Note: Descriptions are shown in the official language in which they were submitted.
CA 02349185 2001-05-29
11158-O1 CA Patent
Method and Device for Sample Introduction of Volatile Analytes
Field of the Invention
[1] The invention relates generally to an interface for use with a
spectroscopic detector
and, in particular to a simple thermal desorption gas introduction interface
for sample
introduction of volatile analytes into an atomic or organic mass spectroscopic
detector.
Background of the Invention
[2] Organometallic species are found in our environment either because they
are
naturally found there or because they have been introduced by anthropogenic
contributions. In general, these species are more toxic than their inorganic
salts - except
organoarsenic compounds - particularly because of their combined hydrophobic
and
lipophilic characteristics making them capable of entering biological cycles
with
detrimental consequences. For instance, methylmercury has attracted
considerable
attention from the scientific community due to its extreme toxicity.
Typically,
methylmercury enters the environment by direct release through abiotic
processes or via
methylation of inorganic mercury in biological systems. The latter process
provides an
alternative route for the bioaccumulation of methylmercury throughout many
food
chains. The resulting biomagnification of methylmercury has dramatic
consequences for
top predators such as humans. The best known example in recent history is the
Minimata
catastrophe in which over one hundred people died and marry more suffered
permanent
disability from high level exposure. Therefore, the need to perform
organometallic
speciation studies has become a topic of growing importance over the last
years.
[3] Generally, in analysis of samples such as, for example, ;>amples of soil,
tissue, water,
fly ash, for trace residues of analytes of interest from matrices it is common
to extract and
then enrich or concentrate the content of the analytes in order to achieve
better detection
capability. Commonly used enrichment methods are: simple concentration of a
dilute
solution containing organic analytes by reducing the content of the solvents;
liquid-liquid
or liquid-solid extraction, generally followed by concentration of the
extracts; gas-solid
extraction or purge and trap methods, generally followed by desorption of
analytes from
1
CA 02349185 2001-05-29
11158-O1 CA Patent
solids or traps; leeching/extracting of analytes from solid samples with an
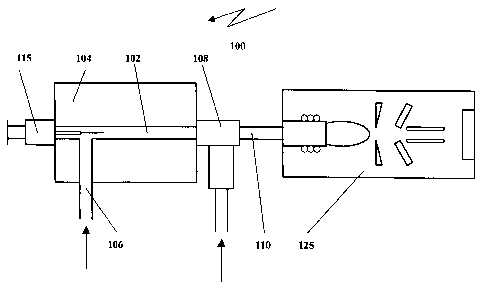
organic
solvent; and supercritical fluid extractions. However, these processes have
one or more of
numerous problems such as being difficult and time consuming, needing large
sample
volumes, having low detection power, or using organic solvents which present
problems
of disposability, toxicity and the like. Some of these common methods are
described in:
[4] M. Garcia, M. L. F. Sanchez, J. E. S. Uria and A. Sanz-:Medel, Mikrochim.
Acta,
1996, 122, 157;
[5] Alli, R. Jaffe and R. Jones, J. High Resolut. Chromatogr. , 1994,17, 745;
[6] E. M. S. Brito and J. R. D. Guimares, Appl. O~gahomet. Chem., 1999,19,
487;
[7] H. Hintelmann, Can. J. Anal. Sci. Spectrosc., 1998, 46, 182;
[8] E. Bulska, D. C. Baxter and B. Allard, Anal Chim. Acta, 1999, 394, 259;
and,
[9] G. Hu, X. Wrang, Y. Wrang, X. Chen and L. Jia, Ahal. .Lett., 1997, 30,
2579,
which are incorporated herein by reference.
[10] Therefore, a solid phase microextraction (SPME) process was recently
developed
by Janusz Pawliszyn to eliminate solvent use. The SPME process is disclosed,
for
example in International Patent (PCT) Publication WO 91/1L5745 of J.
Pawliszyn,
published Oct. 17, 1991, which is incorporated herein by reference. In the
SPME process
a coated or uncoated fiber housed within a needle of a syringe is brought into
contact
with components/analytes in a fluid carrier or headspace above the carrier for
a sufficient
period of time for extraction of the analytes to occur onto tl:~e fiber or
coated fiber.
Subsequently the fiber is removed from the carrier or heads,pace above the
carrier. The
analytes are desorbed from the fiber generally by thermal de;sorption into an
analytical
instrument such as a gas chromatograph (GC) for detection and quantification
of the
analytes. SPME has been shown to be a very useful sample preparation technique
for a
large variety of analytes. Detailed information about the SPME process and its
applications are described in the following references:
2
v i
CA 02349185 2001-05-29
11158-O1 CA Patent
[1l] Z. Mester, R. Sturgeon and J. Pawliszyn, Spectrochim. Acta, Part B, 2001,
56,
233;
[12] Z. Mester, J. Lam, R. Sturgeon and J. Pawliszyn, J. .Anal. At. Spectrom.,
2000,15,
837;
[13] M. Guidotti and M. Vitali, HRC-J. High Resolut. Claormatog~.,1998, 21,
665;
[l4] L. Moens, T. DeSmaele, R. Dams, P. VandenBroech and P. Sandra, Anal.
Chem.,
1997, 69, 1604;
[15] X. Yu, H. Yuan, T. Gorecki and J. Pawliszyn, Anal. Chem., 1997, 71, 2998;
[16] Z. Mester and J. Pawliszyn, Rapid Commun. Mass Spectrom., 1999,13, 1999;
[17] Z. Mester, H. Lord and J. Pawliszyn, J. Anal. At. Spectrom., 2000, 15,
595;
[18] Z. Mester, H. Lord and J. Pawliszyn, J. Chromatogr. A, 2000, submitted
for
publication,
[19] J. Pawliszyn, US Patent 5,496,741, Mar. S, 1996;
[20] J. Pawliszyn et al., PCT Publication WO 99/63335, Dec. 9, 1999;
[21] J. Villettaz et al., PCT Publication WO 99/26063, lVtay. 27, 1999; and,
[22] K. Rasmussen et al., PCT Publication WO 97/2560Ei, July 17, 1997,
which are incorporated herein by reference.
[23] A powerful tool for trace element speciation is an inductively coupled
plasma
mass spectrometer (ICP-MS). The ICP-MS has become an ideal instrument for the
speciation of organometallic compounds in complex environmental samples, which
require the high sensitivity and specifiicity detection provided by the ICP-
MS. Recently,
the ICP-MS has been combined with capillary GC for introducing the analyte to
the ICP-
MS, which is disclosed in the following references:
3
r
CA 02349185 2001-05-29
11158-O1 CA Patent
[24] H. Hintelman, R. D. Evans and J. Y. Villeneuve, J. .Anal. At. Spectrom.,
1995, 9,
619;
[25] T. DeSmaele, P. Verrept, L. Moens and R. Dams, Spectrochim. Acta, Part B,
1995, 11, 1409;
[26] L. Moens, T. DeSmaele, R. Dmas, P. VanDenBroeck and P. Sandra, Anal.
Chem.,
1997, 69, 1604;
[27] N. S. Chong and R. S. Houk, Appl. Spectrosc., 198 i', 41, 66;
[28] G. R. Peters and D. Beauchemin, J. Anal. At. Spectrom., 1992, 7, 965;
(29] W. Kim, M. E. Foulkes, L. Ebdon, S. J. Hill, R. L. Patience, A. G.
Barwise and S.
J. Rowland, J. Anal. At. Spectrom., 1992, 7, 1147;
[30] W. J. Pretorius, L. Ebdon and S. J. Rowland, J. Chromatogr., 1993, 646,
369;
[31] T. DeSmaele, L. Moens, R. Dams, P. Sandra, J. Var~dereycken and J.
Vandyck, J.
Chromatogr. A, 1998, 793, 99;
[32] S. M. Gallus and K. G. Heumann, J. Anal. At Spectrom., 1996, 11, 887;
[33] J. Poehlman, B. W. Pack and G. M. Hieftje, Am. La~~., 1998, 30, C50; and,
[34] M. M. Bayon, M. G. Camblor, J. I. G. Alonso and A. Sanz-Medel, J. Anal.
At.
Spectrom. , 1999, 14, 1317,
which axe incorporated herein by reference.
[35] However, this method needs to couple a relatively bulky and expensive
device -
GC - to the ICP-MS. Furthermore, this coupling is not straightforward. There
are several
limitations, probably the most important one being that the analytes have to
be
maintained in gaseous form during transport from the GC to the ICP-MS,
avoiding any
condensation effect at the interface. Apart from this fact, the effluent from
the GC
requires an aerosol carrier gas to achieve sufficient flow to ~;et the
analytes into the
4
CA 02349185 2001-05-29
11158-O1 CA Patent
central channel of the plasma. Numerous problems still remain to be solved in
the
development of appropriate interfaces able to provide reliable and
reproducible results to
carry out trace element speciation in environmental samples.
[36] It is, therefore, an object of the invention to overconne the drawbacks
of the prior
art by directly coupling SPME with ICP-MS.
[37] It is further an object of the invention to provide a siimple thermal
desorption gas
introduction interface for sample introduction of volatile analytes into an
atomic
spectroscopic detector.
[38] It is another object of the invention to provide an interface of compact
design for
direct placement at the base of the plasma torch.
Summary of the Invention
[39] Fundamental to a system for combining SPME with an atomic spectroscopic
detector is the development of an effective method and device for introducing
an analyte
sampled with the SPME into the atomic spectroscopic detector. The interface
between the
SPME and the atomic spectroscopic detector such as an ICI'-MS serves a dual
function -
liberating the analyte from the extraction phase of the SPM'.E through a
desorption
process and transferring the desorbed analyte to the ICP-MS. Reliable and
reproducible
measurements require a rapid desorption process and a highly efficient
transfer of the
analyte to the atomic spectroscopic detector without substantial loss of
analyte.
[40] In accordance with the present invention there is provided a method for
introducing a volatile analyte into an atomic spectroscopic detector
comprising the steps
of:
providing an injector connected to the atomic spectroscopic detector via a
first port, the
injector comprising a sealed second port for inserting the volatile analyte
bound to an
extraction phase;
providing a heating block surrounding the injector;
heating the injector to a predetermined temperature using th~.e heating block,
wherein the
injector is kept substantially at the predetermined temperature after the same
is reached;
CA 02349185 2001-05-29
11158-O1 CA Patent
providing a carrier gas flow of inert gas into the injector through a third
port of the
injector, the third port being located in proximity to the second port;
providing an auxiliary gas flow of inert gas between the third port and the
atomic
spectroscopic detector;
inserting the volatile analyte into the injector, wherein the volatile analyte
is bound to an
extraction phase, and wherein the extraction phase is inserted through the
seal of the
second port for sealed exposure of the extraction phase within the injector;
exposing the extraction phase of the fiber;
rapidly thermally desorbing the volatile analyte from the extraction phase
through
application of the heat and provision of the carrier gas flow;
transporting the volatile analyte to the atomic spectroscopic detector in a
gas flow
comprising the carrier gas flow and the auxiliary gas flow; <~nd,
retracting the extraction phase after a predetermined time ir.~terval has
elapsed.
[41] In accordance with the present invention there is further provided a
method for
introducing a volatile analyte into an atomic spectroscopic detector
comprising the steps
o~
providing an injector connected to the atomic spectroscopic detector, the
injector
comprising a sealed port for inserting the volatile analyte bound to an
extraction phase;
heating the injector to a predetermined temperature;
providing a carrier gas flow of inert gas;
inserting the extraction phase into the injector through the seal for sealed
exposure of the
extraction phase within the injector;
thermally desorbing the volatile analyte through application. of the heat and
provision of
the carrier gas flow; and,
transporting the volatile analyte to the atomic spectroscopic detector in the
Garner gas
flow.
[42] In accordance with an aspect of the present invention there is provided a
thermal
desorption interface for introducing a volatile analyte into an atomic
spectroscopic
detector comprising:
CA 02349185 2001-05-29
11158-Ol CA Patent
an injector connected to the atomic spectroscopic detector via a first port,
the injector
comprising a sealed second port for sealed insertion of the volatile analyte
bound to an
extraction phase;
a third port interfaced with the injector in proximity of the second port for
provision of an
inert carrier gas;
a heating block surrounding the injector for heating the injector to a
predetermined
temperature and keeping the injector substantially at the predetermined
temperature after
the same is reached, the temperature being sufficient for raI>idly thermally
desorbing the
volatile analyte; and,
a fourth port interfaced between the third port and the atomic spectroscopic
detector for
provision of an auxiliary gas flow of inert gas.
[43] In accordance with the aspect of the present invention there is further
provided a
thermal desorption interface for introducing a volatile analyte into an atomic
spectroscopic detector comprising:
an injector connected to the atomic spectroscopic detector via a first port,
the injector
comprising a sealed second port for sealed insertion of the volatile analyte
bound to an
extraction phase;
a third port interfaced with the injector in proximity of the second port for
provision of an
inert carrier gas; and,
a heating mechanism surrounding the injector for heating the injector to a
predetermined
temperature and keeping the injector substantially at the predetermined
temperature after
the same is reached, the temperature being sufficient for rapidly thermally
desorbing the
volatile analyte.
[44] In accordance with another aspect of the present invention there is
further
provided a thermal desorption interface for introducing a volatile analyte
into an atomic
spectroscopic detector comprising:
a plurality of measurement units movably attached to a transport mechanism for
consecutively moving the plurality of measurement units into a fitted position
with
respect to the atomic spectroscopic detector and for removing the same from
the fitted
position into another position, each measurement unit comprising:
CA 02349185 2001-05-29
11158-Ol CA Patent
an injector comprising:
a first port for connecting to the atomic spectroscopic detector;
a sealed second port for sealed insertion of the volatile anal;yte bound to an
extraction
phase;
a third port in proximity of the second port for provision of an inert carrier
gas; and,
a fourth port interposed between the third port and the first port for
provision of an
auxiliary gas flow of inert gas;
a heating block surrounding the injector for heating the injector to a
predetermined
temperature and keeping the injector substantially at the predetermined
temperature after
the same is reached, the temperature being sufficient for rax>idly thermally
desorbing the
volatile analyte;
a holding mechanism for holding a SPME unit, for moving the same in a linear
fashion
and for moving a plunger of the SPME unit;
a first conduit being interfaced with the third port of the injector of a
measurement unit if
the measurement unit is in the fitted position for provision of the carrier
gas; and,
a second conduit being interfaced with the fourth port of thc; injector of a
measurement
unit if the measurement unit is in the fitted position for provision of the
auxiliary gas
flow.
Brief Description of the Figures
[45] Exemplary embodiments of the invention will now be described in
conjunction
with the following drawings, in which:
(46] Figure 1 is a simplified block diagram of a SPME writ according to the
prior art;
[47] Figure 2a is a simplified block diagram of a thermal desorption interface
according to the invention;
[48] Figure 2b is a simplified flow diagram of a method j:or introducing a
volatile
analyte into an atomic spectroscopic detector according to tlhe invention;
[49] Figure 2c is a simplified block diagram of an embodliment of a thermal
desorption
interface according to the invention;
CA 02349185 2001-05-29
11158-Ol CA Patent
[50] Figure 3 is a simplified diagram illustrating the influence of the
auxiliary gas flow
on the measurement;
[51] Figure 4 is a simplified diagram illustrating a measwrement signal of an
ICP-MS
using the interface according to the invention;
[52] Figure 5 is a simplified block diagram of an embodiment of a thermal
desorption
interface according to the invention;
[53] Figure 6 is a simplified block diagram of another emibodiment of a
thermal
desorption interface according to the invention;
[54] Figure 7 is a simplified block diagram of yet another embodiment of a
thermal
desorption interface according to the invention; and,
[55] Figure 8 is a simplified block diagram of a further embodiment of a
thermal
desorption interface according to the invention.
Detailed Description of Preferred Embodiments
[56] In the following, the invention will be described for sample introduction
of
volatile analytes - in particular volatile metal species - into <tn ICP-MS,
wherein the
analyte is sampled using a SPME sampling process. However, it will become
evident to
persons of skill in the art that the invention is not limited thereto but is
applicable for
introduction of volatile or semi-volatile analytes sampled u:cing any existing
or future
sampling process based on absorption or adsorption of an aaaalyte.
Furthermore, the
invention is applicable for introduction of an analyte into any atomic or
organic mass
spectroscopic detector.
[57] Traditional sample preparation methods are typicall~~ time consuming,
employ
mufti-step procedures having high risk for loss of analytes and use extensive
amounts of
organic solvents. These characteristics make such methods 'very difficult to
automate and
integrate into modern sampling/separation systems. As a consequence, most of
the
analysis time is consumed by sampling and sample preparation. Extensive use of
organic
solvents in analytical laboratories is no longer tolerated bec;~use of the
associated health
risks and disposal concerns. The SPME process overcomes these drawbacks. In
the
SPME process the analyte is attached to the extraction phase deposited on a
fiber either
CA 02349185 2001-05-29
11158-O1 CA Patent
through adsorption or absorption. SPME is used in liquid or gaseous matrices
and
primarily aims for partial or equilibrium extraction of the aJnalyte. The
principal approach
of SPME is the use of a small volume of extraction phase, usually less than 1
,u1. The
extraction phase is a high molecular weight polymeric "liquid" or a solid
sorbent,
typically a high surface area porous material. Fig. 1 illustrai;es
schematically the basic
structure of a commercially available SPME unit. A small diameter fused-silica
fiber 2,
coated with the extraction phase is mounted in a syringe-like device 6 for
protection and
ease of handling. The needle 4 serves to conveniently pierce septa during
sample
extraction and desorption operations. Using a plunger 8 of i:he syringe-like
device 6 the
fiber is extruded from the needle to expose the extraction plhase to the
sample. After the
sampling period the same mechanism is used to retract the ~~ber inside the
needle. During
the extraction and desorption periods the fiber is exposed b;y being outside
the needle,
during transfer of the SPME unit to a desorption apparatus, the fiber end with
the
extraction phase is inside the needle for protection.
[58] Fundamental to a system combining SPME with an atomic spectroscopic
detector
is the development of an effective method and device for introducing an
analyte sampled
with the SPME into the atomic spectroscopic detector. The interface between
the SPME
and the atomic spectroscopic detector such as, an ICP-MS serves a dual
function. First,
the analyte is liberated from the extraction phase of the SP11~IE through a
desorption
process. Second, the desorbed analyte is then transferred to the ICP-MS. For
reliable and
reproducible measurements it is preferable that the desorption process is
rapid and the
transfer highly efficient without substantial loss of analyte.
[59] Referring to Figs. 2a and 2b, a thermal desorption interface 100 and a
desorption
method according to the invention is shown. The thermal de~sorption interface
100
comprises an injector 102 such as a sealed glass-lined splitless GC injector
for interfacing
a SPME unit 115. For example, the injector 102 is sealed with a septum, which
is
penetrated by needle 4 of the SPME unit for inserting fiber :2 into the
injector 102.
Sealing of the injector 102 is necessary in order to prevent a~tnbient air
from entering the
injector during the desorption process. The injector 102 is placed in a heated
block 104
and connected to the base of the ICP torch of an ICP-MS 125 via a Swagelok "T"
108
to
CA 02349185 2001-05-29
11158-O1 CA Patent
and a transfer line 110 comprising, for example, Teflon tubing. The inner
diameter of the
injector 102 and the transfer line 110 is approximately 2 mtn. A volatile
analyte
contained in the exposed extraction phase of the fiber of the; SPME unit 115
is thermally
desorbed into a carrier stream of an inert gas such as Ar, entering the
injector 102 through
conduit 106 and flowing into the ICP-MS. Optionally, the .carrier gas is
heated prior to
entering the injector 102 to increase the speed of the desorption process.
Preferably, the
conduit 106 is connected to the injector at a location of the exposed
extraction phase of
the fiber close to the needle of the SPME unit 115 or at a location of the
needle for
optimum desorption. The heated block 104 comprises, for example, an
electrically heated
A1 block. Preferably, the heated block 104 comprises a thermal insulation for
reducing
heat loss and has a sufficient size in order to keep temperature variations of
the device
during operation at a minimum. Of course, there are numerous other methods for
heating
the injector as is obvious to a person of skill in the art. The heating block
104 provides
enough heat to keep the injector 102 at a temperature high enough for rapidly
releasing
the analyte - typically between 200 °C and 250 °C - and for
heating the injector 102.
Heating of the injector 102 minimizes condensation of the analyte and reduces
interaction
of the analyte with the wall of the injector 102 and the transfer line 110 in
order to
minimize sample loss that is otherwise severe with analytes such as, for
example, methyl
mercury. Optionally, the transfer line 110 is heated separately. An auxiliary
inert gas such
as Ar is introduced via the Swagelok "T" 108 placed betwe<~n the injector 102
and the
transfer line 110 to accommodate a gas flow needed for efficient transfer of
analyte from
the fiber to the plasma and subsequent sampling into the mass spectrometer
125.
Preferably, a same inert gas is used for the carrier gas and the auxiliary
gas.
[60] The carrier gas is heated using a separate heating device - not shown -
prior
entering conduit 106. Preferably, the temperature of the cannier gas does not
exceed the
temperature of the injector 102.
[61] In an alternative embodiment of the thermal desorption interface 100 the
carrier
gas is heated through the heating block 104 while passing through conduit 106,
wherein
the conduit 106 has a sufficient length for heating the carrier gas to a
predetermined
temperature.
11
CA 02349185 2001-05-29
11158-O1 CA Patent
[62] Optionally, also the auxiliary gas is heated in order to prevent
condensation of the
analyte in the transfer line 110. Further optionally, the transfer line 110 is
omitted by
directly connecting the Swagelok "T" to the ICP-MS reducing the transfer
length of the
analyte.
[63] Further optionally, the injector 102 is connected directly to the ICP-MS
omitting
the Swagelok "T" and the transfer line 110, as shown in Fig;. 2c. By
substantially
reducing the distance between the exposed fiber and the ICIP-MS to a minimum
it is
possible to transfer the analyte without provision of an auxiliary gas flow
resulting in a
simpler interface compared to the device shown in Fig. 2a. :However, this is
not a
preferred embodiment because the omission of the auxiliar3r gas flow
substantially
reduces the ability to adjust the gas flow into the ICP-MS.
[64] Variation in the auxiliary gas flow rate effectively alters the sampling
depth in the
plasma. The optimum position is reflecting a balance between atomization -
ionization
processes and subsequent dispersion/neutralization of the malyte. As
illustrated in Fig. 3,
the effect of the auxiliary gas flow shows an optimum.
[65] In a preferred mode of operation the septum sealed injector 102 and the
transfer
line 110 are first flushed with an inert gas, preferably the carrier gas, in
order to provide
contaminant free conditions for reproducible measurements. The injector 102 is
heated to
a predetermined temperature while being flushed with inert gas. Provision of
the inert gas
and heating of the injector 102 substantially removes surface contaminants
attached to the
wall of the injector 102, for example, air molecules adsorbed at the surface.
After
reaching the predetermined temperature, the SPME fiber 2 i.s inserted through
the septum
seal while being still protected by the needle 4. Using the plunger 8 the
extraction-phased
portion of the fiber 2 is then exposed for a predetermined time interval and
is then
withdrawn after elapse of the time interval. The desorption temperature of the
injector
102, the exposure time of the fiber 2 and the temperature anal the flow rate
of the carrier
gas as well as the selection of the carrier gas depend on the sampled analyte,
the amount
of analyte and the type of extraction phase used. Generally, this involves a
calibration
12
CA 02349185 2001-05-29
11158-O1 CA Patent
process, which is performed preferably using the same fiber, which is then
also used for
the measurements.
[66] For example, an applied desorption temperature of 250 °C resulted
in a rapid
release (3 - 4 s transient peak width) for methyl mercury, as~ shown in Fig.
4. An exposure
time of 40 s resulted in a complete clean up of the fiber for samples in a ng
m1-1
concentration range, i. e. all the analyte has been desorbed. This allows
reuse of the fiber
without additional clean up steps. A carrier gas - Ar - flow rate of 35 ml/min
and an
auxiliary gas - Ar - flow rate of 280 ml/min were provided. Test data
generated using the
device and method according to the invention indicate good agreement with
certified
values. In particular, the relative standard deviation is small. due to the
very simple and
reproducible sample handling procedures involved. Furthermore, tests have
shown that
the introduction of analytes using the thermal desorption interface according
to the
invention resulted in measurement data having a very low associated background
or
noise, as shown in Fig. 4.
[67] Referring to Fig. 5 another embodiment of a thermal desorption interface
200
according to the invention is shown. Here, the wall of injector 202 has two
openings for
interfacing conduits 206 and 208 for provision of the carrier gas and the
auxiliary gas
flow. Furthermore, the injector is directly connected to the I:CP-MS 225.
Preferably, the
injector 202 is a sealed glass-lined splitless injector fitted snugly into a
cylindrical
opening of heating block 204. This allows easy removal of the injector 202
after use for
cleaning or disposal. For example, the interface 200 is affixed to the ICP-MS
225. In
preparation of a measurement a sealed injector 202 is inserted into the
heating block 204.
After the measurement the used injector 202 is removed and replaced with a new
or
cleaned and sealed injector 202. Therefore, preparation time; is substantially
reduced
allowing amore efficient use of the ICP-MS 225 and the interface 200.
Optionally, the
carrier gas is heated using heating block 204 or an external :heating device.
Further
optionally, the auxiliary gas is heated too. As is obvious to a person of
skill in the art,
there are numerous materials for producing the injector. Preferably, a
material is selected
that has relatively good heat conductivity and has substantially no
interaction with the
analyte.
13
CA 02349185 2001-05-29
11158-O1 CA Patent
[68] Referring to Fig. 6, a variation 300 of the thermal desorption interface
200
according to the invention is shown. Injector 302 of the thermal desorption
interface 300
comprises conduit 306 for provision of the carrier gas and conduit 308 for
provision of
the auxiliary gas in one mit. All ports 330, 332, 334, and 3:36 of the
injector 302 are
sealed. A manufacturer fills the injector 302 to a predetermined pressure with
an inert gas
such as Ar. In operation the injector 302 is interposed between two heater
block portions
304 A and 304 B held together by, for example, a clamping mechanism 305. Then
needle
like conduits 340, 342 and 344 are inserted through the seals of the ports
334, 336, and
332, respectively, and the interface 300 is ready for operation as disclosed
above but
without the steps of removing contaminants. In order to allow continuous
operation of the
ICP-MS a continuous gas supply, not shown in Fig. 6, is provided to the ICP-MS
during
installation or removal of the injector, as is evident to a person of skill in
the art. Using
the sealed and pre-filled injector 302 ensures reliable and reproducible
measurements by
substantially reducing the risk of having contaminants attached to the walls
of the injector
and the conduits. After use the injector 302 is disposed of and, preferably,
recycled by the
manufacturer. This allows very quick and efficient preparation of measurements
and
leads the way to automation.
[69] Referring to Fig. 7, another embodiment of a thermal desorption interface
400
according to the invention is shown. In order to provide means for automation
of the
measurements the thermal desorption interface 400 comprises a digital signal
processing
unit 420. A processor 424 is connected via a D/A and A/D converter 422 to
heating
elements 405 and 411, respectively, for controlling heating of the heating
block 404 and
the carrier gas. Furthermore, the processor 424 is connected via converter 422
to valves
415 and 417 for controlling the carrier gas flow and the auxiliary gas flow,
respectively.
[70] Optionally, temperature sensors and flow rate measurement sensors are
disposed
for measuring heating block temperature, carrier gas temperature, carrier gas
flow rate
and auxiliary gas flow rate and for providing a feed back to the processor
424. This
provides automated operation of the desorption interface according to
predetermined
values provided through digital port 425 and, preferably stored in memory 426.
For
example, data for defining the above mentioned parameters are stored in the
memory 426
14
CA 02349185 2001-05-29
11158-O1 CA Patent
for measurements of different analytes. An operator of the interface 400 has
then only to
select the analyte to be investigated and the processor 424 regulates the
interface 400
automatically without interference of the operator.
[71] Further optionally, the processor 424 is connected to a detection unit
427 of the
ICP-MS, for supporting calibration processes based on measurement data -
transient
signals - provided by the ICP-MS. For example, this embof.iment allows
optimization of
the gas flow rates of the carrier gas and the auxiliary gas as well as the
temperatures of
the heating block and the carrier gas based on the measured profiles -
transient peak
width and counts per second of the transient signals.
[72] The next step of automation is achieved in the embodiment of a thermal
desorption interface 500 shown in Fig. 8. Here, a plurality of measurement
units 501 is
movably attached to a transportation mechanism 503. Each measurement unit 501
comprises two heater block portions 504 A and 504 B having an injector 502
interposed
therebetween. Furthermore, the measurement unit 501 comprises holding
mechanism 550
attached to one or to both heater block portions for moving a SPME unit 515 in
a linear
fashion and for moving the fiber 2 of the SPME unit 515 by linearly moving
plunger 8.
[73] In operation, a measurement unit 501 is positioned at a predetermined
location
with respect to ICP-MS 525 using the transportation mechanism 503. The
measurement
unit is then moved towards the ICP-MS 525 until conduit 544 is inserted into
the injector
at a predetermined position. Conduits 540 and 542 for provision of carrier gas
and
auxiliary gas are then connected as well as port 552 with connector 553 for
provision of
electrical power for heating the heating block 504 and, optionally, providing
sensed
temperature data to a processor - not shown. Optionally conduits 540 and 542
and
connector 553 are enclosed in one unit and moved as one unit as well. In the
following
step the SPME unit 515 is moved until needle 4 of the SPM:E unit 515 is
inserted into the
injector 502 through seal 560 and the tip of the needle is located at a
predetermined
position within the injector 502. Moving the plunger 8 in a :linear fashion
towaxds the
injector 502 exposes the extraction phase of fiber 2. After predetermined time
interval for
exposing the fiber 2 is elapsed the fiber 2 is retracted. Then., optionally,
the SPME unit
CA 02349185 2001-05-29
11158-O1 CA Patent
515 is retracted. In the following step conduits 540 and 542, as well as
connector 553 are
retracted, followed by the retraction of the measurement unit 501 from conduit
544. The
measurement unit 501 is then free for moving to a parking )position for, for
example,
removal by an operator. The injector 502 is connected to conduits 540, 542 and
544 by
sealed connectors. Alternatively, injector 502 is filled with an inert gas by
a manufacturer
and sealed as shown in Fig. 8. The seals are then penetrated) by needle like
conduits. It is
evident to a person of skill in the art that commercially available
microprocessor
technology allows to program all the functions illustrated above for fully
automated
operation of the thermal desorption interface according to tile invention.
[74] Optionally, the injector is stationary and only the SF'ME units are
movably
attached to a transportation mechanism via a holding mechanism. In operation,
the SPME
unit is positioned at a predetermined location with respect to the injector.
Using the
holding mechanism the SPME unit is moved until the needle of the SPME unit is
inserted
into the injector through the seal and the tip of the needle is located at a
predetermined
position within the injector. Moving the plunger in a linear fashion towards
the injector
exposes then the extraction phase of the fiber.
[75] The thermal desorption interface 500 is highly advantageous for
automating
measurements. For example, one operator prepares a pluraliity of measurement
units 501
and inserts the prepared measurement units 501 into the tra~lsportation
mechanism while
measurements are performed in an automated fashion. After the measurement the
measurement units 501 are provided for removal and prepa'~ation. This allows
continuous
use of the ICP-MS. Furthermore, it allows performing of large numbers of
measurements
which becomes more and more commonplace for monitoring purposes, for example,
monitoring of the air quality at workplaces or in the envirorunent.
[76] The direct coupling of the SPME with ICP-MS via the thermal desorption
interface according to the invention provides a new approach for both the
sampling and
sample introduction of volatile analytes into an atomic spectroscopic
detector. The
compact design of the interface lends itself to direct placement at the base
of the torch of
an atomic spectroscopic detector, significantly minimizing i:he length of the
transfer zone,
16
CA 02349185 2001-05-29
11158-Ol CA Patent
which is particularly important for very reactive analytes such as, for
example,
methylmercury. Furthermore, this technique is applicable for the introduction
of analytes
into any atomic spectroscopic detector as well as organic mass spectroscopic
detectors.
This interface design also offers the possibility for direct introduction of
small amounts
of a sampling phase such as an organic solvent containing volatile analytes
into the
plasma by evaporating the sampling phase and the analyte which are then
transported in a
carrier gas flow. For example, this possibility allows use of liquid-liquid
extraction
techniques for sampling. Particularly attractive is the signifiicant
preconcentration factor
arising from application of the thermal desorption interface with SPME. The
combination
of the sensitive ICP-MS detection with the high efficiency of the sampling and
sample
introduction system also offers a new approach to the passive sampling of
volatile metals
in different environments, for example, use in exposure studies.
[77] Numerous other embodiments of the invention will be apparent to persons
skilled
in the art without departing from the spirit and scope of the invention as
defined in the
appended claims.
1'7