Note: Descriptions are shown in the official language in which they were submitted.
CA 02341554 2001-02-23
Process for the preparation of (2R)-piperidine
derivatives
Description
The present invention relates to a novel process for
the preparation of (2R)-piperidine derivatives of the
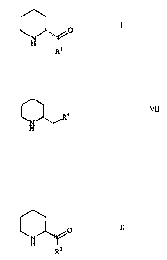
general formulae
..,,. O I
R~
and
VII
in which R1 is hydroxyl, amino or C1_6-alkoxy and R9 is
hydroxyl or amino.
(2R)-Piperidine derivatives of the general formula I,
such as, for example, (R)-pipecolic acid, are important
synthesis units for biologically active compounds, such
as, for example, stimulants of growth hormone secretion
(WO-A-9513069) or anti-anxiety agents (DE-A-37 02 943).
EP-A 686 698 describes a biotechnological process for
the preparation of S-a-aminocarboxylic acids, for
example S-a-pipecolic acid, by reaction of the racemic
RS-a-aminocarboxamides with microorganisms of the
genera Klebsiella and Pseudomonas, the corresponding R-
a-aminocarboxamides also being obtained. For the
preparation of the R-a-aminocarboxamides in pure form,
the S-a-aminocarboxylic acids, however, have to be
removed by relatively complicated processes.
CA 02341554 2001-02-23
- 2 -
A biotechnological process for the preparation of (R)-
pipecolic acid by culturing microorganisms of the genus
Alcaligenes in (RS)-pipecolic acid-containing medium is
also known (Mochizuki, K.; Yamazak:i, Y.; Maeda, H.;
Agric. Biol. Chem. 1988, 52(5), 1113). This process has
the disadvantage that the (R)-pipecolic acid is
obtained as a mixture with (S)-2-aminoadipic acid and
has to be removed from the aminoadipic acid formed
after the preparation.
A process for the preparation of (R)-pipecolic acid
esters is described by R.J. Kazlaus~;as et al (J. Org.
Chem., 1994, 59, 2075-2081). In this process, the
corresponding racemic esters are reacted by means of
isolated stereoselective lipases from Aspergillus niger
to give the desired product. The disadvantage in this
process is that the desired products are in poor
enantiomeric purity and are obtained in low yield. In
addition, very large amounts of enzyme are also
necessary.
JP-A-63 248 393 describes a process for the preparation
of (R)-pipecolic acid starting from racemic pipecolic
acid by means of microorganisms of the genera
Pseudomonas, Kurthia or Alcaligenes. A disadvantage in
this process is the long reaction times.
It is the object of the present invention to eliminate
these disadvantages and to make available a simple
process for the preparation of (2R)-piperidine
derivatives, with which high yields can be achieved and
which is feasible on a large scale.
This object is achieved by the novel biotechnological
process according to Claim 1.
According to the invention, microorganisms are employed
which have the property of being able to utilize a-
CA 02341554 2001-02-23
- 3 -
aminocarboxamides in the form of the racemate or its
optically active isomers of the general formula III
A\
~CH~NHl
NH U
in which A, together with -NH and -CH, is an optionally
substituted 5- or 6-membered saturated heterocyclic
ring, as the only nitrogen source. Microorganisms of
this type are known in the prior ar_t and are already
described in detail in EP-A-686 698.
Preferred microorganisms having the properties
described above are microorganisms of the genera
Klebsiella and Pseudomonas. Microorganisms which are
particularly preferably employed are those of the
species Pseudomonas putida, in particular those of the
species Pseudomonas putida having the designation
DSM 9923, and their functionally equivalent variants
and mutants.
The microorganisms of the species Pseudomonas putida
having the designation DSM 9923 were deposited on
20.04.1995 at the Deutsche Sammlung fur Mikroorganismen
and Zellkulturen GmbH (DSM), Mascherodeweg lb, D-38124
Brunswick, according to the Budapest Convention.
"Functionally equivalent variants and mutants" are
understood as meaning microorganisms which essentially
have the same properties and functions as the original
microorganisms. Variants and mutants of this type can
be formed by chance, for example, by t1V irradiation.
Surprisingly, it has been found that these
microorganisms, or cell-free enzymes thereof, can
degrade the S-isomer in (RS)-piperidine derivatives of
the general formula II
CA 02341554 2001-02-23
- 4 -
0 i(
R
in which R2 is hydroxyl, amino or Cl-s-alkoxy, in the
presence of trace elements and under aerobic conditions
with ring-cleavage and, in the case of the
microorganisms, can utilize it as the only carbon and
energy source. After an adequate reaction period,
customarily after 1 to 48 hours, only a few cyclic S-a-
aminocarboxylic acids or other interfering open-chain
by-products such as S-2-aminoadipic acid or none at all
are then present in the reaction medium. The (2R)-
piperidine derivatives accumulated in the reaction, of
the general formula I
..,,. ~ f
R'
in which R1 is hydroxyl, amino or C1_6-alkoxy, can then
be isolated without difficulties, in particular without
complicated separation processes.
Here and in the following, C1_6-alkoxy has the meaning
of a straight-chain or branched alkoxy group having
from 1 to 6 C atoms. Methoxy, ethoxy, propoxy,
isopropoxy, butoxy, isobutoxy, tert-butoxy, pentaxy and
its isomers and also hexoxy and its isomers may be
mentioned by name.
The enzymes for the cell-free system can be obtained by
technically customary breakdown of the microorganisms.
For this, for example, it is possible to use the
ultrasonic, French press or lysozyme method. These
CA 02341554 2001-02-23
- 5 -
cell-free enzymes can also be immobilized on a suitable
carrier material.
Trace elements are to be understood, for example, as
meaning zinc, manganese, cobalt, copper, nickel,
molybedenum, calcium, magnesium, boron, iron or
sulphur, the optimal composition of the trace elements
being dependent on the microorganism used. Preferably,
zinc, manganese, cobalt, copper, nickel, molybdenum,
calcium, magnesium, boron, iron and sulphur are jointly
employed.
Expediently, the reaction of (RS)-piperidine
derivatives of the general formula II is carried out
with growing microorganisms.
The most preferred substrates of the general formula II
are (RS)-pipecolic acid, ethyl (RS)-pipecolate,
isopropyl (RS)-pipecolate and (RS)-pipecolamide.
Aerobic conditions are understood as meaning culturing
conditions in which the microorganisms are supplied
with oxygen. This can be carried out, for example, in
shaker culture by means of atmospheric oxygen or in
submerse culture by blowing in molecular oxygen or
atmospheric oxygen.
The reaction can be carried out without customary
culturing directly by addition of the microorganisms to
(RS)-piperidine derivatives of the general formula II.
Alternatively, the reaction can be carried out after
customary culturing of the microorganisms using a
suitable carbon and energy source. The carbon and
energy source employed here can be, for example,
sugars, carboxylic acids, sugar alcohols or amino
acids. Sugars which can be used are hexoses such as,
for example, glucose or pentoses. Carboxylic acids
which can be used are di- or tricarboxylic acids and
their salts such as, for example, citric acid or
CA 02341554 2001-02-23
- 6 -
succinate. A sugar alcohol which can be used is a
trihydric alcohol, such as, for example, glycerol. An
amino acid which can be employed is, for example,
glutamate.
Preferably, the reaction is carried out without
customary culturing directly by addition of the
microorganisms to (RS)-piperidine derivatives of the
general formula II.
In the reaction, the microorganisms can utilize the (S)
isomer of the (RS)-piperidine derivative of the general
formula II as the only carbon and energy source and as
the only nitrogen source.
Media which can be used for the process according to
the invention are the technically customary media such
as mineral salt media, for example the mineral salt
medium according to Kulla et al., (Arch. Microbiol.
135, 1-7, 1983) , the media described in Table 1 or low
molar buffers, such as, for example, 10 to 100 mM
phosphate buffer, to which the necessary trace elements
have been added. The process is preferably carried out
in the media as in Table 1.
Expediently, the conditions are chosen such that the S
isomer of the (RS)-piperidine derivatives of the
formula II is preferably utilized as a carbon and
energy source by the microorganisms. Preferably,
besides the compound II, the medium therefore does not
contain any other compounds readily utilizable as a
carbon and energy source.
Expediently, the reaction is carried out with single,
repeated or continuous addition of (RS)-piperidine
derivatives of the general formula II. The addition of
(RS)-piperidine derivatives of the general formula II
is carried out in such a way that the concentration
CA 02341554 2001-02-23
does not exceed 30% by weight, preferably 10% by
weight, particularly preferably 5% by weight.
The pH of the medium is expediently in a range from 4
to 9, preferably from 6 to 8. Expediently, the reaction
is carried out at a temperature from 15 to 80°C,
preferably from 25 to 40°C.
The final products of the general formula I which is
obtained in the reaction depends on the choice of the
starting material and the reaction conditions, in
particular the reaction time and the oxygen supply.
Starting from (RS)-pipecolic acid [(RS)-piperidine
derivative II in which R2 is hydroxyl], only (2R)-
pipecolic acid can be obtained. Starting from (RS)-
pipecolamide [(RS)-piperidine derivative II in which RZ
is amino], both (2R)-pipecolamide and (2R)-pipecolic
acid can be obtained. Starting from an (RS)-pipecolic
acid ester [(RS)-piperidine derivative II in which Rz
is C1_6-alkoxy] , both the (2R) -pipecolic acid ester and
(2R)-pipecolic acid can be obtained.
Whether (2R)-pipecolamide or (2R)-pipecolic acid is
obtained from (RS)-pipecolamide can be controlled, for
example, by means of the reaction time. Thus in the
reaction of (RS)-pipecolamide with the microorganisms
according to the invention, (2R)-pipecolamide first
accumulates, which can then optionally be isolated.
After a longer reaction time, the acid is then formed
from the (2R)-pipecolamide. The reaction can be
monitored analytically, for example
chromatographically. The same applies to the reaction
of (RS) -pipecolic acid esters in which first the (2R) -
pipecolic acid ester and then the acid accumulates.
After a customary reaction time of preferably 1 to 48
hours, the (2R)-piperidine derivatives of the general
formula I, preferably (R)-pipecolic acid, (R)-
CA 02341554 2001-02-23
pipecolamide and ethyl and isopropyl (R)-pipecolates
are preferably obtained in the medium in very high to
quantitative yield.
The (2R)-piperidine derivatives of the general formula
I obtained in this manner can be isolated by customary
working-up methods such as, for example, by removal of
the biomass, acidification, chromatography,
electrodialysis or crystallization.
Alternatively, the course of the conversion reaction
can be regulated by means of the oxygen supply. If, for
example, (R)-pipecolic acid is to be prepared from
(RS)-pipecolamide or (RS)-pipecolic acid ester, the
reaction is expediently carried out under aerobic
conditions until the accumulation of the (R)-
pipecolamide or of the (R)-pipecolic acid ester,
respectively, as described above and the oxygen supply
is then stopped, the acid being formed thereafter.
The reaction of the (RS)-pipecolic: acid derivative
according to formula V
V
0
~3
in which R3 is amino or C1_6-alkoxy, C1_6-alkoxy having
the abovementioned meaning, to give (R)-pipecolic acid
according to formula IV
1V
H
CA 02341554 2001-02-23
_ g _
thus takes place via the (R)-pipecolic acid derivative
according to formula VI
,l.,,, p VI
R3
in which R3 has the abovementioned meaning, which
temporarily accumulates in the medium with increasing
enantiomeric excess and can optionally be isolated. If
the (R)-pipecolic acid derivative according to formula
VI is to be prepared from the (RS)-pipecolic acid
derivative according to formula V, it is isolated after
accumulation. If (R)-pipecolic acid according to
formula IV is to be prepared from the (RS)-pipecolic
acid derivative according to formula V, the reaction is
expediently carried out under aerobic conditions until
the accumulation of the (R)-pipecolic acid derivative
according to formula VI and the oxygen supply is then
stopped.
In order to determine whether the (R)-pipecolic acid
derivative according to formula VI has accumulated in
the medium, the reaction can be monitored analytically,
for example chromatographically. The reaction should be
terminated as soon as the maximum accumulation of the
(R)-pipecolic acid derivative according to formula VI
is achieved.
A further component of the invention is the further
reaction, the reduction, of the (2R)-pipecolir_ acid
derivatives of the general formula VI to give the (2R)-
piperidine derivatives of the general formula VII
VII
,,, .~R.
CA 02341554 2001-02-23
- 10 -
in which R' is hydroxyl or amino.
Customarily, the reducing agent used is an alkali metal
hydride or an alkaline earth metal hydride such as, for
example, lithium aluminium hydride, sodium borohydride,
potassium or sodium aluminium hydride, or magnesium or
calcium borohydride. Lithium aluminium hydride is used
in particular.
The reduction is expediently carried out at a
temperature from 0 to 100°C, preferably at the reflux
temperature of the corresponding solvent.
As known technically, the reduction can be carried out
in a polar organic solvent such as, for example, in an
ether. Suitable ethers are, for example, diethyl ether,
dipropyl ether, tetrahydrofuran or 1,4-dioxane.
After a customary reaction time of 1 to 16 h, the
desired products of the general formula VII, such as
(R)-2-aminomethylpiperidine or (R)-2-hydroxymethyl-
piperidine, can be isolated by known working-up
methods.
Table 1
Medium 1
(NH4) 2504 2. 0 g/1
Na2HP09 2.0 g/1
KH2P09 1.0 g/1
NaCl 2.0 g/1
MgCl2 6H20 0. 4 g/1
CaCl2 2H20 14 . 5 mg/1
FeCl3 6H20 0 . 8 mg/1
ZnS04 7H20 0. 1 mg/1
MnCl24H20 0.09 mg/1
H3BO3 0.3 mg/1
CoCl26H20 0.2 mg/1
CuCl22H20 0.01 mg/1
CA 02341554 2001-02-23
- 11 -
NiCl26Hz0 0.02 mg/1
Na2Mo09 2H20 0. 03 mg/1
FeS04 7H20 2 . 0 mg/1
EDTA-Na22H20 5.0 mg/1
Medium 2
KH2P09 1.3 g/1
NH4C1 5 g/1
(NH4 ) 2SOn 2 g/1
Na2S04 0.25 g/1
MgCl26H20 0.8 g/1
CaCl22H20 0.16 g/1
ZnS04 7Hz0 9 mg/1
MnCl2 4H20 4 mg/1
H3B03 2.7 mg/1
CoCl26H20 1.8 mg/1
CuCl22H20 1.5 mg/1
NiCl26H20 0.18 mg/1
Na2Mo0~ 2H20 0 . 2 mg/1
FeS09 7H20 30 mg/1
EDTA-Na22H20 175 mg/1
Medium 3
Na2S04 0.1 g/1
Na2HP04 2.0 g/1
KH2P09 1.0 g/1
NaCl 2.0 g/1
MgCl2 6H20 0 . 4 g/1
CaCl22H20 14.5 mg/1
FeCl3 6H20 0. 8 mg/1
ZnS04 7H20 0 . 1 mg/1
MnCl24H20 0.09 mg/1
H3BO3 0.3 mg/1
CoCl26H20 0.2 mg/1
CuClz2H20 0.01 mg/1
NiCl26H20 0.02 mg/1
Na2Mo09 2H20 0. 03 mg/1
FeS04 7H20 2 . 0 mg/1
EDTA-Na22H20 5.0 mg/1
CA 02341554 2001-02-23
- 12 -
Examples
Example 1
Preparation of (R)-pipecolic acid from (RS)-pipecolic
acid
1.1 in shaker culture
400 ml of Medium 1 having a concentration of 1.5% of
(RS)-pipecolic acid were introduced into a 1 1
Erlenmeyer flask and treated with Pseudomonas putida
DSM 9923. The batch was incubated at 30°C and 140
revolutions/min on a shaker. After 24 h, the cell-free
supernatant was investigated for the content of (R)-
pipecolic acid at an OD650 of 3.7 using HPLC. (R)-
Pipecolic acid was obtained in a yield of >45% based on
the (RS)-pipecolic acid employed and with an ee value
of >99$ .
1.2 in a fermenter
100 ml of Medium 3 having a concentration of 1% of
(RS)-pipecolic acid were introduced into a 500 ml
Erlenmeyer flask with baffles and treated with
Pseudomonas putida DSM 9923. The batch was incubated
for 16 h at 30°C and 140 revolutions/min on a shaker.
3 1 of Medium 2 having a concentration of 1.5% of (RS)-
pipecolic acid (45 g) were introduced into a 5 1
fermenter and treated with the preculture of
Pseudomonas putida DSM 9923. After 7 h, a further 45 g
of (RS)-pipecolic acid were continuously added as a 10%
solution in the caurse of 5 h. After a further 4 h, the
biotransformation was stopped and the cell-free
supernatant was investigated for the content of (R)-
pipecolic acid using HPLC. (R)-pipecolic acid was
obtained in quantitative yield and with an ee value of
>97$.
CA 02341554 2001-02-23
- 13 -
Example 2
Preparation of (R)-pipecolic acid from ethyl (RS)-
pipecolate
40 ml of Medium 3 having a concentration of 1.5$ of
ethyl (RS)-pipecolate were introduced into a 100 ml
Erlenmeyer flask having baffles and treated with
Pseudomonas putida DSM 9923. The batch was incubated at
30°C and 140 revolutions/min on a shaker. After 26 h,
the cell-free supernatant was investigated for the
content of (R) -pipecolic acid at an OD650 of 2 . 0 using
HPLC. (R)-Pipecolic acid was obtained in a yield of
>45$ based on the ethyl (RS)-pipecolate employed and
with an ee value of >80$. After 44 h, enantiomerically
pure (R)-pipecolic acid was obtained at an OD650 of
3.2.
Example 3
Preparation of (R)-pipecolamide from (RS)-pipecolamide
in shaker culture
40 ml of Medium 3 having a concentration of 1.5$ of
(RS)-pipecolamide were introduced into a 100 ml
Erlenmeyer flask having baffles and treated with
Pseudomonas putida DSM 9923. The batch was incubated at
30°C and 140 revolutions/min on a shaker. After 20 h,
the cell-free supernatant was investigated for the
content of (R)-pipecolamide at an OD650 of 3.0 using
HPLC. (R) -Pipecolamide was obtained i.n a yield of >45$
based on the (RS)-pipecolamide employed and having an
ee value of >99$.
CA 02341554 2001-02-23
- 14 -
Example 4
Preparation of (R)-pipecolamide and (R)-pipecolic acid
from (RS)-pipecolamide in a fermenter
Pseudomonas putida DSM 9923 was cultured in a fermenter
overnight on Medium 3 with 20 g/1 of glutamate. (RS)-
Pipecolamide was added at an OD650 of 14. After 2 h,
according to GC analysis virtually enantiomerically
pure (R)-pipecolamide was present. Pipecolic acid could
not be detected.
A sample of this solution was incubated at 37°C and
140 revolutions/min. in a shaker with exclusion of air.
After 48 h, according to GC analysis enantiomerically
pure (R)-pipecolic acid was present. Pipecolamide could
no longer be detected.
Example 5
Preparation of (R)-pipecolic acid from (RS)-
pipecolamide in shaker culture
40 ml of Medium 3 having a concentration of 1% of (RS)-
pipecolamide were introduced into a 100 ml Erlenmeyer
flask having baffles and treated with Pseudomonas
putida DSM 9923. The batch was incubated on a shaker at
30°C and 140 revolutions/min. After 26 h, the cell-free
supernatant was investigated by GC at an OD650 of 3.9.
(R) -Pipecolic acid with ee >98% was found in about 10%
yield.
Example 6
Preparation of propyl (R)-pipecolate from propyl (RS)-
pipecolate in shaker culture
100 ml of minimum Medium 3 having a concentration of
4 g/1 of propyl (RS)-pipecolate were introduced into a
500 ml Erlenmeyer flask having baffles and treated with
CA 02341554 2001-02-23
- 15 -
Pseudomonas putida DSM 9923. The batch was incubated on
a shaker at 30°C and 140 rpm. After 4.5 h, the cell-
free supernatant was investigated for the content and
enantiomer excess of pipecolic acid and isopropyl
pipecolate at an OD650 of 1.4 using GC. Isopropyl (R)-
pipecolate was obtained enantiomerically pure and in a
yield of 88$ based on isopropyl (R)-pipecolate
employed. A few per cent of enantiomerically pure (R)-
pipecolic acid were found as a by-product.
After separation of the biomass by centrifugation
(20 min at 8000xg), the aqueous solution was evaporated
to one third in vacuo, adjusted to pH 10 using 30$
strength sodium hydroxide solution and immediately
extracted three times with 20 ml of diethyl ether each
time. The combined organic phases were dried using
sodium sulphate and concentrated. By introduction of
hydrochloric acid, 0.195 g of isopropyl (R)-pipecolate
were crystallized as the hydrochloride (80~ yield; ee
>98~ acc. to GC).
Example 7
Preparation of (R)-pipecolic acid from isopropyl (RS)-
pipecolate
100 ml of minimal Medium 3 having a concentration of
4 g/1 of isopropyl (RS)-pipecolate were introduced into
a 500 ml Erlenmeyer flask having baffles and treated
with Pseudomonas putida DSM 9923. The batch was
incubated on a shaker at 30°C and 140 rpm. After 7.5 h,
a sample was taken at an OD650 of 1.4 and incubated
with exclusion of air. After 22 h, the cell-free
supernatant was investigated for the content and
enantiomeric excess of pipecolic acid and isopropyl
pipecolate using GC. (R)-Pipecolic acid was obtained
enantiomerically pure and in a yield of 80o based on
isopropyl (R)-pipecolate employed. A few per cent of
CA 02341554 2001-02-23
- 16 -
enantiomerically pure isopropyl (R)-pipecolate were
found as a by-product.
Example 8
Reduction of (R)-pipecolamide to (R)-2-
aminomethylpiperidine
11.8 g of lithium aluminium hydride were suspended in
500 ml of diethyl ether under a nitrogen atmosphere in
a 2 1 flask. 20 g of (R)-pipecolamide (cf. Ex. 3) were
slowly added at 5°C. The mixture was stirred at room
temperature for 3 h and then under reflux for 5 h.
After cooling to room temperature, 12 ml of water,
12 ml of 15~ strength sodium hydroxide solution and
36 ml of water were slowly added dropwise in
succession. The mixture was stirred at room temperature
until it was snow-white. 120 g of sodium sulphate were
added and the mixture was stirred overnight at room
temperature. The solid was filtered off and washed four
times with 250 ml of diethyl ether each time. The
combined organic phases were evaporated in vacuo.
12.5 g of (R)-2-aminomethylpiperidine were obtained as
a yellowish oil (content: 66% according to GC).
Distillation at 52-53°C/18 mbar yielded 3.6 g (200) of
GC-pure (R)-2-aminomethylpiperidine as a colourless
liquid.
Example 9
Reduction of isopropyl (R)-pipecolate to (R)-2-
hydroxymethylpiperidine
0.2 g of lithium aluminium hydride were suspended in
20 ml of diethyl ether under a nitrogen atmosphere in a
250 ml flask. 90 mg of isopropyl (R)-pipecolate
hydrochloride were slowly added at 5°C. The mixture was
stirred at room temperature for 1 h and then under
reflux for 4 h. After cooling to room temperature,
CA 02341554 2001-02-23
17
0.2 ml of water, 0.2 ml of 15% strength sodium
hydroxide solution and 0.4 ml of water were slowly
added dropwise in succession. The mixture was stirred
at room temperature until it was snow-white. 2 g of
sodium sulphate were added and the mixture was stirred
at room temperature overnight. The solid was filtered
off and washed four times with 20 ml of diethyl ether
each time. The combined organic phases were evaporated
in vacuo. 47 mg of (R)-2-hydroxymethylpiperidine were
obtained as a slightly yellowish oil (content: 95%; ee
>97% according to GC).