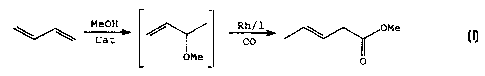Note: Descriptions are shown in the official language in which they were submitted.
CA 02347451 2001-04-17
WO 00/37411 PCT/US99/28247
TITLE OF THE INVENTION
PROCESS F'OR THE PREPARATION OF
BETA-GAMMA UNSATURATED ESTERS
BACKGROUND OF THE INVENTION
1. Field of the Invention:
The present invention relates to a process for the preparation of
beta-gamma unsaturated carboxylic acid esters by catalytic carbonylation
starting with either an allylic butentyl ether or mixture of butadiene and an
alcohol. More specifically but not by way of limitation, the invention relates
to
the use of a rhodium-containing catalyst in combination with an iodide
promoter for the carbonylation of methyl crotyl ether, 3-methoxybutene-1 arid
mixtures thereof to produce methyl-3-pentenoate.
1. Description o f Related Art
1:5 The use of rhodium-containing catalyst with various types of
co-catalysts and promoters for the carbonylation of a variety of saturated and
unsaturated organic compounds is generally known. For example, U.S. Pat.
No. 4,603,020 claims a process for the carbonylation of O-acetyl compounds,
such as acetic anhydride, at 130 to 250 °C using a rhodium catalyst and
an
aluminum accelerator. U.S. P;at. No. 4,625,058 teaches a similar process but
uses boron, bismuth or tertiary amide compounds as accelerators. U.S. Pat. No.
4,642,370 discloses a process for the carbonylation of hydrocarbyl halides
which uses a boron, silicon, aluminum, or zirconium accelerator. U.S. Pat. No.
4,563,309 claims a process far the production of carboxylic acid anhydride of
2~~ the fonmula RC(O)O(O)CCH; by reaction of a methyl carboxylate ester of
formula RC(O)OCH3 with carbon monoxide in the presence of a rhodium
catalyst and a phosphorous-containing ligand.
European patent application 0 428 979 A2 discloses the
carbonylation of allylic butenol.s and butenol esters using a rhodium
catalysts
CA 02347451 2001-04-17
WO 00/37411 PCT/US99/28247
' 2
and a hydrogen bromide or hydrogen iodide promoter under anhydrous
conditions for the production of 3-pentenoic acid.
BRIEF SUMMARY OF THE INVENTION
r~
In view of the above prior art, it has now been discovered that the
an allylic butenyl ether such as an alkyl crotyl ether or a corresponding
mixture '
of butadiene and an alkanol will readily undergo carbonylation directly to a
beta-gamma unsaturated carboxylic acid ester, such as alkyl-3-pentenoate, at
high selectivity and high activity by use of a rhodium-containing catalyst in
the
presence of an iodide-containing promoter.
Thus the present invention provides a process for the
carbonylation of allylic butenyl ether or mixture of butadiene and an alcohol
and the production of beta-gamma unsaturated carboxylic acid ester comprising
the steps of:
(a) reacting an allylic butenyl ether or mixture of butadiene and an
alcohol with carbon monoxide in the presence of a rhodium-containing
catalyst and an iodide-containing promoter; and
(b) recovering a beta-gamma unsaturated carboxylic acid ester.
In one embodiment of the invention the allylic butenyl ether is selected from
the group consisting of methyl crotyl ether, 3-methoxybutene-1 and mixtures
thereof and the beta-gamma unsaturated carboxylic acid ester is
methyl-3-pentenoate. In another embodiment, the carbonylation involves
reacting butadiene and methanol and the beta-gamma unsaturated carboxylic
acid ester is again methyl-3-pentenoate. Preferably, the iodide-containing
promoter is selected from the group consisting of HI, AlI3, SnI4, TiI4, CrI3,
and
CoI2 and the rhodium-containing catalyst is dicarbonylacetylacetonate
rhodium(I).
DETAILED DESCRIPTION OF INVENTION
The process of the present invention involves a rhodium catalyzed
carbonylation of an allylic butenyl ether to produce a beta-gamma unsaturated
CA 02347451 2001-04-17
WO 00/37411 PCT/US99/28247
3
carboxylic acid ester wherein the rhodium-containing catalyst is promoted by
the use of 1-II, HBr or a metal halide salt. The allylic butenyl ether being
earbonylated is either an alkyl crotyl ether, a positional isomer thereof such
as
3-alkoxybutene-1 or mixtures of the same. For purposes of the present
_'i invention, mixtures that produce the allylic butenyl ether or equivalent
in-situ
are equivalent staring materials and such butadiene in the presence of an
alkanol is to be considered an alternate starting material. The following
representative reaction showing; a butadiene in the presence of methanol under
catalytic reaction conditions producing the 3-methoxybutene-1 intermediate (a
positional isomer of methyl crotyl ether) which is then combines with carbon
monoxide to produce methyl pentenoate is illustrative of the overall
carbonylation.
MeOH Rh/1
y C~ / -~ ~ OMe
CO
O
IS
Suitable allylic bu.tenyl ethers are of the form
2
R
C=C H
R1 ~~~ OR4
R3
where one of the groups R', R2, and R3 is methyl and the other two groups are
H and wherein R4 is a C, to C,~, alkyl. It should be appreciated that
acceptable
ailylic compounds includes both cis and trans isomers, other positional
isomers
such as that illustrated by 3-alkoybutene-1 and crotyl ester as well as
mixtures
of various allylic compounds. Also, for purposes of the present invention,
butadiene in combination with a C, to Clo alkyl alcohol leads to in-situ ether
formation and thus total equivalency relative to the use of allylic butenyl
ether
OMe
as starting material for the carbonylation reaction.
CA 02347451 2001 04 17
~'~3~Z8'~~~
4
The reaction can be performed at a temperature in the
range of 40 °C: to about 200 °C. Below 40 °C the reaction
becomes too slow to
be commercially feasible, and above 200 °C the formation of undesirable
products leads to significant yield losses. Preferably the reaction
temperature is
between 90 °C: and 150 °C and most preferably between 90
°C and 150 °C.
Suitable total pressures for the reaction are in the range 0.1724 to
20.65 MPa (2,5 to 3,000 psi.g). Preferably the pressure is between 0.6895 to
13.79 MPa (100 and 2,000 psig) with 1.379 to 6.895 MPa (200 to 1,000 psig)
being most preferred.
i 10 'The source of 'the carbon monoxide (CO) reactant for the present
invention is not crucial. Commercially available grades of carbon monoxide
are acceptable. As such, the carbon monoxide can contain inert impurities such
as carbon dioxide, methane:, nitrogen, noble gasses, and other hydrocarbon
having up to four carbon atoms. Preferably the carbon monoxide also contains
hydrogen typically at about a ten mole percent concentration relative to the
carbon monoxide. At least 1 molar equivalent of carbon monoxide to allylic
butenyl ether or butadiene is needed. Typically, an excess of CO is used. .
Suitable solvents for this process are those which are compatible
with the reactants and the catalyst system under the reaction conditions. Such
solvents include aromatic hydrocarbon solvents, saturated halocarbon solvents,
and mixtures thereof. Carboxylic acid esters and lactones, such as methyl-3-
pentenoate, valerolactone and the like, are also acceptable solvents. Suitable
aromatic hydrocarbon solvents include benzene, toluene, 1,2,4-
trichlorobenzene and other CZ to C4 alkyl substituted benzenes. Suitable
saturated halocarbon solvent's include chlorinated and fluorinated
hydrocarbons
such as methylene chloride, dichloroethane and chloroform as well as the so-
called HCFC's including in particular HCFC-113 (FCC12CF2Cl) and HCFC-
123 (CHC12CF3) or the like. The most preferred solvents are toluene, HCFC-
113 and HCFC-123. Alternatively, the process of this invention where the
starting material is a methoxybutene may be run in the absence of solvent.
-=Panted 13 12 2000~< ,AMENDED SHEET ,,~~s
~. ._u. ., _... . __. ~ ~~::
CA 02347451 2001-04-17
WO 00/37411 PCT/US99/Z8247
S
The rhodium catalyst can be provided from any source or by any
material which will produce rhodium ions under the carbonylation reaction
conditions. Among the materials which can be employed as the source of the
rhodium catalyst are rhodium metal, rhodium salts, rhodium oxides, rhodium
_'~ carbonyl compounds, organorhodium compounds, coordination compounds of
rhodium, and mixtures thereof. Specific examples of such compounds include,
but are not limited to, RhCl3, RI.;, Rh(CO)ZI3, Rh(CO)I;, Rh4(CO),2,
Rh6(CO}16,
Rh(acac)3, Rh(CO)2(acac), Rh(CZH4}z(acac), [Rh(CZH4),Cl]2, [Rh(CO)ZCI]Z,
[Rh(CO)ZBr]Z, Rh(COD)(acac), [Rh(COD)CI]2, RhCI(CO)(PPh3)Z,
Rhz[OZC(CR2)6C'.H3]4, and Rh2(acetate)4, where acac is acetylacetonate, COD is
1,5-cyclooctadie:ne, and Ph is phenyl. Rhodium compounds containing
bidentate phosphorous or nitrogen ligands should be avoided. Preferred
sources of rhodium catalyst include rhodium(I) compounds such as
Rh(CO)Z(acac), [Rh(CO)2C1]Z, [Rh(COD)CI]2, Rh(COD)(acac), and rhodium
iodide compounds such as RhI;; and Rh(CO)~I3. Most preferably the rhodium-
c6ntaining compound is Rh(CO~)Z(acac).
Suitable concentrations of rhodium in the reaction are in the range
of 0.005 to 0.50 % by weight o,f rhodium metal based on the reaction medium.
Preferably the concentration of rhodium is in the range of 0.02 to 0.20 wt%.
Although high reaction rates o~n a per Rh basis can be obtained even at low
concentrations of Rh, it is generally more economical to operate at Rh
concentrations above 0.01 wt°/.. Similarly, Rh concentrations below 2.0
wt%
are preferred to minimize the formation of unwanted by-products.
The rhodium, which may be pre-formed or generated in situ, must
be promoted by HI, HBr or a metal halide, preferably by HI or a metal iodide,
to achieve a satisfactory rate and selectivity to pentenoate ester. Examples
of
suitable promoters are the acid halides as well as halides of Groups IIB,
IIIA,
IIIB, IVA, IVB, VIB, VII, VIII of the periodic table. Preferred promoters are
CA 02347451 2001-04-17
WO 00/37411 PCT/US99/28247
6
those where the halide is iodide, such as but not limited to HI, AII;, SnI4,
TiI4,
CrI3, and CoI2.
The molar ratio of promoter to rhodium can be in the range of
about 1:1 to about 50:1. Although high selectivities to the desired methyl-3-
S penetoate can be obtained even at low promoter to rhodium ratios, the rate
of
formation of methyl-3-pentenoate on a per Rh basis decreases significantly
when the molar ratio of promoter to rhodium is less than 1. This decrease in
reaction rates, coupled with the high cost of rhodium makes it more economical
to use promoter to rhodium ratios greater than 1:1. Similarly, the molar ratio
of
promoter to rhodium must be less than about 50 to obtain reasonable yields of
the unsaturated ester. Preferably, the molar ratio of promoter to rhodium is
between about 10 and about 30.
Reaction times can be varied and depend on choice of reactants,
solvent, catalyst and promoter as well as their respective concentration and
1 S reaction conditions such as temperature and pressure. Residence times of
the
order of about 1 minute to about 20 hours are acceptable.
The reaction of the present invention may be carried out in a
batch or continuous mode. The products can be isolated and recovered by any
of the techniques generally known in the art including by example but not
limited thereto, extraction, distillation or the like.
The following examples are present to further illustrate specific
features and advantages of the present invention and as such are not intended
to
limit the scope of the invention. The conversion data reported is based on
quantitative measurement of the relative amount of the primary or limiting
reactant (e.g., 3-methoxybutene-1 or alternatively butadiene) that is not
consumed by chemical reaction. The selectivity to the desired methyl-
3-pentenoate (M3P) is based on and reported as the amount of methyl ester -
produced relative to the amount of the primary reactant consumed by the
reaction.
CA 02347451 2001-04 17
. yl Y.,i",. . ~~~~ f ~ _
a~ SL.n ~~~ ' ~it~?~.a6.:~1"uw.s.!+~,X.1~ ,..#$5~'
.o '-~.~_.h4aw..~. Lr.~i':
Example 1
3-Methoxybutene-1 Carbonylation to M3P
with Rh Catalyst Promoted by Aluminum Iodide:
.A 120 mL mechanically stirred Hastelloy-C autoclave was
charged with 0.258 grams (0.1 mmole) of dicarbonylacetylacetonate
rhodium(I), 1.63 grams (4.0 mmole} of anhydrous aluminum iodide and 72.1
grams (83.4 mL) toluene. The reaction vessel was pressurized to 13.79 MPa
(400 psig) witlh a 90/10 mixture of CO and hydrogen. The solution was heated
IO to a temperature of 120 °C and the carbonylation reaction was
initiated by
injecting a solution of 8.6 grams (100 mmole) of 3-methoxybutene-1 and 1.0
grams of ortho-dichlorobenzene (ODCB, internal GC standard) in 5 grams of
toluene. The total pressure was then adjusted to 4.826 MPa (700 psig) with the
90/10 CO/HZ. Carbon monoxide and H2 (90/10 ratio) were continuously fed to
the autoclave :from a reservoir so as to maintain the total pressure constant
at
4.826 MPa (700 psig). Samples were removed at intervals for GC analysis on a
DBFFAP 30 M J&W Scientific capillary GC column. The analysis showed
that 62.5% of the methoxybutene charged was converted in the first hour and
the selectivity to methyl-3-pe;ntenoate (M3P; cis and traps isomers) was
93.9%.
After 4 hours the conversion was 98% and the selectivity to M3P was 93%.
The _ only significant by-products were mixed butenes and butadiene (not
separated, 5.2%), valerolactone (1.5%) and 3-pentenoic acid (0.3%). The first
order rate constant for the formation of M3P was 1.02 hr-1, corresponding to a
space-time yield (ST1~ of 734 mmole M3P per liter per hour.
Example 2
Methoxybutene Carbonylation to M3P With Rh Catalyst Promoted by
Aluminum Iodide (Higher Iodide/Rh ratio and Higher Temperature):
The experiment in Example 1 was repeated except that the
3-methoxybutene-1 was replaced with a 70/30 mixture of 1-methoxybutene-2
(methyl crotyl ether) and 3-m~ethoxybutene-1, the iodide to rhodium ratio was
~Pr~nteal 10Y12 2000 AMENDED SHEET '' r
_..~a_.,. .._ .,
CA 02347451 2001-04-17
WO 00/37411 PCT/US99/28247
8
increased from 1'?/ 1 to 18/ 1 and the temperature was increased to
130°C. The
GC analysis showed a methoxy6utene conversion of 85.8% after 30 minutes
and a selectivity to M3P of 82°ro. After 60 minutes the conversion was
97%
r
and the selectivity to M3P was 84.6%. The first order rate constant for the
formation of M3P was 2.96 hr~", corresponding to a space-time yield (STY) of
2,135 mmole M3P per liter per hour.
Example 3
Carbonylation of :3-Methoxybutene-1 using
a Rhodium Catalyst and aqueous HI Promoter:
A 2.5 mL glass lined pressure vessel was charges with 5 mL of a
solution containing 5.9 grams (E~9 mmol) of 3-methoxybutene-1 (3MB1), 0.258
grams (1.0 mmol) of dicarbon,ylacetylacetonate rhodium(I), 1.34 grams (6.0
mmol) of 57% aqueous HI solution, and 1.00 grams of o-dichlorobenzene
(internal gas chromatograph standard) in 100 mL of toluene. The pressure
1 S vessel was freed from air by purging first with nitrogen (twice) and then
with
carbon monoxide containing 10 mol% hydrogen {twice). The vessel was then
pressurized to 500 psig of 90/10 CO/HZ and heated to 120 °C with
agitation for
3 hours. The heat was shut off, i:he pressure vessel was allowed to cool to
room
temperature and the excess gases were vented. The product was analyzed by
gas chromatography on a DBFFAP 30 M J&W Scientific capillary GC column.
The results of the analysis are summarized below:
Before esterificatiun mmol/100 mL
. Butadiene 13.3
Methoxybutenes 0,g
3-Pentnoic acid 9.0
Methyl-3-pentenoate 54.9
Methoxybutene conversion was x)4.0%, selectivity to methyl-3-pentonate (M3P)
was 84.8% and selectivity to 3-pentenoic acid was 13.9%. Product accounting
was 99%.
CA 02347451 2001-04-17
WO 00/3741 I PCT/US99/28247
9
Example 4
In a manner analogous to the procedure employed in Example 3,
an additional nxn was performed except that 0.064 grams of methanol (MeOH)
4
per 100 grams of methoxybutene (2 equivalents per 100 equivalents) was
added. The resulting data for this example as well as the corresponding data
from Example 1 are presented in Table 1.
Example 5
The procedure employed in Example 4 was repeated except that
0.19 grams of .methanol per 1.00 grams of methoxybutene (6 equivalents per
1 ~0 100 equivalents ) was added. The resulting data are presented in Table 1.
Example 4
The procedure employed in Example I was repeated except that
the toluene solvent was replaced with the halocarbon HCFC-123 (CHC12CF3).
The resulting data are presented in Table 1.
I ~> Example 7
The procedure employed in Example 5 was repeated except that
the aqueous HI promoter was replaced with an equivalent amount of AII3 (2
equivalents of AlI3 per g-atom of Rh). The resulting data are presented in
Table 1.
Example 8
The procedure employed in Example 3 was repeated except that
the toluene solvent was replaced with the halocarbon HCFC-113 (FCC12CF2Cl).
The resulting data are presented in Table 1.
Example 9
2-''~ The procedure employed in Example 3 was repeated except that
the toluene solvent was replaced with the halocarbon HCFC-113 (FCCIzCF2Cl)
and the aqueous HI promoter was replaced with an equivalent amount of TiI4 (2
equivalents of TiI4 per g-atom of Rh). The resulting data are presented in
Table 1.
CA 02347451 2001-04-17
WO 00/37411 PCT/US99/28247
Table 1
Example Solvent Iodide MeOH Conversion Selectivity
Promoter per 100 to M3P
meq
(6I/Rh) substrate
5 3 Toluene aq HI 0 94.0 84.8
4 Toluene aq HI 2 93.7 87.0
5 Toluene aq HI 6 94.0 83.5
6 HCFC-113 aq HI 0 95.8 94.8
7 Toluene AlI3 6 80.8 97.2
10 8 HCFC-113 aq HI 0 95.4 91.2
9 HCFC-113 TiI4 2 17.8 67.6
Examples 10-1
S
The procedure employed was repeated
in Example 3 for a
series
of six additional except that iodide
runs the promoter
was varied
and the
resulting product carbonylation
was esterified
with methanol
to determine total
selectivity includingdibasic esters(DBE's;
esters
of adipic
acid,
2-
methylglutaric The results
and ethylsuccinic are summarized
acids). in Table
2.
Table 2
Example Iodide SelectivitySelectivitySelectivity
I/Rh Conversion
Promoter to M3P to M2P DBE's
10 anh', HI 3 33.2. 95.9 0 1.9
11 aq HI 3 22.1 94.9 1.3 1.4
12 aq HI 3 7.2 100 0 0
13 aq HI 3 22.0 94.0 0 1.6
14 AII3 3 97.2 10.1 53.3 36.6
15 aq HI 3 31.2 96.4 0 1.4
* anhydrous HI
in Acetic acid
CA 02347451 2001-04 17
PAP
.~ ~,~~.~~~~:
11
Example 16
Carbonylation of butadiene in the presence of methanol using
a Rhodium Catalyst and aqueous HI Promoter:
A 25 mL glass lined pressure vessel was charges with 5 mL of a
solution containing 5.4 gram:> ( 100 mmol) of butadiene, 1.34 grams (6.0 mmol)
of 57% aqueous HI, 3.2 grams methanol (100 mmole), 0.258 grams (1.0 mmol)
of dicarbonylacetylacetonate rhodium(I}, and 1.00 grams of o-dichlorobenzene
(internal gas chromatograph standard) in 100 mL of toluene. The pressure
vessel was freed from air by purging first with nitrogen (twice) and then with
carbon monoxide containing 10 mol% hydrogen (twice). The vessel was then
pressurized to .3.447 MPa (500 psig) of 90/10 CO/H2 and heated to 120
°C with
agitation for 3 hours. The heat was shut off, the pressure vessel was allowed
to
cool to room temperature and the excess gases were vented. The product
solution was analyzed directly by gas chromatography on a DBFFAP 30 M
J&W Scientific capillary CiC column. The results of the analysis are
summarized below in Table 3.
Examples 17-27
The procedure Employed in Example 16 was repeated for a series
;ZO of eleven additional runs except that the iodide promoter, the
temperature, and
the solvent were varied. The results are summarized in Table 3.
Table
3 s
Example Temp. Solvent Iodide I/RhConversionSelectivity
(C:} Promoter to M3P
16 120 Toluene aq HI 6 59.6 22.0
17 120 Toluene AlI3 6 74.2 46.6
18 120 HCFC-123 aq HI 6 60.6 44.5
19 12U HCFC-123 AlI3 6 83.0 32.7
20 140 Toluene AlI3 6 85.7 58.1
~Prmteci 13 12 200 AMENDED SHEET
CA 02347451 2001-04-17
WO 00/37411 PCT/US99/28247
12
Table 3(continued)
Example Temp. Solvent Iodide I/Rh Conversion Selectivity
21 140 HCFC'-123 AlI3 6 87.5 47.7
22 120 Toluene anh HI 3 62.9 28.0 ,
.'i 23 120 Toluene AII3 3 69.2 39.8
24 120 Toluene SnI4 6 52.6 19.3
25 120 Taluene CrI3 3 53.7 33.0
26 120 Toluene TiI4 3 63.6 41.8
27 120 Toluer,~e aq HI 3 60.5 29.8
1~~ The process of the present invention is useful for the preparation
of beta-gamma unsaturated carboxylic acid esters and in particular the
production of methyl-3-pentenoate by the carbonylation of methyl crotyl ether,
3-methoxybutene:-1 and their mixtures or directly from a mixture of butadiene
and methanol. Such products are useful as difunctional monomers and as
15 intermediates in the synthesis of adipic acid.
Having thus described and exemplified the invention with a
certain degree of particularity, it should be appreciated that the following
claims are not to be so limited but are to be afforded a scope commensurate
with the wording of each element of the claim and equivalents thereof.