Note: Descriptions are shown in the official language in which they were submitted.
CA 02347336 2001-05-31
1
RFC>;PTOR ANTAGONISTS
This is a division of Canadian Patent Application No. 2,172,726 filed
October 28, 1994.
Background of the Invention
Adenosine is an extracellular messenger generated .by all cells in the body.
Adenosine itself, substances that mimic the actions of adenosine, and
substances that
antagonize its actions have important clinical applications. In the heart, an
organ
whose function depends critically on an adequate supply of oxygen, adenosine
regulates the balance between ohygen supply (coronary blood flow) and oxygen
demand (cardiac..work): Adenosine released from working heart cells increases
oxygen supply through coronary dilation and decreases oxygen consumption by
slowing heart rate and modulating [3-adrenergic stimulation. The protective
effects of
adenosine are particularly important when cardiac oxygen supply is limited,
for
example, by coronary artery narrowing.
Several recent reviews describe the adenosine system in detail (Belardinelli,
L.,
J. Linden, RM. Berne [1989] Prog. Cardiovasc. Dis. 32:73-97; Belardinelli, L.,
A.
Pelleg (1990] J. Carrliovasc. ElectrophysioL 1:327-339; Olsson, R.A., J.D.
Pearson
[1990] PhysioL Rev. 70:761-845). The cardiac adenosine system consists of
three
processes: (1) mechanisms for adenosine formation; (2) adenosine receptors and
proteins that couple them to effectors; and (3) mechanisms for the removal of
adenosine. Selective modification of one or more of these systems by means of
drugs
such as adenosine receptor antagonists and adenosine uptake inhibitors can
modify the
actions of adenosine for therapeutic benefit.
Adenosine formation increases when oxygen demand exceeds its supply,
thereby promoting the degradation of adenine nucleotides. The degradation of
adenylates released from nerve terminals along with neurotransmitters and the
CA 02347336 2001-05-31
2
degradation of S-adenosylhomocysteine, a byproduct of methylation reactions,
are
additional sources of adenosine in the heart. Heart muscle and coronary blood
vessel
cells take up very nearly all the adenosine generated in the heart,
reincorporating that
adenosine into the cellular nucleotide pool.
At least two types of receptors mediate the actions of adenosine in the heart.
A, adenosine receptors (A,AR) decrease oxygen consumption, for example, by
slowing
heart rate, and A~ adenosine receptors (AZAR) increase oxygen supply by
causing
coronary vasodilation. The actions of adenosine on cardiac cells are either
direct
(CAMP-independent) or indirect (CAMP-dependent). The direct actions include
the
negative dromotropic effect on the AV node. Those electrophysiological effects
are
the basis of adenosine's anti-arrhythmic properties; adenosine is highly
effective
(>90%) in terminating paroxysmal supraventricular tachycardia (PSVT). The A,AR-
mediated inhibition of agonist-stimulated (but not basal) adenylate cyclase
activity
constitutes the indirect effects of adenosine. Whereas the direct effects of
adenosine
occur in the absence of agents that act through adenylate cyclase, the
indirect effects
reflect the inhibition ~of this enryme when it is stimulated by agents such as
(3-
adrenergic agonists.
A number of pharmacological studies employing receptor-selective agonists
support the idea that A~ARs mediate coronary vasodilation. Although
endothelial cells
contain AZARs and thus could play a role in vasodilation, they are not
essential, for
adenosine acts on coronary smooth muscle cells, causing them to relax.
When adenosine is used as a drug, its side effects are usually transitory, a
reflection o~ its extremely rapid degradation in the body (seconds). The
safety of
adenosine in the diagnosis and treatment of PSVT is now well established. An
important factor which has inhibited the therapeutic development of the
adenosine
analogues is the ubiquitous nature of adenosine's action on a variety of
tissues.
Two kinds of drugs modify the actions of adenosine according to whether they
magnify or attenuate the effects of the nucleoside. Inhibitors of the cell
membrane
nucleoside transporter block the removal of adenosine from the extracellular
space,
thereby increasing its concentration and intensifying its action. Adenosine
uptake
blockers also inhibit the nucleoside transport system in human erythrocytes
and
cardiocyte membranes and potentiate the cardiac actions of adenosine in the
dog.
CA 02347336 2001-05-31
3
Methylxanthines competitively antagonize the binding of adenosine to both the
A,AR and the A2AR. Certain naturally occurring methylxanthines such as
caffeine
and theophylline antagonize the cardiovascular effects of adenosine. For
example, the
administration of adenosine to patients receiving theophylline fails to
produce AV
block or terminate PSVT. However, those methylxanthines are relatively weak
and,
more importantly, are nonselective, antagonizing both the electrophysiological
and
vasodilatory effects of adenosine in laboratory animals and humans.
Theophylline also
ameliorates the non-cardiac effects of adenosine such as flushing, local pain,
and
respiratory stimulation.
Synthetic alkylxanthines, e.g., 8-cyclopentyl-1,3-dipropylxanthine (CPX; see
U.S. Patent Nos. 4,364,922 and 4,980,379), are significantly more potent and
selective
antagonists at the A,AR than are theophylline or caffeine.
Brief Summary of the Invention
The present invention concerns the discovery of certain novel compounds
which can bind to adenosine receptors with surprisingly high affinity,
specificity, and
selectivity. Specifically exemplified herein are xanthine and adenosine
analogues
comprising an epoxide moiety. As explained in more detail herein, these
adenosine
agonists and antagonists have therapeutic utility in a broad range of
applications
including cardiac and renal regulation. Included among these novel compounds
are
both adenosine agonists and antagonists.
In one embodiment of the subject invention, the novel compound known as
1,3-dipropyl-8- {3-oxatri cyclo [3.1.2.02°Joct-6(7)-yl } xanthine,
herein referred to asENX,
is used as an antagonist of adenosine. Advantageously, ENX has been found to
be
uniquely potent, specific, and highly selective for the A, adenosine receptor.
Particular enantiomers of the ENX compound were synthesized and tested for
their
relative activity. Testing of R-and S-enantiomers of ENX revealed advantages
of the
S-enantiomers, namely, potency and selectivity for the A,AR greater than those
of the
racemate or the R-enantiomer. However, the R-enantiomer, by virtue of-its
shorter
biological half life, can be advantageous in defined therapeutic applications
requiring
a short duration of action.
CA 02347336 2001-05-31
4
The subject invention further concerns other xanthines and adenosines
comprising an epoxide moiety in an exocyclic substituent. Further embodiments
of
the invention include compositions and formulations comprising ENX or those
analogues or derivatives which can have therapeutic utility as agonists or
antagonists
of adenosine.
A further aspect of the subject invention is a method for using the disclosed
compounds for modulating the biological activity of adenosine. The compounds,
or
compositions comprising those compounds, can be utilized for their modulating
effect
on adenosine, e.g., as agonists or antagonists of adenosine receptors. The
antagonist
activity of the subject compounds can be utilized in treating conditions where
elevated
levels of adenosine are present; the agonists can be useful where stimulation
of the
adenosine receptor is needed. Such conditions include, but are not limited to,
cardiac,
renal, hepatic, or lung diseases, such as cardiac arrhythmias, renal failure,
liver failure
ascites, and asthma. Modulating adenosine activity can also be used in the
treatment
of maturity onset diabetes.
Brief Description of the Drawings
Figure 1 is a scheme outlining the syntheses of 1,3-dipropylxanthines having
C-8 substituents that contain an epoxide moiety.
Figure 2 is a scheme for the synthesis of adenosine derivatives containing an
epoxide moiety.
Figure 3 shows synthesis of (2R)- and (2S)- and (2S)-endo-5-norbornen-2-
carboxylic acids.
Figures 4A-4D show selective antagonism of the negative dromotropic (S-H
interval prolongation) effect of adenosine (Ado) by ENX. Figures 4A-4B show an
analog record of the prolongation of the S-H interval (A, response, Figure 4A)
and the
increase in coronary conductance (AZ response, Figure 4B) caused by a 3 minute
infusion of adenosine (4 uM) in the absence and presence of 0.4 pM ENX. ENX
inhibited the negative dromotropic effect of adenosine, but did not antagonize
the
coronary vasodilation (increase in coronary conductance) caused by adenosine.
Figures
4C-4D show selective antagonism by ENX (0.4 pM) of the A, receptor-mediated
increase in the S-H interval caused by adenosine (4 pM), but not the Az
receptor
CA 02347336 2001-05-31
s
mediated coronary vasodilation. The values are the mean ~ SEM from five guinea
pig
hearts. The asterisk is indicated by those values significantly different from
adenosine
alone (P<0.05).
Figures SA-SD show a lack of effect of ENX on left ventricular pressure (LVP)
and dP/dt~~. Guinea pig hearts were atrial paced at a constant cycle length of
300
msec and exposed to progressively higher concentrations of ENX, i.e., 2 and
200 pM.
In the same hearts ENX alone caused no significant changes in the stimulus-to-
His
bundle interval (not shown). Identical results were obtained in three other
hearts.
Figure 6 shows the effect of ENX and isobutylmethylxanthines (IBMX) on
phosphodiesterase (PDE) activity in homogenates of DDT,MF-2 cells. The data
for
IBMX, shown as squares in the figure, clearly shows inhibition of
phosphodiesterase
activity. In contrast, phosphodiesterase activity following ENX
administration, shown
as circles in the figure, remained constant and showed no inhibition.
Figure 7 shows the specificity of action of ENX to antagonize the negative
dromotropic effect (S-H prolongation) of adenosine in guinea pig heart. The
effect of
ENX (2 nM, 2 uM) on similar S-H prolongation caused by adenosine (ADO, 4 pM),
magnesium (Mg2+, 3 mM), and carbachol (CCh 0.14 uM) was determined. The height
of each bar graph presents the mem t SEM of 4 experiments. Only the S-H
interval
prolongation caused by adenosine was antagonized by ENX.
Figure 8 shows accumulative urine output in rats intravenously given 0.1 mg/kg
of ENX (racemic) mixture; ENX (R-enantiomer); ENX (S-enantiomer); and a
vehicle
used as a control.
Detailed Description of the Disclosure
The subject invention pertains to novel compounds, and formulations
comprising those compounds, which can advantageously be used as either
agonists or
antagonists at adenosine receptors. Specifically, these compounds either
promote or
antagonize the negative dromotropic, chronotropic, and inotropic effects
mediated by
an A, adenosine receptor (A,AR). In the heart, these compounds can either
promote
or antagonize the negative dromotropic, chronotropic, and inotropic effects
mediated
by A,AR, and in the kidney the antagonists promote dieresis through an A,AR.
CA 02347336 2001-05-31
6
The subject compounds are of two .general types: (1) 1,3-dialkylxanthines
having C-8 substituents that comprise an epoxide (oxiranyl) moiety, and (2)
adenosines
having N-6 substitutents that comprise an epoxide moiety. In a preferred
embodiment
of the subject invention, the xanthine epoxides are 1,3-dialkylxanthines
having an
epoxide moiety covalently bound to the C-8 substituent of xanthine. The
preferred
epoxides of xanthine or adenosine are those having an epoxide moiety as part
of an
exocyclic substituent.
The general structure of one class of 1,3-dialkylxanthines is shown below as
Formula I:
R1 O
(I) ~~2)n-
C
R2
wherein R, and R~ are the same or different, and can be an alkyl group of 1-4
carbons
in length; and n = 0-4. It would also be understood that R, and/or RZ can be a
hydrogen. Compounds which have one of the R-groups as hydrogen and the other R-
group as m alkyl would be epoxides of alkyl xanthine; compounds having both R-
groups as alkyls are epoxides of dialkylxanthine.
The general structure of the 1,3-dialkyl-8-oxatricycloalkylxanthines is shown
below as Formula II:
Rl\N H
N
O~ ~N~~--(CHz)n R
(H) N O
Rz
wherein R, and R= are the same or different, and can be a hydrogen or an alkyl
group
of 1-4 carbons; R3 is either O or an alkyl group of 1-4 carbons; and n = Q-4.
A polymethylene chain 1-4 carbons in length can link the epoxide moiety to
C-8 of 1,3-dialkylxanthine, as in Formula I. The epoxide group can also be
part of
an exocyclic substituent linked to C-8 of the xanthine moiety, either directly
or
CA 02347336 2001-05-31
7
through a (poly)methylene chain 1-4 carbons long, as in Formula II. The
exocyclic
substituent, shown as Formula II, can be a bicycloalkyl group, forming an
oxatricycloalkyl substituent. Other exocyclic epoxide structures ran also be
part of
the compound as would be readily recognized by those skilled in the art having
the
benefit of this disclosure. The bicycloalkyl group can further contain an
alkenyl group
for the formation of a second epoxide moiety.
Figure 1 depicts a general synthesis scheme for the 8-substituted 1,3-
dipropylxanthines.
A preferred embodiment of the subject invention is a compound having the
chemical name 1,3-dipropyl-8-{3-oxatricyclo[3.1.2.OZ~']oct-6(7)-yl}-xanthine,
which is
commonly termed epoxynorbornylxanthine, or ENX. The formula for ENX is shown
as Formula III, below:
(~)
ENX has been demonstrated to have advantageous and unexpected properties as an
adenosine antagonist by its high selectivity and affinity for the A, adenosine
receptor.
Essentially, a patient who has any condition where levels of endogenous
adenosine are, or could become, excessive can benefit from therapeutic use of
the
subject antagonist compound or a composition comprising the compound. For
example, the subject invention pertains to the use of the subject antagonist
compounds
as diuretics or in the treatment of renal failure. In addition, the subject
antagonist
compounds or compositions comprising these compounds can be employed in the
treatment of certain conditions affecting the heart, including
bradyarrhythmias
associated with hypoxia or ischemia (myocardial infarction), sick sinus node
syndrome, and in heart failure, where the positive inotropic effect of the
antagonist can
be advantageous. Other conditions which are recognized as resulting from, ar
affected
by, elevated levels of endogenous adenosine can also be treated with the
subject
adenosine antagonists.
CA 02347336 2001-05-31
s
The high selectivity and affinity for A, adenosine receptor exhibited by the
subject compounds, e.g., ENX, make them particularly useful as diuretics. The
potency of ENX as a diuretic has been demonstrated to be at least as high as
the
potency of furosemide (Lasix), a commonly used diuretic in human and animal
medicine. Thus, it would be understood that ENX could be used in a manner
comparable to the way furosemide is used to produce a diuretic effect in a
patient.
The diuretic activity exhibited by ENX can be exploited in the treatment of
several conditions commonly affecting mammals, especially humans. For example,
congestive heart failure (CHF) is a condition in which diuretics are
extensively used.
Hypertension, often a concurrent condition with CHF, is also regularly treated
with
diuretics. ENX was shown to have comparable diuretic activity and potency as
currently marketed diuretics, e.s., Lasix* used for treatment of such
conditions. Thus,
the subject compounds, especially ENX, can be used in a similar manner for
treatment
of these conditions.
The subject adenosine antagonists can also be indicated as nephroprotecting
compounds. ENX, which has been shown to bind to the A, adenosine receptor, can
be used to block those receptors during the use of contrast agents known to be
nephrotoxic, or can be useful in treatments to counteract the nephrotoxic
effects of
certain antibiotics, e.g., gentamycin, amphotericin, or cyclosporin.
In addition, the subject A, adenosine antagonists, E~.g., ENX, can be useful
for
treatment of the ascites of liver failure. As would be readily understood in
the art,
ENX can be useful with certain modifications of treatment regimens and
indications
for non-transplant patients suffering from liver failure, pre-transplant
patients, or for
transplant patients having hepato-renal syndrome.
The activity as an adenosine A, receptor inhibitor and diuretic indicates that
the subject antagonist compounds, e.g., ENX, also can be used as an analgesic,
especially in the treatment of angina, claudication, and bradyarrhythmias
associated
with ischemia, hypoxia, or reperfusion. Also, the use of exogenously
administered
adenosine in cardiac diagnostic procedures, e.g., imaging of cardiac
vasculature, is
known to produce transitory side effects, including a brief onset of pain. As
this side
effect has been attributed to adenosine's binding to, and stimulation of, the
A~ receptor
(but not the A, receptor), an adenosine antagonist inhibiting the binding of
adenosine
*Trade-mark
CA 02347336 2001-05-31
9
to that A, receptor can be used to counteract the pain experienced by a
patient
undergoing the procedure. The subject compounds, including ENX, selectively
bind
to the A, adenosine receptor, inhibiting the binding of adenosine (and thus
blocking
or counteracting any side effect associated with the binding of adenosine to
the A,
receptor).
Further, the subject antagonist compounds, including ENX, can be used as a
bronchodilator, i.e., an antiasthmatic. ENX has been shown to relax tracheal
smooth
muscle, thus producing bronchodilation. This property is also common to other
much
weaker xanthine derivatives, e.g., theophylline. Such use of the subject
antagonist
compounds as an antiasthmatic treatment suggests that the compound can be
useful
when administered via an inhalation route.
Other routes of administration of the subject compounds can also be used. For
example, it is generally contemplated to administer the compounds according to
the
optimal route indicated for the condition being treated. Thus, the compounds
can be
administered intravenously, pen os, transdermally, etc., and in single or
multiple dosage
regimens, as would be understood by a person of ordinary skill in the art.
It would also be understood by ordinarily skilled artisans that the above-
described uses for the subject compounds can be optimized by using particular
isomers
which demonstrate different biological activities. Having a chiral center, ENX
is
recognized to exist in at least two enantiomeric forms. The'. ENX enantiomers,
namely, the S-enantiomer and the R-enantiomer, have been synthesized as the R-
and
S- isomers of 5-norbomene-2-carboxylic acid by methods available in the art.
See
Poll, T et al. (1985) TetrrrJiedron Lett. 26:3095-3098, and Poll, T. et al.
(1989)
Tetrahedron Lets. 30:5595-5598. The endo-R- and endo-S-enantiomers of ENX are
shown as Formulas IV and V, respectively.
CA 02347336 2001-05-31
5
U
IV V
Studies conducted on the two enantiomers of ENX show that both are selective
for the
A,AR as compared to the A2AR. The S-enantiomer has a longer duration of action
than the R-enantiomer. Although a racemic mixture of the R- and S-enantiomers
can
have the biological activity of either or both isomers, it is now understood
that the S-
and R- isomers can be used separately, as a single enantiomer, to effect
particular
advantageous activities of either enantiomer.
For example, about 80-90% of the biological activity demonstrated by a
racemic mixture of ENX is accounted for by the S-enantiomer. This result is
primarily due to the very short duration of activity by the R-enantiomer as
compared
to the duration of action exhibited by the S-enantiomer. The prolonged action
of the
S-enantiomer can be due to a slower clearance rate in the liver, e.g., slower
metabolic
degradation by enzyme systems such as cytochrome P4so. The S-enantiomer, which
showed slightly increased potency in vitro as compared to the R-enantiomer,
showed
substantially higher potency in vivo, and consequently higher selectivity for
the A,
adenosine receptor as compared to the Az receptor. See Example 4 for specific
data
comparing the selectivity and affinity properties of the S- and R-enantiorners
of ENX.
The advantageous properties, e.g., increased potency (in vitro and in vivo)
and
higher selectivity, as well as the longer duration of action exhibited by the
S
enantiomer, indicates that the S-enantiomer can be very useful as a diuretic
in animals
and humans. In most instances, as those exemplified above, the S-enantiomer
can be
the preferred compound because the length of its duration of activity, which
is more
CA 02347336 2001-05-31
11
than that of the R-enantiomer, can be critical to achieving its effect. In
other words,
the compound must at least cause an effect long enough to accomplish t1e
desired
result.
On the other hand, in instances where short duration of action are desired,
e.g.,
during intravenous infusion of adenosine or onset of myocardial ischemia, when
the
onset of increased adenosine levels is rapid and lasts only for a short period
of time
(on the order of seconds or minutes), an adenosine antagonist having a short
duration
of action, e.g., the R-enantiomer of ENX, can be advantageously used. The
activity
of the ENX R-enantiomer is beneficial for short periods of time. However, the
R-
enantiomer of ENX is rapidly degraded or metabolized. This rapid metabolism
can
prevent complications associated with drug interactions because the
concentrations of
the ENX R-enantiomer are rapidly decreased. Due to its analgesic properties,
the R-
enantiomer of ENX can be administered for the acute pain of angina.
Another application of the subject compounds having a short duration of action
is as an antiasthmatic or bronchodilator. It has been suggested that the high
biological
activity shown for the S-enantiomer of ENX is due to the rapid and selective
metabolism of the R-enantiomer of ENX in the liver. This can be due to a first-
pass
effect exhibited for the R-enantiomer when administered by routes in which the
drug
is degraded by liver enzymes prior to or at about the same time as it reaches
the
appropriate receptors where the pharmacologic effect is induced. However,
certain
other routes of administration can be advantageously used to exploit this
first-pass
effect. For example, the S- and R-enantiomers of ENX have been demonstrated to
be
bronchodilators. Administration of the R-enantiomer alone (or in a composition
comprising the R-enantiomer but not the S-enantiomer) by inhalation
immediately
presents the compound to the appropriate receptors in tle trachea and bronchi
to cause
its action. Any absorbed compound is rapidly eliminated, which reduces
residual
levels of the compound in the body.
Derivatives of adenosine containing an epoxide moiety, particularly those
having an epoxide moiety in an N-6 substituent, can be used as A,AR agonists.
Epoxide derivatives of adenosine agonists can also display high selectivity
for
adenosine receptors. High selectivity for cardiac tissue is also demonstrated.
More
CA 02347336 2001-05-31
12
specifically, N6-substitution of adenosine with epoxycycloalkyl groups can
result in
potent and tissue-selective agonists.
The N6-subregion of the A, adenosine receptor contains chiral recognition
sites
which can be important for the determination of A,/A2 selectivity. The epoxide
can
S be substituted as a cycloalkyl substituent, e.g., cyclopentyl, norbornanyl,
or
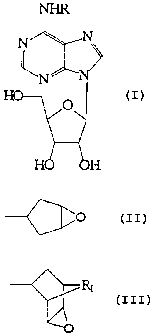
adamantanyl derivative of adenosine. Shown below as Formula VI is an adenosine
epoxide having the epoxide substituent at the N6 position. The epoxide can be
attached as a cyclopentyl or norbomanyl group.
N N~
y
N N
~ HO O
1S
H H
wherein R - -~p
or
~-R1
2S and R, = an alkyl group of 1-4 carbons. The compound can be one of four
isomers:
the 2R-endo, 2R-exo, 2S-endo, or the 2S-exo form.
Biological activity can also be enhanced by modifying other parts of the
cycloalkyladenosine molecule. For example, both 2- and S'-chloro substitutions
of
IV6-cycloalkyladenosines have been used to increase A, selectivity. Figure -2
shows
the steps involved in chemically converting an adenosine molecule or its
derivative
to an adenosine compound comprising an epoxybicycloalkyl group as an N6
substituent. Preferably, dimethyldioxirane is the oxidant used in the
formation of the
CA 02347336 2001-05-31
13
epoxide of the adenosine compound. See Iyer, R.S. et al. (1994) J. Am. Chenr.
Soc.
116:1603-1609. The dimethyldioxirane can be made according to methods and
procedures known in the art. See Murray, R.W., R. Jeyaraman (1985) J. Org.
Che»r.
50:2847-2853; Adam, W. et al. (1991) Chem. Ber. 124:2377.
The subject adenosine agonists can be useful for the treatment of a patient
where stimulation of ArAR is needed. Uses for the subject adenosine agonists
and
compositions comprising those agonists include their use as a functional ~3-
blocker; as
an antiarrhythmic agent for the control of heart rate, including
supraventricular
tachyarrhythmias, catecholamine (CAMP-dependent) supra- and ventricular-
arrhythmias; diabetes type II; and cardioprotection, e.g., decrease infarct
size and
increase tolerance to myocardial ischemia.
The compounds of the subject invention (agonists and antagonists) can be
formulated with a pharmaceutically acceptable carrier into a composition that
can be
administered to a patient who would benefit from the adenosine receptor
agonist or
antagonist properties of the subject compounds or compositions.
Advantageously, dosages of the subject adenosine antagonists for treating post-
resuscitation cardiac arrhythmias can be less than the 0.1-20 mg/kg range
which has
been previously reported for known adenosine antagonists. See U.S. Patent No.
4,980,379. An effective dose can be recognized as the dose at which the
alleviation
of bradycardia and reversal of hemodynamic collapse occurs.
Standard procedures for administration of adenosine antagonists such as
theophylline and aminophylline at effective do::.~ge levels are well
established and are
well known to those skilled in the art. For example, the recommended
therapeutic
range for plasma levels of theophylline for patients with reversible
obstruction of the
airways is from 10-20 pg/ml. The subject compounds, having high selectivity
and
potency, can be useful and effective at known concentrations in the blood.
The above list of treatment uses for the subject compounds or compositions is
by no means exhaustive, and other situations where the subject invention could
be
advantageously employed would be readily recognized by ordinarily skilled-
persons
in this art. For example, it would be readily recognized in the art that other
conditions
which can be treated by reducing the effects of elevated endogenous adenosine
or by
CA 02347336 2001-05-31
14
increasing stimulation of the A,AR can also benefit from the use or
administration of
the subject adenosine antagonists or agonists, respectively.
Following are examples which illustrate procedures, including the best mode,
for practicing the invention. These examples should not be construed as
limiting. All
percentages are by weight and all solvent mixture proportions are by volume
unless
otherwise noted.
Example 1 - Preparation of 8-Epoxyalkylxanthines
Chemistry. The scheme shown in Figure 1 outlines the syntheses of 1,3-
dipropylxanthines having C-8 substituents comprising an epoxide moiety. The
reaction of 5,6-diamino-1,3-dipropyluracil, 1, with an w-alkenoyl halide or an
w-
alkenoyl ester gave an amide 2, which was then cyclized in hot alkali to form
the 8-w-
alkenyl-1,3-dipropylxanthine 3. Oxidation with nr-chloroperbenzoic acid
yielded the
1 S 8-epoxyalkylxanthine 4. Alternatively, the Diels-Alder condensation of 3
with a 1,3-
cycloalkadiene generated an 8-bicycloalkenylxanthine 5. When furan was the
alkadiene the product was the 8-w-{7-oxabicyclo[2.2.1Jhept-2-en-S(6)-
yl)xanthine 5,
which contains both (a) an epoxide moiety and (b) an alkenyl moiety that can
serve
for the formation of a second epoxide moiety. The oxidation of 5 with 2.4
equivalents
of mesa-chloroperbenzoic acid gave the 8-epoxybicycloalkylxanthine 6.
1,3-dinropvl-8-(3-oxatric~clo(3 2 1 02~'loct-6 7)yl}xanthine. A solution of 8-
bicyclo(2.2.1]hept-2-en-5(6)ylxanthine (1.0 g, 3 mmol) and nr-chloroperbenzoic
acid
(0.8 g, 3.6 mmol) in 50 ml CHzCIz was stirred for 24 hours at room
temperature. A
second aliquot of peracid was added and stirring continued for 24 hours.
Evaporation
gave a yellow oil that was purified by preparative reverse phase HPLC on C-18
silica
eluted with a gradient of 70-80% methanol in water. Yield 0.54 g, 52%, mp 149-
150°C.
1_~3-dinronvl-8-(7-oxabicycloj2 2 llhept-2 en 5(6)vl}xanthine. A suspension
of 1,3-dipropyl-8-vinylxanthine (0.4 g, 1.5 mmol) in 50 ml dry THF containing
furan
, (0.22 ml, 3 mmol) was stirred at room temperature. The addition of 1 drop of
TMS
triflate effected solution, and HPLC showed the disappearance of starting
material.
CA 02347336 2001-05-31
Preparative reverse phase HPLC on C-18 silica eluted with a gradient of 50-80%
methanol in water yielded 0.25 g (50%) of product.
Example 2 -Preparation of an Adenosine Derivative Comprisin~poxide Moietv
A compound useful as an adenosine agonist is an adenosine derivative
comprising an oxabicyclo- or oxatricycloalkyl group as an N-6 substituent. A
general
scheme for the preparation of the compound is shown in Figure 2.
N6-endo-~3-oxatric~(3 2 1 0~~°loct 6(7) vl l adenosine. A solution
of N6
(endo-2-norbornene-5-yl)adenosine (0.5 g, 1.4 mmol) in 100 mL dry methanol was
10 cooled to 0-5°C in an ice bath, a solution of dimethyldiaxirane in
acetone (40 mL, 4
mmol) was added; stirring continued for 8 hours in the ice bath and then
overnight at
room temperature. Evaporation of solvent and purification by chromatography
yielded
0.42 g (81%) of a white solid.
15 Example 3 - Use of the Novel Compounds as Adenosine Anta onists
In order to demonstrate the effectiveness of the subject compounds as
adenosine antagonists, the activity of the compounds was compared to known
antagonists. In addition, the specificity, selectivity, and potency of ENX as
an A,
adenosine receptor antagonist, functional and biochemical (radioligand binding
assays)
experiments were carried out on guinea pig isolated hearts, in membranes from
guinea
pig brain, DDT,MF-2, and PC 12 cells. The results of these experiments are
described
below.
1. Functional studies. The functional evidence that an epoxide of
alkylxanthine (ENX) specifically and selectively antagonizes cardiac actions
of
adenosine mediated by A,-adenosine receptor but does not antagonize Az-
adenosine-
receptor mediated coronary vasodilation was obtained in the isolated perfused
guinea
pig heart. The effect of ENX and two other alkylxanthines (NAX and CPX) on the
A,-receptor mediated changes in stimulus-to-His bundle interval (S-H interval;
a
measure of AV nodal conduction) and on the A2 receptor mediated - coronary
vasodilatation were investigated. The potency of ENX, NAX, and CPX to
antagonize
the negative dromotropic (prolongation of S-H interval) of the A, agonist CCPA
and
vasodilatory effect of adenosine are shown in Tables 1 and 2.
CA 02347336 2001-05-31
16
Table 1. Potency of various alkylxanthines to antagonize A, receptor-mediated
cardiac response: results of Schild analysis.
ENX NAX CPX
PAz 8.45 ~ 0.19 8.79 ~ 0.15 8.76 10.02
3.6 nM 1.6 nM 1.7 nM
(1.2-3.9) (1.1-3.2)
(1.6-1.9)
Slope -0.91 ~ 0.06 -0.89 t 0.11 -0.81 ~ 0.03
n 4 3 3
Values are mean f S.E.M. of Uie PAz (-log,°Ke), the equilibrium
dissociation constant Ka, and the slope
of Schild plot. Cardiac response: antagonism of the negative dromotropic
effect of the selective A,
agonist CCPA. The numbers in parentheses arc the minimum and maximum KB
values. n = number of
experiments. Neither the PAS (Ka) nor the slope of Schild plots were
significantly different among the
antagonists.
.....
Table 2. Potency of various alkylxanthines to antagonize AZ receptor-mediated
coronary vasodilation.
ENX NAX CPX
ICso no effect 7.1 ~tM 1.5 uM
(0% at 50 uM) (4.8-9.4) (0.8-2.2)
n 4 3 3
Values are the concentration of antagonist that inhibits 50% (ICs°) of
a maximum coronary vasodilation
caused by adenosine. Values in parentheses are 95% confidence interval of the
IC~° values. n = number
of experiments.
Although all three alkylxanthines were equipotent in antagonizing the A,-
receptor
mediated prolongation of the S-H interval, ENX is far more selective than NAX
and
CPX.
To further demonstrate the selectivity of ENX for A, vs Az receptor,
measurements of A,-receptor mediated S-H interval and Az-receptor mediated
increase
m coronary. conductance were carried out during administration of adenosine
alone and
adenosine plus ENX (Figures 4A-4D). Adenosine (Ado, 4 uM), when administered
alone, produced a significant increase in S-H interval and coronary
conductance.
When adenosine was administered together with ENX (0.4 ~tM), the S-H interval
CA 02347336 2001-05-31
17
prolongation was completely inhibited, whereas the AZ-mediated coronary
vasodilation
remained unaltered. After washout of ENX, a third administration of adenosine
alone
caused a significant prolongation of S-H interval (similar to the first
administration of
adenosine) and increase in coronary conductance. These findings demonstrate
that the
S effects of ENX are reversible and that ENX antagonizes the A,-receptor
mediated S-H
prolongation but not the AZ-receptor mediated increase in coronary conductance
caused
by adenosine. These data also demonstrate the capability of ENX to inhibit
activity
(and thus any side effects) associated with the binding of adenosine to the A,
receptor
while the beneficial pharmacological activity of adenosine stimulation of the
A2
receptor remains unaffected.
To determine whether ENX had a positive isotropic effect, experiments were
conducted to determine its effects on left ventricular pressure (LVP) and its
first
derivative dP/dt, an index of contractility. As illustrated in Figure S, there
were no
significant changes in either LVP or dP/dt of normoxic guinea pig hearts when
these
1 S hearts were exposed to increasing concentrations of ENX (2-200 uM). LVP
and dP/dt
remained constant during the administration of varying concentrations of ENX
and
washout. These results demonstrate the lack of a positive isotropic effect of
ENX.
Consistent with the lack of positive isotropic effect, ENX also did not
inhibit
the enzyme phosphodiesterase (Figure 6). Cells were homogenized in 40 mM Tris
buffer at pH 8.0, and the whole homogenate was used in the enzyme assays. PDE
activity was determined by incubating homogenate (0.4 mg protein) in Tris
buffer
containing 20 mM MgCl2, 4 mM mercaptoethanol, 0.06 mg bovine serum albumin,
0.4
mM cAMP 130 nCi of ['H]CAMP and the indicated concentrations of ENX or IBMX
for 4S min at 30°C. Blank incubations were carried out in parallel
assays without the
homogenate. At the end of the incubation, the suspensions were incubated in a
boiling
water bath for 2 minutes, transferred to an ice-water bath for 2 minutes and
0.1 mg
of snake venom phosphodiesterase was added. The suspensions were incubated far
10 minutes at 30°C, and the adenosine formed was isolated by ion
exchange
chromatography. The control rate of adenosine formed was 220 pmol/mg protein
per
minute. The amount of adenosine formed was linear over the incubation period
used.
Agents that inhibit the enzyme phosphodiesterase are known to produce
positive isotropic effect. The results illustrated in Figure 6 clearly showed
that ENX
CA 02347336 2001-05-31
18
does not inhibit phosphodiesterase, whereas isobutylmethylxanthine (IBMX, a
known
positive isotropic agent) inhibits phosphodiesterase. 'these findings
demonstrate an
advantage of ENX over other alkylxanthines that are known to inhibit
phosphodiesterase, and therefore have the potential to produce a positive
isotropic
action.
Carbachol and MgCl2 were used to test the specificity of antagonism by ENX,
e.g., S-H interval prolongation mediated by adenosine As illustrated in Figure
7, ENX
(2 nM, 2 pM) did not antagonize the negative dromotropic effect of carbachol
or
MgCl2. In contrast, ENX did antagonize the S-H prolongation caused by
adenosine.
In summary, the results of the functional experiments described above
demonstrate that in the heart, ENX is a reversible, specific, and highly
selective
antagonist of adenosine at the A, receptor subtype.
2. Radiolieand binding studies. To determine the binding affinities of an
epoxide of alkylxantline, ENX, and compare to other alhylxanthines (CPX, NAX
and
CPT), radioligand binding experiments were carried out in membranes prepared
from
brain tissue, DDT,MF-2 and PC12 cell lines. The results of these experiments
are
illustrated in Tables 3 and 4. The results summarized in Table 3, below,
indicate that
in brain tissue, ENX is more potent than the other alkylxanthines at the A,AR,
whereas in DDT~MF-2 cell the binding affinity of the alkylxanthines for the A~
receptor are approximately the same. With regard to A2 receptors in PC12 cell
membranes, ENX was markedly less potent than CPX. In addition, the binding
affinity of ENX for the A, receptor, either brain or DD T,MF-2 cells, was
markedly
higher than that at the Ai receptor in PC-12 cell membranes.
CA 02347336 2001-05-31
19
Table 3. Binding affinities of alkylxanthines for the A,- and AZ-adenosine
receptors
in brain, DDT,-MF2 and PC-12 cell membranes
Alkylxanthine
Brain DDT,MFz PC-12
ENX 0.45 ~ 0.02 (5) 0.22 t 0.03 (5) 11,666 ~ 366 (4)
CPX 4.4 t 0.8 (4) 0.13 ~ 0.01 (4) 320 t 40 (3)
NAX 3.8 ~ 0.21 (4) 0.18 t 0.05 (3) _____________________
CPT 4 1.0 t 13.0 (4) _____________________ _____________
A, receptor binding was carried out with f HJCPX in guinea pig forebrain and
cardiac membranes, and in
intact DDT,-MFz cells. AZ receptor binding was carried out with f HJNECA in PC-
12 cell membranes.
Values are mean t SEM of triplicate determinations in each of several (n)
preparations. K; values were
calculated as described in methods. Abbreviations for the alkylxanthines arc
as follows: ENX = 1,3-
dipropyl-8-{3-oxatricyclo[3.1.2.0;'Joel-6(7)-yl)xanthine;CPX= 8-cyclopentyl-
1,3-dipropylxanthine;NAX
= 1,3-dipropyl-8-(3-noradamantyl)xanthine; and CPT = 8-cyclopentyl-1,3-
dimethylxanthine.
Additional radioligand binding studies have been carried out in guinea pig
forebrain (A, receptor) and striatum (A2 receptor) to demonstrate the greater
A,
receptor selectivity of ENX as compared to the previously known adenosine
receptor
antagonists, NAX or CPX. Table 4 shows A, and A, receptor bindine affinities
of
brain tissue expressing At (forebrain) and AZ (striatum) adenosine receptors.
The
results of Table 4 clearly illustrate that ENX is significantly more selective
for A, than
AZ receptors than the other alkylxanthines, NAX and CPX. That is, ENX was 800-
fold
selective for At vs. AZ, whereas NAX and CPX were only 20 and 7.5 fold
selective
for A, vs. A2, respectively. These results of these radioligand binding
studies are fully
consistent with that of the functional studies in guinea pig isolated hearts.
CA 02347336 2001-05-31
Table 4. Binding affinities of alkylxanthines for the At and A~ adenosine
receptor
in brain membranes
Alkylxanthine K~ (~)
A, (forebrain) Az (striatum) Ratio At/Az
S ENX 0.45 t 0.13 360 t 36 800
l.lOt0.lS 226.90 20
CPX 8.4 ~ 3.00 63 t 5.40 ~,S
A, and A= receptor binding was carried out with ('H]CPX and fH]CGS 21,860 in
guinea pig forebrain and
10 striatum, respectively. Values are mean t S.E.M. of triplicate
deterruinations in each of four preparations.
Example 4 - Activities of ENX Enantiomers
The S-enantiomer and R-enantiomer of ENX were synthesized, as described,
and tested for their relative activities and potencies. As shown in Table S,
below, the
1 S lower dissociation constant of the S-enantiomer of ENX suggests slightly
higher
potency (K; 0.98) as compared to the R-enantiomer (Kp=2.1 ).
Table 5. Equilibrium dissociation constants of ENX enantiomers and CPX for rat
brain A, adenosine receptors.
20 Compound K or nM
d K
[3H) CPX 0.49
R-ENX 2.1
S-ENX 0.98
2S
In addition, the S-enantiomer of ENX demonstrated higher binding selectivity
for the
At receptor. See Table 6, below.
CA 02347336 2001-05-31
21
Table G. Potency and selectivity of ENX to antagonize radioligand binding to
rat
brain adenosine A~ and AZ receptors ("ICso"* values)
Ax Selectivity
ENX (racemate) 1.65 nM 2.1 ~tM 1300
S-ENX 1.15 nM 9.0 ~tM 7800
R-ENX 2.70~nM 2.6 ~tM 960
"'ICso' refers to the concentration at which radioligand binding to receptors
was 50% inhibited.
The increased potency of the S-enantiomer is shown in Figure 8. As shown, most
of
the diuretic activity exhibited by ENX as a racemic mixture resided in the S-
enantiomer. Specifically, Figure 8 shows a cumulative urine output measured
for a
period of 2 hours in rats administered 0.1 mg/kg ENX racemate, ENX R-
enantiomer,
and ENX S-enantiomer. It is therefore shown that, as a diuretic, the S-
enantiomer of
ENX is more potent than the R-enantiomer of ENX or a racemic mixture of R- and
S-enantiomers of ENX. The duration of action is also longer for the S-
enantiomer of
ENX. These properties of the ENX S-enantiomer suggest its preferable use as a
long-
lasting diuretic in treating conditions normally calling for administration of
a diuretic.
Standard pharmacologic screening tests showed that the S-enantiomer of ENX
(100
mg/kg per os) relaxed constricted guinea pig tracheal muscle. The S-enantiomer
of
ENX reduced serum cholesterol and heparin precipitating ~i-lipoproteins in
mice after
100 mg/kg per os. Of interest, the observed reduction in HPL/CHOL ratio below
0.92
suggests a possible decrease in atherogenic low density (i-lipoproteins.
Saluretic activity associated with increased urine voltune output was observed
in the hydrated rat at doses of and above 3 mg/kg per os. Moderate kaluretic
activity
was also noted after 30 mg/kg per os in this preparation, suggesting potassium
sparing
diuretic activity.
The R-enantiomer was shown to have activity as an antagonist of adenosine.
Specifically, the R-enantiomer was observed to induce relaxation of
spontaneous tone
in guinea pig trachea. Saluretic activity associated with increased urine
volume output
was observed in the hydrated rat at 10 mg/kg per os of the ENX R-enantiomer.
However, the activity of the ENX R-enantiomer has a very short duration of
action as
CA 02347336 2001-05-31
22
compared to the S-enantiomer. However, that can be useful in treating
conditions that
indicate short-acting treatments.
Example 5 - Synthesis of N6-Substituted Adenosine Derivatives
The subject agonist compounds shown as Structure IV can be synthesized
according to known procedures. For example, the general synthesis scheme for
obtaining these compounds initially involves alkylation of an appropriately
substituted
amine, e.g., a bicyclic amine, with 6-chloropurine riboside. This
straightforward
reaction has been commonly used for the synthesis of N~-substituted
adenosines. See
WO 84 04 882 (1985).
The substituted amine can be functionalized with a double bond which can then
be oxidized to generate the epoxide product. nr-Chloroperbenzoic acid can be
used
for this oxidation reaction. See also Sharpless, K.B., W. Amberg, YL. Bennani,
G.A.
Crispino, J. Hartung, K.-S. Jeong, H.-L. Kwong, K. Morikawa, Z.-M. Wong, D.
Xu,
X.-L. Zhang (1992) J. Org. Chenr. 57:2768-2771; and Kolb, H.C., B.K. Sharpless
(1992) Tetrahedron 48:1015-1030.
An alternative method of generating epoxides is the osmium-catalyzed
dihydroxylation of olefins, which is now well known in view of the discovery
of
phthalazine ligands and that osmate ester hydrolysis is acceleration by
organic
sulfomamides. A simple, one-pot procedure for the conversion of vicinal diols
into
epoxides is known in the art (Kolb, H.C., B.K. Sharpless, supra). This
reaction
proceeds without epimerization via halohydrin ester intermediates. Combination
of
these methods allows epoxides to be obtained from olefins in a stereospecific
fashion.
The substituted amines which can be used for synthesis of the subject
compounds shown as Structure IV are 3-cyclopenten-1-yl amine (for the
cyclopentene
oxide derivative of adenosine) or 5-norbornen-2-yl amine (for the cyclohexene
epoxide
derivative of adenosine). 3-Cyclopenten-1-yl-amine can be synthesized from cis-
1,4-
dichlorobutene and diethyl malonate via a 5-step reaction sequence which is
known
in the art (Murdock, K.C., R.B. Angier [1962] J. Org. Chem. 27:2395-2398).
The synthesis of 5-norbomene-2-yl amine proceeds from 5-norbornene-2-
carboxylic acid, commercially available as a mixture of four isomers, 2R and
2S, each
endo and exo. Conversion of this carboxylic acid to acyl chloride, followed by
CA 02347336 2001-05-31
23
treatment with sodium azide, yields an acyl azide. Curtius rearrangement (loss
of NZ
and migration of the substituent group) and subsequent hydrolysis yields S-
norbornen-
2-yl amine as a mixture of isomers. This reaction sequence can be performed as
a
continuous operation without the isolation of the acyl azide or isocyanate in
the
synthesis of 4-aminocyclohexene. Another variation used for the Curtius
rearrangement involves the preparation of the acyl azide by treatment of the
corresponding acyl hydrazine with nitrous acid. In both cases, the
rearrangement
retains the absolute configuration at the chiral center. The endo and exo
components
can be separated by HI'LC methods known in the art.
The synthesis of the optically pure 5-norbornen-2-yl amines involves the use
of
asymmetric Diels-Alder reactions to obtain intermediate carboxylic acids,
followed by
a Curtius rearrangement as described above. A general scheme for synthesizing
these
compounds is shown in Figure 3.
Example 6 - Uses. Formulations and Administrations
Therapeutic and prophylactic application of the subject compounds, and
compositions comprising them, can be accomplished by any suitable method and
technique presently or prospectively known to those skilled in the art.
Further, the
compounds of the invention have use as starting materials or intermediates for
the
preparation of other useful compounds and compositions. The compounds of the
invention are useful for various non-therapeutic and therapeutic purposes. It
is
apparent from the testing that the compounds of the invention have effective
antiarrhythmic activity. Specifically, they are useful in regulating cardiac
arrhythmia,
including PVST, in animals and humans.
The demonstrated effects of both the agonists and the antagonists on cardiac
chronotropy, dromotropy, and inotropy make them useful therapeutically as
either
stimulants or modulators of cardiac performance, thereby affecting function of
the
heart. For example, the regulation or modulation activity of the subject
compounds
can affect heart rate (chronotropic effect) and impulse conduction
(dromotrapic
effect). The subject compounds can also be used diagnostically to determine
parameters of cardiac function, e.g., as pharmacological reagents useful in
determining
whether adenosine receptors are mediators of dysfunction of the heart or other
organs.
CA 02347336 2001-05-31
24
The subject compounds can also serve as standards for in vitro and in vivo
studies that measure or compare activities of other agonists and antagonists
that act
directly or indirectly through adenosine receptors. As reagents for such
comparisons,
the compounds are valuable pharmacological tools. Their high affinity and
selectivity
for the A, adenosine receptor make them important sources of information about
the
function of those receptors throughout the body.
Other uses for the subject compounds include their use in the characterization
of structure or location of adenosine receptors in organs or tissues. This can
be done
by, for example,' attaching an appropriate label or reporter to the subject
compounds
by standard techniques or procedures known to persons of ordinary skill in the
art.
The labels that are suitable for conjugation to the compounds of the subject
invention
include, but are not limited to, radiolabels (e.g., radioisotopes),
fluorescent labels, and
biotin labels. Radioisotopes that are suitable for labeling the subject
compounds
include Bromine-77, Fluorine-18, Iodine-131, Iodine-123, Iodine-125, Iodine-
126,
Iodine-133, Indium-111, Indium-113m, Gallium-67, Gallium-68, Ruthenium-95,
Ruthenium-97, Ruthenium-103, Ruthenium-1 O5, Mercury-107, Mercury-203, Rhenium-
99m, Rhenium-105, Rhenium-101, Technetium-99m, Tellurium-121m, Tellurium-99m,
Tellurium-125m, Thulium-165, Thulium-167, Thulium-168, and Tritium. The gamma-
emitting Indium species and Technetium-99m are preferred isotopes because
these
isotopes are detectable with a gamma-camera and have favorable half lives for
imaging in vivo. Alternatively, it would be recognized by those of ordinary
skill in
the art that non-radioactive labels, for example, enryme-substrate complexes,
e.g.,
biotin-avidin, horseradish peroxidase-alkaline phosphatase, and the like could
be used.
Also, fluorescent entities suitable for labeling the subject compounds include
fluorescein sodium, fluorescein isothiocyanate, and Texas red sulfonyl
chloride. As
such, the compounds can be used to visualize, in vitro or in vivo, structure
or function
of organs or tissues in which the A, adenosine receptors are present.
A further embodiment of the subject invention involves the use of the
compounds to direct therapeutic compounds to the A, adenosine receptor site.
Because of the specificity of the compounds of the subject invention, they can
be
conjugated to therapeutic compounds in order to direct the therapeutic
compound to
the vicinity of A, adenosine receptor. Also, in the case of compounds of the
subject
CA 02347336 2001-05-31
inventions which have selectivity to a specific type of tissue, such as heart
tissue,
these compounds can be used to direct therapeutic or diagnostic reagents to
those
locations.
The administration of the subject compounds of the invention is useful as an
5 antiarrhythmic agent. Thus, pharmaceutical compositions containing compounds
of
the invention as active ingredients are useful in prophylactic or therapeutic
treatment
of cardiac arrhythmias in humans or other mammals.
The dosage administered will be dependent upon the antiarrhythmic response
desired; the type of host involved; its age, health, weight, kind of
concurrent treatment,
10 if any; frequency of treatment; therapeutic ratio and like considerations.
Advantageously, dosage levels of the administered active ingredients can be,
for
examples, dermal, 1 to about 500 mg/kg; orally, 0.01 to 200 mg/kg; intranasal
0.01
to about 100 mg/kg; and aerosol 0.01 to about 50 mglkg of animal body weight.
Expressed in terms of concentration, the active ingredient of the invention
can
15 be present in the new compositions for use dermally, transdermally,
intranasally,
bronchially, intramuscularly, intravaginally, intravenously, or orally in a
concentration
of from about 0.01 to about 50% w/w of the composition, and especially from
about
0.1 to about 30% w/w of the composition. Preferably, the novel compound is
present
in a composition from about 1 to about 10% and, most preferably, the novel
20 composition comprises about 5% novel compound.
The compositions of the invention are advantageously used in a variety of
forms, e.g., tablets, ointments, capsules, pills, powders, aerosols, granules,
and oral
solutions or suspensions and the like containing the indicated suitable
quantities of the
active ingredient. Such compositions are referred to herein and in the
accompanying
25 claims generically as "pharmaceutical compositions." Typically, they can be
in unit
dosage form, namely, in physically discrete units suitable as unitary dosages
for
human or animal subjects, each unit containing a predetermined quantity of
active
ingredient calculated to produce the desired therapeutic or prophylactic
effect in
association with one or more pharmaceutically acceptable other ingredients,
e.g.,
diluent or carrier.
CA 02347336 2001-05-31
26
Where the pharmaceutical compositions are aerosols, the active ingredients can
be packaged in pressurized aerosol containers with a propellant, e.g., carbon
dioxide,
nitrogen, propane, etc. with the usual adjuvants such as cosolvents, wetting
agents, etc.
Where the pharmaceutical compositions are ointments, the active ingredient can
be mixed with a diluent vehicle such as cocoa butter, viscous polyethylene
glycols,
hydrogenated oils, and such mixtures can be emulsified if desired.
In accordance with the invention, pharmaceutical compositions comprise, as an
active ingredient, an effective amount of one or more non-toxic,
pharmaceutically
acceptable ingredient(s). Examples of such ingredients for use in the
compositions
include ethanol, dimethyl sulfoxide, glycerol, alumina, starch, calcium
carbonate, talc,
flour, and equivalent non-toxic carriers and diluents.
It should be understood that the examples and embodiments described herein
are for illustrative purposes only and that various modifications or changes
in light
thereof will be suggested to persons skilled in the art and are to be included
within
the spirit and purview of this application and the scope of the appended
claims.