Note: Descriptions are shown in the official language in which they were submitted.
CA 02343205 2001-03-06
WO 00/15239 PCT/AU99/00762
-1-
INHIBITION OF TOXIC MATERIALS OR SUBSTANCES USING DENDRIMERS
FIELD OF THE INVENTION
5 This invention relates to inhibition of toxins and other toxic materials or
substances, and in particular it relates to the use of dendrimers as binding
agents for toxic
peptides, proteins, or polyamines and other toxic materials or substances.
BACKGROUND OF THE INVENTION
10
Dendrimers are 3-dimensional polymeric materials of low polydispersity which
are
characterised by a large number of surface terminal groups. In addition the
manner in
which these materials are prepared allows tight control over the size, shape,
and number
and type of surface groups. Dendritic materials have several features that are
useful for
15 use as therapeutic materials: fixed shape which presents a large and
defined surface with
which to interact with biological surfaces and receptors; and the large number
of terminal
groups allow for multiple interactions with the biological targets.
International Patent Applications Nos. PCT/AU95/00350 (WO 95/34595) and
20 PCT/AU97/00447 (WO 98/03573) disclose dendrimers such as a polyamidoamine
or
polylysine dendrimers having a plurality of terminal groups, wherein at least
one of the
terminal groups has an anionic- or cationic-containing moiety bonded or linked
thereto.
The contents of these published International patent applications are
incorporated herein
by reference.
25
The present invention provides the use of dendritic polymers in the inhibition
of
toxic materials or substances, including but not limited to toxins or toxic
peptides such as
snake, scorpion, spider and bee venoms, as well as toxic peptides or other
toxic materials
or substances released during bacterial or viral infection.
30
CA 02343205 2001-03-06
WO 00/15239 PCT/AU99/00762
-2-
SUMMARY OF THE INVENTION
According to the present invention, there is provided a method of prophylactic
or
therapeutic inhibition of a toxic material or substance in a human or non-
human animal
5 patient, which comprises administration to the patient of an effective
amount of a
dendrimer having a plurality of terminal groups wherein at least one of said
terminal
groups has an anionic- or cationic-containing moiety bonded or linked thereto.
Particularly preferred compounds for use in the method of the present
invention
10 are dendrimers having sulfonic acid-containing moieties, carboxylic acid-
containing
moieties, phosphoric or phosphoric acid-containing moieties, boronic acid-
containing
moieties, neuraminic or sialic acid-containing moieties or moieties containing
modified
neuraminic or sialic acid; primary, secondary, tertiary or quaternary amino-
containing
moieties, pyridinium-containing moieties; guanidinium-containing moieties;
amidinium-
15 containing moieties; phenol-containing moieties; heterocycles possessing
acidic or basic
hydrogens; zwitterionic-containing moieties; or mixtures of the above
moieties, linked to
terminal groups thereof.
The compounds used in the method of this invention are referred to herein as
20 polyionic dendrimers, and this term is used throughout this specification
and the claims
which follow to include not only the dendrimers per se, but also their
pharmaceutically or
veterinarily acceptable salts, for example the alkaline metal or alkaline
earth metal salts
such as the sodium, potassium or calcium salts, as well as pharmaceutically
acceptable
anions such as fluoride, chloride, bromide, iodide, citrate, acetate, p-
toluene sulfonate,
25 and the like.
DETAILED DESCRIPTION OF THE INVENTION
Preferred compounds used in accordance with the present invention include
30 polyionic dendrimers of the general formula I:
CA 02343205 2001-03-06
WO 00/15239 PCT/AU99/00762
-3-
I
wherein: I is an initiator core;
Z is an interior branching unit;
n is an integer which represents the number of generations of the
dendrimer; and
A is an anionic- or cationic-containing moiety which may be linked to
interior branching unit Z through an optional linking group X.
Dendrimers are macromolecular highly branched compounds formed by reiterative
reaction sequences starting from an initial, core molecule with successive
layers or stages
being added in successive "generations" to build up a three-dimensional,
highly ordered
polymeric compound. Dendrimers are characterised by the following features: I
an initiator
core(I) which may have one or more reactive sites and be point-like or of
significant size so
as to effect the final topology of the dendrimer; ii layers of branched
repeating units (Z)
attached to the initiator core; iii functional terminal groups (such as
moieties A) attached to
the surface of the dendrimer, optionally through linking groups (such as
linking groups X).
The present invention uses dendritic structures as frameworks for the
attachment of ionic
moieties; the invention is not limited to the spherical dendrimers described
in detail herein
CA 02343205 2001-03-06
WO 00/15239 PCT/AU99/00762
-4-
but can be based on any dendritic structure. The variety of dendrimers in both
shape and
constitution are well known to persons skilled in the art.
The preparation of dendrimers is well known, and is described by way of
example in
5 U.S. Patents Nos. 4,289,872 and 4,410,688 (describing dendrimers based on
layers of lysine
units), as well as U.S. Patents Nos. 4,507,466, 4,558,120, 4,568,737 and
4,587,329
(describing dendrimers based on other units including polyamidoamine or PAMAM
dendrimers). The dendrimers disclosed in these US patents are described as
being suitable
for uses such as surface modifying agents, as metal chelating agents, as
demulsifiers or
10 oil/water emulsions, wet strength agents in the manufacture of paper, and
as agents for
modifying viscosity in aqueous formulations such as paints. It is also
suggested in U.S.
Patents Nos. 4,289,872 and 4,410,688 that the dendrimers based on lysine units
can be used
as substrates for the preparation of pharmaceutical dosages.
15 International Patent Publications Nos. WO 88/01178, WO 88/01179 and WO
88/01180 disclose conjugates in which a dendrimer is conjugated or associated
with another
material such as a carried pharmaceutical or agricultural material. In
addition, International
Patent Publication No. WO 95/24221 discloses dendritic polymer conjugates
composed of
at least one dendrimer in association with a carrier material which can be a
biological
20 response modifier, and optionally a target director. These patent
publications together with
the U.S. patents mentioned above contain a broad disclosure of various
dendrimers and
processes for the preparation thereof, and the disclosure of each of these
publications is
incorporated herein by reference.
25 The term "dendrimer" as used herein is to be understood in its broadest
sense, and to
include within its scope all forms and compositions of these dendrimers as
disclosed in
Patent Publications Nos. WO 88/01178, WO 88/01179 and WO 88/01180. The term
also
includes linked or bridged dendrimers as disclosed in these patent
publications.
30 The preferred dendrimers of the present invention comprise a polyvalent
core
CA 02343205 2001-03-06
WO 00/15239 PCT/AU99/00762
-S-
covalently bonded to at least two dendritic branches, and preferably extend
through at least
two generations. Particularly preferred dendrimers are polyamidoamine (PAMAM)
dendrimers, PAMAM (EDA) dendrimers, poly(propyleneimine) (PPI) dendrimers and
polylysine dendrimers.
In accordance with the present invention, at least one, and preferably a
substantial
number, of the terminal groups on the surface of the dendrimer has an anionic-
or cationic-
containing moiety covalently bonded thereto. The branches of the dendrimer may
terminate
in amino groups or other functional reactive groups such as OH, SH, or the
like, which
subsequently can be reacted with the anionic or cationic moieties. Where the
terminal
groups of the dendrimer are amine groups, the anionic- or cationic-containing
moiety may
be linked to the dendrimer by a variety of functional groups including amide
and thiourea
linkages. Preferred anionic- or cationic-containing moieties which may be
bonded to the
terminal groups of the dendrimer include sulfonic acid-containing moieties,
carboxylic acid-
containing moieties (including neuraminic and sialic acid-containing moieties
and modified
neuraminic and sialic acid-containing moieties), boronic acid-containing
moieties,
phosphoric and phosphonic acid-containing moieties (including esterified
phosphoric and
phosphonic acid-containing moieties) and primary, secondary, tertiary or
quaternary
amino-containing moieties, pyridinium-containing moieties; guanidinium-
containing
moieties; amidinium-containing moieties; phenol-containing moieties;
heterocycles
possessing acidic or basic hydrogens; zwitterionic-containing moieties; or
mixtures of
the above moieties.
Suitable anionic- and cationic-containing moieties which may be bonded or
linked to
the amino or other terminal groups include, by way of example, the following
groups (in
which n is zero or a positive integer, more particularly n is zero or an
integer of from 1 to
20):
CA 02343205 2001-03-06
WO 00/15239 PCT/AU99/00762
-6-
NH(CHZ)"SOj (CH~"SOj Ar(SOj )"
CHZCH(SOj )COOH -CH(SO~~CHzC00H -ArX(CH~y"S03' X = O, S, NH
+ + +
(CH~}"NMe~ Ar(NMe3)" Ar(CHZNMe~)" -
0 O O H 0i'
~SO~Na ~SO~Na ~N~SO Na OOH
[( a
O
S
HN~ HN~ HN~ HN
I SO Na NaOaS I ~ SO~Na I / / Na03S I / ~ S03Na
S03Na
S'' OII O''
Na03S HN ' HN~ HN v ' O
I ~ \ ~ ' (
Na03S ~ / S03Na /
S03Na SOaNa S03Na
O
. o ~+
off
OAc
SOsNa COOH
O 0
NH
O
~AMe3 I /
NHZ
tJMe~
-ArXP(=0)(OR)2 X=0, CHz, CHF, CFZ R=alkyl, aryl, H, Na.
ArXP(=O)(OR')(NRZR') X=O, CH2, CHF, CF2 R'=alkyl, aryl, H, Na R2, R'=alkyl,
aryl
Ar[P(=O)(OR)Z]~ R=alkyl, aryl, H, Na n=1-3
Ar[B(OH)sJ~ n=1-3 -Ar[COOHj~ n=1-3
CA 02343205 2001-03-06
WO 00/15239 PCT/AU99/00762
()
HN ~ HN ~ HN ~ HN ~
I ~ I ~ ~ ~ I
i i i i i
PO(ONa)2
PO(OEt)Z PO(OEt)(ONa) PO(ONa)2 PO(ONa)2
II ~~ S O
HN~ ~~ I
HN
w
I~ I
COONa Na00C ~ COONa I ~ B(pNa)2 - B(ONa)2
HN ~ HN ~ HIVW
O
PO(ONa)2 PO(OEt)(ONa) PO(OEt)2
S S S
HN_ ' HN_ ' HN-
' \ S
~' PO(ONa)2 ~' PO(OEt)(ONa) ~' PO(OEt)2
I N (~
NH N \ N
CHZ y -NR3.
~NHZ
O~ S'O N
3
\NH ~ ~NH N CF
~S ~ H
O
-N'-R-COO'
OH
-N+-R-S03.
-N--NR3+ O
-N+-R-PI/O.
R
'OH
CA 02343205 2001-03-06
WO OO/I5239 PCT/AU99/00762
-g-
R' R= alkyl or arylalkyl; R,, Ri, R3 (which may be
-R~~ same or different) = alkyl or arylalkyl
~R
i
R3
~N'-Alkyi
In addition to the above, various neuraminic or sialic acid-containing
moieties or
modified neuraminic or sialic acid-containing moieties may be bonded or linked
to the
dendrimers in accordance with this invention. These moieties include the
various N- and O-
substituted derivatives of neuraminic acid, particularly N- and O-acyl
derivatives such as N-
acetyl, O-acetyl and N-glycolyl derivatives, as well as moieties in which the
neuraminic acid
group is modified. Suitable modified neuraminic groups include groups which
are
substituted in the 4-position with an amino, amido, cyano, azide or guanidino
group, as well
as unsaturated neuramine acid groups. These moieties may be linked to the
dendrimers
through the 2-, 7-, 9- or 5-NAc positions.
Preferably, in the polyionic dendrimers of the general formula I, n is an
integer of
from 1 to 20 or more, more preferably from 1 to 10. Preferably also, the
dendrimers include
at least three or more terminal groups.
The optional linking groups which may be present to act as a spacer between
the
dendrimer and the moiety A, may consist of an alkyl chain (optionally
substituted or
branched), an alkoxy, polyalkoxy, alkylthio or polyalkylthio chain (optionally
substituted),
or an alkenyl, multiple alkenyl, alkynyl or multiple alkynyl chain (optionally
substituted).
Suitable spacer chains include groups of the formula -(CHZ),"-Z-(CHZ)m-,
wherein Z is -CHZ-,
-CH=CH-, -C---C-, -O- or -S-, and m is an integer of from 1 to 15.
The anionic or cationic dendrimers of this invention may be prepared by
standard
CA 02343205 2001-03-06
WO 00/15239 PCT/AU99/00762
-9-
chemical methods which are well known to persons skilled in this art. Suitable
methods are
described by way of the example in Examples below.
As previously described, the anionic or cationic dendrimers of the present
invention
have been found to inhibit toxic materials or substances. The term "toxic
materials or
substances" as used herein is intended to refer in particular to toxins of
biological (animal,
plant, microbial or viral) origin, including but not limited to animal toxins
or toxic peptides
such as snake, scorpion, spider and bee venoms, toxic polyamines, and toxic
peptides or
other materials or substances released during bacterial infection (such as
bacterial
endotoxins and exotoxins), or during protozoal, fungal or viral infection.
The term "inhibition" is used herein in its broadest sense to include either
full or
partial inhibition or suppression of the toxic effect of the toxic material or
substance in a
human or non-human animal patient. The term is also used to encompass both
prophylactic
and therapeutic treatment.
Thus, in another aspect the present invention provides a pharmaceutical or
veterinary
composition for prophylactic or therapeutic inhibition of a toxic material or
substance in a
human or non-human animal patient, which comprises a dendrimer as broadly
described
above, in association with at least one pharmaceutically or veterinarily
acceptable carrier or
diluent.
The formulation of such compositions is well known to persons skilled in this
field.
Suitable pharmaceutically acceptable carriers and/or diluents include any and
all
conventional solvents, dispersion media, fillers, solid carriers, aqueous
solutions, coatings,
antibacterial and antifungal agents, isotonic and absorption delaying agents,
and the like.
The use of such media and agents for pharmaceutically active substances is
well known in
the art, and it is described, by way of example, in Remington's Pharmaceutical
Sciences,
18th Edition, Mack Publishing Company, Pennsylvania, USA. Except insofar as
any
conventional media or agent is incompatible with the active ingredient, use
thereof in the
CA 02343205 2001-03-06
WO 00/15239 PCT/AU99/00762
- 10-
pharmaceutical compositions of the present invention is contemplated.
Supplementary
active ingredients can also be incorporated into the compositions.
It is especially advantageous to formulate compositions in dosage unit form
for ease
5 of administration and uniformity of dosage. Dosage unit form as used herein
refers to
physically discrete units suited as unitary dosages for the human subjects to
be treated; each
unit containing a predetermined quantity of active ingredient calculated to
produce the
desired therapeutic effect in association with the required pharmaceutical
carrier and/or
diluent. The specifications for the novel dosage unit forms of the invention
are dictated by
10 and directly dependent on (a) the unique characteristics of the active
ingredient and the
particular therapeutic effect to be achieved, and (b) the limitations inherent
in the art of
compounding such an active ingredient for the particular treatment.
In yet another aspect, this invention provides the use of an effective amount
of a
1 S dendrimer as broadly described above in the prophylactic or therapeutic
treatment of, or in
the manufacture of a medicament for prophylactic or therapeutic treatment of a
human or
non-human animal patient by inhibition of a toxic material or substance.
A variety of administration routes are available. The particular mode selected
will
20 depend, of course, upon the particular condition being treated and the
dosage required for
therapeutic efficacy. The methods of this invention, generally speaking, may
be practised
using any mode of administration that is medically acceptable, meaning any
mode that
produces therapeutic levels of the active component of the invention without
causing
clinically unacceptable adverse effects. Such modes of administration include
oral, rectal,
25 topical, nasal, inhalation, transdermal or parenteral (e.g. subcutaneous,
intramuscular and
intravenous) routes. Formulations for oral administration include discrete
units such as
capsules, tablets, lozenges and the like. Other routes include intrathecal
administration
directly into spinal fluid, direct introduction such as by various catheter
and balloon
angioplasty devices well known to those of ordinary skill in the art, and
intraparenchymal
30 injection into targeted areas.
CA 02343205 2001-03-06
WO 00/15239 PCT/AU99/00762
-11-
The compositions may conveniently be presented in unit dosage form and may be
prepared by any of the methods well known in the art of pharmacy. Such methods
include
the step of bringing the active component into association with a carrier
which constitutes
one or more accessory ingredients. In general, the compositions are prepared
by uniformly
and intimately bringing the active component into association with a liquid
carrier, a finely
divided solid carrier, or both, and then, if necessary, shaping the product.
Compositions of the present invention suitable for oral administration may be
presented as discrete units such as capsules, cachets, tablets or lozenges,
each containing a
predetermined amount of the active component, in liposomes or as a suspension
in an
aqueous liquor or non-aqueous liquid such as a syrup, an elixir, or an
emulsion.
Compositions suitable for parenteral administration conveniently comprise a
sterile
aqueous preparation of the active component which is preferably isotonic with
the blood of
the recipient. This aqueous preparation may be formulated according to known
methods
using those suitable dispersing or wetting agents and suspending agents. The
sterile
injectable preparation may also be a sterile injectable solution or suspension
in a non-toxic
parenterally-acceptable diluent or solvent, for example as a solution in
polyethylene glycol.
Among the acceptable vehicles and solvents that may be employed are water,
Ringer's
20 solution and isotonic sodium chloride solution. In addition, sterile, fixed
oils are
conventionally employed as a solvent or suspending medium. For this purpose,
any bland
fixed oil may be employed including synthetic mono-or di-glycerides. In
addition, fatty
acids such as oleic acid find use in the preparation of injectables.
The active component may also be formulated for delivery in a system designed
to
administer the active component intranasally or by inhalation, for example as
a finely
dispersed aerosol spray containing the active component.
Other delivery systems can include sustained release delivery systems.
Preferred
sustained release delivery systems are those which can provide for release of
the active
CA 02343205 2001-03-06
WO 00/15239 PCT/AU99/00762
-12-
component of the invention in sustained release pellets or capsules. Many
types of sustained
release delivery systems are available. These include, but are not limited to:
(a) erosional
systems in which the active component is contained within a matrix, and (b)
diffusional
systems in which the active component permeates at a controlled rate through a
polymer. In
addition, a pump-based hardware delivery system can be used, some of which are
adapted
for implantation.
The active component is administered in prophylactically or therapeutically
effective
amounts. A prophylactically or therapeutically effective amount means that
amount
necessary at least partly to attain the desired effect, or to delay the onset
of, inhibit the
progression of, or halt altogether, the onset or progression of the particular
condition being
treated. Such amounts will depend, of course, on the particular condition
being treated, the
severity of the condition and individual patient parameters including age,
physical condition,
size, weight and concurrent treatment. These factors are well known to those
of ordinary
1 S skill in the art and can be addressed with no more than routine
experimentation. It is
preferred generally that a maximum dose be used, that is, the highest safe
dose according to
sound medical judgement. It will be understood by those of ordinary skill in
the art,
however, that a lower dose or tolerable dose may be administered for medical
reasons,
psychological reasons or for virtually any other reasons.
Generally, daily oral doses of active component will be from about 0.01 mg/kg
per
day to 1000 mg/kg per day. Small doses (0.01-1 mg) may be administered
initially,
followed by increasing doses up to about 1000 mg/kg per day. In the event that
the response
in a subject is insufficient at such doses, even higher doses (or effective
higher doses by a
different, more localised delivery route) may be employed to the extent
patient tolerance
permits. Multiple doses per day are contemplated to achieve appropriate
systemic levels of
compounds.
The active component according to the invention may also be presented for use
in the
form of veterinary compositions, which may be prepared, for example, by
methods that are
CA 02343205 2001-03-06
WO 00/15239 PCT/AU99/00762
-13-
conventional in the art. Examples of such veterinary compositions include
those adapted
for:
(a) oral administration, external application, for example drenches (e.g.
aqueous
or non-aqueous solutions or suspensions); tablets or boluses; powders,
5 granules or pellets for admixture with feed stuffs; pastes for application
to the
tongue;
(b) parenteral administration for example by subcutaneous, intramuscular or
intravenous injection, e.g. as a sterile solution or suspension; or (when
appropriate) by intramammary injection where a suspension or solution is
10 introduced into the udder via the teat;
(c) topical application, e.g. as a cream, ointment or spray applied to the
skin; or
(d) intravaginally, e.g. as a pessary, cream or foam.
Throughout this specification and the claims which follow, unless the context
15 requires otherwise, the word "comprise", or variations such as "comprises"
or "comprising",
will be understood to imply the inclusion of a stated integer or group of
integers but not the
exclusion of any other integer or group of integers.
BRIEF DESCRIPTION OF THE DRAWINGS
20
In the accompanying drawings:
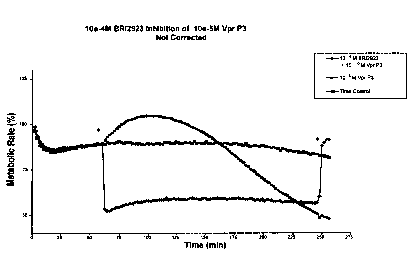
Figures 1 to 4 show the effects of various concentrations of BRI 2923 in
inhibition of the HIV toxic Vpr peptide fraction P3.
25 Further features of the present invention will be apparent from the
following
Examples which are included by way of illustration, not limitation of the
invention. In the
following Examples, PAMAM dendrimers refer to polyamidoamine dendrimers based
on an
ammonia core as detailed in US Patents Nos. 4,507,466, 4,558,120, 4,568,737
and
4,587,329; PAMAM (EDA) dendrimers refer to polyamidoamine dendrimers based on
an
30 ethylene diamine core; and BHAIysXlysylysz dendrimers refer to polylysine
unsymmetrical
CA 02343205 2001-03-06
WO 00/15239 PCT/AU99/00762
-14-
dendrimers based on a benzhydrylamine core and lysine branching units as
described in US
Patents Nos. 4,289,872 and 4,410,688. The polyamidoamine dendrimers PAMAM 1.0,
PAMAM 2.0, PAMAM 3.0, PAMAM 4.0, PAMAM 5.0 or higher generation, PAMAM 4.0
(EDA), and the polylysine dendrimers BHAlyslysz, BHAlyslyszlys4,
BHAlys1ys21ys41ys8
and BHAlyslysZlys41ys81ys,6, BHAlyslysz1ys41ys81ys,61ys32,
BHAlys1ys21ys41ys81ys,61ys3zlys~,,
or higher generations prepared as described in US Patents Nos. 4289872,
4410688,
4507466, 4558120, 4568737 and 4578239 and International Patent Publications
Nos. WO
88101178, WO 88/01179, WO 88/01180 and WO 95/24221 referred to above.
EXAMPLE 1
Reaction of dendritic polymers with 2-acrylamido-2-methyl propane sulfonic
acid to
give sulfonic acid terminated dendrimers.
A PAMAM 1.0
Solid sodium carbonate (0.13g; 1.Ommol) was added slowly to a stirred solution
of
2-acrylamido-2-methyl propane sulfonic acid (0.418; 2.Ommo1) in water (3m1).
After
the evolution of gas had ceased, the pH of the solution was 8Ø A solution of
PAMAM 1.0 (0.12g; 0.33mmo1) in water (lml) was then added to the solution
followed by the addition of four drops of a 40% aq. solution of benzyl
trimethylammonium hydroxide. The solution was then heated under nitrogen at
60°
for three days and then concentrated. The residue was purified by gel
filtration
(Sephadex G 10; water) and then freeze dried to give the sulfonated PAMAM 1.0
dendrimer as an off white solid (0.51g). ~H and t3C nrnr spectra showed a
mixture of
dialkylated and monoalkylated PAMAM 1.0 dendrimer ( ca. 70:30). ~3C nmr (D20):
8 31.0, 31.1, 37.1, 37.7, 41.3, 48.6, 51.5, 53.1, 53.4, 55.6, 56.2, 61.2,
61.5, 178.3,
179.0, 179.8.
CA 02343205 2001-03-06
WO 00/15239 PCT/AU99/00762
-15-
B PAMAM 2.0
PAMAM 2.0 was reacted with 2-acrylamido-2-methyl propane sulfonic acid as
described above. The crude product was purified by gel filtration (Sephadex
G10;
water) and then freeze dried to give an off white solid. ~H and 13C nmr
spectra
S showed a mixture of dialkylated and monoalkylated PAMAM 2.0 dendrimer ( ca.
65:35). '3C nmr (D20): 8 31.0, 31.1, 37.1, 37.7, 41.3, 48.7, 51.5, 53.4, 55.6,
56.2,
61.2, 61.5, 178.4, 179.0, 179.1, 179.6.
When the above reaction was repeated omitting the benzyltrimethylammonium
hydroxide a similar result was obtained.
10
C PAMAM 3.0 BRI2783
PAMAM 3.0 was reacted with 2-acrylamido-2-methyl propane sulfonic acid as
above except that a slight excess of sodium carbonate was used and the
benzyltrimethylammonium hydroxide was omitted. 1H and 13C nmr spectra showed
15 a mixture of dialkylated and monoalkylated PAMAM 3.0 dendrimer ( ca.
50:50). 13C
nmr (D20): 8 31.0, 31.1, 36.9, 37.4, 41.1, 48.6, 51.5, 53.4, 55.7, 56.2, 61.1,
61.5,
178.2, 178.9, 179.0, 179.8.
D PAMAM 4.0 BRI2784
20 PAMAM 4.0 was reacted with 2-acrylamido-2-methyl propane sulfonic acid as
described for PAMAM 3Ø 1H and 13C nmr spectra showed a mixture of
dialkylated
and monoalkylated PAMAM 4.0 dendrimer ( ca. 35:65). 13C nmr (D20): b 31.0,
31.1, 36.9, 37.3, 41.1, 48.5, 51.5, 53.5, 55.7, 56.2, 61.1, 61.5, 178.1,
178.9, 179.0,
179.8.
25
CA 02343205 2001-03-06
WO 00/15239 PCT/AU99/00762
-16-
EXAMPLE 2
Preparation of sodium sulfoacetamide terminated dendrimers.
5 A PAMAM 1.0
A solution of 4-nitrophenyl bromoacetate (0.40g; l.Smmol) in dry DMF (lml) was
added to a stirred solution of PAMAM 1.0 (0.18g; O.Smmol) in DMF (3ml). The
resulting yellow solution was stirred for 20 hours at room temperature, when a
ninhydrin test was negative. The solution was concentrated (30°/
O.lmmHg) to give a
10 yellow oil. This oil was partitioned between water and chloroform and the
aqueous
layer separated and washed with chloroform (2X) and finally with ethyl
acetate. The
aqueous solution was concentrated (35°/ 25mmHg) to give the
bromoacetylated
PAMAM 1.0 dendrimer as a yellow oil (0.36g;100% ). 13C nmr (D20): 8 32.8,
33.3,
43.0, 43.5, 54.4, 174.5, 176.4.
15
A solution of sodium sulfite (0.2g; l.6mmol} in water (lml) was added to a
solution
of the bromoacetylated PAMAM 1.0 dendrimer described above (0.36g; O.Smmol) in
water (Sml) and the solution left to stand at room temperature for eleven
days. The
yellow solution was concentrated to give a yellowish solid (0.60g). 13C nmr
(D20): 8
20 34.4, 43.1, 43.4, 54.0, 61.7, 171.3, 177.2.
The above reaction sequence could be carried out without isolating the
bromoacetylated dendrimer by simply adding the sodium sulfite solution to the
crude
aqueous extract obtained from the first reaction.
25
B PAMAM 2.0
Method 1:
A solution of 4-nitrophenyl bromoacetate (0.18g; 0.7mmol) in dry DMF (lml) was
added to a stirred solution of PAMAM 2.0 (O.IOg; O.lmmol) in DMF (3ml). The
30 resulting yellow solution was stirred for 20 hours at room temperature,
when a
CA 02343205 2001-03-06
WO 00/15239 PCT/AU99/00762
-17-
ninhydrin test was negative. The solution was then added with swirling to
water
(150m1) and the mixture extracted with chloroform (3X) and ethyl acetate. A
solution
of sodium sulfite (0.1 g; O.8mmol) in water ( 1 ml) was added to the crude
bromoacetylated dendrimer solution and the mixture allowed to stand for three
days
5 at room temperature. The yellowish solution was then concentrated to give a
yellow
solid residue, which was purified by gel filtration (Sephadex LH20; water) to
give
the sodium sulfoacetamide terminated PAMAM 2.0 dendrimer (103mg). 13C nmr
(D20}: 8 33.0, 35.7, 36.0, 37.7, 40.3, 43.0, 43.2, 53.4, 53.7, 56.0, 61.6,
171.2, 174.6,
178.5.
10
Method 2:
Solid succinimidyl acetylthioacetate (67mg; 0.33mmo1) was added to a solution
of
PAMAM 2.0 (52mg; 0.05mmo1) in dry DMF (2m1) and the resulting solution stirred
at room temperature for two days. The mixture was then concentrated
(30°/10-3
15 mmHg) to give an oily residue. The residue was partitioned between water
and
chloroform, and the water layer separated and concentrated to give a viscous
oil
(117mg). ~H and 13C nmr showed the oil to be a mixture of the acylated
dendrimer
and N-hydroxy succinimide. Gel filtration (Sephadex G10; water) provide a pure
sample of the acetylthioacetamide terminated PAMAM 2.0 dendrimer (29mg). 13C
20 nmr (D20): b 34.0, 34.2, 37.3, 43.0, 43.1, 43.3, 53.5, 54.0, 56.3, 175.4,
177.2, 177.5.
A solution of the above functionalised dendrimer in 40% aqueous formic acid
(7ml)
was then added to an ice cold freshly prepared solution of performic acid
(l.6mmo1)
in formic acid (2m1). The mixture was stirred for one hour at 0° and
then for twenty
25 hours at room temperature. A small amount of activated charcoal was then
added to
decompose any excess peracid, the mixture stirred for 30 minutes then filtered
and
concentrated to give a viscous oil.
The crude product was dissolved in water, the pH adjusted to 9.0 with aqueous
30 sodium bicarbonate and the material desalted by passage through a column of
CA 02343205 2001-03-06
WO 00/15239 PCT/AU99/00762
-18-
Sephadex G10. A white solid (20mg; ) was obtained after lyophylisation which
was
spectroscopically essentially the same as the material obtained by method 1.
13C nmr
(D20): b 33.0, 38.7, 42.9, 43.0, 43.1, 53.9, 54.3, 56.5, 61.6, 171.2, 176.4,
177Ø
EXAMPLE 3
Preparation of sodium sulfosuccinamic acid terminated dendrimers
A PAMAM 1.0
Solid malefic anhydride (0.11 g; 1.1 mmol) was added to a stirred solution of
PAMAM
1.0 (0.12g; 0.33mmol) in dry DMF (3ml). The mixture became a little warm and
brownish as the anhydride dissolved and the resulting solution was stirred
overnight
at room temperature. The solution was then concentrated (30°/10-4mmHg)
to give a
viscous oil. 1H and 13C nmr (D20) showed complete conversion of the PAMAM 1.0
to the trisamide together with some malefic acid. 13C nmr (D20): b 33.1, 42.8,
43.1,
54.3,135.0,137.1,169.1,171.9,173.3.
The crude trisamide was then dissolved in water (4ml) and solid sodium sulfite
(0.20g; l.6mmol) added. The resulting solution was allowed to stand at room
temperature for four days and then concentrated. 1H and 13C nmr (D20) showed a
1:1 mixture of the regioisomeric sodium sulfosuccinamic acid terminated PAMAM
I .0 dendrimers together with some sulfosuccinic acid. The crude product was
purified by gel filtration (Sephadex G10; water) to afford a sample of the
sodium
sulfosuccinamic acid terminated PAMAM 1.0 dendrimers (10?mg). 13C nmr (D20):
8 33.3, 39.6, 40.0, 42.9, 43.1, 54.0, 67.9, 69.4, 173.8, 176.3, 177.6, 181.8.
B PAMAM 2.0
A mixture of the regioisomeric sodium sulfosuccinamic acid terminated PAMAM
2.0 dendrimers was prepared as described above. 13C nmr PAMAM 2.0 maleamic
acid derivative (D20): b 32.8, 33.0, 38.7, 42.9, 53.8, 54.3, 56.5, 135.2,
136.8, 169.2,
CA 02343205 2001-03-06
WO 00/15239 PCT/AU99/00762
- 19-
171.9, 173.5, 174.6. ~3C nmr PAMAM 2.0 sodium sulfosuccinamic acid derivatives
(D20): 8 37.0, 40.1, 41.1, 43.0, 43.2, 43.9, 53.0, 53.3, 55.5, 68.0, 69.4,
173.8, 177.6,
179.1, 179.5, 179.8, 182.3.
C PAMAM 4.0 BRI6038
Solid malefic anhydride (60mg; 0.6mmol) was added to a stirred solution of
PAMAM
4.0 (5lmg; 0.0lmmol) in dry DMF (2m1). The mixture initially became cloudy but
soon gave a clear solution which was stirred overnight at room temperature.
The
solution was then concentrated (35°/10-' mmHg) to give a viscous oil.
'H and'3C
nmr (D20) showed complete conversion of the PAMAM 4.0 to the polyamide
together with some malefic acid. The crude polyamide was then dissolved in
water
(2m1) and a solution of sodium sulfite (126mg; l.Ommol) in water (2m1} added.
The
resulting solution was allowed to stand at room temperature for two days and
then
concentrated. 1H and ~3C nmr (D20) showed a mixture of the regioisomeric
sodium
sulfosuccinamic acid terminated PAMAM 4.0 dendrimers together with some
sulfosuccinic acid. The crude product was purified by gel filtration (Sephadex
LH20;
water) to afford a sample of PAMAM 4.0 terminated with 24 regioisomeric
sulfosuccinamic acid groups (90mg). ~H nmr (D20): b 2.4-2.6; 2.7-3.1; 3.2-3.4;
3.9-
4Ø~3C nmr (D20): 8 36.2; 39.8; 40.5; 43.0; 43.2; 53.5; 55.8; 68.1; 69.5;
173.8;
177.4; 177.6; 178.7; 182.3.
EXAMPLE 4
Preparation of sodium N-(2-sulfoethyl)succinamide terminated dendrimers
a Preparation of tetrabutylammonium N-(2-sulfoethyl)succinamic acid
Solid succinic anhydride (O.Sg; S.Ommol) was added to a stirred solution of
tetrabutylammonium 2-aminoethylsulfonic acid (1.83g; S.Ommol) in dry
dichloromethane (30m1). The succinic anhydride slowly dissolved and the
resulting
cloudy solution was stirred overnight at room temperature. The mixture was
filtered
CA 02343205 2001-03-06
WO 00/15239 PCT/AU99/00762
-20-
and the filtrate concentrated to give a viscous oil (2.41g). j3C nmr showed
complete
conversion to the desired monoamide together with a small amount of succinic
acid.
Repeated precipitation of the product by dropwise addition of a
dichloromethane
solution to a large excess of diethyl ether gave tetrabutylammonium N-(2-
5 sulfoethyl)succinamic acid as a white solid (1.762g; 76% ), mp 125-
127°C. 1 H nmr
(CDCI3): 8 0.86 (t, 12h, 4xCH3), 1.28 (m, 8H, 4xCH2), 1.50 (m, 8H, 4xCH2),
2.33
(m, 2H, CH2COOH), 2.44 (m, 2H, CH2CONH), 2.76 (m, 2H, CH2NHC0), 3.12 (m,
8H, 4xCH2N), 3.50 (m, 2H, CHZS03-), 7.53 (br t, 1H, NH). 13C nmr (CDC13): 8
13.5 ,19.5, 23.8, 30.1, 30.9, 35.6, 50.0, 58.5, 172.0, 174.1.
10
b Preparation of tetrabutylammonium 4-nitrophenyl N-(2-sulfoethyl)succinamate
A solution of dicyclohexylcarbodiimide (45mg; 0.22mmo1) in dry dichloromethane
( 1 ml) was added to a stirred solution of tetrabutylammonium N-(2-
sulfoethyl)succinamic acid (94mg; O.ZOmmol) in dichloromethane (2m1), and the
15 mixture stirred overnight at room temperature. The resulting suspension was
filtered
and the filtrate concentrated to give the crude active ester, which was used
without
further purification.
A Preparation of sodium N-(2-sulfoethyl)succinamide terminated PAMAM
dendrimers
20
PAMAM 4.0 BRI2786
A solution of the crude tetrabutylammonium 4-nitrophenyl N-(2-
sulfoethyl)succinamate (0.30mmo1) in dry DMF (lml) was added to a stirred
solution
of PAMAM 4.0 (Sl.Smg; O.Olmmol) dissolved in 50% aqueous DMF (3m1) and the
25 resulting yellow solution stirred overnight at room temperature. The
mixture was
then concentrated (35°/10-5 mmHg) and the yellow residue partitioned
between
water and chloroform. The water layer was separated, washed with chloroform
(2X)
and ethyl acetate, and then concentrated to give a yellow oil (134mg). The
crude
product was converted to the sodium salt by passage through a column of
Amberlite
30 IR 120(Na) to yield 85mg of material. This material was further purified by
gel
CA 02343205 2001-03-06
WO 00/15239 PCT/AU99/00762
-21 -
filtration (Sephadex LH20; water) to give the sodium N-(2-
sulfoethyl)succinamide
terminated PAMAM 4.0 dendrimer (45mg). ~3C nmr (D20): 8 33.2, 33.6, 35.5,
39.0,
39.5, 42.8, 43.2, 53.8, 54.1, 54.4, 56.6, 176.5, 176.9, 177.2, 178.9, 179.4.
5 The corresponding PAMAM 1.0 and PAMAM 3.0 (BRI2785) dendrimers
terminated with sodium N-(2-sulfoethyl)succinamide groups were similarly
prepared.
13C ~, p~AM 3.0 derivative (D20): 8 33.4, 35.5, 39.0, 39.5, 42.9, 43.2, 53.8,
10 54.1, 54.3, 56.5, 176.4, 176.9, 177.4, 178.9, 179.4.
t3C ~. pAMAM 1.0 derivative (D20): 8 34.9, 35.5, 39.5, 42.9, 43.1, 53.7, 54.1,
179.0, 179.1, 179.3.
B Preparation of sodium N-(2-sulfoethyl)succinamide terminated polylysine
1 S dendrimers
BHAlyslys21ys41ysglys~6 BRI2789
Trifluoroacetic acid (lml) was added to a suspension of
BHAlys1ys21ys41ys8DBL16
(36.Smg; S.O~mol} in dry dichloromethane (lml) and the resulting solution
stirred at
20 room temperature under nitrogen for two hours and then concentrated. The
residue
was dissolved in dry DMSO (2m1) and the pH adjusted to 8.5 with triethylamine.
A
solution of the crude tetrabutylammonium 4-nitrophenyl N-(2-
sulfoethyl)succinamate (ca. 0.2mmo1) in DMSO (lml) was then added dropwise and
the mixture stirred overnight at room temperature. The yellow solution was
then
25 concentrated (SO°/105 mmI-Ig) and the yellow residue partitioned
between water and
chloroform. The aqueous layer was separated, washed with chloroform (3X) and
ethyl acetate, and then concentrated to give an oil (99mg). The crude product
was
converted to the sodium salt by passage through a column of Amberlite IR
120(Na)
to yield 8lmg of material. This material was further purified by gel
filtration
30 (Sephadex LH20; water) to give the sodium N-(2-sulfoethyl)succinamide
terminated
BHAlyslys21ys41ysglysl6 dendrimer (39mg). 13C nmr (D20): 8 27.0, 32.3, 35.2,
CA 02343205 2001-03-06
WO 00/15239 PCT/AU99/00762
-22-
35.3, 35.6, 35.7, 39.5, 43.5, 54.1, 58.5, 131.5, 132.0, 133.3, 145.1, 177.8,
178.0,
178.4, 178.8, 178.9, 179.2, 179.7, 179.8.
The corresponding BHAlyslys2, BHAlyslys21ys4 (BRI2787) and
BHAlyslys21ys41ysg (BRI2788) terminated with sodium N-(2-
sulfoethyl)succinamide groups were similarly prepared.
13C ~r BHAlyslys21ys41ysg derivative (D20): 8 26.9, 32.3, 35.1, 35.3, 35.6,
35.7,
39.5, 43.5, 54.1, 58.5, 131.6, 131.9, 132.2, 132.3, 133.2, 133.3, 145.0,
145.2, 177.2,
177.8, 177.9, 178.0, 178.2, 178.3, 178.6, 178.7, 178.8, 178.9, 179.2, 179.3,
179.7,
179.8.
13C ~, BHAlyslys21ys4 derivative (D20): b 26.9, 32.3, 35.1, 35.4, 35.7, 35.8,
39.5,
43.5, 54.1, 58.5, 61.8, 131.7, 132.0, 132.2, 132.3, 133.2, 133.3, 145.0,
145.1, 177.3,
178.0, 178.3, 178.4, 178.7, 178.9, 179.0, 179.3, 179.7, 179.8.
13C nmr BHAlyslys2 derivative (D20): 8 26.9, 27.1, 32.2, 32.3, 34.7, 34.8,
35.1,
35.3, 35.6, 35.7, 39.5, 43.4, 54.1, 58.6, 61.8, 131.7, 131.9, 132.2, 132.3,
133.3,
144.9, 145.0, 177.7, 178.4, 178.8, 179.0, 179.3, 180Ø
EXAMPLE 5
Preparation of sodium 4-sulfophenylthiourea terminated dendrimers
A PAMAM 4.0 BRI2791
Solid sodium 4-sulfophenylisothiocyanate monohydrate (SOOmg; 1.96mmol) was
added to a solution of PAMAM 4.0 (300mg; 0.0582mmo1) in water (lOml) and the
resulting solution heated under nitrogen at 53° for two hours and then
cooled. The
solution was concentrated and the yellow solid residue purified by gel
filtration
(Sephadex LH20; water). The pure fractions were combined and freeze dried to
give
the sodium 4-sulfophenylthiourea terminated PAMAM 4.0 dendrimer as a fluffy
white solid (370mg). 1H nmr (D20) : 8 2.28; 2.52; 2.69; 3.15; 3.27; 3.60; 7.32
(d,
CA 02343205 2001-03-06
WO 00/15239 PCT/AU99/00762
- 23 -
J=9Hz); 7.72 (d, J=9Hz). 13C nmr (D20) : b 36.9; 41.1; 43.1; 48.3; 53.6; SS.B;
129.0; 131.1; 144.4; 178.5; 179.1; 184.4.
The corresponding PAMAM 1.0, PAMAM 2.0 (BRI2790), PAMAM 3.0, and
S PAMAM S.0 (BRI2991) dendrimers terminated with 3, 6, 12, and 48 sodium 4-
sulfophenylthiourea groups respectively were similarly prepared.
B PAMAM 4.0 (EDA) BRI6045
Solid sodium 4-sulfophenylisothiocyanate monohydrate (130mg; O.Smmol) was
added to a solution of PAMAM 4.0 (EDA) (69mg; O.Olmmol) in water (4m1) and the
resulting solution heated under nitrogen at S3° for two hours and then
cooled. The
solution was concentrated and the solid residue purified by gel filtration
(Sephadex
LH20; water). The pure fractions were combined and freeze dried to give PAMAM
4.0 terminated with 32 sodium 4-sulfophenylthiourea groups as a fluffy white
solid
1S (I36mg). 1H nmr (D20) : b 2.30; 2.50; 2.70; 3.18; 3.62; 7.35 (d, J=9Hz);
7.72 (d,
J=9Hz). 13C nmr (D20) : 8 36.8; 41.0; 43.1; 48.4; 53.6; SS.7; 128.9; 131.0;
144.3;
178.5; 179.0; 184.5.
C BHAlyslys21ys41ysglysl6 BRI2792
Trifluoroacetic acid (4m1) was added to a suspension of
BHAlys1ys21ys41ysgDBLl6
(0.73g; O.lmmol) in dry dichloromethane (4m1) under nitrogen. A vigorous
evolution
of gas was observed for a short time and the resulting solution was stirred at
room
temperature for two hours and then concentrated. The residual syrup was
dissolved in
water (Sml), the solution passed through a column of Amberlite IRA-401 (OH)
and
2S the filtrate concentrated to give BHAlyslys21ys41ysglysl6 as a viscous oil
(0.49g).
The oil was redissolved in water (Sml) and N,N-dimethyl-N-allylamine buffer
(pH
9.5; 3m1) added. Solid sodium 4-sulfophenylisothiocyanate monohydrate (1.30g;
S.lmmol) was then added and the resulting solution heated under nitrogen at
S3° for
two hours and then cooled. The solution was concentrated and the brownish
solid
residue purified by gel filtration (Sephadex LH20; water). The pure fractions
were
CA 02343205 2001-03-06
WO 00/15239 PCT/AU99/00762
-24-
combined, passed through a column of Amberlite IR 120(Na) and freeze dried to
give the sodium 4-sulfophenylthiourea terminated BHAlyslys21ys41ysglysl6
dendrimer as a fluffy white solid (374mg). 1H nmr (D20) : b 1.40; 1.72; 3.08;
3.42;
4.24; 4.60; 7.30; 7.40 (d, J=9Hz); 7.78 (d, J=9Hz). 13C nmr (D20) : 8 27.3;
32.5;
S 35.9; 43.7; 48.9; 58.6; 63.3; 128.8; 131.0; 143.7; 144.7; 145.1; 177.7;
178.1; 183.8;
185.2.
The corresponding BHAlyslys21ys41ysg, BHAlyslysZlys4lysglys161ys32 (BRI2992),
and BHAlyslys21ys41ysglys~61ys321ys64 (BRI2993) dendrimers terminated with 16,
64, and 128 sodium 4-sulfophenylthiourea groups respectively were similarly
prepared.
EXAMPLE 6
1 S Preparation of sodium 3,6-disulfonapthylthiourea terminated dendrimers
A PAMAM 4.0 B1tI2923
Solid sodium 3,6-disulfonapthylisothiocyanate (160mg; 0.41mmo1) was added to a
solution of PAMAM 4.0 (Slmg; O.Olmmol) in water (3m1) and the resulting
solution
heated under nitrogen at 53° for two hours and then cooled. The
solution was
concentrated and the brown solid residue purified by gel filtration (Sephadex
LH20;
water). The pure fractions were combined and concentrated to give the sodium
3,6-
disulfonapthylthiourea terminated PAMAM 4.0 dendrimer as a brownish solid
(73mg). 1H nmr (D20) : 8 2.30; 2.60; 2.74; 3.20; 3.57; 7.75; 7.86; 8.28. 13C
nmr
(D20) : 8 35.0; 39.9; 43.1; 48.1; 53.8; 56.1; 128.4; 128.6; 129.3; 131.0;
131.3; 136.0;
136.8; 138.2; 145.5; 146.0; 177.2; 177.8; 185.5.
The corresponding PAMAM 2.0 dendrimer terminated with sodium 3,6-
disulfonapthylthiourea groups was similarly prepared.
CA 02343205 2001-03-06
WO 00115239 PCT/AU99/00762
-25-
B PAMAM 4.0 (EDA) BRI6046
Solid sodium 3,6-disulfonapthylisothiocyanate (220mg; 0.57mmol) was added to a
solution of PAMAM 4.0 (EDA) (74mg; O.Olmmol) in water (4m1) and the resulting
solution heated under nitrogen at 53° for two hours and then cooled.
The solution
was concentrated and the brownish solid residue purified by gel filtration
(Sephadex
LH20; water). The pure fractions were combined and concentrated to give PAMAM
4.0 terminated with 32 sodium 3,6-disulfonapthylthiourea groups as a tan solid
(148mg). 1H nmr (D20) : 8 2.30; 2.80; 3.20; 3.54; 7.74; 7.85; 8.25. 13C nmr
(D20)
8 36.0; 40.8; 43.1; 48.3; 53.6; 55.9; 128.5; 129.4; 131.0; 131.3; 136.0;
136.8; 138.3;
145.5; 146.0; 178.2; 185.6.
C BHAlyslys21ys41ysglys~6 BRI2999
Trifluoroacetic acid (2ml) was added to a suspension of
BHAIyslys21ys41ysgDBLl6
(73mg; O.Olmmol) in dry dichloromethane (2m1) under nitrogen. A vigorous
evolution of gas was observed for a short time and the resulting solution was
stirred
at room temperature for two hours and then concentrated. The residual syrup
was
dissolved in water (Sml), the solution passed through a column of Amberlite
IRA-
401(OH) and the filtrate concentrated to give BHAlyslys21ys41ysglysl6 as a
viscous
oil. The oil was redissolved in water (5m1) and N,N-dimethyl-N-allylamine
buffer
(pH 9.5; 3ml) added. Solid sodium 3,6-disulfonapthylisothiocyanate (234mg;
0.60mmol) was then added and the resulting solution heated under nitrogen at
53°
for two hours and then cooled. The solution was concentrated and the brownish
solid
residue purified by gel filtration (Sephadex LH20; water). The pure fractions
were
combined, passed through a column of Amberlite IR 120(Na) and freeze dried to
25 give BHAlyslys21ys41ysglysl6 terminated with 32 sodium 3,6-
disulfonapthylthiourea
groups as a fluffy off white solid ( 119mg). 1 H nmr (D20) : b 1.0-2.0; 3.18;
3.43;
4.31; 7.22; 7.80; 7.89; 8.25. 13C nmr (D20) : b 27.2; 32.4; 35.3; 43.7; 49.0;
58.5;
63.6; 128.4; 129.1; 131.4; 136.1; 136.6; 138.6; 139.0; 145.1; 145.6; 178.4;
184.8;
186.7.
CA 02343205 2001-03-06
WO 00/15239 PCT/AU99/00762
-26-
EXAMPLE 7
Preparation of sodium 4-sulfonapthylthiourea terminated dendrimers
S PAMAM 4.0 BRI2997
Solid sodium 4-sulfonapthylisothiocyanate {180mg; O.Smmol) was added to a
solution of PAMAM 4.0 (5lmg; 0.01mmol) in water (Sml) and the mixture heated
under
nitrogen at 53° for two hours and then cooled. The water was distilled
under reduced
pressure from the resulting suspension and the off white solid residue
purified by gel
10 filtration (Sephadex LH20; water). The pure fractions were combined and
freeze dried to
give the sodium 4-sulfonapthylthiourea terminated PAMAM 4.0 dendrimer as a
fluffy white
solid (60mg). 1H nmr (D20) : b 2.20; 2.60; 3.14; 3.48; 7.23; 7.47; 7.56; 7.77;
7.93 (d,
J=6Hz); 8.56 (d, J=6Hz).13C nmr (D20) : S 35.8; 40.5; 43.1; 48.4; 53.6; 55.9;
127.6; 128.6;
130.3; 131.9; 132.5; 133.5; 134.7; 140.5; 142.7; 177.8; 178.0; 185.4.
IS
EXAMPLE 8
Preparation of sodium 3,5-disulfophenylthiourea terminated dendrimers
20 PAMAM 4.0 BRI6039
Solid sodium 3,5-disulfophenylisothiocyanate (1 l Omg; 0.32mmo1) was added to
a
solution of PAMAM 4.0 (63mg; 0.012mmo1) in water (3m1) and the resulting
solution
heated under nitrogen at 53° for two hours and then Gaoled. The
solution was concentrated
and the brownish solid residue purified by gel filtration (Sephadex G25;
water). The pure
25 fractions were combined and concentrated to give PAMAM 4.0 terminated with
24 sodium
3,5-disulfophenylthiourea groups as an off white solid (I lOmg). 1H nmr (D20)
: b 2.53;
3.08; 3.36; 3.66; 7.90; 7.95. 13C nmr (D20) : 8 34.8; 41.0; 43.1; 48.0; 53.7;
56.2; 124.1;
128.6; 143.5; 148.8; 177.6; 185Ø
CA 02343205 2001-03-06
WO 00/15239 PCT/AU99/00762
-27-
EXAMPLE 9
Preparation of sodium 3, 6, 8-trisulfonaphthylthiourea terminated dendrimers
S PAMAM 4.0 BRI2998
Solid sodium 3, 6, 8-trisulfonaphthylisothiocyanate (250mg; O.Smmol) was added
to
a solution of PAMAM 4.0 (51 mg; 0.01 mmol) and N,N-dimethyl-N-allylamine
buffer (pH
9.5; lml) in water (2m1) and the mixture heated under nitrogen at 53°
for two hours and then
cooled. The mixture was concentrated under reduced pressure to give an orange
solid. The
residual solid was dissolved in water (2ml) and passed through a short column
of Amberlite
IR-120(Na). The filtrate was then concentrated and the residue purified by gel
filtration
(Sephadex LH20; water). The pure fractions were combined and freeze dried to
give the
sodium 3, 6, 8-trisulfonaphthylthiourea terminated PAMAM 4.0 dendrimer as an
off white
solid (102mg). 1H nmr (D20) : 8 2.65; 3.02; 3.30; 3.66; 8.05; 8.42; 8.59;
8.67. 13C nmr
(D20) : 8 33.2; 38.7; 43.2; 43.7; 47.8; 54.0; 54.3; 56.7; 131.0; 131.3; 131.9;
135.9; 138.0;
139.6; 143.8; 144.1; 145.6; 176.2; 176.5; 186Ø
The corresponding sodium 3,6,8-trisulfonaphthylthiourea terminated dendrimer
BHAlys.lyszlys41ys81ys,6 Bl:ZI 7011 was prepared similarly.
EXAMPLE 10
Preparation of sodium 4-(sulfomethyl)benzamide terminated dendrimers
PAMAM 4.0 BltI6040
Solid 4-nitrophenyl 4-(chloromethyl)benzoate (200mg; 0.68mmo1) was added to a
stirred solution of PAMAM 4.0 (70mg; 0.014mmol) in dry DMSO (4m1) and the
resulting
yellow solution stirred at room temperature for two hours. The solution was
then
concentrated (10 4 mmHg; 40°) and the residue extracted with a mixture
of water and
dichloromethane (1:1). The remaining solid material was dissolved in DMSO
(Sml) and a
CA 02343205 2001-03-06
WO 00/15239 PCT/AU99/00762
-28-
solution of sodium sulfite (130mg; lmmol) in water (3m1) added. The slightly
cloudy
mixture that resulted was left to stand for four days, after which time the
addition of more
water (2m1) resulted in the formation of a clear homogeneous yellow solution.
The solution
was then concentrated, first at 25mmHg and 40o then at 10 4mmHg and SOo to
give the
S crude product. The crude product was purified by gel filtration (Sephadex
G25; water) to
give PAMAM 4.0 terminated with 24 sodium 4-(sulfomethyl)benzamide groups
(24mg). 1H
nmr (D20) : b 2.25; 2.66; 3.08; 3.20; 3.33; 3.38; 4.01; 7.40 (br d); 7.62 (br
d). 13C nmr
(D20) : b 36.7; 40.9; 43.0; 43.6; 53.5; 55.5; 61.0; 131.6; 135.0; 137.2;
140.4; 174.5; 178.6;
179.2.
EXAMPLE 11
Preparation of 4-sulfobenzamide terminated dendrimers
PAMAM 4.0 (EDA) BRI6116
Solid potassium N-hydroxysuccinimidyl 4-sulfobenzoate (100mg; 0.3mmol) was
added to a solution of PAMAM 4.0 (EDA) (35mg; O.OOSmmol) in O.1M pH 8.5 borate
buffer (Sml) and the solution stirred at room temperature for two hours. The
resulting milky
solution at this stage had a pH of 4.5. 1 M Sodium carbonate solution ( 1 ml)
was then added
to give a clear solution which was concentrated to give the crude product as a
white solid.
The crude product was purified by gel filtration (Sephadex G25; water) to give
PAMAM 4.0
(EDA) terminated with 32 sodium 4-sulfobenzamide groups (47mg). 1H nmr (D20) :
8 2.25;
2.42; 2.63; 3.05; 3.18; 3.31; 3.38; 7.72 (d, J=8Hz); 7.78 (d, J=8Hz). 13C nmr
(D20) : b 36.0;
40.4; 43.0; 43.7; 53.7; 55.8; 130.2; 132.2; 140.4; 150.1; 173.6; 178.0; 178.5.
CA 02343205 2001-03-06
WO 00/15239 PCT/AU99/00762
-29-
EXAMPLE 12
Preparation of Sodium N-(4-sulfophenyl)propanamide terminated dendrimers
5 PAMAM 4.0 (EDA) BRI6117
Solid sodium N-(4-sulfophenyl)acrylamide (250mg; lmmol) and solid sodium
carbonate (106mg; lmmol) were added successively to a stirred solution of
PAMAM 4.0
(EDA) (78mg; O.OI lmmol) in water (4m1). The resulting solution was stirred
under nitrogen
for four days and then freeze dried to give a fluffy white solid. The crude
product was
10 purified by gel filtration (Sephadex LH20; water to give PAMAM 4.0 (EDA)
terminated
with 64 sodium N-(4-sulfophenyl)propanamide groups (206mg). 13C nmr showed a
faint
trace of what was taken to be mono alkylated terminal amino groups. 1H nmr
(D20) : b
2.10; 2.48; 2.58; 2.79; 3.20; 7.42 (d, J=7Hz); 7.65 (d, J=7Hz). 13C nmr (D20)
: 8 36.5; 37.9;
41.1; 53.4; 55.6; 124.8; 130.9; 143.0; 144.2; 177.4; 178.5.
15
EXAMPLE 13
Preparation of Sodium 4-sulfophenylurea terminated dendrimers
20 PAMAM 4.0 (EDA) BRI6115
A solution of sodium sulfanilic acid (195mg; lmmol) in dry DMSO (3m1) was
added
dropwise to a solution of N,N'- disuccinimidyl carbonate (530mg; 2mmo1) in dry
DMSO
(4ml) and the resulting brownish solution stirred at room temperature for 20
hours. A
solution of PAMAM 4.0 (EDA) (75mg; 0.01 lmmol) in dry DMSO (lml) added and the
25 solution stirred for a further I 8 hours. The solution was then
concentrated under high
vacuum (10 SmmHg; 35°) to give a yellowish semi-solid. The crude
product was dissolved
in DMSO (4ml) and the solution added to 200m1 of well stirred ethyl acetate.
The
precipitated white solid was collected by filtration and washed with ethyl
acetate (2X) and
ether (2X), then dried to give a white powder (275mg). This material was
further purified by
30 gel filtration (Sephadex LH20; water) to give PAMAM 4.0 (EDA) terminated
with 32
CA 02343205 2001-03-06
WO 00/15239 PCT/AU99/00762
-30-
sodium 4-sulfophenylurea groups (106mg). 1H ntnr (D20) : 8 2.31; 2.55; 2.75;
3.19; 7.32
(d, J=9Hz); 7.63 (d, J=9Hz). 13C nmr (D20) : 8 36.3; 40.7; 43.3; 43.8; 53.7;
55.7; 123.3;
130.9; 140.9; 146.0; 161.4; 178.2; 178.6.
5 EXAMPLE 14
Preparation of N,N,N-trimethylglycinamide chloride terminated dendrimers
BHAlyslys21ys41ysglysl6 BRI2922
10 Trifluoroacetic acid (4m1) was added to a suspension of
BHAlyslys21ys41ysgDBLl6
(220mg; 30~mo1) in dry dichloromethane (2m1) and the resulting solution
stirred at room
temperature under nitrogen for two hours and then concentrated. The residue
was dissolved
in dry DMSO (Sml) and the pH adjusted to 8.5 with triethylamine. Solid 4-
nitrophenyl
N,N,N-trimethylglycinate chloride (O.SOg; l.8mmol)was then added and the
mixture stirred
15 overnight at room temperature. The cloudy solution was then concentrated
(50°/10-5 mmHg)
and the residue partitioned between water and dichloromethane. The aqueous
layer was
separated, washed with dichloromethane (3X) and ethyl acetate, and then
concentrated to
give an oil (1.128g). The crude product was purified by gel filtration
(Sephadex LH20;
water) to give the N,N,N-trimethylglycinamide terminated
BHAlyslys21ys41ys81ys16
20 dendrimer (1 l6mg). ~3C nmr (D20): b 25.5, 30.5, 30.8, 33.4, 42.1, 56.5,
57.1, 67.5, 68.1,
166.7, 167.0, 167.1, 176.0, 176.2.
EXAMPLE 15
25 Preparation of 4-Trimethyiammoniumbenzamide terminated dendrimers
PAMAM 4.0 BRI6043
1,1'-Carbonyldiimidazole (85mg; 0.52mmol) was added to a solution of 4-
trimethylammoniumbenzoic acid iodide (154mg; O.Smmol) in dry DMF (4ml) and the
30 mixture stirred at room temperature under argon for two hours. During this
time a white
CA 02343205 2001-03-06
WO 00/15239 PCT/AU99/00762
-31 -
solid separated from the solution. A solution of PAMAM 4.0 (58mg; 0.01 lmmol)
in dry
DMF (2m1) was then added and the mixture stirred overnight at room
temperature. After this
time most of the precipitate had dissolved and a ninhydrin test of the
solution was negative.
The mixture was concentrated (10 4 mmHg; 30°) to give a white solid
residue. The crude
S product was purified by gel filtration (Sephadex LH20; 10% AcOH) to give
PAMAM 4.0
terminated with 24 4-trimethylammoniumbenzamide groups as the acetic acid salt
(89mg).
1H nmr (D20) : S 1.96; 2.65-2.85; 3.25-3.55; 3.64; 7.92. 13C nmr (D20) : b
25.8; 33.1;
33.5; 38.7; 43.1; 43.5; 53.5; 54.1; 56.4; 61.2; 124.8; 133.6; 139.9; 153.2;
173.2; 176.3;
176.8; 182.6.
IO
The corresponding PAMAM 2.0 dendrimer terminated with 6 4-trimethylammonium
benzamide groups was similarly prepared.
EXAMPLE 16
Preparation of 4-(Trimethylammoniummethyl)benzamide terminated
dendrimers
PAMAM 4.0 BRI6044
20 Solid 4-nitrophenyl 4-(chloromethyl)benzoate (150mg; O.Smmol) was added
to a stirred solution of PAMAM 4.0 (52mg; O.Olmmol) in dry DMSO (3m1). The
resulting yellow solution was stirred at room temperature for 20 hours, when a
ninhydrin test was negative (pH ca.8.5). The solution was then concentrated
(10
SmmHg; 400) and the residue shaken with a mixture of water and dichloromethane
( 1:1 ). The insoluble gel-Iike material was collected by filtration, washed
with water
(2X) and dichloromethane (2X), and then air dried. The crude 4-(chloromethyl)-
benzamide terminated dendrimer was dissolved in 25% aq. trimethylamine (20m1)
and the yellow solution left to stand overnight. The solution was then
concentrated,
the residue dissolved in water (Sml) and the solution passed through a column
of
Amberlite IRA-401 (OH). The colourless filtrate was concentrated to give a
viscous
CA 02343205 2001-03-06
WO 00/15239 PCT/AU99/00762
-32-
oil which was purified by gel filtration (Sephadex G10; 10% AcOH) to give
PAMAM 4.0 terminated with 24 4-(trimethylammoniummethyl)benzamide groups
(90mg). 1 H nmr (D20) : b 1.88; 2.65-2.80; 2.98; 3.10-3.60; 7.52 (br d,
J=9Hz); 7.72
(br d, J=9Hz). 13C nmr (D20) : b 26.6; 33.4; 38.8; 43.2; 43.5; 53.6; 53.6;
54.1; 56.8;
62.8; 73.0; 132.1; 135.3; 137.5; 140.0; 176.4; 176.9; 183.6.
EXAMPLE 17
Preparation of N-(2-Acetoxyethyl)-N,N-(dimethylammonium)methyl-
carboxamide terminated dendrimers
PAMAM 4.0
Solid I,1'-carbonyldiimidazole (85mg; 0.52mmo1) was added to a solution of
N-(2-acetoxyethyl)-N-(carboxymethyl)-N,N-dimethylammonium bromide (135mg;
0.5mmol) in dry DMF (3m1) and the resulting solution stirred under nitrogen
for two
hours. A solution of PAMAM 4.0 (60mg; 0.012mmol) in DMF (2ml) was then
added, which caused the immediate formation of a flocculant precipitate which
slowly redissolved. The mixture was stirred for two days and then concentrated
(10
4mmHg; 400) to give a viscous oil. The crude product was purified by gel
filtration
20 (Sephadex G10; 10% AcOH) to give PAMAM 4.0 terminated with 24 N-(2-
Acetoxyethyl)-N,N-(dimethylammonium)methylcarboxamide groups (64mg). 1 H
nmr (D20) : b 1.93; 2.05; 2.70; 3.10-3.60; 3.28; 3.93 (m); 4.14; 4.48 (m). 13C
nmr
(D20) : 8 24.6; 26.2; 33.2; 38.7; 42.8; 42.9; 53.9; 57.4; 62.6; 67.3; 67.5;
168.9;
176.4; 176.8; 177.3; 183.2.
CA 02343205 2001-03-06
WO 00/15239 PCT/AU99/00762
-33-
EXAMPLE 18
Preparation of Guanidino terminated dendrimers
PAMAM 4.0 BRI6042
A solution of PAMAM 4.0 (63mg; 0.012mmol) and methylthiopseudourea
sulfate (170mg; 0.61mmo1) in water (Sml) (pH 10.5) was heated under nitrogen
at
80o for two hours. The solution was then concentrated and the residue purified
by gel
filtration (Sephadex G10; 10% AcOH) to give PAMAM 4.0 terminated with 24
guanidino groups as the acetate salt ( 107mg). 1 H nmr (D20) : 8 2.00; 2.80
(br t); 3.09
(br t); 3.32; 3.45 (br t); 3.60 {br t). 13C nmr (D20) : 8 25.2; 33.2; 33.4;
38.7; 41.2;
42.6; 43.4; 44.7; 53.5; 54.0; 56.3; 176.5; 176.7; 176.9; 181.6.
The corresponding PAMAM 2.0 dendrimer terminated with 6 guanidino groups was
similarly prepared.
EXAMPLE 19
Preparation of 4-([1,4,8,11-tetraazacyclotetradecane]methyl)benzamide
terminated dendrimers
PAMAM 4.0 BRI6041
A solution of 1-(4-carboxyphenyl)methyl-1,4,8,11-tetraazacyclotetradecane
tetra hydrochloride (120mg; 0.25mmo1), N-hydroxysuccinimide (60mg; 0.52mmo1)
and 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (250mg;
l.3mmo1) in pH 7 phosphate buffer (IOmI) was allowed to stand a room
temperature
for one hour and then a solution of PAMAM 4.0 (32mg; 0.006mmol) in pH 7
phosphate buffer (l0ml) added. The mixture was allowed to stand for two days
and
then concentrated. The residue was purified by gel filtration (Sephadex LH20;
10%
AcOH) to give PAMAM 4.0 terminated with ca. 12 4-
CA 02343205 2001-03-06
WO 00/15239 PCT/AU99/00762
-34-
((1,4,8,I1-tetraazacyclotetradecane)methyl)-benzamide groups as determined by
1H
and 13C nmr (80mg). The product was then dissolved in water and passed through
a
column of Amberlite IRA-401 (CI) resin and then concentrated. The residue was
dissolved in water ( 1 ml), concentrated HCl ( 1 ml) added, and the solution
diluted with
ethanol (30m1) to precipitate a white solid. The solid was collected by
filtration
(68mg). Once again IH and 13C nmr showed ca. 50% functionalisation of the
terminal amino groups. IH nmr (D20) : 8 2.17; 2.36; 2.50; 2.78; 2.85; 3.25;
3.40;
3.50; 3.60; 3.62; 4.49; 7.63 (br d); 7.78 (br d). 13C nmr (D20) : b 22.7;
23.1; 33.2;
38.8; 39.9; 40.2; 40.3; 41.0; 41.2; 42.0; 42.9; 43.2; 43.6; 45.5; 46.1; 49.1;
52.2; 53.9;
54.3; 56.6; 62.7; 132.5; 135.7; 137.1; 139.7; 174.3; 176.2; 176.3; 176.7;
177.0;
178.2; 178.5.
EXAMPLE 20
Preparation of 4-Carboxy-3-hydroxybenzylamine terminated dendrimers
PAMAM 4.0 (EDA) BRI6119
Sodium cyanoborohydride (32mg; O.Smmol) was added to a mixture of
PAMAM 4.0 (EDA) (69mg; O.Olmmol), 4-formyl-2-hydroxybenzoic acid (83mg;
O.Smmol), and sodium hydrogen carbonate (42mg; 0.5mmo1) in water (4m1). The
inhomogeneous orange mixture was stirred for four hours at room temperature,
during which time it became homogeneous. The orange solution was then
concentrated and the residue purified by gel filtration (Sephadex LH20; water)
to
give PAMAM 4.0 (EDA) terminated with ca. 32 4-carboxy-3-hydroxybenzylamine
groups (9lmg). IH andl3C nmr (D20) shows mostly mono alkylation but with some
signs of dialkylation of the terminal amino groups, both spectra show broad
peaks.
13C ~, (D20) :b 37.0; 41.1; 50.9; 53.4; 55.5; 55.8; 61.5; 120.9; 122.2; 122.4;
132.3; 132.7; 135.0; 135.8; 163.5; 163.7; 169.0; 178.6; 179.3. IH nmr (D20) :
8
2.20; 2.35; 2.60; 3.15; 3.30; 3.55; 4.25; 6.68; 7.12; 7.55.
CA 02343205 2001-03-06
WO 00/15239 PCT/AU99/00762
-35-
EXAMPLE 21
Preparation of 4-Carboxyphenylamide terminated dendrimers
S PAMAM 4.0 (EDA)
Solid 4-carboxyphenylisothiocyanate (86mg; 0.48mmo1) was added to a
solution of PAMAM 4.0 (EDA) (69mg; O.Olmmol) in water (20m1). The pH of the
resulting cloudy solution was adjusted to 9 with saturated NaHC03 solution and
left
to stir at room temperature for 24 hours. The reaction mixture was then
filtered and
the filtrate concentrated to give a white solid residue, which was purified by
gel
filtration (Sephadex LH20; water) and then freeze dried to give the product as
a
white fluffy solid (68mg).
EXAMPLE 22
Preparation of 3,5-Dicarboxyphenylamide terminated dendrimers
PAMAM 4.0 (EDA)
Solid 3,5-dicarboxyphenylisothiocyanate (1 l2mg; O.Smmol) was added to a
solution of PAMAM 4.0 (EDA) (70mg; 0.01 mmol) in water (Sml). The pH of the
resulting cloudy solution was adjusted to 10 with 1M NaZC03 solution and
heated
under nitrogen at 53° for 2 hours. The reaction mixture was then
filtered and the
filtrate concentrated to give a brownish solid residue, which was purified by
gel
filtration (Sephadex LH20; water) and then freeze dried to give the product as
a pale
brown solid (112mg).
CA 02343205 2001-03-06
WO 00/15239 PCT/AU99/00762
-36-
EXAMPLE 23
Preparation of Sodium 4-Phosphonooxyphenylthiourea terminated dendrimers
PAMAM 4.0 (EDA)
Solid sodium 4-phosphonooxyphenylisothiocyanate (251 mg) was added to a
solution of PAMAM 4.0 (EDA) (69mg; 0.01 mmol) in water (20m1). The resulting
solution (pH 9) was stinted for 24 hours at room temperature under nitrogen.
The
reaction mixture was then concentrated to give a white solid residue, which
was
purified by gel filtration (Sephadex LH20; water) and then freeze dried to
give the
product as a fluffy white solid (86mg).
EXAMPLE 24
Preparation of Sodium 4-(Phosphonomethyl)phenylthiourea terminated
dendrimers
PAMAM 4.0 (EDA)
Solid sodium 4-(phosphonomethyl)phenylisothiocyanate (97mg) was added
to a solution of PAMAM 4.0 (EDA) (69mg; O.Olmmol) in water (30m1). The
resulting solution was stirred for 3 days at room temperature under nitrogen,
maintaining the pH at 8 with periodic addition of saturated NaHC03 solution.
The
reaction mixture was then concentrated to give a white solid residue, which
was
purified by gel filtration (Sephadex LH20; water) and then freeze dried to
give the
product as a fluffy white solid ( 102mg).
CA 02343205 2001-03-06
WO 00/15239 PCT/AU99/00762
-37-
EXAMPLE 25
Preparation of Sodium Ethyl 4-(Phosphonomethyl)phenylthiourea terminated
dendrimers
PAMAM 4.0 (EDA)
Solid sodium ethyl 4-(phosphonomethyl)phenylisothiocyanate (109mg) was
added to a solution of PAMAM 4.0 (EDA) (69mg; 0.01 mmol) in DMF (30m1). The
resulting solution was stirred for 17 hours at room temperature under
nitrogen,
maintaining the pH at 8 with periodic addition of saturated NaHC03 solution.
The
reaction mixture was then concentrated to give a white solid residue, which
was
purified by gel filtration (Sephadex LH20; water) and then freeze dried to
give the
product as a fluffy white solid (30mg).
EXAMPLE 26
Preparation of C"alkyl linked 2-thiosialoside terminated dendrimers
Methyl [(8-octanoic acid N-hydroxysuccinimide ester) 5-acetamido-4,7,8,9-tetra-
O-
acetyl-3,5-dideoxy-2-thio-D-glycero-a-D-galacto-2-nonulopyranosid]orate was
prepared by the following procedure.
To a solution of methyl 5-acetamido-4,7,8,9-tetra-O-acetyl-2-S-acetyl-3,5-
dideoxy-
2-thio-D-glycero-a-D-galacto-2-nonulopyranosonate (Hasegawa et al, 1986)
(100mg.) in dry dimethylformamide (1mI) was added 8-bromooctanoic acid
(4lmg.) and diethylamine (280mg.) and the solution stirred at 20° C for
17 hours.
Solvent was removed under vacuum and the residue partitioned between ethyl
acetate and ice cold 5 % hydrochloric acid. The organic layer was washed with
water, dried over sodium sulphate, and evaporated to give a residue
(130mg.).This
was dissolved in ethyl acetate (Sml.) and N-hydroxysuccinimide (26mg.) and
CA 02343205 2001-03-06
WO 00/15239 PCT/AU99/00762
-38-
dicyclohexylcarbodiimide (46mg.) were added. The mixture was stirred at
20°C for
17 hours then the white precipitate was filtered off. The filtrate was
concentrated
and purified by flash chromatography on silica gel eluting with ethyl acetate.
Fractions containing product were combined and evaporated to give a white foam
97mg. 71 %.
Similarly were prepared:
Methyl ((11-undecanoic acid N-hydroxysuccinimide ester) 5-acetamido-4,7,8,9-
tetra-O-acetyl-3,5-dideoxy-2-thin-D-glycero-a-D-galacto-2-
nonulopyranosid]orate.
Methyl [(acetic acid N-hydroxysuccinimide ester) 5-acetamido-4,7,8,9-tetra-O-
acetyl-3 , 5-dideoxy-2-thio-D-glycero-a-D-galacto-2-nonulopyranosid] orate.
Methyl [(4-butanoic acid N-hydroxysuccinimide ester) 5-acetarnido-4,7,8,9-
tetra-O-
acetyl-3 , 5-dideoxy-2-thio-D-glycero-a-D-galacto-2-nonulopyranos id] orate.
Methyl [(4-methylbenzoic acid N-hydroxysuccinimide ester) 5-acetamido-4,7,8,9-
tetra-O-acetyl-3 , 5-dideoxy-2-thio-D-glycero-a-D-galacto-2-
nonulopyranosid]orate.
A PAMAM [EDA] 4.0 [(8-octanamido)- 5-acetamido-3,5-dideoxy-2-thio-D-
glycero-a-D-galacto-2-nonulopyranosidoic acid]3z BRI 6112
To a solution of the PAMAM [EDA] 4.0 (SOmg.) in dry dimethyl
sulphoxide(4m1.) under an inert atmosphere was added methyl [(8-octanoic
acid N-hydroxysuccinimide ester) 5-acetamido-4,7,8,9-tetra-O-acetyl-3,5-
dideoxy-2-thio-D-glycero-a-D-galacto-2-nonulopyranosid]onate(300mg.)
and the solution stirred for 60 hours at 20°C. The solvent was removed
under vacuum and the residue was dissolved in methanol (2m1.). This
solution was subjected to size exclusion chromatography on Sephadex LH20
CA 02343205 2001-03-06
WO 00/15239 PCT/AU99/00762
-39-
eluting with methanol. On evaporation of solvent, the product, PAMAM
[EDA] 4.0 [methyl [(8-octanamido) 5-acetamido-4,7,8,9-tetra-O-acetyl-3,5-
dideoxy-2-thio-D-glycero-a-D-galacto-2-nonulopyranosid]ovate]32 was
obtained as a white powder. 182mg. 93 %
This was converted to the free sialoside by the following method:
To a solution of PAMAM [ EDA ] 4.0 [methyl [(8-octanamido) 5-
acetamido-4,7, 8, 9-tetra-O-acetyl-3,5-dideoxy-2-thio-D-glycero-a-D-galacto-
2-nonulopyranosid]ovate]32 (182mg.) in dry methanol (3m1.) under argon at
20°C was added a freshly prepared 0.19M solution of sodium methoxide in
methanol (7m1.) and the mixture stirred for 2.5 hours. The solvent was
evaporated and the residue dissolved in water (lOml.) and stirred for 3
hours. This solution was subjected to size exclusion chromatography on
Sephadex LH20 eluting with water. On lyophilisation, the product,
PAMAM [EDA] 4.0 [(8-octanamido)- 5-acetamido-3,5-dideoxy-2-thio-D-
glycero-a-D-galacto-2-nonulopyranosidoic acid]32 was obtained as a pale
lemon powder 110mg. 77%
By a similar procedure were prepared:
PAMAM [EDA] 4.0 [(11-undecanamido)-5-acetamido-3,5-dideoxy-2-thio-
D-glycero-a-D-galacto-2-nonulopyranosidoic acid]32 BRI 6147
PAMAM [EDA] 4.0 [ (acetamido)- 5-acetamido-3,5-dideoxy-2-thio-D-
glycero-a-D-galacto-2-nonulopyranosidoic acid]32 BRI 6121
PAMAM [EDA) 4.0 [(4-methylbenzamido)- 5-acetamido-3,5-dideoxy-2-
thio-D-glycero-a-D-galacto-2-nonulopyranosidoic acid]32 BRI 6120
B BHA lyslys21ys41ys81ys16 [(8-octanamido)- 5-acetamido-3,5-dideoxy-2-thio-
CA 02343205 2001-03-06
WO 00/15239 PCT/AU99/00762
-40-
D-glycero-a-D-galacto-2-nonulopyranosidoic acid]32 BRI 6169
A solution of BHA lyslysZlys41ys81ys,6 (t-Boc)32 (20.3mg.) in a mixture of
trifluoroacetic acid (2m1.) and dichloromethane (2m1.) was stirred at
20°C
for 2 hours then solvent was removed under vacuum. The residue was
dissolved in dry dimethyl sulphoxide (lml.) and di-isopropylethylamine
(25mg.) and methyl [(8-octanoic acid N-hydroxysuccinimide ester) 5-
acetamido-4, 7, 8, 9-tetra-O-acetyl-3 , S-dideoxy-2-thin-D-glycero-a-D-galacto-
2-nonulopyranosid]onate (78mg.) were added. The mixture was stirred
under argon at 20°C for 60 hours then solvent was removed under vacuum.
The residue was dissolved in a freshly prepared O.1M solution of sodium
methoxide in methanol (2.Sml.) and the mixture stirred for 3 hours under
argon at 20°C. The solvent was evaporated and the residue dissolved in
water (1mL) and stirred for 17 hours . This solution was subjected to size
exclusion chromatography on Sephadex LH20 eluting with water. After
lyophilisation ,the product, BHA lyslys21ys41ys81ys,6 [(8-octanamido)- 5-
acetamido-3,5-dideoxy-2-thio-D-glycero-a-D-galacto-2-nonulopyranosidoic
acid]32 was obtained as a white powder 44mg. 86 % .
EXAMPLE 27
Preparation of dendritic sialosides modified in the 4-position of sialic acid
Methyl4-azido-S-acetamido-7,8,9-tri-O-acetyl-2-S-acetyl-3,4,5-trideoxy-2-thio-
D-
glycero-a-D-galacto-2-nonulopyranosonate was prepared by the following
procedure. To a solution of methyl 4-azido-5-acetamido-7,8,9-tri-O-acetyl-2-
chloro-3,4,5-trideoxy-D-glycero-~3-D-galacto-2-nonulopyranosonate (Sabesan,
1994) (Sg.) in dry dichloromethane (150m1.) was added finely powdered
potassium
thiolacetate (5.8g.) and the suspension stirred vigorously at 20°C for
48 hours. The
CA 02343205 2001-03-06
WO 00/15239 PCT/AU99/00762
-41 -
mixture was filtered and evaporated to give a light brown foam (5.2g.). The
required product was isolated by preparative reversed phase HPLC [C,B, 30%
acetonitrile/water] as a white foam 3.9g. 72 % .
5 Methyl [(8-octanoic acid N-hydroxysuccinimide ester) 4-azido-S-acetamido-
7,8,9-
tri-O-acety l-3 ,4, 5-trideoxy-2-thio-D-glycero-a-D-galacto-2-nonulopyranosid]
onate
was prepared by the following procedure.
To a solution of methyl 4-azido-5-acetamido-7,8,9-tri-O-acetyl-2-S-acetyl-
3,4,5-
10 trideoxy-2-thio-D-glycero-a-D-galacto-2-nonulopyranosonate (300mg.) in dry
dimethylformamide (3.Sml.) was added 8-bromooctanoic acid (155mg.} and
diethylamine (1.26m1.} and the solution stirred at 20° C for 17 hours.
Solvent was
removed under vacuum and the residue partitioned between ethyl acetate and ice
cold IO % hydrochloric acid. The organic layer was washed with water, dried
over
15 sodium sulphate, and evaporated to give a yellow foam (385mg.).This was
dissolved in ethyl acetate (20m1.) and N-hydroxysuccinimide (95mg.) and
dicyclohexylcarbodiimide (175mg.) were added. The mixture was stirred at
20°C
for 17 hours then the white precipitate was filtered off. The filtrate was
concentrated and purified by preparative reversed phase HPLC [C18, 30%
20 acetonitrile/water] to give a white foam 340mg. 83 % .
A PAMAM [EDA] 4.0 ( (8-octanamido)- 4-azido-5-acetamido-3,4,5-trideoxy-
2-thio-D-glycero-a-D-galacto-2-nonulopyranosidoic acid]32 BRI 6146
25 To a solution of the PAMAM [EDA] 4.0 (72mg.) in dry dimethyl
sulphoxide (Sml.) under an inert atmosphere was added methyl [(8-octanoic
acid N-hydroxysuccinimide ester) 4-azido-S-acetamido-7,8,9-tri-O-acetyl
3,4,5-trideoxy-2-thin-D-glycero-a-D-galacto-2-nonulopyranosid]onate (318
mg ) and the solution stirred for 60 hours at 20°C. The solvent was
removed
30 under vacuum and the residue was dissolved in methanol (2m1.). This
CA 02343205 2001-03-06
WO 00/15239 PCT/AU99/00762
-42-
solution was subjected to size exclusion chromatography on Sephadex LH20
eluting with methanol. On evaporation of solvent, the product, PAMAM
[EDA] 4.0 [methyl [(8-octanamido) 4-azido-5-acetamido-7,8,9-tri-O-acetyl-
3,4,5-trideoxy-2-thio-D-glycero-a-D-galacto-2-nonulopyranosid]ovate]32 was
obtained as a white foam. 225mg. 81
The free sialoside was obtained by the following method:
To a solution of PAMAM [EDA] 4.0 [methyl [(8-octanamido) 4-azido-5-
10 acetamido-7,8,9-tri-O-acetyl-3,4,5-trideoxy-2-thio-D-glycero-a-D-galacto-2-
nonulopyranosid]ovate]32 (215mg.) in dry methanol (lml.) under argon at
20°C was added a freshly prepared 1M solution of sodium methoxide in
methanol (lml.) and the mixture stirred for 3 hours. The solvent was
evaporated and the residue dissolved in water (2m1.) and stirred for 17
hours. This solution was subjected to size exclusion chromatography on
Sephadex LH20 eluting with water. On lyophilisation, the product,
PAMAM [EDA] 4.0 [(8-octanamido)- 4-azido-S-acetamido-3,4,5-trideoxy-
2-thio-D-glycero-a-D-galacto-2-nonulopyranosidoic acid]3z was obtained as
a fluffy white powder 160mg. 90%
B PAMAM [EDA] 4.0 [(8-octanamido)- 4-amino-5-acetamido-3,4,5-trideoxy
2-thio-D-glycero-a-D-galacto-2-nonulopyranosidoic acid]32 BRI 6149
A slow steam of hydrogen sulphide gas was passed into a solution of
25 PAMAM [EDA] 4.0 [(8-octanamido)- 4-azido-5-acetamido-3,4,5-trideoxy-
2-thio-D-glycero-a-D-galacto-2-nonulopyranosidoic acid]32 (25mg.) in a
mixture of pyridine (40m1. ) and water (20m1. ) at 20°C for 5 days. The
solution was then bubbled with nitrogen for 2 hours to remove excess
hydrogen sulphide. The solution was evaporated to dryness and the residue
taken up in water (5 ml) and filtered through a 0.45~cm. membrane filter to
CA 02343205 2001-03-06
WO 00/15239 PCT/AU99/00762
-43-
remove sulphur. On lyophilisation, the product, PAMAM (EDA] 4.0 [(8-
octanamido)- 4-amino-5-acetamido-3,4,5-trideoxy-2-thio-D-glycero-a-D-
galacto-2-nonulopyranosidoic acid]3z was obtained as a fluffy white powder
23mg. 96
EXAMPLE 28
Preparation of boronic acid terminated dendrimers.
4-Carboxyphenylboronic acid N-hydroxysuccinimide ester
To a solution of 4-carboxyphenylboronic acid (SOOmg.) in dry dimethyl
formamide (Sml) were added N-hydroxysuccinimide (380mg.) and
dicyclohexylcarbodiimide (680mg) The mixture was stirred at 20° C for
64 hours
then the white precipitate was filtered off. The solvent was removed under
vacuum
and the residue dissolve in ethyl acetate (100m1.). This solution was washed
with
water, dried over sodium sulphate and evaporated to give a white solid which
was
crystallised from acetonitrile/water as fine needles 730mg. 92%
PAMAM [EDA] 4.0 [4-benzamidoboronic acid]32 BRI 6160
To a solution of the PAMAM [EDA] 4.0 (69mg.) in dry dimethyl
sulphoxide (Sml) under an inert atmosphere was added 4-carboxyphenylboronic
acid N-hydroxysuccinimide ester (130mg.) and the solution stirred for 65 hours
at
20°C. To the thick slurry was added 1M sodium carbonate solution (lml.)
and the
clear solution stirred an additional 24 hours. The solvent was removed under
vacuum and the residue was dissolved in 10% ammonia solution (Sml.). This
solution was subjected to size exclusion chromatography on Sephadex LH20
eluting
with 10 % ammonia solution . On evaporation of solvent, the product, PAMAM
[EDA] 4.0
[4-benzamidoboronic acid]32 was obtained as a white fluffy solid. 110mg. 94% .
CA 02343205 2001-03-06
WO 00/15239 PCT/AU99/00762
-44-
EXAMPLE 29
Preparation of Sodium 3,6-disulfonaphthylthiourea terminated dendrimers.
S BHAlyslysZlys4lysalys~61ys32
Trifluoroacetic acid (2m1) was added to a stirred suspension of
BHAlyslys21ys41ysg1ys16DBL32 (147mg) in dry dichloromethane (2m1) and the
resulting solution stirred at room temperature under nitrogen for two hours
and
then concentrated. The residue was dissolved in N,N-dimethyl-N-allylamine
buffer
(pH 9.5; Sml) and then solid 3,6-disulfonaphthyl isothiocyanate (400mg) added.
The pH of the mixture was then adjusted to 9.5 by the addition of 1M sodium
carbonate and the solution heated at 53 °C for three hours under
nitrogen. The
reaction mixture was concentrated and the residue redissolved in water and the
solution passed through a column of Amberlite IR 120 (Na). The filtrate was
15 concentrate was concentrated to give the crude product, which was purified
by gel
filtration (Sephadex LH20; water) to give BHAlyslysZlys4lysg1ys161ys32 with 64
sodium 3,6-disulfonaphthylurea groups as a white fluffy solid (I75mg).
EXAMPLE 30
Preparation of Sodium 3,5-Disulfophenylthiourea terminated dendrimers.
BHAlyslys21ys41ysglys161ys32
Trifluoroacetic acid (3m1) was added to a stirred suspension of
25 BHAlys1ys21ys41ys81ys16DBL32 (300mg; 0.02mmo1) in dry dichloromethane (3m1)
and the resulting solution stirred at room temperature under nitrogen for two
hours
and then concentrated. The residue was dissolved in water and the solution
passed
through a column of Amberlite IRA 401 (OH) and the filtrate concentrated to
give
a viscous oil (187mg). The oil was dissolved in a 1:1 mixture of
pyridine/water
(8m1) and solid sodium 3,5-disulfophenyl isothiocyanate (680mg; 2mmol) added.
CA 02343205 2001-03-06
WO 00/15239 PCT/AU99/00762
- 45 -
The resulting solution was heated at 53°C for three hours under
nitrogen. The
solution was then concentrated to give a white solid residue. The crude
product
was purified by gel filtration (Sephadex LH20; water) to give
BHAlys1ys21ys41ys81ys161ys32 with 64 sodium 3,6-disulfophenylurea groups as a
white fluffy solid.
EXAMPLE 31
Preparation of Sodium 3,5-Dicarboxyphenylthiourea terminated dendrimers.
BHAlyslys21ys41ys81ys161ys32 BRI6741
Trifluoroacetic acid (3m1} was added to a stirred suspension of
BHAlyslysz1ys41ys81ys16DBL32 (300mg; 0.02mmo1) in dry dichloromethane (3m1)
and the resulting solution stirred at room temperature under nitrogen for two
hours
and then concentrated. The residue was dissolved in water and the solution
passed
through a column of Amberlite IRA 401 (OH) and the filtrate concentrated to
give
a viscous oil (186mg). The oil was dissolved in a 1:1 mixture of
pyridine/water
(8m1) and sodium 3;5-dicarboxyphenyl isothiocyanate (450mg; 2mmol) added. The
resulting solution was heated at 53 °C for 13 hours under nitrogen. The
solution
20 was then concentrated to give a white solid residue. The crude product was
purified by gel filtration (Sephadex LH20; water) to give
BHAlyslys21ys41ysg1ys,61ys32 with 64 sodium 3,6-dicarboxyphenylurea groups as
a
white fluffy solid.
25 The sodium 3,5-dicarboxyphenylthiourea terminated dendrimer PAMAM 4.0
(EDA) BRI 6195 was similarly prepared.
CA 02343205 2001-03-06
WO 00/15239 PCT/AU99/00762
-46-
EXAMPLE 32
Preparation of Sodium 4-phosphonooxyphenylthiourea terminated dendrimers.
BHAlyslys21ys41ys$1ys161ys32 BRI6181
Trifluoroacetic acid (2m1) was added to a stirred suspension of
BHAlyslys21ys41ys81ys,6DBL32 (147mg; 0.01 mmol) in dry dichloromethane (2m1)
and the resulting solution stirred at room temperature under nitrogen for two
hours
and then concentrated to give a viscous oil. The oil was dissolved in N,N-
dimethyl-N-allylamine buffer {pH 9.5; Sml) and solid 4-phosphonooxyphenyl
isothiocyanate (250mg) added. The pH of the resulting solution was adjusted to
10
with 1M sodium carbonate and the mixture heated at 53°C for three hours
under
nitrogen. The solution was then concentrated to give a white solid residue.
The
residue was redissolved in water and the solution passed through a column of
15 Amberlite IR 120 (Na) and the filtrate concentrated. The residue was then
purified
by gel filtration (Sephadex LH20; water) to give BHAlys1ys21ys41ys81ys,61ys32
with
64 sodium 4-phosphonooxyphenylurea groups as a white fluffy solid (150mg).
EXAMPLE 33
Preparation of Sodium 4-phosphonophenylthiourea terminated dendrimers.
BHAlyslys21ys41ys81ys161ys32
Trifluoroacetic acid (2m1) was added to a stirred suspension of
25 BHAlys1ys21ys41ys81ys16DBL32 (147mg; 0.01 mmol) in dry dichloromethane
(2m1)
and the resulting solution stirred at room temperature under nitrogen for two
hours
and then concentrated to give a viscous oil. The oil was dissolved in N,N-
dimethyl-N-allylamine buffer (pH 9.5; Sml) and solid 4-phosphonophenyl
isothiocyanate {250mg) added. The pH of the resulting solution was adjusted to
9
with saturated sodium bicarbonate solution and the mixture heated at
53°C for
CA 02343205 2001-03-06
WO 00/15239 PCT/AU99/00762
-47-
three hours under nitrogen. The solution was then concentrated to give a white
solid residue. The residue was redissolved in water and the solution passed
through a column of Amberlite IR 120 (Na) and the filtrate concentrated. The
residue was then purified by gel filtration (Sephadex LH20; water) to give
BHAlyslys21ys41ys$1ys161ys32 with 64 sodium 4-phosphonophenylurea groups
BRI 6196 as a white fluffy solid (152mg) after freeze drying.
EXAMPLE 34
Preparation of Sodium 4,6-diphosphononaphthylthiourea terminated
dendrimers .
PAMAM 4.0
A solution of sodium 4,6-diphosphononaphthyl isothiocyanate (165mg) in
water (2m1) was added to a solution of PAMAM 4.0 (Slmg; O.Olmmol) in water
(2ml). The pH of the mixture was adjusted to 9.5 with saturated sodium
bicarbonate solution and the mixture vigorously stirred for one hour at room
temperature and then heated at 53 °C for three hours under nitrogen.
The mixture
was then filtered and the filtrate concentrated to give a brown solid residue.
The
crude product was purified by gel filtration (Sephadex G25; water) to give
PAMAM 4.0 terminated with 24 sodium 4,6-diphosphononaphthylthiourea groups
as a brown solid (8lmg) after freeze drying.
EXAMPLE 35
Preparation of Fluoresceinthiourea terminated dendrimers.
PAMAM 4.0 (EDA)
Solid fluorescein isothiocyanate (188mg) was added to a solution of
PAMAM 4.0 (EDA) (74mg; O.Olmmol) in water (3ml}. Saturated sodium
CA 02343205 2001-03-06
WO 00/15239 PCT/AU99/00762
-48-
bicarbonate solution was added to adjust the pH to 9 and the resulting
homogenous
solution stirred overnight at room temperature and then concentrated. The
orange
residue was purified by gel filtration (Sephadex LH20; water) to give PAMAM
4.0
(EDA) terminated with 21 fluoresceinthiourea groups as a fluffy orange solid
(193mg) after freeze drying.
EXAMPLE 36
Preparation of Sodium (phenyl-3-boronic acid)-thiourea terminated
dendrimers.
PAMAM 4.0 (EDA)
Solid (phenyl-3-boronic acid) isothiocyanate (100mg; O.Smmol) was added
to a solution of PAMAM 4.0 (EDA) (69mg; O.Olmmol) in water (Sml). 1M
sodium carbonate was added to the isothiocyanate dissolved (pH ca.l0). The
mixture was then heated at 53°C for two hours under nitrogen, and then
filtered
and the filtrate concentrated to give a brownish solid residue. The crude
product
was purified by gel filtration (Sephadex LH20; water) to give PAMAM 4.0 (EDA)
terminated with 32 (phenyl-3-boronic acid)thiourea groups as a white fluffy
solid
(87mg) after freeze drying.
EXAMPLE 37
Preparation of Pyridinium dodecyl carboxamido-terminated dendrimers.
PAMAM 2.0 dendrimer. BRI-6807
PAMAM generation 2.0 core (0.0479mmo1; SOmg) was evaporated from a
O.SmI solution in MeOH and then re-dissolved in 10 ml of water. 1-N-
pyridinium
12-dodecanoic acid bromide (0.14g; 0.384mmo1), N-hydroxybenzotriazole hydrate
[HOBT] (52mg; 0.384mmol) ; triethylamine (53~10.384mmo1) and 1-(3-
CA 02343205 2001-03-06
WO 00/15239 PCT/AU99/00762
-49-
diethylaminopropyl-3-ethyl) carbodiimide .HCI [EDC] (74mg; 0.384mmol), were
added to the solution. This reaction mixture was stirred overnight at room
temperature. The volume was reduced to a third under reduced pressure and the
solution was chromatographed on a LH20 column using water as the eluent.
5 Fractions containing the product were collected and pyridinium
dodecylcarboxamide PAMAM 2.0 bromide isolated as a fluffy white solid by
freeze drying.
'H nmr (D20) : b 1.15, 1.45, 1.9, 2.15, 2.75, 2.8, 3.15, 3.35, 3.5, 4.55,
8.05,
8.5, 8,8.
10
PAMAM 4.0 dendrimer. BRI-6809.
PAMAM generation 4.0 core (O.OSmmol; 69mg) was evaporated from a
l.Om1 solution in MeOH and then re-dissolved in 15 ml of water. 1-N-
pyridinium
12-dodecanoic acid bromide (O.I43g; 0. 4mmo1), N-hydroxybenzotriazole hydrate
15 [HOBT] (77mg; 0.4mmol) ; triethylamine (56,u10. 4mmo1) and 1-(3-
diethylaminopropyl-3-ethyl carbodiimide .HCI [EDC] (77mg; 0.4mmo1) were
added to the solution.
This reaction mixture was stirred overnight at room temperature. The volume
was
20 reduced to a third under reduced pressure and the solution was
chromatographed on
a LH20 column using 1 % triethylamine in water as the eluent. Fractions
containing
the product were collected and the pyridinium dodecylcarboxamide PAMAM 4.0
bromide was isolated as fluffy white solid by freeze drying. A small amount of
the
product was reacted with acetic anhydride to confirm the complete capping of
the
25 NH2 end groups of the dendrimer core.
'H nmr (Dz0) : b 1.10, 1.45, 1.9, 2.1, 2.30, 2.5, 2.7, 3.2, 4.5, 8.00, 8.45,
8.80.
CA 02343205 2001-03-06
WO 00/15239 PCT/AU99/00762
-50-
EXAMPLE 38
Preparation of saccharin-terminated dendrimers.
PAMAM 4.0 Dendrimer BRI-6157
To a solution of ethylenediamine core PAMAM 4.0 dendrimer core
(275mg; 39.8uM) in dry dimethyl formamide (25m1) was added 6-(benzosulfimido)
isothiocyanate (400mg; 1.67mM) and the mixture stirred at room temperature for
24 h. The cloudy solution was clarified by the adjustment of the pH with
sodium
carbonate solution to pHlO-10.5. This solution was stirred for a further 24 h
and
volatiles removed on a rotary evaporator. The solution was chromatographed on
a
large Sephadex LH20 column and front fraction collected. The remaining
fractions
were collected and re-chromatographed on a smaller column. The combined front
fractions were evaporated and freeze dried to yield the saccharin-terminated
dendrimer product (450mg; 78%) as a fluffy white solid.
'H nmr (D20) : b 2.20, 2.50 3.23, 3.46, 3.63, 7.52, 7.87.
The saccharin-terminated BHA.Lys.Lys2Lys4.Lyse.Lys,6.Lys32... dendrimer
BRI-6189 was similarly prepared.
EXAMPLE 39
Inhibition of Cobra Venom and Bee Venom Toxin.
A Materials and Methods
Cytosensor Microphysiometer Protocol
The Cytosensor Microphysiometer (Molecular Devices Inc., CA) is a light
addressable potentiometric sensor-based device that can be used to indirectly
measure the metabolic rate of cells in vitro (Farce et al. , 1989; McConnell
et al. ,
CA 02343205 2001-03-06
WO 00/15239 PCT/AU99/00762
-Sl -
1992). Metabolism is determined by measuring the rate of acid metabolite
production from cells immobilised inside a microvolume flow chamber.
Human CEM cells were centrifuged and resuspended in low-buffered serum-
free/bicarbonate-free RPMI 1640 medium (Molecular Devices; hereafter referred
to as "modified medium"). The cells were seeded at a density of 60,000-75,000
cells/capsule onto the polycarbonate membrane (3 ~m porosity) of cell capsule
cups
(Molecular Devices). Cells were immobilised using an agarose entrapment medium
(Molecular Devices). The seeded capsule cups were transferred to sensor
chambers containing the silicon sensor which detects changes in pH (and thus
cellular metabolism). The Cytosensor system used for this set of experiments
contained eight separate chambers for the measurement of acidification rates.
Modified media was pumped across the cells at a rate of 100-120 ~,l/min. Each
cell
chamber was served by fluid from either of two reservoirs.
To measure the acidification rate, flow of the modified media was periodically
interrupted, allowing the accumulation of excreted acid metabolites (lactic
acid and
COZ). In this set of experiments, flow was stopped for 30s, during which time,
a
least squares fit slope to the change in voltage signal over time, the
acidification
rate (measured as ~ V/s), was calculated. This rate data was normalised (using
the
4-5 rate points prior to addition of compound) to allow direct comparison of
the
signals from the four chambers. Measurements of the acidification rate were
made
every 2 min. The chamber was held at 37°C.
Basal acidification rates were monitored (in the absence of any treatment) for
at
least 30 min. After this time, the venoms/peptides were exposed to the cells
at a
range of concentrations for periods of up to 4hrs. A concentration of toxin
showing a pronounced effect on the cells, but less than maximal, was selected
for
testing of inhibition of this toxicity by a range of concentrations of
BRI2923. In all
CA 02343205 2001-03-06
WO 00/15239 PCT/AU99I00762
-52-
experiments, at least one chamber was not exposed to any of the compounds,
providing a negative control.
BRI 2923 was dissolved in water and the solution was pH adjusted to 7.2.
S Concentrations ranging from 100 ~.M to I nM were added to the venom/media
solutions and incubated for periods ranging from 6 min (the minimum incubation
period possible using this equipment) to 1 hr and then introduced to the
cells. All
experiments were repeated in triplicate.
B Results
BRI2923 Inhibition of Cobra Venom.
Crude venom from the forest cobra (Naja malenoleuca) was tested. Cobra
venom added to CEM cells caused an initial increase in cellular metabolism
15 followed by cell death (cell lysis). Cobra venom was particularly damaging
to the
cells causing an initial increase in metabolism of approx. 80% followed
rapidly by
100% cell death within the first 10 minutes. The venoms were initially tested
at
10, 50 and 100 ~,g/ml. The submaximal response from 50 ~cg/ml was selected as
the test dose in most experiments. Two concentrations of BRI2923 (10'~M and
10'SM) were used.
At all incubation periods (6, 30 and 60 mins), 10'SM BRI2923 incubated with 50
~g/ml venom, reduced the initial increase in metabolic rate from approx. 80%
to
approx. S % and delayed the onset of cell death by approx. 15-20 minutes.
10'~M BRI2923, incubated with 50 ~.g/ml venom, blocked both the initial
increase
in metabolic rate as well as the subsequent rapid cell death seen at this
concentration of venom alone. 10'~M BRI2923, incubated with 100 ~,g/ml venom,
also abolished the initial increase in metabolic rate and the cells proceeded
to cell
death after the venom/dendrimer solution was washed out. Snake venoms consist
of
CA 02343205 2001-03-06
WO 00/15239 PCT/AU99/00762
-53-
many toxic components each of which have different modes of toxicity. Cobra
venoms contain a cytotoxic peptide which causes cell lysis in a way similar to
bee
venom toxin, melittin. The amino acid sequence of the cytotoxin isolated from
Naja malenoleuca indicates that this toxin is highly basic (cationic) and
would thus
5 be readily inactivated by polyanionic compounds such as BRI2923. This
electrostatic inactivation as a basis for reduced toxicity is supported by the
experimental finding that an incubation period of 6 minutes gives the same
result as
the longer incubation periods of 30-60 rains used.
BR12923 Inhibition of Melittin (major toxin from bee venom).
Melittin was added to the CEM cells in half log doses ranging from 10-SM
to 10''M. The two highest doses (IO'SM and 5 x 10'6M) caused total cell death
within 15 rains with no initial activation of the cells. 10'6M caused a
transient
increase in cell metabolic rate followed by cell lysis, complete after approx.
1 hr.
15 5 x IO''M caused the lysis curve to shift further to the right and 10-'M
was without
effect. This dose response determination was repeated and 10'~M melittin was
selected as the submaximal concentration to be used with BRI2923. A range of
concentrations of BRI2923 were incubated with 10'6M melittin for 20 rains.
10'°M
and 10'SM BRI2923 completely inhibited the toxic effects of the melittin. 10-
6M
20 BRI2923 completely blocked the melittin toxicity for approx. 30 rains and
during
the final 30 rains of exposure the metabolic rate only fell by approx. 10-15 %
less
than the control cells. 10''M, 5 x 10''M and 10'8M concentrations of BRI 2923
reduced, but did not prevent, toxicity. 10-~lVi BRI2923 had no effect on 10'~M
melittin.
CA 02343205 2001-03-06
WO 00/15239 PCT/AU99/00762
-54-
EXAMPLE 40
Inhibition of HIV (AIDS) Toxin
S Introduction
Vpr is an Human Immunodeficiency Virus (HIV-1) accessory gene product, 14-
lcDa, 96-amino-acid protein. The gene vpr is highly conserved in HIV-1, HIV-2
and the Vpr gene product optimises HIV infection and disease progression.
10 (reviewed by Cullen, 1998; Emerman & Malim, 1998). Among its actions, Vpr
induces apoptosis and cytopathic effects in both human cells and yeast
(Stewart et
al, 1997; Zhao et al, 1996; Macreadie et al, 1996).
The Vpr protein has been fractionated and the peptide sequence which causes
apoptosis has been isolated and designated Vpr P3. Vpr P3 has been found to
15 permeabilise CD4+ T lymphocytes (such as CEM cells) and causes their death
by
apoptosis. The following experiments use this toxic Vpr peptide fraction P3
(Arunagiri et al, 1997).
Method using the Cytosensor Microphysiometer
20
Human CEM cells were centrifuged at 1400 RPM for 7 minutes and resuspended in
modified RPMI 1640 medium. Cells were then immobilised using an agarose
entrapment medium and spotted onto the centre of the polycarbonate membrane of
the capsule cup (Molecular Devices Ltd., CA.). The cells were seeded at a
density of
25 approximately 43,000 - 75,000 cells per capsule cup. The seeded capsule
cups were
transferred to sensor chambers containing the silicon sensor and positioned on
to the
microphysiometer. Modified media was pumped across the cells at a rate of
120~1/min. Each cell chamber was served by fluid from either of two
reservoirs,
which could be alternated using a software command. Cells were allowed to
30 equilibrate within the chambers for 30-60 minutes or until a stable
metabolic rate was
CA 02343205 2001-03-06
WO 00/15239 PCT/AU99/00762
-55-
achieved. In this set of experiments, flow was stopped for 30 s, during which
time, a
least squares fit slope to the change in voltage signal over time, the
acidification rate
(measured as ~V/s), was calculated. This rate data was normalised (using the 4-
5 rate
points prior to addition of compound) to allow direct comparison of the
signals from
5 the four chambers. Measurements of the acidification rate were made every 2
min.
The chamber was held at 37°C.
For all experiments CEM cells were used at 1.3 x 105 to 1.3 x 106 cells/ml
which
corresponds to early to mid log phase growth of these cells.
The cells were set up on the microphysiometer and the Vpr P3 peptide was
perfused
through the cell chambers after an equilibration period of at least 30
minutes. The
concentration range used for Vpr P3 was 10 SM - 2 x 10 SM . Vpr P3 needed to
be in
contact with the cells for upwards of 2 hours for the full effect of the
toxicity to be
apparent.
For experiments testing inhibition of Vpr peptide toxicity by BRI2923, media
solutions of Vpr P3 peptides were made to the appropriate concentration and
the
BRI2923 was added, the solution mixed, and left to equilibrate for 20-30
minutes
prior to placement on the cytosensor. With each inhibition experiment, a
positive
control channel containing the same concentration of the Vpr P3 used to test
BRI2923 inhibition was also run for the same time period as well as a time
control
channel. The Vpr P3 peptide solution and the Vpr P3/BRI2923 solution were in
contact with the cells for a minimum period of 2hr 30min to a maximum period
of
4hr. Also, after washout of the compounds, the metabolic rate of the cells was
monitored for a short time period (minimum of 6 min).
CA 02343205 2001-03-06
WO 00/15239 PCT/AU99/00762
-56-
Results
In the cytosensor system, the VPR P3 peptide tested on its own, causes an
initial
increase in metabolic rate followed by a decline and subsequent cell death due
to
apoptosis of the CEM cells.
In the experiment shown in Figure 1, the VPR peptide P3 (at 10 SM) was pre-
incubated with the compound BRI2923 at 10 4M (Final Volume) in a total volume
of
lml (modified RPMI media) for 30 minutes. Solutions were then diluted to final
volumes (25m1) and perfused through the cells. A large initial drop in
metabolic rate
is seen in the BRI2923/Vpr P3 chamber, due to a combination of the pH
difference of
the 10 4M BRI2923 solution (acidic) and the media (neutral) and an intrinsic
buffering effect of the BRI2923 dendrimer itself. To account for this drop in
metabolic rate, a correction factor has been applied to all results using
BRI2923. The
1 S correction factor used was:
a) the calculated initial drop on the first data point added to all subsequent
data points
and
b) in the same way the calculated washout effect was subtracted from all
points after
the wash.
The corrected graph is shown in Figure 2. This result shows that BRI2923, at
10 4M,
gives complete protection against the toxic effects (apoptosis) induced by VPR
P3 at
10 SM. This experiment was repeated in quadruplicate.
Further concentrations of BRI 2923 were tested against higher concentrations
of Vpr
P3, and the results are shown in Figures 3 and 4. BRI 2923 at 10 SM also
demonstrated inhibition of 2x10 SM Vpr P3-induced apoptosis (experiment in
triplicate). BRI2923 10 6M showed a significant reduction in the effects
produced
by 2x10 SM Vpr P3 (triplicate experiment). 10 ~M BRI2923 on 2x10 SM Vpr P3
toxicity, attenuate to a lesser extent the rate of decline in metabolic rate
(duplicate
experiment).
CA 02343205 2001-03-06
WO 00/15239 PCT/AU99/00762
-57-
Unlike the time controls the cells in the chamber containing BRI2923 did not
show
any time related decline in metabolic rate. Previous cytosensor experiments
using
BRI2923 have also suggested that the dendrimer may confer a cytoprotective
effect in
addition to the inhibition of apoptosis caused by VPR P3.
EXAMPLE 41
Calcein release assay to determine inhibition of cholera toxin.
Materials
Cell Line
~ HPC-8 Human ileocecal carcenoma epithelial cells
Calcein, AM
~ Molecular Probes, catalogue # C-1430
~ C46H46N2~23~ molecular weight 994.87
~ Resuspend 1 mg vial in 100 pl reagent-grade, anhydrous dimethyIsulphoxide
(DMSO) to give a stock concentration of 10 mM.
Toxin
~ Cholera Toxin (Sigma Cat. No. C8052) - A subunit surrounded by five B
subunits.
Inhibitor Dendrimer~s
~ Test dendrimers - BRI2913 and BRI2999.
Method
1. HCP-8 (adherent human ileocecal carcinoma epithelial cell line) in log
phase
growth were trypsinised (trypsin EDTA) in 2ml/75cmz flasks after 2 PBS
(phosphate buffered saline) washes.
2. Cells with trypsin solution were incubated for 5 min.
CA 02343205 2001-03-06
WO 00/15239 PCT/AU99/00762
-58-
3. 20m1 culture medium added to flask to inactivate trypsin.
4. Cell solution transferred to SOmI centrifuge tube and spun at 1200 RPM for
7
min at room temperature (RT). ,
5. Cells washed in PBS and spun at 1200 RPM for 7 min at RT.
6. Cells resuspended in PBS and counted then spun at 1200 RPM for 7 min at
RT and supernatant discarded.
7. Cells resuspended in lml PBS with Spl calcein added and incubated at
37°C
for 45 min.
8. Cells washed in PBS (approx. 5-20m1) and resuspended in PBS to give final
numbers of 5x105 cells/100~,1.
9. 1001 cell solution (5x105 cells per well) added in each well used of a 96
well plate with 50.1 of cholera toxin (dissolved in H20) and 50,1 of
dendrimer solutions (stock solution made to IOmM in DMSO and diluted in
PBS). Each concentration repeated in triplicate.
10. Plate incubated for 2hr at 37 °C in 5 % COZ incubator.
11. Supernatant harvested and fluorometric analysis performed.
Results
Preliminary experiment performed. All results presented are the average
fluorescence intensity of three replicates.
Fluoresence % inhibition
Intensity of cholera
(FI) toxin.
0.3mg/ml cholera 72.1 0%
toxin
100~cM BRI2923 66 g,s%
100~cM BRI2999 59.5 17.5
O.lSmg/ml cholera 65.8 0%
toxin
100~cM BRI2923 61 7. 3
100~cM BRI2999 59.4 9.7
Spontaneous calcein26.8
Release (SR)
inhibition = 100 - (FI toxin + dendrimer/FI toxin x 100)
CA 02343205 2001-03-06
WO 00/15239 PCT/AU99/00762
-59-
References
Arunagiri, C.K., Macreadie, LG., Hewish, D.R. and Azad, A.A. (1997) A C-
terminal
domain of HIV-1 accessory protein Vpr is involved in penetration,
mitochondria) dysfunction
and apoptosis of human CD4 lymphocytes. Apoptosis, 2, 69-76
Cullen, B.R. (1998) HIV-1 auxiliary proteins: making connections in dying
cells. Cell, 93,
685-692.
Emerman, M., and Malim, M.H. (1998) HIV-1 regulatory/accessory genes: keys to
unravelling
viral and host cell biology. Science, 280, 1880-1883.
McConnell, H.K., Owicki, J.C., Parce, J.W., Miller, D.L., Baxter, G.T., Wada,
H.G.
and Pitchford, S. (1992). The cytosensor microphysiometer:biological
applications of
silicon technology. Science, 257, 1901-1912.
Macreadie, LG., Arunagiri, C.K., Hewish, D.R., White, J.F., and Azad, A.A.
(1996)
Extracellular addition of a domain of HIV-1 Vpr containing the aminoacid
sequence motif
H(S/F)RIG causes cell membrane permeabilization and death. Mol. Microbiol.,
19(6),
1185-1192.
Parce, J.W., Owicki, J.C., Kercso, K.M., Sigal, G.B., Wada, H.G., Muir, V.C.,
Bousse, L.J., Ross, K.L., Sikic, B.I. and McConnell, H.M. (1989). Detection of
cell-
affecting agents with a silicon biosensor. Science, 246, 243-247.
Stewart, S.A., Poon, B., Jowett, J.B.M. and Chen, LS. (1997) Human
immunodeficiency virus
type 1 Vpr induces apoptosis following cell cycle arrest. J. Yirol, 71, 5579-
5592.
Zhao, Y., Cao, J., O'Gorman, M.R.G., Yu, M., and Yogev, R. (1996) Effect of
human
immunodeficiency virus type 1 protein R (Vpr) gene expression on basic
cellular function
of fission yeast Schiaoasaccharomyces pombe. J. Virol, 70, 5821-5826.