Note: Descriptions are shown in the official language in which they were submitted.
CA 02350758 2001-05-08
WO 00/31093 PCT/US99/277$0
-1-
BENZYLGLYCOSYLAMIDES AS INHIBITORS OF SMOOTH
MUSCLE CELL PROLIFERATION
10
BACKGROUND OF THE INVENTION
This invention relates to the use of substituted benzylglycosylamides as
smooth muscle cell proliferation inhibitors and as therapeutic compositions
for
treating diseases and conditions which are characterized by excessive smooth
muscle
proliferation such as restenosis.
All forms of vascular reconstruction such as angioplasty and vein bypass
procedures effect a response to injury that ultimately leads to smooth muscle
cell
(SMC) proliferation and subsequently, deposition of profuse amounts of
extrtacellular matrix (Clowes, A. W.; Reidy, M. A. J. Vasc. Surg 1991, 13,
885).
These events are also central processes in the pathogenesis of atherosclerosis
(Raines
E.W.; Ross R. Br. Heart J. 1993, 69 (Supplement), S. 30) as well as transplant
arteriosclerosis (Isik, F.F.; McDonald, T. O.; Ferguson, M.; Yamanaka, E.;
Gordon
Am. J. Pathol. 1992, 141, 1139). In the case of restenosis following
angioplasty,
clinically relevant solutions for controlling SMC proliferation through
pharmacological intervention have remained elusive to date (Hernnan, J. P. R.;
Hermans, W.R.M.; Vos, J.; Serruys P. W. Drugs 1993, 4, 18 and 249). Any
successful approach to selective SMC proliferation inhibition must not
interfere with
endothelial cell repair or the normal proliferation and function of other
cells
(Weissberg, P.L.; Grainger, D.J.; Shanahan C.M.; Metcalfe, J.C. Cardiovascular
Res. 1993, 27, 1191 ).
The glycosaminoglycans heparin and heparan sulfate are endogenous
inhibitors of SMC proliferation, yet are able to promote endothelial cell
growth
(Castellot, J.J. Jr.; Wright, T. C.; Karnovsky, M.J. Seminars in Thrombosis
and
Hemostasis 1987, 13, 489). However, the full clinical benefits of heparin,
heparin
fragments, chemically modified heparin, low molecular weight heparins, and
other
heparin mimicking anionic polysaccharides may be compromised due to other
pharmacological liabilities (excessive bleeding arising from anticoagulation
effects, in
CA 02350758 2001-05-08
WO 00/31093 PCT/US99/27780
-2-
particular) coupled with heterogeneity of the various preparations (Borman, S.
Chemical and Engineering News, 1993, June 28, 27).
WO 96/14325 discloses acylated benzylglycosides as smooth muscle cell
proliferation inhibitors. The compounds of the present invention differ in
that (a) the
carbohydrate posesses an anomeric amide, (b) the substituents on the
carbohydrate
backbone are substantually different and, (c) the activity against smooth
muscle cell
proliferation is greater.
Zehavi, U., in Carbohyd. Res. 1986, 151, 371, disclosed 4-carboxy-2-
nitrobenzyl 4-O-a-D-glucopyranosyl-~i-D-glucopyranoside which is attached to a
polymer for study as an acceptor in the glycogen synthase reaction. The
compounds
of the present invention differ in that (a) the carbohydrate posesses an
anomeric
amide, (b) the substituents on the benzyl groups are different and (c) the use
(smooth
muscle antiproliferation) is different.
Patent numbers US 5,498,775, W096/14324, and US 5,464,827 describe
polyanionic benzylglycosides or cyclodextrins as smooth muscle cell
proliferation
inhibitors for treating diseases and conditions which are characterized by
excessive
smooth muscle proliferation. (i-cyclodextrin tetradecasulfate has been
described as a
smooth muscle cell proliferation inhibitor and as an effective inhibitor of
restenosis
(Reilly, C.F.; Fujita, T.; McFall, R. C.; Stabilito, I. L; Wai-se E.; Johnson,
R. G.
Drcig Development Research 1993, 29, 137). US 5019562 discloses anionic
derivatives of cyclodextrins for treating pathological conditions associated
with
undesirable cell or tissue growth. WO 93/09790 discloses antiproliferative
polyanionic derivatives of cyclodextrins bearing at least 2 anionic residues
per
carbohydrate residues. Meinetsberger (EP 312087 A2 and EP 312086 A2) describes
the antithrombotic and anticoagulant properties of sulfated bis-aldonic acid
amides.
US 4431637 discloses polysulfated phenolic glycosides as modulators of the
complement system. The compounds of the present invention differ from all of
the
prior art in that the compounds (a) are benzylglycosylamides which bear no
structural
resemblance to heparin, sulfated cyclodextrins, or to sulfated lactobionic
acid dimers,
(b) contain no more than two contiguous sugar residues (disaccharide), (c) are
of a
defined structure, (d) and are not sulfated.
CA 02350758 2001-05-08
WO 00/31093 PCTNS99/27780
-3-
DESCRIPTION OF THE INVENTION
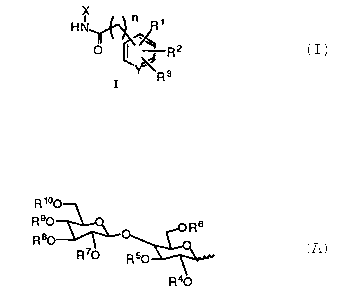
This invention provides benzylglosylamides of formula I
wherein
Y is C or N;
where n is 0 - 3;
X is
HN
C.
O ~ R
R3
I
R ~°O
R90 Rs
R80
R~ R50
R40
R', and R' are each independently, hydrogen, alkyl of 1 to 6 carbon atoms,
halo,
acetyl, phenyl, CF,, CN, OH, NOZ, NH2, alkoxy of 1 to 6 carbon atoms, or
alkoxynitrile of 1 to 6 carbon atoms;
R3 is hydrogen, acylamide of 2 to 6 carbon atoms or alkoxy of 1 to 6 carbon
atoms;
R°, R5, R6, R', and RB are each, independently, hydrogen, acyl of 1 to
6 carbon
atoms, benzyl substituted with R', and R2; or benzoyl substituted with R'
and R2;
R~ and R'° are each, independently, acyl of 1 to 6 carbon atoms, or the
R9 and
R'° groups on the 4' and 6' positions of the maltose may be taken
together
to form a cyclic acetal which may be substituted with alkyl of 1 to 6
carbon atoms, two alkyl groups each having 1 to 6 carbon atoms, pyridine
substituted with R', phenyl substituted with R', benzyl substituted with R',
2-phenylethyl substituted with R', or 3-phenylpropyl substituted with R';
or a pharmaceutically acceptable salt thereof.
CA 02350758 2001-05-08
WO 00/31093 PCTNS99/27780
-4-
Alkyl includes both straight chain as well as branched moieties. Halogen
means bromine, chlorine, fluorine, and iodine. When Y is nitrogen, it is
preferred
that the pyridine carboxamide is pyridine 3-carboxamide.
Pharmaceutically acceptable salts can be formed from organic and inorganic
acids, for example, acetic, propionic, lactic, citric, tartaric, succinic,
fumaric, malefic,
malonic, mandelic, malic, phthalic, hydrochloric, hydrobromic, phosphoric,
nitric,
sulfuric, methanesulfonic, napthalenesulfonic, benzenesulfonic,
toluenesulfonic,
camphorsulfonic, and similarly known acceptable acids. Salts may also be
formed
from organic and inorganic bases, preferably alkali metal salts, for example,
sodium,
lithium, or potassium. Acid addition salts can be prepared when Y is nitrogen
or the
compound of formula I contains a basic nitrogen, and base addition salts can
typically
be prepared when the compound of formula I contains a hydroxyl group.
The compounds of this invention may contain an asymmetric carbon atom and
some of the compounds of this invention may contain one or more asymmetric
centers and may thus give rise to optical isomers and diastereomers. While
shown
without respect to stereochemistry in Formula I, the present invention
includes such
optical isomers and diastereomers; as well as the racemic and resolved,
enantiomerically pure R and S stereoisomers; as well as other mixtures of the
R and S
stereoisomers and pharmaceutically acceptable salts thereof.
Preferred compounds formula I of this invention are those in which
nis0-1;
R', and RZ are each independently, hydrogen, halogen, CF3, OH, NO2, NH2,
methoxy,
butoxy, or butoxynitrile;
R' is hydrogen, acetamide, or methoxy;
R°, R5, R6, R', and R$ are each, independently, hydrogen, an acyl of 1-
6 carbon atoms,
or benzoyl;
R9 and R'° are each, independently, acyl of 1-6 carbon atoms, or the R9
and R'° groups
on the 4' and 6' positions of the maltose are taken together form a
benzylidene ring;
or a pharmaceutically acceptable salt thereof, with all other substituents as
defined
above.
CA 02350758 2001-05-08
WO 00/31093 PCT/US99/27780
-S-
More preferred compounds of formula I are those in which
n is 0;
R', and RZ are each independently, hydrogen or halogen;
R3 is hydrogen;
R', R5, R6, R', and R8 are each, independently, hydrogen, acyl of 1 to 6
carbon atoms,
or benzoyl;
R9 and R'° are each, independently, acyl of 1-6 carbon atoms, or the R9
and R'° groups
on the 4' and 6' positions of the maltose are taken together form a
benzylidene ring;
or a pharmaceutically acceptable salt thereof, with all other substituents as
defined
above.
Specifically preferred compounds of this invention are:
6-Chloro-N-(Hepta-O-acetyl-[3-D-cellobiosyl)-3-pyridinecarboxamide;
N-(4',6'-O -benzylidene-(3-D-cellobiosyl)-6-chloro-nicotinamide;
2-Chloro-piperidine-5-carboxylic acid-(6-O-benzoyl-4',6'-O -benzylidene-1-
deoxy-
/3-D-cellobiosyl)-amide;
(2,6-Dimethoxy-N-(hepta-O-acetyl-(3-D-cellobiosyl)-3-pyridinecarboxamide;
N-(hepta-O-acetyl-~i-D-cellobiosyl)-3-chloro-4-fluoro-benzamide; or
N-(4',6'-O -benzylidene-~i-D-cellobiosyl)-2-chloro-4-flouro-benzamide,
or pharmaceutically acceptable salts thereof.
The compounds of this invention were be prepared according to the following
scheme from commercially available starting materials or starting materials
which
can be prepared using literature procedures. This scheme shows the preparation
of
representative compounds of this invention.
CA 02350758 2001-05-08
WO 00/31093 PCT/US99/27780
-6-
Sohern~ I
oar n ,
0
AcC~PcO NHz + CI, H
J
'f~3
2
glycosidatlon
when F~ = NOZ
H n ~ reduction AcOC~~ ~c H n
Ac0 Pc0 ~ z > Ac Pc0
z
3
~NHz
acylation or
acetolysis sutfonylation
H
HO~' H n R,
HO Ho H acetolysis O~ H n
z ,E
v ~Y~ ~ v i z
3
Rs
1. benzyGdination
2. benzoylation
R'° ORs
RR~O ~ O H n R'
R O RcCY~~ '
R N~~~ z
~a
acylation
R'
s
R O O H n R'
R80 R~ R- Rs z
3
In Scheme I, Y, n, R', R2, R3, R', R5, R6, R7, R8, R9, and R'° are as
defined above.
Thus, the cellobiosyl amine 1 is coupled with a benzoic acid derivative 2 in
the presence of a coupling reagent such as EEDQ, DEC/HOBT, or DCC/HOBT in a
suitable solvent system such as benzene, ethanol, dichloromethane, triethyl
amine at
room temperatures to yield glycoside 3. The glycoside can also be prepared by
coupling the amine 1 to a substituted acid chloride 2 in the presence of
triethyl amine
in a suitable solvent system such as tetrahydrofuran, dichloromethane,
acetonitrile,
and ethyl acetate to yield glycoside 3. When R'is a nitro group, reduction of
the nitro
CA 02350758 2001-05-08
WO 00/31093 PCT/US99/27780
group of 3 can be accomplished with a reducing agent such as stannous chloride
or
iron metal in a polar aprotic solvent such as ethyl acetate or a polar protic
solvent
such as ethanol or methanol at ambient temperature to reflux, or by catalytic
hydrogenation in the presence of a catalyst such as palladium on carbon to
give an
anilino compound 4. Coupling of 4 with an acid chloride or sulfonyl chloride
in the
presence of an amine base such as triethylamine or diisopropylethylamine in an
aprotic solvent such as dichloromethane or tetrahydrofuran at temperatures
ranging
from -20 °C to ambient temperature yields the target compounds 5.
The acetate groups of 3 or 5 can be removed by hydrolysis with a base such as
catalytic sodium methoxide in methanol or aqueous sodium hydroxide in methanol
at
ambient temperature to reflux to yield 6. After hydrolysis of the acetate
groups, the
4' and 6' hydroxy groups of maltose can be reacted with benzaldehyde diemthyl
acetal in the presence of an acid catalyst such as camphorsulfonic acid or
toluene
sulfonic acid in a polar aprotic solvent such as acetonitrile or dimethyl
formamide at
ambient temperature to 70 °C to yield a benzylidene derivative. The 6
hydroxyl
group can be selectively benzoylated in a collidine / tetrahydofuran mixture
at -78 °C
to ambient temperature to yield 7. Reacylation with an acyl anhydride in the
presence
of an amine base such as pyridine or triethyl amine at temperatures ranging
from 0 °C
to ambient temperature to yield 8.
The compounds of this invention are useful as antiproliferative agents. The
following procedures show the evaluation of representative compounds of this
invention in standard pharmacological test procedure which measured ability of
the
evaluated compound to inhibit smooth muscle cell proliferation
Effects of Compounds on Cell Proliferation Usin '_g H Th,ymidine Incorporation
Human and porcine smooth muscle cells were tested in early passage
(generally passage 3 - 7) at sub-confluent conditions. Cultures were grown in
16
mm (24 well) mufti-well culture dishes in medium 199 supplemented with 10%
fetal
bovine serum and 2% antibiotic / antimycotic. At sub-confluence, the cells
were
CA 02350758 2001-05-08
WO 00/31093 PCT1US99/27780
_g_
placed in a defined serum free medium (AIM-V; Gibco) for 24 - 48 h prior to
initiating the experimental protocol.
Although compounds were found to be more effective with longer pre-
incubations, in general, the procedures were initiated with the addition of
compound,
'H thymidine and serum / growth factor to serum deprived synchronized cells
and
results are reported accordingly.
Compounds were added to each well at 50 fold dilution (20 ~L l well) and the
plates were incubated for 24 - 36 h at 37 °C in 5% CO2. Compounds were
initially
dissolved in 50% ethanol and serially diluted into media. Compounds were
routinely
evaluated at concentrations from 1 to 100 ~M. As a control, grade II porcine
intestinal mucosal heparin (sodium salt) was routinely evaluated in all cell
preparations at concentrations from 0.1 to 100 pg/mL.
At the completion of the test procedure, plates were placed on ice, washed
three times with ice cold phosphate buffered saline (PBS) and incubated in ice
cold
10% trichloroacetic acid (TCA) got 30 min to remove acid soluble proteins.
Solution
was transferred to scintillation vials containing 0.4 N HCl (500 ~L,/ vial to
neutralize
NaOH) and each well was rinsed two times with water (500 pL) for a total
volume of
2 mL / vial.
Data was obtained, in triplicate, for both control and experimental samples.
Control (100%a) data was obtained from maximally stimulated cells, as the
result of
growth factor or serum stimulation. Experimental data was obtained from cells
maximally stimulated with growth factor or serum and treated with compound.
Data
are provided below in Table 1 as an ICgp.
Table 1.
Porcine Smooth Muscle
Com ound of Exam Cell
le Anti roliferation IC50
1 3.55 M
2 40% at 50 M
3 20% at 50
4 3.11 M
5 11.67
_6 5% at 50 ~1M
CA 02350758 2001-05-08
WO 00/31093 PCT/US99/27780
-9-
The compounds of this invention are useful in treating or inhibiting diseases
which are characterized by excessive smooth muscle cell proliferation (smooth
muscle cell hyperproliferation). The compounds are particularly useful in
treating
hyperproliferative vascular diseases which are characterized by smooth muscle
cell
hyperproliferation, such as restenosis, which most frequently arises from
vascular
reconstructive surgery and transplantation, for example, balloon angioplasty,
vascular
graft surgery, coronary artery bypass surgery, and heart transplantation.
Other
disease states in which there is unwanted "cellular" vascular proliferation
include
hypertension, asthma, and congestive heart failure. The compounds of this
invention
are also useful as inhibitors of angiogenesis. Angiogenesis
(neovascularization), the
process by which new capillaries are formed, is of principal importance for a
number
of pathological events including chronic inflammation and malignant processes.
The
compounds of this invention are therefore useful as antineoplastic agents.
The compounds of this invention can be formulated neat or with a
pharmaceutical carrier for administration, the proportion of which is
determined by
the solubility and chemical nature of the compound, chosen route of
administration
and standard pharmacological practice. The pharmaceutical carrier may be solid
or
liquid.
A solid carrier can include one or more substances which may also act as
flavoring agents, lubricants, solubilizers, suspending agents, fillers,
glidants,
compression aids, binders or tablet-disintegrating agents; it can also be an
encapsulating material. In powders, the carrier is a finely divided solid
which is in
admixture with the finely divided active ingredient. In tablets, the active
ingredient is
mixed with a earner having the necessary compression properties in suitable
proportions and compacted in the shape and size desired. The powders and
tablets
preferably contain up to 99% of the active ingredient. Suitable solid carriers
include,
for example, calcium phosphate, magnesium stearate, talc, sugars, lactose,
dextrin,
starch, gelatin, cellulose, methyl cellulose, sodium carboxymethyl cellulose,
polyvinylpyrrolidine, low melting waxes and ion exchange resins.
Liquid carriers are used in preparing solutions, suspensions, emulsions,
syrups, elixirs and pressurized compositions. The active ingredient can be
dissolved
or suspended in a pharmaceutically acceptable liquid carrier such as water, an
organic
solvent, a mixture of both or pharmaceutically acceptable oils or fats. The
liquid
CA 02350758 2001-05-08
WO 00/31093 PCT/US99/27780
- 10-
carrier can contain other suitable pharmaceutical additives such as
solubilizers,
emulsifiers, buffers, preservatives, sweeteners, flavoring agents, suspending
agents,
thickening agents, colors, viscosity regulators, stabilizers or osmo-
regulators.
Suitable examples of liquid carriers for oral and parenteral administration
include
water (partially containing additives as above, e.g. cellulose derivatives,
preferably
sodium carboxymethyl cellulose solution), alcohols (including monohydric
alcohols
and polyhydric alcohols, e.g. glycols) and their derivatives, lethicins, and
oils (e.g.
fractionated coconut oil and arachis oil). For parenteral administration, the
carrier
can also be an oily ester such as ethyl oleate and isopropyl myristate.
Sterile liquid
carriers are useful in sterile liquid form compositions for parenteral
administration.
The liquid carrier for pressurized compositions can be halogenated hydrocarbon
or
other pharmaceutically acceptable propellant.
Liquid pharmaceutical compositions which are sterile solutions or suspensions
can be utilized by, for example, intramuscular, intraperitoneal or
subcutaneous
injection. Sterile solutions can also be administered intravenously. The
compounds
of this invention can also be administered orally either in liquid or solid
composition
form.
The compounds of this invention rnay be administered rectally or vaginally in
the form of a conventional suppository. For administration by intranasal or
intrabronchial inhalation or insufflation, the compounds of this invention may
be
formulated into an aqueous or partially aqueous solution, which can then be
utilized
in the form of an aerosol. The compounds of this invention may also be
administered
transdermally through the use of a transdermal patch containing the active
compound
and a carrier that is inert to the active compound, is non toxic to the skin,
and allows
delivery of the agent for systemic absorption into the blood stream via the
skin. The
carrier may take any number of forms such as creams and ointments, pastes,
gels, and
occlusive devices. The creams and ointments may be viscous liquid or semisolid
emulsions of either the oil-in-water or water-in-oil type. Pastes comprised of
absorptive powders dispersed in petroleum or hydrophilic petroleum containing
the
active ingredient may also be suitable. A variety of occlusive devices may be
used to
release the active ingredient into the blood stream such as a semipermeable
membrane covering a reservoir containing the active ingredient with or without
a
carrier, or a matrix containing the active ingredient. Other occlusive devices
are
known in the literature.
The dosage requirements vary with the particular compositions employed, the
route of administration, the severity of the symptoms presented and the
particular
subject being treated. Based on the results obtained in the standard
pharmacological
CA 02350758 2001-05-08
WO 00/31093 PCT/US99/27780
-11-
test procedures, projected daily dosages of active compound would be 0.1 to 10
mg/kg administered parenterally (intravenous preferred), with projected daily
oral
dosage being approximately ten-fold higher. Anticipated intravenous
administration
would last for approximately 5-30 days following acute vascular injury (i.e.,
balloon
angioplasty or transplantation) and for a longer duration for the treatment of
chronic
disorders. Treatment will generally be initiated with small dosages less than
the
optimum dose of the compound. Thereafter the dosage is increased until the
optimum effect under the circumstances is reached; precise dosages for oral,
parenteral, nasal, or intrabronchial administration will be determined by the
administering physician based on experience with the individual subject
treated.
Preferably, the pharmaceutical composition is in unit dosage form, e.g. as
tablets or
capsules. In such form, the composition is sub-divided in unit dose containing
appropriate quantities of the active ingredient; the unit dosage forms can be
packaged
compositions, for example, packaged powders, vials, ampoules, pre filled
syringes or
sachets containing liquids. The unit dosage form can be, for example, a
capsule or
tablet itself, or it can be the appropriate number of any such compositions in
package
form.
The following provides the preparation of representative compounds of this
invention.
EXAMPLE 1
6-Chloro-N-(Hepta-O-acetyl-~3-D-cellobiosvl)-3-pvridinecarboxamide
5tep1
Hepta-O-acetyl-1-[3-cellobiosylamine
Hepta-O-acetyl-1-~i-cellobiosylamine was obtained by the platinum oxide
reduction of the azide prepared by the method of A. Bertho, Justus Liebigs
Ann.
Che»i., 562, 229 (1949).
Step 2
6-Chloro-N-(Hepta-O-acetyl-(3-D-cellobiosyl)-3-pyridinecarboxamide
To a stirred mixture of Hepta-O-acetyl-1-~i-cellobiosylamine, (0.20 g, 0.3147
mmol) and triethyl amine (0.064 g, 0.63 mmol) in dichloromethane (1.5 ml), and
tetrahydrofuran (1.5 ml) was added in one portion 6-chloronicitinoyl chloride
(0.5 g,
CA 02350758 2001-05-08
WO 00/31093 PCT/US99/27780
-12-
0.32 mmol). After 12 h, the reaction was diluted with dichloromethane ( 10 ml)
and
washed successively with water (5 ml), 10% sodium hydroxide (5 ml), and brine
( 10
ml), dried (MgS04) and concentrated. Purification by flash chromatography (50%-
60% EtOAc/petroleum ether gradient) gave the title compound as an off white
solid;
'H NMR (CDC13) b 1.99 (s, 3 H), 2.01 (s, 3 H), 2.04 (s, 6 H), 2.06 (s, 3 H),
2.10 (s, 3
H), 2.12 (s, 3 H), 3.65 - 3.69 (m, 2 H ), 3.79 - 3.81 (m, 2 H), 4.06 (dd, J =
12.3, 2.4,
Hz, 1 H), 4.14 - 4.18 (m, 1 H), 4.38 (dd, J = 12.5, 4.6, Hz, 1 H), 4.50 (d, J
= 11.6, 1
H), 4.51 (d, J = 7.9, 1 H), 4.90 - 4.96 (m, 2 H), 5.07 (apparent t, J = 9.7, 1
H), 5.15
(apparent t, J = 9.2, 1 H), 5.30 - 5.37 (m, 4 H), 7.04 (d, J = 8.6, 1 H), 7.42
(dd, J =
8.3, 0.7, Hz 1 H), 8.02 (dd, J = 8.1, 2.6, Hz, 2 H) 8.73 (dd, J = 7.4, 0.7, Hz
1 H). IR
(KBr) 3400, 1750, 1550, 1245 and 1075 cm-', mass spectrum (+ESI), m/z 775 (M +
H), 797 (M + Na).
EXAMPLE 2
N-(4'.6'-O -benz ly'dene-p-D-cellobiosvl)-6-chloro-nicotinamide
Step 1
6-Chloro-N-((3-D-cellobiosyl)-nicotinamide
To a solution of 6-Chloro-N-(Hepta-O-~3-D-cellobiosyl)-3-pyridine-
carboxamide (1040 mg, 1.34 mmol) in methanol (10 ml) was added 0.075 ml of a
0.34 M solution of sodium methoxide. The reaction was stirred overnight and
quenched with Dowex H+ resin. After 0.5 hr the solution filtered and
concentrated in
vacuo to give the title compound as a white solid, mp 193; 'H NMR (D20-d2) 8
3.19
(t, J = 8.I Hz, 1 H), 3.25 - 3.48 (m, 4 H), 3.58 - 3.62 (m, 4 H), 3.70 - 3.83
(m, 3 H),
4.40 (d, J = 7.9 Hz, 1 H), 5.08 (d, J = 9.2 Hz 1 H), 7.49 (d, J = 8.6 Hz 1 H),
8.09 (dd,
J = 8.3 Hz, 2.4 Hz, 1 H), 8.63 (d, J = 2.0 Hz, 1 H). IR (KBr) 3375, 2900,
1660, 1575
and 1060 cm', mass spectrum (-FAB), m/e 479 (M - H). Anal. Calcd. for
C,$HZSC1N20" ~ HZO C, 43.34; H, 5.46; N, 5.61. Found: C, 43.48; H, 5.55; N,
5.47.
Step 2
N-(4',6'-O -benzylidene-(3-D-cellobiosyl)-6-chloro-nicotinamide
A solution containing 6-Chloro-N-((3-D-cellobiosyl)-nicotinamide (0.33 g,
0.6863 mmoI}, benzaldehyde dimethyl acetal (0.15 ml, 1.0 mmol) and
CA 02350758 2001-05-08
WO 00/31093 PCT/US99/27780
- 13-
camphorsulfonic acid (10 mg, 0.043 mmol) in dimethyl formamide (6 ml) was
heated
at 70 °C. After 4h, the reaction was cooled to ambient temperature and
quenched
with 0.5 ml of a 1N NaOH solution . The solution was concentrated and purified
by
flash chromatography (2, 5 - 10% MeOH/methylene chloride gradient) gave the
title
compound as a white solid, mp 230 °C; 'H NMR (DMSO-d6) 8 3.13 - 3.16
(m, 1 H),
3.34 - 3.45 (m, 7 H), 3.63 - 3.75 (m, 3 H), 4.18 - 4.22 (m, 1 H), 4.51 (d, J =
1.5 Hz, 1
H), 4.55 (d, J = 7.7 Hz, 1 H), 4.62 (apparant t, J = 5.8 Hz, 1 H), 4.98
(apparant t, J =
8.8 Hz, 1 H). 5.19 (d, J = 5.3 Hz, 1 H), 5.38 (d, J = 4.4 Hz, 1 H), 5.35 (d, J
= 5.1 Hz,
1 H), 5.59 (s, 1 H), 7.35 - 7.38 (m, 3 H), 7.42 - 7.45 (m, 2 H), 7.67 (d, J =
8.3 Hz, 1
H), 8.29 (dd, J = 8.3, 2.4 Hz, 1 H), 8.88 (d, J = 2.6 Hz, 1 H), 9.20 (d, J =
8.6 Hz, 1
H). IR (KBr) 3400, 2900, 1650 and1075 cm-', mass spectrum (+FAB), m/e 569 (M +
H). Anal. Calcd. for C~SHZ9C1N20" ~ 1.0 HBO: C, 51.16; H, 5.32; N, 4.77.
Found: C,
51.22; H, 5.26; N, 4.68.
EXAMPLE 3
2-Chloro-nineridine-5-carboxylic acid-l6-O-benzoyl-4',6'-D -benzylidene-1-
deoxv-
J3-D-cellobiosyl)-amide
A solution of N-(4',6'-O -benzylidene-~3-D-cellobiosyl)-6-chloro-
nicotinamide (0.22 g, 0.39 mmol) in dry tetrahydrofuran ( 1.5 ml) and
anhydrous
2,4,6 collidine ( 1.5 ml) was cooled to -40 °C for 0.5 h. Benzoyl
chloride (0.076 ml,
0.507 mmol) was added slowly and the reaction allowed to warm to ambient
temperature overnight. The reaction was diluted with ethyl acetate (30 ml),
and
washed consecutively with 1 N HCl ( 15 ml), saturated aqueous sodium
bicarbonate
( 15 ml), and brine ( 15 ml). The organic layer was dried (MgSO,) and
filtered.
Evaporation and flash chromatography (2, 5 - 10% MeOH/methylene chloride
gradient) gave the title compound as a white solid, mp 260 °C; 'H NMR
(DMSO-d6) S
3.17 - 3.28 (m, 1 H), 3.35 - 3.47 (m, 4 H), 3.55 (dt, J = 8.8, 5.9 Hz, 1 H),
3.64 - 3.71
(m, 2 H), 3.82 - 3.86 (m, 1 H), 4.17 (dd, J = 10.1, 4.2 Hz. 1 H), 4.48 (dd, J
= 12.1,
4.6 Hz, 1 H), 4.57 - 4.65 (m, 2 H), 4.86 (d, J = 3.1 Hz, 1 H), 5.09 (apparant
t, J = 9.0
Hz, 1 H), 5.33 (d, J = 5.3 Hz, 1 H), 5.37 (d, J = 4.6 Hz, 1 H), 5.50 (s, 1 H),
5.57 (d, J
= 5.1 Hz, 1 H), 7.35 - 7.37 (m, 3 H), 7.39 - 7.42 (m, 2 H), 7.55 (t, J = 7.2
Hz, 2 H),
7.63 - 7.69 (m, 2 H), 7.96 (d, J = 8.6 Hz, 2 H), 8.27 (dd, J = 8.3, 2.6 Hz, 1
H), 8.86
CA 02350758 2001-05-08
WO 00/31093 PCT/US99/27780
- 14-
(d, J = 2.6 Hz, 1 H), 9.22 (d, J = 9.0 Hz, 1 H). IR (KBr) 3400, 2900, 1650,
1275 and
1100 cm-', mass spectrum (-FAB), m/z 671 (M - H). Anal. Calcd. for
C,=H33C1NZO,~
1.0 H,,O: C, 55.62; H, 5.10; N, 4.05. Found: C, 55.80; H, 4.99; N, 4.01.
S EXAMPLE 4
12.6-Dimethoxy-N-(he~ta-O-acetyl-~i-D-cellobiosvl)-3-pvridinecarboxamide
To a stirred solution of 2,6-dimethoxy nicitinic acid (0.051 g, 0.26 mmol) in
benzene-ethanol (1:1, v/v, 4 ml) was added in one portion 2-ethoxy-N-carbonyl-
1,2-
dihydroquinoline (0.071 g, 0.29 mmol). After 0.5 h, Hepta-O-acetyl-1-~3-
cellobiosylamine (O.I51 g 0.24 mmol) was added and the mixture was stirred
overnight at room temperature. The solvents were evaporated and the residue
dissolved in methylene chloride. The organic layer was washed successively
with 1
N hydrochloric acid, water, 1 % sodium hydrogencarbonate, and water, dried
(MgS04) and concentrated. Purification by flash chromatography (40%-60%
EtOAc/petroleum ether gradient) afforded the title compound as a white solid,
mp
127 °C; 'H NMR (CDCI,) 8 1.98 (s, 3 H), 1.99 (s, 3 H), 2.01 (s, 3 H),
2.04 (s, 6 H),
2.10 (s, 3 H), 2.12 (s, 3 H), 3.63 - 3.67 (m, 1 H), 3.80 - 3.85 (m, 2 H), 3.96
(s, 3 H),
3.99 - 4.07 (m, 1 H), 4.07 (s, 3 H), 4.17 (dd, J = 12.6, 4.2 Hz, 1 H), 4.37
(dd, J =
12.5, 4.4 Hz, 1 H), 4.44 - 4.47 (m, 1 H), 4.51 (d, J = 7.9 Hz, 1 H), 4.94
(apparant t, J
= 8.1 Hz, 1 H), 5.03 - 5.16 (m, 2 H), 5.33 (t, J = 9.7 Hz, 1 H), 5.39 (t, J =
9.4 Hz, 1
H), 6.41 (d, J = 9.4 Hz, 1 H), 8.34 (d, J = 8.3 Hz, 1H), 8.41 (d, J = 8.8 Hz,
1 H). IR
(KBr) 3400, 2950, 1750, 1245 and 1050 cm', mass spectrum (+FAB), m/e 801 (M +
H), 823 (M + Na). Anal. Calcd. for C~H44C1N2OZO ~ 0.5 H20: C, 50.43; H, 5.60;
N,
3.46. Found: C, 50.56; H, 5.52; N, 3.31.
EXAMPLE 5
N-(he~ta-O-acet~rl-~i-D-cellobiosyl)-3-chloro-4-fluoro-benzamide
The title compound was prepared according to the procedure of Example 1,
Step 2 as a white solid, mp 203 - 205 °C; 'H NMR (CDC13) 8 1.99 (s, 3
H), 2.01 (s, 3
H), 2.04 (s, 6 H), 2.05 (s, 3 H), 2.10 (s, 3 H), 2.13 (s, 3 H), 3.64 - 3.68
(m, 1 H), 3.79
- 3.80 (m, 2 H), 4.05 (dd, J = 12.5, 2.0 Hz, 1 H), 4.14 - 4.19 (m, 1 H), 4.37
(dd, J =
12.5, 4.4 Hz. 1 H), 4.48 - 4.53 (m, 2 H), 4.91 - 4.99 (m, 2 H), 5.05 - 5.17
(m, 2 H),
CA 02350758 2001-05-08
WO 00/31093 PCT/C1S99/Z7780
-15-
5.33 (t, J = 9.2 Hz, 1 H), 5.4U (t, J = 9.2 Hz, 1 H), 6.80 (d, J = 9.0 Hz, 1
H), 7.02 -
7.07 (m, 1 H), 7.14 (dd, J = 8.3, 2.6 Hz, 1 H), 7.67 (dd, J = 8.6, 6.1 Hz, 1
H). IR
(KBr) 3400, 2930, 1750, 1245 and1050 cm-', mass spectrum (-ESI), mlz
789.9/791.9
(M - H). Anal. Calcd. for C33H39CIFNO,B: C, 50.04; H, 4.96; N, 1.77. Found: C,
50.00; H, 4.91; N, 1.85.
EXAMPLE 6
N-(4'.6'-O -benzylidene-~3-D-cellobiosyI)-2-chloro-4-flouro-benzamide
Step 1
N-((3-D-cellobiosyl)-2-chloro-4-iluoro-benzamide
The title compound was prepared according to the procedure of Example 2, Step
1 as
a white solid, mp decomposed 65 °C; 'H NMR (CD30D-d,) s 3.22 - 3.42 (m,
5 H),
3.51 - 3.62 (m, 3 H), 3.67 (dd, J = 11.9, 5.3 Hz, 1 H), 3.83 - 3.90 (m, 3 H),
4.43 (d, J
= 7.9 Hz, 1 H), 5.07 (d, J = 9.2 Hz, 1 H), 7.12 - 7.17 (m, 1 H), 7.29 (dd, J =
8.8, 2.6
Hz, 1 H), 7.61 (dd, J = 8.6, 6.2 Hz, 1 H}, 8.53 (s, 1 H). IR (KBr) 3400, 2930,
1600,
and 1050 cm~', mass spectrum (-FAB), m/z 496/498 (M - H).
Step 2
N-(4',6'-O -benzylidene-~i-D-cellobiosyl)-2-chloro-4-ilouro-benzamide
The title compound was prepared according to the procedure of Example 2,
Step 2 as a white solid, mp 135 - 138 °C; 'H NMR (CD30D-d4) b 3.34 -
3.91 (m, 11
H), 4.28 - 4.31 (m, 1 H), 4.59 (d, J = 7.9 Hz, 1 H}, 5.09 (d, J = 9.2 Hz, 1
H), 5.58 (s,
1 H), 7.16 (dt, J = 8.3, 2.6 Hz, 1 H), 7.29 - 7.52 (m, 4 H), 7.60 (dd, J =
8.8, 5.9 Hz, 1
H), 7.97 - 7.99 (m, 3 H). IR (KBr) 3400, 2900, 1550 and1075 cm~', mass
spectrum
(+FAB), m/e 586 (M + H), 608 (M + Na).