Note: Descriptions are shown in the official language in which they were submitted.
CA 02343521 2001-03-05
1
METHOD FOR PRODUCING Y,S-UrfSATURATED
KETONES BY CARROLL-REACTION
The invention relates to an improved process for preparing
y,S-unsaturated ketones, in particular ineth,ylheptenone,
geranylacetone and farnesylacetone or their dihydro derivatives
or geranylgeranylacetone by a Carroll reaction in the presence of
aluminum catalysts.
A Carroll reaction is taken to mean the chain elongation of an
allyl or propargyl alcohol with acetoacetic.esters or diketene,
with formation of y,8-unsaturated ketones. It~can proceed, ,for
example, according to the following reaction scheme:
aceto- ~ tauto~-
OH
acetylation 0~~ meriz~ation
~O~ ~~O IOH CIO
/ /
Claisen ~ O\~ decarbox~~
rearrangement j!~ ~'I' lation
OH O O
In the key step, therefore, the new acetoac:etic ester of the
unsaturated alcohol, which ester is primara.ly formed from the
allyl or propargyl alcohol and the acetoacetic ester or diketene,
rearranges to form, in a Claisen rearrangement, the 13-keto acid,
which then spontaneously decarboxylates. Initial studies on
Carroll reactions are described in J. Am. C:hem. Soc. 65 (1943)
1992 - 1998.
Since the beginning of the 1950s, this reaction has been employed
in various ways in terpene preparation. For example, the terpenes
2-methyl-2-hepten-6-one, 6,10-dimethyl-5,9-~undecadien-2-one
(geranylacetone) and 6,10,14-trimethyl-5,9,13-penta-
decatrien-2-one (farnesylacetone), which are required as
essential precursors of vitamin A and vitamin E, are prepared on
an industrial scale~by the Carroll reaction.
Thus, for example, GB 695 3i3 discloses Carroll reactions in the
gas phase at from 300 to 600~C with the use of allyl or crotyl
acetoacetate.
US 2,628,250 discloses the preparation of 2-methyl-2-hepten-6-one
from 2-methyl-3-buten-2-of and diketene.
0050/49363
CA 02343521 2001-03-05
2
US 2,660,608 discloses the preparation of
tetrahydrofarnesylacetone from tetrahydronerolidol and diketene.
As catalysts for Carroll reactions, use has been made according
to the process of US 2 795 617 of aluminum alkoxides, in
particular the aluminum isopropoxide of the formula
A1(0-CH(CH3)2)3 in amounts of from 0.8 to 2.5 mold, based on the
alcohol used as starting material.
According to the process of GB 886 353, for Carroll reactions,
use has been made of aluminum complexes containing acetylacetone
or acetoacetic esters, such as aluminum tri(acetylacetonate),
aluminum tri(methylacetoacetate) or aluminum
tri(ethylacetoacetate} as catalysts.
I5
In the case of the previously known Carroll reactions of an
acetoacetic ester with a tertiary vinylcarbinol or propargyl
alcohol, generally, as alkyl acetoacetate, use was made of the
methyl or ethyl esters. The low-boiling primary alcohol (methanol
or ethanol) released in the acetoacetylation of the unsaturated
alcohol was distilled off during the reaction together with the
carbon dioxide formed in the decarboxylation.
The yields achieved in the Carroll reaction using methyl or ethyl
acetoacetate are not yet completely satisfactory for use on an
industrial scale. This is due firstly to the relatively long
reaction times necessary and, secondly, to the fact that, under
the reaction conditions, the unsaturated ketones formed are
hydrogenated to a slight extent to alcohols which are difficult
to remove, which impairs the yields.
It is an object of the present invention, therefore, to shorten
the reaction times for preparing y,8-uns~~turated ketones by
reacting allyl or propargyl alcohols with acetoacetic esters in
an aluminum-catalyzed Carroll reaction a:nd thus to improve the
possibility of a continuous process procedure. It is also an
object of the present invention to further improve the yields.
we have found that this object is achieved by a process described
at the outset. It has now surprisingly been found that when use
is made of acetoacetic esters of tertiary alcohols, such as
tert-butanol, tert-pentyl alcohol (2 met:hylbutan-2-ol) or
dimethylpropylcarbinol (2-methylpentan-2~-ol), instead of methyl.
acetoacetate (AME) or ethyl acetoacetate, the Carroll reactions
proceed to y,~-unsaturated ketones more rapidly and with higher
yields, that is to say forming fewer byproducts. These
improvements are particularly important .if, as starting compound,
0050/49363
CA 02343521 2001-03-05
3
use is made of a higher, and thus more v<~luable, unsaturated
alcohol. This applies especially to the use of
3,7-dimethyl-1,6-octadien-3-of (linalool;),
3,7,11-trimethyl-1,6,10-dodecatrien-3-of (nerolidol),
3,7,10-trimethyl-1,6-dodecadien-3-of (dihydrolinalool) and
particularly to E,E-9,7,11,15-tetramethy:l-1,6,10,14-hexa-
tetraen-3-of (E,E-geranyllinalool).
Although EP 376 859 B1 has already disclosed that, in the
acetoacetylation of nucleophiles, such as alkanols, alkylamines
or alkylthiols, with acetoacetic esters or their derivatives,
good yields can also be achieved if, as acetoacetic esters, use
is made of esters of tertiary alcohols, such as tert-butanol or
tert-amyl alcohol, this does not relate t:o Carroll reactions, but
to the functionalization by acetoacetylat:ion of
low-molecular-weight or polymeric nucleophiles which are
ultimately used as coatings to improve dyeing technology.
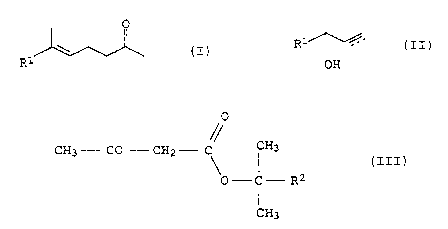
The invention relates to a process for preparing y,b-unsaturated
ketones of the formula I
O
(I)
R
by reacting an allyl alcohol or a proparc_~yl alcohol of the
formula II
R1~_ (II)
OH
where R1 is H or a saturated or unsaturated branched,
unsubstituted or methoxy-substituted hydrocarbon radical having
from 1 to 33 carbons and the dotted line can be a further bond
between the carbons bearing it,
with an alkyl acetoacetate at temperatures of from 150 to 220~C in
an unmodified or modified Carroll reaction in the presence of an
aluminum catalyst and with the alkanol which forms being
distilled off, which comprises making use: of, as alkyl
acetoacetate, an acetoacetic ester of the: formula III
CA 02343521 2001-03-05
0050/49363
4
O
CH3 - CO - CH2 - C ~ H3
(III) r
p-C- R2
CH3
where R2 is an alkyl having from 1 to 4 carbons.
Surprisingly, using the process according to the invention, the
higher y,8-unsaturated ketones can be obtained in a simple manner
and in a continuous process procedure, which is particularly
desirable for processes on an industrial scale, in yields of from
92 to 96% of theory, even if no excess, or only a slight excess,
of one of the reaction components is used. In addition, it is a
great advantage that the process according to the invention can
increase the space-time yields of the presviously known processes.
The process according to the invention is of particular
importance for reacting alcohols of the i'ormula (II), where R1 is
a group of the formula IV
CH;-f-C-CH-CH=-CH_-j- (lU),
X Y
where n is an integer from 1 to 5 and x and y are either both H
or x is methoxy and y is H, or x and y together are an additional
bond between the carbons bearing x and y,, such as
3,7-dimethyl-1,6-octadien-3-of (linalool;i,
3,7-dimethyl-1-octen-3-ol,
3,7,11-trimethyl-1,6,10-dodecatrien-3-of (nerolidol),
3,7,11-trimethyl-1-dodecen-3-ol,
3,7,1I-trimethyl-1,6-dodecadien-3-of (dihydronerolidol) and
3,7,11,I5-tetramethyl-1,6,10,14-(E,E)hexadecatetraen-3-of
(E,E-geranyllinalool).
The use of tert-butyl or tert-amylacetoac:etate is accompanied by
the advantage of a more rapid reaction and the avoidance of
byproducts. The amounts of the reactants used are advantageously
selected in such a manner as to give a molar ratio of alcohol of
the formula II to alkyl acetoacetate of the formula III of from
0.8 to 1.2, preferably from 0.95 to 1.10"
0050/49363
CA 02343521 2001-03-05
Suitable organic aluminum compounds for the process according to
the invention are essentially compounds ~of the formula V
/R4 /R4
AI _~
CH-CO-R5 ~ CH-CO-F~ ~ R-' 3-m-n
m n
where R4 is branched or unbranched alkyl or alkoxy having from 1
to 4 carbons, preferably methyl or ethyl, RS and R6 are branched
or unbranched alkyl or alkoxy having from 1 to 5 carbons,
preferably methyl or 2-butyl, R~ is a branched or unbranched alkyl
having from 1 to 4 carbons and m and n can be integers from 0 to
3, where n+m ~ 3, and aluminum triarylo~s:ylates. Particular
preference is given to liquid aluminum compounds, in particular
aluminum compounds in which R5 is a methyl, R6 is a butyl and the
sum of n+m = 3 and the ratio n/m > 0.3.
The first-mentioned catalysts are therefore lower aluminum
trialkoxides, such as aluminum trimethoxide, aluminum
triethoxide, aluminum triisopropoxide, aluminum tri-sec-butoxide,
and compounds which are formed in the reaction of said aluminum
trialkoxides with stoichiometric amounts of acetylacetonate,
alkyl acetoacetate or alkyl malonate with elimination of alcohol
and transesterification. Examples are aluminum triacetoacetate,
aluminum triacetylacetonate, aluminum monoacetoacetate
diethoxide, aluminum monoacetoacetate diisopropoxide, aluminum
diacetoacetate monoisopropoxide.
Preferably, use is made of the aluminum t:rialkoxides, in
particular aluminum triisopropoxide and aluminum
tri-sec-butoxide. Very particularly preferably, use is made of
mixed aluminum triacetoacetates which are: produced by reacting
aluminum sec-butoxide or aluminum triisopropoxide with methyl
acetoacetate with elimination of 2-butane>1 or isopropanol and
+ v crif.~..v .~...~. .~.f t ~ cthnxv Qr
n " ~ c,. i th the -hpta_npl_
,.rans~st~y at r. h.. m.. . 1 ~re..p : r_,... 2
isopropanol released, where the degree of transesterification is
to be above 30%.
For the purposes of the invention, aluminum triaryloxylates are
the aluminum salts of aromatic hydroxy compounds, such as
aluminum triphenolate, aluminum tricresol.ates, aluminum
trixylenolates, aluminum trinaphtholates, whose aryl radicals can
also be substituted by lower alkyl or alk.yloxy groups, i.e. alkyl
or alkyloxy groups having from 1 to 4 carbons, hydroxyl groups or
0050/49363 ~ 02343521 2001-03-05
6
phenyl. Particularly advantageously, of these, use is made of the
relatively readily accessible aluminum triphenolate.
It is advantageous to use liquid catalysts or solutions of solid
catalysts and to feed these into the reaction vessel in liquid
form.~Thus, for example, use can be made of aluminum trialkoxides
dissolved in alkyl acetoacetate or in a :mixture of alkyl
acetoacetate and an alcohol of the formula II.
The amount of the aluminum compound is generally such that its
concentration in the reaction mixture does not fall below 0.050
by weight of Al and, at the start of the reaction, does not
exceed 6% by weight of A1. Based on alkyl acetoacetate to be
reacted, generally from 0.5 to 5 mold of the aluminum compound
are required. For the aluminum triisopro:poxide preferably used
and the above-described mixed aluminum triacetoacetate prepared
from aluminum sec-butoxide and methyl acetoacetate, amounts of
from about 1 to 3 mold, based on the alkyl acetoacetate to be
reacted, are used, for example.
When use is made of allyl alcohols of the formula II having a
boiling point below 120~C, such as 2-methyl-3-buten-2-ol, it is
particularly advantageous if the Carroll reaction is carried out
in a cyclic carbonate of the formula VI ~or a y-lactone of the
formula VII
R1 \ Rz \ / R1
/"C O, C H C
R \2
C=O C=O
R3\ ~ R3 \
R4 /C O R4 /C O
~vz? ~vII~.
where the radicals R1, R2 and R3 are H, methyl or ethyl,
preferably H or methyl, and R4 is H, methyl, ethyl, isopropyl,
phenyl or methoxymethyl, preferably H or methyl, as solvent.
Suitable cyclic 5-member-ring carbonates of the formula VI are,
in addition to the customary alkylene carbonates, such as
ethylene carbonate, 1,2-propylene carbonate, isabutylene
carbonate and 1,2-butylene carbonate, i.~e. carbonates of the
formula VI where R1 to R4 are H or methyl., or R1 to R3 are H or
methyl and R4 is ethyl, also those where R1 to R3 can additionally
be ethyl and R4 is H, methyl, ethyl, isopropyl, phenyl or
methoxymethyl.
CA 02343521 2001-03-05
0050/49363
The cyclic carbonates used can also be prepared extremely
inexpensively industrially by reacting t:he corresponding alkylene
oxides with COZ. They generally have boi:Ling points which are so
high that, at atmospheric pressure, temperatures of 170~C can be
reached without problems.
Particularly suitable 5-member-ring lactones of the formula VII
are y-butyrolactone and 3-methyl-y-butyrolactone, in particular
y-butyrolactone.
The y-butyrolactones used according to the invention of the
formula VII can also be prepared with ad~Tantage industrially by
dehydrogenating the corresponding butanediols.
The alkanol formed in the reaction attaclts the cyclic carbonates
or lactones, under the reaction conditions, surprisingly so
little that, for example, when use is made of propylene
carbonate, the solvent can be reused for up to 10 reaction cycles
without any clean-up (cf. comparative example lb). The cyclic
carbonate or lactone which is separated off after isolation of
the y,8-unsaturated ketone can be fed into new reaction cycles
without supplementing the catalyst. Residues of unreacted
acetoacetate remain in the solvent and are not lost.
The 5-member-ring carbonates and 5-member-ring lactones are
generally used in amounts of from 50 to :10000 by weight,
preferably from 100 to 500% by weight, based on y,8-unsaturated
ketone formed.
When use is made of alcohols of the formula II which have a
boiling point above 140~C, the Carroll reaction can also
advantageously be carried out without adding significant amounts
of a solvent. This is accompanied by advantages in the workup of
the reaction mixture.
The process according to the invention can be carried out
batchwise and continuously. When it is carried out continuously,
advantageously, the starting compounds and the catalyst are
pumped into a reaction vessel which is provided with a heating
bath and has an attached condensation apparatus for the alcohol
which is eliminated and for discharging t:he carbon dioxide formed
and the reaction product is obtained using an overflow.
Using the process according to the invention, the sought-after
y,8-unsaturated ketones of the formula I can be obtained in a
simple manner in surprisingly high yielda. The tertiary alcohols
0050/49363 ~ 02343521 2001-03-05
eliminated from the acetoacetic ester ca;n be recovered virtually
completely.
Examples
Example la
Carroll reaction of 2-methyl-3-buten-2-o:1 with tert-butyl
acetoacetate in propylene carbonate at 180°C
A mixture of 25.7 g of a 92~ pure 2-methyl-3-buten-2-of and
39.8 g of tert-butyl acetoacetate were [aic] added dropwise in
the course of 2 hours at 170°C to a mixture of 50 ml (45 g) of
1,2-propylene carbonate and 2.8 g of a separately prepared (in
accordance with GB 886 353) aluminum trimethylacetoacetate
catalyst. During the dropwise addition, -there was vigorous
evolution of C02 and low-boilers which were continuously distilled
off (b. p. - 80-85°C). After completion of the dropwise addition,
the mixture was stirred for a further 10 minutes until the
completion of gas evolution and was then cooled. Then, at
approximately 100 mbar, first runnings o:f 2.5 g were distilled
and 30.9 g of a main fraction consisting of 96-98~ pure
2-methyl-2-hepten-6-one. The yield (together with the amount
remaining in the distillation bottom phase) was 92% of theory.
Example lb (Comparative example)
Reaction of 2-methyl-3-buten-2-of with methyl acetoacetate in
1,2-propylene carbonate
a) A mixture of 29.03 g (0.25 mol) of methyl acetoacetate (AME;
purity 98%) and 23.68 g (0.275 mol) of 2-methyl-3-buten-2-of
(MBE; purity 94%) was pumped at 180°C, with stirring, in the
course of 2 hours into a mixture of 45 g of 1,2-propylene
carbonate and 2.8 g of a separately prepared (according to GB
886 353) aluminum trimethylacetoacetate catalyst. During this
time, C02 escaped and 8 g of low-boilers were distilled off,
which low-boilers consisted of about 2/3 of methanol and
about 1/3 of unreacted 2-methyl-3-buten-2-ol. The mixture was
then stirred for a further 30 minutes at 180°C, and then
cooled and, at a reduced pressure of 100 mbar, the desired
2-methyl-2-hepten-6-one was distilled off from the reaction
mixture.
b) The distillation residue produced in this case was again
admixed with the abovementioned amounts of AME and MBE at
180°C in the course of 2 h, the reaction mixture was stirred
0050/49363
CA 02343521 2001-03-05
9
for 30 min at 180~C, then cooled and the resultant
2-methyl-2-hepten-6-one was distilled off therefrom.
c) Procedure b) was repeated a further 8 times. The mean yield
of 2-methyl-2-hepten-6-one over all i0 batches was 88~ of
theory, based on MBE reacted (gas-chromatographic
determination using an internal standard).
Example 2a
Carroll reaction of 3,7-dimethyl-1,6-octadien-3-of (linalovl)
with tert-butyl acetoacetate to form
6,IO-dimethyl-5,9-undecadien-2-one
5.6 g of aluminum trimethylacetoacetate (prepared from aluminum
triisopropoxide similarly to GB 886353) were charged, heated to
180~C and at this temperature in the course of 2 h a homogeneous
mixture of 115.7 g of linalool and 128 g of tert-butyl
acetoacetate was pumped into the reaction vessel. In the course
of this there was spontaneous formation of C02 and tert-butanol,
which was condensed. 51 g of tert-butanol. were isolated.
After completion of the feed, the mixture was stirred for a
further 20 min at an internal temperatures of 180-190~C and was
then cooled. The reaction discharge was distilled at a pressure
reduced to 0.1 mbar. This produced, in two fractions, a total of
139.7 g of 6,10-dimethyl-5,9-undecadien-2-one (geranylacetone),
which is equivalent to a yield of 96g of theory.
Example 2b (Comparative example)
Carroll reaction of linalool with methyl acetoacetate
As in Example 2a), 5.6 g of the aluminum catalyst described there
were charged and a mixture of 115.4 g of linalool and 94 g of
methyl acetoacetate was pumped in at an internal temperature of
180~C. In the course of this there was spontaneous formation of
C02 and methanol, which was condensed. The reaction solution was,
as in Example 2a), stirred for a further 20 min until cessation
of the COZ evolution and was then cooled. Distillation was
performed under reduced pressure, producing 133 g of
geranylacetone. The yield was 91.5% of theory. As byproducts,
1.6 g of 6,10-dimethyl-5,9-undecadien-2-of were formed, which
were not detectable in Example 2a). .
0050/49363 ~ 02343521 2001-03-05
Example 3a
Carroll reaction of 3,7,11,15-tetramethy:l-
i,6,10,14(E,E)hexadecatetraen-3-of (E,E-geranyllinalool) with
5 tert-butyl acetoacetate to form 6,10,14,:L8-tetramethyl-5,9,13,17-
nonadecatetraen-2-one (geranylgeranylacei=one)
2.5 g of aluminum tri-tert-butylacetoacei~ate were dissolved at
40~C in 42.1 g of tert-butyl acetoacetate: and 72.5 g of
10 E,E-geranyllinalool were added to this ai= 20°C. This resulting
mixture was pumped uniformly into a horizontal heated reaction
vessel equipped with an overflow and attached distillation
bridge. The feed rate was set so that the' mean residence time was
10 min. The internal temperature was 190-200°C. During the
reaction, 15 g of tert-butanol distilled off.
After completion of the feed, the reaction discharge was
distilled. This produced 75.9 g of geranylgeranylacetone of a
purity of 98.8x. This is equivalent to a yield of 92°s of theory.
Example 3b (Comparative example)
Carroll reaction of E,E-geranyllinalool with methyl acetoacetate
to form geranylgeranylacetone
2.2 g of aluminum trimethylacetoacetate were dissolved at 60°C in
37.6 g of methyl acetoacetate and the so:Lution was mixed with
87 g of E,E-geranyllinalool. This solution was pumped uniformly
into the horizontal reactor vessel described in Example 3a). The
pumping rate was matched to the available reaction volume to give
a mean residence time of 10 ~ 0.5 min. At an internal temperature
of 190-200°C, the mixture reacted continously to form
geranylgeranylacetone. After completion of the feed, the contents
of the reactor were heated for a further 10 min and then passed
to the cooled reaction discharge. Distil:Lation was performed at
0.3 mbar, producing in the main fraction 84.2 g of
geranylgeranylacetone of a purity of 97.:10. This is equivalent to
a yield of 85% of theory.
Example 4a
Carroll reaction of nerolidol with 2-met:nylbut-2-yl acetoacetate
5.6 g of aluminum trimethylacetoacetate were charged into the
same reaction vessel as described in Example 4a and heated to
190°C. A mixture of 166 g of nerolidol and 139 g of an isopentyl
acetoacetate (purity > 980) prepared in .a manner known per se
0050/49363
CA 02343521 2001-03-05
11
from isopentanol and diketene was added <iropwise uniformly in the
course of 2 h. The reaction temperature was kept by heating at
190-200~C in the course of this. After completion of the feed, the
mixture was stirred for a further 15 min and then cooled. This
produced 212 g of crude material which was distilled at 0.1 mbar.
Two fractions gave 178.7 g of farnesylaceatone. This is equivalent
to a yield of 910 of theory.
Example 4b (Comparative example)
Reaction of nerolidol with methyl acetoac:etate:
5.6 g of aluminum trimethylacetoacetate were charged into a
500 ml reaction vessel equipped with a metering device, paddle
agitator and a 10 cm column having attached reflux condenser and
distillation bridge. The contents were hs~ated to 180-190~C. In the
course of 2 h, a homogeneous mixture of 7166.5 g (0.75 mol) of
nerolidol and 94 g (0.81 mol) of methyl acetoacetate was pumped
in uniformly. The reaction temperature was kept to 180-190~C by
external heating. The methanol formed was condensed. After
completion of the feed, the mixture was stirred for a further
15 min and then cooled. This produced 20F3 g of crude material
which was distilled under greatly reduced pressure. In two
fractions, together, 167 g of farnesylace~tone were isolated. This
is equivalent to a yield of 85% of theory.
Example 5
Carroll reaction of neralidol with 2-methyl-2-pentyl acetoacetate
(isohexyl acetoacetate):
2.8 g of aluminum trimethylacetoacetate were charged into the
reaction vessel described in Example 4a. At a reaction
temperature of 190-200~C, in the course of 1 h, a mixture of 83 g
of nerolidol and 75 g of an isohexyl acet:oacetate (purity > 984)
prepared from isohexanol and diketene were pumped in. After
completion of the feed, the reaction mixture was stirred for a
further 10 min at 200~C and cooled after gas evolution was
complete. This produced 108 g of crude material which were
distilled under greatly reduced pressure.. 88 g of pure
farnesylacetone were isolated in this manner. This is equivalent
to a yield of 90% of theory.
0050/49363
CA 02343521 2001-03-05
12
Example 6
a) Inventive reaction of E,E-geranyllinalool with tert-butyl
acetoacetate
263 ml of a reaction solution consisting of 145 g (0.5 mol)
of E,E-geranyllinalool, 5 g of aluminum tri-tert-butyl
acetoacetate and 83 g (0.525 mol) of tert-butyl acetoacetate
were pumped at a rate of 5.6 ml/min into a magnetically
stirred reaction vessel consisting of a 100 ml three-necked
flask heated to 190 to 200°C by a heating mantle with an
overflow in the center (corresponding to a usable volume of
the flask of 45 ml).
This produced 185 g of crude product and 30.5 g of low
boilers. The crude product comprised 13% unreacted
E,E-geranyllinalool and 79% of the desired
geranylgeranylacetone.
b) Reaction of E,E,-geranyllinalool [sick with methyl
acetoacetate (Comparative example)
234 ml of a reaction solution consisting of 145 g (0.5 mol)
of E,E-geranyllinalool, 5 g of aluminum tri-tert-butyl
acetoacetate and 61 g (0.525 mol) of methyl acetoacetate were
pumped at a rate of 5.0 ml/min into the abovementioned
reaction vessel which was heated to 190 to 200°C.
This produced 199.9 g of crude product and 5.7 g of low
boilers. The crude product comprised 53% unreacted
E,E-geranyllinalool and only 34% of the desired
geranylgeranylacetone.
Comparison of Example 6a with Comparative Example 6b clearly
shows that the reaction with the tert-butyl acetoacetate proceeds
considerably more rapidly (3 to 10 times more rapidly) than the
methyl acetoacetate which has previously been customary for
Carroll reactions.
45