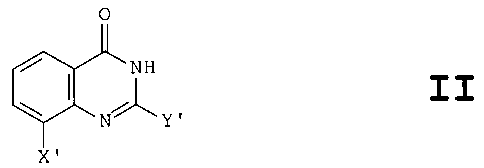Note: Descriptions are shown in the official language in which they were submitted.
y CA 02352592 2001-07-05
1
UINAZOLINONE COMPOUNDS
This application is a division of PCT International
Application No. PCT/GB95/00513 bearing Canadian
Application Serial No. 2,184,747 with the International
Filing Date of March 9, 1995.
The present invention relates to certain
quinazolinone compounds that are of interest as being at
least potentially useful chemotherapeutic agents by
virtue of an ability to inhibit the activity of the
enzyme poly ADP-ribosyltransferase (EC 2.4.2.30), also
known as poly(ADP-ribose) polymerase, commonly referred
to as ADPRT or PARP. In general, the latter
abbreviation, PARP, will be used throughout the present
specification.
BACKGROUND
At least in higher organisms, the enzyme poly ADP-
ribosyltransferase is known to catalyse a transfer of the
ADP-ribose moiety from the oxidized form NAD+ of
nicotinamide adenine dinucleotide to nuclear acceptor
proteins so as to form homo ADP-ribose polymers, and this
process has been implicated in a number of cellular
events- such as, for example, repair of DNA damage,
development of cellular differentiation, transformation
of cells by oncogenes, and gene expression. A common
feature in a number of these processes is the formation
and repair of DNA strand breaks and the stage which
involves the PARP enzyme appears to be that of DNA ligase
II-mediated strand rejoining. In the majority of cases a
role for poly ADP-ribosylation has been implicated by the
use of inhibitors of the PARP enzyme, and this has led to
suggestions that such inhibitors, by interfering with the
intracellular DNA repair mechanism, may have a useful
chemotherapeutic role insofar as they should be able to
modify treatment resistance characteristics and
potentiate or enhance the effectiveness of cytotoxic
drugs in chemotherapy or of radiation in radiotherapy
where a primary effect of the treatment is that of
~
CA 02352592 2001-07-05
2
causing DNA damage in target cells, as for example in
many forms of antitumour therapy.
In this connection, several classes of PARP
inhibitors are already known, including benzamide itself
and various nicotinamide and benzamide analogues,
especially 3-substituted benzamides with small
substituent groups such as 3-amino, 3-hydroxy and 3-
methoxy. PARP inhibitory activity of certain N-
substituted benzamides has also been reported in EP-A-
0305008 wherein it has also been proposed to use these
compounds in medicine for increasing the cytotoxicity of
radiation or of chemotherapeutic drugs.
Regarding this use of benzamides as chemotherapeutic
agents, a number of studies on such compounds that are
known to exhibit PARP inhibitory activity have confIRmed
that they can potentiate the cytoxicity of a range of
antitumour agents in vitro, for example, bleomycin and
methylating drugs. More limited data has further
indicated that such benzamides can also potentiate the
activity of cytotoxic drugs in vivo, although the dose
requIRements have appeared to be rather high (e.g. in the
region of 0.5g kg-1 per dose for 3-aminobenzamide) and
there may be associated problems in preparing
satisfactory pharmaceutical formulations and in avoiding
toxicity limitations. Furthermore, a number of the known
benzamides have also been shown clearly to have potential
as radiosensitizers, increasing for example ionising
radiation-induced tumour cell kill both in vitro and in
vivo, and it is believed that in many cases this effect
is related to these compounds acting as PARP inhibitors
and interfering with DNA repair.
However, notwithstanding the existing data from in vitro
and in vivo studies suggesting that PARP inhibitors have
considerable potential as useful chemotherapeutic agents
which merit further clinical evaluation, for instance in
connection with cancer therapy, currently available known
PARP inhibitors are not considered as yet to be entirely
- CA 02352592 2001-07-05
3
suitable to represent candidate drugs. Accordingly,
there is a need to find and develop a greater range of
compounds having potentially useful PARP inhibitory
properties.
S DISCLOSURE OF THE INVENTION
The present invention identifies a new range or
ranges of compounds of interest as PARP inhibitors that
can be useful in medicine, especially when administered
in conjunction with at least certain cytotoxic drugs or
with radiotherapy for increasing the cytotoxic
effectiveness thereof. In general, the compounds of this
invention as hereinbelow defined include certain
quinazolinones of which at least some may be formed by
molecular rearrangement of related benzamide compounds.
By virtue of their structure in general such compounds
are adapted to act as an alternative substrate to NAD+ for
the PARP enzyme.
More specifically, from one aspect, the invention
resides in the use of a compound as herein defined for
the manufacture of a medical or veterinary preparation
for use in therapy for inhibiting activity of the enzyme
poly(ADP-ribose)polymerase or PARP (also known as ADP-
ribosyl transferase or ADPRT), such enzyme inhibition
constituting an element of a therapeutic treatment, said
compound providing an active PARP enzyme inhibiting agent
and being a quinazolinone compound having the general
structural formula II
0
NH
X'
or a pharmaceutically acceptable salt thereof,
CA 02352592 2001-07-05
4
characterised in that
X' represents hydroxyl, alkyl, alkoxy, or an
optionally substituted aryl (e. g. phenyl) or aralkyl
(e. g. benzyl) group,
S and
Y' represents hydrogen, alkyl or an optionally
substituted aryl (e. g. phenyl) or aralkyl (e. g.
benzyl) group.
The invention also provides for use in therapy as
active pharmaceutical substances quinazolinone compounds
having the general structural formula II (or a
pharmaceutically acceptable salt thereof) in which
X' represents hydroxyl, alkyl or alkoxy
and
Y' represents alkyl or an optionally substituted
aralkyl (e. g. benzyl) group or an optionally
substituted phenyl group other than a phenyl group
having a 4-propoxy substituent or a 2-alkoxy
substituent,
subject to a proviso that
if X' is methyl, Y' is not butyl,
if X' is methoxy, Y' is not methyl or 4-
hydroxyphenyl, and
if X' is hydroxy, Y' is not methyl or ethyl or
phenyl.
The invention further provides novel quinazolinone
compounds having the general structural formula II (or a
pharmaceutically acceptable salt thereof) in which:
X' represents hydroxyl, alkyl or alkoxy
and
Y' represents alkyl or an optionally substituted
aralkyl (e. g. benzyl) group or an optionally
substituted phenyl group other than a phenyl group
CA 02352592 2004-O1-21
having a 4-propoxy substituent or a 2-alkoxy
substituent,
subject to a proviso that
if X' is methyl, Y' is not butyl, isopropyl, phenyl
5 or 2-aminophenyl,
if X' is ethyl, Y' is not 4-hydroxyphenyl,
if X' is methoxy, Y' is not methyl, isopropyl,
4-methylphenyl, 4-hydroxyphenyl or
4-methoxyphenyl,
if X' is ethoxy, Y' is not isopropyl,
if X' is propoxy, Y' is not a halogen substituted
phenyl group, and
if X' is hydroxy, Y' is not methyl or ethyl or
phenyl.
Alkyl groups when present as such or as a moiety in
other groups such as alkoxy will generally be composed of
1-8 carbon atoms, preferably 1-6 carbon atoms, and more
usually 1-4 carbon atoms.
In a preferred embodiment, Y' is phenyl or a phenyl
group having a substituent selected from the group
consisting of -NO2, -NHz, -OH and alkyl.
It has been found that in attempting to prepare
benzoxazole-4-carboxamide compounds having a structural
formula IV
CONH2
0
R5
wherein RS is H, alkyl or an optionally substituted
aralkyl or aryl group, in some methods of preparation
which could be expected to yield the desired compound the
product is liable to undergo a molecular rearrangement
N IV
CA 02352592 2001-07-05
6
(especially if liquid ammonia is used to form the
carboxamide) and an 8-hydroxy quinazolinone derivative is
obtained instead of the expected benzoxazole.
Unexpectedly, it has been found that at least some such
quinazolinone derivatives, which may of course be
prepared by various other methods, possess a potentially
very useful biological activity as PARP inhibitors of
high activity. Examples of such compounds which are of
particular interest include:
(a) 8-hydroxy-2-methylquinazolin-4-[3H]one;
(b) 8-hydroxyquinazolin-4-[3H]one;
(c) 8-hydroxy-2-(4-nitrophenyl)-quinazolin-4-one;
(d) 8-methoxy-2-methylquinazolin-4[3H]-one;
(e) 8-methoxy-2-phenylquinazolin-4[3H]-one;
(f) 8-hydroxy-2-phenylquinazolin-4[3H]-one;
(g) 2,8-dimethylquinazolin-4[3H]-one.
The invention also embraces or extends to methods of
preparing compounds as hereinbefore defined (including
intermediates in some cases) and to the therapeutic use
of such compounds. This includes their use for making
medical or veterinary preparations or pharmaceutical
formulations containing an effective PARP inhibitory
amount of the active compound for administration to a
patient in conjunction with a cytotoxic drug or
radiotherapy in order to increase the cytotoxic
effectiveness of the latter. Such preparations or
formulations may be made up in accordance with any of the
methods well known in the art of pharmacy for
administration in any suitable manner, for example
orally, parenterally (including subcutaneously,
intramuscularly or intravenously), or topically, the mode
of administration, type of preparations or formulation
and the dosage being generally determined by the details
of the associated cytotoxic drug chemotherapy or
radiotherapy that is to be enhanced.
CA 02352592 2001-07-05
7
As indicated, the compounds according to this
invention have at least potential as PARP inhibitors, and
in vitro tests hereinafter described have demonstrated
positive pharmacological activity which it is believed
reflects the activity to be found in vivo in the course
of therapeutic clinical use.
It will be understood that where reference is made
in this specification to compounds of formula II such
reference should be construed as extending also to their
pharmaceutically acceptable salts where relevant. Also,
where any of the compounds referred to can exist in more
than one enantiomeric form, all such forms, mixtures
thereof, and their preparation and uses are within the
scope of the invention.
DESCRIPTION OF EXAMPLES OF PREFERRED EMBODIMENTS
The following examples and descriptions of stages in
synthetic routes of preparation of various preferred
compounds of interest serve to further illustrate the
present invention, but should not be construed in any way
as a limitation thereof.
In the first example (EXAMPLE 1), a method of
preparing an intermediate compound used in EXAMPLE 2 is
described.
~srnrvrpr.~
Methyl 2-(4-Nitrophenyl)benzoxazole-4-carboxylate
(a) 1st stage - Preparation of Methyl 3-hydroxy-2-(N-4-
nitrobenzoyl)aminobenzoate
Methyl 2-amino-3-hydroxybenzoate (0.5g; 2.99mmo1)
was dissolved in m-xylene (40m1) with warming to 60°C.
4-Nitrobenzoyl chloride (0.556m1; 2.99mmo1) was added
dropwise, and this was left to stir for 4 hours. The
solution was cooled to ambient temperature and the m-
xylene removed under reduced pressure. The solid was
dissolved in water (100m1) and the organics extracted
into ethyl acetate (3 x 50m1). The organic fractions
CA 02352592 2001-07-05
were pooled, dried over magnesium sulphate, filtered and
the solvent removed under reduced pressure.
The title product was purified via column chromatography
(1:4 ethyl acetate: petrol as eluent) to yield an orange
solid (490) .
IR . cm-1: 3443 (OH), 2953, 1697, 1649, 1404. M/Z; 316
(15% M+) .
1H: CDC13: b = 3.95 (3H; s; OCH3), 7.25 (1H; t; HS
J=8Hz), 7.31 (1H; dd; H4,; J=6,& 2Hz), 7.67 (1H; dd;
H6,); 8.26 (2H; dd; Hz'; H6' J=2.3Hz); 8.30 (2H; dd;
H3' ; HS' J=2.2Hz) ; 9. 81 (1H; s; OH) ; 12.30 (1H; s;
NH) .
(b) 2nd Stage - Preparation of Methyl 2-(4-nitrophenyl)-
benzoxazole-4-carboxylate
Methyl 3-hydroxy-2-(N-4-nitrobenzoyl)aminobenzoate
(0.1g; 0.34 mmol) was dissolved in m-xylene (20m1), and
to this was added triethylamine (0.033m1; 0.45mmo1) and
pyridinium-4-toluene sulphonate (0.0708; 0.28mmo1). This
was refluxed for 32 hours. The m-xylene was removed
under reduced pressure and the remaining solid dissolved
in water. The organics were extracted into ethyl acetate
(3 x 30m1), dried, filtered and the solvent removed under
reduced pressure to yield a brick red solid (740).
IR . cm 1: , 1726; 1522; 1556, M/Z; 298 (84 0, M+) 267
(100%, -OCH3) , 240 (-CO) 1H: CDCl~: b - 4.01 (3H; s;
OCH3), 7.44 (1H; t; H6 J=8.lHz), 7.76 (1H; dd; H7,;
J=7.2, & 1Hz) , 8. 04 (1H; dd; H5, ) , 8. 35 (2H; dd; Hz' ; H6'
J=2 . 2Hz ) ; 8 . 4 6 ( 2H; d; H3' ; HS' J=2 . 2Hz ) .
EXAMPLE 2
8-Hydroxy-2-(4-nitrophenyl)quinazolin-4-one
(Compound NU1057)
Methyl 2-(4-nitrophenyl)benzoxazole-4-carboxylate
(0.208) obtained as described in Example 1 (2nd stage)
CA 02352592 2001-07-05
9
was dissolved in liquid ammonia (30m1) and sealed in an
autoclave. The reaction mixture was left at 55°C, 20bar
for 20 hours. Under these conditions, the expected 2-(4-
nitrophenyl)benzoxazole derivative apparently rearranged
to give the corresponding quinazolinone derivative. Once
the reaction was complete the ammonia was removed and the
resulting solid recrystallised from boiling ethyl acetate
and petrol ( 84 0 ) .
IR cm 1: , M/Z; 283 (38 M+) 267 (100 0, -NHZ) , 240 (-CO)
0,
1H: CDC13: b 7.35 (1H; dd; H5) , 7.5 (1H; t; H6; ) ,
- 7.7
( 1H; dd; H7 8 . 4 5 d; Hz' ; H6' ) ; 8 . 7 9 ( 2H;
) ( 2H; d; H3' ;
;
HS'), 9.98 (1H; br s; NH), 12.8 (1H; br s; NH).
Similarly, in attempting to prepare benzoxazole-4-
carboxamide, the product underwent a molecular
rearrangement yielding 8-hydroxy-quinazolin-4-[3H]one
(Compound NU1026) which had quite strong PARP inhibitory
activity.
Further examples now follow of the preparation of
more quinazolinone compounds of particular interest.
EXAMPLE 3
8-Methoxy-2-methylquinazolin-4-[3H]-one (Compound NU1063)
nno ~ i-, r"a n
(a) 1st Stage - Preparation of 3-Methoxy-2-nitrobenzamide
A solution of 3-methoxy-2-nitrobenzoic acid (0.1g,
5.1 mmol), thionyl chloride (0.55m1, 7.6 mmol) and
dimethylformamide (0.2m1), in THF (lOml) was stirred for
12 hours at 25°C under nitrogen. Aqueous ammonia (6m1)
was cautiously added and the mixture was stirred for a
further 15 minutes, the solvent was removed under reduced
pressure and the remaining solid was washed with ice-cold
water and collected (0.74g, 750) m.p. 219-222°C
bH (200MHz, d6-DMSO) 4.01 (s, 3H, OCH3); 7.41-7.46 (dd,
1H, Ar-4H); 7.55-7.60 (dd, 1H, Ar-6H); 7.69 7.77 (m, 1H,
Ar-5H); 7.84 (br s, 1H, -NH); 8.31 (br s, 1H, -NH); M/Z
196 (34.3%, M+)
CA 02352592 2001-07-05
Vmax~Cm 1 3350 (br) , 3180 (br) , 3000, 2970, 2920, 2820,
1675.
Elemental analysis: found C 49.03, H 3.93, N 13.97,
C$H$N209 requires C 48 . 98, H 4 . 11, N 14 . 28 0 .
5 (b) 2nd Stage - Preparation of 2-Amino-3-methoxybenzamide
3-Methoxy-2-nitrobenzamide (0.5g, 2.5 mmol) was
dissolved in dry methanol (40m1) and hydrogenated using
palladium-carbon catalyst (80mg). The catalyst was
removed by filtration through Celite, and the residual
10 product (0.35g) was collected and dried (83%) m.p. 145-
147°C
bH (200MHz, d6-DMSO) 3. 88 (s, 3H, -OCH3) ; 6. 40 (br s, 2H, -
NH2); 6.54-6.62 (t, 1H, Ar-5H); 6.96-6.99 (dd, 1H, Ar-4H);
7.23 (br s, 1H, -NH); 7.29-7.33 (dd, 1H, Ar-6H); 7.85 (br
s, 1H, -NH) ~ M/Z 166 (43.8%, M+) Vmax~Cm 1 3480, 3370,
3330, 3150, 2970, 2850, 1680, 1620.
Elemental analysis: found C 57.54, H 5.99, N 16.61,
CeH1aN202 requires C 57.82, H 6.07, N 16.86%.
(c) 3rd Stage - Preparation of 2-N-Acetylamino-3-
methoxybenzamide
To a solution of 2-amino-3-methoxybenzamide (0.5g, 3
mmol) in dry THF (15m1), containing pyridine (0.3m1; 3.9
mmol), was added acetyl chloride (0.2m1, 3.3. mmol) in
THF (2m1) dropwise, and the reaction mixture was stirred
overnight under nitrogen. The solvent was removed under
vacuum and the remaining white slurry washed with aqueous
sodium bicarbonate solution, filtered and washed with
water. The white product (0.198, 31 %) was collected and
dried.
m.p. 243-246°C
~H (200MHz, d6-DMSO) 2.05 (s, 3H, -CH3) ; 3.88 (s, 3H, -
OCH3); 7.14-7.18 (dd, 1H, Ar-4H); 7.21-7.25 (dd, 1H, Ar-
6H); 7.33-7.41 (m, 2H, -NH and Ar-5H; 7.53 (br s, 1H, -
NH); 9.27 (br s, 1H,-NH); M/Z 208 (16.6, M+)
CA 02352592 2001-07-05
11
Vmax/Cm 1 3420, 3240 (br), 3160, 3020, 2980, 2870, 1660.
Elemental analysis: found C 56.98, H 5.38, N 12.78,
C1oH12N203 requires C 57.68, H 5.81, N 13.460.
d) Final Staae - Preparation of 8-Methoxy-2-
methylquinazolin-4-[3H]-one
2-N-Acetylamino-3-methoxybenzamide (0.078, 0.34
mmol) from 3rd stage was dissolved in aqueous sodium
hydroxide solution (2o w/v, 2m1) and the solution was
stirred for 12 hours at 25°C. The reaction mixture was
neutralised with dilute aqueous hydrochloric acid and the
resulting white precipitate that was deposited was
collected by filtration and washed thoroughly with water.
The title compound was recrystallised from ethyl acetate
(0.0438, 670)
m.p. 202-204°C (sublimes).
EXAMPLE 3a
8-Methoxy-2-methylquinazolin-4-[3H]-one (Compound NU1063)
Alternative Method B
To a mixture of 2-amino-3-methoxybenzamide (1.0g, 6
mmol) from the 2nd stage of Example 3 and pyridine
(0.6m1, 7.8 mmol) in dry THF (25m1), was added a solution
of acetyl chloride (0.9m1, 13 mmol) in THF (2m1)
dropwise, and the mixture was stirred overnight under
nitrogen. The solvent was removed under vacuum and the
remaining white slurry was resuspended in 2% aqueous
sodium hydroxide solution and neutralised with aqueous
hydrochloric acid, whereupon a white precipitate formed.
The product was collected by filtration and
recrystallised from methanol-water (0.9158, 80%) m.p.
202-204°C (sublimes)
bH (200MHz, d6-DMSO) 2.43 (s, 3H, -CH3) ; 3. 97 (s, 3H, -
OCH3) ; 7. 37-7. 50 (m, 2H, Ar-6/7H) ; 7. 68-7. 73 (dd, 1H, Ar-
5H) ; ~ C (d6-DMSO) ; 21. 83 (-CH3) ; 56. 05 (-OCH3) ; 114 . 96,
116.99 (Ar-67C); 121.95 (C-CH3); 126.5 (Ar-5C); 140.0 (Ar-
8AC); 153.26 (Ar-8C); 154.33 (Ar-4aC); 162.04 (C=0); M/Z
CA 02352592 2001-07-05
12
190 (96.60, M+) Vmax/Cm 1 3171, 3034, 2903, 1676, 1620,
1574, 1483.
Elemental analysis: found C 62.14, 62.36, H 5.18, 5.29,
N 14.23, 14.36; CloHloNzOz requires C 63.15, H 5.30,
N 14.730.
c~vTnrtnr ~ n
8-Hydroxy-2-methylquinazolin-4-[3H]-one (Compound-NU1025)
A solution of 8-methoxy-2-methylquinazolin-4-[3H]-
one (0.7g, 3.7 mmol) from Example 3 in BBr3 (1.0 M in
CH2C12) 8.4m1, 8.4 mmol) was heated under reflux for 24
hours under nitrogen. Solvents were removed by
distillation under vacuum and the remaining residue was
hydrolysed with sodium hydroxide solution (loo w/v).
Acidification with aqueous hydrochloric acid afforded a
white precipitate, which was removed. The filtrate was
extracted with ethyl acetate (3 x 30m1), dried (MgS09) and
the solvent was removed under vacuum. Recrystallisation
from propan-2-ol-water afforded the target compound (650)
m.p. 253-258°C
~H (200MHz, d6-DMSO) 2.48 (s, 3H, -CH3) ; 7.22-7. 41 (m, 2H,
Ar=6/7H); 7.57-7.62 (dd, 1H, Ar-5H); 9.57 (s, 1H, -OH);
12.26 (s, 1H, -NH); bC (d6-DMSO); 21.72 (-CH3); 115.78,
118.42 (Ar-6/7C); 121.76 (C-CH3)~ 126.54 (Ar-5C); 138.27
(Ar-8aC) 152.58 (Ar-8C); 152.87 (Ar-4aC); 162.05 (C=0);
2S M/Z 176 (100%, M+) : vmax/Cm 1 3320, 3175, 3030, 2900,
2800, 1670.
Elemental analysis: found C 61.39, H 4.54, N 15.88,
C9H8NzOz requires C 61. 36, H 4 . 58, N15. 94 0 .
nvTnrtnT T C
8-Methoxy-2-phenylquinazolin-4-[3H]-one (Compound NU1065)
nrt .. +. Y, .. ,-7 r
(a) 1st Stage - Preparation of 2-N-Benzoylamino-3-
methoxybenzamide
CA 02352592 2001-07-05
13
To a stirred solution of 2-amino-3-methoxybenzamide
(0.5g, 3 mmol) from the 2nd stage of Example 5 in dry THF
(15m1), containing pyridine (0.3m1, 3.0 mmol), was added
benzoyl chloride (0.4m1, 3.3 mmol) in THF (2m1) dropwise.
The reaction mixture was stirred under nitrogen at 25°C.
The solvent was removed under vacuum to afford a white
slurry which was washed with sodium bicarbonate solution,
filtered and washed with water. Recrystallisation from
methanol-water afforded the title compound (0.2g, 410)
m.p. 176-180°C;
bH (200MHz, d6-DMSO) 3.88 (s, 3H, -OCH3); 7.24-7.32 (m,
2H, Ar-4/6H); 7.41-7.49 (m, 2H, -NH, Ar-5H); 7.59-7.73
(m, 4H, -NH, Ph-3'/4'H)~ 8.04-8.08 (dd, 2H, Ph-2'H); 9.85
(s, 1H, -NH); mlz 270 (74.60, M+).
(b) 2nd Staqe - Preparation of 8-Methoxy-2-phenyl
inazolin-4-[3H]-one
2-N-Benzoylamino-3-methoxybenzamide (0.2g, 0.74
mmol) was dissolved in aqueous sodium hydroxide solution
(2% w/v, 2m1) and the solution was stirred at room
temperature for 12 hours. The reaction mixture was
neutralised with hydrochloric acid, and the resulting
white precipitate that formed was collected by filtration
and recrystallised from methanol/water (0.128, 650) m.p.
252-256°C.
EXAMPLE 5a
8-Methoxy-2-phenylquinazolin-4-[3H]-one (Compound NU1065)
Alternative Method B
To a solution of 2-amino-3-methoxybenzamide (1.0g, 6
mmol) (from the 2nd stage of Example 3) and pyridine
(0.6m1, 7.8 mmol) in dry THF (25m1), was added a solution
of benzoyl chloride (0.8m1, 6.6 mmol) in THF (2m1)
dropwise, and the mixture was stirred overnight under
nitrogen. The solvent was removed under vacuum and the
remaining white slurry was resuspended in 2% aqueous
sodium hydroxide solution and neutralised with aqueous
CA 02352592 2001-07-05
14
hydrochloric acid, whereupon a white precipitate formed.
The product was collected by filtration and
recrystallised from methanol-water (1.1g, 750) m.p. 252-
256°C;
bH (200MHz, d6-DMSO) 4.06 (s, 3H, -OCH3); 7.47-7.61 (m,
2H, Ar-4/6H); 7.63-7.69 (m, 3H, Ph-3'/4'H); 7.80-7.85
(dd, 1H, Ar-5H); 8.27-8.32 (m, 2H, Ph-2'H); 12.70 (s, 1H,
-NH) ; M/Z 252 (100%, M+) ; Vmax/Cm 1 3330, 3190, 3170,
3120, 3070, 2950, 2890, 2830, 1660.
Elemental analysis: found C 71.38, H 4.39, N 11.17,
CisHizNzOz requires C 71.42, H 4.79, N 11.10%.
L'Y21MDT ~'
8-Hydroxy-2-phenylquinazolin-4-[3H]-one (Compound NU1068)
A solution of 8-methoxy-2-phenylquinazolin-4-[3H]-
one (0.5g, 2 mmol) from Example 5 or 5a in BBr3 (1.0 M in
CHZClz) (6m1, 6 mmol) was heated under reflux for 24 hours
under nitrogen. Solvents were removed by distillation
under vacuum and the remaining residue was hydrolysed
with sodium hydroxide solution (loo w/v). Acidification
with aqueous hydrochloric acid afforded a white
precipitate, which was removed. The filtrate was
extracted with ethyl acetate (3 x 30m1), dried (MgS04) and
the solvent was removed under vacuum. Recrystallisation
from propan-2-of afforded the target compound (0.187mg,
67 0 )
m.p. 280-284°C;
bH (200MHz), d6-DMSO) 7.73-7.50 (m, 2H, Ar-6/7H); 7.66-
7.72 (m, 4H, Ar-5H, Ph-3'/4'H); 8.51-8.54 (dd, 2H, Ph-
2H); 9.75 (bs, 1H, -OH); 12.60 (bs, 1H, -NH); bC (d6-
DMSO); 116.01, 118.68 (Ar-6/7C); 122.03 (C-Ph); 127.43-
128.76 (Ph-1'/2'/3'/4'C); 137.98 (Ar 8aC); 150.72 (Ar-
8C); 153.31 (Ar-4aC); 162.62 (C=0); M/Z 238 (100%, M+);
Umax/Cm 1 (approx. values) 3380 (br), 3180, 3120, 3050,
2940, 1640.
' CA 02352592 2001-07-05
Elemental analysis: found C 69.54, H 4.05, N 11.46,
C14H10N202 requires C 70.58, H 4.23, N 11760.
L'Y11MDT L' '7
2,8-Dimethylquinazolin-4-[3H]-one (Compound NU1069)
5 To a solution of 2-amino-3-methylbenzamide (0.5g,
3.3 mmol) (prepared by conventional methods) and pyridine
(0.35m1, 4.3 mmol) in dry THF (15m1), was added a
solution of acetyl chloride (0.36m1, 5.0 mmol) in THF
(2m1) dropwise, and the mixture was stirred overnight
10 under nitrogen. The solvent was removed under vacuum and
the remaining white slurry was resuspended in 2o aqueous
sodium hydroxide solution and neutralised with aqueous
hydrochloric acid, whereupon a white precipitate formed.
The solid was collected and recrystallised from methanol-
15 water to furnish the required quinazolinone (0.478, 810)
m.p. 217-220°C;
bH (200MHz, d6-DMSO) 2.44 (s, 3H, -CH3) ; 2.57 (s, 3H, -
CH3); 7.36-7.44 (t, 1H, Ar-6H); 7.68-7.72 (dd, 1H, Ar-7H);
7. 97-8. 0l (dd, 1H, Ar-5H) ; 12.25 (br s, 1H, -NH) ; M/Z 174
2,~ (1000, M+) : Vmax~Cm 1 3325, 3180, 3040, 2990, 2910, 2880,
2795, 1680, 1620.
Elemental analysis: found C 68.76, H 5.57, N 15.90,
CioHloNzO requires C 68.94, H 5.76, N 16.08%.
ASSAY FOR PARP INHIBITORY ACTIVITY
Compounds of the present invention, particularly
those detailed in the preceding Examples, have been
tested in vitro for activity as PARP inhibitors using the
following methods and materials.
In principle, the PARP assay used relies upon
activating endogenous PARP (as hereinafter described) in
cells containing exogenous [32P]-NAD+ introduced therein
by suspending the cells in a solution of [32P]-NAD+ to
which they have been rendered permeable in an initial
pre-treatment step. The poly(ADP-ribose) which is then
- CA 02352592 2001-07-05
16
synthesised by the enzyme can be precipitated by tri-
chloracetic acid (TCA) and the amount of radio-labelled
3zP incorporated therein measured, e.g. using a
scintillation counter, to give a measure of the activity
of the PARP under the particular conditions of the
experiment. By repeating the experiment following the
same procedure, and under the same conditions, in the
presence of each compound to be tested the reduction in
enzyme activity, representative of the inhibitory effect
of the test compound, can then be ascertained from the
reduction, if any, of the amount of [3zP] measured in the
TCA precipitated poly(ADP-ribose).
The results of this assay may be expressed in terms
of percentage inhibition or reduction in activity for one
or more different concentrations of each compound tested,
or it may be expressed in terms of that concentration of
the tested compound which reduces the enzyme activity by
50%, i.e. the ICSO value. Thus, with a range of different
compounds a set of comparative values for inhibitory
activity can be obtained.
In practice, L1210 murine leukaemia cells were used
as the source of the PARP enzyme after being rendered
permeable to exogenous [32P]NAD by exposure to hypotonic
buffer and cold shock. In the preferred technique
developed, which has been found to give exact and
reproducible results, a defined amount of a small
synthetic oligonucleotide, in particular a single strand
oligo-nucleotide having the palindromic sequence
CGGAATTCCG, is introduced into the cell suspension for
activating the PARP enzyme. This oligonucleotide
sequence snaps back on itself to form a double-stranded
molecule with a single blunt end and provides an
effective substrate for activation of PARP. Its
behaviour as a potent activator of the enzyme was
confirmed in the tests carried out.
The experimental protocol adopted, in which a
synthetic oligonucleotide as mentioned above is
CA 02352592 2001-07-05
17
introduced as a specific activator of PARP, discriminates
between PARP and other mono-ADP-ribosyltransferases in
the cells. Thus, introduction of such synthetic
oligonucleotides causes a 5 to 6 fold stimulation in the
S radioactive label incorporated and this is attributable
solely to PARP activity.
Further details of the assay are given below.
__,_
The materials used included the following:
DTT (Dithiothreitol
A 100mM (15.4mg/ml) solution (for use as an anti-
oxidant) was made up, divided into 500u1 aliquots
and stored at -20°C.
Hypotonic buffer:
9mM Hepes (214mg/100m1)
4.5% Dextran (4.5g/100m1)
4.5mM MgCl2 (92mg/100m1)
The above ingredients were dissolved in about
80m1 distilled water, pH was adjusted to 7.8
(NaOH/HC1), the solution was then made up to 100m1
with distilled water, and stored in a refrigerator.
DTT was added to 5mM just before use (50u1/ml).
Isotonic buffer:
40mM Hepes (1.9g/200m1)
130mM KC1 (1.94g/200m1)
4o Dextran (8g/200m1)
2mM EGTA (152mg/200m1)
2.3mM MgCl2 (94mg/200m1)
225mM Sucrose (15.39g/200m1)
The above ingredients were dissolved in about
150m1 distilled water, pH was adjusted to 7.8
(NaOH/HC1), the solution was then made up to 200m1
CA 02352592 2001-07-05
18
with distilled water and stored in a refrigerator.
DTT was added to 2.5mM just before use (25u1/ml).
NAD
NAD was stored as a solid in pre-weighed aliquots at
-20°C. From these, solutions of a concentration of
approximately 6mM (4-4.5mg/ml) were freshly made up
shortly before performing an assay, and the molarity
was checked by measuring the optical density (O. D.)
at 260nm. The stock solution was then diluted with
water to give a concentration of 600uM and a small
amount of 32P labelled NAD was added (e. g. 2-5u1/ml).
Oliaonucleotide
The oligonucleotide having the palindromic sequence
CGGAATTCCG, synthesised by conventional means, was
vacuum dried and stored as pellets in a freezer.
Before use, it was made up to 200ug/ml in lOmM
Tris/HC1, pH 7.8, with each pellet being dissolved
completely in 50m1 of buffer. The solution was then
heated to 60°C in a water bath for 15 minutes, and
allowed to cool slowly to ensure correct
reannealing. After adding 9.5m1 of buffer, the
concentration was checked by measuring the optical
density of a diluted sample at 260nm. The main
solution was then diluted to a concentration of
200ug/ml and stored in 500u1 aliquots in a
freezer, ready for use.
TCA
Solutions of TCA (Trichloroacetic acid) were
prepared at two concentrations. 10% TCA + l00
sodium pyrophosphate, and 1o TCA + 1% sodium
pyrophosphate.
Cells
The L1210 cells used as the source of the PARP
enzyme were maintained as a suspension culture in
RPMI medium + loo foetal bovine serum + glutamine
CA 02352592 2004-O1-21
19
and antibiotics (penicillin and streptomycin).
HEPES and sodium bicarbonate were also added, and
the cells were seeded in 100m1 of medium such that
there would be a concentration of approximately 8 x
105/m1 at the time of carrying out an assay.
Method
The compounds being tested were generally made up as
a concentrated solution in DMSO (Dimethyl sulphoxide).
The solubility of the compound was then checked by adding
a quantity of the DMSO solution to a quantity of the
isotonic buffer, in the required final proportions that
were to be used in carrying out the assay, and after an
interval the solution was examined under a microscope for
any signs of crystals forming.
A desired quantity of the cells, ascertained by
counting with a haemocytometer, was then centrifuged
(1500rpm in a EuropaTM model 24M centrifuge for 5
minutes), the supernatant removed, and the pellets
obtained were resuspended in 20m1 Dul A at 4°C before
centrifuging again at 1500rpm and 4°C. After again
removing the supernatant, the cells were resuspended at a
concentration of 3 x 107 cells/ml in ice cold hypotonic
buffer and left for 30 minutes on ice. Nine volumes were
then added of ice cold isotonic buffer, and the cells,
now rendered permeable to exogenous NAD+, were then used
within the next hour for carrying out an assay. The
permeablisation of the cells may be checked at this stage
by adding duplicate aliquots of cells to an equal volume
of trypan blue, leaving for 5 minutes and then counting
on a haemocytometer.
The assay was then carried out using for convenience
plastic 15m1 conical bottomed assay tubes set up in a
shaking water bath at 26°C which is the optimum
temperature for this enzyme. In a typical assay using
the oligonucleotide solution at a concentration of 5~g/ml
CA 02352592 2001-07-05
and the test compound/DMSO solution at a concentration of
20, and carrying out the assay 'in quadruplicate, there
would then be placed in each assay tube 5u1 of the
oligonucleotide solution, 50u1 of the 600um NAD + [32P]-
5 NAD solution, 8u1 of the test compound/DMSO solution, and
37u1 of water. Prior to the start of the experiment this
"cocktail" would be pre-warmed for 7 minutes at 26°C, as
would be also the cell suspension. The reaction would
then be started by adding 300u1 of the cell suspension.
10 The reaction would be stopped by adding 2m1 of the l00
TCA + 10% sodium pyrophosphate solution.
In addition to the above, six assay tubes would
usually be set up as blanks, these containing the same
ingredients as above but, before adding the cell
15 suspension, TCA solution is added to prevent any reaction
from taking place. This enables corrections to be
applied for any non-specific binding of the labelled
material to the filter used (see below).
After adding the cell suspension at timed intervals
20 to each of the assay tubes, the 10% TCA + 10% sodium
pyrophosphate at 4°C was added to each assay tube exactly
5 minutes after addition of the cell suspension to that
tube. Then, after leaving the tubes on ice for a minimum
time of one hour, the contents of each individual tube
were filtered through an individual filter funnel of a
suction filter apparatus using GF/C filter elements
(rough side up) wetted with loo TCA. After filtering the
contents of each tube and rinsing the filters several
times with to TCA + to sodium pyrophosphate solution, the
filters were carefully removed and dried before being
placed in individual scintillation vials. Four
additional scintillation vials were also set up as
reference standards containing 10u1 of the 600uM NAD +
[32P]-NAD solution, lOml scintillant then being added to
each vial. Counting was carried out for 2 minutes on a [3
counter to obtain measures of the 32P present, and thus
' CA 02352592 2001-07-05
21
the amount of the poly(ADP-ribose) and activity of the
PARP enzyme.
RESULTS OF IN VITRO PARP INHIBITION STUDIES
Apart from applying the PARP enzyme assay in
accordance with the standard procedure outlined above to
a range of compounds which have been made in accordance
with the present invention, for comparison purposes it
was also applied to certain benzamide compounds, in
particular 3-hydroxybenzamide, 3-methoxybenzamide and 3-
aminobenzamide, that are already known to exhibit certain
PARP inhibitory activity. A full tabulated list of the
compounds which have been made and/or studied is
hereinafter presented in TABLE III, together with the
PARP inhibition assay results obtained in different
experiments for different concentrations of the compounds
when tested using the assay hereinabove described.
In reviewing this list, the known PARP inhibitors 3-
aminobenzamide, 3-methoxybenzamide and 3-hydroxybenzamide
may be regarded as reference compounds. Although there
is considerable variation in activity, and in some cases
at least the higher concentrations for aqueous solutions
of the test compounds could not be achieved because of
low solubility, in general the compounds of the present
invention which were tested showed a useful degree of
activity, particularly compounds NU1025, NU1057, NU1063,
NU1068 and NU1069.
In contrast to the results obtained for the
compounds of the present invention, which have in many
cases showed PARP enzyme inhibitory properties that are
well above average and at least comparable with, if not
considerably better than, those of ather known benzamide
PARP inhibitors, various analogous nicotinamide compounds
studied showed no, or very poor, inhibitory activity when
tested in the same manner at similar concentrations.
~ CA 02352592 2001-07-05
22
FURTHER BIOLOGICAL ACTIVITY STUDIES
Again using cultures of the murine leukaemia L1210
cell line, growth inhibition experiments were carried out
to assess the cytostatic effects of the compounds and
clonogenic survival assays were performed to assess
cytotoxicity, especially in relation to use of the
compounds in conjunction with DNA damaging cytotoxic
agents such as cytotoxic antitumour drugs or high energy
radiation. DNA damage and the effect of the PARP
inhibitors on the process of DNA strand break formation
and repair has also been assessed by carrying out DNA
strand break assays and monitoring by alkaline elution in
accordance with published techniques.
By way of example some further details are given
below of studies carried out using the quinazolinone
compounds identified by the reference numbers NU1025 and
NU1057 (derivable by molecular rearrangement of
corresponding benzoxazole compounds) as representative
examples of the PARP inhbiting compounds of the present
invention, and also using for comparison the known PARP
inhibitors 3-aminobenzamide (3AB) and benzamide (BZ)
itself. Results of experiments using the alkylating
agent temozolomide (TM) are also reported, taking this as
a illustrative example of a cytotoxic DNA damaging
antitumour drug, and in some of the studies carried out
gamma ray IRradiation was used to damage the cells.
In the growth inhibition assays, typically the L1210
cells would be seeded at I x 109/m1 in triplicate in 24
well multidishes, and 24 hours later the compounds or
drugs being tested would be added in selected
combinations and concentrations. At this time one set of
replicates would be counted using a Coulter counter (No),
and 48 hours later the remaining samples would be counted
(N1). The percentage (%) growth inhibition of drug-
treated samples could then be estimated. In drug
combination experiments, where evidence of synergistic
effects on cell growth or clonogenicity was being sought,
CA 02352592 2001-07-05
23
a single, fixed concentration of a cytotoxic drug sample,
e.g. temozolomide, would be taken as the control value.
As an illustration of the results obtained, there is
shown in TABLE I at the end of this description the IC5o
values of the above-mentioned PARP inhibitors when used
alone and in conjunction with a fixed concentration
(100uM) of temozolomide, as estimated from the growth
inhibition experiments. Although not shown, it may be
noted that exposure of the cells to TM alone caused
inhibition of cell growth with an ICSO value of 361~25uM.
Also, it was established that co-exposure of the cells to
100uM TM with increasing concentrations of the PARP
inhibitors caused a synergistic increase in growth
inhibition throughout a range of concentrations.
It will be seen from Table I that 10-20 fold higher
concentrations of the compound NU1025 used alone were
required to inhibit cell growth than were required when
the compound was used in conjunction with 100uM TM. For
example, the ICso of NU1025 alone was 0.41mM, and this was
reduced to 0.04mM in the presence of TM. In comparison,
only 2-3 fold differences were obtained with 3AB and BZ,
where there was considerable overlap between the growth
inhibitory effects of the compounds per se and their
effects in conjunction with TM. An identical rank order
was obtained when comparing the effectiveness of the
compounds as PARP inhibitors and their ability to inhibit
cell growth which at least suggests that PARP function is
essential for cell growth.
In the clonogenic survival assays, typically the
L1210 cells were exposed to varying concentrations of TM
~ a fixed concentration of PARP inhibitor for a fixed
time of 16 hours, prior to counting and seeding for
colony formation in 0.12-0.150 agarose in drug-free
medium. After 7-10 days colonies were stained with
0.5mg/ml MTT and counted by eye on a gridded light box.
Survival curves were plotted and typical DEF10 values
obtained are hereinafter given in Table II (DEF10 being
~ CA 02352592 2001-07-05
24
defined as the ratio of the concentration of TM that
reduces survival to loo divided by the concentration of
TM that reduces survival to loo in the presence of a
fixed concentration of PARP inhibitor). Each DEF10 value
in Table II represents the average ratio ~ S.E. (standard
error) derived from the averaged 10% survival for TM
alone (675 ~ 31~M from 22 independent survival curves)
divided by individual loo survival values from at least 3
independent survival curves performed in the presence of
a fixed concentration of inhibitor.
A reasonable correlation was found between growth
inhibitory effects and cytotoxic effects for TM alone
with an ICso value of 361uM ~ 25uM and a LD50 value of 251
~ l3uM respectively, despite differing exposure times (48
hours for growth inhibition and 16 hours for
cytotoxicity). TM has a half life in culture of about 40
minutes, and therefore will exert its full effects well
before the minimum duration of exposure of either
experiment. All compounds potentiated TM cytotoxicity,
but NU1025 produced about the same DEF10 values at very
much lower concentrations than 3AB and BZ respectively.
For example, 50uM NU1025 and 5mM 3AB gave equivalent
DEF10 values of about 4. For NU1025 maximal potentiation
of cytotoxicity was obtained by a concentration in the
range of 50-100uM, and was significant at doses as low as
10~M.
In other clonogenic survival assays gamma ray
irradiation was used to damage the cells. Typically,
L1210 cells (3m1, 4 x 103/m1 in plastic bijoux bottles)
were irradiated at 4°C with varying doses of gamma rays
in the presence or absence of lOmM 3AB or 200uM NU1057
and a final concentration of 2% DMSO. The cells were
then incubated at 37°C for 2 hours in the continued
presence or absence of PARP inhibitor prior to seeding
for colony formation. A significant potentiation of
gamma ray cytotoxicity by NU1057 was observed, with a
DEF10 of 1.l.
CA 02352592 2001-07-05
2S
Repair of potentially lethal damage (PLD) occurs
when cells are held in stationary-phase following
initiation of PLD prior to allowing cell division to take
place. In further typical experiments, L1210 cells were
S allowed to repair gamma ray PLD in the presence or
absence of 3AB or NU1025 as follows. L1210 cells were
maintained in culture until they had attained stationary
phase (>106cells/ml). They were diluted to 1.5 x 105/m1
in conditioned medium from stationary-phase cultures to
prevent further cell division. Replicate 2m1 samples of
cells in plastic bijoux were held on ice prior to and
immediately following 8 Gray gamma ray irradiation. lml
of 3x final concentration of compounds 3AB or NU1025 made
up in conditioned medium from stationary cultures was
1S added (to give final concentrations of 106cells/ml in 1%
DMSO ~ lOmM 3AB or 200uM NU1025) and the cells were
incubated at 37°C for 0, 2 or 4 hours prior to
resuspending in drug-free medium and seeding for colony
formation. Unirradiated stationary phase cultures
incubated at 37°C for 0, 2 or 4 hours with to DMSO ~ lOmM
3AB or 200uM NU1025 were used as appropriate controls for
determining relative cell survival. In the absence of
PARP inhibitor cell survival increased with time allowed
for PLD repair to take place. For example, when seeded
2S immediately after irradiation (no repair) only about 0.20
of the cells survived, but after a 4 hour repair period
this had increased to 0.70. It was observed that both
3A8 and NU1025 blocked this repair.
The cytotoxic effects of the PARP inhibiting
compounds alone has also been investigated. In one set
of experiments, the LD50 values for a 24 hours exposure
of L1210 cells were 14 ~ l.OmM (3AB); 6.0 ~ l.5mM (BZ)
and 1.6 ~ O.lMm (NU1025). The LD50 values differed by
_<3-fold from the ICSO values but maintained the same rank
3S order with respect to their potency as PARP inhibitors.
In agreement with the growth inhibition data there was
_>10-fold difference between the concentration of NU1025
needed to produce maximal potentiation of TM cytotoxicity
CA 02352592 2001-07-05
26
and the concentration needed to produce cytotoxicity per
se.
In respect of DNA strand break assays carried out,
it was found that a 1 hour treatment with TM resulted in
a concentration-dependent increase in the rate of elution
which provides a measure of the extent of DNA strand
breakage. Changes in DNA strand break levels were
detectable at levels of TM as low as 150uM, a
concentration which reduced survival by about 30%. All
the compounds were tested for their ability to produce
strand breaks when used alone. A 24 hour incubation of
cells with 1mM NU1025, and 20mM 3AB or BZ had no
significant effect on DNA strand break levels compared to
untreated cells. However, coincubation for 1 hour of a
fixed concentration of TM (150uM) with increasing
concentrations of all PARP inhibitors tested caused a
progressive increase in the rate of elution (extent of
strand breakage) compared to TM alone.
The results for all the 3 representative compounds
mentioned have been summarised by plotting values of a
parameter related to extent of strand breakage versus
inhibitor concentration. For all the compounds, the
strand breakage increased linearly with increasing
concentration, but values started increasing
significantly for NU1025 at about 100uM, whereas
concentrations above 3mM and 5mM were required to
significantly increase values for BZ and 3AB
respectively. Again, the rank order and potency of the
compounds in the DNA strand break assay demonstrated an
excellent correlation with in vitro PARP inhibitory
potency.
Overall, it is believed that the studies carried out
give clear evidence that the PARP inhibitory character-
istics of compounds of this invention reflects an ability
of these compounds to potentiate the cytotoxicity of DNA
damaging agents such as certain cytotoxic antitumour
drugs and radiation used in radiotherapy. Accordingly,
- CA 02352592 2001-07-05
27
such compounds should be especially useful for
administration in conjunction with such cytotoxic drugs
or radiotherapy to potentiate their effect in the course
of medical treatment as hereinbefore indicated.
Summary
Although the present invention should be regarded
overall as comprising each and every novel feature or
combination of features disclosed herein, the main
aspects of the invention comprise, principally but not
exclusively, broadly the following:-
(i) Novel compounds of formula (II) as defined herein;
(ii) Compounds of formula (II) with substituents as
hereinbefore defined (including salts thereof) for
therapy or for use in medicine and in the
manufacture of medical preparations, useful for
example as PARP inhibitors to be administered in
conjunction with cytotoxic drugs or with
radiotherapy to potentiate the effectiveness of the
latter in treatment of cancer;
(iii)Processes for the preparation of novel compounds of
formula (II) as defined herein, including any novel
intermediate compounds produced in carrying out such
processes;
(iv) Pharmaceutical formulations comprising a compound of
formula (II) as defined herein together with a
pharmaceutically acceptable carrier therein;
(v) Processes for the preparation of a pharmaceutical
formulation as defined in (iv) above, e.g. by
methods referred to herein;
(vi) Quinazolinone compounds of formula (II), possibly
representing molecularly rearranged compounds of
formula (IV), as herein disclosed, for therapy or
for use in medicine and in the manufacture of
medical preparations, useful for example as PARP
CA 02352592 2001-07-05
28
inhibitors to be administered in conjunction with
cytotoxic drugs or with radiotherapy to potentiate
the effectiveness of the latter in treatment of
cancer, and pharmaceutical formulations comprising
said quinazolinone compounds.
CA 02352592 2001-07-05
29
TABLE I
INHIBITOR ICS (mM) t SE ICS (mM) t SE
INHIBITOR ALONE INHIBITOR + IOO~cM
TM
3-AMINOBENZAMIDE 6.7 t 0.2 2.5 0.1
BENZAMIDE 2.5 0.3 0.84 0, t2
NU1025 ~ 0.41 +_ 0.06 ~ 0.04 0.003
TABLE II
INHIBITOR CONCEi~ITRATION DEFT;
1 mM 2.4 0.3
3-A~IINOBENZAMIDE SmM 4.1 0.4
1 mM 4.0 t 0.7
BENZAMIDE 3mM 6.9 0.2
10~M 2.0 0.2
NU1025 ~ SOUM ' 4.0 0.5
100uM 5.1 0.7
CA 02352592 2001-07-05
TABLE III
House Name Structure % Inhibition
Number
10~M 30~M 100uM
Ref 3-hydroxybenzamide O 35 59 81
C~H~N02 ~ NH2
MW = 137 \
OH
Ref 3-methoxybenzamide O 55 78 89
CgH9N02 \ NH2
MW = 151 /
OCH3
Ref 3-aminobenzamide 0 36 63 79
C~HgN202 ' / NH2
MW=136 \
NHz
NU 1025 ~ 8-hydroxy-2-methyl- O 92 92 96.
quinazolin-4-(3HJone 1 ltM = 63
/ ~HN 0.1 uM = 18
C9HgN20Z I ~ O.SIeM = 59
\ N~ CH3 1.O~.M,= 68
M W = 176
OH IC = 0.4 'M
NU1026 8-hydroxyquinazolin-4- p 78 87 95
[3H]one O.S~M = 18
/ ~NH 1.OItM = 38
CgH6N202 \ ~ ~ 2.OlaM = 54
MW = 162 OH lCso = 2 wM
CA 02352592 2001-07-05
w
31
TABLE III (contd.)
House Name Structure % Inhibition
Number
IOM 30pM 100 M
NU1057 8-hydroxy-2- 0 92
(4-nitrophenyl~-
-quinazolin-4-one ~ ~ ICsp = 0.23p.M
i
C14H9N30d
Mw = 283.2
NU I 063 8-methoxy-2- O
methylquinazolin-4[3Hj- ICSQ = 0.78~.M
-one
CloHloN20z \ N~ CHa
MW = 190.2
NU 1065 8-methoxy-2- ~
phenylquinazolin-4[3HJ- ICsp = 4.2~1H
-one ~ ~ ~NH
~C H NO
l5 !2 2 2
OCH3
MW = 252.27
NU1068 8-hydroxy-2- p
phenylquinazolin-4[3Hj- / NN ICso = 0.53~M
-one
C 14H 10N2C2 OH
238.24
NU1069 2,8-dimethylquinazolin- O
4[3FIJ-one ICso = O.ZpM
~NH
Cloth ioN202
'N CH3
174.2
CH3