Note: Descriptions are shown in the official language in which they were submitted.
CA 02360567 2001-07-10
WO 00/43538 PCT/CA00/00047
1
TITLE OF THE INVENTION
Process for the Generation of Oligonucleotide Libraries (OLs)
Representative of Genomes or Expressed mRNAs (cDNAs) and Uses
Thereof
FIELD OF THE INVENTION
The present invention relates to a process for the generation
oligonucleotide librairies (OLs) representative of genomes or expressed
mRNAs (cDNAs) and to the uses thereof. In particular, the present
invention relates to a process for the generation of oligonucleotide
librairies comprising oligonucleotides of uniform length. The present
invention further relates to the uses of these OLs in numerous
biotechnological applications, including the identification and/or
characterization of biological materials, clinical diagnosis (DNA/RNA
level), preparative extraction of specific mRNA (and genes) and genomic
research/mapping.
BACKGROUND OF THE INVENTION
The generation of genomic DNA libraries, or cDNA libraries and the
maintenance, and handling of these libraries are critical procedures in the
field of genomics and/or biotechnology. In classical libraries the relevant
segments of DNA are cloned into vectors, which are maintained and
propagated in particular biological systems (in vivo). Alternatively,
libraries
(in vitro) can be directly constructed from genomic DNA or cDNA. They
contain linkers at the 5' and 3' ends of the DNA which allow PCR
amplification of the library. The information stored in these libraries
CA 02360567 2001-07-10
WO 00/43538 PCT/CA00/00047
2
contains repetitive sequence elements that originated from repetitive
DNA, or high copy mRNAs. This results in a significant redundancy, which
can complicate the use and the outcome of using classical libraries.
Another important feature which reduces the utility of classical libraries is
the heterogeneity in size of the members comprising the library. This
limits the usefulness of classical libraries in subtractive hybridization
procedures (1-2) which are dependent upon the length, complexity and
the redundancy of the libraries, and which therefore are particularly
sensitive to the choice of method and the number of cycles performed.
In fact, one must tailor the hybridization conditions to accommodate the
heterogeneous length and redundancy of stored information in order to
perform subtraction. Thus, the results are more "laboratory-specific" than
library-specific.
A number of diagnostic methods that involve nucleic acid hybridization
have arisen in recent years. Most of them are designed to provide
qualitative information about the presence of a specific sequence motif in
a complex analytical mixture of nucleic acids and use a detection system
based on PCR and/or DNA chip hybridization technologies (3-7). For both
of these technologies, diagnostic oligonucleotides constitute an essential
part of the detection system. These oligonucleotides are primarily chosen
based on the sequence data of the nucleic acids to be detected. In spite
of the power of hybridization to correctly identify a complementary strand,
it does face limitations. In fact, the difference in stability between a
perfectly matched complement and a complement mismatched at only
one base can be as little as 0.5° C (8). This is the fundamental
limitation
to the power of DNA hybridization for specific identification of a cognate
strand. Therefore, the diagnostic power of any chosen oligonucleotide
must be validated using an analytical mixture whose sequence context is
CA 02360567 2001-07-10
WO 00/43538 PCT/CA00/00047
3
not totally known. The problem of adequate probe selection is time and
labour-consuming. On the other hand, the growing complexity of
detection systems based on oligonucleotide technologies requires a fast
selection of a large number of short oligonucleotides.
Akopyants et al (7) performed subtractive hybridization using bacterial
DNAs digested by high-frequency restriction enzymes. The use of such
restriction enzymes tends to generate DNA fragments having a broadly
similar size, about 500 base pairs. However, the uniformity is not
rigorous. Moreover, the library created by these restriction fragments still
contains a significant number of redundant sequences; consequently,
patches of short polymorphism embedded in homologous sequences are
going to be missed when such a library is used.
U.S. Patent No. 5,270,163 (8) teaches a method for the isolation of
nucleic acids using high-affinity nucleic acid ligands. This method has
been termed the SELEX method (Systematic Evolution of Ligands by
Experimental Enrichment) and is based on the use of proteins or small
molecules, but not nucleic acids, as targets. The selection of
oligonucleotides in the SELEX method relies on the three-dimensional
(3D) shape of the oligonucleotides and their fit into the structures of the
target molecules. In contrast to this, the selection of oligonucleotides in
the present invention is based on hybridization with target nucleic acid.
Armour et al (11 ) describes the quantitative recovery of amplifiable
probes hybridised to an immobilised target. The amplifiable probes
consist of PCR or restriction fragments and their technique is meant to
assess the copy number of loci.
CA 02360567 2001-07-10
WO 00/43538 PCT/CA00/00047
4
There thus remains a need for oligonucleotide libraries which allow for the
use of uniform hybridization conditions to perform selection and/or
subtraction while minimizing or eliminating redundant sequences.
Advantageously, these libraries can be used in the selection of highly
informative and target-specific probe libraries. The present invention
seeks to meet these and other needs.
SUMMARY OF THE INVENTION
The procedure described herein results in the generation and selection
of oligonucleotide probes with a high specificity for a given system. These
oligonucleotides cover the entire length of the target DNA, thus increasing
detectability which might be lost in classical oligo-detection systems due
to secondary DNA structure or DNA deletions present in an analyte
mixture. At the same time, they present inexpensive variants of a
multiplex oligonucleotide-detection approach, since they are not required
to be individually synthesized.
More specifically, in accordance with the present invention, there is
provided a process for the generation of oligonucleotide libraries, or OLs.
The present invention teaches a process for generating OLs from
genomic DNAs and cDNAs, and for performing the subtraction of these
libraries.
The present invention further teaches OLs which allow the use of
hybridization conditions which are controllable and reproducible. In
addition, the invention teaches a process for the selection of uniform
length OLs which minimizes or eliminates redundant sequences and
reduces complexity. The result is the production of highly-informative and
target-specific probe libraries.
CA 02360567 2001-07-10
WO 00/43538 PCT/CA00/00047
An object of the present invention is therefore to provide a process for the
generation of oligonucleotide libraries comprising OLs of uniform length
which are self-amplifiable and easily subjected to subtraction.
5
Another object of the invention is to provide OLs which are compatible
with DNA array technology. Indeed, an array of diverse mixtures of
oligonucleotides which show differential hybridization patterns could be
the best choice for the next generation of DNA diagnostics.
Other objects, advantages and features of the present invention will
become more apparent upon reading of the following non-restrictive
description of preferred embodiments thereof, given by way of example
only with reference to the accompanying drawings.
BRIEF DESCRIPTION OF THE DRAWINGS
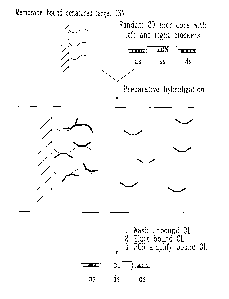
Figure 1: Schematic representation of the experimental procedure for
the preparation of OL. Denatured DNA is bound to the membrane and
hybridized to the random oligonucleotides library in the presence of
blockers. Theses blocking primers disallow the unspecific hybridization
of left and right oligonucleotide arms used for PCR amplification of the
OL. (ss - single-stranded DNA, ds - double-stranded DNA).
Hybridization and PCR amplification of OL are described more
particular below, in the Experimental Methods.
Figure 2: Dot blot hybridization of OL targeted against different genomes.
The first row represents the dot blot hybridization of random probes with
the specified genomic DNA (adenovirus, pBluescript and lambda). The
CA 02360567 2001-07-10
WO 00/43538 PCT/CA00/00047
6
last row shows dot blot hybridization of mixed adenovirus and lambda-
selected OL. The other rows are analytical dot blot hybridizations of
selected OLs with each of the genomes indicated. The procedures of
preparative and analytical hybridization are described in the Experimental
Methods, below.
Figure 3: Specificity and probe distribution of OL generated from
adenoviral genome. (A) The corresponding genome and adenoviral DNA
were run on a 1 % agarose gel stained with ethidium bromide. The type
of restriction enzyme and DNA are indicated on the top of each gel lane.
(B) Southern hybridization of the same gel using adenovirus OL as an
hybridization probe (see Figure 2, row 2). It should be noted that under
the experimental conditions, there was no cross-hybridization with either
lambda or human DNA. (C) The same membrane was stripped and
rehybridized with a OL directed against a 3648 bp-long restriction
fragment. This subset of adenovirus OL was prepared by cutting the
membrane corresponding to the 3648 by band from a similar southern
blot and reamplified by PCR as described in the Experimental Methods,
below. Thus, it is shown that OL specificity may be enhanced by
controlling the choice of targeted DNA fragments in the next round of
selection.
Fi ure 4: The distribution of OL along genomic DNA. The densitrometric
scan of radioactive signal from OL was integrated over total adenoviral
genome (Figure 3, lanes 3 and 5) using Scion Image software (Scion
corporation, Frederick, Maryland). The signal intensity of OL probes
hybridizing to restriction fragments is linearly proportional to the length of
DNA.
CA 02360567 2001-07-10
WO 00/43538 PCT/CA00/00047
7
Figure 5: Subtractive enrichment of OL. (A) The tester OL is presented
by the mixture of two genomes (Adenovirus type 2 and Lambda phage).
The driver OL was produced from the Lambda genome only. The single
stranded (ss) OL from driver DNA was used to pool out the
complementary single stranded mixed tester OL. After removing the
subtracted fraction, the remainder of the mixed OL was used as a probe
in the analytical hybridization step. The mixed OL probes were analyzed
by dot blotting as described (B) before subtractive enrichment and (C)
after subtractive enrichment by hybridization to genomic Adenovirus and
Lambda DNA.
Figure 6: Relative distribution of 20-mers with the different number of
mismatches which do hybridize to targeted DNA. The abscissa shows the
number of mismatches present in the 20-mer, while the y-axis illustrates
the corresponding relative frequencies. The distribution profile was
obtained by calculating the number of combinations for each particular
number of mismatches which are thermostable at 52° C. The y-axis was
normalized to reflect the relative distribution (%) over the total number of
captured oligonucleotides (100%). The majority of n-mers captured after
the first round of selection will be 20-mers with less then 6 mismatches.
This is described further in the Detailed Description, below.
DETAILED DESCRIPTION
The present invention thus provides a process for the generation of
oligonucleotide libraries having the following characteristics:
1 ) A uniform length of about 60 bases, comprising a central segment of
about 20 bases randomly varied to represent all possible combinations,
CA 02360567 2001-07-10
WO 00/43538 PCT/CA00/00047
8
and segments of about 20 bases of a defined sequence flanking the
central segment on each side;
2) A uniform number of copies for each sequence motif (consequently,
there are no differential hybridization kinetics which could originate from
the presence of repetitive DNA); and
3) A melting profile which is characterized by a sharp transition from
double stranded to single stranded (or vice versa) oligonucleotides. This
is a critical advantage in subtractive hybridization procedures.
The use of these OLs enhances the specificity of hybridization to nucleic
acids isolated from various sources, thereby allowing for the preparation
of oligonucleotide mixtures useful in the detection and quantification of
specific nucleic acids or nucleic acid mixtures.
In one particular embodiment, the starting pool of oligonucleotides is
chemically synthesized and consists of a random region of a fixed length
(L), flanked by a constant sequence (primer binding sites, PBS). The
random oligonucleotide pool covers n copies (n=1,2,3...) of all sequence
combinations of length L, i.e. 4', which is a total of 10'2different sequence
motifs for L=20 nucleotides. The basic length of oligonucleotides is long
enough to generate uniform sequence motifs for a particular biological
system. The complexity of the library (10'2) overcomes the complexity of
the template (which is usually between 104-109). The random pool is then
hybridized with a nucleic acid template isolated from any selected source
and the unbound oligonucleotides are washed away under stringent
conditions. The remaining, template-bound oligonucleotides are then
subjected to amplification, using PCR or other methods known to those
of skill in the art and using primers complementary to the constant
CA 02360567 2001-07-10
WO 00/43538 PCT/CA00/00047
9
flanking segments, thus producing a library of oligonucleotides capable
of selectively hybridizing to nucleic acid templates.
The choice of 20-mer for the length of the oligonucleotide library is not
arbitrary but is based on the rationale that the length of the particular
sequence motif should be long enough to be unique for even the most
complex genomes. The L-mer of length L will be, on average, repeated
every 4 power L base pairs. The longer the L, the greater the average
distance between 2 identical sequence motifs of length L will be.
However, this particular combinatorial approach is at best approximative,
and other lengths may be suitable as well. The choice of length will
depend on such factors as the length and/or complexity of the genome
to be detected and compatibility with current nucleic acid amplification
and DNA array technologies.
In another embodiment, the amplification described above is performed
with one of the PCR primers being labelled with biotin, providing means
for purification of the labelled products with streptavidin-labelled
substrates (12) or other similar methods. The amplified mixture of
unlabelled oligonucleotides specific to one template is hybridized with
labelled mixtures of oligonucleotides selected for specificity to one or
several nucleic acid templates, and the unbound material is collected. In
this manner, a mixture of nucleotides which is enriched for nucleic acids
present in the unlabelled library only can be generated.
The process is based on stringent hybridization. Furthermore, high
fidelity hybridization between pools of oligonucleotides and templates
(genomic DNA or cDNA) is the basic mode of transfer of genomic
CA 02360567 2001-07-10
WO 00/43538 PCT/CA00/00047
information into OLs. An efficient subtractive hybridization procedure is
used to accommodate the features of the aforementioned OLs.
The present invention is illustrated in further detail by the following non-
5 limiting example.
~Yennpi ~ ~
Generation of OLs, Use Thereof in Subtractive Hybridization to
Generate Subtractive Oligonucleotides Libraries (SOLs), and Use
10 of OLs or SOLs in Hybridization Experiments
EXPERIMENTAL METHODS
DNA I oligonucleotides
The starting random DNA pool was synthesised by GIBCO BRL
(Burlington, Canada), (RAN), 5'-GCCTGTTGTGAGCCTCCTGTCGAA-
NZO TTGAGCGTTTATTCTTGTCTCCC-3'. The corresponding left and
right arms were (LEFT) 5'-GCCTGTTGTGAGCCTCCTGTCGAA-3' and
(RIGHT) 5'-BioGGGAGACAAGAATAAACGCTCAA-3'. The 5'-end
biotinylated oligonucleotides were used to pool out complement
strands, using BioMag magnetic particles (PerSeptive Biosystems,
Framingham, MA ). During preparative hybridization, the left and right
arms were blocked by (LEFT) 5'-TTCGACAGGAGGCTCACAACAGGC-3'
and (RIGHT) 5'GGGAGACAAGAATAAACGCTCAA-3'. Theses
oligonucleotides are termed 'blockers' in the text.
The following genomic DNA was used to produce OL: Adenovirus DNA
Type 2, (GIBCO BRL), Lambda DNA c1857 ind1 Sam 7 (New England
Biolabs), pBluescript II SK(+) (Stratagene, San Diego, CA). The Human
HeLa DNA used as one control was from Clontech (Palo Alto, CA).
CA 02360567 2001-07-10
WO 00/43538 PCT/CA00/00047
11
Blotting genomic DNA
The genomic DNA was denatured 2-3 minutes at 95° C and cooled on
ice.
The nylon membrane (Hybond-N, Amersham Pharmacia Biotech,
Piscataway, NJ) was blotted with 100 ng of denatured genomic DNA,
dried for 2 minutes on a hot plate and exposed to UV light for 8 minutes.
The prehybridization was done for a minimum of 30 minutes in the
hybridization buffer (7% SDS, 0.25M Na2HP04 pH7.4, 1mM EDTA, pH
8.0 and 10g/L of BSA).
Hybridization and washing of the starting random pool
The preparative hybridization between random core (20N) and targeted
DNA was done with 10 pmoles of starting random pool (RAN). The
random pool was pre-mixed with 100 pmoles (10 times more than RAN)
of LEFT and RIGHT blockers in order to exclude cross-hybridization of left
and right arms with genomic DNA. The oligonucleotide mixture was
heated up to 95°C, cooled at room temperature and added to the
hybridization buffer. The hybridization was done overnight at 50° C.
The
first washing was done with 6X SSC, followed by subsequent 2X SSC
washing at the same temperature as hybridization was done.
Generating OL by PCR
The dot containing the genomic DNA and bound probes was cut out of
the nylon membrane (radius of 2-4mm), soaked in 100 pl Hz0 and heated
to 95°C for 1-2 minutes. The solution containing the denatured probe
(originally RAN) was then collected and passed threw a Sephadex G-50
column in order to eliminate salts and SDS. The PCR was prepared
under standard conditions, typical for SELEX-like amplification of DNA
(10, 13). The RIGHT 5'-end biotinylated primer of the sense strand (the
CA 02360567 2001-07-10
WO 00/43538 PCT/CA00/00047
12
one which did not hybridise with genomic DNA) and LEFT primer of
antisense strand were used in the PCR reaction. The temperature cycles
were 53°C, 72°C, 95°C, each 30 seconds, repeated 20
times.
Probe labelling and hybridization
Before labelling, the PCR reaction mixtures were passed threw Sephadex
G-50 columns. Around 100-200 ng of PCR product was labelled with 50
pmols of yP3z ATP (6000 Ci/mmol, I.C.N. pharmaceuticals, Irvine, CA).
The total amount of probe radioactivity was 300 000 c.p.m. The probe
was added into 0.5 ml of hybridization buffer. The blotting of genomic
DNA was done as described above. Hybridizations were done overnight
at 50°C. The nylon membrane was washed as previously described, and
exposed to Kodak X-GMAT film.
OL labelling and analytical hybridization
The generated OL was tested, using 1 ) the original genomic DNA from
which they were selected (positive control) and 2) using the unrelated
genomic DNA (negative control). The OL labelling, hybridization and
probe washing was done as described, except that hybridization time was
shorter (60 minutes).
Southern blot hybridization
Electrophoresis was performed in a 1 % agarose gel with TBE buffer (80
mM Tris borate, pH 8.0, 2mM Na2EDTA) and stained with ethidium
bromide. One ug of BstEll-digested lambda DNA, 300 ng of adenoviral
DNA and 1 pg of Alul-Hpal-digested human HeLa DNA were run on the
gel according to specifications (all restriction enzymes used in this work
were purchased from New England Biolabs). For Southern hybridization,
DNA was transferred to Nylon membranes by capillary blot procedure
CA 02360567 2001-07-10
WO 00/43538 PCT/CA00/00047
13
following manufacturer's recommendations (Amersham Pharmacia
Biotech). Hybridization was performed as described above with
adenoviral OL. Autoradiographic exposure (using Kodak X-GMAT film)
was done at room temperature, for few hours. Stripping of the membrane
was done by boiling a 1 % SDS solution and pouring it over the Nylon
membrane.
Subtractive enrichment of OL
The tester OL (mixed OL) that reflects the two genomes (Adenovirus type
2 and Lambda) was made by preparing OL from equimolar mixtures of 2
genomes. The driver OL was produced from the lambda genome only.
The production of sense strand (the one which did not hybridize with
genomic DNA) was done using 5'-end biotinylated primer in PCR
reaction. After denaturing PCR product, the biotinylated sense strand was
bound to streptavidin magnetic particles (200 pg, binding capacity > 200
pmols of biotinylated oligonucleotides, Biomag Magnetic Particles,
PerSeptive Biosystems), and pulled-out using a magnet. The
complementary antisense strand was discarded with the liquid phase.
The mixed antisense tester OL (Lambda + Adenovirus DNA) was
produced in the same way. This time, the supernatant with the antisense,
non-biotinylated strand was hybridized overnight at 50°C with 10 times
molar excess of driver Lambda sense stand attached to magnetic beads.
The hybridization buffer was the same as described above but without
SDS. After removing the fraction bound to the magnetic beads, the rest
of the mixture was used in the analytical hybridization step.
RESULTS
The starting random pool of oligonucleotides contains 42° (i.e.
10'2)
different 20-mers. The diversity of the sequence motifs is approximately
CA 02360567 2001-07-10
WO 00/43538 PCT/CA00/00047
14
10~' higher than the diversity of the most complex genomes. A schematic
representation of the procedure for generating OL is presented in Figure
1 and is described in detail in the Experimental Methods, above.
Blockers were used in order to avoid hybridization of the flanking arms to
the targeted genome, and this step was found to be critical to achieve
specificity. The stringency of hybridization conditions eliminates unbound
20-mers, leaving the specific oligonucleotides bound to the membrane via
hybridization of the random core to the genome (Fig. 1 ). This ensemble
of selected oligonucleotides constitutes the OL.
It should be noted that the starting random pool of oligonucleotides
contained about 8 copies of each sequence motif during the first
hybridization step (10-20 pmoles) and that the number of copies of each
particular 20-mer present in the random mixture was smaller than the
number of genome copies.
Figure 2 shows that OLs are able to discriminate genomes with
complexities around 103 to 104. The starting random pool of probes binds
to all three genomes equally (Fig. 2, row 1 ). After one round of selection,
the OL can hybridise specifically towards a single targeted genome (Fig.
2, rows 2, 3 and 4). The OL can be selected against a mixture of two
genomes and the specificity is conserved for both genomes (Fig. 2, row
5).
A Southern blot was performed in order to document the distribution of
adenovirus OL probes along the genome (Fig. 3). There was no apparent
cross-hybridization of adenovirus OL to either HeLa or Lambda DNA (Fig.
3b, lanes 1, 4, 5 and 6). The intensity of radioactive signal over adenoviral
genome generated by adenovirus-specific OL was linearly increasing with
CA 02360567 2001-07-10
WO 00/43538 PCT/CA00/00047
the DNA fragments' length (Fig. 4). Therefore, one could deduce a
uniform distribution of OL throughout the genomic DNA.
In the next step, only the subset of the adenovirus OL bound to the 3648
5 by band in Figure 3b (rows 3 and 6) was reamplified and selected. The
membrane was washed from the original probe and hybridised with the
3648 by subset of the original OL. Figure 3c shows that the specificity of
the OL subset is obtained against the 3648 by band. These data illustrate
the successful increase in the specificity and the reduction in the
10 complexity of the original Adeno-specific OL to that of the 3648 by subset,
using just one additional round of selection.
One round of subtractive enrichment between two oligonucleotide
libraries was performed as schematized (Fig. 5a). The tester OL reflects
15 the two genomes (adenovirus type 2 and lambda phage). The driver OL
was produced from the Lambda genome only. The single stranded (ss)
OL from the driver DNA was used to pool out the complementary single
stranded, mixed, tester OL. After removing the subtracted fraction, the
rest of ssDNA was used as a probe in the analytical hybridization step.
The intensity of hybridization signals between Lambda and Adeno
genomes, before (Fig. 5b) and after (Figure 5c) one round of subtractive
enrichment was shown. It should be noted that further subtraction steps
could be performed by changing the sequence design of flanking arms
between tester and driver OLs, as suggested by recent developments in
subtractive procedures (14).
With reference to Figure 6, the relative distribution of 20-mers with
different numbers of mismatches that hybridized to the targeted DNA was
predicted. The number of 20-mers (N) with (m) number of mismatches
CA 02360567 2001-07-10
WO 00/43538 PCT/CA00/00047
16
(m=0, 1, ..., 20) capable of hybridizing to the target sequence were then
calculated. First, the number of combinations of 20-mers (C) with the
same number of mismatches (m) in the initial random pool of
oligonucleotides that are capable of hybridizing to a specific 20-mer motif
was calculated. Since each full match could be replaced by 3 different
mismatches, the number of combinations must be multiplied by 3m i.e.
C*3"'. Finally, these numbers were adjusted to reflect the sequence-
dependent termostability of 20-mers with (m =0, 1...20) number of
mismatches which hybridised at 52°C using the thermostable fraction of
the binomial distribution for each n-mer population. Duplexes of more
than 7 mismatches are not observed based on this themostability
criterion. Therefore, the majority of 20-mers captured after the first round
of selection will harbour less than 6 mismatches.
A process that generates amplifiable DNA oligonucleotide libraries which
are specific for a given segment of DNA has been described. This
process is akin to random priming, because it is possible to generate
probes without a priori knowledge of the template sequence. One round
of preparative hybridization was enough to produce genome-specific
oligonucleotide libraries (Figs. 2 and 3). The OLs were inferred from
genomes of complexity of 103-104.
The process described herein generates probes with high detection
power. These probes/selected oligonucleotides can contain mismatches.
The notion that introduction of artificial mismatches could increase
detection power of oligonucleotides during single nucleotide
polymorphism (SNP) detection was well documented by Guo et al (6).
However, the prediction of positions and types of mismatches, which
should be introduced to increase detectability of oligonucleotide, remains
CA 02360567 2001-07-10
WO 00/43538 PCT/CA00/00047
17
undefined. Consequently, to enhance oligonucleotide detectability by
introducing (artificial) mismatches, one must search different positions
and types of mismatches along the oligonucletide. Once they are
empirically determined, i.e. tested on 2 different sequence motifs, the
oligonucleotide containing particular mismatches could be used (15).
The present process provides an approach based on differential selection
of thermostable oligonucleotides (i.e. their differential stability), which
are
present in one, but not in the second system. The selection of
oligonucleotides with the highest detectability is inherently present in this
process, i.e. the method suggests a solution to the problem of where and
what type of mismatches should be introduced to increase detection
power of oligonucleotide, or to find the particular oligonucleotide which
best discriminates between 2 sequence motifs which may differ by a
single base.
Without wishing to be bound by any hypothesis, the following provides an
explanation of what is believed to be occurring during the process of the
present invention. Based on calculations, it is expected that the 20-mers
selected in an OL can contain up to 6 mismatches. Nevertheless,
specificity toward a given template was achieved, suggesting that the
presence of these putative mismatches did not interfere with good
discrimination. In other words, mismatch-free hybridization is not critical
for differential detection approach; rather, the relative differences in the
thermodynamical stabilities of the hybridized oligonucleotides appear to
be determinative. The present process uses selection of oligonucleotides
based on this criterion and therefore provides the possibility of
overcoming current technological limitations. In the second and further
rounds of selection, the number of 20-mers both in the targeted genome
and the probe mixture (OL) could be adjusted. Each new round of
CA 02360567 2001-07-10
WO 00/43538 PCT/CA00/00047
18
preparative hybridization (Fig.1 ) and/or subtraction (Fig. 6) could reduce
the complexity of OL, by using the excess OL rather than genomic DNA.
Therefore, the average number of mismatches for each particular 20-mer
will continue to decline until it reaches the sequence-dependent limitation,
but not the concentration-dependent limitation.
In summary, OLs are generated from the template DNAs. These OLs are
used in subtractive hybridization, for example between genomic or cDNA-
based libraries (OL1 and OL2) to make a new Subtractive Oligonucleotide
Library (SOL1/2 and/or SOL2/1)), that is/are specific for one
system/library but not for the other. Oligonucleotides isolated from such
subtractive libraries (SOL) are useful for diagnostic purposes. They can
a) directly serve as highly specific hybridization probes or b) they can be
tested for PCR-specific differential amplification, specific for one, but not
the other biological system.
These libraries (OL or SOL) can be hybridized to oligonucleotide chip
arrays in order to obtain a specific hybridization pattern that is useful for
diagnostic features: each OL produces an image which is specific for the
templated DNA (genome or cDNA). A particular advantage in using OL or
SOL instead of genomic/cDNA libraries is that the hybridization signal is
not dependent on copy number and distribution of particular sequence
motifs. By comparing images of different genomes/cDNA, one can deduce
which oligonucleotides are highly specific for a single genome/cDNA, and
use this or these oligonucleotide(s) as "genome tags". The
oligonucleotides obtained can also be used for specific diagnostic PCR.
CA 02360567 2001-07-10
WO 00/43538 PCT/CA00/00047
19
OLs or SOLs can be inferred from two biologically relevant systems, like
mammalian cells, to detect fine differences in cell cycle, tissue status,
viral
infection, age/development status etc.
Although the present invention has been described hereinabove by way
of a preferred embodiment, it can be modified by one of skill in the art
without departing from the spirit and nature of the subject invention, as
defined more particularly in the appended claims.
CA 02360567 2001-07-10
WO 00/43538 PCT/CA00/00047
LIST OF REFERENCES
1. Diatchenko, L., Lukyanov, S., Lau, Y.F. and Siebert, P.D.
Supression subtractive hybridization: a versatile method for
5 Identifying differentially expressed genes., Methods Enzymol.,
1999, 303, 349-380.
2. Nilssnon, P., Larsson, A., Lundeberg, J., Uhlen, M. and Nygren,
P.A. (1999) Mutation scaning of PCR products by subtractive
10 oligonucleotide hybridization analysis, Biotechniques, 2, 308-
315.
3. Whitcombe, D., Newton C.R. and Little, S. (1998) Curr. Opin.
Biotechnol., 9, 602-608.
4. Watson, A., Mazumder, A., Stewart, M. and Balasubramanian, S.
( 1998) Curr. Opin. Biotechnol. , 9, 609-614.
5. Fredricks D.N. and Relman D.A. (1999) Application of
polymerase chain reaction to the diagnosis of infectious
diseases. Clin Infect Dis., 29, 475-86;
6. Gerhold, D., Rushmore, T. and Caskey, C.T. (1999) Trends
Biochem. Sci., 24, 168-73.
7. Matz M.V. and Lukyanov S.A. (1998) Different strategies of
differential display: areas of application. Nucleic Acids Res., 26,
5537-43.
8. Guo, Z., Qiunghua, L. and Smith, L.M. (1997) Nat. Biotechnology,
15, 331-335.
9. Akopyants et al, Proc. Natl. Acad. Sci. USA 95:13108-13113.
10. U.S. Patent No. 5,270,163 (Gold et an, December 14, 1993:
Methods for Identifying Nucleic Acid Ligands.
11. Armour, J.A., Sismani, C., Patsalis, P.C. and Cross, G. (2000)
Nucleic Acids Res., 28, 605-609.
CA 02360567 2001-07-10
WO 00/43538 PCT/CA00/00047
21
12. Mitchell L.G. and Merril C.R. (1989) Affinity generation of sin le-
stranded DNA for dideoxy sequencing following the polymerise
chain reaction. AnalBiochem., 178, 239-42.
13. Fitzwater, T. and Polsky, B. (1996) Methods Enzymol., 267, 275-
301. Combinatorial Chemistry, A Selex Primer. Academic Press,
New York.
14. Diatchenko L., Lau Y.F., Campbell A.P., Chenchik A., Moqadam F.,
Huang B., Lukyanov S., Lukyanov K., Gurskaya N., Sverdlov E.D.
and Siebert P.D. (1996) Suppression subtractive hybridization: a
method for generating differentially re ulated or tissue-specific
cDNA probes and libraries. Proc. Natl. Acid. Sci., 93, 6025-30;
Luo, J.-H. et al, Suppression subtractive hybridization: a method for
generating differentially regulated or tissue-specific cDNA probes
and libraries. Nucleic Acids Res., 1999, 27, e24.
15. Maldonado-Rodrigues, R., Espinosa-Lara, M., Loyola-Abita, P.
Beattie, W.G. and Beattie, K.L. (1999) Molecular Biotechnology, 1,
13-25; Okano, K., Uematsu, C., Matsunaga, H. and Kambara, H.
(1998), Electrophoresis, 18, 3071-3078.