Note: Descriptions are shown in the official language in which they were submitted.
CA 02361664 2001-08-07
WO 00/53299 PCT/US00/06463
TITLE
SUPPORTED NANOPOROUS CARBOGENIC GAS
SEPARATION MEMBRANE AND PROCESS
FOR PREPARATION THEREOF
FIELD OF THE INVENTION
This invention relates to a supported nanoporous carbon membrane
(SNPCM) exhibiting improved gas separation performance and a novel method
for preparation thereof. The supported nanoporous carbon membrane is formed
by: ultrasonically atomizing a solution of poly(furfuryl) alcohol resin in an
acetone solvent; depositing a thin, uniform layer of the furfuryl alcohol
resin
solution onto a porous support; evaporating the acetone; and pyrolyzing the
furfuryl alcohol resin.
TECHNICAL BACKGROUND -OF THE INVENTION
Inorganic membranes offer potential for high temperature gas separations
and membrane reactor applications. Zeolites, silica, and carbon molecular
sieves
(CMS) are potential candidates for making these membranes. Zeolites have been
difficult to synthesize into crack-free membranes and have been shown to have
poor thermal stability. Silica membranes, synthesized in a clean-room
environment to minimize defects, have achieved an 02/N2 separation factor of
3.9
at 100°C (see De Vos, R.M. and Verweij, H., "High-Selectivity, High-
Flux Silica
Membranes for Gas Separation" Science, 1998 vol. 279, pp. 1710-1711). Carbon
molecular sieves have been prepared by controlled pyrolysis of natural and
synthetic precursors, such as wood and coconut shells (see Vyas S.N.,
Patwardhan, S. R., Vijayalakshmi, S., SriGanesh, K., "Adsorption of gases on
Carbon Molecular Sieves" Journal of Colloidal Interface Science, 1994. 168,
pp. 275-280) as well as synthesized using a variety of different polymeric
resins.
Fitzer, E. and Schaefer, W., "The Effect of Crosslinking on the Formation of
Glasslike Carbons from Thermosetting Resins" Carbon, 1970. vol. 8, p. 353
reported the pyrolysis of polyfiufuryl alcohol to form a glasslike carbon, but
did
not address the issue of forming membranes.
Walker Jr., P. L., "Carbon - An Old but New Material" Carbon, 1972.
vol. 10, p. 369 described a variety of structures and properties for carbon
precursors, such as electrode carbon, glassy carbon, molecular sieves, carbon
black, pyrolytic graphite, and carbon fibers. Polymers like polyvinyl
chloride)
(PVC) are known to form graphitic layers at 1000°C; however, other
polymers
such as poly(acrylonitrile) (PAN) and poly(furfuryl) alcohol (PFA) form a
complex microstructure, with a large concentration of pores in the 3 to
10 angstrom region at much lower temperatures. For gas separation of small
CA 02361664 2001-08-07
WO 00/53299 PCT/US00/06463
molecules this range of microporous diameters is ideal. Foley, H. C.,
"Carbogenic Molecular Sieves: Synthesis, Properties and Applications"
Microporous Materials, 1995. vol. 4, p. 407-433 reported the formation of
carbogenic molecular sieves by pyrolyzing a layer of polyfurfuryl alcohol
which
had been coated (brushed) onto a planar porous metallic support.
Carbon molecular sieves can be thought of as consisting of graphite-like
planes. The carbon planes are stacked similar to graphite and were first
characterized with small-angle X-ray scattering (see Franklin, R. E., "The
Interpretation of Diffuse X-ray Diagrams of Carbon" Acta Crystallographica.,
vol. 3, p. 107, 1950). A hypothetical, ribbon-like structure has been proposed
consisting of disordered planes of carbon atoms (see Jenkins, G. M. and
Kawamura, K., Polymeric Carbons, Cambridge University Press: Cambridge, MA
1976). This structure hypothesis is also corroborated by high resolution
transmission electron microscopy (HRTEM) images combined with fast Fourier
transform (FFT) analysis to determine the spacing between the graphitic layers
(see Kane, M. S., Goellner, J. F., Foley, H. C., DiFrancesco, R., Billinge, S.
J. L.,
Allard, L. F, "Symmetry Breaking in Nanostructure Development of Caxbogenic
Molecular Sieves: Effects of Morpohological Pattern Formation on Oxygen and
Nitrogen Transport" Chemistry of. Materials., 1996. vol. 8 pp. 2159-2171.).
The
disordered structure of carbon molecular sieves results in a pore size
distribution
which can be controlled by varying synthesis parameters such as temperature
and
time (Lafyatis, D. S., Tung, J., Foley, H. C., "Poly(furfuryl) alcohol-Derived
Carbon Molecular Sieves: Dependence of Adsorptive Properties on
Carbonization Temperature, Time and Polyethylene glycol) Additives" Industrial
Engineering for Chemical Research, 1991. vol. 30, pp. 865-873). Due to this
pore
size distribution, all transport mechanisms are present, including bulk,
Knudsen,
surface, and configuration diffusion. The challenge is to minimize the number
of
pores with diameters larger than 20 angstroms, which significantly reduces
both
bulk and Knudsen diffusion and provides a molecular sieving membrane.
Two types of membranes have been synthesized from organic precursors,
which includes unsupported or "hollow" fiber and supported or "asymmetric"
membranes (see Koresh, J. E. and Soffer, A., "Mechanism of Permeation through
Molecular Sieve Carbon Membrane," Journal of the Chemical Society, Faraday
Transactions. I, 1986. vol. 82, pp. 2057-2063). The unsupported membranes have
the disadvantage that they lack significant mechanical strength for practical
application.
Asymmetric membranes have been prepared on a variety of supports,
including porous metals, graphite, ceramics and glasses. Both chemical vapor
2
CA 02361664 2001-08-07
WO 00/53299 PCT/US00/06463
deposition and plasma deposition, as well as conventional coating and dipping
techniques, have been reported.
Carbon membranes supported on macroporous supports such as porous
metals, porous ceramics, porous glasses, or porous composite materials are
believed to be advantageous because the supports may be obtained in a variety
of
sizes, shapes, and porosity, the supports may be readily fabricated and
joined, and
are relatively inexpensive.
BRIEF DESCRIPTION OF THE FIGURES
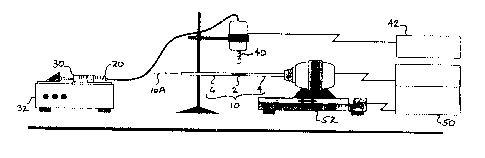
Figure 1 is a pictorial view showing an apparatus for coating a porous
tubular support.
Figure 2 is a pictorial view showing a first apparatus for pyrolyzing the
coating on the porous tubular support.
Figure 2A is a pictorial view showing a second apparatus for pyrolyzing
the coating on the porous tubular support. -
Figure 3 is a table showing the synthesis temperature and carbon coating
results for Examples 1-12.
Figure 4 is a table showing the pure component gas permeances and
separation factors for Examples 1-12.
Figure 5 is a pictorial view, partially in section, showing a membrane
testing module.
Figure 6 is a plot showing the rise in pressure versus time (at
22°C) for
Example 4.
Figure 7 is a plot showing the permeance of nitrogen, oxygen, and helium
gases through Example 4 as a function of core side pressure.
SUMMARY OF THE INVENTION
The invention concerns a nanoporous carbogenic membrane, supported on
a porous substrate, comprised of a plurality of layers of poly(furfuryl)
alcohol
resin pyrolyzed in an inert or reactive atmosphere, each layer being no more
than
10 microns in thickness and having a weight after pyrolysis of no more than
10 milligrams per square centimeter.
The invention further concerns a method of forming a supported
nanoporous carbon membrane, comprising the steps of:
(a) preparing a solution of poly(furfuryl) alcohol resin in an acetone
solvent;
(b) ultrasonically atomizing the solution and depositing a thin,
uniform layer of less than 25 mg per cm2 of the poly(furfuryl) alcohol
resin/acetone solution onto a porous support;
(c) drying the resin/acetone layer by evaporating the acetone;
CA 02361664 2001-08-07
WO 00/53299 PCT/US00/06463
(d) pyrolyzing the poly(furfuryl) alcohol resin in an inert or reactive
atmosphere.
The invention further concerns a carbogenic molecular sieve membrane,
supported on a porous substrate, having improved gas separation performance,
S wherein the membrane is formed by the steps of:
(a) forming a tube assembly by joining one or more segments of
porous tubes between two or more segments of nonporous tubes;
(b) coating the porous segments of the tube assembly with a solution
of poly(furfuryl) alcohol resin in an acetone solvent by ultrasonically
atomizing
the solution into droplets of substantially uniform size between about 0.1 and
10 microns and depositing a thin, uniform layer no more than about 20 microns
in
thickness and having a wet weight of no more than about 25 milligrams per
square
centimeter of the poly(furfuryl) alcohol resin/acetone solution onto the
porous
support;
(c) drying the resin/acetone layer by evaporating the acetone;
(d) pyrolyzing the poly(furfuryl) alcohol resin in an inert or reactive
atmosphere to form a membrane layer which is no more than about 10 microns in
thickness and has a weight after pyrolysis of no more than about 10 milligrams
per square centimeter; and
(e) repeating steps (b)-(d) one or more times to form a mufti-layer
membrane.
Optionally, step (c) may be performed by either preheating the support or
irradiating the coated support with electromagnetic radiation to accelerate
the
evaporation of the acetone and minimize penetration of the coating into the
pores
of the support.
The pyrolyzing step (d) may be performed either in a furnace or by laser
irradiation of the coating. When pyrolyzing the coating in a furnace the
temperature of the furnace is first increased at a predetermined rate and then
the
temperature is held constant within the range from about 150°C to
800°C for a
time ranging from 0 minutes to 480 minutes and then is cooled to room
temperature.
When pyrolyzing the coating with laser irradiation the laser beam may
focussed onto a small area of the tube assembly and the tube assembly rotated
while the laser beam delivery optics traverse the focussed area along the
axial
direction. The laser power output, the size of the focussed area, the rotation
speed
of the tube assembly and the axial traverse rate may be controlled to achieve
the
desired pyrolysis.
4
CA 02361664 2001-08-07
WO 00/53299 PCT/US00/06463
DETAILED DESCRIPTION OF THE INVENTION
Membrane Preparation
The method of preparing the membrane of the present invention comprises
a first step of ultrasonic deposition of PFA onto a suitable porous support.
Metal,
ceramic, glass, or composite material supports have been found suitable for
the
membrane support.
A first embodiment of the membrane uses a metal support, such as
sintered stainless steel (SS304) tubing, such as that manufactured by Mott
Metallurgical Corp., of Farmington, CT. A preferred porous tubing has an
outside
diameter of 6.35 mm with a wall thickness of 1.48 mm and a nominal porosity of
0.2 p,m. The porous tube was first cut to a length of 25 mm using a lathe to
form
a porous segment 2. Solid (i.e., nonporous) stainless steel (SS304) tubing 4,
6
was cleaned and then joined by welding to both ends of the porous tube 2 to
form
a tube assembly 10 (seen in Figure 1 ). Tube assemblies could comprise one or
more porous segments 2 and two or more solid segments 4,6. The tube assembly
10 was typically cleaned for 15 minutes in an ultrasonic bath of
1,1,2-trichlorotrifluoroethane (Freon~ TF) to remove any cutting oils and then
dried in an oven for at least 2 hours at about 120°C. After cleaning,
the tube
assembly was handled in a manner to prevent contamination (such as with
Nitrile~ gloves) and were stored in a dehumidified chamber until coated.
Neat PFA resin, such as that available from Monomer Polymer & Dajac
Laboratories, Inc. (Lot A-1-143) was diluted with reagent grade acetone, such
as
that available from J. T. Baker. A solution 20 of about twenty-five (25)
weight
percent (wt. %) PFA and seventy-five ('75) wt. % acetone was prepared
gravimetrically. The solution 20 was vigorously shaken before use to ensure it
was well mixed. As may be seen in Figure l, a 30 cc syringe 30 was filled with
the solution 20 and was delivered at a rate of 1 cc/min. using a syringe pump
32
(Sage Instruments, Model 355) town atomizer comprising an ultrasonic horn 40.
Although a custom fabricated ultrasonic horn 40 and a Dukane Corporation of
St.
Charles, IL ultrasonic generator 42 were used in the examples reported, a
commercially available ultrasonic atomizer and generator, such as that sold as
model 06-04029 from Sono-Tek Corporation of Milton, NY would also be
suitable. An ultrasonic frequency of forty kilohertz (40 kHz) was used, but
other
frequencies in the range of 20 kHz to 120 kHz are believed suitable.
Ultrasonic
deposition of the solution 20 onto the porous tube segment 2 was used to
overcome the deficiencies of conventional high-pressure gas spraying.
Ultrasonic
deposition can achieve a factor of 102-103 smaller droplet size than gas spray
deposition, and produces a low momentum spray which minimizes penetration of
5
CA 02361664 2001-08-07
WO 00/53299 PCT/I1S00/06463
the droplets into the support. With the present arrangement typical droplet
sizes
from about 10 ~.m to about 100 ~.m were achieved. The syringe pump 32
provides precise control of delivery rate of the solution 20 so that wet film
thickness can be controlled. The ultrasonic horn 40 was positioned about six
millimeters (6 mm) above the porous metal tube assembly 10 and the tube
assembly 10 was rotated about its axis l0A while the ultrasonic horn 40 was
traversed in a direction parallel to the axis l0A of the tube. Using a
commercially
available motor controller 50 and motorized rotation/traverse assembly 52 both
the rotational speed of the tube assembly 10 and the axial traverse rate of
the
ultrasonic horn 40 could be varied to control the deposition rate. A
rotational
speed of one hundred fifty revolutions per minute (150 rpm), and an axial
traverse
rate ranging from 1 to 10 mm/sec was found suitable. Coatings of from about
0.1
to 25 milligrams per square centimeter were achieved. After applying the "wet"
coat, the tube assembly 10 was rotated for about 1-0 minutes in air to allow
the
acetone in the coating to evaporate and then the tube assembly 10 was weighed.
Depending on the axial traverse rate, from about 0.5 to 125 mg of PFA/acetone
could be applied to each porous tube segment 2 using this technique.
The coating step may be performed at substantially room temperature or
the tube assembly 10 may be preheated to a temperature in the range of
30-300 degrees Celsius (°C) preferably 100-300°C. Preheating the
tube assembly
10 causes the acetone to evaporate at a higher rate and limits the penetration
of the
resin/acetone solution into the pores of the porous segment 2. The rate of
evaporation of-the acetone may also be increased by irradiating the coated
tube
assembly 10 during and/or immediately after the coating step with a source of-
electromagnetic radiation 56, such as an infrared source. The tube assembly 10
would typically be continuously rotated during such a drying step.
The pyrolysis of the coating may be performed in a furnace or by laser
irradiation of the coating. In a first pyrolyzing method, as may be seen in
Figure 2, the tube assembly 10 was placed inside a 57 mm diameter quartz tube
60. The tube 60 was fitted with end caps 62, 64 made of Pyrex~ designed to
hold
the coated porous tube segment 2 in the center of the quartz tube 60 and to
allow
the tube assembly 10 to be rotated by a motor drive 66 while being heated. The
quartz tube 60 was placed inside a furnace 65 (LindbergBlue model HTF55322C)
with a temperature controller/timer 65C (Eurotherm model 2416). The quartz
tube 60 was purged at a rate of 100 sccm with scientific grade helium 70
(total
impurities < 1 ppm), such as that available from MG Industries. The quartz
tube
60 was typically purged for 15 minutes to ensure all the air had been removed
before heating. The rate of temperature increase was controlled to about
6
CA 02361664 2001-08-07
WO 00/53299 PCT/US00/06463
5.0°C/min. and both the soak temperature and soak time were controlled
to
predetermined values. Soak temperatures from 150°C to 800°C and
soak times
from 0 to 180 minutes were achieved. Soak times of up to 480 minutes are
believed possible. After the soak time had elapsed the furnace 65 was turned
off
and allowed to cool to room temperature.
Throughout the heating and cooling cycle the tube assembly 10 was
continuously rotated at about 30 revolutions per minute (rpm) to ensure the
coating thereon did not flow during pyrolysis. The pyrolysis protocol followed
established methods used for synthesis of unsupported carbon molecular sieves
such as that described in Lafyatis, D. S., Tung, J., Foley, H. C.,
"Poly(furfuryl
alcohol)-Derived Carbon Molecular Sieves: Dependence of Adsorptive Properties
on Carbonization Temperature, Time and Polyethylene glycol) Additives"
Industrial Engineering for Chemical Research; 1991. vol. 30, pp. 865-873; or
Mariwala, R. K., Foley, H. C., "Evolution of Ultramicroporous Adsorptive
Structure in Poly(furfuryl alcohol)-Derived Carbogenic Molecular Sieves"
Industrial Engineering for Chemical Research, 1994, vol. 33, pp. 607-615. Each
tube assembly 10 was weighed after pyrolysis and typical "dry" carbon weights
ranged from about 0.1 to 50 mg per porous tube segment 2 and carbon yield
ranged from 15-40% depending on the initial "wet" weight and the pyrolysis
conditions. This produced a single-coated nanoporous carbon tubular membrane
1 OM which was formed by cross linking and carbonization of the polymer.
Multiple coats were applied and pyrolyzed before the membrane provided
significant molecular sieving.
In a second pyrolyzing method, as may be seen in Figure 2A, the tube
assembly 10 is placed inside a 57 mm diameter quartz tube 60. The tube 60 is
fitted with end caps 62, 64 made of Pyrex~ designed to hold the coated porous
tube segment 2 in the center of the quartz tube 60 and to allow the tube
assembly
10 to be rotated by a motor drive 66 while being heated. In this second
pyrolyzing method the furnace 65 and associated temperature controller/timer
65C of the first pyrolyzing method are replaced by a continuous wave C02 laser
102 and associated laser beam delivery optics 104. The laser 102 produces an
output beam lO2B. The beam delivery optics 104 comprises a focussing lens
104L and optional beam expanding optics 104B. The beam 102B passes through
the delivery optics 104, through the quartz tube 60 and is focussed by the
lens
104L to irradiate a small area 102P of the coated porous tube segment 2. The
laser power level and the size of irradiated area 102P are controlled to
result in
rapid pyrolysis of the coating on the tube segment 2. A motorized traverse
assembly 152 and associated controller 150 cause the irradiated area 102P to
7
CA 02361664 2001-08-07
WO 00/53299 PCT/US00/06463
traverse along the axis if the tube assembly 10 while motor 66 rotates the
tube
assembly 10. The traverse rate and the speed of rotation of the tube assembly
10,
and thus tube segment 2, relative to the laser beam 102B, and thus the
irradiated
area 102P, are controlled to effect complete pyrolysis of the coating.
A second embodiment of the membrane uses a ceramic support. Porous
alpha alumina supports, coated with gamma alumina, zirconia or titanic, such
at
that manufactured by U.S. Filter Corporation of Deland, Florida and sold under
the trademark Membralox~ have been found suitable for the membrane support.
For this second embodiment a preferred porous tubing has an outside diameter
of
8 mm with a wall thickness of 1 mm and a nominal porosity over the range of
5 ~m to 200 Angstroms. The nominal porosity depends upon on the coating. A
porosity of 5 p,m is typical for the gamma alumina coating, while a porosity
of
200 Angstroms is typical for the zirconia coating. The porous tubes are 250 mm
long and no cleaning was required before coating: The tubes were handled in
such a manner to prevent contamination (such as with Nitrile~ gloves) and were
stored in a dehumidified chamber until coated.
A third embodiment of the membrane uses a porous glass support. Porous
glass, such at that manufactured by Corning, Inc. of Corning, NY under the
trademark Vycor~ has been found suitable for the membrane support. For this
embodiment a preferred porous tubing has an outside diamter of 6 mm with a
wall
thickness of 1 mm and a nominal porosity of 20 to 40 angstroms. Porous tubes
mm long which are attached to 15-20 cm long quartz tubes using a proprietary
heat treatment joining technique are commercially available from Corning. No
cleaning was required before coating. The tubes were kept in deionized water
to
25 prevent contamination and handled with with Nitrile~ gloves.
A fourth embodiment of the membrane uses a carbon composite material
as a support. Porous carbon supports composed of carbon fibers and coated with
a proprietary carbon coating, such as that manufactured by KOCH membrane
systems of New York, NY under the trade name Carbo-CorT"" are believed
suitable for the membrane support. A preferred porous tubing has an outside
diameter of 8 mm with a wall thickness of 1 mm and a nominal porosity of
0.01 ~,m. The porous tubes are 25 to 250 mm long. No cleaning is required
before coating. The tubes are handled in a manner to prevent contamination
(such
as with Nitrile~ gloves) and are stored in a dehumidified chamber until
coated.
A desirable property of the support material is to have porosity less than
about 5 p.m. Finer porosity of the support generally results in thinner
nanoporous
carbon membranes that have corresponding improved flux and selectivity for
separation of small molecules such as gases. When supports are employed that
8
CA 02361664 2001-08-07
WO 00/53299 PCT/US00/06463
have a porosity of greater than about 0.1 p,m, one or more intermediate layers
may
be desirable to further reduce the average pare size of the support before
coating
with polyfurfuryl alcohol. Materials such as titanium dioxide, silica, and
colloidal
graphite suspended in isopropyl alcohol have been applied to the exterior of
the
porous supports to form intermediate layers and thus reduce the average pore
size
(also called support macroporosity) before coating with polyfurfuryl alcohol.
Other materials such as silicon dioxide may also be used as an intermediate
layer.
This intermediate layer minimizes penetration of the polyfurfmyl alcohol into
the
pores and reduces the thickness of the resulting carbon molecular sieving
layer.
This has the added benefit of increasing the permeance of the resulting
membrane
without sacrificing small molecule separation selectivity.
Typically these intermediate coatings would be applied by an ultrasonic
atomization technique, similar to that described in conjunction with the first
embodiment. Optionally, small amounts of polyfurfuryl alcohol may be added to
this intermediate layer material as a binder and then this intermediate layer
may
be fired in a furnace at temperatures between 150 to 800°C. Subsequent
coatings
of polyfurfuryl alcohol in acetone would then be applied and fired to produce
the
molecular sieving layer, as previously described in conjunction with the first
embodiment.
Improved uniformity of membrane layer thickness is believed important.
Such improved uniformity has been achieved by the following method:
(a) preparing a solution of poly(furfuryl) alcohol resin in an acetone
solvent;
(b) as shown in Figure l, positioning an ultrasonic atomizer
comprising the ultrasonic horn 40 at a predetermined distance. f~~om a tube
assembly 10 having an axis 10A, the tube assembly 10 comprising at least one
porous tubular support;
(c) rotating the tube assembly 10 at a predetermined rotation speed
and traversing the ultrasonic horn 40 in a direction parallel with the axis
l0A of
the tube assembly 10 at a predetermined speed, so that the motion of the
ultrasonic horn 40 defines a helical path along the tube assembly, the helical
path
being defined by an axial distance along the tube assembly and a rotational
phase
angle with respect to the tube assembly; the rotational phase angle and the
axial
position of the ultrasonic horn 40 relative to the tube assembly 10 are
controlled
by motor controller 50 and motorized traverse assembly 52;
(d) ultrasonically atomizing the solution 20 and depositing a thin,
uniform layer of no more than about 25 milligrams per square centimeter of the
9
CA 02361664 2001-08-07
WO 00/53299 PCT/US00/06463
poly(furfuryl) alcohol resin/acetone solution onto the porous segments 2 of
the
tube assembly 10;
(e) drying the resin/acetone layer by evaporating the acetone;
(f) pyrolyzing the poly(furfuryl) alcohol resin;
(g) repeating steps (c)-(fJ to form a membrane having a plurality of
successive layers, the rotational phase angle of the ultrasonic horn 40
relative to
the tube assembly 10 being selected to be different for each successive
coating
step (c), causing the helical path of the ultrasonic horn 40 for each
successive
repetition of step (c) to be offset from the previous helical path, so that a
membrane of more uniform thickness is achieved.
It has been found that minimizing the thickness of each layer of the
membrane improves the membrane permeance and minimizes the occurrence of
defects in the membrane. It is believed that defects in the membrane are
related to
areas in the coating which exceed a critical film thickness. The critical film
thickness has been determined empirically to be about 20 +/- 3 microns on
porous
metal supports. It is therefore desirable to employ coating methods that
reduce
the deposited coating thickness and pyrolysis methods that reduce the
thickness of
the pyrolyzed layer.
It has been found that preheating the porous support (tube assembly 10) to
a temperature above ambient before application of the polyfurfmyl alcohol
coating is desirable. Preheating the support to a temperature above
30°C,
preferably from 100 to 300°C, before application of the polyfurfuryl
alcohol
coating facilitates faster evaporation of the acetone and is believed to
produce a
thinner film on the surface of the support with less penetration into the
pores of
the porous support.
It is also believed desirable to pyrolyze the coating as rapidly as possible.
Heating the support and the applied coating with a continuous wave (CVO C02
laser is believed to produce an almost instantaneous pyrolysis of the coating.
It is
believed that this rapid pyrolysis results in a thinner film on the surface of
the
support and minimizes penetration of the coating into the pores of the
support.
Although inert atmospheres have typically been employed in prior art
pyrolysis methods, pyrolysis in a reactive atmosphere is believed to result in
different structural forms of nanoporous carbon having desirable ranges of
pore
dimensions for separation of certain molecular size ranges.
Heating carbon particles in a reducing atmosphere (such as hydrogen) or
an oxidizing atmosphere (such as oxygen, carbon dioxide, and carbon monoxide)
is known to result in different surface chemistries on carbon particles. The
use of
such reactive atmospheres in the pyrolysis step of the present method is
believed
CA 02361664 2001-08-07
WO 00/53299 PCT/US00/06463
to produce different forms of nanoporous carbon structure within the membrane
layers. It is believed that control of the range of pore sizes in the membrane
may
be thus achieved.
Permeation Experiments and Analysis
The tubular membranes l OM were inserted in a module 70 for testing. As
seen in Figure 5, the module 70 consisted of a cylindrical membrane holder 72
with knife-edge flanges 72F on either side and was sealed with copper gaskets
74.
The flanged ends 72F were welded to compression fittings 76 (such as
Swagelok~) each having a ferrule 78. Metal ferrules were not used since the
tube
assemblies 10 would be impossible to remove; therefore, for low temperature
conditions polymer ferrules 78, such as Nylon~ or Teflon~ ferrules, were used
and for high temperature conditions graphite ferrules 78 were used. With
external
compression applied to the outside of the fittings, pressures up to 7000 kPa
could
be maintained with no measurable leakage at 22°C.
Pressure rise time experiments were performed on twelve tubular
membranes (identified as SNPCM l, 2, ..., 12) which were synthesized at 150,
300, 450 and 600°C. Each were held at temperature for 0 to 180 minutes
and
coated two to six times, see Figure 3. Initially, both the core side C and
outer
shell sides S of the membranes were at atmospheric pressure. A probe gas 80
was
introduced on the core side of the supported membrane 1 OM at a pressure of
300 kPa while the shell side pressure rise was continuously monitored using
National Instruments data acquisition hardware 90 (model AT-AO-6/10 and a
model. AIO-16) and LabView~ data acquisition software. The core side and outer
shell side pressures were monitored by MKS Instruments, Inc. model
122BAOSOOOBB pressure transmitters 92 which were accurate to +/- 0.5% of
reading over the range of 0 to 667 kPa. The membrane module 70 was evacuated
and returned to atmospheric pressure (with air) on both the core and outer
shell
side before the introduction of the next probe gas. Typically, the probe gases
were tested in the following order: nitrogen, oxygen, helium, and hydrogen.
All
experiments were conducted at 22°C. Figure 6 is a plot of the data
measured for
example SNPCM 4. Notice the pressure rise time curves are arranged in order of
increasing molecular size. The high sensitivity of the membrane to gas
molecular
size is evident from the fact that there is noticeable separation between
nitrogen
and oxygen, which differ by only 0.2 Angstroms in size.
A simple model was used to describe the unsteady state experiments and
calculate the pure gas permeabilities. (see Rao, M. B. and Sircar, S.,
"Nanoporous
carbon membranes for separation of gas mixtures by selective surface flow"
Journal of Membrane Science, 1993. vol. 85, pp. 253-264; or Acharya, M.,
Raich,
11
CA 02361664 2001-08-07
WO 00/53299 PCT/US00/06463
B. A., Foley, H. C., Harold, M. P., Lerou, J. J., "Metal-Supported Carbogenic
Molecular Sieve Membranes: Synthesis and Applications" Industrial Engineering
of Industrial Research, 1997. vol. 36, p. 2924-2930). A mass balance for the
permeating species from the core to the shell side of the SNCPM can be
written:
_dm _
dt J~M~'~'~ (1)
where m (gm) is the mass of the gas, J is the molar flux (mol/m2.sec), MW
(gm/gmol) is the gas molecular weight, A (m2) is the membrane area, and t
(sec)
is time. The flux across the membrane was defined by:
J = n ' (Pcs - Pss) (2)
where ~' is the gas permeability (mol/m.sec.Pa), A is the membrane thickness
(m),
and Pas and Pss are the pressures (Pa) on the core side and outer shell side
of the
tubular membrane, respectively. Using the ideal gas law to rewrite the mass of
the gas in terms of the shell side pressure, Pss the final expression is
obtained:
dPss A~R~T _~c'
dt Vss ~ A ~ (Pcs Pss) (3)
25
where R (m3.Pa/gmol.K) is the gas constant, T (K) is the temperature, and Vss
(m3) is the shell side volume. Integrating from the initial shell side
pressure, Psso
at time t = 0 to the final shell side pressure, Pss at t = t provides our
final
expression:
Vss IPcs - P ssOl _ ~'
~ In - ~ t
A ~ R ~ T IPcs - Pssl A
(4)
A plot of the left-hand-side of equation (4) versus time should give a
straight line with a slope of ~'/A = ~p which is called the gas permeance. The
permeances are provided in Figure 4 along with the ratio of the permeance with
respect to N2 which provides the ideal separation factors.
It was assumed in the integration of equation 3 that ~o was independent of
pressure. The permeances for SNPCM 4 were measured as a function of pressure
from 300 to 7000 kPa and determined to be substantially independent of
pressure,
3 S see Figure 7.
12
CA 02361664 2001-08-07
WO 00/53299 PCT/US00/06463
Scanning electron microscope (Hitachi 54000 SEM) images taken both of
the exterior surface as well as cross and axial sections of a similarly
prepared
SNPCM at 573 K reveal a defect-free membrane with a uniform radial thickness
of about 16 +/- 3 ~,m. 'The thickness along the axis varied in some areas
possibly
S due to the manual movement of the ultrasonic horn during coating.
Finally, a steady state gas separation experiment was run with SNPCM 4
with air (MG Industries, scientific grade) fed to the core side at a pressure
of
7000 kPa and a flow rate of 150 sccm. A 5 sccm helium purge (MG Industries,
scientific grade) was used as a sweep gas on the shell side of the supported
membrane. Samples were taken manually using a gas tight syringe from the feed
and permeate sides of the membrane and analyzed using a Hewlett Packard gas
chromatograph (model HP5890), a Supelco molecular sieve column (60/80 mesh,
molecular sieve SA, 10' x 1/8", column 256398-10), and a thermal conductivity
detector (TCD). The feed contained 21.0-21.1 vol. % oxygen while the permeate
contained between 41.5-44.0 vol. % oxygen.
The supported nonoporous carbon membrane described herein exhibited
improved gas separation performance. Permeation measurements with pure gases
such as nitrogen (N2), oxygen (02), helium (He), and hydrogen (H2) reveal a
molecular sieving behavior with permeation decreasing with increasing
molecular
size. Gas separation factors ranging up to about 30 for 02/N2; up to about 178
for
He/N2; and up to about 330 for H2/N2 were measured in single gas permeation
experiments at 22°C. The separation factors and permeation values,
which ranged
from 2.7 x 10-14 to 4.1 x 10-8 mol/m2.Pa.sec, were found to depend on the
amount
- of carbon deposited, the pyrolysis temperature, and the pyrolysis soak time.
The
pure component permeance values were found to be independent of pressure from
300 to 7000 kPa indicating shape and size selective effects dominate the
separation. Scanning electron microscope (SEM) images of the surface reveal a
defect-free membrane. A high pressure air feed was continuously separated with
a permeate composition containing 41.5 to 44 volume percent (vol. %) oxygen.
13