Note: Descriptions are shown in the official language in which they were submitted.
CA 02354266 2001-06-07
Hoechst Marion Roussel Deutschland GmbH HMR 1998/L 080 Dr. TH/mk
Description
Preparation having improved therapeutic breadth, comprising nucleotide
synthesis inhibitors
Strongly basic anion exchangers are employed in therapy as
hypolipidemics in heterozygous familial hypercholesterolemia and other
primary hyperlipoproteinemias having a principal proliferation of the LDL
fraction or in chologenic diarrheas. Examples of suitable active substances
which are employed as hypolipidemics are N-(2-aminoethyl)-N'-
[2-[(2-aminoethyl)amino]ethyl]-1,2-ethanediamine polymers with
(chloromethyl)oxirane, which is also called colestipol (Colestid~) or
colestyramine (CAS-No. 11 041-12-6), which is a styrene/divinylbenzene
copolymer. Isoxazole or crotonamide derivatives are described in the
Patent Applications EP 484 223; EP 529 500; US 4 061 767; EP 538 783
or EP 551 230. Compounds which inhibit purine or pyrimidine synthesis are
called nucleotide synthesis inhibitors (Burkhardt and Kalden; Rheumat. Int.
(1997); 17: 85-90), these are, for example, compounds of the formula I
and/or II, brequinar (6-fluoro-2-(2'-fluoro[1 ,1 'biphenyl]-4-yl)-3-methyl-
4-quinolinecarboxylic acid), mycophenolatemofetil (2-morpholinoethyl
(E)-6-(1,3-dihydro-4-hydroxy-6-methoxy-7-methyl-3-oxoisobenzofuran-5-
yl)-4-methyl-4-hexenoate), methotrexate (CAS No. 59-05-02) or mizoribine
(CAS No. 50924-49-7), are absorbed in the intestine of patients after oral
administration and, after a brief increase in the blood levels after
administration (absorption peak), lead to constant high blood levels. The
abovementioned nucleotide synthesis inhibitors are excreted again in the
intestine via the liver and the bile. The excreted compounds mentioned can
partially be absorbed again from the intestine and secreted into the blood.
The compounds mentioned are therefore subject to the enterohepatic
circulation.
In the employment of nucleotide synthesis inhibitors for affecting the
immune system, it was surprisingly found that only brief active effects of
CA 02354266 2001-06-07
2
these substances are needed for the desired action on the immune system.
If blood levels of these substances which lead to active effects are
maintained over a relatively long period, although the side effects increase,
the desired action on the immune system is not increased. By limiting the
active effects to a short period of time, the tolerability of a therapy can be
improved while maintaining the desired pharmacodynamic effects on the
immune system (=improved therapeutic breadth).
In the case of nucleotide synthesis inhibitors which are subject to the
enterohepatic circulation, the duration of action can be reduced by
administering substances which interrupt the enterohepatic circulation.
Owing to interruption of the enterohepatic circulation, the desired action on
the immune system is maintained, but the side effects are drastically
reduced.
The abovementioned nucleotide synthesis inhibitors can also have an
improved therapeutic breadth in their action if compounds which
antagonize the action of the nucleotide synthesis inhibitors are
administered with a displacement in time - i.e. later than the nucleotide
synthesis inhibitors.
The term therapeutic breadth is understood here as meaning a measure of
the tolerability of a pharmaceutical and is essentially the difference
between the lowest dose which still leads to the desired therapeutic effects
and the dose which leads to side effects. The yardstick for the
improvements achieved is, for example, the amount of red blood
corpuscles, hemoglobin content, hematocrit, amount of glutamate
oxaloacetate transaminase, glutamate pyruvate transaminase, alkaline
phosphatase (from bone marrow) or amylase and the weight in comparison
with untreated patients.
The invention therefore relates to a preparation comprising
1 ) at least one compound which essentially prevents the enterohepatic
circulation of the nucleotide synthesis inhibitors or antagonizes the
action of the nucleotide synthesis inhibitors with a displacement in
time, and
CA 02354266 2001-06-07
3
2) at least one nucleotide synthesis inhibitor from the group consisting
of brequinar, mycophenolatemofetil (2-Morpholinoethyl (E)-6-(1,3-
dihydro-4-hydroxy-6-methoxy-7-methyl-3-oxoisobenzofuran-5-yl)-4-
methyl-4- hexenoate), methotrexate, mizoribine and compounds of
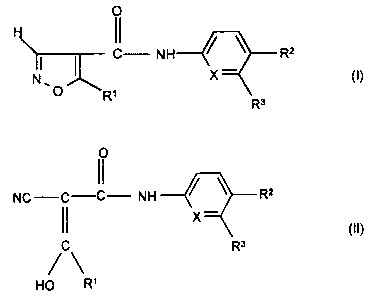
the formulae I and II
0
C- NH / \ Rz
N . X (I)
O R~
R3
O
NC -C-C- NH / \ Rz
X
R3 (II)
C
R~
HO
and/or an optionally stereoisomeric form of the compound of the
formula I or II and/or a physiologically tolerable salt of the compound
of the formula
II, where
R' is a) (C~-C4)-alkyl,
b) (C3-C5)-cycloalkyl,
c) (C2-C6)-alkenyl or
d) (C2-C6)-alkynyl,
R2 is a) -CF3,
b) -O-CF3,
c) -S-CF3,
d) -OH,
e) -N02,
f) halogen,
g) benzyl,
h) phenyl,
i) -O-phenyl,
k) -CN or
I) -O-phenyl, mono- or polysubstituted
by
1 ) (C~-C4)-alkyl,
CA 02354266 2001-06-07
4
2) halogen,
3) -O-CF3 or
4) -O-CH3,
R3 is a) (C~-C4)-alkyl,
b) halogen, or
c) is a hydrogen atom, and
X is a) a -CH group or
b) a nitrogen atom.
A mixture of the nucleotide synthesis inhibitors and compounds of the
formulae I and II or salts of the compounds of the formula II and a mixture
of the compounds which essentially prevent the enterohepatic circulation of
the compound of the formula I or II can also be employed.
The term "compound which essentially prevents the enterohepatic
circulation of the compound of the formula I or II" is understood as
meaning, for example, strongly basic anion exchangers such as colestipol
and colestyramine or active carbon. The term "compounds which
antagonize the action of the nucleotide synthesis inhibitors with a
displacement in time" are understood as meaning compounds such as
uridine, purine, purine nucleotides or pyrimidine nucleotides.
The use of a compound of the formula I and/or II and/or an optionally
stereoisomeric form of the compound of the formula I or II and/or a salt of
the compound of the formula II is preferred, where,
R' is a) methyl,
b) cyclopropyl or
c) (Cs-C5)-alkynyl,
R2 is -CF3 or -CN,
R3 is a hydrogen atom or methyl, and
X is a -CH group,
in combination with at least one compound from the group consisting of
colestipol, colestyramine and active carbon.
CA 02354266 2001-06-07
The use of N-(4-trifluoromethylphenyl)-5-methylisoxazole-4-carboxamide,
N-(4-trifluoromethylphenyl)-2-cyano-3-hydroxycrotonamide, 2-cyano-
3-cyclopropyl-3-hydroxyacrylic acid (4-cyanophenyl)amide or
N-(4-trifluoromethylphenyl)-2-cyano-3-hydroxyhept-2-en-6-ynecarbox-
5 amide in combination with colestyramine is particularly preferred.
The compound of the formula I or II is prepared according to known
processes such as are described in EP 484 223; EP 529 500;
US 4 061 767; EP 538 783 or EP 551 230. The starting substances for the
chemical reactions are known or can easily be prepared by methods which
are known from the literautre.
The terms alkyl, alkenyl and alkynyl are understood as meaning radicals
whose carbon chain can be straight or branched. The alkenyl or alkynyl
radicals can furthermore also contain a number of double bonds or a
number of triple bonds. Cyclic alkyl radicals are, for example, 3- to
5-membered monocyclic systems such as cyclopropyl, cyclobutyl or
cyclopentyl. Salts of the compound of the formula II are, for example,
sodium or lysinium salts which can be prepared as described in European
Patent Application No. EP 0769296.
The preparation according to the invention is suitable, for example, for the
treatment of
- immunological disorders
- inflammatory and cytotoxic processes in connection with gene
therapy interventions
- carcinomatous disorders such as lung cancer, leukemia, ovarian
cancer, sarcoma, Kaposi's sarcoma, meningioma, intestinal cancer,
lymph node cancer, brain tumors, breast cancer, pancreatic cancer,
prostate cancer or skin cancer
- autoimmune disorders such as systemic lupus erythematosus or
multiple sclerosis
- rheumatic disorders
- transplantations or graft-versus-host reactions or host-versus-graft
reactions
CA 02354266 2001-06-07
6
- disorders which are caused by strongly proliferating cells
- psoriasis or atypic dermatitis
- allergy, asthma, urticaria, rhinitis or uveitis
- type II diabetes
- cystic fibrosis, colitis, liver fibrosis or sepsis
- chronic inflammatory disorders such as arteriosclerosis, Crohn's
disease, ulcerative colitis.
The invention also relates to a process for the production of the
preparation, which comprises bringing the nucleotide synthesis inhibitors
and a compound which essentially prevents the enterohepatic circulation
of the nucleotide synthesis inhibitors, or antagonizes the action of the
nucleotide synthesis inhibitors with a displacement in time, into a suitable
administration form with a pharmaceutically suitable and physiologically
acceptable vehicle and, if appropriate, further suitable active compounds,
additives or excipients.
The preparation according to the invention can also include compositions
or combination packs in which the constituents are placed next to one
another and can therefore be used simultaneously, separately or
sequentially on one and the same human or animal body. The sequential
administration of the compound of the formula I and/or II before the
administration of the compound which essentially prevents the
enterohepatic circulation of the compound of the formula I or II is preferred.
To this end, for example, N-(4-trifluoromethylphenyl)-2-cyano-3-
hydroxycrotonamide (called compound 1 in the following) is administered
first. Colestyramine, which essentially prevents the enterohepatic
circulation of the compound I, is administered with a displacement in time,
i.e., for example, 2 hours or 4 hours after the administration of compound 1.
Owing to this time-displaced administration of the compound 1 and
colestyramine, the compound 1 is initially absorbed unhindered from the
digestive tract. After the administration of colestyramine, which is not
absorbed systemically, the compound 1 excreted via the bowel is bound to
colestyramine and can therefore not be reabsorbed again; as a result an
CA 02354266 2001-06-07
7
interruption to the enterohepatic circulation is brought about. Owing to this
measure, the duration of action and the blood level of the compound 1 are
drastically reduced. Despite this drastically reduced blood level, the
activity
in the pathological animal model, such as adjuvant arthritis, is not reduced
by the administration of colestyramine at a low, still just active dose of
approximately 2.5 mg/kg/day of the compound 1. If high doses of
25 mg/kg/day of the compound 1, which already lead to various side
effects, are employed in the same animal model, a clear reduction in the
side effects with retention of the desired actions on the immune system is
observed by means of administration of colestyramine.
The preparation according to the invention can be present as a dose unit in
the form of pharmaceutical forms such as capsules (including
microcapsules), which in general contain no pharmaceutical vehicles),
tablets including coated tablets and pills, or suppositories, it being
possible
when using capsules for the capsule material to assume the function of the
vehicle and for the contents to be present, for example, as a powder, gel,
solution, emulsion or dispersion. It is particularly advantageous and simple,
however, to prepare oral or peroral formulations which contain the
calculated amounts of the active compounds together with any desired
pharmaceutical vehicles using the two active compound components 1 )
(e.g. colestyramine) and 2) (compound of the formula I and/or II). An
appropriate formulation (suppositories) for rectal therapy can also be used.
Transdermal administration in the form of ointments or creams, parenteral
(intraperitoneal, subcutaneous, intramuscular) injection or oral
administration of solutions which contain the combinations according to the
invention is likewise possible. In addition to the active compound,
ointments, pastes, creams and powders can contain the customary
vehicles, e.g. animal and vegetable fats, waxes, paraffins, starch,
tragacanth, cellulose derivatives, polyethylene glycols, silicones, silicic
acid, aluminum hydroxide, talc, zinc oxide, lactose, bentonites, calcium
silicate and polyamide powder or mixtures of these substances. The
tablets, pills or granule bodies can be produced by processes such as
pressing, dipping or fluidized-bed processes or pan coating and contain
vehicles and other customary excipients such as gelatin, agarose, starch
CA 02354266 2001-06-07
8
(e.g. potato, corn or wheat starch), celluloses such as ethylcellulose,
silica,
magnesium carbonate, various sugars such as lactose and/or calcium
phosphates. The coating solution usually consists of sugar and/or starch
syrup and mostly additionally contains gelatin, synthetic cellulose esters,
gum arabic, polyvinylpyrrolidone, pigments, surface-active substances,
plasticizers and similar additives according to the prior art. For the
production of the preparation forms, any customary flow-regulating agent,
lubricant or glidant such as magnesium stearate and release agents can be
used. The preparations preferably have the form of coating/core tablets or
multilayer tablets, the active component 2 being in the coating or in the
core or in one layer, while the active component 1 is in the core, in the
coating or in another layer. The active compound components can also be
present in delayed-release form or adsorbed on release-delaying material
or included in the release-delaying material (e.g. those based on cellulose
or polystyrene resins, e.g. hydroxyethylcellulose). Delayed release of the
active compounds can also be achieved by providing the layer or the
compartment concerned with customary enteric coatings.
A delayed release of the compound which essentially prevents the
enterohepatic circulation of the compound of the formula I or II is preferred.
The dose to be used is, of course, dependent on various factors such as
the living being to be treated (i.e. human or animal), age, weight, general
state of health, the degree of severity of the symptoms, the disorder to be
treated, possible concomitant disorders (if present), the nature of the
concomitant treatment with other pharmaceuticals, or the frequency of the
treatment. The doses are in general administered several times per day
and preferably one to three times per day. The amounts of individual active
compound used are based here on the recommended daily dose of the
respective individual active compound and, in the combination preparation,
should in general be from 10% to 300% of the recommended daily dose,
preferably from 50% to 150%, in particular 80%. Suitable therapy with the
combinations according to the invention thus consists, for example, in the
administration of 1, 2 or 3 individual doses of the preparation consisting of
N-(4-trifluoromethylphenyl)-5-methylisoxazole-4-carboxamide or
N-(4-trifluoromethylphenyl)-2-cyano-3-hydroxycrotonamide in an amount of
CA 02354266 2001-06-07
9
from 2 mg to 250 mg, preferably 5 mg to 150 mg, in particular 10 mg to
50 mg, particularly preferably 10 mg to 20 mg, and colestyramine in an
amount of from 250 mg to 6000 mg, in particular from 1500 mg to 3000 mg.
The preparations according to the invention can furthermore also be
employed together with other suitable active compounds, for example
antiuricopathics, analgesics, steroidal or nonsteroidal antiinflammatories,
platelet aggregation inhibitors, cytokines, cytokine agonists, cytokine
antagonists or immunosuppressant compounds such as cyclosporin A, FK
506 or rapamycin.
CA 02354266 2001-06-07
Example 1
Adjuvant-induced arthritis, modification according to Perper
(Proc. Soc. exp. Biol. Med. 137, 506 (1971))
5 The experimental animals used were male rats of a Lewis strain
(Moellegard, Denmark) having a body weight of from 160 to 210 g. On the
1st day, the animals were injected subcutaneously, into the tail root, with
complete Freund's adjuvant containing a mycobacterium butyricum
suspension in heavy paraffin oil (Difco; 6 mg/kg in paraffin oil; Merck). The
10 compounds N-(4-trifluoromethylphenyl)-2-cyano-3-hydroxycrotonamide and
colestyramine were suspended in carboxymethylcellulose (1 % in water)
and administered orally. The compounds were administered once daily
from the 1st to the 17th day of the experiment; the paw volume and arthritis
index were then determined on the 18th day.
The severity of the disorder was determined by measuring the paw volume
of both hind paws. The measurement was carried out by means of the
water displacement method using a 2060 Plethys monitor (Rhema-
Labortechnik, Hofheim, Germany). The arthritis index was furthermore
determined in the 18th day after injection.
Determination of the arthritis index:
1. ears 0.5 points for each ear on which reddening
occurs and nodules are formed
2. nose 1 point for connective tissue swelling
3. tail 1 point for the emergence of nodules
4. fore paws 0.5 points for each paw on which at least one
inflammation occurs on a joint
5. hind paws 1 point for slight inflammation (swelling)
2 points for a medium-strength inflammation
3 points for a masive inflammatory reaction
On the 1st day, animals of an "arthritis contro" control group were given a
subcutaneous injection, into the tail root, with complete Freund's adjuvant
and were given, however, only the solvent (1% carboxymethylcellulose in
CA 02354266 2001-06-07
11
water). 6 animals in each case were used per dose and in the control
group. Untreated animals were employed as a further "healthy control"
control group. The activity criteria used were the reduction of the increase
in the paw volume and the decrease in the arthritis index, compared with
the untreated control group, and the weight of the animals, in each case in
percent and based on the arthritis control. In the following table,
N-(4-trifluoromethylphenyl)-2-cyano-3-hydroxycrotonamide is described as
compound 1. Colestyramine was administered 4 hours later than
compound 1. Table 1 shows the results obtained.
Table 1
Active Paw volume Arthritis Weight
index
substance (%) (%) (%)
(mg/kg of
live
weight
Healthy control 30
Arthritis 18
control
Colestyramine1000 35 44 8
Compound 2.5 -63 -77 20
1
Compound 2.5 + 1000 -70 -92 20
1 +
Colestyramine
Compound 7.5 -83 -92 18
1
Compound 7.5 + 1000 -73 -95 27
1
Colestyramine
Compound 25 -92 -100 -2
1
Compound 25 + 1000 -72 -100 3
1
Colestyramine
The values shown in the table by a "-' indicate a decrease; a1 otner values
indicate an increase in comparison with the start of the experiment
CA 02354266 2001-06-07
12
The animals treated with the preparation according to the invention showed
a weight increase which, in the case of the amounts 2.5 and 7.5 of
compound 1, came very close to the healthy control and was significantly
better than with compound 1 alone, while the activity of compound 1 was
completely retained.
Example 2
The experimental conditions are analogous to Example 1. The actions of
the compound 1 and colestyramine on the amount of red blood corpuscles
(RBC), hemaglobin content (HGB), hematocrit (HCT), amount of glutamate
oxalacetate transaminase (GOT) and glutamate pyruvate transaminase
(GPT) were determined. Colestyramin was administered 4 hours later than
compound 1. Table 2 shows the results obtained.
Table 2
Active Paw ArthritisGOT GPT RBC HGB HCT Num-
substance volume index (U/I)(U/I)(*106/(g/dl)(%) ber
of
(mg/kg (%) (%) mm3) ani-
of
live weight) mars
Healthy control 50.423.6 7.4 13.1 38.9 4
Arthritis 50.820.3 7.3 13.0 38.0 6
control
Colestyramine1000 10 22 43.525.2 7.6 10.2 31.4 6
Compound 25 -92 -100 85.325.1 3.8 6.1 18.7 6
1
Compound 25 + 1000 -72 -100 52.521.5 6.08 10.2 31.4 5
1
Colestyramine
The values shown in the table by a "-" indicate a aecrease; ail oiner values
indicate an increase in comparison with the start of the experiment
The animals treated with the preparation according to the invention showed
a normalization of the amount of red blood corpuscles (RBC), hemoglobin
content (HGB), hematocrit (HCT), amount of glutamate oxaloacetate
CA 02354266 2001-06-07
13
transaminase (GOT) and glutamate pyruvate transaminase (GPT) which
came very close to the healthy control and was significantly better than with
compound 1 alone, while the activity of compound 1 was completely
retained.
Example 3
The experimental conditions are analogous to Example 1. The actions of
compound 1 and colestyramine on the amount of alkaline phosphatase
(AP) and amylase were determined. Colestyramine was administered 4
hours later than compound 1. Table 2 shows the results obtained.
Table 3:
Active Paw ArthritisAP AmylaseNumber
of
substance volume index (U/I) (U/I) tested
(mg/kg (%) (%) animals
of
live weight)
Healthy control 312.6 3058.3 6
Arthritis 231.5 2251.6 6
control
Colestyramine1000 -60 -46 271.8 2756.6 6
Compound 25 -110 -100 114.8 1306.5 6
1
Compound 25 + 1000 -86 -94 206.6 2783.3 3
1
Colestyramine
The values shown in the table by a "-" indicate a decrease; all other values
indicate an increase in comparison with the start of the experiment
The animals treated with the preparation according to the invention showed
a normalization of the amount of alkaline phosphatase, which came very
close to the healthy control and was significantly better than with compound
1 alone, while the activity of compound 1 was completely retained.
Example 4
A preparation according to the invention consists of a small hard gelatin
capsule which contains 400 mg of colestyramine and a larger hard gelatin
CA 02354266 2001-06-07
14
capsule which contains 20 mg of N-(4-trifluoromethylphenyl )-
5-methylisoxazole-4-carboxamide. The smaller hard gelatin capsule is
completely enclosed by the larger capsule. The filling material employed
between the two capsules is glucose.