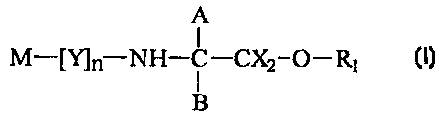Note: Descriptions are shown in the official language in which they were submitted.
CA 02353839 2001-06-04
WO 00/34281 PC'T/AU99/OI073
1
Methotrexate derivatives
TECHNICAL FIELD
The present invention is concerned with derivatives of methotrexate (MTX);
compositions containing the derivative and to methods of treatment involving
use of
these MTX derivatives.
BACKGROUND
MTX can bind to active sites on the enzyme dihydrofolate reductase (DHFR) and
through competitive inhibition block the formation of tetrahydrofolates (THF)
needed in
the biosynthesis of DNA and RNA. MTX also inhibits other folate dependent
enzymes.
The ability of MTX to inhibit nucleic acid synthesis has been used to treat
abnormal cell growth. Since actively proliferating cells are more sensitive to
MTX, it can
be used to selectively impair cancerous cell growth. MTX is a widely used anti-
cancer
25 agent used to treat neoplastic diseases. MTX also acts as an
antiproliferative and
immunoregulator and can be employed in the treatment of psoriasis and
rheumatoid
arthritis.
Oral MTX is currently used for the treatment of moderate or severe arthritis
and
recalcitrant psoriasis. The weekly dose is 2.5 to 25 mg. Use of this drug is
restricted by
its toxicity with the major long term side effect being liver damage. Attempts
to reduce
toxicity and improve efficacy in psoriasis treatment by applying MTX topically
have met
with little or no success, possibly due to either poor dermal penetration or
residence
time of the drug in the skin.
DISCLOSURE OF THE INVENTION
In a first aspect, the present invention provides
A
M-[Y]ri NH-C-CX2-O-R~
B
in which M is methotrexate or an analogue thereof;
A H, CXz-O-R2 or halogen;
B=H, CXz-O-R3 or halogen;
X is independently H or halogen;
n is 0 or greater than or equal to l;
Y is a linker group, and where n is greater than i, each Y is the same or
different;
CA 02353839 2001-06-04
WO 00/34281 PCTIAU99/01073
R1,R2, and R3 are the same ar different and are either hydrogen, substituted
or
unsubstituted methyl, ethyl, a saturated or unsaturated fatty acyl group,
with the proviso that when n is 0 or l, and A is CH2-O-R2 and/or B is CH2-O-
R3,
then R,, RZ and Rz are not selected from a fatty acyl group;
with the proviso that when n is greater than or equal to 2, and A is CH2-O-R2
and/or B is CH2-O-Rz, and at least one of R,, R2 and R3 is a fatty acyl group,
then
-[Y)" is other than -(AA)" or -Y-(AA)"_x-, where AA is an amino acid.
Examples of analogues of methotrexate are described in the prior art. See, for
example, .A.L Jackman ed (1999). "Antifolate Drugs in Cancer Therapy" Humana
Press,
Totowa, New Jersey; Matsuoka, H and Mihara, M (1998). " The synthesis and
biological evaluation of new methotrexate derivatives in rheumatoid arthritis.
Drugs of
the Future 1998. 23(9);1015-1022; and Degraw, J et al. (1995) "New analogs of
methotrexate in cancer and arthritis". Current Medicinal Chemistry 2:630-653,
the
disclosures of which are incorporated herein by reference.
Preferably M is methotrexate.
Examples of linker groups that are useful in the present invention include:
a) a linker with an amino group to M and a carboxyl group to the
NHC(A){B)CXZOR, group, for example, an amino acid or antibiotic.
b) a linker with an amino group to M and a sulphonic acid group to the
NHC(A){B)CXZOR, group, for example, 2-aminoethanesulphonic acid (taurine).
c) a linker with an hydroxyl group to M and a carboxyl group to the
NHC(A){B)CXzOR, group. The linker may be an hydroxy acid. The hydroxy acid may
be
analogue of an amino acid in which the amino group of the amino acid is
replaced by an
hydroxy group. Examples of hydroxy acids include glycolic acid (HOCH2COOH);
lactic
acid (HOCH(CH3)COOH; 4-hydroxy-butyric acid HOCHZCH2CHZCOOH; 6-hydroxy-
caproic acid; 10-hydroxy-decanoic acid; 2-hydroxy-caproic acid; 2-
hydroxybutyric acid;
3-hydroxy-butyric acid, etc. The use of these acids allows linkage of the
Tris, or Tris-
conjugates to the drug via a labile ester bond rather that via the more stable
amide bond.
d) a linker with an hydroxyl group to M and a sulphonic acid group to the
NHC(A){B)CXzOR, group, for example, 2-hydroxyethanesulphonic acid (isethonic
acid).
e) a linker with an hydroxyl group to M and a reactive halide group to the
NHC(A){B)CXZOR, group, for example, 2~chloroethanol.
~ a linker having an hydroxy group and an aidehyde group, for example, p-
hydroxybenzaldehyde,
CA 02353839 2001-06-04
WO 00/34281 PCT/AU99/01073
g} a linker having a halide group and an carboxyl group, for example, 2-
chloraacetic acid;
h) a linker having two reactive halide groups, for example, 1,2-dibromoethane;
and
i) an alkylene oxide, for example, ethyleneoxide.
The NHC(A}(B}CX20R1 group may be attached to the a or y carboxyl group of
the methotrexate compound. Most preferably, the -NHC(A)(B)CX20RI group is
attached to the y carboxyl of the methotrexate compound, optionally by the
linker.
Where Y is an amino acid it may be any suitable amino acid including, natural
and non-natural amino acids and analogues thereof. Preferably the amino acid
is glycine.
Preferably Rl, R2 and R3 are each H.
Preferably n is in the range 0-S.
In a preferred embodiment of the invention, M is methotrexate and -[Y]"- is
-[AA]"-, where AA is an amino acid. Preferably AA is glycine and n is 1. Where
n >_2,
25 each AA need not be the same amino acid.
A preferred methotrexate derivative is methotrexate-y-glycine-TRIS (MTX-y-
GT).
In a second aspect, the present invention provides a pharmaceutical
composition
incorporating a compound of the formula I as defined above and a
pharmaceutically
acceptable carrier or diluent.
In a preferred form of the composition of the invention, M is methotrexate. Y
is
glycine and n is 1.
One preferred methotrexate derivative is methotrexate derivative is
methotrexate-y-glycine-Tris.
Compositions suitable for oral administration include liquid solutions;
capsules,
suspensions, emulsion etc.
The composition of the invention may be in tablet form incorporating the usual
tablet formulation ingredients.
Composition suitable for parenteral administration include aqueous and non-
aqueous injectable solutions or emulsions.
Formulation suitable for topical administration may be in the form of a cream,
gel, paste, salve, poultice, nasal spray, pulmonary aerosol or foam
incorporating the
compound of the present invention.
The composition of the invention may be in the form of suppositories,
pessaries,
tampons, creams, gels, pastes, lotions, implants, patches or foams.
The composition of the invention may be in the form of an implant.
CA 02353839 2001-06-04
WU 00134281 PCT/AU99/01073
4
The composition of the invention may be in the form of a liquid aerosol or
powder for administration to the lung.
The dosage level of the MTX derivative of the present invention in the
pharmaceutical composition may be sufficient to effect a prophylactic or
therapeutic
response in the selected time frame.
The present invention also provides a method of treatment of cancer or a
disease
with an autoimmune component in a subject, the method including administration
of a
prophylactically or therapeutically effective amount of a compound of the
formula I
given above.
Accordingly, in a third aspect, the present invention provides a method of
treatment of a disease with an autoimmune component or a cancer in a subject,
the
method including administration to the subject of an effective amount of a
compound in
accordance with of formula II:
D
M-~ri ~-C~C~-O-Rs
E
wherein:
D=H or CXZ-O-R4 or halogen;
E=H or CX2-O-RS or halogen;
X is as defined above;
n is as defined above;
Y as defined above; and
R4, RS and R~ are the same or. different and either hydrogen, substituted or
unsubstituted methyl, ethyl or fatty acyl groups with either saturated or
unsaturated
bonds.
The disease with an autoimmune component may be any condition where
immunosuppression is called for. For example, the disease with an autoimmune
component may be psoriasis or rheumatoid arthritis.
The disease with an autoimmune component may be inflammatory bowel disease
or Crohn's disease in a subject.
Preferably the compound used in the method of the third aspect is a compound
of
formula I.
The method of treatment of the present invention may be achieved by any
suitable route, for example, oral, parenterai, implant. and topical.
CA 02353839 2001-06-04
WO 001342$1 PCT/AU99/01073
The dosage level of the MTX derivative of the invention depends on the nature
and severity of the condition being treated but generally the administered
amount is
sufFicient to produce a prophylactic or therapeutic effect. Pharmaceutical
compositions
in accordance with the invention may contain the MTX derivative in an amount
in the
range of about 0.00 i to about 90% of the composition. A composition in an
oral form,
for example, a tablet, may contain up to 85% of the MTX derivative, preferably
about 2
to 60% of the oral composition. The concentrations here set forth are provided
by way
of description only and not by way of limitation.
In order that the invention may be more readily understood, the following non-
limiting embodiments are provided.
BRIEF DESCRIPTION OF DRAWINGS:
Figure 1 is a boxplot demonstrating the effect of oral MTX-y-GT, MTX-y-GTP 1
and
MTX-'y-GTP3 in the rat assay of delayed type hypersensitivity footpad
oedema. MTX-'y-GT and MTX-y-GTP1 are both active in this assay as is
MTX. MTX-'y-GTP 1 is statistically more effective than MTX (p=0.021).
Figure 2 shows the effects of MTX and MTX-y-GT in the rat adjuvant-induced
arthritis
model. The results indicate that MTX-y-GT is, at the two lower doses,
equipotent to MTX in delaying the onset of joint inflammation in this model.
Figure 3 shows the suppressing effect of MTX-y-GTP3 on epidermal DNA synthesis
following UVB irradiation. After UVB irradiation mice respond with an initial
suppression of DNA synthesis followed 48h later by a marked increase in DNA
synthesis. MTX-y-GTP3 is at least as active as MTX in suppressing this pulse
of synthesis.
Figure 4 shows the suppressing effect of MTX and MTX-Tris conjugates on
mitotic
activity in epidermal cells following UVB irradiation.
Figure 5 shows Filaggrin expression from the SPA proof of principle trial. The
figure
shows total filaggrin expression following topical MTX-y-GTP3 or MTX
treatment versus the response with betamethasone. Patients in the upper right
hand quadrant responded positively to both betamethasone and 0.5% MTX-y-
GTP3 (indicated by *; patients 1, 2, 4, 6 & 8). Patients 5, 9 and 10 responded
to betamethasone but failed to respond to 0.5% MTX-y-GTP3. MTX (circles)
and 1.0% MTX-y-GTP3 (squares) also showed positive patients in a similar
manner. The correlation between response to betamethasone and MTX-y-
GTP3 suggests that there are two main groups in the trial: responders and non-
responders.
CA 02353839 2001-06-04
WO 00/34281 PCT/AU99101073
6
Figure 6 is a box plot showing the pooled results for filaggrin expression in
the stratum
granulosum of SPA patients treated with oral MTX-y-GTP3 or MTX
compared to betamethasone treatment. There is a statistically significant
effect
of the positive control betamethasone versus its vehicle. The comparison of
0.5% MTX-y-GTP3 with its vehicle gives a borderline significance of 0.08.
Figure 7 shows granularity:parakeratosis ratios for patients treated with
1.0%, 0.5%
MTX-y-GTP3 solution, 0.5% MTX and 0.05% betamethasone valerate cream.
1.0% MTX-y-GTP3 is showing positive results with this early indicator of
plaque resolution.
MODES FOR CARRYING OUT THE INVENTION
Abbreviations:
AICAR phosphoribosylaminoimidazolecarboxamide
APA 4-amino-4-deoxy-N 10-methyipteroic
acid
DAHMP 2,4-Diamino-6-hydroxymethylpteridine
DCC 1,3-dicyclohexylcarbodiimide
DCM dichloromethane
DECP diethyl cyanophosphonate
DMA N,N-dimethyiacetamide
DMAP 4-dimethylaminopyridine
DMF N,N-dimethyl formamide
Et20 diethylether
EtN'Pr2 di-isopropyl ethyl amine
EtOAc ethyl acetate
GT glycine-Tris
GTP 1 glycine-Tris monopalmitate
GTP2 glycine-Tris dipahnitate
GTP3 glycine-Tris tripalmitate
HOAc acetic acid
HOSu N-hydroxysuccinimide
MABA N-methyl aminobenzoic acid
MTX methotrexate
MTX-y-GT methotrexate-y-giycine-Tris
MTX-y-GTP 1 methotrexate-y-glycine-Tris monopalmitate
MTX-y-GTP2 methotrexate-y-glycine-Tris dipalmitate
CA 02353839 2001-06-04
WO 00/34281 PCT/AU99/01073
7
MTX-y-GTP3 methotrexate-y-glycine-Tris tripalmitate
OMe methyl ester
P205 phosphorus pentoxide
PmCI palmitoyl chloride
PPh3 triphenyl phosphine
ItM reaction mixture
RT room temperature
tBu tert Butyl ester
tBuOAc tert Butyl acetate
TFA trifluoroacetic acid
TPPO triphenylphosphine oxide
Z-GT benzyloxycarbonyl glycine-Tris
Z-GTP1 benzyloxycarbonyl glycine-Tris monopalmitate
Z-GTP2 benzyloxycarbonyl glycine-Tris dipalmitate
Z-GTP3 benzyloxycarbonyl glycine-Tris tripalmitate
Preparation of methotrexate-y-gtycine-Tris (MTX-y-GT)
Glycine-Tris (GT)
O
H2N~N OOH
H _
OH
CGH14N2~4
Mol. Wt. 178.19
N-(benzyloxycarbonyl}-glycine-Tris (Z-GT; USP 5,952,499} (1.70 g, 5.44 mmol)
was dissolved in ethanol (60 ml} with warming and stirring. After cooling to
room
temperature by immersion in a cool water bath, palladium-on-carbon ( 10%, 0.14
g) was
added and the system evacuated and filled with hydrogen gas three times. The
mixture
was stirred vigorously under a hydrogen atmosphere at room temperature for 19
h then
f ltered through two glass-fibre (GFIA) filter papers (rinsing with fresh
ethanol (approx.
10 ml)}. The filtrate was evaporated leaving GT (0.97 g, 100%) as a white
solid 1H
n.m.r. (dG-DMSO) 8 2.1, br s, NH2; 3, l,s,NCHzCO; 3.55, s, 3xCH20; 5.0, br s,
3xOH;
7.95, br s, NH.
CA 02353839 2001-06-04
WO 00/34281 PCT/AU99/OI073
8
MTX a tBu-y-GT
O
O
H
N OH
N
H OH
0
N OH
NH2
MTX-a-tB u- y G T
~30~42~1008
M 01. W t.: 670.72
Methotrexate-a-tent-butyl ester (MTX-a-tBu; USP 5,952,499) (0.438 g, 0.86
mmol) was dissolved with stirring, in DMF (4ml). HOSu (0.12 g. 1.03 mmol) was
added
and stirred until dissolution occurred. DCC (0.27 g. 1.29 mmol) was added and
the
resulting mixture stirred at room temperature under nitrogen for 16 h. More
DCC { 100
mg) was added and the mixture stirred for a further haur. GT (0.23 g., 1.29
mmol) in
DMF (1.5 ml) was added and the resulting mixture stirred for 54 h. The mixture
was
filtered through glass wool and the filtrate evaporated. The residue was
radially
chrornatographed an silica gel. Elution with 15% methanol in DCM afforded the
title
compound {0.33 g. S7%) as a yellow solid. 1H n.m.r. (d~-DMSO) 8 1.39, s,
C(CH3)3;
1.96, m, glu-~3-CHz; 2.24, m, glu-y-CH2; 3.21, s, Nme; 3.52, d, J 6 Hz,
3xCH20H; 3.70,
d, J 6 Hz, gly NCH2C0; 4.22, m, glu-a-H; 4.69, t, 3 6 Hz, 3xOH; 4.79, s,
ArCH2n 6.62,
br s, NH2; 6.82, 7.72 AA'BB', J 9Hz, ArH; 7.14, s, tris NH; 7.46, br s, NHZ;
8.10, t, J
6Hz, gly NH; 8.27, d, J 7Hz, glu NH; 8.56, s, H-7, Mass spectrum [electrospray
ionization, positive ion mode (ES(+))] rrrlz 671 {M+1)
CA 02353839 2001-06-04
WO 00/34281 PCT/AI~99/01073
9
MT.Y y~GT
0 COzH 0
N
OH
N HZ ~ N N
H H ON
N ~ N~ N ~ 0 OH
~ -' ~ MTX-y- G T
/ 'N N
NHZ
C 26H 34N 1008
MoI.Wt.:614.62
MTX-a-t-Bu-y-GT (0.459 g, 0.68 mmol) was dissolved, with stirring, in TFA (3
ml). The solution was stirred at room temperature for 1.5 h and the solvent
evaporated
under vacuum. The residue was dissolved in water and the solution pH (approx.
2) was
adjusted to 6 with aqueous sodium bicarbonate solution (lg in 75 ml). Aqueous
HOAc
( 10%) was added to pH 4. The solution was freeze-dried and the residue
purified by
flash chromatography then radial chromatography (twice) on silica gel. Elution
with 1-
3% water in 3:5 methanol:chloroform ai~orded the title compound (0.086 g, 21%)
as a
yellow solid. 1H n.m.r. (ds-DMSO) 8 1.96, m, glu-(3-CH2; 2. i4, m, glu-y-CH2;
3.19, s,
Nme; 3.5-3.7, m, 3xCH20H + 3xCHZOH; 3.95, rn, giy NCH2C0; 4.41, rn, glu-a-H;
4.76, s, ArCH2N; 5.37, br s, NH2; 6.61, br s, NH2; 6.84, 7.62 AA'BB', J 9Hz,
ArH;
7.60, m, glu NH; 7.71, s, tris NH; 8.23, t, 3 6Hz, gly NH; 8.57, s, H-7, ES(+)
mass
spectrum m/z 615 (M+1), 637 (M+Na), 659 (M+2Na).
Preparation of methotrexate-y-slvcine-Tris mono, di and triualmitate ~MTX-y-
GTPls, MTX-y-GTP2, MTX-y-GTP3)
The title compounds can be prepared as described in USP 5,952,499 or via the
following procedures.
CA 02353839 2001-06-04
WO 00/34281 PCT/AU99/01073
Synthesis of 2,4 Diamino-~rhydroxynzetl:ylpteridine (DAHMP)
NH2 NHz
N ''~ N~ OH NH40H N w ~ OH
I ~ ~ I
H2N~N N .HCI H2N~N N
MW 228.65 MW 192.19
5
Ref~. Rosowsky et al, J. Med. Chem., 1985, Vol. 28, No. 5, 660-667.
Reactants:
DAHMP.hydrochloride (69.04g, 0.30mo1)
10 H(~Ac ( 120m1)
H20 ( 1200m1)
Procedure:
DAHMI'.HCI was dissolved {with heating to 80°C) in aqueous acetic
acid. The
solution was filtered through glass wool and cooled to 45°C. The pH was
adjusted to
5.3-5.6 (monitored with pH paper) with concentrated ammonium hydroxide
solution.
RM cooled to RT and filtered. The yellow/caramel coloured solid collected was
washed
twice with water then dried in a vacuum oven over P205 at 80°C
overnight x 2 to afford
the desired DAHMP (free base) (52.94g, 92%) as a golden/tan coloured solid.
4 jj(2,4-Dinmino-6 rteridinyl)ntethylJnzethylan:inoJ-benzoic acid, or 4-nn:ino-
4-
rleoxy NI ~-methylhteroic acid (APA)
CO ZH
NH2 NHZ I
N ~ N~ OH (1 ) PPh3iBr~ N ~ ~ N
I / I
H2N'~ N N (2) MABAI H2N N N
EtN Pr2
MW 192.19 dihydate MW 361.37
Ref: Kralovec et al, J. Med. Chem., 1989, Vol: 32, No. 11, 2426-2431.
Rosowsky et al, J. Med. Chem., 1985, Vol. 28, No. 5, 660-667.
CA 02353839 2001-06-04
WO 00/34281 PCT/AU99/01073
11
Reactants:
DAHMP {15.008; 78mmo1)
Br2 (3eqv, l2.Iml, 37.538, 0.23mo1)
PPh3 (3eqv, 61.448, 0.23mo1)
DMA ( I 87m1)
MABA (13.578, 89mmol)
EtN~Pr2 (3eqv, 40.6m1, 30.18, 0.23mo1)
Procedure:
1U Br2 was added slowly via syringe to an ice-cooled, stirred mixture of PPh3
in
DMA under nitrogen. Solid DAHIVa' was added (nitrogen "breeze" over RM) and RM
stirred at RT for 24h. The amine was added via syringe, followed by solid MABA
(nitrogen "breeze"). RM was allowed to stir at RT for 67.Sh. RM poured into
aqueous
sodium hydroxide solution (0.33M, 125m1), the resulting precipitate filtered
off and the
filtrate adjusted to pH 5.5 with aqueous acetic acid (10% v/v}. The
precipitate was
collected, washed sequentially with water, cold ethanol, and ether, and dried
in a
vacuum oven at 40°C for 4 days to afford a yellow solid {25.798). To
remove the TPPO
and MABA contaminants, the crude product was stirred in 3:2 methanol:ethanol
(SOmI)
at RT for 50h, filtered, washed with Et20 (x2), air-dried and evacuated to
give APA
{24.488, 87%) as a yellow solid.
L-Glutarnic Acirl a tBu-y Me ester
C02H (1}tBuOAc, HCl04 _C02tBu
H2N ~ CO ZMe {2) NaHCO ~ H2N'~ CO 2Me
MW 161.16 MW 217.26
Ref: Rosowsky et al, J. Med. Chem., 1981, Vol. 24, No. 12, 1450-1455.
Reactants:
L-Glutamic acid 5-Me ester (17.548, O.I08mol)
3U tBuOAc (200m1)
70% HC104 ( 10.9m1, 0. I2mmo1)
CA 02353839 2001-06-04
WO 00/34281 PCT/AU99/01073
22
Procedure:
HCI04 was added slowly to a suspension/partial solution of the substrate in
tBuOAc (during acid addition, substrate started to dissolve). RM was stirred
in loosely
stoppered flask at RT for 40h. RM extracted with 0.5N HCI (2 x 200m1, 1 x
150m1).
The aqueous layer was cooled in ice/salt/dry ice bath (> -5°C) and
neutralized with solid
sodium hydrogen carbonate (until pH approx. 8 and no more NaHC03 dissolves)
with
the temperature of the solution being maintained at approx. -4°C. The
mixture was
extracted with ether (RT, 4 x 200m1), the combined organic Layers washed with
brine
( 100m1), dried (MgS04) and evaporated (rotary evaporator, then oil pump,
without
heating) to afford the desired product (10.77g, 45%) as an almost colourless,
viscous oil
which partially solidified on storage at -20°C.
MTX-a tl3it-yLMe ~liester
NH2 , CO~H
N ~ ~ N ~ I
I ~ I
H2N~ N N pih drate MW 361.37
Y
APA
(1) (Et0)2POCN (2) ~02tBu + iPr2NEt
H2N ~' COZMe
r
O C02tBu
NH2 / I H~'/'' CO2Me
N ~ N~ N
i MW 524.59
H2N ~ N N
Ref: (1) Rosowsky et. al, J. Med. Chem., 1981, Voi. 24, No. 12, 1450-1455.
(2) Kralovec et al, J. Med. Chem., 1989, Vol. 32, No. 11, 2426-2431.
2o Reactants:
APA (8.56g, 23.7mmo1)
EtN'Prz (9.Oml, 51.7mmo1)
DMF (460m1)
CA 02353839 2001-06-04
WO 00/34281 PCTIAU99/01073
13
DECD (7.20g, 44.1 mmol)
L-Glutamic Acid a-tBu-y-Me ester (7.26g, 33.4mmol)
EtNiPr2 (8.Sml, 48.8mmo1)
Procedure:
APA (granulated) was added portionwise over 10 minutes to a solution of DECP
and EtN'Pr2 (first portion) in DMF (400m1). The solution was immersed in an
oil bath
{pre-heated to 80°C) far 2 minutes {internal temp. to 34°C). The
flask was removed
from the bath and stirred for 30 rains {internal temp. 22°C). The
second portion of
EtN'Pr2 was added followed by glutamate in DMF (50+lOml rinse). RM was stirred
at
room temperature under nitrogen for 14 h (overnight). A solution of NaHC03
{1.808,
2lmmol) in water (60m1) was added, the mixture stirred for 30 rains and then
evaporated {rotary/oil pump, 45°C). The residue was dissolved in
chloroform {approx.
400m1) and the solution washed with water (300m1) and 5% aqueous sodium
bicarbonate solution {200m1). The organic layer was washed with brine (30m1),
dried
{MgS04) and evaporated. The residue {brown, viscous gum) was filtered through
a
small silica column with 5% methanol in DCM to remove polar impurities and the
resulting product flash chromatographed on silica. gel, eluting with 7-12%
methanol in
DCM to afford the product in three fractions: 1 (trace impurity) {2.17g, 18%);
2 (high
purity) (4.408, 36%); and 3 {trace impurity) (2.77g, 22%) as bright yellow
solids.
MTX a tBu ester
(This is the same intermediate as described in USP 5,952,499 - Compound IV)
O CO ztBu
NH2 ~ I H~C02Me
N ~ N~ N
I ~ I MW 524.59
H2N J~ N N i
Ba(OH)2.8H20
r O CO ~tBu
NH2 ~ I H~COZH
N W N~ N w
H N ~ N N~ I MW 510.56
CA 02353839 2001-06-04
WO 00/34281 PCTIAU99101073
14
Ref: Rosowsky et al, J. Med. Chem., 1981, Vol. 24, No. 12, 1450-1455.
Reactants:
MTX-a-tBu-y-Me diester (5.00g, 9.Smmo1)
Ba(OH)2.8H20 (1.828, 5.8mmo1)
Ethanol (95m1)
Water (110m1)
Na2S04.1OH20 {1.93g, 6.0mmo1) in water (l0ml)
Procedure:
The first three reactants were mixed and stirred at RT in a loosely-stoppered
flask for 19 h. Sodium sulfate in water was added and the mixture stirred for
a few
minutes (BaS04 precipitate seems to form immediately) then filtered through
2xGF/A
filter papers on a No. 3 or 4 glass sinter. The f Itrate was adjusted to pH 5
with 10%
HOAc in water. The resultant suspension (gelatinous precipitate) was
refrigerated for
30m and then the solid collected by filtration using a No. 1 filter paper on a
glass sinter.
The collected solid was air-dried overnight (still on the filter paper in the
funnel) then
evacuated (oil pump) to afford the desired product (4.41g, 91%) as a bright
orange/yellow solid.
CA 02353839 2001-06-04
WO 00134281 PCT/AU99/01073
Z GTP3 (+ Z GTP2 + Z GTPI)
0
O~ N N OH
I H 'I OH
ZGT O OH
PmCI(2eqv)
Et3N
DMF/CH 2C12 (1:4)
r
OPm ~ N OPm
O H~ -OPm + ~ ' O H~ OPm
O OPm ~ O OH
ZGTP3 O ZGTP2
O~ N N OPm
+ ~ H~ OH
/ O OH
ZGTP1
Reactants:
Z-GT (SO.OOg, 0.16mo1)
PmCI (108m1, 97.85g, 0.356mo1)
Et3N ( 100m1, 72.4g, 0.72mo1)
DMF (450m1)
DCM { 1.60 + 0.20L)
Procedure:
Z-GT was dissolved in DMF in a SL, three necked round-bottomed flask. DCM
{1.6L) was added and the stirred solution cooled with an ice-salt bath for lh.
A solution
of PmCI in DCM { 100 ml) was added dropwise over 2h and the bath temperature
maintained at -13 to -10°C The dropping funnel was rinsed with DCM (100
ml). The
RM was stirred at the same temperature for a further 2h and then at 4°C
over the
weekend. The solvent was removed under vacuum (the precipitate that appeared
during
the evaporation was filtered after the removal of DCM in order to remove DMF
efficiently). The residue was extracted with EtOAc (400 ml) then DCM (800 ml)
(EtOAc extract resulted in precipitation on washing the extract with brine or
water).
The combined organic extracts were washed with brine and dried (MgS04).
Solvent
CA 02353839 2001-06-04
WO 00/34281 PCT/AU99/01073
16
removal under vacuum gave the crude product as a yellow wax (133.48g). HPLC
analysis indicated a mixture of Z-GTP3, Z-GTP2 and Z-GTPl in a ratio of 28.8%,
47.4% and 23.8% respectively.
MTX a tliu- y GTP3
O CO 2tBu
NHz ~ ~ H~ CO2H
N ~ N.. N
MW 510.56
H2N ~ N N
GTP3 DCC/DMAP
O C02tBu
GTP3
NH2 N w ~ ' H
N ~ ~N O
H N~ N~ N~ ~ MW 1385.91
2
Reactlnts:
MTX-a-tBu ester (11.00g, 2l.Smmol)
DCC (l.2eqv, S.SOg, 26.7mmol)
GTP3 ( 1. I eqv, 2I .32g, 23.9mmol)
DMAP (0.1 edv, 258mg, 2.1 mmol)
DMF (8bm1)
DCM (340m1)
Procedure:
MTX-oc-tBu ester was dissolved, with stirring, in DMF. DCM (200m1) was
added. DCC was added and the solution stirred for 20 mins. GTP3 in DCM (70m1)
was
added dropwise over 10 rains. DMAP was added and the RM stirred at RT under
nitrogen overnight (24 h). The RM was filtered to remove white solid, urea by-
product
from DCC and concentrated (rotary/oil pump, 50°C) to remove DMF. The
residue was
dissolved in DCM, refiltered and evaporated. The residue was dissolved in
EtOAc/DCM
( I 50/20m1}, washed with brine, dried (MgS04) and evaporated. The residue was
divided into halves and each was flash chromatographed on silica gel, eluting
the first
CA 02353839 2001-06-04
WO 00/34281 PCT/AU99/01073
17
half with 5% methanol in DCM and the second half with 3.7-4.4% methanol in
DCM.
Re-chromatography of the impure fractions afforded the product in two
fractions (bright
yellow solids), (1) (>95%purity) (17.18g, 58%) and (2) (90-95% purity) (4.838,
16%).
Further elution with 8-10% methanol afforded a third fraction containing a
bright yellow
solid by-product.
MT'X y~-GTP3
O C02tBu
GTP3
NH2 -'', ~ H
N ~ N~ N ~ O
H N~ N~ I MW 1385.91
2
TFA
O C02H
GTP3
NH2 N ~ ~ ' H
N ~ ~N O
H N~ N N I MW 1329.81
2
Reactants:
MTX-oc-tBu-y-GTP3 (0.86g, 0.62mmo1)
TFA ( 1.7m1, 22mmo1)
DCM (5ml)
Procedure:
TFA was added dropwise to an ice-cooled, stirred solution of the substrate in
DCM. The RM was allowed to warm to RT then stirred under nitrogen for 5.5 h.
The
RM was placed at 4°C overnight then diluted with DCM (25m1) and
washed with
aqueous, sodium bicarbonate solution (5% w/v, 35m1) (check that pH>7). HOAc
(glacial, 1.8m1) was added and mixture shaken (check that pH~4). The organic
layer was
washed with water (2x25m1) (the emulsion was broken by addition of a little
NaC1 &
methanol & fresh solvents, swirling and warming), dried (MgS04) and
evaporated. The
residue was flash chromatographed on silica gel, eluting with an 8-30% step
gradient
CA 02353839 2001-06-04
WO 00/34281 PCT/AU99/01073
1$
with methanol in DCM {3X4%; 2X5% steps) to afford the product in three
fractions:-
(1) (trace higher Rf by-product, 187mg), (2) (higher purity, 476mg) and (3)
(trace lower
Rf by-product, 79mg). ,All fractions were yellow solids and of quite high
purity. Fraction
(2) -> 97.25% purity (by HPLC). Yield = 742mg, 90%.
MTX-y-GTP 1 and MTX-y-GTP2 can be prepared in a similar manner from Z-
GTP 1 & 2 respectively.
In vitro DHFR Inhibition
Dihydrofolate reductase (DHFR) is a target enzyme of MTX. It catalyses the
following reaction:
DHFR
Dihydrofolate + NADPH + H+ -~ Tetrahydrofoiate + NADp+
Its activity can be measured ifi vitro by following the rate of NADPH
oxidation
~5 at 340nm.
Method:
The inhibition of DHFR by MTX, MTX-y-GT and MTX-y-GTPi was measured
as described by Imbert AM, Pignon T, Lena N (1983) "Enzymatic assay for
methotrexate with a centrifugal analyser (Cobas-Bio)" Clin Chem, 29(6):1317-
1318.
Results:
MTX-y-GT inhibited DHFR in the same concentration range as MTX (Table 1).
Table 1. Inhibitory activity of MTX and MTX-y-GT on DHFR at 2.86 nM
Cam ound % inhibition
MTX 45.5
MTX-y-GT 36.4
The inhibitory activity of MTX-y-GTP 1 was also tested. The concentration
required to cause a 50% inhibition of DHFR was 76nM. This is 16X less active
than
MTX which causes 50% inhibition of DHFR at 4.8nM.
MTX-a-GTP1 did not inhibit DHFR under these conditions.
MTX-y-GTP2 and MTX-y-GTP3 were not tested as their solubility characteristics
are incompatible witlx the assay system.
CA 02353839 2001-06-04
WO 00/34281 PCT/AU99/01073
19
Investigation of MTX-y-GT, MTX-y-GTP1 and MTX-y-GTP3 using the delayed
tyue hypersensitivity (DTH) response in the rat
Delayed type hypersensitivity {DTH) is an immune response which can be used
to assess the action of various therapeutic agents. It can be demonstrated by
sensitising
an individual to a compound, and then on challenge with the same compound,
observing
the development of oedema at the site of challenge. MTX has an
immunosuppressive
effect which reduces oedema, and this reduction in DTH response has been used
to
assess one aspect of the efficacy of MTX-y-GT, MTX-y-GTP 1 and MTX-y-GTP3 .
Methods:
Copenhagen {COP) rats had their abdomens shaved and 100 ~,l of 0.5%
dinitroflurobenzene (DNFB) in acetone was applied daily for 3 days.
Concurrently, the
rats were administered orally either, soy oil, MTX {0.5 mg) or equimolar
amounts of
MTX-y-GT, MTX-y-GTP 1 or MTX-y-GTP3 daily for 5 days. On day 7, rats were
anaesthetised with methoxyflurane and the footpad volume measured immediately
prior
to the addition of 25 p.l of 0.2% DNFB in acetone to the left footpad. Under
anaesthesia, footpad volume was measured at 23h. Piethysmography (the
measurement
of the volume of an object by observed changes in volume after immersion),
using a
laboratory fabricated oncometer, was used to determine the volume of the
footpad. The
nett oedema was calculated by subtracting the volume of the foot at time 0
from time
23h.
Drugs:
MTX (1mg/mI) in Soya oil
MTX-y-GT - (1.4 mgslml) in soya oil - (Batch QY5-64MXS)
MTX-y-GTP1 - (1.88 mglml) in soya oil - (Batch GB4-99 NB00077-p50)
MTX-y-GTP3 - (3 mg/ml) in Soya oil - (Batch QY 4-41 FP)
Control - soya oil
Results:
The nett footpad oedema data is presented in Figure 1.
The mean nett footpad oedema was significantly less in those rats treated with
MTX, MTX-y-GTP 1 and MTX-y-GT, but not MTX-y-GTP3 when compared with
control rats (Table 2). As well, the nett footpad oedema from the MTX-y-GTP 1-
treated
rats was significantly less (p = 0.02 i ) than that in the MTX-treated rats
CA 02353839 2001-06-04
WO 00/34281 PCT/AU99101073
Table 2: Mean, SD and values of data
Treatment n Mean SD p value when
oedema compared to
.....__~__...._ _...~.............___._.._._-_._.~Inl~._
.~......~..~._..._..__ Controls
Control 38 0.363 0.254 -
MTX 8 0.216 0.1 SO 0.044*
MTX-y-GTP3 22 0.327 0.325 0.59
MTX-y-GT 18 0.192 0.237 0.0195*
MTX-y-GTP1 lI 0.055 0.050 <0.0001*
* statistically significant
5 Discussion:
DTH is an irr vivo T cell-dependent immune response manifested as an
inflammatory reaction after antigenic challenge. Thus these results indicate
the
immunosuppressive effects of MTX-y-GT and MTX-y-GTP 1 are at least equal to or
perhaps more effective than MTX.
In vitro Cytotoxicity
The ability of MTX and its Tris-conjugates to inhibit proliferation of
cultured
cells were assessed.
Methods:
The effect of MTX and MTX-Tris-conjugates on cell proliferation was studied
using routine cell culture procedures. The following cell lines were tested:
JURKAT (human acute T-cell leukaemia)
Swiss mouse fibroblast (3T3) cells
CCRF-CEM (CEM) human T-cell leukaemia
JURKAT cells were cultured in the presence of drug for 64 h whilst 3T3 and
CEM cells were cultured for 48h and 96h respectively in the presence of
compound. All
studies were performed in 96-well plates and control wells contained no drug.
Cell
proliferation was then measured using the MTS or MTT detection: (D Marks, L
Belov,
MW Davey, RA Davey, A Kidman. Leukemia Research 16, 1165-1173. "The MTT cell
viability assay for cytotoxicity testing in multidrug-resistant human leukemic
cells."
CA 02353839 2001-06-04
WO 00/34281 PCT/AU99/01073
21
Results:
All conjugates inhibited proliferation but were less toxic to all cell types
tested
than the parent compound, MTX. Results are summarised in the Table 3.
Table 3. Effect of MTX and MTX-Tris conjugates on the proliferation of various
cell types. ICSO = concentration of compound at which cell growth is inhibited
by
50% of control growth.
Com ound ICsa M
3T3 JURKAT CEM
MTX 0.0525 8.2 0.019
MTX-a-GT - - 35.9 1889
MTX-y-GT no effect* - 0.57 30
MTX-y-GTP I - - 0.21 11
MTX-a-GTP2 - 10300 1256 -
MTX-y-GTP2 __ ___ 0.40 (21)
_ 1200 (146)
~ 0.41 {7.8)
~
- not tested; * no effect observed up to 250 nM; (n) Conjugate:MTX ratio.
Reduced activity is likely to be due to the modification of MTX on the a- or Y-
COOH. Itr vivv MTX is polyglutamylated (addition of (glu)") on the y-COOH and
this is
an important aspect of MTX toxicity. It increases intracellular retention and
conseduently results in a sustained block in tetrahydrofolate synthesis via
DHFR. MTX-
y-(glu)" also have greater inhibitory effects on other enzymes involved in DNA
and RNA
synthesis including thymidylate synthetase and AICAR transformylase.
Polyglutamate
formation and accumulation may have unwanted effects ifT vivo as they fozrn in
significant amounts in some organs (kidney and liver) where they may give rise
to
unwanted toxicity due to prolonged intracellular retention.
Some of our conjugates (ie MTX-y-GT, MTX-y-GTP1 and MTX-y-GTP3) are
modified on the y-COOH and are unlikely to be polyglutamylated thus explaining
their
reduced toxicity.
The y-COOH is also involved in cellular uptake and modification may reduce
uptake. In our study, the various MTX-y-conjugates display differences in
their effect on
cell growth. MTX-y-GT is less inhibitory than MTX-y-GTP 1 or 2. It could be
postulated that whilst addition of GT impedes the drug uptake (and therefore
toxicity)
via the folate transport system, this effect may, in part, be over ridden by
the addition of
palmitate groups. It is likely that palmitate-conjugates enter the cell by
endocytosis.
Whilst MTX-y-GT is less toxic to cells than the palmitoylated conjugates, it
is mare
CA 02353839 2001-06-04
WO 00/34281 PCT/AU99/01073
22
inhibitory to DHFR in a biochemical assay (see above). This suggests that it
is the
transport mechanism rather than direct enzyme inhibition that differentiates
between
MTX-y-GT and MTX-y-GTP 1 or 2.
It is widely reported that modification to the a-COOH reduces cell uptake to a
greater extent than modification to the -'y-COOH. Our results are consistent
with this.
Both MTX-a-GT and MTX-GTP2 are much less toxic than MTX-y-GT and MTX-y-
GTP2, respectively.
Overall our results are consistent with literature reports. Some relevant
references are listed:
Rosowsky et al.; J. Med. Chem. (1984) 27, 600-604
Rosowsky et al. J. Med. Chem. ( 1984) 27, 605-609
Rosowsky et al., J. Med. Chem. (1981} 24, 1450-1455
Fan et al. (1991) Biochem. 30, 4573-4580.
Testing of MTX and MTX-w-GT in the rat adjuvant-induced arthritis model.
Doses of 40, I 5 and 5 p.mollkg of MTX have been shown by Baggott and co-
workers (Arthritis Rheum 1998; 41:1407-10) to give dose-dependent inhibition
of
adjuvant induced arthritis in Lewis rats.
A similar model using Dark Agouti (DA) rats has been used to test the effect
of
MTX-y-GT.
Method:
DA rats were injected subcutaneously with Freund's Complete Adjuvant into the
tail base. Arthritis developed 10-I2 days later in untreated rats. Rats were
treated with
PBS (control) or molar equivalent doses ofMTX or MTX-y-GT in PBS orally (by
gavage) once at day 6; six rats per treatment group. The effect of treatment
on arthritis
was monitored daily by assessing body weight and evidence of arthritis, which
was
quantified using an established scoring system. This involves giving a score
from 0 to 4
for each paw (0= no evidence of arthritis or inflammation, 4= global swelling
of the
paw). The theoretical maximum score was 16 but rats were euthanased if a score
greater than 12 was observed. In treated rats, in which mild arthritis
occurred, the
disease was followed beyond the treatment period to determine whether
arthritis was
delayed rather than suppressed or prevented.
CA 02353839 2001-06-04
WO 00134281 PCT/AU99/01073
23
Results:
The results of a single dose of MTX and MTX-y-GT at day 6 are shown in
Figures 1: A to F
Conctusio~~s:
MTX and MTX-y-GT were effective in delaying the onset of inflammation by 2-
4 days. At the doses tested and within experimental error the efl'ects of the
two
compounds were indistinguishable. From the DTH rat footpad results it would be
expected that MTX-y-GTP 1 might also delay the onset of inflammation in this
model.
MTX-y-GTP3 was negative in the DTH assay but shows activity in the human SPA
trial
in the treatment of psoriasis (see below). Its effect on adjuvant-induced
arthritis in rats
remains to be determined.
Toxicity of MTX-Tris Conjugates
Whilst no experiment has been performed that specifically examines toxicity,
observations have been made during experiments examining biological activity
which
indicate that MTX-TRIS conjugates have reduced systemic toxicity compared to
MTX.
The following observations have been noted:
za
1. Experiments examining the effect of daily topical applications over three
consecutive days of vehicle, MTX, MTX-y-GTP2 or MTX-y-GTP3 on UVB-induced
erythema in female Skh-1 mice have been performed. During this study mice
were assessed for toxic signs (dehydration, diarrhoea and anorexia) for 6 days
z5 followed by assessment at necropsy for gross signs of GIT toxicity
(enlarged
fluid-filled intestines). MTX-y-GTPZ and MTX-y-GTP3 appeared much less toxic
than MTX and results are shown in the following table.
CA 02353839 2001-06-04
WO 00134281 PCT/AU99/01073
24
Toxicity results from topical application to Skh-1 hairless mice
TREATMENT SIGNS OF Dg,ATH
GROSS GIT
TOXICITY
vehicle 0/24 0/24
MTX 4 mg/kgi 5/24 8/24
MTX-y-GTP2 9.6 mg/kg1 0/24 0/24
MTX-y-GTP3 11.7 mg/kgs 0/14 0/14
IEquimolar amounts of MTX, MTX-y-GTP2 and MTX-y-GTP3 were used. They
were formulated in oil/water cream:ethyl oleate (4:1) and O.lml was applied to
each
mouse. GIT = gastrointestinal tract.
In many mice treated with MTX, clinical signs of toxicity and at necropsy,
enlarged fluid-filled intestines were prevalent. Histological examination of
these
intestines showed marked microvilli damage and necrotic mucosal cells. In
comparison,
neither GiT damage nor mortality were observed in the mice receiving MTX-y-
GTP2 or
MTX-y-GTP3. In all mice, the liver and kidneys appeared normal.
2. In Delayed Type Hypersensitivity assays studies, Copenhagen rats were
administered orally either soy oil, MTX (0.5 mg) or equimolar amounts of MTX-y-
GT,
MTX-y-GTP 1 or MTX-y-GTP3 daily for 5 days, whilst being concurrently
sensitised by
topical application of DNFB. Following challenge with DNFB and necropsy 24h
later,
the intestines from a representative group of rats were examined for gross
changes and
samples collected for histology.
All MTX-treated rats (rt=7) had an enlarged stomach and intestines filled with
milky fluid. Histology of these GIT samples revealed villi with thickened,
inflammed
cores, separation and sloughing of the epithelium and mucosal necrosis. This
is
indicative of mild to severe damage. Rats receiving MTX-y-GT (n=9) had a
normal GIT
with the exception of one animal which had an enlarged fluid-filled large
intestine.
Macroscopic examination of the GIT from MTX-y-GTP 1 {n=14) and MTX-y-GTP3
CA 02353839 2001-06-04
WO 00/34281 PCT/AU99/01073
(n=14) treatment groups revealed no gross abnormalities. Reduced
histopathology was
displayed in the GIT from rats receiving MTX-Tris conjugates.
In studies examining the effects of MTX and MTX-y-GT on adjuvant-induced
5 arthritis in rats, the following observations were noted:
a) Following a single oral dose of MTX or MTX-y-GT {40 umole/kg), diarrhoea
was
observed 1-2 days later in the MTX group. This was not evident in the MTX-y-GT
and vehicle only groups.
b) In a second experiment, rats were given 3 oral doses, 3 days apart. Toxic
side effects
20 (ruffled fur and ataxia) were evident in rats treated with MTX (5
umolelkg).
Consequently, these animals were euthanased earlier than originally planned.
Post
mortem showed marked changes in the GIT, particularly the small intestine
which
appeared bloated and contracted. Histology showed marked crypt hyperplasia and
loss of villous structure in the mucosa of the jejunum and the ileum. In some
areas
15 there was loss of both crypt and villous epithelium. The mesenteric lymph
nodes of
these rats were also enlarged. None of these effects was seen in the vehicle-
and
MTX-y-GT-treated groups. In addition, peripheral blood samples from these MTX
treated rats showed profound leucopaenia (approx. 2 x 10s/L), due largely to
neutropaenia. Leucocyte counts in the vehicle control and MTX-y-GT groups were
20 not different (approx. 30 x 109/L).
Conclusions:
All MTX-Tris conjugates have reduced toxicity compared with MTX. Similar
results were observed with cytotoxicity experiments (see above).
25 Distribution of MTX-Tris Coniiug~tes in Mice
Introduction:
MTX is an effective oral treatment for a disease with an autoimmune component
such as psoriasis and rheumatoid arthritis. However its long-term use requires
ongoing
liver assessment due to the risk of hepatic atrophy, necrosis and cirrhosis.
MTX also
induces gastrointestinal irritation, renal toxicity and abortion. We have
demonstrated
that MTX-Tris conjugates are biologically active but are less toxic ira vitro
and in vivo
(see above). In addition, the distribution of radiolabelled MTX-Tris
conjugates as well
as MTX, administered topically, orally (p/o) and intravenously (i/v) have been
examined
in mice using liquid scintillation and whole body autoradiography. This was to
determine
if MTX-Tris conjugates (or their breakdown products) accumulate in organs of
concern
CA 02353839 2001-06-04
WO 00/34281 PCT/AU99/01073
zs
for MTX toxicity and if their excretion patterns varied from those of MTX.
Pertinent
results are summarised below.
Results:
1. Mice given [14C]-MTX-y-GT, both i/v and orally, demonstrated a higher level
of
radioactivity excreted in the faeces, but similar amounts in the urine
compared with
animals given ['4C]-MTX. The tissue distribution of [14C] was similar, but at
lower
levels in mice given [14C]-MTX-y-GT. These findings suggest that MTX-y-GT has
greater hepatic clearance than MTX.
2. Following topical application, radiolabel from MTX-y-GTP3 was detected in
the
skin epidermis for longer periods than MTX treated mice. This suggests that
MTX-
y-GTP'~ is retained in the skin to a greater extent than MTX. In addition,
radioactivity is present in the skin from mice given oral ['4C]-MTX-y-GTP 1
and
['H]-MTX-y-GTP3. These findings may have important implications regarding
their
potential use as anti-psoriatic treatments.
3. Following oral administration, both radiolabelled MTX-y-GTP 1 and MTX-y-
GTP3
show significantly reduced levels of counts in organs of concern for MTX
toxicity.
This suggests either decreased absorption or increased hepatic excretion (ie
enterohepatic circulation). However once these conjugates enter the
circulation (ie
2o following i/v administration) they appear to accumulate in the liver,
spleen and lung.
This offers the potential to targeting drugs to specific organs following
their
conjugation to fatty acyl groups via Tris.
4. Radiolabel from ['aC]MTX is mainly excreted in the urine and to a lesser
extent in
the faeces. In contrast, when ['4C]MTX-y-GTP 1 and [14C]MTX-y-GTP3 are
delivered i/v, radiolabel is mainly eliminated in the faeces via hepatic
excretion, with
very low levels detected in the urine.
Discussion:
MTX-Tris conjugates, when given orally, accumulate to a lesser extent than
MTX in organs of concern for MTX toxicity because they either have greater
hepatic
clearance {MTX-y-GT) or low levels of absorption in the formulations tested
(MTX-y-
GTP 1 and MTX-y-GTP3). The distribution patterns may partially explain the
reduced
toxic side effects seen with the conjugates {see Toxicity section above).
Reduced
cellular uptake and polyglutamylation is also likely to contribute (see
Cytotoxicity
Section above). Whilst MTX-y-GTPI and MTX-y-GTP3 may accumulate in the liver,
lung and spleen when administered ilv, we have not assessed toxic side
effects. However
CA 02353839 2001-06-04
WO 001342$1 PCTIAU99/01073
27
it is unlikely that these conjugates are polyglutamylated, which is the
mechanism widely
attributed to MTX toxicity.
Both MTX-y-GTP1 and MTX-y-GTP3 were found in the skin following oral delivery.
This may have implications for their potential use as a therapeutic for
psoriasis.
Clinical trial of MTX-y-GTP3 as a tolvical treatment for usoriasis
Psoriasis is a skin condition that affects 1-2% of people ofEuropean origin.
It is
an inflammatory and epidermal hyperproliferative disorder of unknown aetiology
with a
poor prognosis.
MTX, when taken orally, is an effective treatment for moderate to severe
psoriasis. However its use is associated with immediate gastrointestinal
problems and
long-term liver toxicity. Approximately 6 weeks of treatment are needed before
a
clinical response.
It has been postulated that topically-applied MTX would provide drug at the
affected site and have fewer side-effects. This has been trialed {Weinstein et
al., 1989)
but overall the results were not promising, perhaps due to poor skin
retention. From our
observations it was considered likely that MTX-y-GTP3 would have increased
skin
retention as well as the biological activities of MTX.
Pneclir~ical slz~dies
Preclinical studies indicated that topically-applied MTX-y-GTP3 had biological
activity that may make it suitable for the treatment of psoriasis. It had
equivalent or
better activity to MTX in reducing UVB-induced epidermal hyperproliferation in
hairless
mice (Figure 3) and suppressed UVB-induced epidermal DNA synthesis (Figure 4)
as
determined by the method of Molek et al.(Brit J Derm(1983) 108:25-31).
Intradermal
injection of MTX-y-GTP2 was mare effective than MTX in reducing the growth of
B 16
melanomas (USP 5,952,499). Distribution studies (above) show that topically-
applied
MTX-y-GTP3 is retained in the epidermis of hairless mice for longer periods
than MTX.
MTX-y-GTP3 administered topically or orally is less toxic to hairless mice
than MTX
(See above).
These positive pre-clinical results and the lack of a good animal model for
psoriasis led to a proof of concept trial of topical MTX-y-GTP3 in humans.
Clinical sta~dy design
The Small Plaque Assay (SPA) assesses the anti-psoriatic potential of
compounds over a two week time course. The assay simultaneously tests 6
compounds
CA 02353839 2001-06-04
WO 00/34281 PCTIAU99/01073
28
on a single plaque under occlusion. The position of the test compounds on each
plaque
was randomised to eliminate evaluation bias and any drug interaction effects.
Eleven
patients were studied.
Treatments:
0.5% MTX-y-GTP3 solution (test formulation)
1.0% MTX-y-GTP3 solution (test formulation)
MTX-y-GTP3 vehicle (negative control)
0.5% MTX solution (reference formulation)
MTX vehicle (negative control)
0.05% betamethasone valerate (positive control)
(AIl concentrations of MTX-y-GTP3 solutions are with respect to their MTX
content}.
Schedule
Da Treatment Assessment
.......... ..................-
.._.................................................................._...._....
................_.....~...................._-
._....................................._.......................
..~'..........__.
0 2% salicylic acid clinical & histology
I drugs applied
3 drugs applied
6 drugs applied
8 drugs applied clinical
I0 drugs applied
13 drugs applied
I 5 clinical & histology
29 clinical
Parameters
Approximately 6 weeks of oral MTX treatment are needed before a clinical
response is evident. Therefore it was unlikely that a clinical result would be
seen with
topical MTX-y-GTP3 in the two week duration of the SPA (restricted for ethical
ZO reasons). However, early histological changes consistent with psoriasis
resolution would
be expected with an active agent. Therefore, as well as clinical assessment, a
range of
histopathological and immunohistological markers were assessed as follows:
CA 02353839 2001-06-04
WO 00/34281 PCT/AU99/01073
29
~ Clinical
Infiltration and Erythema on days 0, 8, 1S and 29.
~ Histopatlaology
Parakeratosis Inflammatory inf Itrate
Granularity Acanthosis
Vascular changes Epidermal thickness
Papillomatosis
~ Imnzunohistology
Involucrin ICAM-1
Filaggrin Ki-67+ keratinocytes
CD4 and CD8+ T-cells
CA 02353839 2001-06-04
WO 00134281 PCT/AU99/01073
Patient Selection
Inclusion Criteria Exclusion Criteria
E Male of any ethnic group, I8 to 70 years of Pustular psoriasis,
erythrodermic psoriasis,
age. Females of non-reproductive status inverse psoriasis and/or active
psoriasis
f also allowed. arthritis.
Serious intercurrent illness or active
Chronic plaque type psoriasis vulgaris for at infections requiring treatment,
including
f least 6 months. ~ hepatitis or AIDS.
Known to be HIV positive.
Capable of understanding the purposes and Known hypersensitivity to trial
medication
risks of the trial, giving the impression that components.
she/he is willing to partake in all History of and/or present systemic
assessments for the whole duration and has malignancy.
given written informed consent. Simultaneous participation in any other
investigational drug trial or receiving
Required plaque characteristics: investigational drug 4 weeks prior to start
E ~ On trunk or extremities; not in a flexure of trial.
area. Received any systemic anti-psoriatic
Area at least 5. 5 x 8.3 cm. therapy 4 weeks prior to start of trial.
F ~ Homogeneous appearance (erythema Received any topical anti-psoriatic
therapy
and infiltration). for the plaque tested two weeks prior to
E ~ Severe erythema & infiltration start of trial.
Laboratory values that contra-indicate
participation in the trial.
Any form of substance abuse, psychiatric
disorder or condition which may invalidate
communication with the investigator.
Known to have cirrhosis of the liver or
i other liver disease.
Patients taking medications which may
interact with MTX.
i
v Patients who have a calculated creatinine
clearance < 30 mUmin.
CA 02353839 2001-06-04
WO 00/34281 PCT/AU99/01073
31
Results:
Clinical
The positive control, betamethasone, significantly improved the clinical
appearance of test areas indicating that the trial methodology was preforming
correctly.
Little or no change was observed in the MTX-y-GTP3 or MTX test spot areas over
the
two week period. This was not unexpected.as the time course required for anti-
psoriatic
activity of oral MTX is 6 weeks. No serious side effects or local irritation
were observed
with any test compounds.
Histological
Statistically, betamethasone demonstrated anti-psoriatic activity in all
parameters
except hyperkeratosis and involucrin.
MTX-y-GTP3 treatment had positive effects on filaggrin expression (a marker of
terminal epidermal differentiation), granularity (granular layer is absent or
incomplete in
psoriasis) and parakeratosis (imperfect formation of epidermal horn cells) in
five out of
eleven patients.
~ F~~grin.
Of all the immunohistological markers examined, filaggrin proved to be the
most
powerful indicator of the action of the test drugs. Examination of filaggrin
results
within patients showed a correlation between response to betamethasone and
response to MTX-y-GTP3 solution (Figure 5). MTX-y-GTP3 solution showed the
best filaggrin response at a concentration of 0.5% while 1% MTX-y-GTP3 and
0.5% MTX gave approximately equivalent results. As a group, there were
differences in filaggrin expression in the stratum granulosum with 0.5% MTX-y-
GTP3 solution compared to vehicle. There was a similar response to MTX (Figure
s).
~ Granularity:Parakeratosis Ratio.
The granularity to parakeratosis ratio was calculated to give an index of
therapy - an
increase in the ratio indicates a trend towards psoriasis resolution.
Histopathological
changes in granularity and parakeratosis consistent with early resolution of
psoriasis
were evident with 0.05% betamethasone valerate cream (10 out of 11 patients)
and
1.0% MTX-y-GTP3 solution (5 out of 11 patients) - Figure 7 a) and b).
Treatment
with 0.5% MTX-y-GTP3 and MTX resulted in little response in
granularity:parakeratosis ratio (1 & 2 patients respectively).
CA 02353839 2001-06-04
WO 00/342$1 PCT/AU99/01073
32
Clinical study conclusions
MTX-y-GTP3 demonstrated anti-psoriatic etFects in a number of patients in
comparison to its vehicle as judged by histopathology {granularity and
parakeratosis)
and the keratinocyte maturation marker, filaggrin. This, coupled with its
relatively low
toxicity compared to MTX makes it a potential therapeutic for the topical
treatment of
recalcitrant psoriasis.
It will be appreciated by persons skilled in the art that numerous variations
andlor
modifications may be made to the invention as shown in the specific
embodiments
without departing from the spirit or scope of the invention as broadly
described. The
present embodiments are, therefore, to be considered in all respects as
illustrative and
not restrictive.