Note: Descriptions are shown in the official language in which they were submitted.
CA 02341353 2001-02-20
WO 00/14084 PCT/US99/20306
SUBSTITUTED DIHYDROBENZOPYRANS USEFUL
AS ANTIARRHYTHMIC AGENTS
TECHNICAL FIELD
This invention relates to compounds and pharmaceutical compositions comprising
these
compounds, useful in treating cardiac arrhythmia and/or cardiac fibrillation.
BACKGROUND OF THE INVENTION
The compounds of this invention are active as antifibrillatory and
antiarrhythmic agents.
These compounds exhibit broad efficacy against cardiac arrhythmia and
fibrillation and can be
satisfactorily applied to substantially alleviate and/or prevent arrhythmia
and fibrillation. In
addition, they exhibit a lower incidence of some of the undesirable side
effects than do many
conventional antiarrhythmic therapies. An additional benefit of the compounds
described herein
is that they exhibit both antifibrillatory and antiarrhythmic activity; most
conventional therapies
generally do not exhibit efficacy as antifibrillatory agents. See, e.g.
Coplen, S. E. et al.,
"Efficacy and Safety of Quinidine Therapy for Maintenance of Sinus Rhythm
after
Cardioversion: A meta-analysis," Circulation, Vol. 82, pp. 1106-1116 (1990);
and Echt, D. S. et
al., "Mortality and Morbidity in Patients receiving Encainide, Flecainide, or
Placebo: The
Cardiac Arrhythmia Suppression Trial", N. Engl. J. Med., Vol. 324, pp. 781-788
(1991), both
hereby incorporated by reference herein.
In a healthy, structurally sound heart, the precise, sequential electrical
activation, then
deactivation, of the entire cardiac muscle that occurs unerringly with each
beat is characterized
as normal cardiac rhythm. Arrhythmias are characterized as occurrences of
abnormal electrical
activity that can interfere with normal cardiac rhythm. The abnormal
electrical activity can
interfere with the initiation of, and/or the uniform spread of, the electrical
wave (i.e.
depolarization followed by repolarization of the cardiac muscle) that triggers
the heart to
contract. The disruption of the smooth, cyclical process of cardiac function
associated with
normal cardiac rhythm by the existence of arrhythmias is, in some instances,
life-threatening.
Arrhythmias range in severity from relatively benign (consisting of
asymptomatic and
infrequent premature ventricular complexes [PVCs]) to life-threatening
(consisting of
ventricular fibrillation, and sustained ventricular tachyarrhythmia). For an
excellent review of
arrhythmias and an overview of antiarrhythmic therapy, see, e.g. Bigger,
Thomas J.,
"Antiarrhythmic Treatment: An Overview", Am. J. Cardiol., Vol. 53, pp. 8B-16B,
1984; Reiffel,
J. A. "Prolonging survival by reducing arrhythmic death: pharmacologic therapy
of ventricular
CA 02341353 2001-02-20
WO 00/14084 PCT/US99/20306
2
tachycardia and fibrillation," Am. J. Cardiol.. Vol. 80(8A), pp. 45-55 (1997);
Ganz, L. L;
Antman, E. M. "Antiarrhythmic drug therapy in the management of atrial
fibrillation," J.
Cardiovasc. Electrophys., Vol. 8(10), pp. 1175-89 (1997), all hereby
incorporated by
reference herein. Life threatening arrhythmias are noted as a leading cause of
death
worldwide. For instance, it is estimated that sudden cardiac death resulting
from
venMcular fibrillation kills approximately 400,000-600,000 people in the
United States
each year. U.S. Department of Health and Human Sciences (1985) NCHS Monthly
Vital
Statistics Report 33:8-9.
Arrhythmias are generally classified into two types: I) supraventricular
arrhythmias (for example, atrial fibrillation and flutter) and 2) ventricular
arrhythmias (for
example, ventricular tachyarrhythmia and ventricular fibrillation and
flutter).
Supraventricular arrhythmias are generally not life threatening. Individuals
with
these arrhythmias may experience a wide range of symptoms, from slight to
severe
intensity. These individuals may feel the physical sensation of missed beats,
extra beats,
and/or flutter, may occasionally feel slightly light-headed or dizzy, and may
have shortness
of breath and/or chest pain. Since this situation is, in fact, generally not
life threatening,
more aggressive therapies such as conventional antiarrhythmic drugs sometimes
are not
prescribed, because the side effects usually associated therewith may not be
acceptable for
a non-life-threatening condition. However, the compounds of this invention are
generally
better tolerated than many of the conventional, currently available
antiarrhythmics;
therefore, they would likely be an acceptable therapy for individuals
suffering from
supraventricular arrhythmias and would substantially alleviate the discomfort
these
individuals experience.
Ventricular arrhythmias, on the other hand, are potentially much more serious
and
have been classified into three groups: 1) benign; 2) prognostically-
significant (potentially
lethal); and 3) life threatening (lethal). See, e.g. Morganroth, J. and
Bigger, J. T.,
"Pharmacological management of ventricular arrhythmias after the Cardiac
Arrhythmia
Suppression Trial", Amer. J. Cardiol., Vol. 65, pp. 1497-1503 (1990), hereby
incorporated
by reference herein (Mor ag nroth).
Individuals with benign arrhythmias exhibit very low risk of death, cardiac
scarring,
and heart disease. Benign ventricular arrhythmias are relatively common and
account for
approximately 30% of all ventricular arrhythmias. Benign arrhythmias, such as
premature
ventricular complexes (PVCs), pose minimal risks to individuals and rarely
require
antiarrhythmic therapy. However, the PVCs may be of a frequency or complexity
to
provide alarming symptoms.
CA 02341353 2001-02-20
WO 00/14084 PCT/US99/20306
3
Individuals who exhibit "prognostically-significant" arrhythmias may benefit
from
antiarrhythmic therapy. These individuals generally have suffered a myocardial
infarction
and may have PVCs and/or episodes of non-sustained ventricular
tachyarrhythmia, either
symptomatic or asymptomatic. They may have not immediate, urgent life-
threatening
symptoms, and are not typically in danger of death. They are, however, at a
significantly
greater risk of sudden death than the general populace, and, accordingly,
would be at a
lessened risk of cardiac failure with therapy from the compounds of this
invention. See
Morganroth & Big er at 1498.
Others exhibit sustained ventricular tachyarrhythmia or ventricular
fibrillation
which are life-threatening. These ventricular arrhythmias generally produce
symptoms such
as syncope, heart failure, myocardial ischemia or hypotension. These patients
have the
highest risk of sudden cardiac death and usually the most severe form of
underlying cardiac
disease. (Mor agmoth p. 1498).
The pharmacotherapy of cardiac arrhythmias has over the years employed drugs
with diverse mechanisms of action. These drugs have been classified by their
actions on
the characteristic shape of the cardiac action potential. See Vaughan-
Williams, E. M., "A
classification of antiarrhythmic actions reassessed after a decade of new
drugs." J. Clin.
Pharmacol. Vol. 24, pp. 129-147 (1984). More recently, a classification has
been proposed
on the basis of their individual, characteristic effects on ion channels and
receptor systems
of the myocardium. See "The Sicilian gambit. A new approach to the
classification of
antiarrhythmic drugs based on their actions on arrhythmogenic mechanisms. Task
Force of
the Working Group on Arrhythmias of the European Society of Cardiology,"
Circulation,
Vol. 84 (4), pp. 1831-1851 (1991).
In the Vaughan-Williams scheme, Class I antiarrhythmic drugs act to slow the
upstroke of the cardiac action potential, and slow the conduction velocity of
the electrical
impulse across the heart. These agents act principally by blocking myocardial
Nay
channels.
Class II antiarrhythmic drugs prolong the interval between cardiac action
potentials,
i.e., they slow the rate at which the heart beats. These compounds act by
blocking
catecholamine receptors of the heart.
Class III antiarrhythmic agents prolong the duration of the cardiac action
potential.
This leads to an increase in the refractory period of the heart without a
reduction in the
conduction velocity of the electrical impulse. These compounds act by, for
example,
blocking myocardial potassium currents.
CA 02341353 2001-02-20
WO 00/14084 PCT/US99/20306
4
The pharmacotherapy of cardiac arrhythmias has been undergoing major changes
since 1991, when the Cardiac Arrhythmia Suppression Trials (CAST) showed that
the Class
I antiarrhythmic agents encainide, flecainide, and moricizine were associated
with an
increased risk of sudden cardiac death. As a result of the CAST trials, the
use of Class I
agents is generally avoided in patients who have structural heart disease,
i.e. a prior
myocardialinfarction. See Moreanroth.
Instead, the focus of antiarrhythmic pharmacotherapy has shifted to the use of
Class
III agents. For instance, in the CASCADE clinical trial, the antiarrhythmic
agents d,l-
sotalol and amiodarone were shown to have neutral or slightly beneficial
effects on
mortality. Amiodarone and sotaIol share certain mechanistic features that may
have
contributed to these results, including a mixed Class II and Class III
antiarrhythmic action.
See "Randomized antiarrhythmic drug therapy in survivors of cardiac arrest
(the
CASCADE study)," Am. J. Cardiol.. Vol. 72 (3), pp. 280-7 (1993).
Two major clinical trials (the SWORD trial and the DIAMOND trial) have been
conducted to test whether a pure Class III agent can reduce the incidence of
sudden cardiac
death in patients with depressed left ventricular function following an acute
myocardial
infarction. The SWORD trial was designed to test whether a pure Class III
antiarrhythmic
agent, d-sotalol, in patients with depressed ventricular function following
acute myocardial
infarction or with symptomatic congestive heart failure, would reduce the
incidence of
sudden cardiac death. The trial was terminated prematurely because of excess
mortality in
the d-sotalol-treated group. See Waldo, A. L.; Camm, A. J.; deRuyter, H.;
Friedman, P. L.;
MacNeil, D. "Effect of d- sotalol on mortality in patients with left
ventricular dysfunction
after recent and remote myocardial infarction. The SWORD Investigators.
Survival With
Oral d- Sotalol," Lancet, Vol. 348(9019), pp. 7-I2 (1996).
In the DIAMOND trial, a neutral effect on mortality was observed with the
highly
potent and selective Class III agent dofetilide. See "Dofetilide in patients
with left
ventricular dysfunction and either heart failure or acute myocardial
infarction: rationale,
design, and patient characteristics of the DIAMOND studies. Danish
Investigations of
Arrhythmia and Mortality ON Dofetilide," Clinical CardioloQV, Vol. 20 (8), pp.
704-10
(1997).
Another pure Class III antiarrhythmic agent, ibutilide, has recently gained
marketing authorization for the rapid termination of atrial arrhythmias to
sinus rhythm. The
drug is associated with significant incidence of potentially dangerous
ventricular
arrhythmias, a fact that severely restricts its utility. See Foster, R. H.;
Wilde, M. L;
Markham, A. "Ibutilide : a review of its pharmacological properties and
clinical potential in
CA 02341353 2001-02-20
WO 00/14084 PCT/US99/20306
S
the acute management of atrial flutter and fibrillation," Drues, Vol. 54(2),
pp. 312-330
( 1997).
The compounds of the present invention are Class III antiarrhythmic agents
with
mechanistic features that herald a significant advance over existing
antiarrhythmic agents in
the treatment of cardiac arrhythmias.
The mechanism by which Class III antiarrhythmics typically achieve their
effects
on the cardiac action potential is by inhibiting the efflux of K+ ions from
cardiac myocytes.
The main outward K+ conductance that terminates the plateau phase of the human
cardiac
action potential is termed the delayed rectifier current, IK. Cellular
electrophysiological,
molecular biological, and genetic studies have demonstrated that the IK
current is carried
by two kinetically and molecularly distinct ion channel complexes. They are
named after
their distinct current sub-types, I~ (rapidly activating and deactivating;
carried by the
HERG protein), and IKs (slowly activating and deactivating; carried by the
KvLQTl/minK
protein complex). See Sanguinetti, M. C.; Jiang, C.; Curran, M. E.; Keating,
M. T. "A
mechanistic link between an inherited and an acquired cardiac arrhythmia and
HERG
encodes the IKr potassium channel," Cell, Vol. 81{2), pp. 299-307 (1995) and
Sanguinetti,
M. C.; Curran, M. E.; Zou, A., et al. "Coassembly of KvLQTl minx (IsK)
proteins to form
cardiac IKs potassium channel," Nature, Vol. 384, pp. 80-83 (1996}. Compounds
which
selectively block I~ (such as dofetilide and d-sotalol) have been associated
with an
enhanced risk of proarrhythmia, especially at slow heart rates. See Nair, L.
A., Grant, A. O.
"Emerging class III antiamhythmic agents: mechanism of action and
proarrhythmic
potential," Cardiovasc. Drubs Ther., Vol. 11(2), pp. 149-167 (1997) and
Colatsky, T. J.;
Argentieri, T. M. "Potassium channel blockers as antiarrhythmic drugs,"
DrugYDev. Res.,
Vol. 33(3), pp. 235-49 (1994) and Hondeghem, L. M.; Snyders, D. J. "Class III
antiarrhythmic agents have a lot of potential but a long way to go. Reduced
effectiveness
and dangers of reverse use dependence," Circulation. Vol. 81 (2), pp. 686-90
(1990).
The compounds of the present invention act primarily by the blockade of IKs.
Accordingly, they can terminate atrial and ventricular arrhythmias, as well as
reduce their
frequency and severity. IKs blockers can also reduce the risk of sudden
cardiac death. See
U.S. Pat. 5658901 to Claremon D. A., Liverton N., Selnick H. G., issued 1997
and Selnick,
H. G.; Liverton, N. J.; Baldwin, J. J., et al. "Class III antiarrhythmic
activity in vivo by
selective blockade of the slowly activating cardiac delayed rectifier
potassium current IKs
by (R)-2-(2,4-(1,1,1-Mfluoromethyl})-N-[2-oxo-S-phenyl-1-(2,2,2-
trifluoroethyl)-2,3-
dihydro-1H-benzo[a][1,4]diazepin-3-yl]acetamide," J. Med. Chem., Vol. 40 (24),
pp. 3865-
3868 (1997).
CA 02341353 2001-02-20
WO 00/14084 PCT/US99/20306
6
Since the KvLQTl ion channel and/or the IKs current may have additional
physiological roles in certain other tissues, such as the stimulated
production of gastric acid,
the compounds of the current invention can be used to reduce gastric acid
secretion. This
makes them useful in the treatment of gastric (duodenal) ulcers. See Wargh,
R.; Riedemann,
N.; Bleich, M.; Van Driessche, W.; Busch, A. E.; Greger, R. "The cAMP-
regulated and
2938-inhibited K+ conductance of rat colonic crypt base cells," Pflue~ers
Arch., Vol. 432
(1), pp. 81-88 (1996) and Suessbrich, H.; Bleich, M.; Ecke, D., et al.
"Specific blockade of
slowly activating I(sK) channels by chromanols -- impact on the role of I(sK)
channels in
epithelia," FEBS Letters, Vol. 396 (2-3), pp. 271-5 (1996). The compounds of
the current
invention can also be used for the treatment of diarrhea, see van Kuijck, M.
A.; van Aubel,
R. A. M. H.; Busch, A. E., et al. "Molecular cloning and expression of a
cyclic AMP-
activated chloride conductance regulator: a novel ATP-binding cassette
transporter," Proc.
Natl. Acad. Sci. U.S.A. Vol. 93 (11), pp. 5401-5406 (1996).
The compounds of the present invention contain a heterocyclic ring system
referred
to as a dihydrobenzopyran. Cromakalim (compound 1 below) is a 4-amido-2,2-
dialkyl-3-
hydroxybenzopyran antihypertensive that has shown promise in the treatment of
asthma or
of high blood pressure.
0
NC ~ ,,,OH
O
1
Cromakalim and related 4-amido-2,2-dialkyI-3-hydroxybenzopyrans have been
investigated extensively and summaries of this field are readily available.
See Evans, J. M.;
Hamilton, T. C.; Longman, S. D.; Stemp, G. Potassium Channels and their
Modulators:
From Synthesis to Clinical Experience. Bristol, PA: Tayler & Francis, 1996.
These
compounds exert their pharmacological actions by stimulating the ATP-sensitive
K+
current (IK-ATP) in smooth muscle cells, such as are found in the vasculature
of the
airways or in peripheral blood vessels. Activating IK_ATP results in a
hyperpolarizing
response and eventually the relaxation of these muscle cells and dilation of
the vessel. This
stimulation of an outward K+conductance is the opposite effect that is
characteristic of
Class III antiarrhythmic drugs, which block outward K+currents.
Benzopyrans of structure similar to that of cromakalim have been described in
the
literature:
CA 02341353 2001-02-20
WO 00/14084 PCT/US99/20306
7
U.S. Patent 5,082,858 discloses certain 2,3-dihydro-2,2-dimethyl-3-hydroxy-
benzopyrans. These are said to be antithrombotics and to have effects on the
central
nervous system (CNS), primarily as antidepressants. The compounds are also
said to have
been studied for cardiac effects and they had insignificant or no activity
(see Col. 1 and 8)
European Patent Application 389,861 discloses certain 3-hydroxy-2,2-
dimethyldihydrobenzopyrans that are taught to be potassium channel openers and
activators
of Ig_ATP They are said to have effects like cromakalim, such as vasodilating
and
antihypertensive effects.
U.S. Patent 4,882,353 discloses certain 2,2-dimethyldihydrobenzopyrans that
are
taught to be potassium channel openers and activators of IK_ATP They are said
to have
effects like cromakalim, such as vasodilating and antihypertensive effects as
well as
antithrombotic effects.
U.S. Patent 5,206,252 discloses 4-thiazadolyl-benzopyrans. They are said to
have
effects like cromakalim, such as vasodilating and antihypertensive effects, as
well as
amelioration of urinary incontinence.
Soll, R. M.; Dollings, P. J.; McCaully, R. "N-sulfonamides of benzopyran-
related
potassium channel openers: conversion of glyburide insensitive smooth muscle
relaxants to
potent smooth muscle contractors," Bioorg. Med. Chem. Lett., Vol. 4(5), pp.
769-73 (1994)
describes 6-trifluoromethoxy-4-((l,l,l-trifluoromethyl)sulfonylamino)-
substituted dihydro-
benzopyrans that were evaluated in different tissues and may be K+ channel
blockers,
although the text is not clear.
European Patent 807,629 discloses two different types of 2,3-dihydro-4-
sulfonamidobenzopyrans said to be K+ channel blockers. The reference teaches
on page
11, line 30, that removal of the 3-hydroxy substituent enhances the potency of
action. It is
also taught that the preferred absolute stereochemistry of the 3 and 4
positions is 3S,4R.
See Lohrmann, E.; Burhoff, L; Nitscke, R. B.; et al. "A new class of
inhibitors of cAMP-
mediated Cl secretion in rabbit colon, acting by the reduction of cAMP-
activated K+
conductance," Pflii~ers Arch. - Eur. J. Physiol., Vol 429, 517-530 (1995); and
Suessbrich,
H.; Bleich, M.; Ecke, D.; et al. "Specific blockade of slowly activating IsK
channels by
chromanols - impact on the role of IsK channels in epithelia," FEBS Lett. Vol.
396, 271-
275 (1996). The compounds of the present invention differ from those described
in these
references, by virtue of the surprising and unexpected increase in
pharmacological potency
of 3-hydroxy-4-sulfonamidobenzopyrans when certain substituents are placed in
the 6-
position of the benzopyran. Furthermore, the preferred compounds of the
present invention
have the 3R,4S stereochemistry. This is in direct contrast to the teachings of
Lohrmann et
CA 02341353 2001-02-20
WO 00/14084 PCTlUS99/20306
g
al. and Suessbrich et al., which suggest the preferred stereochemistry is
3S,4R. Applicants
have also discovered that the 3R,4S preference also applies to those compounds
described
by Lohrmann et al. and Suessbrich et al.
The compounds of the present invention differ significantly in their chemical
structure, pharmacological actions and therapeutic utility from certain 2-
spiropiperidinylbenzopyran derivatives, such as MK-499. See Spector, P. S.;
Curran, M.
E.; Keating, M. T.; Sanguinetti, M. C. "Class III antiarrhythmic drugs block
HERG , a
human cardiac delayed rectifier K+ channel: open-channel block by
methanesulfonanilides," Circ. Res., Vol. 78(3), pp. 499-503 (1996) and Lynch,
J. J., Jr.;
Wallace, A. A.; Stupienski, R. F., III, et al. "Cardiac electrophysiologic and
antiarrhythmic
actions of two long-acting spirobenzopyran piperidine class III agents, L-
702,958 and L-
706,000 [MK-499]," J. Pharmacol. Exp. Ther., Vol. 269(2), pp. 541-554 (1994).
These
compounds are known to block IKr, and have been associated with an increased
predisposition to cardiac arrhythmias in animal studies and humans.
SUMMARY OF THE INVENTION
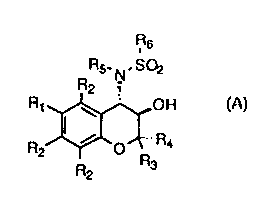
The compounds of this invention are 2,3-dihydro-2,2-dimethyl-3-hydroxy-4-
sulfonamidobenzopyran derivatives of Formula (A):
Rs
R5_ N, S02
R2
R~ ~ OH
R I , O~ R
4
2 R
R 3
2
wherein
(a) Rl is selected from haloalkyl, haloacyl, NR7S02Rg, and
NR7CORg;
(b) each R2 is independently selected from hydrogen, alkyl, halo, alkoxy, and
alkoxyalkyl;
(c) R3 is lower alkyl, or together with R4 forms a spirocycle of from 4 to 7
members;
(d) R4 is lower alkyl, or together with R3 forms a spirocycle of from 4 to 7
members;
(e) RS is selected from hydrogen, alkyl, aryl, arylalkyl, and haloalkyl;
CA 02341353 2001-02-20
WO 00/14084 PCT/US99/20306
9 -_
(f) R6 is selected from alkyl, aryl, cycloalkyl, arylalkyl, haloalkyl, and
alkylamino;
(g) R7 is selected from hydrogen and alkyl; and
(h) Rg is C1-C4 haloalkyl.
This structure also includes any single diastereomer or enantiomer of Formula
(A),
or mixtures of diastereomers or enantiomers of Formula (A). Also included in
the scope of
the present invention is a pharmaceutically-acceptable salt, or
biohydrolyzable amide, ester,
or imide thereof, or any other derivative which is bioconverted to Formula
(A). The
invention further relates to pharmaceutical compositions containing the
compounds of
Formula (A) and methods of delivering these compounds and pharmaceutical
compositions,
for treating arrhythmic disorders and conditions, gastric ulcers, and
diarrhea.
The compounds of this invention are antiarrhythmic agents that exhibit less of
the
undesirable side effects (e.g., pulmonary toxicity, cardiac depression, and
neurological
effects nonspecific to cardiac tissue) associated with many conventional
antiarrhythmic
therapies. In addition the compounds of this invention are useful in the
treatment of
diarrhea and of ulcers.
DETAILED DESCRIPTION OF THE INVENTION
The compounds of the present invention bear a unique substitution pattern and
opposite stereoselectivity than cromakalim-like benzopyrans, which results in
an
unexpected increased potency and selectivity for Igs. Consequently, these
compounds hold
promise as useful antiarrhythmic agents, anti-ulcer agents, and anti-diarrheal
agents.
Furthermore, they are expected to have fewer undesirable side effects than
existing
antiarrhythmic drugs such as amiodarone, dofetilide, or ibutilide.
Definitions and Usage of Terms:
The following is a list of definitions for terms used herein.
"Acyl" or "carbonyl" is described as a radical which could be formed by
removal of the hydroxyl moiety from a carboxylic acid (i.e., R-C(=O)-).
Preferred acyl
groups include (for example) acetyl, formyl, and propionyl.
"Alkenyl" is an unsubstituted or substituted hydrocarbon chain radical having
2
to 15 carbon atoms; preferably from 2 to 10 carbon atoms; more preferably from
2 to 8;
except where indicated. Alkenyl substituents have at least one olefinic double
bond
(including, for example, vinyl, allyl and butenyl).
CA 02341353 2001-02-20
WO 00/14084 PCT/US99/20306
10
"Alkoxy" is an oxygen radical having a hydrocarbon chain substituent, where
the hydrocarbon chain is an alkyl or alkenyl (i.e., -O-alkyl or -O-alkenyl).
Preferred
alkoxy groups include (for example) methoxy, ethoxy, propoxy and allyloxy.
"Alkoxyalkyl" is an unsubstituted or substituted alkyl moiety bearing an
alkoxy
moiety (i.e., -alkyl-O-alkyl). Preferred is where the alkyl has 1 to 6 carbon
atoms
(more preferably 1 to 3 carbon atoms), and the alkoxy has 1 to 6 carbon atoms
(more
preferably 1 to 3 carbon atoms).
"Alkyl" is an unsubstituted or substituted saturated hydrocarbon chain radical
having 1 to I S carbon atoms; preferably from 1 to 10 carbon atoms; more
preferably 1
to 4; except where indicated. Preferred alkyl groups include (for example)
substituted
or unsubstituted methyl, ethyl, propyl, isopropyl, and butyl. A "lower"
hydrocarbon
moiety (e.g., "lower" alkyl) is a hydrocarbon chain comprised of 1 to 6,
preferably
from 1 to 4, carbon chain atoms.
As referred to herein, "spirocycle" or "spirocyclic" or "spiro ring" refers to
a
cyclic moiety sharing a single atom (e.g., carbon) of another ring. Such a
cyclic
moiety may be carbocyclic or heterocyclic in nature. Preferred spirocyclic
ring sizes
include 5-7 membered rings. Preferred heteroatoms included in the backbone of
the
heterocyclic spirocycle include oxygen, nitrogen and sulfur. In addition, the
heteroatom of the heterocycle may be alkyl- or acyl-substituted if valence
allows.
"Alkylamino" is an amino radical having one (secondary amine) or two
(tertiary amine) alkyl substituents (i.e., -NH-alkyl or N-(alkyl)2). For
example,
methylamino (-NHCH3), dimethylamino (-N(CH3)2), methylethylamino (-
N(CH3)CH2CH3).
"Aryl" is an aromatic carbocyclic ring radical. Preferred aryl groups include
(for example) phenyl, indenyl, naphthyl, biphenyl and fluorenyl. Such groups
may be
substituted or unsubstituted.
"Arylalkyl" is an alkyl radical substituted with an aryl group. Preferred
arylalkyl groups include benzyl, phenylethyl, and phenylpropyl. Such groups
may be
substituted or unsubstituted.
"Carbocyclic ring" is an unsubstituted or substituted, saturated or
unsaturated
hydrocarbon ring radical. Carbocyclic rings are monocyclic or are fused,
bridged or
spirocyclic ring systems. Monocyclic carbocyclic rings generally contain 4 to
9 atoms,
preferably 4 to 7 atoms. Polycyclic carbocyclic rings contain 7 to 17 atoms,
preferably
from 7 to 12 atoms. Preferred polycyclic systems comprise 4-,5-,6- or 7-
membered
rings fused to 5-,6-,or 7-membered rings.
CA 02341353 2001-02-20
WO 00/14084 PCT/US99/20306
11
"Cycloalkyl" is a substituted or unsubstituted, saturated hydrocarbon ring
radical comprising from 3 to 6 ring atoms. Examples include cyclohexyl,
cycloheptyl,
cyclobutyl and cyclopropyl.
"Fluoroalkyl" is haloalkyl wherein the only halogen atom contemplated is
fluorine.
"Fused rings" are rings that are superimposed together such that they share
two
ring atoms. A given ring may be fused to more than one other ring. Fused rings
are
contemplated in heteroaryl, aryl and heterocycle radicals or the like.
"Halo", "halogen", or "halide" is a chloro, bromo, fluoro or iodo atom
radical.
Bromo, chloro and fluoro are preferred halides.
"Haloalkyl" is branched or unbranched alkyl radical having one or several halo
substitutents (i.e., -CnH(2n-m)x(m+1 )~ where n = 1 to 15 and m = 0 to 2n).
"Haloacyl" is acyl radical having one or several halo substituents (i.e., -
C(=O)CnH(2n-m)X(m+1)~ where n = 1 to 4, and m = 0 to 2n, e.g. COCF3~ COCF2CF3,
etc.
"Heterocylic ring" is an unsubstituted or substituted, saturated or
unsaturated
ring radical comprising at least one heteroatom (i.e., O, S, or N).
Heterocyclic rings are
monocyclic or are fused, bridged or spirocyclic ring systems. Monocyclic
heterocyclic
rings generally contain 4 to 9 atoms, preferably 4 to 7 atoms. Polycyclic
heterocyclic
rings contain 7 to 17 atoms, preferably from 7 to 12 atoms. Preferred
polycyclic
heterocyclic systems comprise 4-, 5-, 6- or 7-membered rings fused to 5-, 6-,
or 7-
membered rings.
A "pharmaceutically-acceptable salt" is a cationic salt formed at any acidic
(e.g., carboxyl) group, or an anionic salt formed at any basic (e.g., amino)
group.
Many such salts are known in the art, as described in World Patent Publication
87/05297, Johnston et al., published September 11, 1987 (incorporated by
reference
herein). Preferred cationic salts include the alkali metal salts (such as
sodium and
potassium), and alkaline earth metal salts (such as magnesium and calcium) and
organic salts. Preferred anionic salts include the halides (such as chloride
salts),
phosphates, acetates, citrate and maleate salts.
"Biohydrolyzable carbamates" are carbamate derivatives of the compounds of
the invention that do not interfere with the ion channel inhibitory activity
of the
compound, or that are readily converted in vivo by a mammal subject to yield
an active
ion channel blocker.
CA 02341353 2001-02-20
WO 00/14084 PCT/US99/20306
12
A "biohydrolyzable ester" refers to an ester that does not interfere with the
ion
channel inhibitory activity of these compounds or that is readily converted in
vivo by a
mammal to yield an active ion channel Mocker.
"Optical isomer", "stereoisomer", "diastereomer" as referred to herein have
the
standard art recognized meanings (Cf., Hawlev's Condensed Chemical Dictionarx,
I lth
Ed.).
The illustration of specific esters, amides or other derivatized forms of the
Formula (A) compounds is not intended to be limiting. The application of other
useful
protecting groups, salt forms, etc. is within the ability of the skilled
artisan.
Compounds
The present compounds have a structure according to Formula (A):
Rs
R5_ N , S02
R2
R1 ~ OH
R2 I / OT R (A)
4
R R3
2
wherein
(a) RI is selected from haloalkyl, haloacyl, NR~S02Rg, and NR7CORg
(preferably haIoalkyl, NR~S02Rg and NR~CORg, more preferably haloalkyl,
and most preferably trifluoromethyl, pentafluoroethyl and trifluoroacetamido);
(b) each R2 is independently selected from hydrogen, alkyl, halo, alkoxy, and
alkoxyalkyl (preferably hydrogen and halo, more preferably hydrogen);
(c) R3 is lower alkyl, or together with R4 forms a spirocycle of from 4 to 7
members (preferably methyl, ethyl, or together with R4 spiropentyl);
(d) R4 is lower alkyl, or together with R3 forms a spirocycle of from 4 to 7
members (preferably methyl, ethyl, or together with R3 spiropentyl);
(e) R5 is selected from hydrogen, alkyl, aryl, arylalkyl, and haloalkyl
(preferably
alkyl and haloalkyl, more preferably methyl, ethyl, propyl, butyl,
trifluoromethyl, and 1,1,1-trifluorobutyl);
6. R6 is selected from alkyl, cycloalkyl, aryl, arylalkyl, haloalkyl, and
alkylamino
(preferably alkyl and haloalkyl, most preferably methyl, ethyl, propyl, butyl,
and 1,1,1-trifluorobutyl);
7. R7 is selected from hydrogen and alkyl (preferably hydrogen); and
8. Rg is C1-C4 haloalkyl (preferably trifluoromethyl).
CA 02341353 2001-02-20
WO 00/14084 PCT/US99/20306
13 _
Using the examples and this discussion the skilled artisan can generate a
variety of
compounds in a similar fashion, using the guidance of the general synthetic
scheme below.
These steps may be varied to increase yield of desired product. The skilled
artisan will also
recognize the judicious choice of reactants, solvents, and temperatures is an
important
component in successful synthesis. While the determination of optimal
conditions, etc. is
routine, it will be understood that a variety of compounds can be generated in
a similar
fashion, using the guidance of the schemes herein.
The starting materials used in preparing the compounds of the invention are
either
known, made by known methods, or are commercially available.
It is recognized that the skilled artisan in the art of organic chemistry can
readily
carry out standard manipulations of organic compounds without further
direction; that is, it
is well within the scope and practice of the skilled artisan to carry out such
manipulations.
These include, but are not limited to, reduction of carbonyl compounds to
their
corresponding alcohols, oxidations of hydroxyls and the like, acylations,
aromatic
substitutions, both electrophilic and nucleophilic, etherifications,
esterification and
saponification and the like. Examples of these manipulations are discussed in
standard
texts such as March, Advanced Organic Chemistry (Whey), Carey and Sundberg,
Advanced
Organic Chemistry (Vol. 2).
The skilled artisan will readily appreciate that certain reactions are best
carried out
when other functionality is masked or protected in the molecule, thus avoiding
any
undesirable side reactions and/or increasing the yield of the reaction. Often
the skilled
artisan utilizes protecting groups to accomplish such increased yields or to
avoid the
undesired reactions. These reactions are found in the literature and are also
well within the
scope of the skilled artisan. Examples of many of these manipulations can be
found for
example in T. Greene, Protecting-Groups in Or ag nic Synthesis. Of course,
amino acids
used as starting materials with reactive side chains are preferably blocked to
prevent
undesired side reactions.
The compounds of the invention may have one or more chiral centers. As a
result,
one may selectively prepare one stereoisomer (a diastereomer, epimer, or
enantiomer) over
another, for example by chiral starting materials, catalysts or solvents; by
chromatographic
means; by crystallization or distillation; or one may prepare mixtures of
stereoisomers
(mixtures of diastereomers, epimers, or enantiomers) at once. The present
invention
explicitly encompasses either single stereoisomers or mixtures thereof.
In addition, it is recognized that one optical isomer, including diastereomer
and
enantiomer, or stereoisomer may have favorable properties over the other. Thus
when
CA 02341353 2001-02-20
WO 00/t4084 PCT/US99/20306
14
disclosing and- claiming the invention, when one racemic mixture is disclosed,
it is clearly
contemplated that both optical isomers, including diastereomers and
enantiomers, or
stereoisomers substantially free of the other are disclosed and claimed as
well. Indeed, as
discussed above, with respect to preferred compounds, the 3R,4S
stereochemistry is
preferred over the 3S,4R racemate, which is contrary to the teachings of the
prior art
concerning benzopyrans.
Preparation of the compounds
General Reaction Scheme 1
R~ X
R~~H II Rt~H
Tll~ ll R ~ R ~
OH KZCO~ R~ O
I al
~ 2
O p O
R,~~ R3~R, R~ S.NaBH , R~
_ I 2. p-T80H ''~~'~ R a
OH NH R3 ~ Rp O R4
Rp
YI VII IV
R3 0
Rs v OMe
R~ y H VIII Rt H
Rx7 R~ ~0
R~OH R~ O- v 'OMe
Z
IX
~'-N'S~ RS.N.SOp
H
R~ w W [O] R~ ~ O XI R~~~OH
O Ra 3 ~" / / O R3 ~ / / O R3
2 ~z R4 R2 Ra
IV XII
X
CA 02341353 2001-02-20
WO 00/14084 PCT/US99/20306
15 -
Compounds of the present invention can be prepared via synthetic routes well-
established in the literature, and outlined in Scheme 1. Reaction of
appropriately
substituted phenols, I, with a 3-X-alkyne II (where halo or other suitable
leaving group is
X, such as in a 3-chloroalkyne derivative) in the presence of an acid
scavenger such as
K2C03 in a solvent such as acetone or dimethylformamide, provides the
corresponding
propargyl phenyl ethers III, which rearrange under appropriate thermal
conditions (e.g.
50°C-200°C, preferably 150°C) in the presence of an
appropriate base (e.g. aniline,
dimethylaniline, or preferably diethylaniline) in an organic solvent such as
dimethylsulfoxide, acetone, or, preferably, dimethylformamide, to yield the
corresponding
2,2-disubstituted benzopyrans IV. Alternatively, the compounds of general
structure IV are
arrived at by reduction of the 2,2-disubstituted benzopyran-3-ones of general
structure VII
by the action of various reducing agents, including NaBH4, in inert solvents
such as
tetrahydrofuran, and subsequent dehydration by the action of, for example,
para-
toluenesulfonic acid. The requisite 2,2-disubstituted benzopyran-3-ones of
general
structure VII are prepared by either of two general methods. First, by
condensation of
appropriately substituted 2-hydroxyphenyl methyl ketones VI, in the presence
of a
secondary amine, such as pyrollidine, and under removal of H20. See Bergmann,
R. et al.
"Synthesis and antihypertensive activity of 4-(1,2-dihydro-2-oxo-1-pyridyl)-2H-
1-
benzopyrans and related compounds, new potassium channel activators" J. Med.
Chem.
Vol. 33, pp. 492-504 (1990). Second, by reaction of phenols of general
structure I with
acrylate derivatives VIII to provide the esters of general structure IX.
Cyclization to the
2,2-disubstituted benzopyran-3-ones of general structure VII is achieved under
the
influence of strong Lewis or Brensted acids, for example under the action of
polyphosphoric acid or AIC13 in inert solvents such nitromethane.
The 2,2-disubstituted benzopyrans of general structure IV are epoxidized under
a
variety of conditions known to those skilled in the art, for instance by the
method of
Jacobsen. See Lee, N.-H. et al. "Enantiomerically pure epoxychromans via
asymmetric
catalysis." Tetrahedron Letters Vol. 32(38), pp. 5055-8 (1991). The resulting
epoxides X
are reacted with the anion of an appropriately substituted sulfonamides XI to
yield the
target compounds XII. See Lohrmann, E.; Burhoff, L; Nitscke, R. B.; et al. "A
new class of
inhibitors of CAMP-mediated Cl secretion in rabbit colon, acting by the
reduction of
cAMP-activated K+ conductance," Pfliipers Arch. - Eur. J. Ph siol., Vol 429,
517-530
(1995). The requisite sulfonamides XI are conveniently and routinely prepared
by reaction
CA 02341353 2001-02-20
WO 00/14084 PCT/IJS99/20306
16 _
of an appropriate primary amine and an appropriate sulfonyl chloride or
sulfonic acid
anhydride in an inert solvent such as dichloromethane.
General Reaction Scheme 2
Rs Fis
RS.N_$O? RS.N_SOZ
Ri ~ OH SnClz / EtOH HzN ~ OH
O Ra ~ ~ i O Ra
Rz R3 Rz R3
XII XIII
R~ = NOz CFsS02Cl CF3COCI
Fis
H Rs.N_SO2 H R5.N_SO2
F3C~N ~ OH
F3C.S.N ~ OH
O ~ R O ~~~ .~!~~ R4
z ~ a R O R
O z s
Rz XIV R3 XV
NaH, Mel NaH, Mel
Rs ~ Rs
I Rs.N_S~ I Rs.N_SOz
F3C.S.N ~ OH F3C~N ~ OH
02 ~ / O Ra O ~ / O R R4
Rz R3 Rz s
XVI XVII
The preparation of compounds of the present invention which bear a sulfonamido
or trifluoracetamido moiety at the R1 position are illustrated in Scheme 2.
They are
CA 02341353 2001-02-20
WO 00/14084 PCT/US99/20306
17
prepared by further elaboration of compounds XII, where Rl is N02. Thus,
according to
Scheme 2, reduction of the nitro moiety with a mild reducing agent, such as
stannous
chloride in a hydroxylic solvent such as ethanol, provides compounds of
general structure
XIII. These aniline derivatives are reacted with the appropriate acid
chlorides, such as
trifluoracetyl chloride or trifluoromethanesulfonyl chloride, in inert
solvents such as
dichloromethane or tetrahydrofuran, and in the presence of a scavenger amine
such as
triethylamine, to provide amides of general structure XIV or XV. Conversion to
the
corresponding N-methyl derivatives XVI or XVII is accomplished by a routine
NaH / MeI
reaction sequence, in a solvent such as tetrahydrofuran.
The following is a non-limiting list of the phenols that are employed in
Scheme 1.
These phenols are either obtained from commercial sources, or prepared
according to
literature methods:
CA 02341353 2001-02-20
WO 00/14084 PCT/US99/20306
18 _
F3C ~ F3 ~ F3C~ I W
~OH
~ OH NC I ~ OH
CN CI
F3C~ ~ F3C ~ F3C W
I OH ~OH ~OH
N02 CI F
FsC~ ~ Fs ~ O
i ~ ~ / F3C'
OH F ~OH
F OH
F3 \ F3 ' O
FsC
02N OH F OH ~ OOH
F
FaC ~ FsC~ ~ FuF
F3C~
OH
CI OH
OH
02N ~ 02N ~ 02N
i
I OH
CN CI OH F OH
O
02N ~
F3C' \! w
~OH ~ OOH
CI CI
Many of these and similar compounds are available via common commercial
sources, such as Sigma-Aldrich-Fluka Chemical Company; Oakwood Products, Inc.;
Maybridge Chemical Company; Lancaster Synthesis; or others. The preparation of
these
and similar (1,1,1-trifluoromethyl)-bearing phenols are also described in the
synthetic
literature, and can be prepared with ordinary skills and techniques in the art
of synthetic
chemistry. Examples of these materials follow in the examples. The skilled
artisan will
appreciate that suitably substituted starting materials can be chosen and used
to prepare the
compounds of the present invention, using the information provided herein or
available in
the art. The following non-limiting examples illustrate the compounds,
compositions,
and uses of the present invention.
CA 02341353 2001-02-20
WO 00/14084 PCT/US99/20306
I9
Examules
Reagents and solvents are generally used as received from the commercial
supplier.
Reactions are routinely performed under a N2 atmosphere in oven-dried
glassware.
Column chromatography is performed on silica gel (230 - 400 mesh; Merck). Thin
layer
chromatography analysis (TLC) is performed on 250-~1VI pre-coated Merck silica
gel F254
glass-backed plates. Spots are visualized under 254-nm UV light or by staining
with a
spray reagent consisting of 5% phosphomolybdic acid in EtOH.
Examples 1-13
The following Examples 1 through 13 are representative of compounds III in
General Reaction Scheme 1, above.
3-Methyl-3-(4-nitrophenyloxy)-but-1-yne {Example 1). To a stirred mixture of
4-nitrophenol (0.12 mol), dry K2C03 (0.25 mol), KI (0.21 mol), and CuI (2.4
mmol) in dry
dimethyl formamide (DMF) (122 mL) under argon is added 3-chloro-3-methylbut-1-
yne
(0.25 mol). The reaction mixture is heated at 65°C for 4 h. The
reaction is diluted with
water and extracted with 2 x 1 L of hexane. The organic extracts are pooled
and filtered.
The filtrate is washed sequentially with 2N NaOH, 2N HCI, and water. The
organic layer is
dried over anhydrous Na2S04 and filtered. The filtrate is concentrated under
reduced
pressure to leave the title compound, which is used without further
purification.
3-Methyl-3-(4-(1,1,1-trifluoromethyl)phenyloxy)-1-butyne. This compound is
prepared according to Example 1 from 4-(1,1,1-trifluoromethyl)phenol.
3-Methyl-3-(2-nitro-4-(1,1,1-trifluoromethyl)phenyloxy)-1-butyne. This
compound is prepared according to Example 1 from 2-nitro-4-(1,1,1-
trifluoromethyl)-
phenol.
3-Methyl-3-(3-chloro-4-(1,1,1-trifluoromethyl)phenyloxy)-1-butyne. This
compound is prepared according to Example 1 from 3-chloro-4-(I,1,1-
trifluoromethyl)-
phenol.
3-Methyl-3-(3-fluoro-4-(1,1,1-trifluoromethyl)phenyloxy)-1-butyne. This
compound is prepared according to Example 1 from 3-fluoro-4-(1,1,1-
trifluoromethyl)-
phenol.
3-Methyl-3-(2,3-difluoro-4-(1,1,1-trifluoromethyl)phenyloxy)-1-butyne. This
compound is prepared according to Example 1 from 2,3-difluoro-4-(1,1,1-
trifluoromethyl)-
phenol.
CA 02341353 2001-02-20
WO 00/14084 PCT/US99/20306
20
3-Methyl-3-(2-chloro-4-(1,1,1-trifluoromethyl)phenyloxy)-1-butyne. This
compound is prepared according to Example 1 from 2-chloro-4-(1,1,1-
trifluoromethyl)-
phenol.
1-(3-Chloro-4-(2,2-dimethyl-2-propynyloxy)-phenyl)-2,2,2-trifluoro-1-
ethanone. This compound is prepared according to Example 1 from 1-(3-chloro-4-
hydroxyphenyl)-2,2,2-trifluoro-1-ethanone.
3-Methyl-3-{4-(2,2,2-trifluoroethyl)phenyloxy)-1-butyne. This compound is
prepared according to Example 1 from 4-(2,2,2-trifluoroethyl)phenol.
3-Methyl-3-(4-(1,1,1,2,2-pentafluoroethyl)phenyloxy)-1-butyne. This
compound is prepared according to Example 1 from 4-(1,1,1,2,2-
pentafluoroethyl)phenol.
3-Methyl-3-(3-chloro-4-(1,1,1,2,2-pentafluoroethyl)phenyloxy)-1-butyne. This
compound is prepared according to Example 1 from 1-(3-chloro-4-(1,1,1,2,2-
pentafluoroethyl))phenol.
1-(2-Fluoro-4-(2,2-dimethyl-2-propynyloxy)-phenyl)-2,2,2-trifluoro-1-
ethanone. This compound is prepared according to Example 1 from 1-(2-fluoro-4-
hydroxyphenyl)-2,2,2-trifluoro-1-ethanone.
3-Methyl-3-(2,3-dichloro-4-(1,1,1-trifluoromethyl)phenyloxy)-1-butyne. This
compound is prepared according to Example 1 from 2,3-dichloro-4-(1,1,1-
trifluoromethyl)-
phenol.
Examples 14-22
The following Examples 14 through 22 are representative of compounds IV in
General Reaction Scheme 1, above.
3,4-Dihydro-2,2-dimethyl-6-nitro-2H-1-benzopyran (Example 14). A stirred
solution of 3-methyl-3-(4-nitrophenyloxy)-but-1-yne (0.11 mol) and N,N-
diethylaniline (6.8
mmol) in dry DMF (108 mL) is heated at 130°C for 20 h. The reaction is
chilled and
poured over 1 L H20. The aqueous mixture is extracted with 1 x 1 L and 2 x 500
mL of
hexane. The organic extracts are pooled and washed successively with 2 x 1 L
2N NaOH, 1
x 1 L 2N HCI, and 1 x 1 L H20. The organic phase is dried over anhydrous
Na2S04 and
filtered. The filtrate is concentrated under reduced pressure to leave a solid
residue which
is crystallized from hexanes to give the title compound.
2,2-Dimethyl-6-(1,1,1-trifluoromethyl)-2H-1-benzopyran. This compound is
prepared according to Example 14 from 3-methyl-3-(4-(1,1,1-
trifluoromethyl)phenyloxy)-
but-1-yne.
CA 02341353 2001-02-20
WO 00/14084 PCT/US99/Z0306
21 - _
7-Chloro-2,2-dimethyl-6-(1,1,1-trifluoromethyl)-2H-1-benzopyran. This
compound is prepared according to Example 14 from 3-methyl-3-(3-chtoro-4-
(1,1,1-
trifluoromethyl)phenyloxy)-but-1-yne, followed by silica gel chromatography to
separate
the isomeric product 5-chloro-2,2-dimethyl-6-(1,1,1-trifluoromethyl)-2H-1-
benzopyran.
5-Chloro-2,2-dimethyl-6-(1,1,1-trifluoromethyl)-2H-1-benzopyran. This
compound is prepared according to Example 14 from 3-methyl-3-(3-chloro-4-
(1,1,1-
trifluoromethyl)phenyloxy)-but-1-yne, followed by silica gel chromatography to
separate
the isomeric product 7-chloro-2,2-dimethyl-6-(1,1,1-trifluoromethyl)-2H-1-
benzopyran.
2,2-Dimethyl-8-chloro-6-(1,1,1-trifluoromethyl)-2H-1-benzopyran. This
compound is prepared according to Example 14 .from 3-methyl-3-(2-chloro-4-
(1,1,1-
trifluoromethyl)phenyloxy)-but-1-yne.
2,2-Dimethyl-6-trifluoroacetyl-2H-1-benzopyran. This compound is prepared
according to Example 14 from 1-(3-chloro-4-(2,2-dimethyl-2-propynyloxy)-
phenyl)-2,2,2-
trifluoro-1-ethanone.
2,2-Dimethyl-6-vitro-2H-1-benzopyran. This compound is prepared according to
Example 14 from 3-methyl-3-(4-nitrophenyloxy)-but-I-yne.
2,2-Dimethyl-6-(1,1,1,2,2-pentafluoroethyl)-2H-1-benzopyran. This compound
is prepared according to Example 14 from 3-methyl-3-(4-(1,1,1,2,2-
pentafluoroethyl)-
phenyloxy)-but-1-yne.
2,2-Dimethyl-8-fluoro-6-trifluoroacetyl-2H-1-benzopyran. This compound is
prepared according to Example 14 from I-(2-fluoro-4-(2,2-dimethyl-2-
propynyloxy)-
phenyl)-2,2,2-trifluoro-1-ethanone.
Examples 23-28
The following Examples 23 through 28 are representative of compounds X in
General Reaction Scheme 1, above.
(3R,4R)-2,3-Dihydro-2,2-dimethyl-3,4-epoxy-6-vitro-2H-1-benzopyran
(Example 23). A solution of commercial bleach (S00 mL, 5.25% w/w) and aqueous
O.OSM Na2HP04 (100 mL) is brought to a pH of 11.3 with 1 N NaOH. The volume is
adjusted to a total of 636 mL with H20 to give a 0.55 M solution of NaOCI. A
216-mL
portion of this solution is cooled to 0°C with a salt-ice bath, and
vigorously stirred with a
mechanical stirrer. To this is added dropwise via addition funnel a solution
of 3,4-dihydro-
2,2-dimethyl-6-vitro-2H-1-benzopyran (12.0 g, 58.5 mmol) and (R,R)-(-)-N,N'-
bis(3,5-
ditetrabutylsalicylidine-1,2-cyclohexanediaminomanganese III chloride (R,R-
Jacobsen's
catalyst, 1.34 g, 2.11 mmol) in CH2C12 (83 mL). Care is taken to maintain the
reaction
temperature below 0°C. The reaction is monitored by TLC (hexaneBtOAc,
9:1). After
CA 02341353 2001-02-20
WO 00/14084 PCT/US99/20306
22 _
addition is complete, vigorous stirring is continued at 0°C for 16 h.
The reaction mixture is
separated and the organic layer diluted with additional CH2C12 (200 mL), and
the mixture
is filtered through Celite. The filtrate is washed with brine, dried
(Na2SOq/Darco), filtered,
and concentrated under reduced pressure. The residue is taken up in
CH2CI2/hexane (1:9),
slurried on silica gel and filtered. The filtrate is concentrated under
reduced pressure to
provide the title compound.
(3R,4R)-2,3-Dihydro-2,2-dimethyl-3,4-epoxy-6-(1,1,1-trifluoromethyl)-2H-1-
benzopyran. This compound is prepared according to Example 23 from 2,2-
dimethyl-6-
( 1,1,1-trifluoromethyl)-2H-1-benzopyran.
(3R,4R}-7-Chloro-2,3-dihydro-2,2-dimethyl-3,4-epoxy-6-(1,1,1-
trifluoromethyl)-2H-1-benzopyran. This compound is prepared according to
Example 23
from 7-chloro-2,2-dimethyl-6-(1,1,1-trifluoromethyl)-2H-1-benzopyran.
(3R,4R)-5-Chloro-2,3-dihydro-2,2-dimethyl-3,4-epoxy-6-(1,1,1-
trifluoromethyl)-2H-1-benzopyran. This compound is prepared according to
Example 23
from 5-chloro-2,2-dimethyl-6-(1,1,1-trifluoromethyl)-2H-1-benzopyran.
(3R,4R)-2,3-dihydro-2,2-dimethyl-3,4-epoxy-8-fluoro-6-(1,1,1-
trifluoromethyl)-2H-1-benzopyran. This compound is prepared according to
Example 23
from 2,2-dimethyl-8-fluoro-6-(1,1,1-trifluoromethyl)-2H-1-benzopyran.
(3R,4R)-2,3-dihydro-2,2-dimethyl-3,4-epoxy-7-fluoro-6-(1,1,1-
trifluoromethyl)-2H-1-benzopyran. This compound is prepared according to
Example 23
from 2,2-dimethyl-7-fluoro-6-( 1,1,1-trifluoromethyl)-2H-1-benzopyran.
Example 29
The following Example 29 is representative of compounds XII in General
Reaction
Scheme 1, above.
trans-(3R,4S)-N-(3,4-dihydro-3-hydroxy-2,2-dimethyl-6-nitro-2H-1-benzo-
pyran-4-yl)-N-methyl-methanesulfonamide (Example 29). A mixture of sodium
hydride (60% dispersion in oil; 119 mg, 2.98 mmol) in dry tetrahydrofuran
(THF) (2 mL),
is stirred under argon and cooled to 15°C. The mixture is treated with
a solution of N-
methyl-methanesulfonamide (360 mg, 2.98 mmol) in dry THF (2 mL). The reaction
mixture is further chilled to 0°C, and neat trimethylsilyl chloride
(0.23 mL, 1.84 mmol) is
added dropwise. The reaction is allowed to warm to 15°C, and a solution
of (3R,4R)-2,3-
dihydro-2,2-dimethyl-3,4-epoxy-6-nitro-2H-1-benzopyran (219 mg, 0.99 mmol) in
dry THF
(1.5 mL} is added, followed by a solution of tetrabutyl ammonium fluoride (1 M
in THF,
0.99 mL). The reaction mixture is heated at 57°C for 4 h. After cooling
to 20°C, the
CA 02341353 2001-02-20
WO 00/14084 PCT/US99/20306
23 _ ..
reaction is evaporated under reduced pressure. This product is partitioned
between H20
{25 mL) and EtOAC (3 x 50 mL). The organic extracts are pooled, washed with
H20, and
dried over anhydrous Na2S04. The desiccant is filtered off and the filtrate
concentrated
under reduced pressure. Crystallization from MeOH/H20 provides the title
compound.
Example 30
The following Example 30 is representative of compounds XIII in General
Reaction Scheme 2, above.
trans-(3R,4S)-N-(6-amino-3,4-dihydro-3-hydroxy-2,2-dimethyl-2H-1-
benzopyran-4-yl)-N-methyl-methanesulfonamide (Example 30). To a suspension of
trans-(3R,4S)-N-(3,4-dihydro-3-hydroxy-2,2-dimethyl-6-nitro-2H-1-benzopyran-4-
yl)-N-
methyl-methanesulfonamide (1.21 g, 3.51 mmol) in absolute EtOH (15 mL) is
added
SnClz~H20 (11 g, 14 equivalents), and the mixture is heated at 50°C for
5 h. The reaction
mixture treated with CHC13 (20 mL), HZO (20 mL), and saturated aqueous NaHC03
(50
mL), and then filtered through Celite to remove precipitated tin by-products.
The filtrate is
partitioned with isopropanol-CHCl3 (1:5, 60 mL). The organic layer is dried
over MgS04,
filtered, and evaporated to yield the title compound.
Example 31
The following Example 31 is representative of compounds XV in General Reaction
Scheme 2, above.
trans-(3R,4S)-N-(3,4-dihydro-3-hydroxy-2,2-dimethyl-6-trifluoroacetamido-
2H-1-benzopyran-4-yl)-N-methyl-methanesulfonamide (Example 31). To an ice-cold
solution of trans-(3R,4S)-N-(6-amino-3,4-dihydro-3-hydroxy-2,2-dimethyl-2H-1-
benzopyran-4-yl)-N-methyl-methanesulfonamide (233 mg, 0.741 mmol) in CHZC12 (4
mL)
is added trifluoroacetic anhydride (149 mg, 0.708 mmol), followed by slow
addition of
triethylamine (73 mg, 0.72 mmol). The reaction is allowed to warm to ambient
temperature
over a period of 3.5 h. The reaction mixture is partitioned with Hz0 (4 mL),
saturated
NH4C1 (1 mL) and CHZC12 (3 x 3 mL). The organic layers are combined, dried
over
MgS04, and evaporated to yield the title compound.
Using General Reaction Schemes 1 and 2, the chemistry illustrated for
preparing
Examples 1-31, and employing the appropriately substituted sulfonamide, the
following
additional examples of the present invention are prepared with substantially
similar results.
Specifically examples 32 through 74 are prepared according to General Reaction
Scheme 1
and examples 75 through 86 are prepared according to Scheme 2:
CA 02341353 2001-02-20
WO 00/14084 PCT/US99/20306
24
Rs
RS,N_S02
R2
R~ I 5~ OH
R2 ~ a/ O ~ R4 ~A )
R Rs
2
In the following table, the RZ substituents at positions S, 7 and 8 of the
benzopyran
nucleus are all hydrogen, unless otherwise indicated. While Formula (A) and
the table of
substituents does not distinguish between enantiomers, both enantiomers are
represented for
each example.
Example Rl R2 R3 R4 R5 R6 I
32 CF3 H Me Me Me Me
33 CF3 H Me Me Me Et
34 CF3 H Me Me Me n-propyl
35 CF3 H Me Me Me CF3
36 CF3 H Me Me Et Me
37 CF3 H Me Me Et CF3
38 CF3 H Me Me Et Et
39 CF3 H Me Me Et n-propyl
40 CF3 5-Cl Me Me Me CF3
41 CF3 5-Cl Me Me Et Me
42 CF3 5-Cl Me Me n-propyl Et
43 CF3 5-C1 Et Et n-propyl Et
44 CF3 7,8-di-FMe Me Me Me
45 CF3 7,8-di-FMe Me Me Et
46 CF3 7,8-di-FMe Me Me CF3
47 CF3 7,8-di-FMe Me Me n-propyl
48 CF3 7,8-di-FMe Me Et Et
49 CF3 7-Cl Me Me Me Et
50 CF3 7-CI Me Me Et CF3
51 CF3CF2 H Me Me Me Me
i
52 CF3CF2 H Me Me Me Et
CA 02341353 2001-02-20
WO 00/14084 PCT/US99/20306
25
53 CF3CF2 H Me Me Me n-propyl
54 CF3CF2 H Me Me Me CF3
55 CF3CF2 H Me Me Et Me
56 CF3CF2 H Me Me Et CF3
57 CF3CF2 H Me Me Et Et
58 CF3CF2 H Me Me Et n-propyl
59 CF3CF2 8-Cl Me Me Me Me
60 CF3CF2 8-Cl Me Me Me CF3
61 CF3CF2 8-Cl Me Me Me Et
62 CF3CF2 8-CI Me Me n-propyl Me
63 CF3CF2 8-CI Me Me n-propyl CF3
64 CF3CF2 8-CI Me Me n-propyl Et
65 CF3C0 H Me Me Me Et
66 CF3C0 H Me Me Me CF3
67 CF3C0 H Me Me Me n-propyl
68 CF3C0 H Me Me Me iso-propyl
69 CF3C0 H Me Me Me Ph
70 CF3C0 H Me Me Et Me
71 CF3C0 H Me Me Et Et
72 CF3C0 H Me Me Et Ph
73 CF3C0 8-Cl Me Me Et Et
74 CF3C0 8-Cl Me Me n-propyl CF3
75 CF3CONH H Me Me Me Me
76 CF3CONH H Me Me Me CF3
77 CF3CONH H Me Me Me Et
78 CF3CONH H Me Me n-propyl n-propyl
79 CF3CONH H Me Me n-propyl n-butyl
80 CF3CONH H Me Me n-propyl iso-propyl
81 CF3CONH H Me Me Me Ph
82 CF3CONH H Me Me Me 4-CI-Ph
83 CF3CONH H Et Et Me Me
84 CF3CONH H Et Et Me Et
85 CF3CONH H Et Et Me CF3
CA 02341353 2001-02-20
WO 00/14084 PCT/US99/20306
26 ..
86 ~ CF3CONH ~ H ~ Et ~ Et i Me ~ n-propyl
Compositions
Compounds of this invention are useful in treating cardiac arrhythmias and/or
cardiac fibrillation in humans or other mammals. Therefore, this invention
relates to a
method for treating a human or other mammal suffering from cardiac arrhythmia
and/or
cardiac fibrillation which comprises administering to said human or other
mammal a safe
and effective amount of a pharmaceutical composition comprising from I S-90%
of a
compound active ingredient, or mixtures thereof, and from 10-85%
pharmaceutically-acceptable excipients.
The invention compounds can be formulated into pharmaceutical compositions for
use in treatment or prophylaxis of these conditions. Standard pharmaceutical
formulation
techniques are used, such as those disclosed in Remington's Pharmaceutical
Sciences, Mack
Publishing Company, Easton, Pa., latest edition.
A "safe and effective amount" of a Formula (A) compound is an amount that is
effective, to reduce the frequency and severity of cardiac arrhythmias in a
human
subject, without undue adverse side effects (such as toxicity, irritation, or
allergic
response), commensurate with a reasonable benefitlrisk ratio when used in the
manner
of this invention. The specific "safe and effective amount" will, obviously,
vary with
such factors as the particular condition being treated, the physical condition
of the
patient, the duration of treatment, the nature of concurrent therapy (if any),
the specific
dosage form to be used, the carrier employed, the solubility of the Formula
(A)
compound therein, and the dosage regimen desired for the composition.
In addition to the subject compound, the compositions of the subject invention
contain a pharmaceutically-acceptable carrier. The term "pharmaceutically-
acceptable
carrier", as used herein, means one or more compatible solid or liquid filler
diluents or
encapsulating substances which are suitable for administration to a mammal.
The term
"compatible", as used herein, means that the components of the composition are
capable of
being commingled with the subject compound, and with each other, in a manner
such that
there is no interaction which would substantially reduce the pharmaceutical
efficacy of the
composition under ordinary use situations. Pharmaceutically-acceptable
carriers must, of
course, be of sufficiently high purity and sufficiently low toxicity to render
them suitable
for administration to the animal, preferably mammal being treated.
CA 02341353 2001-02-20
WO 00/14084 PCT/US99/20306
27
Some examples of substances which can serve as pharmaceutically-acceptable
carriers or components thereof are sugars, such as lactose, glucose and
sucrose; starches,
such as corn starch and potato starch; cellulose and its derivatives, such as
sodium
carboxymethyl cellulose, ethyl cellulose, and methyl cellulose; powdered
tragacanth; malt;
gelatin; talc; solid lubricants, such as stearic acid and magnesium stearate;
calcium sulfate;
vegetable oils, such as peanut oil, cottonseed oil, sesame oil, olive oil,
corn oil and oil of
theobroma; polyols such as propylene glycol, glycerine, sorbitol, mannitol,
and
polyethylene glycol; alginic acid; emulsifiers, including nonionic
surfactants, such as the
TWEENS' ; wetting agents, such sodium lauryl sulfate; coloring agents;
flavoring agents;
tableting agents, stabilizers; antioxidants; preservatives; pyrogen-free
water; isotonic saline;
and phosphate buffer solutions.
The choice of a pharmaceutically-acceptable carrier to be used in conjunction
with
the subject compound is basically determined by the way the compound is to be
administered.
If the subject compound is to be injected, the preferred pharmaceutically-
acceptable
Garner is sterile, physiological saline, with blood-compatible suspending
agent, the pH of
which has been adjusted to about 7.4.
In particular, pharmaceutically-acceptable carriers for systemic
administration
include sugars, starches, cellulose and its derivatives, malt, gelatin, talc,
calcium
sulfate, vegetable oils, synthetic oils, polyols, alginic acid, phosphate
buffer solutions,
emulsifiers, isotonic saline, and pyrogen-free water. Preferred carriers for
parenteral
administration include propylene glycol, ethyl oleate, pyrrolidone, ethanol,
and sesame
oil. Preferably, the pharmaceutically-acceptable carrier, in compositions for
parenteral
administration, comprises at least about 90°/a by weight of the total
composition.
The compositions of this invention are preferably provided in unit dosage
form.
As used herein, a "unit dosage form" is a composition of this invention
containing an
amount of a Formula (A) compound that is suitable for administration to humans
in a
single dose, according to good medical practice. These compositions preferably
contain from about 0.5 mg (milligrams) to about 500 mg, more preferably from
about
1 mg to about 100 mg, more preferably from about 1 mg to about 30 mg, of a
Formula
{A) compound.
The compositions of this invention may be in any of a variety of forms,
suitable
(for example) for oral, rectal, topical, nasal, ocular or parenteral
administration.
Depending upon the particular route of administration desired, a variety of
pharmaceutically-acceptable carriers well-known in the art may be used. These
CA 02341353 2001-02-20
WO 00/14084 PCT/US99/20306
28
include solid or liquid fillers, diluents, hydrotropes, surface-active agents,
and
encapsulating substances. Optional pharmaceutically-active materials may be
included, which do not substantially interfere with the antiarrhythmic, anti-
diarrheal,
or anti-ulcer activity of the Formula (A) compound. The amount of carrier
employed
in conjunction with the Formula (A) compound is sufficient to provide a
practical
quantity of material for administration per unit dose of the Formula (A)
compound.
Techniques and compositions for making dosage forms useful in the methods of
this
invention are described in the following references, all incorporated by
reference
herein: Modern Pharmaceutics, Chapters 9 and 10 {Banker & Rhodes, editors,
1979);
Lieberman et al., Pharmaceutical Dosage Forms: Tablets (1981); and Ansel,
Introduction to Pharmaceutical Dosage Forms 2d Edition (1976).
Various oral dosage forms can be used, including such solid forms as tablets,
capsules, granules and bulk powders. These oral forms comprise a safe and
effective
amount, usually at least about 5%, and preferably from about 25% to about 50%,
of the
Formula (A) compound. Tablets can be compressed, tablet triturates, enteric-
coated,
sugar-coated, film-coated, or multiple-compressed, containing suitable
binders,
lubricants, diluents, disintegrating agents, coloring agents, flavoring
agents, flow-
inducing agents, and melting agents. Liquid oral dosage forms include aqueous
solutions, emulsions, suspensions, solutions and/or suspensions reconstituted
from
non-effervescent granules, and effervescent preparations reconstituted from
effervescent granules, containing suitable solvents, preservatives,
emulsifying agents,
suspending agents, diluents, sweeteners, melting agents, coloring agents and
flavoring
agents.
The pharmaceutically-acceptable earner suitable for the preparation of unit
dosage
forms for peroral administration are well-known in the art. Tablets typically
comprise
conventional pharmaceutically-compatible adjuvants as inert diluents, such as
calcium
carbonate, sodium carbonate, mannitol, lactose and cellulose; binders such as
starch, gelatin
and sucrose; disintegrants such as starch, alginic acid and croscarmelose;
lubricants such as
magnesium stearate, stearic acid and talc. Glidants such as silicon dioxide
can be used to
improve flow characteristics of the powder mixture. Coloring agents, such as
the FD&C
dyes, can be added for appearance. Sweeteners and flavoring agents, such as
aspartame,
saccharin, menthol, peppermint, and fruit flavors, are useful adjuvants for
chewable tablets.
Capsules typically comprise one or more solid diluents disclosed above. The
selection of
carrier components depends on secondary considerations like taste, cost, and
shelf stability,
CA 02341353 2001-02-20
WO 00/I4084 PCT/US99/20306
29
which are not critical for the purposes of the subject invention, and can be
readily made by
a person skilled in the art.
Peroral compositions also include liquid solutions, emulsions, suspensions,
and the
like. The pharmaceutically-acceptable carriers suitable for preparation of
such
compositions are well known in the art. Typical components of carriers for
syrups, elixirs,
emulsions and suspensions include ethanol, glycerol, propylene glycol,
polyethylene glycol,
liquid sucrose, sorbitol and water. For a suspension, typical suspending
agents include
methyl cellulose, sodium carboxymethyl cellulose, tragacanth and sodium
alginate; typical
wetting agents include lecithin and polysorbate 80; and typical preservatives
include methyl
paraben and sodium benzoate. Peroral liquid compositions may also contain one
or more
components such as sweeteners, flavoring agents and colorants disclosed above.
Such compositions may also be coated by conventional methods, typically with
pH
or time-dependent coatings, such that the subject compound is released in the
gastrointestinal tract in the vicinity of the desired topical application, or
at various times to
extend the desired action. Such dosage forms typically include, but are not
limited to, one
or more of cellulose acetate phthalate, polyvinylacetate phthalate,
hydroxypropyl methyl
cellulose phthalate, ethyl cellulose, Eudragit~~ coatings, waxes and shellac.
Compositions of the subject invention may optionally include other drug
actives.
The compounds of the present invention can be used in combination with a
number of other
pharmacological agents, such as beta-blockers, calcium channel antagonists,
endothelin
antagonists, sodium-hydrogen exchange inhibitors, inotropic agents,
angiotensin-converting
enzyme inhibitors, cholesterol-lowering agents, diuretic agents,
antiplatelette, or
antithrombotic therapy to reduce the frequency and severity of arrhythmic
events. They
could also be used in combination with antiinfective agents to treat
gastrointestinal ulcers.
The compounds of the present invention can be dosed orally or intravenously
after
formulation with appropriate diluents, solvents, or adsorbents.
Other compositions useful for attaining systemic delivery of the subject
compounds
include sublingual, buccal and nasal dosage forms. Such compositions typically
comprise
one or more of soluble filler substances such as sucrose, sorbitol and
mannitol; and binders
such as acacia, microcrystalline cellulose, carboxymethyl cellulose and
hydroxypropyl
methyl cellulose. Glidants, lubricants, sweeteners, colorants, antioxidants
and flavoring
agents disclosed above may also be included.
Methods of Administration:
This invention also provides methods of treating or preventing cardiac
arrhythmias, diarrhea, and gastric ulcers in animals, preferably a mammalian
subject,
CA 02341353 2001-02-20
WO 00/14084 PCTI(JS99/20306
30
by administering a safe and effective amount of a Formula (A) compound to said
subject. The methods of the invention are useful in treating disorders such as
(for
example) atrial tachycardia, atrial flutter, atrial fibrillation, ventricular
tachycardia,
ventricular extrasystole, ventricular fibrillation, Wolfe-Parkinson-White
syndrome,
Long QT Syndrome, or sudden cardiac death.
The Formula (A) compounds and compositions of this invention can be
administered systemically by any method of introducing Formula (A) compound
into
the tissues of the body, e.g., intramuscular, intravenous, intraperitoneal,
subcutaneous,
sublingual, rectal, and oral administration. The Formula (A) compounds of the
present
invention are preferably administered orally or intravenously.
The specific dosage of inhibitor to be administered, as well as the duration
of
treatment, and whether the treatment is administered by the oral or
intravenous route
are interdependent. The dosage and treatment regimen will also depend upon
such
factors as the specific Formula (A) compound used, the treatment indication,
the
ability of the Formula (A) compound to reach minimum efficacious
concentrations in
the heart or the gut, the personal attributes of the subject (such as weight),
compliance
with the treatment regimen, and the presence and severity of any side effects
of the
treatment.
Typically, for a human adult (weighing approximately 70 kilograms), from
about 0.5 mg to about 500 mg, more preferably from about 1 mg to about 100 mg,
more
preferably from about 1 mg to about 30 mg, of Formula (A) compound are
administered per day for systemic administration. It is understood that these
dosage
ranges are by way of example only, and that daily administration can be
adjusted
depending on the factors listed above.
In all of the foregoing, of course, the compounds of the invention can be
administered alone or as mixtures, and the compositions may further include
additional
drugs or excipients as appropriate for the indication.
All references cited herein are hereby incorporated by reference in their
entirety.
As such, they illustrate the state of the art.
While particular embodiments of the subject invention have been described, it
will
be obvious to those skilled in the art that various changes and modifications
of the subject
invention can be made without departing from the spirit and scope of the
invention. It is
intended to cover, in the appended claims, all such modifications that are
within the scope
of this invention.