Note: Descriptions are shown in the official language in which they were submitted.
CA 02336108 2000-12-28
WO 00/004b9 PCT/US99/14758
ALKYL KETONES AS POTENT ANTI-CANCER AGENTS
Field of the Invention
The present invention relates to alkyl ketone compounds effective for treating
tumor cells and particularly effective to induce apoptosis in leukemia cells,
breast
cancer cells, prostate cancer cells, and brain cancer cells.
Background of the Invention
Cancer is a major disease that continues as one of the leading causes of death
at any age. In the United States alone, it is anticipated that more than a
half a million
Americans will die of cancer in 1999. Currently, radiotherapy and chemotherapy
are
two important methods used in the treatment of cancer.
Considerable efforts are underway to develop new chemotherapeutic agents
for more potent and specific anti-cancer therapy, presenting effective and
efficient
cytotoxicity against tumor cells, with minimal interference with normal cell
function.
Accordingly, there is an urgent need for the development and analysis of
novel,
effective anti-cancer agents.
Summary of the Invention
Novel alkyl ketone compounds have been found to be potent cytotoxic agents
with potent activity against cancer cells. For example, certain alkyl ketone
compounds were found to exhibit potent cytotoxic activity, particularly
against
human breast cancer and leukemic cell lines, at micromolar concentrations.
These
compounds were also effective in inhibiting adhesion and invasion by cancer
cells.
Accordingly, the present invention includes novel compounds and
compositions having potent cytotoxic activity. The present invention also
includes
methods for treating tumors by administering to a subject an effective amount
of a
compound of the invention to inhibit growth and/or induce apoptosis of tumor
cells.
Compositions of the invention contain an effective cytotoxic or inhibitory
amount of
a compound.
SUBSTITUTE SHEET (RULE 26)
CA 02336108 2000-12-28
WO 00/00469 PCT/US99/14758
2
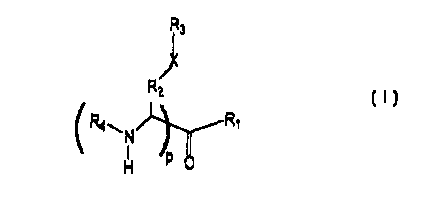
The compounds of the invention have the following formula I:
R3
R
2
R~
p 0
H I
wherein
p is an integer selected from 0 and 1;
XisOorS;
10 R' is H, hydroxyl, (C, - C3o) alkyl, (C, - C3o) alkenyl, (C, - C3o)
haloalkyl, (C1- C3o) diazoalkyl, , -CHz O-C(O)R5, -NR6R~, or -CHz -S-
R9
wherein RS is independently aryl, (C~ - C3o) alkyl, (C~ - C3o)
haloalkyl, (C1- C3o) alkenyl, (C, - C3o) diazoalkyl, (C1- C24) cycloalkyl, or
{C1 - C24) cycloalkenyl,
R6 is independently H, (C~ - C3o) alkyl, (Cl - C3o) haloalkyl, (C, -
C3o) diazoalkyl, (C, - C3o) alkenyl, (C, - C3o) haloalkenyl, (Ci - CZa)
cycloalkyl, or (C~ - C24) cycloalkenyl;
R' is -ORa,
R8 is independently (C, - C3o) alkyl, (C, - C3o) haloalkyl, (Ci - C3o)
diazoalkyl, (C1- C3o) alkenyl, (Cl - C3o) haloalkenyl, (C~ - C24) cycloalkyl,
or (C1- C24) cycloalkenyl;
R9 is independently (C, - C3o) alkyl, (C~ - C3o) haloalkyl, (C~ - C3o)
diazoalkyl, (C~ - C3o) alkenyl, (C~ - C3o) haloalkenyl, (C3 - C24) cycloalkyl,
(Ci - C24) cycloaikenyl, or -Rl°COzH;
R'° is (C1- C3o) alkyl or (C~ - C3o) alkenyl,
R2 is C, or C2; CHZ or CH2CH2
R3 is (C~ - C3o) alkyl, (C, - C3o) haloalkyl, (C~ - C3o) alkenyl, (C~ -
C3o) haloalkenyl, (C, - C24) cycloalkyl, (Cl - Cz4) cycloalkenyl, (C, -
Cz4)aryl, anthroquinonylmethyl, naphthylmethyl, -SRl ~, or -CR12;
SUBSTITUTE SHEET (RULE 26)
CA 02336108 2000-12-28
WO 00/00469 PCT/US99/14758
3
R~ ~ is independently {C1 - C3p) alkyl, (C, - C3o) haloalkyl, (C, -
C3o) alkenyl, or (C, - C3o) haloalkenyl;
R12 is aryl substituted methyl;
R4 is H, -C(O)R~3, or -C(O)-O-R'4;
R13 and R'4 are each independently (C, - C12) alkyl, (C1 - C~Z)
haloalkyl, (C~ - C12) alkenyl, (Cl - C12) haloalkenyl, (C3 - C12) cycloalkyl,
or (C3 - C12) cycloalkenyl; or
a pharmaceutically acceptable acid addition salt thereof.
Preferred compounds of the invention are those where p is the integer 1, Rl
is a haloalkyl, RZ is C,, R3 is a (C, - C22) alkyl, and R4 is acetyl. Most
preferred is
the compound N-Ac-S-dodecyl-Cys chloromethyl ketone (HI-131 ).
Brief Description of the Drawings
Figure 1 A is a graph showing survival of primary cancer cells taken from six
children with leukemia and treated with different concentrations of compound
HI-131 as a function of drug concentration.
Figure 1 B is a graph showing mean survival as a function of drug
concentration from the data of Figure 1 A.
Figures 2A-2F show photographs of apoptosis induced by HI-131 in
treated human Leukemia cells.
Figure 2A. NALM-6 control;Figure 2B. HI-131 treated NALM-6 cells;
Figure 2C. UPN1 control; Figure 2D. HI-131 treated UPN1 cells;
Figure 2E. UPN2 control; Figure 2F. HI-131 treated UPN2 cells.
Figure 3 is a bar graph showing induction of apoptosis by HI-131 in treated
primary leukemic cells and established NALM-6 and MOLT-3 cell lines.
Figure 4 is a bar graph showing inhibition of invasive properties of human
MDA-MB-231 breast cancer cells by HI-131.
Figure 5 is a graph showing inhibition of the invasive properties of human
U373 (glioblastoma) brain tumor cells by HI-131.
Figure 6 is a graph showing inhibition of the adhesion of human MDA-
MB-231 breast tumor cells by HI-131.
Figure 7 is a graph showing inhibition of the adhesion by human U373
(glioblastoma) cells by HI-131.
SUBSTITUTE SHEET (RULE 2fi)
CA 02336108 2000-12-28
WO 00/00469 PCT/US99/14758
4
Detailed Description of the Invention
The present invention includes novel alkyl ketone compounds having potent
activity as cytotoxic agents. The compounds of the invention are useful agents
for
inhibiting growth or inducing apoptosis in tumor cells, for example, leukemia
and
breast tumor cells.
Definitions
All scientific and technical terms used in this application have meanings
commonly used in the art unless otherwise specif ed. As used in this
application, the
following words or phrases have the meanings specified.
As used herein, "alkyl", includes both branched and straight-chain saturated
aliphatic hydrocarbon groups having the specified number of carbon atoms. As a
preferred embodiment, chains of 1 to 22 carbon atoms are included.
As used herein, "alkene", includes both branched and straight chain aliphatic
hydrocarbon groups that have at least one double bond.
As used herein, "alkoxy", includes, saturated and unsaturated, branched and
straight chain aliphatic hydrocarbon groups having a specified number of
carbon
atoms where at least one carbon atom forms a single-bond to an oxygen atom.
As used herein "amine", includes primary, secondary, and tertiary amines.
As used herein "halogen" or "halo" substituent includes fluoro, chloro,
bromo, and iodo.
As used herein, "pharmaceutically acceptable salt thereof' includes an acid
addition salt or a base salt.
As used herein, "pharmaceutically acceptable Garner" includes any material
25 which, when combined with a compound of the invention, allows the compound
to
retain biological activity, such as the ability to induce apoptosis of
leukemia or
breast tumor cells, and is non-reactive with the subject's immune system.
Examples
include, but are not limited to, any of the standard pharmaceutical carriers
such as a
phosphate buffered saline solution, water, emulsions such as oil/water
emulsions,
30 and various types of wetting agents. Compositions comprising such carriers
are
formulated by well known conventional methods (see, for example, Remington's
Pharmaceutical Sciences, Chapter 43, 14th Ed., Mack Publishing Co., Easton,
PA).
"Substituted cycloalkyl" includes cyclic hydrocarbons having substituents
including halo, alkyl, alkenyl, oxyalkyl, oxyalkenyl, haloalkyl, haloalkenyl,
and aryl.
SUBSTITUTE SHEET (RULE 26~
CA 02336108 2000-12-28
WO 00/00469 PCT/US99/14758
"Substituted cycloalkenyl" includes cyclic hydrocarbons having at least one
double bond where substituents include halo, alkyl, alkenyl, oxyalkyl,
oxyalkenyl,
haloalkyl, haloalkenyl, and aryl.
"Substituted aryl" includes aromatic hydrocarbons having substituents
S including hydroxyl, amino, aminomethyl, halo, alkyl, alkenyl, oxyalkyl,
oxyalkenyl,
haloalkyl, haloalkenyl, and aryl.
"Treating" or "Treatment" in the context of this invention means the
prevention or reduction in severity of symptoms or effects of a pathological
condition, including prolonging Iife expectancy. In the context of cancer
therapy,
treatment includes prevention of tumor growth, reduction of tumor size,
enhanced
tumor cell death, and increased apoptosis.
Compounds of the Invention
The novel alkyl ketone compounds of the invention have the general
structure represented by the following formula I:
R3
R2
Rt
p 0
H (I)
wherein
p is 0 or 1;
XisOorS;
Rl is H, hydroxyl, (Cl - C3o) alkyl, (C, - C3o) alkenyl, (Ci - C3o)
haloalkyl, (C, - C3o) diazoalkyl, , -CHZ O-C(O)R5, -NR6R~, or -CHZ -S-
R9,
wherein RS is independently aryl, (Cl - C3o) alkyl, (C~ - C3a)
haloalkyl, (C, - C3o) alkenyl, (C, - C3o) diazoalkyl, (C~ - C24) cycloalkyl,
or
(C~ - CZ4) cycloalkenyl,
SUBSTITUTE SHEET (RULE 26)
CA 02336108 2000-12-28
WO 00/00469 PCT/US99/14758
6
R6 is independently H, (C, - C3o) alkyl, (C~ - C3o) haloalkyl, (Ci -
C3o) diazoalkyl, (C, - C3o) alkenyl, (Cl - C3o) haloalkenyl, (C~ - C2a)
cycloalkyl, or (C, - Cz4) cycloalkenyl;
R' is -ORg,
Rg is independently (C~ - C3o) alkyl, (C, - C3o) haloalkyl, (C~ - C3o)
diazoalkyl, (C, - C3o) alkenyl, (C~ - C3o) haloalkenyl, (C, - Cz4) cycloalkyl,
or (C~ - C24) cycloalkenyl;
R9 is independently (C, - C3o) alkyl, (C, - C3o) haloalkyl, (Cl - C3o)
diazoalkyl, (Ci - C3o) alkenyl, (C1 - C3o) haloalkenyl, (C~ - Cz4) cycloalkyl,
(Ct - C24) cycloalkenyl, or -R'°C02H;
R'° is (C~ - C3o) alkyl or (C, - C3o) alkenyl,
R2 is C, or C2; CH2 or CH2CHz
R3 is (C, - C3o) alkyl, (C, - C3o) haloalkyl, (Ci - C3o) alkenyl, (C, -
C3o) haloalkenyl, (Ci - Cz4) cycloalkyl, (C, - C24) cycloalkenyl, (C, -
C24)aryl, anthroquinonylmethyl, naphthylmethyl, -SRl ~, or -CRia;
Rl' is independently (C1- C3o) alkyl, (CI - C3o) haioaikyl, (C1-
C3o) alkenyl, or (Cl - C3o) haloalkenyl;
R12 is aryl substituted methyl;
R4 is H, -C(O)R~3, or -C(O)-O-R~4;
R~3 and R'4 are each independently (Cl - C~2) alkyl, (C, - CIZ)
haloalkyl, (C1- C,z) alkenyl, (C, - C~2) haloalkenyl, (C3 - C12) cycloalkyl,
or (C3 - C12) cycloalkenyl; or
a pharmaceutically acceptable acid addition salt thereof.
The compounds of formula I are useful for the treatment of cancer,
particularly the treatment of leukemia and breast cancer. In the method of the
SUBSTITUTE SHEET (RULE 26)
CA 02336108 2000-12-28
WO 00/00469 PCT/US99/14758
7
invention, a therapeutic amount of a compound of formula I is administered to
a
patient for the treatment of cancer.
A preferred compound of the invention has the structure of formula II:
13
X
2
R4~HN R1
0
A preferred embodiment of the compound of Formula II is that shown as
having formula III, where X is S, R3 is dodecyl, and R4 is acetyl:
S
0 R2
~ R~
H C' \N
0 (III)
R1 is most preferably chloromethyl; RZ is preferably CH2; R3 is preferably a
C~Z alkyl; R4 is preferably acetyl; and X is preferably S. A most preferred
compound of formula II is N-Ac-S-dodecyl-Cys chloromethyl ketone (HI-131 ).
Another embodiment of the invention is a compound of formula IV:
~SiR2~R1
0
(IV)
SUBSTITUTE SHEET (RULE 26)
CA 02336108 2000-12-28
WO 00/00469 PCT/US99/14758
Preferred compounds of the invention having potent anti-cancer affects are
the following:
S-traps-traps-Farnesyl-mercaptoethyl diazomethyl ketone (HI-83);
N-Boc-S-farnesyl-Cys chloromethyl ketone (HI-124);
S-traps-traps-Farnesyl-2-mercaptoethyl chloromethyl ketone (HI-125);
N-Ac-S-(3-methyl-2-butenyl)-Cys chloromethyl ketone(HI-128);
N-Boc-S-dodecyl-Cys chloromethyl ketone (HI-129);
N-Boc-Gly-S-traps-traps-farnesyl-Cys chloromethyl ketone (HI-130);
N-Ac-S-dodecyl-Cys chloromethyl ketone (HI-131);
N-Ac-S-pentyl-cysteine chloromethyl ketone (H~-224);
N-Ac-S-pentadecyl-cysteine chloromethyl ketone {H~225);
S-Dodecyl-Cys chloromethyl ketone hydrochloride (HI-252);
N-Ac-S-heptyl-cysteine chloromethyl ketone (HI-263);
N-Ac-S-dodecyl-Cys-H (HI-274);
N-Ac-S-methyl-cysteine chloromethyl ketone (H~314);
N-Ac-S-undecyl-cysteine chloromethyl ketone (H~321);
N-Ac-S-dodecyl-Cys diazomethyl ketone (HI-348);
N-Ac-S-trityl-cysteine chloromethyl ketone (HI-350);
N-Ac-S-octyl-cysteine chloromethyl ketone (HI-352);
N-Ac-S-tetradecyl-cysteine chloromethyl ketone (HI-354);
N-Ac-S-hexyl-cysteine chloromethyl ketone (HI-357);
N-Ac-S-butyl-cysteine chloromethyl ketone (HI-363);
N-Ac-S-nonyl-cysteine chloromethyl ketone (HI-364);
N-Ac-S-hexadecyl-cysteine chloromethyl ketone (HI-366);
N-Ac-S-traps-traps-farnesyl-Cys diazomethyl ketone (HI-367);
N-Ac-S-traps-traps-farnesyl-Cys chloromethyl ketone (HI-368);
N-Ac-S-propyl-cysteine chloromethyl ketone (H~-369);
N-Ac-S-decyl-cysteine chloromethyl ketone (HI-371);
N-Ac-S-benzyloxycarbonyl-cysteine chloromethyl ketone (HI-389);
N-Ac-S-2-naphthylmethyl-cysteine chloromethyl ketone (HI-392);
N-9-Fluorenylmethyloxycarbonyl-S-dodecyl-cysteine chloromethyl ketone (HI-
398);
N-Ac-S-allyl-cysteine chloromethyl ketone (HI-419);
N-Ac-S-dodecyl-cysteine bromomethyl ketone (HI-488);
SUBSTITUTE SHEET (RULE 26)
CA 02336108 2000-12-28
WO 00/00469 PCTNS99/14758
9
N-Ac-O-dodecyl-serine chloromethyl ketone (HI-489);
N-Trifluoroacetyl-S-dodecyl-cysteine chloromethyl ketone (HI-490);
N-Benzoyl-S-dodecyl-cysteine chloromethyl ketone (HI-491).
The compounds of the invention are capable of forming bath
pharmaceutically acceptable acid addition and/or base salts. Base salts are
formed
with metals or amines, such as alkali and alkaline earth metals or organic
amines.
Examples of metals used as cations are sodium, potassium, magnesium, calcium,
and the like. Also included are heavy metal salts such as for example silver,
zinc,
10 cobalt, and cerium. Examples of suitable amines are N,N'-
dibenzylethylenediamine, chloroprocaine, choline, diethanolamine,
ethylenediamene, N-methylglucamine, and procaine.
Pharmaceutically acceptable acid addition salts are formed with organic and
inorganic acids. Examples of suitable acids for salt formation are
hydrochloric,
15 sulfuric, phosphoric, acetic, citric, oxalic, malonic, salicylic, malic,
gluconic,
fumaric, succinic, ascorbic, malefic, methanesulfonic, and the like. The salts
are
prepared by contacting the free base form with a sufficient amount of the
desired
acid to produce either a mono or di, etc. salt in the conventional manner. The
free
base forms may be regenerated by treating the salt form with a base. For
example,
20 dilute solutions of aqueous base may be utilized. Dilute aqueous sodium
hydroxide,
potassium carbonate, ammonia, and sodium bicarbonate solutions are suitable
for
this purpose. The free base forms differ from their respective salt forms
somewhat
in certain physical properties such as solubility in polar solvents, but the
salts are
otherwise equivalent to their respective free base forms for the purposes of
the
25 invention.
Cytotoxic Compounds
The compounds of the invention are effective cytotoxic agents, for example,
against tumor cells such as leukemic and breast cancer cells. In the methods
of the
30 invention, the cytotoxic effects of alkyl ketone compounds are achieved by
contacting cells, such as tumor cells, with micromolar amounts of the
inhibitory
compound. By way of example, a particularly useful anti-tumor agent is N-Ac-S-
dodecyl-Cys chloromethyl ketone (HI--131 ) as shown in the Examples below.
SUBSTITUTE SHEET (RULE 26)
CA 02336108 2000-12-28
WO 00/00469 PCT/US99/14758
Tumor Treatment
The compounds of the invention can be used in methods of tumor treatment,
for example, by administering to a subject a compound of the invention in
order to
achieve an inhibition of tumor cell growth, a killing of tumor cells,
induction of
5 apoptosis, and/or increased patient survival time.
The cytotoxic compounds of the invention are suitable for use in mammals.
As used herein, "mammals" means any class of higher vertebrates that nourish
their
young with milk secreted by mammary glands, including, for example, humans,
rabbits, and monkeys.
Apoptosis
Apoptosis, or programmed cellular death, is an active process requiring new
protein synthesis. Typically, the process requires ATP, involves new RNA and
protein synthesis, and culminates in the activation of endogenous
endonucleases that
I 5 degrade the DNA of the cell, thereby destroying the genetic template
required for
cellular homeostasis. Apoptosis is observed in controlled deletion of cells
during
metamorphosis, differentiation, and general cell turnover and appears normally
to be
regulated by receptor-coupled events. For these reasons, apoptosis has been
called
"programmed cell death" or "cell suicide." While every cell likely has the
genetic
20 program to commit suicide, it is usually suppressed. Under normal
circumstances,
only those cells no longer required by the organism activate this self-
destruction
program.
Apoptotic cell death is characterized by plasma membrane blebbing, cell
volume loss, nuclear condensation, and endonucleolytic degradation of DNA at
25 nucleosome intervals. Loss of plasma membrane integrity is a relatively
late event
in apoptosis, unlike the form of cell death termed necrosis, which can be
caused by
hypoxia and exposure to certain toxins and which is typically characterized,
early-on
by increased membrane permeability and cell rupture. As demonstrated in the
Examples, the alkyl ketone compounds of the invention are effective agents for
30 inducing apoptosis in tumor cells.
Administration Methods
The compounds of the present invention can be formulated as pharmaceutical
compositions and administered to a mammalian host, including a human patient,
in a
SUBSTITUTE SHEET (RULE 26)
CA 02336108 2000-12-28
WO 00/00469 PCT/US99/14758
11
variety of forms adapted to the chosen route of administration. The compounds
are
preferably administered in combination with a pharmaceutically acceptable
carrier,
and may be combined with or conjugated to specific delivery agents, including
targeting antibodies and/or cytokines.
The compounds can be administered by known techniques, such as orally,
parentally (including subcutaneous injection, intravenous, intramuscular,
intrasternal
or infusion techniques), by inhalation spray, topically, by absorption through
a
mucous membrane, or rectally, in dosage unit formulations containing
conventional
non-toxic pharmaceutically acceptable carriers, adjuvants or vehicles.
Pharmaceutical compositions of the invention can be in the form of suspensions
or
tablets suitable for oral administration, nasal sprays, creams, sterile
injectable
preparations, such as sterile injectable aqueous or oleagenous suspensions or
suppositories.
For oral administration as a suspension, the compositions can be prepared
15 according to techniques well-known in the art of pharmaceutical
formulation. The
compositions can contain microcrystalline cellulose for imparting bulk,
alginic acid
or sodium alginate as a suspending agent, methylcellulose as a viscosity
enhancer,
and sweeteners or flavoring agents. As immediate release tablets, the
compositions
can contain microcrystalline cellulose, starch, magnesium stearate and lactose
or
other excipients, binders, extenders, disintegrants, diluents and lubricants
known in
the art.
For administration by inhalation or aerosol, the compositions can be prepared
according to techniques well-known in the art of pharmaceutical formulation.
The
compositions can be prepared as solutions in saline, using benzyl alcohol or
other
25 suitable preservatives, absorption promoters to enhance bioavailability,
fluorocarbons or other solubilizing or dispersing agents known in the art.
For administration as injectable solutions or suspensions, the compositions
can be formulated according to techniques well-known in the art, using
suitable
dispersing or wetting and suspending agents, such as sterile oils, including
synthetic
mono- or diglycerides, and fatty acids, including oleic acid.
For rectal administration as suppositories, the compositions can be prepared
by mixing with a suitable non-irritating excipient, such as cocoa butter,
synthetic
glyceride esters or polyethylene glycols, which are solid at ambient
temperatures, but
liquefy or dissolve in the rectal cavity to release the drug.
SUBSTITUTE SHEET (RULE 26)
CA 02336108 2000-12-28
WO 00/00469 PC'flUS99114758
12
Preferred administration routes include orally, parenterally, as well as
intravenous, intramuscular or subcutaneous routes.
More preferably, the compounds of the present invention are administered
parenterally, i.e., intravenously or intraperitoneally, by infusion or
injection. In one
5 embodiment of the invention, the compounds may be administered directly to a
tumor by tumor injection; or by systemic delivery by intravenous injection.
Solutions or suspensions of the compounds can be prepared in water, isotonic
saline (PBS) and optionally mixed with a nontoxic surfactant. Dispersions may
also
be prepared in glycerol, liquid polyethylene, glycols, DNA, vegetable oils,
triacetin
10 and mixtures thereof. Under ordinary conditions of storage and use, these
preparations may contain a preservative to prevent the growth of
microorganisms.
The pharmaceutical dosage form suitable for injection or infusion use can
include sterile, aqueous solutions or dispersions or sterile powders
comprising an
active ingredient which are adapted for the extemporaneous preparation of
sterile
15 injectable or infusible solutions or dispersions. In all cases, the
ultimate dosage form
should be sterile, fluid and stable under the conditions of manufacture and
storage.
The liquid Garner or vehicle can be a solvent or liquid dispersion medium
comprising, for example, water, ethanol, a polyol such as glycerol, propylene
glycol,
or liquid polyethylene glycols and the like, vegetable oils, nontoxic glyceryl
esters,
20 and suitable mixtures thereof. The proper fluidity can be maintained, for
example,
by the formation of liposomes, by the maintenance of the required particle
size, in
the case of dispersion, or by the use of nontoxic surfactants. The prevention
of the
action of microorganisms can be accomplished by various antibacterial and
antifungal agents, for example, parabens, chlorobutanol, phenol, sorbic acid,
25 thimerosal, and the like. In many cases, it will be desirable to include
isotonic
agents, for example, sugars, buffers, or sodium chloride. Prolonged absorption
of
the injectable compositions can be brought about by the inclusion in the
composition
of agents delaying absorption-for example, aluminum monosterate hydrogels and
gelatin.
30 Sterile injectable solutions are prepared by incorporating the conjugates
in
the required amount in the appropriate solvent with various other ingredients
as
enumerated above and, as required, followed by filter sterilization. In the
case of
sterile powders for the preparation of sterile injectable solutions, the
preferred
methods of preparation are vacuum drying and freeze-drying techniques, which
SUBSTITUTE SHEET (RULE 2fi)
CA 02336108 2000-12-28
WO 00/00469 PCT/US99/14758
13
yield a powder of the active ingredient plus any additional desired ingredient
present
in the previously sterile-filtered solutions.
Conjugation to a Targeting Moiety
The compound of the invention can be targeted for specific delivery to the
cells to be treated by conjugation of the compounds to a targeting moiety.
Targeting
moiety useful for conjugation to the compounds of the invention include
antibodies,
cytokines, and receptor ligands expressed on the cells to be treated.
The term "conjugate" means a complex formed with two or more
compounds.
The phrase "targeting moiety" means a compound which serves to deliver the
compound of the invention to a specific site for the desired activity.
Targeting
moieties include, for example, molecules which specifically bind molecules
present
on a cell surface. Such targeting moieties useful in the invention include
anti-cell
15 surface antigen antibodies. Cytokines, including interleukins, factors such
as
epidermal growth factor (EGF), and the like, are also specific targeting
moieties
known to bind cells expressing high levels of their receptors.
Particularly useful targeting moieties for targeting the compounds of the
invention to cells for therapeutic activity include those ligands that bind
antigens or
20 receptors present on the tumor cells to be treated. For example, antigens
present on
B-lineage cancer cells, such as CD 19, can be targeted with anti-CD 19
antibodies
such as B43. Antibody fragments, including single chain fragments, can also be
used. IL4 can also be used to target B-cells. Cancer cells expressing EGF or
IGF
receptors can be targeted with the binding ligand. Other such ligand-receptor
25 binding pairs are known in the scientific literature for specific cancers.
Methods for
producing conjugates of the compounds of the invention and the targeting
moieties
are known.
Useful Dose
30 When used in vivo to kill tumor cells, the administered dose is that
effective
to have the desired effect, such as sufficient to reduce or eliminate tumors.
Appropriate amounts can be determined by those skilled in the art,
extrapolating
using known methods and relationships, from the in vitro data provided in the
Examples.
SUBSTITUTE SHEET (RULE 26)
CA 02336108 2000-12-28
WO 00/00469 PCT/US99/14758
14
In general, the dose of the novel alkyl ketone compounds effective to achieve
tumor cell apoptosis, reduction in tumors, and increased survival time, is 1-
100
mg/kg body weight/dose for a direct targeted administration.
The effective dose to be administered will vary with conditions specific to
each
5 patient. In general, factors such as the disease burden, tumor location
(exposed or
remote), host age, metabolism, sickness, prior exposure to drugs, and the like
contribute to the expected effectiveness of a drug. One skilled in the art
will use
standard procedures and patient analysis to calculate the appropriate dose,
extrapolating from the data provided in the Examples.
10 In general, a dose which delivers about 1-100 mg/kg body weight is
expected to be effective, although more or less may be useful.
In addition, the compositions of the invention may be administered in
combination with other anti-tumor therapies. In such combination therapy, the
administered dose of the alkyl ketone compounds may be less than for single
drug
15 therapy.
EXAMPLES
The invention may be further clarified by reference to the following
Examples, which serve to exemplify some of the embodiments, and not to limit
the
invention in any way.
Example 1
Synthetic procedure for Alkyl Ketones
The methods used to synthesize the alkyl ketone compounds beginning from
readily available starting materials and ending with the desired compounds are
described below.
The compounds contained within Table l, Table 2, Table 3, and Table 6
were synthesized according to Schemes 1, 2, 3, and 4 illustrated below. In
each
30 scheme, a single compound is exemplified and used as a model to generally
describe
the synthesis. The specific synthesis of the other compounds is then discussed
in
detail.
The pathway used to synthesize the remaining compounds contained within
Tables 3 and 6 and the compounds in Tables 4 and S are also described in
detail
3 S below.
SUBSTITUTE SHEET (RULE 26)
CA 02336108 2000-12-28
WO 00/00469 PCT/US99/14758
IS
All chemicals were purchased from Aldrich Chemical Company (Milwaukee,
Wisconsin) and used directly for synthesis without further purification.
Anhydrous
tetrahydrofuran was dried over sodium and distilled immediately prior to use.
Column chromatography was performed using 230-400 mesh silica gel obtained
from the Merck Company, with eluant as noted in the experimental procedure.
COMPOUNDS SYNTHESIZED ACCORDING TO SCHEME 1
The compounds (3a-a and 4a-a in Table 1; 3f, 4f, 3g, and 4g in Table 2)
were prepared by the pathway exemplified in Scheme 1 for N-Ac-S-farnesyl-Cys
chloromethyl ketone (HI-368) (4a).
Scheme 1
Farnesyl
SH ~S i) NMM, i-BuOCOCI
Farnesyl-Br THF, -15 °C
Acs H Acs OH >
NH3, MeOH H O ii) CH2N2, Et20, 0 °C
2a
Farnesyl Famesyl
S HCI S
ANN .., N EtOAc Ac'N CI
H O 2 0°C H O
3a 4a
General description of the synthetic pathway illustrated in Scheme 1
The first step of scheme 1 was isoprenylation of the thiol group of N-Ac-
Cys-OH by reaction of the appropriate isoprenyl bromide in a 4 M solution of
20 ammonia in methanol according to the method of Brown and co-workers. This
step
was carried out in the presence of ethyl acetate when N-Ac-Cys-OH was
dodecylated in order to solvate the bromododecane. in the second step, the S-
alkylated acid (2a) was activated as the mixed anhydride derivative using
isobutylchloroformate and converted to the diazomethyl ketone (3a) by
treatment
with diazomethane. In the last step, the diazomethane was converted to the
chloromethyl ketone (4a) with HCl in ethyl acetate at 0 °C for 10
minutes.
SUBSTITUTE SHEET (RULE 26)
CA 02336108 2000-12-28
WO 00/00469 PCT/US99/14758
16
The chloromethyl compounds in Tables 1 and 2, 4a-4g, were made from the
analogous diazomethyl compounds, 3a-3g, by replacement of the diazomethyl
group
with a chloromethyl group. The specific synthesis of each two member analogous
group will therefore be considered together.
Specific methods used to synthesize the analogous pain of diazomethyl
compounds using Scheme 1
10 1. N-Ac-S-traps-traps-farnesyl-Cys diazomethyl ketone (HI-367)(3x)
and
N-Ac-S-traps-traps-farnesyl-Cys chloromethyl ketone (HI-368)(4x)
1 S Traps-traps -farnesyl bromide (1.57 g, 1.49 mL, 5.5 mmol) was added to
N-Ac-Cys-OH (1) (0.82 g, S mmol) in 4 M ammonia in methanol (35 mL) at 0
°C.
The reaction was stirred at 0 °C for 3 h then at room temperature for 1
h. The
solvent was removed under reduced pressure and the residue partitioned between
1
butanol and water. The butanol layer was dried (MgS04) and the solvent removed
20 under reduced pressure. The residue was redissolved in methanol and washed
with
hexane. The methanol was then removed under reduced pressure to give N-Ac-S-
traps-traps-farnesyl-Cys-OH (2a).
The N-Ac-S-traps-traps-farnesyl-Cys-OH (2a) (1.84 g, 5 mmol)
produced in the previous step was dissolved in dry THF (30 mL) and cooled to -
25 15 °C. 4-methyl morpholine (0.51 g, 0.55 mL, 5 mmol) and iso-butyl
chloroformate (0.68 g, 0.65 mL, S mmol) were added to the solution. The
mixture
was stirred at -15 °C for S minutes before being filtered by gravity
into a solution of
diazomethane in ethanolic ether (11 mmol, 30 mL) cooled in an ice bath. The
resulting solution was stirred in ice for 3 h. Excess diazomethane was purged
with
30 nitrogen gas and the reaction mixture was washed with 5% sodium bicarbonate
solution and water, dried over MgS04 , and then the solvent was removed under
reduced pressure. The product was purified by chromatography on silica gel (10-
50% ethyl acetate in hexane) to give N-Ac-S-traps-traps-farnesyl-Cys
diazomethyl ketone (3a).
35 A solution of HCl in ethyl acetate (1 M, 2 mL, 2 mmol) was added to a
solution of the previously synthesized N-Ac-S-traps-traps-farnesyl-Cys
diazomethyl ketone (3a) (1.57 g, 1 mmol) in ethyl acetate (10 mL) cooled in an
ice
SUBSTITUTE SHEET (RULE 26)
CA 02336108 2000-12-28
WO 00/00469 PCT/US99/14758
17
bath. The reaction mixture was stirred at 0 °C for 5 to 10 min until
the starting
material was consumed by TLC. The solvent was then removed under reduced
pressure and the residue purified by chromatography on silica gei ( 1:3 ethyl
acetate:hexane) to give N-Ac-S-traps-traps-farnesyl-Cys chloromethyl ketone
{4a).
2. N-Ac-S-traps-geranyl-Cys diazomethyl ketone (HI-122)(3c) and
N-Ac-S-traps-geranyl-Cys chloromethyl ketone (HI-127)(4c)
N-Ac-S-traps-geranyl-Cys-OH (2c) was prepared as described above for
N-Ac-S-traps-traps-farnesyl-Cys-OH (2a) except that traps-geranyl bromide
was used instead of farnesyl bromide.
N-Ac-S-traps-geranyl-Cys diazomethyl ketone (HI 122) (3c) was prepared
as described above for N-Ac-S-traps-traps-farnesyl-Cys diazomethyl ketone (3a)
except that N-Ac-S-traps-geranyl-Cys-OH (2c) (2.99 g, 10 mmol) was used
1 S instead of N-Ac-S-traps-traps-farnesyl-Cys-OH (2a). The crude material
produced was purified by chromatography on silica gel (0-67% ethyl acetate in
hexane) to give N-Ac-S-traps-geranyl-Cys diazomethyl ketone (HI 122) (3c).
N-Ac-S-traps-geranyl-Cys chloromethyl ketone (HI 127) (4c) was
prepared as described above for N-Ac-S-traps-traps-farnesyl-Cys chloromethyl
ketone (4a) except that N-Ac-S-traps-geranyl-Cys diazomethyl ketone (3c) (0.10
g, 0.3 mmol) was used instead of N-Ac-S-traps-traps-farnesyl-Cys diazomethyl
ketone (3a). The crude material produced was purified by chromatography on
silica
gel (0-40% ethyl acetate in hexane) to give N-Ac-S-traps-geranyl-Cys
chloromethyl ketone (HI 127) (4c).
3. N-Ac-S-(3-methyl-2-butenyl)-Cys diazomethyl ketone (HI-123){3d)
and
N-Ac-S-(3-methyl-2-butenyl)-Cys chloromethyl ketone (HI-128)(4d)
30 N-Ac-S-(3-methyl-2-butenyl)-Cys-OH (2d) was prepared as described
above for N-Ac-S-traps-traps-farnesyl-Cys-OH (2a) except that 4-bromo-2-
methyl-2-butene was used instead of farnesyl bromide.
N-Ac-S-(3-methyl-2-butenyl)-Cys diazomethyl ketone (3d) was prepared
as described above for N-Ac-S-traps-traps-farnesyl-Cys diazomethyl ketone (3a)
except that N-Ac-S-(3-methyl-2-butenyl)-Cys-OH (2d) (0.69 g, 3 mmol) was
SUBSTITUTE SHEET (RULE 26)
CA 02336108 2000-12-28
WO 00/00469 PCT/US99/14758
18
used instead of N-Ac-S-traps-traps-farnesyl-Cys-OH (2a). The crude material
was purified by chromatography on silica gel (0-5% methanol in methylene
chloride) to give N-Ac-S-(3-methyl-2-butenyl)-Cys diazomethyl ketone (3d).
N-Ac-S-(3-methyl-2-butenyl)-Cys chloromethyl ketone (4d) was
prepared as described above for N-Ac-S-traps-traps-farnesyl-Cys chloromethyl
ketone (4a) except that N-Ac-S-(3-methyl-2-butenyl)-Cys diazomethyl ketone
(3d) (0.30 g, 1.2 mmol) was used instead of N-Ac-S-traps-traps-farnesyl-Cys
diazomethyl ketone (3a). The crude product was purified by chromatography on
silica gel (0-50% ethyl acetate in hexane) to give N-Ac-S-(3-methyl-2-
butenyl)-Cys chloromethyl ketone (4d).
4. N-Ac-S-dodecyl-Cys diazomethy! ketone (HI-34$) (3e) and
N-Ac-S-dodecyl-Cys chloromethyl ketone (HI-131) (4e)
15 N-Ac-Cys-OH (1.62 g, 9.9 mmol) and bromododecane (2.5 g, 10 mmol)
were dissolved in a mixture of ethyl acetate (30 mL) and methanol (20 mL). A
solution of NH3 (4.2 M, 100 mL) in methanol was added at 0 °C and the
reaction
mixture was allowed to stir overnight at room temperature. The solvent was
then
removed under reduced pressure and the residue was partitioned between ethyl
20 acetate and 1 M HCI. The organic layer was dried over MgSOa and the solvent
removed under reduced pressure to give N-Ac-S-dodecyl-Cys-OH (2e).
N-Ac-S-dodecyl-Cys diazomethyl ketone (3e) was prepared as described
above for N-Ac-S-traps-traps-farnesyl-Cys diazomethyl ketone (3a) except that
N-Ac-S-dodecyl-Cys-OH (2e) (0.96 g, 2.9 mmol) was used instead of N-Ac-S
25 traps-traps-farnesyl-Cys-OH (2a). The crude product was purified by
chromatography on silica gel ( 1:3 ethyl acetate/hexane) to give N-Ac-S-
dodecy!-
Cys diazomethyl ketone (3e).
N-Ac-S-dodecyl-Cys chloromethyl ketone (4e) was prepared as described
above for N-Ac-S-farnesyl-Cys chloromethyl ketone (4a) except that N-Ac-S-
30 dodecyl-Cys diazomethyl ketone (3e) (0.19 g, 0.5 mmol) was used instead of
N-
Ac-S-traps-traps-farnesyl-Cys diazomethyl ketone (3a). N-Ac-S-dodecyl-Cys
chloromethyl ketone (4e) was obtained after removal of solvent.
SUBSTITUTE SHEET (RULE 26)
CA 02336108 2000-12-28
WO 00/00469 PCTNS99/14758
19
5. N-Boc-S-farnesyl-Cys diazomethyl ketone (HI-82) (3b) and
N-Boc-S-farnesyl-Cys chloromethyl ketone (HI-124) (4b)
These compounds were prepared using published literature procedures
starting from L-cysteine.
6. S-traps-traps-FarnesyI-mercaptoethyl diazomethyl ketone (HI 83) (3f)
and
S-traps-traps-Farnesyl-2-mercaptoethyl chloromethyl ketone (HI 125)
(4f)
10 3-(S-traps--traps-Farnesyl)-mercaptopropionic acid (2f) was prepared as
described above for N-Ac-S-traps-traps-farnesyl-Cys-OH (2a) except that 3-
mercaptopropionic acid was used instead of N-Ac-Cys-OH (1).
S-traps-traps-Farnesyl-mercaptoethyl diazomethyl ketone (3f) was
prepared as described above for N-Ac-S-traps-traps-farnesyl-Cys diazomethyl
15 ketone (3a) except that 3-(S-traps-traps-Farnesyl)-mercaptopropionic acid
(2fJ
was used instead of N-Ac-S-traps-traps-farnesyl-Cys-OH (2a). The crude
material was purified by chromatography on silica gel (1% MeOH in CHCI3) to
give
S-traps-traps-Farnesyl-mercaptoethyl diazomethyl ketone (3fj.
S-traps-traps-Farnesyl-2-mercaptoethyl chloromethyl ketone (HI 125)
20 (4fj was prepared as described above forty-Ac-S-traps-traps-farnesyl-Cys
chloromethyl ketone (4a) except that S-traps-traps-Farnesyl-mercaptoethyl
diazomethyl ketone (3f) (0.52 g, 1.6 mmol) was used instead of N-Ac-S-trans-
trans-farnesyl-Cys diazomethyl ketone (3a). The crude product was purified by
chromatography on silica gel (0-10% ether in hexane) to give S-traps-trans-
25 Farnesyl-2-mercaptoethyl chloromethyl ketone (HI 125) (4f).
7. S-traps-traps-Farnesyl-mercaptomethyl diazomethyl ketone (HI 84)
(3g) and
S-traps-traps-Farnesyl-mercaptomethyl chloromethyl ketone (HI 126)
30 (4g)
S-traps-traps-Farnesyl-mercaptoacetic acid (2g) was prepared as described
above for N-Ac-S-traps-traps-farnesyl-Cys-OH (2a) except that mercaptoacetic
acid was used instead of N-Ac-Cys-OH ( 1 ).
S-traps-traps-Farnesyl-mercaptomethyl diazomethyl ketone (3g) was
35 prepared as described above for N-Ac-S-traps-traps-farnesyl-Cys diazomethyl
ketone (3a) except that S-traps-traps-Farnesyl-mercaptoacetic acid (2g) (0.71
g,
SUBSTITUTE SHEET (RULE 26)
CA 02336108 2000-12-28
WO 00/00469 PCT/US99/14758
2.4 mmol) was used instead of N-Ac-S-traps-traps-farnesyl-Cys-OH (2a). The
crude material was purified by chromatography on silica gel (0-100% CHC13 in
hexane) to give S-traps-traps-Farnesyl-mercaptomethyl diazomethyl ketone
5 S-traps-traps-Farnesyl-mercaptomethyl chloromethyl ketone (4g) was
prepared as described above for N-Ac-S-traps-traps-farnesyl-Cys chloromethyl
ketone (4a) except that S-traps- traps-Farnesyl-mercaptomethyl diazomethyl
ketone (3g) (0.58 g, 1.8 mmol) was used instead of N-Ac-S-traps-traps-farnesyl-
Cys diazomethyl ketone (3a). The crude product was purified by chromatography
on
10 silica gel (0-10% ether in hexane) to give S-traps-traps-Farnesyl-
mercaptomethyl chloromethyl ketone (4g).
COMPOUNDS SYNTIiESIZED ACCORDING TO SCHEME 2
The synthesis of compounds 8 and 9 in Table 1 were prepared by the
15 pathway exemplified in Scheme 2.
Scheme 2
Dodecyl Dodecy~
S
SH Dodecyll3r S i) BoozO, EtOAc
OCI~ N~, MeOH, EtOAc H2 OCI-~ ii) NaOH, MeOH Boy OH
O O O
g 7
Dodec I
i) NMM,i-BuOCOCI Dodecyl i Y
THF, -15°C S HCI S
> -->
ii) CH,zN2, Et20, 0°C B°~N I EtOAc HCI ~ HZ I
iii) HCI, EtOAc, 0 °C H O O
g 9
General description of the synthetic pathway illustrated in Scheme 2
The first step in scheme 2 was the dodecylation of cysteine methyl ester in a
25 mixture of ethyl acetate and methanol. This was followed by Boc-protection,
ester
hydrolysis and conversion in turn to the chloromethyl ketone (8). S-Dodecyl-
Cys
chloromethyl ketone hydrochloride (9) was prepared from N-Boc-S-dodecyl-Cys
SUBSTITUTE SHEET (RULE 26)
CA 02336108 2000-12-28
WO 00/00469 PCT/US99/14758
21
chloromethyl ketone (8), by simply deprotecting the Boc group in a saturated
solution of HCl in ethyl acetate.
Specific methods used to synthesize compounds using Scheme 2
8. N-Boc-S-dodecyl-Cys chloromethyl ketone (HI-129) (8) and
S-Dodecyl-Cys chloromethyl ketone hydrochloride (HI-252) (9)
Ethyl acetate (30 mL) was added to a mixture of Cys-OCH3 hydrochloride
(1.71 g, 10 mmol) and bromododecane (2.5 g, 10 mmol) followed by addition of a
solution of NH3 (6.4 M) in methanol. The resulting cloudy suspension was
allowed
to stir overnight at room temperature and then the insoluble material was
filtered off.
The solution was concentrated and ethyl acetate (100 mL) was added. The
solution
was subsequently washed with water (3 x 40 mL) and dried over Na2S04 to obtain
S-Dodecyl-Cys-OCH3 (6).
Di-tert-butyl dicarbonate ( 1.7 mL, 7.4 mmol) was added to a solution of the
previously synthesized S-dodecyl-Cys-OCH3 (6) (2.11 g, 7 mmol) in ethyl
acetate
at 0 °C . The reaction mixture was allowed to stir for 4 h at room
temperature. N-
Boc-S-dodecyl-Cys-OCH3 was obtained after removal of solvent.
A solution of NaOH (3 M, 3.6 mL, 10.8 mmol) was added to a solution of
N-Boc-S-dodecyl-Cys-OCH3 (3.42 g, 8.5 mmol) in methanol (80 mL) at 0
°C.
The reaction mixture was allowed to stir overnight at room temperature and
then the
solvent was removed under reduced pressure. Water (50 mL) was added to the
residue and the aqueous solution, whose pH value was adjusted to 5, was washed
with hexane (2 x 30 mL) and extracted with ethyl acetate (3 x 30 mL). The
combined ethyl acetate fractions were dried over anhydrous Na2S04 and the
solvent
removed under reduced pressure to give N-Boc-S-dodecyl-Cys-OH (7).
N-Boc-S-dodecyl-Cys diazomethyl ketone was prepared as described
above for N-Ac-S-traps-traps-farnesyl-Cys diazomethyl ketone (3a) except that
N-Boc-S-dodecyl-Cys-OH (7) (0.89 g, 2.3 mmol) was used instead of N-Ac-S-
SUBSTITUTE SHEET (RULE 26)
CA 02336108 2000-12-28
WO 00/00469 PCT/US99/14758
22
traps-traps-farnesyl-Cys-OH (2a). The crude product was purified by
chromatography on silica gel ( 1:2 ethyl acetate/hexane) to give N-Boc-S-
dodecyl-
Cys diazomethyl ketone.
N-Boc-S-dodecyl-Cys chloromethyl ketone (8) was prepared as described
above for N-Ac-S-traps-traps-farnesyl-Cys chloromethyl ketone (4a) except that
N-Boc-S-dodecyl-Cys diazomethyl ketone (0.56 g, 1.4 mmol) was used instead of
N-Ac-S-traps-traps-farnesyl-Cys diazomethyl ketone (3a). N-Boc-S-dodecyl-
Cys chloromethyl ketone (8) was obtained upon removal of solvent.
The N-Boc-protected chloromethyl ketone (8) (0.21 g, 0:5 mmol) was
dissolved in ethyl acetate (20 mL) and cooled in an ice bath. It was then
saturated
with dry HCl gas and the solution was stirred at room temperature until the
starting
material had disappeared by TLC (approx. 2 h). The solvent was removed under
reduced pressure and the product, S-Dodecyl-Cys chloromethyl ketone
hydrochloride (9), was obtained after recrystallization from ether.
COMPOUNDS SYNTHESIZED ACCORDING TO SCHEME 3
The compounds 12 and 13 in Table 1 were prepared by the pathway
exemplified in Scheme 3.
SUBSTITUTE SHEET (RULE 26)
CA 02336108 2000-12-28
WO 00/00469 PCT/US99/14758
23
Scheme 3
~ amesyl iamesy~
SH Famesyl-Br iS i) Boc-Gly-OH, EDC, HOBt O S
-~. 1 H
HZ CH3 NH3, MeOH H2~OCH3 ii) NaOH, MeOH Bo~N N OH
O 'OI H O
10 11
i) NMM, i-BuOCOCI ~ amesyl Famesyl
THF, -15 °C ~ iS HCI, EtOAc, 0 °C O S
N~ ~ N~,
ii) CH2N2, EtzO, 0 °C god N~N2 Boc~ N I
H 0 H O
i2 13
5 General description of the synthetic pathway illustrated in Scheme 3
The first step in scheme 3 was the farnesylation of cysteine methyl ester
according to the method of Brown et al. The farnesylated cysteine methyl ester
was
then coupled with N-Boc-Gly-OH using EDC/HOBt. The ester was hydrolyzed to
the acid (11) and the chloromethyl ketone prepared via the diazomethyl ketone
(12).
Specific methods used to synthesize compounds using Scheme 3
9. N-Boc-Gly-S-traps-traps-farnesyl-Cys diazomethyl ketone (HI-401)
(12) and
15 N-Boc-Gly-S-traps-traps-farnesyl-Cys chloromethyl ketone (HI-130)
(13)
A solution of ammonia (3.4 M, 80 mL) in methanol was added to a mixture
of Cys-OCH3 hydrochloride (1.72 g, 10 mmol) and traps-traps-farnesyl bromide
(2.85 g, 10 mmol) in methanol (30 mL) at 0 °C. The reaction mixture was
allowed
to stir overnight at room temperature and then the solvent was removed under
reduced pressure. The remaining residue was dissolved in ethyl acetate (100
mL)
which had been washed with HCl (0.3 M), water, NaHC03 (6%) and dried over
Na2S04. The solvent was then removed under reduced pressure to give S-trans-
traps-Farnesyl-Cys-OCH3 (10).
DMF (30 mL) was added to a mixture of N-Boc-Gly-OH (1.3 g, 7.5 mmol),
S-traps-traps-Farnesyl-Cys-OCH3 (10) (1.28 g, 7.5 mmol) and HOBt (0.6 g, 4.4
SUBSTITUTE SHEET (RULE 2fi)
CA 02336108 2000-12-28
WO 00/00469 PCT/US99/14758
24
mmol). The solution was cooled to 0 °C before the addition of EDC (1.52
g, 8.2
mmol) and the reaction mixture was stirred overnight at room temperature. The
solvent was then removed under reduced pressure. The resulting residue was
dissolved in ethyl acetate (60 mL) which had been washed with HC1 (0.2 M),
water,
NaHC03 (6%) and dried over Na2S04. Removal of the solvent under reduced
pressure gave a yellow oil that was purified via chromatography on silica gel
(1:1
ethyl acetate/hexane) to give pure N-Boc-Gly-S-traps-traps-farnesyl-Cys-OCH3
10 N-Boc-Gly-S-traps-traps-farnesyl-Cys-OCH3 (1.25 g, 2.52 mmol) was
dissolved in methanol (70 mL) and then a solution of NaOH (3 M, 2.5 mL, 7.56
mmol) at 0 °C was added. The solution was stirred overnight at the same
temperature. The solvent was then removed and the residue was dissolved in
water
(SO mL) that was subsequently acidified to pH 5. The aqueous solution was
15 extracted with chloroform, the combined organic fractions were dried over
anhydrous Na2S04 , and the solvent was removed under reduced pressure to give
N-
Boc-Gly-S-traps-traps-farnesyl Cys-OH (11).
N-Boc-Gly-S-traps-traps-farnesyl-Cys diazomethyl ketone (12) was
prepared as described above for N-Ac-S-traps-traps-farnesyl-Cys chloromethyl
20 ketone (4a) except that N-Boc-Gly-S-traps-traps-farnesyl-Cys-OH (11) (0.39
g,
0.8 mmol) was used instead of N-Ac-S-traps-traps-farnesyl-Cys diazomethyl
ketone (3a). The crude material was purified by chromatography on silica gel
(1:3
ethyl acetate/hexane) to give N-Boc-Gly-S-traps-traps-farnesyl-Cys
diazomethyl ketone (12).
25 N-Boc-Gly-S-traps-traps-farnesyl-Cys chloromethyl ketone (13) was
prepared as described above for N-Ac-S-traps-traps-farnesyl-Cys chloromethyl
ketone (4a) except that N-Boc-Gly-S-traps-traps-farnesyl-Cys diazomethyl
ketone (0.21 g, 0.41 mmol) was used instead ofN-Ac-S-traps-traps-farnesyl-Cys
diazomethyl ketone (3a). N-Boc-Gly-S-traps-traps-farnesyl-Cys chloromethyl
30 ketone (13) was obtained upon removal of solvent.
SUBSTITUTE SHEET (RULE 2G)
CA 02336108 2000-12-28
WO 00100469 PCT/US99/14758
COMPOUNDS SYNTHESIZED ACCORDING TO SCHEME 4
The compounds 4h-4y in Table 3 and the compounds in Table 6 were
5 prepared by the pathway exemplified in Scheme 4.
Scheme 4
R R
SH S i) NMM, i-BuOCOCI
R-Br, NH3 THF, -78 °C S
Acs OH Acs OH
MeOH, EtOAc H ii) CH2N2, Et20 ANN CI
O O iii) HCI, EtOAc, 0°C H O
2h-y 4h-y
Synthesis of the straight chain alkyl ketone derivatives (Tables 3 and 6)
The straight chain alkyl ketone derivatives (4h-y) were synthesized by a
modification of the standard literature procedure. Previously, the standard
15 conditions for making diazomethyl ketones were used, but a closer study of
the
dodecyl derivative, N-Ac-S-dodecyl-Cys chloromethyl ketone (4e), showed
significant formation of the methyl ester as a side-product. Presumably, the
mixed
anhydride intermediate either could not completely form or was hydrolyzed back
to
the acid before the diazomethane could react with it. Conducting the mixed
20 anhydride formation at -78 °C increased the stability and longevity
of the mixed
anhydride and improved yield. Adding diazomethane directly to the solution at -
78 °C without filtration led to over a three-fold improvement in yield
from 15 to
55% for the dodecyl derivative, N-Ac-S-dodecyl-Cys chloromethyl ketone (4e),
and gave a satisfactory yield of product for the majority of cysteine
derivatives
25 reported herein.
10. Specific methods used to synthesize the compounds of Table 3 and Table 6
The appropriate 1-bromoalkane (11 mmol) was added to a solution ofN-
Ac-Cys-OH (1.63 g, 10 mmol) in methanol (15 mL) and ethyl acetate (15 mL) that
30 had been cooled in an ice bath. This was followed by the addition of a
solution of
ammonia in methanol (4M, 50 mL). The resulting solution was allowed to slowly
warm to room temperature and was stirred overnight. The solvent was then
removed
under reduced pressure and the residue partitioned between ethyl acetate and 1
M
SUBSTITUTE SHEET (RULE 26)
CA 02336108 2000-12-28
WO 00/00469 PCT/US99/14758
26
HC1. The layers were separated and the organic layer was dried over anhydrous
MgS04. The solvent was removed under reduced pressure to give the desired N-
Ac-S-alkyl-cysteine compound. An analytical sample of this could be obtained
by
recrystallization from ethyl acetate/hexane but was otherwise of sufficient
purity to
be used in further reactions.
NMM (0.20 g, 0.22 mL, 2 mmol) and isobutyl chloroformate (0.27 g, 0.26
mL, 2 mmol) was added to the desired N-Ac-S-alkyl-cysteine (2 mmol) in
anhydrous THF (20 mL) that had been cooled to -78 °C. The solution was
then
stirred at -78 °C for 20 min. A solution of diazomethane in ethanolic
ether ( 10 mL)
10 was carefully added and the solution allowed to slowly warm to room
temperature.
Further portions of diazomethane solution were added until a yellow color
persisted.
The solution was diluted with ether, washed with water and sodium bicarbonate
solution and then dried over anhydrous MgS04. The solvent was removed under
reduced pressure to yield the crude diazomethyl ketone compound.
15 The diazomethyl ketone was dissolved in ethyl acetate (20 mL) and cooled in
an ice bath. A solution of HCl in ethyl acetate (2M, 2 mL) was added and the
solution stirred in ice for 5 min until no more diazomethyl ketone could be
observed
by TLC. The solvent was removed under reduced pressure and the residue
purified
by chromatography on silica gel (ethyl acetate/hexane) to give the pure
20 chloromethyl ketone compounds in Table 3 and Table 6.
11. Synthesis of the alkyl ketone compounds in Table 4
Synthesis of the compounds having variable R~ groups shown in Table 4 was
either by the method described above for HI-131, 348,and 208 or as follows.
The
25 bromomethyl ketone (HI-488) was synthesised from the diazomethyl ketone
using
HBr in a similar fashion to the chloromethyl ketone. The benzoyloxymethyl
ketone
(HI-508) was made starting from either the bromomethyl ketone or the
chloromethyl
ketone by displacement of the halogen with benzoic acid in the presence of
potassium fluoride in DMF. The aldehyde (HI-274) was made from the acid (HI-
30 208, 2e) via formation of the Weinreb amide (HI-267) by activation of the
acid as
its' mixed anhydride followed by coupling with N,O-dimethylhydroxylamine. The
aldehyde was then synthesized by reduction of the Weinreb amide using LiAlH4.
The thiomethyl ketones (HI-269, 302, 399, 365 & 273) were made by displacement
SUBSTITUTE SHEET (RULE 26)
CA 02336108 2000-12-28
WO 00/00469 PCT/US99/14758
27
of the halogen of the bromo or chloromethyl ketones with the appropriate thiol
in the
presence of potassium carbonate in DMF.
12. Synthesis of the alkyl ketone compounds in Table 5
5 The compounds varying the R4 substituent (Table 5) were made either as
noted above for HI-252, 131, and 129 or as follows. The majority of these
compounds were made by reacting the appropriate acyl chloride or anhydride
with
the free amine compound (HI-252) in the presence of triethylamine. The
dimethylaminobenzoyl derivative (HI-268) required activation of
10 dimethylaminobenzoic acid as its mixed anhydride prior to reaction with the
free
amine. The serine derivative (HI-266) was synthesized by reaction of Boc-Ser-
OH
with sodium hydride and 1-bromododecane in DMF to give the N-Boc-O-
dodecyl-Ser-OH and then with isobutylchloroformate/diazomethane and HCl in
ethyl acetate at 0 °C as in the chemistry noted above. The acetyl
serine derivative
15 (HI-489) was synthesized from HI-266 by removal of the Boc group in
saturated
HCl in ethyl acetate followed by acetylation using acetic anhydride in
dichloromethane in the presence of triethylamine.
20 Example 2
Characterization of synthesized compounds
NMR spectra were recorded using a 300 MHz Varian instrument and the
25 chemical shifts reported are in ppm based on tetramethylsilane as the
internal
standard. Chemical shifts for'3C NMR are referenced to the chloroform peak at
77.0 ppm. Melting points were done using a Fisher-Jones apparatus and are
uncorrected. Fourier Transform Infra-red spectra were recorded on a FT-Nicolet
model Protege 460 instrument. GC/MS analysis was done using a Hewlett-Packard
30 GC/MS model 6890 with an HP5973 electron impact mass spectrometer. In
addition, a Hewlett-Packard Matrix-Assisted Laser Desorption Ionization Time
of
Flight (MALDI-TOF) spectrometer model G2030A was used with cyano hydroxy
benzoic acid as the matrix. Data are shown below.
SUBSTITUTE SHEET (RULE 26)
CA 02336108 2000-12-28
WO 00/00469 PCT/U599/14758
28
N-Ac-S-traps-traps-farnesyl-Cys-OH (2a) Yellow oil. Yield 99%, 1.83
grams. 'H NMR (DMSO-d6) 8 1.54 (s, 6H), 1.61 (s, 3H), 1.62 (s, 3H), 1.84 (s,
3H),
1.97 (m, 8H), 2.60 (dd, J = 8.5, 13.7 Hz, 1 H), 2.79 (dd, J = 5.0, 13.7 Hz, 1
H), 3.13
(m, 2H~, 4.35 (m, 1 H), S.OS (m, 2H), 5.14 (t, J = 7.8 Hz, 1 H), 8.22 (d, J =
8.2 Hz,
1H); ~ C NMR (CDC13) 8 172.5, 169.5, '138.7, 134.8, 130.9, 124.3, 123.8,
120.4,
52.1; 48.8, 32.2, 29.0, 26.4, 26.1, 25.8, 22.6, 17.8, 16.1, 16.0; IR (KBr)
3334, 2966,
2922, 2854, 1732, 1659, 1543, 1441, 1377, 1219 cm 1.
N-Ac-S-traps-geranyl-Cys-OH (2c) Clear oil. Yield 97%, 2.92 g. ~H
NMR (CDC13) 8 1.60 (s, 3H), 1.65 (s, 3H), 1.68 (s, 3H), 2.07 (m, 4H), 2.10 (s,
3H),
2.95 (ABX, J = 3.6, 5.7, 13.2 Hz, 2H), 3.18 (d, J = 6.9 Hz, 2H), 4.72 (m, 1
H), 5.07
(m, 1 H), 5.20 (t, J = 7.9 Hz, 1 H), 6.72 (br, 1 H), 9.95 (br, 1 H); 13C NMR
(CDC13) &
177.1, 139.9, 131.6, 123.7, 119.4, 52.6, 39.7, 33.0, 30.1, 29.8, 26.5, 25.8,
23.0, 20.9,
17.8, 16.2; IR (KBr) 3307, 2968, 2857, 261 l, 1716, 1646, 1550, 1417, 1377,
1241
cm ~; MS (MALDI-TOF) 323 (M + H + Na).
N-Ac-S-(3-methyl-2-butenyl)-Cys-OH (2d) Clear viscous oil. Yield
83%, 0.96 g.'H NMR (CDCl3) 8 1.66 (s, 3H), 1.73 (s, 3H), 2.07 (s, 3H), 2.95
(ABX,
J = 4.6, 6.2, 13.9 Hz, 2H), 3 .17 (d, J = 7.7 Hz, 2H), 4.65 (m, 1 H), 5.20 (t,
J = 7.9 Hz,
1H), 7.28 (br, 1H), 8.25 (br, 1H); 13C NMR (CDCl3) 8 174.1, 171.2, 136.1,
119.7,
52.7, 33.1, 30.1, 25.9, 25.8, 22.9, 17.8; IR (KBr) 3271, 3078, 2966, 2930,
2874,
1722, 1651, 1615, 1557, 1427, 1377, 1299, 1213, 1107, 1031 cm ~.
N-Ac-S-dodecyl-Cys-OH (2e) White solid. Yield 98%, 3.23 g. m.p. 82-
84 °C. 1H NMR (CDC13) 8 0.88 (t, J= 6.6 Hz, 3H), 1.26 (s, 20H), 1.57
(m, 2H), 2.09
(s, 3H~, 2.55 (t, J = 7.4 Hz, 2H), 3.03 (m, 2H), 4.75 (m, 1H), 6.40 (d, J =
7.1 Hz,
1H); 1 C NMR (CDC13) 8 168.3, 47.3, 28.8, 28.1, 27.3, 25.0, 24.9, 24.8, 24.6,
24.2,
18.4, 18.1, 9.6; iR (KBr) 3334, 2922, 2850, 1703, 1622, 1562, 1470, 1416,
1377,
1315, 1257, 1244 cm '; MS (MALDI-TOF) 332.7 (M + 1).
3-(S-traps-traps-Farnesyl)-mercaptopropionic acid (2f) Clear oil.
Yield 100%, 1.54 g. 1H NMR (CDC13) 8 1.60 (s, 6H), 1.66 (s, 3H), 1.68 (s, 3H),
2.03
(m, 8H), 2.60 (t, J = 6.8 Hz, 2H), 2.72 (t, J = 6.8 Hz, 2H~, 3.17 (d, J = 7.7
Hz, 2H),
5.09 (m, 2H), 5.22 (t, J = 7.3 Hz, 1 H), 8.77 (br, 1 H); 1 C NMR (CDC13) 8
162.9,
139.0, 135.2, 131.2, 124.2, 123.7, 120.0, 60.4, 39.75, 39.65, 36.8, 35.3,
31.7, 29.4,
26.8, 26.5, 26.1, 25.8, 17.8, 16.2, 16.1; IR (ICBr) 3179, 2969, 2920, 2855,
1710,
1595, 1436, 1381, 1307 cm 1.
S-traps-traps-Farnesyl-mercaptoacetic acid (2g) Clear oil. Yield 97%,
1.43 g. 1H NMR (CDC13) 8 1.60 (s, 6H), 1.66 (s, 3H), 1.68 (s, 3H), 2.03 (m,
8H),
3.19 (s, 2H), 3.30 (d, J= 7.7 Hz, 2H), 5.09 (m, 2H), 5.21 (m, 1H), 8.95 (br,
1H); 13C
NMR (CDC13) 176.5, 140.7, 135.3, 131.2, 124.2, 123.5, 118.9, 39.7, 39.6, 32.0,
30.0, 26.8, 26.4, 25.8, 17.8, 16.12, 16.09; IR (ICBr) 3323, 2959, 2925, 2875,
1705,
1'605, 1436, 1381, 1227, 1073, 1033 cm ~.
SUBSTITUTE SHEET (RULE 2fi)
CA 02336108 2000-12-28
WO 00/004b9 PCT/L3S99/14758
29
N-Ac-S-traps-traps-farnesyl-Cys diazomethyl ketone (HI 367) (3a)
Yellow oil. Yield 51%, 1 g. 'H NMR (CDC13) 8 1.60 (s, 6H), 1.67 (s, 3H), 1.68
(s,
3H), 2.03 (m, 8H), 2.05 (s, 3H), 2.82 (ABX, J = 6.3, 6.6, 14.0 Hz, 2H), 3.20
(m,
2H), 4.64 (m, 1 H), 5.09 (t, J = 6.9 Hz, 2H), 5.22 (t, J = 7.7 Hz, 1 H), 5.58
(s, 1 H),
6.51 (d, J = 7.7 Hz, 1H); '3C NMR (CDC13) 8 191.8, 169.7, 139.9, 135.3, 131.2,
124.2, 123.6, 119.5, 60.3, 55.2, 54.9, 39.65, 39.55, 33.1, 30.1, 26.7, 26.3,
25.7, 23.1,
17.7, 16.1, 16.0; IR (KBr) 3290, 3057, 2966, 2920, 2854, 2108, 1651, 1533,
1441,
1375 cm '; GC/MS 363 (M - N2).
10 N-Ac-S-traps-geranyl-Cys diazomethyl ketone (HI 122) (3c) Yellow
oil. Yield 45%, 1.45 g. 'H NMR (CDC13) 8 1.60 (s, 3H), 1.67 (s, 3H), I.68 (s,
3H),
2.05 (m, 7H), 2.82 (ABX, J = 6.0, 6.3, 13.8 Hz, 2H), 3.20 (m, 2H), 4.64 (m,
1H),
5.08 {m, 2H), 5.22 (t, J= 7.8 Hz, 1H), 5.59 (s, 1H), 6.56 (d, J= 7.2 Hz, 1H);
'3C
NMR (CDC13) 8 191.8, 169.7, 139.8, 131.6, 123.7, 119.5, 65.8, 55.3, 55.0,
39.6,
15 33.1, 30.2, 29.7, 26.5, 25.8, 23.2, 17.8, 16.2; GC/MS 295 (M - N2); IR
(KBr) 3300,
3061, 2965, 2924, 2856, 2107, 1732, 1651, 1538, 1455, 1373, 1260, 1111 cm ';
MS
(MALDI-TOF) 296.5 (M + H - Nz).
N-Ac-S-(3-methyl-2-butenyl)-Cys diazomethyl ketone (HI 123) (3d)
20 Yellow oil. Yield 50%, 0.38 g. 'H NMR (CDC13) 8 1.68 (s, 3H), 1.75 (s, 3H),
2.05
(s, 3H), 2.82 (ABX, J= 6.0, 6.3, 13.5 Hz, 2H~, 3.18 (m, 2H), 4.64 (m, 1H),
5.21 (m,
1H), 5.59 (s, 1H), 6.58 (d, J= 7.5 Hz, 1H); ' C NMR (CDCI3) b 191.7, 169.6,
136.1, 119.7, 55.3, 54.9, 52.7, 52.6, 51.8, 37.7, 33.2, 30.3, 30.1, 25.7,
25.6, 23.1,
19.1, 17.8; GC/MS 227 (M - NZ); IR (KBr) 3296, 3062, 2970, 2928, 2108, 1745,
25 1667, 1548, 1441, 1373, 1145, 1039 cm '; MS (MALDI-TOF) 278.6 (M + Na),
256.3 (M + H), 250.4 (M + Na - N2), 228.3 (M + H - N2).
N-Ac-S-dodecyl-Cys diazomethyl ketone (HI 348) (3e) Off-white solid.
Yield 15%, 0.16 g. m.p. 45-48 °C. 'H NMR (CDC13) 8 0.88 (t, J= 6.7
Hz, 3H),
30 I.25 (m, 18H), 1.61 (m, 2H), 2.05 (s, 3H), 2.55 (t, J= 7.4 Hz, 2H), 2.88
(d, J= 6.3
Hz, 2H), 4.62 (m, 1 H), 5.59 (s, 1 H), 6.40 (d, J = 7.1 Hz, 1 H); '3C NMR
(CDC13) 8
191.8, 169.8, 55.4, 55.0, 34.1, 32.9, 32.8, 31.9, 29.6, 29.5, 29.4, 29.3,
29.2, 28.8,
28.2, 23.2, 22.7, 14.14; IR (KBr) 3287, 3068, 2919, 2853, 2126, 1655, 1547,
1383,
1168, 725 cm '.
35
S-traps-traps-Farnesyl-mercaptoethyl diazomethyl ketone (HI 83) (3fj
Yellow oil. Yield 45%, 0.45 g. 'H NMR (CDC13) 8 1.60 (s, 6H), 1.67 (s, 3H),
1.68
(s, 3H), 2.03 (m, 8H), 2.58 (t, J = 7.2 Hz, 2H), 2.75 (t, J = 7.2 Hz, 2H),
3.17 (d, J =
7.7 Hz, 2H), 5.09 (m, 2H), 5.24 (t, J= 7.3 Hz, IH), 5.29 (s, 1H); '3C NMR
(CDC13)
40 8 139.1, 135.2, 131.2, 124.2, 123.6, 120.0, 68.1, 51.8, 39.7, 39.6, 38.7,
34.8, 30.4,
29.8, 29.4, 26.8, 26.5, 26.0, 25.8, 17.8, 16.2, 16.1; IR (KBr) 3095, 2967,
2922,
2855, 2105, 1715, 1645, 1445, 1368, 1318 cm '; MS (MALDI-TOF) 357 (M + Na).
S-traps-traps-Farnesyl-mercaptomethyl diazomethyl ketone (HI 84)
45 (3g) Yellow oil. Yield 47%. 0.36 g.'H NMR (CDCI3) 8 1.60 (s, 6H), 1.66 (s,
3H),
1.68 (s, 3H), 2.03 (m, 8H), 3.20 (m, 4H), 5.09 (m, 2H), 5.20 (t, J = 7.8 Hz, 1
H), 5.77
(s, 1H); '3C NMR (CDC13) 8 140.6, 135.3, 131.2, 124.2, 123.6, 118.9, 54.1,
47.2,
39.8, 39.7, 38.8, 31.9, 30.0, 39.8, 26.8, 26.6, 26.4, 25.8, 17.8, 16.2, 16.15;
IR (KBr)
SU8ST1TUTE SHEET (RULE 2B)
CA 02336108 2000-12-28
WO 00/00469 PCT/US99/14758
3107, 2966, 2923, 2854, 2105, 1724, 1639, 1449, 1355 cm ~; MS (MALDI-TOF)
343 (M + Na).
N-Ac-S-traps-traps-farnesyl-Cys chloromethyl ketone (HI 368) (4a)
5 Pale yellow solid. Yield 55%, 0.22 grams, m.p. 59-61 °C. 1H NMR
(CDCl3) 8 1.56
(s, 6H), 1.64 (s, 6H), 2.01 (s, 3H), 2.04 (m, 8H), 2.85 (ABX, J = 5.8, 6.6,
13.9 Hz,
2H), 3.14 (m, 2H}, 4.31 (s, 2H), 4.85 (m, 1 H}, 5 .05 (t, J = 6.9 Hz, 2H},
5.17 (t, J =
7.7 Hz, 1H), 6.36 (br, 1H); C NMR (CDC13) b 199.9, 170.0, 140.5, 135.4, 131.3,
124.2, 123.5, 119.2, 60.4, 55.3, 47.3, 39.7, 39.6, 31.9, 29.9, 26.7, 26.4,
25.7, 22.9,
10 17.7, 16.2, 16.0; IR (KBr) 3300, 3061, 2926, 2852, 2825, 1738, 1633, 1541,
1448,
1421, 1371, 1286, 1078 cm ~; GC/MS 363 (M - HCl).
N-Ac-S-traps-~eranyl-Cys chloromethyl ketone (HI 127) (4c) Yellow
oil. Yield 27%, 0.03 g. H NMR (CDC13) b 1.61 (s, 3H), 1.67 (s, 3H), 1.68 (s,
3H),
15 2.05 (s, 3H), 2.07 (m, 4H), 2.88 (ABX, J= 5.9, 6.3, 13.7 Hz, 2H), 3.18 (m,
2H), 4.34
(m, 2H), 4. 89 (q, J = 6.3 Hz, 1 H), 5 .07 (m, 1 H), 5 .21 (t, J = 7.8 Hz, 1
H) ~ C NMR
(CDCl3) 8 199.8, 169.9, 140.4, 131.8, 123.6, 119.2, 55.3, 47.3, 39.7, 39.6,
32.0,
30.0, 29.8, 26.5, 25.8, 23.0, 17.8, 16.3; IR (KBr) 3342, 2970, 2926, 2859,
1741,
1732, 1664, 1651, 1538, 1446, 1379, 1301, 1084 cm 1; GC/MS 295 (M - Cl), 205
20 (farnesyl).
N-Ac-S-(3-methyl-2-butenyl)-Cys chloromethyl ketone (HI 128) (4d)
Off-white solid. Yield 56%, 0.18 g. ~H NMR (CDCl3) S 1.68 (s, 3H), 1.76 (s,
3H),
2.06 (s, 3H), 2.89 (ABX, J= 6.0, 6.5, 13.8 Hz, 2H), 3.17 (m, 2H), 4.3413m,
2H), 4.89
25 (q, J= 6.3 Hz, 1H), 5.20 (t, J= 7.8 Hz, 1H), 6.32 (d, J= 5.5 Hz, 1H); C NMR
(CDCl3) b 199.8, 169.9, 136.8, 119.4, 55.4, 47.3, 32.1, 30.2, 25.8, 23.0,
17.9; IR
(KBr) 3302, 3060, 2976, 2958, 2927, 2853, 1738, 1635, 1541, 1423, 1371, 1286,
1216, 1078 cm 1; GC/MS 227 (M - Cl).
30 N-Ac-S-dodecyl-Cys chloromethyl ketone (HI 131) (4e) Pale yellow
solid. Yield 100%, 0.18 g. m.p. 73-74 °C. ~H NMR (CDC13) 8 0.88 (t, J=
6.7 Hz,
3H), 1.26 (m, 18H), 1.57 (m, 2H), 2.46 (s, 3H), 2.54 (t, J= 7.3 Hz, 2H~, 2.94
(ABX,
J = 6.0, 6.3, 13.9 Hz, 2H), 4.3 5 (m, 2H), 4.91 (m, 1 H), 6.31 (br, 1 H); 1 C
NMR
(CDCl3) 8 195.2, 165.3, 50.8, 42.7, 29.6, 28.4, 28.2, 27.3, 25.1, 25.0, 24.9,
24.83,
24.8, 24.7, 24.5, 24.1, 18.3, 18.1, 9.5; IR (KBr 3302, 2924, 2854, 1738, 1660,
1537, 1456, 1377, 1261, 1165, 1095, 1040 cm ~; MS (MALDI-TOF), 364.9 (M +
1 ), 328.9 (M - Cl).
S-traps-traps-Farnesyl-2-mercaptoethyl chloromethyl ketone (HI 125)
(4f) Yellow oil. Yield 68%, 0.36 g. 1H NMR (CDCl3) 8 1.60 (s, 6H), 1.67 (s,
3H),
1.68 (s, 3H), 2.03 (m, 8H), 2.74 (t, J = 6.6 Hz, 2H), 2.89 (t, J = 6.6 Hz,
2H~~ 3.18 (d,
J = 7.5 Hz, 2H), 4.10 (s, 2H), 5.09 (m, 2H), 5.23 (t, J = 7.8 Hz, 1 H); C NMR
(CDCl3) b 200.9, 139.2, 135.3, 131.2, 124.2, 123.6, 120.0, 48.3, 40.1, 39.7,
39.6,
29.8, 26.8, 26.5, 25.8, 24.8, 17.8, 16.2, 16.1; IR (KBr) 2964, 2925, 2852,
1726,
1441, 1379, 1351, 1110, 1077 cm ~; GC/MS 342 (M+), 307 (M - Cl), 205
(farnesyl);
MS (MALDI-TOF) 342.6 (M+).
SUBSTITUTE SHEET (RULE 26)
CA 02336108 2000-12-28
WO 00/00469 PCT/US99/14758
31
S-traps-traps-Farnesyl-mercaptomethyl chloromethyl ketone (HI 126)
(4g) Clear oil. Yield 17%, 0.10 g. ~H NMR (CDCl3) 8 1.60 (s, 6H), 1.66 (s,
3H),
1.68 (s, 3H), 2.03 (m, 8H), 3.16 (d, J = 7.8 Hz, 2H), 3.34 (s, 2H), 4.34 (s,
2H), 5.09
(m, 2H), 5.18 (m, 1H); '3C NMR (CDC13) 8 197.2, 140.9, 135.3, 131.1, 124.1,
123.5, 118.6, 46.2, 39.7, 39.6, 38.4, 37.4, 37.1, 32.0, 31.8, 29.8, 26.7,
26.5, 26.3,
25.3, 17.7, 16.2, i6.1; IR (KBr) 3444, 2965, 2926, 2859, 1732, 1712, 1664,
1446,
1384, 1243, 1108 cm '; GC/MS 328 (M+), 294 (M - Cl), 205 (farnesyl).
S-Dodecyl-Cys-OCH3 (6) Light yellow oil. Yield 61%, 1.85 g. 'H NMR
(CDCl3) 8 0.88 (t, J = 6.6 Hz, 3H), 1.26 (s, 20H), 2.53 (t, J = 7.5 Hz, 2H),
2.76 (m,
1H), 2.91 (m, 1H), 3.67 (m, 1H), 3.75 (s, 3H); IR (KBr) 3381, 2924, 2854,
1743,
1466, 1437, 1196, 1175 cm ~; GC/MS 303 (M+).
N-Boc-S-dodecyl-Cys-OCH3 Clear oil. Yield 100%, 2.82 g. 'H NMR
(CDC13) 8 0.88 (t, J = 7.1 Hz, 3 H), 1.25 {s, 20H), 1.45 (s, 9H), 2.51 (t, J =
7.5 Hz,
2H), 2.95 (d, J = 5.2 Hz, 1H), 3.76 (s, 3H), 4.52 (m, IH), 5.37 (m, 1H); IR
(ICBr)
2977, 2932, 1814, 1759, 1062 cm '; GC/MS 403 (M+), 286, 215, 57.
N-Boc-S-dodecyl-Cys-OH (7) Yellow oil. Yield 80%, 2.65 g. ~H NMR
(CDC13) 8 0.88 (t, J = 6.6 Hz, 3H), I .24 (m, 20H), I .45 (s, 9H), 2.52 (t, J
= 7.4 Hz,
2H), 2.96 (d, J = 4.9 Hz, 2H), 4.50 (m, 1H), 5.38 (br, 1H); '3C NMR (CDCI3) 8
175.0, 156.0, 80.0, 53.0, 34.5, 34.1, 32.9, 32.6, 32.0, 29.7, 29.6, 29.4,
29.2, 28.8,
28.2, 22.8, 14.0; IR (KBr) 3328, 2922, 2852, 1719, 1503, 1368, 1172, 1052 cm
'.
N-Boc-S-dodecyl-Cys diazomethyl ketone Light yellow oiI that solidified
upon standing. Yield 59%, 0.56 g.'H NMR (CDCl3) 8 0.88 (t, J= 6.7 Hz, 3H),
1.26
(s, 18H), 1.46 (s, 9H), 1.58 (m, 2H), 2.53 (m, 2H), 2.89 (m, 2H), 4.30 (m,
1H), 5.61
(br, 1H); IR (KBr) 3323, 2924, 2853, 2108, 1718, 1647, 1499, 1369, 1168 cm ';
MS
(MALDI-TOF) 386.5 (M - N2).
N-Boc-S-dodecyl-Cys chloromethyl ketone (HI 129 (8) Clear oil that
solidified upon standing. Yield 100%, 0.57 g. m.p. 43-49 °C. H NMR
(CDC13) 8
0.88 {m, 3H), 1.26 (s, 18H), 1.45 (s, 9H), 1.57 (m, 2H), 2.53 (m, 2H), 2.93
(m, 2H),
4.37 (s, 1H), 4.60 (m, 1H), 5.37 (br s, 1H);'3C (CDCl3) 8 172.5, 130.8, 56.9,
53.2,
52.5, 47.4, 34.5, 33.2, 32.9, 32.8, 32.0, 29.7, 29.6, 29.5, 29.4, 29.2, 28.8,
28.6, 28.3,
28.0 22.8, 14.2; IR {ICBr) 3363, 2926, 2854, 1713, 1497, 1468, 1367, 1252,
1167
cm .
S-Dodecyl-Cys chloromethyl ketone hydrochloride (HI 252) (9) Silvery-
white solid. Yield 81%, 0.14. m.p. 118-119 °C (decomp.).'H NMR (DMSO-
d6) b
0.84 (t, J = 6.7 Hz, 3H), 1.23 (m, 18H), 1.51 (m, 2H), 2.57 (t, J = 7.3 Hz,
2H), 2.96
(dd, J = 6.9, 14.4 Hz, 1 H), 3.09 (dd, J = 5.8, 14.4 f-Iz, 1 H), 4.44 (t, J =
6.2 Hz, 1 H),
4.84 (m, 2H), 8.58 (br, 3H); '3C NMR (DMSO-db) S 193.2, 51.4, 43.9, 27.3,
27.1,
26.2, 24.9, 24.6, 24.4, 24.0, 18.0, 9.8; IR (KBr) 2953, 2922, 2852, 1738,
1470, 1385,
1149 cm ~ .
SUBSTITUTE SHEET (RULE 26)
CA 02336108 2000-12-28
WO 00/00469 PCT/US99/14758
32
S-traps-traps-Farnesyl-Cys-OCH3 (10) Clear oil. Yield 68%,2.31 g. 'H
NMR (CDCl3) 8 1.59 (s, 6H), 1.67 (s, 6H), 2.08 (m, 8H), 2.68 (dd, J= 7.7, 13.5
Hz,
1H), 2.89 (dd, J= 4.8, 13.5 Hz, 1H), 3.19 (m, 2H), 3.61 (m, 1H), 3.75 (s, 3H),
5.08
(m, 2H), 5.21 (t, J= 7.9 Hz, 1H); IR (KBr) 3348, 3376, 3314, 2978, 2922, 2860,
S 1746, 1684, 1452, 1395, 1209, 1168, 843 cm '; GC-MS 339 (M+), 270, 202, 135,
81, 69.
N-Boc-Gly-S-traps-traps-farnesyl-Cys-OCH3 Yellow oil. Yield 61 %,
2.27 g. 'H NMR (CDC13) b 1.45 (s, 9H), 1.60 (s, 6H), 1.66 (s, 3H), 1.68 (s,
3H),
1.94-2.09 (m, 8H), 2.81 (ABX, J = 5.1, 6.2, 13.9 Hz, 2H), 3.16 (m, 2H), 3.76
(s,
3H), 3.85 (t, J = 5.5 Hz, 2H), 4.80 (m, 1 H), 5.10 (m, 2H), 5.18 (m, 2H), 6.86
(br d, J
= 7.3 Hz, 1H); MS (MALDI-TOF), 496.5 (M~.
N-Boc-Gly-S-traps-traps-farnesyl Cys-OH (11) Clear oil. Yield 61%,
0.74 g. 'H NMR (CDC13) 8 1.45 (s, 9H), 1.60 (s, 6H), 1.65 (s, 6H), 1.95-2.20
(m,
8H), 2.80-3.08 (m, 2H), 3.10-3.27 (m, 2H), 3.70-3.90 (m, 2H), 3.95-4.10 (m,
1H),
4.75-4.95 (m, 1 H), 5.08 (m, 2H), 5.20 (m, 1 H), 5.41 (s, 1 H), 7.10 (m, 1 H);
IR
(KBr), 3338, 2979, 2930, 1725, 1660, 1525, 1241, 1172 cm '; MS (MALDI-TOF),
505.3 (M + Na+)
N-Boc-Gly-S-traps-traps-farnesyl-Cys diazomethyl ketone (III 401)
(12) Yellow oil. Yield 53%, 0.21 g.'H NMR (CDC13) b 1.46 (s, 9H), 1.60 (s,
6H),
1.67 (s, 6H), 1.97-2.10 (m, 8H), 2.85 (m, 2H), 3.19 ~m, 2H), 3.82 (m, 2H),
4.63 (m,
1H), 5.07-5.24 (m, 3H), 5.68 (s, 1H), 6.93 (m, 1H), 3C NMR (CDC13) 8 169.3,
140.1, 135.4, 131.4, 130.9, 128.8, 124.2, 123.1, 119.4, 80.6, 68.0, 65.9,
55.2, 54.8,
52.8, 48.3, 44.5, 39.7, 33.0, 31.7, 31.0, 30.2, 29.7, 28.3, 26.7, 26.4, 25.7,
23.5, 17.7,
16.2, 16.0; IR (KBr), 3318, 3084, 2983, 2916, 2859, 2105, 1669, 1510, 1369 cm
'.
N-Boc-Gly-S-traps-traps-farnesyl-Cys chloromethyl ketone (HI 130)
(13) Yield 100%, 0.22 g. 'H NMR (CDC13) b 1.45 (s, 9H), 1.60 (s, 6H), 1.68 (s,
6H), 1.96-2.09 (m, 8H), 2.81-2.97 (m, 2H), 3.10-3.24 ~m, 2H), 3.82 (m, 2H),
4.33
{s, 2H), 4.89 (m, 1H), 5.07-5.22 (m, 3H), 6.94 (m, 1H), 3C NMR (CDC13), b
199.7,
169.9, 156.0, 140.4, 135.3, 131.2, 124.2, 123.5, 123.1, 119.2, 111.5, 109.7,
80.5,
60.3, 55.4, 53.1, 51.8, 47.4, 44.2, 41.1, 39.6, 39.6, 31.8, 29.9, 28.2, 26.7,
26.4, 25.7,
21.0, 17.7, 16.1, 16.0, 14.1; IR {KBr), 3309, 2987, 2925, 1695, 1514, 1175 cm
';
MS (MALDI-TOF), 379.4 (M - Boc - Cl).
N-Ac-S-methyl-cysteine chloromethyl ketone (4h, HI 314) Yellow oil.
Yield 10%. 'H NMR (CDC13) 8 2.04 (s, 3H), 2.11 (s, 3H), 2.98 (ABX, J = 6.0,
6.3,
14.0 Hz, 2H), 4.36 (m, 2H), 4.87 (m, 1H), 6.41 (br d, 1H);'3C NMR (CDC13) 8
16.1,
22.9, 35.0, 47.4, 55.0, 170.3, 200.0; GC/MS 209 (M+); IR {KBr) 3278, 2922,
1741,
1659, 1537, 1429, 1209 cm '.
N-Ac-S-ethyl-cysteine chloromethyl ketone (4i, HI 315) Yellow solid.
Yield 45%. 'H NMR (CDCl3) s 1.24 (t, J = 7.4 Hz, 3H), 2.03 (s, 3H), 2.54 (q, J
=
7.3 Hz, 2H), 2.93 (ABX, J= 6.0, 6.3, 14.0 Hz, 2H), 4.34 (m, 2H), 4.88 (m, 1H),
6.36
(br d, 1H); '3C NMR (CDCl3) b 10.0, 18.3, 22.0, 27.9, 42.7, 50.7, 165.5,
195.4;
GC/MS 194 (M-CHZCH3); IR (KBr) 3299, 2927, 2872, 1740, 1659, 1537, 1535,
1425, 1371 cm '.
SUBSTITUTE SHEET (RULE 26)
CA 02336108 2000-12-28
WO 00/00469 PCT/US99/14758
33
N-Ac-S-~ropyl-cysteine chloromethyl ketone (4j, HI 369) Yellow semi-
solid. Yield 20%. H NMR (CDCl3) 8 0.96 (t, J= 7.3 Hz, 3H), 1.57 (m, 2H), 2.03
(s,
3H), 2.50 (t, J= 7.3 Hz, 2H), 2.91 (m, 2H), 4.33 (m, 2H), 4.77 (m, 1H), 6.32
(br d,
1H); ~3C NMR (CDC13) 8 13.2, 22.6, 22.7, 32.5, 34.5, 47.6, 55.5, 170.6, 200.0;
GC/MS 238 (M+1) ; IR (KBr) 3020, 2976, 2933, 1740, 1676, 1518, 1423, 1215 cm
i
N-Ac-S-butyl-cysteine chloromethyl ketone (4k, HI 363) Yellow solid.
Yield 65%. m.p. = 75-76 °C.'H NMR (CDC13) 8 0.90 (t, J= 7.1 Hz, 3H),
1.38 (m,
2H), 1.52 (m, 2H), 2.04 (s, 3H), 2.53 (t, J = 7.3 Hz, 2H), 2.98 (ABX, J = 6.0,
6.3,
14.0 Hz, 2H), 4.28 (m, 2H), 4.93 (m, 1H), 6.21 (br d, 1H); 13C NMR (CDCl3) b
14.2,
22.9, 31.4, 32.4, 32.9 47.4, 55.4, 170.1, 200.0; GC/MS 215 (M-HCl); MS
(MALDI-TOF) 250 (M+); IR (KBr) 3425, 3020, 1732, 1651, 1537, 1466 cm 1.
N-Ac-S-pentyl-cysteine chloromethyl ketone (41, HI 224) Yellow solid.
Yield 41% yield. m.p. = 74-75 °C. ~H NMR (CDCl3) 8 0.86 (t, J = 6.8
Hz, 3H),
1.2-1.4 (m, 4H), 1.55 (m, 2H), 2.04 (s, 3H), 2.52 (t, J= 7.5 Hz, 2H}, 2.93
~ABX, J=
6.0, 6.3, 13.5 Hz, 2H), 4.32 (m, 2H), 4.89 (m, 1H), 6.26 (br d, 1H), 3C NMR
(CDCl3) b 14.1, 22.7, 22.9, 28.7, 29.2, 29.6, 31.9, 32.9, 47.4, 55.4, 170.1,
200.0;
GC/MS 266 (M+1); MS (MALDI-TOF) 266 (M+1); IR (KBr) 3296, 2918, 2850,
1738, 1660, 1539, 1464 cm ~.
N-Ac-S-hexyl-cysteine chloromethyl ketone (4m, HI 357) Yellow solid.
Yield 44%. m.p. = 75-77 °C. ~H NMR (CDC13) 8 0.87 (t, J= 6.7 Hz, 3H),
1.2-1.4
(m, 6H), 1.56 (m, 2H), 2.05 (s, 3H), 2.53 (t, J = 7.4 Hz, 2H), 2.93 (ABX, J =
5.8,
6.0, 13.9 Hz, 2H), 4.35 (m, 2H), 4.89 (m, 1H), 6.36 (br d, 1H);'3C NMR (CDCl3)
b
14.0, 22.5, 22.9, 28.4, 29.4, 29.6, 31.3, 32.8, 47.4, 55.4, 170.1, 200.0;
GC/MS 243
(M-HCl); IR (KBr) 3304, 3053, 2951, 2926, 2870, 1738, 1639, 1537, 1425, 1371,
1283 cm ~.
N-Ac--S-heptyl-cysteine chloromethyl ketone (4n, HI 263) Yellow solid.
Yield 19%. m.p. = 77-81 °C. 'H NMR (CDCl3) 8 0.87 (t, J= 6.6 Hz, 3H),
1.2-1.4
(m, 8H), 1.54 (m, 2H), 2.03 (s, 3H), 2.51 (t, J = 7.3 Hz, 2H), 2.92 (ABX, J =
5.8,
6.3, 13.7 Hz, 2H), 4.32 {m, 2H), 4.88 (m, 1H), 6.25 (br d, 1H); 13C NMR
(CDCl3) 8
14.1, 22.5, 22.9, 28.6, 28.7, 29.4, 31.6, 32.7, 32.8, 47.4, 55.4, 170.1,
200.0; GC/MS
259 (M+1-Cl); MS (MALDI-TOF) 294 {M+1); IR (ICBr) 3306, 2918, 2922, 1737,
1641, 1537, 1466, 1372, 1283, 1134 cm 1.
N-Ac-S-octyl-cysteine chloromethyl ketone (40, HI 352) Yellow solid.
Yield 30% yield. m.p. = 76-77 °C. ~H NMR (CDCl3) b 0.88 (t, J = 6.9
Hz, 3H),
1.2-1.4 {m, l OH), 1.55 (m, 2H), 2.05 (s, 3H), 2.53 (t, J= 7.4 Hz, 2H), 2.93
(m, 2H),
4.35 (m, 2H), 4.87 (m, 1H), 6.36 (br d, 1H); 13C NMR (CDCl3) 8 14.0, 22.5,
28.4,
29.3, 31.3, 32.7, 32.9, 47.3, SS.3, 170.0, 199.7; GC/MS 271 (M-HCl); IR (KBr)
3300, 3055, 2918, 2852, 1738, 1639, 1537, 1425, 1371, 1283 cm 1.
SUBSTITUTE SHEET (RULE 26)
CA 02336108 2000-12-28
WO 00/00469 PCTNS99/14758
34
N-Ac-S-nonyl-cysteine chloromethyl ketone (4p, HI 364) Yellow solid.
Yield 35%. m.p. = 78-81 °C. 1H NMR (CDC13) b 0.86 (t, J= 6.5 Hz, 3H),
1.2-1.4
(m, 12H), 1.58 (m, 2H), 2.06 (s, 3H), 2.56 (t, J = 7.3 Hz, 2H), 2.98 (ABX, J =
6.1,
6.3, 13.8 Hz, 2H), 4.36 (m, 2H), 4.86 (m, 1H), 6.41 (br d, 1H); 13C NMR
(CDCl3) 8
14.1, 22.6, 22.9, 28.7, 29.1, 29.2, 29.4, 31.8, 32.7, 32.8, 47.4, 55.4, 170.2,
200.0;
GC/MS 285 (M-HCl); MS (MALDI-TOF) 322 (M+1); IR (KBr) 3302, 2920, 2850,
1738, 1643, 1537,
1394 cm ~.
N-Ac-S-decyl-cysteine chloromethyl ketone (4q, HI 371) Yellow solid.
Yield 10%. m.p. = 80-81 °C. 'H NMR (CDCl3) 8 0.86 (t, J = 6.7 Hz, 3H),
1.2-1.4
(m, 14H), 1.59 (m, 2H), 2.06 (s, 3H), 2.56 (t, J= 7.3 Hz, 2H), 2.98 (ABX, J=
6.0,
6.3, 13.7 Hz, 2H), 4.28 (m, 2H), 4.93 (m, 1H), 6.21 (br d, 1H); 13C NMR
(CDCl3) b
14.1, 22.7, 22.9, 28.7, 29.2, 29.3, 29.4, 29.5, 31.9, 32.8, 47.4, 55.4, 170.1,
200.0; MS
(MALDI-TOF) 301 (M-Cl); IR (KBr) 3302, 2920, 2850, 1736, 1635, 1539, 1467
cm .
N-Ac-S-undecyl-cysteine chloromethyl ketone (4r, HI 321) Yellow
solid. Yield 25%. m.p. = 85-86 °C. 'H NMR (CDC13) 8 0.86 (t, J = 6.7
Hz, 3H),
1.2-1.4 (m, 16H), 1.59 (m, 2H), 2.06 (s, 3H), 2.56 (t, J = 7.3 Hz, 2H), 2.98
(ABX, J
= 6.0, 6.3, 13.7 Hz, 2H), 4.28 (m, 2H), 4.93 (m, 1H), 6.21 (br d, 1H); ~3C NMR
(CDC13) 8 14.1, 22.7, 22.9, 28.7, 29.2, 29.3, 29.4, 29.5, 29.6, 31.9, 32.8,
32.9, 47.4,
55.4, 170.1, 200.0; GC/MS 313 (M-Cl); MS (MALDI-TOF) 313 (M-Cl); IR (KBr)
3305, 2918, 2850, 1738, 1651, 1537, 1466 cm 1.
N-Ac-S-tridecyl-cysteine chloromethyl ketone (4s, HI 323) Yellow
solid. Yield 22%. m.p. = 83-84 °C. 'H NMR (CDC13) 8 0.87 (t, J = 6.3
Hz, 3H),
1.2-1.4 (m, 20H), 1.59 (m, 2H), 2.06 (s, 3H), 2.56 (t, J= 7.2 Hz, 2H), 2.98
{ABX, J
= 5.8, 6.0, 14.0 Hz, 2H), 4.28 (m, 2H), 4.93 (m, 1H), 6.21 (br d, 1H); '3C NMR
(CDCl3) 8 14.1, 22.7, 23.0, 28.8, 29.2, 29.4, 29.4, 29.5, 29.6, 31.9, 32.8,
32.9, 47.3,
55.4, 170.1, 200.0; GC/MS 341 (M-HCl); MS (MALDI-TOF) 379 (M+2); IR (KBr)
3307, 2916, 2850, 1737, 1651, 1537, 1466, 1402, 1282 cm ~.
N-Ac-S-tetradecyl-cysteine chloromethyl ketone (4t, HI 354) Yellow
solid. Yield 32%. m.p. = 79-80 °C. 'H NMR (CDCl3) 8 0.88 (t, J = 6.7
Hz, 3H),
1.2-1.4 {m, 22H), 1.57 (m, 2H), 2.06 (s, 3H), 2.51 (t, J= 7.3 Hz, 2H), 2.94
(ABX, J
= 6.0, 6.3, 13.9 Hz, 2H), 4.35 (m, 2H), 4.90 (m, 1H), 6.30 (br d, 1H); '3C NMR
(CDCl3) S 14.1, 22.7, 22.9, 28.7, 29.2, 29.35, 29.4, 29.5, 29.6, 29.7, 31.9,
32.8, 32.9,
47.3, 55.4, 170.1, 200.0; GC/MS 355 {M-HCl); IR (KBr) 3302, 3259, 2916, 2848,
1738, 1660, 1537, 1464, 1373, 1317 cm ~.
N-Ac-S-pentadecyl-cysteine chioromethyl ketone (4u, HI 225) Yellow
solid. Yield 25%. m.p. = 86-89 °C. ~H NMR (CDC13) 8 0.86 (t, J = 6.5
Hz, 3H),
1.2-1.4 (m, 24H), 1.55 (m, 2H), 2.03 (s, 3H), 2.51 (t, J= 6.2 Hz, 2H), 2.9
~3(ABX, J
= 6.0, 6.3, 14.0 Hz, 2H), 4.34 (m, 2H), 4.88 (m, 1H), 6.33 (br d, 1H), C NMR
(CDCl3) 8 14.1, 22.7, 23.0, 28.8, 29.0, 29.2, 29.4, 29.5, 29.7, 30.7, 31.9,
32.2, 32.8,
32.9, 47.3, 55.4, 170.1, 200.0; GC/MS 369 (M-HCl); MS (MALDI-TOF) 407
(M+2); IR (KBr) 3307, 3124, 2916, 2850, 1738, 1651, 1533, 1487, 1404, 1284 cm
~.
SUBSTITUTE SHEET (RULE 26)
CA 02336108 2000-12-28
WO 00/00469 PCT/t1S99/14758
N-Ac-S-hexadecyl-cysteine chloromethyl ketone (4v, HI 366) Yellow
solid. Yield 18%. m.p. = 87-88 °C. ~H NMR (CDCl3) 8 0.86 (t, J = 6.7
Hz, 3H),
1.2-1.4 (m, 26H), 1.55 (m, 2H), 2.03 (s, 3H), 2.51 (t, J= 7.3 Hz, 2H), 2.98
(ABX, J
= 5.8, 6.0, 14.0 Hz, 2H), 4.32 (m, 2H), 4.93 (m, 1H), 6.26 (br d, 1H); '3C NMR
(CDC13) 8 14.2, 22.7, 23.0, 28.8, 29.0, 29.2, 29.4, 29.5, 29.7, 31.9, 32.8,
32.9, 47.3,
55.4, 170.1, 200.0; GC/MS 383 (M-Cl); MS (MALDI-TOF) 382 (M-HCl); IR
(KBr) 3259, 2916, 2848, 1740, 1660, 1539, 1471, 1432, 1134 cm ~.
N-Ac-S-octadecyl-cysteine chloromethyl ketone (4w, HI 370) Yellow
10 solid. Yield 78%. m.p. = 84-86 °C. 'H NMR (CDCl3) b 0.88 (t, J = 6.7
Hz, 3H),
1.2-1.4 (m, 30H), 1.57 (m, 2H), 2.06 (s, 3H), 2.53 (t, J= 7.4 Hz, 2H), 2.94
(ABX, J
= 5.8, 6.3, 13.7 Hz, 2H), 4.35 (m, 2H), 4.90 (m, 1H), 6.31 (br d, 1H); '3C NMR
(CDC13) b 14.2, 22.7, 23.0, 28.8, 29.2, 29.35, 29.4, 29.5, 29.6, 29.7, 31.9,
32.8, 32.9,
34.4, 47.3, 55.4, 170.0, 200.0; IR (KBr) 3313, 2916, 2850, 1738, 1651, 1537,
1466,
15 1385, 1281, 1254 cm 1.
N-Ac-S-eicoyl-cysteine chloromethyl ketone (4x, HI 226) Yellow solid.
Yield 11 %. m.p. = 84-89 °C. ~H NMR (CDCl3) b 0.87 (t, J = 6.6 Hz, 3H),
1.2-1.4
(m, 34H), 1.55 (m, 2H), 2.04 (s, 3H), 2.52 (t, J= 7.9 Hz, 2H), 2.92 (ABX, J=
6.0,
20 7.7, 14.0 Hz, 2H), 4.32 (m, 2H), 4.92 (m, 1H), 6.28 (br d, IH); 13C NMR
(CDCl3) 8
14.2, 22.7, 23.0, 28.7, 29.2, 29.4, 29.5, 29.6, 29.7, 31.9, 32.8, 32.9, 47.3,
55.4, 170.0,
200.0; MS (MALDI-TOF) 477 (M+2); IR (KBr) 3307, 2918, 2850, 1736, 1662,
1541, 1464, 1261, 1097 cm I.
25 N-Ac-S-docosyl-cysteine chloromethyl ketone (4y, HI 322) Yellow
solid. Yield 15%. m.p. = 95-97 °C. iH NMR (CDCl3) 8 0.88 (t, J = 6.5
Hz, 3H),
1.2-1.4 (m, 38H), 1.55 (m, 2H), 2.04 (s, 3H), 2.52 (t, J= 7.8 Hz, 2H), 2.92
(ABX, J
= 6.0, 6.3, 14.0 Hz, 2H), 4.32 (m, 2H), 4.92 (m, 1H), 6.28 (br d, 1H); ~3C NMR
(CDC13) 8 14.2, 22.7, 23.0, 28.8, 29.2, 29.4, 29.5, 29.7, 31.9, 32.8, 32.9,
47.3, 55.4,
30 170.1, 200.0; MS (MALDI-TOF) 493 (M+2-Cl+Na); IR (ICBr) 3259, 2918, 2848,
1740, 1660, 1537, 1471, 1261, 1099 cm ~.
N-Ac-S-allyl-cysteine chloromethyl ketone (HI-419) Pale yellow solid:
35 1H NMR (CDC13) b 2.02 (s, 3H), 2.85 (m, 2H), 3.12 {d, 2H), 4.31 (m, 2H),
4.87 (m,
1 H), 5.12 (dd, 1 H), 5.16 (s, 1 H}, 5.74 (m, 1 H), 6.32 (br, 1 H).
N-Ac-S-t-butyl-cysteine chloromethyl ketone (HI-349) Pale yellow
solid: ~H NMR (CDCl3) 8 1.24 (s, 9H), 1.97 (s, 3H), 2.90 (dd, 2H), 4.27 (m,
2H),
4.87 (m, 1H), 6.32 (br, 1H); MS (EI) m/z 251 (M+).
N-Ac-S-2-methylpropyl-cysteine chloromethyl ketone (HI-391) Pale
yellow solid: ~H NMR (CDCl3) 8 0.94 (m, 6H), 1.74 (m, IH), 2.01 (s, 3H), 2.40
(d,
2H), 2.87 (t, 2H), 4.33 (m, 2H), 4.83 (m, 1 H), 6.51 (d, 1 H); MS (EI) m/z 252
(M+).
N-Ac-S-2,2-dimethylpropyl-cysteine chloromethyl ketone (HI-421)
Pale yellow solid: 1H NMR (CDC13) 8 1.32 (s, 9H), 2.06 (s, 3H), 3.15 (m, 2H),
3.45
(m, 2H}, 4.34 (s, 2H), 4.94 (m, 1 H), 6.42 (d, 1 H); MS (EI) m/z 227 (M -
'Bu).
SUBSTITUTE SHEET (RULE 26)
CA 02336108 2000-12-28
WO 00/00469 PCT/US99/14758
36
N-Ac-S-3-methylbutyl-cysteine chloromethyl ketone (HI-387) Pale
yellow solid: IH NMR (CDCl3) 8 0.89 (d, 6H), 1.44 (m, 1H), 1.62 (m, 2H), 2.04
(s,
3H), 2.53 (t, 2H), 2.93 (m, 2H), 4.34 (m, 2H}, 4.88 (m, 1H), 6.30 (d, 1H); MS
(EI}
m/z 229 (M - CI).
N-Ac-S-2-ethylbutyl-cysteine chloromethyl ketone (HI-390) Pale
yellow solid: 'H NMR (CDCI3) 8 0.83 (m, 6H), 1.35 (m, SH), 2.03 (s, 3H), 2.51
{d,
2H), 2.90 (m, 2H), 4.32 (d, 2H), 4.87 (m, 1H), 6.34 (d, 1H); MS (EI) m/z 243
(M
Cl}.
N-Ac-S-cyclopropylmethyl-cysteine chloromethyl ketone (Hi-507)
Pale yellow solid: 1H NMR (CDC13) b 0.57 (d, 4H), 0.95 (m, 1H), 2.03 (s, 3H),
2.45
(d, 2H), 2.97 (m, 2H), 4.33 (d, 2H), 4.89 (m, 1H), 6.34 (d, 1H).
N-Ac-S-cyclobutylmethyl-cysteine chioromethyl ketone (HI-385) Pale
yellow solid: ~H NMR (CDC13) 8 1.83 (m, 7H), 2.02 (s, 3H), 2.58 (d, 2H), 2.89
(m,
2H), 4.32 (d, 2H), 4.84 (m, 1H), 6.32 (d, 1H); MS (EI) m/z 227 (M - Cl).
N-Ac-S-cyclohexylmethyl-cysteine chloromethyl ketone (HI-386) Pale
yellow solid: ~H NMR (CDC13) 8 0.90 (m, 2H), 1.25 (m, 4H), 1.41 (m, 4H), 1.70
(m,
1H), 2.04 (s, 3H), 2.40 (d, 2H}, 2.89 (m, 2H), 4.34 (m, 2H), 4.86 (m, 1H),
6.35 (d,
1 H).
N-Ac-S-benzyl-cysteine chloromethyl ketone (HI-251) Pale yellow
solid: ~H NMR (CDC13) 8 1.98 (s, 3H), 2.82 (m, 2H), 3.70 (s, 2H}, 4.08 (d,
2H), 4.82
(m, 1 H), 6.18 (d, 1 H), 7.29 (m, SH).
N-Ac-S-4-methoxybenzyl-cysteine chloromethyl ketone (HI-349) Pale
yellow solid: 'H NMR (CDC13) 8 1.91 (s, 3H), 2.78 (m, 2H}, 3.62 (s, 3H), 3.73
(s,
2H), 4.12 (d, 2H), 4.74 (m, 1 H), 6.07 (d, 1 H), 6.80 (m, 2H), 7.18 (m, 2H);
MS (EI)
m/z 279 (M - CI).
N-Ac-S-benzyioxycarbonyl-cysteine chloromethyl ketone (HI-389)
Pale yellow solid: 'H NMR (CDCl3) 8 1.98 (s, 3H), 3.36 (m, 2H}, 4.31 (d, 2H),
5.01
(m, 2H), 5.42 (m, 1H), 6.43 (d, 1H), 7.35 (m, SH).
N-Ac-S-diphenylmethyl-cysteine chloromethyl ketone (HI-418) Pale
yellow solid: 'H NMR (CDCl3) 8 2.00 (m, 4H), 3.37 (m, 2H), 4.34 (d, 2H}, 5.02
(m,
1H), 6.53 (d, 1H}, 7.30 (m, lOH).
N-Ac-S-trityl-cysteine chloromethyl ketone (HI-350) Pale yellow
solid: 'H NMR (CDCI3) 8 1.92 (s, 3H), 2.73 (m, 2H}, 3.89 (s, 2H), 4.40 (m,
1H),
5.82 (d, 1 H), 7.21 (m, 1 SH).
SUBSTITUTE SHEET (RULE 26)
CA 02336108 2000-12-28
WO 00/00469 PCT/US99/14758
37
N-Ac-S-2-naphthylmethyl-cysteine chloromethyl ketone (HI-392)
Pale yellow solid: 'H NMR (CDC13) 8 1.89 (s, 3H), 2.81 (m, 2H), 3.84 (s, 2H),
4.13
(d, 2H), 4.83 (m, 1 H), 6.10 (d, 1 H), 7.43 (m, 2H), 7.66 (s, 1 H), 7.72 (m,
4H).
N-Ac-O-dodecyi-serine chloromethyl ketone (HI-489) White solid: ~H
NMR (CDCI3) 8 0.88 (t, 3H), 1.26 (m, 20H), 2.05 (s, 3H), 3.41 (t, 2H), 3.57
(m, 1H),
3.88 (m, 1H), 4.29 (d, 2H), 4.90 (m, 1H), 6.32 (d, 1H).
N-Boc-O-dodecyl serine chloromethyl ketone (HI-266) Pale yellow
solid: 1H NMR (CDCl3) 8 0.88 (t, 3H), 1.26 (m, 20H), 1.46 (s, 9H), 3.41 (t,
2H),
3.50 (m, 1 H), 3.57 (dd, 1 H), 4.37 (d, 2H), 4.56 (m, 1 H), 5.21 (d, 1 H); MS
(EI) m/z
357 (M - Cl).
N-Propyloxycarbonyl-S-dodecyl-cysteine chloromethyl ketone (HI
413) Pale yellow solid: 'H NMR (CDCl3) 8 0.92 (m, 6H), 1.25 (m, 20H), 1.60 (m,
2H), 2.52 (t, 2H), 2.91 (d, 2H), 4.01 (t, 2H), 4.45 (s, 2H), 4.66 (m, 1 H),
5.48 (d, 1 H);
MS (EI) mla 371 (M - Cl).
N-Benzyloxycarbonyl-S-dodecyl-cysteine chloromethyl ketone (HI
320) Pale yellow solid: ~H NMR (CDC13) 8 0.86 (t, 3H), 1.27 (m, 20H), 2.49 (t,
2H), 2.91 (d, 2H), 4.32 (s, 2H), 4.70 (m, 1H), 5.19 (s, 2H), 5.59 (d, 1H),
7.32 (s,
SH); MS (EI) m/z 419 (M - CI).
N-9-Fluorenylmethyloxycarbonyl-S-dodecyl-cysteine chloromethyl
ketone (HI-398) Pale yellow solid: 1H NMR (CDC13) 8 0.87 (t, 3H), 1.24 (m,
18H), 1.52 (m, 2H), 2.52 (t, 2H), 2.93 (m, 2H), 4.22 (t, 1 H), 4.28 (s, 2H),
4.46 (m,
2H), 4.68 (m, 1 H), 5.59 (d, 1 H), 7.3 S-7.77 (m, 8H).
N-3-Dimethylaminobenzoyl-S-dodecyl-cysteine chloromethyl ketone
(HI-268) Pale yellow solid: iH NMR (CDCl3) 8 0.94 (t, 3H), 1.26 (m, 20H), 2.54
(t, 2H), 3.01 (d, 2H), 3.93 (d, 6H), 4.13 (d, 2H), 4.68 (m, 1 H), 5.53 (d, 1
H), 7.00 (dd,
1H), 7.40 (m, 3H).
N-Ac-S-dodecyl-cysteine bromomethyl ketone (HI-488) Pale yellow
solid: 'H NMR (CDC13) 8 0.84 (t, 3H), 1.22 (m, 18H), 1.53 (m, 2H), 2.02 (s,
3H),
2.50 (t, 2H), 2.92 (m, 2H), 4.12 (s, 2H), 4.89 (m, 1 H), 6.44 (d, 1 H).
N-Ac-S-dodecyl-Cys-N(OCH3)-CH3 (HI-267) Pale yellow oil: 'H
NMR (CDCl3) 8 0.87 (t, 3H), 1.25 (m, 20H), 2.02 (s, 3H), 2.51 (t, 2H), 2.82
(m, 2H),
3.22 (s, 3H), 3.79 (s, 3H), 5.17 (m, 1H), 6.38 (d, 1H); MS {EI) m/z 314 (M
N(OCH3)CH3).
N-Ac-S-dodecyl-Cys-H (HI-274) White solid: 1H NMR (CDC13) 8 0.88
(t, 3H), 1.26 (m, 18H), 1.58 (m, 2H), 2.09 {s, 3H), 2.54 (t, 2H), 3.00 (m,
2H), 4.63
(m, 1 H), 6.37 (d, 1 H), 9.65 (s, 1 H).
SUBSTITUTE SHEET (RULE 26)
CA 02336108 2000-12-28
WO 00/00469 PCT/US99/14758
38
N-Ac-S-dodecyl-Cys-CH2-SPh (HI-269) Pale yellow solid: ~H NMR
(CDC13) 8 0.88 (t, 3H), 1.25 (m, 18H), 1.53 (m, 2H), 1.97 (s, 3H), 2.46 (t,
2H), 2.2
(m, 2H), 5.16 (m, 1H), 5.43 (s, 2H), 6.16 (d, 1H), 7.33 (s, SH).
N-Ac-S-dodecyl-Cys-CH2 S-2-naphthyl (HI-302) Pale yellow solid:
1H NMR (CDCI3) 8 0.81 (t, 3H), 1.16 (m, 18H), 1.39 (m, 2H), 1.84 (s, 3H), 2.36
(t,
2H), 2.77 (m, 2H), 5.18 (m, 1H), 5.59 (s, 2H), 6.08 (d, 1H), 7.43 (s, 3H),
7.73 (m,
4H).
N-Ac-S-dodecyl-Cys-CHZ S-CHZCH2COZH (HI-273) White solid: 'H
NMR (CDCI3) 8 0.88 (t, 3H), 1.26 (m, 18H), 1.33 (m, 2H), 2.07 (s, 3H), 2.54
(t, 2H),
2.77 (m, 4H), 2.97 (m, 2H), 3.48 (m, 2H), 5.02 (m, 1 H), 6.44 (d, 1 H), 8.04
(br, 1 H).
Example 3
~totoxicit~of alkyl ketone compounds
The cytotoxicity of the alkyl ketone compounds against tumor cells was
evaluated in leukemic cells, breast cancer cells, prostate cancer cells, and
brain
cancer cells.
Cytotoxicity Assay
Cytotoxicity of various compounds against tumor cells was performed
using the MTT (3-[4,5-dimethylthiazol-2-ylJ-2,5-diphenyl tetrazolium bromide)
assay (Boehringer Mannheim Corp., Indianapolis, IN). Unless otherwise
specified,
all cell lines were obtained from the American Type Culture Collection (ATCC).
Briefly, exponentially growing cells were seeded into a 96-well plate at a
density
of 2.5 x 104 cells/well and incubated for 36 hours at 37°C prior to
drug exposure.
On the day of treatment, culture medium was carefully aspirated from the wells
and
replaced with fresh medium containing the indicated compound at concentrations
ranging from 0.1 to 250 p,M. Triplicate wells were used for each treatment.
Human leukemic cell lines (NALM-6, MOLT-3) glioblastoma cells
(U373) and human breast tumor cell lines (BT20 and MDA-MB-231) were
obtained from the American Type Culture Collection and maintained as a
continuous cell line in Dulbecco's modified Eagles's medium supplemented with
10% fetal bovine serum and antibiotics.
SUBSTITUTE SHEET (RULE 26)
CA 02336108 2000-12-28
WO 00/00469 PCT/US99/14758
39
The cells were incubated with the various compounds for 24-36 hours at
37°C in a humidified 5% C02 atmosphere. To each well, 10 ~l of MTT (0.5
mg/ml
final concentration) was added and the plates were incubated at 37°C
for 4 hours to
allow MTT to form formazan crystals by reacting with metabolically active
cells.
The formazan crystals were solubilized overnight at 37°C in a solution
containing
10% SDS in 0.01 M HCI. The absorbence of each well was measured in a
microplate reader (Labsystems) at 540 nm and a reference wavelength of 690 nm.
To translate the OD s4o values into the number of live cells in each well, the
OD s4o
values were compared to those on standard OD s4o - versus - cell number curves
generated for each cell line. The percent survival was calculated using the
formula:
live cell number [test]
Survival = X 100
live cell number [control]
The ICso values were calculated by non-linear regression analysis and are
shown below in Tables 1 - 6.
Table 1. Structure and activities of S-alkyl cysteine diazo and chloromethyl
ketone derivatives against Nalm-6 (B-lineage ALL), Molt-3 (T-
lineage ALL), BT-20 (breast cancer), PC-3 {prostate cancer) and
U-373 (glioblastoma) cell lines.
S
R. N X
H O
No. HI R~ ~ R3 ~ R~ ~ ICso (1~1)
No. ~ ~ ~ ~ Nalm-6 Molt-3 BT-2(
B-lineage ~ T-lineage ~ Breast ~ Prostate ~ Glioblas
ALL ALL Cancer Cancer -toma
12401 Boc-trans,trans-FamesylCH=N s1.3 84.5 9s.1 90.6 >100
3b82 Boc traru,trans-FarnesylCH=N 49.8 s0.1 >100 >100 >100
3a367 Ac trans,trans-FamesylCH=N 30.3 32.2 >100 >100 >100
3c122 Ac trans-GeranylCH=N >100 >100 >100 >100 >100
SUBSTITUTE SHEET (RULE 26)
CA 02336108 2000-12-28
WO 00/00469 PCT/US99/14758
3d123 Ac 3-Methyl-2-butenylCH=N > l00 > 100 > > 100 >
l00 100
3e348 Ac Dodecyl CH=N 15.4 22.9 97.9 > 100 >
100
13130 Boc-trans,trans-FamesylCHz- 12.9 17.5 >I00 >100 71.3
4b124 Boc trans.trans-FamesylCHz- 10.7 7.7 >100 >100 >l00
4a368 Ac trans.trans-FamesylCHz- 3.0 1.4 55.8 26.8 61.3
4c127 Ac trarra-GeranylCHz- >l00 >100 >100 >100 >100
4dl28 Ac 3-Methyl-2-butenylCHz- 12.6 7.9 25.7 47.8 >
I00
4e131 Ac Dodecyl CHz- 2.0 10.9 10.0 22.1 35.1
8 129 Boc Dodecyl CHz- 15. 15.5 48.9 64.6 >
I 100
9 252 H.HCDodecyl CHz- 17.7 12.5 >I00 >100 >(00
Table 2. Structure and activities of farnesylthio methyl ketone derivatives
against Nalm-6 (B-lineage ALL), Molt-3 (T-lineage ALL), BT-
5 20 (breast cancer), PC-3 (prostate cancer) and U-373
(glioblastoma) cell lines.
O
/ / / S X
n
No.HI n R~ ICso
Number (IBM)
Nalm-6Molt-3BT-20 PC-3 U-373
-
B-lineageT-lineageBreastProstateGliobla
ALL ALL CancerCancer stoma
3f84 I CH=Nz > 100 60.3 > 100 > 100 >
100
3g83 2 CH=Nz 53.5 6.8 >100 >100 >100
4t126 l CHz-CI84.3 >100 >100 >100 >100
4g125 2 CHz-Cl40.7 35.5 >100 >l00 >100
is
Table 3. Comparison of the effect of the S-alkyl chain length upon the
activities of cysteine chloromethyl ketone derivatives against
Nalm-6 (B-lineage ALL) and Molt-3 {T-lineage ALL), BT-20
(breast cancer), PC-3 (prostate cancer) and U-373 (glioblastoma)
cell lines
SUBSTITUTE SHEET (RULE 26)
CA 02336108 2000-12-28
WO 00/00469 PCT/US99/14758
41
X
I
O S
H3C' _ N CI
H
O
No. ~ HI ~ R3 ~ ICso (ItM)
No. Nalm-6 Molt-3 BT-20 PC-3 U-373
B-lineageT-lineageBreastProstateGlioblastoma
ALL ALL CancerCancer
4h314 Methyl30.3 80.8 > 100 > I > 100
00
4I315 Ethyl52.8 99.9 96.4 41.2 >100
4j369 Propyl6.9 8.0 >100 37.7 97.7
4k363 Butyl41.4 5.6 >100 >100 86.9
41224 Pentyl5.8 5.4 89.5 >100 >100
4m357 Hexyl3.3 0.7 >100 25.1 91.9
4n263 Heptyl4.8 2.5 84.9 31.3 62.6
40352 Octyl5.6 4.1 58.8 35.4 >100
dp364 Nonyl7.3 6.7 > 100 96.3 88.2
4q371 Decyl4.7 3.4 >100 >100 86.2
4r321 Undecy1.7 3.0 99.1 64.7 56.7
I
4e131 Dodecy2.0 10.9 10.0 22.1 35.1
I
4s323 Tridecy>l00 >t00 >100 >100 >100
I
4t354 Tetrade8.7 8.8 >I00 54.8 >100
cyl
4u225 Pentad8.9 8.6 >l00 >t00 >100
ecyl
4v366 Hexade16.0 17.3 >100 >100 >100
cyl
4w370 Octade>100 >100 >100 >100 >100
cyl
4x226 Eicosyl>100 >100 >100 >100 >100
4y322 Docosy>100 >100 >100 >100 >l00
I
5
SUBSTITUTE SHEET (RULE 26)
CA 02336108 2000-12-28
WO 00/00469 PCT/US99/14758
42
Table 4. Examination of the effect of changing R1 of the molecule.
S
O
H3C II N X
J~H O
HI R~ ICS
No. (~M)
Nalm-6 Molt-3BT-20 PC-3 0373
131 CHZ-Cl 2.0 2.3 10.0 22.1 35.1
348 CH=NZ 15.4 22.9 97.9 > 100 >
100
488 CHZ Br 1.3 3.19 33.83 39.6 22.5
208 OH
508 CHZ O-CO-Ph
267 N(OCH3)CH3 50.1 > 100 > 100 >
100
274 H 12.6 13.1 22.8 >
100
269 CHz S-Ph >100 >100 >100 >100 >100
302 CHz S-2-naphthyl>100 >100 >100 >100 >100
399 CHi S-C6F5 91.2 40.8 > 100 91.6 >
100
365 CHZ S-trityl 93.1 97.3 >100 >100 >100
273 CHz S-CHZCHZ 44.4 50.3 20.06 > 100 >
l00
COZH
SUBSTITUTE SHEET (RULE 26)
CA 02336108 2000-12-28
WO 00/00469 PCT/US99/14758
43
Table 5. Examination of the effect of changing X from S to O and altering R4.
R. N
H
S
H1 ~ R° ~ X ICso (pM)
No. Nalm-6 Molt-3 BT-20
252 H.HCI S 17.7 12.5 >l00 >100 >100
131 Acetyl S 2.0 2.3 10.0 22.1 35.1
489 Acetyl O 3.8 15.3 > 100 > I 00 > l 00
490 Trifluoroacetyl S 32.6 41.3 >100 >100 >100
129 t-Butyloxycarbonyl S I5.1 15.5 48.9 64.6 >100
266 t-Butyloxycarbonyl O 64.5 49.9 37.1
319 Ethyloxycarbonyl S 77.6 84.2 >100 >I00 >100
413 Propyloxycarbonyl S
320 Benzyloxycarbonyl S 13.5 >100 >100 >100 >100
398 9- S 25.7 32.3 >100 >100 >100
Fluorenylmethyloxycarbonyl
491 Benzoyl S 5.4 6.5 >100 52.1 >100
268 3-Dimethylaminobenzoyl S 96.1 >I00 >100 >100
Table 6. An examination of the effects of incorporating rings and branched
chains
into R3.
10
X
l
S
O
H3C"N CI
H ~
SUBSTITUTE SHEET (RULE 26)
CA 02336108 2000-12-28
WO 00/00469 PCT/US99/14758
44
HI R' ICso
No. (1tM)
Nalm-6Molt-3BT-20 PC-3 U373
419 Allyl 4.3 7.3 55.7 >!00 56.7
400 5-Hexenyl
349 t-Butyl 71.0 71.9 >! 00 > 100 >
l00
391 2-Methylpropyl
421 2,2-Dimethylpropyl
388 t-Butylthio
387 3-Methylbutyl
390 2-Ethylbutyl
507 Cyclopropylmethyl
385 Cyclobutylmethyl
386 Cyclohexylmethyl
251 Benzyl
351 4-Methoxybenzyl60.17 4.75 73.1 43.8 45.6
389 Benzyloxycarbonyl1.2 3.5 49.3 23.4 54.4
418 Diphenylmethyl
350 Trityl 10.0 26.4 88.8 52.2 87.1
392 2-Naphthylmethyl2.6 2.9 40.4 36.6 4L8
420 2-Anthraquinonylmethyl
5 The data shown in Table I suggest that substitution at the R1 position with
a
chloromethyl group is better than substitution with a diazomethyl group.
Placement
of the dodecyl and farnesyl groups at the R3 position produced compounds
having
the greatest cytotoxicity.
The effect changing the length of R2 are reported in Table 2. Omission of the
sidegroup caused the cytotoxicity of the compounds to decrease. Additionally,
compounds where R2 comprises two carbon atoms were more cytotoxic than
compounds
A series of compounds was prepared with different aliphatic chain lengths in
the R3 position to determine the effect of chain length on cytotoxicity. As
shown in
Table 3, compounds with chain lengths of about 5 to 15 were the most effective
anti-cancer agents. Preferred lengths were chain lengths of about 11 to 12.
SUBSTITUTE SHEET (RULE 26)
CA 02336108 2000-12-28
WO 00/00469 PCT/US99/14758
Various groups were tested in the R' position to further define effective anti-
cancer compounds. The results are reported in Table 4. Substitution of a
bromomethyl group at the R' position produced the most effective compound,
consistent with Table I where chloromethyl was the most effective compound.
The effect of changing of X and R4 on cytotoxicity was examined and
reported in Table S. The data show that a small acetyl substituent as R4
produces a
potent cytotoxicagent, as both absence of acetyl and substitution with a
larger group
caused a loss in potency. Substitution of R4 with benzoyl produced an
effective
compound. The effect of changing X to O is more complex, with the derivatives
10 being comparable in some cell lines but different in others. The ether
compounds
may offer advantages in terms of stability, despite their lower cytotoxic
potency.
Various substitutions of R3 were analyzed to determine the effects of branched
and ring structures on cytotoxicity. The results demonstrate that ringed and
branched
structures are effective anti-cancer compounds.
Example 4
Cytotoxicity of HI-131 in Primary Cancer Cells
The cytotoxicity of HI-131 against primary cancer cells was evaluated in
leukemia cells taken from six children (Figure 1 A), using the MTT assay
described
for Example 3. The cells were exposed to HI-131 at concentrations ranging from
0
to 50 ~M. Percent survival was calculated as described for Example 3 and
plotted
against the HI-131 concentration used in the experiment. A composite
concentration
survival curve was then prepared from the data (Figure 1 B).
The results clearly show a dose dependent cytotoxic effect of HI-131 in
primary cancer cells taken from all six patients.
Example 5
HI-131 Induces Apoptosis of Leukemia Cells
The ability of HI-131 to induce apoptosis cells was evaluated in human
leukemia in NALM-6 cells and primary leukemic cells from 2 patients. Cells
were
treated with 50 uM compound HI-131 for 24 hours. After incubatin, the cells
were
SUBSTITUTE SHEET (RULE 26)
CA 02336108 2000-12-28
WO 00/00469 PCTNS99/14758
46
harvested and analyzed for apoptosis by in situ TUNNEL analysis and confocal
laser
scanning microscopy as described in Sudbeck et al., 1999, Clin. Cancer Res.,
5:1589-82. Controls were treated with vehicle alone.
The data are shown in Figures 2A-2F. Controls (Figures 2A, 2C, and 2E)
failed to induce apoptosis. In contrast treatment with HI-131 (Figures 2B, 2D,
and
2F) greatly induced apoptosis in the NALM-6 and primary leukemic cells.
Induction of apoptosis was further evaluated in primary leukemic cells and
established NALM-6 and MOLT-3 cell lines following treatment with HI-131.
Data are shown in Figure 3.
Primary leukemia cells treated with vehicle alone typically displayed an
apoptotic rate of approximately 25%, with one sample exhibiting the much
higher
rate of about 70%. The rate of apoptosis in the control treated NALM-6 and
MOLT-3 cell lines was much lower at about 5%. The rate of apoptosis in all but
one
of the primary leukemic cell samples increased dramatically after treatment
with HI-
131 with an approximately 4-fold induction of apoptosis being typical. HI-131,
induced apoptosis was much more dramatic in NALM-6 and MOLT-3 cells, with
an approximately 20-fold induction of apoptosis.
Example 6
HI-131 Inhibits Tumor Cell Invasion
The ability of HI-131 to inhibit invasion by MDA-MB-231 breast cancer
cells and U373 glioblastoma cells was evaluated.
Cell Invasion Assay
The in vitro invasiveness of MDA-MB-231 breast cancer cells and U373
glioblastoma cells was assayed using a previously published method which
employs
Matrigel-coated Costar 24-well transwell cell culture chambers ("Boyden
chambers") with 8.0-p,m-pore polycarbonate filter inserts (Albini, et al.,
1987,
Cancer Res., 47:3239-3245). The chamber filters were coated with 50 ~g/ml of
SUBSTITUTE SHEET (RULE 26)
CA 02336108 2000-12-28
WO 00/00469 PCTNS99/14758
47
Matrigel matrix, incubated overnight at room temperature under a laminar flow
hood
and stored at 4°C. Matrigel matrix is made up of several components of
the
extracellular matrix (ECM), including collagens, laminin and proteo-glycans.
On the day of the experiment, the coated inserts were rehydrated with 0.5 ml
serum-free DMEM containing 0.1% bovine serum albumin for 1-2 hours. To study
the effects of HI-131 on invasiveness of glioblastoma and breast cancer cells,
exponentially growing cells were incubated overnight with HI-131 at various
concentrations ranging from 1 p,M to 25 pM and 2.5 ~M to 25 ~M, respectively.
The
cells were trypsinized, washed twice with serum-free DMEM containing BSA,
counted and resuspended at 1 x 105 cells/ml. 0.5 ml cell suspension containing
5 x
104 cells in a serum-free DMEM containing HI-131 or vehicle was added to the
Matrigel-coated and rehydrated filter inserts. Next, 750 p.l of NIH fibroblast
conditioned medium was placed as a chemoattractant in 24-well plates and the
inserts were placed in wells and incubated at 37°C for 48 hours. After
the
incubation period, the filter inserts were removed, the medium was decanted
off and
the cells on the top side of the filter that did not migrate were scraped off
with a
cotton-tipped applicator. The invasive cells that migrated to the lower side
of the
filter were fixed, stained with Hema-3 solutions and counted under microscope.
Five to 10 random fields per filter were counted to determine the mean (tSE)
values
for the invasive fraction. The invasive fractions of cells treated with HI-131
were
compared to those of DMSO treated control cells and the percent invading
relative to
the control was determined using the formula:
Invading = 100 * Number of Adherent Drue Treated Cells
Number of Adherent Control Cells
Each treatment condition was evaluated in duplicate in 3 independent
experiments. ICSO values were calculated by non-linear regression analysis
using
Graphpad Prisin Software Version 2.0 (Graphpad Software Inc., San Diego, CA).
Results
As shown in Figures 4 and S, MDA-MB-231 breast cancer cells and U373
glioblastoma cells were highly invasive in Matrigel-coated Boyden chambers. HI-
SUBSTITUTE SHEET (RULE 26)
CA 02336108 2000-12-28
WO 00/00469 PCT/US99/14758
48
131 inhibited the invasion of both MDA-MB-231 breast cancer cells and U373
glioblastoma cells through the Matrigel matrix in dose-dependent fashion.
Example 7
HI-131 Inhibits Adhesion of Cancer Cells
The ability of HI-131 to inhibit adhesion of MDA-MB-373 and U373
Glioblastoma cells was evaluated.
During the multistep process of tissue invasion, tumor cells initially adhere
to
the extracellular matrix proteins via cell surface integrin receptors and then
gain
migratory capacity to enter the surrounding tissues. ECM proteins such as
laminin,
fibronectin, and type IV collagen are thought to play an important role in
tumor cell
attachment and migration. Laminin, fibronectin and collagen have been found in
the
basal lamina of blood vessels and in the glial limitans externs in the brain
that
promote the adhesion and invasion of tumor cells in situ (Carbonetto, 1984,
Trends
Neurosci., 7:382-387; Rutkaet al. J. Neurosurg., 69:155-170; Venstrom, et al,
1993, FASEB J., 7:996-1003). The effects of these ECM proteins on integrin-
mediated U373 glioblastoma and MDA-MB-231 cell adhesion was examined.
Cell Lines
A human brain tumor cell line derived from an adult patient with
glioblastoma, U-373 MG (Cat. #HTB-17) and MDA-MB-231 breast cancer cells
(Cat. #HTB-26) were obtained from American Type Culture Collection (ATCC,
Manassas, VA) and maintained in liquid culture using DMEM supplemented with
10% fetal bovine serum and antibiotics. Fibroblast conditioned medium was used
as
a source of chemoattractant in vitro invasion assays. Conditioned medium was
prepared as described previously (Albini, et al., 1987, Cancer Res., 47:3239-
3245).
For the preparation of this conditioned medium NIH/3T3 embryonic fibroblasts
(ATCC cat. #CRL-1658) were grown to 80% confluency in DMEM medium
supplemented with 10% FBS and cultured for 24 hours in serum-free medium
containing 0.5 pg/ml bovine serum albuminutes The culture supernatants were
collected, centrifuged at 1000 x g for 15 minutes to remove cellular debris
and used
as conditioned medium.
SUBSTITUTE SHEET (RULE 26)
CA 02336108 2000-12-28
WO 00/00469 PCT/US99/14758
49
Adhesion Assays
In vitro adhesion assays were performed to (a) study the baseline adhesive
properties of U373 glioblastoma and MDA-MB-231 breast cancer cells and (b)
evaluate the effects of HI-131 on the adhesive properties of these cells. The
plates
for the adhesion assays were precoated with the extracellular matrix proteins
laminin, fibronectin or type IV collagen (each at a final concentration of 1
p,g/ml in
PBS) overnight at 4°C and dried. On the day of the experiment, the
wells were
rehydrated and blocked with 10% bovine serum albumin in PBS for 1 hour at room
temperature and used for the adhesion assays, as described below.
To study the effects of HI-131 on glioblastoma and breast cancer cell
adhesion, exponentially growing cells in DMEM were incubated with the compound
HI-131 or with genistein at concentrations ranging from 10 pM to 50 ~M and 5
~,M
to 25 p,M respectively for 16 hours in a humidified 5% COz atmosphere. DMSO
(0.1 %) was included as a vehicle control. After treatment, cells were
detached from
the flasks with 0.05% trypsin (Life Technologies) resuspended in DMEM,
incubated
at 37°C for 2 hours to allow them to recover from the trypsinization
stress and
examined for their ability to adhere to plates precoated with ECM proteins.
In adhesion assays, cells were centrifuged, washed twice with serum-free DMEM,
counted and resuspended in serum-free DMEM to a final concentration of 2.5 x
105
cells/ml. One hundred ~1 of the cell suspension containing 2.Sx 104 cells were
added to each well and cells were allowed to adhere for 1 hour at 37°C
in a
humidified S% COZ atmosphere. The adherent fraction was quantitated using MTT
(3-(4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide) assays. In
brief,
after washing the wells, 10 p.l of MTT (0.5 mg/ml final concentration)
(Boehringer
Mannheim Corp., Indianapolis, IN) was added to each wel! and the plates were
incubated at 37°C for 4 hours to allow MTT to form formazan crystals by
reacting
with metabolically active cells. The formazan crystals were solubilized
overnight at
37°C in a solution containing 10% SDS in 0.01 M HC1. The absorbance of
each
well was measured in a microplate reader (Labsystems) at 540 nm and a
reference
wavelength of 690 nm. To translate the OD54o values into the number of cells
in
each well, the OD54o values were compared to those on standard ODs4o-versus-
cell
number curves generated for each cell line. The adherent fraction of cells
treated
SUBSTITUTE SHEET (RULE 26)
CA 02336108 2000-12-28
WO 00/00469 PCT/US99/14758
SO
with HI-131 was compared to the DMSO-treated control cells and the percent
adhesion relative to the control was determined.
Each treatment condition was evaluated in duplicate in 3 independent
experiments. The ICSo values were calculated by non-linear regression
analysis.
S
Results
As shown in Figures 6 and 7, control U373 glioblastoma cells adhered to
plates precoated with laminin, fibronectin, or type IV collagen about equally.
Similar results were obtained with MBA-MD-231 breast cancer cells. Treatment
with HI-131 resulted in a dose-dependent loss of adhesion in both glioblastoma
and
breast cells (See Figures 6 and 7).
All publications, patents, and patent documents described herein are
incorporated by reference as if fully set forth. The invention described
herein may
1 S be modified to include alternative embodiments. All such obvious
alternatives are
within the spirit and scope of the invention, as claimed below.
SUBSTITUTE SHEET (RULE 26)