Note: Descriptions are shown in the official language in which they were submitted.
CA 02347280 2001-05-31
i
_ 1
1
NOVEL PROCESS FOR THE PREPARATION OF NUCLEOTIDES
1. Field of the Invention
The present invention relates to a novel process
for the preparation of hydroxyphosphonylmethoxypropyl
nucleosides, and novel intermediates produced therein.
2. Background Art
Nucleoside analogs possessing a 3-hydroxy-2-
(phosphonylmethoxy)propyl (HPMP) side chain have been
reported as potent antiviral compounds having a broad
spectrum of activity. Examples of compounds belonging
to this class include HPMP-adenine (HPMPA), HPMP-
guanine (HPMPG) and HPMP-cytosine (HPMPC). HPMP-
substituted nuc;le~sides contain a chiral center and it
has been postulated.that the biological activity may
reside in one enantiomer and not the other. It is
therefore desirable to develop a synthetic method
which will preferentially yield the active enantiomer
using readily available and inexpensive starting
materials.
Bronson et al (J. Med. Chem., 1989, 32:1457)
reported the synthesis of (S)-HPMPC which involves the
coupling of cytosin~a with 3-O-benzyl-2-O-[(diethyl-
phosphonyl) methyl] -:3-O- (methylsulfonyl) glycerol,
followed by subsequent deprotection to afford the
product. The glycerol starting material is derived
from chiral (R)-glycerol acetonide.
Holy et al (Co:ll. Czech. Chem. Comm., 1989,
54:2470) reported the synthesis of (S)-HPMPC by
reacting (R)-glycerol acetonide tosylate with
4-methoxy-2-pyrimid:inone, the resultant product is
CA 02347280 2001-05-31
2
then converted to 1.-[(2,3-dihydroxy)propyl]cytosine.
The latter compound. is reacted with
chloromethylphosphanyl dichloride, and the product is
converted to (S)-HPMPC by base catalyzed
rearrangement.
Glycerol acetonide was also used in the synthesis
of (S)-HPMPA (Webb, Nucleosides and Nucleotides, 1989,
8:619) and HPMPG ('ferry et al, Antiviral Res., 1988,
10:235). These procedures all require the use of the
expensive chiral glycerol acetonide as starting
material, and involve multi-step process requiring
chromatographic purifications of intermediate
compounds.
The reaction of glycidol with adenine, cytosine
or uracil to form the 2,3-dihyroxypropyl substltute3
nucleosides was reported by Ueda et al, J.
Heterocyclic Chem., 1971, 8:827. The reaction of
(~)-glycidol with thymine or 5-fluorouracil was
reported by Seiter et al, Bull. Chem. Soc. Jpn., 1973,
46:1572. The prior art does not disclose or suggest
the process of the present invention for the
preparation of HPMP-nucleotides which offers marked
improvement over previously known methods.
SUMDZARY OF THE INVENTION
The present invention provides a novel and
improved process for the preparation of
hydroxyphosphonomethoxypropyl (HPMP) nucleoside
antiviral compounds. The process of the instant
invention comprises the steps of reacting an
optionally substitutedvpurine or pyrimidine base with
an optionally substituted glycidol; if glycidol is
used in the previous step, protecting the primary
CA 02347280 2001-05-31
3
hydroxy group of th.e intermediate thus formed;
reacting this product with a methanephosphonate
derivative; and removing the various protecting groups
to afford the final product.
The instant process starts with readily available
purine and pyrimidine bases, and glycidol. The
process offers advantages in economies of both
material and labor costs by virtue of eliminating the
need for isomer separations and subsequent
chromatographic purifications; and unlike prior art
processes, the instant process is suitable for large
scale synthesis of the final products. Furthermore,
the process is stereospecific and, starting with a
chiral glycidol, produces the products without
racemization.
DETAILED DESCRIPTION OF THE INVENTION
The present process for the preparation of HPMP-
type nucleoside antiviral compounds is shown in
Scheme I.
CA 02347280 2001-05-31
4
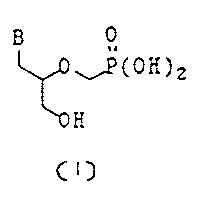
Scheme I
eI~
J] H
~0
~H
H
t;~~a) '
( I I la]
bese
B --~ B ~- protect i on
0
~R
B
(iib) H 1) Base H' 0
0 ~P(ORZ)2
~R~ z) L-CHZP(ORZ)Z ~ 1
'O R
(Illb] (IV]
(V]
Deprotection
B 0
~vP(OH)2
H
<I] ~ _. .
In Scheme I, B is a purine or a pyrimidine base;
B' is a purine or pyrimidine base or a protected
purine or pyrimidine base; L is a conventional leaving
group; R' is a hydroxy protecting group; and RZ is an
alkyl group having 1-5 carbon atoms.
"Purine or pyrimidine base" includes, but is not
limited to, adenine, guanine, thymine, uracil,
cytosine, xanthine, hypoxanthine, 8-bromoguanine,
8-chloroguanine, 8--aminoguanine, '8-hydrazinoguanine,
8-hydroxyguanine, 8-methyiguanine, 8-thioguanine,
2-aminopurine, 2,6--diaminopurine, 5-ethylcytosine,
5-methyicytosine, 5-bromouracil, 5-iodouracil,
5-ethyluracil, 5-propyluracil, 5-vinyluracil, and
5-bromovinyluracil. .
CA 02347280 2001-05-31 '
"Protected purine or pyrimidine" refers to a
purine or pyrimidine base in which functional groups
that may interfere with the desired reaction have been
blocked by a group stable under basic uc~nditions. For
5 example, the 4-amino group of cytosine may be blocked
by the benzoyl group.
"Leaving group" includes, but is not limited to,
halides such as chloride, bromide, and iodide;
mesylate; and tosyl.ate. "Alkyl" includes both
straight and branched carbon chains. "Hydroxy
protecting group" includes, for example, trityl,
allyl, and benzyl groups.
In Scheme I, t:he first step involves the
preparation of a compound of formula (IIIb). A purine
or a pyrimidine bay>e B' is first treated with a base
in order to generate the corresponding anion. The
base is not particularly restricted and may be
selected from metal. hydrides such as sodium and
potassium hydrides, metal carbonates such as sodium
and potassium carbonates, and metal alkoxides such as
potassium t-butoxide; preferably the base is used in a
catalytic amount.
Where the puri.ne or pyrimidine base contains 1 or
more functional groups that may be reactive to form .
undesired products under the reaction conditions of
the present process, for example, the 4-amino group of
cytosine and adenine and the 2-amino and 4-oxo groups
of guanine, such functional groups may be blocked
using the protecting group commonly employed in
nucleoside chemistry. For example, the 4-amino group
of adenine and cytosine may be protected by benzoyl;
the 4-oxo and 2-amino groups of guanine may be
protected by the triphenylmethyl group. The selection
CA 02347280 2001-05-31
6
of methods for introducing and subsequent removal of
such protecting groups are well known to one of
ordinary skill in 'the pertinent art.
The anion B'' generated in situ is reacted with
glycidol (IIa) to generate the 2,3-dihydroxy
nucleoside of formula (IIIa). The primary alcohol of
the compound of formula (IIIa) is blocked prior to the
addition of the phosphonate group. For the present
process, however, it is preferred that the glycidol
reactant is one in which the primary alcohol is
protected, i.e. a compound of formula (IIb). The
reaction of a prot~=_cted glycidol with B' consistently
gives the corresponding product of formula (IIIb) in
higher yields than reactions in which unprotected
glycidol is used. The hydroxy protecting group may
be; fox: example, t:riphenylmethyl-type where the phenyl
groups are unsubst:ituted or 1 or more of the phenyl
groups are substituted, for example with methoxy; or
allyl, benzyl, and the like. Preferably, the hydroxy
protecting group i:a one selected from the group of
triphenylmethyl type compounds.
The reaction :LS carried out in an inert dipolar
aprotic organic so:Lvent such as dimethylformamide,
N-methyl-2-pyrrolidinone (NMPO), dimethyl sulfoxide,
and hexamethyl pho:aphoramide at a temperature that .
favors the formation of the desired products;
generally, the reacaion temperature is elevated and
may be from about X50°C to about 150°C. Preferably,
the reaction is carried out at about 100°C to about
120°C. 'The starting materials B' and the glycidol are
used in molar equi~ralent or one or the other reactant
may be used in a s:Light excess, e.g., up to about 2
equivalents relati~re to the other. Preferably, B' is
. , :~cw~u.~.,~;'
CA 02347280 2001-05-31
7
employed in excess in an amount up to about 1.3
equivalent of the glycidol.
The second step of the present process involves
the introduction of the methanephosphonate moiety to
the secondary hydroxy group of a compound of formula
(IIIb). Prior to carrying out this step, if B'
contains an unprotected functional group, this may be
optionally protected. For example, the 4-amino group
of cytosine may be converted to the corresponding
dimethylformamidino derivative upon treatment with
N,N-dimethylformamide or an acetal thereof.
Thus, a compound of formula (IIIb) is first
treated with a base to generate the corresponding
alkoxide anion. The base may be a metal hydride, for
example sodium hydride, p~~tassium hydric~~ or lithium
hydride; and metal a~lkoxides, for example, potassium
t-butoxide or sodium,~methoxide and the like. The
reaction mixture containing the alkoxide anion is then
treated with the methanephosphonate LCH2P(O)(ORZ)2 (IV)
wherein L is a leaving group and RZ is an alkyl group
containing 1-5 carbon atoms as previously defined to
provide the protected HPMP nucleoside of formula (V).
L is preferably selected from the group consisting of
p-toluenesulfonate (tosylate), methanesulfonate
(mesylate), and trifluoromethanesulfonate (triflate);
and R2 is preferabl~r an alkyl group having from 1-3
carbon atoms, e.g., methyl, ethyl, n-propyl, and
isopropyl.
The third step of the process involves the
removal of the phosphonic protecting group, i.e. R2,
the hydroxy protecting~group, and if present, any
protecting groups on the purine or pyrimidine base.
The phosphonate may be converted to the parent acid by
.... , .. ;7':~..~;~~~;:.i...:
CA 02347280 2001-05-31
treatment with a trialkylsilyl halide such as
trimethylsilyl bromide or trimethylsilyl iodide, and
optionally followed by the addition of water. Methods
to be employed for the removal of the hydroxy
protecting group, and if present, protecting groups on
the purine or pyrimidine base will of course depend on
the nature of the protecting group; examples of
typical deprotecting techniques include acid or base
catalyzed hydrolysis, hydrogenation, or metal mediated
deprotection.
In a preferred embodiment of the present process,
the reaction sequence is conveniently carried out from
the starting material to the end product without
isolating and purifying the intermediate compounds
formed. The elimination of the need for costly and
lai~or intensive isolation and purification of
intermediates represent a marked improvement over
prior processes. Another advantage of the present
invention is that t:he stereochemistry of the glycidol
reactant is maintained throughout the process such
that end product having the desired stereo
configuration is obtained without racemization.
The process of the present invention, while
adaptable to the synthesis of a wide variety of HPMP
substituted purine ~~nd pyrimidine bases, is especially
applicable to the s~~nthesis of
hydroxyphosphonometlzoxypropyl cytosine (HPMPC);
particularly (S) -HP2~IPC. A preferred embodiment of the
present~,process suii~able for the preparation of
(S)-HPMPC is illust~_ated in Scheme II.
S
v~.'j7L.-.; ~~r~;
CA 02347280 2001-05-31
a
9
Scheme II
0 0 0
II II II
NHCPh NHCPh NHCPh
N%~ 1) Base N~ 1) base N~~
z ) o ' o~.~ o o~N~ a
H ~Tr ~H 2) TsOCHZP(OEt)Z .,p~p(OEt)z
"OTr ~Tr
(VI) (VII) (VIII)
n.
0 0
II II
NHCPh NHCPh
(I) NH40H N%', ~ BrSirie.~ N
0 ~ 0
0 ~."OvP(OH)2 0 N ..l~~,iP(OEt)2
'OH ~H
(X) (IX)
In Scheme II, Tr~ is triphenylmethyl and Ts is
tosyl. N4-Benzoylc.~tosine (VI) is converted to its
anionic form by treatment with a base in an aprotic
polar organic solvent at elevated temperature;
suitable bases are for example sodium hydride,
potassium t-butoxide, potassium or sodium carbonate,
and the like; suitable solvents are for example
dimethylformamide, N-methyl-2-pyrrolidinone, dimethyl
sulfoxide, hexamethylphosphoramide, and the.like; and
typical reaction temperature ranges from about 70°C to
about 150°C. Subsequently, (S)-[(triphenylmethoxy)-
methyl]axirane is added to the above reaction solution
and thersolution maintained at the elevated
temperature to effect the formation of (S)-N4-benzoyl-
N~-[(2-hydroxy-3-tr:iphenylmethyl)propyl]-cytosine
(vII) .
CA 02347280 2001-05-31
a
The above obtained diprotected (2,3-dihydroxy)-
propyl cytosine is treated with a metal hydride, e.g.
sodium hydride, at ice bath temperature, and then
treated with dieth~~l tosyioxv~~et:hylphosphonate to
5 provide the compound (S)-N4-benzoyl-N~-[[(2-diethyl-
phosphonylmethoxy)--3-triphenylmethyl]propyl]cytosine
(VIII) .
Next, the trit:yl protecting group is removed to
10 provide the compound of formula (IX) by treating the
above obtained compound (VIII) with an acidic medium,
for example with h~~drochloric acid at about 0-5°C. A
wide range of other acids may be employed to
accomplish this step, and examples include, acetic
acid, formic acid, trifluoroacetic acid, zinc bromide,
acidic ion exchangE: resins, to name but a few.
Suitable reaction temperature, and time may be readily
ascertained by a person skilled in the art.
Following detritylation, the resulting compound
(X) is treated with a trialkylsilyl halide such as
trimethylsilyl bromide at room temperature to convert
the diethyl phosphonate to the phosphoric acid. This
latter compound is then treated with a base such as
ammonium hydroxide to remove the benzoyl protecting
group to afford the: desired end product (S)-HPMPC.
Another preferred process for the preparation of
(S)-HPMPC is illustrated in Scheme III.
CA 02347280 2001-05-31
0
11
Scheme III
NHZ NHz N ~(CH3)Z
N~ i) 8ase N HC(OCH,),N(CH,), N I
Or~~ 2 ) 0
H ~Tr l nH ~H
1Y~~00Tr 1'~Tr
(XI) (XII)
i) Hase
0
II
2) TsOCHZP(OEt)Z
NHZ 1N Z1(CH3)Z
(I) BrSilie~ N~ , H N' l 0
O~N J ~I O~N~J II
~O~P(OEt)Z ~~,~P(OEt)2
bH Tr
(XIY) (XIII)
~n Scheme III,, cytosine is coupled with (S)- ° -
[(triphenylmethoxy;~methyl]oxirane in the presence of a
base such as one pI-eviously enumerated to give
cytosine derivatives of formula (XI). The 4-amino
group of compound (XI) is then converted to the
corresponding dimet:hyl formamidine derivative (XII)
upon treatment with dimethylformamide or an acetal
thereof. Compound (XII) is subjected to base promoted
alkylation with diethyl tosyloxymethylphosphonate as
previously describe=d to provide compound of formula
(XIII). Compound (XIII) is deprotected in a acidic
medium and the product thereof is treated with e.g.
trimethylsilyl brornide to.afford (S)-HPMPC.
Irk another pre=ferred process for the preparation
of (S)-HPMPC, as i7Llustrated in Scheme IV, cytosine
derivative of_formula (XI) is treated with a base,
followed by diethy7L tosyloxymethylphosphonete to
afford the compound of formula (XV). The latter
compound is treated with an acid to remove the trityl
CA 02347280 2001-05-31 ".,.,.,.
0
12
protecting group and affords the compound of formula
(XIV) which is con~rerted to (S)-HPMPC as previously
described.
Scheme IV
NH2
1. base N
H'
~xl~ 0 i 0~~ 0 -'' CxIV~
2. TsOCH2P~0Et)z ~.,,,.O~.~OEt~2
OTr
~xv~
Another aspect. of the present invention concerns
novel intermediates in the synthesis of (S)-HPMPC.
These include compounds of formulas (VII), (VIII),
to (Ix), (x), (xII), I;xiII), and (xv).
The process oi° this invention is illustrated in
greater detail by t:he following examples which are not
to be construed to limit the scope of the invention in
any manner.
Preparation I. (~~-Triphenylmethoxymethyloxirane
Trityl chloride (18.816 g, 0.067 mol) was added
to a stirred solution of (~)-glyc~idol (5 g, 0.067 mol)
and triethylamine 1;13.84 g, 0.137 mol) in anhydrous
methylene chloride (54 ml). After 15 hours of
stirring, at room temperature, the reaction solution
was washed with wager (2 x 10 ml) and brine (20 ml).
The organic phase was evaporated after drying over
anhydrous NaZS04 to give a yellow foam which was
CA 02347280 2001-05-31
a
13
purified on silica gel (5% EtOAc in hexane) to afford
the title compound (17.64 g, 82.6%) as a solid.
~H NMl~ (C:OC:13) : 2.62 (dd, J = 2.4 and 5.2 Hz,
1H), 2.77 (t, J = ~E.5 Hz, iH), 3.08-3.18 (m, 2H),
3.29-3.80 (m, 1H), 7.20-7.38 (m, 3H), 7.45-7.52 (m,
2H) .
Preparation II. (:>)-Triphenylmethoxymethyloxirane
A 5 L 3-neck round bottom flask was charged with
trityl chloride (1;13.8 g, 0.48 mol) and methylene
chloride (400 ml) . It was cooled to 0°C under NZ and
treated with triethylamine (70.7 g, 0.70 mol). After
an hour of stirring at 0°C, a solution of (R)-glycidol
(88% ee, 37.03 g, 0.5 mol) in methylene chloride (100
'ml) was adcieu over 0.75 hour. The resulting solution
was allowed to warm~:-to ambient temperature and was
stirred for 3 hour~s.'p It was then filtered, and the
filtrate was washed with water (2 x 500 ml) and brine
(2 x 500 ml). The organic phase was dried over MgS04
and concentrated tc> a foam, which on crystallization
from isopropyl alcohol gave the title compound (116.2
g, 76.5%) as an off-white powder.
[a]p = -6.01 (C = 1, MeOH).
Example 1. Preparation of (~)-N'-[(2,3-dihydroxy)-
propyll-cytosine
Cy.,tosine (0.5f. g, 4.95 mmol) , (~)-glycidol (0.404
g, 5.45 mmol), and anhydrous potassium carbonate (5
mg, 0.04 mmol) in dry DMF (6 ml) were stirred at 71°C
for 3 hours. Glycidol'(4) was totally reacted
according to TLC of the reaction mixture. The DMF was
distilled off under high vacuum, and the resulting
CA 02347280 2001-05-31
14
yellowish thick liquid was absorbed on silica gel (3
g). This was placEad on top of a silica gel column,
which was eluted with 20% MeOH in ethyl acetate to
give a mixture (0.!40 g) of the title compound =.nd a
polymer derived from glycidol. Crystallization from
ethanol afforded the title compound (0.44 g, 52.3%) as
a solid.
MP: 169°-71°C.
LJV: ~,~X 274 nm (E = 8,083) .
~H NMR (DMSO-cl6): 3.11-3.47 (m, 3H), 3.55-3.75
(m, 1H), 3.88 (dd, J = 3.3 and 13.3 Hz, 1H), 4.71 (t,
J = 5.8 Hz, 1H), 4.95 (d, J = 5.3 Hz, 1H), 5.61 (d, J
- 7.1 Hz, 1H), 7.00 (bd, J = 23.7 Hz, 2H), 7.44 (d, J
- 7.1 H2, 1H).
Analysis calcdfor C~HiiN303~ 0.5H20:
C, 43.30; H, 6.23; N, 21.63
Found: C, 43.33; H, 5.92; N, 21.38
Example 2. Preparation of (S)-N1-[(2,3-dihydroxy)-
propyl]-cytosine
Reaction of cytosine (2.2 g, 19.8 mmol) with (R)-
glycidol (88% ee, 1.51 ml, 22.8 mmol) in the presence.
of anhydrous potassium carbonate (40 mg, 0.289 mmol)
in dry DMF (20 inl) at 72°C for 5'hours, as described
in Example 1, furnished the title compound (88% ee) in
43.1% yield.
CA 02347280 2001-05-31
Example 3. Preparation of (~)-N'-[(2-hydroxy-3-
triphenylmethoxy)propyl]cytosine by
tritylation of (~)-N'-[(2,3-dihydroxy)-
propyi'~cytosine
5
(a) using 1. J_ eq. of cLlycidol
A mixture of cytosine (0.55 g, 4.95 mmol), (~)-
glycidol (0.362 ml, 5.46 mol), and anhydrous potassium
10 carbonate (5 mg) in dry DMF (5 ml) was stirred at 71°C
for 3 hours. It w~~s cooled to room temperature and
treated with DMAP 1;0.031 g, 0.25 mmol), dry pyridine
(0.783 g, 9.9 mmol), and trityl chloride (1.48 g, 5.2
mmol). The resulting reaction mixture was stirred at
15 80°C for 3 hours and at room temperature for 17 hours.
It was diluted with ethyl acetate (60 ml), washed with
saturated sodium~bicarbcr.ate (2 x 15 ml), water (15
ml) , and brine (15 xnl) , and dried over MgS04. The
ethyl acetate was Evaporated to give a crispy foam
(1.98 g), and purification by chromatography over
silica gel (10-15% methanol in ethyl acetate)
furnished the title: compound as a crystalline solid
(0.74 g, 35%).
MP: 227°-228"C.
W: ~1~X 274 nm (e = 7, 149) .
~H NMR (DMSO-d6): 2.81-2.98'(m, 2H), 3.26-3.42
(m, 1H), 3.85-3.97 (m, 1H), 4.02 (dd, J = 4.7 and 14.2
Hz, 1H) , 5.23 (d, ,:r = 5.8 Hz, 1H) , 5.54 (d, J = 7.1
Hz, 1H), 7.93 (bd, 2H), 7.1-7.29 (m, 16H).
(b) using 1.F~ eq: of qlycidol
CA 02347280 2004-O1-21
16
Cytosine (0.275 g, 2.48 mmol), (~)-glycidol
(0.281 g, 3.7 mmol), and anhydrous potassium carbonate
(2.5 mg, 0.018 mmol) in dry DMF (2.5 ml) were stirred
at 70°C for 1.5 hours. The DMF was distilled under
reduced pressure. Proton magnetic resonance (PMR)
~f the resulting solid showed
that it contained (~)-N'-[(2,3-dihydroxy)propyl]-
cytosine and cytosine in 89:11 ratio.
The above solid was dissolved in dry pyridine (4
ml) and trityl chloride (0.602 g, 2.14 mmol) and DMAP
(13 mg) were added successively at room temperature.
After 3 hours of stirring at 85°C, followed by work-up
as described in Example 3 (a), su ra, a foamy solid
(0.88 g) was obtained. Crystallization from methylene
chloride and toluene gave the title compound (0.360 g
34%). The mother liquor was concentrated and purified
by silica gel chromatography (10% methanol in ethyl
acetate) to give the title compound (60 mg, 5.7%)'and 1
(~)-N'-[[2-[(2-hydroxy-3-triphenylmethoxy)-propyloxy]-
3-triphenylmethoxy]propyl]cytosine (hereinafter
referred to as the dimer) (10 mg, 0.80).
(c) using 2 eg of alycidol
The above experiment was repeated using 2 eq. of
(~)-glycidol to provide the title compound in 39.60
yield and the dimer in 1.6o yield.
Example 4. Preparation of (~)-N~=[(2-hydroxy-3-
triphenylmethoxy)propyl]cytosine by
reaction of cytosine and
(~)-triphenylmethoxymethyloxirane
CA 02347280 2001-05-31
17
( a ) us inct a c:ata lyt is amount of NaH
Cytosine (80 mg, 0.72 mmol) was added to a
stirred suspension of 80% r~3H (4 mj, 0.13 mmol) in dry
DMF (3 ml). After an hour at room temperature,
(~)-trityloxymethy7.-oxirane (0.19 g, 0.6 mmol) was
added to the reaction mixture, and stirring continued
at 106°C for 5 hours. The reaction was completed as
indicated by TLC of: the reaction mixture. It was
cooled, and the DMF was distilled off under vacuum.
The resulting solid was partitioned between ethyl
acetate (20 ml) and water (2 ml). The organic phase
was separated, washed once again with water (5 ml),
and dried over NaZS04. Evaporation of the ethyl
acetate gave the brown solid (0.28 g) which was
crystallized from methylene chloride-toluene (2 ml and
30 ml) to furnish t:he title compound (0.21 g) in 81.7%
yield.
(b) using onE: equivalent of NaH
Carrying out t:he above reaction using
(~)-trityloxymethyl.oxirane (0.190 g, 0.6 mmol),
cytosine (67 mg, 0.6 mmol), and sodium hydride (80~,
18 mg, 0.6 mmol) in anhydrous DMF (4 ml) furnished the
title compound (60 mg) in 23.3% yield.
c. usinc~KZC03 instead of NaH
Following the procedure described above, the
title compound was obtained in 82% yield from
(~)-trityloxymethy~.oxirane (0.19 g, 0.6 mmol) and
cytosine (0.08 g, 0.72 mmol) in the presence of
potassium carbonate: (10 mg, 0.072 mmol) in anhydrous
DMF (3 ml) .
CA 02347280 2001-05-31
18
Example 5. Preparation of (~)-N4-benzoyl-N~-
[(2-hyd.roxy-3-triphenylmethoxy)-
propyllcytosine
(a) usin~:auivalent of N4-benzoylcytosine
Treatment of N'4-benzoylcytosine (0.388 g, 1.803
mmol) with (~)-trit.yloxymethyloxirane (0.571 g, 1.805
mmol) in the presence of 80% sodium hydride (12 mg,
0.4 mmol), according to the procedure of Example 4
(a), gave the title: compound as a crystalline solid in
72.9% yield after chromatography over silica gel using
hexane-EtOAc (1:3).
MP: 105°-7°C.
Ui': ~,~X 259 nm (e = 23,500) , 306 nm (e = . .
10,380).
~H NMR (CDC13):: 3.05-3.18 (m, 1H), 3.21-3.33 (m,
1H), 3.66-3.90 (m, 1H), 4.2 (bS, 1H), 4.35 (d, J =
13.6 Hz, 1H), 7.13-7.72 (m, 15H), 7.88 (d, J = 7.5 Hz,
1H), 8.73 (bS, 1H).
Analysis calcd. for C33H29N304=
C, 74.56; H, 5.50; N, 7.90
Found: C, 74.02; H, 5.67; N, 7.63.
(b) using 1.2 equivalents of N4-benzoylcytosine
(~)-Tityloxymethyloxirane (0.762 g, 2.41 mmol)
was reacted with N4-benzoylcytosine (0.621 g, 2.89
mmol) in the presence of 80% sodium hydride (16 mg,
0.53 mmol) in dry I)MF,'as described above, to obtain
the title compound in 85% yield.
CA 02347280 2001-05-31 ... . , .. . .. .
19
Example 6. Preparation of (S) -N4-benzoyl-N~-
[(3-allyloxy-2-hydroxy)propyl]
cytosine
To sodium hydride (80%, 12 mg, 0.4 mmol) stirring
in anhydrous DMF (4.5 ml) at room temperature was
added N4-benzoylcyt«sine (0.466 g, 2.17 mmol). The
reaction mixture was stirred for an hour and treated
with (S)-allyloxymethyl-oxirane (0.206 g, 1.8 mmol).
It was then heated at 105°C for 6 hours, cooled, and
concentrated in vacuo. The resulting orange red gummy
material was treated with water (5 ml) and ethyl
acetate (20 ml). It was stirred for 5 minutes, and
the insoluble solid (0.145 g, 31.1% of recovery) was
collected by filtration and identified as
N4-benzoylcytosine. The filtrate was transferred into
a separatory funnel, and the ethyl- acet_at-a l~.yer was
separated. It was washed with water (3 x 5 ml), dried
over NaZS04, and evaporated to obtain 0.423 g of pale-
yellow solid. Slurrying of this material in diethyl
ether gave the title compound (0.331 g) in 55.7%
yield. The ether filtrate was evaporated, and the
resulting light greenish gummy material was purified
by flash chromatography on silica gel (0-5% MeOH in
EtOAc) to furnish the title compound (20 mg) in 3.6%
yield.
MP: 139°-41°f.
[a]o = -55.06 (C = 1.155, MeOH).
W: .1~X 259 nm (e = 21,500), 305 nm (e =
10, 120) .
CA 02347280 2001-05-31
'H NMR (CDC13): 3.4-3.56 (m, 2H), 3.77 (dd, J =
7.6 and 13.5 Hz, 1:H), 3.98 (d, J = 5.7 Hz, 3H), 4.16-
4.25 (m, 1H), 4.28 (dd, J = 2.7 and 13.5 Hz, 1H),
5.10-5.25 (m, 2H), 5.79-5.92 (m, 1H), 7.39-7.62 (m,
5 4H) .
Analysis calcd. for C~7H~9N304:
C, 61.97; H, 5.85; N, 12.79
Found: C, 61.82; H, 6.05; N, 12.77
Example 7. Preparation of (~) -N4-benzoyl-N~-[ (3-
allyloxy-2-hydroxy)propyl]-
cytosine
The title compound was prepared in 39.5% yield,
following the procedure of Example 6, from
(~)-ailyloxyn~ethyloxirane (9.02 g, 0.079 mol),
N4-benzoylcytosine ('20.40 g, 0.095 mol), and 80% sodium
hydride (0.526 g, 0.018 mol) in dry DMF (241 ml).
Example 8. Preparation of (S)-N4-benzoyl-N~-[(3-
benzyloxy-2-hydroxy)propyl]-
cytosine
(R)-benzyloxymethyloxirane (0.296 g, 1.8 mmol) in
dry DMF (0.5 ml) was added to sodium salt of
N4-benzoylcytosine,, prepared from N4-benzoylcytosine
(0.388 g, 1.8 mmol) and 80% sodium hydride (0.012 g,
0.4 mmol) in dry DMF (4 ml) at room temperature for an
hour, and stirred at 110°C for 6 hours. The reaction
was complete as confirmed by HPLC of the reaction.
Most of the DMF was distilled off under reduced
pressure. The resulting gummy product was partitioned
. between ethyl acetate '( 2 0 ml ) and water ( 5 ml ) . The
ethyl acetate layer was separated, washed with water
(3 x 10 ml), dried over NazS04, and evaporated to give
CA 02347280 2001-05-31
0
21
a yellow product (0.595 g). Trituration with ethyl
acetate furnished 'the title compound (0.392 g) in
57.3%.yield. The mother liquor was concentrated and
chromatographed on silica gel (ethyl acetate) to gig,°c
the title compound (50 mg, 7.3%).
MP: 138°C.
[a]p = -49.19 (C = 1.425, MeOH).
UV: ~.~X 259 nm (E = 22,440), 306 nm (e =
10, 220) .
~H NMR (CDC13): 3.45-3.58 (m, 3H), 3.79 (dd, J =
7.1 and 13.3 Hz, 2:H), 4.16-4.33 (m, 2H), 4.51 (s, 2H),
7.2-7.6 (m, 8H), 7.69 (d, J = 7.2 Hz, 1H), 7.89 (d, J
- 7.4 Hz, 2H), 8.91 (bS, 1H). .- -
Analysis calcd:' for C2~H2~N304~ 0.9H20:
C, 63.76; H, 5.81; N, 10.62
Found: C, 63.97; H, 5.50; N, 10.63
Example 9. Preparation of (~)-N4-benzoyl-N~-[(3-
benzyloxy-2-hydroxy)propyl]-
cytosine
Treatment of (~)-benzyloxymethyloxirane (6.0 g,
0.0365 mol) with N4-benzoylcytosine (9.446 g, 0.0439
mol) in the presence of sodium hydride (80% pure,
0.263 g, 8.1 mmol) in dry DMF (85 ml), according to
the procedure of Example 8, afforded the title
compound (8.2 g) in 59.2% yield.
MP: 144°-6°C.
CA 02347280 2001-05-31
22
Example 10. Preparation of (~)-N~-[(2-diethyl-
phosph.onylmethoxy-2-triphenylmethoxy)-
t~ropyl.1 cytos ire
One Pot Synthesis: A mixture of cytosine (0.134
g, 1.21 mmol) and 8~0% sodium hydride (8 mg, 0.27 mmol)
in anhydrous DMF (3~ ml) was stirred at room
temperature. After 1 hour, (~)-trityloxymethyloxirane
(0.38 g, 1.2 mmol) was added in 1 portion, and
stirring was continued for 5 hours at 105°C. The
formation of (~)-N~-[(2-hydroxy-3-trityloxy)propyl]-
cytosine was noted by its HPLC.
The above homogenous reaction solution was cooled
in an ice bath and successively treated with 80%
sodium hydride (0.7_00 g, 3.3 mmol) and diethyl
tosyluxymethylphosi~licnatc-: (25% pure, 0.682 g, 1.8
mmol) . After beinc~~-stirred at 0°C for 0.5 hour and at
room temperature for'~15 hours, a few drops of ethanol
were added to quench excess sodium hydride. The
solvent was removed under reduced pressure, and the
resulting orange residue was partitioned between ethyl
acetate (30 ml) and water (5 ml). The organic phase
was separated and washed with saturated sodium
bicarbonate (10 ml) and brine (10 ml). After drying
over Na2S04, the ethyl acetate was evaporated to give
the orange-colored product (0.550 g), which was
purified by chromai~ography on silica gel (10-15% MeOH
in CHZC12) to furnish the title compound (0.26 g,
37.4%) as a foamy solid.
~H NMR (CDC13): 1.25 (t, J = 7 Hz, 6H), 2.97-3.12
(m, 1H), 3.33 (dd,~J = 3 and 10.5 Hz, 1H), 3.55-3.68
(m, .2H), 3.84-3.96 (m,'2H), 3.96-4.24 (m, 5H), 5.63
(d, J = 6.9 Hz, 1H), 7.0-7.6 (m, 18H).
CA 02347280 2001-05-31 '";,.,,.:.
23
Example 11. Preparation of (~)-N'-[(2-dimethyphos-
phonyl.methoxy-3-triphenylmethoxy)-
propryllcytosine
Repeating the experiment of Example 10, using
dimethyl tosyloxymethylphosphonate instead of
diethylphosphonate afforded the title compound in
15.6% yield.
~H NMR (CDC13);; 3.0-3.1 (m, 1H), 3.27 (dd, J =
2.8 and 10.5 Hz, 1H), 3.51-4.26 (m, lOH), 4.17 (dd, J
- 3.0 and 13.6 Hz, 1H), 5.69 (d, J = 6.9 Hz, 1H), 7.0-
7. 66 (m, 18H) .
Example 12. Preparation of (~)-N'-[(2-diethylphos-
' phony7methoxy,-3-hydroxy~propyl]cytosine
Treatment of cytosine (0.249 g, 2.24 mmol) with
(~)-trityloxymethy7Loxirane (0.590 g, 1.87 mmol) in the
presence of a catalytic amount of 80% sodium hydride
(13 mg, 0.44 mmol) followed by in situ alkylation of
the intermediate w_Lth diethyl tosyloxymethylphos-
phonate (85.1% purea, 1.06 g, 2.80 mmol) in the
presence of 80% sodium hydride (0.099 g, 3.3 mmol),
according to the procedure of Example 10, gave the
crude (~)-N'-[(2-diethylphosphonylmethoxy-3-
trityloxy)propyl]cytosine (1.267 g).
To the above nucleotide, 80% acetic acid (20 ml)
was added and stirred at 95°C for 3 hours. Water (20
ml) was added to the reaction which was then cooled to
-0°C. 'The precipii~ated trityl alcohol was collected
by filtration. The filtrate was evaporated, and the
resulting thick product was co-distilled with water (3
x 30 ml) and with toluene (3 x 30 ml) to remove acetic
acid. It was then applied on a silica gel column
CA 02347280 2001-05-31
24
which, on elution with 15% MeOH in CHzCl2, afforded the
title compound (0.:147 g, 23.5%) as a gummy material.
Further elution of the column with 20% MeOH in CHZC12
gave (~)-N'-[(2,3-dihydroxy;p~-opyl]~ytosine (20.2 ntg,
6%) .
0
~H NMR (MeOH-f~,4): 1.30 (t, J = 7.1 Hz, 3H), 1.31
(t, J = 7.1 Hz, 3H;~, 3.5-3.63 (m, 1H), 3.67-3.92 (m,
4H), 3.97-4.2'2 (m, 6H), 5.85 (d, J = 7.2 Hz, 1H), 7.53
(d, J = 7.2 Hz, 1H;1.
Example 13. Preparation of (~)-N'-[(2-diethylphos-
phony:Lmethoxy-3-hydroxy)propyl]cytosine
via formamidine method
A mixture of i~he sodium salt of cytosine,
obtained from cyto:~ine (0.134. g, 1.21 mol) and 80%
sodium hydride (8 mg, 0.27 mmol) in dry DMF (3 ml) at
room temperature for''1 hour, and (~)-trityloxymethyl-
oxirane (0.38 g, 1.2 mmol) was stirred at 110°C for 5
hours. The resulting solution of 12 was cooled to
room temperature, and DMF dimethyl acetal (0.286 g,
2.4 mmol) was added in 1 portion. It was then stirred
at 85°C for 1.5 hours and concentrated under reduced
pressure to ca. 1 ml of the crude dimethylformamidine
derivative of 11. This and diethyl tosyloxyZnethyl-
phosphonate (0.909 g, 2.4 mmol) in anhydrous DMF (3 .
ml) was cooled to 0°C and treated with 80% sodium
hydride (64 mg, 2.:L3 mmol). The~resulting yellow
reaction mixture was stirred at 0°C for 1.5 hours and
at room temperatures for 14 hours. The crude product
obtained after work-up is a mixture of (~)-N~-[(2-
diethylphosphonylmeathoxy-3-trityloxy)propy]cytosine
and its N4-dimethylformamidine derivative. This
mixture was dissolved in 80% acetic acid (11 ml) and
refluxed for 3 hours. After work-up, the yellowish
CA 02347280 2004-O1-21
gummy product (0.693 g) was obtained which, on
purification by chromatography on silica gel (15o MeOH
in EtOAc), afforded the title compound (0.175 g) in
43.60 yield.
5
Example 14. Preparation of (~)-N~-[(2-ethylhydro-
genphosphonylmethoxy-3-hydroxy)propyl]-
cytosine
10 2N Sodium hydroxide solution (4.5 ml) was added
to (~)-N'-[(2-diethylphosphonylmethoxy-3-hydroxy)-
propyl]cytosine (0.230 g, 0.69 mmol). TLC of the
reaction after 1.25 hours at room temperature showed
that starting material was completely consumed. The
15 reaction was acidified with Dowex~50 x 8 (H+) and
filtered. The resin was washed with 20 ml of water.
The combined filtrate was evaporated to provide the
title compound (0.15-3 g) in 77.3% yield.
20 ~H NMR (Dz0): 1.26 (t, J = 7.1 Hz, 3H), 2.52-2.68
(m, 2H), 3.75-4.0 (m, 6H), 4.21 (dd, J = 2.8 and 14.1
Hz, 1H), 6.19 (d, J = 7.6 Hz, 1H), 7.88 (d, J = 7.6
Hz, 1H) .
25 MS: molecular ion (m/e) for C~oH~8N306P,308~ 1011:
Found: 308.1009
Example 15. Preparation of (S)-N4-benzoyl-N~-((2-
hydroxy-3-triphenylmethoxy)-
propyllcytosine
(a) To N4-benzoylcytosine (100.1 g, 0.47 mol) in
dry DMF (1,000 ml) at 100°C under N2 was added 800
sodium hydride (3.0 g, 0.10 mol) in 1 portion, and the
slurry was stirred for 0'.25 hour. (S)-trityloxy-
methyloxirane (88o ee, 125.1 g, 0.40 mol) was added
* trade-mark
CA 02347280 2001-05-31 ., .
26
and further stirred at 110°C for 4 hours. The
reaction was compls~ted as confirmed by its HPLC. The
reaction mixture w<is filtered and used in the
subsequent reaction without further purificdvion. The
filtrate contained a 90% in solution yield of the
title compound based on HPLC.
(b) In a separate experiment, the crude product
obtained from the above reaction was purified by
chromatography on :silica gel (3-5 % MeOH in CHZC12) and
provided analytica:Lly pure title compound.
MP: 105°-7°C.
W: ~,~X 259 nm ( E = 23, 500) , 306 nm
10, 380) .
~H NMR (CDC13)::-,3.05-3.18 (m, 1H), 3.21-3.33 (m,
1H), 3.66-3.90 (m, 1H), 4.2 (bS, 1H), 4.35 (d, J =
13.6 Hz, 1H), 7.13-7.72 (m, 15H), 7.88 (d, J = 7.5 Hz,
1H), 8.73 (bS, 1H).
Analysis calm. for C33H29N304:
C, 74.56; H, 5.50; N, 7.90
Found: C, 74.02; H, 5.67; N, 7.63
(c) The reaction described in (a) supra was also
repeated using solvents and conditions to afford the
title compound.
(1) NaH, NMPO, 70°-80°C for 2 hours followed by
100°-104° for 3.5 hours.
(2) NaH, 18-crown-6, DMF, 103°C, 5 hours.
CA 02347280 2001-05-31 ~.. .....
27
(3) NaH, benzyltriethylammonium chloride, DMF,
70°C for 6 hours, 105°C for 4 hours.
(4) KOL(CH3)3, i~MF, 70°C for 16 hours followed by
105°C for 8 hours.
Example 16. Preparation of (S)-N4-benzoyl-
[(diet:hylphosphonylmethoxy-3-
triphe:nylmethoxy) prop,yl l cytosine
(a) A solution of the crude (S)-N4-benzoyl-N~-
[(2-hydroxy-3-tripr~enylmethoxy)propyl]cytosine in DMF,
obtained in Examples 15 (a), was placed in a 5 L 3-neck
round bottom flask and cooled to 0°C. 80% sodium
hydride (32.4 g, 1.06 mol) was added in 2 portions,
and an exotherm of 8~C was noted. Immediately,
diethyl tosyloxymet:hylphosphonate (80% pure, 215.6 g,
0.54 mol) was addect,~~and the reaction was completed
after 6 hours of st:irring. The reaction was diluted
with ethyl acetate (2 L), quenched with water, washed
with water (2 x 1 h) and saturated NaHC03 (1 L), dried
over MgS04, and concentrated to afford crude title
compound (230.1 g) with 2% of (S)-trityloxymethy-
loxirane, as indicated by its proton NMR spectrum.
This crude product was used in the next procedure
without further purification.
(b) In a separate experiment, a small amount of
the crude product was purified by column
chromatography on :oilica gel (1-3~ MeOH in CHZClz) to
provide an analytical sample of the title compound.
~H NMR (DMSO-d6) : ' 1. 14 (t, J = 7 Hz, 3H) , 1. 16
(t, J = 7 Hz, 3H), 2.94-2.98 (m, 1H), 3.24-3.31 (m,
CA 02347280 2001-05-31
28
1H), 3.58-4.09 (m, 9H), 7.23-7.63 (m, 19H), 7.98 (d, J
- 7 Hz, 3H), 11.19 (bS, 1H).
Analysis calcd. for C38H4oN30~P~ 0.5Hz0:
C, 66.07; H, 5.98; N, 6.08
Found: C, 65.96; H, 5.84; N, 6.09
Example 17. Preparation of (S)-N4-benzoyl-N~-[(2-
diethylposphonylmethoxy-3-hydroxy)-
propyl]cytosine
(a) Hydrogen chloride gas was bubbled into a
solution of the crude (S)-N4-benzoyl-N'-[(2-
diethylphosphonyl-3-triphenylmethoxy)propyl]cytosine
(230.1 g, obtained from Example 16) in methylene
chloride (1.2 L) at 0°-5°C until starting material was
consumed as determined by HPLC (ca 10 minutes). Water
(500 ml) was added,:. and the resulting 2-phase mixture
stirred rigorously for 5 minutes. The organic phase
was separated and extracted with 10% hydrochloric acid
(2 x 250 ml). The combined aqueous solution was
cooled to 0°-5°C, adjusted to pH = 8 with 40~ sodium
hydroxide solution, and then extracted with CHZC12 (2 x
500 ml). The combined CHZCLZ solution was dried over
MgS04 and concentrated in vacuo to give crude title
compound (96.2 g), as a viscous oil, in 55% yield from
(S)-trityloxymethoxy-oxirane after 3 steps.
~H NMR (DMSO-d6): 1.16 (t, J = 7 Hz, 3H), 1.18
(t, J = Hz, 3H), 3.44-3.57 (m, 2H), 3.68-3.80 (m, 3H),
3.88-4.01 (m, 5H), 4.13 (dd, J = 8 and 17 Hz, 1H),
4.88 (t, J = 6 Hz, 1H), 7.27 (br d, J = 7 Hz, 1H),
7.49 (t, J = 7 Hz, 2H), 7.60 (t, J = 7 Hz, 1H), 7.98
(d, J = 7 Hz, 3H), 11.:18 (br s, iH).
Analysis calc~d. for C~9H26N307P~ 0.5H20:
C, 50.89; H, 6.07; N, 9.37
.., . .. ..."s.. ... ..., . . _~..a...u~
CA 02347280 2001-05-31
29
Found: f., 50.99; H, 6.03; N, 9.32
(b) Detrityla.tion was carried out with the
following reagents and conditions to afford the title
compound in moderate to excellent yields:
(1) 80% acetic acid, 75°C, 45 minutes.
(2) 80% acetic acid, 100°C, 30 minutes.
(3) 80o acetic acid, 60°C, 3 hours.
(4) 80o formic acid, 0°-5°C, 30 minutes.
(5) 95-97% formic acid, room temperature, 5
minutes.
(6) Trifluoroacetic acid, n-butanol or
isopropylY alcohol, or CH2Clz, 22 hours.
(7) ZnBrZ, CHzClz, room temperature, l0 minutes-
3 hours"
(8) Amberlyst 15*(H+), MeOH, 50°C, 24 minutes.
(9) Amberlyst 15 (H+) activated by HC1/MeOH
wash, 50°C, 6.5 hours.
(10) Dowex 5t) x 8 (H'') activated by HC1/MeOH.
Example. 18 . Preparation of ( S) -N4-benzoyl-N'- [ ( 3-
hydro:~y-2-phosphonylmethoxy)-
t~ropy:L~ cytos ine
A solution of (S)-N~-[(2-diethylphosphonylmethoxy-
3-hydroxy)propyl-N'']-benzoylcytosine (188 g, 0.428 mol)
*Trademark
CA 02347280 2001-05-31
in methylene chloride (1.2 L) at room temperature
under argon was treated with bromotrimethylsilane (200
ml, 1.52 mol), and the resulting mixture was stirred
fuL 18 hours. It was then concentrated in vacuo to a
5 residue which was :redissolved in methylene chloride
(500 ml) and reconcentrated to furnish the crude
persilylated title compound (289 g) as a tan foam.
This material was 'used in the next step without
further purification. An analytical sample of the
10 title compound was prepared by treating the crude foam
with water from which the desired title compound
crystallized.
~H NMR (DMSO-d6): 3.45-3.81 (m, 6H), 4.11 (dd, J
15 - 4 and 13 Hz, 1H), 7.26 (d, J = 7 Hz, 1H), 7.49 (t, J
- 7 Hz, 2H), 7.61 (t, J = 7 Hz, 1H), 7.98 (d, J = 7
I-Iz, 2H) , 8.04 (d, J = 7 Hz, 1H) .
._
Analysis calcd-' for C~SH~$N307P~ 0.5H20:
20 C, 45.93; H, 4.84; N, 10.71
Found: C, 46.04; H, 4.67; N, 10.71
Example 19. Preparation of (S)-N'-[(3-hydroxy-2-
phosphonylmethoxy)propyl]cytosine
The crude persilyated (S)-N4-benzoyl-N~-[(3-
hydroxy-2-phosphors.ylmethoxy)propyl]cytosine (289 g),
obtained from the previous example, was dissolved on
conc NH40H (850 ml) and stirred ~t~room temperature for
4 hours. The aqueous reaction mixture was extracted
with CH2C12 (2 x 600 ml) to remove most of the
benzamide and then, filtered and concentrated in vacuo
until the pH of th,e aqueous solution was neutral. The
concentrated solut.ion~was diluted with water to a
volume of 800 ml, and ethanol (600 ml) was added. The
product was precipitated by adjusting the pH to 3.0
CA 02347280 2001-05-31
31
with careful addition of conc HCl (65 ml). The
resulting thick slurry was stirred at room temperature
for 1 hour and them stored at 0°-5°C for 16 hours.
The solid product was collected by filtration, washed
with water ethanol (2:1, 2 x 150 ml), and dried to
constant weight in vacuo at 40°C to give (S)-HPMPC
(105 g) in 78% yield from 22S after 2 steps. This
material contained 5% of the undesired (R)-isomer as
determined by chiral HPLC. Two crystallizations of
the crude product iby adjusting an aqueous slurry to pH
- 6 with 40o NaOH ;solution, followed by
reprecipitation with conc HC1 to pH = 3, reduced the
level of the undesired (R)-isomer to 2.4, a 90% weight
recovery.
MP: 260°C (decomp).
[a)o = -86.65 (C = 0.40, Hz0) .
~H NMR (D20): d 3.59-3.67 (m, 2H), 3.79-3.94 (m,
4H), 4.20 (dd, J = 3 and 14 Hz, 1H), 6.17 (d, J = 8
Hz, 1H) , 7.90 (d, ~T = 8 Hz, 1H) .
Analysis calcd. for C$H~4N306~ 2Hz0:
C, 30.4F3; H, 5.75; N, 13.33
Found: C, 30.30; H, 5.70; N, 13.25
Example 20. Preparation of (~)-N~-[(3-hydroxy-2-
~hosptionvlmethoxy) propyll~tosine
Synthesis of (;~)-HPMPC was achieved in 42.4%
yield after 5 steps (Examples 15, 16, 17, 18, and 19)
starting from (~)-t:rityloxymethyloxirane and
N4-benzoylcytosine.
CA 02347280 2001-05-31
32
Example 21. Preparation of (R)-N'-[(3-hydroxy-2-
phost~r.onvlmethoxy~ propel l cytosine
'l~he title compound (R)-HPMPC was prepared from
(S)-glycidol (88% e:e) and N4-benzoylcytosine, following
the method described for (S)-HPMPC.
Example 22. Preparation of (~)-[(3-hydroxy-2-
phosphonylomethoxy)prowl]uracil
A solution of (~)-N'-[(2-diethylphosphonyl-3-
hydroxy)propyl]cytosine (0.228 g, 0.68 mol) in 2N
sodium hydroxide (4.5 ml) was heated at 82°C for 60
hours. The reaction was complete as indicated by its
HPLC. It was acidified with Dowex 50 x 8 (H'') form at
room temperature and filtered, and the resin was
washed wits water (30 ml). Evaporation of the
filtrate afforded t:he title compound (0.157 g, 82.4%)
as a solid.
~H NMR (DZO): 3.16-3.29 (m, 1H), 2.55-4.17 (m,
9H), 5.87 (d, J = T.9 Hz, iH), 7.72 (d, J = 7.9 Hz,
1H ) .
*Trademark