Note: Descriptions are shown in the official language in which they were submitted.
CA 02340168 2001-02-08
1
N-SUBSTITUTED AZABICYCLOHEPTANE DERIVATIVES,
PRODUCTION AND USE THEREOF
The invention relates to novel N-substituted azabicycloheptane
derivatives, their preparation and use for controlling diseases.
Exo-6-phenyl-3-azabicyclo[3.2.0]heptane deriviates have
interesting properties as potential neuroleptics (WO 94/00458,
WO 95/15312). Of particular importance in this connection are the
observed high affinities for D4 and 5-HT2 receptors.
The most interesting substance from the above class of compounds
with high D4/5-HTZA affinity and good selectivity versus Dz is
(+)-(1S,5R,6S)-exo-3-[2-[6-(4-fluorophenyl)-3-azbicyclo[3.2.0]-
heptan-3-yl]ethyl]-1H,3H-quinazoline-2,4-dione (= substance A),
which is a potential neuroleptic. However, there is an upper
limit to dosage of substance A due to prolongations which occur
in the QT interval in the cardiac [sic) ECG.
We have now found substances with better properties.
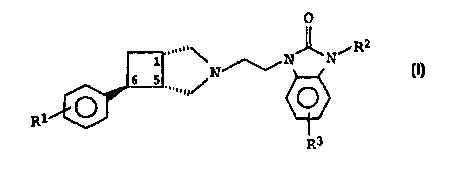
The invention relates to N-substituted 3-azabicyclo[3.2.0]-
heptane derivatives of the formula I
O
R2
.....,,\ ~N~Ni
6 5 N I,
R
R3
in which
R1 is fluorine or chlorine,
R2 is C1-C3-alkyl or cyclopropyl, and
R3 is hydrogen, fluorine or chlorine,
and the salts thereof with physiologically tolerated acids.
CA 02340168 2001-02-08
la
Preferred compounds are those in which
R1 is chlorine, preferably in the p position,
R2 is methyl or cyclopropyl and
R3 is hydrogen.
CA 02340168 2001-02-08
0050/49277
2
The following compounds are to be mentioned as particularly
preferred:
(+)-(1S,5R,6S)-exo-1-[2-[6-(4-chlorophenyl)-3-azabicyclo[3.2.0]-
heptan-3-yl]ethyl]-3-methyl-2H-1,3-dihydrobenzimidazol-2-one,
(+)-(1S,5R,6S)-exo-1-[2-[6-(4-chlorophenyl)-3-azabicyclo[3.2.0]-
heptan-3-yl]ethyl]-3-ethyl-2H-1,3-dihydrobenzimidazol-2-one, and
(+)-(1S,5R,6S)-exo-1-[2-[6-(4-fluorophenyl)-3-azabicyclo[3.2.0]-
heptan-3-yl]ethyl]-3-cyclopropyl-2H-1,3-dihydrobenzimidazol-
2-one.
The compounds of the formula I according to the invention can be
prepared by reacting a compound of the formula II
R2
N~N~
Nu ~ II,
R3
in which R2 and R3 have the abovementioned meanings, and in Nu is
a nucleofugic leaving group, with a 3-azabicyclo[3.2.0]heptane
derivative of the formula III as (+)-(1S,5R,exo-6S) enantiomer
1 ..,."~~ NH
6 5 , ~ III,
Rl O
in which R1 has the abovementioned meaning, and converting the
compound obtained in this way where appropriate into the acid
addition salt with a physiologically tolerated acid.
Suitable and preferred nucleofugic leaving groups for Nu are
halogen atoms, in particular bromine or chlorine.
The reaction expediently takes place in the presence of an inert
base such as triethylamine or potassium carbonate as acid
acceptor in an inert solvent such as a cyclic saturated ether, in
particular tetrahydrofuran or dioxane, or a benzenoid hydrocarbon
such as toluene or xylene.
CA 02340168 2001-02-08
0050/49277
3
The reaction generally takes place at temperatures from 20 to
150~C, in particular from 80 to 140~C, and is generally complete
within 1 to 10 hours.
The compounds of the formula I according to the invention can be
either recrystallized by recrystallization [sic] from
conventional organic solvents, preferably from a lower alcohol
such as ethanol, or purified by column chromatography.
The free 3-azabicyclo(3.2.0]heptane derivatives of the formula I
can be converted in a conventional way into the acid addition
salt with a pharmacologically suitable acid, preferably by
treating a solution with one equivalent of the appropriate acid.
Examples of pharmaceutically suitable acids are hydrochloric
acid, phosphoric acid, sulfuric acid, methanesulfonic acid,
sulfamic acid, malefic acid, fumaric acid, oxalic acid, tartaric
acid or citric acid.
The compounds according to the invention have valuable
pharmacological properties. They can be used as neuroleptics (in
particular atypical), antidepressants, sedatives, hypnotics, CNS
protective agents or agents for treating cocaine dependency. It
is possible for several of the types of action mentioned to occur
in combination in a compound according to the invention.
Z5
The substances are characterized in particular by a very high and
selective affinity for the dopamine D4 and serotonin 2A receptors.
The prolongations of the QT interval measured using the model of
the guinea pig papillary muscle are negligibly small. This means
that the novel substances are well tolerated even at high
dosages.
The invention accordingly also relates to a therapeutic
composition which has a content of a compound of the formula I or
its pharmacologically suitable acid addition salt as active
ingredient in addition to conventional carriers and diluents, and
to the use of novel compounds for controlling diseases.
The compounds according to the invention can be administered in a
conventional way orally or parenterally, intravenously or
intramuscularly.
The dosage depends on the age, condition and weight of the
patient and on the mode of administration. The daily dose of
active ingredient is usually between about 1 and 100 mg/kg of
CA 02340168 2001-02-08
0050/49277
4 ,
body weight on oral administration and between 0.1 and 10 mg/kg
of body weight on parenteral administration.
The novel compounds can be used in conventional solid or liquid
pharmaceutical forms, e.g. as uncoated or (film-coated) tablets,
capsules, powders, granules, suppositories, solutions, ointments,
creams or sprays. These are produced in a conventional way. The
active ingredients can for this purpose be processed with
conventional pharmaceutical aids such as tablet binders, bulking
agents, preservatives, tablet disintegrants, flow regulators,
plasticizers, wetting agents, dispersants, emulsifiers, solvents,
release-slowing agents, antioxidants and/or propellant gases (cf.
H. Sucker et al.: Pharmazeutische Technologie, Thieme-Verlag,
Stuttgart, 1978). The administration forms obtained in this way
normally contain the active ingredient in an amount of from 1 to
99~ by weight.
The substances of the formula II and III required as starting
materials for synthesizing the compounds according to the
invention are known (WO 94/00458; Heterocycles 40 (1), 319-330
(1995); J. Heterocyclic Chem. 18, 85-89 (1981)) or can be
synthesized from analogous starting materials using the
preparation methods described in the literature.
The following examples serve to illustrate the invention:
A Preparation of the starting materials
a) 1-(a-Phenylvinyl)-2H-1,3-dihydrobenzimidazol-2-one
21.6 g (200 mM [sic]) of o-phenylenediamine and 37 ml
(214 mM [sic]) of ethyl benzoylacetate in 75 ml of
4-tert-butyltoluene with the addition of a spatula tip of
p-toluenesulfonic acid were refluxed with a water trap at
a bath temperature of 200~C under nitrogen for 1 h, and
the liberated water was separated off. After cooling,
80 ml of acetonitrile were added to the reaction mixture
and, after stirring in an ice bath, the solid was
filtered off with suction and washed with cold
acetonitrile. 39.5 g (840) of product were isolated,
melting point 167-169~C.
1-(a-Phenylvinyl)-6-chloro-2H-1,3-dihydrobenzimidazol-2-one
can be prepared in an analogous manner (starting material:
4-chloro-1,2-diaminobenzene).
CA 02340168 2001-02-08
0050/49277
b) 1-(a-Phenylvinyl)-3-methyl-2H-1,3-dihydrobenzimidazol-
2-one
40 rnl of 10~ strength sodium hydroxide solution, 0.2 g of
5 benzyltriethylammonium chloride and 1.76 ml (18.5 mM
[sic]) of dimethyl sulfate were added to 3.5 g (14.8 mM
[sic]) of 1-(a-phenylvinyl)-2H-1,3-dihydrobenzimidazol-
2-one in 70 ml of toluene, and the mixture was stirred at
60~C for 2 h. The toluene phase was then concentrated,
and 3.65 g (99~) of product were isolated as an oil with
sufficient purity for the next reaction.
The following were prepared in an analogous manner:
1-(a-phenylvinyl)-3-ethyl-2H-1,3-dihydrobenzimidazol-
2-one (alkylating agent: diethyl sulfate) and
1-(a-phenylvinyl)-3-n-propyl-2H-1,3-dihydrobenzimidazol-
2-one (alkylating agent 1-bromopropane), 1-(a-phenyl-
vinyl)-3-methyl-6-chloro-2H-1,3-dihydrobenzimidazol-
2-one (starting material: 1-(a-phenylvinyl)-6-chloro-
2H-1,3-dihydrobenzimidazol-2-one) and 1-(2-chloroethyl-
3-methyl-5-chloro-2H-1,3-dihydrobenzimidazol-2-one [sic]
(starting material: 1-(2-chloroethyl-5-chloro-2H-
1,3-dihydrobenzimidazol-2-one [sic]).
c) 1-Methyl-2H-1,3-dihydrobenzimidazol-2-one
3.65 g (14.6 mM [sic]) 1-(a-phenylvinyl)-3-methyl-2H-1,3-
dihydrobenzimidazol-2-one were dissolved in 30 ml of
ethanol and, after addition of 60 ml of 10~ strength
hydrochloric acid and of 10 ml of concentrated
hydrochloric acid, stirred at 80~C for 2 h. The mixture
was then concentrated, and ice was added to the remaining
aqueous solution. The precipitated solids were stirred
while cooling in an ice bath, filtered off with suction
and washed with water. 1.7 g (79~) of product were
isolated.
The following were prepared analogously: 1-(2-chloro-
ethyl)-5-chloro-2H-1,3-dihydrobenzimidazol-2-one
(starting material: 1-(2-chloroethyl)-3-(a-phenylvinyl)-
5-chloro-2H-1,3-dihydrobenzimidazol-2-one) and 1-methyl-
5-chloro-2H-1,3-dihydrobenzimidazol-2-one (starting
material: 1-(a-phenylvinyl)-3-methyl-6-chloro-
2H-1,3-dihydrobenzimidazol-2-one).
CA 02340168 2001-02-08
0050/49277
6
d) 1-(2-Chloroethyl)-3-methyl-2H-1,3-dihydrobenzimidazol-
2-one
1.7 g (11.5 mM [sic]) of 1-methyl-2H-1,3-
dihydrobenzimidazol-2-one in 40 ml of acetonitrile were
mixed with 1.6 g (11.5 mM [sic]) of finely powdered
potassium carbonate and 2.9 ml (35 mM [sic]) of
1-bromo-2-chloroethane and refluxed for 14 h. After
cooling, the solids were filtered off with suction,
washing with acetonitrile, and then the filtrate was
concentrated. 2.3 g (95~) of product were isolated as an
oil which slowly crystallized, melting point 87-89~C.
The following were prepared analogously: 1-(2-chloro-
ethyl)-3-(a-phenylvinyl)-5-chloro-2H-1,3-dihydrobenz-
imidazol-2-one (starting material: 1-(a-phenylvinyl)-
6-chloro-2H-1,3-dihydrobenzimidazol-2-one) and
1-(2-chlproethyl)-3-methyl-6-chloro-2H-1,3-dihydro-
benzimidazol-2-one (starting material: 1-methyl-5-chloro-
2H-1,3-dihydrobenzimidazol-2-one).
e) (+)-(1S,5R,6S)-exo-6-(4-Chlorophenyl)-3-azabicyclo-
[3.2.0]heptane
The (+) enantiomer was isolated by the method described
in Heterocycles 40 (1), 326 (1995).
f) 1-(a-Phenylvinyl)-3-cyclopropyl-2H-1,3-dihydrobenz-
imidazol-2-one
1.35 g (45 mM [sic]) of sodium hydride (80 percent) were
added in portions to 10.0 g (42.4 mM [sic]) of
1-(a-phenylvinyl)-2H-1,3-dihydrobenzimidazol-2-one in
80 ml of dimethylformamide with thorough stirring, and
the reaction mixture was then stirred for 2 h.
Subsequently 8.0 ml (100 mM [sic]) of cyclopropyl bromide
were added, and the reaction mixture was transferred into
a 0.3 1 stirred autoclave which was then heated at 200~C
for 10 h. After cooling and concentration in a rotary
evaporator, the residue was partitioned between methylene
chloride and water, acidifying with 10 percent
hydrochloric acid. The aqueous phase was extracted once
more with methylene chloride. After drying and
concentration of the organic phase, 12.4 g of crude
product were isolated and were purified by column
chromatography (silica gel, mobile phase methylene
chloride). Yield 3.6 g (31g) of adequate purity.
CA 02340168 2001-02-08
0050/49277
7
B Preparation of the final products
Example 1
(+)-(1S,5R,6S)-exo-1-(2-[6-(4-Chlorophenyl)-3-azabicyclo-
[3.2.0]heptan-3-yl]ethyl]-3-methyl-2H-1,3-dihydrobenz-
imidazol-2-one tartrate x 2H20
2.5 g (12.1 mM [sic]) of (+)-(1S,5R,6S)-exo-6-(4-
chlorophenyl)-3-azabicyclo[3.2.0]heptane in 60 ml of xylene
were mixed with 2.55 g (12.1 mM [sic]) of
1-(2-chloroethyl)-3-methyl-2H-1,3-dihydrobenzimidazol-2-one
and 1.7 g (12.1 mM jsic]) of finely powdered potassium
carbonate and refluxed for 20 h. The mixture was then
concentrated in a rotary evaporator, and the residue was
partitioned between water and methyl tert-butyl ether at
pH 10. The aqueous phase was extracted once more with methyl
tert-butyl ether, and then the combined organic phases were
concentrated. The crude product was purified by column
chromatography (silica gel, mobile phase methylene
chloride/methanol 97/3. 3.3 g (71~) of product were isolated
as an oil which was dissolved in 200 ml of ether and
converted with l.4 g (9.3 mM [sic]) of tartaric acid,
dissolved in ethanol, into the tartrate (melting point 107 to
109~C). [a]o = + 55.4 (EtOH)
Elemental analysis C22H24N30C1 x C4H606 x 2H20
calculated C 54.97 H 6.03 N 7.39 O 25.34 C1 6.24
found C 54.9 H 5.8 N 7.1 O 25.5 C1 6.2
The following were prepared in an analogous manner:
2. (+)-(1S,5R,6S)-exo-1-[2-[6-(4-Chlorophenyl)-3-azabicyclo-
[3.2.0]heptan-3-yl]ethyl]-3-ethyl-2H-1,3-dihydrobenzimidazol-
2-one x HC1, melting point 174 to 176~C, [a]p = + 67,7 (EtOH)
3. (+)-(1S,5R,6S)-exo-1-[2-[6-(4-Chlorophenyl)-3-azabicyclo-
[3.2.0]heptan-3-yl]ethyl]-3-n-propyl-2H-1,3-dihydrobenz-
imidazol-2-one x HCl, melting point 178 to 180~C, [a]p =
+66.4 (EtOH)
4. (+)-(1S,5R,6S)-exo-1-[2-[6-(4-chlorophenyl)-3-azabicyclo-
[3.2.0]heptan-3-yl]ethyl]-3-methyl-5-chloro-2H-1,3-dihydro-
benzimidazol-2-one x HC1, melting point 101 to 103~C, [a]D =
+92.3 (EtOH)
CA 02340168 2001-02-08
0050/49277
8
5. (+)-(1S,5R,6S)-exo-1-[2-[6-(4-chlorophenyl)-3-azabicyclo
[3.2.0]heptan-3-yl]ethyl]-3-methyl-6-chloro-2H-1,3-dihydro-
benzimidazol-2-one x HC1, melting point 230 to 232~C, (a]D =
+77.2 (EtOH)
6. (+)-(1S,5R,6S)-exo-1-[2-[6-(4-chlorophenyl)-3-azabicyclo-
[3.2.0]heptan-3-yl]ethyl]-3-cyclopropyl-2H-1,3-dihydro-
benzimidazol-2-one x HC1, melting point 109 to 111~C, [a]D =
+73.5 (EtOH)
7. (+)-(1S,5R,6S)-exo-1-[2-[6-(4-fluorophenyl)-3-azabicyclo-
[3.2.0]heptan-3-yl]ethyl]-3-cyclopropyl-2H-1,3-dihydro-
benzimidazol-2-one x HC1, melting point 121 to 123~C, [a]D =
+64.5 (EtOH)
20
30
40