Note: Descriptions are shown in the official language in which they were submitted.
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
LENTIVIRAL VECTORS
This application is a continuation-in-part of 08/935,312,
filed September 22, 1997, which is incorporated by reference
to the extent that it does not directly conflict with the
teachings of the present application.
This application is also a nonprovisional of Serial No.~
60/086,635, filed May 26, 1998, which likewise is incorporated
by reference.
Mention of Government Grant
The inventions disclosed herein may have arisen in part
from work done under one or more U.S. government grants,
including NIH grant No. P50 HL-59412. Consequently, the U.S.
Government may have certain rights in the inventions.
FIELD OF THE INVENTION
The present invention relates to improved lentivirus
derived packaging and transducing vectors useful for the
expression of genes at high levels in eukaryotic cells. The
improved vectors are safer, yet permit increased efficiency of
packaging the recombinant viral genome and increased long-term
gene expression.
BACKGROUND OF THE INVENTION
1. Gene Transfer; Gene Therapy
Viral vectors transduce genes into target cells with high
efficiencies owing to specific virus envelope-host cell
receptor interaction and viral mechanisms for gene expression.
Consequently, viral vectors have been used as vehicles for the
transfer of genes into many different cell types including
whole embryos, fertilized eggs, isolated tissue samples, and
cultured cell lines. The ability to introduce and express a
foreign gene in a cell is useful for the study of gene
expression and the elucidation of cell lineages (J. D. Watson
et al., Recombinant DNA, 2d Ed., W.H Freeman and Co., NY
[1992], pp. 256-263). Retroviral vectors, capable of
integration into the cellular chromosome, have also been used
for the identification of developmentally important genes via
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
2
insertional mutagenesis (J. D. Watson et al., supra, p. 261).
Viral vectors, and retroviral vectors in particular, are also
used in therapeutic applications (e. g., gene therapy), in which
a gene (or genes) is added to a cell to replace a missing or
defective gene or to inactivate a pathogen such as a virus.
In view of the wide variety of potential genes available
for therapy, it is clear that an efficient means of delivering~~
these genes is sorely needed in order to fulfill the promise
of gene therapy as a means of treating infectious, as well as
l0 non-infectious diseases. Several viral systems including
murine retrovirus, adenovirus, parvovirus (adeno-associated
virus), vaccinia virus, and herpes virus have been developed
as therapeutic gene transfer vectors (For review see, A.W.
Nienhuis et al., Hematology, Vol. l6:Viruses and Hone Marrow,
N.S. Young (ed.), pp. 353-414 [1993]).
Factors affecting viral vector usage include tissue
tropism, stability of virus preparations, genome packaging
capacity, and construct-dependent vector stability. In
addition, in vivo application of viral vectors is often limited
by host immune responses against viral structural proteins
and/or transduced gene products.
One of the key issues in human gene therapy is the
toxicity and safety to the treatment subjects. Gene therapy
applications in humans have met with problems associated with
the host immune responses against the gene delivery vehicles
or the therapeutic gene products. Viral vectors (e. g.,
adenovirus) which co-transduce several viral genes together
with the therapeutic genes) are particularly problematic. For
example, readministration is necessary for adenovirus vectors
because of the transient nature of viral gene expression. As
such, a host immune response to the vector or the therapeutic
gene product may be detrimental (B.C. Trapnell and M.
Gorziglia, Curr. Op. Biotechnol., 5:617-625 [1994]; and S.K.
Tripathy et al., Nature Med., 2:545-550 [1996]).
Although MLV vectors have not been reported to induce
cytotoxicity and do not elicit strong host immune responses,
lentiviral vectors such as HIV-1 which carry several
immunostimulatory gene products have the potential to cause
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/115I6 .
3
cytotoxicity and induce strong immune responses in vivo. The
latter are known to induce strong cell-mediated immune
responses upon transient exposure (M. Clerici et al., J. Inf.
Dis., 165:1012-1019 [1992]; M. Clerici et al., J. Amer. Med.
Assoc. , 271 :42-46 [1994] ; L.A. Pinto et al. , J. Clin. Invest . ,
96:867-876 [1995]; and S. Rowland-Jones et al., Nature Med.,
1:59-64 [1995]). However, this may not be a concern for~I
lentiviral derived transducing vectors, as the latter need not
encode any viral genes in the transducing vector.
Of course, in some instances, the purpose of the vector
is to provoke a clinically useful immune response against an
encoded protein.
Another important issue related to the lentiviral vector
usage is that of possible cytopathogenicity upon exposure to
some cytotoxic viral proteins. Exposure to HIV-1 proteins may
induce cell death or functional unresponsiveness in T cells (N.
Chirmule et al., J. Virol., 69:492-498 [1995]; C.J. Li et al.,
Science 268:429-431 [1995]; J.D. Lifson et al., Science
232:1123-1127 [1986]; I.G. Macreadie et al., Mol. Microbiol.,
19:1185-1192 [1996]; and T. Nosaka et al., Exp. Cell. Res.,
209:89-102 [1993]). During the development of the present
invention, it was observed that direct gene transfer into
tissue culture cells by the calcium-phosphate DNA
co-precipitation method could induce more than 80o cell death
which is caused mainly by necrosis and a residual percentage,
approximately 2-4%, by programmed cell death
A final concern is the possibility of generating
replication-competent, virulent virus by recombination.
Safety concerns have prompted much effort towards the
development of non-viral vector systems, such as
liposome-mediated gene transfer, naked DNA injections and gene
gun technology. However, all of these non-viral gene transfer
methods lack the ability to allow permanent integration of
foreign genes into the host cell chromosomes, and are
relatively inefficient. For long term expression of
therapeutic genes in target cells, efficient means of
transduction and genome integration are essential.
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516 .
4
2. Retroviruses; Retroviral Vectors
The term "retrovirus" is used in reference to RNA viruses
that utilize reverse transcriptase during their replication
cycle. The retroviral genomic RNA is converted into double-
s stranded DNA by reverse transcriptase. This double-stranded
DNA form of the virus is capable of being integrated into the
chromosome of the infected cell; once integrated, it is"
referred to as a "provirus . " The provirus serves as a template
for RNA polymerase II and directs the expression of RNA
molecules which encode the structural proteins and enzymes
needed to produce new viral particles. At each end of the
provirus are structures called "long terminal repeats" or
"LTRs." The LTR contains numerous regulatory signals including
transcriptional control elements, polyadenylation signals and
sequences needed for replication and integration of the viral
genome.
There are several genera included within the family
Retroviridae, including Cisternavirus A, Oncovirus A, Oncovirus
B, Oncovirus C,~Oncovirus D, Lentivirus, and Spumavirus. Some
of the retroviruses are oncogenic (i.e., tumorigenic), while
others are not. The oncoviruses induce sarcomas, leukemias,
lymphomas, and mammary carcinomas in susceptible species.
Retroviruses infect a wide variety of species, and may be
transmitted both horizontally and vertically. They are
integrated into the host DNA, and are capable of transmitting
sequences of host DNA from cell to cell. This has led to the
development of retroviruses as vectors for various purposes
including gene therapy.
Retroviral vectors derived from the amphotropic Moloney
murine leukemia virus (MLV-A), use cell surface phosphate
transporter receptors for entry and then permanently integrate
into proliferating cell chromosomes. The amphotropic MLV
vector system has been well established and is a popular tool
for gene delivery (See e.g., E.M. Gordon and W. F. Anderson,
Curr. Op. Biotechnol., 5:611-616 [1994); and A.D. Miller et
al., Meth. Enzymol., 217:581-599 [1993]).
Other retroviruses, including human foamy virus (HFV) and
human immunodeficiency virus (HIV) have gained much recent
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516 .
attention, as their target cells are not limited to dividing
cells and their restricted host cell tropism can be readily
expanded via pseudotyping with vesicular stomatitis virus G
(VSV-G) envelope glycoproteins (See e.g., J.C. Burns et al.,
5 Proc. Natl. Acad. Sci. USA 90:8033-8037 [1993]; A.M.L. Lever,
Gene Therapy. 3:470-471 [I996]; and D. Russell and A. D.
Miller, J. Virol., 70:217-222 [1996]). However, a useful~~
lentiviral vector system has not been well established, mainly
because of the lack of sufficient studies on lentiviral
vectorology and safety concerns.
While many viral vector systems are available, virtually
all of the current human gene therapy trials use retroviral
vectors derived from the amphotropic Moloney murine leukemia
virus (M-MuLV), such as pLNL6 (Genbank M63653), see Baker, et
al. , J. Virol. 61:1639 (1987) , for gene transfer (see also A.D.
Miller and C. Buttimore, Mol. Cell. Biol., 6:2895 [1986]).
Among the vectors known in the art, special note may be taken
of Chang, USP 5,693,508 (1997) which discloses retroviral
vectors contining chimeric MoMLV/CMV-IE/HIV-TAR LTRs. The
elements essential to the retroviral vector system are viral
structural proteins Gag, Pol and Env, the long terminal repeats
(LTR), the reverse transcription templates including primer
binding site (PBS) and polypurine tract (PPT), and the
packaging signals (psi [~]). The MLV-A vector system is
comprised of a packaging cell line expressing Gag, Pol and Env,
and a vector construct containing LTRs, PBS, PPT and the
packaging signal sequences. Up to 8 kbp of foreign sequences
can be inserted into the MLV vector and packaged into virus
particles. The commonly used amphotropic MLV packaging cell
lines such as PA317, PG-13, ~-CRIP, GP-AM12 and FLY-A13 produce
105-107 transducing units per ml after vector DNA transfection
(F. -L. Cosset et al . , J. Virol . , 69 : 7430-7436 [1995] ; H. Kotani
et al., Human Gene Ther., 5:19-28 [1994]; J.S. Lam et al.,
Human Gene Ther., 7:1415-1422 [1996]; D. Markowitz et al., J.
Virol., 62:1120-1124 [1988]; A.D. Miller and F. Chen, J.
Virol., 70:5564-5571 [1996]).
The M-MuLV system has several advantages: 1) this specific
retrovirus can infect many different cell types; 2) established
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
6
packaging cell lines are available for the production of
recombinant M-MuLV viral particles; and 3) the transferred
genes are permanently integrated into the target cell
chromosome. The established M-MuLV vector systems comprise a
DNA vector containing a small portion of the retroviral
sequence (the viral long terminal repeat or "LTR" and the
packaging or "psi" [~] signal) and a packaging cell line. The
gene to be transferred is inserted into the DNA vector. The
viral sequences present on the DNA vector provide the signals
necessary for the insertion. or packaging of the vector RNA into
the viral particle and for the expression of the inserted gene.
The packaging cell line provides the viral proteins required
for particle assembly (D. Markowitz et al., J. Virol., 62:1120
[198s] ) .
The vector DNA is introduced into the packaging cell by
any of a variety of techniques (e. g., calcium phosphate
coprecipitation, lipofection, electroporation, etc.). The
viral proteins produced by the packaging cell mediate the
insertion of the vector sequences in the form of RNA into viral
particles which are shed into the culture supernatant. The M-
MuLV system has been designed to prevent the production of
replication-competent virus as a safety measure. The
recombinant viral particles produced in these systems can
infect and integrate into the target cell but cannot spread to
other cells . These safeguards are necessary to prevent the
spread of the recombinant virus from the treated patient and
to avoid the possibility of helper virus-induced disease (A. D.
Miller and C. Buttimore, supra; and D. Markowitz et al.,
supra) .
After selection, producer cell clones can be established
to generate 104-106 transducing units per ml. Increased
transduction efficiencies may be achieved by modification of
the transduction protocols through means such as repetitive
infection steps, cocultivation with the producer cell line,
centrifugation, and modification of the culture conditions
using growth factors and fibronectin etc. (H. Kotani et al.,
Human Gene Ther., 5:19-28 [1994]; and T. Moritz et al., Blood
88:855-862 [1996] ) .
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516 .
7
Despite these advantages, existing M-MuLV-based retroviral
vectors are limited by several intrinsic problems: 1) they do
not infect non-dividing cells (D. G. Miller et al., Mol. Cell.
Biol., 10:4239 [1990]); 2) they produce only low titers of the
recombinant virus (A.D. Miller and G.J. Rosman, BioTechn., 7:
980 [1989]; and A.D. Miller, Nature 357: 455 [1992]); 3) they
express foreign proteins at low levels and often get "turned-~~
off" or inactivated after integration (A. D. Miller, Nature
357: 455 [1992]); (4) the instability of the enveloped virus
particles, as it is both difficult to concentrate in vitro and
difficult to manipulate in vi vo (A. D. Miller, Nature
357:455-460 [1992]); 5) the MLV LTR activity is also known to
be suppressed in embryonal cells (P.M. Challita et al., J.
Virol., 69:748-755 [1995]; and T.P. Loh et al., J. Virol.,
62:4086-4095 [1988]); and 6) long term expression after viral
integration is often restricted by transcription repression,
likely due to DNA methylation (J. Boyes and A. Bird, Cell
64:1123-1134 [1991]; and M. Szyf et al., Mol. Cell. Biol.,
10:4396-4400 [1990] ) .
The low production of recombinant virus produced by the
M-MuLV system (e. g., 106/ml) compared to the adenoviral system
(up to lOlz/ml) means that human cells are infected at a very
low efficiency. This low efficiency is particularly
problematic when the target cell type is represented at very
low numbers in the tissue to be infected. Although the
hematopoietic stem cell is a preferred target for gene therapy
in a large number of disorders, these cells are present at very
low frequencies. For example, totipotent embryonic stem cells
have been reported to occur at a frequency of 10-4 to 10-6 in
bone marrow (B. R. Glick and J.J. Pasternak, Molecular
Biotechnology, American Society for Microbiology, Washington,
D.C., p. 412 [1994]). Thus, the low titer produced by
existing M-MuLV vector systems is highly problematic for stem
cell infection.
The promoter present in the M-MuLV LTR is quite weak
compared with other viral promoters such as the human
cytomegalovirus immediate early (CMV-IE) enhancer/promoter.
In order to increase expression of the genes carried on the
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
8
retroviral vector, internal promoters possessing stronger
activities than the M-MuLV promoter have been utilized.
However, the inclusion of an internal promoter to drive the
expression of the inserted gene does not always lead to
increased levels of expression (D. Robinson et al., Gene
Therapy 2:269 [1995]). Apparently, the activity of the
internal promoter is significantly decreased because of ~~
interference from the upstream M-MuLV promoter (i.e.,
transcriptional read-through interference). .The dual
transcription-unit construct is, however, a common feature in
almost all M-MuLV vectors.
To create an improved retroviral vector suitable for a
wide variety of gene expression studies and gene therapy
applications, the clinically approved gene therapy vector pLNL6
has been modified to allow synthesis of high basal levels of
mRNA, and increased packaging efficiency (See e.g., co-pending
U.S. Patent Appln. Ser. No. 08/336,132, and PCT/US95/14576, to
Chang, herein incorporated by reference). However, other
limitations remain.
Given these limitations, it is clear that improved vector
systems are urgently needed to provide a means of delivering
and expressing genes efficiently in mammalian cells,
particularly human cells. Improved vectors will aid the study
of gene expression and development and are necessary if the
promise of gene therapy is to be realized.
The major limitation in the use of the simple retroviral
vectors in gene transfer is that use of the MLV-based vector
is restricted to dividing cells. This led to the development
of the present invention, in which lentiviruses, capable of
infecting non-dividing cells are provided.
3. Lentiviruses; Lentiviral Vectors
As used herein, the term "lentivirus" refers to a group
(or genus) of retroviruses that give rise to slowly developing
disease. Viruses included within this group include HIV (human
immunodeficiency virus; including HIV type 1, and HIV type 2),
the etiologic agent of the human acquired immunodeficiency
syndrome (AIDS); visna-maedi, which causes encephalitis (visna)
CA 02333481 2000-11-27
WO 00/00600 ~ PCT/US99/11516 .
9
or pneumonia (maedi) in sheep, the caprine arthritis-
encephalitis virus, which causes immune deficiency, arthritis,
and encephalopathy in goats; equine infectious anemia virus,
which causes autoimmune hemolytic anemia, and encephalopathy
in horses; feline immunodeficiency virus (FIV), which causes
immune deficiency in cats; bovine immune deficiency virus
(BIV), which causes lymphadenopathy, lymphocytosis, and~~
possibly central nervous system infection in cattle; and simian
immunodeficiency virus (SIV) , which cause immune deficiency and
encephalopathy in sub-human.primates. Diseases caused by these
viruses are characterized by a long incubation period and
protracted course. Usually, the viruses latently infect
monocytes and macrophages, from which they spread to other
cells. HIV, FIV, and SIV also readily infect T lymphocytes
(i.e., T-cells).
Lentivirus virions have bar-shaped nucleoids and contain
genomes that are larger than other retroviruses. Lentiviruses
use tRNAlYs as primer for negative-strand synthesis, rather than
the tRNAPr° commonly used by other infectious mammalian
retroviruses. The lentiviral genomes exhibit homology with
each other, but not with other retroviruses (See, Davis et a1 . ,
Microbiology, 4th ed., J.B. Lippincott Co., Philadelphia, PA
[1990] , pp. 1123-1151) . An important factor in the disease
caused by these viruses is the high mutability of the viral
genome, which results in the production of mutants capable of
evading the host immune response. It is also significant that
they are capable of infecting non-dividing cells.
Lentiviruses including HIV, SIV, feline immunodeficiency
virus (FIV) and equine infectious anemia virus (EIAV) depend
on several viral regulatory genes in addition to the simple
structural gag-pot-env genes for efficient intracellular
replication. Thus, lentiviruses use more complex strategies
than classical retroviruses for gene regulation and viral
replication, with the packaging signals apparently spreading
across the entire viral genome. These additional genes display
a web of regulatory functions during the lentiviral life cycle .
For example, upon HIV-1 infection, transcription is
up-regulated by the expression of Tat through interaction with
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/I1516 .
an RNA target (TAR) in the LTR. Expression of the full-length
and spliced mRNAs is then regulated by the function of Rev
which interacts with RNA elements present in the gag region and
in the env region (RRE) (S. Schwartz et al., J. Virol.,
5 66:150-159 [1992]). Nuclear export of gag-pol and env mRNAs
is dependent on the Rev function. In addition to these two
essential regulatory genes, a list of accessory genes,~~
including vif, vpr, vpx, vpu, and nef, are also present in the
viral genome and their effects on efficient virus production
10 and infectivity have been demonstrated, although they are not
absolutely required for virus replication (K. and F.
along-Staal, Microbiol. Rev., 55:193-205 [1991]; R.A.
Subbramanian and E. A. Cohen, J. Virol. 68:6831-6835 [1994];
and D. Trono, Cell 82:189-192 [1995]}.
HIV-1 virions contain 60 % protein and 2 % nucleic acid.
The genome consists of two molecules of linear positive-sense
single stranded RNA (held together by hydrogen bonds to form
a dimer) . Even within a single virion, these molecules need not
be identical. Hence, genetic variation can occur through
recombination between the two viral RNAs of a single virion.
The HIV-1 genome is about 9.7 kb in length. Many HIV-1
proviral genome sequences have been sequenced in their
entirety. The sequence GenBank M19921, LOCUS HIVNL43, Human
immunodeficiency virus type 1, NY5/BRU (LAV-1} recombinant
clone pNL4-3, 9709 by ss-RNA, is used as a reference sequence
in this discussion. The construction of pNL4-3 has been
described in Adachi,A., Gendelman,H.E., Koenig,S., Folks, T.,
Willey,R., Rabson,A. and Martin,M.A., Production of acquired
immunodeficiency syndrome-associated retrovirus in human and
nonhuman cells transfected with an infectious molecular clone,
J. Virol. 59, 284-291 (1986). pNL4-3 is a recombinant
(infectious) proviral clone that contains DNA from HIV
isolates NY5 (5' half) and BRU (3' half). The site of
recombination is the EcoRI site at positions 5743-5748. The
final sequence is set forth in Dai,L.C., Littaua,R.,
Takahashi, K. and Ennis, F.A. , Mutation of human immunodeficiency
virus type 1 at amino acid 585 on gp41 resultis in loss of
killing by CD8+ A24-restricted cytotoxic T lymphocytes, J.
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
11
Virol. 66, 3151-3154 (1992).
For several reasons, the HIV-1 genome has a high mutation
rate. First,~there can be recombination between the two RNAs
of a single virion. Secondly, a single cell can be infected
by more than one viral particle simultaneously, and
recombination occur between the two viral genomes. Finally,
the HIV reverse transcriptase has a high frequency of ~~
misincorporation (:1700 to 1:4000). The replication error rate
for HIV is such that each newly synthesized HIV genome carries
on average approximately one mutation. For all of these
reasons, there is not one HIV-1 sequence, but rather a family
of closely related sequences. Different HIV-1 sequences may
be identified even in different samples isolated from a single
individual. The degree of genetic variation observed is
phenomenal--up to 20% within an infected individual. This is
essentially due to remorseless cycles of viral replication,
most probably due tochronic activation of the immune system.
It can be estimated that the number of variants in existence
worldwide must be in excess of 10(14)-10(18), and given the
nature of RNA viruses even more novel variants should emerge.
HIV-1's are currently divided into two genetic groups
based on phylogenetic reconstruction using DNA sequences. The
majority of these sequences fall into the M (major) group,
while a smaller, but growing, number of sequences are
classified as O (outlier) . Most HIV-1 strains from around the
world can be placed into one of nine nucleotide
sequence-defined Glades; these Glades have been given the
letter designations A through I. However, more than a dozen
HIV-1 strains isolated from patients have now been shown to
have chimeric genomes in that their gag and env genomic regions
cluster with different
Glades. Interclade recombination is relatively easy to
demonstrate because strains from different Glades typically
differ substantially in their nucleotide sequence identities.
For example, the env gene sequences of HIV-1 strains of
different Glades may differ by 20% or more. As might be
expected, interclade HIV-1 recombinants have most often been
detected in geographic regions where two or more Glades are
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
12 ,
prevalent. At least 17 HIV Glades have now been reported in
humans: nine HIV-1 Glades in the major grouping (A through I),
three HIV-1 group O group "outlier" Glades, and five HIV-2
Glades. An additional three lentiviruses are known in nonhuman
primate species (African green monkeys, mandrils, and Syke's
monkeys). Thus the potential gene pool for primate lentivirus
recombination is on the order of 20, e.g., 20 gag genes and 20~~
pol genes . The current HIV-1 Glades may have arisen in part
through past recombination between some of these genes. Viable
recombinants between SIV and HIV ("SHIV" strains) have been
genetically engineered in research laboratories..
The principal elements of the HIV-1 genome are set forth
below, in the 5' to 3' direction. For further information, see
Vaishnav and Wong-Staal, Ann. Rev. Biochem., 60: 577-630
(1991). The positions of each element are given according to
the Genbank numbering of the complete genome sequence (M19921}
cited above. That means that the numbering begins with the
first base of the 5' LTR, not with the cap site. The exact
positions will vary from strain to strain, and some elements
are better defined than others. Note that some genetic
elements overlap, and that two (Tat and Rev) are interrupted.
For a compilation of numerous sequences and alignments, at both
the nucleic acid and amino acid levels, for many lentiviruses
and othe retroviruses, see the HIV Sequence Database at
http://hiv-web.lanl.gov.
5' LTR (1-634)
Each end of the DNA provirus contains the so-called long
terminal repeats (LTRs) . The 5' LTR and 3' LTR regions are
essentially identical in the wild-type HIV-1 genome. These LTRs
are 634-by non-coding sequences, located at the extreme 5' and
3' ends of the proviral genome, that contain enhancer and
promoter regions. The LTRs consist of three distinct coding
regions, U3, R, and U5, which can be subdivided into the
separate enhancer and promoter regions. The U3 region is 450,
the R sequence 100 and the U5 region some 85 nt long.
Transcription initiates at the first base of the R region in
the 5' LTR, and polyadenylation occurs immediately after the
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516 .
13
last R region base in the 3' LTR. The primary transcript is
thus about 600 bases shorter than the provirus.
The U3 region includes several features of interest: the
integration attachment site (att) at the tar 5' end, the
S promoter TATA box ( a segment of DNA, located approximately 19
27 base pairs upstream from the start point of eukaryotic
structural genes, to which RNA polymerase binds), promoter~~
(SP1) regions (promoter binding site for RNA polymerase and
reverse transcriptase), the kappa-enhancer (contains two
imperfect 11-by repeats, GGGACTTTCC), and IL-1 and IL-2
homologous enhancers.
The R region (454-550) contains the transcription
initiation site, the TAR (Tat-activating) region and the poly
A signal (-AATAAAA-); the latter is significant only in the 3'
LTR). The primary transcript corresponds to bases 455 to
9626.
The U5 region contains a polyA downstream element and a
second integration attachment site at the 3' end. These are
significant only in the 3' LTR.
PBS
Immediately downstream of the 5' LTR is the primer binding
site (637-651) for minus-strand DNA synthesis, called the RNA
cap. The primer binding site is complementary Lo the 3' end
of a Lys transfer RNA.
5' Leader (L)
The 5' leader (L) , the untranslated region between the
primer binding site and the initiation codon for gag, has two
elements worthy of note.
The first is the major 5' splice donor (SD) site (the
splice point is at 748) which is used for the processing of
full-length genomic RNA to subgenomic mRNA for the syntheses
of various viral proteins. The major splice donor site is so
called because it acts as the donor site during splicing of the
vif, vpr, tat, rev, vpu-env and nef subgenomic RNAs (The Gag
Pol polyprotein is translated from genomic RNA.) There are
also minor splice donor sites in the vicinity of the first exon
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
14
of the rev gene.
The other is the major packaging signal (psi) (651-669)
which serves as a contact point for the Gag nucleocapsid (Ncp7)
protein to bind the RNA and to incorporate it into virus
particles. Note that one can define an extended packaging
signal extending into the gag gene, to about 820.
The 5' leader also contains a sequence which participates ~~
in the dimer-linkage structure of 70S RNA. This DLS overlaps
with the major packaging signal.
A secondary structure model of the leader, and the 5' end
of gag, was prepared by Baudin, et al., J. Mol. Biol., 229:
382-97 (1993).
Structural Genes
The gag gene encodes a polyprotein (55kDa) (CDS 790..2292)
which is cleaved by the viral protease (see pol) to yield
various core and nucelocapsid proteins. The gag coding region
extends from the ATG initiation codon at nucleotide 337 to
nucleotide 1837 relative to the RNA cap site. The polyprotein
is translated from unspliced viral RNA. The precursor Gag
protein is cleaved by protease to produce p17 (the major matrix
MA protein, involved in membrane anchoring, env interaction,
and nuclear transport of viral core), p24 (the core capsid CA
protein), p7 (the nucleocapsid NC protein, which binds RNA),
and p6 (which binds Vpr). A pair of zinc finger motifs in the
NC protein binds to the major packaging signal in the viral
RNA.
The gag gene is believed by some authors to contain one
or more minor packaging signals.
The pol gene (CDS est. 2085..5096) codes for a large
polyprotein which is a precursor to the virion proteins
providing the viral enzyme functions: protease, reverse
transcriptase, and integrase. The gag and pol genes overlap
by 241 nucleotides, and are in different reading frames. A
slippage sequence in or upstream of the gag-pol overlap region
induces an occasional ribosomal frameshift at a frequency
(about 5%) which ensures that Gag proteins are made in large
amounts and Pol proteins in small amounts. Initially, a gag-
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516 .
pol fusion protein (p190) is created as a result of the
ribosomal frameshift, which does not interrupt translation.
The viral protease cleaves Gag from Pol, and further digests
Gag and Pol to separate the various mature proteins. In the
5 case of Pol, the cleavage products are protease (p10), reverse
transcriptase (p50), Rnase H (ply) and integrase (p31).
Roughly 50 0 of the RT remains linked to Rnase H as a single ~~
polypeptide (p66). The principal functional form of RT is
actually a heterodimer of p66 and p50. All pol gene products
10 are found within the capsid of free HIV-1 virions.
Reverse transcriptase is responsible for the synthesis of
double-stranded DNA from the viral RNA. Activity of RT is
localized to the N-terminus. RT in HIV has an extremely high
error rate, 1/1700 nucleotides. At the 3' end of the pol
15 coding region is the coding region for viral
endonuclease/integrase. Integrase functions to integrate the
proviral DNA in the host genome.
The env gene (CDS 6221..8785) is located at the 3' end of
the genome. It encodes the envelope protein gp160, some of
which is cleaved to yield the envelope proteins gp120 and gp4l.
Both function in cell recognition on the outer envelope of a
released virus. The C-terminus of gp120 interacts with the
viral receptor CD4 of human T lymphocytes to facilitate the
viral entry into the host cell. Only a 12 amino acid sequence
in gp120 is necessary for binding to CD4; the rest of the
protein is mutable. The gp120 polypeptide contains nine
conserved intrachain disulfide bridges and, within this
scaffolding, folds into five globular domains (I-V) . There are
five hypervariable regions (Vl-V5) whose sequences vary
especially widely among HIV-1 isolates.
Regulatory Genes
The tat gene (CDS 5830..6044, 8369..8414) encodes Tat, a
trans-activating protein, the most important activator of of
the LTR promoter region. Three functional domains have been
identified: an amino terminal amphipathic helix, a cluster of
seven cysteine residues, and a stretch of basic amino acids
involved in nuclear localization. It is known that
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
16
conservative mutations of the acidic amino acids of the
amphipathic helix are tolerated. Tat mediates the 5' LTR by
interacting with its R region, in a segment termed the "TAR"
(trans-activating response) element (bases 436-497). The "TAR"
- 5 element forms a stable stem loop structure that interacts with
the Tat protein to prevent premature termination of
transcription initiation. Tat is reported in the literature~I
to be absolutely essential for HIV transcription and
consequently for viral replication.
The rev gene (CDS 5969..6044, 8369..8643) encodes Rev,
another transactivator. Rev is phosphorylated at serine
residues, but serine substitution mutants which are not
phosphorylated are fully active. The amino terminal 20 amino
acids and the carboxy terminal 25 amino acids are known to be
dispensable. There are two important domains, a stretch of
basic amino acids, which is involved in nuclear localization
and in interaction with RRE RNA, and a leucine-rich region,
presumed to be involved in transactivation, whose leucines are
intolerant of mutation. Rev is a protein whose target is
termed RRE (Rev-response element), on the env protein coding
region of the mRNA. Interaction of Rev with the RRE region
apparently allows for transport of unspliced RNA from the
nucleus to the cytoplasm. RRE (7758-7992) is an RNA secondary
structure element. Proviruses lacking Rev function remain
transcriptionally active but fail to generate new viral
particles.
Accessory Genes
The nef gene (CDS 8787..9407) encodes Nef, and overlaps
the env gene and the 3' LTR. Nef may be involved in signal
transduction, although this is controversial. There has also
been speculation that Nef down-regulates viral expression. The
Nef protein does not appear to be essential to the HIV life
cycle in tissue culture.
The vif gene (CDS 5041..5619) encodes Vif, the virion
infectivity factor. Vif-deficient mutants are typically much
less efficient than wild type HIV at cell-free (as opposed to
cell-to-cell) virus transmission. It is not a virion component
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516 .
17
and the mechanism by which it affects infectivity is unclear.
The vpr gene (CDS 5559..5849) encodes Vpr, a virion
protein which accelerates the replication and cytopathic effect
of HIV-1 in CD4+ T-cells. About 100 copies of Vpr are
associated with each virion.
The vpu gene (CDS 6061..6306) encodes Vpu. The vpu gene
encodes part of a polycistronic transcript which also includes~~
the env gene. Vpu is a cytoplasmic protein which is thought
to facilitate assembly and/or release of viral particles.
PPT (bases 9059-9075)
Immediately upstream from the 3' LTR is the polypurine
tract vital to initiation of positive-strand DNA synthesis.
3 'LTR (9076..9709)
The 3' LTR is identical to the 5' LTR, but is
significantly mainly by virtue of its poly-A signal
(9602..9607), and the "R" repeat sequence (9529..9626) allowing
RT jumping during DNA synthesis.
Infectivity
HIV-1 infects activated and resting lymphocytes,
terminally differentiated monocytes and neuronal cells through
cellular receptors and co-receptors such as CD4, chemokine
receptors and galactosyl ceramide (J.M. Harouse et a1 . , Science
253:320-323 [1991]; and R.A. Weiss, Science 272:1885-1886
[1996]). The restricted lentiviral host cell tropism can be
expanded by pseudotyping the virus particles with broadly
tropic viral envelope proteins from human T cell leukemia virus
type I (HTLV-I), amphotropic MLV envelope protein or the
vesicular stomatitis virus G glycoprotein (J. C. Burns et al.,
Proc. Natl. Acad. Sci. USA. 90:8033-8037 [1993]; N.R. Landau
et a1 . , J. Virol . , 65 : 162-169 [1991] ; K.A. Page et al. , J.
Virol., 64:5270-5276 [1990]; and D. H. Spector et al., J.
Virol., 64:2298-2308 [1990]). Alternatively, a CD4 receptor
can be introduced into target cells by adenovirus transduction
before HIV vector transduction in a two-step transduction
protocol (K. Miyake et al., Human Gene Ther., 7:2281-2286
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
18
[1996]). Naldini et al. have demonstrated that HIV-1 vectors
pseudotyped with MLV-A or VSV-G envelope could produce up to
X 105 transducing units/ml of vectors capable of infecting
nondividing cells such as macrophages and terminally
5 differentiated neurons (L. Naldini et a1. , Science 272 :263-267.
(1996] ) .
Infection of nondividing cells by lentiviruses such as~~
HIV-1 is mediated by the nuclear localization signal (NLS) in
the Gag MA protein (M.I. Bukrinsky et al., Nature 365:666-669
[1993]). Efficient viral entry and integration into
non-dividing cells may also require some of the accessory gene
products such as Vpr (T. M. Fletcher et al., EMBO J.,
15:6155-6165 [1996]; and N.K. Heinzinger et al., Proc. Natl.
Acad. Sci. USA. 91:7311-7315 [1994]).
Cytotoxicity
One difficulty related to HIV vector development
encountered during the development of the present invention is
the cytotoxicity of many HIV gene products to human cells. In
particular, it has been difficult to establish continuous cell
lines expressing the essential structural proteins Gag, Pol and
Env for particle assembly. Cell lines expressing Tat, Rev, Nef
have been established. However, expression of Gag, Rev and Vpr
has been shown to induce cytopathoiogy, cell death and cell
cycle arrest in human cells (See, M. Emerman, Curr. Biol..,
6:1096-1103 [1996]; G. Miele and A. M. L. Lever, Gene Ther.,
3:357-361 [1995]; and T. Nosaka et al., Exp. Cell. Res-,.
209:89-102 [1993]}. The development of a tightly inducible
system was favored for a lentiviral packaging cell line (H. Yu
et al., J. Virol., 70:4530-4537 [1996]). HIV-1 Vpr also
induces apoptosis in human cells. The expression of VSV-G
protein induces syncytium formation which again is problematic
for establishing a packaging cell line.
Other Safety Issues
Unlike other retroviruses, the lentiviruses are able to
infect non-dividing cells. Hence, lentiviral vectors have the
potential to overcome this limitation of prior vectors systems.
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
19
However, there is an understandable concern as to the safety
of lentiviral vectors, especially those derived from HIV-1.
The foremost safety consideration is the risk that either
packaging vector and transducing vector will recombine, either
with themselves or with defective virus endogenous to the host
cell genome, to produce a replication-competent, infectious
lentivirus, in particular, replication-competent HIV (RC-HIV). ~~
While the vector constructs are replication-defective, the risk
of generating RC-HIV is increased with the DNA co-transfection
procedure, when a high frequency of recombination events can
occur at both DNA and RNA levels. Thus, the packaging
constructs and the transducing vectors of lentiviruses could
potentially recombine and generate replication-competent
viruses (RCV) as do the MLV vectors during co-transfection.
However, the chances of generating RCV are reduced if multiple
recombination steps are necessary, and if the key envelope gene
of HIV-1 is deleted.
Due to the restricted tissue tropism of the native
lentiviral env gene, lentiviral vectors were developed that use
a pan-tropic envelope gene such as amphotropic MLV env or
VSV-Gs. This reduced the possibility of producing a wild-type
lentiviral RCV (e. g., an HIV-1 Env-trophic virus). However,
it is still possible that an RCV could be generated via
recombination with these pan-tropic env genes or endogenous
retrotransposon env genes. The fact that human genomes carry
numerous human endogenous retroviral sequences (HERVs) further
increases the probability of generating a fortuitous
recombinant RCV (T. P. Loh et al., J. Virol., 62:4086-4095
[1988]). For example, a recent study demonstrated that a
member of the HERV family encodes a protein resembling the
lentivirus rev gene product with a nucleolar localization
signal, a putative RNA binding domain, and a sequence similar
to the Rev effector domain consensus sequence (R. Lower et a1 . ,
J. Virol., 69:141-149 [1995]).
Some human tissues and cell lines such as the placenta,
syncytiotrophoblasts, brain, differentiated U-937 cells,
teratocarcinomas, and the mammary carcinoma T47D cells have
been shown to express complete human endogenous retrovirus env
CA 02333481 2000-11-27
WO 00/00600 PCf/US99/11516
gene and release retrovirus-like particles. These endogenous
retroviruses may form defective particles which lack
infectivity. Although the possibility of generating a
recombinant RC-HIV with an HERV env gene is low, it is worth
5 examining.
Discussion of Particular Lentiviral Vector Systems
Page, et al., J. Virol., 64: 5270-6 (1990) prepared a
noninfectious transducing vector HIV-gpt in which the env gene
was replaced with SV-gpt, and a helper vector providing either
10 the HIV-1 gp160 env gene (the HXB2-env vector) or the
amphotropic MLV env gene (the SV-A-MLV-env vector).
Shimada, et al., J. Clin. Investig., 88: 1043-7 (1991)
describes a recombinant HIV-1 gene transfer system employing
two vectors. The packaging vector has a CMV promoter, and an
15 insertion mutation in the packaging signal. The transducing
vector replaces part of gag, and all of pol, with a reporter
gene cassette. The vector system uses wild type HIV-1 Env
proteins to target CD cells. It is worth noting that Shimada
et al. state that sequences upstream of gag AUG are important
20 for gag expression, implying that they cannot be modified.
Corbeau, et al, Proc. Nat. Acad. Sci. (USA), 93: 14070-5
(1996) constructed an HIV-1 derived packaging vector by
deleting the major packaging signal (37 nucleotides, starting
from 6 nt downstream of the 5' major splice donor site to 7
nucleotides upsteam of the beginning of gag) . The genome, which
was derived from HIV-1-MN-ST.1 because of its high efficiency
of infection in both monocytes and T cells, was otherwise
intact. Their transducing vector had the components LTR-gag-
RRE-reporter gene (SL3-gpt)-env-LTR. Titers of 10E5
transducing units (TU)/mL were reported.
Corbeau et al. suggest that the first 500 nt of the gag
gene may be directly or indirectly involved in the binding of
the viral RNA to the nucleocapsid of the virion, and that a
stretch within the env gene, including the RRE, also contains
a packaging signal.
Corbeau et al. also criticize prior vectors. They
attribute the alleged deficiencies of these vectors to the
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
21
truncation of the vpr gene from the packaging vector, and/or
to the deletion of gag and/or env sequences which may contain
additional packaging signals from the transducing vector.
Akkina, et al., J. Virol., 70: 2581-5 (1996) demonstrated
that an HIV-1 based retroviral vector containing the firefly
luciferase reporter gene can be pseudotyped with a broad host
range VSV G envelope glycoprotein. The luciferase gene~~
replaced the HIV-1 nef gene. The authors suggested that such
a vector should be able to infect CD34+ hematopoietic
progenitor cells with high. efficiency.
Markowitz, et al, J. Virology 62: 1120-4 (1988) had
suggested that viral genes could be separated onto two
different plasmids, to provide a safer packaging line for gene
transfer. Markowitz et al. Placed the gag and pol genes of MLV
on one plasmid, and the env gene on the other. The plasmids
had deletions of the 3'LTRs and the packaging signal as well.
Hence, to generate intact retrovirus, there would need to be
several recombination events. Markowitz' strategy was adapted
to HIV-1 by Naldini et al., as described below.
Naldini, et al., Science, 272: 263 (1996) describes a
lentiviral vector-based system for gene delivery. There are
three vectors in the system. The first packaging vector
(pCMV~R9) provides the HIV gag, pro, pol, vif, nef, tat, rev,
and vpr genes, but the env and vpu genes, and the packaging
signal, were inactivated. (A later paper, cited below, makes
it clear that the env gene was inactivated by insertion of a
linker containing multiple stop codons.) The human
cytomegalovirus (CMV) immediate early promoter was substituted
for the 5' LTR, while the 3' LTR was replaced with a polyA site
from the human insulin gene. The major splice donor site was
preserved. A second packaging vector was used to broaden the
tropism of the vector system. In one variant, this vector
expressed the amphotropic envelope of Moloney leukemia virus
(MLV), under control of the MLV LTR, and in the other, it
expressed the G glycoprotein of vesicular stomatitis virus
(VSV) under the direction of the CMV promoter. (The
alternative Env protein was the only expression product of the
second vector.) The final element of the system was a
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
22
transducing vector (pHR'), providing, in order, the 5'LTR, the
major splice donor site, the major packaging signal, nearly 350
base pairs of gag, the env sequence encompassing the RRE
element, a splice acceptor site, an internal CMV promoter, a
reporter gene (luciferase or beta-galactosidase) , and a 3' LTR.
Naldini et al., Proc. Nat. Acad. Sci. (USA), 93: 11382-8
(1996) discuss the use of VSV-G-pesudotyped lentiviral vector~~
particles to achieve "long-term" expression of a transgene in
adult rat brains injected with the particles. The packaging
l0 vector differs from that described above in that 1.4 kbp was
deleted from the env gene, downstream of the functional vpu
gene, and replaced with an inframe stop codon. See also Blomer
et al., J. Virol., 71: 6641-9 (Sept. 1997).
In their transducing vectors, the env-RRE and part of gag
sequences are still there. The entire 3' LTR was also there.
Miyoshi, et al., J. Virol., 72:8150-57 (Oct. 1998) created
a self-inactivating HIV-1 vector. The U3 region of the 5' LTR
was replaced with the CMV promoter, and self-inactivation was
accomplished by deleting 133 by in the U3 region of the 3' LTR,
including the TATA box and binding sites for transcription
factors Spl and NF-kappa B. The deletion is transferred to the
5' LTR after reverse transcription and integration in infected
cells, resulting in the transcriptional inactivation of the LTR
in the proviruses.
Vector systems similar to those of Shimada or Naldini have
been constructed which are derived from FIV, see Poeschla, et
al., Nature Med., 4:354-7 (1998), or HIV-2, see Poeschla, et
al., Proc. Nat. Acad. Sci. (USA), 93:11395-9 (1996).
White, et al., J. Virol., 73:8232-40 (1999) describes HIV-
1/SIV chimeric vectors, with VSV-G pseudotyping.
Olsen, Gene Therapy, 5:1481-7 (1998) has described
lentiviral vectors derived from equine infectious anemia virus.
The vector system comprised packaging vector pEV53
(gag+pol+tat+rev+env-), pseudotyping vector pCI-VSV-G, and
transducing vector pECX. In pEV53, there were partial
deletions in env (736 nt from SU coding region and 168 nt from
TM coding region). The PPT and 3' LTR were also deleted, but
a BGH polyA was provided. The 5' sequences, including the RNA
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516 .
23
encapsidation (packaging) signal, were left intact. In pECX,
the CMV enhancer/promoter was substituted for the U3 domain of
the 5' LTR. The vector provided all cis-acting sequences
required to support reverse transcription and integration, and
a cloning site for inserting foreign genes such as beta-
galactosidase gene (lacZ) or puromycin-N-acetyl transferase
dominant selectable marker gene (puro).
As pointed out by Olsen, EIAV has a relatively simple
genome organization. Hence, it was not clear that his results
could be extrapolated to HIV, SIV or FIV. Also, Olsen's
vectors~exhibited a level of transfection about 100-fold lower
than with MLV vectors.
Sodroski, USP 5,654,195 (1997) describes a hybrid virus
in which the 5' DNA segment encodes functional SIV or HIV-2
gag, pol, pro, vif, and vpx proteins, and the 3' DNA segment
encodes functional HIV-1 env, tat and rev proteins, and a
functional SIV or HIV-2 nef protein. The 5' and 3' LTRs are
from SIV or HIV-2.
Sodroski, USP 5,665,577 (1997) discloses an HIV vector
which comprises the gag, pol and env genes but lacks the HIV
major packaging signal, identified therein as
AAAAATTTTGACTAGCGGA. When introduced into a eukaryotic host
cell, these express the structural proteins to form HIV virions
that do not contain sufficient HIV RNA to result in a
replication-competent HIV virion.
See also, Uberla, W098/39463 and Kingsman, W098/17815.
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
24 .
SUMMARY OF THE INVENTION
The present invention contemplates attenuated
lentiviruses, and improved viral packaging and transducing
vectors derived from lentiviruses, especially HIV-1, and useful
for the delivery of nonlentiviral genes to target cells. It
also contemplates the use of these vectors in delivering
transgenes to target cells, especially nondividing cells, in..
organisms, especially humans.
Packaging vectors
The packaging vectors (HP) of the present invention differ
from those known previously in that they contain less in the
way of lentiviral sequences from a single lentivirus, and hence
present a reduced risk of recombination. In particular, the
packaging vectors of the present invention are characterized
by either the use of a modified but functional major splice
donor site, substantially incapable of serving as a site for
homologous recombination, or by the complete omission of the
major splice donor site. In a preferred embodiment, the
modified major splice donor site is modified so that it is
substantially identical to the major splice donor site of a
non-lentiviral retrovirus, especially that of Rous Sarcoma
Virus (RSV) .
Preferably, other non-essential sequences, such as the
accessory genes, of the source lentivirus are also deleted in
the course of the construction of the packaging vector.
Preferably, in the S' LTR region of the packaging vector, the
wild-type promoter and enhancer are replaced with a
nonhomologous promoter (and, optionally, a nonhomologous
enhancer) .
These changes likewise serve to reduce the risk of generating
replication-competent virus through recombination with the
transducing vector or a defective provirus endogenous to the
host or target cell.
Preferably, the 5' LTR promoter is an tightly inducible
promoter, so that expression of Gag, Pol and Env proteins is
under the control of the biologist. This, together with the
inactivation of certain accessory genes, tends to reduce
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
cytotoxicity.
Preferably, the Gag and Pol functions are encoded by one
vector and the Env functions (preferably, a non HIV-1-like
envelope protein) by another vector.
5 Preferably, gag expression is enhanced by the operable
linking of the gag gene to a Kozak sequence.
Transducing Vectors
In a preferred embodiment, the transducing vector (FV)
likewise is characterized by a functional major splice donor
10 site which differs from that of its source lentivirus. In the
latter case, its major splice donor site need not be identical
to that of the packaging vector(s). The modification should
leave a functional packaging signal, too.
Preferably, it likewise has a strong nonlentiviral
15 promoter/enhancer in place of the normal 5' LTR.
Preferably, the gag (except for packaging signals) and pol
sequences are deleted. Desirably, the env sequences are
deleted to the extent that this can be done without a
substantial loss in yield.
20 While there may still be regions of sequence identity
between the packaging and transducing vectors which are
sufficiently long to present a meaningul risk of homologous
recombination, a characteristic of the preferred vector system
is that homologous recombination alone, among only the
25 packaging and transducing vectors, cannot create a recombinant
virus which possesses, simultaneously, a functional packaging
signal, a functional major splice donor site, and a gag AUG,
even if the recombined virus possesses a 5' promoter/enhancer
and genes otherwise encoding equivalents of the Gag, Pol and
Env proteins. The first region of significant homology is in
the gag gene, after the initiation codon. Hence, if the
recombinant virus derives a functional packaging signal and a
functional major splice donor site from the transducing vector,
it will lack the gag AUG, since it can crossover to the
packaging vector only after the AUG. Contrariwise, if it has
the 5' sequence of the packaging vector through the gag AUG,
it will lack a functional packaging signal and a functional
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
26
major splice donor site. Of course, a replication-competent
virus could still be generated by nonhomologous recombination,
or by further recombination with a defective endogenous
retrovirus.
Certain speculative vector systems are also described
herein which further increase safety.
While the invention is not so limited, the embodiments not ~~
already disclosed in 08/935,312 are of particular interest.
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
27
BRIEF DESCRIPTION OF THE DRAWINGS
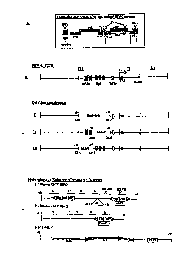
Figure lA is a simplified schematic illustration showing
the HIV-1 genomic structure.
Figure 1B is a simplified schematic illustration of the
HIV-1 LTR.
Figure 1C provides simplified schematic illustrations of
three HIV-1 LTR deletion constructs.
Figure ID provides simplified schematic illustrations of
three heterologous enhancer/promoter inserts (human CMV IE(a),
human CMV IE(b), and Mo-MLV).
Figure 2 is a graph showing the reverse transcriptase
activity of a representative attenuated recombinant HIV-1 tat
mutants over time (days post-infection).
Figures 3A-3C show the organization of the HIV-1 genome
and a series of HIV-1 mutants containing LTR, tat, and nef
mutations.
Figure 4 shows replication efficiencies of several HIV-1
recombinants carrying heterologous genes.
Figure S shows an HIV-1 transducing vector diagram for the
HIV packaging construct 1-del.env (pHP-ldl).
Figure 6 shows cell-free RT activity (cpm/~,L) measured in
duplicate using supernatant of cell cultures transfected with
various packaging vectors.
Figure 7 shows seven pHP-1-derived packaging vector
constructs.
Figure 8 shows six pTV-derived transducing vector
constructs.
Figure 9A shows a pTV~-derived construct.
Figure 9B shows a pTV~-derived construct.
Figure 10 shows the Gag processing rates of wild-type HIV-
infected MT4 compared with tat-C HIV chronic high producing
cells.
Figure 11 shows kinetics of retro- and lentiviral
transgene expression in three different human cell lines.
Celss were transduced with 105 to of pTVnCMV-nlacZ or
pMFGnlacZ and propagated for long-term study. At different
passage times as indicated, cells were collected and stained
for (3-galactosidase activity to determine the percentage of
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
28
positive cells.
Figure 12 illustrates the possible cross-over to generate
RCV from co-transfection of pHP-d1.28 and pTV-dl.CMVnlacZ.
Fig. 12A similarly illustrates possible crossover with the same
packaging vector and a different transducing vector, pTVp.
Figures 13A provides a schematic showing a portion of the
wild-type HIV-1 sequence, as well as the tat-B (wild-type~~
sequence provided in SEQ ID N0:4; the tat-B sequence is
provided in SEQ ID N0:20).
Figure 13B provides a.schematic showing a portion of the
wild-type HIV-1 sequence, as well as the nef-A mutations and
nef-B mutations (wild-type sequence provided in SEQ ID NOS:5
and 6). The nef-B mutations are shown in SEQ ID NOS:18 and
19). The nef-A sequence is the same as the wildtype sequence
for the sequence shown starting at base 9001 (i . e. , SEQ ID NO: 6
represents the sequences for both wild-type and nef-A). For
the sequence shown starting at base 8781, the nef-A sequence
is the same as the nef-B sequence shown in SEQ ID N0:5 (i.e.,
SEQ ID N0:5 represents the sequences for both nef-A and nef-B
in the sequence shown starting at base 8781).
Figure 14 compares (A) HIV genome structure with that one
embodiment (B) of an HP/TV vector system. Att, integrase
attachment site; SD, splice donor; ~, packaging signal; ppt,
polypurine tract. In pHPd120, 28 nucleotides in env was
deleted as elsewhere described.
Figure 15. Mutagenesis and PCR primers and their relative
locations on pTV. Various primers for PCR mutagenesis are set
on the pTV map as shown at the top. The location and direction
of these primers are also depicted.
Figure 16. Experimental approach diagram. For vector
production and titration, five plasmids were co-transfected
into TE671 cells as shown to the left. Human growth hormone
plasmid pXGHS was used as transfection control. To examine RNA
expression of pTV vectors, pCMVrev, pCEP4tat, and pXGH5 were
co-transfected.
Figure 17 provides the sequence of a portion of the wild-
type HIV-1 sequence, as well as the tat-B (wild-type sequence
provided in SEQ ID N0:4), and tat-A (SEQ ID N0:16), tat-B (SEQ
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516 .
29
ID N0:20), and tat-C (SEQ ID N0:17).
Figure 18 compares the strucutres of pHP, wt HIV-1 and
pTV~.
Figures 19A-19J show the structures of HIV-1 and numerous
transducing vector variants, together with the viral titers
relative to pTV~ set at unity. The locations of the SD, the
gagAUG, and various known or potential packaging signals (stem- ~~
loops) are indicated.
HIV-1 and its packaging signals (A) compared to (A) pTVn
CMVnlacZ and derivatives: (B) gag-env deletion mutants, (C)
packaging signal mutants nAUG, nSD, nSD/AUG, (D) env mutants,
(E) dlRRE (and packaging vector PHP-EFgp), (F) combination
mutants, (G) 5' U3 modifications, (H) 3'U2 modifications (The
size of U3 deletion is indicated and the nucleotide numbers on
the map are based on pNL4-3; for example, NFkB and Spl binding
sequences are from nt 9393 to nt 9489; USE: upstream element),
(I) 3' U5 modifications, (J) 5' U5 deletion.
For Figs. 19(G), (H), the titer of wild type pTVnCMVnlacZ
was arbitrarily set at 1.00 with standard errors (n=4). The
actual titer vaule of the wild type pTV construct is 7.3 x 105
+/-0.2.
In Fig. 19(I), The 3' U5 was deleted by PCR mutagenesis
as described in Materials and Methods (pTVDCMVnlacZdl3'U3#lU5).
In pTVDCMVnlacZdl3'U3#lUSpA, bGH polyadenylation sequence was
inserted at the Hind III site in the 3' R. The same
modifications were introduced into the 5' U3 replaced pTV
vectors. Relative titers to the control pTVDCMVnIacZ were
determined and shown to the right with standard errors (n=4).
The actual titer value of the wild type pTV construct was 7.3
x 105 +/- 0.2.
In Fig. 19 (J) , 5' U5 was deleted by PCR mutagenesis as
described in Materials and Methods and the sizes of deletion
are indicated (D62, D50, and D35). Relative titers to
pTVDCMVnIacZ are presented with standard errors (n=4). The
actual titer value of the wild type pTV construct was 7.3 x 10'
+/- 0.2..
Figure 20 is a table setting forth the relative titers for
the transducing vectors of Figs. 19A-19C, but further
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
indicating the number of constructs tested in each sample
group, the standard error, and the paired P value.
Figure 21 is a table comparing wild-type HIV-1, pHP and
pTV 5' sequences.
5 Figure 22 schematically presents the detection of proviral
DNA after lentiviral vector transduction.
Figure 23 Analyses of 5' splice site and SL2 deletion~~
mutants. Schematic illustration of the four stem-loop
structure of the HIV packaging signal, SD mutations, and
10 relative vector efficiencies. The relative vector titer of
each mutant was determined by normalizing against that of pTV
which was 7.3 ~ 0.2 x 105 tu/ml and is arbitrarily set at 1.00.
(B) Quantitative analyses of viral titer, cytoplasmic full-
length viral RNA, virion RNA, and packaging efficiency of SD3,
15 SD4, and SD1* mutants vs. wt pTV. For easy comparison, the
vector titer, cytoplasmic full-length RNA, packaged virion RNA,
and packaging efficiency are all normalized against those of
pTV which are set at 1.00.
Figure 24 Analyses of gag AUG and SL4 mutants. (A)
20 Schematic diagram of gag AUG and 5' gag mutants in comparison
to a previously reported mutant, gag/env.dl5* (4) , and relative
vector titers. (B) Quantitative comparison of viral titer,
cytoplasmic unspliced RNA, packaged virion RNA, and packaging
efficiency.
25 Figure 25. Analyses of vector functions of combination
mutations in SD, SA, gag AUG, gag, and env. (A) Schematic
diagram of pTV mutant constructs and their relative titers.
(B) Comparison of viral titer, cytoplasmic full-length RNA,
packaged virion RNA, and packaging efficiency of the
30 combination pTV mutants.
Figure 26 Currently preferred HP/TV lentiviral vector
system (a) the wild type HIV-1 diagram and the known locations
of packaging signals and CRS/INS regulatory elements, (b) pHP
has the entire 5' LTR deleted except for some sequences in the
U3 (HIV-1, pNLA-3, nt.288-318) plus the TAR sequence, and the
rest of HIV-1 sequence starts from gag AUG to nt.8784 at the
AUG of nef gene. The most important safety feature is two
deletion regions in the env gene, d1.695 and d1.28. We have
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
31
also made mutations in vpu, vpr, vif in pHP. The most advanced
pTVdI.SIN transducing construct has similar 5' U3 sequence to
pHP 5' U3, and d1.35 in the 5' U5, the intact 5' untranslated
leader containing packaging signals, and 40 nt of gag sequence
but with mutations in the splice donor site (SD1) and gag AUG
(TAG). The rest of HIV genome was deleted in this pTV up to
the PPT at the 3' end. At the 3' end, only 24 nt attL of U3~I
and R regions of HIV are kept.
Figure 27 An advanced (theoretical) HP/TV vector system.
In the packaging vector, we have replaced the 5' LTR and TAR
sequences completely with EF1 alpha enhancer/promoter and HIV
sequence starts from gag AUG and ends at the end of pol. We
will need to insert a CTE-like element to ensure gag-pol
expression in different types of cell lines. Certain cell
types, such as TE671 cells may not need CTE.
The pTV construct has only about 550 nt of HIV sequence
left, including TAR (5'R), 35 nt deleted U5, PBS, leader
sequence containing SL1-SL4 packaging signal and 40 nt of gag
sequence, the 3' end PPT, 24 nt att L of 3' U3, and R sequence.
The payload of this vector is more than 9 kp in theory.
Figure 28 Possible cross-over between pHP and pTV of our
Fig. 26 version. The 5' cross-over may occur in the 40 nt of
gag sequence through homologous recombination or, legitimate
recombination as shown in solid arrow, (or in the 5' TAR
region, but must go back from the 40 nt of gag) to pick up gag-
pol and the rest of the genome sequence; and the 3' (dashed
arrow) cross-over lacks sequence homology; so it has to be non-
homologous recombination (illegitimate recombination). The
result is a non-functional defective RCV.
Note: The titers reported in various examples and figures
for the same construct may vary depending on which runs are
averaged together. Generally, the most inclusive titer data
is on the figures.
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
32
DETAILED DESCRIPTION OF THE PREFERRED EMBODIMENTS OF THE
INVENTION
The present invention relates to attenuated and/or
replication-defective lentiviruses, and to packaging and
transducing vectors derived in whole or in part from a
lentivirus.
In particular, this invention relates to a recombinant~~
HIV-1 vector system with multiple safety features based on a
packaging/helper construct (pHP) and a transducing vector
construct (pTV). The possibility of producing replication
competent virus (RCV) was carefully examined. The transduction
efficiency of the HP/TV vector and a conventional MLV vector
was studied using different human cell types including TE671
(muscle) , 293T (kidney) , HepG2 (liver) , neuronal stem cells and
primary CD34 hematopoietic progenitor cells and nonhuman
primary rat neural and muscle cells. Transduction efficiency
was assayed over short and long duration in tissue culture .
The safety, expression kinetics, duration and integration
status of various lentiviral HP/TV vector systems are
presented.
The Len ti virus
A "source" or "original" lentivirus is a wild-type
ientivirus from which an attenuated and/or repiication-
defective lentivirus is derived, or which is used as a starting
point, during construction of the packaging or transducing
vector, for the preparation of one or more of the genet-is
elements of the vector. The genetic element may be employed
unchanged, or it may be mutated (but not beyond the point where
it lacks a statistically significant sequence similarity to the
original element). A vector may have more than one source
lentivirus, and the different source lentiviruses may be, e.g. ,
HIV-1 and HIV-2, or HIV and SIV, and so forth.
One may also speak of a "source" or "original" gene,
genetic element or protein for a vector gene, genetic element
or protein. (The term "genetic element" includes but is not
limited to a gene.)
The cognate lentivirus is the wild-type lentivirus with
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
33 ,
which the vector in question has the greatest percentage
sequence identity at the nucleic acid level. Normally, this .
will be the same as the source lentivirus. However, if a
source Ientivirus is extensively mutated, it is conceivable
that the vector will then more closely resemble some other
lentivirus. It is not necessary that the cognate lentivirus
be the physical starting point for the construction; one may~~
choose to synthesize a genetic element, especially a mutant
element, directly, rather than to first obtain the original
element and then modify it:
One may also speak of a "cognate" protein, gene, or
genetic element (e. g., splice donor site or packaging signal).
When referring to a cognate protein, percentage sequence
identities are of course determined at the amino acid level.
The term "cognate" lentivirus may be difficult to
interpret in the extreme case, i.e., if all lentiviral genetic
elements have been replaced with surrogate non-lentiviral
genetic elements. In this case, the preferred source HIV-1
strain mentioned previously is arbitrarily considered to be the
cognate lentivirus.
HIV type 2 (HIV-2) is known to be less pathogenic than
HIV-1 in humans, and HIV-2 infection is associated with natural
protection against HIV-1 infection. Simian immunodeficiency
virus (SIV) also infects human cells; however, it is unclear
whether it can cause AIDS in humans. Thus, both HIV-2 and SIV
may be better candidates than HIV-1 for developing lentiviral
vectors. It may be advantageous to derive both the packaging
and transducing vectors from a lentivirus other than HIV-1, or
to derive one from HIV-1 and the other from a lentivirus other
than HIV-1. Use of different sources for the two vectors
reduces the risk of homologous recombination to generate RCV,
and use of a source other than HIV-1 reduces the health risk
if recombination, homologous or otherwise, occurs.
Applicant' s preliminary work was with HIV-1 derived vectors and
attenuated viruses because much more is known about the HIV-1
genome and proteins than about those of other lentiviruses.
However, in the long run, it may be preferable to derive the
instant vector system from a less threatening lentiviral source
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516 .
34 .
virus, such as HIV-2 or SIV.
The term "replication" as used herein in reference to a
virus or vector, refers not to the normal replication of
proviral DNA in a chromosome as a consequence of cell
reproduction, or the autonomous replication of a plasmid DNA
as a result of the presence of a functional origin of
replication, but rather to the completion of a complete viral.
life cycle wherein infectious viral particles containing viral
RNA enter a cell, the RNA is reverse transcribed into DNA, the
DNA integrates into the host chromosome as a provirus, the
infected cell produces virion proteins and assembles them with
full length viral genomic RNA into new, equally infectious
particles.
The term "replication-competent" refers to a wild-type
virus or mutant virus that is capable of replication, such that
replication of the virus in an infected cell result in the
production of infectious virions which, after infecting
another, previously uninfected cell, causes the latter cell to
likewise produce such infectious virions . The present
invention contemplates the use of replication-defective virus.
As used herein, the term "attenuated virus" refers to any
virus (e. g., an attenuated lentivirus) that has been modified
so that its pathogenicity in the intended subject is
substantially reduced. Preferably, the virus is attenua~ed
to the point it is nonpathogenic from a clinical standpoint,
i.e., that subjects exposed to the virus do not exhibit a
statistically significant increased level of pathology relative
to control subjects.
The present invention contemplates the preparation and use
of an attenuated lentivirus. In some embodiments, the
attenuated lentivirus is selected from the group consisting of
attenuated mutants of human immunodeficiency virus type 1,
human immunodeficiency virus type 2, feline immunodeficiency
virus, simian immunodeficiency virus, visna-maedi, caprine
arthritis-encephalitis virus, equine infectious anemia virus,
and bovine immune deficiency virus. Thus, the attenuated virus
may be an attenuated HIV-1, attenuated HIV-2, attenuated SIV,
or a virus comprised of portions of more than one lentiviral
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
species (e.g., a hybrid, comprised of portions of HIV-1 and
HIV-2, or HIV-1 and SIV, etc.)
A reference virus is a virus whose genome is used in
describing the components of a mutant virus. For example, a
5 particular genetic element of the mutant virus may be said to
differ from the cognate element of the reference virus by
various substitutions, deletions or insertions. It is note
necessary that the mutant virus actually be derived from the
reference virus.
10 The preferred reference HIV-1 was mentioned previously.
For HIV-2, see LOCUS HIV2ROD, 9671 by ss-RNA, Human
immunodeficiency virus type 2, isolate ROD, complete proviral
genome, ACCESSION M15390, see Clavel,F., Guyader,M.,
Guetard,D., Salle,M., Montagnier,L. and Alizon,M, Molecular
15 cloning and polymorphism of the human immunodeficiency virus
type 2, Nature 324, 691-695 (1986).
The preferred reference SIV sequence is LOCUS SIVMM239,
13068 by ss-RNA, a Simian immunodeficiency virus isolated from
a macaque, isolate 239 (Macaca mulatta Mm239-82); complete
20 proviral genome and flanking sequence, GenBank ACCESSION
M33262, see Regier,D.A. and Desrosiers,R.C., The complete
nucleotide sequence of a pathogenic molecular clone of simian
immunodeficiency virus, AIDS. Hum. Retroviruses 6, 1221-1231
(1990) .
25 The preferred reference RSV sequence is Genbank
locus/accession ## AF052428, 9396 by DNA, the Rous sarcoma
virus strain Schmidt-Ruppin B, complete genome.
Len ti viral Vector System
The present invention contemplates a gene amplification
30 and transfer system comprising a transducing vector (TV), one
or more compatible packaging vectors (HP), and a suitable host
cell, the transducing vector and at least one packaging vector
being derived from a lentivirus, which allow (1) transfection
of the packaging vectors into the host cell to form a packaging
35 cell line which produces essentially (packaging vector RNA)-
free viral particles, (2) transfection of the transducing
vector into the packaging cell line, (3) the packaging of the
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
36
transducing vector RNA by the packaging cell line into
infectious viral particles, and (4) the administration of the
particles to target cells so that such cells are transduced
and subsequently express a transgene carried by the transducing
vector.
Either the particles are administered directly to the
subject, in vivo, or the subject's cells are removed, infected ~~
in vitro with the particles, and returned to the body of the
subject.
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516 .
37
The basic characteristics of the packaging vector and the
transducing vector are summarized in the following table:
Lentiviral Genetic Packaging Vector Transducing Vector
Element
5' LTR U3: preferably deleted preferably deleted'
integration (not in mRNA
attachment site transcript)
(att)
5' LTR U3: preferably replaced as for packaging
promoter/enhancer by any sufficiently vector, but must be
strong functional in the
heterologous packaging cells;
promoter (opt. preferably CMV or
incl. enhancer) EF-la
functional in the promoter/enhancer,
packaging cell line or an inducible
promoter
5' LTR R: TAR site may be deleted may be replaced
with an alternative
R of another
retro~irus (Note 1)
5' LTR R: Poly-A preferably deleted see above
5' LTR U5 preferably deleted may be at least .
partially deleted
5' LTR U5-3' att preferably deleted functional att
site required
L: PBS preferably deleted may be replaceable
with a mutant PBS
or with the PBS of
another retrovirus
if reverse
transcriptase also
replaced
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516 .
38
L: major splice replaced with an may be mutated,
donor site alternative splice e.g., GGTG to.GCAG
donor site, the RSV or GGGG to reduce
site is preferred homology to source
(see text); or sequence and retain
deleted totally packaging function"
and 5' poly A
suppression
L: major packaging inactivated functional
signal packaging signal
required; need not
be wild-type
L: genomic RNA inactivated functional DLS
dimer linkage site required; need not
be wild-type
L: region upstream preferably insert no strong
of gag initiation Kozak sequence preference, but
codon mutations may be
introduced if they
do not
substantially
interfere with
packaging signal
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
39
gag a gene or genes the gene is
encoding proteins inactivated vis-a-
substantially vis production of
identical to the functional Gag
wild-type Gag proteins, e.g., by
proteins is/are replacing the
required; the initiation codon
identity or with a stop codon
similarity is (e. g., TAG) and/or
preferably deleting at least
sufficient so that part of the gene,
the protein retains but the minor
the nuclear packaging signal in
translocation bases 1-40 of the
function, and (if gag gene is
the target cell is preferably retained
a nondividing cell) in functional form
the function of
entering the
nucleus of
nondividing cells
pol a gene or genes inactivated,
encoding ccmpatible preferably by
protease, deletion
integrase, and
reverse
transcriptase
is/are required
(see text)
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
env a gene encoding a inactivated,
retroviral preferably by
envelope protein, deletion of part of
or a chimera of an the gene; the RRE
envelope protein region is
and one or more preferably ,.
foreign binding retained in
moieties, is functional form to
required. maintain its
packaging and
nuclear transport
functions. The
entire env gene
including RRE can
be deleted if other
cis-repressive
sequence (CRS) in
gag, pol and 5'
splice sites are
deleted, viz, pTV
dl.SD1/gag/env/RRE.
tat may be deleted or preferably
otherwise inactivated,
inactivated preferably by
deletion
rev may be deleted or preferably
otherwise inactivated,
inactivated if preferably by
INS's in gag and deletion
pol inactivated,
and RRE in env
inactivated
vif preferably deleted preferably deleted
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
41
vpr may be deleted; may preferably deleted
inactivate its cell
cycle arrest
function and
maintain its
nuclear transport
function
vpx (HIV-2) preferably deleted; preferably deleted
may inactivate its
cell cycle arrest
function and
maintain its
nuclear transport
function
vpu preferably deleted preferably deleted
nef preferably deleted preferably deleted
except for the PPT
and attL sites
PPT preferably deleted preferable to have
a functional PPT;
may be possible to
replace with the
PPT of another
retrovirus
3' LTR-U3 enhancer-promoter preferably deleted,
region preferably except that a
deleted functional att site
is required
3'LTR - R preferably deleted may be replaced
and replaced by a with the R region
functional, non-HIV of another
polyA site retrovirus {see
Note 1)
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
42
3'LTR-U5 preferably replaced may be deleted; if
with any polyA it contains a polyA
signal compatible element, it may be
with host cell preferable to
retain that element
at least
Notes: (1) In the transducing vector, The 5'LTR and 3 'LTR R
regions must be sufficiently identical so that ssDNA jumping
by the reverse transcriptase will occur. (2) there can be more
than one packaging vector, carrying separate structural genes.
For example, one vector can encode gag and pot functions, and
another vector, env functions. (3) If "may be deleted" or
"preferably deleted", partial deletion is acceptable but
complete deletion preferred, unless otherwise stated.
The packaging vectors and transducing vectors of the
present invention are each replication-incompetent viruses.
Moreover, the vectors chosen for incorporation into a given
vector system of the present inventionare such that it is not
possible, without further mutation of the packaging vectors)
or transducing vector, for the cotransfected cells to generate
a replication-competent virus by homologous recombination of
the packaging vectors) and transducing vector alone.
In many embodiments, the two vector constructs, pHP,
(which directs the synthesis of necessary viral proteins for
virion assembly), and pTV, (which serves as a gene transducing
vehicle for foreign gene delivery), were derived from a LTR-
modified recombinant HIV-1 plasmid pNL4-3.
For example, pHP-1 contains a recombinant cytomegalovirus
immediate early (CMV-IE) enhancer/promoter HIV-TAR element
which replaces the 5' LTR of pNL4-3. The entire untranslated
5' leader sequence, nef, and the 3' LTR of pNL4-3 were also
deleted in pHP-1. The HIV-1 5' untranslated leader sequence
was replaced with an artificial 59 by Rous sarcoma virus (RSV)
major splice donor sequence containing a mutated RSV gag AUG
and a preferred eukaryotic translation initiation sequence (-
CCACCATG-) for the HIV-1 Gag synthesis. The nef gene in pNL4-3
was replaced by the bacterial gpt selective marker gene, and
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516 .
43
the 3' LTR was replaced by the SV40 polyadenylation signal.
To prevent RCV production, the env gene was deleted by Ba131
exonuclease digestion at the NheI site. Two variants of env-
deleted pHP-1 were generated and analyzed, pHP-ldl.2 (with a
2-nt deletion) and pHP-1d1.28 (with a 28-nt deletion). An
alternative pHP construct, pHP-VSVG, was generated by inserting
the VSV-G envelope gene between the env AUG and the NheI site~~
in gp120, and introducing mutations in vpr, tat, and vpu.
The transducing constructs (pTV) were made from pNL4-3 by
deleting sequence extending from the middle of gag to the
middle of env. A reporter gene cassette containing a
heterologous enhancer/promoter directing a reporter gene was
inserted in the nef region of the pTV vector.
The HP/TV vector system has gone through extensive RCV
testing and has been approved for use in a biosafety level II
laboratory employing a level III standard operating protocol.
The level III protocol was approved by the Biosafety Committees
at the University of Alberta and the University of Florida. The
lentiviral vector tissue culture and animal studies were
performed in a level II+/III laboratory using protocols
approved by the Animal Warfare Committees and the Biosafety
Committees at the University of Alberta and at the University
of Florida .
The most preferred system includes a transducing vector
featuring the dl.SDl/gag/env/RRE deletion in combination with
the 3' U3 mutation of pTVdI.CT-CMVnlacZdl3'U3#lUSpA and 3'
mutations of pTV d1.35-d1.3'U3U5pA. With this most favored
construct, we will have most of HIV sequnce deleted from the
pTV transducing vector, which include the 5' major splice site
(Splice donor site, SD) all env sequence (including RRE
deletion) deleted, most of gag deleted except for the first 40
nucleotide, most of 5' U3, 3' U3 and 3' U5 deleted, and part
of the 5' U5 deleted (35 nucleotide deletion). This will
generate a vector with less than 550 nucleotides of HIV
sequence. The entire HIV genome is about 9.5 to lOkb.
Therefore, we improved the vector system to be able to
accomodate more than 9 kb of insert (i.e., a payload of 9 kb
or so). This is a major improvement on both safety and
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
44 ,
capacity of the lentiviral vector system. Thus, the advantages
are 1. improved safety for less HIV sequence homology; 2.
increased payload; 3. SIN feature; 4. Rev and possibly Tat
independent (Tat may be needed for high efficiency gag-pol
synthesis.
The transducing vector with least homology but with
optimal titer (i.e. no less than 50% of the reference ~~
transducing vector pTV nCMVnlacZ titer) is the one indicated
above. However, we can take out more. The 5' US can be
deleted further as shown in (d1.62) which drop the titer down
to 28%, and the gag/env.dl.7 construct with deletion of SL4 in
the packaging signal including deletion of gag AUG and the
first 40 nucleotide, which drops the titer down to 26%.
Our preferred packaging vector is one without all HIV
sequences except gag-pol ORF and regulatory elements (such as
CTE replacing RRE/Rev function) necessary for gag-pol
synthesis.
Our preferred pseudotyping vector is pHEF-VSVG, but we
have interest in envelope proteins from other viruses, such as
ebola virus.
Packaging Signal
As used herein, the term "packaging signal" or "packaging
sequence" refers to sequences located within the retroviral
genome or a vector which are required for, or at least
facilitate, insertion of the viral or vector RNA into the viral
capsid or particle. The packaging signals in an RNA identify
that RNA as one which is to be packaged into a virion. The
term "packaging signal" is also used for convenience to refer
to a vector DNA sequence which is transcribed into a functional
packaging signal. Certain packaging signals may be part of
a gene, but are recognized in the form of RNA, rather than as
a peptide moiety of the encoded protein.
The major packaging signal is the signal having the
predominant effect on whether viral RNA is inserted into the
particle. This signal is located in the 5' leader region
(spanning the SD site and the gag AUG) of the wild-type
lentiviral genome. It is not equivalent to the conventional
site of the MLV vectors, in that the latter alone allows
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
efficient MLV vector packaging.
There are also minor packaging signals with a lesser
effect on packaging efficiency. Several studies have shown
that many sequences in HIV-1, including LTR, TAR, RRE, and in
5 the 5' and 3' gag ORF, the pol ORF, and in the sequences
flanking the RRE, contribute to efficient genome packaging,
pointing to the complex nature of HIV-1 packaging signals (See~~
e.g., A. Aldovini and R. A. Young, J. Virol., 64:1920-1926
[1990); J.F. Kaye et a~., J. Virol., 69:6593-6599 [1995]; A.
10 Lever et al., J. Virol., 63:4085-4087 [1989]; J. Richardson et
al., J. Virol., 67:3997-4005 [1993]).
Earlier studies of the HIV packaging signal demonstrated
that a 46 nt (751-796) stem-loop structure derived from the
splice donor site to the 5' gag coding region is sufficient to
15 allow packaging of a heterologous Sendai virus RNA but the
efficiency was not determined and the location of the insertion
was critical to the stem-loop conformation. See Hayashi T,
Shioda T, Iwakura Y, Shibuta H. RNA packaging signal of human
immunodeficiency virus type 1. Virology 1992; 188:590-9. They
20 further showed that the 46 nt sequence must be inserted in the
5' end of the Sendai RNA to serve as a packaging signal;
inserting in the midst of the Sendai RNA destroyed the
packaging signal. Secondary structure analysis showed that
several stem-loop structrual domains can be identified in the
25 5' untranslated leader region and in the 5' gag coding region.
See Baudin F, Marquet R, Isel C, Darlix JL, Ehresmann B,
Ehresmann C. Functional sites in the 5' region of human
immunodeficiency virus type 1 RNA form defined structural
domains. J Mol Biol 1993; 229:382-97. McBride et al. further
30 showed that the packaging signals in the 5' end of the HIV
genome include TAR and four stem-loops from upstream of the
major 5' splice donor site extending into the first 7 amino
acid codons in the gag coding region. See McBride MS,
Panganiban AT. The human immunodeficiency virus type 1
35 encapsidation site is a multipartite RNA element composed of
functional hairpin structures. J. Virol. 1996; 70:2963-2973;
McBride MS, Schwartz MD, Panganiban AT. Efficient encapsidation
of humna immunodeficiency virus type 1 vectors and further
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
46
characterization of cis elements required for encapsidation.
J. Virol. 1997; 71:4544-4554. Parolin et al. demonstrated that
up to 653 nt in the gag coding region can enhancer RNA
packaging efficiency. See Parolin C, Dorfman T, Palu G,
Gottlinger H, Sodroski J. Analysis in human immunodeficiency
virus type 1 vectors of cis-acting sequences that affect gene
transfer into human lymphocytes. J. Virol. 1994;,68:3888-3895.~~
Luban and Goff showed that the first 40 nt of gag coding
sequence is strongly influential on the packaging function, see
l0 Luban J, Goff SP. Mutational analysis of cis-acting packaging
signals in human immunodeficiency virus type 1 RNA. J. Virol.
1994; 68:3784-3793, and the Goff group further reported that
the HIV-1 packagng signal requires the very 5' edge of the RNA
and sequences downstream of the 170th nt of gag or sequences
in pol, see Berkowitz RD, Hammarskjold M-L, Helga-Maria C,
Rekosh D, Goff SP. 5' regions of HIV-1 RNAs are not sufficient
for encapsidation: implications for the HIV-1 packaging signal.
Virology 1995; 212:718-723. Their studies indicate that for
efficient packaging function, the four stem-loop structure may
not be sufficient. Instead, the packaging signal as well as
its sequence context consist of the entire packaging signal.
This is consistent with the study of Kaye et al. who have
reported that the RRE and env sequences, although not essential
to render RNA packaging, may have a positive effect on
enhancing the packaging efficiency, see Kaye JF, Richardson JH,
Lever AML. cis-Acting sequences involved in human
immunodeficiency virus type 1 RNA packaging. J. Virol. 199.5;
69:6593-6599. This latter group also reported that the mutation
of the gag AUG is detrimental to RNA packaging. It is thus
clear that the packaging signal of HIV is not as simple as MLV
and RSV.
A further reason for including the major packaging signal
in a transducing vector is because it overlaps with the dimer
linkage sequence (DLS) which is also essential for genome
packaging (See, J.L. Clever et al., J. Virol.,
70:5902-5908[1996]; J.-C. Paillart et al., J. Virol.,
70:8348-8354 [1996); and J.-C. Paillart et al., Proc. Natl.
Acad. Sci. USA. 93:5572-5577 [1996]).
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
47
The key distinction between a packaging vector and a
transducing vector is that in the packaging vector, the major
packaging signal is inactivated, and, in the transducing
vector, the major packaging signal is functional. Ideally, in
the packaging vector, all packaging signals would be
inactivated, and, in the transducing vector, all packaging
signals would be functional. However, countervailing ~~
considerations, such as maximizing viral titer, or inhibiting
homologous recombination, may render such constructs less
desirable.
Using a precise quantitative assay for vector function,
we have found that the 5' major splice donor site, the gag AUG
and the extended gag sequences are dispensible for the
packaging of a functional HP/TV vector. The highly conserved
sequences essential to HIV replication (the SD and gag AUG, and
additional coding sequence) have now been deleted from the pTV
vector which has greatly improved the safety of the HP/TV
vector system and totally eliminated the possibility of
generating RCV via homologous recombination at the gag region.
Packaging System; Packaging Vectors; Packaging Cell Line
A packaging system is a a vector, or a plurality of
vectors, which collectively provide in expressible form all of
the genetic information required to produce a virion which can
encapsidate suitable RNA, transport it from the virion-
producing cell, transmit it to a target cell, and, in the
target cell, cause the RNA to be reverse transcribed and
integrated into the host genome in a such a manner that a
transgene incorporated into the aforementioned RNA can be
expressed. However, the packaging system must be substantially
incapable of packaging itself. Rather, it packages a separate
transducing vector which is described below. The general
abbreviation for a packaging vector in this specification is
HP or pHP.
In the case of an HIV-1 vector, the packaging system will
provide functional equivalents of the gag, pol and env genes
as discussed below. One may use a single vector which
provides all three genes (a "GPE" vector) , or a two vector
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516 .
48
system wherein one vector provides the gag-pol genes (a "GP"
vector) and the other vector (an "E" vector) provides the env
gene. In theory, a three vector system ("G", "P", and "E"
vectors) is possible if one is willing to construct distinct
gag and pol genes on separate vectors, and operably link them
to different regulatable promoters (or one to a regulatable and
the other to a constitutive promoter) such that their relative~~
levels of expression can be adjusted appropriately.
The vector or vectors which together compose the packaging
system are called the packaging and pseudotyping vectors.
A pseudotyping vector is one which encodes an Env-
equivalent function, e.g., VSV-G, but not a lentiviral env.
A packaging vector is any vector providing at least one of Gag,
Pol or Env, or an equivalent of at least one of Gag or Pol.
A vector may be both a pseudotyping vector (providing, e.g.,
VSV-G) and a packaging vector (providing Gag and Poly, but
normally these functions are separated. A packaging system
need not include a pseudotyping vector but must include at
least one packaging vector.
A packaging cell line is a suitable host cell transfected
by a packaging system which, under achievable conditions,
produces viral particles. As used herein, the term "packaging
cell lines" is typically used in reference to cell lines that
express viral structural proteins ( e. g. , gag, pot and env) , but
do not contain a packaging signal. For example, a cell line
has been genetically engineered to carry at one chromosomal
site within its genome, a 5'-LTR-gag-pot-3'-LTR fragment that
lacks a functional psi' sequence (designated as Opsi), and a
5'-LTR-env-3'-LTR fragment which is also opsi located at
another chromosomal site. While both of these segments are
transcribed constitutively, because the psi' region is missing
and the viral RNA molecules produced are less than full-size,
empty viral particles are formed.
If a host cell is transfected by the packaging vectors)
alone, it produces substantially only viral particles without
the full-length packaging vector Preferably less than l00 of
the viral particles produced by the packaging cell contain full
length packaging vector-derived RNA. However, since the
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516 .
49
packaging vector lacks a functional primer binding site, even
if these particles infect a new cell, the packaging vector RNA
will not be reverse transcribed back into DNA and therefore the
new cell will not produce virion. Thus, by itself, the
packaging vector is a replication-incompetent virus.
Preferred packaging vectors are identical or at least
substantially identical, to one or more of packaging vectors ~.
disclosed in the examples, such as pHP-1, pHP-d1.2 and pHP-
d1.28, pHP-VSVG, pHP-CMV, pHP-CMVdeI.TAR/SD, pHP-CMV-EFla
intron, and pHP-EF, or are hybrids thereof.
The pHP construct was made by first replacing the 5' LTR
with the CMV-TATA-TAR chimeric promoter, obtained from the
BbrPI to HindIII fragment of the chimeric LTR containing CMV
IE promoter-TATA box and TAR seqence, which was derived from
a recombinant HIV-1 LTR as described previously, see Chang L-J,
Zhang C. Infection and replication of Tat-minus human
immunodeficiency viruses: genetic analyses of LTR and tat
mutants in primary and long-term human lymphoid cells . Virology
1995; 211:157-169; Chang L-J, McNulty E, Martin M. Human
immunodeficiency viruses containing heterologous
enhancer/promoters are replication competent and exhibit
different lymphocyte tropisms. J Virol 1993; 67:743-752, then
deleting the rest of the 5' leader sequence extending from the
i~indIII site in the end of TAR region to the gag AUG using a
synthetic oligonucleotide containing a splice donor site of
Rous sarcoma virus and a conserved Kozak sequence -CCACC-
adjacent to the gag AUG. The Kozak sequence serves to increase
the translational efficiency. The gag-pol coding sequence is
kept intact. Alternatively, the conserved reverse
transcriptase (RT) domain of the pol sequence is replaced with
RSV RT domain by PCR amplification and cloning. The vif, vpr,
vpu and env genes were mutated by site-specific mutagenesis to
eliminate the AUG initiation codon and some of the coding
sequence but not affecting Gag-Pol or Tat/Rev syntheses. The
tat coding sequence can also be mutated as described below
either by inserting multiple stop codons (e. g. tat-B mutant)
or by deleting the initiation AUG codon and part or all of the
coding sequence (e.g. tat-C mutant) because the- pHP can be
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
tat-independent. A rev independent gag-pol construct can also
be made by mutating the nuclear retention signals in the
gag-pol coding region as indicated by Schneider R, Campbell M,
Nasioulas G, Felber BK, Pavlakis GN. Inactivation of the human
5 immunodeficiency virus type 1 inhibitory elements allows
Rev-independent expression of Gag and Gag/protease and particle
formation. J. Virol. 1997; 71:4892-4903, and in the env coding~~
region including the RRE element. In this rev-independent pHP
construct, the rev open reading frame is mutated by removing
10 the intiation codon AUG and deleting the coding sequence. The
3' nef-PPT-LTR of HIV-1 was entirely deleted from the nef
initiation AUG codon which was mutated to contain a new HindIII
site and replaced with a selective marker gene gpt and an SV40
polyadenylation signal.
15 In some embodiments, the packaging cell and/or cell line
contains a transducing vector. The packaging cell line will
package the transducing vector into infectious particles. Such
a cell line is referred to herein as a "transgenic virion
production cell line".
20 It is contemplated that packaging may be inducible, as
well as non-inducible. In inducible packaging cells and
packaging cell lines, lentiviral particles are produced in
response to at least one inducer. In non-inducible packaging
cell lines and packaging cells, no inducer is required in order
25 for lentiviral particle production to occur.
The packaging vectors necessarily differ from wild-type,
replication-competent lentiviral genomes by virtue of the
inactivation of at least one packaging signal of the cognate
wild-type genome. More than one packaging signal may be
30 inactivated. Preferably, the only lentiviral genes provided by
the packaging vector are those encoding structural, or
essential regulatory, proteins.
Ancillary vectors
These encode nonvirion proteins, like tat, but are not
35 packaging, transducing or pseudotyping vectors.
Transducing Vectors
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
51 ,
A transducing vector is an expression vector which bears
an expressible nonlentiviral gene of interest and includes at
least one functional lentiviral packaging signal, so that,
after said transducing vector is transfected into a packaging
cell line, the transducing vector is transcribed into RNA, and
this RNA is packaged into an infectious viral particle. These
particles, in turn, infect target cells, their RNA is reverse~~
transcribed into DNA, and the DNA is incorporated into the host
cell genome as a proviral element, thereby transmitting the
gene of interest to the target cells.
As used herein, the term "transduction" refers to the
delivery of a gene (s) using a viral or retroviral vector by
means of infection rather than by transfection. In preferred
embodiments, retroviral vectors are transduced. For example,
an anti-HIV gene carried by a retroviral vector can be
transduced into a cell through infection and provirus
integration. Thus, a "transduced gene" is a gene that has been
introduced into the cell via lentiviral or vector infection and
provirus integration. In preferred embodiments, viral vectors
(e. g., "transducing vectors") transduce genes into "target
cells" or host cells).
It may be convenient to classify transducing vectors as
follows:
Generation 0 pTV: pTV vectors containing non-replication
essential genes or genetic elements. (e.g vectors previously
reported by Naldini et al. and Shimada et al.
Generation 1 pTV: pTV vectors with deletions of all the
accessory genes and non-replication essential genetic elements
(e. g. vif, vpr, vpu, nef, NF-kB/Spl)
Generation 2 pTV: pTV vectors with deletions of
replication-essential genetic elements (e. g., gag AUG, SD site,
env sequences, RRE, TAR, such elements are also missing on pHP)
Generation 3 pTV: pTV vectors with substitutions of
vector-essential genetic elements (complementary substitutions
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516 .
52
are also present on pHP).
Preferred transducing vectors are identical or
substantially identical to the transducing vectors disclosed
in the examples, such as the GO and G1 vectors, including pTV~,
pTV~100, pTV~140, pTV.~.nlacZ, and pTV~CMV-nlacZ-hyg-dl.SmaI,
pTV~, pTV~-X, pTV~EFnlacZ, pTVOEFGFP, pTV~CMV-X, pTV~CMVnIacZ,
pTV~SVneo, pTV~SVhyg, pTV~CMV-GFP, pTV~CMV-nlacZ, and~~
pTVOCMV-nlacZ-hyg, or the various G2 and G3 vectors, or hybrids
thereof. However, it is not intended that the present
invention be limited to these specific transducing vectors.
For example, the "pTV~-X, " indicates that the vector may be
comprised of "pTV~" in combination with any gene ("X"). Thus,
the present invention encompasses transducing vectors that are
suitable for use in the present invention that are linked to
any gene of interest (or a "marker gene" or "reporter gene,"
used to indicate infection or expression of a gene).
One preferred transducing vector pTV is made of a chimeric
CMV-TATA-TAR-U5/att-PBS-packaging signal-mutated SD-portion of
gag-portion of env-mutated nef-PPT-U3/att-R-U5 which exhibits
packaging function like the wild type HIV. The U5 sequence was
mutated such that all of it was deleted except for the 3' 24
nt att site. The 5' chimeric promoter is derived from the
NF-kB/Spl deleted CMV-TATA construct of the HIV LTR mutant
described previously which directs transcription at the native
HIV transcriptional initiation site. The TAR is in the R
region which can be mutated at both ends to maintain the
repetitive function of the R but significantly different from
the wild type HIV R. Alternatively, the R sequence can be
replaced with RSV R so it is completely different from HIV R
sequence. Alternatively, the PBS can be modified to become RSV
PBS such that the chimeric pHP RT (gag-RSV-RT-pol) can initiate
minus-strand DNA synthesis using the appropriate tRNA primer.
The packaging signal will have conserved stem-loop secondary
structure as described by McBride et al. as SL1 to SL4 but with
mutations in SD (GGTG to GCAG or GGGG) and gag AUG (replaced
with ACC or UAG) We showed that the latter mutations have
minimal effect on packaging efficiency. The mutant SD/gagAUG
pTV RNA genome is packaged into transducing particles at near
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516 .
53
70o wild type efficiency.
In preferred embodiments, the vectors of the present
invention are capable of "high efficiency transduction." This
is intended to encompass transducing vectors capable of
transduction at a level of at least 105/mI, although in
particularly preferred embodiments, the vectors are capable of
transduction levels of up to 101°/ml. As used herein, the term~~
"low efficiency transduction" refers to transducing vectors
capable of transduction at levels less than or equal to 103/mI.
As used herein, the term "long-term transduction" refers
to vectors that are capable of remaining transduced in host or
target cells for time periods that are longer than those
observed with other vectors. For example, the present
invention provides lentiviral vectors that are capable of
remaining transduced for at least 120 days, more preferably at
least one year, most preferably for the life of the subject or
the necessary time course of treatment. Long-term gene
transduction and high efficiencies of transduction of human
cells by the HIV vectors of the present invention were compared
with the conventional MLV vector (See, Table 5). The duration
of expression is a function of the choice of promoter and the
target cell type, more so than the choice of vector.
The term "stable transduction" or "stably transduced"
refers to the introduction and integration of zoreign DNA into
the genome of the transducted cell. The term "stable
transductant" refers to a cell which has stably integrated
foreign DNA into the genomic DNA.
The term "transient transduction" or "transiently
transduced" refers to the introduction of foreign DNA into a
cell where the foreign DNA fails to integrate into the genome
of the transducted cell. The foreign DNA persists in the
nucleus of the transducted cell for several days. During this
time the foreign DNA is subject to the regulatory controls that
govern the expression of endogenous genes in the chromosomes.
The term "transient transductant" refers to cells which have
taken up foreign DNA but have failed to integrate this DNA.
In some preferred embodiments, the target and/or host
cells of the present invention are "non-dividing" cells. These
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516 .
54
cells include cells such as neuronal cells that do not normally
divide . However, it is not intended that the present invention
be limited to non-dividing cells (including, but not limited
to muscle cells, white blood cells, spleen cells, liver cells,
eye cells, epithelial cells, etc.).
In particularly preferred embodiments, the vector and the
vector progeny are capable of transducing a plurality of target ~~
cells so as to achieve vector titers of at least 105 cfu/ml.
The preferred multiplicity of infection (MOI) would be at least
one ( i . a . , one hit on average per cell ) , more preferably at
least two.
Preferably, vector titers are at least 100, more
preferably at least 25%, still more preferably at least 50%,
of (a) wild-type reference lentivirus, and/or (b) the titer of
pTVnCMVnlacZ.
Adaptations for HIV-2 and SIV Derived Vectors
Based upon the experiments conducted during development
of the HIV-1 vector system, HIV-2 and SIV vector systems may
be developed (pH2P and pSIVP). To establish a lentiviral
vector based on HIV-2 or SIV, the 5' LTR and the untranslated
leader sequences of HIV-2ROD and SIVmac239 may be replaced with
the recombinant HP-1 enhancer/promoter and a synthetic leader
sequence with or without a splice donor site, both obtainable
from the pHP vectors. The 3' LTR may be replaced by the SV40
polyadenylation signal. The nef and env genes may bath be
deleted from the vector. The expression of vpx is preferably
included in the HIV-2/SIV packaging cells because it has been
shown that the HIV-2/SIV vpx (or SIVagm vpr) is necessary and
sufficient for nuclear import function and does not inhibit
cell cycle progression as does vpr. The VSV-G envelope gene
is preferably expressed from a separate expression vector.
Previous studies suggested that SIV or HIV-2 genomes can
be assembled into the HIV-1 particles, indicating that the
packaging signals of SIV or HIV-2 can be recognized by HIV-1
nucleocapsids. Thus, one may construct a hybrid vector which
is essentially an HIV-1 derived vector with SIV or HIV-2
packaging signals (from 3' of the PBS to the extended gag
CA 02333481 2000-11-27
WO 00/00600 PCT1US99/11516
55 .
sequences). These HIV-2 and SIV transducing vectors (pTV2 and
pTVS) may be tested in co-transfection experiments using pH2P
or pSIVP.
Alternatively, one may construct transducing vectors
wherein the lentiviral genetic elements are derived solely from
HIV-2 (pTV2) and SIV (pTVS) . However, instead of using
modified LTRs, a strong heterologous promoter is preferably ~~
used and the transcription initiation site is placed at the
beginning of the R-U5 sequence. Sequences in gag-pot and env
genes are deleted and the major SD and the gag AUG are mutated.
A CMV-driven reporter gene cassette such as the
CMV-IE-nlacZ-IRES-hyg from the pTV~-nlacZ-hyg vector may be
inserted in the nef ORF of the HIV-2 and the SIV vectors. The
3' LTR resembles the native LTR but with a deletion in the U3
except for the 5'att site.
For adaptation to other lentiviruses, one may identify and
modify the analogous genetic elements. For FIV, see, e.g.,
Elder and Phillips, Adv. Virus Research, 45:225-243.
Genetic Elements of the Packaging and Transducing Vectors
These are discussed in detail below.
Modified Major Splice Donor Sites
A splice donor site is a sequence which directs the
splicing of one exon to another exon. Typically, the first exon
lies 5' of the second exon, and the splice donor site
overlapping and flanking the first exon on its 3' side
recognizes a splice acceptor site flanking the second exon on
its 5' side. Splice donor sites have a characteristic
consensus sequence represented as (A/C) AG GURAGU (R=purine).
See Jackson, Nucleic Acids Res., 19: 3715-98 (1991). The first
three bases of the splice donor consensus are the last three
bases of the exon.
A splice acceptor site is a sequence which acts in
conjunction with a splice donor site, so that the intron
separating the two sites is removed. The characteristic splice
acceptor site is YYYYYYYYYYNYAG (Y=pyrimidine, N=any).
In a preferred embodiment, the HIV-1 major SD is replaced
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
56
with the cognate RSV SD, or a mutated form thereof. The
preferred mutant synthetic RSV SD sequence is split into two
parts with an AgeI site (ACCGGT) inserted in place of the RSV
gag AUG site:
Synthetic RSV SD: (sequence derived from RSV, Gene Bank ACCESSION # AF052428,
is
underlined and in bold)
~'-AGCTTGGTCGCCCGGTGGATCAAGACCGGTAGCCGTCATAAAGGTGATTTCGTCGGATC-3'
(Agel).
The original RSV SD:
S'-ATTCTGGTCGCCCGGTGGATCAAGCATGGAAGCCGTCATAAAGGTGATTTCGTCCGCGT-3'
The HIV-1 LTR consensus A 5' leader sequences (5'sj is in bold and underlined,
the
construct was made from HIV-1NL4-3, Access # M19921):
5'-GGCTTGCTGAGGTGC--?CACAGCAAGAGGCGAGAG----CGGCGACTGGTGAGTAC
GCC-??AAATTTT-3'
l5The entire 5' leader sequence of HIV-1 consensus A:
GCCTTGAG?TGCTT?AAGTA-GTGTGTGCCCGTCTG?TT?T?TGACTCTGGTAACTAGAGATCCCT
CAGACCACT?TAGACTGTGT--AAAA.ATCTCTAGCAGTGGCGCCCGAACAGG?????????????
???GACTCGAAAGCGAAAG-----------------------TTCCAGAGAAG?----TCTCTCG
ACGCA?-GGACTCGGCTTGCTGAGGTGC--?CACAGCAAGAGGCGAGAG----CGGCGACTGGT
20GAGTACGCC-??AAATTTT??-GACTAGCGGAG------GCTAGAAGGAGAGA?A
For reference, the corresponding HIV-2 and SIV sites are as
follows:
HIV-2ROD 5' splice junction: (ACCESSION # M15390)
255'-caaaaactgtagccgaaagggcttgctatcctacctttagacag2t agaagattgtgggag-3'
SIV 239 (ACCESSION # M33262 M61062-M61093):
acggcgtgaggagcgggagaggaagaggcctccggttgcagc~taagtgcaacacaaaaaa
gaaatagctgtcttttatccaggaaggggtaataagatag agtgggagatg
The artificially engineered splice donor (SD) site from
30 Rous sarcoma virus (RSV) in the pHP-1 construct, a site that
is unrelated to HIV sequences, was found to work like the wild-
type SD site (i.e., allowing partition of spliced tat and rev,
and unspliced gag-pol mRNAs into the cytoplasm). This is a
critical factor in some embodiments of the present invention
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516 .
57 ,
(i.e., the replacement of the HIV SD site with the RSV SD
site), as the native leader sequences and the major splice
donor site must both be deleted from the HP constructs to
decrease the probability of homologous recombination with the
transducing vectors (TV).
The splicing junction sequences have been previously
studied, see Ezzell C. Eukaryotic mRNA processing. The Journal~~
of NIH Research, 1995; 7:101-104; Mount SM. AT-AC introns: an
ATtACk on dogma. Science 1996; 271:1690-1692. In our previous
l0 studies, we showed that the first tat coding exon contains
positive and negative splicing regulatory elements and the
splicing signals can be hundreds of nucleotides away from the
splice junciton sites. Amendt BA, Hesslein D, Chang LJ,
Stoltzfus CM. Presence of negative and positive cis-acting RNA
splicing elements within and flanking the first tat coding exon
of human immunodeficiency virus type 1. Mol Cell Biol 1994;
14:3960-3970. Therefore, the success of inserting a functional
splice site in the leader region of HP construct using an
oligonucleotide sequence containing a small number of
nucleotide sequences from RSV 5' splice junction site was
surprising.
The splice donor site in the packaging constructs is used
solely for the expression of tat and rev genes downstream and
serves to stabilize the gag-pol transcript. It is possible
that tat and rev functions can be provided in trans and the
5' splice donor site can be totally eliminated. For example,
an SV40 promoter with a replication origin can be used in the
packaging constructs and the DNA can be transfected into a
SV40 large T antigen expressing cell lines such as COS7 cells
(African green monkey kidney cells expressing SV40 T Ag).
Modified Packaging Signals
The packaging signal is of course inactivated in the
packaging vectors. In the transducing vectors, a functional
packaging signal is required, but need not be identical to the
source signal.
The packaging signals have a secondary structure; they
may be mutated so as to alter the primary sequence while
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
58
substantially retaining the secondary structure. Applicant
has found it possible to mutate the HIV-1 major packaging
signal by replacing GGTG with GCAG or GGGG.
Lentiviral packaging signals may be replaced with
nonlentiviral packaging signals, or functional mutants thereof,
such as the cognate packaging signal of another virus, such
as RSV or MLV. If so, it will generally be necessary to make~~
corresponding mutations in the Gag nucleocapsid protein so that
it recognizes the new packaging signal. Thus, one could make
a chimera of the Gag nucleocapsid protein and the cognate
nucleocapsid protein of the other virus.
Finally, in the case of the packaging vector(s), one may
delete the HIV-1 major packaging signal altogether.
Structural Genes/Proteins
The terms "Gag protein" and "Gag proteins" refer to any
or all proteins, respectively, encoded by the gag gene,
including both the ultimate virion proteins and their
precursors (i.e., proteins which are processed intracellularly
into the ultimate virion proteins.) The terms "Pol protein(s)"
and "Env protein(s)" are analogously defined. These terms can
be further modified by "-like" or "-equivalent" as elsewhere
defined.
As noted above, the structural virion genes are the gag,
pol and env genes. At least one, and preferably all of these
genes is inactivated in the transducing vector. The only part
of gag or env necessary to keep is the part that play essential
roles in packaging. We have preliminarily identified the first
39 nucleotides of the gag coding sequence excluding the
initiation codon and the RRE in the beginning of gp41 coding
region of the env sequence are essential to keep. However,
site-specific mutagenesis can be performed to further change
these sequences to introduce stop codons in the gag gene and
in the env gene and to kill the RRE function of interacting
with Rev. This latter changes can further improve the safety
of the HP/TV vector system.
However, the packaging vectors must collectively provide
genes encoding the functions of the gag, pol and env genes in
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516 .
59
order to provide a functional virion. Nonetheless, these
genes may differ from the source genes by silent and other
functional mutations.
Silent Mutations
These may be made almost freely throughout the gene. The
only areas where caution is required is where the choice of ~~
sequence has regulatory significance, e.g., the slippage region
in gag-pol, or the RRE region in env. In some instances, such
as in the case of an INS element, it may actually be desirable
l0 to inactivate the regulatory element. In other instances, the
regulatory element may be useful, and only silent mutations
which leave it functional are desirable.
Functional Mutations
These are mutations which affect the amino acid sequence
of one or more of the encoded polypeptides, but which do not
substantially abolish the relevant biological activity of the
affected polypeptide(s).
The comments which follow apply not only to mutation of
lentiviral proteins, but also to mutation of naturally
occurring, nonlentiviral proteins which are acting as the
equivalent of a lentiviral protein. For example, instead of
using wild-type VSV G protein in place of HIV-1 gp120, one may
use a functional mutant of VSV G protein.
As explained below, while the result of a mutation is not
absolutely predictable, some mutations are clearly more likely
to be tolerated than others.
The accuracy of these predictions is dependent in part on
whether a 3-D structure for the protein is known, whether
homologous proteins (i.e., functional mutants, naturally
occurring or otherwise) have been sequenced, and whether the
biologically relevant binding sites of the protein have been
identified.
The tremendous natural variation of the HIV-1 genome
suggests that it is quite tolerant of multiple mutations in
many genes. The following specific guidance is offered:
A general source of 3D structures is the Protein Data
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
Bank, which is searchable on the Internet.
Gag: The overall sequence variability of Gag proteins in
HIV-1 isolates is more than 20%. With deliberate mutations, it
is likely that a higher degree of sequence variation can be
5 tolerated. The 3D structure of the nucleocapsid protein of
Gag, complexed to human cyclophilin A, has been determined;
Gamble, et al., Cell, 87: 1285-94 (1996); see also PDB
structures latv and lncp. Mutational analysis reveals that
the zinc finger domains in the NC protein play important roles
10 in RNA encapsidation and HIV infectivity. Mizuno, et al., AIDS
Res. Hum. Retroviruses, 12: 793-800 (1996). . Charged amino
acids have also been shown to be involved in RNA packaging and
infectivity. Poon, et al., J. Virol., 70: 6607-16 (1996).
Mutational studies have also been made of the CA proteins,
15 using deletion mutants and chimeras. Carriere, et al., J.
Virol., 69: 2366-77 (1995). For structure-function
relationships in general, see Wills, AIDS, 5: 639-54 (1991).
3D structures are available for the CA (PDB lafv) and MA (PDB
lhiw) proteins.
20 Pol: The overall sequence variability among HIV-1
isolates is more than 20%; in the protease domain, a
variability of more than 40o has been observed. The 3D
structure of the reverse transcriptase, complexed to an
inhibitor, is known, see Kohlstaedt, et al. , Science, 256 : 1783-
25 90 (1992), and a structural model for the protease has been
proposed, see Pearl and Taylor, Nature, 329: 351-4 (1987). The
polymerase and protease functional domains have been studied
by mutagenesis, see Loeb et al., Nature, 329: 351-4 (1989); Le
Grice, et al, EMBO J., 10: 3905-11 (1991). For 3D structures,
30 see also PDB entries lhnv and lrtl (RT), litg (integrase), and
lhvk (protease).
Env: For the Env proteins gp120 and gp4l, the overall
sequence variability among HIV-1 isolates exceeds 600. For 3D
structures, see PDB entries laik (gp41) and lacy (gp120
35 fragment) For discussion of functional domains, see Moulard M,
Challoin L, Canarelli S, Mabrouk K, Darbon H, Retroviral
envelope glycoprotein processing: structural
investigation of the cleavage site, Biochemistry 1998 Mar
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516 .
61
31;37(13):4510-4517.
Pseudotyping studies have shown that the Env proteins can be
entirely replaced by the retroviral but non-lentiviral MoMLV
Env proteins, or the unrelated VSV-G protein.
The envelope proteins encoded by the packaging vector may
be lentiviral or non-lentiviral proteins. The advantage of a
non-lentiviral protein is that it can confer on the produced~~
particles the ability to bind to a cell surface receptor of a
class of cells not normally infected by the lentivirus. An
example of a non-lentiviral.envelope protein of interest is the
vesicular stomatitis virus (VSV) G protein. VSV-G pseudotyped
particles are rigid and can be concentrated more than 1000-
fold. They also bind to different cells than those bound by
HIV-1 gp120 typed particles.
Where one of the packaging vectors encodes a non
lentiviral envelope protein, it is referred to as an envelope
pseudotyping vector. In preferred embodiments, the
pseudotyping vector is selected from the group consisting of
pHEF-VSVG, pHEF.A-env, Gibbon ape leukemia virus env, and MLV
Amphotropic env.
Alternative Env proteins: The Env proteins of HIV-1 may
be replaced with Env proteins of other lentiviruses, of
nonlentiviral retroviruses, of nonretroviral viruses, or with
chimeras of these proteins with other peptides or proteins.
Examples are the Env proteins of VSV (G protein), the
hemagglutinin protein of influenza virus, the surface antigen
(S and preS) of hepatitis B virus, and the Env protein of RSV.
These modifications increase the range of cells which can be
transduced with HIV-1 derived vectors.
Particular tissues or organs normally infected by the
source virus of the envelope protein may be targeted, e.g.,
HBV preS-Ag... Liver
Respiratory syncytium virus (RSV) ... lung and
respiratory tracts
Herpes simplex virus.... central nerves system
Ebola virus.... broad host cell tropism
HCV.... liver, spleen.
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516 .
62
We used HN and F of Sendai virus by PCR amplification of
the viral RNA and cloned it into expression vector pHEFla.
Upon co-transfection of this pseudotyping vector with pHP and
pTV, we obtained vector titer on TE671 cells about 10% of the
VSV-G pseudotyped vector system. It is possible that this
pseudotyped virus may have increased efficiency on lung
epithelial cells. °
Chimeric Env Proteins : A chimera may be constructed of an
env protein and of a ligand that binds to a specific cell
surface receptor in order to target the vector to cells
expressing that receptor. Examples are chimeras including
FLA16 (a 6 a.a. peptide which binds integrin receptors),
erythropoietin (which binds the erythropoietin receptor}, human
heregulin (which binds the EGF and related receptors), and
stromal cell derived-factor (SDF-1) (which binds to CXCR4
chemokine receptor of CD34 cells).
Alternatively, the chimera could include an antibody
variable light or heavy domain, or both domains joined by
suitable peptide linker (a so-called single chain antibody).
Such an antibody domain could target any desired cell surface
molecule, such as a tumor antigen, the human low-desnity
lipoprotein receptor, or a determinant on human MHC Class I
molecules.
Derivatized Env Proteins: Virions may be chemically,
enzymatically or physically modified after production in order
to alter their cell specificity. Examples of modifications
include chemical or enzymatic addition of a ligand which would
be recognized by a cell surface receptor (e.g., addition of
lactose so that the virions will transduce human hepatoma cells
which express asialoglycoprotein receptors), or incubation of
the virus with a biotinylated antibody directed against the
vector's Env protein, followed by addition of a streptavidin-
linked ligand recognized by the cell-surface receptor. A
heterobispecific antibody could be used to link the virion's
Env protein to such a ligand, too.
Regulatory Genes
The vector system may provide the regulatory proteins, or
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
63
surrogates therefor, or wholly omit them. If Tat or Rev
equivalents are provided, the corresponding genes may be placed
on the transducing vector, or on the same or different helper
vector (s) . These genes need not be placed on the packaging
vectors. Again, silent mutations may be made almost freely.
Functional mutation of Tat and Rev should be feasible.
For the 3D structure of Rev, see PDB entry lrpv; for RRE, see~~
letf and letg; for Tat, see ltiv; for TAR, see lkis.
Tat is a transcriptional factor which acts to bind the
polII transcription elongation complex and increases the
processivity of transcription.The amino acid sequence of Tat
is highly conserved amongst different HIV-1 strains with more
than 80% homology. Mutational analysis has shown that the
functional domain is in the first coding exon because deletion
of the second exon does not affect its transactivation
function. The N-terminal domain is highly charged and
contains a long stretch of basic amino acids such as arginine
which is the characteristic of RNA binding domain. Tat has
been shown to bind to the TAR sequence at the loop of a
stem-loop structure in the 5' end of the genome. In addition
to its transcriptional activation function, Tat has also been
shown to enhance reverse transcription and in our laboratory,
we have shown that Tat can enhance gag protein precursor
processing. Therefore, the muitipie functions of Tat may
indicate that it may be required for high titer vector
production. However, Tat may be substituted with different
lentiviral transactivators to avoid recombination of HIV
sequences.
Rev is also a transcriptional regulator which acts at a
post transcriptional step in the nucleus to enhance the export
of RRE-containing RNA to the cytoplasm. Its amino acid
sequence is highly conserved amongst different HIV-1 strains.
Human T cell leukemia virus type 1 (HTLV-1) encodes a similar
protein named Rex. Rex and Rev share low sequence homology
(less than 400} but have similar functions. Mutational
analysis have shown that rev function requires both coding
exons. Rev binds to RRE in env and interacts with cellular
proteins in the nucleus to mediate the nuclear export of the
CA 02333481 2000-11-27
WO 00/00600 PC'T/US99/11516 .
64
RRE-containing transcripts. The function of Rev is
dispensable if RRE and the inhibitory sequences in the gag-pol
and env are mutated.
Although TAR and RRE are known for their functions in
mediating Tat and Rev interaction with the viral RNA, these
two RNA elements may have other functions unrelated to Tat and
Rev interaction which may be important for gene transfer~~
vector function. It is possible that RRE or TAR may contain
minor packaging signal to enhance viral RNA encapsidation.
The example of RRE mutation on vector function is presented
later.
With regard to complete deletion, Tat and Rev have been
reported to be absolutely required for viral replication in
vitro or in vivo Vaishnav YN, Wong-Staal F. The biochemistry
of AIDS. Ann Rev Biochem 1991; 60:577-630; Greene WC.
Regulation of HIV-1 gene expression. Annu Rev Immunol 1990;
8:453-475.
However, a small element from the Mason-Pfizer monkey
virus genome can make human immunodeficiency virus type 1
expression and replication Rev-independent, Bray M, Prasad S,
Dubay JW, et al. A small element from the Mason-Pfizer monkey
virus genome makes human immunodeficiency virus type 1
expression and replication Rev-independent. Proc Natl Acad Sci
Dsa 1994; 91:1256-1260, and this strategy has been used to
develop a rev-independent HIV vector system, see Srinivasakumar
N, Chazal N, Helga-Maria C, Prasad S, Hammarskjold M-L, Rekosh
D. The effect of viral regulatory protein expression on gene
delivery by human immunodeficiency virus type 1 vectors
produced in stable packaging cell lines. J. Virol. 1997;
71:5841-5848.
Also, we have reported that HIV tat mutants with stop
codon mutations or deletions in the tat open reading frame can
still infect human lymphocytes and macrophages, Chang L-J,
Zhang C. Infection and replication of Tat-minus human
immunodeficiency viruses: genetic analyses of LTR and tat
mutants in primary and long-term human lymphoid cells . Virology
1995; 211:157-169. The requirement for Tat transactivation of
HIV-1 LTR can be diminished if the LTR enhancer promoter
CA 02333481 2000-11-27
WO 00/00600 PCTNS99/11516
65 .
elements are replaced with a chimeric CMV-IE-HIV LTR. Robinson
D, Elliott JF, Chang L-J. Retroviral vector with a
CMV-IE/HIV-TAR hybrid LTR gives high basal expression levels
and is upregulated by HIV-1 Tat. Gene Therapy 1995; 2:269-278.
LTR and tat mutants of HIV-1 have been shown to have diminished
replication phenotypes (See e.g. , L. -J. Chang et a1. , J Virol. , ~~
67:743-752 [1993]; L.-J. Chang and C. Zhang, Virol.,
211:157-169 [1995]; and J.C. Leonard et al., J Virol.
63 :4919-4924 [1989] ) .
Accessory Genes
The accessory proteins of HIV-1 may have important
functions in viral pathogenesis, see Trono D. HIV accessory
proteins: leading roles for the supporting cast. Cell 1995;
82:189-192; but they are dispensible for viral replication in
tissue culture. We and others have shown that the accessory
genes are not essential to the creation of functional packaging
and transducing vectors, i.e., they may be completely deleted.
Hence, it is unnecessary to consider in detail the guidance
offered by the art as to which mutations of the accessory
proteins might be functional. Of course, if one chooses to
retain an accessory gene, such guidance can be found in the
literature on, e.g., sequences of HIV-1 isolates.
In general, it is preferable to delete all lentiviral
accessory genes when constructing the transducing vector, in'
order to reduce the risk of homologous recombination to form
RCV. However, certain accessory genes, such as vpr or vpx, may
increase transduction efficiency of nondividing cells, in which
case there is a countervailing advantage to retaining them in
a form in which they encode functional protein. If so, silent
mutations, and other functional mutations, may be introduced
to reduce the risk of homologous recombination without loss of
gene function.
Other Genetic Elements
In the packaging vectors (pHP-likes), the 5' LTR can be
totally eliminated but a functional promoter will be needed
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516 .
66
to drive RNA transcription and gag-pol gene expression.
Preferably, a strong enhancer/promoter, e.g., one at least 50%
as strong as the wild-type 5' LTR, will be used to replace the
5' LTR.
Tat may be needed for high efficiency of Gag-Pol
synthesis. In this case, HIV-1 TAR sequence may be retained
in the 5' end for enhanced promoter function. In the~~
transducing vectors (pTV-likes), the necessary functions for
vector production in the 5' LTR are the repetitive sequence R,
which serves as annealing sequence for minus-strand DNA
jumpting to the 3' R, and the attachment site (att) in the 3'
end of U5 adjacent to the PBS for provirus integration. The
R can be made different from the native HIV R but have the
same mutated R in the 3' end. The att site is necessary for
integrase recognition and binding and therefore cannot be
changed (unless one can be coordinated with a corresponding
change in the gag-encoded integrase).
Preferably the lentiviral promoter /enhancer elements of
the 5' LTR are replaced with a nonlentiviral promoter/enhancer
in at least one (a) the packaging vectors or (b) the
transducing vector. Both the HP 5' LTR and TV 5'LTR
promoter/enhancers may be replaced with the same or with
different promoter/enhancers, e.g., CMV IE in one and EF-la in
the other.
In the 5' leader region, no HIV functional elements are
necessary for the packaging construct. However, for the
transducing vector, several elements are needed, in an order
from 5' to 3' including PBS, packaging signal, and dimer
linkage sequence (DLS). HIV uses lysine tRNA PBS which may
be mutated to a different retroviral PBS such as histidine
tRNA or proline tRNA of RSV or MLV. However, a coupled change
in the RT domain which recognizes the corresponding PBS will
also be needed. The packaging signal for HIV RNA has been
shown to include different areas in the genome. It is
possible that site-specific mutations can be made to change
the primary sequence but maintain the secondary structure.
The major 5' splice donor site and the gag AUG have been shown
by others to be essential for genome packaging. However, we
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
67
have demonstrated that both the SD and the gag AUG can be
mutated and the modified transducing vector can still be
packaged in high efficiency (see examples below). The DLS is
not well defined. However, both primary sequence and
secondary structure may be necessary for a functional DLS
which overlaps the packaging signal between SD and the gag
AUG. "
In one embodiment, the packaging vector replaces the HIV-
1 SD with an RSV SD. The splicing junction sequences have been
previously studied. Ezzell.C. Eukaryotic mRNA processing. The
Journal of NIH Research 1995; 7:101-104; Mount SM. AT-AC
introns: an ATtACk on dogma. Science 1996; 271:1690-1692. In
our previous studies, we showed that the first tat coding exon
contains positive and negative splicing regulatory elements and
the splicing signals can be hundreds of nucleotides away from
the splice junction sites. Amendt BA, Hesslein D, Chang LJ,
Stoltzfus CM. Presence of negative and positive cis-acting RNA
splicing elements within and flanking the first tat coding exon
of human immunodeficiency virus type 1. Mol Cell Biol 1994;
14:3960-3970.
In the case of the R region, the R regions of the
transducing vector may be replaced with a functional R from
another source, e.g., RSV. The MLV R is longer than that of
RSV and therefore is less desirable. It is believed that
almost any sequence of similar length to the HIV R region would
work if it appears in both ends of the vector genome, allowing
RT to jump and anneal.
We also believe that it may be possible to use an
arbitrary sequence for the major splice donor site, provided
it was compatible with the splice acceptor sites, or the latter
were modified to be compatible.
Coordinated Mutations
In a number of instances, a mutation of one genetic
element is preferably complemented by a mutation of another
genetic element in the same or a different vector:
(a) transducing vector PBS and packaging vector RT;
(b) transducing vector packaging signal, or dimer linkage
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516 .
68
site, and packaging vector Gag nucleocapsid protein;
(c) packaging vector Rev deletion, and, in transducing
vectors, inactivation of INS's in gag and pol, and RRE;
(d) vector tat and vector TAR, and
(e) transducing vector att site and packaging vector gag
integrase protein.
Wild-Type, Mutant and Surrogate Proteins
From time to time this specification has cause to
characterize a protein, or a gene encoding a protein, as being
(a) identical to a naturally occurring protein; (b) a mutant
which is substantially similar in amino acid sequence to the
naturally occurring protein, and retaining a substantial
portion of the biological function of the naturally occurring
protein, or (c) substantially dissimilar in amino acid sequence
to the naturally occurring protein, but nonetheless capable of
substituting for the naturally occurring protein. It is
convenient to develop a concise r_erminology for the various
possibilities.
Hence, the term "wild-type" X implies that the protein is
identical to a naturally occurring form of protein X.
The term X "-like" protein implies that the protein is
either identical to X, or is a mutant as described in (b)
above. The precise scope of this Lerm wil:1 vary depending on
how narrowly X is defined. If the reference is to an "HIV-1
gp120-like protein", the amino acid sequence and biological
activity of the X-like protein will be compared to that of the
HIV-1 gp120 proteins. If it is to a "lentiviral Env-like
protein", it will be compared to that of the most similar of
the lentiviral envelope proteins. And so forth.
A mutant is more likely to be considered substantially
identical to a reference protein if (a) the overall sequence
identity is within the natural range of variation of homologous
proteins (e.g, of all HIV-1 gp120 variants, if the referent is
a particular gp120); (b)most or all of the sites of mutation
are sites of high variability in that family; and (c) most or
all of the substitutions, especially at low variability sites,
are at least semiconservative, and more preferably conservative
CA 02333481 2000-11-27
WO 00/00600 PCT/US99111516
69 -
or highly conservative substitutions in general, or favored
by experimental data.
The term "X-like protein mutant" implies that the protein is
not identical to X.
The term "X-equivalent protein" includes all of the
possibilities (a)-(c) above. Possibility (a) is excluded by
the phrasing "X-equivalent protein mutant". Possibilities (a)~r
and (b) are both excluded by the phrasing "X surrogate".
Similarly, we may speak of a gene encoding a "wild-type
X", an "X-like protein", an "X-like protein mutant", an "X
equivalent protein", an "X-equivalent protein mutant", or an
"X-surrogate". The gene may encode a precursor of the protein
in question, rather than the mature protein per se.
Similar terminology applies to genetic elements other
than genes.
If not otherwise defined for use in the particular
context in question, a mutant is considered substantially
similar in sequence ito a reference sequence if it is at least
50% identical with the reference sequence, with percentage
identity being calculated by the default procedure set forth
below.
Inactiva Lion
This invention contemplates that certain genetic elements
of the lentiviral genome will be substantially inactivated to
render the genome more suitable (e.g., safer) for use as a
vector in the delivery of therapeutic genes to a patient. The
inactivation may, but need not be, absolute. Preferably, the
level of inactivation is at least 50%, more preferably at least
9%, still more preferably at least 950, most preferably at
least 990.
There are two fundamental methods of inactivating a
protein. First, one may delete the corresponding gene so that
the protein is simply not produced. Secondly, one may alter
the corresponding gene so that the expressed polypeptide is a
nonfunctional mutant.
A gene is substantially inactivated if it is mutated so
that it substantially is no longer capable of being transcribed
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516 .
70 .
and translated into a polypeptide retaining a substantial
amount of the objectionable biological activity of the
originally encoded polypeptide. A gene may be inactivated by
(1) completely deleting it, (2) removing its initation codon
so that it is not translated, (3) inserting one or more stop
codons into the coding sequence, preferably immediately after
or in place of the initiation codon, so as to stop translation~~
prior to production of a functional polypeptide, (4) inserting
or deleting a number of bases, other than a multiple of three,
so as to cause a frameshift, and thus the production of an
erroneous polypeptide sequence downstream of the frameshift
mutation, (5) inserting or deleting one or more whole codons,
at either end or internally, so that a nonfunctional
polypeptide (or a polypeptide of substantially different
function) is encoded, (6) making one or more base substitutions
(point mutations) in the gene which alter the encoded amino
acid, so that a nonfunctional polypeptide is encoded, or (7)
a combination of any of (2) to (6) above.
Preferably, if there is no reason to preserve any of the
sequence of the gene, it is inactivated by completely deleting
it. In the lentiviral genome, some genes overlap, so that it
is not possible to inactivate one gene by complete deletion
without inactivating the overlapping gene. And some genes
contain regulatory elements (like RRE in envy, and so cannot
be completely deleted without loss of a regulatory function.
Hence, in these circumstances, methods (2)-(6) above are
appropriate. If point mutations are employed, preferably these
are multiple mutations.
This specification explains how to identify which
mutations of a gene are likely to be functional. By
disregarding such advice, one may obtain inactivating
mutations.
A genetic element other than a gene is inactivated if it
is mutated so that it is no longer capable of performing its
normal biological function. Normally, this means that it is
mutated so that it is no longer recognized by a nucleic acid-
binding protein. In some cases, the protein recognizes the
primary structure (nucleotide sequence) of the genetic element,
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
71
in other cases, it recognizes a secondary structure arises from
the folding of the strand. A single nucleic acid strand can
fold upon itself to bring complementary regions into proximity;
these are then held together by hydrogen bonding between the
complementary G : C or A: T bases . The HIV maj or packaging signal
is an example of a genetic element recognized on the basis of
its secondary structure. (Of course, the secondary structure~~
of a genetic element is a consequence of its primary sequence . )
The secondary structure of a nucleic acid sequence may be
predicted by conventional methods, such as those of Tinoco, and
one or more segments predicted to have a secondary structure
(e. g., a stem loop) deleted or modified until an acceptable
level of inactivation is obtained.
Complete deletion is the preferred method of
inactivation, if a genetic element is not at all desirable .
However, it is possible that a genetic element A which is to
be inactivated lies between two genetic elements B and C which
are to be retained, and which preferably are at a particular
distance from each other, or that all or part of the sequence
forming the genetic element to be inactivated is also part of
another genetic element which is to be retained. In these
situations, complete deletion is not desirable. If so, single
or multiple insertions, deletions or substitutions, whether
consecutive or nonconsecutive in Lhe primary sequence, may be
used to alter the sequence sufficiently so that the
objectionable genetic element is inactivated without
substantial adverse consequences.
A vector may be said to comprise an inactivated genetic
element even if the inactivation is accomplished by completely
deleting the element, so it is not present in the vector; the
phrase then indicates that at least one of the differences
between the vector and a source (or cognate) lentivirus is that
the element in question, which is missing in the former, is
found in the latter.
Where this specification refers to "deletion" of a
genetic element, but does not specify complete deletion, it
should be taken to include a partial deletion (even a single
nt) if sufficient to accomplish inactivation of the element.
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
72
However, in general, unless said to be impossible, complete
deletion should be considered a more preferred embodiment of
any preferred "deletion". Moreover, in the case of partial
deletions, unless otherwise indicated, the deletion is at least
50, 10%~~ 15%, 20%, 250, 300, 350, 40%, 450, 500, 550, 600, 700,
75%, 800, 85%, 90%, or 95%. With the higher values being most
preferred. °'
Replacement
In some cases, a genetic element performs an essential
function, but one nonetheless would like to modify it to reduce
the risk of homologous recombination. In that case, one may
replace it with an equivalent element which has the same
function but which differs in nucleotide sequence.
While it is possible to randomly mutate a genetic element
and screen for mutants which preserve function (this has been
done with promoters, operators, and genes encoding DNA-binding
proteins, as in the work of Hecht, Reidhaar-Olsen, and Ladner),
it may be more expedient to use a replacement element which
exists in nature, for example, as an element of a nonlentiviral
2G retrovirus, a virus other than a retrovirus, or even of a cell.
This application has looked to RSV as a preferred source
or replacement element because it is on nonhuman and indeed
nonmammaiina (avian) origin, and hence less likely co do harm
to human or other mammalian cells.
However, MLV is another potential source of replacement
elements.
It should be noted that a replacement element may be
substantially, but not exactly, identical to a natural element .
For example, we modified the RSV major SD element.
Homologous Recombination
Homologous recombination is the formation of a hybrid of
two sequences, wherein the point of crossover between one
sequence and the other lies at a region of significant length
wherein the two sequences are substantially identical. This
region is called a region of homology.
Preferably, the packaging vectors) and transducing
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516 .
73
vector are chosen so that the frequency of homologous
recombination between them is less than that experienced with
the vector systems of Naldini, et al., Science, 272: 263 (1996)
or of Corbeau, et al., PNAS USA 93:14070-5 (1996). Preferably,
if homologous recombination so occurs, it is not enough by
itself to form a replication-competent virus.
It should be noted that mutations which inactivate a gene~I
do not necessarily prevent that gene from being a site for a
crossover event, and, conversely, mutation of a gene in one
vector to eliminate homology with a corresponding gene in
another vector will not necessarily inactivate the gene. For
example, insertion of a stop codon does not prevent crossover
within the untranslated downstream sequence, and silent
mutations may be used to destroy homology, without affecting
the nature of the encoded polypeptide. Of course, if a gene
is entirely deleted, it is both inactivated and incapable of
participating in homologous recombination.
The probability of a recombination occurring between the
packaging vector and the transducing vector increases as the
number, length and degree of identity of the two sequences
increases. Cell GeneSys, W091/06667, which relates to the
deliberate induction of homologous recombination of a
transgene, states that homologous sequences as short as 14
bases may provide for homologous recombination, but that its
preferred flanking sequences are at least about 150 bp. They
cite Rubnitz and Subramani, Mol. And Cell. Biol., 4:2253-8
(1984), as describing the minimum amount of homology required
for homologous recombination in mammalian cells, and Kim and
Smithies, Nucleic Acids Res., 16: 8887-8903 (1988) as
describing a PCR-based assay for homologous recombination.
Chappel, USP 5,272,071, states that it has been suggested that
the minimum requirement for homologus recombination is 25 base
pairs, citing Ayares et al., ONAS USA 83:5199-5203 (1986). It
has been reported in studies of bacterial recombination that
a 10% divergence in sequence between two incompletely
homologous sequences reduces the frequency of recombination
between them by a factor of about 40. See Shen and Huang,
Genetics, 112: 441-457.
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516 -
74 .
Watt, et al., PNAS USA 82:4768-72 (1985) states that, for
E. Coli recombination, a minimum of about 20 base pairs of
completely homologous segment is required for significant
recombnation, that there is an exponential increase in
frequency over the range 20-74 bp, and a linear increase in
frequency with length for longer perfectly homologus segments.
There was a 100-fold increase in recombination frequency from ~~
30 by to 150 by homology. Other relevant papers include
Smokik-Utlaut, et al. , Mol. Cell . Biol. , 3 :1204-11 (1983} ; Watt
et al., PNAS USA 82:4768-72 (1985).
Homologus recombination begins with a hybridization step,
and it is therefore worthwhile to consider studies of
hybridization probes. Typically, even perfectly homologous
probes must be 15-20 bases long to exhibit reasonable
specificity against the mammalian genome. A published formula
relating Tm (deg C) to, inter alia, probe length, indicates
that the Tm should decrease according to the term (600/N),
where N is the probe length. Moreover, each 1% divergence in
sequence is expected to reduce Tm by 1-2 deg C. Finally, the
Tm increases with increasing GC content, and hence it is more
helpful to eliminate GC than AT pairs.
Therefore, the likelihood of recombination can be
decreased by minimizing the lengths of identical segments in
the two vectors. In practice, this means that it is preferable
to (1) completely delete any genes or other genetic elements
which do not substantially contribute to the functionality of
the vector (packaging or transducing) in question, (2) whem a
function of a genetic element does make such a contribution,
replace it, where possible, in one of the vectors (or
differently in both vectors) with a genetic element which is
different in sequence but similar in function, and
substitutable for the original element (e. g., the cognate
element in another strain or Glade, in another lentivirus, in
another retrovirus, in another virus, or in a microbial cell,
or a functional mutant of the original element or a cognate
element of another organism). In some instances, such as the
PBS and the RT, Rev and RRE, and Tat and TAR, it may be
necessary to make coordinated changes in two or more genetic
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516 .
elements.
In the case of a gene, silent mutations may be introduced
so as to reduce its sequence identity with the original gene,
even though both the mutated and original genes encode the same
5 protein. In many codons, the third base of the codon is a
wobble position in which any of 2-4 different bases can appear
without alteration of the encoded amino acid. While there are~~
a few codons (Met and Trp) which do not allow silent alteration
of the third base, these are counterbalanced by the codons
10 which allow some variation of the first base, too. Thus, one
can reasonably expect to be able to make silent alterations to
perhaps one-third of the total sequence, with the alterations
being fairly evenly distributed. Care must be taken that such
silent mutations do not substantially interfere with any
15 important regulatory element of the gene, such as the slippage
region in gag-pol, unless a functional substitute for that
regulatory element is provided.
In the case of a genetic element which is recognized by
virtue of its secondary structure, paired bases may be
20 identified and swapped, i.e., if a G at position 1 pairs to a
C at position 10, forming part of a stem, position 1 may be
changed to C and position G to C. Indeed, it may also be
possible to replace G:C with A:T, and A:T with G:C, pairs,
although such changes are less certain to be tolerated in view
25 of the difference in strength between the two interactions.
In case of a genetic element which is recognized by
virtue of its primary sequence, it is relatively rare that the
recognition is absolutely specific, that is, only one
functional primary sequence exists. The specificity of the
30 recognition may be explored by combinatorial mutagenesis, e.g.,
as taught by Reidhaar-Olsen and Ladner, in a system which
screens or selects for recognition of the mutated element.
Alternatively, if the gene encoding the binding protein is
accessible to manipulation, e.g., it is a viral or host cell
35 gene, one may attempt to modify that gene so that a modified
binding protein is produced, either in place of or in addition
to the wild-type protein, and the cognate recognition sequence
may then be modified as well for recognition by the mutant
CA 02333481 2000-11-27
WO 00/00600 PCT/US99111516
76 .
protein. It may be possible to simultaneously explore
mutations of both the binding protein and the target sequence
by combinatorial mutagenesis. (These methods are of course
applicable, although less urgently, to the case of genetic
elements recognized by virtue of their secondary structure,
too . )
Default Definition of Percentage Sequence Identity
For the purpose of this specification and claims, unless
otherwise stated, the percentage sequence identity between two
sequences is to be determined by (1) aligning~to maximize the
local similarity score (as hereafter defined) between the two
sequences, and (2) expressing the number of identical aligned
pairs as a percentage of (a) the total length of the overlap
region, including nulls (gaps), or (b) the original length of
the shorter sequence, whichever of (a) or (b) is larger.
The two sequences are to be aligned by a rigorous (linear
programming) based local alignment algorithm in which the
overall similarity score for a given alignment is obtained by
summing the pairwise alignment scores, for each aligned pair
of bases or amino acids, and a gap penalty for each gap
introduced into either sequence in an attempt to improve the
overall similarity score for the alignment. The pairwise
alignment scores are derived from a 20x20 scoring matrix for
amino acids and a 4x4 scoring matrix for nucleotides. The gap
penalties are a linear combination of a gap initiation penalty
imposed for the first null of a given gap, and a gap extension
penalty for each additional null added to that gap. Only
internal gaps will be penalized. The alignment must be
statistically significant (chance expectation < 0.001 as
elsewhere defined) in order to be considered.
In the case of an amino acid sequence alignment, the
scoring matrix will be the PAM250 matrix, in the form wherein
the scores range from +17 to -8; the gap initiation penalty
will be -12; and the gap extension penalty will be -4.
For nucleotide sequence alignments, the scoring matrix
will be an identity matrix in which all identities are scored
6 and non-identities are scored zero, the gap initiation
CA 02333481 2000-11-27
WO 00/00600 PCTNS99/11516
77
penalty will be -12, and the gap extension penalty will be -4.
Functional Mutation
In certain instances, we have indicated that it is
required, or at least desirable, that the function of a
particular lentiviral gene be retained. This does not mean
that the gene cannot be mutated, or even that the mutations be~~
limited to silent mutations. Functional mutations, which
substantially preserve the relevant biological activity of the
corresponding protein(s), are permissible.
Most residues of a protein can tolerate some degree of
mutation. Mutations may take the form of single or multiple
substitutions, insertions, or deletions. Preferably,
insertions or deletions are directed to the termini of the
molecule, or to surface loops or interdomain boundaries.
Preferably, internal insertions and deletions are of no more
than five residues, absent evidence (such as an example in a
homologous protein) that a larger internal insertion or
deletion could be tolerated.
There is no preferred maximum with respect to an
insertion at a terminus, which is more aptly termed an
"addition" or "fusion". It is routine to fuse one protein to
another to facilitate expression, or to provide a fusion
protein which has the combined biological activities of its
components. A fusion protein may be useful as a precursor,
which can be cleaved to liberate an active protein, even if the
fusion protein itself lacks activity.
With regard to deletion at a terminus, more aptly termed
"truncation", the purpose of the modification is important.
It is routine to extensively truncate a protein when one is
interested only in its immunological properties. One may
abstract from a protein an epitope as small as five amino
acids, and use it by itself to elicit a T cell response, or
conjugated to copies of itself or to an immunogenic carrier to
elicit a B cell response. When it is a biological activity
which must be preserved, the limits on truncation may be more
stringent.
Preferably, after considering substitutions, and any
CA 02333481 2000-11-27
WO 00/00600 PC1'/US99/11516
78
internal deletions and insertions, the mutant is at least 50%,
more preferably at least 800, identical in sequence to the
original protein.
A protein is more likely to tolerate a mutation which
(a) is a substitution rather than an insertion or
deletion;
(b) an insertion or deletion at the. termini, thane
internally, or, if internally, at a loop or an
interdomain linker;
(c) affects a. surface residue rather than an
interior residue;
(d) affects a part of the molecule distal to the
binding site;
(e) is a substitution of one amino acid for another
of similar size, charge, and/or hydrophobicity; and
(f) is at a site which is subject to substantial
variation among a family of homologous proteins to
which the protein of interest belongs.
These considerations can be used to design functional mutants
of lentiviral proteins, and of naturally occurring
nonlentiviral surrogates of lentiviral proteins.
The preferred mutants are those which comprise an amino
acid sequence which is
(I) at least 50o identical in amino acid sequence with
the corresponding amino acid sequence of a first reference
protein, after the mutant protein and the first reference
protein are aligned to maximize local similarity as hereafter
defined, and
(II) which differs from the corresponding amino acid
sequence of the first reference protein solely by one or more
of the following mutations:
(A) conservative substitutions as hereafter defined,
(B) nonconservative substitutions at positions shown to
be tolerant of at least one nonconservative substitution by
one or more of the following criteria:
(1) retention of at least l0a of the biological
activity of the first reference protein in a
CA 02333481 2000-11-27
WO 00/00600 PCTNS99/11516 .
79
mutant which differs from the first reference
protein by a single alanine substitution at
such position,
(2) existence of a second reference protein which
is a member of a recognized protein family to
which the first reference protein also
belongs, and having at least 10% of the~~
biological activity of the first reference
protein, which differs from said first
reference protein by a nonconservative
substitution at such position,
(3) existence of a known 3D structure for the
first reference protein, or of a predicted 3D
structure on the homology between the first
reference protein and a second reference
protein of known 3D structure, on the basis
of
which such position is known or predicted to
lie on the surface of the protein, or
(4) retention of at least 10% of the biological
activity of the first reference protein in a
second mutant protein which differs from said
first reference protein at least by a
nonconservative amino acid substitution at
such position,
(B) a truncation
or extension at
the amino terminal,
(C) an in ternal deletion or insertion of residues where
(1) the residues lie within an interdomain region
of the first reference protein,
(2) the residues correspond to a loop of the first
reference protein, or
(3) the internal deletion or insertion corresponds
to a difference between the first reference
protein and a homologous second reference
protein.
If the first reference protein is an HIV-1 protein, the
second reference protein is any corresponding lentiviral
protein. In some instances, it may be appropriate to consider
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
the possible second reference proteins as including
corresponding nonlentiviral proteins, especially retroviral
proteins, as well.
Preferably, for the framework residues, and more
5 preferably for the whole chain, the predicted or experimentally
determined 3D structure of the modified protein has a main
chain (Ca-carbon) conformation whose root-mean-square deviation ~~
from the predicted or experimentally determined 3D structure
of the original protein is preferably less than 5A, more
10 preferably less than 3A, still more preferably less than 2A,
most preferably less than 1A.
"Conservative modifications" are defined as
(a) conservative substitutions of amino acids as
hereafter defined; and
15 (b) single or multiple insertions or deletions of
amino acids at the termini, at interdomain
boundaries, in loops or in other segments of
relatively high mobility.
Preferably, except at the termini, no more than about
20 five amino acids are inserted or deleted at a particular locus,
and the modifications are outside regions known to contain
binding sites important to activity.
Conservative substitutions are herein defined as
exchanges within one of the following five groups:
25 I. Small aliphatic, nonpolar or slightly
polar residues:
Ala, Ser, Thr (Pro, Gly)
II. Polar, negatively charged residues, and
their amides
30 Asp, Asn, Glu, Gln
III. Polar, positively charged residues:
His, Arg, Lys
IV. Large, aliphatic, nonpolar residues:
Met, Leu, Ile, Val (Cys)
35 V. Large, aromatic residues:
Phe, Tyr, Trp
Residues Pro, Gly and Cys are parenthesized because they
have special conformational roles. Cys participates in
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
81
formation of disulfide bonds. Gly imparts flexibility to the
chain. Pro imparts rigidity to the chain and disrupts a
helices. These residues may be essential in certain regions
of the polypeptide, but substitutable elsewhere.
"Semi-conservative substitutions" are defined herein as
being substitutions within supergroup I/II/III or within
supergroup IV/V, but not within a single one of groups I-V.
If a substitution is not conservative, it preferably is semi-
conservative. Highly conservative substitutions are Arg/Lys,
Asp/Glu, Met/Ile/Leu/Val, and Phe/Tyr/Trp.
Surface vs. Interior Residues
Charged residues almost always lie on the surface of the
protein. For uncharged residues, there is less certainty, but
in general, hydrophilic residues are partitioned to the surface
I5 and hydrophobic residues to the interior. Of course, for a
membrane protein, the membrane-spanning segments are likely to
be rich in hydrophobic residues.
Surface residues may be identified experimentally by
various labeling techniques, or by 3-D structure mapping
techniques like X-ray diffraction and NMR. A 3-D model of a
homologous protein can be helpful.
r3inding Site Residues
Residues forming the binding site may be identified by
(1) comparing the effects of labeling the surface residues
before and after complexing the protein to its target, (2)
labeling the binding site directly with affinity ligands, (3)
fragmenting the protein and testing the fragments for binding
activity, and (4) systematic mutagenesis (e. g., alanine-
scanning mutagenesis) to determine which mutants destroy
binding. If the binding site of a homologous protein is known,
the binding site may be postulated by analogy.
Protein libraries may be constructed and screened that a
large family (e.g. , 108) of related mutants may be evaluated
simultaneously.
3-D Structure Determination
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516 .
82
The determination of the 3-D structure of a protein can
provide considerable guidance to one seeking to modify that
protein for a useful purpose, or at least to avoid inactivating
modifications. If the full 3-D structure is known, the
practitioner knows which residues are on the surface and which
are on the interior, which residues have side chains pointing
toward the outside, which residues are packed closely together ~~
and which are not, where the chain is flexible and where it is
constrained, which residues are in secondary structures, which
residues are brought into. proximity as a result of chain
folding, and which may be interacting in the form of H-bonding
and salt bridges.
A protein may be modified at an interior residue, a
surface residue distant from the binding site of interest, or
at a surface residue which is part of, or close enough to
affect, the binding site of interest.
Mutations at surface residues are more likely to be
tolerated than at internal residues. Mutations at the latter
positions have greater potential to destabilize the protein,
thereby, by denaturing the protein, affecting all of its
binding activities. Mutation at a surface residue may have no
effect on binding activity at all, or it may affect some
activities but not others. In any event, they are unlikely to
denature the protein.
The principal methods of determining the complete 3-D
structure of a nrotei n arP x-ra~, ,.Y~,~~-~, ~ ~..~.,~~,.. __~ "~,~r
spectroscopy.
Amino acid-specific chemical affinity labels may be used
to ferret out which residues are in fact exposed. The most
useful labels are likely to be those which react with charged
residues, as those are most likely to appear on the surface.
Sample labels include the following:
Amino Acid Affinity Label
Asp, Glu diazo compounds (with nonionized AA) or
epoxides (with ionized AA)
Lys 2, 4, 6-trinitrobenzene sulfonic acid;
acetic, succinic, malefic and citraconic
anhydrides
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516 .
83
Arg cyclohexanedione, hydrazine
Labeled and unlabeled protein are then separately
subjected to a fragmentation reagent such as cyanogen bromide,
pepsin, papain, chymotrypsin, trypsin or iodosobenzoic acid.
The peptides resulting from cleavage of the labeled protein are
compared to those derived from the native protein, using two-
dimensional electrophoresis. Peptides that have altered~~
mobility are sequenced, and modified amino acids are
determined.
Surface residues may also be identified by means of
photoaffinity labels which, upon exposure to light, form highly
reactive intermediates, e.g. nitrenes and carbenes. These
species are capable of insertion into C-H bonds, and therefore
can react with any accessible amino acid. For this reason,
photoaffinity labeling has been used to study membrane
topography. Some proteins lie at the periphery of the
membrane, others are integral to it. To identify a protein at
the membrane surface, a label is used which ideally is
indiscriminate, so that any accessible component would be
labeled, and which is itself membrane impermeant. Of course,
such a reagent will not only identify a membrane surface
protein, but also the exposed amino acids of any soluble
protein.
Another example of a nor~specific labeling reagent is
tritium. A folded protein may be tritiated (by hydrogen
exchange with tritiated water), denatured, and fragmented, and
the fragments sequenced and tested for the presence of tritium
(which is radioactive).
All of these labeling methods may also be used to
determine whether residues, besides being on the surface, are
part of a binding site. The distribution of label obtained
when free protein is labeled is compared with that obtained
when the complexed protein is labeled. Since in the complex,
the binding partner occludes the binding site residues of the
binding protein, binding site residues should be labeled in the
free protein and not in the complexed protein.
Prediction of 3D Structure
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516 .
84
The most accurate method for the prediction of a protein
structure is model building from a protein or proteins of known
structure that have been identified as homologous from sequence
analysis. Surprisingly, proteins with very little detectable
sequence identity can still fold into very similar structures.
The coordinates of protein structures can be obtained
from Protein Data Bank or the Cambridge Crystal Structure Data
Centre. Sequence databases include the Protein Identification
Resource (National Biomedical Research Foundation), GENBANK
(Los Alamos National Laboratory), EMBL (European Molecular
Biology Laboratory) and SBASE (International Center for Genetic
Engineering and Biotechnology). Derived alignment databases,
in which 3D structure and amino acid sequence have been
correlated, include NRL-3D (U. S. Naval Research Lab), HSSP
(EMBL), 3D-ALI (EMBL), FSSP (EMBL), and the Overington database
(J. P. Overington, Pfizer Central Research). For complete
addresses see Table 2 in Johnson et al., Crit. Rev. Biochem.
& Mol. Biol. 29(1):1-68 (1994).
The basic approach is to (1) identify related sequences
and structures; (2) identify structurally equivalent residues;
(3) model structurally conserved regions (SCRs); and (4) model
structurally variable regions (SVRs). The model of the SCRs
acts, to a greater or lesser degree, as a constraint in the
modeling of the SVRs. Because she core residues are usually
more structurally conserved than surface residues, they are
usually modeled first. For similar reasons, helices and
strands are usually modeled before loops. Typically, the main
chain (Ca atom) conformation is determined first, and then the
side chain conformations. Modeling steps may be iterated to
arrive at successively improved approximations of the true
structure. Typically, the predicted structures are more
accurate for protein cores than for protein loops.
It is not necessary that more than one 3-D structure be
available for model building. However, if the 3-D structures
of two or more homologous proteins are known, the accuracy of
the model can be improved. Preferably, the 3-D structures are
"weighted" to reflect teh relatedness of the homologous protein
to the protein of interest. One popular scheme is to weight
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
by the square of the percentage sequence identity.
Moreover, information regarding homologous substructures
of nonhomologous proteins may be used in addition to, or even
in lieu of, a 3-D structure of a homologous protein. See Jones
5 and Thirup, Curr. Comm. Molec. Biol., :75-76 (1986); Unger,
et al., Proteins, 5:335 (1989); Claesseus, et al., Protein
Eng., 4:335 (1989); Levitt, et al., J. Mol. Biol., 226:507~~
(1992) for the building of models by combining "spare parts"
from different proteins.
10 It is not necessary for a molecular biologist to be an
expert in protein modeling, as several programs exist which
automate the modeling process. These include COMPOSER (Tripos
Associates).
If a 3-D structure is available for the binding partner,
15 as well as for a binding protein of interest, molecular
modeling software may be used to predict potential binding
sites, or to predict the effect of a proposed mutation on a
binding site, by attempting to "dock" the binding partner to
the site. See, e.g., Guruprasad, et al., Protein Eng., 9:849
20 56 (1996); Constantino and Pelliccian, J. Med. Chem. 39:3998-
4006 (1996).
Surface Residues
In general, within families of proteins of similar
sequence and function, surface residues are more likely to vary
25 than are interior residues. This is most likely because the
surface residues are unlikely to be involved in interactions
with other residues which are necessary to maintain the overall
conformation of the protein.
Some surface residues are directly involved in the
30 binding surface by which a protein exercises a particular
binding activity. Mutation of such residues is likely to
affect binding; however, it is not necessarily undesirable to
make such mutations. For example, mutation of the binding site
of a serine protease can alter what is bound, as opposed to
35 simply rendering the protein inactive altogether.
The most reliable method of identifying the surface
residues of a protein is to determine the protein's 3-D
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
86
structure by X-ray diffraction. Even an incomplete 3D
structure can be useful in designing mutants. Residues with
high mobility, as evidenced by a higher than average
crystallographic thermal factor, are those least susceptible
to destabilizing mutations. See Alber, et al., Biochemistry,
26:37-54-8 (1987).
Interior Residues
Although many amino acid substitutions can be made at
surface positions with no adverse effects, substitutions at
internal positions tend to be severely destabilizing. Within
families of homologous proteins, the most conserved residues,
apart from functional amino acids, are those which are buried.
The main contribution to the free energy of protein
folding, and hence to protein stability, comes from burying
hydrophobic side chains in the interior, thereby shielding them
from solvent. Packing densities are typically high. In
general, the ability of a protein to tolerate mutations which
alter the volume of core residues is dependent more on the net
change in the total core residue volume, then on the magnitude
of the individual residue volume changes. In other words, an
increase in the volume of one core position can compensate for
a decrease in the volume of another core position. Preferably,
the net change in the total core residue volume is not more
than 10%, more preferably, not more than 50. See Lim and
Sauer, Nature, 339:31-36 (1989); Lim et al., Biochemistry,
31:4324-33 (1992).
In the absence of evidence to the contrary, all residues
identified as interior residues may be assumed to be part of
a single core . However, if it is likely that the protein folds
to form several distinct cores, the above-stated volume
conservation rule should be applied separately to each core.
Amino acids differ in terms of their propensity to be
buried residues. The following table shows, for each residue,
the percentage which were in buried positions, based on a study
of the 3D structures of a collection of unrelated proteins:
Amino Acid % Buried
Gly 36
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
87
Ala 38
Val 54
Ile 60
Leu 45
Phe 50
Pro 18
Ser 22
Thr 23
CYs 48
Met 40 "
Tyr 15
Trp 27
His 17
Asn 12
Gln
Asp 15
Glu 18
LYs 3
Arg 1
The makeup of the buried core of a protein is dependent,
not only on the propensity of each amino acid, if present, to
be buried, but also on the overall frequency of occurrence of
that amino acid in the protein. The most commonly buried
residues are, in descending order, Val, Gly, Leu, Ala, Ile and
Ser.
Lim et al., Biochemistry, 31:4324-33 (1992) reported that
replacing a single hydrophobic amino acid (Leu, Val) in the
protein core with a hydrophilic amino acid (Asn, Gln) prevented
the complete toiding of the protein and destroyed biological
activity.
Buried Cys, (-S-S- form), Asp, Gly, His, Pro, and Trp are
more than 70% likely to be unchanged in a homologous protein.
Therefore, if these residues occur in a buried position in the
protein of interest it is preferable to leave them unchanged.
Their conservation is probably explainable as follows: Cys
(disulfide bonding), Asp (charged but rigid side chain), Gly
(chain flexibility), His (charged and uncharged states both
available at physiological pH), Pro (unique chain geometry),
and Trp (largest side chain).
The other residues, with the exception of Met, are 40-600
likely to left unchanged when buried (Met is unchanged only 26 0
of the time, but it is 250 likely to be replaced by Leu).
The following buried residue substitution probabilities
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
88
exceed 10%:
Ala~Val, Glu-~Gln, Phe-~Leu, Ile->Leu, Ile-jVal, Lys~Arg,
Leu~Ile, Leu->Val, Met-~Leu, Met-jVal, Asn-Asp, Asn~Ser,
Arg~Lys, Arg~Gln, Ser-jAla, Thr~Ser, Val-~Ile, Val~Leu,
Tyr-~Phe , Cys ( - SH ) -~A1 a .
These further substitutions have probabilities in the 5-
10% range:
Ala-->Ser, Asp~Asn, Glu~Arg, Glu~Val, Phe-~Ile, Phe--jVal,
Phe~Tyr, His-~Val, Leu-jPhe, Met-~Ala, Met-jIle, Gln-jGlu,
Gln-His, Gln~Met, Ser~Gly, Ser-jThr, Thr~Val, Val~Ala,
Trp~Phe, Tyr-~Leu, Cys(-SH)~Ser.
See Overington, et al., Protein Science, 1:216-226
(1992), Table 5.
The most consistent exchange groups appear to be (Arg,
Lys), (Leu, Ile, Met, Val, Phe), and (Ser, Thr, Ala). However,
Ala and Val appear to be fairly promiscuous.
In general, therefore, it is preferable to avoid mutating
buried residues at all. However, if they are mutated, one
should limit the overall change in the volume of the core, and
most preferably should limit the mutation to the replacement
of one residue with another whose typical substitution
probability exceeds zero, more preferably is at least 5%, and
most preferably at least 10%. Mutation of buried Cys(-S-S),
Asp, Gly, His, Pro and Trp should be avoided, absent
justification by other evidence. The safest core mutations are
exchanges of one hydrophobic amino acid for another, and of Arg
for Lys (or vice versa) .
Nonetheless, judicious mutation at internal residues may
be used to improve protein stability. Such mutations could
introduce additional stabilizing interactions (hydrogen bonds,
ion pairs) compatible with the native structure, or could
reduce the mobility of nearby interacting groups by replacing
smaller amino acids with larger ones, or linear side chains
with branched or aromatic ones. See Alber, et al.,
Biochemistry, 26:3751-8 (1987).
Identification of Naturally Occurring Homologous Proteins
The most useful information for determining which
residues are safely mutatable is knowledge of the sequence of
CA 02333481 2000-11-27
WO 00/00600 PCT/L1S99/11516 .
89
proteins of similar sequence which have similar activity. The
sequences of these homologous proteins may then be aligned, and
residues which are not conserved are more likely to be safely
mutatable. The degree of confidence which one has as to the
tolerance of a residue to mutation is a function of the degree
of variation of amino acid type at that site among the protein
family, as well as the extent to which all of the proteins in~I
the family, despite their differences, retain the desired
activity.
While studies of homologous proteins are useful in
identifying sites which, by virtue of their variability, are
likely to be tolerant of mutation, it is less certain that
sites which are strongly conserved are necessarily invariant.
According to Shenkin, et al., (1991), random mutagenesis
studies indicate that "proteins are able to accommodate, both
structurally and functionally, a far greater variety of
mutations than occur naturally".
Homologous proteins are those which are similar in
structure to the protein of interest, to a statistically
significant degree, and which perform the same or an analogous
biological function. Examples are human growth hormone and human
prolactin, and human alpha globin and human myoglobin. When
homologous proteins occur in nature, the similarities may imply
that they have a common evolutionary origin, and the time of
origin may be estimated by calculating the number of mutations
which would give use to the observed sequence divergence and
dividing by the mutation rate.
Cognate proteins are homologous proteins, expressed in a
different species of organism, which perform the same
biological function as that performed by the protein of
interest, although they may differ in activity, specificity,
timing of expression, etc. Examples of cognate proteins are
human and fish growth hormones, or human and other vertebrate
alpha (or beta) globins.
The possession of the cDNA or genomic DNA (the "starting
DNA") encoding the protein of interest (the "starting protein")
greatly facilitates the isolation of homologous proteins. For
the use of probes to identify homologous genes in other
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
species, see, e.g., Schwinn, et al., J. Biol. Chem., 265:8183-
89 (1990) (hamster 67-by cDNA probe vs. human leukocyte genomic
library; human 0.32kb DNA probe vs. bovine brain cDNA library,
both with hybridization at 42°C in 6xSSC); Jenkins et al., J.
5 Biol. Chem., 265:19624-31 (1990) (Chicken 770-by cDNA probe vs.
human genomic libraries; hybridization at 40°C in 50% formamide
and SxSSC); Murata et al., J. Exp. Med., 175:341-51 (1992)
(1.2-kb mouse cDNA probe v. human eosinophil cDNA library;
hybridization at 65°C in 6xSSC); Guyer et al., J. Biol. Chem.,
10 265:17307-17 (1990) (2.95-kb human genomic DNA probe vs.
porcine genomic DNA library; hybridization at 42°C in 5xSSC).
Identification of Homologous DNA
Similarly, similar genetic elements may be defined, in
addition to or in place of definition by percentage identity,
15 by their ability to hybridize.
When a genetic element is desired to be similar to a
reference element, it preferably will specifically hybridize
under the relaxed conditions set forth above, and more
preferably under stringent conditions (wash temperature which
20 is not more than 15°C, more preferably not more than 10°C,
below the Tm of a perfect duplex of the reference element) .
When a genetic element is desired to be different from a
reference element, if preferably will not specifically
hybridize under stringent conditions, and more preferably not
25 even under relaxed conditions.
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516 .
91
Alignment of Homologous Sequences
In order to derive guidance from the sequences of
homologous proteins, it is necessary to identify which proteins
are homologous and to align the sequence of the protein of
interest with that of the homologous proteins. Such alignment
is guided by calculating a homology or alignment score for each
possible alignment, and determining the highest such score fore
each pair of potentially homologous proteins. Homologous
proteins are distinguished from nonhomologous proteins by
having an alignment score which is significantly higher.
Global alignment algorithms (e.g., Pearson's ALIGN and
ALIGNPS programs) consider both complete sequences in generating
similarity scores for a given alignment, and, in general, allow
"gapping". They are most appropriate when the sequences are
known or expected to be similar over their entire length. The
most popular form are those which do not penalize end-gaps.
Local alignment algorithms (e. g., Pearson's LALIGN)
search for similar fragments of two sequences, and, in general,
do not allow gaps. They are useful in locating common
subdomains between long sequences that otherwise share little
similarity.
Scoring of Mismatches
A mismatch occurs when the amino acid residues at the
same site in two different aligned amino acid sequences are
different. Several systems are used in the art for scoring
matches (identities) and mismatches.
The simplest, the Identity Matrix, gives a score of one
to each match and zero to each mismatch. The other schemes
give more weight to "similar" though nonidentical pairings of
residues.
The Genetic Code Matrix (GCM) scores amino acid
similarity based on the maximum number of common nucleotide
bases (which can range from 3 to 0) between their closest
matching representative codons. The original scoring system
awarded 3 points for three common bases, 2 for 2, etc., (i.e.,
a 3/2/1/0 system) and used a gap penalty of -3. Feng, et al.
(1985) reported that best results are obtained with a 4/2/1/0
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516 .
92
system and a gap penalty of -4. The file Codaa.mat
accompanying FASTA uses the system 6/2/-2/-6.
A Mutation Data Matrix (MDM) scores amino acid similarity
on the basis of the frequency of the exchange of the two amino
acids in question between two members of a family of homologous
proteins, or of a member and the inferred ancestor of that
protein. It is customary for an MDM to take into account the ~~
apparent evolutionary distance, too. Thus, it calculates the
probability that one residue will be mutated into another
residue in a specified unit of a evolutionary time. To
calculate this matrix, proteins of known sequences are
clustered into families of homologous proteins, a phylogenetic
type is constructed for each protein, and an ancestral sequence
inferred the amino acid exchanges which apparently occurred
between each modem sequence and the ancestral sequence are
tailied, and the minimum number of base changes which could
explain those exchanges are calculated. The assumption is made
that mutations are strictly Markovian processes. The basic
unit of molecular evolution expressed in an MDM is the
"accepted point mutation" (PAM). In sequence analysis, the
most commonly used MDM is the 250 PAM matrix, i.e., one
characterizing the amino acid exchanges that would be expected
to occur between sequences separated by 250 PAMs.
The Structural Similarity Matrix weighs pairings
according to the similarity of the amino acids in size,
hydrophilicity, and/or other structural measures.
Hybrid matrixes have also been devised.
It is important that the mutation matrix values and gap
penalties be scaled so that identities have appropriate
positive scores relative to the gap penalties.
In FASTA, the standard protein matrix is the Dayhoff PAM
250 matrix. In BLAST, the default matrix is the PAM120 matrix,
which is more selective.
Scoring of "Gaps"
The justification for "gapping" is that the introduction
of a gap can improve the apparent homology between two
sequences so extensively that there is no reasonable doubt that
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
93
the gap reflects evolutionary history.
In general, overhangs (terminal gaps) are not counted.
The simplest method of scoring for internal gaps is to count
each deleted residue as a simple mismatch. Another approach
is to impose an initiating gap penalty and an extension gap
penalty. Typically, if the reward for an identity is R, the
cost of initiating a gap is at least R and more usually at ~~
least 2*R. Another approach is to set the initiation cost at
1.5 times the most negative as pair in the matrix, which is -S
in pam 250. The extension cost is typically 1/3 to 1/20 the
initiation cost. The default penalties associated with the
pam250 matrix in FASTA are -12 for the first deletion and -4
for each additional consecutive deletion.
Statistical Significance
Two random amino acid sequences (of equimolar amino acid
composition) would have, an average, an identity of 5% if
gapping is not allowed. If gapping is permitted, two random
sequences can be 10-20% identical. In general, if two
sequences are longer than 100 residues, and are more than 25%
identical after suitable gapping, it is likely that they are
genuinely related, i.e., that the similarity is not due to
chance. The "twilight zone" is 15-25% identity.
The statistical significance of an alignmen~ may be
determined by comparing the alignment score obtained when the
two authentic sequences are aligned, with the mean and standard
deviation of the alignment scores obtained when both sequences
are repeatedly randomized, and each "jumbled" sequence in one
set is aligned with the jumbled sequences in the other set.
The score of the authentic sequence is then expressed as so
many standard deviations above the mean of the jumbled group.
See, e.g., Doolittle, Science, 214:149 (1981); Lipman and
Pearson, Science, 277:1435 (1985). However, since similarity
scores are distributed according to the extreme value
distribution, not the normal distribution, the extreme value
statistical form of the z-value should be used. See Altschul,
et al., Nature Genetics, 6:119-129 (1994).
The program PRSS, which is part of the FASTA package, may
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516 .
94 '
be used to evaluate the statistical significance of a local
alignment, e.g., with LALIGN.
Lipman and Pearson are of the opinion that the z value of
the optimized alignment similarity score obtained with FASTP
should be evaluated as follows: >3, possibly significant; >6,
probably significant; >10, significant. Doolittle used a more ,
stringent identity matrix scoring system and considered a score
of 3.0 S.D. or more to be significant.
A second approach to determining significance of a
particular alignment is to compare the alignment score for that
alignment with the mean and standard deviation of the alignment
scores for the alignments of the query sequence with all
sequences in a sequence library. Once again, a z value is
calculated. See Wilbar and Lipman Proc. Nat. Acad. Sci. USA
80:726-30 (1983). Most of these sequences will be unrelated
to the query sequence. Of course, the choice of sequences in
the library will reflect the interests of the scientific
community, e.g., it will tend to favor the sequences of those
organisms which are most closely studied, e.g., humans, fruit
flies, S. cerevisiae, and E. coli.
Measures of Variability
The variability index (Vk) is a simple method of
quantifying the degree of variation of amino acid residues at
a particular aligned site. It is the ratio of the number of
different amino acid types which appear at the position, to the
fraction of the time which the site is occupied by the most
common of these types. Wu, et al., J. Exp. Med. 132:211-49
(1970) . Vk ranges from 1 to 400 for proteins. Preferably,
mutations are directed to sites having a variability index
which is within the upper 50%, more preferably the upper 20%,
of all sites of the protein.
A more sophisticated approach involves calculating the
informational entropy of the site. This is
s=-E P; log2P; (for all Pi>0)
where there i different amino acid residues appearing at the
site, and P; is the fraction of the total number of aligned
sequences in which residue i appears at the site . See Shenicin,
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
95 -
et al., Proteins: Structure, Function and Genetics, 11:297-313
(1991). If only one residue appeared at a site, the entropy
would be zero. If all 20 genetically encoded residues appeared
with equal frequency, the entropy would be -logz ( . 05) , or about
4.32. The informational entropy is less likely to ~~jump~~ than
the variability index when a new sequence is added.
Preferably, mutations are directed to sites which have an "
informational entropy S greater than 1.0, more preferably
greater than 2Ø (A related measure, VS, is defined as 6 x
2S; it ranges from 6 to 120 for proteins. The factor 6 was
chosen empirically to make the Vk and VS scales roughly
comparable.
Shenkin et al. (1991) reported that for all
immunoglobulin light chains, Vk ranged from 1 to 96, S from 0
to 3.4792, and Vs from 6 to 66.91.
The two methods described above do not take into account
either the normal equivalency of the different residues (the
appearance of both Arg and Gly at a site is more revealing of
tolerance than the appearance of both Arg and Lys) or the
degree of relationship between the source organisms (the
conservation of a residue between human and chimpanzee should
be less significant than the conservation of the same residue
between human and fruit fly). Various weighting schemes can
be used to adjust for these subtleties.
Mutagenic.Analysis of Binding Sites
Binding sites may also be identified by mutagenesis
strategies designed to locally perturb the protein. One such
strategy is alanine scanning mutagenesis. In this technique,
all non-alanine residues of the protein (or of a region of the
protein suspected to contain the binding site are replaced,
one-by-one, with alanine, yielding a collection of single
substitution mutants. Alanine is used because (1) it is the
most common amino acid residue in proteins, (2) it has a small
side chain, and therefore is not likely to sterically hinder
other residues, and (3) its side chain (-CH3) does not form H-
bonds, but is not especially hydrophobic. Cunningham and Wells
(1989) conducted an Ala scanning mutagenesis study of residues
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
96
2-19, 54-74, and 167-191 in hGH. A total of 62 Ala mutations
were produced. Of these, fourteen mutants could not be
produced in quantities sufficient for affinity testing.
Presumably, these mutations globally destabilized the protein,
rendering it vulnerable to proteolysis. Eleven mutants
seemingly enhanced binding, although it is unclear which
improvements were significant. Of the remaining 37 mutants,
only four impaired binding by 10-fold or more, and only nine
by 5-fold or more. See generally Genentech, W090/04788.
For other uses of Ala=scan mutagenesis, see Yu, et al.,
J. Mol. Biol., 249:388-97 (1995) (complete scan of a single
disulfide derivative of the 58-residue protein BPTI}; Allen,
et al., Nature, 327:713 (1987) (Ala-scan of residues 52-61 of
hen egg white lysozyme); Ruf, et al., Biochemistry, 33:1565-72
(1994) (Ala-scan of residues other than Gly, Pro and Cys;
multiple Ala mutants examined first, then single Ala mutants);
Williams, et al., J. Biol. Chem., 270:3012-6 (1995) (Ala-scan
in insulin receptor of (1) charged amino acids, (2) aromatic
residues, and (3) residues adjacent to (1) or (2), other than
prolines, cysteines, or potential N-linked glycosylation
sites); Kelly and O'Connell, Biochemistry, 32:6828-35 (Ala-scan
of antibody CDR). Ala-scanning mutagenesis may be applied to
all residues of a protein, or to residues selected on some
rational basis, such as amino acid type (e.g., charged and
aromatic residues), degree of variability in a homologous
protein family, or relevance to function as shown by homologue-
scanning mutagenesis.
Preferably, further mutations (especially nonconservative
mutations) are made at sites where an alanine substitution does
not worsen the activity of interest by more than 20-fold, more
preferably, by more than 10-fold, even more preferably, by more
than 5-fold, still more preferably, by more than 2-fold. Most
preferably, mutations are made at sites at which an alanine
substitutions improves activity.
Preferably, if multiple mutations are made, the expected
(additive) effect of the mutations is one which does not worsen
the activity more than 10-fold, more preferably, by more than
5 fold, still more preferably, by more than two fold. Most
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
97
preferably, the expected effect is to improve activity. The
expected effect of a conservative substitution is the effect
of that mutation as a single substitution if known, or
otherwise neutral. The expected effect of a nonconservative
substitution is the effect of that mutation as a single
substitution if known, or otherwise the effect of a single
substitution of a different residue of the same exchange group "
as the actual replacement residue, if known, or otherwise the
effect of a single Ala substitution.
Another approach is homologue-scanning mutagenesis. This
involves identifying a homologue which can be distinguished in
an activity assay from the protein of interest, and screening
mutants in which a segment of the protein of interest is
replaced by corresponding segments of the homologue (or vice
versa). If the replacement alters the activity of the modified
protein, the segment in question presumably contributes to the
observed difference in activity between the protein of interest
and the homologous protein, and comparison of the interchanged
segments helps to explain the character of the binding site
involved in that activity. For example, segments of prolactin,
which does not bind the GH receptor, have been used to replace
segments of growth hormone, which does. If a substitution
disrupts GH binding, it implies that the replaced segment was
part of the GH receptor binding site, and one may then focus
on how the replaced and replacing segments differ. See
W090/04788.
If a residue is determined to be a part of the binding
site, one may prepare all possible single substitution mutants
of that site.
Multiple Mutation
It is possible to incorporate two or more tolerable
mutations into a protein.
Generally speaking, as a first approximation, it is
reasonable to assume that the effect of two mutations will be
additive in nature. See Wells, Biochemistry, 29:8509-17
(1990); Sandberg and Terwilliger, Proc. Nat. Acad. Sci. (USA),
90:8367-71 (1993); Gregoret and Sauer, Proc. Nat. Acad. Sci.
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
98
(USA), 90:4240-50 (1993); Schreiber and Fersht, J. Mol. Biol.,
248:478-86 (1995); Lowman and Wells, J. Mol. Biol. 234: 564-78
(1993); Lawman, et al., J. Biol. Chem., 266:10982-8 (1991);
Lin, et al., Proc. Nat. Acad. Sci. (USA), 91:10265-9 (1994);
Venkatachalam, et al., J. Biol. Chem., 269:23444-50 (1994);
Akasako, et al., Biochemistry, 34:8115-22 (1995); Behravar, et
al., Eur. J. Biochem., 198:589-92 (1991); Lin, et al., Proc. ~.
Nat. Acad. Sei. (USA), 91:10265-9 (1994); Zuckerman, et al.,
Proc. Nat. Acad. Sci. (USA), 89:4505-9 (1992). Gregoret, et
al., Proc. Nat. Acad. Sci. (USA), 90:4246-50 (1993) assumed
that, under selective conditions, the frequency of occurrence
of a mutation in an active mutant was an indication of whether
the mutant conferred resistance, and found that an additive
model (multiplying the mutational frequencies of a pair of
single Ala substitution mutants) was about 90% effective in
predicting the activity class of a binomial (multiple Ala
substitution) mutant.
The most common reason for combining mutations is to
benefit from their additive or synergistic effect in
combination. For example, if a mutation has both favorable and
unfavorable activities, it may be possible to combine it with
a second mutation that neutralizes the unfavorable activity of
the first mutation.
One use of multiple mutation is to achieve, by combining
mutations which individually have a small but favorable effect
on activity, a mutant with a more substantial improvement in
activity. It is not necessary that the mutations be strictly
additive; it is sufficient that they be at least partially
additive for the combination to be advantageous. See Blacklow,
et al., Biochemistry, 30:8470-6 (1991) (improved catalytic
effectiveness of triosephosphate isomerase); Akasako, et al.,
Biochemistry, 34:8115-22 (1995) (multiple thermostabilizing
mutations in ribonuclease HI); Lowman et al., J. Biol. Chem.,
266:10982-8 (1991) (HGH-receptor binding properties of human
placental lactogen improved about 500-fold by five
simultaneous, mutations, with "reasonably additive" effects);
Lowman and Wells, J. Mol. Biol., 234:564-78 (1993) (HGH-
receptor binding properties of HGH improved about 400-fold by
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/I1516
99
combination of 15 substitutions. Sandberg and Terwilliger,
Proc. Nat. Acad. Sci. (USA), 90:8367-71 (1993), reported that
there was only a weak correlation between changes in DNA
binding protein stability and changes in DNA binding affinity,
and hence that it was possible to combine mutations so as to
selectively change one property without changing the other.
In binomial Ala-scanning mutagenesis, one constructs a
library in which, at each position of interest of a given
protein molecule, the residue is randomlv either the native
residue, or Ala. See Gregoret and Sauer, Proc. Nat. Acad. Sci.
(USA), 90:4246-50 (1993). If it is feasible to screen a
library of 101° mutants, then the combined effects of up to 30
different Ala substitutions (2z'~10~°) can be studied in one
experiment. It should be noted that the Ala:non-Ala ratio at
each position may be, but need not be equal. The choice made
for this ratio will determine the degree of substitution will
predominate, according to a binomial distribution.
If the protein is too large for all sites of interest to
be sampled by binomial Ala-scanning mutagenesis in a single
experiment, one may divide the protein into segments and
subject each segment in turn to such mutagenesis, and then, as
a cross-check, similarly mutate one residue from each segment.
Design of Chimeric Proteins
The term "chimera" implies a protein which is a hybrid of
two or more different parental proteins which are associated
with two or more different organisms.
Functional chimeras may be identified by a systematic
synthesize-and-test strategy. It is not necessary that all
theoretically conceivable chimeras be evaluated directly.
One strategy is described schematically below. We divide
the aligned protein sequences into two or more testable units.
These units may be equal or unequal in length. Preferably, the
units correspond to functional domains or are demarcated so as
to correspond to special features of the sequence, e.g.,
regions of unusually high divergence or similarity, conserved
or unconserved regions in the relevant protein family or the
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516 .
100
presence of a sequence motif, or an area of unusual
hydrophilicity or hydrophobicity. Let "1" represent a unit of
the protein 1, and "2" a corresponding unit of protein 2. If
there are five units (the choice of five instead of two, three,
four, six, ten, etc. is arbitrary), we can synthesize and test
any or all of the following chimeras, which
will help us rapidly localize the critical regions:
(a) progressive C-terminal substitution of exogenous
sequence for host sequence, e.g.,
1 1 1 1 1
1 1 1 1 2
1 1 1 2 2
1 1 2 2 2
1 2 2 2 2
2 2 2 2 2
(b) progressive N-terminal substitution of exogenous
sequence for host sequence
1 1 1 1 1
2 1 1 1 1
2 2 1 1 1
2 2 2 1 1
2 2 2 2 1
2 2 2 2 2
(c) dual terminal substitutions, e.g.,
2 2 2 2 2
1 2 2 2 1
1 1 2 1 1
1 1 1 1 1
and
1 1 1 1 1
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
101
2 1 1 1 2
2 2 1 2 2
2 2 2 2 2,
and
(d) single replacement "scans," such as
2 1 1 1 1
1 2 1 1
I
1 1 2 1
1
1 1 1 2
1
1 1 1 1
2
and
1 2 2 2 2
2 1 2 2 2
2 2 1 2 2
2 2 2 1 2
2 2 2 2 1
Based on the data these tests provide, it may appear
that, e.g., the key difference between the exogenous and host
sequences vis-a-vis, say, display on the host cell membrane,
is in the fifth unit. One can then subdivide that unit into
subunits and test further, e.g.
2 2 2 2 (11)
2 2 2 2 (12)
2 2 2 2 (21)
2 2 2 2 (22)
where the parenthesized entries refer to the two subunits into
which the original fifth unit was subdivided
Transgene
The transgene is a gene encoding a polypeptide which is
foreign to the lentivirus(es) from which the vector is
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516 .
102
primarily derived, and which has a useful biological activity
into the organism which is ultimately infected with the
transducing vector in its virion-packaged form.
The transgene may be identical to a wild-type gene, or it
may contain one or more mutations. The transgene may be
derived from genomic DNA, cDNA, synthetic DNA, or a combination
thereof. Intronless "minigenes", which are normal genes from ~~
which introns have been removed, have been especially popular.
Intron-containing genes may be employed, but they may be
inserted into the vector in the reverse orientation if removal
of the introns is not desired. Silent mutations may be
introduced to facilitate gene maipulation, to avoid undesirable
secondary structure in the mRNA, to inhibit recombination, r_o
control splicing, etc. Nonsilent mutations alter the encoded
protein, and may be either gratuitous, or aimed at beneficially
altering the biological activity of the protein.
One example of a transgene is a remedial gene. As used
herein, the term "remedial gene" refers to a gene whose
expression is desired in a cell to correct an error in cellular
metabolism, to inactivate a pathogen or to kill a cancerous
cell. For example, the adenosine deaminase (ADA) gene is the
remedial gene when carried on a retroviral vector used to
correct ADA deficiency in a patient.
uhe applications of transgenes include the following:
--cell marking: for some purposes, it is useful to follow cells
after they have been introudced into a patient.
--anti-pathogen or anti-parasite: anti-pathogen genes or anti
parasite can be introduced into a host infested, or especially
vulnerable to infestation, by the pathogen or parasite in
question.
--genetic disease: an inherited genetic defect may be
ameliorated by supplying a functional gene.
It is not necessary that the endogenous gene be repaired
by homologous recombination. Monogenetic genetic diseases are
of particular interest. Suitable approaches include providing
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
3 ''
genes encoding the enzyme adenosine deaminase (ADA), especially
to hematopoietic stem cells so as to provide longterm treatment
of ADA deficiency; and correcting familial hypercholesterolemia
with a vector encoding the low density lipoprotein (LDL)
5 receptor.
Gene therapy has been used to successfully correct inborn
errors of metabolism using existing vector systems. For
example, the adenosine deaminase gene has been introduced into
peripheral blood lymphocytes and cord blood stem cells via
10 retroviral vectors in order to treat patients with severe
combined immunodeficiency due to a lack of functional adenosine
deaminase (K.W. Culver et al. , Human Gene Ther. , 2 : 107 [1991] ) .
Partial correction of familial hypercholesterolemia has been
achieved using existing retroviral vectors to transfer the
receptor for low density lipoproteins (LDL) into hepatocytes.
However, it was estimated that only 5% of the liver cells
exposed to the recombinant virus incorporated the LDL receptor
gene with the vector utilized (M. Grossman et al., Nat. Genet.,
6:335 [1994]).
A number of single-gene disorders have been targeted for
correction using gene therapy. These disorders include
hemophilia (lack of Factor VIII or Factor IX), cystic fibrosis
(lack of cystic fibrosis transmembrane regulator), emphysema
(defective a-1-antitrypsin), thaiassemia and sickle cell anemia
(defective synthesis of /3-globin), phenylketonuria (deficient
phenylalanine hydroxylase) and muscular dystrophy (defective
dystrophin) (for review see A.D. Miller, Nature 357:455
[1992]). Human gene transfer trials have been approved far a
number of these diseases.
The molecular genetics of cystic fibrosis (CF) has been
studied and gradually understood in recent years.
Approximately 70% of the CF patients carry a single amino acid
deletion ((F508) mutation in one of the two nucleotide-binding
domains in the CF transmembrane regulator (CFTR) protein
[Miller, 1993 #535]. Other forms of genetic mutations in the
CFTR genes have also been identified. This rich genetic
information makes CF an ideal gene therapy candidate.
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
104 '
The target cells for CF patients are undifferentiated,
proliferating and differentiated, non-proliferating lung
epithelial cells. It is hoped that both of the dividing and
non-dividing lung epithelial cell types can be efficiently
targeted by VSV-G pseudotyped lentiviral vectors carrying a
wild type CFTR cDNA.
CF patients have CFTR mutations which leads to basic chloride
flux defect in the respiratory ciliated epithelial cells. This
CFTR dysfunction causes chronic infection and inflammation of
the respiratory tract and leads to high morbidity and mortality
in CF patients. The CFTR cDNA gene transfer by adenoviral
vectors or liposomes has demonstrated partial correction of the
defective CFTR channel activity in the nasal epithelium of CF
patients. An important indication that CFTR dysfunction in CF
patients could be treated by gene therapy protocols came from
the study of Johnson et al. who demonstrated that
overexpression of CFTR which numerically corrected 6-10% of a
mutant CF epithelial monolayer resulted in a bioelectric
phenotype similar to sheets of 1000 corrected cells. In a
recent study, Dorin et al. further showed in a mouse model that
50 of the normal level of CFTR gene expression resulted in a
correction of the chloride ion transport defect up to 50% of
normal level and obtained 100 o survival . These studies suggest
that gene therapy may offer great benefits to CF patients even
if only partial correction of CFTR gene function is achieved
with the current gene transfer tools.
--cancer: cancers may be treated with vectors carrying genes
which express cancer antigens, or immunomodulatory proteins,
and thereby stimulate an immune response against the cancer
cells, or which express a normal tumor suppressor gene to
replace the function of a mutated, tumor-prone gene, such as
a p53 mutant.
In addition to replacement of defective genes, it has
been proposed that viral vectors could be used to deliver genes
designed to stimulate immunity against or to otherwise destroy
tumor cells. Although the integration of therapeutic genes
into tumor cells_is not required for cancer gene therapy
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516 .
105
application in most cases, sustained expression of the
therapeutic genes in tumor cells may be required, for example,
to elicit a long lasting in vivo anti-tumor immunity.
Gene therapy, originally developed for treating inherited
and acquired diseases by introducing therapeutic genes to
somatic cells, has great potential for cancer treatment. With
the rapid advances in molecular medicine and gene delivery
technology during the past decade, gene therapy approaches have
brought excitement and new hopes to fighting cancers.
Currently, more than 700 of approved clinical trial gene
therapy protocols worldwide are designed for treating cancers.
The list is growing rapidly because of the ineffectiveness of
conventional cancer treatments, especially to those late stage,
metastatic cancers. There are three major components to be
considered in the design and development of a gene therapy
regimen: the therapeutic genes, the mode of gene delivery (ex
vivo or in vivo), and an appropriate preclinical study model
for the assessment of the therapeutic efficacy. Various
therapeutic genes have been utilized in cancer treatments. The
common examples include: (1) genes that are capable of changing
the cellular sensitivity to chemo- or radiation therapy in
cancer patients either to sensitize tumor cells, or to minimize
the damage of chemotherapy to normal cells such as the
hematopoietic stem cells, (2) genes that interfere with
proliferating tumor cell cycle by either replacing the mutated
genes (i.e. tumor suppresser genes and apoptotic genes), or
inactivating the oncogenes to prevent further tumor
development, and (3) genes that can augment a systemic
anti-tumor immunity in cancer patients; this can be
accomplished by the injection of modified tumor infiltrating
lymphocytes (TIL) or immunomodulatory gene-modified tumor
cells, or by the modification of antigen presenting cells
(APC). Retroviral vectors containing genes encoding tumor
necrosis factor (TNF) or interleukin-2 (IL-2) have been
transferred into tumor-infiltrating lymphocytes in patients (A.
Kasid et al., Proc Natl Acad Sci USA. 87:473-477 [1990]; and
S.A. Rosenberg, Human Gene.Therapy 5: 140 [1994]). It is
postulated that the secretion of TNF or IL-2 stimulates a
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516 .
106
tumor-specific immune response resulting in the destruction of
the tumor or the recruitment of effective tumor infiltrating
lymphocytes from nearby lymph nodes. Other proposed anti-tumor
gene therapy strategies include the delivery of toxin genes to
the tumor cell.
Applications of antisense genes or antisense
oligonucleotides in inhibition of oncogenes and modulation of "
growth factors have the potential to reduce the mortality of
cancer, in particular, human leukemia (For review see, A.M.
Gewirtz, Stem Cells 3:96 [1993]; and L. Neckers and L.
Whitesell, Amer. J. Physiol., 265: L1 [1993)).
--HIV: vectors may be used to deliver transgenes which protect
susceptible cells against HIV by synthesizing proteins,
antisense RNAs, or ribozymes that block HIV binding and entry,
reverse transcription, integration, or replication. Of course,
the transgenes must be regulated so they do not interfere with
the packaging of the transducing vector.
Selectable and Screenable Markers
A vector may contain one or more selectable or screenable
markers. Such markers are typically used to determine whether
the vector has been successfully introduced into a host or
target cell. A selectable marker is a gene whose expression
substantially affects whether a cell will survive under
particular controllable conditions. A selectable marker may
provide for positive selection (cells with the marker are more
likely to survive), negative selection (cells with the marker
are less likely to survive}, or both (the choice of
environmental condition dictating whether positive or negative
selection occurs).
Selectable markers include those which confer antibiotic
resistance (or sensitivity), the ability to utilize a
particular nutrient, and resistance (or sensitivity) to high
(or low) temperature. Suitable selectable markers include the
bacterial neomycin and hygromycin phosphotransferase resistance
genes, which confers resistance to 6418 and hygromycin,
respectively, the bacterial gpt gene, which allows cells tog
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/I1516
107
row in a medium containing mycophenolic acid, xanthine and
aminopterin; the bacterial hisD gene which allows cells to grow
in a medium lacking histidine but containing histidinol; the
multidrug resistance gene mdr; the hprt and HSV thymidine
kinase genes, which allow otherwise hprt- or tk- cells to grow
in a medium containing hypoxanthine, amethopterin and
thymidine, and the bacterial genes conferring resistance to ~~
puromycin or phleomycin. Positive or negative selection may
require the use of a particular strain of host cell for the
selection to be effective.
Screenable markers are genes which encode a product whose
presence is readily detectable, directly or indirectly, but
which do not necessarily affect cell survival. The green
fluorescent protein (GFP) is an example. Any cell surface
protein not native to the host cell can be used as an
immunoscreenable marker. Transformed cells may be segregated
out by using a fluorescent antibody to the protein and a cell
sorter. Many enzyme-encoding genes are useful as screenable
markers, especially those encoding enzymes which can act upon
a substrate to provide a colored or luminescent product. The
luciferase and beta-galactosidase genes have been especially
popular.
A dominant marker encodes an activity which can be
detected in any eukaryotic cell line. Examples of dominant
selectable markers include the bacterial aminoglycoside 3'
phosphotransferase gene (also referred to as the neo gene)
which confers resistance to the drug 6418 in mammalian cells,
the bacterial hygromycin G phosphotransferase (hyg) gene which
confers resistance to the antibiotic hygromycin and the
bacterial xanthine-guanine phosphoribosyl transferase gene
(also referred to as the gpt gene) which confers the ability
to grow in the presence of mycophenolic acid. Other selectable
markers are not dominant in that their use must be in
conjunction with a cell line that lacks the relevant activity.
Examples of non-dominant selectable markers include the
thymidine kinase (tk) gene which is used in conjunction with
tk- cell lines, the CAD gene which is used in conjunction with
CAD-deficient cells and the mammalian hvpoxanthine-guanine
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
108
phosphoribosyl transferase (hprt) gene which is used in
conjunction with hprt-cell lines.
A review of the use of markers in mammalian cell lines is
provided in Sambrook, J. et al., Molecular Cloning: A
Laboratory Manual, 2nd ed., Cold Spring Harbor Laboratory
Press, New York [1989] pp.16.9-16.15.
Regulation of Gene Expression
The transgene(s) of the transducing vector, and the
markers) and viral genes yor replacements) of the packaging
and transducing vectors, are expressed under the control of
regulatory elements.
As used herein, the term "regulatory element" refers to
a genetic element which controls some aspect of the expression
of nucleic acid sequences. For example, a promoter is a
regulatory element which facilitates the initiation of
transcription of an operably linked coding region. Other
regulatory elements are splicing signals, polyadenylation
signals, termination signals, etc. (defined infra). A
constitutive promoter is one which is always active at
essentially a constant level.
Transcriptional control signals in eukaryotes comprise
"promoter" and "enhancer" elements. Promoters and enhancers
consist of short arrays of DNA sequences that interact
specifically with cellular proteins involved in transcription
(T. Maniatis et al., Science 236:1237 [1987]). Promoter and
enhancer elements have been isolated from a variety of
eukaryotic sources including genes in yeast, insect and
mammalian cells and viruses (analogous control elements, i.e.,
promoters, are also found in prokaryotes). The selection of
a particular promoter and enhancer depends on what cell type
is to be used to express the protein of interest. Some
eukaryotic promoters and enhancers have a broad host range
while others are functional in a limited subset of cell types
(for review, see, S.D. Voss et al., Trends Biochem. Sci.,
11:287 [1986]; and T. Maniatis et al., supra [1987]). For
example, the SV40 early gene enhancer is very active in a wide
variety of cell types from many mammalian species and has been
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/I1516
109
widely used for the expression of proteins in mammalian cells
(R. Dijkema et al., EMBO J. 4:761 [1985]). Two other examples
of promoter/enhancer elements active in a broad range of
mammalian cell types are those from the human elongation factor
1a gene (T. Uetsuki et al., J. Biol. Chem., 264:5791 (1989];
D.W. Kim et al., Gene 91:217 [1990]; and S. Mizushima, and S.
Nagata, Nuc. Acids. Res., 18:5322 [1990]) and the long terminal "
repeats of the Rous sarcoma virus (C. M. Gorman et al., Proc.
Natl. Acad. Sci. USA 79:6777 [1982]) and the human
cytomegalovirus (M. Boshart et al., Cell 41:521 [1985]).
As used herein, the term "promoter/enhancer" denotes a
segment of DNA which contains sequences capable of providing
both promoter and enhancer functions (i.e., the functions
provided by a promoter element and an enhancer element, see
above for a discussion of these functions). For example, the
long terminal repeats of retroviruses contain both promoter and
enhancer functions. The enhancer/promoter may be "endogenous"
or "exogenous" or "heterologous." An "endogenous"
enhancer/promoter is one which is naturally linked with a given
gene in the genome. An "exogenous" or "heterologous"
enhancer/promoter is one which is placed in juxtaposition to
a gene by means of genetic manipulation (i.e., molecular
biological techniques) such that transcription of that gene is
directed by the linked enhancer/promoter.
A regulatable promoter is one whose level of activity is
subject to regulation by a regulatory molecule. An inducible
promoter is one which is normally substantially inactive, but
which is activated by the binding of an inducer to an operator
site of the promoter. A repressible promoter is one which is
normally active, but which is substantially inactivated by the
binding of a repressor to an operator site of the promoter.
Similar terminology applies to enhancers.
The inducer or repressor molecules are typically
expressed only in particular tissues, at a particular
developmental stage, or under particular environmental
conditions (e.g. , damage to the cell, infection, overproduction
of a metabolite, absence of a nutrient, etc.) . In the
absence of an inducer an inducible promoter may be inactive or
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516 .
110
may produce a low level of The level of activity in the
presence of the inducer will be higher than the basal rate. A
tightly inducible promoter is one whose basal level of activity
is very low, e.g., less than 10 0 of its maximum inducible
activity.
Different promoters may have different levels of basal
activity in the same or different cell types. When two "
different promoters are compared in a given cell type in the
absence of any inducing factors, if one promoter expresses at
a higher level than the other it is said to have a higher basal
activity.
The activity of a promoter and/or enhancer is measured by
detecting directly or indirectly the level of transcription
from the element(s). Direct detection involves quantitating
the level of the RNA transcripts produced from that promoter
and/or enhancer. Indirect detection involves quantitation of.
the level of a protein, often an enzyme, produced from RNA
transcribed from the promoter and/or enhancer. A commonly
employed assay for promoter or enhancer activity utilizes the
chloramphenicol acetyltransferase (CAT) gene. A promoter
and/or enhancer is inserted upstream from the coding region for
the CAT gene on a plasmid; the plasmid is introduced into a
cell line. The levels of CAT enzyme are measured. The level
of enzymatic activity is proportional to the amount of CAT tZNA
transcribed by the cell line. This CAT assay therefore allows
a comparison to be made of the relative strength of different
promoters or enhancers in a given cell line. When a promoter
is said to express at "high" or "law" levels in a cell line
this refers to the level of activity relative to another
promoter which is used as a reference or standard of promoter
activity.
Efficient expression of recombinant DNA sequences in
eukaryotic cells requires expression of signals directing the
efficient termination and polyadenylation of the resulting
transcript. Transcription termination signals are generally
found downstream of the polyadenylation signal and are a few
hundred nucleotides in length. The term "poly A site" or "poly
A sequence" as used herein denotes a DNA sequence which directs
CA 02333481 2000-11-27
WO 00100600 PCT/US99/11516
111
both the termination and polyadenylation of the nascent RNA
transcript. Efficient polyadenylation of the recombinant
transcript is desirable as transcripts lacking a poly A tail
are unstable and are rapidly degraded. The poly A signal
utilized in an expression vector may be "heterologous" or
"endogenous . " An endogenous poly A signal is one that is found
naturally at the 3' end of the coding region of a given gene
in the genome. A heterologous poly A signal is one which is
one which is isolated from one gene and placed 3' of another
gene. A commonly used heterologous poly A signal is the SV40
poly A signal. The SV40 poly A signal is contained on a 237
by Bam HI/Bc1 I restriction fragment and directs both
termination and polyadenylation (J.Sambrook et al., supra, at
16.6-16.7).
The cytomegalovirus immediate early promoter-enhancer
(CMV-IE) is a strong enhancer/promoter. See Boshart M, Weber
F, Jahn G, Dorsch-Hasler K, Fleckenstein B,
Schaffner W. A very strong enhancer is located upstream of an
immediate early gene of human cytomegalovirus. Cell 1985;
41:521-530. For its incorporation into HIV-1 derived viruses,
see Chang L-J, McNulty E, Martin M. Human immunodeficiency
viruses containing heterologous enhancer/promoters are
replication competent and exhibit different lymphocyte
tropisms. J Virol 1993; 67:743-752.
Another strong promoter-enhancer for eukaryotic gene
expression is the elongation factor 1 alpha promoter enhancer.
Kim DW, Uetsuki T, Kaziro Y, Yamaguchi N, Sugano S. Use of the
human elongation factor 1a promoter as a versatile and
efficient expression system. Gene 1996; 91:217-223; Mizushima
S, Nagata S. pEF-BOS, a powerful mammalian expression
vector. Nucleic Acids Res. 1990; 18:5322.
The internal promoter for a transgene may be the promoter
native to that transgene, or a promoter native to the target
cell (or viruses infecting the target cell), or another
promoter functional in the target cell.
The preferred promoters and enhancers are those
exhibiting tissue or cell type sepecificity which can direct
the transgene expression in the target cells at the right
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516 .
112
time(s). For example, a promoter to control human
preproinsulin must be operable under control of carbohydrate
in the liver. An example of such a promoter is the rat S-14
liver-specific promoter.
Promoters (and enhancers) may be naturally occurring
sequences, or functional mutants thereof, including chimeras
of natural sequences and mutants thereof. For example, a ~I
tissue-specific, development-specific, or otherwise regulatable
element of one promoter may be introduced into another
to promoter.
Chen et al, Proc. Nat. Acad Sci USA 93: 10057-62 (1996)
placed a VSV G gene under the control of a tetracycline-
inducible promoter and also expressed a fusion of the ligand
binding domain of the estrogen receptor to a chimeric
transcription factor, tTA, obained by fusing the tet repressor
(tetR) and the activation domain of HSV virion protein 16.
For the ability to replace the endogenous 5' LTR
promoters and enl-lancers with heterologous ones, such as CMV
immediate-early enhancer-promoter, see Chang, et al., J.
Virol., 67: 743-52 (1993). Vector; Transfection of Vectors
As used herein, the term "vector" is used in reference to
nucleic acid molecules that can be used to transfer nucleic
acid ( a . g. , DNA) segment ( s ) from one cel l to another . The term
"vehicle" is sometimes used interchangeably with "vector." It
is intended that any form of vehicle or vector be encompassed
within this definition. For example, vectors include, but are
not limited to viral particles, plasmids, transposons, etc.
The term "transfection" as used herein refers to the
introduction of foreign DNA into eukaryotic cells.
Transfection may be accomplished by a variety of means known
to the art including but not limited to calcium phosphate-DNA
co-precipitation, DEAE-dextran-mediated transfection,
polybrene-mediated transfection, electroporation,
microinjection, liposome fusion, lipofection, protoplast
fusion, retroviral infection, and biolistics.
Vectors may contain "viral replicons "or "viral origins
of replication." Viral replicons are viral DNA sequences which
allow for the extrachromosomal replication of a vector in a
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
113
host cell expressing the appropriate replication factors.
Vectors which contain either the SV40 or polyoma virus origin
of replication replicate to high copy number (up to 104
copies/cell) in cells that express the appropriate viral T
antigen. Vectors containing the replicons from bovine
papillomavirus or Epstein-Barr virus replicate
extrachromosomally at low copy number 0100 copies/cell).
Expression vector
The term "expression vector" as used herein refers to a
recombinant DNA molecule containing a desired coding sequence
and appropriate nucleic acid sequences necessary for the
expression of the operably linked coding sequence in a
particular host organism. Nucleic acid sequences necessary for
expression in prokaryotes usually include a promoter, an
operator (optional), and a ribosome binding site, often along
with other sequences. Eukaryotic cells are known to utilize
promoters, enhancers, and termination and polyadenylation
signals. In some embodiments, "expression vectors" are used
in order to permit pseudotyping of the viral envelope proteins .
Host Cells
The host cell is a cell into which a vector of interest
may be introduced and wherein it may be replicated, and, in the
case of an expression vector, in which one or more vector-based
genes may be expressed.
It is not necessary that the host cell be infectable by
the transducing vector virions of the present invention.
Indeed, it is preferable that they not be so infectable, so the
hos cells do not bind the virions and thereby reduce the vector
production titer. This can be achieved by choosing (or
engineering) cells which do not functionally express the
receptor to the vector particle envelope protein.
Target Cells and Organisms
The transducing vector may be administered to a target
organism by any route which will permit it to reach the target
cells. Such route may be, e.g., intravenous, intramuscular,
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
114 -
subcutaneous, or, with an enteric coating, oral.
Alternatively, target cells may be removed from the organism,
infected, and they (or their progeny) returned to the organism.
Or the transducing vector may simply be administered to target
cells in culture.
The target cells into which the transgene is transferred ,
may be any cell which the transducing vector, after packaging
into a virion, is capable of infecting, and in which the
control sequences governing expression of the transgene are
functional. Generally speaking, it will be a eukaryotic
cell, preferably a vertebrate cell, more preferably a cell of
a mammal or bird. If a mammal, the mammal will preferably
belong to one of the orders Artiodactyla (e. g., cows, pigs,
goats, sheep), Perissodactyla (e. g., horses), Rodenta (e. g.,
rats, mice), Lagomorpha (e. g., rabbits), Carnivora (e. g., dogs,
cats) or Primata (e.g., humans, apes, monkeys, lemurs). If a
bird, it will preferably be of the orders Anseriformes (e. g.,
ducks, geese, swans) or Galliformes (e. g., quails, grouse,
pheasants, turkeys, chickens). Most preferably it will be a
human cell.
The cells in question may be dividing or non-dividing
cells. Non-dividing cells of particular interest include
neuronal cells and astrocytes. Dividing cells of particular
interest include hematopoietic stem cells, muscle cells, white
blood cells, spleen cells, liver cells, epithelial cells and
eye cells.
TE671, HepG2, HeLa, 293T, and MT4 are of particular
interest for experimental studies.
TE671 rhabdomyosarcoma cells can be induced to differentiate
into muscle cells by HIV-1 Vpr. HepG2 hepatoma, HeLa cervical
carcinoma, 293T human kidney carcnoma and MT4 lymphoma cells
are all transformed by HTLV-I human T cell leukemia virus type
I. MT4 cells are very susceptible to wild-type HIV-1 NL4-3 and
hence have been used as indicator cell for RCV.
Miscellaneous Definitions
As used herein, the term "endogenous virus" is used in
reference to an inactive virus which is integrated into the
chromosome of its host cell (often in multiple copies) , and can
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
115 '
thereby exhibit vertical transmission. Endogenous viruses can
spontaneously express themselves and may result in
malignancies.
The term "gene" refers to a DNA sequence of a vector or
genome which comprises a coding sequence and which is operably
linked to one or more control sequences such that, in a
suitable host cell, under suitable conditions, a biologically ~.
active gene product, or a gene product which is a precursor of
a biologically active molecule, is produced which is encoded
by the coding sequence. This gene product may be a
transcriptional product, i.e., a messenger RNA, as in the case
of an antisense RNA or a ribozyme. Or it may be a
translational product, i.e., a polypeptide (the term
"polypeptide" as used herein includes oligopeptides), which is
either biologically active in its own right, or further
processed by the cell to generate one or more biologically
active polypeptide products. In the case of retroviruses,
where the genome is RNA, the term "gene" also refers to the RNA
sequence of the retroviral genome which a suitable host cell
reverse transcribes into a DNA sequence which acts as a gene
in the classic sense.
Depending on context, the term "gene" may refer to the
DNA sequence encoding a single mRNA transcript, or only to that
portion of the DNA sequence which is ultimately expressed as
a single polypeptide chain.
In the vectors of the present invention, each gene may be
constructed from genomic DNA, complementary DNA (DNA reverse
transcribed from mRNA), synthetic DNA, or a combination
thereof . The gene may duplicate a gene which exists in nature,
or differ from it through the omission of introns (noncoding
intervening sequences), a so-called mini-gene, silent mutations
(i.e., mutations which do not alter the amino acid sequence of
the encoded polypeptide), or translated mutations (i.e.,
mutations which do alter that sequence). In the latter case,
the mutations may be functional mutations (ones which preserve
at least a substantial portion of at least one of the
biological activities or functions of the encoded polypeptide)
or nonfunctional (inactivating) mutations.
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516 .
116
As used herein, the term "transcription unit" refers to
the segment of DNA between the sites of initiation and
termination of transcription and the regulatory elements
necessary for the efficient initiation and termination. For
example, a segment of DNA comprising an enhancer/promoter, a
coding region and a termination and polyadenylation sequence
comprises a transcription unit.
Assays
From time to time, one may wish to ascertain various
information concerning the packaging and transducing vectors
of the present invention.
One might like to know whether the vectors have become
established in the cell; whether particular vector genes have
integrated into the genome; whether the packaging cell line is
producing viral proteins; whether those viral proteins are
being assembled into viral particles; whether, in the absence
of the transducing vector, those viral particles are
essentially free of RNA, especially lentiviral RNA (e. g.,
packaging vector RNA); whether recombination occurs between the
packaging vector and the transducing vector, or between these
two vectors and defective retroviruses endogenous to the host
(or target) cell; whether such recombination, if any, produces
replication-competent virus; whether recombinant virus is
packaged by the packaging cell line; the efficiency with which
the packaging cell line packages the transducing vector into
the viral particles; whether the transducing vector-containing
viral particles are infectious vis-a-vis the target cells;
whether the latter particles are cytotoxic to the target cells;
whether the latter particles are immunogenic to the target
organism; whether infected target cells themselves produce
viral RNA-containing particles, infectious or otherwise; and
the level and duration of expression of the transgene in the
target cells.
The successful establishment of the packaging or
transducing vector in the host (or target) cell may be verified
by selecting for the presence of a selectable marker, or
screening for the presence of a screenable marker, carried by
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
117
the vector. The integration of the relevant packaging or
transducing vector genes may be determined by collecting
genomic DNA, amplifying the gene of interest by PCR, and
detecting the amplified sequence with a suitable hybridization
probe. The production of viral proteins may be detected by an
immunoassay; the sample may be a cell lysate or a cell
supernatant . An immunoassay by itself cannot determine whether ~.
the viral proteins are produced in functional form, although
there is greater assurance of this if the antibody used is
directed to a conformational epitope, or is an activity-
neutralizing antibody. One may alternatively detect the
appropriate messenger RNA by means of a hybridization probe.
The functionality of the produced Gag and Env protein may
be determined by examining the cell lysate or supernatant for
the presence of viral particles; these may further be examined
for proper morphology by means of an electron microscope. It
is also possible that antibodies could be used which bind to
the formed viral particles, but not to gp120 or gp41 by itself .
The functionality of the Pol reverse transcriptase may be
determined by assaying the viral particles for RT activity.
The functionality of the Pol integrase is apparent only in
assays which examine whether RNA from viral particles is
integrated into the target cell.
viral particles produced by the packaging ceii line may
be collected and assayed for total RNA content. If more
specific information is desired as to the nature of any
packaged RNA, a suitable hybridization probe may be employed.
In an infectivity assay, the vector is introduced into a
first culture of susceptible cells. Then, either a second
culture is layered onto the first, so that infectious particles
may travel by cell-to-cell contact, or the second culture is
exposed to the supernatant of the first culture. The cells of
the first and second culture are examined for a least one of
the following indicia: RT activity, p24 Gag antigen expression,
production of viral particles, and cytotoxic effects. The
stringency of the assay is dependent on the susceptible of the
cells to infection and to cytotoxicity, and the time allowed
for the recombination and spread of the virus in the first and
CA 02333481 2000-11-27
WO 00/00600 PC'T/US99/11516
118
second cultures. Typically, the infectivity of the vector or
vector system will be compared with that of a wild-type,
unattenuated, replication-competent lentivirus.
Animal studies may be used to ascertain the
immunogenicity and pathogenicity of the vector system.
Some of these assays are described in greater detail "
below.
Measurement of Infectivity 'of Packaging Vector per se
The ability of a packaging vector to generate
transmissible virus, as opposed to defective virus, may be
measured. One method is described by Mann, et al., Cell, 33:
153-9 (1983). The packaging vector and its wild-type
counterpart are independently transfected into suitable host
cells, and reverse transcriptase activity in the culture
supernatants is assayed over a period of days or weeks. A
rapid increase in RT activity over 24-48 hrs is indicate of
gene expression after transient transfection. A continued
increase is indicative of the efficient spread of virus from
the initially transfected cells to the remaining cells on the
plate.
A slow or delayed increase could be indicative of either
a steady but attenuated spread oz virus, or to generation of
competent virus by mutation, or by recombination with a
cellular sequence capable cf providing the missing function.
To differentiate these possibilities, one may use various
dilutions of culture supernatants from cells previously
transfected (days or weeks before) with the vector (or with the
control virus), use them to infect fresh cells, and monitor RT
activity in the latter. If the latter cells develop high
levels of RT activity, it suggests that nondefective virus was
present in the transferred culture supernatant.
Measurement of Packaging Efficiency
The packaging efficiency of a packaging cell line in the
presence or absence of the packageable transducing vector may
be measured in a variety of ways. One method is described by
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
119
Mann, et al., Cell, 33: 153-9 (1983). In esssence, total
cellular RNA is purified from the culture supernatant of the
test and control cell lines, and viral RNA is extracted from
purified viral particles released from the test and control
cell lines. The two virion preparations are normalized by
reference to their reverse transcriptase activity just prior
to RNA extraction. The purified RNAs are probed with a virus-
specific hybridization probe (e.g., a plasmid containing the
entire viral genome) in a slot-blot assay, and the amount of
viral RNA in the particles and in the cells is thereby
quantified.
It is not unusual for the packaging efficiency of a
packaging cell line to be less than 1 o that of a host cell
infected by wild-type virus.
Measurement of Packaging Specificity
It is also desirable that the packaging cell line be able
to efficiently package the highly defective transducing vector
into viral particles, and bud the particles into the culture
supernatant (in vitro) or extracellular environment (in vivo)
without also budding helper virus (the packaging vectors).
One method of measuring this packaging specificity is
described by Mann, et al., Cell, 33: 153-9 (1983). In essence,
the transducing vector is transfected in~o the packaging
(helper) cell line. After 24 hours, the culture supernatants
are used to infect fresh potential host cells (reporter cells) .
Two days later, selection pressure for the transferred gene is
applied, and 8-10 days later, the transferred gene-positive
colonies or, cells are counted. In addition, one determines the
reverse transcriptase activity of the supernatant colelcted
from the packaging cell lines, and the reverse transcriptase
activity of the fresh cells. A transducing vector-specific
packaging cell line will produce a high transfer gene activity
and a low reverse transcriptase activity in the reporter
cells. In addition, the reporter cells will not produce
reporter gene-positive colony-forming units (cfus).
Measurement of Helper Activity
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
120
The ability of a packaging vector to provide all viral
functions required in trans may be assayed by cotransfecting
host cells with the packaging vector (or control virus) and
with a reporter vector carrying a selectable reporter gene.
After 24 hours, culture supernatants of the transfected cells
are used to infect a second plate of host cells . Selection
pressure for the reporter gene is applied, and reporter-
positive colonies are counted. If the helper activity is of
wild-type magnitude, the count for the packaging vector should
be of the same order of magnitude as that for the control
virus, and no reporter activity should be detectable in the
second plate when the reporter vector or the control wild-type
virus expressing all viral functions is transfected into the
host cells of the first plate by itself.
Measurement of Generation of Replication-Competent Virus
Several sensitive assays are available for the detection
of RCV in the present lentiviral vector systems. These
include: (1) co-cultivation with a sensitive cell line such as
MT4, AA2 or PBLs; (2) the CD4 HeLa MAGI cell assay which relies
on Tat transactivation of an integrated LTR-lacZ gene; and (3)
a sensitive immunohistochemical staining method for the
detection of HIV antigen expression at the individual cell
ievei. As described in the Examples below, the latter method
was modified and developed for the characterization of
"Tat-minus" HIV-1 infection, although all three methods are
suitable for the routine titration of infectious HIV-1.
RC-HIV can also be studied in an in vivo model by
transduction of humanized SCID/beige mice. In the latter
model, a long in vivo incubation time can be performed,
mimicking the situation that exists in a human clinical trial.
In addition, the possibility of generating HIV/HERV
recombinants may be carefully tested using an artificially
constructed HIV/HERV-env recombinant.
Virion Stability
Since one class of the therapeutic agents of the present
invention would be the packaged transducing vectors, the
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516 .
121
stability of the packaged transducing vectors under adverse
conditions, especially those which might be encountered during
storage, is of interest. Thermostability may be ascertained
by subjected them to elevated (e. g., 37 deg. C) or depressed
(e. g., 4 deg. C) temperatures for various periods of time
(e. g., 2, 4, 6 or 8 hrs., or overnight), or to a number (e. g.,
1-6) freeze-thaw cycles, and determining the number of
infectious particles remaining as a percentage of the number
of such particles prior to treatment. See Burns et al. 1993.
Assays for Immunogenicity
A preferred method for determinining whether the
contemplated vectors, or their gene products, could elicit an
immune response in a subject involves evaluating cell-mediated
immunity (CMI) using either an immunocompetent mouse model or
a a humanized SCID/beige mouse model.
Using a modified hu-PBL-SCID mouse reconstitution
protocol, an in vivo model for evaluating CMI against HIV-1 in
humans has been developed. SCID/beige mice lacking T, B and
natural killer (NK) cell functions are severely
immunodeficient. This strain of mice can be successfully
reconstituted with fresh human peripheral blood lymphocytes
(PBLs), and exhibits functional human naive, memory and
activated T cell markers for more than 2-3 months (See e.g.,
copending U.S. Patent Appln. Ser. Nos. 08/848,760, and
08/838,702, both of which are herein incorporated by
reference). In these experiments, spleen and peripheral blood
lymphocytes were harvested 38 days after reconstitution from
reconstituted SCID/beige mice, and red blood cells were lysed
prior to incubation with anti-mouse 2Kd, anti-human CD45,
anti-human CD3, anti-human CD4 and anti-human CD8 labeled
antibodies. Reconstituted human lymphoid cell populations in
the spleen and in the peripheral blood of the SCID/beige mice
can reach up to 50-80o and 5-120, respectively.
~'or the immune response study, mice repetitively injected
with the viral vectors will be analyzed. Their sera will be
assayed for Ab response to viral antigens, such as p24 Gag or
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
122
the pseudotype env (e. g., VSV-G). For cell-mediated immune
response study, the mouse splenocytes will be isolated and an
in vitro assay for cellular immunity will be performed as
described below. T cell response to recall antigen is normally
characterized by the production of interferon gamma (IFN~y) .
This assay requires activation of lymphocytes with the test
Ags, such as Gag p24 or Gag-Pol or VSV-G env proteins of the ~~
vector.
Upon activation, the Thl lineage of T cells produce
interferon gamma (IFN-g) and the measurement of IFN-g
production has been shown to be a reliable assay for CMI.
Thus, to determine CMI against HIV-1 using the in vivo
humanized SCID/beige mouse model, a sensitive ELISPOT assay for
the detection of IFN-g producing cells was developed. With the
computer assisted imaging system integrated into this protocol,
the ELISPOT method was shown to be very convenient and more
sensitive than the conventional limiting dilution assay for the
determination of the effector T cell precursor frequency. This
in vivo model and the ELISPOT assay system were developed for
the evaluation of in vivo CMI after lentiviral gene transfer.
(See, e.g., PCT/US98/06944).
EXAMPLES
The following examples serve to illustrate certain
preferred embodiments and aspects of the present invention and
are not to be construed as limiting the scope thereof.
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
123
Abbreviations
In the experimental disclosure which follows, the
following abbreviations apply: RCR (replication-competent
retrovirus); RCV (replication-competent virus); WT (wild-type);
PBL (peripheral blood lymphocyte); M (molar); mM (millimolar);
~M (micromolar); mol (moles); mmol (millimoles); ~,mol
(micromoles); nmol (nanomoles); g (gravity); gm (grams); mg ~~
(milligrams); ~.g (micrograms); pg (picograms); L (liters); ml
(milliliters); ~.1 (microliters); cm (centimeters); mm
(millimeters); ~.m (micrometers); nm (nanometers); hr (hour);
min (minute); msec (millisecond); 'C (degrees Centigrade); AMP
(adenosine 5'-monophosphate); cDNA (copy or complimentary DNA);
DTT (dithiothreitol); ddH20 (double distilled water); dNTP
(deoxyribonucleotide triphosphate); rNTP (ribonucleotide
triphosphate); ddNTP (dideoxyribonucleotide triphosphate); by
(base pair); kb (kilo base pair); TEM (transmission electron
microscope); SEM (scanning electron microscope); TLC (thin
layer chromatography); tRNA (transfer RNA); nt (nucleotide);
VRC (vanadyl ribonucleoside complex); RNase (ribonuclease);
DNase (deoxyribonuclease); poly A (polyriboadenylic acid); PBS
(phosphate buffered saline) ; OD (optical density) ; HEPES (N- [2-
Hydroxyethyl]piperazine-N-[2-ethanesulfonic acid]); HBS (HEPES
buffered saline); SDS (sodium dodecyl sulfate); Tris-HCl
(irisiHydroxymethyl]aminomethane-:hydrochloride); rpm
(revolutions per minute); ligation buffer (50 mM Tris-HCl, 10
mM MgCl" 10 mM dithiothreitol, 25 ug/ml bovine serum albumin,
and 26 ~M NAD+, and pH 7.8); EGTA (ethylene glycol-bis(/3-
aminoethyl ether) N, N, N', N'-tetraacetic acid}; EDTA
(ethylenediaminetetracetic acid); ELISA (enzyme linked
immunosorbant assay); ELISPOT (enzyme-linked immunosorbent spot
assay); LB (Luria-Bertani broth: ZO g tryptone, 5 g yeast
extract, and 10 g NaCl per liter, pH adjusted to 7.5 with 1N
NaOH); superbroth (12 g tryptone, 24 g yeast extract, 5 g
glycerol, 3.8 g KH2P04 and 12.5 g, KzHP04 per liter); DMEM
(Dulbecco's modified Eagle's medium); ABI (Applied Biosystems
Inc., Foster City, CA); Amersham (Amersham Corporation,
Arlington Heights, IL); ATCC (American Type Culture Collection,
Rockville, MY); AIDS Research and Reference Reagent Program
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516 .
124
(AIDS Research and Reference Reagent Program of the National
Institutes of Health, Bethesda, MD); Beckman (Beckman
Instruments Inc., Fullerton CA); BM (Boehringer Mannheim
Biochemicals, Indianapolis, IN); Bio-101 (Bio-101, Vista, CA);
BioRad (BioRad, Richmond, CA); Brinkmann (Brinkmann Instruments
Inc. Wesbury, NY); BRL, Gibco BRL and Life Technologies
(Bethesda Research Laboratories, Life Technologies Inc.,"
Gaithersburg, MD); CRI (Collaborative Research Inc. Bedford,
MA); Eastman Kodak (Eastman Kodak Co., Rochester, NY);
Eppendorf (Eppendorf, Eppendorf North America, Inc., Madison,
WI); Falcon (Becton Dickenson Labware, Lincoln Park, NJ); IBI
(International Biotechnologies, Inc., New Haven, CT); ICN (ICN
Biomedicals, Inc . , Costa Mesa, CA) ; Invitrogen ( Invitrogen, San
Diego, CA); New Brunswick (New Brunswick Scientific Co. Inc.,
Edison, NJ); NEB (New England BioLabs Inc., Beverly, MA); NEN
(Du Pont NEN Products, Boston, MA); Nichols Institute
Diagnostics (Nichols Institute Diagnostics, San Juan
Capistrano, CA); Pharmacia (Pharmacia LKB Gaithersburg, MD);
Promega (Promega Corporation, Madison, WI); Stratagene
(Stratagene Cloning Systems, La Jolla, CA) ; UVP (UVP, Inc. , San
Gabreil, CA); USB (United States Biochemical Corp., Cleveland,
OH); Taconic (Taconic, Germantown, NY); and Whatman (Whatman
Lab. Products Inc, Clifton, NJ).
Sources
Unless otherwise indicated, all restriction enzymes were
obtained from New England Biolabs and used according to the
manufacturers directions. Unless otherwise indicated,
synthetic oligonucleotides were synthesized using an ABI DNA
synthesizer, Model No. 391.
In the following Examples, non-attenuated HIV strains
used-include the NL4-3 HIV-1 strain, HIV-1 primary isolates
covering the different HIV Glades (e. g., 92RW008, 92HT593,
etc. ) , the ROD strain of HIV-2, and the SIVmac239 strain of
SIV, all of which are available from the AIDS Research and
Reference Reagent Program.
Methods
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516 .
125
Plasmid DNA construction. HIV-1 LTR and tat mutations were
constructed as described previously (Chang et a1.1993; Chang
and Zhang, 1995). Cloned HIV proviruses with heterologous
enhancer/promoters were constructed by ligating three fragments
from an HIV-1 molecular clone HIVNL4-3(Adachi et a1.1986), two
fragments isolated from the U3-R-CAT plasmids containing o,
inserted heterologous enhancer/promoters and the BamHI plus
PstI digested pT7T318U vector. The proviral segments used in
the ligation were as described before (Chang et a1.1993). The
structures of the reconstructed HIV proviral DNAs were verified
by extensive restriction enzyme mapping, and the LTR regions
were checked by nucleotide sequencing.
RT assay and p24 ELISA for the detection of HIV gag and pol
products. RT assays detect functional reverse transcriptase
activity which were performed as described below. The
supernatants from transfected cells were spun in a microfuge
at 3000 rpm for 5 min before being added to the reaction
mixture. Supernatants from virus infections were removed from
cultures after the cells had settled. Each reaction mixture
contained 10 ml of supernatant and 50 ml of RT cocktail (60 mM
Tris-HCl, pH 7.8, 75 mM KC1, 5 mM MgCl2, 0.1% Nonidet P-40, 1mM
EDTA, 5 mg/ml poly rA and 0.16 mg/ml oligo-dT) and was
incubated at 37oC for 1 h. The radioactive producLS generated
in the CAT and RT assays were quantitated by using a Fuji
phosphoimager. The results obtained were comparable to those
derived by scintillation counting. p24 antigen is derived from
p55 gag precursor. The p24 antigen expression was quantified
using a commercial ELISA kit from Coulter (Coulter Corp.,
Hialeah, FL).
RT-PCR and sequencing of the packaged viral genomic RNA.
Cell-free particles, present in the supernatants of vector
producing cells, were harvested (100 microl), centrifuged at
top speed for 5 min in a microcentrifuge at room temperature,
and filtered through a 0.45 mm-pore-size Eppendorf spin filter.
The particles present in the filtrate was dissociated by
vortexing in the presence of an equal volume of 8 M LiCl,
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
126
placed on dry ice for 20 min, transferred to a -20oC freezer
for at least 2 h, and centrifuged at top speed in a
microcentrifuge at 4o C for 20 min. The RNA pellet was then
rinsed with 70% ethanol, dried briefly under vacuum,
resuspended in water and reverse transcribed by using an
appropriate primer and the RiboClone cDNA Synthesis System ,
(Promega) for the synthesis of the first DNA strand. A
control reaction excluding the reverse transcriptase was
performed in parallel. The cDNA was amplified by PCR using the
polymerase and reagents obtained from Perkin Elmer Cetus; 5'
and 3' primers (0.1 micromole each) were added to a reaction
mixture containing the cDNA (1/20 of the RT product) and
amplified for 30 cycles under the following conditions: 94oC
for 1 min, 58oC for 1 min and 72oC for 3 min. The product
obtained was then subjected to asymmetric PCR amplification
(i.e., two primers at 10:1 molar ratio) to generate single
stranded DNA for sequencing as described by Meltzer et al.
(39). Excess primers were removed with a centricon 100
filtration device (Amicon) after each amplification step.
Nucleotide sequencing was performed using Sequenase and
protocols supplied by USB.
Immunofluorescent and immunohistochemical staining.
For immunofiuorescenL staining, non-adherent cells were
attached to the surface of a microscope cover glass (12 mm
circle, Fisher Scientific, Pittsburgh, PA) which had been
pretreated with poly-D-lysine (1 mg/ml, Sigma) at room
temperature for 10 min. The attached cells were washed with
phosphate buffered saline (PBS) three times, fixed in cold
acetone and methanol (1:1) for 5 min, washed three times in
PBS, and incubated in blocking (20 % FBS, 0 .1% TritonX100 in
PBS) solution for 30 min. An HIV patient's serum was used as
the primary antibody, which was diluted at 1:2000 in blocking
solution, and the cells were incubated at room temperature for
1 h or at 4oC overnight with constant shaking. After washing
in PBS 4 times for 5 min each, the cells were incubated with
normal goat or sheep antisera (1:200 dilution) at room
temperature for 30 min to block non-specific binding. The
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
127
secondary antibody was FITC-labeled goat anti-human IgG (Fab
specific, Sigma Chemical Company, St. Louis, MO). After
staining, the cover glass was washed four times in PBS and
examined using a fluorescent microscope. For direct
immunohistochemical staining, a peroxidase-linked sheep
anti-human Ig (Amersham) was used as the secondary antibody.
Alternatively, a biotinylated sheep anti-human antibody ~I
(Amersham) was used at 1:2000 dilution and incubated at room
temperature for 1 h. The latter step provided a more sensitive
method for detection of low level of HIV antigens which was
described in detail elsewhere (Chang and Zhang, 1995).
RNA and protein analyses.
Northern analysis was performed as previously described
(Robinson et a1.1995). For protein analysis, cells were lysed
in a buffer containing 50 mM Tris pH 7.4, 300 mM NaCl, 0.5%
Triton X100, 1% (v/v) aprotinin and 1 mM PMSF at 4oC for 10 min
and freeze-thawed once. Virus particles were collected by
centrifugation in a refrigerated micro centrifuge in a small
volume (200 microliters) at 23, 000 g for 1 hr. The supernatant
was carefully removed and to the pellet, 20 ml of SDS sample
buffer (final 2°s SDS, 5o glycerol, 0/OOlo BPB, 0.5o NP-40) was
added and the denatured protein was resolved by polyacrylamide
gel electrophoresis (PAGE) as described previously (Chang et
a1.1990).
For Western blot analysis, the protein was transferred to
a 0.2 micron nitrocellulose filter, stained with Ponceau S to
identify the molecular weight marker, and blocked with l00
dried milk in TBS-T (Tris-buffered saline with 0.3% Tween 20)
at room temperature for 30 min to 1 hr. After washed briefly
at room temperature, the blot was placed into a "seal-a-meal"
bag and incubated with an AIDS patient's serum (diluted at
1:2,000, or a rabbit polyclonal anti-Vpr antibody at 1:1,000,
or a monoclonal anti-Nef antibody at 1:1000) in TBS-T
containing 2% dry milk at 4oC overnight. After four washes
with TBS-T, the blot was blocked with normal goat sera (the
same species as the secondary Ab) at 1:200 dilution in a
shallow tray or in a bag at room temperature for 30 min. The
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
128
blot was then transferred to a second bag containing a horse
radish peroxidase (HRP) conjugated goat anti-human (or goat
anti-rabbit, or goat anti-mouse) antibody and incubated at room
temperature for 1 hr. The blot was washed four times in TBS-T
and developed using the chemiluminescence ECL immunodetection
reagents from Amersham. The blot was then exposed to a
hyperfilm (Amersham) normally for 1 min and developed.
Cells and culture conditions
HeLa (human cervical carcinoma) and HepG2 (human
hepatoma) cells were obtained from ATCC, Rockville, Maryland.
TE671 (human rhabdomyosarcoma) and 293T (transformed human
primary embryonal kidney) cells were kindly provided by Dr.
Takeuchi (Chester Beatty Laboratories, obtained from ECACC,
England) and Dr. H. Goldstein (Albert Einstein College of
Medicine, N. Y.), respectively. H9, CEM, MT4, C8166 and AA2
were obtained from NIH AIDS Research and Reference Reagent
Program. Maintenance of the continuous human lymphoid cell
lines H9, CEM, MT4, AA2 and the primary human PBLs were as
described (Chang et a1.1993). The Molt3 and THP-1 were
obtained from the American Type Culture Collection (Rockville,
MD). HeLa clone HL3T1, C8166 and U937 cells were kindly
provided by G. Pavlakis, K.-T. Jeang, and K. Peden,
respectively. HeLa CD4+ clones 1022 and HT-6C (Chesebro and
Wehrly, 1988) were obtained through the AIDS Research and
Reference Reagent Program, Division of AIDS, NIAID, NIH, from
Dr. Bruce Chesebro.
HeLa, HepG2, TE671 and 293T cells were propagated in
Dulbecco's modified Eagle's medium (DMEM) supplemented with 10%
fetal bovine serum (FBS) (Gibco Canada), penicillin and
streptomycin.
The macrophage culture was prepared from
HIV-sero-negative donors by adherence of PBLs to plastic flasks
as described previously with minor modifications (Hassan, et
al. 1986). PBLs was prepared using lymphocyte separation
medium (Organon Teknika Corp., Durham, NC) by density gradient.
The PBLs were resuspended in RPMI 1640 medium supplemented with
20o heat-inactivated human serum. Approximately 5 x 107 PBLs
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516 .
129
were attached to a T-75 flask and incubated overnight at 37oC.
The next day cells were washed three times with phosphate
buffered saline and the attached cells were incubated with
0.02% EDTA in PBS for 5-10 min. The cells were collected with
a cell scraper and plated onto a 48-well plate at 5 x 104 cells
per well. The viability approached 100% as determined by ,
trypan blue staining. The initial monocytes were characterized
by Wright's staining and the mature macrophages by both
Wright's staining and microscopic examination.
Animals
Sprague Dawley rats (180-200 gram of body weight) were
purchased from the Health Science and Laboratory Animal Service
(HSLAS) at the University of Alberta.
Plasmid construction and site-directed mutagenesis
The tat-A, tat-B and tat-C site-directed mutations were
generated by the "Megaprimer" method of Sarkar and Sommer
(Sarkar and Crissman, 1990) using the following mutagenic
oligos:
5'-GAATTGGGTGTCGACATAGCGGCCGCTTGTACCAATTGCTATTG-3',
5'-GGTACAAGCAGTTTAAGGCTAACTTCCTGGATGCTTCC-3', and
5'-CGACAGAGGAGAGCAAGAAACGGCGCCTCGCGTAGCTAGCGG-3',
respectively.
A fragment containing the tat mutation [EcoRI-SacI (260
nt)] generated by PCR mutagenesis was used to construct the
full-length two LTR HIV plasmids. Construction of the tat-A arid
tat-C mutations have been described elsewhere (Dimitrov, et al.
1993; Amendt, et al. 1994). The dl.Sp1/CMV tat-B
macrophage-tropic virus was made by replacing the EcoRI to
BamHI fragment in a T-cell tropic construct (pNL4-3, Adachi,
et al. 1986) with the same fragment from a macrophage-tropic
construct (pNLAD8, kindly provided by Eric Freed). Sequences
of the PCR fragment and its flanking region in the final
constructs were verified by DNA sequencing.
Transfection and Northern analysis
HeLa cells. were transfected using the original
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516 .
130
Ca3(P04)2-DNA co-precipitation procedure with modifications
(Graham and van der Eb, 1973) . In brief, HeLa cells were split
into 6-well plates 20 h prior to transfection. The plasmid DNA
was in 90 ml of ddH20 and mixed with 10 ml of 2.5 M CaCl2
(Mallinckrodt) in a polycarbonate tube. To the DNA mixture,
a 100 ml of BES-buffered solution (50 mM "
N,N-bis[2-hydroxyethyl]-2-aminoethanesulfonic acid
[Calbiochem], 280 mM NaCl, 1.5 mM Na2HP04, pH 6.95) was added
dropwise. The solution was allowed to sit at room temperature
for 45 min to 1 hr before being added to the 2 ml growth
culture (pH. 7.1). After adding the DNA, the culture was
maintained in a 3% COz incubator at 37oC overnight. For the
CAT assay, HeLa cells were transfected with 3 mg of CAT plasmid
in the presence or absence of 0.1 mg of a tat plasmid pSVtat
(Peterlin et a1.1986) or pCEP-tat (Robinson et al. 1995). For
the assay of Tat function using HL3T1 cells, transfection was
done using 10 mg of DNA of different HIV-1 constructs. To
generate virus stocks, HeLa cells were transfected with 10 mg
of cloned HIV-1 plasmids and virus was harvested, filtered
through a 0.45 m filter (MILLEX-HV, Millipore Products
Division, Bedford, MA) and frozen at -80oC for later use. All
transfections were performed in the presence of a control human
growth hormone plasmid pXGHS (Nichols Institute Diagnostics).
Northern analysis of viral RNA was done as described previously
(Chang et a1.1993) and analyzed using a phosphoimager (Fuji,
BAS1000) .
Quantitative Immunostaining of HIV-infected cells
Adherent cells were washed with phosphate buffered saline
(PBS) three times, fixed in cold acetone and methanol (1:1) for
2 min, washed three times in PBS, and incubated in blocking
solution (20% FBS, 0.1% TritonX100 in PBS) for 30 min.
Non-adherent cells were attached to the surface of a 24-well
plate which had been pretreated with poly-D-lysine (1 mg/ml,
Sigma) at room temperature for 10 min. We used an HIV patient
serum which was diluted at 1:2000 in a blocking solution
containing 20% FBS, 0.1% TritonX100 and 2% dry milk in PBS as
the first antibody and the incubation was done at room
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
131
temperature for 1 h or at 4oC overnight with constant shaking.
After being washed in PBS for 5 min 4 times, the cells were
incubated with a 1:200 dilution of normal sheep antisera at
room temperature for 30 min to block non-specific signals. The
secondary antibody was a biotinylated sheep anti-human antibody
(Amersham) which was used at 1:2000 dilution and incubated at
room temperature for 1 h. The cells were washed four times in
PBS-Tween 20 (0.3%) and incubated in the ultra-sensitive ABC
staining solution (containing avidin and biotinylated
l0 horseradish peroxidase, Pierce Chemical Co.) at room
temperature for 30 min. After four more washes in PBS-Tween
20, the cells were incubated in 3, 3'-Diaminobenzidine
tetrahydrochloride (DAB) solution (Sigma) containing 0.3% NiCl2
for 2-3 min. The reaction was stopped by washing cells with
tap water for 1-2 min. Cell staining was scored under an
inverted microscope and photographed. To reduce background
staining, both the primary and the secondary antisera were
preabsorbed with fixed human PBLs. Pretreatment of fixed cells
with 0.01% H2O2 at room temperature for 5 min essentially
eliminated all nonspecific background signals. The percentages
of positive cells were determined by taking the average of more
than three representative counts of 1,000 or 10,000 cells.
Genomic and Hirt DNA preparation and Southern and PCR analyses .
A modified protocol was used which allowed simultaneous
preparation of genomic and Hirt DNA. Motmans, et al.,
BioTechniques, 23:1044-6 (1997). Briefly, cells were washed
three times with PBS and re suspended in 250 ~,1 25 mM Tris-HC1
pH 8.0 buffer containing 50 mM glucose, and 10 mM EDTA. The
resuspended cells were incubated at room temperature for 5 min,
and then lysed in 200 ~.1 lysis buffer containing 200 mM NaOH
and to SDS on ice for 5 min. The lysate was neutralized by
adding 150 ~,l potassium acetate (5 M, pH 4.8) . Cell debris and
chromosomal DNA (in pellet) were removed by centrifugation at
10,000 x g for 5 min. The supernatant containing the Hirt DNA
was loaded onto a QIAprep Spin Columns and centrifuged for 1
min. Columns were washed to remove residual endonucleases and
salts, and the DNA was eluted with 100 ~.1 distilled water
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516 .
132
(75°C) by centrifugation at 10,000 x g for 1 min. The pellet
containing the genomic DNA was processed using a Qiagene
genomic DNA harvesting kit starting from the proteinase
digestion step according to the manufacturerOs instructions.
Southern analysis was performed using standard protocols
as described by Maniatis et al., Molecular Cloning: A
Laboratory Manuarl (1989) and a modified hybridization "
procedure as described previously Chang, et al., Virol,
211:157-69 (1995). PCR analysis of unintegrated lentiviral
proviral DNA was performed using the following two sets of
nested primers flanking the LTR of the circular lentiviral
proviral DNA: 5'-ACG ACT CCT GGA GCC CG- (3' end of the lacZ
gene) and 5'-ACA AGG CAG CTG TAG ATC TTA GCC- (5' end of poly-
purine tract (PPT) of HIV-1); 5'-ACT TTC GCT TTC AAG TCC C-
(upstream of primer binding site) and 5'-ACT GAC GCT CTC GCA
CCC AT- (downstream of gag AUG). The amplified products from
the one-LTR lentiviral proviral circular DNA will be 715 by and
from the two-LTR proviral circular DNA will be 1351 bp. For
the detection of MLV unintegrated DNA, we used the following
two sets of nested primers flanking the LTR of the circular MLV
priviral DNA: 5'-AAC CAG CCA TCG CCA TC- (3' of lacZ), 5' ACG
ACT CCT GGA GCC CG- (3' of lacZ) or 5'- AAA AGA TTT TAT TTA GTC
TCC AG- (5' end of PPT of MLV); 5'ACT AGA CAA TCG GAC AGA C-
(outside of U5) and 5' -TCG TCT CCT ACC HGA ACC- (our.side of
U5). The amplified products from the one-LTR MLV proviral DNA
will be 733 by or 1195 bp. Two-LTR circles were infrequently
amplified from both HIV and MLV vector transduced cells.
Introduction to Examples 1-9
The ability of lentiviruses to infect non-dividing cells
such as macrophages and neurons makes them good candidates for
use as gene transfer tools. However, the complicated genome
organization and regulation of viral gene expression, as well
as the concerns of possible spread of AIDS with HIV-derived
vectors, have hindered the wide dissemination of lentiviral
vector technology. Using attenuated HIV-1 constructs, we have
successfully generated a recombinant lentiviral vector gene
transfer system that is efficient and safe. Efficient
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
133 -
synthesis of HIV-1 Gag-Pol requires the activation of LTR by
Tat and the interaction of Rev-RRE to mediate nuclear export
of mRNA, whereas the accessory gene functions of vif, vpr, vpu
and nef have been shown to be dispensable for viral
replication, as well as for vector function in tissue culture.
Both tat and rev genes are functional in the gag-poI packaging
construct pHP.
In pHP, several cis-elements essential to viral
replication have been deleted, including both the 5' and the
3' LTRs, the 3' PPT and the entire 5' leader sequences except
for TAR. A 59 by artificial RSV splice donor sequence has been
inserted into pHP which supports tat and rev mRNA splicing.
The RSV gag AUG is located in the 59 by artificial leader
sequence 5' to the RSV SD site. To prevent interference with
the use of the downstream HIV gag AUG, the RSV gag AUG was
mutated in pHP. Although it was not clear if mutation in the
RSV gag AUG might affect the RSV SD function, the expression
of functional tat and rev genes by pHP indicates that the 5'
RSV gag AUG mutation does not interfere with the RSV SD
function.
Compared with wt HIV-1, the modifications in pHP had
little effect on viral RT synthesis, nor did it diminish vector
titer. Interestingly, although wt HIV-1 exhibited higher RT
activity than pHP when co-transfected with a pTV vector, wt
HIV-1 produced less infectious vectors than pHP, possibly due
to interference of the wt genome with vector genome packaging.
In fact, the HP/TV vector system consistently produced 3=5
times more vector than was obtained from wt HIV/TV co-
transfections. Western blot analyses showed that HP produced
Gag at levels similar to that of wild type HIV-1. This result
is consistent with the results of our previous studies in which
we found that the CMV-TAR chimeric promoter exhibits high
transcriptional activity and remains Tat responsive.
We examined RCV production via recombination of HP/TV by
testing env-deleted HP constructs in co-transfection
experiments. By coupling the co-culture method with
immunostaining, we were able to detect very low amounts of RCV.
Using this assay, RCV was easily detected in cultures co
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516 .
134
transfected with the original pHP-1 construct which contains
a wild type env gene, but not in cultures co-transfected with
pHP-1d1.2 which has a 2-nt deletion between the C5 and the C6
domains of env gp120. The absence of RCV production in these
cultures was likely due to the lack of env function rather than
the lack of recombination because a recombinant virus lacking "
nef but containing a reporter gene could still be generated via
a double genetic cross-over between HP and TV. Additional
mutations were introduced in pHP and pTV to eliminate
homologous recombination which have effectively eliminated the
possibility of RCV production.
Naldini et al . have compared transduction ef f iciencies of
an HIV-1-based vector versus an MLV-based vector using both
proliferating and growth-arrested HeLa cells and 208F rat
fibroblasts. The results of their studies show that the HIV-
based vectors infect G1/S and G2 arrested HeLa cells and GO
arrested 208F cells more efficiently than the MLV-based vector.
In our studies, we showed that the HP/TV vector transduced
growth-arrested or terminally differentiated cells efficiently
in tissue culture including primary rat and human neuronal
cells. 4Ve have also demonstrated efficient transgene
expression in vivo in rat muscles after intramuscular injection
with the HP/TV vectors but not with the retroviral vectors.
in addition, we tested three different human cell lines, TE671,
293T and HepG2, and compared short-term and long-term
transduction using both an HIV-1-based HP/TV vector and an MLV-
based MFG vector. Our results showed that the HP/TV vector
expressed the bacterial nlacZ transgene at higher efficiencies
than the MFG vector in all three cell lines, but that
expression of the lentivirally transduced nlacZ gene gradually
decreased after multiple passages. After more than 35
passages, the expression level of the HP/TV nlacZ transgene was
similar to that observed for the MLV nlacZ transgene . Southern
analyses of TE671 genomic DNA suggested that the HP/TV nlacZ
transgene was integrated into the target cells, but was lost
from the cultures upon repeated passages. Southern analyses
of 293T and HepG2 genomic DNA revealed that the transgene was
still present in these cells even after 49 passages, a result
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/I 1516
135 '
suggesting that the decrease in HP/TV nlacZ transgene
expression may have been due to loss of promoter activity.
Hirt DNA analyses indicated that unintegrated lentiviral
proviral DNA could persist in transduced cells for more than
4-5 passages, but that after 40 passages it could not be
detected in any of the three cell types examined. In contrast,
no MLV-derived unintegrated proviral DNA was detected as early
as passage 3 in all three cell types. Together, these data
suggest that lentiviral vectors are more efficient than MLV
vectors possibly attributable to the prolonged presence of
unintegrated lentiviral proviral DNA which drives lentiviral
transgene expression. Stevenson et al. reported that
unintegrated HIV-1 DNA can serve as a template for HIV-1
antigen synthesis, whereas Sakai et al. reported opposite
results with an HIV-1 integrase mutant. Our data show that
high level lentiviral vector transgene expression coincides
with the presence of unintegrated proviral DNA which is
consistent with Stevenson's observation. It is conceivable
that the internal CMV-IE promoter in the unintegrated HP/TV
proviral DNA is active but the 5' LTR promoter is not because
the latter requires Tat transactivation.
We have observed differentiation of TE671 cells into
muscle cells after transduction with vpr+ but not with vpr-
HP/TV vectors. Thus, it is also possible that the loss of
integrated pTV gene in TE671 culture was due to the
disappearance of transduced and differentiated TE671 cells with
time. On the other hand, pTV contains a CMV-IE
enhancer/promoter which has been shown to frequently be
inactivated after integration. Promoter inactivation could
therefore have contributed to loss of HP/TV gene expression.
Interestingly, in a separate study, we observed increased
transgene expression with time, even after passage 30, in TE671
cells transduced with lentiviral vector carrying a human growth
hormone gene under control of different internal promoters,
CMV-IE and human elongation factor la promoters (Iwakuma and
Chang, unpublished?. Thus, the expression phenotype of a
transgene is strongly influenced by the characteristics of the
transduced cell, the internal promoter as well as the transgene
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516 .
136
itself. We then further modified of the HP/TV vector system
to improve both the safety and the long term efficacy of this
lentiviral vector system.
Example lA: Construction of Attenuated Recombinant HIV-1
Constructs
As described below, several modified HTV-1 constructs
which exhibit reduced cytopathic effects in tissue culture were
chosen for use in the development of the present invention.
HIV-1 LTR Mutants. Investigation of virus attenuation
was essential to the understanding of viral pathogenesis, the
development of preventive vaccines, and development of a safe
lentiviral vector system. For production of a safe HIV vector,
attenuated mutant molecular constructs of HIV-1 were viewed as
better starting materials than wild-type constructs.
One approach to developing these attenuated constructs
was establishing mutations in the LTRs of HIV-1. For example,
the function of HIV-1 LTR enhancer/promoter elements has been
studied using recombinant LTRs containing heterologous
enhancer/promoters (See, Figure 1). After deleting the
regulatory elements including the NF-kB, Splbinding sites,
and/or the TATA box, and inserting a minimal cytomegalovirus
enhancer element, delayed replication kinetics has been
observed in some CD4+ human lymphoid cell lines (See e.g., L.-
J. Chang et al., J Virol., 67:743-752 [1993]) . However, these
LTR mutations do not severely affect the replication of the
full-length HIV-1 constructs in tissue culture. Although NF-kB
and Spl binding sites in the HIV-1 LTR are not absolutely
required for viral replication and pathogenicity in vivo, a
correlation of LTR mutations with low viral load and prolonged
asymptomatic state has been observed for isolates of long term
survivors of HIV-1 infection.
It was also found that several LTR deletion mutants
containing a cytomegalovirus enhancer element were capable of
attenuating HIV-1 (i.e., the mutants were capable of infecting
human lymphocytes with reduced cytopathic effects when the tat
gene also was deleted) . Instead of killing the entire culture,
infection with these LT1~ and tat mutants led to rapid cell
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516 .
137
recovery and establishment of persistent infection. The
replication efficiency was not markedly affected by these
mutations. By mutating the tat gene, it was also found that
the recombinant LTRs (CMV-IE-HIV-LTR) exhibited increased basal
levels of promoter activity which could support virus
replication without Tat (L.-J. Chang, and C. Zhang, Virol.,
211:157-169 [1995]; and D. Robinson et al.; Gene Ther.,
2:269-278 [1995]). These different HIV-1 mutant constructs
were useful for the development of lentiviral vectors.
Replication-Competent tat-Minus Mutants. LTR mutants
with kB/Spl or Spl deletion and CMV-IE enhancer/promoter
insertion have been shown to replicate with delayed kinetics
in human lymphocyte culture, including primary PBLs (peripheral
blood lymphocytes) and macrophages (L.-J. Chang et al., J
Virol., 67:743-752 [1993]; and L.-J. Chang and C. Zhang,
Virol., 211:157-169 [1995]). As they still exhibit cytopathic
effects in culture and thus may be pathogenic in vivo, these
constructs are not safe for vaccine use in the present form.
The tat gene was also a target, as it is a gene that is
essential for efficient HIV-1 replication. HIV-1 Tat has been
implicated in the induction of Kaposi's sarcoma, repression of
MHC Class I gene promoter, induction of functional
unresponsiveness of T cells, modulation of monocyte function,
induction of IL-10 expression, potenciating TNF-induced NF-kB
activation and cytotoxicity, and sensitizing T cells to
Fas-mediated apoptosis (L.-J. Chang et al., J Virol.,
67:743-752; N. Chirmule et al., J Virol. 69:492-498 [1995]; B.
Ensoli et al., Nature. 371:674-680 [1994]; T.K. Howcroft et
al., Science. 260:1320-1322 [1993]; R.M. Lafrenie et al., J.
Immunol., 156:1638-1645 [1996]; M.O. Westendorp et al., Nature
375:497-500 [1995]; and M.O. Westendorp et al., EMBO J.,
14:546-554 [1995]). To examine whether Tat could be
dispensable during HIV-1 replication, a series of tat mutants
(two stop-codon mutants, tat-A & B, and a deletion mutant
tat-C) were investigated (See, Figure 13A) . In Figure 13A, the
dashes (i.e., ----) indicate bases that are shared with the
wild-type sequence, while slashes, (i.e., ////) indicate bases
that are deleted in the mutant sequence, but are present in the
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
138
wild-type sequence.
Mutant constructs containing both LTR and tat mutations
were established. These LTR/tat double mutants were generated
using the LTR mutant constructs which exhibited enhanced
transcriptional activity after inserting heterologous enhancer
elements. The recombinant LTR (CMV-IE-HIV-LTR), which has been
shown to exhibit increased basal level of promoter activity,
can support HIV-1 replication without Tat (L.-J. Chang and C.
Zhang, Virol., 211:157-169 [1995]; D. Robinson et al., Gene
Therap., 2:269-278 [1995]).
During the development of the present invention, it was
determined that the tat-C mutant is more defective than the
tat-A and -B mutants, and the dl.Sp1/CMV tat-B double mutant
is more defective than the dl.Sp1/CMV LTR mutant or the
dl.Sp1/CMV tat-A double mutant reported previously (L.-J.
Chang and C. Zhang, Virol., 211:157-169 [1995]). The
dl.Sp1/CMV tat-B double mutant infects human lymphoid cell
lines with delayed kinetics and exhibited reduced cytopathic
effects.
In addition, this double mutant HIV-1 infected primary
human PBLs poorly and replicated in primary macrophage culture
with reduced kinetics. Based on these results, these already
attenuated HIV-1 constructs, dl.Spl/CMV tat-B and dl.Sp1/CMV
tat-C, were chosen for HIV vector development.
Attenuated LTR/tat Double Mutants. The phenotypes of
the LTR/tat mutants were further characterized in human
lymphoid cell culture. The tat-A or tat-B LTR double mutants
(Spl deleted and CMV-IE enhancer inserted) infected human MT4
cells with slightly reduced cytopathic effects. Further, these
mutants exhibited delayed replication kinetics when compared
with wild-type HIV-1. On the other hand, when cells were
infected with the tat-C LTk mutant (Spl/CMV mutant), the
cytopathic effect was not so apparent and interestingly, the
infected culture recovered rapidly and a persistent infection
was established (See, "C1" and "C2, " in Figure 2 and "chrl" and
chr2" in Table 1. In this table, "chr." indicates chronic
infection, while the 1 and 2 indicate that the experiment was
repeated twice, i.e., the "1" refers to the results of the
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516 .
139
first experiment, and the "2" refers to the results of the
second experiment?. In this table, the first column lists the
cell line used and the virus used to infect the cells. For
example, "MT4/mock" means that MT4 cells were tested without
infection with HIV-1 virus (i.e., it was a control). °WT"
refers to wild-type virus.
Immunofluorescent staining of cells in the persistent "
culture using an HIV-1 patient's sera showed that every cell
was infected. Continuous output of attenuated infectious virus
from these cultures was illustrated by a titration assay on CD4
HeLa cells, and the virus particles were visualized by electron
microscopy (TEM and SEM). The persistently infected culture
produced large quantities of fully assembled HIV particles.
Virions produced from these high producer cells are tat-minus
and exhibit greatly diminished infectivity. No cytopathic
effect has been observed when they were further passed onto
human lymphocyte cultures. Interestingly, some cultures
recovered from wild-type HIV-1 infection after long term
passage also became persistently infected (See, Table 1, AA2/WT
[chr.] 'and Molt3/WT [chr.]). It is possible that the latter
persistent cultures were survivors of mutant HIV-1 infection
(e. g., vpr-minus).
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
140 '
Table 1. Viability and Doubling Time of
Tat +/- HIV-1 Infected Cultures
Doubling Time ( 2
Cell Line/Virus % Viability ( 50)
hrs)
MT4/(mock) 88 40
MT4/WT (acute) 0 -a
MT4 / to t-A
(dl.Spl/CMV) 0
MT4 / to t -B
(D1 . Spl/CMV) 0
MT4/tat-C (chr.l} 97 35
MT4/tat-C (chr.2} 86 32
AA2/WT (chr.) 73 n.d.b
Molt3/WT (chr.) 80 n.d.
a"-," No survivors; bn.d., not determined.
HIV-1 LTR/tat/nef Triple Mutants. Prolonged
asymptomatic survival of macaques infected with a nef-deleted
SIV strain SIVmac239 suggested that the nef gene is a
pathogenesis factor (H.W. Kestler et al., Cell 65:651-662
[1991]). Evidence to strongly support this suggestion came
from studies of a cohort of long term survivors infected with
HIV-1 through blood transfusion from a single donor in
Australia. All the survivors were found to carry HIV-1 strains
with multiple deletions in nef and in the U3 region of the 3'
LTR (N.J. Deacon et al., Science 270:988-991 [1995]).
The LTR/tat-minus HIV-1 constructs were further modified
by mutating the nef gene. To generate nef mutations, site
specific mutagenesis was performed in the nef ORF to destroy
its initiation codon, and a HindIII restriction site was
generated (-AAGCTT-, nef-A mutant). Also, an additional stop
codon was inserted in the nef ORF upstream of the polypurine
tract (PPT) in the nef-A mutant, to generate a more defective
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516 .
141
nef-minus mutant (nef-B mutant, see below). The nucleotide
sequence of pNL4-3 (HIV-1) from 9001 to 9031 (WT) was 5'-
CTCAGGTACCTTTAAGACCAATGACTTACAA-3' (SEQ ID N0:2), while the
nef-B mutant sequence generated by site-specific mutagenesis
was 5'-CTCAGGTACCTTTAAGACTCTAGATCTAGAA-3' (SEQ ID N0:3).
Figure 13B provides a schematic showing a portion of the wild-
type HIV-1 sequence, as well as the nef-B mutations (Figure ~~
13B; wild-type sequence provided in SEQ ID NOS:5 and 6). The
nef-A mutations are also shown in this Figure 13B. As
indicated in this Figure, the nef-A and nef-B mutations contain
the same mutations in the sequence shown starting at base 8781
(i . e. , SEQ ID NO: 5 corresponds the the nef-A sequence and nef-B
sequence for this stretch of bases) . The nef-A sequence is the
same as the wild-type sequence for the sequence shown starting
i5 at base 9001 (i.e., SEQ ID N0:6 represents the sequences for
both wild-type and nef-A).
Since it is the non-syncytiurn-inducing, rather than the
syncytium-inducing isolates of HIV-1 that are preferentially
transmitted during primary infection, the T cell-tropic env
gene of the LTR/tat/nef mutant was also substituted with a
macrophage-tropic env (HIVADA). A schematic diagram of these
HIV-1 mutants is shown in Figure 3. These infectious molecular
clones are further modified and attenuated by mutating other
accessory genes including vpr, vif and vpu, as well as the U3
transcriptional regulatory elements NF-AT, NRT-1, USF and
TCF-la. A safe HIV-1 vector construct is developed from these
attenuated HIV-1 LTR/tat/nef mutant constructs with a total
deletion of U3 except for the att site.
Additional packaging and transducing vectors derived from
mutant HIV-1 LTR, tat and nef constructs established during the
development of the present invention were generated and tested
for vector function.
Based on the results of experiments with the HIV-1
vectors, HIV-2 and SIV vectors will be constructed using two
molecular clones, HIV-2ROD and SIVmac.
Continued experiments will establish an inducible
packaging cell line using the tetracycline (TET-OFF) inducible
system.
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516 .
142
Example 1B: Replication-Competent HIV-1 Vectors Carrying
Heterologous Foreign Genes
Earlier reports of HIV-1 vector systems demonstrated
difficulties in generating high vector titers. This was likely
due to multiple modifications in the viral genome during vector
construction and the lack of a full understanding of the ,
packaging mechanisms of HIV-1. In addition, vector titers are
often construct-dependent. To analyze the ability of HIV-1
vectors carrying heterologous genes to express them at high
levels, several ~~replication-competent" HIV-1 vectors
containing different foreign genes which were inserted in the
nef open reading frame (ORF) in the 3' end of the viral genome
were constructed.
The nef gene has been shown to play an important role in
viral pathogenesis (Z. Du et al., Cell 82:665-674 [1995]; B.D.
Jamieson et al., J. Virol., 68:3478-3485 [1994]). Thus, it was
considered to be safer to delete the nef allele from the
lentiviral vector system to produce useful vectors. Since the
nef gene of HIV is dispensable for viral replication in tissue
culture, and since the nef ORF does not overlap with other
genes, a foreign gene can be inserted into the nef ORF without
inactivating the virus.
Figure 4 shows a comparison of the replication
efficiencies of recombinant HIV-1 constructs carrying
heterologous foreign genes. In these experiments, TE671 cells
were transfected with plasmid DNA; 48 hours later, culture
supernatants were used for the in vitro RT (reverse
transcriptase) assay. Virus titer (i.e., transduction
efficiency) was determined by infecting CD4 HeLa MAGI cells,
and blue cell foci were counted under an inverted microscope
after X-gal staining. The MAGI cells carry an integrated
LTR-lacZ gene which can be transactivated by transduced HIV-1
Tat (J. Kimpton and M. Emerman, J Virol., 66:2232-2239.30
[1992]). The two scales in this Figure are numerically
identical.
In addition, reporter genes including human T cell
receptor CD8, T cell costimulator B7-2 (B70), the bacterial
hygromycin-B-phosphotransferase (hyg),
CA 02333481 2000-11-27
WO 00/00600 PCTlUS99/11516 .
143
neomycin-phosphotransferase (neo), xanthine-guanine
phosphoribosyltransferase (gpt), puromycin-resistant gene, and
histidinol dehydrogenase (hisD) with or without an internal
promoter (SV40) were inserted into the nef ORF at the new
HindIII site or a downstream XhoI site in the nef-A mutant.
These heterologous HIV-1 vector constructs were assessed by
transfecting human TE671 cells, and quantitatively measuring ~~
viral RT expression and transduction efficiencies on a human
CD4 cell line. Transduction efficiency was determined by
counting the blue nucleated cell foci after X-gal staining.
Two independent transfections were done. Representative
results are shown in Figure 4 (the standard deviation is not
shown). An insertion of up to 1.5 kb of nucleotide sequences,
such as B70 and SV-his, seemed to have no effect on RT
production. Furthermore, the infectivity of HIV-SVhis is as
high as wild-type HIV-1.
However, it was surprising to find that the nef-B
mutation appeared to have an adverse effect on RT production
(See, nefB tested in duplicate, Figure 4). The cause of this
adverse effect is unclear ( i . e. , it may have been caused by
interference with packaging or reverse transcription of the RNA
genome), although an understanding of this mechanism is not
required in order to use the present invention. Several
vectors derived from the nef-B mutant construct showed the game
deficiency and thus were reconstructed. A good correlation
between RT activity and virus titer was observed in this study,
except for pHP-1, which is a packaging vector construct lacking
the HIV-1 packaging signals (see below).
These early experiments led to some embodiments of the
methods of the present invention for manipulation of the HIV-1
genome for gene expression. For example, it appeared that
HIV-1 can sustain extensive changes in the enhancer and
promoter region. Indeed, the replacement of the entire U3,
except for att, can be tolerated. Partial substitution of the
intron region for the regulatory genes ( tat and rev) in the env
ORF with foreign sequences can affect the splicing efficiency
of the singly-spliced messages, although the nearest splice
acceptor site is almost 1 kb away ( See e. g. , B . A. Amendt et
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
144
al., Mol. Cell. Biol., 14:3960-3970 [1994]). These results
suggested that: 1) a modified LTR with reduced homology to
wild-type HIV-1 could be used in the vector design; and 2)
deletion of the env sequence might interfere with expression
of the tat and rev regulatory genes. In HIV-1 vector system,
the env gene function may be deleted and replaced by the VSV-G ,
envelope gene. As indicated herein, in some cases, it may be
desirable to provide additional tat and rev functions for
efficient Gag-Pol synthesis. Although an understanding of the
mechanisms) involved is not necessary in order to use the
present invention, the study of heterologous replication
competent HIV-1 constructs indicated that insertion of foreign
sequences in the nef ORF is well tolerated and has minimal
effects on viral replication. These advantages led to the
development of various embodiments of the lentiviral vector
systems of the present invention.
Example 1C: Construction of HIV-1 Packaging Vectors
In this Example, HIV-1 packaging and transducing vectors
were constructed. Two packaging plasmids, "pHP-1" and
"pHP-VSVG," containing HIV-1 env and VSV-G envelope gene
respectively, were constructed. Figure 7 is a structural
diagram of seven different pHP vector constructs, including
pHP-1 and pHP-VSVG.
In this Example, attenuated HIV-1 constructs were
modified to produce the "pHP-1" expression vector capable of
synthesizing all viral structural proteins, but lacking the
packaging signal function. This vector included a strong
promoter (in preferred embodiments, it is preferably not a
native HIV-1 LTR), the gag-pot gene, the RRE element, the tat,
and the rev gene. The RRE-Rev interaction is of great
importance to the efficient synthesis of the Gag-Poi protein.
This dependency may be compensated for if the INS' s are deleted
and RRE is replaced by a surrogate regulatory element such as
the CTE of the Mason-Pfizer monkey virus.
Two approaches to designing the vectors were considered,
namely 1) dissecting down the wild-type genome while carefully
monitoring vector titers following each modification step, and
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516 .
145
2) starting with an over-simplified, inefficient vector
construct and building back to restore wild-type function
gradually. The goal was to achieve the best efficiency of
vector production yet have the vector remain
replication-defective to minimize the chance of generating a
replication-competent recombinant HIV-1 (RC-HIV). To achieve
this, the expression construct pHP-1, which contained a
modified 5' HIV-1 LTR, a novel major splice donor site derived
from RSV, the entire gag-pol-env, vif, vpr, vpu, tat, and rev
genes, a selectable gpt marker gene, and an SV40
polyadenylation signal as shown in Figure 5 was developed.
The wild type HIV-1 genome contains genetic elements in
the 5' to 3' order:
5'LTR(TJ3RU5)-PBS-Psi-SD-gag-pol-vif-vpr-tat-rev-vpu-env-nef-
PPT-3'LTR (U3RU5),
and the pHP construct contains from 5' to 3'~
a chimeric CMV-TAR promoter sequence-gag-pol-tat-rev-PPT-SV40
polyA signal.
pHP-1 lacks the native HIV-1 U3 TATA box, the primer binding
site (PBS), poiypurine tract (PPT), 3' LTR and most oz the
untranslated 5' leader sequences including the conventional
retroviral packaging signal (~) and the major HIV-1 splice
donor (SD) site. pHP-1 contains all HIV structural and
accessory genes except for the nef gene and thus is capable of
expressing the vast majority of the viral proteins, and also
contains the bacterial gpt gene. pHP-1 provides a provirus
capable of mimicking HIV-1 infection in terms of the viral
proteins expressed yet this virus cannot be packaged into viral
particles.
Further mutations introduced by derivatives of the pHP-I
provirus, including deletion in the env and in the 5' LTR, vpr,
vif, and vpu, greatly reduce the possibility that wild-type HIV
will be produced by recombination. Thus, pHP-1 and its
derivatives provide excellent HIV packaging vectors. Examples
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516 .
146
of th pHP-derived packaging vectors include: pHP-dl.Vpr, pHP-
Vpr/ala/leu. PHP-dl.env/Vpu I, and pHP-dl.env/Vpu II.
pHP-1 was constructed as follows. First, the
Tat-responsive enhancer promoter CMV-TATA-TAR fragment
(approximately 400 bp} was isolated from dl.kB/Spl-CMV-TATA-TAR
HIV (Chang et al., J. Virol. 67:743 [1993]) by BhrpI-HindIII "
digestion, and cloned into EcoRV-BamHI digested pSP72 (Promega,
Madison WI, USA) via a linker providing HindIII and BamHI
cohesive sites which contains a modified gag AUG with Kozak
translation initiation context (-CCACCATG-) and a major splice
donor site of derived from Rous sarcoma virus and containing
a mutated RSV gag AUG. See Chang, et al., J. Virol. 53:969-72
(1985). This linker was formed by annealing the following
oligonucleotides:
5'-AGC TTG GTC GCC CGG TGG ATC AAG ACC GGT AGC CGT CAT AAA GGT
GAT TTC GTC G-3' (SEQ ID N0:9) and
5'-GAT CCG ACG AAA TCA CCT TTA TGA CGG CTA CCG GTC TTG ATC CAC
CGG GCG ACC A-3' (SEQ ID NO:10).
This first subclone was called pSP-CMV-TAR-SD.
Secondly, the gag coding sequence for the pHP-1 construct
was obtained by PCR from pNL4-3 (a full-length HIV-1 piasmid)
using a 5' primer
(5'-CGG GAT CCA CCA TGG GTG CGA GAG CGT C-3' [SEQ ID NO:11])
and a 3' primer downstream of the SphI site in the gag gene
(5'-ATC CTA TTT GTT CCT GAA GG-3' [SEQ ID N0:12]).
The PCR product was digested with BamHI-SphI (.-660 bp) and this
fragment was ligated with BamHI-SphI digested chimeric
pSP-CMV-TAR-SD to obtain pSP-CMV-TAR-SD-dl. gag.
Next, a poly-A minus subclone of pHP-dl.pA was
constructed by ligating the following three fragments: a 1112
by HpaI-SphI fragment isolated from pSP-CMV-TAR-SD-dl. gag
(contains the promoter-TAR-SD-dl. gag), a 7922 by SphI-XhoI
fragment (dl.gag-pol-env-gpt) of pNLgpt, and a plasmid vector
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
147
backbone provided by EcoRV-XhoI digested pBS-KS(-) (Stratagene,
LaJolla, CA, USA) . pNLgpt was generated by cloning the E. coli
xanthine-guanine phosphoribosyltransferase (gpt) genefrom pMSG
(Pharmacia) into the nef open reading frame (ORF) of pNL4-3
between the nef AUG and the XhoI site.
Lastly, pHP-1 was made by the following ligation: ,
NotI-XhoI (9059 bp) of pHP-dl.pA containing
dl.CMV-TATA-TAR-SD-gag-pol-env-gpt, a 422 by poly-A site from
XhoI-PstI digested pREP9 (Invitrogen), and NotI-PstI digested
pBS-KS(-). The sequence of pHP-1 (12,479 kb) is provided in
SEQ ID N0:13; this sequence begins at the promoter of the half-
BbrPI site from pNL4-3 (an HIV clone available from the AIDS
Research and Reference Reagent Program; the sequence of this
recombinant clone is shown in Genbank Accession No. M19921).
Additional mutations of pHP-1 to generate pHP-1d12 and pHPl-
d1.28 are described above (See also, Figure 5).
Several additional HP constructs were also made
("pHP-VSVG," "pHP-CMV," "pHP-EF," "pHP-CMVnTAR/SD,", "pHP-CMV-
EFla-intron", "pHP-dl.Vpr", "pHP-Vpr/ala/leu", "pHP-dl.env/Vpu
I", and "pHP-dl.env/Vpu II"), each with additional changes
( See, Figure 7 ) .
pHP-VSVG was derived from pHP-1, with the HIV-1 env gene
being replaced by the VSV-G gene, and with wild-type vpr and
tat, or the vpr and tat genes mutated by site-specific
mutagenesis.
pHP-CMV was derived from pHP-1 with the promoter being
replaced by the cytomegalovirus immediate early promoter
(CMV-IE) and the tat, rev, env, vpr and vpu deleted.
pHP-CMVnTAR/SD was derived from pHP-CMV, with the TAR and
RSV SD deleted. In other words, this construct lacks any major
SD site.
pHP-CMV-EFla -intron was derived from pHP-CMVnTAR/SD,
with an insertion of the EFla-intron between the promoter and
the Gag AUG.
pHP-EF was derived from pHP-CMVnTAR/SD by replacing the
CMV-IE promoter-enhancer and the synthetic SD site with the
human elongation factor la (EFla )'s promoter and enhancer-
containing intron (the latter being of course proceeded by a
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/1I516
148
splice donor site). The intron-containing EFla has been shown
to be a stronger promoter than the CMV-IE promoter. The TAR
sequence was also deleted. It also contains a poliovirus-
derived internal ribosomal entry site (IRES)and the vpr gene.
The expression of Vpr may increase the vector transduction
efficiency in non-dividing cultures.
These constructs were tested for their expression of HIV-
1 proteins.
Both packaging constructs (i.e., pHP-1 and pHP-VSVG) used
a recombinant CMV/HIV-TAR as promoter and a synthetic major
splice donor site. No sequence homology was observed with the
HIV-I genome between TAR (in the 5' end of the RNA) and the gag
AUG in these two constructs. A BamHI site was generated near
the gag AUG for the purpose of inserting recombinant HIV-2 and
SIV gag-pot sequences in subsequent experiments.
The pHP-VSVG construct with vpr and tat mutations lacks
vpr and tat genes, and the VSV-G gene is substituted for the
env gene exactly at the env AUG by PCR mutagenesis. These two
constructs were the first two packaging plasmids tested.
The construction of these pHP-1 derivatives is described
in greater detail below. The three .pHP-CMV derivatives were
tested, and found to be inefficient in synthesizing HIV
proteins, indicating that the pHP-1, pHP-VSVG and pHP-1 dl
derivatives are the preferred embodiments of the efficient HIV
vector system of the present invention.
pHP-VSVG. This clone was made to delete the HIV-1 env
gene and replace it with the VSV-G gene, as well as delete the
HIV-1 vpr and tat genes. It was constructed by combining the
following four pieces of DNA fragments: 1) the recombinant LTR
(dl.kB/Spl-CMV-TATA-HIV-TAR) gag-pol from NotI to EcoRI
fragment of pHP-1; 2) a fragment from HIV-1 with deletion in
the C-terminal of Vpr and the N-terminal of Tat by PCR using
the following two primers:
5' -TAA GAA TTC TAG TAG GTT GCT TTC ATT GCC-3' (SEQ ID
N0:14), and
3'-CTT CTC CTT CAC TCT CGA GTG ATC ACT GTC TTC TGC TCT
TTC-3' (SEQ ID N0:15),
with the second sequence encompassing the env AUG with a
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
149
new XhoI and a BclI site); 3) the VSV-G gene fragment cut by
SalI-XbaI from pBS-VSV-G (obtained from Tom Hobman of the
University of Alberta); and 4) the 3'-env-gpt-SVpA and plasmid
vector backbone from NheI-NotI digested pHP-1.
The differences between the final pHP-VSVG construct and
the pHP-1 vector are that the region between the 3' end of the
vpr coding region and the 5' end of the tat first exon has been
deleted, and a portion of the HIV-1 gp120 env gene has been
substituted with VSV-G gene in the pHP-VSVG construct.
pHP-CMV. This clone was derived from pHP-1, with the 5'
recombinant LTR replaced by a CMV-IE enhancer-promoter and the
entire env, tat, vpu, rev, vpr, nef deleted, but with the vif
gene remaining intact. This clone was constructed by ligation
of the following 3 pieces of DNA: 1) the vector pcDNA3.IZeo(+)
from Invitrogen cut with Nhel-XhoI; 2) the TAR/SD-gag-pot from
pHP-1 digested with XbaI-EcoRI; and 3) the RRE element from
pBS-RRE digested with EcoRI-XhoI. pBS-RRE was constructed by
ligating BglII (nt. 7611) to HindIII (nt. 8131) of pNL4-3 of
HIV-1 with BglII-HindIII digested pBS-EF. pBS-EF was from the
PCRed EFla enhancer promoter cloned into pBS(-).
pHP-CMV-eTAR/SD: This clone is the same as pHP-CMV
except that the 5' TAR and splice donor site are deleted. This
construction was made by ligating the following two fragments:
1) a 702 by fragment of MIuI-BamHI digested pcDNA3.IZeo(+j
containing the CMV enhancer; and 2) the vector containing
MluI-BamHI digested pHP-CMV which has deleted TAR and contains
the RSV splice donor site.
pHP-CMV-EFla-intron. This clone is similar to
pHP-CMV-n.TAR/SD but with an intron from human EF-la gene
inserted between the CMV promoter and the gag AUG. It was made
by ligating the following three DNA fragments: 1) pHP-1
BamHI-EcoRI zragment containing gag-pol and vif; 2) the
MluI-EcoRI of pcDNAZeonlacZ-RRE containing the vector backbone
of pcDNA3.IZeo(+), HIV-1 RRE and part of the CMV promoter; and
3) the rest of the CMV enhancer promoter was obtained from
BamHI-MluI digested pcDNAZeoHGHP2EF, a pcDNAZeo3.1(+) vector
containing EFla intron and the human growth hormone gene. The
hGH cloned sequence is available from GenBank. See co-pending
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516 .
150
U.S. Patent Appln. Ser. No. 08/848,760 (incorporated by
reference) for additional information regarding this construct.
pHP-1 dl2 and pHP-1 d128: To further mutate pHP-1 for
safety reasons (as discussed below), portions of the env gene
were deleted by Ba131 excision. To generate HIV-1 env
deletions, pHP-1 was digested at the unique restriction enzyme "
site NheI in the env gene, and treated with Ba131 exonuclease
for 1, 2 or 5 minutes. The digested product was self-ligated
after T4 DNA polymerise treatment. The self-ligated plasmid
DNA was then transformed into competent E. coli DHSa and from
a pool of more than 48 deletion mutants, two clones (pHP-1d1.2
and pHP-1d1.28), were selected, sequenced and used in this
study.
pHP-1d1.2 and pHP-ld1.28 have 2 and 28 nucleotide
deletions in the env gene respectively (See, Figure 5). We
have further modified pHP-1 d1.28 to produce various
derivatives.
First, the vpr gene was mutated by site-specific
mutagenesis so it retains the nuclear localization function but
loses its cell cycle arrest function. Specifically, a mutation
was made at amino acid 30, changing from ala to leu, as
described in Mahalingam S, Ayyavoo V, Patel M, Kieber-Emmons
T, Weiner DB.Nuclear import, virion incorporation, and cell
cycle arrest/differentiation are mediated by distinct
functional domains of human immunodeficiency virus type 1 Vpr.
J Virol 1997; 71:6339-47.
The env/vpu was also mutated by site-specific mutagenesis
to delete the env initiation AUG codon and part of the vpu
reading frame.
These mutations were first made individually and then
combined.
Construction of G1 (generation one, deletion of vpu, vif, vpr)
and G2 (deletion of tat, rev) pHP vectors based on pHP-d1.28
(nef deleted and env partially deleted):
1. Construction of env AUG and Vpu deletion mutant
pHP-dl . env/vpu I and pHP-dl . env/vpu I I : the mutant I has a long
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516 .
151
deletion from nt 6I95 at the vpu amino acid codon 45 to nt.
6330 at the 38th amino acid codon of env gene and a stop codon
TAA inserted in the sequence. The primer used was a forward
primer: -GTTAATTGATAGACTAGTCTAATATGGGGTACCTG- .
The mutant II has a small deletion from nt. 6216 at vpu a.a.
codon 52 to nt. 6237 at vpu a.a. codon 59 and env a.a. codon ,
6 which also has a stop codon mutation TAA at the vpu a. al.
codon 50. Note that although these mutations are G1
mutations, they are made into the G2 vector pHP-dl . 28 backbone .
2 . Construction of vpr mutants of pHP : two vpr mutants were
made, one with frameshift mutation which inactivate the entire
vpr function and the other with one amino acid substitution
which inactive the cell cycle arrest function of vpr but not
the nuclear localization function which can assist transduction
of non-dividing cells:
pHP-dl.vpr is a frame-shift mutant which was made by
EcoRI digestion at nt . 5745, near vpr amino acid codon #62, and
resulted in a 4 by insertion which caused a frameshift.
pHP-vpr/ala/leu: this is a point mutation which has
changed alanine to leucine at a.a. #30. This mutation deleted
the cell cycle arrest function but not the nuclear localization
function of vpr as reported before [Zhang, 1997 #3492;
I~iahalingam, 1997 #3791]. The primer used for mutagenesis is:
-CCTAGGAAAATGTCTAACTAGTTCACTCTTAAGTTCCTC-. Note that although
these mutations are G1 mutations, they were made with the G2
vector pHP-d1.28 backbone.
3. Combination of dl.env/vpu I and dl.env/vpuII with
vpr/ala/leu mutations to generate pHP-vpr/env/vpu I and
pHP-vpr/env/vpu II. These two combination mutants were made
by ligating the two designated mutant fragments, SpeI (in gag)
to EcoRI (in vpr) containing the vpr mutation, and EcoRI to
NdeI (in env) containing the env/vpu mutation, into NdeI and
SpeI digested pHP-d1.28.
Note that although these mutations are G1 mutations, they are
made into the G2 vector pHP-d1.28 backbone.
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
152
4. Construction of a G2 vector, pHP-dl.NdeI, which has deletion
of the following genes, vif/vpr/tat/rev/env/vpu : this mutant
was made by deleting nt. 5122 to nt. 6399 via NdeI digestion
of pHP-d1.28, and resulted in a packaging vector with all the
accessory gene functions deleted and the env gene partially
deleted. This Ndel-NdeI deletion also included the 3' splice "
sites SA4, SA5, SA6 and SA7, used for the syntheses of vpr,
tat, rev, vpu and env.
Example 1D: Construction of HIV-1 Transducing Vectors (TV)
Two families of transducing vectors were constructed. In
the pTV~ vectors, the major packaging signal was modified
relative to the source HIV-1 signal. In the pTV~ vectors, the
source major packaging signal was used.
Figure 8 provides a diagram of six HIV-1 transducing
vectors, in which the vector backbone is derived from pNL4-3
and different LTRs. The IRES element shown in this Figure was
derived from poliovirus, which could allow bicistronic gene
expression.
To engineer a packaging signal for the construction of
HIV-1 transducing vectors (TV), an artificial HIV-1~ sequence
using four synthetic oligonucleotides was synthesized, which
comprised sequences between the PBS and the gag AUG (referred
to as "100" or "PAK100") and sequences extending into the gag
ORF (referred to as "140" or "PAK140"). These synthetic
HIV-1~ sequences contained a mutated SD site (three nucleotides
changed in PAK100 and PAK140, GAGTA ->CATTC) and a mutated gag
AUG (HindIII and BamHI sites inserted upstream of gag AUG in
both; PAK100 stopped just upstream of gag AUG; PAK 140 changed
gag AUG to UAG and second codon from GGT to GCC) to avoid
possible adverse effects in gene expression. PAK100 and PAK140
both started at nt 690 of provirus, i.e., 5' base of U3=1. The
synthetic ~ signals were cloned into the pTV~ vector as shown
in Figure 8, which is comprised of two recombinant LTRs
("dl.kB-CMV/HIV-TAR!'), the PBS and 5' leader sequences, an
SV40-driven neo resistance gene, and the 3' PPT.
The packaging efficiencies of pTV~100 and pTV~140 (Figure
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
153
8, constructs 1 and 2) were tested in a co-transfection
experiment . HeLa cells were transfected with pHP-1 and pTV~y100
or pTV~140 and 48 hours later, the culture supernatants were
harvested and used to transduce CD4 HeLa cells (not VSV-G
pseudotyped). 6418 resistant colonies were counted 10 days
later. As a control, HeLa cells were transfected with wild-
type HIV-1 DNA; 48 hours later, the culture supernatant was /~
used to infect CD4 HeLa cells. The titer of the wild-type
HIV-1 was determined by a sensitive immunohistochemical
staining method using anti=Gag p24 mAb as described by Chang
and Zhang (L.-J. Chang and C. Zhang, Virol., 211:157-169
[1995]). Results of this study showed that both pTV~100 and
pTV~140 were packaged at a very low efficiency (approximately
3 logs of magnitude less than the wild-type HIV-1).
This result indicated that additional HIV-1 sequences are
needed to improve the packaging function of pTV~100 and
pTV~140. Therefore, more HIV-1 sequences, including an
additional gag sequence and an RRE element, were cloned into
pTV~140. One such example is shown in Figure 9A
(pTV~+CMV-nlacZ-hyg). Again, the pTV~+ was not packaged
efficiently, indicating the splice donor site and Gag AUG
mutations in pTV~100 and pTV~140 are detrimental to HIV
packaging. While the tested pTV~s cannot be used as efficient
transducing vectors, pTV~s can be efficiently packaged and
transduced, as shown below.
Thus, site-specific mutagenesis was performed to change
1-2 nucleotides in the splice donor site, and the Gag AUG in
pTV~s using primers:
5'-GCGGCGACTGGGGAGGACGCCAA-3' (SEQ ID N0:7) and
5'-GAAGGAGAGAGTTGGGTGCGAG-3' (SEQ ID N0:8),
to generate pTV~SM vectors.
It is desirable to avoid sequence homology between the
packaging construct and the transducing vector construct so as
to reduce the probability of recombination. However,
cotransfection with additional accessory genes such as vpr, nef
and vpu may also help to increase the vector titer and the
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
154 '
transduction efficiency. Inclusion of accessory genes in the
transducing vector does not increase the probability of
recombination, provided that such genes are omitted from the
packaging vectors. The homology between the preferred pHP and
pTV constructs is sufficiently low so that recombination was
not detected in our study. ,
In order to generate a replication-competent HIV-1, the
major SD site, the gag AUG and the env sequences must be
restored, because they are deleted from the modified pHP and
pTV constructs.
In an alternative approach for the construction of an
efficient transducing vector the wild-type genome was gradually
deleted (pTV~). In this embodiment, the two
replication-competent HIV-1 vectors, "HIV-1-SVneo" and
"HIV-1-SVhyg" (See, Figure 4) were used as a starting point.
These two constructs are nef-minus, and exhibited up to 50-70%
of the wild-type HIV-1 replication efficiency. A deletion was
made starting from the middle of the gag ORF to the middle of
the env ORF. This did not delete the RRE element.
First, pTV~SVneo was created by digesting pNL-SVneo with
NheI (with a site located in the middle of the env gene), and
SpeI (with a site in the middle of the gag gene) , and then
self-ligating it to delete the gag-pol-env, and vif, vpu, vpr,
tat, and rev genes. pNLSV neo was created by inserting SVneo
(an SV40 promoter-driven neomycin-resistant gene) at a HindIII
site generated by site-specific mutagenesis, inbetween the nef
AUG and the unique XhoI site in the 5' region of nef in pNL4-3.
pTVOCMVnIacZ was made by digesting pTV~SVneo with XhoI
and Kpnl, which deleted SVneo and part of the nef sequences
near the 5' end of the PPT of HIV-1. The product was then
ligated with a SalI-KpnI fragment containing CMV-nlacZ sequence
from pcDNAzeo-nlacZ. pcDNAzeo-nlacZ was generated by
inserting nlacZ of pSP72nlacZ into pcDNA3.lzeo(+).
The nuclear lacZ gene was generated by fusing a nuclear
localization signal of SV40 large T antigen to the N-terminus
of the bacterial lacZ gene that was obtained from the
pBlueBacHisA vector (Invitrogen) using PCR mutagenesis. The
oligonucleotide primer 5' - CCC GGG TCT AGA AGC TTC CAC CAT GCC
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/I ISI6 .
155
TAA GAA GAA ACG AAA GAT CGA TCC CGT CGT TTT ACA ACG TCG-3',
which contains a favored eukaryotic translation initiation
codon (underlined), was used in the PCR procedure.
The two deletion vectors, "pTV~SVneo" and "pTV~SVhyg,"
(See, Figure 8, constructs 3 and 4) were examined for their
transduction efficiencies in cotransfection experiments.
Three additional pTV~ vectors were also constructed, each
containing a different reporter gene: CMV-GFP (green
fluorescent protein, pTVOCMV-GFP), CMV-nlacZ (pTV~CMV-nlacZ)
and CMV-nlacZ-hyg (pTV~CMV-nlacZ-hyg), as illustrated in Figure
8 (See, Figure 8, constructs 5 and 6, as well as Figure 9B).
pTVnEFGFP was generated by replacing the CMVnlacZ gene of
pTVnCMVnlacZ withthe human elongation factor la promoter of
pHEF kindly provided by D Denny plus a GFP reporter gene.
pTVnEFGFP with nlacZ gene. The VSV-G envelope expression
plasmid pHEF-VSVG was constructed by inserting a PCR amplified
VSV-G fragment containing a favored translational initiation
codon into the EcoRI site of pHEF (kindly provided by D.
Denny). The mutagenesis site and flanking sequences were
confirmed by DNA sequencing.
Example 2
Detection of Synthesis of HIV-1 Proteins by Packaging Cell
Lines
Western Blot analysis:
In this Example, Western analyses of HIV-1 proteins in
HeLa cells transfected with various vector constructs were
tested. In this Example, cell lysates were prepared and
analyzed by Western blotting and compared with a wild-type
HIV-1 construct (pNL4-3), in order to determine the level of
viral proteins synthesized by pHP-1 and pHP-VSVG (with and
without Tat), in comparison with wild-type HIV-1. In this
Example, the Western blots were performed using serum obtained
from an HIV-infected individual, and methods known in the art
(See e.g., Ausubel et a1. (eds.) Short Protocols in Molecular
Biology, 2d ed., John Wiley & Sons, New York, NY [1992], pp.
10.33-10.35).
CA 02333481 2000-11-27
WO 00!00600 PCT/US99/11516 .
156
The results showed that the level of viral proteins
synthesized by pHP-1 was similar to that of the wild-type
pNL4-3. Similar results were obtained when pHP.l.dl constructs
were used.
These results indicated that in the absence of Tat, the
recombinant LTR of pHP-VSVG is inactive. Thus, it is likely ,
that the TAR element in the LTR down-regulates transcriptional
elongation. These results led to the generation of an
inducible packaging cell line using the pHP-VSVG construct as
described in Example 4.
Reverse Transcriptase Activity
RT and vector titer analyses of HP/TV vs. wt HIV-1. To
examine the efficiency of gene expression, we first compared
viral reverse transcriptase (RT) production. HeLa cells were
transfected with either wt HIVNL4-3 , pHP-1, pHP-1d1.2, or pHP-
1d1.28 and the culture supernatant was harvested for RT assay
48 h later. Figure 6 shows that the levels of RT synthesis by
the pHP-1 derived constructs were comparable to those observed
for wt HIV-l; however, the vpr/tat-deleted pHP-VSVG produced
only minute amounts of RT even when co-transfected with tat
and vpr. Transfected HeLa cell lysates were also analyzed to
see if the supernatant RT activity correlated with
intraceliuiar Gag synthesis. The results showed that the
amount of Gag protein produced by pHP-1 was similar to that
produced by wt HIV-1 infected cells or transfected cells.
Interestingly, although tat and/or vpr co-transfection did not
markedly boost RT production of pHP-VSVG, it substantially
increased synthesis of Gag protein as measured by Western blot .
The data for a preliminary run is shown below:
Packaging construct *RT (cpm/~,L) exp.l / exp.2
pNL4-3 12,094 / 9,120
pHP-1 8,400 / 8,678
pHP-1d1.2 7,250 / 4,682
pHP-1d1.28 6,436 / 8,682
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
157 '
*RT, two independent transfection and assay results are shown;
the RT
background activity of 50 cpm/uL was not subtracted.
The expression of Gag-Pol function indicates that tat and rev
are functional. Thus, the artificially engineered splice donor
(SD) site in the pHP-1 construct, which is unrelated to HIV
sequences, works like the wild-type SD site (i.e., allowing
partition of spliced and unspliced mRNAs into the cytoplasm).
The packaging vector pHP-d1.28 expressed RT at 50-90% of
the wild type level, indicating that the mutations in pHP-dl.
did not substantially affect the synthesis and function of
Gag-Pol.
Comments
Analyses of RNA expression and packaging function by pHP-
CMV and pHP-EF were performed in order to compare these
vectors directly with the wild-type HIV-1. These experiments
showed that pHP-CMV and pHP-EF do not express Gag-Pol protiens
at high efficiencies, indicating that the pHP-1-derived vectors
have important viral sequences that are necessary for ef f icient
vector production. pHP-VSVG did not express HIV-1 proteins
unless the Tat transactivating protein is also present. Thus,
although expression of VS'v-G and Gag may be cytotoxic, an
inducible packaging cell line could be established using
pHP-VSVG without a tat plasmid.
PHEF-VSVG: human elongation factor 1 alpha promoter
driven VSV-G envelope and pHEF-A-env: EFla promoter driven
amphotropic MLV envelope, were also constructed in our
laboratory and shown to express high levels of envelope
proteins; better than the CMV-IE promoter driven construct.
It should also be noted that overexpression of Gag-Pol
may not increase the vector titer because earlier studies have
shown that overexpression of Gag-Pol induces protease
activation and prevents virus assembly and budding (V.
Karacostas et al., Virol., 193:661-671 [1993]; J. Park and C.
D. Morrow, J. Virol., 65:5111-5117 [1991]). The present
examples describe vectors that produce measurable amounts of
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
158
Gag-Pol (e.g., pHP-1, pHP-ldel, and pHP-VSVG), as well as
vectors that do not express detectable amounts of Gag-Pol
(e. g., pHP-CMV and its derivatives). The latter require
further mutation to be useful as vectors.
EXAMPLE 3
Requirement for HIV-1 Tat for Efficient Gal-Pol Protein
Processing
In this Example, the requirement of Tat for efficient
Gag-Pol processing and HIV-1 vector production in certain
packaging systems was demonstrated.
1. Western Analysis of Tat' and Tat-
HIV-1 Particles and Infected Cells
Virus pellets ("P") and cell lysates ("L") were prepared
from Tati (tat WT) and Tat- (tat- B and tat-C) virus-infected
cells, and the protein contents were separated by a 10% SDS
protein gel, and detected in Western analysis using AIDS
patient's serum. The signals were amplified using the Amersham
ECL chemiluminescence kit.
The Western analysis showed that the two tat-minus mutant
HIV constructs (tat-B and tat-C) derived from chronically
infected cell lines can replicate at high efficiency and
synthesize viral proteins at wild type efficiency.
2. Gag Processing Deficiency of Tat-minus HIV-1
Demonstrated by Metabolic Labeling of Chronically
Infected Cells
WT or tat-minus HIV-1 chronically infected cultures were
metabolically labeled with 3H-leucine overnight,
immunoprecipitated with pooled HIV patient sera, and analyzed
by SDA-PAGE (10%). The relative ratio of Gag p55:p24 is shown
at the bottom. Processing of the envelope gp160 to gp120 was
not significantly different between different samples. The'H-
labeled protein bands were quantified using a phosphoimager
(BAS1000).
We found that HIV polyprotein processing requires Tat
function. In the absence of Tat, there is a high GaG p55/p24
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516 .
159
ratio compared with wild type.
3. Cells Chronically Infected with Tat-minus HIV-1 are
Deficient in Gag Protein Processing Demonstrated by
Pulse-Chase Metabolic Labeling
The same number of viable cells (3 x 106) was used in
each lane of a 10% SDS-PAGE gel system. Cells were labeled
with medium containing bands for HIV-1 Env gp120, and Gag p55
and p24 . A Fuj i phosphoimager was used for quantitation of Gag
p55 and p24 of WT-infected MT4 and tat-C chronic high producer.
In Figure 10, the resultant decrease of p55 and increase of p24
(p55, p24 / pulse-labeled p55) with time (P, 2, 4, 6, 8 hours)
were shown and plotted. In Figure 10, the solid curves
demonstrate efficient processing of p55 of HIV~,~_3 with steady
increase of p24 and decrease of p55; the dashed curves
demonstrate that the amounts of p55 and p24 are not
significantly changed with time in the tat-C high producer
cells, indicating a deficiency in Gag processing.
4. Tat Enhances Gag Processing in HeLa cells
HeLa cells were transfected with plasmid DNA encoding
HIV-1 Gag, Rev, Tat, HTLV Tax/Rex, SIV Tat, or HIV Tat exon 1.
The results clearly demonstrate that Tat enhances p55 to p24
Gag processing. The effect of Tat on Gag processing is TAR
independent as GagTAR- construct which has TAR deletion is also
sensitive to this Tat effect. This function of Tat resides in
the exon 1 which can be partially restored by SIVTat and HTLV
Tax/Rex.
5. Tat Enhances Gag Processing From the pHP-VSV-G
Packaging Construct
TE671 cells were transfected with plasmids as described
above. Cell lysates were harvested 24 hours after DNA removal
and analyzed by SDS-PAGE and Western blotting as described
using anti-p24 MAb. The result indicated that Gag processing
is enhanced by the presence of Tat.
EXAMPLE 4
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
160
Generation of an Inducible Packaging Cell Line
In this Example, an inducible packaging cell line was
generated using the pHP-VSVG, and its derivative construct.
First, pHP-VSVG was linearized and transfected into human TE671
cells by electroporation, together with a selective marker.
After selection, individual cell clones were tested for Gag-Pol "
expression by direct extracellular RT assay in the presence or
absence of a transfected tat plasmid. The expression of VSV-G
protein was detected by immunohistochemical staining.
Briefly, the pHP-VSVG linearized by digestion with NotI,
and transfected into the TE671 cells along with pSV2-neo ( i . e. ,
with G-418 as the selectable marker). Transfection was
accomplished by electroporation, using methods known in the
art.' Transfected cells were grown in 1 mg/ml of 6418 culture
in DMEM containing loo FBS. The induced gag-poI Gag-Pol
expression was then determined by direct extracellular RT
assays with and without transfected tat plasmid. HIV-1 Gag and
RT expression were detected by p24 antigen ELISA or RT {See,
co-pending U.S. Patent Appln. Ser. Nos. 08/791,994 and
08/838,702; See also, L.J. Chang and C. Zhang, Virol.,
211:157-169 [1995]; and L.J. Chang et al., J Virol., 67:743-752
[1993] ) .
The expression of Gag-Pol in this inducible cell line
still requires Tat function. To make a user-friendly packaging
cell line, vpr and tat genes can also be expressed by an
inducible promoter. The vpr gene is included because of its
function in promoting transduction of nondividing cells. Vpr
is a virion-associated protein, and the vpr gene is therefore
assigned to the packaging vector so that equivalents of Vpr,
like those of Gag, Pol and Env, are produced only in the
packaging cell line. A tetracycline-inducible expression vector
(a TET-OFF system, suppression of expression in the presence
of tetracycline or doxycycline) has been chosen for this
purpose. An inducible tat-vpr expression vector has been
constructed into the pcDNA3.1/Zeo plasmid with genes arranged
in the following order:
-tetOP-tat-IRES-vpr-IRES-tetR.VPl6-SVpA-(inverted tk-zeo-pA).
Preliminary studies of this construct showed co-expression of
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/I1516 .
161
Tat and Vpr in the absence of tetracycline or doxycycline,
indicating that the two internal ribosomal entry site (IRES)
are functional. However, even in the presence of tetracycline
or doxycycline, this inducible construct still expresses Tat
function, indicating a leaky expression of the tetR.VPl6. As
a result, this construct was only used for coexpression of Tat,
and Vpr in the co-transfection experiments.
A second construct, -tetOP-tat-P2-vpr-SVpA-(inverted
tk-zeo-pA), which is up-regulated by a separate tetR.VPl6
expression plasmid, has been constructed and used to generate
an inducible cell line.
tetOP-tat-P2-vpr-P2-tetR-VP16-SVpA-(inverted tk-zeo-pA) is a
clone that expresses HIV-1 Tat and Vpr and the tet tTA operon
inducer tetR-VP16 which was made by ligation of the following
fragments: tetOP, HIV-1 Tat, internal ribosomal entry site
(IRES) P2, HIV-1 Vpr, IRES P2, tetR-VP16, and the vector pREP9
with EBNA1 gene sequence deleted. The two tTA plasmids were
obtainable from Display Systems Biotechnology, Inc. (now
distributed by Clontech). This clone is auto-inducible by the
removal of tetracycline or doxycycline (2-10 microgram/ml) from
the culture media (a Tet-OFF system)(See, M. Gossen and H.
Bujard, Proc. Natl. Acad. Sci. USA. 89:5547-5551 [1992]).
As these plasmids use different selective markers (neo,
zeo, and nyg) it was possible Lo co-select Lhem in Lhe same
cell. However, a large number of cell clones had to be
screened before a stable inducible packaging cell line could
be established.
EXAMPLE 5
Internal CMV-IE in pT~T~CMVnlacZ Promoter Exhibits Higher
Promoter Activity Than Native CMV-IE
In this Example, the expression of the reporter lacZ gene
from the pTV-~CMVnIacZ was compared with pcDNAnlacZ (i. e. , CMV-
IE promoter-driven), 48 hours after transfection of TE671
cells. TE671 cells were transfected with 5 ~.g of pcDNA3-nlacZ
or pTV~CMVnlacZ, as described above. Following transfection
and growth, cells were fixed and stained for ,Q-galactosidase
actvity, as described below.
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516 .
162
The beta-galactosidase activity was detected by the
following protocol as published by Kimpton and Emerman (J.
Kimpton and M. Emerman, J Virol., 66:2232-2239 [1992]).
Briefly, cells were fixed in culture plate at room temperature,
with 1% formaldehyde (1.33 ml of 37.6% for final 50 ml) and
-0.2% glutaraldehyde (0.4 ml of 25% for final 50 ml) in PBS for
5 minutes. The cells were then washed three times with PBS,
and incubated with 500 ~l ddHzO containing 4 mM potassium
ferrocyanide (100 ~,l of 0.4 M for final 10 ml), 4 mM potassium
ferricyanide (100 ~.l of 0.4 M) , 2 mM MgCl2 (20 /C1 of 1 M) , 0.4
mg/ml X-Gal (200 ~.1 of 20mg/ml) at 37°C for SO minutes to
several hours. The blue-staining (i.e., ~i-galactosidase
positive) cells were counted under an inverted microscope.
These results indicated greater expression by the pTV~CMVnlacZ
vector, as compared with the pcDNA3-nlacZ. Table 2 shows the
results, with more "+" indicating the presence of a relatively
greater number of blue-staining cells.
Table 2. ~3-Galactosidase Activity
Plasmid Cells Stained (Blue)
Mock -
pcDNAnlacZ ++
pTV~CMVnIacZ +++
EXAMPLE 6
Production Efficiency of Transducing Vectors
As safety is a major concern with HIV-derived vectors,
the HP/TV vector system has been developed to minimize the
possibility of homologous recombination and RCV production.
To examine vector efficacy and RCV production, human
rhabdomyosarcoma TE671 cells were co-transfected with one of
the pHP constructs, pHP-1 or pHP-VSVG, the envelope expression
plasmid pHEF-VSVG, and one of the three pTV constructs,
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
163
pTV~SVneo, pTVOSVhyg, or pTVOCMV-nlacZ (Fig. 7). Since tat was
deleted in pHP-VSVG, a tat plasmid was cotransfected with pHP-
VSVG. The resulting vector titer was determined by titration
on TE671 cells using transfected culture supernatants. The
reporter genes, neo, hyg, and lacZ, were assayed by selecting
resistant cell colonies using either 6418, hygromycin B, or by
colorimetric staining for ,Q-galactosidase activity,
respectively. For the detection of RCV, the transfected TE671
cells were co-cultured with MT4 cells, one of the most
susceptible cell lines to HIV-1NL4-3 infection. After co-
cultivation, MT4 cells were monitored for syncytium formation
and culture supernatant was subjected to HIV-1 reverse
transcriptase (RT) assay. To increase the sensitivity of
detecting RCV, fresh MT4 cells were added to the transfected
culture weekly, and one month after co-culture, MT4 cells were
fixed and immunostained using anti-HIV antibodies as described
in the Material and Method. Results of this experiment are
summarized in Table 2A. HP/TV co-transfection successfully
produced viral vectors from all three pTV reporter constructs
(SV-neo, SV-hyg and CMV-nlacZ) suggesting that the transducing
vectors could be packaged into virus particles. However, RCV
was detected in all transfected cultures except for pHP-VSVG.
To generate RCV from co-transfections of pHP-1 and pTV~, at
least two homologous genetic cross-overs must occur, one in gag
and the other in env. These results demonstrate that
recombinant RCVS could arise from co-transfection of these
vectors and that the co-culture and immunostaining RCV assay
provide a sensitive means for detecting RCVs.
CA 02333481 2000-11-27
WO 00/00600 PCT/US99111516
164
Table 2A. HP/TV vector transduction and RCV assays.
pHP pTV Additional Genes Reporter* RCV*
pHP-1 pTVOSVneo VSV-G + +
pTVOSVhyg VSV-G + +
pTVOCMV-nlacZ VSV-G + +
pHP-VSVG pTVnCMV-nlacz VSV-G, tat + -
"Reporter genes neon or hyg= of pTVO were assayed by infecting HeLa cells
with 50 ul and 100 ul of culture supernatant followed with 6418 or
hygromycin B selection 24 hr after transduction, or for nlacZ reporter
gene, by infecting HeLa cells with 20, 50 and 100 ul of culture
supernatant and stained with X-gal 48 hr .later as described in the
Materials and Methods. pHP-1/pTV co-transfection produced >10'/ml
vector and pHP-VSVG/pTV produced <102/ml vector; however, the titer was
not precisely determined due to the generation of RCV.
*Replication-competent virus (RCV), assayed by co-culturing with MT4
cells for up to 30 days; RCV was detected by monitoring cytopathic
effects on MT4 cells, RT and MAGI assays.
Example 6A: Production Efficiency of VSV-G and pHEF-VSVG
Pseudo typed TVe and TV~Y Vectors
Ex. 6A.1
In this experiment, VSV-G pseudotyped vectors were
produced and the target cells were CD4-minus human cell
lines. pHP-VSVG (which is both a packaging and a
pseudotyping vector) was co-transfected with a pTV~ plasmid
and a tat plasmid (pCEP4tat) into TE671 cells. Culture
supernatant was harvested 48 hours later. Tat was included
to transactivate both pHP-VSVG and pTV~. The production of
virus was confirmed by RT assay, and expression of HIV-1
p24 and VSV-G was confirmed by immunohistochemical
staining. Virus produced from the transfected cells were
harvested without further concentration, and used to infect
TE671 cells. After selection with either 6418 or
hygromycin for 7-10 days, cell colonies were counted under
an inverted microscope.
Culture supernatants were harvested 24 hours after
removal of transfection solution. HIV RT activity was
detected by an in vitro RT assay and vector titers were
determined by transduction and beta-galactosidase assay of
TE671 cells 48 hours later. The VSV-G pseudotyped
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516 .
165
pTV~SVneo and pTVOSVhyg both produced transducing titers up
to 103/ml without further concentration. This titer was
increased to 105/ml without concentration, when pHP-d1.2 or
pHP-d1.28 were co-transfected with pHEP-VSV-G. This result
indicated that pHP-VSVG does not function efficiently.
Ex. 6A.2
Therefore, we adopted a different (pure) pseudotyping
vector, pHEF-VSVG. This is just a eukaryotic expression
vector, with the VSVG gene under human EF1-alpha promoter
control. It is not derived from a lentivirus.
To further compare vector production efficiencies of
pHP and wt HIV-1, TE671 or HeLa cells were co-transfected
with a packaging vector (pHP-1, pHP-ldl.2 or wt pNL4-3), a
transducing vector pTV~CMVnlacZ, and the envelope construct
pHEF-VSVG. The results are summarized in Table 3. The pHP
packaging vectors produced RT at 30-40% of the wt level in
TE671 cells but produced near wt level in HeLa cells.
Interestingly, the vector titer assay indicated that HP/TV
co-transfections using either pHP-1 or pHP-1d1.2 produced
2-3 times more transducing vector than the wt HIVNL4-3/TV
co-transfection. Further, the results obtained from TE671
transfections confirmed this result (Table 3A). Packaging
constructs pHP-1, pHP-ldi.2 and pHP-ldi.28 produced vector
titers 3-7 times higher than the wt construct HIV-1NL4-3 ~ No
RCV was detected from co-transfection of pTV with either
pHP-1d1.2 or pHP-1d1.28 using our RCV assay.
Table 3 also showns results for experiments with
transducing vector pTV~CMV-nlacZ-hyg-dl.SmaI. They showed
very low titers.
CA 02333481 2000-11-27
WO 00/00600 PCTNS99/11516 .
166
v
~ U
_
* 00 00 0~,
~
~
E
Nw ~ d~ k k
~ X ~
G dl N Ln l~ ~ 1J
d' lfl ~ ~S
f., p ~ N ,-I [-i
N 111
O
U _ G
'""~O O o o o o
~ a O O
- ~ ~
o
~ ~k ~ X ~ ~ ~
~ x
o o xE
o ,o
,
U . . . . . . Q,
~ . .
f~ M M M M U
d~ d~
H H
O m
A
rt5 .u .~ ~ y..r w
O
~ ~ ~ a
i ~ i i
~ U U U U
'b j
C2~ f.2~ Or f.~.~ H -~
N
E~ ~ O
~ H ' H r~
U ~ ~ ~ ~ ~ ~ ~ ~ ~
p N ~ N r~ N t6
U E U ~ U E ~ O
U U U
''(~4 ~ N 4 ~ N 4 ~ N ~ H
U rtS Cn c0 CO rtS U1
G ~, ~ U E-~ ~ U H ~
O G ~ ~ ,-a G
~ i~ ~ C1 ~ fl ''~ H U
b '~ ~
~ , ~ .~ ~
~
O H
l~ 4-.I
'~ Q.'
v' ~ ~ a
b a
w o ~ ~ ~ ~ ~ ~ ~ ra
a~
~ x x x x x
~
. x ~'
s.~ a~ a~ a, a~ w
M ~
~, a~
ar
N N 47
'Lf
G ' ~..~ er
U
H -~ ~ ~ ~ .-~ ~ ~ p
~ ,
~H
~ ~ O
a p, pa *
v
a ~ a a
s ~ ~
a~ a~ a. s
a
m
b
G
..
in o
CA 02333481 2000-11-27
WO 00/00600 ,~ 6~ PCTNS99/11516
N
C
N
N R
O U
t
N N
U
-p O
C H
_
CO
_N O
C3 C
~L C
CC
C_ ,p
N
U C
NN
~
~
_ R
_
G Li
T
L
N
R
L
U CS
3
o ~ U
o
o o 3
x
X 7G 7G ="
X
y .-. - N O O
I~ M N y ~r
w
U j
~ ~ U
v
_ ~
2
~
.r d-
;n
O~~ .r l~ U
V1 d' y
N
U T
Lr fA L '~
R _
a.~ Q
x o z
L
_
4- p O
~ "r .,.. C
'C3 ~ ~.r
~
~, r.
" r
_N
a
u
7 ~ > 7 ' E-~-
~
4 4
0 N
4 d
U cG V ~ U " ~ ;
V H 7 LL~ ~, LL ' c'~c
Q.
L
G ..... N N ~' ~ ~ y
U
bD ~ M b '~ v ~ C~ ca
_ n.
~
.Q'~ ~'J G. p ~ E-~
UU ..~ Cl.
_ U p
~ ~ ~ .,.. ~
~ a
E ~ . n. n. ~ .R Q
.
0
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516 .
168
Example 6C: Production efficiency of Second Generation
Constructs
Comparison of vector titer of different pHP packaging
constructs using pTVdeltaCMVnlacZ or pTVdeltaEFnlacZ as
transducing reporter gene:
Methods: TE671 cells were co-transfected with pHP construct
(8 microgram per well in a 6-well plate), pTV construct (8
microgram per well), pHEFVSV-G (5 microgram per well as
envelope pseudotype) with a tat expression plasmid pCEP-tat
(0.5-1 microgram) and a rev expression plasmid pCMV-rev
(0,.5-1 microgram). The tat and rev expression plasmids
were included because we have shown that they could enhance
the vector titers for most of the pHP constructs and they
were necessary for pHP-dl.NdeI which has a tat and rev
deletion and for pHP-VSVG which has a tat deletion.
We have shown that the original construct pHP-d1.28 (a
Gl construct) expressed RT at 50-90% of the wild type level
indicating that the mutations in pHP-dl. did not affect the
synthesis and function of Gag-Pol. The preliminary
relative titers of different pHP mutants are shown below:
(all included a co-transfected pTV reporter transgene)
pAP-di.28 (env, nef deletion, relative titer: 1.00);
pNL4-3 (wild type HIV-1 control which in fact produce less
vector than pHP-d1.28, relative titer: 0.40);
pHP-VSVG (vpr, tat, env and nef deletion, relative titer .
0.014); pHP-dl.env/vpu I (vpu, env, nef deletion, relative
titer: 0.43); pHP-dl.env/vpu II (vpu, env, nef deletion,
relative titer: 0.38); pHP-dl.vpr (vpr, env, nef deletion,
relative titer: 0.85); pHP-vpr/ala/leu (vpr funtional
mutation, env, nef deletion, relative titer: 0.85);
pHP-vpr/env/vpu I (vpr functional mutation and vpu, env,
nef deletion, relative titer: 0.24);
pHP-vpr/env/vpu II (vpr functional mutation and vpu, env,
nef deletion, relative titer: 0.50)
pHP-dl.NdeI (vif, vpr, tat, rev, vpu, env, and nef
deletion, relative titer: 0.006).
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
169
Thus, as more and more of the essential genes were deleted,
such as tat and rev, and seqeunces such as major splice
acceptor sites SA4 at nt. 5390, SA5 at nt. 5777, SA6 at nt.
5960, and SA7 at nt. 5976 and the 5' of env coding
sequence, the vector efficiency gradually or drastically
decreased. Nevertheless, the date showed that the second
generation pHP construct such as pHP-vpr/env/vpu II can be
made with relative titer still at 50% level of the
pHP-d1.28 and this is about the same efficiency as using a
wild type HIV-1 as the packaging vector (pNL4-3 titer = 40%
of pHP-d1.28).
In theory, a G2 pHP construct should contain only gag-pol
open reading frames and the RRE regulatory sequences such
as the pHP-CMV, pHP-CMVdel.TAR/SD, pHP-CMVEFla-intron, or
pHP-EF constructs (although the vif gene is still present
in all of them). However, these constructs exhibited
reduced levels of gag-pol activity as shown by the
following summary table:
methods: TE671 cells were transfected with 5 microgram of
each test HP plasmid and 0.5 microgram of pCEPtat (except
for one construct, pHP-CMVEFla-intron, we tested both with
and without Tat) and 1 microgram of pCMVrev. The culture
supernatant was harvested and p24 level was determined by
ELISA as described before.
(The relative level of p24 shown with pHP-d1.28 set at
1.00)
pHP-1 (1.00)
pHP-d1.28 (1.00)
pHP-VSVG (0.008)
pHP-dl.vpr (0.34)
pHP-dl.env/vpu I (0.43)
pHP-dl.env/vpu II (1.41)
pHP-dl.NdeI (0.007)
pHP-CMV (0.05)
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
170
pHP-CMVdel. TAR/SD (0.03)
pHP-CMVEFla-intron (0.21, with Tat))
pHP-CMVEFla-intron (0.04, without Tat)
pHP-EF (0.27)
It was thus shown that deleting TAR in the 5' LTR as seen
in pHP-CMVEFIa-intron did not make the pHP construct
Tat-independent, suggesting that Tat has alternative
effects on gag-pol expression besides promotor
transactivation via TAR. In addition, the EFla enhancer
promoter and intron construct exhibited the highest level
of p24 expression suggesting that the EF-la promoter is a
better choice than the CMV promoter in later pHP
modifications .
EXAMPLE 7
Production of RC-HIV
To improve the safety, the env gene was deleted in pHP
and two deletion constructs were generated, pHP-1d1.2 and
pHP-1d1.28. In order to determine whether an RC-HIV
recombinant could be generated, the transfected human
rhabdomyosarcoma TE671 cells (ATCC CRL 8805) were co-
cultured with the human lymphoma cell line MT4. MT4 cells
axe an riTLV-1 transformed human CD4+ lymphoma ceii line,
that are very sensitive to HIV-1 infection. These cells
are available from the National Institutes of Health AIDS
Reagents and Reference Program. Uninfected MT4 cells were
added into the co-culture every week during these
experiments. The wt HIV-1 plasmid pNL4-3 was included for
comparison.
In this Example, it was found that the pHPl packaging
construct, but not the env-deleted constructs pHP-1d1.2 (2
nt deletion) and pHP-1d1.28 28 nt deletion), produced
replication-competent HIV-1 (RCV) after co-transfection
with pTV plasmid. Infectious virus was detected from
pHP+pTV~CMVnlacZ MT4 co-culture in 8 days. In addition, no
infectious virus was detected from pHP.dl.2 or
pHP.d1.28+pTV~CMVnlacZ MT4 co-culture in 60 days (See,
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516 .
171
Table 4, below).
The 28 nt env deletion in pHP-1d1.28 did not
negatively affect virus titer which was consistently
greater than 105 transducing units/ per ml of vector
supernatant 24 hr following transfection of either TE671 or
293T cells (Table 4A). To minimize the possibility of
recovering env function, further deletions have been made
in the env gene without affecting vector efficiency.
TE671 cells were co-transfected with pHP+pTV+pHEF-VSV
G as shown in the Table below (Table 4), and the culture
supernatants were harvested 48 hours after DNA removal for
RT assay and vector titer was determined as described
before.
In the co-culture RCV assay, MT4 lymphoblastoid cells
were added to the HP/TV transfected cells 48 h after
transfection. Fresh medium was added to the co-cultures
every 3 days at which time two thirds of the culture media
was discarded. Fresh MT4 cells were added to the cultures
once a week. The transfected cells were co-cultured with
the human MT-4 lymphoblastoid cell line, which is very
sensitive to HIV-1 infection, for up to 2 months. The
culture supernatants were harvested at different time
points after co-culture.
The supernatant from the co-culture was assayed for
HIV-1 RT activity and for infectious RCV by passage onto
CD4+HeLa cells or uninfected MT4 culture. Infection of
CD4+HeLa cells was examined by anti-p24 immunohistochemical
staining using pooled AIDS patients' sera. At different
time points, MT4 cells in the co-culture were collected and
immunostained using a monoclonal anti-p24 antibody for the
detection of HIV-1 Gag antigen as described previously,
Chang, Virol., 211:157-169 (1995). The assay also looked
for infection of MT4 cells by cytopathic effects of RCV.
Syncytium formation was observed under an inverted
microscope.
A very sensitive assay which would detect cell-cell
transmission of poor replicative virus was also used.
After four months of co-culture, the MT4 cells were removed
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516 .
172
and added to fresh MT4 cells and further cultured for 4
days. The co-cultured MT4 cells were fixed and
immunostained with HIV patients' sera.
The results showed that both pHP-1d1.2 and pHP-1d1.28
were incapable of producing RC-HIV. In sum, these results
indicated that pHP-1 transfected cultures produced ,
replication-competent HIV-1 after 8 days of co-culture.
However, no RCV was detected after a 60-day co-culture for
either pHP-ldl.2 or pHP-1d1.28 cotransfection. The vector
titers produced by pHP-ldl.2 and pHP-1d1.28 were as high as
that produced by pHP-1. The 28 nt deletion vector pHP-.
1d1.28 was shown to be as efficient as pHP-1, and did not
produce RCV, based on the sensitive HIV infection assay.
Thus, the deletion does not affect vector production
efficiency and the env-deleted pHP constructs are safe for
vector production without generating RCV.
Figure 12 illustrates the possible cross-over between
pHP-d1.28 and pTV-dl.CMVnIacZ, to generate RCV during co-
transfection. These results clearly indicate that the
recombinants are not infectious, due to the deletion in env
and the LTR mutation, and requires two homologous
recombination events.
CA 02333481 2000-11-27
WO 00/00600 1 ~3 PCT/US99/11516
~a
fIf H
bx
o"
.r.,
3
~ 'p
v
O
C1. U '~ <.
+~
H [-~ U1
x
a,
3 y~ -,~
w
~ + + ~ rt,~
O o + +
U to + + N O U
U
N
+ (~ . ~-~I . ~-i
1~ oD * t-l '
U w~ N + +
x ~, ~;
? >'' -~ ~ O 3
ao + + + ~ ~ U w
x A +
a
(li 1~ .IJ .~1 'H O U
(a ~ 1.~ N
-O ~ m..~ ~ U ~ ~ E-
0 -1 ~ W W W
~ U C~ C~
O w ~ .u
O ~ ,
V c~J~ °~° ~ U
-~-1 U X-1 ~ N ~ N a N ~ N ~ ~' ~ O ~ U
W ~ O U ~ U ~ U ~ U rtf U to O
~ U 4 ~ 4 ~ 4 ro 4 ~ ~ ~ ~ ~ H
C~-~ ~ E~-~ ~ E-~ ~ H ~ rti p ~ ~ U7 z3
O
r-1
~~ pa~,~ 3
~.~1 b N W N
fly U7 CA C~/j r-I ~ p U ~ -r-I
A p ~ i i i i O
'~ :. .' Ga C~-~ fsa f~ r~j ~-1 ,~ _ 't-~" ~
x x x x
Qr ~ of fIS ~.,' U7 U (a 1J
i-I w-1 ~ W-I U
~'~ O N N
w ~ ~ ~ U U7 w
N 0 + U
-r~ '~ M O ri ,-i ~ N U) ~ ~ ~ ~ ~ -r-I
.b ~ . ~, .~ .,~ En ~ 1~ O
zo a~ c~ a,b ~o~~~.~~
a~ u° °' v x ,~
~ + ~N~ uu
* . .~~~ ~,.
w
0
CA 02333481 2000-11-27
WO 00/00600 1'4 PCT/US99/11516
N 00 U]
d' ri
f~
.t~ Tf r1
ca N S.~
1~ l~
I
al
O O
x x
3 .,~..
. r~
('~1N ~ y~ 'Tf
N J.-1''~
~
U
H i1,4)
'b
r-I
\ _
t31 S-a
d' 00 lf1 U
U) ..., ~
r-i~ lf1~c1 U ,-~
C7
U a ~ W
H
N N I
~ U U W rt5O
N x
r-Iri ~ ''(~la
O u~
~ U U l~ l~
W r1
'J''J E
..r ca
E'' C1~~ -~
r~ .u
.~.~-r-I
-
r~ N N i~r
3 ~ -~
O w ~ ~ O
I I c~
p ~ N rtS
C~ w r~ ~C
R~ f~ '~ H
O
W
x p ~ .~ ~ ._
., 3 ~ ~ 'O
b .~, o, w
~r ~ ~ x cn
N ~ N H y..~
H U --
m
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
175
EXAMPLE 8
Transduction of Target Cells
Example SA: In Vitro Transduction of Mitomycin-C-Treated
Human Cells
To demonstrate that the HP/TV vectors are capable of
transducing non-proliferating cells, TE671 or HeLa cells
were treated with mitomycin C to arrest the cell division
cycle and were then transduced with pTV~CMV-nlacZ viral
vector.
In this Example, two cell cultures were transduced
with HP-TV and observed for its transduction efficiency.
To arrest cell cycle, TE671 or HeLa cells were treated with
the DNA synthesis inhibitor, mitomycin C, at 10 ~.g/ml for
4 hours, trypsinized and plated into a 6-well culture
plate. Cell cycle arrest was monitored by propidium iodide
staining and FACS analysis. The cells were transduced with
HP-TV HIV vector carrying a nlacZ marker gene in the
presence of 4-8 /cg/ml polybrene in a total volume of 0.5 ml
for 2-3 hours and fed with growth media (DMEM containing
10% FBS). After 48 hours, the expression of the transduced
lacZ gene was detected by X-gal staining as described
above. The results indicated that the HP-TV vector was
capable of afficientiy transducing mitomycin-C-trea~.ed,
non-dividing human cells. This was further confirmed in
numerous assays; we rountinely use mitomycin C-treated
cells and normal dividing cells for vector stock titration.
The HP/TV lentiviral vectors transduce cells with
different efficiencies depending on the cell cycle stage at
the time of transduction. To demonstrate this, TE671 was
treated with 5 microgram/ml of mitomycin C in DMEM growth
media for 2.5 hr and the treated cells were transduced with
the pTVdeltaEFnlacZ vector and 48 h later, the transduction
efficiency was determined by x-gal staini assay. The
result demonstrated that cells were most efficiently
transduced at 24-48 hr after mitomycin C treatment, at
which time, the cells were arrested at S or G2/M phases.
At later stage, when cells entered high chromosomal content
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516 .
176
(>4N) stage the transduction efficiency became reduced.
This result suggests that although HP/TV lentiiral vector
transduces post-mitotic cells, the efficiency of gene
transduction is still dependent on the cell cycle stage.
EXAMPLE 8B: In Vitro Transduction of Primary Neuronal Cells
To demonstrate that the HP/TV vectors are capable of
transducing non-proliferating cells, TE671 or HeLa cells
were treated with mitomycin C to block cell division and
then transduced with pTVnCI~IV-nlacZ viral vector.
In this Example, rat neuronal cells were isolated from
the brains of Fisher rats according to the method of Ure et
a1. (Ure et al., Develop. Biol., 154:388-395 [1992]).
The cells were grown in culture medium containing L15C02
(GIBCO, Grand Island, New York), containing 200 ng/ml 2.5
S nerve growth factor (NGF), 2.55 rat serum, 1 mg/ml
ascorbic acid, and 10 ~.M cytosine arabinose (Sigma), to
inhibit divisions of non-neuronal cells.
In addition to rat neuronal cells, human neurons and
astrocytes were obtained from differentiated embryonal
1
neural stem cells provided by Neurospheres, Ltd (Calgary,
Alberta, Canada). These cells were infected with the HP-TV
vectors carrying the nlacZ reporter gene as described
above. Briefly, cells were incubated in culture media
containing the HP-TV vector. After two hours of
incubation, conditioned media (i.e., supernatant medium
harvested from cultured neuronal cells after 24 hours of
culture) were added, and the culture continued to incubate
for five days. The cells were then fixed with formaldehyde
and glutaraldehyde, and incubated with X-gal substrate as
described in the (3-galactosidase assay described above. In
vivo transduction of rat muscle was performed using a 25
gauge insulin syringe. pTV~CMVnlacZ vector, 2-5 x 105
tu/ml, was prepared from transfected TE671 and used for
intramuscular injection. Each injection site received 200
~1 of unconcentrated vector or 20 ~1 of 30-50x concentrated
vector.
The results indicated that the HP-TV vector
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
177
efficiently transduces primary neuronal cells obtained from
rat brains, and human neuronal stem cells (neurons and
astrocytes); both were post-mitotic, terminally
differentiated cells.
EXAMPLE 8C: In Vivo Transduction of Muscle Cells
In this Example, the HP-TV HIV vector was used to
transduce muscle cell in vivo. The hind-legs of mice CB-17
SCID/beige mice (Taconic) were intramuscularly injected
with 50-100 ~.l of vectors carrying the nlac2 reporter gene
as unconcentrated (105/ml) or microcentrifuge concentrated
(30 x 105/ml) stocks in the presence of 4 ~.g/ml of
polybrene. The mice were sacrificed two days later and the
injected tissue was prepared for frozen section and for ~i-
galactosidase analysis. The results showed that HP-TV
vector transduced muscle cells efficiently in vivo. In
particular, tissues exposed to the concentrated vector
stock were transduced at near 1000 efficiency at the site
of injection. It was also noticed that microcentrifuge
concentration increased the infectious virus titer, but not
in proportion to the fold of concentration. In contrast,
injections with retroviral vectors did not produce
reproducible positive results.
Interestingly, when human TE671 cells were transduced
in vitro with high titer HP and pTVnEFGFP vectors,
differentiated muscle cells expressing GFP reporter gene
was observed; it is possible that the virion-associated Vpr
was present at concentration high enough to induce
differentiation of TE671 into muscle cells.
Example 8D: Miscellaneous In vitro Transduction
1. Human TE671 cells were transduced with lentiviral
vector pTVnEFGFP three times (3-5 MOI) and incubated for 4-
5 days. The GFP gene expression was detected directly
under a Zeiss Axiovert 25 inverted fluorescent microscope
with x10 Fluar objective lens.
In contrast, injections with retroviral (MLV) vectors
did not produce reproducible positive results.
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516 .
178 '
2. We found that K562, a human chronic myelogenous
leukemic cell line, can be efficiently transduced (>95%) as
measured by flow cytometry analysis of GFP expression
following tranduction with VSV-G pseudotyped HP/TV vectors,
and transgene expression remained stable for over three
months. In constrast, KG1, an acute myelogenous leukemic
cell line, displayed only -5% transduction efficiency using
the same vector construct. PCR analysis of integrated
proviral DNA suggests that the reduced level of
transduction in KG1 correlates with the low level of
proviral DNA, which can be caused either by steps limiting
viral entry, proviral synthesis and/or integration in KG2
cells.
CA 02333481 2000-11-27
WO 00!00600 PCT/US99/11516 .
179
Example 8E: HIV Vectors are More Efficent than MLV Vectors
The efficacy of gene therapy vectors is often judged
by their transduction efficiency as wel as long-term
stability.
In this Example, HIV vectors were compared with the
standard MLV vectors commonly in use. The results obtained
in these experiments indicated that HIV vector is more
efficient than the MLV vector.
Generation of vectors
The MLV-derived vectors were generated by transfecting
the packaging cell line PA317 with pMFGnlacZ which contains
the same recombinant nlacZ gene as the one cloned into
pTVOCMVnlacZ. The HP/TV vectors were generated by co-
transfecting HeLa or TE671 cells with the pHP, pTV, and
pHEF-VSVG plasmids. A modified calcium phosphate DNA
transfection protocol was performed as previously described
lo. The transfection efficiency, normally ranging from 50-
90%, was determined by X-gal staining or by a
radioimmunoassay for human growth hormone when the XGH5
plasmid was included in the transfection procedure (Nichols
Institute Diagnostics). The transfected cells normally
produce retro- or lenti-viral vectors with titers ranging
Lrom 10=-105 transducing units (tu) per mi. The VSV-G
pseudotyped vectors were routinely concentrated 30-50 times
by centrifugation in a table-top microfuge (21,000 g) for
2.5-3 h at room temperature.
Vector transduction and titration. Virus supernatants were
harvested 24, 48, and 72 hr following addition of DNA by
low speed centrifugation (12008 for 5 min) or by filtration
using a 0.45 ~,m low-protein binding filter to remove cell
debris from transfected culture. No reduction in titer was
observed between 24-48 h, but a one-log reduction in titer
was frequently observed in harvests carried out at 72 h.
The supernatants were aliquoted and stored at -80°C until
use. Retroviral vector was titered on HeLa or TE671 cells
and lentiviral vector was titered on mitomycin C-treated
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516 .
180
TE671 cells (5 ~g/ml for 2.5 h). To titer the vector,
cells were infected with diluted virus stocks at low
multiplicity of infection (MOI) in a small volume of growth
medium containing polybrene (8 ~.g/ml). Cultures were grown
for 3-4 hr, supplemented with additional growth medium, and
then incubated for a final period of 48 hr before staining.
A minimum of two different dilutions were examined for each
titration sample. To detect the transduced cells, the
cultures were washed twice with phosphate buffered saline
(PBS), and fixed at room temperature with 1% formaldehyde
and 0.2% glutaraldehyde (Sigma) in PBS for 5 min. After
three additional washes with PBS, the cells were incubated
at 37°C in PBS or distilled water containing 4 mM K-
ferrocyanide, 4 mM K-ferricyanide, 2 mM MgCl2 and 0.4 mg/ml
X-Gal overnight. The transduced cells stained with X-Gal
were examined with an inverted microscope the next day.
The high background beta-galactosidase activity in HepG2
cells, primary cultures and tissues can be eliminated by
increasing the pH of the incubation buffer. The efficacy
of gene therapy vectors is often judged by their
transduction efficiency as well as long-term stability.
Discussion
To directly compare lentivirai and retroviral vectors,
a combined short-term/long-term in vitro study was carried
out. Three different human cell types, TE671, 293T and
HepG2 cells, were transduced with either retroviral vector
MFGnIacZ, that was produced from PA317 packaging cells and
contained a MLV LTR-driven nlacZ gene, or lentiviral vector
pTV~CMVnlacZ, that was generated from co-transfection with
pHP-1d1.28 and contained a CMV-IE promoter-driven nlacZ
gene. About 105 transducing units of vector (approximately
1 moi) were used for each transduction in a total of three
rounds of transduction. Transduced cultures were grown
until confluent (3-5 days), trypsinized, counted, and
plated into 6-well culture plates. Twenty four hours after
plating, the cells were sampled for lacZ assay and the
percentage of cells transduced was determined. The results
CA 02333481 2000-11-27
WO 00/00600 PCTNS99/11516
181
of this short-term study showed that the lentiviral vectors
transduced all three types of human cells 3 to 10 times
more efficiently than did the MLV vectors (Table 5).
For the long-term assays, the transduced cells were
continuously propagated without selection. At different
passage times, the percentage of nlacZ-expressing cells and
the expression kinetics were determined (Fig. 11).
For the longterm study, cells were passaged for 48
days, and stained for (3-galactosidase activity again.
The results showed that the stability of transgene
expression varied as a function of cell type. Expression
of the HP/TV vector, which was driven by the CMV-IE
promoter, decreased with time in all three cell types,
whereas MLV transgene expression gradually decreased in
293T cells but not in TE671 and HepG2 cells. The results
showed that in the long term culture, the HP+TV HIV vectors
exhibited gene expression stability.
Table 5 below, shows a direct comparison of the
transduction efficiences observed at 48 hours and 48 days.
As previously mentioned, TE671 are rhabdomyosarcoma cells,
293T are kidney cells, and HepG2 are hepatoma cells. In
this table, the numbers indicate the percent of cells
transduced after one passage or multiple passages (for the
48 hour samples, the cells were Lransduced three times and
propagated once, before staining for ~3-galactosidase
activity as previously described in Example 6.
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
182
Table 5. Comparison of Long-Term
Transduction Efficiencies
Transduction
Cell Efficiencies
of
MLV vs.
HIV nlacZ
Vectors
Lines
48'Hours 48 Days: ..
MLV HIV MLV 'HIV
HepG2 3 1.5 29 8 15 0.2 27 4
TE671 20 4 62 2 12 2 45 2
293T 7 0.5 46 2 1.2 0.2 13 3
Example 8F: Gene transduction into CD34+ human
hematopoietic procursor cells
Human CD34 hematopoietic progenitor cells are a slow-
dividing cell population which is known to be difficult to
transduce with conventional retroviral vectors.
Gene transfer into the human hematopoietic stem cells
(HSCs) has encountered with problems of vector transduction
efficiency and long term expression stability. See
Barranger JA. Hematopoietic stem cell gene transfer. Gene
Therapy 1996; 3:379-380; Brenner MK. Gene transfer to
hematopoietic cells. N. Engl. J. Med. 1996; 335:337-339.
Amphotropic MLV vectors transduce mouse HSCs quite
efficiently but human HSCs poorly due to the low level of
cell surface MLV-env receptor expression; see Orlic D,
Girard LJ, Jordan CT, Anderson SM, Cline AP, Bodine DM.,
The level of mRNA encoding the amphotropic retrovirus
receptor in mouse and human hematopoietic stem cells is low
and correlates with the efficiency of retrovirus
transduction. Proc. Natl. Acad. Sci. USA 1996;
93:11097-11102; Sabatino DE, Do BQ, Pyle LC, et al.
Amphotropic or gibbon ape leukemia virus retrovirus.binding
and transduction correlates with the level of receptor mRNA
in human hematopoietic cell lines, Blood Cells Mol Dis
1997; 23:422-33; and possible cis-repressive elements in
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516 .
183
the MLV LTRs.Challita PM, Skelton D, E1-Khoueiry A, Yu XJ,
Weinberg K, Kohn DB, Multiple modifications in cis elements
of the long terminal repeat of retroviral vectors lead to
increased expression and decreased DNA methylation in
embryonic carcinoma cells. J Virol 1995; 69:748-755. In
particular, transduction of HSCs in clinical trials has
been very difficult. See Dunbar CE. Gene transfer to
hematopoietic stem cells: implications for gene therapy of
human disease. Annu Rev Med 1996; 47:11-20.
Adeno-associated virus vector has been demonstrated capable
of transducing hematopoeitic stem cell-derived erythroid
cells but only works at extremely high titer. See Nienhuis
AW, Bertran J, Hargrove P, Vanin E, Yang Y. Gene transfer
into hematopoietic cells. Stem Cells 1997; 1:123-34. To
overcome the problem with low amphotropic MLV env receptor
on CD34 cells, infectious HIV-1 constructs have been
pseudotyped with vesicular stomatitis G envelope proteins
(VSV-G) and shown to infect CD34 cells quite efficiently.
See Akkina R, Walton RM, Chen ML, Li Q-X, Planelles V, Chen
ISY. High-efficiency gene transfer into CD34+ cells with a
human immunodeficiency virus type-1-based retroviral vector
pseudotyped with vesicular stomatitis virus envelope
glycoprotein G. J. Virol. 1996; 70:2581-2585. However,
for obvious safety reasons, such replication-competent
HIV-1 constructs would never be used in gene therapy
application.
The HP/TV vector efficiently transduces actively
dividing human cell lines including TE671
(rhabdomyosarcoma), 293T (kidney carcinoma) HepG2
(hepatoma), and HeLa (cervical carcinoma) cells.
Non-dividing and terminally differentiated cells such as
mitomycin C-treated TE671 or HeLa cells, neruons,
monocyte-derived macrophages and muscles can also be
efficiently transduced by the HP/TV vectors. In contrast,
transduction of metabolically quiescent human peripheral
blood lymphocytes or bone marrow mobilized blood CD34 stem
cells with lentiviral vectors have not been reported, and
in our experience, transduction of these cells with viral
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516 .
184
vectors including AAV, retroviral vectors or lentiviral
vectors is extremely inefficient, probably because in the
absence of growth factor activation these cells have very
low metabolic enzyme and transcriptional activities, and
accordingly, viral integration and gene expression do not
proceed efficiently. ,
Nevertheless, we have demonstrated transduction of
human CD34 derived hematopoietic precursor cells with the
HP/TV vectors carrying either nuclear lacZ or green
fluorescent protein (GFP) reporter gene. This has been
demonstrated using pTV vector containing human elongation
factor la (EFla) promoter as an internal promoter possibly
because EFla promoter has very high transcriptional
activity even in quiscent human hematopoietic precursor
cells.
To demonstrate transduction of HSC-derived precursor
cells, human peripheral blood lymphocytes (PBLs) were
collected from patients treated with G-CSF
(granulocyte-colony stimulating factor) to mobilize bone
marrow stem cells and purified through an anti-CD34
antibody affinity column (CellPro, Bothel, WA, USA). The
collected C34+ cells were washed 2-3 times with RPMI medium
containing loo fetal bovine serum (FBS) without growth
factor supplements, centrifuged at 800 g for 5 min, and
resuspended in the same growth medium supplemented with 50
ng/ml human flt3 ligand, 50 ng/ml human c-kit ligand and 50
ng/ml human IL-3 at 1 x 105 cells/100 microliter.
To prepare HP/TV vectors, TE671 cells were transfected
with pHP-1d1.28 (8 microgram/well), one of the lentiviral
reporter vectors pTVdI.EFnlacZ or pTVdI.EFGFP (8
microgram/well), pHEF-VSVG (5 microgram/well) and pCEP-tat
(0.2 microgram/well) plasmid DNA in a 6-well culture plate,
and 48 hr after DNA was added, culture supernatant was
collected and centrifuged at 1000 g for 5 min. The clear
supernatant was stored at -80oC for future use. The human
CD34 cells were transduced 2-3 times with TV vectors at a
multiplicity of infection (MOI) of 5-10, i.e., at MOI 10,
approximately 10E5 cells were transduced with 10E6
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
185
infectious units (iu) of pTV vectors in a final volume of
100 ul in DMEM or RPMI growth medium supplemented with 8
microgram/ml of polybrene for 3-4 h each time. The 10E6 iu
of pTV vectors were prepared from two ml of vector stocks
containing 5x105 iu/ml which can be concentrated 30-40 fold
in a microfuge spun at 20,800 g at room temperature for
90-120 min. The transduced CD34 cells could be maintained
in RPMI supplemented with growth factors for 1-4 days
before they were plated into semi-solid methylcellulose
colony assay media. The plated hematopoietic precursor
cells grew and formed colonies in 2-4 weeks and the
expression of transduced nlacZ and GFP genes were assayed
by X-gal colorimetric staining and observed under an
inverted fluorescent microscope.
For the X-gal staining, the reaction substrate was
prepared in phosphate buffered saline adjusted to pH 8.5
using 150 mM Tris containing 4 mM K-ferrocyanide, 4 mM
K-ferricyanide, 2 mM MgCl2, 0.8 mg/ml X-Gal. One ml of the
x-gal substrate was added to each 30 mm dish containing
HSC-derived colonies and the dish was incubated at 37 deg.
C in a 5o C02 incubator for 24-72 hr. The total colonies
and the dark blue-stained colonies were counted under an
inverted microscope. The GFP expression was observed
directly under an inverted fluorescent microscope. The
expression efficiency of transduction was determined to be
less than 1% at 3-4 weeks after CD34 cells were plated.
However, after 5-6 weeks, the efficiency of expression of
the transgene (e.g. GFP gene) increased to more than 20%.
To determine the efficiency of transduction of the CD34
cells by the pTV vector, the colonies formed in
methylcellulose agar were individually picked up and the
genomic DNA extracted and subjected to polymerase chain
reaction (PCR) using primers specific to the pTV vector.
Twenty out of the twenty colonies picked were positive
for pTV sequence, suggesting that the transduction
efficiency had been near 100%.
This study suggests that the CD34 cells can be
efficiently transduced by the VSV-G pseudotyped HP/TV
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
186 '
vectors, at least at MOI of 5-10, but gene expression is
delayed and the level of expression is very low.
We expect that the expression level can be increased
by using a stronger promoter, such as human EF1 alpha
promoter. In addition, targeted delivery, e.g., with an
SDF1/coat protein chimera, may compensate for low
expression.
Example 8G
Prolonged presence of lentiviral but not retroviral
un-integrated proviral DNA in transduced cells. To
determine if the HP/TV transgene was integrated, extra
chromosomal (Hirt) and chromosomal DNA was harvested from
the lentiviral vector-transduced cells and analyzed by
Southern blotting. The nlacZ sequence was detected at
passage 7 in the chromosomal DNA of TE671 and HepG2 cells,
but not in the Hirt preparations (lanes 2 & 3). To further
investigate the causes of the reduction of transgene
expression, we compared the chromosomal and Hirt DNA
harvested from early and late passages of the transduced
culture by Southern analysis. The results of these
analyses revealed that similar amounts of nlacZ DNA were
present in early (passage 3) and late passages (passage 40)
of 293T cells and of HepG2 cells (passage 6 vs. passage 49)
even though the expression kinetics indicated that nlacZ
expression in transduced 293T and HepG2 cells gradually
decreased with time. In contrast, in TE671 cells the
integrated DNA was lost in late passages [passage 40, vs.
passage 4], a result consistent with the observed decrease
in nlacZ expression with time.
To see if the presence of unintegrated proviral DNA in
the short and long term cultures could explain the
transient high expression phenotype of lentiviral vectors,
a more sensitive assay was performed. Hirt DNA was
harvested from early and late passages of cells transduced
with either HIV or MLV vectors and analyzed by PCR using
nested primers that specifically amplify one- or two-LTR
un-integrated proviral DNA circles. One-LTR proviral DNA
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
187
was detected in all three HP/TV vector transduced cultures
from early passages [TE671, 293T and HepG2 passage 3-5] but
not from late passages (passage 36-44). Further
amplification of the first round nested PCR products did
not reveal the presence of lentiviral proviral DNA in the
late passages of cells (not shown). In contrast, MLV
proviral DNA was not detected either from early or from
late passages of transduced TE671, HepG2 or 293T cells via
a sensitive nested PCR assay using two sets of nested
primers (passage 3-39 of MLV transduced cells).
Example 9
An innovative Short-term Stromal-type HP/TV Producer Cells
(SSPC)-- a novel protocol for efficient HSC transduction
with HP/TV vectors:
Retroviral vectors transduce HSCs poorly due to
reasons including low number of receptors on HSCs, low
vector titers, and possible blocks to reverse transcription
after entry, see Sinclair AM, Agrawal YP, Elbar E, Agrawal
R, Ho AD, Levine F. Interaction of vesicular stomatitis
virus-G pseudotyped retrovirus with CD34(+) and
CD34(+)CD38(-) hematopoietic progenitor cells. Gene Therapy
1997; 4:918-927. Protocols to improve transduction
efficiency have been developed for retroviral gene transzer
into HSCs, for examples, coating culture plates with
fibronectin fragment FN30/35, see Moritz T, Dutt P, Xiao X,
et al. Fibronectin improves transduction of reconstituting
hematopoietic stem cells by retroviral vectors: evidence of
direct viral binding to chymotryptic carboxy-terminal
fragments. Blood 1996; 88:855-862, or adding a pretreatment
step using medium containing 5 ng/ml of anti-TGF-beta for
10-20h, see Hatzfeld A, Batard P, Panterne B, Taieb F,
Hatzfeld J. Increased stable retroviral gene transfer in
early hematopoietic progenitors released from quiescence.
Human Gene Therapy 1996; 7:207-213., and applying
centrifugal force during infection to increases the
reversible binding of virus to the cells, see Bahnson AB,
Dunigan JT, Baysal BE, et al. Centrifugal enhancement of
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516 .
188 '
retroviral mediated gene transfer. J Virol Methods 1995;
54:131-43. These protocols may or may not improve the poor
transduction efficiencies of lentiviral vectors on HSCs as
we showed in the preliminary studies. Co-culturing target
cells with retroviral producer cells has been shown to
improve retroviral transduction efficiency. To improve the
efficiency of transducing HSCs with lentiviral vectors, a
modified protocol is proposed which combines the growth
factor stimulation step with the lentiviral producer cell
co-culture step. This protocol will also eliminate the
vector concentration step which involves the use of a
ultracentrifuge. The cells used for lentiviral production,
TE671, can be modified to express human IL-3, SCF, and flt3
ligand via cDNA co-transfection for the purpose of
supporting long term culture and transduction of
CD34+/CD38- HSCs. Alternatively, freshly prepared human
stromal cells can be modified to become lentiviral vector
producer cells by co-transfection using HP/TV vectors plus
pHEF-VSV-G or pHEV-GALV-env (Gibbon ape leukemia virus)
constructs.
Thus, TE671 (or other human cell line) transfectants
expressing human IL-3, SCF, and flt3 ligand via
transfection, or freshly prepared human stromal cells are
co-transfected with HP/TV vector plus pHEF-VSV-G or
pHEV-GALV-env (Gibbon ape leukemia virus) constructs and
24-48 hr later, or when the cells become 100% confluent,
the transfected cells were treated with mitomycin C (5
microgram/ml) for 2.5 hr, washed and refed with RPMI growth
media.
Human IL-3 cDNA was amplified using primers:
-TTTCTAGACCACCATGAGCCGCCTGCCCGTCC- and
-AAGGATCCCTAAAAGATCGCGAGGCTC-,
per Otsuka T, Miyajima A, Brown N, et al. Isolation and
characterization of an expressible cDNA encoding human
IL-3. Induction of IL-3 mRNA in human T cell clones. J.
Immunol. 1988; 140:2288-2295.
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
189
Human SCF cDNA was amplified using primers:
-TTTCTAGACCACCATGAAGAAGACACAAACTTG- and
-AAGGATCCTTACACTTCTTGAAACTC-,
per Martin FH, et al., Primary structure and functional
expression of rat and human stem cell factor DNAs. Cell
1990; 63:203-211. "
Human flt3 ligand cDNA was amplified using primers:
-TTTCTAGACCACCATGA.CAGTGCTGGCGCCAG- and
-AAGGATCCTCAGTGCTCCACAAGCAGC-,
per Lyman SD, James L, Johnson L, et al. Cloning of the
human homologue of the murine flt3 ligang: a growth factor
for early hematopoietic progenitor cells. Blood 1994;
83:2795-2801.
CA 02333481 2000-11-27
WO 00!00600 PCT/US99/11516 .
190
Example 100
G2 Transducing Vectors
Towards construction of G2 pTV:
Gag AUG, SD, gag coding sequence, env coding sequence,
RRE and gag/env/RRE deletion mutants: To see if the highly
conserved packaging signal, i.e. sequences spanning the gag
AUG and the 5' major splice donor, can be changed without
affecting packaging function of pTV, the following mutants
were constructed and tested for cytoplasmic RNA synthesis
(exported from nucleus), and packaging function by virion
RNA slot-blot assay, and transduction functions by vector
titration.
A. Mutant construction:
All mutants were made by the megaprimer site-specific
mutagenesis method described before or by direct DNA
molecular cloning.
A-1: 5' splice site (SD at nt. 744) and Gag AUG (at nt.
790) mutations (Fig. 19C). The two gag AUG mutants and the
two SD mutants were made using primers containing the
mutation sequences as listed below:
l.pTVdeltaAUGl: -CTC TCG CAC CGG TCT CTC TCC TTC-
2.pTVdeltaAUG2: -CTC TCG CAC CCT ACT CTC TCC TTC-
3.pTVdeltaSDl: -GGC GGC GAC TGC AGA GTA CGC CAA-
4.pTVdeltaSD2: - GGC GGC GAC TGG GGA GTA CGC CAA-
A-2: Gag/Env coding sequence mutations (Fig. 19B). pTV
has a gag-pol-env deletion from nt. 1507-7250. The series
of additional gag coding sequence mutants feature further
deletions, as indicated, which were made by site-specific
mutagenesis using primers designed to delete specific
lengths of gag coding region as shown in Fig. and described
below:
1. pTVgag/env dl. l, deletion of 180 bp, from nt 7430-7611.
using the following primer:
-CTC CAG GTC TGA AGA TCT TTG ACC CTT CAG TAC TC-
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
191
2. pTVgag/env d1.2, deletion of 361 bp, from nt 7250-7611.
using the following primer:
-CTC CAG GTC TGA AGA TCT ACT AGT AGT TCC TGC TAT G-
3. pTVgag/env dl. 3 deletion of 591 bp, from nt. 1277-1507
and nt.7250-7611 using primer:
-CTC CAG GTC TGA AGA TCT GCC TTC TCT TCT ACT ACT-
4. pTVgag/env dl. 4 deletion of 824 bp, from nt. 1044-1507
and nt. 7250-7611 using primer:
-CTC CAG GTC TGA AGA TCT GAG GAC TGC TAT TGT ATT-
5. pTVgag/env dl. 5 deletion of 1039 bp, from nt. 829-1507
and 7250-7611 using primer:
-CTC CAG GTC TGA AGA TCT CTA ATT CTC CCC CGC TT-
A3. Env coding sequence and splice acceptor 8 (SA8 at nt.
8369) and SA9 (at nt. 8515) mutations (Fig. 19D). The
series of env mutants, some of which contained splice
acceptor site 8 & 9 deletion, were made by Ba131 deletion
at the BamHI site at nt. 8465 and six deletion mutants were
isolated and sequenced and their deletions were confirmed
as follows:
1. pTVenv dl. l, BamHI 2'-12, from nt 8375-8559, between
RRE and the CMV promoter but SA8 site (splice acceptor site
8 at nt. 8369) is intact.
2. pTVenv d1.2, BamHI 2'-6: from nt 8355-8586, between RRE
and the CMV promoter.
3. pTVenv d1.3, BamHI 2'-8: from nt 8315-8586, between RRE
and the CMV promoter.
4. pTVenv d1.4, BamHI 5'-3 from nt. 8160-8604, between
RRE and the CMV promoter.
5. pTVenv d1.5, BamHI 5'-8 from nt. 8215-8730, between RRE
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
192
and the CMV promoter.
6. pTVenv d1.6, BamHI 5'-10 from nt. 8214-8785, between
RRE and the CMV promoter.
A. 4. RRE and RRE/gag/env deletion mutations.
The RRE deletion mutant and the RRE/gag/env deletion
mutant were constructed using the following methods and
primers:
1. RRE deletion mutant, deletion pTVdI.RRE (Fig. 19E) : a
primer flanking both end of RRE with the following seqeunce
w a s a s a d t o c o n s t r a c t R R E - d 1 .
AACCCCAAATCCCCATTCCCACTGCTCTTTTT. The first round PCR
generated a 1.3 kbp product which was used as megaprimer to
amplify a 2.3 kbp fragment which was digested with SphI and
NotI sites for cloning into pTV vectors. The SphI-NotI
1350bp was ligated with SphI and NotI-XbaI 4025 by and
XbaI-SphI 7332 by of pTVDnlacZ to generate the RRE deletion
mutant.
2. RRE/gag/env deletion mutant, pTVdl.gag/env/RRE (Fig.
19F) : This deletion starts from gag nt . 829 to env nt . 8785
which was constructed using t:~ree =ragmenL ligation
approach. The three fragments are: BssHII to BglII 125 by
of pTV gag d1.5 containing 5' leader-gag-env, BglII to XbaI
4016 by from pTVnCMVnlacZ, and XbaI to BssHII 6600 by from.
pTVDnlacZ as plasmid backbone.
A. 5. Combination of SD1 (GGTG to GCAG)/ gag AUG (AUG to
TAG) or SD1/env coding sequence/SA deletion, or SD1/
RRE/gag/env deletion mutations. To make generation 2
pTVs, deletion of more essential sequences such as the SD
site coupled with gag AUG, or gag or env coding sequences
in the pTV constructs will make the vector system even
safer. Surprisingly, in some cases, the combination of
mutations did not further decrease vector titer, instead,
the combination of mutations increased vector titers (see
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516 .
193
below) .
a). pTVdeltaSDl/AUG2: this mutant was made by
site-specific mutagenesis using the existing AUG2 primer:
-CTC TCG CAC CCT ACT CTC TCC TTC- (AUG to TAG) and using
the pTVdeltaSD1 as backbone.
b). pTVdeltaSDl/envdl.6 (Fig. 19F): this mutant was made by
restriction enzyme digestion and isolation of DNA fragments
containing either the SD1 mutation or the env d1.6 mutation
and ligated with the pTVdeltaCMVnlacZ backbone.
c). pTVdeltaSD1/dl.gag/env/RRE (Fig. l9F): this mutant
was made by megaprimer mutagenesis as described before
using the SD1 primer: -GGC GGC GAC TGC AGA GTA CGC CAA-
and a primer resided in the CMV-IE promoter downstream of
the dl.gag/env/RRE region. The amplified fragment
containing both SD1 mutation and the dl.gag/env/RRE
sequence was ligated with two fragments obtained from
pTVdeltaCMVnlacZ to generate pTVdeltaSD1/dl.gag/env/RRE.
Results and Discussion: The preliminary results of analyses
of vector RNA, packaging function and vector titer are
summarized in the table below:
(full-length/spliced RNAs; Virion RNA levels;
relative titers)
Control pTVdeltaCMVnlacZ: (+++++/+++++; +++++; 1.00)
l.pTVdeltaAUGl: (++++/++++; ++; 0.35) translation void,
stead-state RNA less than wt.
2.pTVdeltaAUG2: (++++/++++; ++; 0.72) translation void,
stead-state RNA less than wt.
3.pTVdeltaSDl: (++/-; ++; 0.98) less RNA made and less
detected in virions, but wt titer.
4.pTVdeltaSD2: (++/-; ++; 0.85) less RNA made and less
detected in virions, but wt titer.
5. pTVgag/env dl. l: (+++++/+++++; ++++++; 1.08)
6. pTVgag/env d1.2: (+++++/+++++; +++++; 0.90)
7. pTVgag/env d1.3: (+++++/+++++; +++++; 0.81)
8. pTVgag/env d1.4: (+++++/+++++; +++++; 0.94)
CA 02333481 2000-11-27
WO 00/OOb00 PCT/US99/1151b
194
9. pTVgag/env d1.5: (+++++/+++++; +++++; 0.65)
10. pTVenv dl. l: (++/-; +++; 0.48) no spliced RNA.
11. pTVenv d1.2: (n. d.; n.d.; 0.65) no spliced RNA.
12. pTVenv d1.3: (++/-; +++; 0.47) no spliced RNA.
13. pTVenv d1.4: (++/-; +++; 0.60) no spliced RNA.
14. pTVenv d1.5: (n. d.; +++; 0.64) no spliced RNA.
15. pTVenv d1.6: (+++/-; +++; 0.44) more full-length RNA
but less titer than other env dl..
16. pTVdI.RRE: (++/+++++;+; 0.10) detected 20% virion RNA
but less titer. 17. pTVdl.gag/env/RRE: (++/-; +; 0.02)
detected 20% virion RNA but much less titer.
18. pTVdeltaSD1/AUG2: (n. d.; ++; 0.61)
19. pTVdeltaSD1/env d1.6: (n. d.; +++++; 1.00)
20. pTVdeltaSDl/dl.gag/env/RRE: (n. d.; ++++; 0.30) detected
80% virion RNA but less titer.
footnote: +++++ represents 1000 level with each "+"
representing 200, "-" representing undetected; n.d., not
determined.
Later results are shown on the figures.
The preliminary results showed that:
1. Northern analyses of cytoplasmic RNA indicated that
neither the gag AUG mutants nor the gag deletion mutants
have much detrimental effects on mRNA synthesis and the
transduction functional analysis showed, as determined by
vector titration of vectors on TE671 cells, that one of the
two gag AUG mutants (AUG2), and all of the gag coding
sequence mutants exhibitedhave unno significant effects on
vector titers (less than 350% reduction compared with
original pTV vector). However, the gag AUG1 mutant
pTVdeltaAUGl showed more reduction one of the gag AUG
mutants showed more detrimental effects on vector titer
compared with wild type construct (350 of the wild type
vector level).
2. Analyses Northern analyses of cytoplasmic polyA+ RNA
indicated that the two SD mutants expressed less amount of
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
195
the full-length vector RNA. However, the titration study
showed that both SD mutants showed and the env deletion
series have little to no significant effects on vector
titers compared with wild type vector, suggesting that the
two SD mutations either enhanced the RNA packaging
function, or enhanced the efficiency of the transgene
expression. "
3. Northern analyses of cytoplasmic polyA+ RNA showed that
the env conding sequence deletion mutations (SA mutations
as well) have some minor effects (50%) on steady-state
level of RNA synthesis which correlated well with the
vector titer results. Therefore, this region of env and
the SA sites are dispensable for vector construction. The
env deletion series, all have the splice acceptor function
deleted but maintained an intact SD site, exhibited less
cytoplasmic mRNA and less virion RNA than wild type which
correlated well with the titer data. Interestingly, the
pTVenv d1.6 mutant exhibited more full-length RNA but less
titer than other env dl. suggesting that a minot packaging
signal may reside in the region between nt 8730 and nt
8785.
4. Northern analyses of cytoplasmic polyA+ RNA showed that
the RRE mutant and the RRE/gag/env mutant were both were
suppressed in full-length cytoplasmic RNA synthesis,
suggesting that the deletedse sequences are necessary for
RNA nuclear export. However, the titer study showed that
the RRE/gag/env mutant is more defective than the RRE
mutant although both the latter mutants expressed similar
levels ofless full-length RNA in the cytoplasm, suggesting
that the RRE/gag/pol sequence has additional
functionseffects on vector packaging or transduction
efficiency. Interestingly, the levels of virion RNA
detected in the RRE mutants did not correlate with the
titer reduction, suggesting that the RRE sequence has
additional functions besides nuclear transport of the
full-length RNA. This finding indicates that RRE is
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516 .
196
necessary not only for vector RNA export to the cytoplasm,
but also for high vector transduction efficiency.
5. The observation that the internal CMV promoter activity
was affected when the SD site was mutated suggesting that
the splicing machinary has some effects on the internal
enhancer/promoter function, possibly through interfering
with transcriptional factor binding to the CMV-IE
enhancer/promoter elements.
A.6., Construction of 3' U3 deletion mutants and assay for
vector titer.
To generate U3 deletion in the vector system, both the
5' U3 and the 3' U3 will be deleted except for the att site
in the 3' U3 region which is needed for provirus
integration. The 5' U3 was deleted using the same
CMV-TATA-HIV-TAR promoter as illustrated in the
construction of pHP-1. The 3' U3 was deleted by megaprirner
directed site-specific mutagenesis. We established 5
different deletion mutants as described below:
a.pTVdl.kB/Spl: this construct was made using a kB/Spl
deleted HIV-1 construct as reported by Chang eL al. 1993
(J. Virology) to replace the 3' LTR of pTVdeltaCMVnlacZ.
The kB/Spl deleted HIV-1 construct was digested with KpnI
(in the nef region of the genome, nt. 9005) and NgomI
(NaeI, nt. 10349) and ligated with KpnI to NotI and NotI to
NgomI fragments from pTVdeltaCMVnlacZ to generate
pTVdl.kB/Spl.
b. pTV-U3d1.1, pTV-U3d1.2, pTV-U3d1.3, and pTV-U3d1.4 (Fig.
19H) were made by megaprimer mutagenesis to generate
deletions from nt.9098-9528 (entire U3 deletion to the
beginning of R except for the 5' 24 nt att site),
nt.9154-9528 (the 5' sequence of U3 from 9098-9154 was
retained), nt. 9098-9512 (the extended 5' TAR sequence in
the U3 is retained), and nt. 9154 to 9512 (both S' and 3'
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
197
extra sequences in the U3 were retained). These mutants
were made using the following primers:
primer U3d1.1: -GTCTAACCAGAGAGACCCTGGGAGTGAATTAGCCCTTC-
primer U3d1.2: -GTCTAACCAGAGAGACCCCAGGGAAGTAGCCTTGTG-
primer U3d1.3: -CCAGTACAGGCAAAAAGCTGGGAGTGAATTAGCCCTTC
primer U3d1.4: -CCAGTACAGGCAAA.P.AGCCAGGGAAGTAGCCTTGTG
and using a 5' primer annealed to the EcoRI site of the
n 1 a c Z g a n a . 5 ' R I p r i m a r
-GTCTAACCAGAGAGACCCTGGGAGTGAATTAGCCCTTC-
and a 3' primer next to the NgoMI site:
3' NgoMI primer: -ATAGAACTCCGTTCTCC-
The PCR amplified fragment was digested with EcoRI and
NgoMI and ligated into EcoRI and NgoMI digested
pTVdeltaCMVnlacZ to generate the four U3 mutants.
Results and Discussion:
The relative vector titer of these mutants was determined
by co-transfection with pHP-d1.28 and pHEF-VSVG as
described above and the transfected culture supernatant was
harvested 48 hr later and used to infect TE671 and 48 hr
after infection, the lacZ gene expression was assay by
x-gal staining and the blue nucleated cells were counted.
The relative vector titer was shown with the
pTVdeltaCMVnlacZ set at 1.00.
Table: (relative vector titer)
1. pTVdeltaCMVnlacZ: (1.00 +/- 0.00)
2. pTVdl.kB/Spl: (1.00 +/- 0.10)
3. pTV-U3d1.1: (0.80 +/- 0.24)
4. pTV-U3d1.2: (0.91 +/- 0.24)
5. pTV-U3d1.3: (1.22 +/- 0.06)
6. pTV-U3d1.4: (0.84 +/- 0.27)
Summary: The results showed that the 3' U3, except for the
att site, can be deleted from the transducing vector pTV
construct without affecting vector titer. The 5' U3
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516 .
198
deletion had no effect on vector promoter function as shown
in the past. The elimination of U3 sequence from the
vector system greatly improved the safety of our HP/TV
vector system because U3 is an essential HIV replication
element and may play important pathogenesis roles during
viral infection.
Therefore, in combination, we have deleted the
following HIV-1 essential elements, U3, SD, gag AUG,
gag-pol, env, tat, rev and 3' SA sites, and all the
accessory genes from the pTV construct. To generate a RCV
from our HP/TV vector system, a non-homologous
recombination must occur at the gag AUG site to bring the
pTV leader sequence into pHP gag-pol and to cross back to
pTV at the 3' env/RRE region which is about 1106 nt (nt .
7250-8355) and into the inserted reporter gene cassette and
the 3' U3-deleted LTR. Although the overlap in the env
region including the RRE is still quite long, the
recombined product will be lacking 5' U3, SD, gag AUG, env,
all accessory genes, and 3' U3. This recombinant will not
be replication competent and will not exhibit any viral
function.
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516 .
199
Example 101
Mutations of 5' splice site down-regulate cytoplasmic RNA
expression but only have moderate effects on vector titer.
We first mutated the concensus SD sequence in pTV by PCR
site specific mutagenesis (primers listed in Table 101).
Mutations of SD from GGTG to GCAG (SD1) or to GGGG (SD2)
resulted in a moderate decrease (10-300) in vector titer
(Fig. lA) . To determine the mutational effects of SD on
pTV RNA expression, TE671 cells were transfected with 20 ~.g
of pTV, 2 ug of pCEP4 tat; 4 ~,g of pCMVrev, and 0.2 ~,g of
pXGH5 which encodes human growth hormone as transfection
control. Forty hours post-transfection, the cells were
lysed and cytoplasmic poly (A)' RNA was harvested for
Northern analyses as previously described (10). The wt pTV
produced four different sizes of RNA: full-length (F),
short intron-spliced (ss), a CMV promotor driven nlacZ
transcript (CMV) which co-migrated with a spliced RNA
species (s), and large intron-spliced (ls) RNA. The SD
mutations (SD1 Gnd SD2), as predicted, abrogated splicing.
However, minute amounts of ss RNA, due to the use of a
cryptic 5' splice site as previously reported (36), were
detected. It is noteworthy that mutations of SD reduced
expression of cytoplasmic full-length RNA by more than 70%
compared with wt vector, possibly due Lo decreased
stability of the un-spliced RNA or pre-termination of
transcription. It has been reported that the 5' SD imposes
a suppressive effect on activation of the polyadenylation
site in the 5' LTR (1, 2) and thus mutations of SD may
activate 5' polyadenylation. The decline in genomic RNA
expression (70%), however, did not correlate with the
reduction in vector titer (10-30%). This could be
explained by the following two possibilities. First, the
amount of full-length RNA is always in excess of viral
packaging requirement and thus is not a determining factor
of vector titer. Second, the elimination of spliced RNA by
SD mutation abrogated its interference in genomic RNA
packaging and thus resulted in indirect enhancement of
packaging of full-length RNA. In any event, our results
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
200 .
demonstrated that SD itself is not directly involved in HIV
packaging and can be mutated in the vector system.
CA 02333481 2000-11-27
WO 00/00600 201 PCT/US99/11516
w
o~o r
V'10N
V' rlN O N
r1 W n-Irirl
-1
VI . I I ~
M ~ . I t'.
I 1 M M
I I M I
M 1-1JJ1JI
I roU .u~ M
U 1~roro1~I
a~ U U 1JU
U ~ U aItr1
ro 1.~ro1.~U t~
10l011 U 1.~1JU 1J
l11lf1b7 U U roU 1!
r !~ro tJ1~1~U U
I I U 1~1~U U U
A-1b1U tr)J-1U
M M 1J ro1J1JU (J1
I t U ~ U U 1JW
roroU b71.1ro1JU
U U tr7~ U R71~ U
u
b)tn. rob~tr~U U
U U \ \ \ \ \
roro~
E U
b,~ ~ .u~ . ~ s
U
roro~
d7trIX31CTlU
tr1b1tr1tr1tr1J-I
a ~ a ~ a a ro
O
U 01tJttr1b101tr1tr1U
b1bltrb1
~ ~
O fJ7~
1J
~ U U U U U ro
?.I . tn u1>.nu n u1
N ~ lJ7I I I I I I
1J E I I N O O opO tp
wi M M N N N N N O
t"1 ~ ~-I M M lD l0l0l0l0O
o w r r.r r r t~r m
O
N
U
O
4-1
O
.
N
U
S-I O O O O -~N
O r~ rlrlriO 01
-,-j ~ ~J10 l01Dl0r-IQ1
FCc7r r ~ n ~ r
O U C7 I I
. ~ ~ 00lf1I 01
M .-i~ d~o m
C7C74 a a a a a
N
QJ
LJl
1.I rwlN M V~u1
b b b 'z1
U
ro v v v v
tr,rnrna,rnb
0
r-1
a~
E
.r.,
.p
fa W n-1N N d'f1 ~ pp
l 10
I
H
L(1 O
r-~
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
202
Example 102
Most of gag and the entire env sequences except RRE
are dispensable for optimal lentiviral vector function.
The gag and env sequences contain potential packaging
S signals, splice sites, and CRS/INS elements. To examine
their effects on vector functions, a series of deletions in
gag and env were generated and tested. An array of 180,
361, 591, 824, and 1039 by deletions in gag and env
(upstream of RRE) was made by PCR site-specific mutagenesis
(Table 101). Deletions of.env 5' to RRE and most of gag 3'
to the first 255 nt coding sequence in pTV had no
significant effects on vector titer (gag/env, d1.1 to d1.4,
Fig. 19A). Deletion of gag up to the first 40 nt led to a
30% decrease in vector titer (d1.5, Fig. 19A). Northern
analyses showed that these mutants expressed cytoplasmic
RNA at levels similar to that of the wt pTV (not shown).
Therefore, deletions of 5' env and 3' gag did not
significantly affect RNA expression and vector titer.
Further deletion of gag 5' coding sequences (gag/env.dl.5)
had a mild effect on vector titer (Fig. 19A) although it
did not affect viral RNA expression, suggesting a role of
the gag 5' 255 nt in RNA packaging. These results are
consistent with previous observations that the first 21-653
nt of gag is important for RNA packaging (4, 13, 20, 28,
34). To further dissect the effects of env sequences on
vector function, six more deletions (env.dl.l to d1.6) in
env 3' to RRE, which contains two 3' splice acceptor sites.
(SA), were generated by Ba131 exonuclease digestion (Fig.
19D). The deletion of 3' env SA resulted in an overall
~40o reduction in vector titer (Fig. 19D) and Northern
analyses showed reduction in full-length genomic RNA and
abrogation of the spliced 'ss' RNA species. Therefore, the
sequences of env SA appear to have moderate effects on
full-length RNA expression as well as vector function.
Example 103
Deletion of RRE markedly diminishes vector titer. To
determine the effects of RRE on lentiviral vector function,
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516 .
203
precise RRE deletion was made by PCR mutagensis (Table 1;
Fig. 19E). The RRE deletion alone led to a marked
reduction in vector titer (>90%). Interestingly, Northern
analyses indicated that the RRE deletion had only moderate
effects on vector RNA expression in TE671 cells. This is
inconsistent with previous observations that removal of RRE
from HIV usually abolishes cytoplasmic expression of full-
length viral RNA (24, 34). It is possible that the
recombinant vector backbone might contribute to this
phenotype. Alternatively, the specific cell type (TE671)
we used fcr the production of vectors might be different
from the ones used by others. To clarify these issues,
three different cell lines, TE671, HaLa, and 293, were
transfected with different vector constructs, pTV, dl.RRE,
or pHP-EFgp (a pHP mutant helper construct with rev
deletion, Fig. 19E) in the presence or absence of a rev
plasmid. Cytoplasmic and nuclear RNA was harvested and
analyzed by Northern blotting. The results showed that
pHP-EFgp did not transport unspliced RNA into the cytoplasm
of the transfected cells in the absence of rev. When rev
was present, the full-length, unspliced RNA species were
detected in the cytoplasm of all three cell types,
suggesting that a similar rev phenotype can be seen with a
less-truncated HIV vector construct. Interestingly, when
RNA of the wt pTV construct was examined, differences in
RNA partition ratio were observed in different cell types.
In the presence of Rev, HeLa cells exhibited the highest.
ratio of cytoplasmic unspliced RNA to spliced RNA (2.38),
while 293 cells exhibited the lowest (0.19). However, both
HeLa and 293 cells showed the loss of full-length RNA in
the cytoplasm when rev was absent. TE671 cells, in
contrast, did not exhibit an obvious rev effect when
transfected with either pTV or dl.RRE (TE671, 0.58 vs. 0.68
for pTV and 0.44 vs. 0.44 for pTVdl.RRE). Therefore, the
specific RNA partition phenotype of the RRE mutant in TE671
is due to both the cell type and the specific pTV construct
we used. Despite the presence of similar amounts of
cytoplasmic full-length RNA in TE671 cells transfected with
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
204
wt pTV and the dl.RRE mutant, the titer of the RRE mutant
was less than l00 of that of the wt pTV, suggesting an
alternative effect of RRE on vector function independent of
the known RRE effect on cytoplasmic viral RNA expression.
Example 104
The combination of SD, SA, RRE and gag-env CRS
mutations restores vector titer to 50~ of the wt level.
The above results suggest that most of the HIV regulatory
elements in pTV, except for RRE, have only moderate effects
on vector function. To examine whether these regulatory
elements could be deleted all together, we combined
different mutations and analyzed their effects. The
combination of SD/SA mutation generate a vector with near
wt level of titer (dl.SD1/env.dl.6, Fig. 19F), which
apparently repaired the defect of the env.dl.6 (SA)
mutation (Fig. 19D). When RRE mutation was included, the
resulted dl.SD1/env.dl.6/RRE mutant exhibited vector titer
close to zero (0.3% of wt level, Fig. 19F). Interestingly,
further removal of gag and env sequences resulted in a
mutant, dl.SD1/gag/env/RRE, which showed vector titer close
to 50% of the wt level. This restored vector function was
again diminished to about 10 of the wt level when the SD
mutation was reverted to wL (di.gag/env/RRE, Fig. 19F),
again suggesting a negative effect of splice site (SD/SA)
disparity. Northern analyses demonstrated that
when both SD and SA were deleted (dl.SD1/env.dl.6, lane 2,-
Fig. 4B), the full-length viral RNA expression was reduced
by about 50o but the vector titer maintained at 90o wt
level. Further deletion of RRE from this mutant
(dl.SD1/env.dl.6/RRE) did not reduce RNA expression to a
great extent, but the vector function was markedly
diminished (~0.3o wt level, Fig. 19F). This result
corroborated well with the observed effects of pTV dl.RRE
mutant. Surprisingly, when most of the gag (except for the
first 40 nt) and the entire env CRS/INS including RRE in
the vector were deleted, a two-fold increase in full-length
viral-RNA expression compared to wt vector was observed
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516 .
205
(dl.SD1/gag/env/RRE) with accompanied increase in vector
titer (--50% wt level, Fig. 19F) . When the mutated 5' SD
was reverted to generate dl.gag/env/RRE, the expression of
the full-length viral RNA was again suppressed to about 400
of the wt level and the vector function was again abolished
(-to wt level, Fig. 19F). Therefore, only when all the
CRS/INS regulatory elements, including SD, most of gag, the
entire env, and RRE were deleted, can the optimal vector
function be restored.
In summary, we have demonstrated that deletion of
essential HIV regulatory elements, including splice sites,
gag, env, and RRE, from the HP/TV vector system is possible
without significant loss of vector titer. Together with
further deletion of LTR elements in pTV from a separate
study (22), we have minimized the HIV sequences in pTV to
less than 550 bp. This has effectively increased
lentiviral vector payload to more than 9 kb because the HIV
genome size is approximately 9700 nt. These modifications
have effectively reduced the sequence homology between pHP
and pTV and thus greatly improved the safety of this vector
system. Future modifications of remaining lentiviral
sequences will require the substitution of native viral
essential elements with alternative functional elements
while preserving necessary functions zor ezficient vector
production and gene transduction.
References for Examples 100-104
1. Ashe, M. P., P. Griffin, W. James, and N. J.
Proudfoot. 1995. Poly(A) site selection in the HIV-1
provirus: inhibition of promoter-proximal polyadenylation
by the downstream major splice donor site. Genes Dev
9:3008-25.
2. Ashe, M. P., L. H. Pearson, and N. J. Proudfoot. 1997.
The HIV-1 5' LTR poly (A) site is inactivated by U1 snRNP
interaction with the downstream major splice donor site.
Embo J 16:5752-63.
3. Barksdale, S. K., and C. C. Baker. 1995. The human
immunodeficiency virus type 1 Rev protein and the Rev-
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516 .
206
responsive element counteract the effect of an inhibitory
5' splice site in a 3' untranslated region. Mol Cell Biol
15:2962-71.
4. Berkowitz, R. D., M. L. Hammarskjold, C. Helga-Maria,
D. Rekosh, and S. P. Goff. 1995. 5' regions of HIV-1 RNAs
are not sufficient for encapsidation: implications for the
HIV-1 packaging signal. Virology 212:718-23.
5. Berthold, E., and F. Maldarelli. 1996. cis-acting
elements in human immunodeficiency virus type 1 Rnas direct
viral transcripts to distinct intranuclear locations. J
V i r o 1 7 0 . 4 6 6 7 - 8 2 .
6. Borg, K. T., J. P. Favaro, and S. J. Arrigo. 1997.
Involvement of human immunodeficiency virus type-1 splice
sites in the cytoplasmic accumulation of viral RNA.
Virology 236:95-103.
7. Brighty, D. W., and M. Rosenberg. 1994. A cis-acting
repressive sequence that overlaps the Rev-responsive
element of human immunodeficiency virus type 1 regulates
nuclear retention of env mRNAs independently of known
splice signals. Proc Natl Acad Sci U S A 91:8314-8.
8. Chang, D. D., and P. A. Sharp. 1990. Messenger RNA
transport and HIV rev regulation. Science 249:614-5.
9. Chang, D. D., and P. A. Sharp. 1989. Regulation by
HIV Rev depends upon recognition of splice sites. Cell
59:789-95.
10. Chang, L.-J., and C. Zhang. 1995. Infection and
replication of Tat- human immunodeficiency viruses:
genetic analyses of LTR and tat mutations in primary and
long-term human lymphoid cells. Virology 211:157-69.
11. Chang, L.-J., V. Urlacher, T. Iwakuma, Y. Cui, and
J. Zucali. 1999. Efficacy and safety analyses of a
recombinant human immunodeficiency virus type 1 derived
vector system. Gene Therapy (in press).
12. Clavel, F., and J. M. Orenstein. 1990. A mutant of
human immunodeficiency virus with reduced RNA packaging
and abnormal particle morphology. J Virol 64:5230-4.
13. Clever, J., C. Sassetti, and T. G. Parslow. 1995.
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
207
RNA secondary structure and binding sites for gag gene
products in the 5' packaging signal of human
immunodeficiency virus type 1. J Virol 69:2101-9.
14. Clever, J. L., D. A. Eckstein, and T. G. Parslow.
1999. Genetic dissociation of the encapsidation and
reverse transcription functions in the 5' R region of
human immunodeficiency virus type 1. J Virol 73:101-9.
15. Clever, J. L., and T. G. Parslow. 1997. Mutant human
immunodeficiency virus type 1 genomes with defects in RNA
dimerization or encapsidation. J Virol 71:3407-14.
16. Cochrane, A. W., K. S. Jones, S. Beidas, P. J.
Dillon, A. M. Skalka, and C. A. Rosen. 1991.
Identification and characterization of intragenic
sequences which repress human immunodeficiency virus
structural gene expression. J Virol 65:5305-13.
17. Das, A. T., B. Klaver, and B. Berkhout. 1998. The 5'
and 3' TAR elements of human immunodeficiency virus sxert
effects at several points in the virus life cycle. J
Virol 72:9217-23.
18. Das, A. T., B. Klaver, B. I. Klasens, J. L. van
Wamel, and B. Berkhout. 1997. A conserved hairpin motif
in the R-U5 region of the human immunodeficiency virus
type 1 RNA genome is essential for replication. J Virol
71:2346-56.
19. Emerman, M., and M. H. Malim. 1998. HIV-1
regulatory/accessory genes: keys to unraveling viral and
host cell biology. Science 280:1880-4.
20. Frankel, A. D., and J. A. Young. 1998. HIV-1:
fifteen proteins and an RNA. Annu Rev Biochem 67:1-25.
21. Hammarskjold, M. L., H. Li, D. Rekosh, and S.
Prasad. 1994. Human immunodeficiency virus env expression
becomes Rev-independent if the env region is not defined
as an intron. J Virol 68:951-8.
22. Iwakuma, T., Y. Cui, and L.-J. Chang. Improved self-
inactivating lentiviral vectors with U5 and U3
modifications. Virology (Submitted).
23. Kafri, T., U. Blomer, D. A. Peterson, F. H. Gage,
and I. M. Verma. 1997. Sustained expression of genes
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
208
delivered directly into liver and muscle by lentiviral
vectors. Nat Genet 17:314-7.
24. Kaye, J. F., J. H. Richardson, and A. M. Lever.
1995. cis-acting sequences involved in human
immunodeficiency virus type 1 Rna packaging. J Virol
69:6588-92.
25. Lever, A., H. Gottlinger, W. Haseltine, and J.
Sodroski. 1989. Identification of a sequence required for
efficient packaging of human immunodeficiency virus type
1 RNA into virions. J Virol 63:4085-7.
26. Luban, J., and S. P. Goff. 1994. Mutational analysis
of cis-acting packaging signals in human immunodeficiency
virus type 1 RNA. J Virol 68:3784-93.
27. Maldarelli, F., M. A. Martin, and K. Strebel. 1991.
Identification of posttranscriptionally active inhibitory
sequences in human immunodeficiency virus type 1 RNA:
novel level of gene regulation. J Virol 65:5732-43.
28. McBride, M. S., and A. T. Panganiban. 1997. Position
dependence of functional hairpins important for human
immunodeficiency virus type 1 RNA encapsidation in vivo.
J Virol 71:2050-8.
29. McBride, M. S., M. D. Schwartz, and A. T.
Panganiban. 1997. Efficient encapsidation of human
immunodeficiency virus type 1 vectors and further
characterization of cis elements required for
encapsidation. J Virol 71:4544-54.
30. Naldini, L., U. Blomer, F. H. Gage, D. Trono, and I.
M. Verma. 1996. Efficient transfer, integration, and
sustained long-term expression of the transgene in adult
rat brains injected with a lentiviral vector. Proc Natl
Acad Sci U S A 93:11382-8.
31. Naldini, L., U. Blomer, P. Gallay, D. Ory, R.
Mulligan, F. H. Gage, I. M. Verma, and D. Trono. 1996. In
vivo gene delivery and stable transduction of nondividing
cells by a lentiviral vector. Science 272:263-7.
32. Nasioulas, G., A. S. Zolotukhin, C. Tabernero, L.
Solomin, C. P. Cunningham, G. N. Pavlakis, and B. K.
Felber. 1994. Elements distinct from human
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516 .
209
immunodeficiency virus type 1 splice sites are
responsible for the Rev dependence of env mRNA. J Virol
68:2986-93.
33. Olsen, H. S., A. W. Cochrane, and C. Rosen. 1992.
Interaction of cellular factors with intragenic cis-
acting repressive sequences within the HIV genome.
Virology 191:709-15.
34. Parolin, C., T. Dorfman, G. Palu, H. Gottlinger, and
J. Sodroski. 1994. Analysis in human immunodeficiency
IO virus type 1 vectors of cis-acting sequences that affect
gene transfer into human lymphocytes. J Virol 68:3888-95.
35. Powell, D. M., M. C. Amaral, J. Y. Wu, T. Maniatis,
and W. C. Greene. 1997. HIV Rev-dependent binding of
SF2/ASF to the Rev response element: possible role in
Rev-mediated inhibition of HIV RNA splicing. Proc Natl
Acad Sci U S A 94:973-8.
36. Purcell, D. F., and M. A. Martin. 1993. Alternative
splicing of human immunodeficiency virus type 1 mRNA
modulates viral protein expression, replication, and
infectivity. J Virol 67:6365-78.
37. Schwartz, S., M. Campbell, G. Nasioulas, J.
Harrison, B. K. Felber, and G. N. Pavlakis. 1992.
Mutational inactivation of an inhibitory sequence in
human immunodeficiency virus type 1 results in Rev-
independent gag expression. J Virol 66:7176-82.
38. Schwartz, S., B. K. Felber, and G. N. Pavlakis.
1992. Distinct RNA sequences in the gag region of human
immunodeficiency virus type 1 decrease RNA stability and
inhibit expression in the absence of Rev protein. J Virol
66:150-9.
39. Vicenzi, E., D. S. Dimitrov, A. Engelman, T. S.
Migone, D. F. Purcell, J. Leonard, G. Englund, and M. A.
Martin. 1994. An integration-defective U5 deletion mutant
of human immunodeficiency virus type 1 reverts by
eliminating additional long terminal repeat sequences. J
Virol 68:7879-90.
Example 200
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516 .
210
Similar to all retroviruses, lentiviral LTR contains
enhancer/promoter elements, the left integration attachment
site (attL) in U3, polyadenylation signal in R, and part of
the polyadenylation signal and the right integration
attachment site (attR) in U5. In addition, lentiviral LTR
contains a Tat-interacting TAR sequence overlapping R
region, which is essential for viral replication. In the
present study, we investigated the possibility of deleting
most of the U3 and U5 sequences in the LTRs of pTV to
generate an improved lentiviral SIN vector.
Here, we have extensively modified long terminal
repeats (LTRs) of pTV to generate a safer lentiviral vector
system. The 5' U3 was replaced with a truncated
cytomegalovirus (CMV) immediate early (IE) enhancer/TATA
promoter and the 3' U3 (except for the integration
attachment site) was also deleted. These modifications
resulted in a vector with 80~ wild type vector efficiency.
Further deletion of 3' U5 impaired the vector function;
however, this problem was solved by replacing the 3' U5
with bovine growth hormone polyadenylation (bGHpA)
sequence. The pTV vector containing all these
modificaitons including 5' promoter. substitution, 3' U3
deletion, and the substitution of 3' U5 with bGHpA
exhibited self-inactivating (SIN) phenotype azter
transduction, transduced both dividing and non-dividing
cells at similar efficiencies, and produced vector titers
twice as high as that of the wild type construct . Thus,
both safety and efficacy of the HP/TV vector have been
improved by these LTR modifications. Further deletion of
5' U5 impaired vector efficiency, suggesting that the 5' U5
has critical roles in vector function.
MATERIALS AND METHODS
Cell cut ture
TE671 (human rhabdomyosarcoma) cells were obtained
from ECACC, England. Cells were maintained in Dulbecco's
modified Eagle's medium (DMEM, Mediatech) supplemented with
loo fetal bovine serum (FBS, Gibco BRL) and 100 units/ml of
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516 .
211
penicillin-streptomycin (Gibco BRL).
Plasmid constructions
pTVnCMVnlacZ, pHPd120, pHEF-VSVG, and pCEP4tat were
constructed as described previously. Replacement of 5' U3
with a truncated CMV-IE enhancer/TATA promoter was
accomplished by replacing Stu I to Nsi I of pTVnCMVnlacZ
with Pme I to Nsi I of CMV-IE (a)/TATA-HIV-1NL4-3 (Chang et
al., 1993). To generate pTVnCMVnlacZdl3'U3kB/Spl, a DNA
fragment from dl.kB/Spl-HIV-1NL4-3 (Adachi et al., 1986;
Chang et al., 1993; Hunninghake et al., 1989) digested with
Kpn I and NgoM I was cloned into pTVnCMVnlacZ. To
construct pTV mutants by PCR mutagenesis, the primers
listed in Fig. 15 were used. For 3' U3 deletions, primers
"a" and "c~f" were used in the first round of PCR, using
pTVnCMVnlacZ as a template. The amplified products were
purified by Microcon 50 (AMICON) and used as a mega-primer
to pair with 3' primer "b" for the second round PCR using
pTVnCMVnlacZ as template. The amplified fragments were
digested witr. EcoR I and NgoM I and cloned into
pTVnCMVnlacZ. To generate pTVnCMVnlac2d13'U3#1U5,
pTVoCMVnlacZdl3'U3#1 was used as template and primer "g"
was used as 3' primer in the first round PCR. The rest of
the cloning steps were similar to that of the 3' U3
deletion construction. To generate 5' U5 deletions,
primers "h" and "j~l" were used for the first round PCR
with pTVnCMVnlacZ as templates. The products were used as.
mega-primers to pair with 3' primer "i" in the second round
PCR. The final PCR products were digested with BspE I and
Nsi I, and cloned into pTVnCMVnlacZdl3'U3#lUSpA. All PCR
amplified sequences were verified by DNA sequencing.
pTVnCMVnlacZdl3'U3#lUSpA was constructed by digesting
pcDNA3.1/Zeo (+) (Invitrogen) with Hind III and NgoM I and
a 512 by DNA fragment containing bGH polyadenylation
sequence was isolated and cloned into pTVDCMVnlacZdl3'U3#1
digested with the same enzymes.
DNA transfection A modified calcium phosphate DNA
transfection protocol was performed as previously described
CA 02333481 2000-11-27
WO 00/00600 PC1'/US99/11516
212
(Chang and Zhang, 1995; Chen and Okayama, 1987).
Transfection efficiency was determined by a
radioimmunoassay for human growth hormone that was produced
by a co-transfected plasmid pXGH5 (Nichols Institute
Diagnostics). Supernatants of transfected culture were
collected and used for both human growth hormone and virus
titration assays. Vector production and titration to
produce lentiviral vectors, 10 mg of pTV, 10 mg of pHPd120,
and 5mg of pHEF-VSVG were co-transfected with 1 mg of a
tat-encoding plasmid pCEP4tat and 0.1 mg of pXGH5 into
TE671 cells (5x105) in each well of a 6-well plate the day
before transfection. Media were changed 18-24 hr after DNA
was added, and the following day virus was harvested by
filtration using 0.45 mM low-protein binding filters
(Millex-HV, Millipore) to remove cell debris. The
supernatants were stored at -80°C in aliquots until use.
For vector titration, two different dilutions of
supernatants were used to infect 4x104 of TE6'71 cells
plated in 24-well plates in the presence of 8 mg/ml of
polybrene. After 3- 4 hr of infection, fresh media was
added, and 48 hr after infection, culture was stained with
X-gal substrate as previously described (Chang et al.,
1999). Virus titer was determined by counting the blue
nucleated cells and relative vector titer to the wild type
control was presented after normalization for transfection
based on the control human growth hormone expression.
Lentiviral transduction of irradiated TE671 cells TE671
cells were irradiated with 20,000 rad and maintained in a
5% COZ incubator for 28 hr before transduction. The
irradiated cells were tarnsduced with lentiviral vectors at
different multiplicity of infection and 48 hr later, cells
were assayed for the lacZ reporter gene expression by X-gal
staining, and the transduction efficiency was determined.
Hirt DNA preparation
A modified protocol was used for simultaneous
preparation of genomic and Hirt DNA (Motmans et al., 1997).
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516 .
213
Briefly, cells were resuspended in a 250 ml buffer
containing 50 mM glucose, 25 mM Tris-HC1 (pH 8.0), and 10
mM EDTA after washing with PBS, and incubated at room
temperature for 5 min. Then cells were lysed with 200 ml
lysis buffer containing 200 mM NaOH and 1% SDS on ice for
5 min. The lysate was neutralized by adding 150 ml of 5 M
potassium acetate (pH 4.8), and cell debris and chromosomal
DNA were pelleted by centrifugation at 10,000 g for 5 min.
The supernatant containing Hirt DNA was treated with
proteinase K, extracted with phenol and chloroform, and
followed by ethanol precipitation.
PCR analysis of unintegrated proviral intermediates
Approximately 100 ng of Hirt DNA was used as template to
detect proviral intermediates using a nested PCR method.
Hirt DNA was purified from cells infected with lentiviral
vectors for 12 hr. For the first round of PCR, a 5' primer
in lacZ, 5'-ACG ACT CCT GGA GCC CG-3', and a 3' primer in
gag, 5'-TGT GTT GAA TTA CAG TAG AAA AAT TCC CCT C-3', were
used. For the second round PCR, another 5' primer in lacZ
downstream of the first lacZ primer, 5'-GGC GGA ATT CCA GCT
GAG-3', and another 3' primer in gag upstream of the first
3' primer, 5'-ACT GAC GCT CTC GCA CCC AT-3', were used.
Annealing temperature was 58oC and PCR reaction was
performed for 30 cycles for both rounds.
Cytoplasmic poly (A)+ RNA purification and Northern-
blotting
TE671 cells were seeded into a 6-well plate at 5X105
cells per well one day before transfection. To drive the
expression of packageable full length mRNA from a pTV
plasmid, a Rev-expression plasmid, pCMVrev, which is
important for the nuclear export of genomic RNA, was co-
transfected with the pTV plasmid. Cells were transfected
with 20 mg of pTV or its derivatives, 1 mg of pXGH5 for
transfection control, and 4 mg of pCMVrev with or without
2 mg of pCEP4tat per well. Media were changed the next day
and the following day, cytoplasmic poly (A)' RNA was
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
214
harvested for Northern analyses as previously described
(Chang and Zhang, 1995; Robinson et al., 1995). The RNA
blot was hybridized with a lacZ probe and re-hybridized
with a chicken b-actin probe to normalize the RNA amounts.
Before re-hybridization, the first probe was stripped off
by boiling the blot in ddHzO containing 0.1% SDS for 5 min.
Recovery of integrated lentiviral vectors
The cells infected by virus derived from pTVoCMVnlacZ
and pTVnCT-CMVnlacZdl3'U3#lUSpA were cultured for 33 days
(passage 10). These cells were transfected with 15 mg of
pHPd120, 10 mg of pHEF-VSVG, 2 mg of pCEP4tat, and 0.1 mg
of pXGH5 plasmids as described above. After 48 hr, 20 ml,
100 ml, and 200 ml of the transfection supernatants,
containing 8 mg/ml of polybrene, were used to infect TE671
cells for vector titration as described above.
Example 201
5' U3 replacement with a heterologous enhancer/promoter
Based on previous studies of chimeric HIV-1 LTRs
(Chang et al., 1993), we replaced the U3 of the 5' LTR with
a truncated CMV-IE enhancer/TATA promoter and generated
pTVnCT. This replacement eliminates the entire 5' U3 of
HIV-1 except for 25 nucleotides upstream of the NFkB
binding sites. When assayed for vector efficiency and
compared with that of the prior construct pTVn, pTVnCT
exhibited vector titer close to wild type level. To
examine the chimeric promoter activity, poly (A)+
cytoplasmic RNA was purified 42 hr after co-transfection of
the pTV DNA with a Rev-expression vector, pCMVrev, in the
presence or absence of pCEP4tat, followed by Northern
blotting and hybridization using a lacZ probe. The results
showed that both the wt and the mutant constructs expressed
unspliced, full-length mRNA in the absence of Tat and both
were Tat-responsive.
Example 202
3' U3 deletion to generate a SIN vector
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
215
Retroviral vectors with 3' U3 mutations are self-
inactivated (SIN) after reverse transcription (Bishop,
1983; Hwang et al., 1997; Miyoshi et al., 1998, 1998; Olson
et al., 1994; Temin, 1990; Zufferey et al., 1997). Self-
inactivation is due to the conversion of 3' U3 to the end
of the provirus (DNA form of retrovirus) during reverse
transcription. Therefor, if you mutate or delete the
enhancer/promoter in the 3' LTR (i.e. elements in the U3,
enough to kill the promoter activity, e.g. mutate the TATA
box or deletion of the entire U3 except for the integrase
attachment site near the 5' end of U3), you self-
inactivate. To see if the 3' U3 region in pTV can be
deleted, a series of 3' U3 deletions were made by PCR
mutagenesis using primers shown in Fig. 5. We first
deleted NFkB and Spl binding sequences in the 3' U3 as
described in Materials and Methods. This mutation did not
alter virus titer when compared with the wild type pTV (D97
bp, pTVnCMVnlacZdl3'U3kB/Spl, Fig. 19b). This encouraged
us to delete more of the 3' U3 sequence. Further deletions
were made to include most of the U3 sequence with or
without the flanking 9098-9154, and 9512-9528 regions
(according to the numeric system of HIV-1 plasmid pNL4-3).
These mutations were generated by PCR mutagenesis using
primers "c~f" listed in Fig. 15. In all of these mutants,
the left integrase attachment site (attL, 24 nt) was
retained which is essential for the integration function of
the pTV vector (Reicin et al., 1995). These mutants were-
compared with wt pTV by co-transfection with pHP in TE671
cells, and the relative vector efficiency was determined.
The results are summarized in Fig. 19G. The longest 3' U3
deletion did not appear to have any notable effects on
vector titer (pTVnCMVnlacZdl3'U3#1). To verify this, the
3' U3 deletion #1 (D431 bp) was introduced into the
chimeric construct pTVnCT-CMVnlacZ to generate pTVnCT-
CMVnlacZdl3'U3#1. The titer assay again confirmed that
this large U3 deletion did not have a notable effect on
vector efficiency. Interestingly, we observed that the two
deletion constructs (pTVoCMVnlacZdl3'U3#1 and #2), which
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
216
lack an "upstream element" (USE), always produced lower
titers than those with USE (pTVnCMVnlacZdl3'U3#3 and #4),
which suggests a positive role of USE on vector function
(Fig. 19G).
To test thisTo see if the U3 deleted pTV vectors could
be properly reverse transcribed and maintain the U3
deletion in the provirus, we examined the provirus
formation in the transduced cells. TE671 cells were
transduced with 3' or both 5'/ 3' U3-deleted or wild type
U3 vectors, and extrachromosomal DNA was purified.
Proviral intermediates in the Hirt preparation were
amplified by PCR using a nested primer set as described in
Materials and Methods. The PCR primers were designed to
specifically amplify unintegrated intermediates (circular
proviral DNA) but not the co-transfected plasmid DNA or the
linear pre-integration intermediates (Fig. 22). The
results showed that a distinct band of 984 by representing
the product of unmodified one-LTR proviral DNA circles was
detected for both pTVeCMVnlacZ and pTVnCT-CMVnlacZ. In
contrast, the size of the U3 deleted proviral DNAs of
pTVeCMVnlacZdl3'U3#1 (3' U3 deleted) and
pTVnCT-CMVnlacZdl3'U3#1 (both 5' and 3' U3 deleted) 1 was
shifted down to 552 bp, which was consistent with the
expected deletion size of U3. Southern analysis of the PCR
amplified products did not detect wild type U3 signal in
provirus of the U3 deleted vector constructs. This result
demonstrates that both the single and the double U3 deleted
constructs can be properly reverse transcribed after
transduction.
Example 203
Substitution of 3' U5 with a heterologous polyadenylation
signal increased vector titer
To delete more LTR sequences from pTV, the 3' U5
was further modified. We first deleted the entire 3' U5
and generated pTVDCMVnlacZdl3'U3#lU5 using a mutagenesis
primer "g" listed in Fig. 15. This deletion led to a
severe loss of vector function as determined by vector
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
217
titration assay (12% of wild type level, Fig. 19I).
Northern analysis showed marked reduction of genome sized
mRNA to undetectable level. These results suggested a
possible defect in polyadenylation and/or stability of
mRNA. Accordingly, we substituted U5 with a heterologous
polyadenylation sequence derived from bovine growth hormone
(bGH) gene and generated a new pTV construct,
pTVDCMVnlacZdl3'U3#lUSpA. Analyses of this construct
showed an improved vector function almost twice as high as
that of the wild type. This was confirmed when the 3' U5
of the previously modified pTVDCT-d13'U3#1, which had 5'
and 3' U3 modifications, was also replaced with the bGHpA
and tested (pTVDCT-CMVnlacZdl3'U3#lUSpA, (Fig. 19I).
To see if these modified vectors can efficiently
infect non-dividing cells, we blocked the cell cycle of
TE671 cells by g-irradiation (20,000 rad) and then
transduced these irradiated cells with different LTR
modified vectors. The transduction efficiencies of these
vectors were studied in both dividing and non-dividing
cells and compared with that of the wild type pTV. The
titer of wild type pTV on irradiated cells was determined
to be 0.94 +/- 0.05 in relation to wild type pTV on
dividing cells which was arbitrarily set at 1. In
addition, the titers of both Lhe 3' U3/U5- ar~d the 5'U3-
3'U3/U5-modified vectors on irradiated cells were
comparable to that on non-irradiated cells. These results
confirmed that LTR-modified lentiviral vectors transduced.
both dividing and non-dividing cells at similar
efficiencies.
To examine the SIN phenotype of the pTV vectors with
3' U3/U5 deletion, a provirus recovery experiment, as
illustrated in the Fig. 16 diagram, was performed. TE671
cells were transduced with wild type pTV and the modified
d13'U3#lUSpA vectors and continuously cultured for 33 days
till passage 10. The long-term propagation of the
transduced cells would reduce the level of contaminating
carryover transfection DNA and the extrachromosomal
proviral DNA. The percentages of transduced cells in these
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516 .
218
passage 10 cultures were determined by X-gal staining to be
50o and 55% for pTVeCMVnlacZ and for
pTVnCMVnlacZdl3'U3#lUSpA, respectively. To recover
packageable proviral RNA, these cultures were transfected
with the lentiviral helper construct pHPd120, the
pseudotype env construct pHEF-VSVG and pCEP4tat to enhance
LTR transactivation, and 48 hr later, the culture
supernatants were harvested and assayed on TE671 cells.
The average of four repeated assays is shown in Table 203.
The cells transduced by pTVnCMVnlacZ released more than 104
per ml of infectious vectors, whereas cells transduced by
pTVaCMVnlacZdl3'U3#lUSpA released zero infectious unit of
viral vector. This result suggests that there is no LTR-
derived full-length RNA present in cells transduced with
pTVnCMVnlacZdl3'U3#lUSpA and that the provirus derived from
the SIN vector does not have a transcriptionally active LTR
after integration.
Example 204
The 5' U5 is critical to optimal vector function
To further improve this vector system, we attempted to
delete the 5' U5 sequence by PCR mutagenesis using primers
"j~l" as listed in Fig. 15. A pTV mutant with the entire
5' U5 deleted except for tile 24 nt attR was first made and
examined. The titration assay showed that this U5 deletion
construct, pTVDCMVnIacZdlS'U5e62-3'U3U5pA, exhibited only
300 of the wild type vector function (e62, Fig. 19J). When
the 5' 12 and 27 nt of U5 were added back to the e62
construct to generate the e50 and e35 pTV mutants,
respectively, the titration assay showed that the e50
construct restored 20% of vector function. Interestingly,
the e35 construct restored vector efficiency close to the
level of wild type pTVnCMVnlacZ. Nevertheless, as compared
with its parental construct pTVeCMVnlacZdl3'U3#IUSpA, the
e35 vector function was still reduced by 500.
Discussion of Examples 200-204
In this study, we sequentially deleted U3 and U5
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516 .
219 ,
sequences of both LTRs of a lentiviral transducing vector
construct, pTV, to generate an improved SIN vector. pHP
provides helper viral gene functions and pTV produces
packageable genome. pHP does not have any LTR sequence
because the 5' LTR had been replaced with a chimeric CMV-IE
enhancer/promoter and the 3' LTR had been replaced with an
SV40 polyadenylation signal. The LTRs in pTV, however,
have potential risk of generating replication competent HIV
upon recombination with pHP. Thus, deleting LTRs in pTV
would eliminate all native HIV LTR sequences in the vector
system and greatly improve the safety of the HP/TV vector
system. The replacement of both pHP and pTV LTRs with the
same CMV IE enhancer/promoter, however, may not be
desirable because it increases the possibility of
generating a recombinant virus carrying a heterologous CMV-
IE promoter. Nevertheless, this can be avoided by
replacing the pHP or the pTV promoter with a heterologous
promoter. The replacement of 5' U3 with a truncated CMV-IE
enhancer/TATA promoter did not affect vector efficiency and
this chimeric promoter appears to act like the native HIV-1
promoter in TE671 cells. Others have reported similar
replacement with a full-length CMV-IE enhancer and
illustrated a tat-independent promoter function using a
different lentiviral vector system (Kim et al., 1998;
Miyoshi et al., 1998). Although the truncated CMV-IE
enhancer/TATA promoter in pTVDCT-CMVnlacZ appears to have
slightly higher basal promoter activity than that of the
native HIV LTR in the absence of Tat as demonstrated in
repeated Northern analyses, Tat is still required for high
vector efficiency in our system.
It appears that except for the first 24 nt of 3' U3
which contains the left integrase attachment site (attL),
all U3 sequences could be deleted without affecting vector
function. This result is in agreement with the studies of
Miyoshi et a1. and Zufferey et al. (Miyoshi et al., 1998;
Zufferey et al., 1998). However, we found that the
deletion of USE in the 3'U3 had a mild effect on vector
titer (-20% reduction). This could be due to the
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
220
ineffectiveness of poly (A) processing, because the
interaction of a 160-kD subunit of cleavage and
polyadenylation specificity factor (CPSF) with USE is known
to enhance poly (A) processing (Gilmartin et al., 1992;
Gilmartin et al., 1995; Valsamakis et al., 1991). The
GU-rich or U-rich element in U5 is known to be associated
with the cleavage stimulation factor (CstF), which is
important for modulating RNA polyadenylation (Kelley, 1995;
MacDonald et al., 1994). This could explain why deletion
of 3' U5 led to a severe reduction in vector titer and in
mRNA synthesis. This defect could be overcome by replacing
sequence downstream of polyadenylation signal, AATAAA, with
bGH poly (A) sequence. The resulting vector produced two
times the titer of the parental pTVDCMVnlacZ, which is more
than 1 x 106/ml.
Further deletion of 5' U5 of pTV is less tolerable.
Since 5' U5 has been shown to have multiple roles including
packaging, reverse transcription, and integration, the
defect of the 5' U5 deleted vector may be multi-factorial
Das et al., 1997; Huang et al., 1998; Vicenzi et al.,
1994). Vicenzi et a1. have reported that the middle part
of 5' U5 could be deleted without affecting HIV-1
replication, while deletions of either 5' or 3' one third
impaired virus replication (Vicenzi et al., 1994). Results
of the three pTV 5' U5 deletion constructs are consistent
with those earlier observations.
In summary, we have generated a modified lentiviral-
vector system with most of the native HIV LTR sequences
deleted without affecting its function of transducing non-
dividing cells. The LTR-deleted vector exhibited vector
efficiency similar to or better than that of the parental
vector. It is believed that the entire HIV 5' R region
could be replaced with the R region of another retrovirus,
such as the RSV R region.
References for Examples 200-204
Adachi, A., Gendelman, H.E., Koenig, S., Folks, T., Willey,
R., Rabson, A., and Martin, M.A. (1986). Production of
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
221
acquired immunodeficiency syndrome-associated retrovirus in
human and nonhuman cells transfected with an infectious
molecular clone. J. Viroll. 59, 284-291.
Ashe, M.P., Griffin, P., James, W., and Proudfoot, N.J.
(1995). Poly(A) site selection in the HIV-1 provirus:
inhibition of promoter- proximal polyadenylation by the
downstream major splice donor site. Genes. Dev. 9, 3008-
3025.
Ashe, M.P., Pearson, L.H., and Proudfoot, N.J.. (1997). The
HIV-1 5' LTR poly(A) site is inactivated by U1 snRNP
interaction with the downstream major splice donor site.
EMBO J. 16, 5752-5763.
Bishop, J.M. (1983). Cellular oncogenes and retroviruses.
Annu. Rev. Biochem. 52, 301-354.
Burns, J.C., Friedmann, T., Driever, W., Burrascano, M.,
and Yee, J.K. (1993). Vesicular stomatitis virus G
glycoprotein pseudotyped retroviral vectors: concentration
to very high titer and efficient gene transfer into
mammalian and nonmammalian cells. Proc. Natl. Acad. Sci.
USA 90, 8033-8037.
Carroll, R., Lin, J.T., Dacquel, E.J., Mosca, J.D., Burke,
D.S., and St Louis, D.C. (1994). A human immunodeficiency
virus type 1 (HIV-1)-based retroviral vector system
utilizing stable HIV-1 packaging cell lines. J. Viroll. 68,
6047-6051.
Chang, L.-J., Urlacher, V., Iwakuma, T., Cui, Y., and
Zucali, J. (1999). Efficacy and safety analyses of a
recombinant human immunodeficiency virus derived vector
system. Gene Ther. (in press).
Chang, L.-J., McNulty, E., and Martin, M. (1993). Human
immunodeficiency viruses containing heterologous
enhancer/promoters are replication competent and exhibit
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
222
different lymphocyte tropisms. J. Virol. 67, 743-752.
Chang, L.-J. and Zhang, C. (1995). Infection and
replication of Tat- human immunodeficiency viruses: genetic
analyses of LTR and tat mutations in primary and long-term
human lymphoid cells. Virology 211, 157-169.
Chen, C. and Okayama, H. (1987). High-efficiency
transformation of mammalian cells by plasmid DNA. Mol.
Cell. Biol. 7, 2745-2752.
Cui, Y., T. Iwakuma and L.-J. Chang (1999). The
contributions of viral splice sites and cis-regulatory
elements on lentiviral vector functions. J. Virol. {in
press)
Das, A.T., Klaver, B., Klasens, B.I., van Wamel, J.L., and
Berkhout, B. (1997). A conserved hairpin motif in the R-U5
region of the human immunodeficiency virus type 1 RNA
genome is essential for replication. J. Virol. 71, 2346-
2356.
Gilmartin, G.M., Fleming, E.S., and Oetjen, J. (1992).
Elctivation of HIV-1 pre-mRNA 3' processing in vitro
requires both an upstream element and TAR. EMBO J. 11,
4419-4428.
Gilmartin, G.M., Fleming, E.S., Oetjen, J., and Graveley,
B.R. (1995). CPSF recognition of an HIV-1 mRNA 3'-
processing enhancer: multiple sequence contacts involved in
poly(A) site definition. Genes. Dev. 9, 72-83.
Gordon, E.M. and Anderson, W.F. (1994). Gene therapy using
retroviral vectors. Curr. Opin. Biotechnol. 5, 611-616.
Huang, Y., Khorchid, A., Gabor, J., Wang, J., Li, X.,
Darlix, J.L., Wainberg, M.A., and Kleiman, L. {1998). The
role of nucleocapsid and U5 stem/A-rich loop sequences in
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516 .
223
tRNA(3Lys) genomic placement and initiation of reverse
transcription in human immunodeficiency virus type 1. J.
Viroll. 72, 3907-3915.
Hunninghake, G.W., Monick, M.M., Liu, B., and Stinski, M.F.
(1989). The promoter-regulatory region of the major
immediate-early gene of human cytomegalovirus responds to
T-lymphocyte stimulation and contains functional cyclic
AMP-response elements. J. Viroll. 63, 3026-3033.
Hwang, J.J., Li, L., and Anderson, W.F. (1997). A
conditional self-inactivating retrovirus vector that uses
a tetracycline-responsive expression system. J. Viroll. 71,
7128-7131.
Kafri, T., Blomer, U., Peterson, D.A., Gage, F.H., and
Verma, I.M. (1997). Sustained expression of genes delivered
directly into liver and muscle by lentiviral vectors. Nat.
Genet. 17, 314-317.
Kaul, M., Yu, H., Ron, Y., and Dougherty, J.P. (1998).
Regulated lentiviral packaging cell line devoid of most
viral cis- acting sequences. Virology 249, 167-174.
Keller, W. (1995). No end yet to messenger RNA 3'
processing! Cell 81, 829-832.
Kim, V.N., Mitrophanous, K., Kingsman, S.M., and Kingsman,
A.J. (1998). Minimal requirement for a lentivirus vector
based on human immunodeficiency virus type 1. J. Viroll.
72, 811-816.
MacDonald, C.C., Wilusz, J., and Shenk, T. (1994). The 64-
kilodalton subunit of the CstF polyadenylation factor binds
to pre-mRNAs downstream of the cleavage site and influences
cleavage site location. Mol. Cell. Biol. 14, 6647-6654.
Martin, M., Noel, D., and Piechaczyk, M. (1997). Towards
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516 .
224
efficient cell targeting by recombinant retroviruses. Mol.
Med. Today 3, 396-403.
Miller, A.D., Miller, D.G., Garcia, J.V., and Lynch, C.M.
(1993). Use of retroviral vectors for gene transfer and
expression. Methods Enzymol. 217, 581-599.
Miyoshi, H., Blomer, U., Takahashi, M., Gage, F.H., and
Verma, I.M. (1998). Development of a self-inactivating
lentivirus vector. J. Viroll. 72, 8150-8157.
Miyoshi, H., Takahashi, M., Gage, F.H., and Verma, I.M.
(1997). Stable and efficient gene transfer into the retina
using an HIV-based lentiviral vector. Proc. Natl. Acad.
Scii. USA. 94, 10319-10323.
Motmans, K., Thirion, S., Raus, J., and Vandevyver, C.
(1997). Isolation and quantification of episomal expression
vectors in human T cells. Biotechniques 23, 1044-1046.
Naldini, L., Blomer, U., Gage, F.H., Trono, D., and Verma,
I.M. (1996a). Efficient transfer, integration, and
sustained long-term expression of the transgene in adult
rat brains injected with a lentiviral vector. Proc. Nati.
Acad. Sci. USA 93, 11382-11388.
Naldini, L., Blomer, U., Gallay, P., Ory, D., Mulligan, R.,
Gage, F.H., Verma, I.M., and Trono, D. (1996b). In vivo
gene delivery and stable transduction of nondividing cells
by a lentiviral vector. Science 272, 263-267.
Olson, P., Nelson, S., and Dornburg, R. (1994). Improved
self-inactivating retroviral vectors derived from spleen
necrosis virus. J. Viroll. 68, 7060-7066.
Poeschla, E., Corbeau, P., and Wong-Staal, F. (1996).
Development of HIV vectors for anti-HIV gene therapy. Proc.
Natl. Acad. Sci. USA 93, 11395-11399.
CA 02333481 2000-11-27
WO 00/00600
PCT/US99/11516 .
225
Poeschla, E.M., Wong-Staal, F., and Looney, D.J. (1998).
Efficient transduction of nondividing human cells by feline
immunodeficiency virus lentiviral vectors. Nat. Med. 4,
354-357.
Reicin, A.S., Kalpana, G., Paik, S., Marmon, S., and Goff,
S. (1995). Sequences in the human immunodeficiency virus
type 1 U3 region required for in vivo and in vitro
integration. J. Viroll. 69, 5904-5907.
Robinson, D., Elliott, J.F., and Chang, L.J. (1995).
Retroviral vector with a CMV-IE/HIV-TAR hybrid LTR gives
high basal expression levels and is up-regulated by HIV-1
Tat. Gene Ther. 2, 269-278.
Srinivasakumar, N., Chazal, N., Helga-Maria, C., Prasad,
S., Hammarskjold, M.L., and Rekosh, D. (1997). The effect
of viral regulatory protein expression on gene delivery by
human immunodeficiency virus type 1 vectors produced in
stable packaging cell lines. J. Viroll. 71, 5841-5848.
Temin, H.M. (1990). Safety considerations in somatic gene
therapy of human disease with retrovirus vectors. Hum. Gene
~i~her. 1, lli-123.
Valsamakis, A., Zeichner, S., Carswell, S., and Alwine,
J.C. (1991). The human immunodeficiency virus type 1
polyadenylylation signal: a 3' long terminal repeat element
upstream of the AAUAAA necessary for efficient
polyadenylylation. Proc. Natl. Acad. Sci. USA 88, 2108-
2112.
Verma, I.M. and Somia, N. (1997). Gene therapy -- promises,
problems and prospects [news). Nature 389, 239-242.
Vicenzi, E., Dimitrov, D.S., Engelman, A., Migone, T.S.,
Purcell, D.F., Leonard, J., Englund, G., and Martin, M.A.
(1994). An integration-defective U5 deletion mutant of
human immunodeficiency virus type 1 reverts by eliminating
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516 .
226
additional long terminal repeat sequences. J. Viroll. 68,
7879-7890.
Zufferey, R., Dull, T., Mandel, R.J., Bukovsky, A., Quiroz,
D., Naldini, L., and Trono, D. (1998). Self-inactivating
lentivirus vector for safe and efficient In vivo gene
delivery. J. Viroll. 72, 9873-9880.
Zufferey, R., Nagy, D., Mandel, R.J., Naldini, L., and
Trono, D. (1997). Multiply attenuated lentiviral vector
achieves efficient gene delivery in vivo. Nat. Biotechnol.
15, 871-875.
Examt~le 300 Analysis of Stem Loops SL2 and SL4
To improve the safety and payload of lentiviral
vectors, we have previously developed a modified lentiviral
transducing vector with deletions of all the env sequences
including the Rev-responsive element, most of the gag
sequences except for the first 40 nt, and mutation of the
major splice donor site (SD). In this study, we further
investigated the functional significance of the second and
fourth stem-loop SL2 and SL4, including SD and the first 40
nt of gag respectively, of the four stem-loop packaging
signal using a simplified ientiviral vector system.
Partial or complete deletion of SL2 resulted in a greater
than 50% decrease in vector titer, whereas removal of all
gag sequences including SL4 led to an 80% decline in vector
titer. These results indicated that both SL2 and SL4 are
important for lentiviral vector function. Interestingly,
combination of SD point mutation in SL2 and deletion of the
entire gag including SL4 and env from the transducing
vector still preserved the function of lentivirai vectors
with efficiency as high as 10' transducing units per ml.
Therefore, it is possible to further minimize the primary
lentiviral packaging signal sequences and improve the
safety and payload of this simplified lentiviral vector
system.
We have previously minimized the viral sequences in
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
227
the transducing lentiviral vectors down to 40 nt of 5' gag
with a SD mutation in the 5' UTR in addition to
modifications of both LTRs (5, 9). Although most HIV
sequences have been removed from this simplified
transducing vector, the existence of a native 5' UTR and
the overlapping 40 nt of gag between the helper vector and
the transducing vector still poses safety concerns in ~~
therapeutic applications.
As one of the essential elements in HIV replication,
the 5' UTR possesses the primer binding site (PBS) and the
conventional packaging signal (~Y) spanning from 3' of PBS
to the first 40 nt of gag. This ~ region has been
demonstrated to form a four stem-looped secondary structure
(SL1-SL4) (2, 4, 7), and SL1 and SL3 have been shown to be
more critical than SL2 and SL4 to HIV genome packaging (4,
11-13). In addition, our previous studies demonstrated
that mutations in the SD, which is located in SL2, did not
affect genomic RNA packaging (5). To examine whether these
SL sequences can be further deleted, we analyzed the roles
of SL2 and SL4 in our lentiviral vector system.
The SL2 hairpin structure is required for efficient genome
packaging
Our previous studies demonstrated chat a poin~
mutation in the HIV SD abrogated most cf the RNA splicing
without diminishing genome packaging and vector function
(5). We further examined whether the SD and the adjacent.
cryptic splice site in SL2 could be deleted without
interfering with vector function. By PCR site specific
mutagenesis using primers listed in Table 1, a partial and
a complete SL2 deletion mutants were constructed and
studied (~SD3 and OSD4 Fig. 23A). These pTV transducing
vector mutants were assayed for vector efficiency by co-
transfection with a helper construct pHP into human TE671
cells as described previously (3). The relative vector
titer was determined by titration of virus on TE671 cells
using ~i-galactosidase reporter gene assay and normalized
against that of the wt vector pTV which is set at 1.00. In
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516 .
228
contrast to the near wt phenotype of the SD point mutation
(SD1), deletions of SD, which also disrupted the SL2
hairpin structure, led to a 60% decrease in vector titer
(Fig. 23). Further analyses of cytoplasmic RNA by Northern
blotting revealed that both SD3 and SD4 mutants completely
abolished splicing as expected. Similar to the SD1 mutant,
the cytoplasmic genomic RNA (F) of SD3 and SD4 mutants was
also down-regulated by 800 (Fig. 23A and 23B). This down-
regulation of genomic RNA expression was likely due to a
decrease in RNA stability. and/or pre-termination of RNA
transcription, as was demonstrated by others (1, 5, 13).
Despite of the similar levels of cytoplasmic RNA for all
three SD mutants, the vector titers of SD3 and SD4 were
about 500 of the SD1 level (Fig. 23A and 23B).
To further delineate the mutational effects of SD1,
SD3 and SD4, we examined virion RNA by slot blot analyses.
Virion RNA was pelleted by centrifugation at 14,000 rpm fer
3 h. To eliminate plasmid DNA contamination in the virion
RNa preparation, the viral pellet was treated with two
rounds of DNase I, proteinase K, and phenol/chloroform
extraction before used for slot blot hybridization. The
amount of packaged virion RNA was analyzed on slot-blots
and quantified using a Fuji phosphoimager. Genomic RNa
packaging efficiency is defined as the ratio of relative
amounts of packaged virion RNA to the corresponding
cytoplasmic genomic RNA. As summarized in (Fig. 23B), SD3
and SD4 mutants packaged much less virion RNA than did SD1.
It is apparent that the 50a decrease in vector titer of SD3
and SD4 compared with SD1 was caused by similar decrease in
packaging efficiency (Fig. 23B). While this result appears
to be different from previous reports that SL2 is not
essential for gag binding and packaging (12, 13), it is in
agreement with a recent study demonstrating that
destruction of SL2 stem pairing significantly reduced viral
replication as well as packaging (8). Therefore, the
conserved splice site sequence of the SD is not critical
for virion packaging, but the entire SL2 hairpin is
important for maintaining optimal packaging function.
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
229
Gag AUG and the first 40 nt of gag are important for
lentiviral RNA packaging
Located in SL4, the first 40 nt of gag has been shown
to be important for HIV packaging (11). Using our
lentiviral vector system, we examined the contributions of
gag AUG, the SL4 stem-loop, and its down-stream G-rich
region to lentiviral vector function and packaging.
Mutations of gag AUG (PCR primers shown in Table 300)
resulted in a 30-50o reduction, whereas deletion of the
purine-rich region either by itself or in combination with
deletion of the SL4 hairpin structure (Table 300 for PCR
primers) resulted in an SOo decline in vector titer (Fig.
24). About half of the 80o decrease was attributed to the
deletion of the SL4 structure (comparing gag/env.dl7 and
gag/env.dl5 in Fig. 24. Northern analyses revealed that
mutation of gag AUG or deletion of the entire gag did not
suppress cytoplasmic genomic RNA expression. Thus, the
marked decrease in vector titer of the G-rich region
deletion (gag.dl6) or along with SL4 deletion (gag.dl7) was
due to a greater than 70% reduction in their packaging
efficiencies (Fig. 24B). Mutation of gag AUG, however,
caused about 40-50% decrease in packaging efficiencies
(Fig. 24B). The importance of SL4 (the first 40 nt of gag)
to genome packaging and the SL4 purine-rich region to viral
replication have been reported under different systems (8,
11, 13). The contribution of gag AUG to packaging,
however, has been unclear. Unlike the observation of
Richardson (16) that gag AUG mutations did not affect
genome packaging, our gag AUG mutations resulted in about
40-50% reduction in packaging efficiency (Fig. 24B).
Interestingly, it has been reported that when
nucleotides upstream of gag AUG were mutated to disrupt gag
translation, a severe defect in genome packaging occurred,
even in the presence of a helper construct for gag
production (11).
Combination of mutations in SD, gag, env, and RRE still
produces a viable transducing vector.
CA 02333481 2000-11-27
WO 00/00600 PCTNS99/11516
230
To further examine whether extensive mutations in
other regions of the transducing vector (5) could be
combined with mutations in the packaging signal region
without profound loss of vector function, several
combination mutants were constructed and analyzed. The
combination of gag AUG mutation (TAG) with any of the
splice site mutants (5' splice site SD1 or 5' and 3' splice
sites SD1/env.dl.6) caused further decrease in vector titer
(Fig. 25). Similarly, introducing gag AUG mutation into
SDI/gag.dl5/env/RRE, which has most of gag, the entire env
and RRE deleted, also reduced vector function (Fig. 25).
Northern analyses indicated that inclusion of the AUG-TAG
mutation did not further affect the genomic RNA expression.
Thus, the further decrease in vector titer after the
introduction of AUG mutation is consistent with the
observed AUG-TAG mutational effects on packaging function
(Fig. 24B and 25B).
Further deletion of the first 40 nt of gag from
SD1/TAG/gag.dl5/env/RRE to generate mutant
SD1/gag.dl7/env/RRE resulted in a pTV construct with
complete deletion of HIV gag and env sequences.
Surprisingly, this vector still exhibited vector function,
albeit at a reduced level (up to 105 tu/ml, Fig. 25).
Northern analyses indicated that this construct had more
than two-fold increase in cytoplasmic genomic RNA
expression compared to the wt pTV construct. Thus, the
drastic reduction in vector titer was likely due to the
reduced packaging efficiency [Fig. 25]. These results
further support that the stem-loop structure of SL4 and its
down-stream purine-rich sequences are important for
lentiviral genome packaging.
In conclusion, we showed that both SL2 and SL4 in the
primary lentiviral packaging signal are important for the
vector genome packaging function. We also demonstrated
that it is possible to delete all of the HIV gag and env
sequences in the lentiviral transducing vector, which
further reduced sequence overlap between the transducing
pTV and the helper pHP constructs. These modifications
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
231
should further relieve safety concerns over the lentiviral
vector system.
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516 .
232
N
r
M ~
O~
00 h M
N 1f1i i
l~ N Cn
~ M JJ
M ~ U L
M U U
tr1~ b 01
U 1~U 1J
1~ U b1U
t'-C'U U U U
~ rU b11.~~
~U U U 1~
U 1~l.!b1
M Ma1 U U t0
U U p11.~
a 1.~b7 U rt5rtS
U ~U t31tr1l..i
U U~ U U U
1~ Ub1 \ U t0
U L\ (J1\ \
U~ t~1~
U i.~1J ~ U U
1~ UX37.!-~t~1.~
Ubl yJtr1U1
b1 itt0r~
~
S-iU UiJ ~ ~ (
77
E to Ut~ U U U
-'iU c01J 1.1J..1
tn aro b~tr~tn
A' u u'~ u ' tr~
a
N . c . .,
a ,
1~ W U 1W U U U
C
O 1~ UU a7r..iy
c0 U 1~~ U U U
J-~ v i UCn U i i
U - ii i
E W n ,. m n
N m num n
N ~i i aD00
En CJ~O N10 M N N
(1~ N OD Ol0 i~l010
C4 * c~0f~ f'l'~l~
ri M
.,-1
z
v
~, ~
o v ~
;
U \ ,
0
.
~
W JJ
~ Cl'
1J [~ N
a E U H
1
0 0
U ~ iOp M rirl
O
w H H~ r r U
~
N ~rlr.~r~i i i i v
v w M o ~
tn tr~ ~ N
1a o~ rt cor ~ oor.
i~ cn tn rna a a a
v
m
'
v
v b i
a c~ > > ~n
a ~ H ~ ~ ~n
rt ~ ~ \ \
v ~ G
a
a,~
c~
v
0
0
M v C
v
LL ri NM ~ iD H
L(1
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516 .
233
References for Example 300
1. Ashe, M. P., L. H. Pearson, and N. J. Proudfoot.
1997. The HIV-1 5' LTR poly(A) site is inactivated
by U1 snRNP interaction with the downstream major
splice donor site. Embo J 16:5752-63.
2. Baudin, F., R. Marquet, C. Isel, J. L. Darlix, B.
Ehresmann, and C. Ehresmann. 1993. Functional sites
in the 5' region of human immunodeficiency virus
type 1 RNA form defined structural domains. J Mol
Biol 229:382-97.
3. Chang, L.-J., V. Urlacher, T. Iwakuma, Y. Cui, and
J. Zucali. 1999. Efficacy and safety analyses of a
recombinant human immunodeficiency virus type 1
derived vector system. Gene Therapy (in press).
4. Clever, J., C. Sassetti, and T. G. Parslow. 1995.
RNA secondary structure and binding sites for gag
gene products in the 5' packaging signal of human
immunodeficiency virus type 1. J Virol 69:2101-9.
5. Cui, Y., T. Iwakoma, and L.-J. Chang. 1999. The
contributions of viral splice sites and cis-
regulatory elements to lentiviral vector function. J
Virol (in press).
6. Gasmi, M., J. Glynn, M. J. Jin, D. J. Jolly, J. K.
Yee, and S. T. Chen. 1999. RequiremenLS for
efficient production and transduction of human
immunodeficiency virus type 1-based vectors. J Virol
73:1828-34.
7. Harrison, G. P., and A. M. Lever. 1992. The human
immunodeficiency virus type 1 packaging signal and
major splice donor region have a conserved stable
secondary structure. J Virol 66:4144-53.
8. Harrison, G. P., G. Miele, E. Hunter, and A. M. L.
Lever. 1998. Functional analysis of the core human
immunodeficiency virus type 1 packaging signal in a
permissive cell line. J Virol 72:5886-96.
9. Iwakuma, T., Y. Cui, and L.-J. Chang. Improved
self-inactivating lentiviral vectors with both U3
and U5 modifications. Virology (Submitted).
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516 .
234
10. Kim, V. N., K. Mitrophanous, S. M. Kingsman, and A.
J. Kingsman. 1998. Minimal requirement for a
lentivirus vector based on human immunodeficiency
virus type 1. J Virol 72:811-6.
11. Luban, J., and S. P. Goff. 1994. Mutational analysis
of cis-acting packaging signals in human
immunodeficiency virus type 1 RNA. J Virol 68:3784-
93.
12. McBride, M. S., and A. T. Panganiban. 1996. The
human immunodeficiency virus type 1 encapsidation
site is a multipartite RNA element composed of
functional hairpin structures [published erratum
appears in J Virol 1997 Jan;71(1):858]. J Virol
70:2963-73.
13. McBride, M. S., and A. T. Panganiban. 1997. Position
dependence of functional hairpins important for
human immunodeficiency virus type 1 RNA
encapsidation in vivo. J Virol 71:2050-8.
14. Miyoshi, H., U. Blomer, M. Takahashi, F. H. Gage,
and I. M. Verma. 1998. Development of a self-
inactivating lentivirus vector. J Virol 72:8150-7.
15. Mochizuki, H., J. P. Schwartz, K. Tanaka, R. O.
Brady, and J. Reiser. 1998. High-titer human
immunodeficiency virus type 1-based vector systems
for gene delivery into nondividing cells. J Virol
72:8873-83.
16. Richardson, J. H., L. A. Child, and A. M. Lever.
1993. Packaging of human immunodeficiency virus type
1 RNA requires cis-acting sequences outside the 5'
leader region. J Virol 67:3997-4005.
17. Zufferey, R., D. Nagy, R. J. Mandel, L. Naldini, and
D. Trono. 1997. Multiply attenuated lentiviral
vector achieves efficient gene delivery in vivo. Nat
Biotechnol 15:871-5.
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
235
Example 1001 Cystic Fibrosis
This example is a mixture of preliminary studies and
hypoyhetical experiments.
1. Studv of the effects of HIV-1 Vpr on human
bronchial epithelial cells. A key to long term therapy
in CF is to avoid repeated vector administration and to
transduce CFTR functional gene into undifferentiated,
proliferating airway epithelial cells. HIV-1 Vpr is
virion-associated which participates in the nuclear
translocation of the preintegration core. Vpr has also
been shown to arrest cell cycle and cause cell
differentiation. Thus, on one hand, Vpr may be necessary
to improve transduction and expression efficiencies of
HIV vectors but on the other hand, its presence may block
proliferation. To see if HIV-1 Vpr has any biological
effects on human bronchial epithelial cells, two human
airway epithelial cell lines, IB3-1 and BEAS-2B, will be
transfected with a vpr expression eukaryotic vector,
pHEF-vpr (driven by a strong human elongation factor la
promoter). The differentiation and cytotoxic effects of
Vpr will be evaluated after transfection. Transfected
cells will be monitored by a co-transfected green
fluorescent protein (GFP) marker. Morphologic and
functional features of differentiated epithelial cells
will be characterized as described [Engelhardt, 1995
#3704]. The apoptotic effects of Vpr on epithelial cells
will be examined by Hoechst dye staining. This
preliminary examination of Vpr function in human
epithelial cells will be useful for later evaluation of
various HIV-1 vector constructs and their effects on
airway epithelial cells.
2. Transduction of human respiratory epithelia_1_
cells in vitro. We have transduced IB3-1 cell line, which
is a human CF bronchial epithelial cell line (CFTR
genotype is d1F508/W1282X), with HIV-1 vector
pTV~CMV-nlacZ. These cells retain all of the ion channel
and cytokeratin expression characteristics of bronchial
epithelial cells.
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
236
In this preliminary transduction, vectors with or
without HIV-1 Vpr were prepared using two different HIV-1
packaging pHP constructs, one with wild type vpr gene and
the other with an in-frame stop codon mutation. Both
vectors transduced I83-1 at expected efficiencies.
Our preliminary studies with human rhabdomyosarcoma
cell line TE671 indicate that repeated transduction with
high titer (>105 transducing units/ml) of Vpr+ HIV-1
vectors causes TE671 differentiation into muscle cells.
To see if this is true to human airway epithelial cells,
IB3-1 cells will be transduced with high titer Vpr+ or
Vpr- pTV~CMV-nlacZ vector. The transduced culture will
be monitored for differentiation, apoptosis, and
proliferation. The nuclear lacZ gene expression will also
be quantitatively recorded with time. If expression of
Vpr induces differentiation of human airway epithelial
cells, these differentiated cells will be used as target
cells for Vpr+ and Vpr- HIV vector transduction. The
latter experiment will answer whether HIV vector can
efficiently transduce differentiated airway epithelial
cells.
3. Transduction of mouse lung tracheal epithelial
cells. A recent study by Goldman et al. suggests that
HIV vector does not transduce well-di=zerenLia~ed
bronchial epithelium xenografts. In contrast, with
poorly differentiated xenografts, substantial
transduction was observed. It is not clear whether
lentiviral vector can efficiently transduce airway
epithelial cells or whether the presence of HIV-1 Vpr has
an effect on such transduction. We propose to transduce
mouse lungs via intracheal instillation with either Vpr+
or Vpr-pTV~CMV-nlacZ vector, VSV-G pseudotyped and
concentrated at 107-108 transducing units/ml. The mouse
lungs will be studied at 1 week, 8 weeks, or 6 months
after transduction. The nuclear beta-galactosidase
expression will be detected by X-gal staining. The
transduced airway epithelial cells will be collected, and
un-integrated and integrated proviral DNA will be
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
237
harvested by a Hirt method coupled with a genomic DNA
extraction protocol used routinely in our laboratory. If
the pilot study demonstrates long term gene transduction
in adult mouse lungs, further study will be performed in
neonatal rabbit to evaluate whether lentiviral vector
gene expression persists throughout the alveolar phase of
lung development as described by Rubenstein et al.
(1997) .
3A. Generation of different pTV-CFTR HIV vectors
Although the study of pTV~.CMV-nlacZ showed that the
internal CMV promoter is a strong promoter after HIV
vector transduction, some vector constructs, such as
those carrying reporter genes such as GFP or placenta
alkaline phosphatase (PLAP) exhibited undetectable amount
of gene products in our preliminary studies. Therefore,
the optimal CFTR HIV vector will have to be empirically
established. pTV constructs containing either CMV or'
human elongation factor la internal promoter will be used
to generate CFTR vectors. The upstream HIV major splice
donor site and the gag AUG initiation codon have both
been deleted without affecting vector titers. These
different CFTR HIV vector constructs will be generated
and used to transduce IB3-1 epithelial cells. The
expression oz apical CFTR will be immunostained with a
monoclonal antibody MATG1031 (Demolombe, 1996), specific
to the first extracellular loop sequence of the CFTR
protein which is absent in IB3-1 cells. The level of
expression will be determined under a confocal
microscope.
4. Functional study of pTV-CFTR HIV vector transduction
of CF respiratory epithelial cells IB3-1 epithelial
cells will be transduced with high titer pTV-CFTR, using
pTV~CMV-nlacZ as control, and 2, 7, 30, and 60 days
later, the 36C1- isotope tracer efflux profiles will be
assayed in the presence and absence of forskolin,
CPT-cAMP, and IBMX (to increase intracellular cAMP). A
statistically significant increase in the rate of efflux
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
238
in the presence of increased CAMP as compared with the
basal rate is indicative of functional CFTR expression.
Once the optimal CFTR construct is chosen based on the
expression study, the electrochemical properties of the
transduced CFTR gene will be studied in CF patient's
respiratory epithelial cells.
All of the amplified fragments have XbaI site at the
5' end and BamHI site at the 3' end so they are digested
with XbaI and BamHI and cloned into XbaI and BamHI
digested vector pHEF. HSCs are then added on top of the
TV producers (stromal cell transfectants) and co-cultured
for 1-5 days (or longer) to allow infection of HSCs by
the vectors via direct contact with producer cells. This
protocol greatly improve the transduction efficiency of
HSCs with the lentiviral HP/TV vector (up to more than
50%) which can be determined by the methylcellulose
colony (LTC-IC, long term culture initiating colony
assay). See Glimm H, Kiem HP, Darovsky B, et al.
Efficient gene transfer in primitive CD34+/CD381o human
bone marrow cells reselected after long-term exposure to
GALV-pseudotyped retroviral vector. Hum Gene Ther 1997;
8:2079-86.
Example 1002 Retro- and lenzi-viral transduction and in
vitro culture ofestablished and primary breast cancer
cells.
Like Example 1001, this example is a mixture of
preliminary studies and planned experiments.
The ultimate goal of gene therapy for treating
breast cancer is to generate a cancer vaccine to prevent
the growth of abnormal breast or mammary tumors. A
cancer vaccine can be generated by genetically modifying
cancer cells using appropriate gene therapy vectors. For
therapeutic purposes, efficient in vitro or in vivo
delivery of therapeutic genes to the cancer targets is
essential. We have tested two breast cancer cell lines,
MCF7 and MDA468, for retro- and lentiviral gene transfer.
Since many tumor cells lose major histocompatibility
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516 .
239
complex (MHC) surface markers which are essential to
immune recognition, we first examined these molecules by
FACS analyses. MCF7 cells expressed neither class I or
class II MHC molecules, whereas MDA468 expressed class I
strongly but not class II MHC. It is known that
cytokines such as interferon gamma (IFN-g) induces or
up-regulates cellular class I or II MHC expression. To
test this, we constructed three retroviral (non-
lentiviral) vectors encoding IFN-g, GM-CSF and IL-12
(bicistronic A and B chains of human IL-12),
respectively, and transduced both cell lines with all
three retroviral vectors and re-examined the cell surface
MHC expression following transduction. The results
showed that there was an up-regulation of both class I
and II MHC expression for MCF7 but not for MDA468 cells
following transduction of all three cytokine genes. To
see whether lentiviral vector could transduce these two
breast cancer cell lines, cells were transduced with a
green fluorescent protein (GFP) reporter vector,
pTVnEF-GFP, and the GFP expression was analyzed by FACS
48-72 hr after transduction. Following the lenti-TV
transduction, more than 500 of MDA468 and MCF7 cells
expressed the lentiviral GFP gene as illustrated by the
green fluorescent shift of the Lransduced cells.
To see how efficiently primary breast tumor cells
could be transduced with retro- or lentiviral vectors, a
total of 8 primary breast cancer specimens were processed
by mincing, digesting with collagenase, hyaluronidase,
and DNase, and passing through a stainless steel mesh.
Single cell suspension and small chunks of tumor tissue
were plated onto Falcon Primaria tissue culture flasks or
regular tissue flasks coated with collagen. Six of the
eight specimens attached to the flask and expanded into
small colonies. Five of the six samples had been
cultured for longer than one month and three of the six
for longer than two months. The overall growth rate of
the breast cancer cells was slow in vitro. Nevertheless,
we were able to keep them in culture long enough for the
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516 .
240
gene transduction study. Using conventional MLV-derived
retroviral vectors, we observed poor transduction
efficiencies in the primary breast tumor culture. In
contrast, the primary breast tumor cells showed GFP
expression after they were transduced with the lentiviral
GFP reporter vector. Although preliminary, these results
demonstrated the feasibility and certain advantages of
lentiviral vectors over retroviral vectors for the
transduction of primary breast cancer cells.
Establishment of a scid/beiQe mouse human tumor
model.
In vivo study of human tumors requires appropriate
immunodeficiency animals so to minimize xenograft
rejection. Nude mice and scid/scid mice have both been
used in such xenograft studies but the human tumor take
rate is often less than satisfactory in these animals
likely due to the remaining immune functions within these
animals which can reject foreign tissues. Alternative
strains of scid mice have been considered for the in vivo
human tumor transplantation study. It has been reported
that the SCID/beige mice, lacking all the T, B and
natural killer (NK) cell functions, are severely
immunodeficient. To test if this strain of mice are
suitable for human tumor engraftment, we injected
different human tumor cells into this strain of mice and
studied the success rate for engraftment. The results
showed that all the human tumor lines tested including
breast tumor, melanoma, hepatoma, and glioblastoma, were
successfully engrafted into the scid/beige mice. In
addition, most of the tumors were tangible within one
week. The same~melanoma tumor line was reported to have
a 60% tumor take rate in the scid/scid mice. The breast
cancer cell line MCF7 has been reported to be engraftable
only in the presence of estrogen in scid/scid mice. We
have successfully engrafted MCF7 in the scid/beige mice
without external supplies of estrogen. Therefore, the
scid/beige mice might be useful as an in vivo human
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516 .
241
breast cancer model.
In addition to the use of established tumor cell
lines, engraftment of primary human tumors into
scid/beige mice was also studied. The study, however,
was restricted to primary melanoma, glioblastoma and
hepatoma. Our results demonstrated that all three
surgical melanoma tumors were successfully engrafted in
the scid/beige mice, although at a slower growth rate
than that of the established melanoma cell lines. The
engraftment of primary glioblastoma and hepatoma in the
scid/beige mice has not been successful, i.e. no palpable
tumor detectable in 3 months. The success rate of
primary human tumor engraftment largely depends on the
condition and the stage of the cancer cells obtained at
surgery, as well as the tissue or cell types. As our
sample size is still small, no firm conclusion can be
made before more primary tumor specimens are examined.
Comparing transduction efficiencies of lentiviral
vs. retroviral vectors in breast cancer cell lines and
primary breast tumor culture.
Experimental design and methodology: Viral vectors
including retroviral vectors pMFG-nlacZ, pMFG-GFP and
Ientiviral vectors pTVDEFnlacZ, pTVDEFGFP, carrying
either nlacZ or GFP reporter gene cassette, will be
prepared by DNA co-transfection using relevant packaging
plasmids, pHEF-gag-pol for the MLV vector, and pHP for
the lentiviral vector. These vectors will be pseudotyped
with VSV-G envelope protein by cotransfection with a
VSV-G plasmid and the vector titer will be determined on
human HeLa or TE671 cells. Both established breast
cancer cell lines and primary tumor specimens will be
used for this study. We plan to culture 20 primary
breast cancer specimens plus the two breast tumor cell
lines, MCF7 and MDA468. The primary tumor tissues will
be processed as described above and transduced after
plated out in tissue culture for 24 h. The cells will be
transduced three times using an infectious dose between
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
242
104-106/ml with vectors carrying either nlacZ or GFP
reporter gene. Transduction efficiency will be
determined using the X-Gal colorimetric staining method
for nlacZ expression or using a fluorescent microscope or
FAGS flow cytometry for GFP expression. For long term
expression study, the transduced cells will be maintained
in culture for six months and assayed for gene expression
and stability. Gene integration will be determined by
Southern analysis. The percentage of transduced cells
will be determined after each 5-10 passages. Long term
study of the primary culture will depend on their
culturing efficiency. The surgical tumor specimens will
also be transduced with viral vectors as soon as they are
processed into single cell suspension. The efficiency of
transduction can be improved by using concentrated vector
preparations as the VSV-G pseudotyped lenti- or
retro-viral vectors can be concentrated to 10E8 - 10E9
transducing units/ml by ultracentrifugation. The
transduction efficiency will be determined at different
time points after the surgical tissues being cultured.
This study will determine whether it is possible to
generate gene-modified tumor cells and whether or not the
transgene will continue to express at high efficiency.
Targeted transgene expression in breast cancer cells
using lenti viral vectors.
Rationale: A major concern with human gene therapy
practice has been the specificity of the transgene to be
accurately delivered to the target tissue. The
therapeutic genes can be engineered to contain tissue
specific enhancer/promoter so to restrict its expression
in specific cells or tissues. The DF3/MUC1 gene has been
shown to contain breast cancer specific promoter by
Kovarik et al. and Manome et al. Using adenoviruses
containing the DF3 promoter, Chen et al. have further
demonstrated the tissue specificity of DF3/MUC1 promoter
using the Ad-tk gene transduction approach. The
generation of a breast tumor specific lentiviral vector
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516 .
243
with increased specificity and possibly improved
expression efficiency will be useful for future breast
cancer gene therapy application. We propose to test the
DF3 tumor specific promoter using the lentiviral vector
in this study.
Experimental design and methodology: To generate a
breast tumor specific lentiviral vector the DF3 promoter,
from nucleotide -725 to +31 in the DF3/MUC1 gene, will be
amplified from chromosomal DNA prepared from MCF7 tumor
cells by using the following two primers:
5' primer - ATA AGA ATG CGG CCG CTA AGT GA AAT TTC TTC
CC-, and
3' primer - CTA GCT AGC GGA AGA AAG AGA CGG-.
The amplified DNA will be cloned into pTVDEFnIacZ and
pTVDEFGFP using the Not I and Nhe I restriction sites to
replace the EF-la promoter to generate pTVDDF3nIacZ and
pTVDDF3GFP. Transduction efficiencies of these newly
constructed vectors will be compared with those of the
CMV or the EF-la driven vectors in breast tumor cell
lines. The tissue specificity will be determined by
comparing reporter gene expression in the breast tumor
cells with non-breast tumor cell lines such as HeLa and
TE671 cells and normal human tissues like foreskin
fibroblasts. Tissue specific expression will be
quantitatively determined by Northern analysis of the
transgene mRNA, and by reporter gene assay.
Assessing lentiviral gene transduction in vivo following
intratumoral injection.
Rationale: In vivo gene transduction using
lentiviral vectors have been successfully demonstrated in
rat brains, eyes, and lungs 47-49. Using the HP/TV
lentiviral vector system, we have demonstrated efficient
in vivo transduction of muscles of rats. Our preliminary
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
244
studies also showed that the HP/TV vectors transduced
tissue culture cell lines such as HeLa, TE671 and HepG2
cells more efficiently than the MFG retroviral vectors
both short term and long term (more than 6 months).
Transduction via intratumoral injection using
retroviral vectors have been very inefficient. It is
possible that the center of the solid tumor may grow
slowly or stop growing totally and therefore retroviral
transduction becomes inefficient. Since the HP/TV
vectors transduce non-dividing cells, they may also
transduce solid tumors more efficiently than retroviral
vectors. To prove this, we will evaluate the in vivo
transduction efficiency of the HP/TV lentiviral vectors
using the human breast tumor scid/beige mouse model.
Experimental design and methodology: We will first
use the s.c. established breast tumors in scid/beige mice
for the intratumoral injection. The mice will be
injected s.c., on both flanks, with 5x106 MCF7 (or MDA)
cells on each side. It normally takes two weeks to
establish palpable solid breast tumor in the mice. Once
palpable, the tumor nodules will receive three times of
vector injection, either unconcentrated (usually 105
tu/ml) or concentrated (106-107 tu/ml), one time each day
for 3 days. For side-by-side comparison, one frank of
the mice will receive HP/TV vectors and the other
retroviral vectors. The mice will be terminated after
three days and the injected tumors will be fixed in 4%
paraformaldehyde and sectioned for reporter gene assay.
The initial short term study will determine the
efficiency of transduction using different concentrations
of vectors. Once the short term expression is
demonstrated, long term study will be initiated. The
breast tumor will be established in mice and vectors will
be injected and at different time intervals, from 1 week
to 2 months, mice will be terminated for analysis.
To see if the lentiviral vectors can transduce
primary tumors in vivo, scid/beige mice implanted with
tumors in the mammary fatpad will be injected with either
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516 .
245
retro- or lentiviral vectors and the mice will be
sacrificed at different time points. The analyses of
reporter gene transduced tumors will determine the
efficiency of transduction. At the same time,
micrometastasis of the primary breast tumors to other
organ sites will be studied.
These in vivo studies will determine the efficiency
of intratumoral transduction of lentiviral vectors.
Further experiments will test the breast tumor-targeting
vectors pTVDDF3nIacZ and pTVDDF3GFP. These vectors
should exhibit tumor specific gene expression and
therefore the surrounding normal tissue should have
minimal transgene expression. To determine the
specificity of these DF3 vectors, both the breast tumor
cell lines and the primary tumors established in the
scid/beige mice will be transduced and studied. Tumor
specific gene expression will be examined carefully in
the metastasis sites to evaluate the vector specificity.
For quantitative determination of viral transduction
efficiency, in situ hybridization will be performed on
tissue section using biotinylated DNA probes. The
efficiency of DNA integration will be compared with the
relevant transgene expression to determine if all
transgenes are expressed in the target tissue.
2 5 A11 publ i ca ti ons and pa ten is men ti oned in the above
specification are herein incorporated by reference.
Various modifications and variations of the described
method and system of the invention will be apparent to
those skilled in the art without departing from the scope
and spirit of the invention. Although the invention has
been described in connection wi th specific preferred
embodiments, it should be understood that the invention
as claimed should not be unduly limited to such specific
embodiments. Indeed, various modifications of the
described modes for carrying out the invention which are
obvious to those skilled in molecular biology or related
fields are intended to be within the scope of the
following claims.
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
246
Summarv of Preferred Embodiments
The preferred embodiments of the invention include
but are not limited to
1. A packaging vector comprising a nucleotide
sequence encoding Gag and Pol proteins of a reference
lentivirus, said packaging vector differing from said
reference lentivirus at least in that "
(a) its major splice donor site is either deleted,
or if provided, while functional, differs in sequence
from that of said reference lentivirus sufficiently so
that said major splice donor site is not a potential site
for homologous recombination between said packaging
vector and
said reference lentivirus, and
(b) it lacks a functional major packaging signal,
which vector, after introduction into a suitable host
cell, is capable of causing such cell, either through
expression from said vector alone, or through co-
expression from said vector and a second vector providing
for expression of a compatible envelope protein, to
produce packaging vector particles comprising functional
Gag and Pol proteins and having a normal or a pseudotyped
envelope, where said particles are free of the RNA form
of said packaging vector as a result of (b) above,
where said cell, as a result of said expression or
co-expression, produces particles encapsulating the RNA
form of a transducing vector possessing a compatible and
functional packaging signal if said transducing vector is
introduced into said cell.
2. The vector of embodiment 1 in which the
reference lentivirus is HIV-1.
3. The vector of embodiment 1 in which the
reference lentivirus is HIV-2.
4. The vector of embodiment 1 in which the
reference lentivirus is SIV.
5. The vector of embodiment 1 which encodes one or
more envelope proteins.
6. The vector of embodiment 1 which does not encode
CA 02333481 2000-11-27
WO 00/00600 PCTNS99/11516 .
247
a functional envelope protein.
7. The vector of embodiment 1 wherein the major
splice donor site of said vector differs in sequence from
that of any lentivirus major splice donor site
sufficiently so that said major splice donor site is not
a potential site for homologous recombination between
said packaging vector and said lentivirus. '
8. The vector of embodiment 7 wherein the major
splice donor site of said vector is substantially
identical to the RSV splice donor site.
9. The vector of embodiment 1 which comprises a
sequence encoding lentivirus Env proteins.
10. The vector of embodiment 1 which comprises a
sequence encoding the VSV-G envelope protein.
11. The vector of embodiment 1 which further
differs from said reference lentivirus in that at least
portions of at least one gene selected from the group
consisting of the env, vpr, vif, and v~u genes of said
reference lentivirus is or are deleted.
12. The vector of embodiment 1 which lacks the
native primer binding site of said reference lentivirus.
13. The vector of embodiment 1 which lacks the
native polypurine tract of said reference lentivirus.
14. The vector of embodiment 1 which lacks a
functional nef gene.
15. The vector of embodiment 1 which further
differs from said lentivirus in that the 5' LTR has been
modified.
16. The vector of embodiment 1 in which the 5' LTR
is a chimera of a lentivirus LTR and a CMV
enhancer/promoter.
17. The vector of embodiment 1 comprises a tat gene
and a TAR sequence.
18. The vector of embodiment 1 which comprises a
rev gene and an RRE element.
19. The vector of embodiment 1 which further
differs from the reference lentivirus in that at least a
portion of the tat gene and the TAR sequence are deleted.
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516 .
248
20. The vector of embodiment 1 which further
differs from the reference lentivirus in that at least a
portion of the env gene and the RRE element are deleted.
21. A packaging cell which comprises the packaging
vector of embodiment 1 and is suitable for production of
lentivirus-like particles.
22. The cell of embodiment 21, which further
comprises a pseudotyping vector.
' 23. The cell of embodiment 21 which further
comprises a transducing vector which by itself is
incapable of coding for expression of infectious
transducing vector particles, but which cell, as a result
of the expression of genes of said packaging vector,
packages the RNA form of said transducing vector into
infectious transducing vector particles.
24. The cell of embodiment 21 where said
transducing vector further comprises a remedial gene.
25. The cell of embodiment 21 wherein packaging is
inducible.
26. A method of a producing a transducing vector
comprising a remedial gene, in the form of an infectious
particle, which comprises
(a) transfecting a cell with a packaging vector
according to embodiment 1, and, if said packaging vector
does not itself provide for expression of a compatible
envelope protein, a pseudotyping vector which does
provide expression, so said cell is capable of producing
packaging vector particles,
(b) transfecting said cell with a transducing
vector comprising said remedial gene, and a functional
packaging signal, but which by itself is incapable of
causing a cell to produce transducing vector particles,
and
(c) causing the cell to produce infectious
transducing vector particles comprising said transducing
vector in RNA form, said Gag and Pol proteins, and said
envelope protein.
27. A method of delivering a remedial gene to
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516 .
249
target cells which comprises producing the particles by
the method of embodiment 26 and then (d) infecting the
target cells with an effective amount of the particles of
step (c) .
28. The method of embodiment 27 in which the cells
are nondividing cells.
29. The method of embodiment 27 in which the target
cells are cells in a target mammal.
30. A kit comprising a packaging vector according
to embodiment 1 and a transducing vector comprising a
functional and compatible packaging signal, said
transducing vector being incapable by itself of causing a
cell transfected by said tranducing vector to encapsulate
the RNA form of said transducing vector into a
lentivirus-like particle.
31. The kit of embodiment 30, said packaging vector
comprising a gene encoding a compatible envelope protein.
32. The kit of embodiment 30, further comprising a
pseudotyping vector comprising a gene encoding a non-
lentiviral envelope protein incorporatable into said
particles.
33. The packaging vector of embodiment 1 in which
the major splice donor site is a modified RSV major
splice donor site corresponding to the splice donor site
included in SEQ ID N0:9 and SEQ ID NO:10.
34. The packaging vector of embodiment 1 in which
the reference lentivirus is HIV, SIV, FIV or EIAV.
35. The packaging vector of embodiment 16 in which
the reference lentivirus is HIV, SIV, FIV or EIAV.
36. The packaging vector of embodiment 1 in which
the reference lentivirus is a primate lentivirus.
37. The packaging vector of embodiment 16 in which
the reference lentivirus is a primate lentivirus.
38. The vector of embodiment 1 in which the dimer
3S linkage site is inactivated.
39. The vector of embodiment 1 in which the gag AUG
is operably linked with a kozak sequence.
40. The vector of embodiment 1 in which the major
CA 02333481 2000-11-27
WO 00100600 PCT/US99/11516 .
250
splice donor site is deleted.
41. The vector of embodiment 1 in which rev is
inactivated and the INS's in QaQ and pol are likewise
inactivated.
42. The kit of embodiment 32 wherein, in the
packaging vector, rev is inactivated and the INS's in cLact
and pol are likewise inactivated, and RRE in env, if
present.
43. The vector of embodiment 1 in which net is
deleted.
44. The vector of embodiment 1 in which at least
part of the 3' LRT U3 region is deleted.
45. The vector of embodiment 1 in which the 3' LTR
R region is replaced by a functional, non-lentiviral,
poly A site.
46. The vector of embodiment 1 in which the 5' LTR
U5 region is replaced with a functional poly A signal.
47. The vector of embodiment 1 in which the 5' LTR
R region is deleted.
48. The vector of embodiment 1 in which the 5' LTR
U5 region is deleted.
49. The vector of embodiment 1 in which the 5' LTR
U3 integration attachment site is deleted.
50. H reference lentivirus-derived transducing
vector which by itself is incapable of coding for
expression of infectious transducing vector particles,
but comprising a packaging signal capable of interacting
with lentiviral virion proteins expressed in a cell in
which said vector resides to cause the RNA form of said
transducing vector to be packaged into infectious
transducing vector particles, said transducing vector
differing from its reference lentivirus by one or more
modifications in the 3' LTR and/or the R or U5 region of
the 5' LTR.
51. The vector of embodiment 50 which further
comprises a remedial gene operably linked to a promoter
functional in mammalian cells.
52. The packaging vector of embodiment 1, selected
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
251
from the group consisting of vectors substantially
identical to
pHP-1
pHP-VSVG
pHP-CMV
pHP-CMVnTAR/SD
pHP-CMV-EFla-intron
pHP-EF
pHP-1 dl2 ,.
pHP-1 d128
pHP-dl/env/vpuI
pHP-dl/env/vpuII
pHP-dl.vpr
pHP-vpr/ala/leu
pHP-vpr/env/vpuI
pHP-vpr/env/vpuII
pHP-dl.NdeI
53. A transducing vector selected from the group
consisting of vectors substantially identical to
pTV~Y100
pTV~Yl4 0
pTV~+CMV-nlacZ-hyg
pTVe
pTVnSM
' pTVnSVneo
pTVnCMVIacZ
pcDNA zeo-nlacZ
pTVeCMV-GFP
pTVeCMV-nlacZ
pTVnCMV-nlacZ-hyg
pTVnEFnlacZ
pTVnCMV-GRF
pTVdI.EFnlacZ
pTVdI.EFGFP
pTVnAUGl
pTVnAUG2
pTVnSDl
pTVnSD2
pTVgag dl.l
pTVgag d1.2
pTVgag d1.3
pTVgag d1.4
pTVgag d1.5
pTVenv dl.l
pTVenv d1.2
pTVenv d1.3
pTVenv d1.4
pTVenv d1.5
pTVenv d1.6
pTV dl.RRE
pTV dl.gag/env/RRE
pTV eSDl/AUG2
pTV eSDl/env d1.6
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
252
pTV oSDI/dl.gag/env/RRE
pTV dl.kB/Spl
pTV U3 dl.l
pTV U3 d1.2
pTV U3 d1.3
pTV U3 d1.4
CA 02333481 2000-11-27
WO 00/00600 253 PCT/US99/11516
O
o m "
00
W n
v
~ ~ ro
o : . .r.,
r v _ ro .
,
h
W .
a
O
M ~ O (!~ -rl N
ro
i ,~ Q -r-I ~ .~-1
i v
tl? H U
d~
'~ ~
z
W .t~
-.-I
a. ~ ~ N ~ _
-
v ~ -,~ _ o a - ,-, m
~ u~
z > v; x
0
x v~ '
~ ~ b '
.~
a ~ ~ o ~ .~i ~~
U ~
~' _
a y ~ ~ u~
3 '~
'~ ~ , ~n
x a~ vi ~ ~
x .~ q o
~ u~
.C7 '~ v
U H ~ U
bi ~
.~
v
-
N ~ ~ ~ ~ ~
~
C2. ~> v- . ~ ~
~
x U ~ ~ ~
~
E ' -~ ~ f~
~1
U7 U O H
~
~ ~ 1 _ f~ r-I v (a -rl 01
v r--i
~
, ~ - ~ ~ '-~ J.J ~ 4-I p1
w . !l~ r-i
~ .~ ~ w ~ v w r~ v .~ ~
~--s w
v
. ., ~ ~ O ~ '
. ~ ~
?~ ~ > x - 3 .~ 0
r~ m t
~ ~ U ~ N ~ .,~ ~ u7
w ~r
O
m
UI ?~ ?~ ro v ~ ~ a ~ o v~,
~ d' rnwW
v Ua U ~~ v ~r v -- ~ .~~
~ ~ .~
oo,
s~~ ~z ~~
v a, -~ ra N o, ~ -~ U
w ~ ~ .x ~n ~
v ~
-.-~ o
v
--
O -.~ -~I O ~ ~ ,-~ O -- O x O
' ~ ~ U O ~
c~
C!~
U
O~ U U I~ b'.~'..~ ~ I~- 00 -~-i ..C,
w N v ~ .("., I11
-'-~i r-1 C~
~
'
t~
O . -,...~.,..~pa U U -- o~ o0 fIJ ,
.-, ~ ro ~ ~ pq _ ttf Q,
ro r-I
~ N
N
~-I
t~ v w w ~ ~ ~ N N ~ ~ ~r E
O .~ y~ O E ~ cn
~ E N N O O ~ ~ ~- O ~ ~ U1 >.,
(~ ~ O v _ ~ -~ O
cd CJ~
t1'
a1
H o ro ro ~~~~ w o ,~ . --v v.~ ~
U ~ .
1,
v
H ~ o O b~ o E a~ W ~, 3 cn ~
~ ro m
H v ~ ~ ~ . yn ~ . ro w ~ Ei
.u v _ ~
,...i
W 31 ~ ~ U1 ~" Ul U v O O
~., (Y, !Lf r-I N
rl
in E E ~ u~ O ~ - tn .~ u~ U
c~ r.~ ~
FC m
tl
E E f-i -~ O .-i tn v U
M u ~ ~ N ~ ~ U ~ U~
z ,n ~ , -~1 mo
N .L2
-~ M
. . .~ ~ -.~ o -.~ ,..~ x
.,~ m ra N ~n . o v o
-~ ~
~n cn
. o
~
M
v .p v ~ r~ U .~.~ ~ r-, .~ o
~ -.~ ~ oo ca a -~ s~
~ a v s~ ~
ro
x
o
~
M
a ~ ~ ~, ~ v -~, ,~ ~n ~
~ ~ m .~ ~ ,-~ .~ a -~
~ - ..~ ~ o,
N o ~
~r ~
v
o,
pa ~ a3 ~ -- ro v ~ ao ~ -- . .
o, o ~ U .~ ~- .x ~ ~ O
co u~ a
,.-i
--
~
r-i
r-,
m
~ ~ ~ ~ ~ r~ O w N U f~ -~ .u
~ U S~ .~ ~ ~ N
N -~
U
~
o
H Q ~ ".~ v '~ ~.I L: lp .('', '~~
H ~-1 N -rH ~ ~'.,~ ~ ~ ~
l'~1 N
fLS
~
v
Ul
xx U~ x x>~~,~rx w-~ .~h~Na~ ~Ma~ rn vh~,~3zUao
rn s~
z
0 0 ~ win aww~n aw~n aww~n
aw
~zU~ ~Urx ~zUrx
zo ~z
w zHzowzzow zHzowzH
~ H
w wx a c~awxac~wxa c~awxarxa
~ W 3 W ~ H ~ q rx D ~1 c~
~ H H ~ E-~ E-~
P~ E~ ~ ~1
E-~
O W W O W W p
~ ~ H O H O W
W C~
H
O U W W ~ E-~ ~ ~ W ~ W ~C ~ ~ Cs.
W q O ~C E-~ E~ FC E-~
~ ~
are ~z xu~ o rx
~ ~
H H N N M
M
CA 02333481 2000-11-27
WO 00/00600 254 PCT/US99/11516 .
v
vp M
J-~ 1'i V~ f~ M
zn a a ~ ~ '
~~ ,~~~ b
-~
i v A '~ ~ a H -~, ~,
v o ~ ~ z
H
a v H
, ci
r, ~n .~ ~n ~, x ca ~ s.~ ~ a, ~ v
~n w v u~ ,~
o x a H >~ v v -~ x v v v v ~ v zs s~,
H
v -~ ~d v o rn ~ ~ v E v E t~ U o
~ x -~I
U ~ ra ~ ,~ .-i t~ _ ,~ _ ,.>~ cd
H ~ ~ c(S O ~ U
L~ 1..1 ~ fa 1J b1 .~", ~, .!J Ul -rl
O ,~. H J.-~ ~ N flj
U7
v ~ 4.a v ~ ~ ~ x o o v ~ ~ E cn
E .x o ~ ,~ w ~, m -~ ~n ~ w v >~ r~
~a w tr o o
v
r G a rt ~ O u~ v 3 .~ .o O .~ v
~ O ~ -~
.~.
m ~ ~ rn rn v ~ -- v O ~ O ~ m m -~ v
-~, rn
~ U x s~ x s~ s~ E v
v
U7U ra c~ N t~ -.-~ ~ p ~ O E O 'd ~I
T3 ~-1 ~ ~ _ E c>~ ~
w-1 ,~ v ~ ..>~ Ti -~-Is~ t~ w-I O
O O v U -~ -r1 -~
O
.C~ .IJ .N O U~ U7 -rl -~-I -~-I 1J 1-I 1~ U
~ i~ .~..) U1 U N tJ1
~
al N U N ~-I l..l ra rti ~ W -rl
Cf' 1.~ S-Wa.-In'S !~ s~
vH,~ o ~I v v~ 3 0 3 0 ~ o-~I cn v
E z7 ~ H E-, ~ y~ ~ ,-.If.~, p., cn v v -~I
m .u cn O ~d
O N O H C1'L~ H v fa '., ~ N ~I >~ t71 U
N U7 O
b ,-~ ~n ~ -~ H rd ..~ .~ m ~ -r-I s~
v . m v U
~
4-I-rl U 'Lj 'LJ U H ,.f.;H Q H O 4l E fij 4-I
' Z31
J C.' N >-..' N (fj 'Z7 ''[~ H r--I .i~, N
(U E-~ ''O H .C.' f>3
r-I >:.,'' rl
N O rl la 1.~ O >~ ~ N H p H U .a..i U 'C3-
U1 ~ U (d O
U ~-I rU -~ (1, E-~ O U U >~ ,~ R.~ O
co ~ rti ctS
f.1 ~I -~ U~ U7 ~ J~ ~ N ~ N i~ 4-1 O >~ f.2,
ao ~ O J.~ -r1 U
v o~ -~I 4-a N -~ t~ N .~ N ~ ~ O N O ~ O
r~ >~
?~ r ~ r-I H ~I m E ~ ~ ~ ~r N U~ O E U
~-I w v t~ ~ U r-I ,.C
L31r-1 O fa (1,' S-I Q) ~ f-1 ~ S-i O ~ 01 U7 E
~ fLS O ri Lfl a N .i..l
v z1 E ~I .~ O O ~ tr v b v 23 v 3 w s~ -.~
u!~ ~ v b~ d'
~ O !~ U U rd ao
~ ~
s cn m G w H !~ n N m
-~ ~ ~ N W ~
h S~ - W ~-I r-~I w ra w
O w O H r w
r~ -~ a~ ~I
x ~ ~-I ~ ~n ~ o -~I _ -,~ _ .~ .~ a o, W
~n -~, x _
v V~ tl7 v O U7 a..yl1 N ~''ON ~'b U co -.-i
~ O ~
v N ~n ~ .~ -,~ .~, ~ ~ >~ -- v -~I o .-, ~ rx
~ v ~ -~ ~ v t~
U -.~ o ~n .~ rn o o -_ o -- v ~ m ~a x N
s~ ~n -~I v b .u . ~ to
~
o ~ ~ w ~ v H ..ca o o mn ~-mn s~ r>~ ~ ~ = a
~ v -.~ v
r~
va v~z cn ~Ia ra rn~r Uao U~ rrs~ U o. a a
R,, rrs~ u~ o
U U -~ W ~ U o~ ov O ~ O -.1\ E
O ~ E-~
tr t~ v m tn ~n -~I r~ m ~-I v cn .u >~ m u~
v ~ ~ ~ b w x o
v rx
u~
v . v u-I v >~ a v -~I H -- H -- ~ .,~ -~ o -r-I.
zs ~ rt ~ ~ o - a o, ,r,
.,~ ' _
N U1 ;~ >;: J.-1 O ri H O H O S-1 J-~ tn rl ~.,"II
ffS .("., O 'b 1J G~. O d~ tl1
II
ct~'i~ ~-rl (~ -r! '~ (a H t~ H l~ rl .--.O .l-J(a .
ilk U ~ (~ r-W (~ (~ . l~ l'~N
-.
v
al4-I N ~"' r-1 1J O N ri r-I U ,'~ (?r(~ t311.1
tI) U1 U) _ U E 01 l~
,~ 1J
O N cn O rt U a .N i~ rl ~-I ~ p; O U ~I 0
-~I . ~ cn >~ E ~
p~ 0
N ,~ ~ U1 S~ ~ r>i p., c>~ rtS om..~ +.~O O W
.~ N a~ pp W O O ~I n
.~..~
pp
,~y M ~'., -r1 -rl ~I ~., ~,' '~,' fa a \ \
1J rl ',~ J..1 H -rl ~,.," (Y., rl V-1H H cr
~.1 \
Cdr i to ,.Q , O 4-I ~ , -~I ~ r~
N H cti '. ~ m w
~
O O a1 ~ >~ ~ E 5 ~ rn b~ eT O ~ ~-I ~
v -~I H !~ O E-~ -~ cn
s~
~-I
cnU a ..~ H O N ~I ~ -~ ~r -~ O
~ O -~ d~ -~-I ~ r.>r 5 v
h N
z ~ x v v w ~ m ~ ,~ ~ ~ v w v
a v ~I v ~ -~ ~
t~ c~ E v .~ cn v ..~ o ~ o rl E E cn 0
.u E C~ ~ b b
E
~ H O E ~ 1..~ m ~ U~ ~-I ~-I O ~ w
a.-> -~ O -~I ~
~-I
v v Fc v U o v ra -~ ~n v v v v U o v >
U v u~ m
o
v r-I .~ v ~I 4-I 4-I .s~ U N 4.-IN N
H U ~ ..~ ,~ O O ,s~ ~ N v
~ U v 4-a
cnU z H s~ w o o cn .u H -- H -- ~, -- ~, ~,
~ ~ .~ ~, ~, ~, ~, ~, ~,
--
v
i
U r~
~ U
z a
w
w m
x
x
H
0
w
U
w
'n o ~n o m
ri r-~ N N l'~1 M
CA 02333481 2000-11-27
WO 00/00600 PCT/L1S99/11516
255
waw~~aUa waw~~aU~xc~
xa a.rxHxa rxa wrxHx~na~
w~'~x~H~r~ ~ w~~xaH~r.~aa
awxaHxHZ ~ awxaHxHw~~
wx rxxUxc~ v wx~xxUxrxuH
c~n~~3a~oax _a~ ~H3azAx Hu
~c~~3awa~xH
>a~a~azxxa ~ ~xzwuz~z~uH~
~aHa~Hzw~~x
H~~wHo~z~ n ~ a HHp;wHpIzG4aC7
xrxww~~~ww ~ v = xxa~w3~~~wU
xau~zrxax~a ~ a= ~ xacnzrxaxaxH
axHHxaaH~n ~ v v o axHHxaaaaw
H x U' 1) ~I t77 U1 x ~ ~ H ~ H H w ,.~ I-I
~UawHw~s~w ,~ ~s s~ ~UaWHw~rxaa
a~xxwHHaa b ~, ~ a~xxwHHwNH
xa~a~~Hwcn >~ v-~ U xa~ac~~NwHo~
x~~~~~~c rx o~.-a v x~~~~ ~ are
u~aawza,~a ~ m s~ c~~aaw~Zw~w~
uHu Hxza~n »,v ca., uHu Hxzawa
wHa a~~Hc~~ -~, > ~n ~,ra ~~~rxu~u
= c~za~ww>wa ~ ~ v ~ r~za~r~w~ww~
z = ~ a~zazaaHa~ m-~~ -~ a~zazaaNu~x
x ~ o r~aaxxcncnU~ ra v r~aaxxcnv~U~H
E ~ H U7 ~ ~7 H ~ ~ ~1 a ~-I ~.-~I .u H cn a L7 E-W ~7 E-~ f~
~ xrxHu~ol xw ro,sa,J o xxHu a~axc~c~
U o ~ waa,~~ww~x ~ ~ ~, wa~,~~ww~au
-~' ~ v 3w~HU~~Caa ov v a. 3w~r-~u axH
v xw zH axH ~n v~ ~, xwzzH~d~x a
o _~ ~ a~naaaa~uw o 3 0 ,-~ av~aaaa~u~u
s.~ aurn~wHxww .~ o a. o aucn~wHrxw~H
wH~Nax xa--. -~,r-, >~ wH~Nax~x~~
rn ~ ~ c~aaza~~ux.~ >~~Ix c~aaza~ uw
z -~I uacnaHr~xUao~ o U ~ ua~naHwxUV~w
rx v cW n z ~1 W rx C7 x ~n o ~-I u.~ -~ o cn cn z ~1 cn rx C~ x a x
~= awzaHaa~3w~n ~ .~ -~= ar~zaHar~3cn3
O oo ~ pr ~7 W E-~ ~ C7 U Ei ~ a~ 3 u~ ~ ~ OI C7 c~ E~ ~ C7 U z rx
>.~,~v~aNNC~~uc~H ~ ~~o ~,~cnaHrlu~uuzc~
v v a~~~ar~~c~a~xH~r, ,~o v w~ aa~~a~xaw
uHcxc~xw~o N ~ w~u uHCxc~xxa
rlN~a~wawaxwo v ~ ~ ~N~a~wr~warxr~z
O M C7 H U! U7 W U W L7 N N II7 1J p M (7 H U7 U7 ~L U C1~
~ a~rn~aaauw~~r~ ._ goo U w rn~aaauw~~5w
rxa~rxxH rx~~ ~ao ~s,~ ~ rxaa;xxHxaw
~1 ~I II CJ1 (~ II U U7 l1~ l~ ~ U fs, 00 O .-~ N S~ II ~ [a II U Cn W !3a ~ U
a a
.I~ raH,~>;~~wwc.7~zcno Q,m ~,.~ raH ~u~wwc~~zw~
~luw owx~xawxwN ~ ~.u m ~uw owx~xawxrxH
_ _ -~~nxw~c~w~w ~ rn~a r» ra= _ -~u~xa~zcxwr~c~r~
lD fa l0 f($ N (~ 1~ II II 1J [-i Cn H QI H a H Cl, ~ ([f ~ ~ ,1,~ II II 1-1 [-
i CIA H QI H ,a H [-i Q
N.uc~ >JO, rnu~.uw r»wx~uaHxwo:~"~ v o u~,~4..a rawx»aHxw~C
~= N= ~U vrlazwwa~ur~o,= = rJ o ~U v,-lazwa~a~c~~nc~
o, mn n N a >~ ~ ~I ~naaw~Ht~wE-~c~ a a ~~ ~ ~ ~ u~ao~w~Hc~w E
~ v . v ~ v ov ~ ~uwv~HCnwxw--v v v ~ o~ ~t ~t~wcnH~na~x ca
~b o mwwwa~HC~uw_s~ ~~ ~ ezs o mwwr~a~HC~u~a
In o~r oo v o ~,~ ~~H~~~az~ o rn~ ~ °' ° ~b~~H~~~aza~
u1 ~ d~ ~ a~ ~1 U f.~''C? ~
~r~.~wwwxwx~~r~cn-~-,w mw.wxwx~~r.~ww
c~
U
!~
rd
1J
O
E
~ 4 U
'rv o m o m
'~ r.I N N M M
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
256
Hw3xaaxr~~~cnHxrx= wax or
wHc~a~nHc~~c~xuxacnr~ xr~H
~uaa~a3~~nc~r~~xAw xa~a a
aawx~AHw aH~Nwxn xax z
aaazx~H HC~ax~~a ~~a,
~ a
~~rxa~nHwc~~cx wax =
x
H w z a 3 E-
x w cn H
rC H ~n
x w 4 pn
x3~wuawuz~ x~ r~~na
3x
wwawHH~a~
x~ w~ x~~ rx~n
wA~Ha~~Hwc~w~a zu rx w~
~
Harxxx~Hwaa~~uaA xa wu
aHHNac~xwc~~xaHHr~ c~x~ xz
wrxc~xHxaxv~aaxHxc~ ~~a
3xwxo~>wr~waaxH~ Hey.
azHazw~aazuaxH ~,
wa
xawc~>wwaa~xwaaa ~Ha w~
>wzHNxH~~~a~wxx xcn~n ma
xxzw3aa~aHu~xac~ x~u x x
wwHa~nHo~axucnHZa~ xua a H
uacnaawxxHxr~w~wa tax W >
r~~w~nxrx~5~x auz wc~
~a~nxrx
~ rxax, a
~aHUwzaaa
HuwHH
c~x~>wwzwaww~HorH xxc~ arx
wxw~a~Hw w~xw~Hx ~~r~ w~
x 3 ~ a H 3 H a a a x ~
> a x N ~ ~
axHC~Hw~z c~
a ~
x
~~a,o~H zx
xH~r~wac~~a Hw wx ~u -~
Hcn~~
acnx~awxw z~w~o~Hr~ xu ut~ wu
LL Ca C~.' E-i
~ ~ ~ ~ ~ Ci,
W W W
H W
~xwaHwrx3 Hxar~zC~~ ~xw~
Hxa~~w~w awzw~~ ac
a a~w -~,
wxxHxN~a~HxwHa,Hx nH
H H (~ ~
W a ~,
H a x A
~ H (Y.,
H
a~~aHwwu~wHCnc~c7WCa ~cxt~ o~w off
cnww4x HH ~
a mr
~m
~c HUrx a,a u~
H~aa~a~x~~a
~w~z
~ H Hax ua voo
a~wcnzwwaaxw = HHc~ = as ~,
~a~nn ~
wNwa~HZZaH
x
~a o ~,c~a
~ a~Hx
zz~xuax ~rxH3 NaazH Nwa u~
H~H
a~~x~~w w~az~c~c~~ ~r3xrxx ~rwH o
H w a w
3 w H oo x ao ~ rx M
rx r~ H
m a
cn
~
~C
H
uz~waHw r~~xa Nzr~~a, Notes a~oo
c~w~wwua~ ~~C
Na~H~u~
Mwc~~nx MwH ,~
Hr~r~awc~~r~ ~wx~uxw rn~awz rn~r~ .~
ac~aHw,~Hw~cnwx ~_ ~.= ~_ -~= a
xc~ a~cn~ w
~H~xwaa3uwxr~~xx a ~a n ~,q o n
a Hwr~ n ~
axHHxx~H ~-~H ~ n,H H~
cnww w~ ~xuw ~c~ ,~
acn~~auHw~xwa~za s
=
~~a~ = ~~w ~x
zHCncnxa3xc~aaxHxa~~.-~ o>r~w o~ ._
ra= _ ~ ~_ _ 0 ."
-~wwN~, .
,3~0
xxxuxaaaar~ .u a ~ ~o
HNHxH-~ a ~u~~a~r a,.u n r~.~
~c~xaxxc~Hxaa~u~nw~~ cn a ~H~
~
.~~-.~ > cn,~~ wM"
HWawr~~awH~r~a ~~a~~,= ~a~nxx~o r~r~~ u,
m v~,Hac~~, = m a~~c~~, = ao=
HxxHa,xc~awaHa w ~ ~ ~ ~ ~ ~ a ~,
~ cnxx~ ~n~ ~ u
. ~ ~
a
~
wa~HHaxr~Hc~a x ob x a~ ow .
~ a . ~wH~n x aH ~--v
E a~ . ~ o
~ z
c ~s o ~ zs o .u
~ ~ it ~ ra ~
-~ ~ x w z a w M ~
zH q 3 w a, b
~n a ~
~
ww~ccnw~ax~C
uaH
~
~
c~> o ~,,p a~ o s~.~ o-~
~ o ~,~aa~n ~~~ a~
o
awc7z~wo ~ a~zs~v~C~a,m rn~ ~,zi~H~ a o
tn tnu
HH~~~~H~
wuxx3HwNC~~caa~aHr~~, www~a,~ wwx~, w~w
ra
i
a ~ -.~ o
a
m o u~ o ~n o m
r-I r-1 N N M
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
257
o, ~wzw~~~;axa~
x~, A ~~w~wrxcnc~worz
~~v~uz
cnzH
a ~ z
c~a 3
c~AU~ HwHaa
~~ZZx~nzaz H
x
~'
xHrxuau~
U aa
~3Hw
~~, ~ H Hcnxaw
~
~
~wH
~nurxr~a
~
_ ~z~w
azrxxwv~
zwww~~a
~
z
o a
a
~
w w~ H ~ xw~~wzwuN
cn
wHH~~uw~nwa3
z~ ~ ~ H ~xaHwH~a~n
~~rxwcnxc~w~zrar
_ ~ ~uc~c~c~wuzz
w ~ H . ~-I U
a cn r~
z H a ~C
a O~ 3
H cnaa r~z~r~aa~
~
u~ ~~ H o a, ~ ~w way
H u~Ha w~~H
~ rn awzHa
zr~ ar-~ = HaH~a~x
Hw ~a xHN
rna ~~ :7wz~~rx-~u
q G~
v ~ a
~
aw ~ zHHv~c~~~
cnu
a r~ v ca aa~az
H a,HwxaH
~ ~azu~
uwa~aa
a,a a~ Hq ~o HHxcnwc~a~3xax
N uwaHrxar~~~c~c~
rxa
Hw ~,~ ~a aaHH~za~cn
~nN as
H~ -~I a. xu Hrxuxc~Hwau
~a ~~ Hx v rn z~ u~xH~nr~,v~~u
~x
~ uxxcnNv~~naxH~
a
o . ~HaHUxax~a~-
x~~nzarxxHzHu
w p., rti
~ 3 ~ f~
~ E-~ f~
CY
q ~ a
~ .
o ~ a u
~
w ~~ qm H~zHurxcna
~c~ v rn
x
a s = cnrx = H~ awu~zHw~r-~a
' ~
'~= axw~
~w H N~Cw zNaoua
~
o, ~~a ,-, cnM~ ~HH a~xHU~A
~Ha rn Nx Z rxzHu~x
war~r~
~4a~o ~rcn~ o~rw Hxau r~zHC~Ha
~
~~z v~c Nw~ v.~.~ooxr~~xw u:~r~n~~n~
o n "
C1~ ~
a ~''~ a ~ , N ~ ~
U ~ C7
z Cra c!a
p,' U'
Q.,'
b1 ~l ~ H = O -r1 M
E ~~W fx W CL
~ E-~ H Ot
~ Cs-~
W
r--I rn~~xwuww
~
_ _ WH
_ (Y., W .-1 c c >
11 ~ C~ II t') ~-1 (I 1
J.J W x -
C~ ~ w
II n a z ~
~ a
O
f[S 1~ v H .S~', II ~ C~ II II ~ N l~
H Cl~ E-i W II f~ Cn
(.," JJ ~2, H c
(Z,' ~ 1J ~"
l0 CJ~ '
H '
" U
I
~
, ,
C~ _ ~-1 L1: _ ~-I ~ W ~
L~ ~ ~ H (~' o w ~,
o l O
P.' w E-~ E-~
' r~i z U
G4 H
= ~-1 w
r~ W o
~ t~ x
'~
z C/1 Cn
'~'
y' (2
'
~a ~ ra= _ -~ICncn~o~ ra= _ .~Iz~n.
' u 1 a .
a v -
il uxw -
~xo, ,
~ ca= v=
-~u3o~za~cuza~C
. a~ II a ~c~~o~~ n ~ n
~w . > ~, ~r~a~zaHwzwa
~arx~ u~.., ~r~~
~I m~~ r>~HaM
. v ~", o~,..,a~nxu~aHZaa
v = Iv v~-Iolyo= to v.-Ir~oor>~
v.-Ixo, = Iv= v~
NHAxHxcn~7r~o~
m a ~ ~ ~, r~ n ~ ~ ~, n ~ ~ a
x rx E-~ . cn w ' S
~
~ m E -~
a v o 't~ ~' v o v x f~ v o 'b H
x ~ UI ar ' r~ ~
a
H
a
3
c~
c7
~
v x ~, H
o ~b o Icarxcn~l~b o I~s~~ s~~ o~ Ira ~
W H
c~ H
~ [-a
[~
H
W
H
H
~w~n~Cx~cnrxc~u
~.u v o s~.n ~ww~ov o s~.~ v o ~ o.~
s~~-~I rno a,zs~~>o ~,wN ~wxHwr~zzr~~~
a~b~cn ~o o rnu
o v~,HN
, s~~~t ~x~cnuz3wa
w,. W W a ~
W
H
-r-,
W
y
a
~o
~ W W 3 pt
H rar ~1
x 3 a
E-
A
U U
° ~ ° m o in
H H ~ N N M M
CA 02333481 2000-11-27
WO 00/00600 PCTNS99/11516 .
258
xAa H
W
= H
Hxza
~
~ C
'J
a~~x xaW ~U~rnrnrn.~~a~Urnrn~~a~a
U ~ U rn ~ U ~ b7 U .mU
01 b1 rti U
cd U U U b1 U tT .u U U
ra3 U ra rn rn
U U ~ U U .i, c~f U rn
U r~ to ~ b1 rn
U ~ rn U U ~ U 1~ ~ U rn
U U rn b1
z CIA x ~ d1 rtf r>J U ~ ~ (15
C7 LZ, ~ ~ .u b1 rn ~ t~
3
f~
~ rn rn rn U U rn U rt
G4 ~ a W t~ U rn ~ r~
W =
C7
H
U ~ ~ b1 rn rti U r>J rn
FC Cs.. rn U b7 Id c0 ~.~
~ U
i.~ t31 c~ ~ .u rn U U
~ x W ra cd ca U rn rti b1
Z '
L1 tff it b1 b
L7 1 it b1 tJ1 U rti b1 b1
~ tr1 rn ~ ra
a L7 a y 71 U rIi b1 U X11 rd rtS
f~ a fs ~ r~ rn rtS U rif +.~
a
H
v~~ax
~~~w rn~.rJ~ ~a ~o rnrn~.~ rn~s
~ rnrnr=~
~:
W
L~
r.~ r~ b1 cU U to U r ra
3 U ra rn ~ ~ .t~
W
~
H
b1 rn td b1 .N U b7 ~-~
~ rti td U .t~ ~ .~ ra
l~ W ~ rn ctf rn rn rt b11~
0, C7 a U c~ r U U .r.~ U
fx
f.~
U rn U b1 U ~ ~ rn ~ ~
~ rtS rn ra r>j r~
J--~ (a l~1 1.1 .iJ i~
Vl V W
~l
y'Jl ~
'1 J
.
l
V
(~
(a U r
S
H f
' cci 1~ 1 ~ r(f 1~ U rn
U ~ ~ rn rtS
- C51 U rn U ~ ~ rn U b1
~ U U ~ rti rd
w wa a ~~y,x ~ ~ rn~ rn~a~ rn.rJ ra U
U rn~ rn~
W W a ~ ~ rn U to U U rn U rtf
a ~ w ~ rn b1 ~ b1 U
~7
z fx p rn U t71 U U b1 t71 rn ~ ~
t1~ ,, -~ U U U ~ 171 rtS
f~ ~ ~ ~
H ~
x U riS ca U td rtS .t~ U
v~ ~ ca b11~ rt1 rn r
a
~
W W ~ C7 ~ ~ U ra t311~ b1 U .i, U
~ P: rn rn U cd ~7 rd
R;
l~
rd U rn rtS U U rd rn rn
.~'', .1, ~ t~ rti c>i U ~
rn of 1J b1 U 1
I CJl rn
~
-
H p 1
bl bl bl (~ ~1 cti
t~ 1~
rt3 U
U
z ,, ~., ~ ~ .
fx q .
Cl~ d5 b1 ~1 U 1~ rd b1 U ra
C7 .~.~
a b1 rtf rn r>3 U U rn ~
a ~' z - r,S rn U rtS U rtf c>3
W ~
W
1-W o ra U t ~ U (a U U U .U
~ CT1 U bl .1.~ iT3
W _ (~ C1, ~,, rti r U U rn rn b1 rti
U ~ a N U .1~ ~ rn U 1.~ U
~ W
~
W d~ x O ~ U .1~ U U rti .U rn 1.~
a W C~ U bl ..3 U r>j ra (tS
'~ a
H
H
cn
H
N U7 U ~ t~S U c>3 U U U b1 b1 rd
W W 1~ U t~ .r.~ ~ (~
~
Q) b1 ctf 1.~ 'd rti rn .1~
J-1 ra U ~ rn b1 b1 (a
~ b7 U rn 1! U U rn JJ
x N U' M of U U fIS b1 ',,j' 1
~ OI C7 '
la W
FC
n
w ~"~ C7 d Q' ~ ~ S al !d r~ .~.~ U U U rn
3 FC rn ~ 1~ rn U it
a
~-7 rtS rd ~ ~ 1 ~ cIf rn rn
W E-W J rn U rtS U .4.W ra
7
- 1 (1J U .1J .iJ 1~ rn rn
H a xo = r-1 lD 1~ (tj U (d JJ rn rn (L$
H nwqil ~ oM ~arnrnrnrn~n~~~t,~Um~r
az
v a.N ~ rn~,~ ra it rn+~ U rnrt~~
Ri 1J N ~ rnrn
~, H ~,'
,~ ~, H
C7 E-~
U J~ fIS 1J (a (Ij .1..1
zaa~C ~zw ou3x v J-J (a J-.) rn f[f U fff
= a rn
zC7cncnr-u-I r= _ ~ ~~Z U.1..1 U U CJ1U Ol~ ~s
H ,~ aao, rnrnrnra rnrn
~ ~
C7
z
o
N
~ II N o (x U rtS ~ ~1 i31 cd rn U
W II ~ U ~ ~ ~ rd t71 cd rn
a C.~ Cn l0 fY, 1J t71 1J (a
~ o . lfl ~ IJ 1
:~ U7 1J ~ rtf rn U 1
d~ 4..1
~; ca 'Z,
a ,7
L~ (~'7
.
fx I U N ~ c a1 .
C7 r-~ c to 7171 b7 U rn tfj
a H LL
41 41 =
=
bl JJ U U rn U rn 1.J rn
a >~ ~ II JJ 1J rn rn U U
W ~ U1
q x
II II
~
II tf1 rn U bl r(i .!~ rtS rn
3 O T3 O Q) U b1 t77 rn rtS rn bl.U
~ ?C >~ ~
R; Ot a
OI N
aJ
I 1.~ U 1..1 ~ rn yyl d1
W TS O a~ t~ N ~ rti WJl U 1J b'1
H I r ~ H
H Ew R:
cl) a ~o
l~ ~
~
~ U tJ1 rn (a rn c~ rt
~ O 3-r N O o O U ~ U ~ rn rn rn
~ .R ~ ~
CIA z fY.
3 C!7
m t~ O
v
.i.
3 U f.~a'O t!1 W D ~ C71 r~ ~ aS U U .~.~
H .t~ ~ bl ~ U ~1 Cn ~
!x U7 d
~ E-~
t~ o ~
rn
> td 1~ ~ ~ 1.J rn U rn U
C-~ o~ \ o~ rtf rn U bl rn ra
C~ \
f~
OI
ao
\
\
\
\
\
cn
U7
z
o~
\
cd U N rn td rn ~ to ~
~ cU U1 rn .u .u
.~.WS rn rd ~ U ~ ~ U r~
~ ca ( rti .t~
H U ~ b1 b1 tr1 U .u .U Ol
U ~ rd b7 rn rn
N b7 U ~ r>j rtS ~ U ~ c>i
~ .t~ rd U ( rn
v
rLS M al b1 ~ b1 t5t U b1 rtf
rn b7 U tr1 rn rd ~
rn ~ 2S lIf U (~ fi5 rn rn U
t11 .U U fli fI$ rn J--I
N b1 ra ~ U ca tT1 ~ b1 rtf
~..yl U rd b1 c rtf
rn U ~ ~ ~ U rn ~ U ~ r>i
r~ to ~1 rtf
I U1 U1 rf~ b) cr3 b1 t711~
U U rn rn rn fIS b1 tI$
~U~~rtirtf~Urnbl..~aSUUca~U
d1 ~ ~ rl r-~ rl r-W -1 r-I rl
r1 f-1 r-I '-i r1 r-I
r~ r1
C~ r~ l0 N 00 d' O lfl N 00 ~
O l0 N 00 d~
U
r1 r-I N c'~) M V' d' tn
lD l0 I~ l~ 00
H
W C7
Cn H
P(7 ~
O U1 O L(1 O Lf1
H N N M M
CA 02333481 2000-11-27
WO 00/00600 259 PCT/US99111516 .
1~ (tS U cd U (a cd U ~tS ~ rtf ~ (d b1 b1 U 01 r~ td U U tr1 U t51 rti ra 1.~
rtS ~ b1 ~ U b1 td U ~
C51 U U cd ca cd ra U U U U (a ~ .t~ ~ tJ1 ~ ra t~1 .u U ~ U ~ rti .t, ~1 rti
ca .i.~ ~ W.J aJ (a ~
1J JJ (~ U ~ fa ~1 b1 U d'1 U J-J tJ7 fQ 1.1 ~1 (LS fff t51 f~ f31 t71 U fa
tT1 (i~ .L~ 1J (~ 1J (~f ~ (~ a...t U U
U ra U t51 tr ~ c~ rti t-~ rt U b1 ~ tT1 (~ U cIS U W tJ1 rt1 bl U Ol tll U U
bl rtS rtS t~ bl b7 ~ U U
b1 t31 rd t~ td ~ U U rtS .1.~ ~ .i..1 r~ b1 U U ~ b1 tJ7 c0 U cIS r~ rt t31 ~
~ rIf ~ (~f .u rtf b'1 rtS .U b1
b1 ~cf t51 U U tn ~ b1 U rd (~ ~ t~ .~.~ ~ U c~ b1 ~ rtS c~ U ~1 U t31 b'1 rtS
U rd (a U ra ~ U ~ rt
ra ~ cd rd b1 ~ cd r~ b1 U l.m~ U .t~ rtS rd U ~ cd b1 ~ bi U b1 Ch t71 rtf U
b'1 rtS ra ~1 U t11 U 01
(~ 1.~ (a U 1.1 ~ 1-~ ~ 1.) t~ U bl rtf ~ (~ ~l U tJ7 ra X51 U (~ bl (~ rtS U
f~ ctf ca lfS .L.l bl f~ 1~ bl f~
b7 U t~ ~1 ~l J .l.l U .tJ b1 U r6 bl ref t~ C31 ttS 1~ r~ bl U r~ Rf bl .t~
~1 trl .1.~ (a U CIl bl ~ U ~ ~
tff fd (i3 (31 r~ r0 rt3 +J (iS J-1 (a fa f~ (LS t~ ~1 1J 1J tr1 (~ X71 b1 U
bl c~ 1J (a b1 b1 JJ rtf J.~ CJl ~6 b1 J,
U rtS ~ cLS .1~ U U cd b1 t77 U ~..~ b1 c~ ~ tT U cd C11 ~ rd c~S ~1 to ~1 U
rtS y ra i.~ U U rti .t~ .i.J U
b1 ~1 rt5 ~ X51 C71 U rd rfl U b1 td rIS ~ b1 ~7 r(f U 1-~ bl U b7 U i.~ .t~ ~
U U 1W d 1.l U ra t~ 1~
rd ~d .~ rd ~ ~ cIi ctf ~ rti ~ ra t~ rd 1-r t71 rtS U .1.~ ~ rtf ~ ~ ca rt5
(~ ~1 rIS fa rif bW ..~ ~ ~ r~
U f~ fIS (a U (~ U tTl (~ b'1 f~ .1, 1J .t-~ J--1 .i..l U 1WT1.W U U J.~ 1~
l13 rtf 1~ X71 tJl rtt (~ tWa ~1 U (~
r0 C57 ~1 r~ U ~ ~ t51 t71 bt t6 ~ ~ b1 tIi L71 U ~ 1.~ .~ U t~ tW ~ rtf ca ~
tiS 1~ rtf cd ((f 1~ cd rtS r~
b1 !t3 rCS tJ1 U rtS .t~ b7 Cn b1 ~ ca U ~1.i..~ ~ ~ a..r U .u rIi U U b1 ~ ~
~ CJ1 ~ c~ t~ t31 b1 rti ~ rd
r U ~ rG ~'~ .u ~% t37 tT G1 rti rt U rd U ~1 ru v'1 ~1 r~ r71 U ~ r ra r ra
.~ ~ "~ c~ r~ ~ byT cn
b7.u c~ ra ~..~ .u .i.y7l~ ~ U ca U t71 di b1 rti ~ b1 ra rd U U .t~ U ~ 1.~ ~
U ~ to ra U ~1 rtS U
(fS (~ LJ11J ~1 ~J (71 U1 (($ fIf (tf 1J t~f rt3 bl tT1 (a CT7 bW ..~ U 1~ (fS
1J U c~ f~ (tS U bl f~S U J-~ (~ U (d
i-yl Ol b1 ~ U .U 1..~ tiS U U td rtS ~ rtf ra U rtS ra U ~7 U U t~ r~ U U
.i..~ ~ c~ b1 ~.ma U ~ b1
t31 c~ ra rct rt rct t31 t5'1 U rti c~ tT U cd U ca rt1 ctS CJ7 rti rtf .i..r
.N rti 1.i ~ U t71 rtS . r~ U ~ t~ (a .L~
r~ ~ti ca rd cIf U c~ rti cU bl cd b'1 r~ ~ ~ ra ~ ~ tn is ~ ra U cd U ca U
~..r b1 N cti rt ~! ~ rtf
1~ U rLS rd ra b1 U U U (~ 1~ .N ca U tr1 ~11-~ 1-W ra rtf U ~11W1 rtf U .~ (~
bl rd c~ rtf U c~ U
U ld U rti U rtf U to C11 C31 t~ t~ rtS 1~ .i-~ 1~ CJl t31 c(S U U ~ rtS U bl
~ ~.r tT ~ f6 ~ ~1 ~Q ca ~7 ~l
U b1 ~ U U ~ ra U ~ r~ ~1 rd rtt ~ t.W a cd t~ b7 r~ .t~ ~ U ~1.t~ aS b1 cti U
~ c~ c~ ~ t31 rtS (IS
b1 (~ rfS rd CJ1 U J..~ cd U b1 b'1 ~1 U U rtS U t~ U b1 ~11~ U t~ N rti b'11~
~ ~ to U b1 b7 rtS 1.J .1.~
tr1 U U rti to U rtf rt3 tT rtS ~ CJ1 U b1 ~7 CJ1 U rtf ctS rtS U rti U nS U1
rtS U ~ td ~ U to ra ra ~ t31
LJ 1.i bl rCl U rtS to ca its ~7 c~ b1 to td rtS rd U b1 t~ b1 1~ b1 U b1 c~ U
U to ~ b1 tT ra U tti rtf 1.1
U J.-1 1.1 tIS 1J U 1~ J-W~ 1.J X11.) ZJl f~ U U bl (~ U ctS 11 rd U ~l rtf
1.1 ri$ U .1.W11-1 JJ ra .iJ tJl ~
U U tJt U CT1 ~ b1 U tT ~S b1 U b1 U U ra ~ r~ U ~1 ~ bl ~..~ ~ Ol rtS U U ~ ~
+.~ rt3 t~ ~ C51 td
.u U .~ b~ b1 r~ ra C51 tT1 trt rtS U 0.T b'1 U b1 tT r0 U ~ ~ rtS ~ rti b1 b1
r0 U b7 rt ~ ~6 rd ~t b1 U
to U ~1 to rd ~ rcS .~ rti rd tW -J cIi ~ rtf ~ 1.~ X71 U U rtf c~ U cd tIi ~
.t~ .~.~ ra rt r~ ~r ra r~ rtS U
~ 1.~ ~ b'1 U (a b1 (a b7 U rtf rtf U t71 rtS rt3 rt5 rtf bl rtS rt b1 U1 ~ U
rt5 U .N ra td 1~ .m 1.~ 1~ U .u
~ f~ ~ r~ U U rd U i~ U rtf c~ ctS U t31 ~1 to rti bl.u t31 b7 r~ ~ U ~t U ~
r~ t51 ~1 U ~ ~ U .v.~
J..l U ~ r~ b1 U U U rf5 b1 rtf 1J b1 U U .1..) U tIf ~1 tr1 b1 b1 .~..) fa b1
b'1 r~ 1~ r~ (a ~ ffS CJ1 U fa t~
b1 U 1W1 to Ol U cd rtf b7 U ca rti ~1 t31 N C51 U cd ~..~ bl b1 .t~ r~ .L.~
ca b1 rtt U U U cd b1 U U .u
rtf ((i U t11 U tJ1 U ~ U (~ ttf C~Tl (d (tf .U rtS r~ U U 1.~ (tS t~ 1.~ U
1.~ U t51 (tS U dS U Ch U~ U t~ rt
~ fLS t~ (ti tT1 (~ 1J U rd ~'11.~ ~1 (d rti ~ rd tT1 liS U L U b1 .1..1 ([j
fa rtj U 1J .t~ ftf rtS J.J CT rtj
tJ7 U U t57 rt U rt3 ~ rtS U b1 ~ rtS ca t~ b7 b1 ctf .u Cn U .u U tJ1 ~ b1
.i..~ .~.i ~1 C3'1 U b1 U i..~ rtS U
U ctS U rtf U .J~ U rt U it b1 rtf U rtf ~ ra ~ tn ~.J rti is is 1~ b1 rtS tr
t31.~.yl r-~ tii rd fJ7 b7 tr1 b1
1J .l..l 1J 1J (a t~ .1-1 .J..) U U rd CJl (~ .N (~ cLS U LJl cd rtS bl trl
c~f ~7 bl t~ fa .U 1J 1.J .U U 1J ca 1J !Ii
+.~ U t77 (~ r~ U +~ ~ ca b1 U (ti b1 U U tJ1 b1 rtS to ca c~ rtS ~ ZJ1 ~ ~ ~
U rti ~ r~ ~ U U rd U
(IS ~1 (d bl (ti c~ ~ 1.1 ~1 .!J 1~ (d ~1 .1.~ U (a ZJl 1J (d tTl (lS U bl ~1
tU t31 .1..1 (a ~Jl (a (~ 1~ U 1.J (~ U
~1 ~ U ra tr1 .~ U ~ rtS ~ .t~ ~ .W U r.i .t~ a.~ ~ ca .t~ t5l.u 1.~ ra rd (~
ra U ~ td U ~ ~ ~ U r6
U U ~ ca ~1 ~1 tr tJ1 Cn t~ U cd U to t31 U ~ r-~ ca ra t31 a..~ U .t~ td rtS
.N bl .1J ra rtf to b ~.~ cd - U
~ rtf rtS 1.~ n3 b1 ~1 rtS r~ ~1 U ~ .u rtS b1 l6 ~ .i.~ U rt b1 U ttf (~ tJ1
cfS b1 ~ rti l71 .u b1 U 1J ni ~
~1.i..~ c~ U ~.W d rtS rd U U cd .t~ ~ .N U .i.~ ~ U r~ ~ ~1 ra b7 (a U ca U
r~ ~ ~ ni (IS ~ rd r~
bl ~1 fLf bl (a (!S (tS U ~ U ~ ~ fti ftS .I~ rd tT1 (~ Z71 U (a (lS tJl ca
(If .L.f U bl bl 1.1 U 1~ U (~ J..~ al
c~ b1 rd fIS U (tt tJ1 rti !IS b1 ~ U U t~ CJ~ b1 rti U ra U U X11 b1 (~ b1 ca
(~ (71 ~1 ~1 U t51 U J-~ (~ b1
U ~ rti ca nS U .u b1 tr1 .v-i t31 ~ ~ U t71 ~1 i.~ rt rtS rd ~ rtS ra ~ .~..~
~ ~ ~ ~ rtS rtS ~ ~ r6
U U rff .~.~ ~1 U b1 U .U tr1 n3 (~ cIi ~ ~ ~7 c~ b1 rt5 U U b1 cd .~ t~ ~ t71
~ U 1.~ td ~ ~ U b1 r~
b1 rW .-~ U 1-~ CJ7 to U tr7 ra .i.~ i~ U U U ~ ~ ra U r0 U ra U rti ~ .r..y1
c~ U .u ra ~ rLS tJl rti td
c~ .i~ rti U U tJ1 ca U .t~ U U ca U .tJ U tJ1 U ra (d Cn U to fIS cd rd .1.~
~ b1 ~ r~ r~ rtf rtf .u ~ t57
~1 (a rd ~1 ~1 CJl tJl fd f(S bl (d 07 rd 1J tt$ b1 X71 (i$ U tJl i..l b1 b7 U
i.~ b1 .IJ (ZS fff U rff fIi ~1 (fl U t31
b1 cI3 11 fi$ t~ b1 (IS U (LS 1.~ 1.1 f~3 1-W~ ca rti r~ ~J1 ~'1 r~ .t-~ (a r~
rLf b1 01 (a U t~ b1 tJ1 (~ bl b1 U J-~
CJ7 ~ rtS r~ U rtf 1~ U r~ rtf U b1 U C51 rtS U ctf ctf b1 rti U U a.-r U rtf
t~ W..~ ~ rtf ~ ~ rtS U ~ rd
r~U~bltnUbl~lUU(~CJtUU~IUrtUb1t71UUrtS.uUbIUU~JrdUtni.~bl~~1
U (fS r~ U1 rt U r~i r~ b1 ~1 (fS rd U U ~ rt 1J (t$ (fS tTl b1 ffj Ol bl fI3
~1 CJl fa .t-1 f~ U fa l.J ~1 rtS bl
bl b1.i.~ ni U t~ .i..yl .u ~ tJ1 t~ b1 ni U b1 ~ ~ c~ rc3 b1 U U U .1.~ ~ Mf
t31 rtS ciS t71 ~1 ~ tJ1 U b1
(~ r~ .~.~ (i$ t71 U b1 b1 rIS ~1 ~1 b1 fiS ZJ1 ca b1 U tIS bl l~ 1.~ U U 1J
(a 1.1 (fS 1~ (iS 1.1 01 Ch ttf .1J U (~
~ r-1 v-1 r-i r-I r-i rl r-I n-I r-W -1 ~-I W --I r--I r-1 r-i r! r-I r-1 ~ r~
rW -1 i--I r-I r~ ~ '-I '-i ri ri r-i r~ r-~ r-i
O l0 N 00 ~ O l0 N 00 d' O l0 N o0 d~ O lfl N CO d~ O lD N 00 ~ O l0 N a0 ~ O
l0 N 00 ~ O
01 01 O O ri N N M M d~ Lfl lfl l0 lD (~ Op Op p~ ~ O ,--~ ,--1 N N M d~ V'
Ll7 Lfl lD [~ l~ Cp 00 01 O
f-1 ~-1 r1 r~ T"'W -1 ~-1 ~--I f-I N N N N N N N N N N N N N N N N M
lf1 O Lf1 O Lf1 O Lf1
ri n1 N M M
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516 .
260
~ ra tr1 rt3 ~ b1 rt'f ccS rti b1 ~ ra ffS r~ ~ ~ C11 tJ1 1-~ t3t CT1 U c~ tn
rtf ~ rd U r~ ra rd ra ~ rIS ~ bl
rtS b1 b1 U J.~ ca r1W71 rU X11 ca b1 C51.U t31 ca rtf rd b1 r~ 1~ b1 n3 ra rt
l W ti ra Ol .t~ U rd 1J U (d tT1
b'7 .r-~ .t~ ra a-.~ U a..W tS rd ca rff bl rI5 U U tJl U rcS b1 c~ U ~ b1 U ~
b7 ~ rd bl U C71 ~ rtf r(S ~ ~1
t77 W (a b1 Uf fI5 U U (I$ tr7 (fS .u bt rd fa b1 U tti b1 ~1 U (iS C3l U b1
1.1 01 b1 b1 (a r~ b1 t71 U 1~ (IS
rti b1 t71 tJ1 b1 rtf ~ rtS b1 rtf ~ U r~ rcS c~ ~ ra r0 .t~ r6 r~ U rt ~ rtf
U ra ~ b1 r~ U rtf t77 rd a ~
.u ~ tJ7 r~ rtS .u .~ ~ ~ U .t~ U .N rti t~ U ra rti ra rli U U rd ~ U U b1 U
tJ1 rtf cd ~ tJ1 r~ ~ U
b1 U ~.pU U U rti rtf U N .u rli ~ t51 U t~ U tr b1 U U t71 tTt rIf U t31 ~
t31 p1 U ~1 tJ7 rt ~..y1 rd
~ ~ W r~ ~ t~ t31 r~ .7~ rtS td U b1 tJ1 U ~ r~ b1 cti U ra rtS rtf ra ~1 U r~
t71 r~ ra ~1 ra U .u t31 U
c~ rW ..r ra C51 U rti rtf rd ~..~ rtS U rc5 b1 U rd U rd ~ U ~ rti ~ ~ tTl U
rt3 rtt r~ 1..~ t71 U ~ rd .i.~ U
J..~ U U 07 r~ b'1 CJl bl rd .U r~ bl U (a U tTl C71 rd b1 b1 U bl J-.1 d1 1J
~1 U U (IS r~ (Li ~1 (1S bl U U
b1 r~ U rti rd rfS ~1 ra r6 rtS .u r~ U ~ U b7 ra r0 rtf t51 U ~1 ~ r~ rd t31
1W r~ U U D'~ U b1 rt3 ~
~ rd ~ U U aS b1 U rtS r~ U rtS ra rtf ~ ra r~ r~ r~ rtS ra tJ1 r~ rtS ~ r~ bl
rtS a1 rd rti b1 :.~ rti b1 rtS
~ U ~ U ~ rIi ctf b7 rtS U U U ~ ra tr1 U U rt5 U rd r~ ~1 U b7 r(5 rd rtS b1
rIS ~ ~ b1 rtf C51 t31 U
.u rtS r6 t31 ~1 U U rti r..mtS 1-~ t31 C31 U .t~ b1 ~ rt3 ra l i ~ rIf yf r~
rtf 1~ rtf ra r~ rtS +~ rd Ol 1-~ rtS r~
r~ b1 U ~ ~1 U rtf 1~ ~ rtf U b1 b1 U ~ 1.~ 1J .U r(S rd ~ ra U U b1 .1.~ rti
b1 rli U r~ ra rIi ~ r~ U
C57 ra U U ~ rIf r~S rU ~ (li ca .U 1.~ bl C51 ~ r~ ca a1 ~ 1.~ rd rLS C51 b1
X51 U rI1 rd ra 1.~ b7 r0 b1 .1.~ r~3
1-1 t'J11~: v1 .1.J 1u r~ ~'G r~ t'J1 X71 J.J r~ rfS rfS 11 U rG 1~ rG 1~ n 1~
n ~~ n iJ r~ 1J ct~ 1J .~..1 r6 Vl U 1J
r~ ~ U ~ rt1 ~7 rtW .J U 1~ r~ ra t~ U ra U tW ..W ~ .U rtS 1..~ CS'1 r~ U U U
.u .1-~ U .u a1 U ra b'1 al
bt U U ~1 r~ CJl C511J U bl fL5 1J 1.1 b1 (f3 ~7 rt .tJ rid fIf b1 U 1J (71
b~1 ra U b7.I-~ b1 ZT7 CJ1 t71 U ra rtf
b1 r~ r~ r~ X57 fJ1 rI3 U tr1 1~ cU C11 ~1 b1 fti rt3 r~ X51 r~ r~ ~ ~11.~ rIS
rtf ~ .t.y71 .U r~ X17 r~ r~ rtf rd t~
.t~ rLS rt ~ .u tTt U ~ rt5 rtf b1 r~ r~ b1 rd ~ ~1 ra b1 tT1 tT ra rtS ~ .v-~
U r~S ra rd ~ ~1 rcS ra tT rt tT
(~ ~1 ~J1 (a .t~ rtS b7 bl bl tJl X11 CIl lli trl U U tJ1 U ~1 b7 (a U CJ1 r~
11 la fti U Iti ca U r~ ftf (If tJt (LS
U tJ1 rd ~ rtS ~ t71 rt5 rd 1-~ b1 r~ 01 ~ ra .u Ol b1 ra 1..~ t~ .u ra ~ rtS
~ U rd U r~ ~ b1 rt ~ ~!1 U
rd r~ ffi U N .i.~ .I-y~ rd rtS tJl U 1~ ra bl ra 1~ ra t31 rt5 U ~ ~ rt3 rt5
bl rti CJl r~ .1~ l..y'1 ~ ~1 rt3 1.J
.~1 ~1 rd U f(i U U rB U r~ .t~ c'~ tJl a1 r~ U J-1 b1 t11 trl t31 J.~ U JJ
rtS (TS 1-~ bl U ra 1~ 1~ ra rIf !t'S 1~
ra ra b1 171 ca 1~ ra ra ~ ~r c~ tr1 r0 ra t~ 1.~ rrf cd ~1 rd ~1 ra p1 ca rtf
U U rd U U ~r U rd ra U Cr
ft$ 1~ fa fa X71 1J r~ U rd U 1l tll 1..1 1.~ ~1 .1J U 1J 1J 1.1 1~ fIS r~ bl
J-1 U U .1~ 1J rd fa J-1 b) U 1J (IS
U ra U U Cn U fJ7l~ ~ r~ r~ .U 1.~ t31 C71 rd b1 rU r~ .u r~ ra U b7lm~ U rd
rd b1 ttS U r~ b11.~ rtf
r~ ra .u ra r71 rtf rd rff .U U rtf b1 U .i.~ r~ rIi ~ rd r~ .t~ rtf ~ b1.u U
U y .r.~ U rd ra U rti ~ .u ~
N rd rW ..~ t~ rd ~ U ~ ra .t~ ~1 U rd b1 U rW ..~ b1.u U rd b1 bW .u ~ .1.~
.~ .u t~ t~ U ~ rtS ra
U CJ1 tJ1 rd U U ra U tT1 1.~ U .1~ rtS b1 a..> r6 C71.U b1 tT L~ rti U .~.~
ca to 1-1 Z71 U U U ~1 1.1 r(S
U rt ra rd ctf ~ tJ1 U rd U r~ r0 1.~ U U rIi to rd r~ rrs rtS ~ ~ U U .r-~ U
ra rIS ra .l.yl t71 ~ ra ra
~ U rti U 1.~ tr1 rtf rr5 rIi U 1.1 U U .U (~ ra r~ rtS rIf 1~ b1 ~l ~ U ~ ra
X71 r~ ~ ~ .N crs .t~ b1 1.~ ra
(IS c~ rd r~ L1 1J bl C51 U Ol fa C3T U 1-1 CJl U U U ra U r~ .1..J rte ~11~
rd bl ra bl rti ra ra ~11.J tJl fLi
U r~ rtf ~1 r~ ra rd W ..~ ~ U r~ U ~ .u r~ rff ~ U ra tJl U ra r~ U U +~ b1
r~ r~ rd t~ rd 1~ ~ U
~.J ~7 r~ t37 rIf J-~ U ra rt3 bl bl r~ U U U ~ U bl rtS 1-> C51 bl t71 1~ rd
bl +~ r~ U X71 ra U ~ ra r~ U
bl r~ r~ .i.~ rtf ~ t17 t~ t~ b1 ct5 CT1 rti cd ctS J-~ .U ra U b1 1~ rd b7
CJ1 ~ rd .~.~ ra bl c~ ra CT1 t31 ~1 tW ..)
r~ ~ U ra tr1 r~S r~ ~ ra b1 rd tn tma rti b1 U U r~ r~ ~ U rtf L ra U rtf ~
rJ1 rti t~ rti rW ..~ rtf ttf
a..1 r6 ra rI~ rti ct5 r~ rtS U rtf (S ~ rd rIT +~ rd rd ~ U r~ rti U U ra U
b1 r~ ~ ~ rIS U U rd rtf U ra
c~ U bl (a U U t5'1 1-~ rtS ra ~ .~ r~ ~1 al X17 bl bl bl rIf rti bl U U L77
CJl bl ~ cd ~ !IS U cLf ~1 U 1 ~
U b1 r~ s.~ r~ b1 ra b1 t71 ~ rti r~ U rtf .t~ .i.~ r~ rd ra t11 .i.~ rtf U .1-
~ r~ 1..~ bl rd rt b1 .i.~ r~ ra .t~ r~ U
fiS rtS U fa J-1 Z31 r~ 1.~ t711J U U 1J U f(f U U 1J U bl ~771~ b11~ U r1~ r~
fa r0 b7lJ b7lJ ~1 U r~
~ U U t71 ra rd b7 b1 .t~ rd ra rtS ~1 tT 1.~ rtf ra 1.~ U rti c~ t71 r~ b1
rIS rif U b1 U t77 rtf tJ7 rtS ~1 rti 1~
r6 b1 ca .u U r~ r~ rd rd r~ U rd .t~ rd ra tJ1 b1 b7 rt U U rd ~ rtf rtS U
C37 rti r~ rtS r~ rtS U rd cU t31
U b1 U U r~ .t~ U b1 ra tr1 U rt! ~ b1 b1 ctf .u rd .i.~ .u rti t~ b1 U ra ra
rtS rd .t-~ U r~ r0 rt3 U r~ rIf
U b1 fti U tT ~1 r!It ~l U ci5 b1 b1 ~ C71 b1 rd tJ7 ~1 01 r~ U t~ rJ (71 ~1
b1 rtf (d ~1 bl U rtS b7 ~1 rti ra
.1.~ rd U ~ ~ rti r~ .~.~ U r~ rlS b1 tT rtf rt tJ1 rti rt tT rtf .N r~ U ra
rd ra U ~ r~ .i..r rtf Ch ~ bl ~ rti
ra ~ U rd c~ ra 1~ r6 b1 U ~ rtf r~ .tJ U ra ~ U b'1 b1 rti ra rd ~ ra U .u
rtf U b1 r~ b1 tJ1 ~ b1 b1
ra ca rtf U rd ra U U C!1 b1 rtS nS b1 r~ b1 U rti ~ .t~ CJ11~ ~t b7 b7 U rtS
r~ ra b1 rtf ra 1J rtf rtf rd .u
rd rt3 ~ U U .tW a ra r0 .~ rd rii bl rtf ra 1.~ U rd r~ .t~ rtS rti r~ b1
t711-~ C51 b1 rti U rd ~ ~ b1 r~ rti
rd ra .~ 1~ ~J ~ U 1.W a rtS n5 ra bl .t~ rtf rd r~ rd U U r~ ra .t~ .~.~ b1
ra bl ~ U ~ (t3 .t~ rd ra ~J ~
U C31.i..~ U tT r~ U b1 U ~ ra U ~ ~ rlf rt r~ bl b1 b1 rd ra bllWT U b1 ca
rtS ~ U ~1 rt5 U rtf U
fli r~ (i3 (L$ 1J tJ1 rd ~1 a1 (Li r~i (fS bl U rd fa (fS .I~ dl .L.! tJl fIS
(a U r6 fl$ ~? yJ b1 ~1 fa r~ 1~ (tS tTl r~
r~ 1.~ b'1 rtf U a1 ~ U b1 rd ca .u rtS U tJ1 U ~ 01 ~ b1 c~ U rd 1~ U ~ U U
ra bl s.~ U rti ra r~ U
rd .i..~ b1 t11 rtf tJ1 t11 U tr1 rt3 U rt b1 U t31 ra b1 rt1 U ~ t37 b1 U r~
rti b1 t31 .N r~ b1 ~ U t31 ra ra 1.~
td U X11.1 d1 r~ fLS fa (71 rd (tj U .iJ r~ r~ fa fa a1 U U 1J ([j rI3 1-~ bl
ra bl rfJ 1.J bl r~ 1.J r[~ !~f ra rtf
C71 ra b1 r6 b1 U ~ r~ rti b'1 .t~ U U rt3 ~1 U rd riS rt5 ~ rd .t~ t71 .l.ma
U b1 rtS ~ b1 r~ ri5 ra 01 r~ U
r6 b11~ 1J .i.J b1 d1 CJ1 U X77 b'1 U U ~11~ to C51 ra i.3 b1 U ~1 t31 b1 CJ1
r~ 1W31 U C51 b1 CJ7 U ~1 b1 r~3
tJ y711..~ U .N ra ra tn td rti rtS ~ rff rd U rtS ~ U b7 rtS rIS t~ rtf rti
rti ~ rd ~ ~ rd ra rt ~ Ot ~1
~ U ~1 C71 ~1 rtf r~ c~ tU U U t~ ~ rtS ~ ra ~ US t~ ~ ra ~ c~ r~ U rtS t51
.i.~ ra .i.W ~ 01 ~ N t~ rtf
~ r-I r-i r~ W-I '-I r-I rl r-I r-I t--I rl n-i r~ r-1 r-I r~ ~ r-1 rl r-I ~-i
r-I '-I r~ r-I v-i rl rl v--i r-I r-1 r-1 r~1 r-I
l0 N OD d' O lD N OD ~ O lD N ~ d~ O l~ N CO ~ O l0 N 00 ~ O l~ N DO d' O lD N
~ d' O l0
Or--lrlNMMd'd'Lr1l0l01~(~CO0101OOriNNMMd'111LrllOlDl~CO0001010~-1rl
MMMMMMMMMMMMMMMM V~cr~~ V~~~~~~~,~,~,~,~,d,~,
O ~ O ~ O In
rl ~-.i N N M M
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
261
1~ 1J ffJ U ~Jl U rt b1 U (d b11~ rt f(S 1J b7 c~f 1J (~ (~ ~1 rt 1J .iJ (a U
U rt rt ffS ~ 1~ U 1J U rd
cd U 1~ rtf rt U 011. t71 b1 rtS b1 b11~ .~ ~1 1.~ ~ Ol tIS tJ1 b1 171 rt b1
rti 1.1 rt U ~1 ca rt b11~ U U
U U ~ ~ rt r6 rt rt ~.y7 U ~ ~ tT t31 c~f ~I ~t L71 .1~ ~ b11~ rtf ~ ~ rt .v.~
rt rt b1 r.~ rt ra U rt
b1 U .u ~ rt L51 rt ~ U ca .v..i ~ rt .i.~ ~ U ca t-W ~ r~ .i.~ td CJl b1 (Jl
rU ~ bt U rtf ~ b7 rd r0 (ff U
tT aS U r~ ~ rt ~ U ~ ~1 b1 t-~ ~1 rti .t~ ~ (a rti rtf U l.J b1 ~..i ~ ~ rIi
~ ~ rtS bl U U U rt t51 rd
~ b'I b1 U rt cd .r..~ ~ rtS rt (~ t7~ U U ~ b1 b1 tT ~ rt rt .u U ~1 ctS .N
tJt rt ~ ca CT1 U rt U ~1 ~
U r6 1~ rd tJ7 b1 U to U U ca cff rt rti rtf rd W .t~ b1 1-W r0 ~ ~7 ~..W ~ ca
rtS ~1 t~ c~ .u b1 b1 bl U
rtf ~ U CJl rtf rt rt .i.~ td rd ~ c~ b1 ~ U ~ b1 t~ .~..1 b1 bl U U tJl rt d1
rt rti .u 01 U to ccS rt C71 t~
b1 b1 ~..r b1 U U ~ rt rt tn tT U U ~1 b1 cti 01 rt b1 tr1 rt3 bl (~ U U rt rd
rti rt3 rt rt3 tW1.u ra .u
rd ~d rtS ca rt U b1 U U U rt U ~1 ra ra rti rt 1.~ .i.~ cd ~ .i.~ U ~1 (Jl
1.~ U .1-~ rtS U ~1 bl 1.~ U bl rtf
~1 c~ ~ U rt U t31 rI3 tr1 ~ U b1 (~ ~ ~ td rt ctf b1 b1 b1 rt U rd rt cd U U
U CJ1 b1 rt ra tJ1 b'1 rt
(d U tJ1 b7 rt U rt rt ~ U U .u U b1 rtS rt t31 tr t~ rt rt ~1 U ~ ca rt .u .u
c0 rt to rt t~ rt to ~
rt ttf ~ t0 rtf b1 b1 U U rt ~ .i-~ c~ rd rti r~ ~ tT U U rt t~ U tTl U U ~1 ~
~ ~ .v.~ ctS rtS ~ U b1
rt U U rd U rt rt rt ~ .L.W (5 rt ZT1 rd 1~ t71 ~ tr1 U rt C11 U rt rtf rt U ~
rti ~1 U ra US (~ ccf ~ 1~
C31 rt .1~ U U rt ~ b11~ t~ U U N .U rt ra rt 1~ ~ 1~ rt ~ rtS +-~ U rt U ~1
rt ~..~ rt rt rt bl U U
U .tJ .iJ c~ U .t~ b1 rt b1 t51 .u tJt b'1 ra ~ t71 ~ .~ rt rt rt .I, b'1 ~1
.~ U U U ~1 ~J t31 b7 c~ U
Gl bl W G rtf r .v..5 ru ~ U n'S .t~ ~Jl cIf 1~ rt (tf U Ol bl U v'1 .t~ cG W
75 'vl Gl r ru (u cfS W iJ .~.y
bl ca .U ~-1 (!f tIS ~ .1.~ b'1 ~1 (lS .t~ U U rtf U t11 U ~ .i~ rt rt cff 1-~
U rti ~7 .tJ U U U rt ~ rt tI3 U
ca 1.J 1~ rt rt bl U 1J rt ZT) ~1 U bl tr7 rt rt 1J ~ ~1 td U 1.1 rt (d 1J 1-~
ra .t, ~1 bl U U d.J rt rt .!-1
U rt CJ7 rt .U b1 bl U rt rt bl X31 fd rt U b1 cti U b7 1~ U CJl rt rt rt ca U
.v.~ rt bl to rt rt rt U ~
rtf ~ ~ t?1 to ~ rt rt .tW ..~ ~ ~r tIi rff ~7 t~ rtf bl ~ r~ U CJ1 ~ U ~ ~
rtf .i~.~,., U a ra .err rt rt Lr ~
1J rt1 .L.> 1~ (d rt Z71 rt rt ca U 1l tJl (a rt fa U 1J fff U rt 1J JJ U (~
rt U CT71J rt rt rtS r~ JJ (~S 11
rt t71 ca ~11.J b1 rt 1-~ rt cp U ~7 ra U 1~ 1~ t71 tW .J bl (6 t~ tJl fIS rLf
~t rt b1 C71 c(S rIf rt c6 1~ ~ 1~
U rd tn ~ ~ rt t-~ rt ~..y1 U ~ rt .t~ b~ ~ b1 U .~ rt U U ~ .u U ~ ~ .~ ~ rtS
~ U rt m..~ r~
b1 b1 i.~ C51 f~S U U U ni rt tJ1 ~1 ~1 (~ rt r~ .i~ U rtS rt U rti tr7 rt ~.~
.i.~ .~ U rt .u U ca b1 U .v.W d
~ n6 .i.~ b1 U ca rt U U U rt rt b~ .u a,.~ ~ t51 rd .u ~ U b'1 ~ rt ~ U rt
t31 rti ~ b1 rt b1 rtS .i.~ b1
U to ~ rt b1 b7 U .i.~ U b1 ~7 rd rt U bt (a rt U U 1~ U 1J rti .IJ .u rt U b1
~ Q1 +~ U rt U U b1
~ rt ~ ~ rt b1 cti U 01 rt rt cU U ~ rd ~ U b1 ~ U ra rt U U U ca .i.~ U U t~
rL3 rt ~ U ~ b1
t51 t!S (~ U U rt 01 ~1 rt .1~ .I~ ca b1 U 1.~ rt rt b7 ~1 ~1 C71 rt U rt 1.~
rt tr1 U rd U rt ca (~ tT r(S b1
tJ1 rtS .t~ U b1 b1 CJ1 b~ rIi rt U rt b7 ~ ~ to t31 b1 ~ ~ rt rd t31.~ U ~ rt
U .t~ 01 U U rt ~ rt rt
~7 b1 U t.ma c6 .~ ~ t77 U rt ~ 1-~ 1.~ t~f C51 rt b1 U rtf U nS rtS rtS b7 (~
U U bl ~ ca U U fa 1~
~ rti U rtS U rt ctS 1.~ ~ b1 ca ~ ~ t31 tn td fn ~ U ~ U b1 rt ~ U .W U ~ rt
rt rd rt ~ C51 ra
~ ~1 b1 ~ r~ b1 t31 rt tJ1 U 1.~ ~ U rt rt ~ rt rt .y.~ U U ~1.~1 rt rt .t~ U
b1 U rtS U t11 .I-~ b1 U X31
rd011..~~UI~.tWlrtl..lUrtU(~.t~rtUrtbll~U.l~Urtrt~rt~UU(iScaZllJ..lrt~
.t-1 .1-~ U bl bl U ra bl bl tr1 U .4.) .1.J U b11~ bl bl ~J7 c~ ((S bl U b1
(d U U 1~ fl5 1.) U td 1.J ([$ fIS 1.J
rf3 rt 1~ rt b1 ca rt rt b1 ~ .u U U t~ rd t~ to CJ1 ~1 U +~ .~ bl rt rt .N rt
rt b1 U ~ U 1~ (~ rt ra
U rt rd t~ .u rt U ~ td b1 td b1.t~ rt ~ rt b1 ~ ~ ~ ~1 rt rt ~ rt ~ rt ~ X11
rt ~ rd t~ rt rt rt
cd CJ1 U rt ~ rt (~ U U b1 CJ71~ ~ U ~ 01 rt tT C511-~ .U U rt b1 rt rt 1.~ t~
rt rt t~ ~ N rt t~ .U
rd rt .u .tJ U 011J U tr1 b'1 c0 .W U 1-1 rt rd rt ~71.~ l71 ~T1 (~ rii .J.~
.U U C31 rt U U ra b1 U U rt 1~
U ~ ~ tn ca tr1 rt t~ ~7 l.J ~ rt ~t U U .u r~ tn t~ .1 ~ t~ rt U ~ rt tT ~ U
rt ~ tci ~ b1 b1 rt ~
cd to rff U +~ td U 1~ ~1 ~.yJ1 rt C31 rtf ~ rt bl b1 c~ .N U .t-~ .W .4~ b1
JJ .i.~ rt ca rt U J-1 rt ca .1J t~
ttf U ra cd b1 ~ U ~ ~ rt r~ U r~S b1 rt5 U rt tT ra .u U .t~ ca rt rd rt b1 ~
is .i-~ U td b1 bl (cS bt
~! U .i.y71 rt ~..~ b1 .u ~ ra U U r~ ~ t~ ~ ~ .u rt .t~ ~1 .J.~ ~ bl tn ~ r~
r~S ~1 t7~ b1 to rd ra c~ rt
~ .N U t71 U ~1 r6 ca U tr1 U rt ~ U ~ rt rtS ~ rt cd a.-1 ~ tr7 .i.~ r~ t0 t~
U .u ca ~ ~ b1 J.~ rtS U
rt U (~ c~ rt t~ t11 U td cd Cn ~ U t~ rd rt rtS b1 rti ~ rt c~ U7 U b1 rtS
a..~ U rd ~ U ~ t~ al tT rd
~ .1.~ rd 1-W .t t71 b1 ttf ~ U rti al U b1 a..W .U cd b1 U U ccf ~ ~ tJ1 b1
r~ U td b1 ~ rt b~ rti tJ1 U
tJl tJl U 1-~ U fiS b1 t51 t~ 1.1 t11 .J.J ~1 (~ f~ t71 U ail (d 1.~ (~ tLS t~
U (IS cd X11..1 U tf3 (d (a (a 1J 1J bl
~ ra U rt ~ ~ ~ r~ b1 i.1 ~1.U rt U rt rt c~ t71 U U U rt ~..i bl rt rc1 .u .u
i.~ U b1 rt U s~ i.~ U
.t~ tr1 rti ~ U U t31 t~ b1 ~ ~ U rt t~ ~ ~ tn .u ref rt ca b1 ~ .t~ c~ rt .~
rt .i..i U rt b1 U ~ .~ rt
b1 b1 (d 1J U rt 1J b1 (a rt ~7 rt (a rt (fS rd bllJ U U rte .l.J d1 b1 b1 b1
rt t31 t57 1~ t~ (~ U ti! r
r6 b1 (~ U to b7 U b1 b'1 U cd ~ rd 01 .u U ~ ~ U U U U b'1 rtS rti rt ~7 U U
t77 ~ ~ C31 rd cd ~
rt ~ U U ~1 t~ rti ~ U U cd U U rt U U rd .1~ tJ1 c~S b1 td rt rd b1 U rt U
rtS rt ca b1 b1 rt U is
rt U b1 rd ~t .~.~ U U rt r-~ ~1 rd rtS U U ~ tJ1 U .u U bl b1 U rt b7 b'1 .u
t11 b7 U ra ~ b~ ~ ca ~
1~ ~ ra .u r~ ~ U t11 .t-~ rtf rtS td b1 .N ra al ~..~ rt b1 U tJl ~ .~ ~-i ~
.~ ~ rat cIS rt3 ~ ~ U bl bl c~ i-~
U f7t .N rt ~..~ U t51 rtf .tW ..~ rt r~ ~ cti ra b1 ~ U rtS rt3 ~ t51 (a .u
to ~1 1..~ b1 r~ ~ti b1.~.md ~ ~1 cit
b1 b1 U cd C37 U bl ~ U .N U to rt U U .a.~ ~ b7 ~ rtS .u .N rtf t~ ~ ~1 U .u
t~ (~ ~ rt t17 ~ rt cd
b1 U ra 01 to ~7 f31 rtf ~ ~1 .t~ U ~ b1 b1 rt fd b1 U .N rt ~ b1 ~ ra CT1 ~
rt b1 ~ U rt ra ~1 ~
c~ ~ rt bl cd U ra .t~ rtS b1 rt U .t-~ U ~ ~ .N U .U C51 tJl r~ ca rt rt r~
rti ~ rtf bl t11 r~ U U r~ t57
tT a.~ 01 ca r~ U ra b1 rt 1~ ~ U 1.~ b1 rt b1 .N .i.~ U ra .r-W Ii ~ ~ t31 1~
U U ~ ~ ~ U U rti rtf to
r-i ~ r-I r-i r-I r~ rl r-I r-i i-I f-1 r-I ri r~ ~ r-I r~i r-i r-I r~ ri r-I
r~ t-i ~-I r-i r-I r-I '--i '-1 r--1 r~ r-1 r~ r-1 ~-I
N 00 ~ O l0 N 00 d~ O l0 N 00 d' O l0 N Op ~ O l0~ N OD ~ O l0 N CO ~ O l0 N
CO d~ O lO N
N N M d' d~ Lfl IIl l0 L~ L~ CO 00 01 O O rl rl N ("1 f~ cr V~ Lf7 l0 l0 C~ I~
OD 01 01 O O rl N N ('~1
L!'1 lfl L!~ Lfl Ln l(1 111 Lfl Ln Ll7 Lfl U1 l.n lD l0 lfl 10 l0 l0 lfl l0
lfl lD lD lfl l0 l0 l0 lfl lD I~ C~ L~ I~ l~ l~
Lf1 O tf1 O tI1 O tf1
N N M M
CA 02333481 2000-11-27
WO 00/00600 PCT/US99/11516
262
ra rtS .u tT1 ~ r6 U ~ U td U ~ U ~ ~ ~ tr1 ~ rt5 tJ1 ra 1~ rd t31 01 ra U rt
U Cr ra b1 U +.~ ~
tT b1 cd td ~7 b1 ~ b1 rtf b1 U rt .u ~ CJ1 rff rd b1 U ~1 rtf t71 U ~ ctf U
rd (a ~r rti t~ .i.~ ~ U b7 a1
~1 ~1 +.~ U rd td U rd U b1 LT1 b1 ca 1.~ ~ .i.~ U rti U Ot t11 ~1 ra .t~ tr1
c~ U .~ ra U rtf td .~.r b1 rt r-~
U fa fIS 1W.J b7 tT J-1 1~ ~ 1.~ b1 I~ b'1 J-1 yJ (~j .~J (~ ~1 ~ tT1 ~Jl fd
b1 (IS c~ 1J bl U bl U 1! 1J Cfl U
(~ U rtf r-~ ~1 cd rd ~ U ~ b1.i.yl ~ !a t11 b1 ~ .t~ l0 c~S rd rti b1 b11~ U
.u 1.~ b1 ra ~ t~ ~ t71 ~
U t11 (~ U rtS b1 rd .u r~ c0 ~ U rtS to X71 (tS U rtS U U rtf ~ rtS 1~ .U U ~
~ ~ ~1 U ~ U U b7 U
~ tJ1 U .u (a rd tJ7 rti td ~..~ U U rd U ~ ~1 U b1 b7 ~ .u (a ~ t71 b1 bl b1
~ U tr1 U U b1 ~1 LTW
ca .7.~ .i.W ~ cIS U b1 trl U U b1 ca trl C57 ra rti U U rtS U U ~1 ~ .U LJl
rtS ~ U U rt5 b1 U ra rti t~ U
~ ~1 cli m ~ ~1 rtf ~ tn U ~ t3~ .u ~1 r~ ~ nS ~ U rtf (6 rti ~ tn bl U U rti
a..~ U ra ~t ~ tn U t~
rtS 1.~ U rd rti ~ U U ~ rd U ~r ~ ~11..~ ra C71 ~ ~ tr1 ~ U ~1 ~ b1 Ln U U rt
U 01 rtf U U rtS b1
(IS (a .a-1 bl J..l b7 ~1 J~ 1J ~ rt td (a t~ ctf t~ LJ1 (a .u bl t51 rd (tf J-
~ 1~ (d 1.J U .LJ tIf .t~ b1 U 11 b1 ~1
(a U .7~ U ca b1 to CJ7 b1 rtS U U rt t~ U 01 CJ7 U U b7 01 b7 (~ b1 rtf U
.i.J b'1 rd U J..r .u b1 rti ~1 t71
U t~ tTl U (~ J-~ U .t-) iJ tJl U f~ 1.~ tli 1-l .l..l bl U .~1 ~J (6 a7 bl to
bl (~ ~ fa tJl rti tJl bl U U ~1 1.1
~ rIf 1.~ .U cd b7 tJl l.~ U r~ (~ r~ t~ ~ .lJ ~1 r~ ~-~ U U U b1 t~ ttS td 1J
.i..1 ~ t~ U t~ .u U rtS U U
~1 (a ra CJ1 ~ rd r~ rt3 .u ra U ~ U .N rtS rt5 tJ7 rti U .1.-~ .u tT ~ tIS U
ttf b1 ~ ra rti U U C51 b1 b'1 rti
b1 a..»1 ~1 (a C37 tT ~ c~ rti b1 ra U r~ l.J ~ U b1 C~7 t~ b1 fIi U rti b1
tti tJ1 U U 1..~ U U ~ J-~ C57 .~
'Jl f3 cu ~'11.~ .u ~l.u U x',11.7 (LS 1u ~1.~1 (~ U ru .u U (G 'vl U (G (u J
~1.1_i fIS J w iJ ii iJ ~ ~1
ca 1J .i~ 1J (IS (iS tr1 (d fJ1 b711 ~1 U (~ (~ ~ U U t71 fCj ~Jl .1-~ (d U U
b11.~ rd Cft cIf .L~ r~ cIf b11~ .
fa .4~ 1J tt~ JJ ZJ71.1 (d ftS 1J 11 bl (IS JJ (~ U 1J rd 1J (j~ tT) U J.~ 1.~
(J7 (~ t71 ~1 (U r~ t71 U b11~ U U
tJl J-W a c~ .U r~ 1~ U U ~ rtf .U U ~ cU J-~ ~ b7 U ~7 1~ ~Jl rtS C71 td .t~
~ rIS (~ b7 rl3 r~ ~.~ U U U
.i.~ rd !If U c0 U1 U td rtS t~ U ~r ~ t~ rd .u ttf rtf U ~1 t~ rtf U ~1 U b1
rti a.-y7 ca .~ r~ a..~ ra tJl C71
U (d C6 rd ca to ~ trl r~ U .i..r 1~ .J.~ n'S ~ L7 U bt b1 1~ rd 1-~ to ~ U
rtS ~1 Cll t~ U U 1~ ~1 n3 tI1 ~t
(d U U (LS trl fd 1~ ca U t11 U N ~ al (fS U U (LS c0 C511J bl U ~ tJl f~ (d ~
~ bl tJl b1 bl b11~ 1-1
~ ca rtS U ~ to ~1 U al ~1 ca b1 rd b1.i.~ rtS U U b1.t~ tJ1 ca U r~ ~ U rti U
~ b1 rtS ~ b1 rti b1 ~
~l td CJl c~ t71 U bl U U l.Wa fa !J 1~ (a y.! .1..J (a t71 J-1 (Cf U tJl la
t31 to trl bl U N n3 1J (l5 (ff U 1J
R3 (~ 01 ~ (d b1 bl ~ 01 U ~ U 1J .1~ t~ ~t U b'7 U fa U tr1 U U 1~ U t17 t~ U
a.t U U f51 U bl t~
b1 fa .iJ .i~ (~ 0311-1 trl b1 U ca (t$ U rd ~11..r U (a tr1 tT7 c~ f~ .rJ b1
U 1J (~f U U (IS (a Z71 ~1 .1..1 ~1 U
t71 JJ b'1 to 05'1 rtS l..r tti ~ ~ b1 t~ b'1 .t~ b1 U ~ b'1 .r.~ CJ7 1.~ 1.~
:J 071 b1 td ~7 b1.1~ b1 1..1 rtS .u ~ rcS ~7
1~ (d rtS ~ r~ rd U U ~ r~ tn trl cd ~ ~ b7 U ~ U rti U rti r~ 1J rt n3 tn bl
U ~ U U ~1 U b1 ~
.u ~ U .u b1 U U rtS .u tIi .u ~1 rtS ~ ~1 ~ U Di ~r ~1 U U .u tn b1 U rtf ~
t~ U tr7 U rtS rt tT U
U ~1.t~ b1 b1 U t~ ~ i.y1 U ~ U r~ 1. ~ ~ U t~ t~ U 1~ U U ~1 U b1 ~ ~ U U
i.m~ t11 ~ tn tT
a'S rtS rt ~1 ~ rd .1~ tJ1 (a (a .u U ra dl U 1.~ ~ ~1 b1 rti U bl bl tn (a (d
rtS U .u U t37 U ~ tTl rt rti
.t~ U U .U ~ U b1 ~1 .i.~ to U ~ U to b1 ~ b1 t1'S U ca ~ ra rtf 1.~ b1 tJ1
rtS r~ .u ~r b1 ~ .U ~ U U
b1 k31 U b1 fa U ~ U U U b1 U rtS (a tJl lJ c~ ~1 rd 1.~ rt5 U Z31 ~ to tr1 U
~ r~ 1.~ .i.WIS .t~ ~J7 U b1
cd ~ U b7 rd U ~ ra b1 CJ11-~ .t~ .t~ U .t~ tn U b1 Cn b7 rti rt rtS t11 ~1 t~
to .1~ n6 U rd t37 b1 tJ1.r.ma
1J f~ 1-~ d.~ U rtf 1.W b1 t311J r~ 1~ ca .U cI5 .1~ .Lyl l~ b1 U 1~ (~ 1.J
f~S U U .i..~ f~ tJ1 c~S .~.J U l.~ r~
(tS U U ca fa U U 1-~ b1 rd b1 rtf c~ r~ ra 1J 1~ CfT bl lJ b1 U cIi (~ rti U
tJ1 (tS U 1..1 b1 t31 b7 U .~.J a..~
(~ U U t31 t11 b1 ~1 U rcf U b1 ~ r~ is rrt rtS ~ fT ~ ~ .u ~1 i~ ~ r~ td r~
t~ tJ1 U ~ rti rt W U trt
1-1 U U rt3 ZJ) ca rtf bl CJl U CJl ~.J U L! (~ (a ~1 rti U bl C'f1 10 J~ ca
d1 ftf f(S .t-1 bl ~7 1~ ~1 (~ (a ra 1J
1J .i~ U ~l t71 ~! Z71 U 1.1 .1.! bl fIi (a ((S U ZJl U rti 1.~ .1J tr1 fa U
y.! (a (~f bl tti b7 LJ7 .IJ b1 Ol U tr1 f~
~ U b1 rff rti b1 b1 ra U U ~ ra rtS ~ r0 rtS ~ ~7 rtf ~ .~ rtf b1 U clf ttf
cd rd c~ ra U ~ rtS ~1 b1 U
tJl (13 1l t6 bl tlf (IS trl b1 b1 J, .t~ 1-1 t77 rIS (d rti (~ .~.) U 1.1 U
rtf b1 (~ (a .tJ U ra r~ U ctS b1 J-1 b1 ~
Z31 U r~ U 1~ bllJ 1J 1J (If .L.f dllJ rtt ~..W..J 1J ttS 1.J 1.) (~ 1J U J-~
b1 ~7 U U b1 U rtf c~ cti U t111~
.~.) cLS 1J (13 (if ~J1 ft$ ca 1J U (If (a fa (a (LS J~ 1J b1 U U s~ U b1 .IJ
b1 (~ ~1 (d b1 ftS X17 J-WJ1 b1 .I, U
J-~ U b1 to 1-1 to fa rlf 1.~ to b1 L31 rt rt U J..1 fd of (1S td ca bl rtf t~
b'1 L1 b1 Z31 1J U .~..1 cIf to rlS U b1
U .u ~ .v.~ ~ r~ ~'1 U of rti b1 ~1 cd (a ni 031 U rtf U ~ rtf .~.~ ra ~ ra U
.i.~ c~ U cd U rtS b1 fJ~ t31 ~
c~ r~ ~ ~ b1 ~ b7 1.~ c~ ra Z711..~ b1 Cr rtf b1 U b7 tn t~ rtS .~ U rtS r1i U
U rtS ~ U rtS U ~ rtS U ca
+~ ~ rtS r~ U r~ 01 b1 U U b1 t~ ~ to ~ r~ (0 to rtS U U U ~ t31 1-~ chi U
.i.~ b1 r~ U U U tJl U bl
tT U U t~ b1 U ~ U rtf ~ ~ bi b7 ~ 1.~ ~ U ~..~ .u rt 1~ rti l..r bl b1 ~1
~m..~ is U U b1 U U .t~ tI3
t~ td b1 U t31 U ~l cd r~ rti U ra ca U 1.~ bl 1.~ r~ 1.~ Cn U rtf 1~ rd 1J ra
.t~ ~ b1 U 1~ b1 U U ~ U
.t~ U rtf b1 ~ rti ca U 01 U U .i..~ U t31 t17 tJ1.r..~ rt U ~ U .N ra ca U b1
bl U ~1 rti ra rtf rtS U t~ ~.~
~ r~ (a .N ~1 rd U ~1 ni tJ1.~ U cd rf3 b7 ~..~ ~ b1 U ~1 U .i.~ .u rtS b1 U
.t~ U b1 W ..~ (31 b~ b1 U U
(a trl c~ U 01 ~'1 b1 1-~ U bl U ~1 b1 U t~ .~l 1J ZJ1 1.) ~S ~1 ~ b1 b1 J..1
1.~ 1J r~ CJ7 U ra rd 1.~ 077 fly U
1.~ 1~ rtS b1 (~ 1J df U bl ~1 ~1 .I.WI U ~.1 U (If fIi (Ii b1 (~ ~..~ td a1 U
U U 1J rt3 t~ 071 (a ni .t-W1 U
a..~ b1 ~1 ~1 tr11~ b1 b1 ra Z71 c0 rtS b'1 ~ ca ~1 b1 ~ t51 .r.J rti U rtf
.1..1 U J-~ ~1 tr1 c~ ca rte t77 b1 bl b1 b1
1.~ cLS ~1 b1 ~ ~ rtf tT U .iJ U (~ t~ ~ r~ b'1 b7 b1 bl ..! b1 CJl t17 ~ b1
r~ ~ CTl ~ b1 U rt Gl U ~1 f~
bl (~ r~ t~ 1J c~ N b"! tJl U c~ Z7l bl (IS b1 (a C7~ U U U trl J..1 (a U ~ 1J
U (a t~ ~ 1J 1J .LJ fd c~ tr7
1J b1 1J U U rd ca J..1 (IS J-~ fti X51 (~ ca LTl ~1 to U ftS b1 ~'1 b1 iJ tJ1
1J b1 tr1 U 051 t~ b1 071 (d U c~f U
U b1 ~1 ~ U ~ rti r~ U CJ1 U 1~ b1 U 1J X71 U U rtS U 1~ ~ ra b1 (a ctS 1~ .u
cIS tJ1 b1 tr1 ca .N U b1
(IS (d tIi ~l ca (Ii fIS J-WJ1 (a 1~ .t-Wl ~1 t71 ca Ol b1 b1 U ~T11.~ 1.~ bl
CT U rti U ra tJ tT1 r1S bl fa (fS trl
r-I r-1 r-I r-I r-I r-I r-I r1 ri r-1 r-I r-1 r~ r-1 r-! rW --1 r-1 r-i '-1 r-
I r-I ~ r-1 v-1 r-I r-1 r-W -i r~ r-1 r-1 r-I t--I r~ r-I
~ cr O lp N 00 ~ O lD N 00 V~ O l0 N 00 ~ O l0 N 00 ~ O l0 N CO d' O l0 N pp ~
O ~p N CO
M cr lI7 L!1 l~ lC: L~ d0 00 01 01 O r-1 ~ N N M cr V' 111 L!1 lD h I~ CO 00
01 O O r-1 rl N M M ~' V~
l~ L~ I~ L~ l~ I~ L~ l~ I~ L~ C~ 00 OD 00 a0 00 a0 CO CO ~ CO CO QO 00 O 00 00
01 a1 01 01 01 01 01 01 01
O ~ O ~ O tf1
N N M M
CA 02333481 2000-11-27
WO 00/00600 263 PCTNS99/11516 .
U b1
U b1
t111~
ra U
U
U b~
~J7 JJ
J~ bl
U 1~ cc3
rd ~1 U
U .i.~ b1
U 1.~ r6
U 011-
.v 1J U
t~ U .t~
b1 ~ U
~1 CJl ,1,
CJl U tfS
rd U rti
1~ U rtf
U ~1 (0
r~ .u b1
c~ L~1 t71
U tJ1 b1
bl ~ ~
bl ~1 ~1
U .U U
1J tJ1 JJ
U td b1
1J (a t~
U U ~..~
X51 1, 1-1
b1 U .u
~7 bl U
~l.t~ U
1~ tJ1 U
U ca Ia
U b1 ~1
bl ~ rd
rtS .N U
C51 U 1.~
.~ U U
U t31 U
~ t~ U
1J 1~
t11 U rd
r~ CJl b1
U rt rtS
U rd b1
r~ rd cd
~1 JJ 1.~
rt rtS U
.u c~ rtS
~-1 U rt3
cr o to
o~ 01 0~