Note: Descriptions are shown in the official language in which they were submitted.
CA 02333756 2000-11-30
WO 99/62902 PCT/EP99/03648
N-BENZODIOXANYLMETHYL-I-PIPERIDYL-METHYLAMINE
COMPOUNDS HAVING AFFINITY FOR 5-HT RECEPTORS
The present invention relates to novel therapeutic agents which have affinity
for 5-HT~A and/or a~ and/or D2 receptors, to processes for their preparation,
to
pharmaceutical compositions containing them and to their use in the treatment
of
central nervous system disorders, for example depression, anxiety, psychoses
(for
example schizophrenia), tardive dyskinesia, Parkinson's disease, obesity,
hypertension, Tourette's syndrome, sexual dysfunction, drug addiction, drug
abuse,
cognitive disorders, Alzheimer's disease, senile dementia, obsessive-
compulsive
behaviour, panic attacks, eating disorders and anorexia, cardiovascular and
cerebrovascular disorders, migraine, non-insulin dependent diabetes mellitus,
hyperglycaemia, constipation, arrhythmia, disorders of the neuroendocrine
system,
stress, prostatic hypertrophy, drug-induced extrapyramidal symptoms and
spasticity.
In W093/17017 there are described [(benzodioxanyl, benzofuranyl and
benzopyranyl)alkylamino]alkyl substituted 2-pyrimidinyl compounds which have
vasoconstrictor activity. These compounds are claimed to be useful in treating
conditions related to vasodilation.
In W095/07274 compounds of formula I
A Rz U-Q-T
~R>>
\ B R3 t
Ra
and pharmaceutically acceptable salts thereof in which A is methylene or -O-;
B is
methylene or -O-; and g is 0, 1, 2, 3 or 4; R~ represents, halo, optionally
substituted
alkyl, optionally substituted alkoxy, optionally substituted alkylthio,
hydroxy, acyloxy,
hydroxymethyl, cyano, alkanoyl, alkoxycarbonyl, optionally N-substituted
carbamoyl,
carbamoylmethyl, sulphamoyl or sulphamoylmethyl, an amino group optionally
substituted by one or two alkyl groups, or two adjacent R~ groups together
with the
carbon atoms to which they are attached form a fused bent ring; R2 is H, alkyl
or
alkoxy; R3 and Ra, which are the same or different, are H, or alkyl; U is an
alkylene
chain optionally substituted by one or more alkyl; Q represents a divalent
group of
formula Ila, Ilb or Ilc
CA 02333756 2000-11-30
WO 99/62902 PCT/EP99/03648
2
Rs
X
N V~ ~ N Ila
X'
-N-V'-N- lib
Rs
/X
N ~V-N- Ilc
\X ~'
in which V is a bond or an alkylene chain optionally substituted by one or
more alkyl;
VN is an alkylene chain optionally substituted by one or more alkyl; X is a
bond or an
alkylene chain and X' is an alkylene chain, provided that the total number and
carbon
atoms in X and X' amounts to 3 or 4; R5 is H, or alkyl; and T represents an
optionally
substituted aromatic group which optionally contains one or more N atoms,
provided
that T is not 2-pyrimidinyl when A is -O-; are disclosed as having utility in
the
treatment of central nervous system disorders, for example depression,
anxiety,
psychoses (for example schizophrenia), tardive dyskinesia, Parkinson's
disease,
obesity, hypertension, Tourette's syndrome, sexual dysfunction, drug
addiction, drug
abuse, cognitive disorders, Alzheimer's disease, senile dementia, obsessive-
compulsive behaviour, panic attacks, eating disorders and anorexia,
cardiovascular
and cerebrovascular disorders, non-insulin dependent diabetes mellitus,
hyperglycaemia, constipation, arrhythmia, disorders of the neuroendocrine
system,
stress, prostatic hypertrophy, and spasticity.
Surprisingly it has been found that certain compounds selected from the
general disclosure of W095107274, but not specifically named or exemplified
therein,
exhibit increased activity and also increased selectivity compared to the
compounds
exemplified in W095I07274.
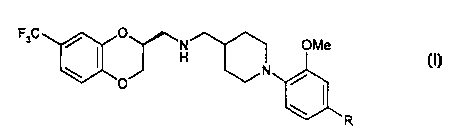
The present invention provides compounds of formula I
/ ~ O N OMe
H I
\ N ~ I
O
R
CA 02333756 2000-11-30
WO 99/62902 PCT/EP99/03648
3
including pharmaceutically acceptable salts thereof in which R represents H or
F.
Specific compounds of the present invention are
(S)-(-)-1-[1-(4-fluoro-2-methoxyphenyl)piperid-4-yl]-N-(7-trifluoromethyl-2,3-
dihydro-
1,4-benzodioxin-2-ylmethyl)methylamine
and
(S)-(-)-1-[ 1-(2-methoxyphenyl)piperid-4-yl]-N-(7-trifluoromethyl-2, 3-dihydro-
1,4-
benzodioxin-2-ylmethyl)methylamine
and pharmaceutically acceptable salts thereof.
The compounds of the present invention are advantageous over compounds
known in the prior art because of their selectivity in receptor binding assays
and their
superior oral activity.
Compounds of formula I may exist as salts with pharmaceutically acceptable
acids. Examples of such salts include hydrochlorides, hydrobromides,
sulphates,
methanesulphonates, nitrates, maleates, acetates, citrates, fumarates,
tartrates [eg
(+)-tartrates, (-)-tartrates or mixtures thereof including racemic mixtures],
succinates,
benzoates and salts with amino acids such as glutamic acid. Compounds of
formula
I and their salts may exist in the form of solvates (for example hydrates).
Certain compounds of formula I and their salts may exist in more than one
crystal form and the present invention includes each crystal form and mixtures
thereof. Certain compounds of formula i and their salts may also exist in the
form of
solvates, for example hydrates, and the present invention includes each
solvate and
mixtures thereof.
The present invention also includes pharmaceutical compositions containing
a therapeutically effective amount of a compound of formula I or a salt
thereof
together with a pharmaceutically acceptable diluent or carrier.
CA 02333756 2000-11-30
WO 99/62902 PCT/EP99/03648
4
As used hereinafter, the term active compound denotes a compound of
formula I or a salt thereof. In therapeutic use, the active compound may be
administered orally, rectally, parenterally or topically, preferably orally.
Thus the
therapeutic compositions of the present invention may take the form of any of
the
known pharmaceutical compositions for oral, rectal, parenteral or topical
administration. Pharmaceutically acceptable carriers suitable for use in such
compositions are well known in the art of pharmacy. The compositions of the
invention may contain 0.1-99% by weight of active compound. The compositions
of
the invention are generally prepared in unit dosage form. Preferably the unit
dosage
of active ingredient is 1-500 mg. The excipients used in the preparation of
these
compositions are the excipients known in the pharmacist's art.
Compositions for oral administration are the preferred compositions of the
invention and these are the known pharmaceutical forms for such
administration, for
example tablets, capsules, syrups and aqueous or oil suspensions. The
excipients
used in the preparation of these compositions are the excipients known in the
pharmacist's art. Tablets may be prepared by mixing the active compound with
an
inert diluent such as calcium phosphate in the presence of disintegrating
agents, for
example maize starch, and lubricating agents, for example magnesium stearate,
and
tableting the mixture by known methods. The tablets may be formulated in a
manner
known to those skilled in the art so as to give a sustained release of the
compounds
of the present invention. Such tablets may, if desired, be provided with
enteric
coatings by known methods, for example by the use of cellulose acetate
phthalate.
Similarly, capsules, for example hard or soft gelatin capsules, containing the
active
compound with or without added excipients, may be prepared by conventional
means and, if desired, provided with enteric coatings in a known manner. The
tablets and capsules may conveniently each contain 1 to 500 mg of the active
compound. Other compositions for oral administration include, for example,
aqueous
suspensions containing the active compound in an aqueous medium in the
presence
of a non-toxic suspending agent such as sodium carboxymethyl- cellulose, and
oily
suspensions containing a compound of the present invention in a suitable
vegetable
oil, for example arachis oil.
CA 02333756 2000-11-30
WO 99/62902 PCT/EP99/03648
The active compound may be formulated into granules with or without
additional excipients. The granules may be ingested directly by the patient or
they
may be added to a suitable liquid carrier (for example water) before
ingestion. The
granules may contain disintegrants (for example a pharmaceutically acceptable
5 effervescent couple formed from an acid and a carbonate or bicarbonate salt)
to
facilitate dispersion in the liquid medium.
Compositions of the invention suitable for rectal administration are the known
pharmaceutical forms for such administration, for example, suppositories with
cocoa
butter or polyethylene glycol bases.
Compositions of the invention suitable for parenteral administration are the
known pharmaceutical forms for such administration, for example sterile
suspensions
or sterile solutions in a suitable solvent.
Compositions for topical administration may comprise a matrix in which the
pharmacologically active compounds of the present invention are dispersed so
that
the compounds are held in contact with the skin in order to administer the
compounds transdermally. A suitable transdermal composition may be prepared by
mixing the pharmaceutically active compound with a topical vehicle, such as a
mineral oil, petrolatum andlor a wax, for example paraffin wax or beeswax,
together
with a potential transdermal accelerant such as dimethyl sulphoxide or
propylene
glycol. Alternatively the active compounds may be dispersed in a
pharmaceutically
acceptable cream or ointment base. The amount of active compound contained in
a
topical formulation should be such that a therapeutically effective amount of
the
compound is delivered during the period of time for which the topical
formulation is
intended to be on the skin.
The compounds of the present invention may also be administered by
continuous infusion either from an external source, for example by intravenous
infusion or from a source of the compound placed within the body. Internal
sources
include implanted reservoirs containing the compound to be infused which is
continuously released for example by osmosis and implants which may be (a)
liquid
such as a suspension or solution in a pharmaceutically acceptable oil of the
CA 02333756 2000-11-30
WO 99/62902 PCT/EP99/036A8
6
compound to be infused for example in the form of a very sparingly water-
soluble
derivative such as a dodecanoate salt or ester or (b) solid in the form of an
implanted
support, for example of a synthetic resin or waxy material, for the compound
to be
infused. The support may be a single body containing all the compound or a
series
of several bodies each containing part of the compound to be delivered. The
amount
of active compound present in an internal source should be such that a
therapeutically effective amount of the compound is delivered over a long
period of
time.
In some formulations it may be beneficial to use the compounds of the
present invention in the form of particles of very small size, for example as
obtained
by fluid energy milling.
In the compositions of the present invention the active compound may, if
desired, be associated with other compatible pharmacologically active
ingredients.
The pharmaceutical compositions containing a therapeutically effective
amount of a compound of formula 1 or a salt thereof may be used to treat
depression,
anxiety, psychoses (for example schizophrenia), tardive dyskinesia,
Parkinson's
disease, obesity, hypertension, Tourette's syndrome, sexual dysfunction, drug
addiction, drug abuse, cognitive disorders, Alzheimer's disease, senile
dementia,
obsessive-compulsive behaviour, panic attacks, eating disorders, anorexia,
cardiovascular and cerebrovascular disorders, migraine, non-insulin dependent
diabetes mellitus, hyperglycaemia, constipation, arrhythmia, disorders of the
neuroendocrine system, stress, prostatic hypertrophy, drug-induced
extrapyramidal
symptoms and spasticity in human beings. Whilst the precise amount of active
compound administered in such treatment will depend on a number of factors,
for
example the age of the patient, the severity of the condition and the past
medical
history and always lies within the sound discretion of the administering
physician, the
amount of active compound administered per day is in the range 1 to 1000 mg
preferably 5 to 500 mg given in single or divided doses at one or more times
during
the day.
CA 02333756 2000-11-30
WO 99/62902 PCT/EP99/0364$
7
The ability of compounds of formula I to interact with 5-hydroxytryptamine (5-
HT) receptors has been demonstrated by the following test which determines the
ability of the compounds to inhibit tritiated ligand binding to 5-HT receptors
in vitro
and in particular to 5-HT~A receptors.
Hippocampal tissue from the brains of male Charles River CD rats weighing
between 150-250 g were homogenised in ice-cold 50 mM Tris-HCI buffer (pH 7.7)
when measured at 25°C, 1:40 w/v) and centrifuged at 30,000 g at
4°C for 10
minutes. The pellet was rehomogenised in the same buffer, incubated at
37°C for 10
minutes and centrifuged at 30,000 g at 4°C for 10 minutes. The final
pellet was
resuspended in 50 mM Tris-HCI buffer (pH 7.7) containing 4 mM CaCl2, 0.1 % L-
ascorbic acid and 10 ~.M pargyline hydrochloride (equivalent to 6.25 mg wet
weight
of tissue/ml) and used immediately in the binding assay. Aliquots (400 ~I;
equivalent
to 2.5 mg wet weight of tissue/tube) of this suspension were added to tubes
containing the ligand (50 ~.I; 2 nM) and distilled water (50 ~I; total
binding) or 5-HT
(50 ~I; 10 ~M; non-specific binding) or test compound (50 ~l; at a single
concentration of 106 M or at 10 concentrations ranging from 10'"-10'3 M). The
ligand
was [3H]8-hydroxy-2-(dipropylamino)tetralin ([3H]8-OH-DPAT) and the mixture
was
incubated at 25°C for 30 minutes before the incubation was terminated
by rapid
filtration.
The filters were washed with ice-cold Tris-HCI buffer and dried. The filters
were punched out into vials, scintillation fluid added and radioactivity
determined by
liquid scintillation counting. The percentage displacement of specific binding
of the
tritiated ligand was calculated for the single concentration (10~ M) of test
compound.
Displacement curves were then produced for those compounds which displaced
>_50% of specific binding of the tritiated ligand at 10'6 M using a range of
concentrations of the compound. The concentration which gave 50% inhibition of
specific binding (ICso) was obtained from the curve. The inhibition
coefficient Ki was
then calculated using the formula
I C5o
K; _
1+([iigand]/Kp)
CA 02333756 2000-11-30
WO 99/62902 PCT/EP99/03648
8
in which (ligand] is the concentration of the tritiated ligand used and Kp is
the
equilibrium dissociation constant for the ligand.
The ability of compounds of formula I to interact with adrenoceptor binding
sites has been demonstrated by the following test which determines the ability
of the
compounds to inhibit tritiated ligand binding to adrenoceptors in vitro and in
particular
a~-adrenoceptors.
Whole cortical tissue from the brains of male Charles River CD rats weighing
between 150-250 g were homogenised in ice-cold 50 mM Tris-HCI, pH 7.6 (at
25°C;
1:40 w/v) and centrifuged at 1000 g at 4°C for 10 minutes. The
supernatant was
centrifuged at 30,000 g at 4°C for 10 minutes. The pellet was
rehomogenised in
50 mM Tris-HCI, pH 7.6 (1:40 w/v) and centrifuged at 30,000 g at 4°C
for 10 minutes.
The final pellet was resuspended in 50 mM Tris-HCI, pH 7.6 (equivalent to 12.5
mg
wet weight of tissuelml) and used immediately in the binding assay. Aliquots
(400 fit;
equivalent to 5 mg wet weight of tissue/tube) of this suspension were added to
tubes
containing the ligand (50 ~I; 0.1 nM} and distilled water (50 ul; total
binding) or
phentolamine (50 pl; 5 pM; non-specific binding) or test compound (50 ~.I; at
a
single concentration of 10-6M or at 10 concentrations ranging from 10'" -
10~3M). The
ligand was [7-methoxy-3H]prazosin and the mixture was incubated at 30°C
for 30
minutes before the incubation was terminated by rapid filtration.
The filters were washed with ice-cold Tris-HCI buffer and dried. The filters
were punched out into vials, scintillation fluid added and radioactivity
determined by
liquid scintillation counting. The percentage displacement of specific binding
of the
tritiated ligand was calculated for the single concentration (10~ M) of test
compound.
Displacement curves were then produced for those compounds which displaced
>_50% of specific binding of the tritiated ligand at 10~ M using a range of
concentrations of the compound. The concentration which gave 50% inhibition of
specific binding (IC5o) was obtained from the curve. The inhibition
coefficient Ki was
then calculated using the formula
CA 02333756 2000-11-30
WO 99/62902 PCT/EP99/03648
9
ICSo
K;
1+([ligand]/Ko)
in which [ligand] is the concentration of the tritiated ligand used and Kp is
the
equilibrium dissociation constant for the ligand.
The ability of compounds of formula I to interact with dopamine receptors has
been demonstrated by the following test which determines the ability of the
compounds to inhibit tritiated ligand binding to dopamine receptors in vitro
and in
particular to the D2 dopamine receptors.
Striatal tissue from the brains of male Charles River CD rats weighing
between 140-250 g were homogenised in ice-cold 50 mM Tris-HCI buffer (pH 7.7
when measured at 25°C) and centrifuged at 40,000 g for 10 minutes. The
pellet was
resuspended in Tris salts buffer (50 mM Tris-HCI buffer containing 120 mM
NaCI,
5 mM KCI, 2 mM CaCl2 and 1 mM MgCl2 with the addition of 6 mM ascorbic acid;
pH 7.7 when measured at 25°C), and again centrifuged at 40,000 g for 10
minutes.
The final pellet was stored at -80°C. Before each test the pellet was
resuspended in
Tris salts buffer (equivalent to 2 mg wet weight of tissue/ml). Aliquots (720
~.I;
equivalent to 1.44 mg wet weight of tissue/tube) of this suspension were then
added
to tubes containing the ligand (40 ul; 1 nM) and Tris salts buffer (40 wl;
total binding)
or spiroperidol (40 pl; 10 nM; non-specific binding) or test compound (40 ul;
at a
single concentration of 10'6M or at 6 concentrations ranging from 10'"-10~'M).
The
ligand was tritiated (S)-sulpiride and the mixture was incubated at 4°C
for 40 minutes
before the incubation was terminated by rapid filtration.
The filters were washed with ice-cold Tris-HCI buffer and dried. The filters
were punched out in to vials, scintillation fluid added and were left for
about 20 hours
before being counted by scintillation spectrophotometry. The percentage
displacement of specific binding of the tritiated ligand was calculated for
the single
concentration (10's M) of test compound. Displacement curves were then
produced
over a range of concentrations for those compounds which displaced >_50% of
specific binding of the tritiated ligand at 10-6M. The concentration which
gave a 50%
CA 02333756 2000-11-30
WO 99/62902 PCT/EP99/03648
inhibition of specific binding (IC5o) was obtained from the curve. The
inhibition
coefficient Ki was then calculated using the formula
I Cso
5 K; _
1 +([ligand]/Kp)
in which [ligand] is the concentration of the tritiated ligand used and Kp is
the
10 equilibrium dissociation constant for the ligand.
The Ki values obtained in the above tests for 5-HT~A, a~ and D2 binding for
each of the final products of Examples 1, 2, and Comparative ExampIeA are
given in
Table I below. It is clear from these data that compounds of the present
invention
have significantly less affinity for the a~ adrenoceptor than compounds
previously
described. This is important as it is known in the art that a~ receptor
antagonism
mediates serious side-effects such as hypotension, sedation and sexual
dysfunction.
Thus, compounds which interact preferentially with Dz and 5-HT~A receptors
whilst
having less interaction with a~ receptors are advantaged.
TABLE 1
Example Ki (nM) value
Number for
5-HT~A D2 a~
1 23 65 183
2 31 54 404
A 22 44 53
Antagonism of A~omorphine-Induced Climbing in Mice
Groups of 10 male mice weighing 18-35 g (max. range 10 g) were treated
with test compound or control vehicle by po administration. 30 minutes later,
mice
were injected subcutaneously with apomorphine (0.88 mg/kg). Immediately after
the
apomorphine injection the mice were placed in the test cages and the climbing
CA 02333756 2000-11-30
WO 99/62902 PCT/EP99/03648
11
behaviour of each mouse was assessed at 10 and 20 minutes on a simple 0-2
ranking scale.
The ED5o values (dose causing 50% of the control score) for the test
compounds and 95% confidence limits were calcuiated. EDSO values are
calculated
as free base equivalents and are given in Table 2 alongside Comparative
Example
A. The compounds of the present invention are more potent orally than
compounds
previously described. More potent compounds are advantaged as they are less
likely to induce systemic toxicological effects on organs which are not the
therapeutic
target.
TABLE 2
Example ~ EDSO ( mg/kg )
- 1
2 1.8
A 5
Processes for the preparation of compounds of formula I will now be
described. These processes form a further aspect of the present invention. The
processes are preferably carried out at atmospheric pressure, at a temperature
in the
range 0-200°C, preferably in the range 20-150°C. The
substituents are as defined
for formula I above unless otherwise stated.
Compounds of formula I may be prepared by reacting a compound of formula
F3C / O
~Z II
O
in which Z is a leaving group, for example toluene-4-sulphonyloxy, with a
compound
of formula III
CA 02333756 2000-11-30
WO 99/62902 PCT/EP99/03648
12
HZN ~ Me
N / ~ 111
R
in which R is as previously defined, optionally in the presence of a suitable
solvent or
mixture of solvents, for example a hydrocarbon, eg toluene, or a polar
solvent, eg
dimethylformamide, or mixtures thereof, optionally in the presence of a base,
for
example potassium carbonate at a temperature in the range of 0 - 250°C.
The invention is illustrated by the following Examples which are given by way
of example only. The final product of each of these Examples was characterised
by
one or more of the following procedures: gas-liquid chromatography; high
performance liquid chromatography; elemental analysis, nuclear magnetic
resonance
spectroscopy and infrared spectroscopy.
Example 1
Part A
a) A solution of 5-fluoro-2-nitrophenol (25.0 g) in dry dimethylformamide
(100 ml) was added dropwise with stirring over 1 hour to a suspension of
sodium
hydride (7.0 g of a 60% dispersion in mineral oil) in dry dimethylformamide
(250 ml)
under nitrogen. The mixture was stirred for 1 hour and then iodomethane (10.0
ml)
was added dropwise to the mixture. The mixture was then stirred and heated on
a
steam bath for 3.5 hours. Further iodomethane (2.5 ml) was added and the
mixture
was stirred at 95-100°C for 1 hour and then allowed to cool overnight.
Concentrated
aqueous ammonia solution (15.0 ml; S.G. 0.880) was added dropwise and the
mixture was stirred for 15 minutes. The mixture was poured into water (1.5 l)
basified
with aqueous sodium hydroxide solution (5M) and extracted with dichloromethane
(4 x 400 mi). The combined organic extracts were washed with water, brine,
dried,
filtered and evaporated to give a liquid which was dissolved in ether (500
ml),
washed with water, brine, dried, filtered and evaporated to give an oil which
was
again dissolved in ether and the previous washing and drying process repeated
to
give an oil which was triturated with petroleum ether, b.p. 60-80°C,
and allowed to
CA 02333756 2000-11-30
WO 99/62902 PCT/EP99/03648
13
stand overnight. The solid produced was collected by filtration and dried in
vacuum
to give 4-fluoro-2-methoxynitrobenzene. The structure was confirmed by ~H nmr.
b) A solution of 4-fluoro-2-methoxynitrobenzene (23.7 g) in industrial
methylated
spirits (400 ml) was hydrogenated at ambient temperature using 10% palladium
on
charcoal as the catalyst. After the theoretical uptake of hydrogen, the
mixture was
filtered to remove the catalyst and the filtrate was evaporated to give 4-
fluoro-2-
methoxyaniline as an oil.
c) A mixture of 4-fluoro-2-methoxyaniline (5.7 g) and 4-carbamoyl-1-(2,4-
dinitrophenyl)pyridinium chloride (22.4 g, prepared as described in
W095/07274)
and methanol (350 ml) was stirred and boiled under reflux for 2 hours. The
mixture
was cooled in an ice-water bath and filtered. The filtrate was evaporated
under
reduced pressure to give a solid which was triturated with boiling acetone
(900 ml).
The mixture was allowed to coot and the product collected by filtration and
washed
with acetone to give 4-carbamoyl-1-(4-fluoro-2-methoxyphenyl)pyridinium
chloride.
d) The product from c) (13.2 g), ammonium formate (26.6 g) and 10% palladium
on charcoal (6.6 g) were stirred under nitrogen whilst industrial methylated
spirits
(250 ml) was added. The mixture was stirred vigorously and boiled under reflux
under nitrogen for 3.5 hours with sublimed ammonium formate being washed back
into the reaction vessel with water. The mixture was allowed to cool then
filtered
through a filtration aid which was washed with industrial methylated spirits,
ethyl
acetate and water. The filtrate was evaporated under reduced pressure and the
residue was partitioned between water (1 I) and ethyl acetate (500 ml). The
mixture
was made strongly alkaline with 5M sodium hydroxide solution. The aqueous
layer
was separated and extracted with ethyl acetate (3 x 500 ml). The combined
ethyl
acetate extracts were dried, filtered and evaporated to give 1-(4-fluoro-2-
methoxyphenyl)piperidine-4-carboxamide.
e) The product from d) (10.4 g) was suspended in tetrahydrofuran (300 ml) and
added in portions with stirring to a suspension of lithium aluminium hydride
(3.4 g) in
tetrahydrofuran (100 ml) under nitrogen in an ice-water bath. The mixture was
stirred
in an ice-water bath for 1.5 hours then stirred at ambient temperature for 19
hour.
CA 02333756 2000-11-30
WO 99/62902 PCT/EP99/03648
14
Water (20 ml) was added cautiously to the reaction mixture followed by 5M
sodium
hydroxide solution (20 ml) and water (40 ml). The mixture was stirred for 1
hour then
filtered through a filtration aid, washing the aid with ether (700 ml). The
filtrate was
separated and the organic phase was dried, filtered and evaporated to give a
gum
which was triturated with dichloromethane (100 ml) and filtered to remove some
solid. The filtrate was evaporated to give an oil which was purified by flash
column
chromatography on silica using methanol followed by methanol/triethylamine
(10:1 )
as the mobile phase. Appropriate fractions were collected, combined and
evaporated to give 1-(4-fluoro-2-methoxyphenyl)piperidine-4-methylamine.
Part B
a) Hexamethylenetetramine (47.5 g) was added portionwise to a stirred solution
of 4-triffuoromethylphenol (50 g) in trifluoroacetic acid (680 ml) and the
mixture was
heated at reffux temperature for 24 hours. After cooling, water (355 ml) was
added
followed by aqueous sulphuric acid (50% v/v, 190 ml) and the reaction was
stirred at
ambient temperature for 4 hours. The acidic aqueous phase was extracted with
diethyl ether (3 x 500 ml). The combined organic extracts were washed with
hydrochloric acid (5M, 3 x 500 ml) then water (500 ml) and dried over
magnesium
sulphate. The solvent was removed under reduced pressure and the residue
purified
by column chromatography on silica eluting with a 4:1 mixture of petroleum
ether
(b.p. 40-60°C) and ethyl acetate. The appropriate fractions were
combined and the
solvent removed under reduced pressure to give 5-trifluoromethyl-2-
hydroxybenzaldehyde (25 g) as a light pink solid.
b) A mixture of (R)-glycidyl 4-toluenesulphonate (24 g), 5-trifluoromethyl-2-
hydroxybenzaldehyde (20 g) and potassium carbonate (16 g) in dimethylformamide
(550 ml) was stirred and heated at 60°C for 72 hours. After cooling,
brine (1.5 I) was
added and the resultant mixture extracted with ether {4 x 500 ml). The
combined
ether extracts were washed with brine (2 x 500 ml), then water (500 ml) and
dried
over magnesium sulphate. The residue was purified by flash column
chromatography on silica eluting with a 3:1 mixture of petroleum ether (b.p.
40-60°C)
and ethyl acetate to give (R)-5-trifluoromethyl-2-(2,3-
epoxypropoxy)benzaldehyde
(18.7 g) as a yellow oil.
CA 02333756 2000-11-30
WO 99/62902 PCT/EP99/03648
c) A mixture of the product from the previous reaction (18.7 g) and 3-
chloroperoxybenzoic acid (57-86%, 48.7 g) in dichloromethane (1 I) was heated
under reflux for 24 hours then allowed to cool to ambient temperature. The
mixture
5 was washed with saturated aqueous sodium bicarbonate (3 x 700 ml), water
(2 x 700 ml) and brine (700 ml), then dried over magnesium sulphate. The
solvenl
was evaporated to give crude (R)-5-trifluoromethyl-2-(2,3-epoxypropoxy)phenyl
formate (16.7 g).
10 d) A mixture of the product from the previous reaction (16.7 g),
tetrahydrofuran
(220 ml) and a saturated aqueous potassium carbonate solution (175 ml) was
stirred
vigorously at ambient temperature for 24 hours. Water (500 ml) was added and
the
organic phase was removed. The aqueous phase was extracted with ethyl acetate
(3 x 300 ml) and the combined organic extracts were dried over magnesium
15 sulphate. The solvent was removed under reduced pressure and the residue
purified
by flash column chromatography on silica eluting with a 4:1 grading to 1:1
mixture of
petroleum ether (b.p.40-60°C) and ethyl acetate. Appropriate fractions
were
combined and the solvent was removed under reduced pressure to give (S)-7-
trifluoromethyl-2,3-dihydro-1,4-benzodioxin-2-ylmethanol (12 g) as a yellow
oil.
e) A solution of 4-toluenesulphonyl chloride (9.6 g) in dichloromethane (60
ml)
was added dropwise to a solution the product from the previous reaction (10.7
g) and
4-dimethylaminopyridine (6.7 g) in dichloromethane (90 ml) between 0-
5°C. The
mixture was stirred at ambient temperature for 4 hours then allowed to stand
for 18
hours. The solution was washed with dilute hydrochloric acid (5M, 2 x 300 ml)
dried
over magnesium sulphate and the solvent was removed under reduced pressure to
afford (R)-7-trifluoromethyl-2,3-dihydro-1,4-benzodioxin-2-ylmethyl 4-toluene-
sulphonate (15.5 g) as a white solid.
Part C
The 4-toluenesuiphonate from Part B (e) (4.3 g), the methylamine from Part A
(e) (2.4 g), potassium carbonate (2.4 g), toluene (80 ml) and
dimethylformamide
(30 ml) were stirred and boiled under reflux for 24 hours. The mixture was
allowed to
CA 02333756 2000-11-30
WO 99/62902 PCT/EP99/03648
16
cool, poured into water (1.5 I) and then extracted into .ethyl acetate (3 x
350 ml). The
combined organic extracts were extracted with 5M hydrochloric acid (3 x 250
ml).
The combined acidic extracts were basified with 5M sodium hydroxide solution
and
extracted with ethyl acetate (3 x 250 ml) and these organic extracts were
dried,
filtered and evaporated to give an oil. This oil was dissolved in ether (100
ml) and
dried over potassium carbonate. The mixture was filtered and evaporated to
give an
oil which was purified by flash column chromatography on silica using ethyl
acetate/
petroleum ether, b.p. 60-80°C {9:1) as the mobile phase. Appropriate
fractions were
collected, combined, and evaporated to give an orange oil which was dissolved
in
ether (100 ml) and hydrogen chloride gas was bubbled through the solution for
15
minutes. A precipitate which formed was collected by filtration and dried
under
vacuum at 80°C for 23 hours to give (S)-(-)-1-[1-(4-fluoro-2-
methoxyphenyl)piperid-4-
yl]-N-(7-trifluoromethyl-2,3-dihydro-1,4-benzodioxin-2-ylmethyl)methylamine
dihydrochloride, m.p. 230-231°C.
Example 2
A mixture of 1-[1-(2-methoxyphenyl)piperid-4-yl]methylamine (3.3 g, prepared
as described in W095/07274), (R)-7-trifluoromethyl-1,4-benzodioxan-2-ylmethyl
4-
toluene sulphonate (3.0 g), potassium carbonate (2.5 g), toluene (22 ml) and
dimethylformamide (10 ml) was boiled under reflux for 18 hours with stirring.
The
mixture was cooled, poured into water {100 ml) and extracted with ethyl
acetate. The
combined organic extracts were washed with dilute hydrochloric acid (2 x 200
ml)
and the acidic extracts were basified with concentrated sodium hydroxide
solution
and then extracted with ethyl acetate (200 ml). These organic extracts were
dried,
filtered and evaporated to give an oil which was purified by flash column
chromatography on silica using ethyl acetate/methanol (95:5) as the mobile
phase to
give an oil which was treated with hydrogen chloride gas as described in
Example 1
to give (S)-(-)-1-[1-(2-methoxyphenyl)piperid-4-yl]-N-(7-trifluoromethyl-2,3-
dihydro-
1,4-benzodioxin-2-ylmethyl)methylamine dihydrochloride, m.p.243-245°C
with
decomposition.
CA 02333756 2000-11-30
WO 99/62902 PCT/EP99/03648
17
Comparative Example A
(S)-(-)-N-(7-Chloro-1,4-benzodioxan-2-ylmethyl)-1-(1-(2-methoxyphenyl)piperid-
4-
yl)methylamine (alsa known as (S)-(-)-N-(7-chloro-2,3-dihydro-1,4-benzodioxin-
2-
methyl)-1-(1-(2-methoxyphenyl)piperid-4-yl)methylamine) was prepared as
described
in WO 95/07274.