Note: Descriptions are shown in the official language in which they were submitted.
CA 02356632 2001-06-08
WO 00/34417 PCTIUS98/26134
-1-
1 DEHYDROCYCLIZATION PROCESS WITH
2 DOWNSTREAM DIMETHYLBUTANE REMOVAL
3 FIELD OF THE INVENTION
4 The present invention is directed to dehydrocyclization using a highly
selective dehydrocyclization catalyst, and particularly to dimethylbutane
6 removal from the recycle to a highly selective dehydrocyclization catalytic
7 zone.
8 BACKGROUND OF THE INVENTION
9 In the process of the present invention aromatics are formed from feed
hydrocarbons by dehydrocyclization. Other reactions may take place in the
11 reaction zone, and the reaction step may more generally be referred to as
12 reforming. Thus, in the reaction step of the process of the present
invention,
13 besides the dehydrocyclization or aromatization reaction, other reactions
can
14 occur, such as dehydrogenation, isomerization, hydroisomerization,
cyclization and hydrocracking. The main reaction is dehydrocyclization to
16 _ form aromatics from paraffins.
17 U.S. Patent No. 4,104,320, which was granted August 1, 1978, discloses that
18 it is possible to dehydrocyclize paraffins to produce aromatics with high
19 selectivity using a monofunctional nonacidic catalyst. Preferably, the
catalyst
comprises a type L zeolite. The type L zeolite preferred in the U.S. Patent
21 No. 4,104,320 process are those having exchangeable cations of which at
22 least 90% are sodium, lithium, potassium, rubidium or cesium. The catalyst
23 used in U.S. Patent No. 4,104,320 also contains at least one Group VIII
noble
24 metal (or tin or germanium). In particular, catalysts having platinum in an
L zeolite, wherein potassium in the L zeolite has been exchanged to replace a
26 portion of the potassium with rubidium or cesium, are claimed in the '320
CA 02356632 2001-06-08
WO 00/34417 PCT/US98/26134
-2-
1 patent to achieve exceptionally high selectivity for n-hexane conversion to
2 benzene. As disclosed in U.S. Patent No. 4,104,320, the L zeolite used is
3 typically synthesized in the potassium form. A portion, usually not more
than
4 80%, of the potassium cations can be exchanged so that other cations,
preferably rubidium or cesium, replace the exchangeable potassium.
6 After U.S. Patent No. 4,104,320 in 1978, an important step forward was
7 disclosed in U.S. Patents Nos. 4,434,311; 4,435,283; 4,447,316; and
8 4,517,306, all of which patents were granted in 1984 and 1985. These
9 patents describe dehydrocyclization catalysts comprising a large-pore
zeolite
exchanged with an alkaline earth metal (barium, strontium or calcium,
11 preferably barium) and wherein the catalyst contains one or more Group VIII
12 metals, most preferably platinum. An essential element in the catalyst of
13 these patents is the alkaline earth metal. Especially when the alkaline
earth
14 metal is barium, and the large-pore zeolite is L zeolite, the catalysts of
these
patents were found to provide higher selectivities than the corresponding
16 alkali exchanged L zeolite catalyst disclosed in U.S. Patent No. 4,104,320.
17 These platinum on L zeolite catalysts referred to in the previous two
18 paragraphs, whether in the potassium form, or other alkali metal form, or
in
19 the alkaline earth metal exchanged form, are substantially "nonacidic".
These
nonacidic catalysts have been referred to as "monofunctional" catalysts.
21 Such nonacidic, monofunctional catalysts are highly selective for forming
22 aromatics via dehydrocyclization of paraffins.
23 Having a highly selective catalyst, commercialization seemed
straightforward.
24 However, that was not the case. It was found that the high selectivity
L zeolite catalysts containing a Group VIII metal were unexpectedly
26 susceptible to sulfur poisoning at ultra low levels of sulfur in the feed.
U.S.
27 Patent No. 4,456,527 discloses this discovery. Specifically, it was found
that
28 the concentration of sulfur in the hydrocarbon feed must be reduced to
ultra
CA 02356632 2001-06-08
WO 00/34417 PCT/US98/26134
-3-
1 low levels, preferably less than 50 parts per billion, to achieve acceptable
2 stability, i.e., long run length, for the catalyst when used in the
3 dehydrocyclization process.
4 With the progress of U.S. Patent No. 4,456,527, more attention was given to
the process arrangement under which the overall dehydrocyclization process
6 to produce aromatics was carried out.
7 U.S. Patents Nos. 4,648,961 and 4,650,565 disclose processes using a
8 highly selective dehydrocyclization catalyst wherein a paraffins rich feed
is
9 contacted with the catalyst to form aromatics, then the aromatics are
separated from the reaction zone effluent by means of a solvent extraction
11 step, or via molecular sieve separation, and a raffinate paraffins rich
stream
12 from the solvent extraction or molecular sieve separation step is recycled
to
13 the dehydrocyclization reaction zone.
14 U.S. Patent No. 4,594,145 and Reissue Patent 33,323 disclose a process
using a highly selective dehydrocyclization catalyst wherein a paraffins rich
16 feed is contacted with the catalyst to form aromatics, then aromatics are
17 separated from the reaction zone effluent, and the remaining paraffins are
18 recycled to the reaction zone.
19 U.S. Patents Nos. 4,568,656 and 4,595,668 disclose use of highly selective
dehydrocyclization catalyst wherein the Group VIII metal component of the
21 catalyst is highly dispersed on a zeolite L support. These two U.S. patents
22 state (see Col. 16, line 38, of the `668 patent): "Since the catalyst is
23 monofunctional and does not promote isomerization without cyclization, feed
24 compounds such as dimethylbutanes are not effective."
Another process oriented patent using a highly selective dehydrocyclization
26 catalyst is European Patent 335,540. This patent discloses a process
CA 02356632 2001-06-08
WO 00/34417 PCT/US98/26134
-4-
1 wherein a hydrocarbon feed is (a) separated into a first fraction comprising
C5
2 minus hydrocarbons and dimethylbutanes, and a second fraction comprising
3 C6 plus hydrocarbons; (b) separating the second fraction into (i) a light
4 fraction comprising not more than 10% by volume dimethylbutanes, the light
fraction being selected from a C. fraction, a C,, a C. fraction, a C6-C,
fraction,
6 C; C8 fraction, CB-CB and a fraction consisting essentially of C. and C$
7 hydrocarbon; and (ii) a heavy fraction; and (c) dehydrocyclizing the light
8 fraction under dehydrocyclization conditions in the presence of a
9 monofunctional catalyst. Thus, according the EP 335,540 process,
dimethylbutanes are removed from the hydrocarbon fresh feed prior to
11 dehydrocyclization of the fresh feed to form aromatics.
12 SUMMARY OF THE INVENTION
13 According to the present invention, a process is provided for
14 dehydrocyclization, which process comprises:
(a) contacting fresh paraffins rich feed hydrocarbon containing 0.1 to
16 20 wt. % dimethylbutanes with a highly selective dehydrocyclization
17 catalyst in a reaction zone under dehydrocyclization reaction conditions
18 to convert paraffins to aromatics and obtain an aromatics rich effluent;
19 (b) separating aromatics from the effluent to obtain a paraffins rich
raffinate;
(c) removing dimethylbutanes from the raffinate to obtain a raffinate of
21 reduced dimethylbutane content; and
22 (d) recycling the raffinate of'reduced dimethylbutane content to the
reaction
23 zone.
CA 02356632 2001-06-08
WO 00/34417 PCT/US98/26134
-5-
1 Preferably, the highly selective dehydrocyclization catalyst is a nonacidic
2 catalyst.
3 The catalyst preferably includes a Group VIII metal, most preferably
platinum,
4 and preferably includes a unidimensional crystalline aluminosilicate. The
term
"unidimensional" means that the pores or channels in the crystals run
6 substantially only in one direction along the length or c-axis of the
crystals.
7 Preferred aluminosilicates are zeolite L, zeolite omega, ZSM-10 and
8 mordenite.
9 Most preferably, the aluminosilicate is zeolite L. Zeolite L is
unidimensional.
In the process of the present invention, a centrally important feature is the
11 separation of a major portion of dimethylbutanes downstream of the
12 dehydrocyclization reaction zone. Thus, the dimethylbutanes are not
13 separated primarily from the fresh feed to the process. Rather, in the
present
14 invention, the dimethylbutanes are separated primarily from the effluent
from
the dehydrocyclization reaction. The downstream dimethylbutane removal
16 may be carried out after aromatics are separated from the reaction zone
17 effluent, or prior to aromatics separation; but, in either case, downstream
of
18 the dehydrocyclization reaction zone.
19 The aromatics separation from the reaction step effluent preferably is done
by
solvent extraction, distillation or molecular sieve extraction. Most
preferably,
21 the aromatics separation is done by solvent extraction. This aromatics
22 separation step results in an aromatics rich product stream, and a
paraffins
23 rich raffinate stream.
CA 02356632 2001-06-08
WO 00/34417 PCT/US98/26134
-6-
1 In the process of the present invention, dimethylbutanes preferably are
2 separated from the raffinate stream. Then the raffinate is recycled to the
3 reaction step.
4 Among other factors, the present invention is based on my conception of
removing the dimethylbutanes primarily downstream of the dehydrocyclization
6 reaction step rather than removing dimethylbutanes from the fresh feed to
the
7 reaction zone. Further, the present invention is based on my finding that
this
8 unusual point of dimethylbutanes removal results in surprising advantages in
9 terms of yield and operating expense for the highly selective
dehydrocyclization process.
11 According to one embodiment of the present invention, as described above,
12 downstream dimethylbutane removal is carried out after aromatics are
13 removed from the effluent from the dehydrocyclization step.
14 According to another embodiment of the present invention, dimethylbutanes
are removed downstream of the dehydrocyclization reaction zone but prior to
16 treatment of the reaction zone effluent to separate aromatics from
paraffins.
17 Thus, according to this embodiment, a process is provided for
18 dehydrocyclization of hydrocarbon which comprises:
19 (a) contacting fresh paraffins rich feed hydrocarbon containing 0.1 to
20.0 wt. % dimethylbutanes with a highly selective dehydrocyclization
21 catalyst in a reaction zone under dehydrocyclization reaction conditions
22 to convert paraffins to aromatics and obtain an aromatics rich effluent;
23 (b) removing dimethylbutanes from the aromatics rich effluent to obtain a
24 paraffins-aromatics mixture of reduced dimethylbutane content;
CA 02356632 2001-06-08
WO 00/34417 PCT/US98/26134
-7-
1 (c) separating aromatics from the paraffins-aromatics mixture to obtain an
2 aromatics lean raffinate; and
3 (d) recycling the raffinate to the reaction zone.
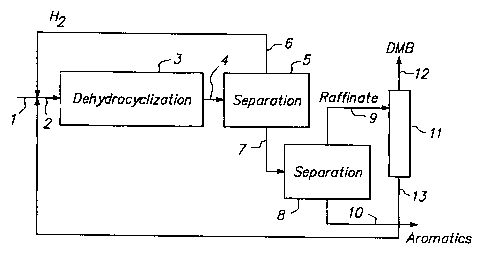
4 BRIEF DESCRIPTION OF THE DRAWING
The drawing is a simplified schematic process flow diagram illustrating a
6 preferred embodiment of the present invention, wherein dimethylbutanes are
7 removed downstream from the dehydrocyclization reaction zone.
8 DETAILED DESCRIPTION OF THE INVENTION
9 Referring to the simplified drawing in more detail, fresh feed is introduced
to
the dehydrocyclization reaction zone via lines 1 and 2. Suitable feeds are
11 nonaromatic organic compounds, preferably naphtha boiling range feedstocks
12 such as CB-C,o, more preferably Cg Cg, paraffins. Because
dehydrocyclization
13 is the key reaction in reaction zone 3, the feedstock preferably is CB or
higher
14 nonaromatic organic compounds. Examples of preferred Ce-C8 paraffinic
feedstock components include n-hexane, n-heptane and n-octane.
16 The naphtha feedstock may be derived directly from crude petroleum by
17 distillation, or may be indirectly derived from a petroleum feedstock by
18 separation of a naphtha boiling range stream from the effluent from
19 hydrocracking, coking or catalytic cracking. Suitable naphtha feedstocks
may
boil between about 60 C and 220 C. Preferably, the naphtha feedstock is
21 predominantly (more than one half by weight) paraffins, and most preferably
22 is C6-C8 paraffins. Ce paraffins, especially n-hexane and also
23 methylpentanes, are particularly preferred feedstocks.
CA 02356632 2001-06-08
WO 00/34417 PCT/US98/26134
-8-
1 As discussed in more detail below, these feedstocks almost invariably
contain
2 dimethylbutanes, as the dimethylbutanes boil close to hexanes. Table 1
3 below lists the boiling points of hydrocarbons in the C5-C, paraffin range.
4 TABLE 1
Boiling Points of Selected
6 C5-C7Hydrocarbons
F C
2-Methylbutane (isopentane) 82 28
n-Pentane 97 36 C5 H12
Cyclopentane 120 49 C5 H,o
2,2-Dimethylbutane 121.5 50
2,3-Dimethylbutane 136 58
2-Methylpentane 140.5 60 Cs H14
3-Methylpentane 145.9 63
n-Hexane 156 69
Methylcyclopentane 161 72 Cg H12
Cyclohexane 177 81 CB H12
2,2,3-Trimethylbutane 178 81
2,2-Dimethylpentane 174.5 79
2,3-Dimethylpentane 194 90
2,4-Dimethylpentane 177 80
3,3-Dimethylpentane 187 86 C, H1e
2-Methylhexane 194 90
3-Methylhexane 197 92
3-Ethylpentane 200 93
n-Heptane 209 98
7
CA 02356632 2001-06-08
WO 00/34417 PCT/US98/26134
-9-
1 As can be seen from Table 1, dimethylbutanes boil near n-hexane, and also
2 near methylpentanes, both of which are preferred feedstocks for the
3 dehydrocyclization reaction zone.
4 The net fresh feed to the process of the present invention will contain
dimethylbutanes, typically from 0.1 to 20 wt. % dimethylbutanes, preferably
6 0.3 to 18%, more preferably 0.5 to 18%, and most preferably 1.0 to 15%,
7 based on weight of the net or fresh hydrocarbon feed to the process.
8 As dimethylbutanes are not desired in the dehydrocyclization reaction zone
3,
9 the prior art teaches removal of dimethylbutanes from the fresh feed. The
separation of dimethylbutanes from the fresh feed can be done by distillation,
11 as taught by the previously cited EP 335,540.
12 However, in the process of the present invention, dimethylbutanes are not
13 primarily removed from the fresh feed to reaction zone 3. In the present
14 invention, dimethylbutanes are primarily removed downstream in the process,
after aromatics have been formed in the dehydrocyclization reaction zone.
16 This downstream removal of dimethylbutanes is further described below.
17 The term "fresh feed" is used herein to mean the net hydrocarbon feed to
the
18 process which has not been passed through the dehydrocyclization zone.
19 In the present invention, reaction zone 3 uses a dehydrocyclization
catalyst
which has a high selectivity to form aromatics. The term "selectivity" as used
21 in the present invention is defined as a percentage of moles of acyclic
22 hydrocarbons converted to aromatics compared to moles converted to
23 aromatics and cracked products. Thus, percent selectivity may be defined by
24 the following formula:
CA 02356632 2001-06-08
WO 00/34417 PCTIUS98/26134
-10-
1 100 x moles of
2 acyclic hydrocarbons
3 i.e., Selectivity = converted to aromatics
4 moles of
acyclic hydrocarbons
6 converted to aromatics
7 and cracked products
8 Isomerization of paraffins and interconversion paraffins and
9 alkylcyclopentanes having the same number of carbcin atoms per molecule
are not considered in determining selectivity.
11 The selectivity for converting acyclic hydrocarbons to aromatics is a
measure
12 of the effectiveness of the dehydrocyclization reaction step in converting
13 acyclic hydrocarbons to the desired and valuable products: aromatics and
14 hydrogen, as opposed to the less desirable by-products, such as products
from hydrocracking in the dehydrocyclization reaction zone.
16 Highly selective catalysts produce more hydrogen than less selective
17 catalysts because hydrogen is produced when acyclic hydrocarbons are
18 converted to aromatics, whereas hydrogen is consumed when acyclic
19 hydrocarbons are converted to cracked products. Increasing the selectivity
of
the process increases the amount of hydrogen produced (more
21 aromatization) and decreases the amount of hydrogen consumed (less
22 cracking).
23 Preferred catalysts for use in reaction zone 3 are highly selective
24 dehydrocyclization catalysts, particularly catalysts comprising a Group
VIII
metal dispersed on a unidimensional crystalline aluminosilicate. The highly
26 selective dehydrocyclization catalyst preferably is a nonacidic catalyst,
and
27 preferably is a monofunctional catalyst.
CA 02356632 2001-06-08
WO 00/34417 PCT/US98/26134
-11-
1 Preferably, the catalyst used in reaction zone 3 contains one or more
2 Group VIII metals, for example, nickel, iridium, rhodium, palladium, rhenium
or
3 platinum. The most preferred Group VIII metals for use in the
4 dehydrocyclization catalyst are iridium, palladium and platinum, most
especially, platinum. The amount of Group VIII metal in the catalyst
6 preferably is between 0.1 and 5 wt. % based on the total catalyst, more
7 preferably 0.3 to 2 wt. %.
8 The Group VIII metal or metals may be introduced to the catalyst such as the
9 zeolite component of the catalyst, by synthesis, impregnation, or ion
exchange carried out in aqueous solution of an appropriate salt. When it is
11 desired to introduce two Group VIII metals into the zeolite component of
the
12 catalyst, the operation may be carried out simultaneously or sequentially.
13 By of way of example, platinum can be introduced by impregnating the
zeolite
14 with an aqueous solution of tetrammine (II) nitrate, chloroplatinic acid,
chloroplatinous acid, diamino-platinum or tetrammineplatinum (II) chlorite. In
16 an ion exchange process, platinum can be introduced by using cationic
17 platinum complexes such as tetrammineplatinum (Il) nitrate.
18 Preferably, an inorganic oxide is used as a carrier to bind the zeolite
19 containing the Group VIII metal and provide added strength and integrity
for
the catalyst as a particle. The carrier can be a natural or synthetically
21 produced inorganic oxide or a combination of inorganic oxides. Typical
22 - inorganic oxides supports which can be used include clays, alumina and
silica
23 in which acidic sites are preferably exchanged by cations which do no
impart
24 strong acidity (such as sodium, potassium, rubidium, cesium, calcium,
strontium or barium).
CA 02356632 2001-06-08
WO 00/34417 PCT/US98/26134
-12-
1 Preferred unidimensional aluminosilicates for use in the dehydrocyclization
2 catalyst are L zeolite, omega zeolite, ZSM-10, cancrinite and mordenite.
3 L zeolite is especially preferred for the catalyst used in reaction zone 3.
4 Type L zeolites are synthetic zeolites. A theoretical formula is
M9/n[Al02)9(Si02)27] in which M is a cation having the valency n.
6 The real formula may vary without changing the crystalline structure; for
7 example, the mole ratio of 65 silicon to aluminum (Si/Al) may vary from 1.0
to
8 3.5.
9 Although there are a number of cations that may be present in zeolite L, in
one embodiment, it is preferred to synthesize the potassium form of the
11 zeolite, i.e., the form in which the exchangeable cations present are
12 substantially all potassium ions. The reactants accordingly employed are
13 readily available and generally water soluble. The exchangeable cations
14 present in the zeolite may then conveniently be replaced by other
exchangeable cations, as will be shown below, thereby yielding isomorphic
16 form of zeolite L.
17 In one method of making zeolite L, the potassium form of zeolite L is
prepared
18 by suitably heating an aqueous metal aluminosilicate mixture whose
19 composition, expressed in terms of the mole ratios of oxides, falls within
the
range:
CA 02356632 2001-06-08
WO 00/34417 PCT/US98/26134
-13-
1 K20/( K20 + Na20) From about 0.33 to about 1
2 (K20 + Na20)/Si02 From about 0.35 to about 0 5
3 SiO2/A12O3 From about 10 to about 28
4 H20/( K20 + Na20) From about 15 to about 41
The desired product is hereby crystallized out relatively free from zeolite of
6 dissimilar crystal structure.
7 The potassium form of zeolite L may also be prepared in another method
8 along with other zeolite compounds by employing a reaction mixture whose
9 composition, expressed in terms of mole ratios of oxides, falls within the
following range:
11 K20/( K20 + Na20) From about 0.26 to about 1
12 (K20 + Na20)/Si02 From about 0.34 to about 0 5
13 SiO2/A1203 From about 15 to about 28
14 H20/( K20 + Na20) From about 15 to about 51
It is to be noted that the presence of sodium in the reaction mixture is not
16 critical to the present invention.
17 When the zeolite is prepared from reaction mixtures containing sodium,
18 sodium ions are generally also included within the product as part of the
19 exchangeable cations together with the potassium ions. The product
obtained from the above ranges has a composition, expressed in terms of
21 moles of oxides, corresponding to the formula:
22 0.9-1.3[(1-x)K20,xNa2O]: A1203:5.2-6.9 Si02:HZO
23 wherein "x" may be any value from 0 to about 0.75 and "y" may be any value
24 from 0 to about 9.
CA 02356632 2001-06-08
WO 00/34417 PGT/US98/26134
-14-
1 In making zeolite L, representative reactants are activated alumina, gamma
2 alumina, alumina trihydrate and sodium aluminate as a source of alumina.
3 Silica may be obtained from sodium or potassium silicate, silica gels,
silicic
4 acid, aqueous colloidal silica sols and reactive amorphous solid silicas.
The
preparation of typical silica sols which are suitable for use in the process
of
6 the present invention are described in U.S. Pat. No. 2,574,902 and U.S. Pat.
7 No. 2,597,872. Typical of the group of reactive amorphous solid silicas,
8 preferably having an ultimate size of less than 1 micron, are such materials
as
9 fume silicas, chemically precipitated and precipitated silica sols.
Potassium
and sodium hydroxide may supply the metal cation and assist in controlling
11 pH.
12 In making zeolite L, the usual method comprises dissolving potassium or
13 sodium aluminate and alkali, viz., potassium or sodium hydroxide, in water.
14 This solution is admixed with a water solution of sodium silicate, or
preferably
with a water-silicate mixture derived at least in part from an aqueous
colloidal
16 silica sol. The resultant reaction mixture is placed in a container made,
for
17 example, of metal or glass. The container should be closed to prevent loss
of
18 water. The reaction mixture is then stirred to ensure homogeneity.
19 The zeolite may be satisfactorily prepared at temperatures of from about 90
C
to 200 C, the pressure being atmospheric or at least that corresponding to
21 the vapor pressure of water in equilibrium with the mixture of reactants at
the
22 higher temperature. Any suitable heating apparatus, e.g., an oven, sand
23 bath, oil bath or jacketed autoclave, may be used. Heating is continued
until
24 the desired crystalline zeolite product is formed. The zeolite crystals are
then
filtered off and washed to separate them from the reactant mother liquor. The
26 zeolite crystals should be washed, preferably with distilled water, until
the
27 effluent wash water, in equilibrium with the product, has a pH of between
28 about 9 and 12. As the zeolite crystals are washed, the exchangeable cation
CA 02356632 2008-09-05
-15-
1 of the zeolite may be partially removed and is believed to be replaced by
2 hydrogen cations. If the washing is discontinued when the pH of the effluent
3 wash water is between about 10 and 11, the (K20 + Na20))/AI203 molar ratio
4 of the crystalline product will be approximately 1Ø Thereafter, the
zeolite
crystals may be dried, conveniently in a vented oven.
6 Zeolite L has been characterized in "Zeolite Molecular Sieves" by Donald W.
7 Breck, John Wiley & Sons, 1974, as having a framework comprising
8 18 tetrahedra unit cancrinite-type cages linked by double 6-rings in columns
9 and crosslinked by single oxygen bridges to form planar 12-membered rings.
These 12-membered rings produce wide channels parallel to the c-axis with
11 no stacking faults. Unlike erionite and cancrinite, the cancrinite cages
are
12 symmetrically placed across the double 6-ring units. There are four types
of
13 cation locations: A in the double 6-rings, B in the cancrinite-type cages,
C
14 between the cancrinite-type cages, and D on the channel wall. The cations
in
site D appear to be the only exchangeable cations at room temperature.
16 During dehydration, cations in site D probably withdraw from the channel
17 walls to a fifth site, site E, which is located between the A sites. The
18 hydrocarbon sorption pores are approximately 7 to 8 Angstroms in diameter.
19 A more complete description of these zeolites is given, e.g., in U.S. Pat.
No. 3,216,789 which, more particularly, gives a conventional description of
21 these zeolites. U.S. Pat. No. 3,216,789 shows a type L zeolite useful in
the
22 present invention.
23 Zeolite L differs from other large pore zeolites in a variety of ways,
besides
24 X-ray diffraction pattern.
One of the most pronounced differences is in the channel system of zeolite L.
26 Zeolite L has a one-dimensional channel system parallel to the c-axis,
while
27 most other zeolites have either two-dimensional or three-dimensional
channel
CA 02356632 2008-09-05
-16-
1 systems. Zeolites A, X and Y all have three-dimensional channel systems.
2 Mordenite (Large Pore) has a major channel system parallel to the c-axis,
and
3 another very restricted channel system parallel to the b-axis. Omega zeolite
4 (Mazzite) has a one-dimensional channel system.
Another pronounced difference is in the framework of the various zeolites.
6 Zeolite L has cancrinite-type cages linked by double-six rings in columns
and
7 crosslinked by oxygen bridges to form planar 12-rings. Zeolite A has a cubic
8 array of truncated octahedra, beta-cages linked by double-four ring units.
9 Zeolites X and Y both have truncated octahedra, beta-cages linked
tetrahedrally through double-six rings in an arrangement like carbon atoms in
11 a diamond.
12 Mordenite has complex chains of five-rings crosslinked by four-ring chains.
13 Omega has a fourteen-hedron of gmelinite-type linked by oxygen bridges in
14 columns parallel to the c-axis.
ZSM-10 is constructed from columns of alternating cancrinite-type cages and
16 double-six rings. In ZSM-10, there are two one-dimensional 12-ring pore
17 systems which run parallel to the c-axis. One 12-ring channel is apparently
18 identical to the undulating channel in the zeolite L framework, the other
is
19 apparently identical to the 12-ring channel in the framework of offretite.
Thus,
ZSM-10 is believed to have two distinct, parallel, 12-ring channel systems.
21 ZSM-10 is described in "ZSM-10: Synthesis and Tetrahedral Framework
22 Structure", Zeolites, 16, 4, 236-244 (April 1996). The synthesis of ZSM-10
is
23 also described in U.S. Patent No. 3,692,470.
CA 02356632 2001-06-08
WO 00/34417 PCT/US98/26134
-17-
1 Various factors have an effect on the X-ray diffraction pattern of a
zeolite.
2 Such factors include temperature, pressure, crystal size, impurities, and
type
3 of cations present. For instance, as the crystal size of the type L zeolite
4 becomes smaller, the X-ray diffraction pattern becomes broader and less
precise. Thus, the term "zeolite L" includes any zeolites made up of
6 cancrinite cages having an X-ray diffraction pattern substantially similar
to the
7 X-ray diffraction patterns shown in U.S. Pat. No. 3,216,789.
8 Type L zeolites are conventionally synthesized largely in the potassium
form,
9 i.e., in the theoretical formula given previously, most of the M cations are
potassium. The M cations are exchangeable, so that a given type L zeolite,
11 e.g., a type L zeolite in the potassium form, can be used to obtain type
12 L zeolites containing other cations, by subjecting the type L zeolite to
ion
13 exchange treatment in an aqueous solution of appropriate salts. However, it
14 is difficult to exchange all of the original cations, e.g., potassium,
since some
exchangeable cations in the zeolite are in sites which are difficult for the
16 reagents to reach.
17 As previously mentioned, preferably the catalyst used in reaction zone 3 is
18 nonacidic. Nonacidity can be achieved by use of alkaline metals or alkaline
19 earth metals in the zeolite component, for example, as described in U.S.
Patents Nos. 4,104,320 and 4,435,311.
21 The term "nonacidic" is used in contrast to the acidic dual function
catalysts
22 such as platinum on halided alumina, or platinum rhenium on halided
23 alumina, or platinum on an ammonium exchanged (and then calcined) zeolite.
24 For examples of these dual function acidic reforming/dehydrocyclization
catalysts, see U.S. Patent No. 3,006,841; U.S. Patent No. 3,415,737; and
26 U.S. Patent No. 3,783,123 (such as Example 16, pertaining to an ammonium
27 exchanged zeolite support).
CA 02356632 2001-06-08
WO 00/34417 PCTIUS98/26134
-18-
1 Although halides, particularly chloride, have been used in the past to
achieve
2 a measure of acidity in platinum alumina dual function reforming or
3 dehydrocyclization catalysts, it should be noted that halides may be
involved
4 in the preparation of nonacidic highly selective dehydrocyclization
catalysts,
particularly when the support is an aluminosilicate such as zeolite L. Recent
6 catalysts which involve use of halides in making nonacidic Group VIII metal-
L
7 zeolite catalysts are exemplified by U.S. Patent No. 4,681,865; U.S. Patent
8 No. 5,073,652; U.S. Patent No. 5,196,631; and U.S. Patent No. 5,294,579.
9 All of these latter cited patents exemplify nonacidic, monofunctional
dehydrocyclization catalysts which have selectivity for converting paraffins
to
11 aromatics.
12 Referring again to the drawing, in reaction zone 3, the feed is contacted
with
13 the highly selective dehydrocyclization catalyst under dehydrocyclization
14 reaction conditions to convert paraffins to aromatics. The term "highly
selective dehydrocyclization catalyst" is used herein to refer to catalysts
16 which, in the conversion of n-hexane to aromatics under dehydrocyclization
17 reaction conditions as described hereinbelow, result in a selectivity for
18 aromatics of at least 40% by weight, preferably at least 50%, more
preferably
19 at least 70%, and most preferably at least 80% selectivity for conversion
of
n-hexane to aromatics, on a per pass (excluding recycle conversion) basis.
21 Preferably the highly selective dehydrocyclization catalyst is used in the
22 present invention under reaction conditions, such as those described
23 - hereinbelow, effective to achieve per pass conversion of paraffins to
24 aromatics and other hydrocarbons of at least 50 wt. %, more preferably at
least 60%, and most preferably at least 70%. The yield of desired aromatics
26 product, on a per pass basis, is the per pass conversion times the
selectivity.
CA 02356632 2001-06-08
WO 00/34417 PCT/US98/26134
-19-
1 Suitable reaction conditions include a temperature between 400 C and
2 600 C, so that the highly selective dehydrocyclization reaction occurs with
3 acceptable speed and selectivity.
4 Preferably, the dehydrocyclization is carried in the presence of hydrogen at
a
pressure adjusted so as to favor the reaction thermodynamically and limit
6 undesirable hydrocracking reactions by kinetic means. The pressures used
7 preferably vary from 1 atmosphere to 500 psig, more preferably 50 to
8 200 psig. The molar ratio of hydrogen to hydrocarbons preferably is from 1:1
9 to 10:1, more preferably 2:1 to 6:1.
The liquid hourly space velocity of the hydrocarbons through the catalyst bed
11 in reaction zone 3 is preferably between 0.3 and 5, more preferably between
12 0.5 and 2Ø
13 As the dehydrocyclization reaction is endothermic, the combined fresh feed
14 and recycle feed, and recycled hydrogen, are heated in one or more furnaces
prior to introduction to the catalytic reaction zone. The furnaces are not
16 shown as a separate feature in the simplified block flow diagram. Also, the
17 reaction zone shown in the simplified diagram is not split into a series of
18 reforming or dehydrocyclization reactor vessels, although preferably the
19 dehydrocyclization reaction is carried out in a series of reactor vessels.
In
addition, the simplified diagram does not show heat exchange steps.
21 Effluent from reaction zone 3 is passed to separation zone 5, which may
22 comprise a series of separation steps. Hydrogen is recycled from separation
23 zone 5 via line 6 back to reaction zone 3.
24 Product aromatics rich material is withdrawn via line 7 from separation
zone 5
and is passed to separation zone 8. In separation zone 8, aromatics are
26 separated and withdrawn as product via line 10. Nonaromatics are withdrawn
CA 02356632 2001-06-08
WO 00/34417 PCT/US98/26134
-20-
1 via line 9. The separation of the aromatics from the nonaromatics in zone 8
2 may be done by extraction using a solvent, distillation, or by use of
molecular
3 sieves. In any of these separation means, for convenience, we use the term
4 "raffinate" to refer to the paraffins rich stream separated from the
aromatics
product.
6 The term "rich in paraffins" is used to mean more than 50% by weight
7 paraffins. Preferably, the raffinate contains more than 80 wt. % paraffins.
8 Use of molecular sieves for separating product aromatics from the paraffins
9 rich raffinate can be done by passing the aromatics and paraffins through a
bed of molecular sieves. The molecular sieves adsorb the normal paraffins
11 and some of the isoparaffins present, but not the aromatics. To cause such
a
12 separation, the molecular sieve should have an effective pore diameter of
13 from 4.5 to 5.5 Angstroms. Examples of such molecular sieves are
silicalite,
14 L zeolite, A zeolite, X zeolite, Y zeolite, offertite and ZSM-5 zeolite,
with
cations properly used to tailor the size of the zeolite opening to accommodate
16 the desired separation.
17 Alternatively, the separation in zone 8 can be carried out by distillation
to
18 separate a "raffinate" stream which is rich in paraffins from the aromatic
19 product.
Most preferably, the paraffins are separated from aromatics in zone 8 by
21 solvent extraction, that is, the aromatics are absorbed into a solvent and
22 thereby extracted from the reaction zone effluent aromatics-paraffins
stream.
23 The extracted aromatics can be separated from the solvent by distillation.
24 Solvents that can be used in such a solvent extraction method include
phenol, sulfolane and N-formyl morpholine. Preferred solvent extraction
26 means useful for the present process, particularly including extractive
CA 02356632 2008-09-05
-21-
1 distillation, are described further in U.S. Patent No. 5,401,365.
2 After the aromatics removal step, dimethylbutanes remaining in the paraffins
3 rich stream may range from 5 to 50 wt. %, preferably 10 to 40 wt. %, and
4 more preferably 15 to 35 wt. %, and most preferably 20 to 30 wt. %, based on
total weight of the raffinate stream (paraffins rich stream).
6 In accordance with this preferred embodiment of the present invention, prior
7 to recycle of the paraffinic raffinate to reaction zone 3, dimethylbutanes
are
8 separated from the raffinate. Preferably, the dimethylbutanes are separated
9 by distillation. We have found that the overall dehydrocyclization process
is
surprisingly efficient when dimethylbutanes are removed primarily
11 downstream versus removing most of the dimethylbutanes upstream of the
12 dehydrocyclization reaction zone. The dimethylbutanes are removed from
13 separation zone 11 via line 12.
14 After dimethylbutanes removal from the above-described paraffins rich
stream, the dimethylbutanes remaining preferably is from 0.01 to 15 wt. %,
16 more preferably from 0.1 to 10 wt. %, and most preferably from 1 to 10 wt.
%
17 of the paraffins rich stream.
18 Preferably, the downstream dimethylbutanes removal of this embodiment is
19 carried out to remove 70 to 99.8% of the dimethylbutanes from the paraffins
rich stream, more preferably 75 to 99%, and most preferably from 80 to 95%
21 of the dimethylbutanes, based on the total weight of the paraffins rich
stream.
22 The drawing shows downstream dimethylbutanes removal after aromatics are
23 separated from the recycle paraffins.
CA 02356632 2001-06-08
WO 00/34417 PCT/US98/26134
-22-
1 According to another preferred embodiment of the present invention,
2 dimethylbutanes are removed downstream of the dehydrocyclization reaction
3 zone but prior to treatment of the reaction zone effluent to separate
aromatics
4 from paraffins. Thus, according to this embodiment, a process is provided
for
dehydrocyclization of hydrocarbon which comprises:
6 (a) contacting fresh paraffins rich feed hydrocarbon containing 0.1 to
7 20.0 wt. % dimethylbutanes with a highly selective dehydrocyclization
8 catalyst in a reaction zone under dehydrocyclization reaction conditions
9 to convert paraffins to aromatics and obtain an aromatics rich effluent;
(b) removing dimethylbutanes from the aromatics rich effluent to obtain a
11 paraffins-aromatics mixture of reduced dimethylbutane content;
12 (c) separating aromatics from the paraffins-aromatics mixture to obtain an
13 aromatics lean raffinate; and
14 (d) recycling the raffinate of to the reaction zone.
In this preferred embodiment, dimethylbutanes in the paraffins-aromatics
16 mixture prior to downstream dimethylbutanes removal may range from 0.1 %
17 to 20% by weight, preferably 0.5% to 15%, more preferably 1% to 12%, and
18 most preferably 2% to 10%, based on the weight of the mixture.
19 Remaining dimethylbutanes in the paraffins-aromatics mixture after
dimethylbutane separation may range from 0.01 % to 8%, preferably from
21 0.05% to 6%, more preferably from 0.1 % to 4%, and most preferably from
22 0.2% to 1%, based on the weight of the mixture.
23 Preferably, the downstream dimethylbutane removal is carried out by
24 distillation. The distillation step may be carried out in one or more
distillation
CA 02356632 2001-06-08
WO 00/34417 PCT/US98/26134
-23-
1 columns, using conventional distillation techniques conducted in accordance
2 with the dimethylbutanes removal requirements of the present invention. In
3 the embodiment where downstream dimethylbutane removal is carried out
4 prior to aromatics separation, the preferred distillation separation step
removes 60-99.5% of the dimethylbutanes from the aromatics rich effluent,
6 more preferably 70-99%, most preferably 80-90% of the dimethylbutanes in
7 the aromatics rich effluent. It may be noted that the removal referred to in
this
8 paragraph is based on feed to the dimethylbutanes downstream removal
9 step, not on the fresh feed. Percent removal based on fresh feed is the
basis
of the next two paragraphs.
11 In the present invention, for any of the embodiments, dimethylbutanes are
12 removed primarily downstream of the highly selective dehydrocyclization
step.
13 The term "primarily" is used to mean at least 50% based on the total
14 dimethylbutanes in the fresh feed, exclusive of that portion of the
dimethylbutanes that are effectively removed from the system due to cracking
16 or other dimethylbutane conversion reactions in the dehydrocyclization
17 reaction step. Usual amounts of dimethylbutanes removal by conversion in
18 the dehydrocyclization reaction zone is 10% to 50%, more typically 15% to
19 45% based on dimethylbutanes content in the fresh feed.
Preferably, at least 50% of the dimethylbutanes that are removed by
21 distillation or other physical separation (as opposed to removal by
reactive
22 conversion to other mate(als in the reaction zone) are removed by
23 downstream removal, more preferably at least 60% of the dimethylbutanes
24 are removed from the feed to the dehydrocyclization step by downstream
removal. This relatively high percent removal is in contrast to that portion
of
26 dimethylbutanes which may be removed upstream, for example, from the
27 fresh feed when the fresh feed, alone or with recycle paraffins, is
28 prefractionated to remove C5 and other hydrocarbons. Downstream removal
CA 02356632 2001-06-08
WO 00/34417 PCT/US98/26134
-24-
1 refers to downstream of the reaction zone. Percent removal of
2 dimethylbutanes, unless otherwise indicated, is based on the dimethylbutanes
3 in the fresh feed.
4 To achieve relatively high percentage dimethylbutanes removal downstream
of the reaction zone, as is required by the present invention, the percent
6 removal of dimethylbutanes from the feed to the downstream dimethylbutane
7 removal process must be high. Thus, as previously indicated, preferably the
8 percent dimethylbutanes removal based on the paraffins in the feed to the
9 downstream dimethylbutanes removal step is at least 70%, and more
preferably at least 80%.
11 Referring again to the drawing, prior to the dehydrocyclization step, the
fresh
12 feed, or the combined fresh feed and recycle paraffins, such as the
raffinate
13 shown by line 13, can be distilled to remove C5 hydrocarbons. The C5 is not
14 suitable feed for the dehydrocyclization reaction step. The
prefractionation
step, which is a preferred step to remove C5 , is not shown in the drawing.
16 The prefractionation of the fresh feed or the combined fresh feed and
recycle
17 raffinate stream can remove dimethylbutanes along with removal of C5- and
18 other hydrocarbons. However, according to the present invention, as
19 previously indicated, the amount of dimethylbutanes removed by such
upstream prefractionation or distillation is less than 50% of the total
21 dimethylbutanes, based on the fresh feed, preferably less than 40% of the
22 dimethylbutanes removed by distillation or other physical separation.
CA 02356632 2001-06-08
WO 00/34417 PCT/US98/26134
-25-
1 EXAMPLES
2 The first configuration of this example is one where raffinate, from an
3 aromatics extraction unit following a highly selective dehydrocyclization
4 reforming reaction step, is combined with fresh feed, and then distilled to
remove C5- and a minor portion of the dimethylbutanes. Then the distilled
6 fresh feed and raffinate is fed to the dehydrocyclization step. This is
referred
7 to as the base case.
8 In a second case, in accord with the invention, raffinate from the aromatics
9 extraction unit is distilled to remove a major portion of the
dimethylbutanes,
and then the distilled raffinate, along with fresh feed, are combined and
11 passed to the dehydrocyclization step.
12 In both cases, the fresh feed is composed of the following components: C5-,
13 dimethylbutanes (DMBs), 2-methylpentanes, 3-methylpentanes,
14 methylcyclopentane, n-hexane, cyclohexane. In this example, the
dimethylbutanes portion of the feed contained 18% 2,2-dimethylbutane and
16 82% 2,3-dimethylbutane. The relative feed compositions for the two cases
17 are shown below, on a liquid volume percent basis. (Weight percent for
these
18 type feeds would be close to liquid volume percent.)
19 Feed Component Base Case Invention
C5- 0.1 0.1
21 dimethylbutanes 21.5 7.3
22 2-methylpentane 76.4 67.8
23 3-methylpentane 61.0 62.0
24 methylcyclopentane 40.2 43.7
n-hexane 100.0 100.0
26 cyclohexane 9.8 10.3
CA 02356632 2001-06-08
WO 00/34417 PCT/US98/26134
-26-
1 The per pass conversion in the highly selective dehydrocyclization step in
this
2 example is shown below, as a percent by weight of each feed component.
3 The catalyst used was nonacidic PtL zeolite.
4 dimethylbutanes 29.8
2-methylpentane 80.8
6 3-methylpentanes 84.9
7 methylcyclopentane 90.5
8 n-hexane 95.0
9 cyclohexane 98.6
The benzene yield for the two processes are shown below in pounds of
11 benzene per pound of feed to the reaction zone:
12 Base Case Invention
13 0.65 0.68
14 The yield of benzene in the invention case was 3% greater on a feed basis
compared to the base case. This is a substantial economic advantage for the
16 preferred case, because the feedstock cost is a major portion of the total
cost
17 of making product aromatics. Distillation costs were also surprisingly
18 advantageous for the invention case versus the base case.