Note: Descriptions are shown in the official language in which they were submitted.
CA 02337999 2000-12-28
WO 00/00202 PCT/US99/14923
METHOD FOR INHIBITING C-JUN EXPRESSION
USING JAK 3 INHIBITORS
The protooncogene c jun is the cellular counterpart of the v jun
oncogene of avian sarcoma virus 17. C; jun expression is activated in response
to
a diverse set of DNA-damaging agents including ara-C, W radiation,
topoisomerase II inhibitors, alkylating agents, and ionizing radiation. As an
immediate early response gene that is rapidly induced by pleiotropic signals,
c jun may have important regulatory functions for cell cycle progression,
proliferation, and survival. See Ryder, K., Lau, L. F., and Nathans, D. "A
gene
activated by growth factors is related to the oncogene v-jun," Proc 1V'atl
Acad Sci
USA. 85: 1487-1491,1988; Schutte, J., Viallet, J., Nau, M., Segal; S.,
Fedorko,
J., and Minna, J. ' jun-B inhibits and c fos stimulates the transforming and
traps-activating activities of c-jun, Cell. 59: 987-997,1989; Neuberg, M.,
Adamkiewicz, J., Hunter, J. B., and Mueller, R. "A fos protein containing the
Jun leucine zipper forms a homodimer which binds to the AP-1 binding site,"
Nature. 341: 589-590,1989; Mitchell, P. J. and Tjian, R. "Transcriptional
regulation in mammalian cells by sequence-specific DNA binding proteins,"
Science. 245: 371-378,1989; Bohmann, D., Bos, T. J., Admon, T., Nishimura,
R., Vogt, P. K., and Tijian, R. "Human protooncogene c-jun encodes a DNA
binding protein with structural and functional properties of transcription
factor
AP-1," Science. 238: 138b-1392, 1988; Kharbanda, S. M., Sherman, M. L., and
Kufe, D. W. "Transcriptional regulation of c jun gene expression by
arabinofuranosylcytosine in human myeloid leukemia cells," J Clin invest. 86:
1517-1523,1990; Rosette, C. and Karin, M. "Ultraviolet light and osmotic
stress: activation of the JNK cascade through multiple growth factor and
cytokine receptors," Science. 274: 1194-7,1996; Rubin, E., Kharbanda, S.,
Gunji, H., and Kufe, D. "Activation of the c jun protooncogene in human
myleloid leukemia cells treated with etoposide;" Molecular Pharmacology. 39:
697-701,1991; Dosch, J. and Kaina, B. "Induction of c-fos, c-jun, junB and
junD mRNA and AP-1 by alkylating mutagens in cells deficient and proficient
CA 02337999 2000-12-28
WD 00100202 PCTIUS99I14923
z
for the DNA repair protein 06-methylguanine-DNA methyltransferase (MGMT)
and its relationship to cell death, mutation induction and chromosomal
instability," Oncogene. l3: 1927-35,1996; Chae, H. P., Jarvis, L. J., and
Uckun,
F. M. "Role of tyrosine phosphorylation in radiation-induced activation of c
jun
S protooncogene in human lymphohematopoietic precursor cells," Cancer Res. 53:
447-51,1993; and Karin, M., Liu, Z.-G., and Zandi, E. "AP-1 function and
regulation," CuYrent Opinion in Cell Biology. 9: 240-246,1997.
C jun encodes the nuclear DNA-binding protein, JLJN, that
contains a leucine-zipper region involved in homo- and heterodimerization: JUN
protein dimerizes with another JIJN protein or the product of c fos gene and
forms the activating protein-i (AP-1) transcription factor. JUN-JUN
homodimers and JCTN-FOS heterodimers preferentially bind to a specific
heptameric consensus sequence found in the promoter region of multiple growth
regulatory genes. Alterations of c;jun protooncogene expression can therefore
1S modulate the transcription of several growth-regulators affecting cell
proliferation and differentiation. See Ryder, K., Lau, L. F., and Nathans, D.
"A
gene activated by growth factors is related to the oncogene v-jun," Proc Natl
Acad Sci USA. 85: 1487-1491,1988; Neuberg, M., Adamkiewicz, J., Hunter, J.
B., and Mueller, R. "A fos protein containing the Jun ieucine zipper forms a
homodimer which binds to the AP-1 binding site," Nature. 341: 589-590,1989;
Karin, M., Liu, Z.-G., and Zandi, E. "AP-1 function and regulation," Current
Opinion in Cell Biology. 9: 240-246,1997; Angel, P., Allegretto, E. A., Okino,
S. T., Hattori, K., Boyle, W. J., Hunter, T., and Kaxin, M. "Oncogene jun
encodes a sequence-specific trans-activator similar to AP-1," Nature. 332:
2S 166-170,1988; and Musti, A. M., Treier, M., and Bohmann, D. "Reduced
ubiquitin-dependent degradation of c-Jun after phosphorylation by MAP
kinases," Science. 275: 400-402,1997.
C jun plays a pivotal role in Ras-induced transformation and has
also been implicated as a regulator of apoptosis when de novo protein
synthesis
is required. C; jun induction is required for ceramide-induced apoptosis and
stress-induced apoptosis after UV exposure or other forms of DNA damage.
This induction is thought to be triggered by activation of JUN-N-terminal
kinases (JNKs) (also known as stress-activated protein kinases) which leads to
CA 02337999 2000-12-28
WO 00/00202 PCTIUS99/14923
3
enhanced c; jun transcription by phosphorylation of JUN at sites that
increases its
ability to activate transcription. Ectopic expression of a dominant-negative
c;jun
mutant lacking the N terminus or a dominant-negative 3NK kinase abolishes
stress-induced apoptosis. See Karin, M., Liu, Z.-G., and Zandi, E. "AP-1
S function and regulation," Current Opinion in Cell Biology. 9: 240-246,1997;
Collotta, F., Polentarutti, N., and Mantovani, A. "Expression and involvement
of
c-fos and c-jun protooncogenes in programmed cell death induced by growth
factor deprivation in lymphoid cell lines," J. Biol. Chem. 267: 18278-18283,
1992; Harn, J., Babij, C., Whitfield, J., Pfarr, C. M., Lallemand, D., Yaniv,
M.,
and Rubin, L. L. "A c-Jun dominant negative mutant protects sympathetic
neurons against programmed cell death," Neuron. 14: 927-939,1995; Verheij,
M., Bose, R., Lin, X. H., Yao, B., Jarvis, W. D., Grant, S., Birrer, KM. J.,
Szabo,
E., Zon, L. L, Kyriakis, J. M., Haimovitz FA., Fuks, Z., and Kolesnick, R. N.
"Requirement for ceramide-initiated SAPKl3NK signalling in stress-induced
1S apoptosis," Nature. 380: 7S-9,1996; Hibi, M., Lin, A., Smeal, T., Minden,
A.,
and Karin, M. "Identification of an oncoprotein- and UV-responsive protein
kinase that binds and potentiates the c-Jun activation domain," Genes Dev. 7:
2135-48, 1993; Derijard, B., Hibi, M., Wu, I. H., Barrett, T., Su, B., Deng,
T.,
Karin, M., and Davis, R. J. "JNKI: a protein kinase stimulated by UV light
arid
Ha-Ras that binds and phosphorylates the c-Jun activation domain," Cell. 76:
1025-37,1994; and Chen, Y. R., Wang, X., Ternpleton, D., Davis, R. J., and
Tan, T. H. "The role of c-Jun N-terminal kinase (JNK) in apoptosis induced by
ultraviolet C and gamma radiation. Duration of JNK activation may determine
cell death and proliferation," JBiol Chem. 271: 31929-36;1996.
2S Protein tyrosine kinases (PTK) play important roles in the
initiation and maintenance of biochemical signal transduction cascades that
affect proliferation and survival of B-lineage lymphoid cells. Oxidative
stress
has been shown to activate BTK, SYK, and Src family PTK. It is known that
PTK activation precedes and mandates radiation-induced activation of cyun
protooncogene expression in human B-lineage lymphoid cells (Chae, H. P.,
3arvis, L. J., and Uckun, F. M. Cancer Res. 53: 447-S1, 1993). However, the
identity of the PTK responsible for radiation-induced c;jun activation is not
yet
known. See Uckun, F. M., Waddick, K. G., Mahajan, S., Jun, X., Takata, M.,
CA 02337999 2000-12-28
WO OOI00202 PCTI~1S99I14923
4
Bolen, J., and Kurosaki, T. "BTK as a mediator of radiation-induced apoptosis
in
DT-40 lymphoma B cells," Science. 273: 1096-100,1996; Kurosaki, T.
"Molecular mechanisms in B cell antigen receptor signaling," Curr Opin
Immunol. 9: 309-18,1997; Uckun F.M, Evans W.E, Forsyth C.J, Waddick K.G,
T-Ahlgren L., Chelstrom L.M, Burkhardt A., Bolen J., Myers D.E. "Biotherapy
of B-cell precursor leukemia by targeting genistein to CD19-associated
tyrosine
kinases." Science 267:886-891,1995; Myers D.E., Jun X., Waddick K.G.,
Forsyth C., Chelstrorn L.M., Gunther R.L., Tumer N.E, Bolen J., Uckun F.M.
"Membrane-associated CD19-LYN complex is an endogenous p53-independent
and bci-2-independent regulator of apoptosis in human B-lineage lymphoma
cells." Proc Nat'1 Acad Sci USA 92: 9575-9579,1995; Tuel Ahlgren, L., Jun,
X., Waddick, K. G., Jin, J., Bolen, J., and Uckun, F. M. "Role of tyrosine
phosphorylation in radiation-induced cell cycle-arrest of leukemic B-cell
precursors at the G2-M transition checkpoint," Leuk Lymphoma. 20: 417-26,
1996; Qin, S., Minami, Y., Hibi, M., Kurosaki, T., and Yarnamura, H.
"Syk-dependent and -independent signaling cascades in B cells elicited by
osmotic and oxidative stress," JBiol Chem. 272: 2098-103,1997; Saouaf, S. J.,
Mahajan, S., Rowley, R. B., Kut, S., Fargnoli, J., Burkhardt, A. L., Tsukada,
S.,
Witte, O. N., and Bolen, J. B. "Temporal differences in the activation of
three
classes of non-transmembrane protein tyrosine kinases following B cell antigen
receptor surface engagement," Proc Natl Acad Sci USA. 91: 9524-28,1994;
Law, D. A., Chan, V. F. W., Datta, S. K., and DeFranco, A. L. "B-cell antigen
receptor motifs have redundant signalling capabilities and bind the tyrosine
kinases PTK72,Lyn and Fyn," Curr Biol. 3: 645-57,1993; Hibbs, M: L.,
Tarlinton, D. M., Armes, J., Grail, D., Hodgson, G., Maglitto, R., Starker, S.
A.,
and Dunn, A. R. "Multiple defects in the immune system of Lyn-deficient mice,
culminating in autoimmune disease," Cell. 83: 301-3 i 1,1995; Aoki, Y.,
Isselbacker, K. J., and Pilaff, S. "Breton tyrosine kinase is tyrosine
phosphorylated and activated in pre-B lymphocytes and receptor-ligated B
cells,"
Proc Natl Acad Sci USA. 91: 10606-10609,1994; Jegloff, L. S. and Jongstra
Bilen, J. "Cross-linking of the IgM receptor induces rapid translocation of
IgM-associated Ig alpha, Lyn, and Syk tyrosine kinases to the membrane
skeleton, Jlmmunol. 159: 1096-106,1997; Thomis, D. S., Gurniak, C. B., Tivol,
CA 02337999 2000-12-28
WO 00/00202 PCT/US99/14923
E., Sharpe, A. H., and Berg, L. J." Defects in B lymphocyte maturation and T
lymphocyte activation in mice lacking Jak 3," Science. 270: 794-797,1995;
Nosaka, T., Van Deursen, J. M., Tripp, R.A., Thierfelder, W. E., Witthuhn, B.
A., McMickle, A. P., Doherty, P. c., Grosveld, G. C., and Ihle, J. N.
"Defective
lymphoid development in mice lacking Jak 3," Science. 270: 800-802,1995.
U.S. Patent Application Serial Number 09/087,479 (entitled
Quinazolines For Treating Brain Tumor; filed 28 May 1998) discloses
hydroxyquinazoline derivatives that exhibit potent cytotoxicity against human
glioblastoma cells (i.e. brain tumor cells): Because JAK-3 is not known to be
present in these glioblastoma cells, the cytotoxic activity of the
hydroxyquinazoline derivatives is not believed to result from inhibition of
JAK-
3 activity. Additionally, the cytotoxic activity of the hydraxyquinazoline
derivatives is not known to result from the inhibition of c-jun activation.
There is currently a need for therapeutic agents and methods that
are useful for preventing or reducing cell damage that results from exposure
to
radiation and chemical agents that cause DNA-damage. There is also a need for
chemical agents as well as in vitro and in vivo methods that can be used to
further investigate the biological pathways associated with DNA-damage that
results from exposure to radiation or chemical agents.
The invention provides a method comprising inhibiting c jun
expression in cells (e.g. rnammalian or avian) by contacting the cells (in
vitro or
in vivo) with a substance that inhibits the activity of 3anus family kinase 3
(JAK-3).
The invention also provides a therapeutic method for preventing
or treating a pathological condition in a mammal (e.g. a human) wherein c-jun
activation is implicated and inhibition of its expression is desired
comprising
administering to a mammal in need of such therapy, an effective amount of a
substance that inhibits the activity of JAK-3.
The invention also provides navel compounds of formula I as
well as processes and intermediates useful for their preparation.
CA 02337999 2000-12-28
WO 00/00202 PCT/US99/14923
6
The invention also provides substances that are effective to inhibit
JAK-3 for use in medical therapy (preferably for use in treating conditions
that
result from exposure to radiation or to chemical agents that cause DNA
damage),
as well as the use of substances that inhibit JAK-3 for the manufacture of a
S medicament for the treatment of a condition that is associated with exposure
to
radiation, or to chemical agents that cause DNA damage.
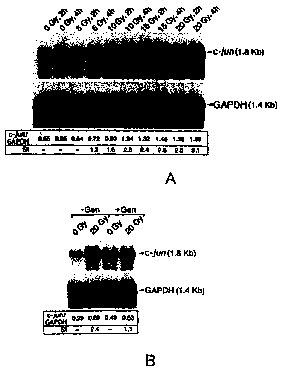
Figure 1. Radiation-induced c-jun activation in wild-type
DT-40 lymphoma B-cells. [A]. pac r ~~for in ~ .. .'on a~~~iu_n mRNA.
DT-40 chicken cells were irradiated at the indicated doses (0,10,15,20 Gy).
Total
RNA was extracted after a 2 hours or 4 hours post-irradiation time period. RNA
(20 mg) was loaded on a Northern gel and transferred by capillary blotting to
a
nylon membrane. The Northern blot was hybridized with a 32P labeled chicken
1S c jun probe (top panel) or a chicken GAPDH probe (bottom panel). The inset
shows the values for the c;junIGAPDH transcript expression ratios as
determined
with a Bio Rad Storage Phosphor Imager and corresponding SI values [B].
F~~g t~f'tho PTK inhibitor g~,rr rain on indt~ctian '~f ~ own m~A_. Cells were
treated with 30 mg/ml of genistein for 24 hours at 37°C prior to
exposure to 20
Gy ionizing radiation. c jun expression levels were determined as in [A].
Figure 2. Radiation-induced activation of c-jun in BTK'
DT-40 cells. Two representative experiments (shown in [A] and [B]) showing
induction of c ; jun mRNA expression by ionizing radiation in wild type (WT)
and BTK DT-40 cells. Poly (A)'" RNA was isolated from non-irradiated cells as
2S well as irradiated cells {20 Gy, with a 2 hours post-radiation recovery
period).
Northern blots of 2 mg of poly (A)+ were hybridized with c jun probe (top
panel), (-actin probe (middle panel in [A] only), and GAPDH probe (bottom
panel). The inset below each panel shows the relative expression of c jun
normalized for RNA load {c jun/GAPDH ratio) and SI (fold induction over
non-irradiated controls).
Figure 3. induction of c juh mRl~IA expression by ionizing
radiation in wild type and mutant DT-40 cell lines. DT-40, BTK' DT-40,
SYK DT-40 (shown in [A]), as well as LYN' DT-40 and LYN' SYK DT 40
CA 02337999 2000-12-28
WO 00/00202 PCT/US99114923
7
cells (shown in [B]) were irradiated with 20 Gy and poly (A)+RNA {in [A]) or
total RNA (in [B]) was harvested after a 2 hour recovery period. RNA from
non-irradiated cells was used as a control. Northern blots containing 2 mg of
poly (A)+ {in [A]) or 20 mg of total RNA (in [B]) from each cell line were
hybridized with both ~2 P labeled c jun probe(top panel) and GAPDH probe
(bottom panel). The insets below the panels show the relative expression of
c jun normalized for RNA loading (c;junl GAPDH ratios) as well as the SI (fold
induction over non-irradiated controls).
Figure 4. JAK 3 Inhibitors. [A]. Structures of JAK-3
inhibitors. [B] Specificity of JAK-3 inhibitors. Sf21 cells infected with
baculovirus expression vectors for JAK-1 JAK-2 or JAK-3 were subjected to
irnmunoprecipitation with anti-3AK antibodies. JAK-1 (shown in B.1), JAK-2
(shown in B.2) and JAK-3 (shown in B.3 and B.4 which illustrate results from 2
independent experiments) immune complexes were treated with I% DMSO
I S (vehicle control = CON), Compound 1, or Compound 2 for 1 hour prior to hot
kinase assays, as described (20,22). Both compounds inhibited JAK-3 when used
at 10 ~,g/ml whereas they did not inhibit JAK-1 or JAK-2 even at 75 wg/ml [C].
EMSAs of 32Dc22-IL-2R(3 cells. Compound 1(100 {g/ml) and Compound 2
(100 (g/ml) inhibited IL-2 triggered JAK-3-dependent STAT activation but not
IL-3-triggered 3AK-1/JAK-2-dependent STAT activation in 32Dc1 I-IL-2R~3
cells.
Figure 5. Effects of a JAK 3 inhibitor on cyun induction in
irradiated DT-40 cells. Cells were treated with the quinazoline derivative
4-(3'-Bromo-4'-hydroxyl-phenyl)-amino-6,7-dimethoxyquinazoline (100
mg/ml) for 24 hours at 37 °C prior to exposure to 20 Gy ionizing
radiation.
c jun expression levels were determined as outlined in Figures 1-3.
As used herein, the term "inhibit" means to reduce by a
measurable amount, or prevent entirely; and the phrase "inhibit c jun
activation"
includes the inhibition of RNA production and the inhibition of the production
of
the protein encoded by the RNA.
CA 02337999 2000-12-28
WO 00!00202 PCT/IJS99/14923
8
Applicants examined the potential involvement of BTK, SYK and
LYN in radiation-induced c jun activation, using DT-40 chicken lymphoma
B-cell clones rendered deficient for these specific PTK by targeted gene
disruption. It was found that BTK plays no role in radiation-induced c-jun
activation. Similarly, neither LYN nor SYK are required for activation of c
jun
after radiation exposure. However, their participation may influence the
magnitude of the c;jun response. .It was unexpectedly discovered, however,
that
an inhibitor of Janus family kinase 3 (JAK-3) abrogated radiation-induced c
jun
activation.
C-jun expression can be activated by exposure to chemical agents
that damage DNA such as ara-C, a topoisomerase II inhibitors, or alkylating
agents. C jun activation can also result from exposure to ultraviolet
radiation or
ionizing radiation. According to the invention, inhibitors of JAK-3 can be
used
to inhibit c-jun expression resulting from exposure to radiation or exposure
to
chemical agents.
The methods of the invention can be carried out in vitro. Such in
vitro methods are also useful for studying the biological processes associated
with cell response to DNA damaging agents. The methods of the invention can
also be carried out in vivo. Such methods can also be used to study the
biological processes associated with cell response to DNA damaging agents, as
well as for treating pathological conditions in mammals (e.g. humans) that
result
from exposure to DNA-damaging agents.
Pathological conditions that result from exposure to DNA-
damaging agents include conditions that result from oxidative stress, such as
tissue or organ (e.g. heart, liver, or kidney) damage, inflammation; and hair
loss;
as well as the negative effects that are produced by oxygen free radicals
during
chemotherapy. Oxidative stress may result from exposure to external agents, or
may result from internal processes. Thus, JAK-3 inhibitors are also useful for
treating conditions resulting from the action of internally generated oxygen
free
radicals, such as aging and amyelotrophic lateral sclerosis (ALS).
According to the invention, the JAK-3 inhibitors may be
administered prophylactically, i.e. prior to exposure to the DNA-damaging
CA 02337999 2000-12-28
WO 00/00202 PCT/ITS99/14923
9
agent, or the JAK-3 inhibitors may be administered after exposure to the DNA
damaging agent.
The 3AK-3 inhibitors useful in the methods of the invention
include all compounds capable of inhibiting the activity of JAK-3, it being
well
lrnown in the art how to measure a compounds ability to inhibit JAK-3, for
example, using standard tests similar to the test described hereinbelow in
Example 2 under the heading "Effects of a JAK-3 inhibitor on radiation-induced
c jun activation in DT40_cells."
JAK-3 inhibitors that are useful in the methods of the invention
include compounds of formula I:
(I)
wherein
X is HN, R"N, S, O, CHZ, or R~ 1CH;
Rl t is hydrogen, (C,-C4)alkyl; or (C,-C4)alkanoyl;
Rl-R8 are each independently hydrogen, hydroxy, mercapto, amino, vitro,
(C,-C4)alkyl, (C,-C4)alkoxy, (C,-C4)alkylthio, or halo; wherein two adjacent
groups of R,-RS together with the phenyl ring to which they are attached may
optionally form a fused ring, for example forming a naphthyi or a
tetrahydronaphthyl ring; and further wherein the ring formed by the two
adjacent
groups of Rl-RS may optionally be substituted by 1, 2, 3, or 4 hydroxy,
mercapto,
amino, vitro, (C,-C4)alkyl, (Cf-C4)alkoxy, (Ci-C4)alkyithio, or halo; and
Rg and Rlo are each independently hydrogen, (C,-CQ)alkyl, (C,-C4)alkoxy,
halo, or (C,-C4)alkanoyl; or R9 and R,o together are methylenediaxy; or a
pharmaceutically acceptable salt thereof
CA 02337999 2000-12-28
WO 00100202 PCT/US99/14923
The following definitions are used, unless otherwise described:
halo is fluoro, chloro, bromo, or iodo. Alkyl, alkanoyl, etc. denote both
straight.
and branched groups; but reference to an individual radical such as "propyl"
embraces only the straight chain radical, a branched chain isomer such as
5 "isopropyl" being specifically referred to. (Cr-C4)Alkyl includes methyl,
ethyl,
propyl, isopropyl, butyl, iso-butyl, and sec-butyl; {Cj-C4)alkoxy includes
methoxy, ethoxy, propoxy, isopropoxy, butoxy, iso-butoxy, and sec-butoxy; and
(C,-C4)alkanoyl includes acetyl, propanoyl and butanoyl.
A specific group of compounds are compounds of formula I
10 wherein Rl-R5 are each independently hydrogen, rnercapto, amino, vitro, (C,-
C4)alkyl, (C,-C4)alkoxy, (C,-C4)alkylthio, or halogen.
Another specific group of compounds are compounds of formula
I wherein R9 and Rlo are each independently hydrogen, (C,-C4)alkyl, halo, or
{C~-C4)alkanoyi; or R9 and R,a together are methylenedioxy; or a
pharmaceutically acceptable salt thereof.
JAK-3 inhibitors that are useful in the methods of the invention
also include compounds of formula I as described in U.S. Patent Application
Serial Number 09/087,479 (entitled Quinazolines For Treating Brain Tumor;
filed 28 May 1998).
Preferred JAK-3 inhibitors include 4-(4'-hydroxylphenyl)-amino-
6,7-dimethoxyquinazoline and 4-{3'-bromo-4'-hydroxylphenyl)-
amino-6,7-dimethoxyquinazoline; or a pharmaceutically acceptable salt thereof.
Substances that inhibit JAK-3 ("the Substances)") can be
formulated as pharmaceutical compositions and administered to a mammalian
host, such as a human patient in a variety of forms adapted to the chosen
route of
administration, i.e., orally or parenterally, by intravenous, intramuscular,
topical
or subcutaneous routes.
Thus, the Substances may be systemically administered; e.g.,
orally, in combination with a pharmaceutically acceptable vehicle such as an
inert diluent or an assimilable edible earner. They may be enclosed in hard or
soft shell gelatin capsules, may be compressed into tablets, or may be
incorporated directly with the food of the patient's diet. Fox oral
therapeutic
administration, the Substance may be combined with one or more excipients and
CA 02337999 2000-12-28
WO 00/00202 PCTIUS99114923
11
used in the form of ingestible tablets, huccal tablets, troches, capsules,
elixirs,
suspensions, syrups, wafers, and the like. Such compositions and preparations
should contain at least 0.1% of the Substance. The percentage of the
compositions and preparations may, of course, be varied and may conveniently
be between about 2 to about 60% of the weight of a given unit dosage form. The
amount of Substance in such therapeutically useful compositions is such that
an
effective dosage level will be obtained.
The tablets, roches, pills, capsules, and the like may also contain
the following: binders such as gum tragacanth, acacia, corn starch or gelatin;
excipients such as dicalcium phosphate; a disintegrating agent such as corn
starch, potato starch, alginic acid and the like; a lubricant such as
magnesium
stearate; and a sweetening agent such as sucrose, fructose, lactose or
aspartame
or a flavoring agent such as peppermint, oil of wintergreen, or cherry
flavoring
may be added. When the unit dosage form is a capsule, it may contain, in
addition to materials of the above type, a liquid carrier, such as a vegetable
oil or
a polyethylene glycol. Various other materials may be present as coatings or
to
otherwise modify the physical form of the solid unit dosage form. For
instance,
tablets, pills, or capsules may be coated with gelatin, wax, shellac or sugar
and
the like. A syrup or elixir may contain the active compound, sucrose or
fructose
as a sweetening agent, methyl and propylparabens as preservatives, a dye and
flavoring such as cherry or orange flavor: Of course, any material used in
preparing any unit dosage form should be pharmaceutically acceptable and
substantially non-toxic in the amounts employed. In addition, the Substance
may be incorporated into sustained-release preparations and devices.
The Substances may also be administered intravenously or
intraperitoneally by infusion or injection. Solutions of the Substance can be
prepared in water, optionally mixed with a nontoxic surfactant. Dispersions
can
also be prepared in glycerol, liquid polyethylene glycols, triacetin, and
mixtures
thereof and in oils. Under ordinary conditions of storage and use, these
preparations contain a preservative to prevent the growth of microorganisms.
The pharmaceutical dosage forms suitable for injection or
infusion can include sterile aqueous solutions or dispersions or sterile
powders
comprising the Substance which are adapted for the extemporaneous preparation
CA 02337999 2000-12-28
WO 00/00202 PCT/US99/14923
12
of sterile injectable or infusible solutions or dispersions, optionally
encapsulated
in liposomes. In all cases, the ultimate dosage form must be sterile, fluid
and
stable under the conditions of manufacture and storage. The liquid carrier or
vehicle can be a solvent or liquid dispersion medium comprising, for example,
S water, ethanol, a polyol (for example, glycerol, propylene glycol, liquid
polyethylene glycols, and the like), vegetable oils, nontoxic giyceryl esters,
and
suitable mixtures thereof. The proper fluidity can be maintained, for example,
by the formation of liposomes, by the maintenance of the required particle
size in
the case of dispersions or by the use of surfactants. The prevention of the
action
of microorganisms can be brought about by various antibacterial and antifungal
agents, for example, parabens, chlorobutanol, phenol, sorbic acid, thimerosal,
and the like. In many cases, it will be preferable to include isotonic agents,
for
example, sugars, buffers or sodium chloride. Prolonged absozption of the
injectabie compositions can be brought about by the use in the compositions of
agents delaying absorption, for example, aluminum monostearate and gelatin.
Sterile injectable solutions are prepared by incorporating the
Substance in the required amount in the appropriate solvent with various of
the
other ingredients enumerated above, as required, followed by filter
sterilization.
In the case of sterile powders for the preparation of sterile injectable
solutions,
the preferred methods of preparation are vacuum drying and the freeze drying
techniques, which yield a powder of he active ingredient plus any additional
desired ingredient present in the previously sterile-f ltered solutions.
For topical administration, the Substances may be applied in pure
form, i.e., when they are liquids. However, it will generally be desirable to
administer them to the skin as compositions or formulations, in combination
with a dermatologically acceptable carrier, which may be a solid or a liquid.
Useful solid carriers include finely divided solids such as talc,
clay, mierocrystalline cellulose, silica, alumina and the like. Useful liquid
carriers include water, alcohols or glycols ar water-aicohol/glycol blends, in
which the Substances can be dissolved or dispersed at effective levels,
optionally
with the aid of non-toxic surfactants. Adjuvants such as fragrances and
additional antirnicrobial agents can be added to optimize the properties for a
given use. The resultant liquid compositions can be applied from absorbent
CA 02337999 2000-12-28
WO 00/00202 PCT/US99/14923
13
pads, used to impregnate bandages and other dressings, or sprayed onto the
affected area using pump-type or aerosol sprayers.
Thickeners such as synthetic polymers, fatty acids, fatty acid salts
and esters; fatty alcohols, modified celluioses or m~dified mineral materials
can
also be employed with liquid carriers to form spreadable pastes, gels,
ointments,
soaps, and the like, for application directly to the skin of the user.
Examples of useful dermatological compositions which can be
used to deliver the Substances to the skin are known to the art; for example,
see
Jacquet et al. (U.S. Pat. No. 4,608,392), Geria (IJ.S. Pat. No. 4,992,478),
Smith
et al. {U.S. Pat. No. 4,559,157) and Wortzman (U.S. Pat. No. 4,$20,508}.
Useful dosages of the compounds of formula I can be determined
by comparing their in vitro activity, and in vivo activity in animal models.
Methods for the extrapolation of effective dosages in mice, and other animals,
to
humans are known to the art; for example, see U.S. Pat. No. 4,938,949.
Generally, the concentration of the Substance in a liquid
composition, such as a lotion, will be from about 0.1-25 wt-%, preferably from
about 0.5-10 wt-%. The concentration in a semi-solid or solid composition such
as a gel or a powder will be about 0.1-5 wt-%, preferably about 0.5-2.5 wt-%.
The amount of the Substance required for use in treatment will
vary not only with the particular salt selected but also with the route of
administration, the nature of the condition being treated and the age and
condition of the patient and will be ultimately at the discretion of the
attendant
physician or clinician.
In general, however, a suitable dose will be in the range of from
about fl.5 to about 100 mg/kg, e.g., from about 10 to about 75 mglkg of body
weight per day, such as 3 to about 50 mg per kilogram body weight of the
recipient per day, preferably in the range of 6 to 90 mglkg/day, most
preferably
in the range of 15 to 60 mg/kg/day.
The Substance is conveniently administered in unit dosage form;
for example, containing 5 to 1000 mg, conveniently 10 to 750 mg, most
conveniently, 50 to 500 mg of active ingredient per unit dosage form.
Ideally, the Substance should be administered to achieve peak
plasma concentrations of from about 0.5 to about 75 ~M, preferably, about 1 to
CA 02337999 2000-12-28
WO 00100202 PCT/US99114923
14
50 wM, most preferably, about 2 to about 30 uM. This may be achieved, for
example, by the intravenous injection of a 0.05 to 5~/o solution of the
Substance,
optionally in saline, or orally administered as a bolus containing about l-100
rng
of the Substance. Desirable blood levels may be maintained by continuous
infusion to provide about 0.01-5.0 mg/kglhr or by intermittent infusions
containing about 0.4-15 mg/kg of the Substance.
The Substance may conveniently be presented in a single dose or
as divided doses administered at appropriate intervals, for example, as two,
three,
four or more sub-doses per day. The sub-dose itself may be further divided,
e.g.,
into a number of discrete loosely spaced administrations; such as multiple
inhalations from an insufflator or by application of a plurality of drops into
the
eye.
The invention will now be illustrated by the following non-
limiting Examples.
CA 02337999 2000-12-28
WO 00/00202 ' PCT/US99/14923
F.~sa~l~l. Chemical synthesis and Characterization of JAK-3 Inhibitors
5
Melting points are uncorrected. 'H NMR spectra were recorded
using a Varian Mercury 30fl spectrometer in DMSO-d6 or CDC13. Chemical
shifts are reported in parts per million (ppm) with tetramethylsilane (TMS) as
an
internal standard at zero ppm. Coupling constant (~ are given in hertz and the
10 abbreviations s, d, t, q, and m refer to singlet, doublet, triplet, quartet
and
multiplet, respectively. Infrared spectra were recorded on a Nicolet PROTEGE
460-IR spectrometer. Mass spectroscopy data were recorded on a FINNIGAN
MAT 95, VG 7070E-HF G.C. system with an HP 5973 Mass Selection Detector.
UV spectra were recorded on BECk:MAN DU 7400 and using MeOH as the
15 solvent. TLC was performed on a precoated silica gel plate (Silica Gel KGF;
Whitman Inc). Silica gel (200-400 mesh, Whitman Inc.) was used for all column
chromatography separations: All chemicals were reagent grade and were
purchased from Aldrich Chemical Company (Milwaukee, Wis) or Sigma
Chemical Company (St. Louis, MO).
The common synthetic precursor
4-chloro-6,7-dimethoxyquinazoline (7), used for preparing compounds (1) and
(2), was prepared using literature procedures as illustrated in Scheme 1.
CA 02337999 2000-12-28
WO 00100202 PCT/US99/14923
16
O O
CH3O ~ ~ OH SOC CH3O ~ ~ ~~2 CuSO4
CH30 \ N02 OOH CH30 \ N02 NaBH4
(3) {~)
O O
CH30 ~ ~ ~2 HCOZH CH30 ~ ~ NH POCK
CH30 \ NH ~ CH30 \ NJ
2
(5) (6)
C1
CH30 ~ ~N
CH30 \ ~ NJ
(~)
Scheme 1
4,5-Dimethoxy-2-nitrobenzoic acid (3) was treated with thionyl
chloride and then reacted with ammonia to give
4,5-dimethoxy-2-nitrobenzamide (4) as described by F. Nomoto et al. Chem.
Pharm. Bull. 1990, 38, 1591-1595. The vitro group in compound {4) was
reduced with sodium borohydride in the presence of copper sulfate (see C.L.
Thomas Catalytic Processes and Proven Catalysts Academic Press, New York
(1970)) to give 4,5-dimethoxy-2-arninobenzamide (5) which was cyciized by
refluxing with formic acid to give 6,7-dimethoxyquinazoline-4(3H)-one (6).
Compound (6) was refluxed with phosphorus oxytrichloride to provide the
common synthetic precursor {'~.
Compounds 1 and 2 (Figure 4) were prepared from the common
synthetic precursor (7) and the requsite aniline as follows.
CA 02337999 2000-12-28
WO 00/00202 PCT/US99/14923
17
4-(4'-Hydroarylphenyl)-amino-b,7-dimethoxyquinazoline (1). A
mixture of 448 mg (2 mmol) of 4-chloro-6,7-dimethoxy-quinazoline (7) and 2.5
mmol of 4-hydroxyaniline in 20 ml of alcohol {EtOH or MeOH) was refluxed
for 8 hours. After cooling triethylamine was added to basify the solution, and
the solvent was concentrated to give material that was recrystallized from DMF
to give compound (1); 84.29%; m.p. 245.0- 248.0 °C; 'H NMR {DMSO-d6): &
11.21(s, 1H, -NFi), 9.70(s, IH, -OH), 8.74(s, IH, 2-H), 8.22(s, 1H, 5-H),
7.40(d,
2H, J= 8.9 Hz, 2',6'-H), 7.29(s, 1H, 8-H), 6.85{d, 2H, J= 8.9 Hz, 3',5'-H),
3.98(s, 3H, -OCH3), 3.97(s, 3H, -OCH3). LJV(MeOH) ~,",~(e): 203.0, 222.0 ,
251.0, 320.0 nm. IR(KBr)um~: 3428, 2836, 1635, I516, 1443, 1234 crri'.
GC/MS m/z 298 (M++1, 100.00), 297(M~, 26.56), 296( M+-1, 12.46).
4-(3 '-Bromo-4 '-hydroxylphenyl)-amino-6, 7-dimethoxy-
quinazoline (2). A mixture of 448 mg (2 mmol) of 4-chloro-6,7-dimethoxy-
quinazoline (7) and 2.5 mmol of 3-bromo-4-hydroxyaniline in 20 mI of alcohol
(EtOH or MeOH) was refluxed for 8 hours. After cooling, triethylamine was
added to basify the solution, and the solvent was concentrated to give
material
that was recrystallized from DMF to give compound {Z); 89.90%; m.p.
233.0-233.5 °C; 'H NMR(DMSO-d6): 8 10.08{s, 1H, -NH), 9.38(s, 1H ; -
OH),
8.40{s, 1H, 2-H }, 7.89(d, 1H, JZ.,S.= 2.7 Hz, 2'-H}, 7.75(s, 1H, 5-H),
7.55(dd,
1 H, J 5.,6. = 9.0 Hz, J Z.,6. = 2.7 Hz, , 6'-H), 7. I4(s, 1 H, 8-H), 6.97{d,
1 H, J 5..6- _
9.0 Hz, 5'-H), 3.92(s, 3H, -OCHER, 3.90(s, 3H, -OCH3). UV(MeOH)~,m~(e ):
203.0, 222.0 , 250.0, 335.0 nm. IR(KBr)u",~: 343i(br}, 2841, 1624, 1498, 1423,
1244 czri'. GC/MS mlz 378( M++2, 90.68), 377{M'~ +1,37.49), 376(M+,100.00),
360(M~3.63), 298(18.86), 282 (6.65).
CA 02337999 2000-12-28
WO 00/00202 PCT/US99/14923
18
Exanngle2.. Biological Screening
MATERIALS AND METHODS
Cell Lines. The establishment and characterization of
BTK-deficient, SYK-deficient, and LYN-deficient clones and reconstituted
SYK-deficient cell Iines of DT-40 chicken lyrnphoxna B-cells were previously
reported. The culture medium was RPMI 1640 (Life Technologies;
Gaithersburg, MD), supplemented with 1% chicken serum (Sigma; St. Louis,
MO), 5% fetal bovine serum (Hyclone, Logan,UT) and 1%
penicillin-streptomycin (Life Technologies). See Uckun, F. M., Waddick, K. G.,
Mahajan, 5., Jun, X., Takata, M., Bolen, J., and Kurosaki, T. Science. 273:
1096-100,1996; Kurosaki, T. Curr Opin Immunol. 9: 309-18,1997; Kurosaki;
T.; Johnson, S. A., Pao, L., Sada, K., Yamamura, H., and Cambier, J. C. J.
Bxp.
Med. 182: 1815-1823,1995; and Dibirdik L, Kristupaitis D., Kurosaki T.,
Tuel-Ahlgren L., Chu A., Pond D., Tuong D., Luben R., Uckun F.M. J. Biol.
Chem. 273(7), pp:4035-4039;199$.
Use of PTK Inhibitors. Cells (2 x 106/ml) were treated for 24
hours at 37 °C with either (1) the PTK inhibitory isoflavone genistein
(Calbiochem, La Jolla, CA) at 111 mM (30 mglml) concentration or (2) the
Janus family kinase, 3 (JAK-3)-specific PTK inhibitor
4-(3'-bromo-4'-hydroxyphenyl)-amino-6,7-dimethoxyquinazoline,
C,6,H,4Br(N3O3), kindly provided by Dr. Xing-Ping Liu, Alexander and Parker
Pharmaceutical Inc., Roseville, MN) at 270 mM (100 mg/ml) prior to radiation
in order to assess the effects of these agents on radiation-induced c ; jun
activation.
Irradiation of cells. Cells (2 x 10~ cells/ml) in plastic tissue
culture flasks were irradiated with 10-20 Gy at a dose rate of 4 Gy/min during
Iog phase growth and under aerobic conditions using a'3'Cs irradiator (J.L.
Shephard, Glendale, CA, as previously described by Tuel Ahlgren, L., Jun, X.,
Waddick, K. G., Jin, J., Bolen, J., and Uckun, F. M. "Role of tyrosine
phosphorylation in radiation-induced cell cycle-arrest of Ieukemic B-cell
precursors at the G2-M transition checkpoint," Leuk Lymphoma. 20: 417-26,
1996; and Uckun, F.M., Jaszcz, W., Chandan Langlie, M., Waddick, K.G., Gajl
CA 02337999 2000-12-28
WO 00/00202 PCT/US99/14923
19
Peczalska, K. and Song, C.W. "Intrinsic radiation resistance of primary
clonogenic blasts from children with newly diagnosed B-cell precursor acute
lymphoblastic leukemia," J Clin Inves. 91:1044-1051,1993. In some
experiments, cells were preincubated with PTK inhibitors for 24 hours prior to
irradiation.
c jun probe. A 506 basepair (bp) c jun probe was obtained by
polymerase chain reaction (PCR) amplification of chicken genomic DNA.
Primer sequences were determined based upon the sequence of chicken c; jun
(GenBank accession code CHKJUN). Two primers: 5'-ACTCTGCACC
CAACTACAACGC-3' (SEQ. lD NO: 1) and 5'-CTTCTACCGT
CAGCTTTACGCG-3' (SEQ ID NO: 2) were used for amplification.
Amplification was performed with a mix of Taq poiymerase and a proof reading
polymerise (eLONGase:?'aq polymerise plus Pyrococcus species GB-D
polymerise, Gibco BRL, Grand Island, N~ on an thermocycler, Ericomp Delta
II cycles, using a hot start. PCR products were subsequently cloned into the
cloning vector, PCR 2.1 (Invitrogen, San Diego, CA). An insert of the proper
size (506 basepair) was identified as chicken c;jun by sequence analysis using
PRISM dye terminator cycle sequencing (AmpliTaq~ DNA Polyrnerase, FS) and
analyzed on an automated sequences, ALF express sequences (Pharmacia
Biotech, Piscataway, N~. A 538 base pair chicken glyceraldehyde 3-phosphate
dehydrogenase (GAPDH) probe was generated by reverse transcription and
subsequent PCR amplification (RT-PCR) frarn chicken RNA with the following
primers: 5'-AGAGGTGCTGCCCAGAACATCATC-3' {SEQ ID NO: 3) and
5'-GTGGGGAGACAGAAGGGAACAGA-3' (SEQ ID NO: 4). A 413 by
chicken B-actin probe was generated by RT-PCR amplification from chicken
RNA with the following primers: 5'-GCCCTCTTCCAGCATCTTTCTT-3'
(SEQ ID NO: 5) and 5'-TTTATGCGCATTTATGGGTT-3' {SEQ ID NO: 6}.
The amplified cDNAs were cloned into PCR 2.1.
RNA isolation and Northern blot hybridization analysis.
Total RNA was extracted from approximately 2.5 x 107 cells with Trizol
Reagent, a monophasic solution of phenol and guanidine isothiocyanate as
described by Chorncznski, P. and Sacchi, N. "Single-step method of RNA
isolation by guanidinium-thiocyanate-phenol-chloroform extraction," Anal.
CA 02337999 2000-12-28
WO 00/00202 PCT/(JS99/14923
Biochem. 162: 156-159,1987. Poly (A)+ RNA was isolated directly from 1-3 x
10g cells with an Invitrogen Fast Trak 2.0 mRNA isolation kit. In brief, cells
were lysed in a sodium dodecyl sulfate (SDS) Iysis buffer containing a
proprietary mixture of pmteases. The Iysate was directly incubated with oligo-
dT
5 for absorption and subsequent elution of poly (A)+ RNA.
Two micrograms of poly (A)+ or 20 micrograms of total RNA
were denatured in formaldehyde/formamide loading dye at 65° prior to
loading
onto a 1% agarose-formaldehyde denaturing gel: Transcript sizes were
determined relative to RNA markers of 0.5-9 kb. The gels were stained with
10 Radiant Red in H20 to check loading and integrity of RNA prior to transfer.
The
RNA was subsequently transferred to positively charged nylon membrane with
20X standard sodium citrate(SSC) transfer buffer (1XSSC= 0.15 M sodium
chloride-0.01 S M sodium citrate) by downward capillary transfer. The c jun
fragment was radiolabeled by random priming with [( 32P]-dCTP {3000 Ci/mM)
15 [Amersham, Arlington Heights, IL] (40). Northern blots were hybridized
overnight at 42 °C in prehybridization/hybridization solution (50%
formamide
with proprietary blocking and background reduction reagents; Ambion, Austin,
TX) for 16-24 hours and unbound probe was removed by washing to a final
stringency of 0.1% SDS, O.1XSSC {65 °C). The blots were analyzed both
by
20 autoradiography and using the BioRad Storage Phosphor Imager System
(BioRad, Hercules, CA) for quantitative scanning. The blots were subsequently
stripped in boiling 0. i % SDS, and then rehybridized with a chicken GAPDH
andlor chicken ((3-actin probe to normalize for loading differences.
RESULTS AND DISCUSSION
Exposure of DT-40 chicken lymphoma B-cells to ionizing
radiation activates the c jun protooncogene. Exposure of human lymphoma
B-cells to 10-20 Gy-rays results in enhanced c jun expression with a maximum
response at 1-2 hours (Chae, H. P., Jarvis, L. J., and Uckun, F. M. Cancer
Res.
53: 447-51,1993). It has also been reported that ionizing radiation triggers
in
DT-40 chicken lymphoma B-cells biochemical and biological signals similar to
those in human lymphoma B-cells (Uckun, F. M., Waddick, K. G., Mahajan, S.,
Jun, X., Takata, M., Bolen, J:, and Kurosaki, T. Science. 273: 1096-100,1996).
CA 02337999 2000-12-28
WO 00/00202 PCT/US99/14923
21
In order to determine if DT-40 chicken lymphoma B-cells show a similar c ; jun
response to ionizing radiation, DT-40 cells were irradiated with 5,10,15 or 20
Gy and examined total RNA harvested from cells 2 or 4 hours after radiation
exposure for expression levels of 1.8 kb chicken c jun transcripts by
quantitative
Northern blot analysis. As shown in Figure lA, radiation exposure increased
the
level of c;jun transcripts in a dose-and time-dependent manner without
significantly affecting the GAPDH transcript levels with a maximum stimulation
index (Sn has determined by comparison of the c-jun/GAPDH ratios in
non-irradiated versus irradiated cells] of 3.1, 4 hours after 20 Gy. In seven
additional independent experiments, the stimulation index for 20 Gy ionizing
radiation at 2 hours after radiation exposure ranged from 2.4 to 3.8 {mean (
SE =
2.9 ~ 0.4).
The role of PTK in radiation-induced activation of c jun
expression in chicken lymphoma B cells was examined next, since PTK
inhibitors were shown to prevent radiation-induced c jun activation in human
lymphoma B-cells. As shown in Figure 1B, ionizing radiation did not
significantly enhance c;jun expression levels in DT-40 cells treated with the
PTK-inhibitory isoflavone, genistein (stimulation index=1. I ) indicating that
activation of a PTK is required for radiation-induced c jun expression in
chicken
lymphoma B cells as well. These findings established DT-40 chicken lymphoma
B-cells as a suitable model to further elucidate the molecular mechanism of
radiation-induced c jun activation.
Cytoptasmic protein tyrosine ltinases BTK, LYN, and SYK
are not required for radiation induced c jun activation. BTK is abundantly
expressed in lymphoma B-cells and its activation has been shown to be required
for radiation-induced apoptosis of DT-40 cells (Uckun, F. M., Waddick, K. G:,
Mahajan, S., Jun, X., Takata, M., Bolen, J., and Kurosaki, T. Science. 273:
1096-100,1996). DT-40 cells rendered BTK-deficient by targeted disruption of
the BTK genes do not undergo apoptosis after radiation exposure. Therefore, we
set out to determine if BTK could be the PTK responsible for radiation-induced
c jun activation as well, by comparing the levels of c jun induction in
BTK-deficient (BTK~ versus wild-type DT-40 cells. Contrary to our
expectations, 20 Gy ionizing radiation did not fail to induce c jun expression
in
CA 02337999 2000-12-28
WO 00/00202 PCT/US99114923
22
BTK-deficient DT-40 cells in any of the three independent experiments
performed. The stimulation indices ranged from 1.6 to 3.9 (mean t SE = 2.4 t
0.5) (Figure 2). Thus, ionizing radiation-induced increases in c jun
transcript
levels do not depend upon the presence of BTK.
Since SYK is also abundantly expressed in DT-40 cells and is
rapidly activated after ionizing radiation, we next examined if SYK might be
the
PTK responsible for radiation-induced increases in c;jun transcript levels. As
shown in Figure 3A, 20 Gy ionizing radiation enhanced c jun expression in
SYK' DT-40 cells rendered SYK-deficient by targeted gene disruption even
though the stimulation indices observed in five independent experiments were
lower than from those in wild-type cells (1.9 ~ 0.2, vs 2.9 ~ 0.4, p <0.01).
Thus,
SYK is not required for radiation-induced c;jun activation in DT-40 cells but
it
may participate in generation of an optimal signal.
DT-40 cells express high levels of LYN but do not express other
1 S members of the Src PTK family, including BLK, HCK, SRC, FYN, or YES at
detectable levels (see Uckun, F. M., Waddick, K. G., Mahajan, S., Jun, X.,
Takata, M., Bolen, J., and Kurosaki, T. Science. 273: 1096-100,1996; Kurosaki,
T., Johnson, S. A., Pao, L., Sada, K., Yamamura, H., and Cambier, J. C. "Role
of
the Syk autophosphorylation site and SH2 domains in B cell antigen receptor
signaling," J. Exp. Med.182: 1815-1823,1995; and Takata, M., Homma, Y., and
Kurosaki, T. "Requirement of phospholipase C-y2 activation in surface
immunoglobulin M-induced B cell apoptosis.," JExp Med. 182: 907-914,1995.
Since it has previously been demonstrated that SRC family PTK are essential
for
W-stimulated increases in c jun expression, we postulated that the predominant
SRC-family member, LYN, might mediate radiation-induced c;jun expression
in DT-40 cells. To test this hypothesis, we examined the ability of ionizing
radiation to activate c-jun expression in DT-40 cells rendered LYN-deficient
by
targeted gene disruption. LYN-deficient (LYNN cells showed enhanced c jun
expression after irradiation, however the stimulation indices were lower than
those in wild-type DT-40 (Figure 3B). Since LYN and SYK have been shown to
cooperate in the generation of other signals in B-cells (see Kurosaki, T.
"Molecular mechanisms in B cell antigen receptor signaling," Curr Opin
Immunol. 9: 309-18,1997), the ability of ionizing radiation to induce c;jun
CA 02337999 2000-12-28
WO 00/00202 PCTlUS99/14923
23
expression in LYN SYK DT-40 cells, generated by targeted disruption of the
syk gene in LYN' deficient DT-40 cells was examined. As shown in Figure 3B,
LYN' SYK DT-40 cells showed elevated c jun transcript levels after
irradiation,
indicating that the c jun response does not depend an either of these PTK,
either
alone or in cooperation. Similar to SYK, LYN is not required for
radiation-induced c jun activation in DT-40 cells but it may participate in
generation of an optimal response.
Interestingly, in four independent experiments, we observed
higher baseline expression levels of c;jun in SYK DT-40 cells than in wild-
type
DT-40 cells (Range: I .4 - 2.3-fold, mean ~ SE = 1.6 t 0.2-fold), suggesting
that
Syk may be involved in regulation of baseline c jun levels. To further explore
this possibility, we compared c jun levels in SYK cells to those of SYK cells
reconstituted with wild-type or kinase domain mutant (K~ syk gene. We
observed that reconstitution with wild-type syk reduced the higher baseline
expression levels of c jun in SYK cells, whereas reconstitution with a K syk
failed to reduce c jun levels {data not shown). These results implicate SYK as
a
negative regulator of c jun expression. This novel function of SYK seems to
depend on its kinase domain.
Effects of a JAK 3 inhibitor on radiation-induced c jun
activation in DT40-cells. B-cell signal transduction events direct fundamental
decisions regarding cell survival during periods of oxidative stress. A better
understanding of the dynamic interplay between B-cell signaling pathways is
needed to determine how vital decisions are dictated during intracellular
oxidation changes: STAT proteins (signal transducers and activators of
transcription) are a family of DNA binding proteins that were identified
during a
search for interferon (IFrT) a- or g-stimulated gene transcription targets.
There
are presently seven STAT family members. The JAK family of cytoplasmic
protein kinases were originally demonstrated to also function in IFN
signaling,
and are now known to participate in a bmad range of receptor-activated signal
cascades. Different iigands and cell activators employ specific JAK and STAT
family members. The basic model for STAT activation suggests that in
unstimulated cells, latent farms of STATs are predominantly localized within
the
cytoplasm. Ligand binding induces STAT proteins to associate with
intracellular
CA 02337999 2000-12-28
WO 00/00202 PCT/US99/14923
24
phosphotyrosine residues of transmembrane receptors. Once STATs are bound
to receptors, receptor-associated JAK kinases phosphorylate the STAT proteins.
STAT proteins then dimerize through specific reciprocal SH2-phosphotyrosine
interactions and may form complexes with other DNA-binding proteins. STAT
complexes translocate to the nucleus and interact with DNA response elements
to enhance transcription of target genes. Signaling events regulating
apoptotic
responses have been shown to utilize STAT proteins. Notably, a recent study
demonstrated JAK activation by tyrosine phosphorylation in cells that are
exposed to reactive oxygen intermediates; which in-turn lead to tyrosine
phosphorylation and activation of STAT-1, STAT-3 and STAT-6.
After establishing that LYN, BTK, and SYK kinases are not
required for radiation-induced c-jun activation, we set out to determine if c-
jun
activation is functionally linked to the JAK-STAT pathway. To this end, we
examined the effects of a JAK-3 inhibitory novel quinazoline derivative on c-
jun
expression levels in irradiated DT-40 cells: To identify a potent JAK-3
specific
inhibitor, the effects of two novel quinazoline derivatives on the enzymatic
activity of JAK-1, JAK-2, and JAK-3 were examined using Sf21 cells that were
infected with baculovirus expression vectors for these kinases, using standard
methods (Figure 4). Infected cells were harvested, JAKs were
immunoprecipitated with appropriate antibodies (anti-JAK-1: (HR-785), cat#
sc-277, rabbit polyclonal IgG affinity purified, 0.1 mglml, Santa Cruz
Biotechnology; anti-JAK-2: (C-20)-G, cat # sc-294-G, goat polyclonal IgG
affinity purified, 0.2 mglml, Santa Cruz Biotechnology; anti-3AK-3: (C-21),
cat
# sc-513, rabbit polycional IgG affinity purified, 0.2 mg/ml, Santa Cruz
Biotechnology), and kinase assays were performed following a 1 hour exposure
of the immunoprecipitated 3aks to the quinazoline compounds, as described by
Uckun, F. M., Waddick, K. G., Mahajan, S., Jun, X., Takata, M., Bolen, J., and
Kurosaki, T. Science. 273: 1096-100,1996; Uckun F.M, Evans W.E, Forsyth
C.J, Waddick K.G, T-Ahlgren L., Chelstrom L.M, Burkhardt A., Bolen J., Myers
D.E. Science 267:886-891,1995; and Myers D.E., Jun X., Waddick K.G.,
Forsyth C., Chelstrom L.M., Gunther R.L., Tumer N.E, Bolen J., Uckun F.M.
Proc Nat'1 Acad Sci USA 92: 9575-9579,1995; and Tuel Ahlgren, L., Jun, X.,
CA 02337999 2000-12-28
WO 00/00202 PCT/US99/14923
Waddick, K. G., Jin, J., Bolen, J., and Uckun, F. M. Leuk Lymphoma. 20:
417-26, 1996.
As shown in Figure 4B, both compounds inhibited JAK-3
(Figures B.3 and B.4) but not JAK-1 (Figure B.1) or JAK-2 (Figuxe B.2) (Figure
5 4D). Electrophoretic Mobility Shift Assays {EMSAs) were performed to
examine the effects of both compounds on cytokine-induced STAT activation.
Specifically, 32Dc11lIL2R~i cells (gift from James Ihle, St. Jude Children's
Research Hospital) were exposed at 8 x lOb/ml in RPMI supplemented with FBS
to the JAK-3 inhibitors at a final concentration of 10 ~,g/ml in 1% DMSO) for
1
10 hour and subsequently stimulated with IL2 or IL3 as indicated. Cells were
collected after 15 minutes and resuspended in lysis buffer (100 mM Tris-HCl pH
8.0, 0:5% NP-40, 10% glycerol, 100 mM EDTA, 0.1 mM NaV03, 50 mM NaF,
150 mM Nacl, 1 mM DTT, 3 (glml Aprotinin, 2 glml Pepstatin A, 1 (g/ml
Leupeptin and 0.2 mM PMSF). Lysates were precleared by centrifugation for 30
15 minutes. Cell extracts (approximately 10 g) were incubated with 2 p,g of
poly(dI-dC) for 30 minutes, followed by a 30 minute incubation with i ng of
poly nucleotide kinase-32P labeled double stranded DNA oligonucleotide
representing the IRF-1 STAT DNA binding sequence (Santa Cruz
Biotechnology, Santa Cruz, CA). Samples were resolved by nondenaturing
20 PAGE and visualized by autoradiography. As shown in Figure 4C; both
compounds inhibited the JAK-3-dependent STAT activation after stimulation
with IL-2, but they did not affect the JAK 1/JAK-2-dependent STAT activation
after stimulation with IL-3. Compound 2 was selected for further experiments
designed to examine the effects of JAK-3 inhibition on radiation-induced c-jun
25 activation.
As shown in Figure 5; ionizing radiation failed to induce c jun
expression in DT-40 cells treated with the JAK-3 inhibitor: This demonstrates
that JAK-3 inhibitors are capable of inhibiting radiation induced c jun
expression.
in untreated cells, c-jun expression is induced by exposure to
DNA-damaging chemical agents and by exposure to radiation. Thus, c-jun
expression is an early marker of cellular respanse to such DNA-damaging
agents. It has been shown that compounds that inhibit JAK-3 are capable of
CA 02337999 2000-12-28
WO 00/00202 PCT/US99/14923
26
inhibiting the expression of c jun. Accordingly, JAK-3 inhibitors may be
useful
to prevent or treat diseases or conditions that result from exposure to DNA-
damaging agents.
JAK-3 maps to human chromosome 19p12-13.1. A cluster of
genes encoding protooncogenes and transcription factors is also located near
this
region. JAK-3 expression has been demonstrated in mature B-cells as well as
B-cell precursors. 3AK-3 has also been detected in ieukemic B-cell precursors
and lymphoma B-cells. The physiological roles for JAK-3 have been borne out
through targeted gene disruption studies in mice, the genetic analysis of
patients
with severe combined immunodeficiency, and biochemical studies of JAK-3 in
cell lines. A wide range of stimuli result in JAK-3 activation in B-cells,
including interleukin 7 and interleukin 4. The B-cell marker CD40
constitutively associates with JAK-3 and ligation of CD40 results in JAK-3
activation which has been shown to be mandatory for CD40-mediated gene
expression. Constitutive activity of JAK-3 has been observed in v-abl
transformed pre-B cells and coimmunoprecipitations show that v-abl physically
associates with JAK-3 implicating JAK-3 in v-abl induced cellular
transformation. See Ihle, J. N. "3anus kinases in cytokine signalling," Philos
T'rans R Soc Lond B Biol Sci 351:159-66,1996; Leonard, W. J. "STATs and
cytokine specificity," Nat Med 2:968-9,1996; Levy, D. E. "The house that
JakIStat built," Cytokine Growth Factor Rev 8:81-90, 1997; Riedy, M.C. et al.
"Genomic sequence, organization, and chromosomal localization of human
JAK-3," Genomics 37, 57-61,1996; Safford, M.G., Levenstein, M., Tsifrina, E.,
Amin, S., Hawkins, A.L., Griffin, C.A., Civin, C.I. and Small, D. "JAK-3:
expression and mapping to chromosome 19p12-13.1" [published erratum appears
in Exp Hematol 1997 JuI;25(7):650]. Exp Hematol 25, 374-86,1997; Kumar, A.,
Toscani, A., Rane, S. and Reddy, E.P. "Structural organization and chromosomal
mapping of JAK-3 Iocus," Oncogene 13, 2009-14,1996; Hoffman, S.M., Lai,
K.S., Tomfohrde, J., Bowcock, A., Gordon, L.A. and Mohrenweiser, H.W.
"3AK-3 maps to human chromosome 19p12 within a cluster of proto-oncogenes
and transcription factors," Genomics 43, 109-111,1997; Tortolani, P.J. et al.
"Regulation of JAK 3 expression and activation in human B cells and B cell
malignancies," Jlmmunol 155, 5220-6,1995; Shade, N., Dadi, H.K., JJ, O.S.
CA 02337999 2000-12-28
WO 00/00202 PCT/US99/14923
27
and Roifman, C.M. "JAK-3 activation in human lymphocyte precursor cells,"
Clin Exp Immunol 108, SS2-6,1997; Gurniak, C.B. and Berg, L.J. "Marine
JAK-3 is preferentially expressed in hematopoietic tissues~and lymphocyte
precursor cells," Blood 87, 3151-60,1996; Rolling, C., Treton; D., Beckmann,
S P., Galanaud, P. and Richard, Y. "JAK-3 associates with the human
interleukin 4
receptor and is tyrosine phosphorylated following receptor triggering,"
Oncagene 10, 1757-61,1995; Rolling, C., Treton, D., Pellegrini, S:; Galanaud,
P. and Richard, Y. "IL4 and IL 13 receptors share the gamma c chain and
activate
STATE, STAT3 and STATS proteins in normal human B cells," FEES Lett 393,
S3-6,1996; Hanissian, S.H. and Geha, R.S. "3AK-3 is associated with CD40 and
is critical for CD40 induction of gene expression in B cells," Immunity b,
379-87,199'7; Danial; N.N., Pernis, A. and Rothman, P.B. "3ak-STAT signaling
induced by the v-abl oncogene," Science 2b9, 1875-7,1995.
1 S Summary
Exposure of B-lineage lymphoid cells to ionizing radiation
induces an elevation of c jun protooncogene mRNA levels. This signal is
abrogated by protein tyrosine kinase (PTK) inhibitors, indicating that
activation
of an as yet unidentified PTK is mandatory for radiation-induced c jun
expression. Experimental evidence shows that the cytoplasmic tyrosine kinases
BTK, SYK and LYN are not required for this signal. Lymphoma B-cells
rendered deficient for LYN, SYK or both by targeted gene disruption showed
increased c jun expression levels after radiation exposure, but the magnitude
of
the stimulation was lower than in wild-type cells. Thus, these PTK may
2S participate in the generation of an optimal signal. Notably, inhibitors of
Janus
family kinase 3 (JAK-3) abrogated radiation-induced c;jun activation. This
suggests that JAKs are important regulators of radiation-induced c jun
activation, and that JAK-3 inhibitors are useful for preventing or treating
diseases or conditions that result from chemical-induced or radiation-induced
cyan activation.
All publications, patents, and patent documents are incorporated
by reference herein, as though individually incorporated by reference. The
invention has been described with reference to various specific and preferred
CA 02337999 2000-12-28
WO 00/00202 PCT/US99/14923
28
embodiments and techniques. However, it should be understood that many
variations and modifications may be made while remaining within the spirit and
scope,of the invention.
ii
CA 02337999 2000-12-28
WO 00/00202 PCT/US99/14923
1
SEQUENCE LISTING
<110> Hughes Institute, et al.
<120> Method for inhibiting c-jun expression
using JAK-3 inhibitors.
ifl <130> 957.OO1W01
<150> US 60/091150
c151> 1998-06-30
1$ <160> 6
cl?0> FastSEQ for Windows Version 3.0
<210> 1
ZQ <211> 22
<212> DNA
<213> Gallus gallus
<400> 1
25 actctgcacc caactacaac gc 22
<210> 2
c211> 22
<212> DNA
3U <213> Gallus gallus
<400> 2
cttctaccgt cagctttacg cg 22
<210> 3
<211> 24
<212> DNA
<213> Gallus gallus
4a <400> 3
CA 02337999 2000-12-28
WO 00100202 PCT/US99l14923
2
agaggtgctg cccagaacat cats 24
<21.0> 4
<211> 23
$ <212> DNA
<213> Gallus gallus
<400> 4
gtggggagac agaagggaac aga 23
~0
<210> 5
<211> 22
<212> DNA
<213> Gallus gallus
IS
<400> 5
gccctcttcc agcatctttc tt 22
<210> 6
2~ <211> 20
<212> DNA
<213> Gallus gallus '
<400> 6
25 tttatgcgca tttatgggtt 20