Note: Descriptions are shown in the official language in which they were submitted.
CA 02339888 2001-03-19
60412-20310
1
ENZYMATICALLY ACTIVE RECOMBINANT GLUCOCEREBROSIDASE
This application is divided out of Canadian Patent
Application No. 2,006,709, which was filed on December 27,
1989.
Background of the Invention
This invention relates to expression of enzymatically
active recombinant glucocerebrosidase.
Gaucher's disease is an autosomal recessive lysosomal
storage disorder characterized by a deficiency in a lysosomal
enzyme, glucocerebrosidase (~~GCR"), which hydrolyzes the
glycolipid glucocerebroside. In Gaucher's patients, deficiency
in this enzyme causes the glycolipid glucocerebroside, which
arises primarily from degradation of glucosphingolipids from
membranes of white blood cells and senescent red blood cells,
to accumulate in large quantities in lysosomes of phagocytic
cells, mainly in the liver, spleen and bone marrow. Clinical
manifestations of the disease include splenomegaly,
hepatomegaly, skeletal disorders, thrombocytopenia and anemia.
Current treatments for patients suffering from this
disease include administration of analgesics for relief of bone
pain, blood and platelet transfusions, and in severe cases,
splenectomy. Joint replacements may be necessary for patients
who experience bone erosion. Brady, 1966, 275 New England
Journal of Medicine 312, proposed enzyme replacement therapy
with GCR as a treatment for Gaucher's disease. However,
Furbish et al., 1978, 81 Biochem. Biophys. Research
Communications 1047, observed that infused human placental GCR
does not reach the site at which it is active, namely lysosomes
of cells of the reticuloendothelial system, but rather is taken
up by hepatocytes. Furbish et al., 1981, 673 Biochem. Biophys.
CA 02339888 2001-03-19
60412-2031
1a
Acta 425, improved delivery of human placental GCR to
phagocytic cells by treating the GCR sequentially with
neuraminidase, I3-galactosidase and
CA 02339888 2001-03-19
- 2 -
f3-N-acetylhexosaminidase, and demonstrated that the
treated GCR was taken up more efficiently by rat Kupffer
cells than untreated protein.
Sorge et al., 1985, 82 Proc. Nat'1. Acad. Sci.,
USA 7289, and Tsuji et al., 1986, 261 J. Biol. Chem. 50
describe cloning and sequencing of a gene encoding human
GCR.
Summary of the Invention
In general, in one aspect, the invention
to features recombinant enzymatically active GCR produced
by a eukaryotic cell. The term "recombinant GCR"
("rGCR") is used herein to mean any GCR produced from
genetically manipulated GCR-encoding nucleic acid
inserted into a cell, such as, e-q., an insect cell, a
yeast cell, or a mammalian cell such as, e.q., a CHO
cell. The nucleic acid is generally placed within a
vector, such as a plasmid or virus, as appropriate for
the host cell; for expression in an insect cell, for
example, the nucleic acid can be placed within an insect
20 virus such as, e-q., a baculovirus. "Insect cell", as
that term is used herein, means any living insect cell,
such as, eq., a Dipteran or Lepidopteran cell, present
within a living insect or .in tissue culture. "Mammalian
cell", as that term is used herein, means any living
25 mammalian cell, such as, e-q., a rodent cell, or a
primate cell, present within a living mammal or in
tissue culture. The term "enzymatically active" is
used herein with respect to recombinant GCR to mean that
the rGCR is able to hydrolyze a glucocerebroside, and
3o can cleave the low molecular substrate .
4-methyl-umbelliferyl-f3-D-glucoside with an activity of
at least 106 units per milligram of rGCR.
In a second aspect, the invention features
recombinant enzymatically active GCR having at least one
CA 02339888 2001-03-19
- 3 -
exposed mannose residue, wherein the GCR is capable of
specifically binding with a human mannose receptor protein.
The term "GCR having at least one exposed mannose
residue" means that the GCR is glycosylated and at least one
of the carbohydrate groups attached to the GCR has a
carbohydrate chain terminating with a mannose residue.
Preferably, the exposed mannose residue is readily available
to bind with a mannose receptor protein, the exposed mannose
receptor being positioned external to the GCR in its three
dimensional configuration. A GCR that is "capable of
specifically binding with a human mannose receptor protein" as
that term is used herein is a GCR that is specifically
recognized by the receptor protein at an exposed mannose
residue.
In preferred embodiments, the rGCR has an amino acid
sequence with at least 95% homology to an amino acid sequence
of a primate GCR, e.q., of a human GCR; the rGCR has at least
two exposed mannose residues; the rGCR includes a carbohydrate
moiety having between 3 and 9 mannose residues; preferably the
mannose residues are arranged in a Man3 to Man9 structure
(referred to as Man3GlcNAc2, etc.}; the receptor protein
naturally occurs in a phagocytic cell; and the GCR is produced
within an insect cell such as, e.a., a Dipteran or a
Lepidopteran cell, or within a yeast cell, or within a
mammalian cell such as, e.a., a CHO cell.
Examples of a variety of carbohydrate moietes having
between 3 and 9 mannose residues arranged in a Man3 to Man9
60412-2031
CA 02339888 2001-03-19
- 4 -
structure are discussed below. The term "Man3 to Mang
structure", as used herein, refers to arrangements of mannose
residues such as are discussed below and their structural
isomers.
In a third aspect, the invention features a
eukaryotic cell containing genetically manipulated nucleic
acid, capable of expression in the cell, encoding
enzymatically active rGCR capable of specifically binding with
a human mannose receptor protein.
The invention further comprises a mammalian cell
isolated from said mammal comprising nucleic acid encoding
enzymatically active glucocerebrosidase capable of
specifically binding with a human mannose receptor protein
said cell being capable of secreting said enzymatically active
glucocerebrosidase into a culture medium when said cell is
cultured in said medium.
In preferred embodiments, the nucleic acid is a
vector including DNA encoding an amino acid sequence having at
least 95o homolog~~ to an amino acid sequence of a naturally
occurring GCR; most preferably having 95% homology to an amino
acid sequence of a naturally occurring primate GCR such as,
e.a., a human GCR.
In other preferred embodiments, the nucleic acid is
DNA lacking at Ieast 500 of the region that is present in a
naturally occurring GCR gene between the promoter of the GCR
coding sequence and the ATG start site of the gene; more
preferably the nucleic acid is the DNA present in the plasmid
60412-2031
CA 02339888 2001-03-19
- 5 -
pVL941.GCRD21, or in the plasmid pAc373.GCR2.2; and the cell
is an insect cell transformed with such plasmids.
In other preferred embodiments, the nucleic acid is
DNA present in the plasmid pGB20, or in the plasmid pGB37, or
in the plasmid pGB42; and the cell is a mammalian cell,
preferably a Chinese hamster ovary cell, transformed with any
of such plasmids or cotransformed with glasmid pGB34 and any
of such plasmids.
In other preferred embodiments, the cell containing
the CGR-encoding nucleic acid is an insect cell, a yeast cell,
or a mammalian cell.
In a related aspect, the invention features a living
insect including an insect cell containing the CGR-encoding
nucleic acid, or a living mammal including a mammalian cell
containing the GCR-encoding nucleic acid, as described above.
In another aspect, the invention features a method
for producing enzymatically active rGCR, including steps of
introducing rGCR-encoding nucleic acid into a eukaryotic cell,
causing the cell to express and secrete the rGCR into a
culture medium, and purifying the rGCR from the culture
medium. Expressed rGCR that is retained by the cell can be
purified from an extract of the cell; expressed rGCR that is
secreted by the cell into the surrounding medium can be
purified directly from the medium.
Preferably the pH of the culture medium is between
about 6.5 and 7.2, especially between about 6.6 and 6.8. The
culture medium preferably also contains 02 in an amount below
60412-2031
CA 02339888 2001-03-19
- 5 a --
saturation and sufficient to maintain the cells, especially
between about 20~ and 30~ saturation.
In preferred embodiments, the method includes
culturing the cell in vitro, or growing the cell in vivo
within a living eukaryotic organism, such as a living insect
or mammal.
The invention provides enzymatically active rGCR in
a form that is specifically recognized by human mannose
receptor proteins. The rGCR of the invention is suitable for
administration to a human suffering from Gaucher's disease
using a standard enzyme replacement protocol. The invention
also provides enzyme which is free from viral or bacterial
agents commonly found in human tissues such as, for example,
human placenta, from which GCR is conventionally derived. In
addition, the rGCR of the invention is secreted in large
amounts from the cells in which it is produced into the
surrounding medium, frorn which it is readily purified.
The invention further provides a commercial package
comprising a pharmaceutically effective amount of an
enzymatically active recombinant glucocerebrosidase derived
from a culture of an isolated mammalian cell together with
instructions for use thereof in an enzyme replacement protocol
in a patient suffering from Gaucher's disease.
The invention also provides use of an enzymatically
active recombinant glucocerebrosidase derived from a culture
of an isolated mammalian cell in preparation of a medicament
50412-2031
CA 02339888 2001-03-19
- 5b -
for use in an enzyme replacement protocol in a patient
suffering from Gaucher's disease.
Other features and advantages of the invention will
be apparent from the following description of the preferred
embodiments thereof, and from the claims.
Description of the Preferred Embodiments
The drawings will first briefly be described.
Drawings
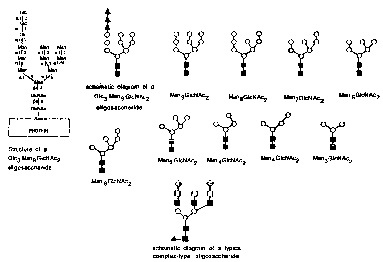
Fig. 1 is a diagrammatic representation of various
forms of mannose-terminating carbohydrate moiety
50412-2031
CA 02339888 2001-03-19
- 6 -
of rGCR and of a typical complex-type carbohydrate
moiety; circles represent mannose residues, black
triangles represent glucose residues, black squares
represent N-acetylglucosamine residues, open squares
represent galactose residues, diamonds represent
N-acetyl neuraminic acid residues, shaded triangles
represent fucose;
Fig. 2 is a schematic representation of the
spatial~relationships in various clones of the GCR gene
to among the GCR encoding regions, the 5' and the 3'
noncoding regions and the polyhedrin regulatory
sequences;
Fig. 3 is a schematic representation of
construction of a deletion version of GCR (with a 5'
noncoding region removed and a portion of the 3'
noncoding region removed) inserted within a plasmid,
namely plasmid pGB9.D21;
Fig. 4 is a schematic representation of
construction of plasmid pVL941.GCRD21;
Fig. 5 is a schematic representation of
construction of plasmid pAc373.GCR2.2;
Fig. 6 is a schematic representation of
construction of plasmids pGB9.D21C and pGB9.D21C1;
Fig. 7 is a schematic representation of
construction of plasmid pGB20.
Fig. 8 is a schematic representation of
construction of plasmid pG834.
Fig. 9 is a schematic representation of
construction of plasmids pGB37 and pGB42.
Glucocerebrosidase GCR
As defined above, GCR has an enzyme activity
which causes hydrolysis of a glucocerebroside. This
invention includes all enzymes having such activity, a
non-limiting example of which is the enzyme found within
CA 02339888 2001-03-19
-
human placenta. A gene encoding this enzyme activity
has been cloned as described above, and its DNA sequence
is known. In the present application, applicants
provide examples of use of this cloned DNA to cause
production of rGCR having a structure suitable for
therapeutic use in humans.
There follows a description of examples of
insertion of human GCR-encoding DNA into insect vectors.
and of expression of the DNA by insect cells, and
l0 examples of insertion of human GCR-encoding DNA into
mammalian vectors and expression of the DNA by mammalian
cells. These examples are not limiting to this
invention and those skilled in the art will recognize
that applicants have enabled practice of this invention
commensurate with the breadth of the appended cl.a.ims .
GCR Expression in Insect Cells
Example 1: Insect Vector, pAc373.GCR2.2
In general, in order to insert a GCR gene
within an insect virus, the gene is first inserted into
a transfer vector, for example, pAc373, and then insect
cells such as, for example, SF9 cells, are co-infected
with the transfer vector together with wild-type viral
DNA to allow recombination of the transfer vector and
the viral DNA to produce a desired recombinant virus
encoding GCR.
Referring to Figs. 2 and 5, transfer vector
pAc373 (obtained from Dr. M. Summers, Texas A & M
University System, College Station, Texas) has a BamHI
cloning site to allow positioning of genes under the
control of polyhedrin regulatory signals. Otherw
promoters are also suitable in this invention, such as,
e-q., the p10 promoter of Baculovirus. Plasmid pGCS-2Kb
contains all of the coding sequence of GCR, including
approximately 160 by non-coding 5' and 500 by noncoding
CA 02339888 2001-03-19
-
3' sequences, inserted into a unique EcoRI site of
pBR322 (Sorge et al., 82 Proc. Natl. Acad. Sci. USA
7289, 1985). This plasmid was obtained from Dr. E.
Beutler (Scripps Clinic and Research Foundation,
LaJolla, CA) and the gene was cloned into pAc373 in the
following manner. Plasmid pGCS-2Kb was cleaved with
EcoRI and the ends blunt-ended with T4 polymerase.
Bc~III linkers were added and the 2.2 kb GCR containing
fragment was purified. The BglII ends are compatible
l0 with BamHI ends and allowed cloning into the BamHI site
of pAc373. Recombinants containing the GCR gene in the
correct orientation were identified by standard
restriction analysis. The resulting plasmid was named
pAc373.GCR2.2.
Transfer of the 2.2 kb fragment coding for GCR
to the genome of the multiple nuclear polyhedrosis virus
Autoqrapha californica (AcMNPV) isolate E2 (obtained
from Dr. M. Summers, Texas A & M University System,
College Station, TX) was accomplished by co-transfecting
the Spodoptera fruqiperda cell line SF9 (a clonal
- isolate of Spodoptera fruqiperda IPBL-Sf21-AE cells;
American Type Culture Collection accession number
CRL1711, obtained from Dr.,M. Summers, Texas A & M
University System, College Station, TX) with wild type
virus DNA and pAc373.GCR2.2 prepared by standard
procedures. Briefly, SF9 cells were cultured in TNM-FH
medium (Hink, 266 Nature 466, 1970) containing 10% fetal
calf serum at a temperature of 27°C ~ 1°C. Cells were
cultured either in monolayer or in suspension. Cells
were subcultured approximately 2-3 times a week. When
grown in suspension, flask spinners were stirring at
50-60 rpm. The procedure for transfecting SF9 cells
with viral DNA, or cotransfecting with a mixture of
viral DNA and transfer vector DNA, was a modification of
CA 02339888 2001-03-19
_ g _
the calcium phosphate technique of Graham et al., 52
Virology 456, 1973, described by Burand et al., 101
Virology 286, 1980, and Carstens _et al., lol Virology
311, 1980. Briefly, a 25 cm2 flask was seeded with 2
x 106 cells. After 1-2 hours the flask was aspirated,
and 0.75 ml of Grace's medium containing 10% fetal calf
serum was added to the adherent cells. 0.75 ml of 25 mM
Hepes, pH 7.1, 140 mM NaCl, 125 mM CaCl2, containing 1
ug of viral DNA (with or without 2 ug transfer vector
DNA) was then added slowly. The flask was incubated for
4 hours at 27°C, after which the transfection mixture
was removed and replaced with 5 ml of TNM-FH containing
10°s fetal calf serum. For the production of the
recombinant virus encoding the 2.2kb GCR fragment from
pGCS-2kb, 1 x 106 cells were plated in a 9 cm2 dish,
after which the cells were transfected with 2 ug of
pAc373.GCR2.2 and 1 ug of Autoqrapha californica closed
covalent circular DNA ("cccDNA"). After cells are
co-transfected with wild-type virus DNA and transfer
vector DNA, a small percentage of the resulting
transfectants produce recombinant virus, in which the
polyhedrin gene is no longer expressed. These are
identified by visually inspecting a sufficient number of
viral plaques generated by~infecting adherent cells with
the supernatant harvested from the transfected flask 48
hours after the transfection. One hour after the
infection with the transfection supernatant, an agarose
overlay (1.5% low-melting SeaPlaque in growth medium) is
applied. After 4-6 days, the plates are inverted and
viewed under a dissecting microscope with low-angle
incoming light, to discriminate wild-type from
recombinant plaques. In this example, the same
procedure was followed 4 days after transfection, and
the medium was collected and serially diluted (103 to
CA 02339888 2001-03-19
1~ -
105). The serial dilutions (1 ml volume) were used to
infect subconfluent cells (5 x 106 in 100 mm dishes).
After infection the cells were overlayed with
low-melting agar. Six days post infection the plates
were inspected for recombinant plaques.
Appropriate plaques were picked, subjected to
two more rounds of plaque-purification, and used for the
propagation of virus stock. A more detailed description
of the plaque identification procedure is provided by
l0 Summers et al., A Manual of Methods for Baculovirus
Vectors and Insect Cell Culture Procedures, Texas
Agricultural Experiment Statioh Bulletin 1555 (1987).
Eight plaques having an appearance that
suggested a lack of polyhedrin expression were selected
and further plaque-purified. Virus from three plaques
(designated 3-1-1, 3-2-1, 3-3-1) were further propagated
and cells infected with these three isolates were
analyzed for GCR expression.
The following assays were performed to ensure
that sufficient rGCR having sufficient enzyme activity
is produced by the selected clones. First, cell lysates
of adherent cells which were separately infected with
the three above described.isolates were prepared and
analyzed for the presence of GCR-specific DNA sequences.
Cells were lysed in 0.5 M NaOH, the lysate was
neutralized with 10 M NH4Ac, and the solution was
passed through nitrocellulose using a dot-blot apparatus
(BioRad). The nitrocellulose sheet was processed
according to~standard DNA hybridization procedures, and
probed with the 2.2 kb GCR fragment (32P-labeled) from
pGCS-2kb. Cells infected with viral isolates 3-2-1 and
3-3-1 contained GCR sequences, whereas cells infected
with 3-1-1 and cells infected with wild-type virus did
not. The structure of one such clone encoding a
CA 02339888 2001-03-19
- 11 -
GCR-encoding sec-iuence is shown in Fig. 2, where the
GCR-encoding DNA is inserted within the partially
deleted polyhedrin gene.
Two assays were employed for determination and
quantification of the enzymatic activity of the
recombinant human glucocerebrosidase. 'Three days after
ir.~eczion, the cells were lysed in 50 mM Na-phosphate ~H
6.8/1, deoxycholate, and these lysates were used in the
enzymatic assays. The first assay is based on the
LO hydrolysis of the low molecular-weight substrate
4-methyl-umbelliferyl-(3-glucoside (4MU-GIc). Although
several other glucosidases are capable of cleaving this
substrate and releasing the fluorescent aglycon, at low
cH aid in the presence of taurocholate (an inhibitor of
ss're=al other non-specific glucosidases) this assay is
us2rul for determining the level of e:~pression of rGCR.
'1'hi~ expression was compared with the level of 4MU-Glc
acti-rity in SF9 cells infected with wild-type virus.
':h~ assay was performed at pH 5.9, in 100 ~-nM potassium
~hosprate buffer containing 0.15% Triton X-100 and
0.125% taurocholate, essentially as described by Suzuki,
138 Dteth. Enzymol. 749, 1987. rGCR having an activity
of at least 106 U/mg is useful in this invention.
For a glucocerebroside-specific assay
[14C]-glucocerebroside (Brady et al., 1965, 240 J.
Biol. Chem. 39) was used. After incubation of the
enzyme preparation with this radioactive substrate, the
mixture was precipitated with 5% TCA. The amount of
glucose (14C-radioactivity) remaining in the
3o supernatant provides a direct measure of
glucocerebrosidase activity.
Lysates derived from cells infected with
isolate 3-1-1 did not show any activity above background
levels with the radioactive substrate, whereas the
CA 02339888 2001-03-19
- 12 -
lysates from cells infected with 3-2-1 and 3-3-1 showed
substantial activity.
An immunological assay was also used to
determine the amount of GCR expressed by SF9 cells.
Generally, this assay involved the following procedure.
In a 24-well plate, cells were seeded at a density of 5
x 105 cells/well. Cells were infected at a
multiplicity of infection of 10, and 3 days
post-infection, the medium was harvested and cells lysed
in buffer (10 mM Tris, pH 7.2, 150mM NaCl, 5mM EDTA, 10%
DOC, 0.1% Triton X-100, 0.1% SDS, and 0.020 NaN3).
GCR was immunoprecipitated using a polyclonal rabbit
antibody, and the precipitated protein was subjected to
SDS-PAGE. After electrophoresis, the protein was
transferred to nitrocellulose, and the resulting western
blot was probed with the same antibody. The amount of
bound antibody was determined using biotinylated protein
A and alkaline phosphatase conjugated streptavidin. By
comparison with known quantities of human
placenta-derived GCR, it was estimated that, for both
active isolates, 106 cells produced approximately
2-4 ug of rGCR.
Example 2: Insect Vector,.pVL941.GCRD21
In order to optimize the expression of the GCR
gene in insect cells, excess non-coding sequence from
the 5' and 3' ends of the gene were removed. The start
codon for GCR should be placed as close t.o the
polyhedrin gene regulatory sequences as possible in
order to minifnize sequence- or distance-associated
negative effects on expression of GCR. This procedure
minimizes or eliminates message destabilizing sequences
at the 3' and 5' end of the GCR gene. To this end,
referring to Figs. 2, 3 and 4, we constructed the
plasmid pGB9.D21 (Fig. 3), which carries a deletion
CA 02339888 2001-03-19
- 13 -
derivative, GCR.D21, bounded by Bc~II sites. This Bc~lII
cassette carrying the GCR gene has 5 bases at the 5' end
and 290 bases at the 3' end between the cloning (BqlII)
ends and the GCR coding sequences, compared to 160 and
500 bases, respectively, in the original cDNA pGCS-2kb
with EcoRI cloning ends. The stepwise construction of
pGB9.D21 is summarized in Fig. 3. Briefly, the GCR cDNA
clone, pGCS-2kb, was cleaved approximately 290 base
pairs from the 3' end of the GCR coding sequence with
l0 SacI and the ends blunt-ended with T4-DNA polymerase. A
second cut, approximately 160 by from the 5' end of GCR,
with EcoRI created a 2.0 kb fragment containing the GCR
coding sequence. This fragment was purified and ligated
to HincII and EcoRI cut pGB3 to form pGB9. _E. coli
plasmid pGB3 was constructed from pBluescript SK+
(Stratagene, La Jolla, CA) by independently blunting the
Sacl and A~aI sites with T4 polymerase and ligating in
BglLI linkers. The resulting plasmid, pGB3, contains
BglII linkers at either end of its multiple cloning site
and retains lacZ activity in the host XL-1 Blue. pGB9
was digested with BstXI, EcoRI, and then with
exonuclease III and mung bean nuclease to remove excess
5' non-coding sequence between the Bc~lII site and the 5'
end of the GCR coding sequence. After this treatment
the ends were religated, and the plasmids were used to
transform E. coli XL-1 blue. Transformants were
screened using restriction analysis, and then DNA
sequencing was used to determine the lengths of the
deletions. Using this approach we identified pGB9.D21,
which has only 5 by between the BglII site and the start
codon of GCR.
The shortened version of the GCR-encoding
fragment, GCR.D21, was then cloned into a transfer ~
vector, pVI,941, as shown in Fig. 4. Transfer vector
CA 02339888 2001-03-19
- 14 -
pVL941 (obtained from Dr. M. Summers, Texas A & M
University System, College Station, Texas) is very
similar to pAc373, as it contains a BamHI cloning site
for the positioning of genes under the control of the
polyhedrin regulatory signals. The main difference
between the two vectors is that the polyhedrin
regulatory sequence of pAc373 extends only to position
-8 relative to the polyhedrin translation start codon,
whereas in pVL941 the entire 5' non-coding regulatory
sequence of the polyhedrin gene is present. This
difference may result in an increase in the expression
of genes placed under the control of the regulatory
sequence.
Transfer of the GCR cDNA fragment spanning the
-6 to +1850 domain (see Fig. 2) to the AcMNPV genome was
accomplished by co-transfecting SF9 cells with wild-type
virus DNA and pVL941.GCRD21. The procedures followed
for cotransfection and for subsequent isolation of a
viral plaque resulting from a recombinant,
_ 20 non-polyhedrin coding virus, were as described above for
pAc373.GCR2.2. Upon infection with this recombinant
virus, cells produced material that cross-reacted with
the polyclonal anti-GCR antibody, and that was
enzymatically active as determined by the 4-MU-glucoside
and [l4Clglucosylcerebroside assays.
Production of rGCR in insect cells
To produce rGCR in sufficient amount to be
useful, uninfected insect cells are grown and then
infected by standard procedure with one of the above
recombinant viruses and the resulting protein is
purified as follows. SF9 cells are grown either
adherently or in suspension culture, in a spinner flask,
e-q., a Bellco 500 ml flask operated at 50-60 rpm. The
culture medium is, e.g., TNM-FH (Hink, 266 Nature 466,
CA 02339888 2001-03-19
- 15 -
1970) supplemented with 10% fetal calf serum. The SF9
cells can be adapted to lower fetal calf serum
concentrations, as described by Tramper et al., 469 Ann.
NY Acad. Sci. 279, 1986. Alternatively, it is possible
to grow cells in serum-free medium, as described, e.q.,
by Roder, 69 Naturwissenschaffen 92, 1982; or to grow
insect cells encapsulated in microcapsules to allow
growth to high cell densities, and facilitate large
scale insect cell culture, e-q., as described by King et
al., 10 Biotechnology Letters 683, 1988.
Purification of rGCR produced in insect cells
rGCR was purified from the culture medium of
SF9 cells infected with recombinant virus as follows.
Three days post infection the medium (containing the
rGCR) was separated from the cells by centrifugation of
the cell suspension (15 minutes, 2000 rpm in a Sorvall
RC3). After centrifugation the medium was subjected to
hydrophobic chromatography on a butyl substituted matrix
e-q., TSK-gel, Toyopearl Butyl 650C (Nest Group,
Southboro, MA). A column of 25 ml (1.6 x 12 cm) is
sufficient for at least 500 ml of culture medium. The
column was washed with 50 mM citrate buffer pH 5.0,
containing 20% ethylene glycol, and fractions were
eluted with an ethylene glycol gradient from 20 to 80%.
The fractions were collected and assayed for GCR
activity using the 4MU-glucoside assay described above.
In a typical experiment, a recovery of 85% was
obtained. The partially purified GCR was then subjected
to a second hydrophobic chromatography step, on a
phenyl-substituted matrix, e-g., TSK-gel, Toyopearl .
Phenyl 6505 (Nest Group, Southboro, MA). The GCR
solution was brought to 50 mM citrate, pH 5.0, 20%
ethylene glycol, and loaded onto a column equilibrated
in the same buffer. Under these conditions the GCR
CA 02339888 2001-03-19
- 16 -
adsorbed to the phenyl-substituted matrix.
Subsequently, the column was washed with 50 mM citrate
pH 5.0 to remove ethylene glycol. The GCR was then
eluted with an ethanol gradient (in 50 mM citrate) from
0 to 50% and collected as described above. This
procedure yielded a GCR preparation virtually free of
other proteins. The resulting GCR was then subjected to
oligosaccaride structural analysis, as described below.
Expression of GCR in Insects
To obtain expression of rGCR in whole insects
the procedure of Maeda et al., 1985, 315 Nature 592, can
be used. Briefly, a recombinant BombYx mori nuclear
polyhedrosis virus (BmNPV) is generated encoding rGCR.
This recombinant virus, obtained as extracellular virus
from cultured cells, is used to infect silkworm larvae
by injection into the body cavity of the silkworms. The
rGCR protein is recovered from the haemolymph.
Alternatively,. since BmNPV is a baculovirus similar to
Autoqrapha californica multiple nuclear polyhedrosis
virus with respect to lifecycle, mode of replication,
polyhedrin production and DNA homology, a similar
procedure can be used with recombinant AcMNPV harboring
a GCR gene. The recombinant virus harvested from
infected cultured SF9 cells is used to infect permissive
insects, such as larvae of Spodoptera fru iperda, or
Trichoplusia ni by injection of the virus into the
insect. The recombinant virus is allowed to replicate
in the infected insect, which then produces rGCR in
place of polyhedrin. After a suitable period, the
recombinant protein is harvested from the insect.
In another method, the insect can be infected
by orally administering virus encoding rGCR. The
infectivity of AcI~lPV depends on its target cell, as
well as on the viral form. Nuclear polyhedrosis viruses
CA 02339888 2001-03-19
- 17 -
occur in two forms, known as extracellular virus and
occluded virus. The latter form is produced late in
infection, and consists of multiple virus particles
entrapped in polyhedra. Polyhedra are large,
proteinaceous structures which are formed in the nucleus
of the infected cell by the deposition of the major
structural virus-encoded protein polyhedrin, thus
embedding many virus particles. After cell or insect
death these polyhedra remain infective when administered
orally. The virus particles released in the midget of
an orally infected insect by hydrolysis of the embedding
polyhedra are capable of infecting the midget cells.
Secondary infection of the other organs of the insect
then follows. Polyhedra-embedded, occluded virus
particles, mixed in the dietary intake, are an ef~ective
way of infecting large numbers of insects. A procedure
for producing recombinant occluded virus is described by
Emery et al., 1987, I Protein Engineering 359. Briefly,
a fragment encoding the polyhedrin promoter toge~her
with a GCR gene downstream of that promoter is cloned
into a plasmid representing an AcMNPV DNA restriction
fragment encoding polyhedrin and its promoter, and
sufficient sequence 5' and 3' of the gene to allow
recombination after co-transf ection. The cloning site
in this plasmid is upstream (but in the opposite
orientation) of the natural polyhedrin promoter present
in that plasmid. The resulting transfer vector has,
therefore, both the normal polyhedrin gene and a GCR
gene, each with.its own copy of the polyhedrin
transcriptional machinery. Co-transfections of
S~odo~tera frugi~erda cells with this vector together
with infectious, polyhedrin-negative AcMNPV DNA yields
recombinant virus which encodes both polyhedrin and
GCR. The two forms of this recombinant virus can thus
CA 02339888 2001-03-19
- 18 -
be obtained. The occluded form is useful as described
above for infecting insect larvae orally.
GCR Expression In Mammalian Cells
.. Optimization Recombinant GCR Gene Cassette.
To optimize expression of GCR in mammalian
cells, we further modified the GCR.D21 III cassette
containing the gcr gene. In general, with reference to
Fig. 6, the modifications were made using
oligonucleotide directed mutagenesis (described
generally in Kunkel, 1986, 82 Proc. Natl. Acad. Sci.
U.S.A. 488-492; Wang et al., 1989, 7 Biotechniques, '
1000-1010) to alter the nucleotide sequence near the GCR
translation start to match the consensus sequence
(CCACCATGG) for optimal translation in mammalian cells
(as described in Kozak, 1986, 44 Cell 283-292); and to
delete the excess sequence 3' of the gcr.D2lC stop
codon. The excess 3' sequence deletion removes
potential message destabilizing sequences, and permits
modulation of DHFR expression relative to GCR from
bicistronic vectors.
Vector constructions
A bicistronic gcr-dhfr expression vector for
CHO cells was constructed as shown in Fig. 7 from the
vector pSV2-dhfr (described in Subramani et al., 1981,
9 Molecular and Cellular Biology 854-864). This vector,
pGB20, contained gcr.D2lC followed by dhfr under the
control of the SV40 early promoter. DHFR expression
depends upon ribosomes translating the dhfr after
translating gcr, which is located adjacent at the 5' end
of the dhfr message. This bicistronic arrangement
reduced levels of expression of DHFR relative to GCR. A
resulting gain in GCR expression relative to DHFR can be
realized after stable transfectants have been selected
using methotrexate.
CA 02339888 2001-03-19
- 19 -
A second series of bicistronic gcr expression
vectors were constructed having the following
characteristics: 1) transcription of gcr driven by a
SV40 enhancer-Adenovirus major late promoter (Ad MLP)
combination; 2) transcription of gcr in a bicistronic
arrangement with dhfr; 3) termination and
polyadenylation signals from SV40; and 4) a minimal 2 kb
segment of DNA containing a prokaryotic origin and
ampicillin resistance gene, but lacking the "poison"
sequences which can be deleterious in mammalian cells
(Lusky and Botchan, 1981, 293 Nature 79-81).
The base vector, pGB34, was constructed from
pSV2dhfr as shown in Fig. 8. In a first step "poison"
sequences between the PvuII and MaeII sites which might
adversely affect expression in the final vector were
removed. The resulting plasmid carries the SV40
enhancer (72 by repeat), the pBR322 replication origin
and the ampicillin resistance coding gene (bla) within a
minimal fragment (2 kb) between the FokI and EcoRI
sites. In a second step the 2 kb FokI-EcoRI fragment
combined with a fragment from pDHFRIII (described in
Berkner and Sharp, 1985, 13 Nucleic Acids Research
841-857) containing the Ad-MLF and the dhfr gene and the
SV40 polyadenylation signal. Excess sequence between
the polyadenylation site and the pBR322 sequences were
removed and the PstI cloning site was changed to BamHI
by standard procedures using BamHI linkers. The
resulting construct, pGH34, carries very little sequence
in excess of that required for efficient production of
recombinant proteins in mammalian cells. Versions of
this vector were constructed using either gcr.D2lC or
gcr.D21C1 (shown in Fig. 6) for expression of GCR in CHO
cells, as shown in Fig. 9. These constructs express
DHFR at different levels, thus modulating DHFR selection
CA 02339888 2001-03-19
- 20 -
and the relative amplification of GCR expression. These
constructs can be used by themselves as bicistronic
amplifiable gcr expression vectors; alternatively, the
more widely used scheme of cotransfection using pGB34
can be used.
Transfectinq Mammalian Cells
Any of several procedures can be used for
introducing the vector DNA into mammalian cells,
including calcium phosphate transfection and
to electroporation (described generally in F.M. Ausubel _et
al., eds., 1989, Current Protocols in Molecular Biology,
pages 9.1.1-9.1.4 and 9.3.1-9.3.2, respectively, Wiley &
Sons, New York), Lipofectin'" transfection (as
described by the manufacturer, Bethesda Research
Laboratories, Gaithersburg, MD), protoplast fusion (as
described, e-q., in Sandri-Goldin et al., 1981, 1 Mol.
Cell. Biol. 743-52, or polybrene transfection.
Selection of recombinant colonies is performed by
incubating the cells in culture medium devoid of
2o ribonucleosides and deoxyribonucleosides, containing
dialyzed fetal calf serum.
Amplifications
To increase expression of rGCR the cells can be
subjected to an amplification procedure similar to the
one described in F.M. Ausubel et al., eds., 1989,
Current Protocols in Molecular Biology, pages
9.9.1-9.9.6, Wiley & Sons, New York), which is similar
to the procedure described in R.J. Kaufman _et al., 1982,
159 J. Cell. viol. Hiol. 601-21. In general, higher
levels of expression result from higher levels of
methotrexate resistance. Other amplification procedures
can be utilized as well, by using other amplifiable
genes instead of the dhfr gene. Examples are the ,
ornithine decarboxylase gene, the adenosine deaminase
CA 02339888 2001-03-19
- 21 -
gene, and others, as well as combinations thereof, known
to persons of ordinary skill. Examples have been
reviewed by R.J. Kaufman in J. Setlow, Ed., 1989,
9 Genetic Engineering 155-198, Plenum Press, New York.
In order to get amplification using other genes, the
dhfr gene in the vectors described above is replaced by
the gene of choice, e-q., the ornithine decarboxylase
gene or the adenosine deaminase gene; after transfection
with such a vector, amplification can be obtained using
l0 the appropriate selective medium.Assa s
Enzymatic activity of recombinant GCR expressed
in Chinese hamster ovary cells was measured using
4-methyl-umbelliferyl-B-D-glucoside as a substrate.
Enzymatic hydrolysis of this substrate generates a
fluorescent product, which is quantitated using a
spectrofluorometer. Details of this procedure are
described in Methods of Enzymology, Vol. L, pp. 478-79,
1978. The assay is caried out under conditions in which
other, non-GCR glucosidase activities are partially
inhibited, i.e., by using a phosphate buffer, pH 5.9,
0.125 % taurocholate, 0.15 % Triton X-100.
Another way to establish the presence of
recombinant GCR is by using a polyclonal antibody raised
in rabbits against a preparation of glucocerebrosidase
purified from human placenta. The medium or the cell
lysate from GCR producing cells can be subjected to
SDS-polyacrylamide gel electrophoresis, after which
proteins are transferred from the gel to
nitrocellulose. The presence of rGCR on nitrocellulose
is established by probing with the antibody, which is
detected by standard techniques using the biotinylated
protein A-alkaline phosphatase-conjugated streptavidin
technique, or biotinylated goat-anti-rabbit IgG ,
antibody-alkaline phosphatase-conjugated streptavidin,
CA 02339888 2001-03-19
- 22 -
or any other standard method for detection of antibody
on nitrocellulose. Details of such procedures have been
described in, for example, E. Harlow and D. Lane, Eds.,
Antibodies, a Laboratory Manual, Cold Spring Harbor
Laboratory, NY, 1988.
Another way of determining production of rGCR
by CHO cells is by labeling the growing cells in vivo
with [35S]methionine, and then harvesting the medium and
lysing the cells, e.g., in a buffer containing 50 mM
citrate pH 6.5, 1% sodium cholate, at intervals after
labeling. The lysate and medium are then
immunoprecipitated using the po~lyclonal antibody
according to standard procedures. The polyclonal
antibody can be used in various forms, such as, eq., in
the form of antiserum, or purified on protein A-agarose
or protein G-agarose, or affinity-purified over a matrix
onto which glucocerebrosidase has been immobilized. A
higher purity of the antibody generally results in a
lower background signal. Alternatively, a monoclonal
antibody against GCR can be used. Examples of these
detection procedures have been described in, far
example, E. Harlow and D. Lane, Eds., Antibodies, a
Laboratory Manual, Cold Spring Harbor Laboratory, NY,
1988.
These detection methods demonstrated expression
of rGCR in CHO cells. In a typical run, cells that were
transfected with one of the vectors described above,
either using the dicistronic approach or the
co-transfection procedure, expressed rGCR at levels
between 1-10 mg/L, varying with cell density, culture .
conditions and cell support matrix. The rGCR is found
both intracellularly and in the medium. The
intracellular rGCR is sensitive to endoglucosaminidase H ,
and to endoglucosaminidase F, indicating that the
CA 02339888 2001-03-19
- 23 -
carbohydrate chains on the protein are most likely of
the so-called high-mannose type, and thus the
intracellular rGCR is particularly useful for treatment
of Gaucher's disease, as described below. The rGCR
recovered from the medium is larger in molecular weight,
and is resistant to endoglucosaminidase H.
Carbohydrate Structure of rGCR
To determine whether a rGCR produced as
described above has been correctly glycosylated to be
l0 useful for treatment of Gaucher's disease the following
procedure can be performed to determine the carbohydrate
structure, and in particular the mannose configuration
within the carbohydrate structure, of the rGCR.
Generally, the carbohydrate should contain at least one
exposed mannose and preferably has a Man3-Man9
structure, or a structure with the same functional
features as a carbohydrate having a Man3-Man9
structure (e. Q., other sugar residues can be substituted
for one or more of the sugar residues shown). There are
generally four oligosaccharide moieties in human
placental GCR. Recombinant GCR having these moieties
present in a Man3-Man9 structure has greater
affinity than unglycosylated GCR for the mannose
receptor in humans. The more mannose residues per
oligosaccharide moiety, and the more such moieties, the
greater this affinity. Recombinant GCR according to
this invention has at least one such oligosaccharide
with at least one exposed mannose, but preferably has
two, three, or four such oligosaccharides each with a
Man3-Man9 structure. '
Analysis of oligosaccharides derived from rGCR
generally followed the procedure described by Hirani
_et al., 162 Anal. Biochem. 485, 1987. Asn-linked ~
oligosaccharides were released from SDS-denatured rGCR
CA 02339888 2001-03-19
- 24 -
(loo ug) by incubation with N-glycanase enzyme (80
units) at 37°C for 18 hours. The completeness of the
reaction was judged by SDS-polyacrylamide gel
electrophoresis. The liberated oligosaccharides were
recovered in the supernatant after precipitating the
protein with ethanol (75% v/v) and radiolabeled at the
reducing end by treatment with sodium borotritide.
Labeled oligosaccharides were analyzed by a combination
of high performance liquid chromatography (hplc) and
l0 exoglycosidase digestion.
The degree of sialylation was determined by
analyzing the tritium-labeled o-ligosaccharides by hplc
on a Micropak AX-10 column pre-equilibrated in 25 mM
KH2P04, titrated to pH 4.0 with phosphoric acid. GCR
with high sialic acid content is unlikely to useful in
this invention, but GCR with a low sialic acid content,
i.e., uncharged GCR, is more likely to have exposed
mannose moieties. The column was eluted with the same
buffer for 15 minutes and then for 30 minutes using a
linear gradient of 25 mM KH2P04, pH 4.0, to a final
concentration of 500 mM KH2P04,pH 4Ø In this
elution protocol, oligosaccharides eluted from the
column in characteristic positions depending upon the
number of attached sialic acid residues. The Asn-linked
oligosaccharides derived from recombinant GCR consisted
primarily (95%) of neutral species. Such neutral
species are potentially useful in this invention.
The size of each neutral and/or desialylated
oligosaccharide was analyzed by hplc using a Micropak
AX-5 column. The column was pre-equilibrated with .
acetonitrile:water (65:35) and elution was performed
using a 60 minute gradient in which the water content of
the solvent increased at the rate of 0.5%/minute. This
procedure fractionates oligosaccharides according to
CA 02339888 2001-03-19
- 25 -
size. The column was calibrated with oligosaccharide
standards having the structures shown below. Analysis
of the oligosaccharides derived from rGCR obtained from
infected SF9 cells showed that they consisted of a
single species with a retention time similar to
Man3GlcNAc(Fuc)GlcNAc~T'
Gal-GIcNAc~
to (NeuAc)o-4 Gal-GIcNAc/M~~
Gal-GIcNAc%M~,Man-GIcNAc-GIcNAc~
Gal-GIcNAc
Man
Man-GIcNAc-GIcNAc ~.
Man
Fuc
Man ~
M~~Man-GIcNAc-GIcNAc~.
A presence of exppsed mannose groups is readily
determined by treatment of these oligosaccharides with
25 mannosidases which specifically remove mannose groups.
Such treatment, and the analysis of the results, is
performed by standard procedures.
In another method for determining the
usefulness of'an rGCR, the ability of the rGCR to bind
3o to and be taken up by macrophages is measured. This
targe~ing of rGCR to macrophages is mediated by the
Man/GlcNAc receptor and can be determined using
thioglycollate-elicited peritoneal macrophages obtained ,
from mice, as described by Stahl et al., 93 J. Cell
CA 02339888 2001-03-19
- 26 -
Biol. 49, 1982. Briefly, mice (25-30 g, C57 strain) are
injected.intraperitoneally with 1-1.5 ml thioglycollate
broth (Difco, Detroit, MI). After 3-4 days the mice are
sacrificed by suffocation in C02 and the peritoneal
cavity rinsed with phosphate buffered saline. The cells
are pelleted by centrifugation (500 g, 10 min),
resuspended in DME (GIBCO, Grand Island, NY) containing
l0°s fetal calf serum, and plated in 96-well tissue
culture plates. After 90 min. non-adherent cells are
washed away, and the adherent macrophages are incubated
for specified time periods, ranging from 0 to 180
minutes in culture medium containing specified
quantities of rGCR, ranging from 0 to 20 ug in 200 ul at
a temperature of 37°C, in the absence and presence of
yeast mannan (2-l0 mg/ml). After incubation, the medium
containing excess rGCR is removed, the cells are washed
several times and then lysed, and the amount of rGCR
taken up by the cells is determined in the cell lysate.
The amount taken up in the presence of yeast mannan is
non-specific uptake. The difference between the two
values is the amount taken up specifically via the
Man/GlcNAc receptor. No uptake occurs when the
experiment is done at 4°C,.but the rGCR binds to the
cell surface. In this way, Man/GlcNAc receptor-specific
binding of rGCR can be determined.
Use
The rGCR of this invention is useful for
therapeutic treatment of Gaucher's disease by providing
a therapeutic'amount of the rGCR. By therapeutic amount
is meant an amount of rGCR which will cause significant
alleviation of clinical symptoms of Gaucher's disease.
Such rGCR must be post-translationally modified, as
described above, to provide a carbohydrate structure ,
which will target to human mannose receptors.
CA 02339888 2001-03-19
- 27 -
Generally, such rGCR has at least two carbohydrate moieties
each having a Man3-Man9 structure, and such rGCR represents at
least 50% of the rGCR provided in the therapeutic composition.
For example, provision of between 10 and 500 milligrams per 70
kg patient per month to provide that patient with between 0.25
and 3 grams rGCR over one year period. The rGCR is provided
in the form of a pharmaceutical composition in any. standard
pharmaceutically acceptable carrier, such as physiological
saline, and is administered by any standard procedure, for
example by intravenous injection.
Deposit
An E. coli strain (DH5a) harboring the plasmid
pVL941.GCRD21 was deposited on December 22, 1988 with the
American Type Culture Collection and was assigned ATCC
accession number 67,866. An E. coli strain DHSalpha/pGH42
harboring the plasmid pGB42 was deposited on December 21, 1989
with the American Type Culture Collection and was assigned
ATCC accession number 68194.
Applicants and their assignee, Genzyme Corporation,
acknowledge their responsibility to replace these cultures
should they die before the end of the term of a patent issued
hereon, 5 years after the last request for a culture, or 30
years, whichever is the longer, and their responsibility to
notify the depository of the issuance of such a patent, at
which time the deposit will be made irrevocably available to
the public.
60412-2031
CA 02339888 2001-03-19
s
- 27a -
Other Embodiments
Other embodiments are within the following claims.
For example, rGCR having an appropriate
60412-2031
CA 02339888 2001-03-19
- 28 -
carbohydrate structure can be produced by introducing
GCR-encoding DNA into any vertebrate or invertebrate
eukaryotic cell, and treating that cell during its
growth with inhibitors of carbohydrate processing such
as deoxy-mannojirimycin, swainsonine, castanospermine,
deoxy-nojirimycin, N-methyl-deoxy-nojirimycin, or their
equivalent inhibitors. These inhibitors act to inhibit
specific steps in the conversion of
Glc3Man9GlcNAc2 to smaller species shown in the
1o figure, thus, providing a greater number of exposed
mannose residues.