
material so that the battery shown in Fig. 2 was
manufactured by the process similar to that according to
Example 48.
Comparative Example 54
The lithium-copper-cobalt oxide prepared by
Comparative Preparation Method 54 was used as the
positive electrode active material so that the battery
shown in Fig. 2 was manufactured by the process similar
to that according to Example 48.
Evaluation of Performance of Lithium Secondarv Battery
The performance of lithium secondary batteries
according to Examples and Comparative Examples was
evaluated. The evaluation was performed by a charge and
discharge cycle test under the following conditions with
respect to the cycle life of the batteries according to
Comparative Examples.
The conditions for the cycle test were made as
follows: the charge and discharge was performed by 0.2C
(electric current which was 0.2 times capacity/time),
CA 02331602 2001-10-25
-227-
pause for 30 minutes and a cut-off voltage of 1.0V was
applied. A charging/discharging apparatus HJ-1O1M6
manufactured by Hokuto Electric was used. The
charge/discharge test was commenced at discharge, the
battery capacity was evaluated by the quantity of the
third discharge and the cycle life was evaluated by the
number of cycles when the battery capacity had
deteriorated to 60% or less.
The lithium batteries using the positive electrode
active materials respectively according to the present
invention and the comparative examples, that is, the
battery capacities and cycle life of the examples of the
present invention and the comparative examples were
evaluated as shown in Table 12 while making the
performance of the battery according to the comparative
example to be a reference value of 1.
As can be understood from Table 12, the comparisons
made between Examples 46 to 55 and Comparative Examples
46 to 54 resulted that the use of the batteries
according to the present invention enabled the capacity
of the battery to be enlarged and the cycle life to be
lengthened.
CA 02331602 2001-10-25
-228-
Table 12
Capacity Cycle life
of
Lithium Battery Battery (Present
Manufactured
(Present Invention/
Invention/ Comparative
Comparative Comparative Example)
Examples Examples Example)
Example 46 Comparative 2.3 4.g
Example 46
Example 47 Comparative 3.1 3.2
Example 47
Example 48 Comparative 2,1 1.7
Example 48
Example 49 Comparative 2.0 1.4
Example 49
Example 50 Comparative 4.1 2,g
Example 50
Example 51 Comparative 3.2 2.1
Example 50
Example 52 Comparative 2.4 1.4
Example 51
Example 53 Comparative 2.3 1.5
Example 52
Example 54 Comparative 1.7 2.0
Example 53
Example 55 Comparative 2.2 ~.
Example 54
Further, combinations of the embodiments of the
present invention will enable further improved secondary
battery to be obtained.
According to the present invention, if dendrite of
lithium or zinc grows at the time of charge, short
circuits between the negative electrode and the positive
electrode can be prevented. Therefore, a lithium
CA 02331602 2001-10-25
-229-
secondary battery, a nickel zinc secondary battery and
an air zinc secondary battery exhibiting a long
charge/discharge cycle life can be manufactured.
Further, the metal lithium can be used as the negative
electrode active material. Therefore, a secondary
battery exhibiting a high energy density can be
manufactured while improving safety.
Although the invention has been described in its
preferred form with a certain degree of particularly, it
is understood that the present disclosure of the
preferred form can be changed in the details of
construction and the combination and arrangement of
parts may be resorted to without departing from the
spirit and the scope of the invention as hereinafter
claimed.