Note: Descriptions are shown in the official language in which they were submitted.
CA 02358895 2001-07-18
WO 00/46225 PCT/US00/00886 -
-1-
PYRIDOBENZODIAZEPINE AND PYRIDOBENZOXAZEPINE
CARBOXYAMIDE VASOPRESSIN AGONISTS
This invention concerns benzoheterocyclic carboxyamides, particularly
pyridobenzodiazepine and pyridobenzoxazepine carboxyamides, which act as
vasopressin Vz agonists, as well as methods of treatment and pharmaceutical
compositions utilizing these compounds.
BACKGROUND OF THE INVENTION
Vasopressin (antidiuretic hormone, ADH) a nonapeptide hormone and
neurotransmitter, is synthesized in the supraoptic nuclei of the hypothalamus
of the
brain and transported through the supraoptico-hypophyseal tract to the
posterior
pituitary where it is stored. Upon sensing an increase in plasma osmolality by
brain
osmoreceptors or a decrease in blood volume or blood pressure (detected by the
baroreceptors and volume receptors), vasopressin is released into the blood
circulation and activates vasopressin V1~ receptors on blood vessels causing
vasoconstriction to raise blood pressure; and vasopressin VZ receptors of the
nephron
of the kidney causing reabsorption mainly of water and to a lesser degree
electrolytes,
to expand the blood volume (Cervoni and Chan, Diuretic Agents, in Kirk-Othmer,
Encyclopedia of Chemical Technology, 4th ed., Wiley, Volume 8, 398-432,
(1993)).
The existence of vasopressin in the pituitary was known as early as 1895
(Oliver and
Schaefer, J. Physiol. (London), 18, 277-279, (1895)). The determination of the
structure and the total synthesis of vasopressin were accomplished by du
Vigneaud
and coworkers in 1954 (du Vigneaud, Gish and Katsoyannis, J. Am. Chem. Soc.,
76,
4751-4752, (1954)).
CA 02358895 2001-07-18
WO 00/46225 PCT/US00/00886 -
-2-
The actions of vasopressin V1~ receptors are mediated through the
phosphatidylinositol pathway. Activation of vasopressin Vla receptors causes
contraction of the smooth muscle of the blood vessels to raise blood pressure.
The
actions of the vasopressin VZ receptors are mediated through activation of the
adenylate cyclase system and elevation of intracellular levels of cAMP. The
activation of vasopressin VZ receptors by vasopressin or vasopressin-like
(peptidic or
non-peptidic) compounds increases water permeability of the collecting ducts
of the
nephron and permits the reabsorption of a large quantity of free water. The
end result
is the formation and excretion of a concentrated urine, with a decrease in
urine
volume and an increase in urinary osmolality.
Vasopressin plays a vital role in the conservation of water by concentrating
the urine at the site of the collecting ducts of the kidney. The collecting
ducts of the
kidney are relatively impermeable to water without the presence of vasopressin
at the
receptors and therefore, the hypotonic fluid formed after filtering through
the
glomeruli, passing the proximal convoluted tubule, the loops of Henle, and the
distal
convoluted tubules, will be excreted as dilute urine. However, during
dehydration,
volume depletion or blood loss, vasopressin is released from the brain and
activates
the vasopressin VZ receptors in the collecting ducts of the kidney rendering
the ducts
very permeable to water; hence, water is reabsorbed and a concentrated urine
is
excreted. In patients and animals with central or neurogenic diabetes
insipidus, the
synthesis of vasopressin in the brain is defective and therefore, they produce
no or
very little vasopressin, but their vasopressin receptors in the kidneys are
normal.
Because they cannot concentrate the urine, they may produce as much as 10
times the
urine volumes of their healthy counterparts and they are very sensitive to the
action of
vasopressin and vasopressin V~ agonists. Vasopressin and desmopressin, which
is a
peptide analog of the natural vasopressin, are being used in patients with
central
diabetes insipidus. Vasopressin VZ agonists are also useful for the treatment
of
CA 02358895 2001-07-18
WO 00/46225 PCT/US00/00886 -
-3-
nocturnal enuresis, nocturia, urinary incontinence and temporary delay of
urination
whenever desirable.
Vasopressin, through activation of its V~~ receptors, exerts vasoconstricting
effects so as to raise blood pressure. A vasopressin V~~ receptor antagonist
will
counteract this effect. Vasopressin and vasopressin agonists release factor
VIII and
von Willebrand factor so they are useful for the treatment of bleeding
disorders, such
as hemophilia. Vasopressin and vasopressin-like agonists also release tissue-
type
plasminogen activator (t-PA) into the blood circulation so they are useful in
dissolving blood clots such as in patients with myocardial infarction and
other
thromboembolic disorders (Jackson, "Vasopressin and other agents affecting the
renal
conservation of water", in Goodman and Gilman, The Pharmacological Basis of
Therapeutics, 9th ed., Hadman, Limbird, Molinoff, Ruddon and Gilman Eds.,
McGraw-Hill, New York, pp. 715-731 (1996); Lethagen, Ann. Hematol. 69, 173-180
(1994); Cash et al., Brit. J. Haematol., 27, 363-364 (1974); David, Regulatory
Peptides, 45, 311-317 (1993); Burggraaf et al., Cli. Sci., 86, 497-503
(1994)).
The following prior art references describe peptidic vasopressin antagonists:
Manning et al., J. Med. Chem., 35, 382 (1992); Manning et al., J. Med. Chem.,
35,
3895 (1992); Gavras and Lammek, U.S. Patent 5,070,187 (1991); Manning and
Sawyer, U.S. Patent 5,055,448 (1991); Ali, U.S. Patent 4,766,108 (1988);
Ruffolo et
al., Drug News and Perspectives 4(4), 217 (May 1991); Albright and Chan, Curr.
Pharm. Des. 3(6), 615 (1997). Williams et al., have reported on potent
hexapeptide
oxytocin antagonists [J. Med. Chem., 35, 3905 (1992)] which also exhibit weak
vasopressin antagonistic activity in binding to V~ and V, receptors. Peptidic
vasopressin antagonists suffer from a lack of oral activity and many of these
peptides
are non-selective antagonists since they also exhibit partial agonist
activity.
CA 02358895 2001-07-18
WO 00/46225 PCT/US00/00886 -
-4-
Non-peptidic vasopressin antagonists have recently been disclosed. Albright
et al. describe tricyclic azepines as vasopressin and oxytocin antagonists in
U.S.
Patent 5,516,774 (1996); tetrahydrobenzodiazepine derivatives as vasopressin
antagonists are disclosed in J.P. 0801460-A (1996); Ogawa et al., disclose
benzoheterocyclic derivatives as vasopressin and oxytocin antagonists, and as
vasopressin agonists in WO 9534540-A; and Venkatesan et al., disclose
tricyclic
benzazepine derivatives as vasopressin and oxytocin antagonists in U.S. Patent
5,521,173 (1996).
As mentioned above, desmopressin (1-desamino-8-D-arginine vasopressin)
(Huguenin and Boissonnas, Helv. Chim. Acta, 49, 695 (1966)) is a vasopressin
agonist. The compound is a synthetic peptide with variable bioavailability. An
intranasal route is poorly tolerated and an oral formulation for nocturnal
enuresis
requires a 10-20 fold greater dose than the intranasal administration.
Albright et al. broadly disclose a subset of tricyclic pyrido benzodiazepine
and
pyridooxazepine indole ca~~hovyamides of the present application, as t% ~
and/or V,
vasopressin receptor antagonists and oxytocin receptor antagonists in L;.S.
Patent
5,512,563 (1996); U.S. Patent 5, 686.445 ( 1997); U.S. Patent 5.736.538 (
1998); EP
640592 A 1 ( 1.995); WO 97/47624 A 1; and WO 97/47625 A 1, ziater czlia.
Compounds of general structure 16b in Scheme 4 of the above applications.
are taught by Alhright et al. to possess vasopressin and oxytocin receptors
antagonist
properties.
CA 02358895 2001-07-18
WO 00/46225 PCT/US00/00886 -
-5-
R~
R2
N
wherein Y= N or O; R'= H, or lower alkyl (Cl-C3).
However, the above indole carboxyamides of general structure 16b, have been
found unexpectedly to be vasopressin ~', receptor a~ronists in vivo, and thus
possess
different biological profile and clinical utility from those originally
disclosed. Thus.
rather than having an aquarctic effect, they unexpectedly cause reabsorption
of water,
i.e. they reduce urine volume and increase urine osmalality.
The compounds of this invention are non-peptidic and have a good oral
bioavailability. They are vasopressin VZ receptor agonists, and as such they
promote
reabsorption of water. They demonstrate no vasopressin V,, receptor agonist
effects
and, thus, do not raise blood pressure. In contrast, the prior art compounds
(except
some in WO 9534540-A) are described as vasopressin antagonists at both the V,~
and
V~ receptors.
16b, Scheme 4 (Albright et al.)
CA 02358895 2001-07-18
WO 00/46225 PCT/US00/00886
-6-
SUMMARY OF THE INVENTION
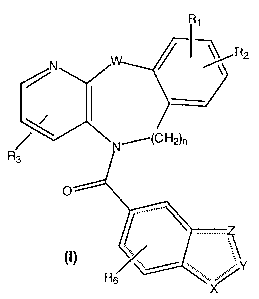
This invention relates to novel and known compounds selected from those of
formula (I):
~~R2
~~C H2)n
R3
( ) /, ... ",..Y
Rs
wherein:
X, Y and Z independently, are selected from a group consisting of O, S, CH,
CH2, N, or NR4;
W is NR5 or O;
R~ and RZ are independently, hydrogen, straight chain alkyl (C,-C6), branched
chain alkyl (C3-C~), cycloalkyl (C3-C~), alkoxyalkyl (CZ-C~), halogen,
straight or
branched chain alkoxy (C,-C6), hydroxy, CF3, or perfluoroalkyl (CZ-C6);
R3 is hydrogen or a straight chain alkyl group (C~-C6), branched chain alkyl
(C3-C~), cycloalkyl (C3-C~), alkoxyalkyl (CZ-C,), or hydroxyalkyl (C,-C6);
R~ is selected from hydrogen, or lower alkyl (C,-C~); and
RS is independently selected from hydrogen, acyl (CZ-C6), straight chain alkyl
(C,-C6), or branched chain alkyl (C3-C,);
R~ is selected from hydrogen or halogen;
CA 02358895 2001-07-18
WO 00/46225 PCT/US00/00886 -
7_
or a pharmaceutically acceptable salt thereof.
Among the preferred moieties represented in Formula (I) by the structure:
are the following:
~ / /
N ~ \~~ O ~ \~~ O
R5 R5 R5
/ S / N
N and \/~ N
R5 R5
Among the preferred compounds of this invention are:
(5,11-Dihydro-pyrido [2,3-b][1,5] benzodiazepin-10-yl)-(1-methyl-1H-indol-
5-yl)-methanone;
Benzo[1,3]dioxol-5-yl-(5,11-dihydro-pyrido [2,3-b][1,5] benzodiazepin-10-
yl)-methanone;
(2,3-Dihydro-benzofuran-5-yl)-(5,11-dihydro-pyrido [2,3-b][1,5] benzodiazepin-
10-
yl)-methanone;
CA 02358895 2001-07-18
WO 00/46225 PCT/US00/00886
-g_
Benzo[2]oxa[1,3]diazol-5-yl-(5,11-dihydro-pyrido [2,3-b][1,5]
benzodiazepin-10-yl)-methanone;
Benzothiazol-6-yl-(5,11-dihydro-pyrido [2,3-b][1,5] benzodiazepin-10-yl)-
methanone; and
( 1-Methyl-1 H-indol-5-yl)-( 11 H-5-oxa-4,10-di aza-dibenzo [a,d]cyclohepten-
10-yl)-methanone;
It is understood by those practicing the art that some of the compounds of
this
invention depending on the definition of R1, R2, R3, R4, and RS may contain
one or
more asymmetric centers and may thus give rise to optical isomers and
diastereomers.
The present invention includes such optical isomers and diastereomers; as well
as the
racemic and resolved, enantiomerically pure R and S stereoisomers and
pharmaceutically acceptable salts thereof, which possess the indicated
activity.
Optical isomers may be obtained in pure form by standard procedures known to
those
skilled in the art. It is also understood that this invention encompasses all
possible
regioisomers, and mixtures thereof which possess the indicated activity. Such
regioisomers may be obtained in pure form by standard separation procedures
known
to those skilled in the art.
The pharmaceutically acceptable salts are those derived from such organic and
inorganic acids as: citric, lactic, acetic, tartaric, succinic, malefic,
malonic,
hydrochloric, hydrobromic, phosphoric, nitric, sulfuric, methanesulfonic, and
similarly known acceptable acids.
Also according to the present invention there are provided methods of
treating, preventing or alleviating disorders which are remedied or alleviated
by
vasopressin receptor agonist activity. These methods of inducing vasopressin
agonism in a mammal include, but are not limited to, methods of treating,
preventing
or alleviating diabetes insipidus, nocturnal enuresis, nocturia, urinary
incontinence,
CA 02358895 2001-07-18
WO 00/46225 PCT/US00/00886 -
-9-
bleeding and coagulation disorders, and for inducing temporary delay of
urination,
whenever desirable, in humans or other mammals, which comprises administering
to
a human or other mammal an effective amount of a compound or a pharmaceutical
composition of this invention.
The present invention accordingly provides a pharmaceutical composition
which comprises a compound of this invention in combination or association
with a
pharmaceutically acceptable carrier or excipient. In particular, the present
invention
provides a pharmaceutical composition which comprises an effective amount of a
compound of this invention and a pharmaceutically acceptable carrier.
The compositions are preferably adapted for oral administration. However,
they may be adapted for other modes of administration, for example, parenteral
administration for patients suffering from coagulation disorders.
In order to obtain consistency of administration, it is preferred that a
composition of the invention is in the form of a unit dose. Suitable unit dose
forms
include tablets, capsules and powders in sachets or vials. Such unit dose
forms may
contain from 0.1 to 1000 mg of a compound of the invention and preferably from
2 to
50 mg. Still further preferred unit dosage forms contain 5 to 25 mg of a
compound of
the present invention. The compounds of the present invention can be
administered
orally at a dose range of about 0.01 to 100 mg/kg or preferably at a dose
range of 0.1
to 10 mg/kg. Such compositions may be administered from 1 to 6 times a day,
more
usually from 1 to 4 times a day. The compositions of the invention may be
formulated with conventional excipients, such as a filler, a disintegrating
agent, a
binder, a lubricant, a flavoring agent and the like. They are formulated in
conventional manner, for example, in a manner similar to that used for known
antihypertensive agents, diuretics and (3-blocking agents.
CA 02358895 2001-07-18
WO 00/46225 PCT/US00/00886
- 10-
Also according to the present invention there are provided processes for
producing the compounds of the present invention.
PROCESS OF THE INVENTION
The compounds of the present invention of general formula (I) may
conveniently be prepared according to the process shown in Scheme 1.
Scheme 1
z
.,.. Y
,;,.,Y
~6 G
R~
I -~ R2
R
i
I~ R -
~2
R/ ~ / (C H2)n
~(C Hen O
R3 3 H
""~ ...........z
R'
s
Thus, a pyridobenzodiazepine (benzoxazepine) of formula (3, wherein V~ is O
or NRS , and R~, R2, R3, and n are as defined above) is treated with an
appropriately
activated heteroaryl carboxylic derivative of formula (2) to provide the
desired
n6
CA 02358895 2001-07-18
WO 00/46225 PCT/US00/00886
-11-
compounds of formula (I) wherein R1, R2, R3, R4, R5, R6, X, Y, Z, W and n are
as
defined above.
The heteroaryl carboxylic acids of general formula (1) may be activated as
their acid halides, preferably the chloride (2, J= Cl), and reacted with the
pyridobenzodiazepine (benzoxazepine) of formula (3) in the presence of an
inorganic
base such as potassium carbonate in a polar, aprotic solvent such as N,N-
dimethylformamide; or an organic base such as 4-dimethylamino pyridine in an
aprotic solvent such as dichloromethane or tetrahydrofuran at temperatures
ranging
from -40° C to 50°C.
Alternatively, the acylating species of formula (2) can be a mixed anhydride
of the corresponding carboxylic acid, such as that prepared by treating said
acid with
2,4,6-trichlorobenzoyl chloride in an aprotic organic solvent such as
dichloromethane, according to the procedure of Inanaga et al., Bull. Chem.
Soc. Jpra.,
52, 1989 (1979). Treatment of the mixed anhydride of general formula (2) with
the
pyridobenzodiazepine (benzoxazepine) of formula (3) in an aprotic solvent such
as
dichloromethane and in the presence of an organic base such as 4-
dimethylaminopyridine at temperatures ranging from 0° C to the reflux
temperature
of the solvent, yields a compound of formula (I) wherein R~, R" R3, R4, R5,
R6, X, Y,
Z, W and n are as defined above.
Alternatively, the activation of the carboxylic acids of general formula (1)
can be carried out by reacting said acids with other peptide coupling reagents
known
to those skilled in the art, in an organic aprotic solvent such as
dichloromethane,
tetrahydrofuran, N,N-dimethylformamide, or the like, at temperatures ranging
from -
40°Cto120°C.
CA 02358895 2001-07-18
WO 00/46225 PCT/US00/00886 -
-12-
The activating reagent for the carboxylic acids of formula (1) is ultimately
chosen on the basis of its compatibility with the RQ and RS groups, and its
reactivity
with the tricyclic pyridobenzodiazepine (benzoxazepine) of formula (3).
The carboxylic acid intermediates (1) of Scheme 1 are either available
commercially, or are known in the art, or can be readily prepared by
procedures
analogous to those in the literature for the known compounds.
The compounds of general formula (I) wherein RQ is other than hydrogen; and
W is NRS, and RS is other than hydrogen; and Rl' R~, R3, X, Y, Z and n are as
defined
above, can be prepared by alkylation or acylation of a compound of formula (I,
wherein W is NH, and R4 is other than hydrogen ) of Scheme 1, as outlined in
Scheme 2 R
Ri R5 i
R2
R2
, iru ~.,
IC'.4-I..ln R
R3
Z
(I) (I)
x-
(W = NH, R4 is not H) (W = N~~ R4 is not H)
Scheme 2
CA 02358895 2001-07-18
WO 00/46225 PCT/US00/00886
-13-
Thus, the compound of formula (I, W is NH, and RQ is not hydrogen ) of
Scheme 1 are alkylated by treatment with a base such as sodium (or potassium)
hydride and an alkylating agent such as an alkyl halide, preferably an alkyl
chloride
(bromide or iodide) in an aprotic solvent such as N,N-dimethylformamide or
tetrahydrofuran at temperatures ranging from 0°C to 80°C to
yield compounds of
formula (I) wherein W is NRS and RS is alkyl, R4 is other than hydrogen, and
Rl, R2,
R3, X, Y, Z and n are as defined above.
Alternatively, the compounds of formula (I, W is NH, and RQ is other than
hydrogen) of Scheme 1 are acylated by treatment with a carboxylic acid halide
or a
carboxylic acid anhydride in the presence of an amine base such as pyridine or
a
trialkylamine such as triethylamine in an aprotic solvent such as
dichloromethane or
with no addition of solvent when pyridine is used as the base, at temperatures
ranging
from 40°C to ambient, to yield compounds of formula (1) wherein W is
NRS and RS
is acyl, R4 is other than hydrogen, and R,, R~, R3, X, Y, Z and n are as
defined above.
The subject compounds of the present invention were tested for biological
activity
according to the following procedures.
Vasopressin VZ Agonist Effects of Test Compounds in Normal Conscious Water-
Loaded Rats:
Male or female normotensive Sprague-Dawley rats (Charles River
Laboratories, Inc., Kingston, NY) of 350-500 g body weight were supplied with
standard rodent diet (Purina Rodent Lab. Chow 5001) and water ad libitum. On
the
day of test, rats were placed individually into metabolic cages equipped with
devices
to separate the feces from the urine and containers for collection of urine. A
test
compound or a reference agent was given at an oral dose of 10 mg/Kg in a
volume of
CA 02358895 2001-07-18
WO 00/46225 PCT/US00/00886 -
- 14-
10 mL/Kg. The vehicle used was 20% dimethylsulfoxide (DMSO) in 2.5%
preboiled corn starch. Thirty minutes after dosing the test compound, rats
were
gavaged with water at 30 mL/Kg into the stomach using a feeding needle. During
the test, rats were not provided with water or food. Urine was collected for
four
hours after dosing of the test compound. At the end of four hours, urine
volume was
measured. Urinary osmolality was determined using a Fiske One-Ten Osmometer
(Fiske Associates, Norwood, MA, 02062) or an Advanced CRYOMATIC
Osmometer, Model 3C2 (Advanced Instruments, Norwood, MA). Determinations of
Na', K' and Cl- ion were carried out using ion specific electrodes in a
Beckman
SYNCHRON EL-ISE Electrolyte System analyzer. The urinary osmolality should
increase proportionally. In the screening test, two rats were used for each
compound.
If the difference in the urine volume of the two rats was greater than 50%, a
third rat
was used.
The results of this study are shown in Table 1.
Table 1
Example Urine Volume Changes in UrinaryRat Type'
(% decrease) Osmolalit
1 74 159 CD
2 61 247 CD
3 2~) 54 CD
4 13 CD
5 31 78 CD
6 21 3 5 CD
" Percent decrease in urine volume vs. control at a dose of 10 mg/Kg
'' Osmolality changes expressed as percent of control at a dose of 10 mg/Kg
' Rat model used: Sprague-Dawley (CD)
The following non-limiting examples further illustrate the invention.
CA 02358895 2001-07-18
WO 00/46225 PCT/US00/00886 -
-15-
Example 1
(5,11-Dihydro-pyrido f2,3-b1f1,51 benzodiazepin-10-yl)-(1-methyl-1H-indol-5-
yl)-methanone
Step A. 1-Methyl-indole-5-carboxylic acid methyl ester
Under an atmosphere of nitrogen, a solution of indole-5-carboxylic acid
methyl ester (2.5 g, 14.3 mmol) in dry tetrahydrofuran (20 mL) was added
dropwise
to a stirred slurry of hexane-washed potassium hydride (1.63 g, 14.3 mmol, 35%
in
oil). When the hydrogen evolution ceased, iodomethane (1.3 mL, 21.5 mmol) was
added to the stirred solution. After an additional 30 minutes at room
temperature, the
precipitate was filtered and washed with diethyl ether. The filtrate was
concentrated
in vacuo and the residue triturated with hexane to provide the title compound
as a
yellow solid (2.6 g).
NMR (CDC13, 400 MHz): 8 3.82 (s, 3H), 3.93 (s, 3H), 6.58 (dd, 1H), 7.10 (d,
1H),
7.32 (d, 1H), 7.92 (dd, 1H), 8.39 (s, 1H)
MS (EI, m/z): 189 [M]+, 158, 130
Step B. 1-Methyl-indole-5-carboxylic acid
A solution of 1-methyl-indole-5-carboxylic acid methyl ester of Step A (2.5 g,
13.2 mmol) in ethanol (40 mL) containing 2.SN aqueous NaOH (3:1, v/v) was
heated
at reflux for one hour. The reaction mixture was concentrated in vacuo, and
the
residue partitioned between diethyl ether and 1N HCI. The organic layer was
washed
with brine, dried over sodium sulfate, and evaporated to dryness to provide
the title
compound as an off-white solid (1.82 g).
NMR (DMSO-db, 300 MHz): b 3.82 (s, 3H), 6.58 (dd, 1H), 7.42 (d, 1H), 7.48 (d,
1H), 7.75 (d, 1H), 8.22 (s, 1H), 12.38 (broad s, 1H)
CA 02358895 2001-07-18
WO 00/46225 PCT/US00/00886 -
-16-
Step C. (5,11-Dihydro-pyrido[2,3-b][1,5]benzodiazepin-10-yl)-(1-methyl-1H-
indol-5-yl)-methanone
Under anhydrous conditions, 2,4,6-trichlorobenzoyl chloride was added in one
portion to a stirred solution of equimolar amounts of 1-methyl-indole-5-
carboxylic
acid (0.327 g, 1.87 mmol) of Step B, and triethylamine in dry dichloromethane
(25-
50 mL). When anhydride formation was complete, the 6,11-dihydro-5H-pyrido[2,3-
b][1,5]benzodiazepine (0.519 g, 2.8 mmol) and N,N-dimethylaminopyridine were
added to the clear solution. Stirring was continued until the reaction was
complete
(TLC). The reaction mixture was diluted with dichloromethane, washed with
saturated aqueous sodium bicarbonate and brine, and dried over sodium sulfate.
Removal of the solvent and flash-chromatography of the residue (on silica gel
Merck-
60, hexane-ethyl acetate 4:1) provided the title compound as a white solid
(0.260 g),
m.p. 147-148°C, upon recrystallization from diethyl ether.
NMR (DMSO-db, 400 MHz): 8 3.70 (s, 3H), 4.06 (broad d, 1H), 5.62 (broad d,
1H),
6.30 (s, 1H), 6.48 (t, 1H), 6.50 (d, 1H), 6.73 (m, 1H), 6.89 (d, 1H), 7.05 (t,
1H), 7.20
(d, 1H), 7.33 (m, 2H), 7.43 (s, 1H), 7.51 (broad s, 1H), 8.14 (m, 1H), 9.56
(s, 1H)
MS (EI, m/z): 354 [M]', 158
Example 2
Benzo f1,31dioxol-5-yl-(5,11-dihydro-pyrido f2,3-b1f1,51 benzodiazepin-10-yl)-
methanone
Prepared from piperonylic acid (0.332 g, 2 mmol) and 6,11-dihydro-5H-
pyrido [2,3-b][1,5] benzodiazepine (0.398 g, 2 mmol) in a manner essentially
identical to that of Example 1. The title compound was obtained as a white
solid
(0.400 g), m.p. 205-207°C, upon recrystallization from diethyl ether.
CA 02358895 2001-07-18
WO 00/46225 PCT/US00/00886 -
-17-
NMR (DMSO-db, 400 MHz): 8 4.06 (broad d, 1H), 5.54 (broad d, 1H), 5.97 (s,
2H),
6.57-6.75 (m, 6H), 7.07 (t, 1H), 7.30 (d, 1H), 7.51 (broad s, 1H), 8.09 (m,
1H), 9.56
(s, 1H)
MS (EI, m/z): 345 [M]+, 196, 181, 149
Anal. Calcd. for CZOH15N3OZ: C 69.56; H 4.38; N 12.17. Found: C 69.10; H 4.58;
N
12.04
Example 3
(2,3-Dihydro-benzofuran-5-yl)-(5,11-dihydro-pyrido f2,3-b1f1,51 benzodiazepin
10-yl)-methanone
Prepared from 2,3-dihydro-benzofuran-5-carboxylic acid (0.328 g, 2 mmol)
and 6,11-dihydro-5H-pyrido[2,3-b][1,5]benzodiazepine (0.398 g, 2 mmol) in a
manner essentially identical to that of Example 1. The title compound was
obtained
as an off-white solid, m.p.188°C, upon recrystallization from diethyl
ether.
NMR (DMSO-db, 400 MHz): 8 3.05 (m, 2H), 4.06 (broad d, 1H), 4.47 (t, 2H), 5.60
(broad d, 1H), 6.51 (d, 1H), 6.60 (m, 2H), 6.75 (m, 2H), 7.07 (m, 2H), 7.31
(d, 1H),
7.50 (broad m, 1H), 8.09 (m, 1H), 9.54 (s, 1H)
MS (EI, m/z): 343 [M]+, 196, 181, 147
Anal. Calcd. for CZIHI~N30z: C 73.45; H 4.99; N 12.24. Found: C 73.15; H 5.18;
N
11.91
Example 4
Benzof2loxaf1,31diazol-5-yl-(5,11-dihydro-pyrido f2,3-b1f1.51 benzodiazepin-10
yl)-methanone
Under a nitrogen atmosphere, an equimolar mixture of benzofurazan-5-
carbonyl chloride (0.5 g, 2.75 mmol), 6,11-dihydro-5H-pyrido[2,3-
b][1,5]benzodiazepine (0.54 g, 2.75 mmol) and potassium carbonate in N,N-
CA 02358895 2001-07-18
WO 00/46225 PCT/IIS00/00886
-18-
dimethylformamide (10 mL) was stirred at room temperature for 1.5 hours. The
reaction mixture was partitioned between water and ethyl acetate. The organic
phase
was washed with water and brine, and dried over sodium sulfate. The solution
was
filtered through a thin pad of silica gel Merck-60, and the filtrate
evaporated in
vacuo. The residual oil was crystallized from diethyl ether to provide the
pure title
compound as a yellow solid (0.495 g), m.p. 193-194°C.
NMR (DMSO-db, 400 MHz): 8 4.21 (d, 1H), 5.56 (d, 1H), 6.54 (t, 1H), 6.83 (m,
2H), 7.07 (t, 1H), 7.16 (d, 1H), 7.34 (d, 1H), 7.62 (d, 1H), 7.80 (s, 1H),
7.89 (d, 1H),
8.14 (m, 1H), 9.69 (s, 1H)
MS (EI, m/z): 343 [M]+, 196
Example 5
Benzothiazol-6-yl-(5,11-dihydro-pyrido f2,3-b1f1,51 benzodiazepin-10-yl)
methanone
Prepared from benzothiazole-6-carbonyl chloride (0.55g, 2.78 mmol) and
6,11-dihydro-5H-pyrido[2,3-b][1,5]benzodiazepine (0.53 g, 2.7 mmol) in a
manner
essentially identical to that of Example 4. The title compound was obtained as
a white
solid (0.200 g), m.p. 237°C (with sintering at 233°C).
NMR (DMSO-db, 400 MHz): 8 4.17 (d, 1H), 5.60 (d, 1H), 6.47 (t, 1H), 6.61 (d,
1H),
6.77 (m, 1H), 7.03 (t, 1H), 7.10 (d, 1H), 7.32 (d, 1H), 7.60 (d, 1H), 7.84 (d,
1H), 8.06
(s, 1H), 8.12 (m, 1H), 9.40 (s, 1H), 9.62 (s, 1H)
MS (EI, m/z): 358 [M]', 196, 181, 162
CA 02358895 2001-07-18
WO 00/46225 PCT/US00/00886
-19-
Example 6
(1-Methyl-1H-indol-5-yl)-(11H-5-oxa-4,10-diaza-dibenzofa,dlc c~pten-10-yl-
methanone
Under anhydrous conditions, 2,4,6-trichlorobenzoyl chloride was added in
one portion to a stirred solution of equimolar amounts of 1-methyl-indole-5-
carboxylic acid (0.124 g, 0.71 mmol) of Example 1, step B and triethylamine in
dry
dichloromethane (25-50 mL). After the anhydride formation was complete, 6,11-
dihydro-5H-pyrido[2,3-b][1,5]benzodiazepine (0.141 g, 0.71 mmol) and N,N-
dimethylamino pyridine were added to the clear solution. Upon completion of
the
reaction (TLC), the mixture was diluted with dichoromethane, washed with
saturated
aqueous sodium bicarbonate and brine, and dried over sodium sulfate. The
residue
obtained upon evaporation of the solvent, was flash chromatographed on silica
gel
Merck-60 first with dichloromethane-ethyl acetate 4:1, and then hexane-ethyl
acetate.
The pure title compound was obtained as a white solid (0.045 g).
NMR (DMSO-d6, 400 MHz): 8 3.71 (s, 3H), 5.11 (broad s, 2H), 6.38 (d, 1H), 6.91
(m, 2H), 7.05 (d, 1H), 7.22 (m, 2H), 7.27 (d, 1H), 7.32 (m, 2H), 7.56 (s, 1H),
8.24
(m, 1H)
MS (EI, m/z): 355 [M]', 158
Example 7
(6-Bromo-benzofl,3ldioxol-5-yl)-(5,11-dihydro-pyrido f2,3-b1f1,51
benzodiaze~in-10-xl)-methanone solvate with 0.23 dieth l~ ether
Prepared from 6-bromo-1,3-benzodioxole-5-carboxylic acid (0.150 g, 0.61
mmol) and 6,11-dihydro-5H-pyrido[2,3-b][1,5]benzodiazepine (0.119 g, 0.61
mmol)
in a manner essentially identical to that of Example 1. The title compound was
CA 02358895 2001-07-18
WO 00/46225 PCT/US00/00886
-20-
obtained as a white solid (0.075 g), m.p. 249-250C, upon recrystallization
from
diethyl ether.
NMR (DMSO-db, 400 MHz): 8 4.10 (d, 1H), 5.38 (d, 1H), 5.99 (s, 2H), 6.58 (t,
1H),
6.80 (m, 2H), 7.02 (t, 2H), 7.24 (d, 1H), 7.34 (d, 1H), 7.56 (d, 1H), 8.08 (d,
1H), 9.50
(s, 1H)
MS (EI, m/z): 423 [M]+, 344, 227
Anal. Calcd. for CZOHZ6BrNz02 + 0.23 CZH50: C 56.94; H 3.72; N 9.52. Found: C
56.57; H 3.61; N 9.40.