Note: Descriptions are shown in the official language in which they were submitted.
WO 00/47612 CA 02362541 2001-08-10 pCT~S00/03665
STRUCTURE-BASED SCREENING TECHNIQUES FOR DRUG DISCOVERY
This application is a continuing application of U.S.S.N.s 60/120.009, filed
Febnaary 11, 1999 and of
60/131,674, filed April 29, 1999, each of which are expressly incorporated by
reference.
FIELD OF THE INVENTION
The invention relates to novel non-naturally occurring cell surface receptor
analogs, ligand analogs
and nucleic acids encoding them. The invention further provides methods for
screening of ligand
analogs and bioactive agents capable of modulating the signaling activity of a
non-naturally occurring
cell surface receptor analog or capable of binding to a non-naturally
occurring cell surface receptor
analog.
BACKGROUND OF THE INVENTION
Cytokines and hormones are secreted proteins that bind to cell surface
receptors and activate cellular
differentiation and proliferation through a cascade of intracellular signaling
events. They include
insulin, erythropoietin (EPO), granulocyte colony stimulating factor (GCSF),
thrombopoietin (TPO),
human growth hormone (hGH), vascular endothelial growth factor (VEGF),
angiostatin, endostatin,
insulin, the interleukins, and the interferons. In general, each cytokine has
a specific cell-surface
receptor. These receptors comprise three major domains: an extracellular
portion that binds the
cytokine, a transmembrane domain to anchor the receptor in the membrane, and
an intracellular
signaling domain that is activated by cytokine binding.
Cytokines generally have at least two binding sites, and sometimes three, for
the receptors.
2 0 Monomeric receptors are brought together by cytokine binding, to result in
the formation of a receptor
oligomer. This oligomer is the biologically active form and is necessary for a
variety of intracellular
receptor signaling events.
From a commercial perspective, cytokines are used to treat millions of
patients for anemia, cancer,
diabetes, neurological and growth disorders. However, cytokines are generally
large molecules that
must be administered by intravenous or subcutaneous injection. Accordingly,
the pharmaceutical
WO 00/47612 CA 02362541 2001-08-10
PCT/LJS00/03665
industry has been highly motivated to develop small molecule replacement that
can be taken orally.
Thus, there is enormous commercial interest in finding cytokine-mimetic drugs
that could eliminate the
need for injection and lower the cost of producing the recombinant proteins.
However, a variety of technical barriers have prevented the discovery and
commercialization of small
molecule mimics for cytokines. The development of small molecule cytokine
mimics is blocked by the
difficulty in reconstituting the biologically relevant receptor structure, a
precisely oriented receptor
oligomer. So far it has not been possible to reconstitute the active receptor
oligomer for use in in vitro
drug screening assays, aiming to isolate cytokine mimetics. Current screening
approaches utilize
receptor molecules in random orientations, not as functional dimers or
trimers, thereby screening for
receptor affinity rather than activity. Cell-based assays have recently been
developed that present
receptors in a 'natural' manner, however, these assays are difficult to use
and limited in screening
power for high throughput screening.
De novo protein design has received considerable attention recently, and
significant advances have
been made toward the goal of producing stable, well-folded proteins with novel
sequences. Efforts to
design proteins rely on knowledge of the physical properties that determine
protein structure, such as
the patterns of hydrophobic and hydrophilic residues in the sequence, salt
bridges and hydrogen
bonds, and secondary structural preferences of amino acids. Various approaches
to apply these
principles have been attempted. For example, the construction of a-helical and
p-sheet proteins with
native-like sequences was attempted by individually selecting the residue
required at every position in
2 0 the target fold (Hecht et al., Science 249:884-891 (1990); Quinn et al.,
Proc. Natl. Acad. Sci USA
91:8747-8751 (1994)). Alternatively, a minimalist approach was used to design
helical proteins, where
the simplest possible sequence believed to be consistent with the folded
structure was generated
(Regan et al., Science 241:976-978 (1988); DeGrado et al., Science 243:622-628
(1989); Handel et
al., Science 261:879-885 (1993)), with varying degrees of success. An
experimental method that
relies on the hydrophobic and polar (HP) pattern of a sequence was developed
where a library of
sequences with the correct pattern for a four helix bundle was generated by
random mutagenesis
(Kamtekar et al., Science 262:1680-1685 (1993)). Among non de novo approaches,
domains of
naturally occurring proteins have been modified or coupled together to achieve
a desired tertiary
organization (Pessi et al., Nature 362:367-369 (1993); Pomerantz et al.,
Science 267:93-96 (1995)).
3 0 Though the correct secondary structure and overall tertiary organization
seem to have been attained
by several of the above techniques, many designed proteins appear to lack the
structural specificity of
native proteins. The complementary geometric arrangement of amino acids in the
folded protein is the
root of this specificity and is encoded in the sequence.
2
WO 00/47612 CA 02362541 2001-08-10 pCT~S00/03665
Several groups have applied and experimentally tested systematic, quantitative
methods to protein
design with the goal of developing general design algorithms (Hellinga et al.,
J. Mol. Biol. 222: 763-785
(1991); Hurley et al., J. Mol. Biol. 224:1143-1154 (1992); Desjarlaisl et al.,
Protein Science 4:2006-
2018 (1995); Harbury et al., Proc. Natl. Acad. Sci. USA 92:8408-8412 (1995);
Klemba et al., Nat.
Struc. Biol. 2:368-373 (1995); Nautiyal et al., Biochemistry 34:11645-11651
(1995); Betzo et al.,
Biochemistry 35:6955-6962 (1996); Dahiyat et al., Protein Science 5:895-903
(1996); Dahiyat et al.,
Science 278:82-87 (1997); Dahiyat et al., J. Mol. Biol. 273:789-96; Dahiyat et
al., Protein Sci. 6:1333-
1337 (1997); Jones, Protein Science 3:567-574 (1994); Konoi, et al., Proteins:
Structure, Function and
Genetics 19:244-255 (1994)). These algorithms consider the spatial positioning
and steric
complementarity of side chains by explicitly modeling the atoms of sequences
under consideration. In
particular , W098/47089, and U.S.S.N. 09/127,926 describe a system for protein
design; both are
expressly incorporated by reference.
A need still exists for a method of screening for cytokine mimetics. Thus, it
is an object of the present
invention to provide non-naturally occurring cell surface receptor analogs,
capable of binding naturally
occurring ligands, such as cytokines. It is a further aspect of this invention
to provide nucleic acids
encoding the receptor analogs and methods of using the receptor analogs for
screening cytokine
mimetics.
SUMMARY OF THE INVENTION
In accordance with the objects outlined above, the present invention provides
non-naturally occurring
2 0 cell surface receptor analogs, also termed "cell surface receptor analogs"
(e.g. the proteins are not
found in nature) comprising amino acid sequences that are less than about 95 -
97% identical to the
extracellular domains of corresponding naturally occurring cell surface
receptors. The non-naturally
occurring cell surface receptor analogs have at least one biological property
of a naturally occurring
cell surface receptor; for example, the non-naturally occurring cell surface
receptor analog binds the
2 5 natural ligand for the naturally occurring cell surface receptor. Thus,
the invention provides non-
naturally occurring cell surface receptor analogs with amino acid sequences
that have at least about
5% amino acid substitutions, deletions and/or insertions in their
extracellular domain as compared to
the naturally occurring cell surface receptor.
Further, the present invention provides non-naturally occur-ing ligands, also
termed "ligand analogs"
3 0 (e.g. the proteins are not found in nature) comprising amino acid
sequences that are less than about
95 - 97% identical to corresponding naturally occurring ligands. The ligand
analog have at least one
biological property of a naturally occurring ligand; for example, the ligand
analog binds the natural
WO 00/47612 CA 02362541 2001-08-10 pCT~S00/03665
receptor for the naturally occurring ligand. Thus, the invention provides
ligand analogs with amino acid
sequences that have at least about 5% amino acid substitutions, deletions
and/or insertions when
compared to the corresponding naturally occurring ligand.
In a further aspect, the present invention provides receptor analog conformers
that have three
dimensional backbone structures that substantially correspond to the three
dimensional backbone
structure of a naturally occurring cell surface receptor. The amino acid
sequence of the conformer and
the amino acid sequence of the naturally occurring cell surface receptor are
less than about 95%
identical with respect to the extracellular domain. In one aspect, at least
about 90% of the non-
identical amino acids are in a core region of the conformer. In other aspects,
the conformer has at
least about 100% of the non-identical amino acids in a core region.
In another aspect, the present invention provides receptor analog conformers
that comprise a three
dimensional backbone structure of an extracellular domain that substantially
con-esponds to the
corresponding three dimensional backbone structure of a naturally occurring
cell surface receptor
complexed with its natural ligand. The amino acid sequence of the conformer
and the amino acid
sequence of the naturally occurring cell surface receptor, complexed with its
natural ligand, are less
than about 95% identical with respect to the extracellular domain. In one
aspect, at least about 90% of
the non-identical amino acids are in a core region of the conformer. In other
aspects, the conformer
has at least about 100% of the non-identical amino acids in a core region.
In a further aspect, the present invention provides ligand analog conformers
that comprise a three
2 0 dimensional backbone structure that substantially corresponds to the
corresponding three dimensional
backbone structure of a naturally occurring ligand. The amino acid sequence of
the conformer and the
amino acid sequence of the naturally occurring ligand, are less than about 95%
identical. In one
aspect, at least about 90% of the non-identical amino acids are in a core
region of the conformer. In
other aspects, the conformer has at least about 100% of the non-identical
amino acids in a core
2 5 region.
In a further aspect, the invention provides recombinant nucleic acids encoding
the non-naturally
occurring cell surface receptor analogs, the ligand analogs, expression
vectors comprising the
recombinant nucleic acids, and host cells comprising either the recombinant
nucleic acids or the
recombinant nucleic acids and expression vectors.
3 0 In an additional aspect, the invention provides methods of producing the
non-naturally occurring cell
surface receptor analogs and the ligand analogs of the invention comprising
culturing host cells
comprising either the recombinant nucleic acids or the recombinant nucleic
acids and expression
4
CA 02362541 2001-08-10 pCT~S00/03665
WO 00/47612
vectors under conditions suitable for expression of the nucleic acids. The
proteins may optionally be
recovered.
In a further aspect, the invention provides for eukaryotic cells, prokaryotic
cells, viruses and solid
supports displaying a non-naturally occurring cell surface receptor analog.
In an additional aspect, the invention provides methods of screening for
ligand analogs comprising the
step of adding a candidate ligand to a non-naturally occurring cell surface
receptor analog of the
invention and determine the binding of said candidate ligand to said receptor
analog.
In an additional aspect, the invention provides methods of screening for
ligand analogs comprising the
step of adding a candidate ligand to a non-naturally occurring cell surface
receptor analog of the
invention and determine the signaling activity of said receptor analog.
In a further aspect, the invention provides methods of screening for bioactive
agents modulating the
binding of a ligand analog to a receptor analog comprising the steps of (i)
adding a ligand analog to a
non-naturally occurring cell surface receptor analog, (ii) adding a candidate
bioactive agent and (iii)
determine whether said candidate bioactive agent modulates the binding of said
ligand analog and
said non-naturally occurring cell surface receptor analog.
In a further aspect, the invention provides methods of screening for bioactive
agents modulating the
binding of a figand analog to a receptor analog comprising the steps of (i)
adding a ligand analog to a
non-naturally occurring cell surface receptor analog, (ii) adding a candidate
bioactive agent and (iii)
determine whether said candidate bioactive agent modulates the signaling
activity of said non-naturally
2 0 occurring cell surface receptor analog.
BRIEF DESCRIPTION OF THE DRAWINGS
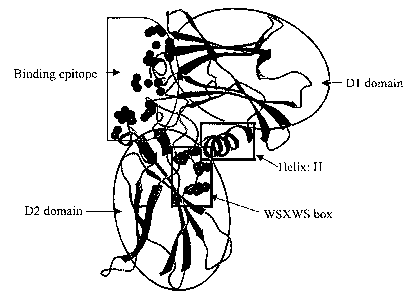
Figure 1 illustrates the structure of one erythropoietin receptor (EPOR)
monomer in the EPO-EPORZ
complex. The side chains within 4.5 A of EPO are shown as spheres in the
binding epitope region,
and the highly conserved residues are shown in the WSXWS box. The D1 and D2
domains are
2 5 indicated by the oval regions and the N-terminal helix is indicated by
Helix: H.
Figure 2 illustrates the structure of the binding interface between human
growth hormone receptor
(hGHR) with its ligand (spheres in upper portion of Figure) within 5A and the
contact interface between
the two receptor monomers (spheres in lower portion of Figure).
5
CA 02362541 2001-08-10
WO 00/47612 PCT/US00/03665
Figure 3 illustrates a homology model for GCSFR, the granulocyte colony
stimulating factor receptor
(indicated in light grey) vs. the NMR structure of its fragment (indicated in
dark grey).
Figure 4 illustrates the structure of the tumor necrosis factor receptor
(TNFR) trimer.
Figure 5 illustrates on the left side x-ray structures of EPO receptors in
complex with ligands, such as
the naturally occurring erythropoietin: EPO-EPOR2; EMP1, a weak agonist: (EMP1-
EPOR)z; and
EMP33, an antagonist: (EMP33-EPOR)z. At the right, 2-dimensional schematic
drawings of EPO
receptors in complexes are shown (the ligands are not shown). Two 7-beta-
strand domains, D1 and
D2, and the N-terminal helix (H) are shown. The orientation (angle) between
the two receptor
monomers is different for each complex, and the separation between the two
monomers and relative
position of the a-helix in the EMP1 and EMP33 complexes differs from that of
the EPO complex.
Figure 6 illustrates in row A) an in vitro screen for EPO mimics using
bivalent antibodies (Wrighton et
al., Science 273:458-64 (1996)]. This approach suffers from poorly controlled
EPOR dimerization; the
bioactive dimer conformation (shown in brackets) is not favored (in
equilibrium with large ensemble of
inactive conformations. In row B), an approach using coiled coil fused to an
EPOR is shown. Using a
coiled coil approach with adjustable linker length between the coiled coil and
e.g. the D2 domain of
EPOR allows more control over dimerization and stabilization of dimer
orientation. Using e.g., PDA
design, the bioactive conformation will be strongly favored.
Figure 7 illustrates schematically the coiled coil motif being fused to the D2
domain of a coupled
receptor, such as EPOR, leading to dimerization and the coiled coil motif
being fused to the D2 domain
2 0 of an uncoupled receptor, such as TNFR, leading to trimerization.
Figures 8A, 8B, 8C, and 8D illustrate the sequence of EPOR and the residues
targeted by PDA
design. The amino acid sequence shown in the query corresponds to the
extracellular domain of
human EPOR. The signal sequence of the EPOR precursor (amino acid residues 1-
24 in GenBank
accession #P19235) has been taken off. Thus, the 225 amino acid sequence shown
corresponds to
amino acid residues 25-249 of GenBank accession #P19235. The amino acid
sequence shown in the
subject corresponds to the sequence used in the PDA design and differs from
the query by not
including amino acid residues 1- -9 and 221-225 of the query. PDA sites and
elbow PDA sites,
indicated by asterisks, refer to the sites used in the PDA design. The design
was done first on
domains D1 and D2 and the sequences were generated using d1 and d2 alone or in
combination d12.
3 0 EPOR-PDA sequences are based on the structures of (1) the EPOR with an EPO
mimetic peptide at
2.8A resolution: 1ebp, (EPOR + EMP1)2 dimer complex; (2) the EPOR with EPO at
2.8A resolution:
1 blw - -( also called 1 cn4); and (3) EPOR with EPOR at 1.9A resolution: 1
eer. d 1 211 or d2 211
6
WO 00/47612 CA 02362541 2001-08-10 pCT~S00/03665
means PDA on the left arm of EPOR dimer (amino acid residues 1-211); d1 422 or
d2 422 means
PDA on the right arm of EPOR dimer (amino acid residues 212-422); d1 and d2
without appendix
mean PDA on EPOR dimer; ew refers to PDA on EPOR dimer; ew1 refers to PDA on
the left arm (1-
211 ); ew2 refers to PDA on the right arm (212-422).
DETAILED DESCRIPTION OF THE INVENTION
The present invention is generally directed to novel methods of screening for
ligand analogs. Briefly,
the invention may be described as follows. A receptor/ligand pair is chosen,
and the receptor is
modeled in an active conformation. The receptor analogs, provided herein, are
stable receptor
complexes held in a biologically active conformation similar to the structure
of the corresponding
naturally occurring receptors complexed with their cognate ligands. Creating
such a structural mimic
of an active, naturally occurring receptor combines the benefits of simple
affinity-based screening with
those of accurate but more complicated cell-based activity screening into a
simple screening
technique. Thus, as detailed further below, the receptor analogs of the
invention may be used for high
throughput screening.
Accordingly, the present invention provides non-naturally occurring cell
surface receptor analogs.
By "non-naturally occurring" or "synthetic" herein is meant an amino acid
sequence or a nucleotide
sequence that is not found in nature; that is, an amino acid sequence or a
nucleotide sequence that
has been intentionally modified by man in the laboratory. Accordingly, by
"naturally occurring° or "wild
type" or grammatical equivalents, herein is meant an amino acid sequence or a
nucleotide sequence
2 0 that is found in nature and includes allelic variations; that is, an amino
acid sequence or a nucleotide
sequence that has not been intentionally modified by man in the laboratory.
By "cell surface receptor", "cell membrane receptor", "receptor" or
grammatical equivalents herein is
meant a proteinaceous molecule that has an affinity for a ligand. Included
within this definition are
proteinaceous molecules that are capable of being displayed on the surface of
a cell, membrane or
2 5 virus. In general, cell surface receptors have three components: an
extracellular domain, which binds,
as outlined above, the ligand, a transmembrane domain, to anchor the receptor;
and an intracellular
domain that usually is involved in signaling. Receptors may be exposed to the
intracellular
compartment (e.g., when located on cellular membranes, such as nuclear
membranes, endoplasmatic
reticulum membranes, mitochondrial membranes, etc.) or to the extracellular
environment (e.g., when
3 0 located on the surface of a cell or on the surface of a virus).
7
CA 02362541 2001-08-10 pCT~S00/03665
WO 00/47612
Receptors appear to fall into two general classes: type 1 and type 2
receptors. Type 1 receptors have
generally two identical subunits associated together, either covalently or
otherwise. They are
essentially preformed dimers, even in the absence of ligand. The type 1
receptors include the insulin
receptor and the IGF (insulin like growth factor) receptor. The type-2
receptors, however, generally
are in a monomeric form, and rely on binding of one ligand to each of two or
more monomers, resulting
in receptor oligomerization and receptor activation. Type-2 receptors include
the growth hormone
receptor, the leptin receptor, the LDL (low density lipoprotein) receptor, the
GCSF (granulocyte colony
stimulating factor) receptor, the interleukin receptors including IL-1, IL-2,
IL-3, IL-4, IL-5, IL-6, IL-7, IL-
8, IL-9, IL-11, IL-12, IL-13, IL-15, IL-17, etc., receptors, EGF (epidermal
growth factor) receptor, EPO
(erythropoietin) receptor, TPO (thrombopoietin) receptor, VEGF (vascular
endothelial growth factor)
receptor, PDGF (platelet derived growth factor, A chain and B chain) receptor,
FGF (basic fibroblast
growth factor) receptor, T-cell receptor, transferrin receptor, prolactin
receptor, CNF (ciliary
neurotrophic factor) receptor, TNF (tumor necrosis factor) receptor, Fas
receptor, NGF (nerve growth
factor) receptor, GM-CSF (granulocyte/macrophage colony stimulating factor)
receptor, HGF
(hepatocyte growth factor) receptor, LIF (leukemia inhibitory factor), TGFa/(3
(transforming growth
factor a/p) receptor, MCP (monocyte chemoattractant protein) receptor and
interferon receptors (a, p
and y). Further included are T cell receptors, MHC (major histocompatibility
antigen) class I and class
II receptors and receptors to the naturally occurring ligands, listed below.
Accession numbers for naturally occurring cell surface receptors are readily
available. For example,
2 0 amino acid sequences for the human erythropoietin receptor (EPOR) are
available under P19235 and
ZUHUR. -Nucleotide sequences encoding human EPOR are available under NM
000121, M60459,
and M34986. Amino acid sequences for the human tumor necrosis factor receptor
(TNFR) are
available under AAA36753, AAA36754, and AAA36756. Nucleotide sequences
encoding human
TNFR are available under M60275, M63121, and M58286. Amino acid sequences for
the human
2 5 growth hormone receptor (GHR) are available under AAA52555 and AAC50653.
Nucleotide
sequences encoding human GHR are available under AH002706 and U60179.
The invention provides non-naturally occurring cell surface receptor analogs.
By "non-naturally
occurring cell surface receptor analog", or "cell surface receptor analogs" or
"receptor analog", or
grammatical equivalents thereof, herein is meant a cell surface receptor
having an amino acid
3 0 sequence or a nucleotide sequence that is not naturally occurring. The
receptor analogs and nucleic
acids of the invention are distinguishable from naturally occurring cell
surface receptors. Accordingly,
by "naturally occurring cell surface receptor", or "wild type receptor", or
grammatical equivalents
thereof, herein is meant a cell surface receptor having an amino acid sequence
or a nucleotide
sequence that is naturally occurring.
8
CA 02362541 2001-08-10
WO 00/47612 PCT/US00/03665
In a preferred embodiment, the receptor analogs are naturally occurring human
receptor conformers.
By "conformer" herein is meant a protein that has a protein backbone 3D
structure that is virtually the
same but has significant differences in the amino acid side chains. That is,
e.g., the receptor analogs
of the invention define a conformer set, wherein all of the extracellular
domains of the receptor analogs
share a backbone structure and yet have sequences that differ by at least 3-5%
when compared to the
corresponding sequence of the naturally occurring human cell surface receptor.
"Backbone" in this
context means the non-side chain atoms: the nitrogen, carbonyl carbon and
oxygen, and the a-
carbon, and the hydrogens attached to the nitrogen and a-carbon. To be
considered a conformer, a
protein must have backbone atoms that are no more than 2 P, from the naturally
occurring human cell
surface receptor structure, with no more than 1.5 A being preferred, and no
more than 1 A being
particularly preferred. In general, these distances may be determined in two
ways. In one
embodiment, each potential conformer is crystallized and its three dimensional
structure determined.
Alternatively, as outlined below, the sequence of each potential conformer is
run in the PDA program
to determine whether it is a conformer.
In a preferred embodiment, the extracellular domain of a receptor analog has
an amino acid sequence
that differs from the sequence of a corresponding naturally occurring receptor
by at least 3% of the
residues. That is, the extracellular domains of receptor analogs of the
invention are less than about
97% identical to the corresponding amino acid sequences of naturally occurring
cell surface receptors.
Accordingly, a receptor analog comprises an extracellular domain that is
preferably less than about
2 0 97%, more preferably less than about 95%, even more preferably less than
about 90% and most
preferably less than 85% identical to the corresponding amino acid sequence of
a naturally occurring
cell surface receptor. In some embodiments the homology will be as low as
about 75 to 80%. For
example, based on the sequence corresponding to the potential extracellular
domain of the human
erythropoietin receptor, comprising amino acids 25-250 (accession number
#P19235), a receptor
2 5 analog has at least about 6-7 residues that differ from the naturally
occurring receptor sequence (3%),
with receptor analogs having from 11 different residues (about 5%) to upwards
of 34 different residues
(about 15%) being preferred. In some instances, the extracellular domains of
receptor analogs have 3
or 4 different residues when compared to the corresponding naturally occurring
human cell surface
receptor sequence. Preferred receptor analogs have 10-24 different residues
with from about 10 to
3 0 about 14 being particularly preferred (that is, 4-7 % of the extracellular
domain is not identical to that of
a naturally occurring human cell surface receptor).
In a preferred embodiment, the receptor analog comprises amino acid
substitutions within the
extracellular domain(s), intracellular domains) and/or transmembrane regions)
when compared to the
corresponding sequence of a naturally occurring cell surface receptor. In this
embodiment one or
9
CA 02362541 2001-08-10
WO 00/47612 PCT/US00/03665
more amino acids within one or more domains of the naturally occurring cell
surface receptor are
substituted by one or more amino acids to generate the receptor analog.
In another preferred embodiment, the receptor analog comprises amino acid
insertions within the
extracellular domain(s), intracellular domains) and/or transmembrane regions)
when compared to the
corresponding sequence of a naturally occurring cell surface receptor. In this
embodiment one or
more amino acids within one or more domains of the naturally occurring cell
surface receptor are
inserted to generate the receptor analog. Insertions usually are on the order
of from about 1 to 20
amino acids, although considerably larger insertions may be tolerated.
In another preferred embodiment, the receptor analog comprises amino acid
deletions within the
extracellular domain(s), intracellular domains) and/or transmembrane regions)
when compared to the
corresponding sequence of a naturally occurring cell surface receptor. In this
embodiment one or
more amino acids within one or more domains of the naturally occurring cell
surface receptor are
deleted to generate the receptor analog. Deletions usually range from about 1
to 20 amino acids,
although in some cases considerably larger deletions may be tolerated.
In a preferred embodiment, the receptor analog comprises a portion of the
extracellular domain of a
naturally occurring cell sun'ace receptor. The term "portion", as used herein,
with regard to a protein
refers to a fragment of that protein. This fragment may range in size from 10
amino acid residues to
the entire amino acid sequence minus one amino acid.
The receptor analogs of the invention are distinguishable from naturally
occurring cell surface
2 0 receptors, however, they exhibit at least one biological function of a
naturally occurring cell surface
receptor. By "biological function" or "biological property" of a receptor
analog or grammatical
equivalents thereof, herein is meant any one of the properties or functions of
a naturally occurring cell
surface receptor, including, but not limited to: (1 ) ability to bind a ligand
(which may be a naturally
occurring ligand or a ligand analog, as further defined below); (2) ability to
be displayed on the surface
of a cell or on the surface of a virus; (3) ability to oligomerize; (4)
ability to signal. In addition,
depending on the bilogical role of the ligand, additional biological functions
may be present, including,
but not limited to (1) upon ligand binding, the ability to stimulate cell
proliferation, particularly of
hematopoietic stem cells; (2) upon ligand binding, the ability to inhibit cell
proliferation, particularly of
cancerous cells; (3) upon ligand binding, the ability to induce apoptosis,
particularly of cancerous cells;
3 0 and (4) upon ligand binding, the ability to treat disease.
Included within the definition of a receptor analog is a hybrid cell surface
receptor analog. By "hybrid
cell surface receptor analog" or "hybrid receptor analog" or grammatical
equivalents herein is meant a
CA 02362541 2001-08-10 pCT~S00/03665
WO 00/47612
receptor analog comprising individual domains derived from more than one
naturally occurring cell
surface receptor.
Thus, in a preferred embodiment, a hybrid receptor analog of the invention
comprises (i) an
extracellular domain similar to that of a first naturally occurring cell
surface receptor, as described
herein, and (ii) an intracellular domains) and/or transmembrane region that
islare similar to a domain
found in a second and/or third naturally occurring cell surface receptor. For
example, a receptor
analog of the instant invention may comprise (i) the extracellular domain of
an erythropoietin receptor,
(ii) the transmembrane region of a thrombopoietin receptor and (iii) the
intracellular domain of a
granulocyte colony stimulating factor receptor. Another example includes a
receptor analog,
comprising (i) the extracellular domain and transmembrane domain of an
erythropoietin receptor and
(ii) the intracellular domain of a granulocyte colony stimulating factor
receptor. Numerous
combinations of these individual domains are within the scope of this
invention.
Also included within the definition of receptor analogs are chimeric single
chain receptor analogs. As
outlined above, type-2 receptors are in a monomeric form, and generally rely
on the binding of a ligand
to the respective monomers to form an active receptor complex that is capable
of signaling. As known
in the art, these activated receptor complexes generally comprise identical
monomeric subunits,
comprising identical extracellular domains, identical transmembrane domains
and identical intracellular
(cytoplasmic) domains. In this embodiment of the invention, the receptor
analog comprises a three
dimensional extracellular structure resembling the three dimensional structure
of an activated receptor
2 0 complex comprising at least two monomers. However, the receptor analog
comprises only one single
amino acid chain with the capability to display the similar active
extracellular domain as a naturally
occurring receptor. The receptor analog, described in this embodiment, is
referred to as "chimeric
single chain receptor analogs. Accordingly, such a chimeric single chain
receptor analog is encoded
by single gene.
2 5 A chimeric single chain receptor analog mimics an active receptor complex.
In a preferred
embodiment, and as further outlined below, such a chimeric single chain
receptor analog is used to
screen for candidate bioactive agents capable of binding to it. In one
preferred embodiment,
screening for a candidate bioactive agent is performed without the addition of
a naturally occurring
ligand or ligand analog.
3 0 Also included within the definition of receptor analogs are chimeric cell
surface receptor complexes.
By "chimeric cell surface receptor complex" or grammatical equivalents, herein
is meant a receptor
complex, comprising at least two receptor analog monomers, wherein each
receptor analog monomer
comprises a different amino acid sequence and wherein each monomer sequence is
derived from the
11
WO 00/47612 CA 02362541 2001-08-10 pCT~S00/03665
same corresponding naturally occurring cell surface receptor. For example, one
receptor analog
monomer is derived from the human EPO receptor and comprises, with respect to
the naturally
occurring sequence, amino acid exchanges in the extracellular domain, e.g.,
within positions 25-120.
The second receptor analog monomer is also derived from the human EPO
receptor, however,
comprises amino acid exchanges within positions 121-250. In the above example,
both receptor
analog monomers are derived from the same naturally occurring cell surface
receptor, comprise
different extracellular domain sequences and identical transmembrane and
intracellular domain
regions. Generally, in this embodiment, at least two receptor analog monomers,
generated as
described herein, comprise a three dimensional extracellular structure
resembling the three
dimensional structure of an activated receptor complex. However, in contrast
to a naturally occurring
activated receptor complex, which comprises at least two identical naturally
occurring receptor
monomers, the chimeric cell surface receptor complex of the invention
comprises at least two different
receptor analog monomers. Each individual receptor analog monomer, herein
designated as
monomer "A" and monomer "B", is designed to comprise an optimized amino acid
sequences, allowing
each monomer to specifically interact with another monomer. Based on the
interaction between
monomer "A" and monomer "B" a three dimensional structure is obtained, which
resembles that of a
corresponding naturally occurring activated cell surface receptor complex. In
some embodiments, for
example for trimeric receptors, such as TNFR, an optional monomer "C" may be
included. By
"optimized amino acid sequence" herein is meant an amino acid sequence that
best fits, for example,
2 0 the mathematical equation of the computational PDA process.
In one aspect of the above embodiment, receptor analog monomers "A" and "B"
are designed that
they do not strongly form oligomers with the same monomer, i.e. monomer "A"
does not strongly
multimerize to form an oligomeric "A" complex. Instead, as a consequence of
the respective amino
acid side chain exchanges, receptor analog monomer "A" preferably oligomerizes
with receptor analog
monomer "B" and vice versa. In another aspect of this embodiment, a receptor
analog monomer is
designed to strongly interact with a naturally occurring cell surface receptor
monomer to form a
chimeric complex, resembling the three dimensional structure of naturally
activated receptor complex.
As such, the receptor analog monomer competes with the naturally occurring
cell surface receptor
dimerization. Accordingly, as will be appreciated by those in the art, in
order to express the chimeric
3 0 cell surface receptor complexes of the invention, usually at least two
genes, one encoding monomer
"A", and the second gene encoding monomer "B", are expressed in a host cell.
Optionally a third
gene, encoding monomer "C" or a naturally occurring cell surface receptor is
also expressed in the
same cell or within the same virus. However, as is known in the art,
expression of polycistronic genes
and/or polycistronic mRNAs is an option to simultaneously express more than
one protein in the same
3 5 cell or within the same virus.
12
CA 02362541 2001-08-10
WO 00/47612 PCT/US00/03665
As described above, the receptor analog of the invention have the capability
to bind a ligand. By
"ligand" herein is meant a molecule capable of binding to a receptor.
Upon binding of a ligand, a receptor may undergo a process called receptor
activation. By "receptor
activation" or grammatical equivalents herein is meant the biological function
associated with ligand
binding to a receptor. As will be appreciated by those in the art, this will
vary widely depending on the
identity of the ligand and receptor. For example, as a result of ligand
binding, cell surface receptors
undergo conformational changes or multimerize into oligomeric receptor
complexes or both. As a
consequence of these events receptors become phosphorylated, or associate with
a cellular protein,
which then results in phosphorylation of either the receptor, the cellular
protein, or yet another
molecule. In this way signaling is accomplished.
In one aspect of the invention, a ligand capable of binding to a receptor
analog, is a naturally occurring
ligand. By "naturally occurring ligand" or "wild type ligand" or grammatical
equivalents, herein is meant
a ligand that is naturally occurring.
Naturally occurring ligands include but are not limited to, those with known
structures (including
variants), including cytokines IL-lra, IL-1, IL-1a, IL-1b, IL-2, IL-3, IL-4,
IL-5, IL-6, IL-8, IL-10, IFN-(3,
INF-y, IFN-a-2a; IFN-a-2B, TNF-a; CD40 ligand (chk), human obesity protein
leptin, GCSF, BMP-7,
CNF, GM-CSF, MCP-1, macrophage migration inhibitory factor, human
glycosylation-inhibiting factor,
human rantes, human macrophage inflammatory protein 1~3, hGH, LIF, human
melanoma growth
stimulatory activity, neutrophil activating peptide-2, CC-chemokine MCP-3,
platelet factor M2,
neutrophil activating peptide 2, eotaxin, stromal cell-derived factor-1,
insulin, IGF-I, IGF-II, TGF-(31,
TGF-X32, TGF-X33, TGF-a, VEGF, acidic-FGF, basic-FGF, EGF, NGF, BDNF (brain
derived
neurotrophic factor), CNF, PDGF, HGF, GCDNF (glial cell-derived neurotrophic
factor), EPO, other
extracellular signaling moieties, including, but not limited to, hedgehog
Sonic, hedgehog Desert,
hedgehog Indian, hCG; coagulation factors including, but not limited to, TPA
and Factor Vlla.
Accession numbers for naturally occurring ligands are readily available from
NCBI, as described
above. For example, amino acid sequences for the human erythropoietin are
available under
NP_000790, AAF23134 and AAF23132. Nucleotide sequences encoding human
erythropoietin are
available under AH009005, AH009003, and M11319. Amino acid sequences for the
human tumor
necrosis factor and human tumor necrosis factor beta (lymphotoxin) are
available under AAD18091,
3 0 and BAA02139, respectively. Nucleotide sequences encoding human tumor
necrosis factor and
human tumor necrosis factor beta (lymphotoxin) are available under AF129756
and D12614,
respectively. Amino acid sequences for the human growth hom~one are available
under NP 000506,
13
CA 02362541 2001-08-10 pCT/US00/03665
WO 00/47612
AAC42099 and CAA00065. Nucleotide sequences encoding human growth hormone are
available
under NM 000515, M36282 and AA00469.
In another embodiment, the ligand of the instant invention is a non-naturally
occurring ligand that is
distinguishable from a naturally occurring ligand. Accordingly, by "non-
naturally occurring ligand" or
"ligand analog" or grammatical equivalents thereof, herein is meant a ligand
that is not naturally
occurring.
In one preferred embodiment, the ligand analogs of the invention define a
conformer set, wherein all of
the domains of the ligand analog share a backbone structure and yet have
sequences that differ by at
least 3% when compared to the corresponding sequence of the naturally
occurring ligands. That is,
the ligand analogs of the invention are less than about 97% identical to the
corresponding amino acid
sequences of naturally occurring ligands. Accordingly, a ligand analog
comprises an amino acid
sequence that is preferably less than about 97%, more preferably less than
about 95%, even more
preferably less than about 90% and most preferably less than 85% identical to
the corresponding
amino acid sequence of a naturally occurring ligand. In some embodiments the
homology will be as
low as about 75 to 80%. For example, a ligand analog, comprising 225 amino has
at least about 6-7
residues that differ from the naturally occurring ligand (3%), with ligand
analogs having from 11
different residues (about 5%) to upwards of 34 different residues (about 15%)
being preferred. In
some instances, the domains of ligand analogs have 3 or 4 different residues
when compared to the
corresponding naturally occurring human ligand sequence. Preferred ligand
analogs have 10-24
2 0 different residues with from about 10 to about 14 being particularly
preferred (that is, 4-7 % of the
amino acid sequence is not identical to that of a naturally occurring human
ligand).
A ligand analog of the invention exhibits at least one biological function of
a naturally occurring ligand.
By "biological function of a ligand analog" or "biological property of a
ligand analog" or grammatical
equivalents thereof, herein is meant any one of the properties or functions of
a naturally occur-ing
2 5 ligand, including, but not limited to: (1 ) ability to bind a naturally
occurring receptor; (2) ability to bind a
receptor analog; (3) ability to be secreted from a cell or a virus; (4)
ability to oligomerize. In addition,
depending on the biological role of the ligand, additional biological
functions may be present, including,
but not limited to (1)the ability to stimulate cell proliferation after
binding to a receptor, particularly of
hematopoietic stem cells; (2) the ability to inhibit cell proliferation after
binding to a receptor,
3 0 particularly of cancerous cells; (3) the ability to induce apoptosis after
binding to a receptor, particularly
of cancerous cells; and (4) the ability to treat disease.
The cell surface receptors and the ligands may be from any number of
organisms, with cell surface
receptors and ligands from mammals being particularly preferred. Suitable
mammals include, but are
14
CA 02362541 2001-08-10 pCT~S00/03665
WO 00/47612
not limited to, rodents (rats, mice, hamsters, guinea pigs, etc.), primates,
farm animals (including
sheep, goats, pigs, cows, horses, etc) and in the most preferred embodiment,
from humans (this is
sometimes referred to herein as naturally occurring human cell surface
receptor). As will be
appreciated by those in the art, receptor analogs based on naturally occurring
cell surface receptors
and ligand analogs based on naturally occurring ligands from mammals other
than humans may find
use in animal models of human disease. As will be further appreciated by those
in the art, a human
cell surface receptor may bind a ligand from mammals other than humans.
The receptor analogs of the invention are proteins. By "protein° herein
is meant at least two covalently
attached amino acids, which includes proteins, polypeptides, oligopeptides and
peptides. The protein
may be made up of naturally occurring amino acids and peptide bonds, or
synthetic peptidomimetic
structures, generally depending on the method of synthesis. Thus "amino acid",
or "peptide residue",
as used herein means both naturally occurring and synthetic amino acids. For
example, homo-
phenylalanine, citrulline and noreleucine are considered amino acids for the
purposes of the invention.
"Amino acid" also includes imino acid residues such as proline and
hydroxyproline. The side chains
may be in either the (R) or the (S) configuration. In the preferred
embodiment, the amino acids are in
the (S) or L-configuration. Stereoisomers of the twenty conventional amino
acids, unnatural amino
acids such as a,a-disubstituted amino acids, N-alkyl amino acids, lactic acid,
and other
unconventional amino acids may also be suitable components for proteins of the
present invention.
Examples of unconventional amino acids include, but are not limited to: 4-
hydroxyproline, y-
2 0 carboxyglutamate, e-N,N,N-trimethyllysine, e-N-acetyllysine, O-
phosphoserine, N-acetylserine, N-
formylmethionine, 3-methylhistidine, 5-hydroxylysine, w-N-methylarginine, and
other similar amino
acids and imino acids. If non-naturally occurring side chains are used, non-
amino acid substituents
may be used, for example to prevent or retard in vivo degradations. Proteins
including non-naturally
occuring amino acids may be synthesized or in some cases, made recombinantly;
see van Hest et al.,
FEBS Lett 428:(1-2) 68-70 May 22 1998 and Tang et al., Abstr. Pap Am. Chem.
S218:U138-U138 Part
2 August 22, 1999, both of which are expressly incorporated by reference
herein.
In a preferred embodiment, the ligand analogs are proteins.
For the remainder of the description of this invention, if not explicitly
otherwise noted, receptor
analogs) and ligand analog(s), are collectively referred to as "analog
protein(s)." It should be further
3 0 noted that unless otherwise stated, all positional numbering within the
sequence of an analog protein
is based on the sequence of the corresponding naturally occurring protein and
in particular, to the
human sequences. That is, as will be appreciated by those in the art, an
alignment of the naturally
occurring protein and the analog protein can be done using standard programs,
as is outlined below,
with the identification of "equivalent" or "homologous" positions between the
two proteins.
CA 02362541 2001-08-10
WO 00/47612 PCT/US00/03665
Homology in this context means sequence similarity or identity, with identity
being preferred. As is
known in the art, a number of different programs can be used to identify
whether a protein (or nucleic
acid as discussed below) has sequence identity or similarity to a known
sequence. Sequence identity
and/or similarity is determined using standard techniques known in the art,
including, but not limited to,
the local sequence identity algorithm of Smith & Waterman, Adv. Appl. Math.,
2:482 (1981), by the
sequence identity alignment algorithm of Needleman & Wunsch, J. Mol. Biol.,
48:443 (1970), by the
search for similarity method of Pearson & Lipman, Proc. Natl. Acad. Sci.
U.S.A., 85:2444 (1988), by
computerized implementations of these algorithms (GAP, BESTFIT, FASTA, and
TFASTA in the
Wisconsin Genetics Software Package, Genetics Computer Group, 575 Science
Drive, Madison, WI),
the Best Fit sequence program described by Devereux et al., Nucl. Acid Res.,
12:387-395 (1984),
preferably using the default settings, or by inspection. Preferably, percent
identity is calculated by
FastDB based upon the following parameters: mismatch penalty of 1; gap penalty
of 1; gap size
penalty of 0.33; and joining penalty of 30, "Current Methods in Sequence
Comparison and Analysis,"
Macromolecule Sequencing and Synthesis, Selected Methods and Applications, pp
127-149 (1988),
Alan R. Liss, Inc.
An example of a useful algorithm is PILEUP. PILEUP creates a multiple sequence
alignment from a
group of related sequences using progressive, pairwise alignments. It can also
plot a tree showing the
clustering relationships used to create the alignment. PILEUP uses a
simplification of the progressive
alignment method of Feng & Doolittle, J. Mol. Evol. 35:351-360 (1987); the
method is similar to that
described by Higgins & Sharp CABIOS 5:151-153 (1989). Useful PILEUP parameters
including a
default gap weight of 3.00, a default gap length weight of 0.10, and weighted
end gaps.
Another example of a useful algorithm is the BLAST algorithm, described in
Altschul et al., J. Mol.
Biol., 215, 403-410, (1990) and Karlin et al., Proc. Natl. Acad. Sci. U.S.A.,
90:5873-5787 (1993). A
particularly useful BLAST program is the WU-BLAST-2 program which was obtained
from Altschul et
al., Methods in Enzymology, 266:460-480 (1996); http://blast.wustlledu/blast/
README.html]. WU-
BLAST-2 uses several search parameters, most of which are set to the default
values. The adjustable
parameters are set with the following values: overlap span =1, overlap
fraction = 0.125, word threshold
(T) = 11. The HSP S and HSP S2 parameters are dynamic values and are
established by the program
itself depending upon the composition of the particular sequence and
composition of the particular
3 0 database against which the sequence of interest is being searched;
however, the values may be
adjusted to increase sensitivity.
An additional useful algorithm is gapped BLAST as reported by Altschul et al.,
Nucl. Acids Res.,
25:3389-3402. Gapped BLAST uses BLOSUM-62 substitution scores; threshold T
parameter set to 9;
the two-hit method to trigger ungapped extensions; charges gap lengths of k a
cost of 10+k; X~ set to
16
WO 00/47612 CA 02362541 2001-08-10 pCT~S00/03665
16, and X9 set to 40 for database search stage and to 67 for the output stage
of the algorithms.
Gapped alignments are triggered by a score corresponding to -22 bits.
A % amino acid sequence identity value is determined by the number of matching
identical residues
divided by the total number of residues of the "longer" sequence in the
aligned region. The "longer"
sequence is the one having the most actual residues in the aligned region
(gaps introduced by WU-
Blast-2 to maximize the alignment score are ignored).
In a similar manner, "percent (%) nucleic acid sequence identity" with respect
to the coding sequence
of the analog proteins identified herein is defined as the percentage of
nucleotide residues in a
candidate sequence that are identical with the nucleotide residues in the
corresponding nucleotide
sequence encoding the naturally occurring protein. A preferred method utilizes
the BLASTN module of
WU-BLAST-2 set to the default parameters, with overlap span and overlap
fraction set to 1 and 0.125,
respectively.
The alignment may include the introduction of gaps in the sequences to be
aligned. In addition, for
sequences encoding analog proteins, which contain either more or fewer amino
acids than the
corresponding naturally occurring proteins, it is understood that in one
embodiment, the percentage of
sequence identity is determined based on the number of identical amino acids
in relation to the total
number of amino acids. Thus, for example, sequence identity of sequences
shorter than the
sequence encoding the naturally occurring protein, is determined using the
number of amino acids in
2 0 the shorter sequence, in one embodiment. In percent identity calculations
relative weight is not
assigned to various manifestations of sequence variation, such as, insertions,
deletions, substitutions,
etc.
In one embodiment, only identities are scored positively (+1) and all forms of
sequence variation
including gaps are assigned a value of ."0", which obviates the need for a
weighted scale or
2 5 parameters as described below for sequence similarity calculations.
Percent sequence identity can be
calculated, for example, by dividing the number of matching identical residues
by the total number of
residues of the "shorter" sequence in the aligned region and multiplying by
100. The "longer"
sequence is the one having the most actual residues in the aligned region.
Thus, analog proteins of the present invention may be shorter or longer than
the amino acid
3 0 sequences of the corresponding naturally occurring cell surface receptors.
Thus, in a preferred
embodiment, included within the definition of analog proteins are portions or
fragments thereof.
Fragments of analog proteins are considered receptor analogs or ligand analogs
if a) they share at
17
CA 02362541 2001-08-10
WO 00/47612 PCT/US00/03665
least one antigenic epitope; b) have at least the indicated homology; c) and
preferably have biological
activity as defined herein.
As is more fully outlined herein, any of the receptor analog variations may be
combined in any way to
form additional novel receptor analogs, novel chimeric cell surface receptor
complexes and novel
hybrid cell surface receptor analogs comprising at least two different
receptor analogs.
In addition, analog proteins can be made that, for example, comprise an
epitope or purification tag, or
other fusion sequences, etc., as outlined below. For example, the analog
proteins of the invention
may be fused to other therapeutic proteins such as IL-11 or to other proteins
such as Fc or serum
albumin for pharmacokinetic purposes. See for example U.S. Patent No.
5,766,883 and 5,876,969,
both of which are expressly incorporated by reference.
Analog proteins may also be identified as being encoded by analog protein (AP)
nucleic acids. In the
case of the nucleic acid, the overall homology of the nucleic acid sequence is
commensurate with
amino acid homology but takes into account the degeneracy in the genetic code
and codon bias of
different organisms. Accordingly, the nucleic acid sequence homology may be
either lower or higher
than that of the protein sequence, with lower homology being preferred.
In a preferred embodiment, an AP nucleic acid encodes an analog protein. As
will be appreciated by
those in the art, due to the degeneracy of the genetic code, an extremely
large number of nucleic
acids may be made, all of which encode the analog proteins of the present
invention. Thus, having
identified a particular amino acid sequence, those skilled in the art could
make any number of different
2 0 nucleic acids, by simply modifying the sequence of one or more codons in a
way which does not
change the amino acid sequence of the analog protein.
In one embodiment, the nucleic acid homology is determined through
hybridization studies. Thus, for
example, nucleic acids which hybridize under high stringency to a nucleic acid
encoding a naturally
occurring protein or its complement and encode an analog protein is considered
an analog protein
gene.
High stringency conditions are known in the art; see for example Sambrook et
al., Molecular Cloning:
A Laboratory Manual, 2d Edition, 1989, and Short Protocols in Molecular
Biology, ed. Ausubel, et al.,
both of which are hereby incorporated by reference. Stringent conditions are
sequence-dependent
and will be different in different circumstances. Longer sequences hybridize
specifically at higher
3 0 temperatures. An extensive guide to the hybridization of nucleic acids is
found in Tijssen, Techniques
in Biochemistry and Molecular Biology--Hybridization with Nucleic Acid Probes,
"Overview of principles
18
WO 00/47612 CA 02362541 2001-08-10 pCT/US00/03665
of hybridization and the strategy of nucleic acid assays" (1993). Generally,
stringent conditions are
selected to be about 5-10°C lower than the thermal melting point (Tm)
for the specific sequence at a
defined ionic strength and pH. The Tm is the temperature (under defined ionic
strength, pH and nucleic
acid concentration) at which 50% of the probes complementary to the target
hybridize to the target
sequence at equilibrium (as the target sequences are present in excess, at Tm,
50% of the probes are
occupied at equilibrium). Stringent conditions, e.g. are those in which the
salt concentration is less
than about 1.0 M sodium ion, typically about 0.01 to 1.0 M sodium ion
concentration (or other salts) at
pH 7.0 to 8.3 and the temperature is at least about 30°C for short
probes (e.g. 10 to 50 nucleotides)
and at least about 60°C for long probes (e.g. greater than 50
nucleotides). Stringent conditions may
also be achieved with the addition of destabilizing agents such as formamide.
In another embodiment, less stringent hybridization conditions are used; for
example, moderate or low
stringency conditions may be used, as are known in the art; see Sambrook,
supra, Ausubel, supra,
and Tijssen, supra.
The analog proteins and nucleic acids of the present invention are
recombinant. As used herein,
"nucleic acid" may refer to either DNA or RNA, or molecules which contain both
deoxy- and
ribonucleotides. The nucleic acids include genomic DNA, cDNA and
oligonucleotides including sense
and anti-sense nucleic acids. Such nucleic acids may also contain
modifications in the ribose-
phosphate backbone to increase stability and half life of such molecules in
physiological environments.
The nucleic acid may be double stranded, single stranded, or contain portions
of both double stranded
2 0 or single stranded sequence. As will be appreciated by those in the art,
the depiction of a single
strand ("Watson") also defines the sequence of the other strand ("Crick");
thus the sequence depicted
in Figure 1 also includes the complement of the sequence. By the term
"recombinant nucleic acid"
herein is meant nucleic acid, originally formed in vitro, in general, by the
manipulation of nucleic acid
by endonucleases, in a form not normally found in nature. Thus an isolated AP
nucleic acid, in a linear
form, or an expression vector formed in vitro by ligating DNA molecules that
are not normally joined,
are both considered recombinant for the purposes of this invention. It is
understood that once a
recombinant nucleic acid is made and reintroduced into a host cell or
organism, it will replicate non-
recombinantly, i.e. using the in vivo cellular machinery of the host cell
rather than in vitro
manipulations; however, such nucleic acids, once produced recombinantly,
although subsequently
3 0 replicated non-recombinantly, are still considered recombinant for the
purposes of the invention.
Similarly, a "recombinant protein" is a protein made using recombinant
techniques, i.e. through the
expression of a recombinant nucleic acid as depicted above. A recombinant
protein is distinguished
from naturally occurring protein by at least one or more characteristics. For
example, the protein may
19
WO 00/47612 CA 02362541 2001-08-10 pCT/US00/03665
be isolated or purified away from some or all of the proteins and compounds
with which it is normally
associated in its wild type host, and thus may be substantially pure. For
example, an isolated protein
is unaccompanied by at least some of the material with which it is normally
associated in its natural
state, preferably constituting at least about 0.5%, more preferably at least
about 5% by weight of the
total protein in a given sample. A substantially pure protein comprises at
least about 75% by weight of
the total protein, with at least about 80% being preferred, and at least about
90% being particularly
preferred. The definition includes the production of an analog protein from
one organism in a different
organism or host cell. Alternatively, the protein may be made at a
significantly higher concentration
than is normally seen, through the use of a inducible promoter or high
expression promoter, such that
the protein is made at increased concentration levels. Furthermore, all of the
analog proteins outlined
herein are in a form not normally found in nature, as they contain amino acid
substitutions, insertions
and deletions, with substitutions being preferred, as discussed below.
Also included within the definition of analog proteins of the present
invention are amino acid sequence
variants of the analog protein sequences outlined herein. That is, an analog
protein may contain
additional variable positions as compared to a starting analog protein. These
variants fall into one or
more of three classes: substitutional, insertional or deletional variants.
These variants ordinarily are
prepared by site specific mutagenesis of nucleotides in the DNA encoding an
analog protein, using
cassette or PCR mutagenesis or other techniques well known in the art, to
produce DNA encoding the
variant, and thereafter expressing the DNA in recombinant cell culture as
outlined above. However,
2 0 variant analog protein fragments having up to about 100-150 residues may
be prepared by in vitro
synthesis using established techniques. Amino acid sequence variants are
characterized by the
predetermined nature of the variation, a feature that sets them apart from
naturally occurring allelic or
interspecies variations.
Directed molecular evolution can be used to create analog proteins with novel
function and properties.
There is a wide variety of methods known for generating and evaluating
sequences. These include,
but are not limited to, sequence profiling (Bowie and Eisenberg, Science
253:164-70, (1991)), rotamer
library selections (Dahiyat and Mayo, Protein Sci 5:895-903 (1996); Dahiyat
and Mayo, Science .
278:82-7 (1997); Desjarlais and Handel, Protein Science 4:2006-2018 (1995);
Harbury et al, Proc.
Natl. Acad. Sci. U.S.A. 92:8408-8412 (1995); Kono et al., Proteins: Structure,
Function and Genetics
3 0 19:244-255 (1994); Hellinga and Richards, Proc. Natl. Acad. Sci. U.S.A.
91:5803-5807 (1994)); and
residue pair potentials (Jones, Protein Science 3:567-574, (1994)).
In a preferred embodiment, the analog proteins, are designed using a method
termed "Protein Design
Automation", or PDA, that utilizes a number of scoring functions to evaluate
sequence stability. PDA
was previously described in W098/47089 and U.S.S.N. 09/127,926, both of which
are expressly
3 5 incorporated by reference in their entirety. PDA is a computational
modeling system that allows the
W~ 00/47612 CA 02362541 2001-08-10 pCT~S00/03665
generation of extremely stable proteins without necessarily disturbing the
biological functions of the
protein itself. In this way, novel receptor analogs and ligand analogs and
their nucleic acids are
generated, that can have a plurality of mutations in comparison to their
naturally occurring
counterparts and yet retain significant activity.
The computational method used to generate and evaluate the analog proteins of
the invention is briefly
described as follows. In a preferred embodiment, the computational method used
to generate the
primary library is Protein Design Automation (PDA), as is described in
U.S.S.N.s 60!061,097,
60/043,464, 60/054,678, 09/127,926 and PCT US98/07254, all of which are
expressly incorporated
herein by reference. Briefly, PDA, which can be applied to any protein, can be
described as follows. A
known protein structure is used as the starting point. The residues to be
optimized are then identified,
which may be the entire sequence or subsets) thereof. The side chains of any
positions to be varied
are then removed. The resulting structure consisting of the protein backbone
and the remaining side
chains is called the template. Each variable residue position is then
preferably classified as a core
residue, a surface residue, or a boundary residue; each classification defines
a subset of possible
amino acid residues for the position (for example, core residues generally are
selected from the set of
hydrophobic residues, surface residues generally are selected from the
hydrophilic residues, and
boundary residues may be either). Each amino acid can be represented by a
discrete set of all
allowed conformers of each side chain, called rotamers. Thus, to arrive at an
optimal sequence for a
backbone, all possible sequences of rotamers must be screened, where each
backbone position can
2 0 be occupied either by each amino acid in all its possible rotameric
states, or a subset of amino acids,
and thus a subset of rotamers.
Two sets of interactions are then calculated for each rotamer at every
position: the interaction of the
rotamer side chain with all or part of the backbone (the
°singles° energy, also called the
rotamer/template or rotamer/backbone energy), and the interaction of the
rotamer side chain with all
other possible rotamers at every other position or a subset of the other
positions (the "doubles"
energy, also called the rotarner/rotamer energy). The energy of each of these
interactions is
calculated through the use of a variety of scoring functions, which include,
but are not limited to, the
energy of van der Waal's forces, the energy of hydrogen bonding, the energy of
secondary structure
propensity, the energy of surface area solvation and the electrostatics. Thus,
the total energy of each
3 0 rotamer interaction, both with the backbone and other rotamers, is
calculated, and stored in a matrix
form.
The discrete nature of rotamer sets allows a simple calculation of the number
of rotamer sequences to
be tested. A backbone of length n with m possible rotamers per position will
have m" possible rotamer
sequences, a number which grows exponentially with sequence length and renders
the calculations
21
CA 02362541 2001-08-10 pCT~S00/03665
WO 00/47612
either unwieldy or impossible in real time. Accordingly, to solve this
combinatorial search problem, a
"Dead End Elimination" (DEE) calculation is performed. The DEE calculation is
based on the fact that
if the worst total interaction of a first rotamer is still better than the
best total interaction of a second
rotamer, then the second rotamer cannot be part of the global optimum
solution. Since the energies of
all rotamers have already been calculated, the DEE approach only requires sums
over the sequence
length to test and eliminate rotamers, which speeds up the calculations
considerably. DEE can be
rerun comparing pairs of rotamers, or combinations of rotamers, which will
eventually result in the
determination of a single sequence which represents the global optimum energy.
Once the global solution has been found, a Monte Carlo search may be done to
generate a rank-
ordered list of sequences in the neighborhood of the DEE solution. Starting at
the DEE solution,
random positions are changed to other rotamers, and the new sequence energy is
calculated. If the
new sequence meets the criteria for acceptance, it is used as a starting point
for another jump. After a
predetermined number of jumps, a rank-ordered list of sequences is generated.
In addition, as will be
appreciated by those in the art, a Monte Carlo search may be done from a DEE
run that is not
completed; that is, a partial DEE run that has a number of sequences may be
used to generate a
Monte Cario list.
As outlined in U.S.S.N. 09/127,926, the protein backbone (comprising (for a
naturally occurring
protein) the nitrogen, the carbonyl carbon, the a-carbon, and the carbonyl
oxygen, along with the
direction of the vector from the a-carbon to the p-carbon) may be altered
prior to the computational
2 0 analysis, by varying a set of parameters called supersecondary structure
parameters.
Once a protein structure backbone is generated (with alterations, as outlined
above) and input into the
computer, explicit hydrogens are added if not included within the structure
(for example, if the structure
was generated by X-ray crystallography, hydrogens must be added). After
hydrogen addition, energy
minimization of the structure is run, to relax the hydrogens as well as the
other atoms, bond angles
and bond lengths. In a preferred embodiment, this is done by doing a number of
steps of conjugate
gradient minimization (Mayo et al., J. Phys. Chem. 94:8897 (1990)) of atomic
coordinate positions to
minimize the Dreiding force field with no electrostatics. Generally from about
10 to about 250 steps is
preferred, with about 50 being most preferred.
The protein backbone structure contains at least one variable residue
position. As is known in the art,
3 0 the residues, or amino acids, of proteins are generally sequentially
numbered starting with the N-
terminus of the protein. Thus a protein having a methionine at it's N-terminus
is said to have a
methionine at residue or amino acid position 1, with the next residues as 2,
3, 4, etc. At each position,
the wild type (i.e. naturally occurring) protein may have one of at least 20
amino acids, in any number
22
CA 02362541 2001-08-10
WO 00/47612 PCT/US00/03665
of rotamers. Each analog protein residue can differ from the naturally
occurring protein at an
equivalent position. This is called a variable residue position. By "variable
residue position" herein is
meant an amino acid position of the protein to be designed that is not fixed
in the design method as a
specific residue or rotamer, generally the wild-type or naturally occurring
protein residue or rotamer.
In a preferred embodiment, all of the residue positions of the protein are
variable. That is, every amino
acid side chain may be altered in the methods of the present invention.
In an alternate preferred embodiment, only some of the residue positions of
the protein are variable,
and the remainder are "fixed", that is, they are identified in the three
dimensional structure as being a
particular amino acid in a set conformation. In some embodiments, a fixed
position is left in its original
conformation (which may or may not correlate to a specific rotamer of the
rotamer library being used).
Alternatively, residues may be fixed as a non-wild type residue; for example,
when known site-directed
mutagenesis techniques have shown that a particular residue is desirable (for
example, to eliminate a
proteolytic site or alter the active site), the residue may be fixed as a
particular amino acid.
Alternatively, the methods of the present invention may be used to evaluate
mutations de novo, as is
discussed below. In an alternate preferred embodiment, a fixed position may be
"floated"; the amino
acid at that position is fixed, but different rotamers of that amino acid are
tested. In this embodiment,
the variable residues may be at least one, or anywhere from 0.1% to 99.9% of
the total number of
residues. Thus, for example, it may be possible to change only a few (or one)
residues, or most of the
residues, with all possibilities in between.
2 0 In a preferred embodiment, residues which can be fixed include, but are
not limited to, structurally or
biologically functional residues. For example, residues which are known to be
important for biological
activity, such as the residues which form the binding site for a binding
partner (ligand/receptor,
antigen/antibody, etc.), phosphorylation or glycosylation sites which are
crucial to biological function,
or structurally important residues, such as disulfide bridges, metal binding
sites, critical hydrogen
2 5 bonding residues, residues critical for backbone conformation such as
proline or glycine, residues
critical for packing interactions, etc. may all be fixed in a conformation or
as a single rotamer, or
"floated".
Similarly, residues which may be chosen as variable residues may be those that
confer undesirable
biological attributes, such as susceptibility to proteolytic degradation,
dimerization or aggregation sites,
3 0 glycosylation sites which may lead to immune responses, unwanted binding
activity, unwanted
allostery, undesirable biological activity but with a preservation of binding,
etc.
23
WO 00/47612 CA 02362541 2001-08-10 pCT/US00/03665
In a preferred embodiment, each variable position is classified as either a
core, surface or boundary
residue position, although in some cases, as explained below, the variable
position may be set to
glycine to minimize backbone strain.
In one embodiment, only core residues are variable residues; alternate
embodiments utilize methods
for designing analog proteins containing core, boundary and surface variable
residues; core and
surface variable residues; core and boundary variable residues; surface and
boundary variable
residues; as well as surface variable residues alone, or boundary variable
residues alone. In general,
preferred embodiments do not utilize surface variable residues, as this can
lead to undesirable
antigenicity; however, in applications that are not related to therapeutic use
of the analog proteins, it
may be desirable to alter surface residues. Any combination of core, surface
and boundary positions
can be utilized.
The classification of residue positions as core, surface or boundary may be
done in several ways, as
will be appreciated by those in the art and outlined in W098I47089, hereby
incorporated by reference
in its entirety. In a preferred embodiment, the classification is done via a
visual scan of the original
protein backbone structure, including the side chains, and assigning a
classification based on a
subjective evaluation of one skilled in the art of protein modeling.
Alternatively, a preferred
embodiment utilizes an assessment of the orientation of the Ca-C(3 vectors
relative to a solvent
accessible surface computed using only the template Ca atoms. In a preferred
embodiment, the
solvent accessible surface for only the Ca atoms of the target fold is
generated using the Connolly
2 0 algorithm with a probe radius ranging from about 4 to about 12A, with from
about 6 to about 10A being
preferred, and 8 A being particularly preferred. The Ca radius used ranges
from about 1.6P, to about
2.3A, with from about 1.8 to about 2.1 P, being preferred, and 1.95 A being
especially preferred. A
residue is classified as a core position if a) the distance for its Ca, along
its Ca-Chi vector, to the
solvent accessible surface is greater than about 4-6 A, with greater than
about 5.0 A being especially
preferred, and b) the distance for its C~3 to the nearest surface point is
greater than about 1.5-3 A, with
greater than about 2.0 A being especially preferred. The remaining residues
are classified as surface
positions if the sum of the distances from their Ca, along their Ca-Cp vector
, to the solvent accessible
surface, plus the distance from their Chi to the closest surface point was
less than about 2.5-4 A, with
less than about 2.7 P, being especially preferred. All remaining residues are
classified as boundary
3 0 positions.
Once each variable position is classified as either core, surface or boundary,
a set of amino acid side
chains, and thus a set of rotamers, is assigned to each position. That is, the
set of possible amino acid
side chains that the program will allow to be considered at any particular
position is chosen.
Subsequently, once the possible amino acid side chains are chosen, the set of
rotamers that is
24
WO 00/47612 CA 02362541 2001-08-10 pCT~S00/03665
evaluated at a particular position can be determined. Thus, a core residue
will generally be selected
from the group of hydrophobic residues consisting of alanine, valine,
isoleucine, leucine,
phenylalanine, tyrosine, tryptophan, and methionine (in some embodiments, when
the a scaling factor
of the van der Waals scoring function, described below, is low, methionine is
removed from the set),
and the rotamer set for each core position potentially includes rotamers for
these eight amino acid side
chains (all the rotamers if a backbone independent library is used, and
subsets if a rotamer dependent
backbone is used). Similarly, surface positions are generally selected from
the group of hydrophilic
residues consisting of alanine, serine, threonine, aspartic acid, asparagine,
glutamine, glutamic acid,
arginine, lysine and histidine. The rotamer set for each surface position thus
includes rotamers for
these ten residues. Finally, boundary positions are generally chosen from
alanine, serine, threonine,
aspartic acid, asparagine, glutamine, glutamic acid, arginine, lysine
histidine, valine, isoleucine,
leucine, phenylalanine, tyrosine, tryptophan, and methionine. The rotamer set
for each boundary
position thus potentially includes every rotamer for these seventeen residues
(assuming cysteine,
glycine and proline are not used, although they can be). Additionally, in some
preferred embodiments,
a set of 18~naturally occurring amino acids (all except cysteine and proline,
which are known to be
particularly disruptive) are used.
Thus, as will be appreciated by those in the art, there is a computational
benefit to classifying the
residue positions, as it decreases the number of calculations. It should also
be noted that there may
be situations where the sets of core, boundary and surface residues are
altered from those described
2 0 above; for example, under some circumstances, one or more amino acids is
either added or
subtracted from the set of allowed amino acids. For example, some proteins
which dimerize or
multimerize, or have ligand binding sites, may contain hydrophobic surface
residues, etc. In addition,
residues that do not allow helix capping" or the favorable interaction with an
a-helix dipole may be
subtracted from a set of allowed residues. This modification of amino acid
groups is done on a residue
by residue basis.
In a preferred embodiment, proline, cysteine and glycine are not included in
the list of possible amino
acid side chains, and thus the rotamers for these side chains are not used.
However, in a preferred
embodiment, when the variable residue position has a ~ angle (that is, the
dihedral angle defined by 1)
the carbonyl carbon of the preceding amino acid; 2) the nitrogen atom of the
current residue; 3) the a-
3 0 carbon of the current residue; and 4) the carbonyl carbon of the current
residue) greater than 0°, the
position is set to glycine to minimize backbone strain.
Once the group of potential rotamers is assigned for each variable residue
position, processing
proceeds as outlined in U.S.S.N. 09/127,926 and PCT US98/07254. This
processing step entails
analyzing interactions of the rotamers with each other and with the protein
backbone to generate
WO 00/47612 CA 02362541 2001-08-10 pCT/US00/03665
optimized protein sequences. Simplistically, the processing initially
comprises the use of a number of
scoring functions to calculate energies of interactions of the rotamers,
either to the backbone itself or
other rotamers. Preferred PDA scoring functions include, but are not limited
to, a Van der Waals
potential scoring function, a hydrogen bond potential scoring function, an
atomic solvation scoring
function, a secondary structure propensity scoring function and an
electrostatic scoring function. As is
further described below, at least one scoring function is used to score each
position, although the
scoring functions may differ depending on the position classification or other
considerations, like
favorable interaction with an a-helix dipole. As outlined below, the total
energy which is used in the
calculations is the sum of the energy of each scoring function used at a
particular position, as is
generally shown in Equation 1:
Equation 1
Etota~ = nEvdw + nEas + nEh_bondin9 + nEss + nEelec
In Equation 1, the total energy is the sum of the energy of the van der Waals
potential (E"dW), the
energy of atomic solvation (Eas), the energy of hydrogen bonding (Eh_bo~ing),
the energy of secondary
structure (E~) and the energy of electrostatic interaction (Ee~e~). The term n
is either 0 or 1, depending
on whether the term is to be considered for the particular residue position.
As outlined in U.S.S.N.s 60/061,097, 60/043,464, 60/054,678, 09/127,926 and
PCT US98107254, any
combination of these scoring functions, either alone or in combination, may be
used. Once the scoring
functions to be used are identified for each variable position, the preferred
first step in the
2 0 computational analysis comprises the determination of the interaction of
each possible rotamer with all
or part of the remainder of the protein. That is, the energy of interaction,
as measured by one or more
of the scoring functions, of each possible rotamer at each variable residue
position with either the
backbone or other rotamers, is calculated. In a preferred embodiment, the
interaction of each rotamer
with the entire remainder of the protein, i.e. both the entire template and
all other rotamers, is done.
However, as outlined above, it is possible to only model a portion of a
protein, for example a domain of
a larger protein, and thus in some cases, not all of the protein need be
considered.
In a preferred embodiment, the first step of the computational processing is
done by calculating two
sets of interactions for each rotamer at every position: the interaction of
the rotamer side chain with the
template or backbone (the "singles" energy), and the interaction of the
rotamer side chain with all other
3 0 possible rotamers at every other position (the "doubles" energy), whether
that position is varied or
floated. It should be understood that the backbone in this case includes both
the atoms of the protein
structure backbone, as well as the atoms of any fixed residues, wherein the
fixed residues are defined
as a particular conformation of an amino acid.
26
CA 02362541 2001-08-10
WO 00/47612 PCT/US00/03665
Thus, "singles" (rotamer/template) energies are calculated for the interaction
of every possible rotamer
at every variable residue position with the backbone, using some or all of the
scoring functions. Thus,
for the hydrogen bonding scoring function, every hydrogen bonding atom of the
rotamer and every
hydrogen bonding atom of the backbone is evaluated, and the EHB is calculated
for each possible
rotamer at every variable position. Similarly, for the van der Waals scoring
function, every atom of the
rotamer is compared to every atom of the template (generally excluding the
backbone atoms of its own
residue), and the E"dW is calculated for each possible rotamer at every
variable residue position. In
addition, generally no van der Waals energy is calculated if the atoms are
connected by three bonds
or less. For the atomic solvation scoring function, the surface of the rotamer
is measured against the
surface of the template, and the Eas for each possible rotamer at every
variable residue position is
calculated. The secondary structure propensity scoring function is also
considered as a singles
energy, and thus the total singles energy may contain an E~ term. As will be
appreciated by those in
the art, many of these energy terms will be close to zero, depending on the
physical distance between
the rotamer and the template position; that is, the farther apart the two
moieties, the lower the energy.
For the calculation of "doubles" energy (rotamer/rotamer), the interaction
energy of each possible
rotamer is compared with every possible rotamer at all other variable residue
positions. Thus,
"doubles" energies are calculated for the interaction of every possible
rotamer at every variable
residue position with every possible rotamer at every other variable residue
position, using some or all
of the scoring functions. Thus, for the hydrogen bonding scoring function,
every hydrogen bonding
2 0 atom of the first rotamer and every hydrogen bonding atom of every
possible second rotamer is
evaluated, and the EHe is calculated for each possible rotamer pair for any
two variable positions.
Similarly, for the van der Waals scoring function, every atom of the first
rotamer is compared to every
atom of every possible second rotamer, and the E~dW is calculated for each
possible rotamer pair at
every two variable residue positions. For the atomic solvation scoring
function, the surface of the first
2 5 rotamer is measured against the surface of every possible second rotamer,
and the Eas for each
possible rotamer pair at every two variable residue positions is calculated.
The secondary structure
propensity scoring function need not be run as a "doubles" energy, as it is
considered as a component
of the "singles" energy. As will be appreciated by those in the art, many of
these double energy terms
will be close to zero, depending on the physical distance between the first
rotamer and the second
3 0 rotamer; that is, the farther apart the two moieties, the lower the
energy.
Once the singles and doubles energies are calculated and stored, the next step
of the computational
processing may occur. As outlined in U.S.S.N. 09/127,926 and PCT US98/07254,
preferred
embodiments utilize a Dead End Elimination (DEE) step, and preferably a Monte
Carlo step.
27
WO 00/47612 CA 02362541 2001-08-10 pCT/US00/03665
The computational processing results in a set or library of optimized protein
sequences. These
optimized protein sequences are generally, but not always, significantly
different from the wild-type
sequence from which the backbone was taken. That is, each optimized protein
sequence preferably
comprises at least about 3-10% variant amino acids from the starting or wild-
type sequence, with at
least about 10-15% being preferred, with at least about 15-20% changes being
more preferred and at
least about 30% changes being particularly preferred.
The cutoff for the optimized protein sequences is then enforced, resulting in
a set of sequences
forming a library of optimized protein sequences. As outlined above, this may
be done in a variety of
ways, including an arbitrary cutoff, an energy limitation, or when a certain
number of residue positions
have been varied. In general, the size of the library will vary with the size
of the protein, the number of
residues that are changing, the computational methods used, the cutoff applied
and the discretion of
the user. In general, it is preferable to have the library be large enough to
randomly sample a
reasonable sequence space to allow for robust screening. Thus, libraries that
range from about 50 to
about 10'3 are preferred, with from about 1000 to about 10' being particularly
preferred, and from
about 1000 to about 100,000 being especially preferred.
In a preferred embodiment, although this is not required, the library
comprises the globally optimal
sequence in its optimal conformation, i.e. the optimum rotamer at each
variable position. That is,
computational processing is run until the simulation program converges on a
single sequence which is
the global optimum. In a preferred embodiment, the library comprises at least
two optimized protein
2 0 sequences. Thus for example, the computational processing step may
eliminate a number of
disfavored combinations but be stopped prior to convergence, providing a
library of sequences of
which the global optimum is one. In addition, further computational analysis,
for example using a
different method, may be run on the library, to further eliminate sequences or
rank them differently.
Alternatively, as is more fully described in U.S.S.N.s 60/061,097, 60/043,464,
60/054,678, 09/127,926
and PCT US98/07254, the global optimum may be reached, and then further
computational
processing may occur, which generates additional optimized sequences in the
neighborhood of the
global optimum.
In addition, in some embodiments, library sequences that did not make the
cutoff are included in the
library. This may be desirable in some situations to evaluate the library
generation method, to serve
3 0 as controls or comparisons, or to sample additional sequence space. For
example, in a preferred
embodiment, the wild-type sequence is included.
The present invention utilizes a variety of methods to generate analog
proteins, in particular receptor
analogs, e.g., analogs with an "activated" conformation. In a preferred
embodiment, the analog
proteins of the invention are designed by Protein design Automation (PDA).
Protein design using PDA
3 5 utilizes a three dimensional structure of the target protein, e.g. a
natural ligand or a natural receptor.
28
WO 00/47612 CA 02362541 2001-08-10 pCT~S00/03665
Evidence from structural and mutagenesis studies both indicate that x-ray
crystal structures of e.g.,
cytokine receptors in complex with their natural cytokine ligands are indeed
the optimal conformations
for the receptor complexes.
Known protein structures can be obtained from the National Center for
Biotechnology Information
(NCBI) at e.g. www ncbi.nlm.nih.gov/structure. The NCBI Structure Group
maintains MMDB, a
database of macromolecular 3D structures, as well as tools for their
visualization and comparative
analysis. MMDB, the Molecular Modeling Data Base, contains experimentally
determined biopolymer
structures obtained from the Protein data Bank (PDB). Thus, e.g., accession
number 1 EER provides
the crystal structure of human erythropoietin complexed to its receptor at 1.9
Angstroms; accession
number 1CN4 provides erythropoietin complexed with the extracellular domains
of the erythropoietin
receptor; and accession numbers 1EBA and 1EBP provide the structures of
complexes between the
extracellular domain of the erythropoietin receptor an inactive peptide or
agonist peptide, respectively.
Several regions can be designed in order to constrain and stabilize, e.g., a
cytokine receptor complex
in its optimal conformation without disrupting ligand binding: (1) the
interface between the two
monomers in the complex (inter-monomer interface); (2) the angle between
different domains within a
receptor monomer such as D1 and D2 (intra-monomer interface); (3) domain D1;
(4) domain D2; (5)
the conserved WSXWS box; and (6) the N-terminal helix (see Figure1). The
conformations of these
regions vary significantly in the presence and absence of a ligand, agonist or
antagonist. Owing to the
character of the different cytokine receptor structures, the PDA strategy
employed is dependent on the
2 0 structure of the respective cytokine receptor.
In one embodiment, protein design is used to constrain the inter-monomer
interface as well as the
intra-monomer interface between domains D1 and D2. This "Type-I Design" is
applicable to coupled
receptors with direct inter-monomer contact of the respective extracellular
domains (ECD), including,
but not limited to those found in the human growth hormone receptor (hGHR) and
the erythropoietin
receptor (EPOR). Also, within this embodiment are coupled receptors that are
constrained only in
their inter-monomer interface and receptors that are constrained only in their
intra-monomer interface
between domains D1 and D2.
In a preferred embodiment the receptor analog is an erythropoietin receptor
(EPOR) analog. Complex
structure between EPO, EPO mimetics and EPOR have been published and
coordinates have been
3 0 released (e.g., 1eer; see above and Figure 1). These structures and the
currently available receptor
dimer structure in complex with EMP1, a peptide agonist, shows that the two
receptor monomers
contact each other across a small inter monomer interface. The contact
residues are two arginine and
one leucine residues (Arg155, L175 and 8178) in 1ebp. In leer, the contact
residues are Asp133 and
29
CA 02362541 2001-08-10
WO 00/47612 PCT/US00/03665
Ser135. There are at least four targeting sites on EPOR for protein design:
(1) the interface between
two receptors, (2) the interface between domain D1 and D2, (3) domain D1, and
(4) domain D2.
These sites are distant from the ligand binding sites. The different design
strategies that follow may
be employed to stabilize the complete EPOR and/or the extracellular domain
(ECD) of EPOR,
comprising residues 1-225. The ECD is the fragment of EPOR solved in x-ray
structure; it binds EPO
with the same affinity as the intact EPOR.
In a preferred embodiment, protein design comprises the inter-monomer
interface between EPO
receptors. At least three contacting residues of each EPOR and nearby
positions are designed to
reconfigure the interface between the two receptors and engineer hydrogen
bonds to fix the orientation
between the receptors. In one aspect of this embodiment, the reconfiguration
of the interface between
receptors is done by introducing a disulfide linkage. In yet another aspect of
this invention,
reconfiguration of the interface between receptors is done using other known
crosslinking agents as
outlined below. The contact residues may vary depending on whether EPOR is
bound by its naturally
occurring ligands, EPO, or by an EPO mimetic. Thus, conformational specific
cross-linking is possible
within different EPOR and EPOR analog complexes.
In one aspect of this embodiment, the receptor analog is designed based on the
structure of EPOR
complexed with an EPO mimetic peptide. Herein the three contact residues are
Arg155, Leu175 and
Arg178 (see 1ebp in Figure 8).
In another aspect of this embodiment, the receptor analog is designed based on
the structure of
EPOR complexed with EPO. Herein the contact residues are Asp133, and Ser135
(see leer in Figure
8).
In another preferred embodiment, protein design comprises the stabilization of
domains D1 and D2,
either alone or in combination. In one aspect of this embodiment, the amino
acid residues chosen for
design include, but are not limited to one or more of the following amino acid
residues of EPOR within
D1: Trp40, Tyr53, Phe55, Tyr57, Leu69, Va179, Phe81, Leu85, Leu96, Leu98,
Va1100, and Tyr109 or
within D2: Leu127, A1a129, Va1138, Leu140, Trp142, Tyr156, Va1158, Va1160,
I1e174, Leu183, Tyr192,
Phe194, Va1196, A1a198, GIy207, and Leu218 (see Figure 8). Positions are
chosen that should either
not contact the residues involved in ligand-receptor interaction or should not
significantly reduce the
ligand-receptor interaction.
3 0 !n one aspect of this embodiment, protein design comprises stabilization
of domain D1 alone.
Positions are chosen that should either not contact the residues involved in
ligand-receptor interaction
or should not significantly reduce the ligand-receptor interaction. In one
aspect of this embodiment,
WO 00/47612 CA 02362541 2001-08-10 pCT/US00/03665
the contact residues include, but are not limited to one or more of the
following amino acid residues of
EPOR: Trp40, Tyr53, Phe55, Tyr57, Leu69, Va179, Phe81, Leu85, Leu96, Leu98,
Va1100, and Tyr109
(see Figure 8).
In another aspect of this embodiment, protein design comprises stabilization
of domain D2 alone.
Positions are chosen that should either not contact the residues involved in
ligand-receptor interaction
or should not significantly reduce the ligand-receptor interaction. In one
aspect of this embodiment,
the contact residues include, but are not limited to one or more of the
following amino acid residues of
EPOR: Leu127, A1a129, Va1138, Leu140, Trp142, Tyr156, Va1158, Va1160, I1e174,
Leu183, Tyr192,
Phe194, Va1196, A1a198, GIy207, and Leu218 (see Figure 8).
In another preferred embodiment, protein design comprises the conserved WSXWS
box. Positions
are chosen that should either not contact the residues involved in ligand-
receptor interaction or should
not significantly reduce the ligand-receptor interaction. In one aspect of
this embodiment, the contact
residues include one or more of the following amino acid residues of EPOR:
Trp209, Sei210, A1a211,
Trp 212, and Ser213.
In another preferred embodiment, protein design comprises the stabilization of
the N-terminal helix.
Positions are chosen that should either not contact the residues involved in
ligand-receptor interaction
or should not significantly reduce the ligand-receptor interaction. In one
aspect of this embodiment,
the contact residues include, but are not limited to one or more of the
following amino acid residues of
EPOR: Phe11, A1a15, Leu17, Leu18, A1a19, Phe29, Va137, Phe39 (see Figure 8).
In a preferred embodiment, positions designed comprising (1) the inter-monomer
interface between
EPO receptors; (2) the interface between domain D1 and D2, (3) domain D1; (5)
domain D2; (6) the
WSXWS box and (4) the N-terminal helix are combined to give a combined protein
design for both
interfaces. Any combination of designed positions, either individually or in
groups is within the scope
of the invention.
In another preferred embodiment, disulfide bonds are designed to link the two
receptor monomers at
inter-monomer contact sites. In one aspect of this embodiment the two
receptors are linked at
distances < 5A. In another aspect of this embodiment, the linkage occurs
between dimerization
motifs, such as coiled coil, fused to the receptor analog. Suitable amino acid
residues for linkage are
Arg155, Leu175 and Arg178 (see 1ebp complex; Figure 8) and Asp133 and Ser135
(see leer
3 0 complex; Figure 8).
31
W~ 00/47612 CA 02362541 2001-08-10 pCT~S00/03665
In a preferred embodiment, a dimeric coiled-coil [designed by e.g., PDA to
increase stability and
specificity, e.g., see Dahiyat et al., Protein Science 6:1333-7 (1997)] is
linked to the ECD of EPOR to
assist dimer assembly; the linkage can be designed so that the angular
register of the EPOR dimer
favors the optimal conformation and the linker length and composition can be
designed to complement
the receptor structure. Coiled-coil motifs may be added to other receptor
analogs and ligand analogs
of the invention.
In a preferred embodiment, the coiled coil motif comprises, but is not limited
to one of the following
sequences: RMEKLEOKVKELLRKNERLEEEVERLKQLVGER, based on the structure of GCN4;
AALESEVSALESEVASLESEVAAL, and LAAVKSKLSAVKSKLASVKSKLAA, coiled-coil leucine
zipper
regions defined previously (see Martin et al., EMBO J. 13(22):5303-5309
(1994), incorporated by
reference). Other coiled coil sequences from e.g. leucine zipper containing
proteins are known in the
art and are used in this invention. See, for example, Myszka et al., Biochem.
33:2362-2373 (1994),
hereby incorporated by reference).
In another preferred embodiment, the analog receptor includes a linker. For
example, the coiled coil
motif may be fused to a receptor analog via a linker. By "linker", "linker
sequence", "spacer", tethering
sequence" or grammatical equivalents thereof, herein is meant a molecule or
group of molecules
(such as a monomer or polymer) that connects two molecules and often serves to
place the two
molecules in a preferred configuration, e.g., so that a ligand can bind to a
receptor with minimal steric
hindrance. In one aspect of this embodiment, the linker is a peptide. Useful
linkers include glycine-
2 0 serine polymers (including, for example, (GS)~, (GSGGS)~ (GGGGS)~ and
(GGGS)~, where n is an
integer of at least one), glycine-alanine polymers, alanine-serine polymers,
and other flexible linkers
such as the tether for the shaker potassium channel, and a large variety of
other flexible linkers, as will
be appreciated by those in the art. Glycine-serine polymers are preferred
since both of these amino
acids are relatively unstructured, and therefore may be able to serve as a
neutral tether between
2 5 components. Secondly, serine is hydrophilic and therefore able to
solubilize what could be a globular
glycine chain. Third, similar chains have been shown to be effective in
joining subunits of recombinant
proteins such as single chain antibodies.
In another preferred embodiment, the analog receptor is a human growth hormone
receptor (hGHR)
analog. hGHR has a large interface buried between two receptors upon
dimerization (Figure 2). The
3 0 interface is formed by the same residues of each receptor determined by
the approximate 2-fold
symmetry of the receptor in the complex. In this embodiment, the interface
between these two
receptors is designed generating a single sequence for the designed receptor
analog. Further
included within this embodiment are hGHR analogs comprising designed intra-
monomer sites to
stabilize the conformation of each monomer. The contact residues between
ligand and receptors are
32
CA 02362541 2001-08-10
WO 00/47612 PCT/US00/03665
far away from the inter-monomer contact sites between the two receptors and as
such, binding of the
ligand is not compromised.
In another preferred embodiment, the receptor analog is a tumor necrosis
factor receptor (TNFR)
analog. For TNFR, there is little or no interference between receptors in the
trimer structure (Figure
4).
In uncoupled receptors that do not contact each other in the x-ray structure,
including, but not limited
to TNFR, the interface between D1 and D2 domains is stabilized, constraining
each monomer in its
active conformation. Thus, in this preferred embodiment, the interface between
D1 and D2 is
designed, e.g., by PDA to rigidify the orientation between them. Positions are
chosen that either do
not contact the residues involved in ligand-receptor interaction or do not
have a negative effect on
ligand-receptor interaction.
In a preferred embodiment, protein design is used to enhance the stability and
specificity of protein
oligomerization motifs, in particular, dimeric/trimeric coiled-coil proteins.
These oligomerization motifs
are used to assemble the receptor monomers into the functional active
oligomerization state by fusing
e.g. the PDA-designed motif to the receptor. A major goal is to stabilize the
coiled-coil trimeric motif
to maximize the oligomerization of TNFR and to prevent incorrect
oligomerization, such as dimers and
tetramers. Protein design such as PDA can be used for this purpose because a
trimeric coiled-coil x-
ray structure is available. Although this "Type-II Design" approach is not as
direct as the "Type-I
Design" approach, the entropically constrained receptor complex is still far
better suited for methods of
2 0 screening for ligand analogs and bioactive agents.
In a preferred embodiment, this "Type-II Design° approach is used to
introduce a coiled-coil
trimerization domain fused to the TNFR. In one aspect of this embodiment, the
coiled-coil
trimerization domain is fused to the carboxy terminus of TNFR (see Figure 7).
In another preferred embodiment, the coiled-coil (designed e.g., by PDA with
the increased stability
2 5 and specificity) is fused to the extracellular fragment of TNFR to assist
the assembly of TNFR trimer;
the linkage should be registered to favor the TNFR trimer in an optimal
conformation. Positions are
chosen that either do not contact the residues involved in ligand-receptor
interaction or do not have a
negative effect on ligand-receptor interaction.
In a further aspect of this embodiment, a linker sequence between receptor
monomer and attachment
3 0 point of the coiled coil is designed to control the relative spacing and
orientation between them. The
33
CA 02362541 2001-08-10
WO 00/47612 PCT/US00/03665
orientation and position of the linker segment between the oligomerization
domain and the receptor
are optimized to favor the desired receptor orientation (Figure 6 and Figure
7).
In another preferred embodiment, the receptor analog is a tumor necrosis
factor receptor II (TNFR-II)
receptor analog. In this embodiment, exemplified by TNFR-II, when no x-ray
structure information for
the respective receptor/ligand complex is available, the present invention
provides a 'Type-III Design"
approach, wherein homology modeling performed by e.g., PDA in combination with
oligomerization-
assisted receptor assembly is used to design functional receptor complexes.
This approach can be
applied to design other cytokine receptors that share sequence homology with
existing cytokine
receptor x-ray structures.
In a preferred embodiment, modeling of the TNFR-II receptor (P75 kD) sequence
is performed onto
the TNFR-I receptor (P55 kD) structure. TNFR-II receptor (P75 kD) belongs to
the same class of
receptor family as TNFR-I receptor (P55 kD).
In a preferred embodiment, this TNFR-II receptor model structure is used to
guide mutagenesis
experiments to identify ligand binding sites on the TNFR-II receptor.
In one aspect of this embodiment, modeling of the GCSF receptor sequence is
performed onto the
hGHR structure. GCSF receptor belongs to the same class of receptor family as
hGHR. The resulting
model structure is strikingly similar to the NMR structure of a GCSF receptor
fragment containing
WSXWS motif (Figure 3).
In another embodiment, the x-ray structure obtained for a natural ligand, a
natural receptor or a natural
2 0 receptor complexed with its natural ligand from one species is used to
design the corresponding
human analogs. In one aspect of this embodiment, protein design, such as PDA
has been used
successfully to design human GCSF analogs based on a structural model of the
bovine GCSF x-ray
structure.
Except for residues involved in ligand binding, most residues on the receptor
do not interact directly
2 5 with the ligand. However, most receptor residues are structurally
important for scaffolding the
fibronectin type III (Fn3) domain that presents the binding epitopes. Thus, in
another embodiment,
protein design, such as PDA is used to redesign amino acid residues to
stabilize the Fn3 scaffold as
well as the relative orientation between the D1 and D2 domains.
34
CA 02362541 2001-08-10
WO 00/47612 PCT/US00/03665
The computational processing results in a set of optimized analog proteins.
These optimized analog
protein sequences are generally significantly different from the wild-type
naturally occurring cell
surface receptor sequence from which the backbone was taken.
Structurally defined analog proteins, in particular the receptor analogs,
designed by e.g. PDA, are
experimentally tested and validated in in vivo and in in vitro assays, as
described further below, for
e.g., by examining binding affinity to natural ligands and to high affinity
agonists and/or antagonists. In
addition to cell-free biochemical affinity tests, quantitative comparison are
made comparing kinetic and
equilibrium binding constants for the natural ligand to the naturally
occurring receptor and to the
receptor analogs. The kinetic association rate (Ko~) and dissociation rate
(Koff), and the equilibrium
binding constants (Kd) can be determined using surface plasmon resonance on a
BIAcore instrument
following the standard procedure in the literature (Pearce et al.,
Biochemistry 38:81-89 (1999);
incorporated by reference). For most receptors described herein, the binding
constant between a
natural ligand and its corresponding naturally occurring receptor is well
documented in the literature.
Comparisons with the corresponding naturally occurring receptors are made in
order to evaluate the
sensitivity and specificity of the receptor analogs. In particular, binding
affinity to natural ligands and
agonists is expected to increase relative to the naturally occurring receptor,
while antagonist affinity
should decrease. Receptor analogs with higher affinity to antagonists relative
to the non naturally
occurring receptors may also be designed by e.g. PDA.
The analog proteins and AP nucleic acids of the invention can be made in a
number of ways. As will
2 0 be appreciated by those in the art, it is possible to synthesize proteins
using standard techniques well
known in the art. See for example Wilken et al., Curr. Opin. Biotechnol. 9:412-
26 (1998), hereby
expressly incorporated by reference.
Alternatively, and preferably, the proteins and nucleic acids of the invention
are made using
recombinant techniques. In a preferred embodiment, when combinations of
variable positions are to
be made, the nucleic acids encoding the analog proteins are made using a
variety of combinatorial
techniques. For example, "shuffling" techniques such as are outlined in U.S.
Patent Nos. 5,811,238;
5,605,721 and 5,830,721, and related patents, all of which are hereby
expressly incorporated by
reference.
In a preferred embodiment, multiple PCR reactions with pooled oligonucleotides
is done. In this
3 0 embodiment, overlapping oligonucleotides are synthesized which correspond
to the full length gene.
Again, these oligonucleotides may represent all of the different amino acids
at each variant position or
subsets.
CA 02362541 2001-08-10
WO 00/47612 PCT/US00/03665
In a preferred embodiment, these oligonucleotides are pooled in equal
proportions and multiple PCR
reactions are performed to create full length sequences containing the
combinations of variable
positions.
In a preferred embodiment, the different oligonucleotides are added in
relative amounts corresponding
to a probability distribution table; that is, different amino acids have
different probabilistic chances of
being at a particular position. Thus, for example, if out of 1000 sequences, a
given amino acid position
has valine 35% of the time, leucine 26% of the time, and isoleucine 31 % of
the time, the multiple PCR
reactions will result in full length sequences with the desired combinations
of variable amino acids in
the desired proportions.
The total number of oligonucleotides needed is a function of the number of
positions being mutated
and the number of mutations being considered at these positions:
(number of oligos for constant positions) + M1 + M2 + M3 +... Mn = (total
number of oligos required)
where Mn is the number of amino acids considered at position n in the
sequence.
In a preferred embodiment, each overlapping oligonucleotide comprises only one
position to be varied;
in alternate embodiments, the variant positions are too close together to
allow this and multiple
variants per oligonucleotide are used to allow complete recombination of all
the possibilities. That is,
each oligo can contain the codon for a single position being varied, or for
more than one position being
varied. The multiple positions being varied must be close in sequence to
prevent the oligo length from
2 0 being impractical. For multiple variable positions on an oligonucleotide,
particular combinations of
variable residues can be included or excluded in the library by including or
excluding the
oligonucleotide encoding that combination. The total number of
oligonucleotides required increases
when multiple variable positions are encoded by a single oligonucleotide. The
annealed regions are
the ones that remain constant, i.e. have the sequence of the reference
sequence.
Oligonucleotides with insertions or deletions of codons can be used to create
a library expressing
different length proteins. In particular computational sequence screening for
insertions or deletions
can result in secondary libraries defining different length proteins, which
can be expressed by a library
of pooled oligonucleotide of different lengths.
In a preferred embodiment, error-prone PCR is done. See U.S. Patent Nos.
5,605,793, 5,811,238,
and 5,830,721, all of which are hereby incorporated by reference. This can be
done on the optimal
sequence or on top members of the analog protein set. In this embodiment, the
gene for the optimal
36
W~ 00/47612 CA 02362541 2001-08-10
PCT/US00/03665
analog protein sequence found in the computational screen can be synthesized.
Error prone PCR is
then performed on the optimal sequence gene in the presence of
oligonucleotides that code for the
variable residues at the variant positions (bias oligonucleotides). The
addition of the oligonucleotides
will create a bias favoring the incorporation of the variations in the
secondary library. Alternatively,
only oligonucleotides for certain variations may be used to bias the library.
In a preferred embodiment, gene shuffling with error prone PCR can be
performed on the gene for the
optimal sequence, in the presence of bias oligonucleotides, to create a DNA
sequence library that
reflects the proportion of the variations. The choice of the bias
oligonucleotides can be done in a
variety of ways; they can chosen on the basis of their frequency, i.e.
oligonucleotides encoding high
variation frequency positions can be used; alternatively, oligonucleotides
containing the most variable
positions can be used, such that the diversity is increased; if the analog
protein set is ranked, some
number of top scoring positions can be used to generate bias oligonucleotides;
random positions may
be chosen; a few top scoring and a few low scoring ones may be chosen; etc.
What is important is to
generate new sequences based on preferred variable positions and sequences.
Similarly, a top set of
analog proteins may be "shuffled" using traditional shuffling methods or
overlapping oligonucleotide
methods.
Using the nucleic acids of the present invention which encode an analog
protein, a variety of
expression vectors are made. The expression vectors may be either self-
replicating
extrachromosomal vectors or vectors which integrate into a host genome.
Generally, these
2 0 expression vectors include transcriptional and translational regulatory
nucleic acid operably linked to
the nucleic acid encoding the analog protein. The term "control sequences"
refers to DNA sequences
necessary for the expression of an operably linked coding sequence in a
particular host organism.
The control sequences that are suitable for prokaryotes, for example, include
a promoter, optionally an
operator sequence, and a ribosome binding site. Eukaryotic cells are known to
utilize promoters,
polyadenylation signals, and enhancers.
Nucleic acid is "operably linked" when it is placed into a functional
relationship with another nucleic
acid sequence. For example, DNA for a presequence or secretory leader is
operably linked to DNA
for a polypeptide if it is expressed as a preprotein that participates in the
secretion of the polypeptide;
3 0 a promoter or enhancer is operably linked to a coding sequence if it
affects the transcription of the
sequence; or a ribosome binding site is operably linked to a coding sequence
if it is positioned so as to
facilitate translation.
In a preferred embodiment, when the endogenous secretory sequence leads to a
low level of secretion
of the naturally occurring protein, a replacement of the naturally occurring
secretory leader sequence
37
CA 02362541 2001-08-10
WO 00/47612 PCT/US00/03665
is desired. In this embodiment, an unrelated secretory leader sequence is
operably linked to an AP
encoding nucleic acid leading to increased protein secretion. Thus, any
secretory leader sequence
resulting in enhanced secretion of the analog protein, when compared to the
secretion of the naturally
occurring protein and its secretory sequence, is desired. Suitable secretory
leader sequences that
lead to the secretion of a protein are know in the art.
In another preferred embodiment, a secretory leader sequence of a naturally
occurring protein or a
analog protein is removed by techniques known in the art and subsequent
expression results in
intracellular accumulation of the recombinant protein.
Generally, "operably linked" means that the DNA sequences being linked are
contiguous, and, in the
case of a secretory leader, contiguous and in reading phase. However,
enhancers do not have to be
contiguous. Linking is accomplished by ligation at convenient restriction
sites. If such sites do not
exist, the synthetic oligonucleotide adaptors or linkers are used in
accordance with conventional
practice. The transcriptional and translational regulatory nucleic acid will
generally be appropriate to
the host cell used to express the fusion protein; for example, transcriptional
and translational
regulatory nucleic acid sequences from Bacillus are preferably used to express
the fusion protein in
Bacillus. Numerous types of appropriate expression vectors, and suitable
regulatory sequences are
known in the art for a variety of host cells.
In general, the transcriptional and translational regulatory sequences may
include, but are not limited
to, promoter sequences, ribosomal binding sites, transcriptional start and
stop sequences,
2 0 translational start and stop sequences, and enhancer or activator
sequences. In a preferred
embodiment, the regulatory sequences include a promoter and transcriptional
start and stop
sequences.
Promoter sequences encode either constitutive or inducible promoters. The
promoters may be either
naturally occurring promoters or hybrid promoters. Hybrid promoters, which
combine elements of
more than one promoter, are also known in the art, and are useful in the
present invention. In a
preferred embodiment, the promoters are strong promoters, allowing high
expression in cells,
particularly mammalian cells, such as the STAT or CMV promoter, particularly
in combination with a
Tet regulatory element.
In addition, the expression vector may comprise additional elements. For
example, the expression
3 0 vector may have two replication systems, thus allowing it to be maintained
in two organisms, for
example in mammalian or insect cells for expression and in a procaryotic host
for cloning and
amplification. Furthermore, for integrating expression vectors, the expression
vector contains at least
38
CA 02362541 2001-08-10
WO 00/47612 PCT/US00/03665
one sequence homologous to the host cell genome, and preferably two homologous
sequences which
flank the expression construct. The integrating vector may be directed to a
specific locus in the host
cell by selecting the appropriate homologous sequence for inclusion in the
vector. Constructs for
integrating vectors are well known in the art.
In addition, in a preferred embodiment, the expression vector contains a
selectable marker gene to
allow the selection of transformed host cells. Selection genes are well known
in the art and will vary
with the host cell used.
A preferred expression vector system is a retroviral vector system such as is
generally described in
PCT/US97101019 and PCT/US97I01048, both of which are hereby expressly
incorporated by
reference.
In a preferred embodiment, components of an expression vector, such as the
genes encoding the
receptor analog, the ligand analog, or libraries of candidate ligands reside
on one vector. Alternatively,
in another embodiment, particularly when e.g., multiple and/or different
monomers of a receptor
analog are expressed within the same cell (as described herein), the genes
encoding these
monomers, the ligand analogs, or libraries of candidate ligands may reside on
more than one
expression vector. As will be appreciated by those in the art, all
combinations are possible and
accordingly, as used herein, this combination of components, contained within
one or more vectors,
which may be retroviral or not, is referred to herein as a "vector
composition".
The AP nucleic acids are introduced into the cells, either alone or in
combination with an expression
2 0 vector. By "introduced into " or grammatical equivalents herein is meant
that the nucleic acids enter
the cells in a manner suitable for subsequent expression of the nucleic acid.
The method of
introduction is largely dictated by the targeted cell type, discussed below.
Exemplary methods include
CaP04 precipitation, liposome fusion, lipofectin~, electroporation, viral
infection, etc. The AP nucleic
acids may stably integrate into the genome of the host cell (for example, with
retroviral introduction,
2 5 outlined below), or may exist either transiently or stably in the
cytoplasm (i.e. through the use of
traditional plasmids, utilizing standard regulatory sequences, selection
markers, etc.).
The analog proteins of the present invention are produced by culturing a host
cell transformed either
with an expression vector containing nucleic acid encoding an analog protein
or with the nucleic acid
alone, under the appropriate conditions to induce or cause expression of the
analog protein. The
3 0 conditions appropriate for analog protein expression will vary with the
choice of the expression vector
and the host cell, and will be easily ascertained by one skilled in the art
through routine
experimentation. For example, the use of constitutive promoters in the
expression vector will require
39
CA 02362541 2001-08-10
WO 00/47612 PCT/US00/03665
optimizing the growth and proliferation of the host cell, while the use of an
inducible promoter requires
the appropriate growth conditions for induction. In addition, in some
embodiments, the timing of the
harvest is important. For example, the baculovirus used in insect cell
expression systems is a lytic
virus, and thus harvest time selection can be crucial for product yield.
Appropriate host cells include yeast, bacteria, archaebacteria, fungi, and
insect and animal cells,
including mammalian cells. Of particular interest are Drosophila melangaster
cells, Saccharomyces
cerevisiae and other yeasts, E. coli, Bacillus subtilis, SF9 cells, C129
cells, 293 cells, Neurospora,
BHK, CHO, COS, Pichia Pastoris, etc.
In a preferred embodiment, the analog proteins are expressed in mammalian
cells. Mammalian
expression systems are also known in the art, and include retroviral systems.
A mammalian promoter
is any DNA sequence capable of binding mammalian RNA polymerase and initiating
the downstream
(3') transcription of a coding sequence for the fusion protein into mRNA. A
promoter will have a
transcription initiating region, which is usually placed proximal to the 5'
end of the coding sequence,
and a TATA box, using a located 25-30 base pairs upstream of the transcription
initiation site. The
TATA box is thought to direct RNA polymerase II to begin RNA synthesis at the
correct site. A
mammalian promoter will also contain an upstream promoter element (enhancer
element), typically
located within 100 to 200 base pairs upstream of the TATA box. An upstream
promoter element
determines the rate at which transcription is initiated and can act in either
orientation. Of particular use
as mammalian promoters are the promoters from mammalian viral genes, since the
viral genes are
2 0 often highly expressed and have a broad host range. Examples include the
SV40 early promoter,
mouse mammary tumor virus LTR promoter, adenovirus major late promoter, herpes
simplex virus
promoter, and the CMV promoter.
Typically, transcription termination and polyadenylation sequences recognized
by mammalian cells are
regulatory regions located 3' to the translation stop codon and thus, together
with the promoter
elements, flank the coding sequence. The 3' terminus of the mature mRNA is
formed by site-specific
post-translational cleavage and polyadenylation. Examples of transcription
terminator and
polyadenlytion signals include those derived form SV40.
The methods of introducing exogenous nucleic acid into mammalian hosts, as
well as other hosts, is
well known in the art, and will vary with the host cell used. Techniques
include dextran-mediated
3 0 transfection, calcium phosphate precipitation, polybrene mediated
transfection, protoplast fusion,
electroporation, viral infection, encapsulation of the polynucleotide(s) in
liposomes, and direct
microinjection of the DNA into nuclei. As outlined herein, a particularly
preferred method utilizes
retroviral infection, as outlined in PCT US97/01019, incorporated by
reference.
CA 02362541 2001-08-10
WO 00/47612 PCT/US00/03665
As will be appreciated by those in the art, the type of mammalian cells used
in the present invention
can vary widely. Basically, any mammalian cells may be used, with mouse, rat,
primate and human
cells being particularly preferred, although as will be appreciated by those
in the art, modifications of
the system by pseudotyping allows all eukaryotic cells to be used, preferably
higher eukaryotes. As is
more fully described below, a screen can be set up such that the cells exhibit
a selectable phenotype
in the presence of a bioactive peptide. As is more fully described below, cell
types implicated in a wide
variety of disease conditions are particularly useful, so long as a suitable
screen may be designed to
allow the selection of cells that exhibit an altered phenotype as a
consequence of the presence of a
peptide within the cell.
Accordingly, suitable cell types include, but are not limited to, tumor cells
of all types (particularly
melanoma, myeloid leukemia, carcinomas of the lung, breast, ovaries, colon,
kidney, prostate,
pancreas and testes), cardiomyocytes, endothelial cells, epithelial cells,
lymphocytes (T-cell and B
cell) , mast cells, eosinophils, vascular intimal cells, hepatocytes,
leukocytes including mononuclear
leukocytes, stem cells such as haemopoetic, neural, skin, lung, kidney, liver
and myocyte stem cells
(for use in screening for differentiation and de-differentiation factors),
osteoclasts, chondrocytes and
other connective tissue cells, keratinocytes, melanocytes, liver cells, kidney
cells, and adipocytes.
Suitable cells also include known research cells, including, but not limited
to, Jurkat T cells, NIH3T3
cells, CHO, Cos, etc. See the ATCC cell line catalog, hereby expressly
incorporated by reference.
In one embodiment, the cells may be additionally genetically engineered, that
is, contain exogenous
2 0 nucleic acid other than the AP nucleic acid.
In a preferred embodiment, the analog proteins are expressed in bacterial
systems. Bacterial
expression systems are well known in the art.
A suitable bacterial promoter is any nucleic acid sequence capable of binding
bacterial RNA
polymerase and initiating the downstream (3') transcription of the coding
sequence of the analog
2 5 protein into mRNA. A bacterial promoter has a transcription initiation
region which is usually placed
proximal to the 5' end of the coding sequence. This transcription initiation
region typically includes an
RNA polymerase binding site and a transcription initiation site. Sequences
encoding metabolic
pathway enzymes provide particularly useful promoter sequences. Examples
include promoter
sequences derived from sugar metabolizing enzymes, such as galactose, lactose
and maltose, and
3 0 sequences derived from biosynthetic enzymes such as tryptophan. Promoters
from bacteriophage
may also be used and are known in the art. In addition, synthetic promoters
and hybrid promoters are
also useful; for example, the tac promoter is a hybrid of the trp and lac
promoter sequences.
41
CA 02362541 2001-08-10
WO 00/47612 PCT/US00/03665
Furthermore, a bacterial promoter can include naturally occurring promoters of
non-bacterial origin that
have the ability to bind bacterial RNA polymerase and initiate transcription.
In addition to a functioning promoter sequence, an efficient ribosome binding
site is desirable. In E.
coli, the ribosome binding site is called the Shine-Delgarno (SD) sequence and
includes an initiation
codon and a sequence 3-9 nucleotides in length located 3 -~ 11 nucleotides
upstream of the initiation
codon.
The expression vector may also include a signal peptide sequence that provides
for secretion of the
analog protein in bacteria. The signal sequence typically encodes a signal
peptide comprised of
hydrophobic amino acids which direct the secretion of the protein from the
cell, as is well known in the
art. The protein is either secreted into the growth media (gram-positive
bacteria) or into the
periplasmic space, located between the inner and outer membrane of the cell
(gram-negative
bacteria). For expression in bacteria, usually bacterial secretory leader
sequences, operably linked to
the PA nucleic acid, are preferred.
In a preferred embodiment, the analog proteins of the invention are expressed
in bacteria and
displayed on the bacterial surface. Suitable bacterial expression and display
systems are known in
the art (Stahl and Uhlen, Trends Biotechnol. 15:185-92 (1997); Georgiou et
al., Nat. Biotechnol. 15:29-
34 (1997); Lu et al., Biotechnology 13:366-72 (1995); Jung et al., Nat.
Biotechnol. 16:576-80 (1998);
all of which are expressly incorporated by reference).
2 0 The bacterial expression vector may also include a selectable marker gene
to allow for the selection of
bacterial strains that have been transformed. Suitable selection genes include
genes which render the
bacteria resistant to drugs such as ampicillin, chloramphenicol, erythromycin,
kanamycin, neomycin
and tetracycline. Selectable markers also include biosynthetic genes, such as
those in the histidine,
tryptophan and leucine biosynthetic pathways.
These components are assembled into expression vectors. Expression vectors for
bacteria are well
known in the art, and include vectors for Bacillus subtilis, E. coli,
Streptococcus cremoris, and
Streptococcus lividans, among others.
The bacterial expression vectors are transformed into bacterial host cells
using techniques well known
in the art, such as calcium chloride treatment, electroporation, and others.
42
WO 00/47612 CA 02362541 2001-08-10 pCT/US00/03665
In one embodiment, analog proteins are produced in insect cells. Expression
vectors for the
transformation of insect cells, and in particular, baculovirus-based
expression vectors, are well known
in the art.
In a preferred embodiment, analog proteins are produced in yeast cells. Yeast
expression systems
are well known in the art, and include expression vectors for Saccharomyces
cerevisiae, Candida
albicans and C. maltosa, Hansenula polymorpha, Kluyveromyces fragilis and K.
lacfis, Pichia
guillerimondii and P. pastoris, Schizosaccharomyces pombe, and Yarrovvia
lipolytica. Preferred
promoter sequences for expression in yeast include the inducible GAL1,10
promoter, the promoters
from alcohol dehydrogenase, enolase, glucokinase, glucose-6-phosphate
isomerase, glyceraldehyde-
3-phosphate-dehydrogenase, hexokinase, phosphofructokinase, 3-phosphoglycerate
mutase,
pyruvate kinase, and the acid phosphatase gene. Yeast selectable markers
include ADE2, HIS4,
LEU2, TRP1, and ALG7, which confers resistance to tunicamycin; the neomycin
phosphotransferase
gene, which confers resistance to 6418; and the CUP1 gene, which allows yeast
to grow in the
presence of copper ions.
In a preferred embodiment, the analog proteins of the invention are expressed
in yeast and displayed
on the yeast surface. Suitable yeast expression and display systems are known
in the art (Boder and
Wittrup, Nat. Biotechnol. 15:553-7 (1997); Cho et al., J. Immunol. Methods
220:179-88 (1998); all of
which are expressly incorporated by reference). Surface display in the ciliate
Tetrahymena
thermophila is described by Gaertig et al. Nat. Biotechnol. 17:462-465 (1999),
expressly incorporated
2 0 by reference.
In one embodiment, analog proteins are produced in viruses and are expressed
on the surface of the
viruses. Expression vectors for protein expression in viruses and for display,
are well known in the art
and commercially available (see review by Felici et al., Biotechnol. Annu.
Rev. 1:149-83 (1995)).
Examples include, but are not limited to M13 (Lowman et al., (1991)
Biochemistry 30:10832-10838
(1991); Matthews and Wells; (1993) Science 260:1113-1117; Stratagene); fd
(Krebber et al., (1995)
FEBS Lett. 377:227-231); T7 (Novagen, Inc.); T4 (Jiang et al., Infect. Immun.
65:4770-7 (1997);
lambda (Stolz et al., FEES Lett. 440:213-7 (1998)); tomato bushy stunt vines
(Joelson et al., J. Gen.
Virol. 78:1213-7 (1997)); retroviruses (Buchholz et al., Nat. Biotechnol.
16:951-4 (1998)). All of the
above references are expressly incorporated by reference.
3 0 In addition, the analog proteins of the invention may be further fused to
other proteins, if desired, for
example to increase expression.
43
WO 00/47612 CA 02362541 2001-08-to pCT/US00/03665
In one embodiment, the AP nucleic acids, proteins and antibodies of the
invention may be labeled. By
"labeled" herein is meant that a compound has at least one element, isotope or
chemical compound
attached to enable the detection of the compound. In general, labels fall into
three classes: a) isotopic
labels, which may be radioactive or heavy isotopes; b) immune labels, which
may be antibodies or
antigens; and c) colored or fluorescent dyes. The labels may be incorporated
into the compound at
any position.
Once made, the analog proteins may be covalently modified. One type of
covalent modification
includes reacting targeted amino acid residues of an analog protein with an
organic derivatizing agent
that is capable of reacting with selected side chains or the N-or C-terminal
residues of an analog
protein. Derivatization with bifunctional agents is useful, for instance, for
crosslinking an analog
protein to a water-insoluble support matrix or surface for use in the method
for purifying anti- analog
protein antibodies or screening assays, as is more fully described below.
Commonly used crosslinking
agents include, e.g., 1,1-bis(diazoacetyl)-2-phenylethane, glutaraldehyde, N-
hydroxysuccinimide
esters, for example, esters with 4-azidosalicylic acid, homobifunctional
imidoesters, including
disuccinimidyl esters such as 3,3'-dithiobis(succinimidylpropionate),
bifunctional maleimides such as
bis-N-maleimido-1,8-octane and agents such as methyl-3-[(p-
azidophenyl)dithio]propioimidate.
Other modifications include deamidation of glutaminyl and asparaginyl residues
to the con-esponding
glutamyl and aspartyl residues, respectively, hydroxylation of proline and
lysine, phosphorylation of
2 0 hydroxyl groups of seryl or threonyl residues, methylation of the "-amino
groups of lysine, arginine, and
histidine side chains [T.E. Creighton, Proteins: Structure and Molecular
Properties, W.H. Freeman &
Co., San Francisco, pp. 79-86 (1983)], acetylation of the N-terminal amine,
and amidation of any C-
terminal carboxyl group.
2 5 Another type of covalent modification of the analog protein included
within the scope of this invention
comprises altering the native glycosylation pattern of the corresponding
naturally occurring protein.
"Altering the native glycosylation pattern" is intended for purposes herein to
mean deleting one or
more carbohydrate moieties found in the naturally occurring protein, and/or
adding one or more
glycosylation sites that are not present in the naturally occurring protein.
Addition of glycosylation sites to a analog protein may be accomplished by
altering the amino acid
sequence thereof. The alteration may be made, for example, by the addition of,
or substitution by, one
or more serine or threonine residues to the naturally occurring protein (for O-
linked glycosylation
sites). The analog protein amino acid sequence may optionally be altered
through changes at the
DNA level, particularly by mutating the DNA encoding the analog protein at
preselected bases such
that codons are generated that will translate into the desired amino acids.
44
CA 02362541 2001-08-10 pCT~S00/03665
WO 00/47612
Another means of increasing the number of carbohydrate moieties on the analog
protein is by
chemical or enzymatic coupling of glycosides to the polypeptide. Such methods
are described in the
art, e.g., in WO 87/05330, published September 11, 1987, and in Aplin and
Wriston, CRC Crit. Rev.
Biochem., pp. 259-306 (1981).
Removal of carbohydrate moieties present on the analog protein may be
accomplished chemically or
enzymatically or by mutational substitution of codons encoding for amino acid
residues that serve as
targets for glycosylation. Chemical deglycosylation techniques are known in
the art and described, for
instance, by Hakimuddin et al., Arch. Biochem. Biophys., 259:52 (1987) and by
Edge et al., Anal.
Biochem., 118:131 (1981). Enzymatic cleavage of carbohydrate moieties on
polypeptides can be
achieved by the use of a variety of endo-and exo-glycosidases as described by
Thotakura et al., Meth.
Enzymol., 138:350 (1987).
Another type of covalent modification of an analog protein comprises linking
the analog protein to one
of a variety of non-proteinaceous polymers, e.g., polyethylene glycol,
polypropylene glycol, or
polyoxyalkylenes, in the manner set forth in U.S. Patent Nos. 4,640,835;
4,496,689; 4,301,144;
4,670,417; 4,791,192 or4,179,337.
Analog proteins of the present invention may also be modified in a way to form
chimeric molecules
comprising an analog protein fused to another, heterologous polypeptide or
amino acid sequence. In
one embodiment, such a chimeric molecule comprises a fusion of an analog
protein with a tag
polypeptide which provides an epitope to which an anti-tag antibody can
selectively bind. The epitope
2 0 tag is generally placed at the amino-or carboxyl-terminus of the analog
protein. The presence of such
epitope-tagged forms of an analog protein can be detected using an antibody
against the tag
polypeptide. Also, provision of the epitope tag enables the analog protein to
be readily purified by
affinity purification using an anti-tag antibody or another type of affinity
matrix that binds to the epitope
tag. In an alternative embodiment, the chimeric molecule may comprise a fusion
of an analog protein
2 5 with an immunoglobulin or a particular region of an immunoglobulin. For a
bivalent form of the
chimeric molecule, such a fusion could be to the Fc region of an IgG molecule.
Various tag polypeptides and their respective antibodies are well known in the
art. Examples include
poly-histidine (poly-his) or poly-histidine-glycine (poly-his-gly) tags; the
flu HA tag polypeptide and its
antibody 12CA5 [Field et al., Mol. Cell. Biol., 8:2159-2165 (1988)]; the c-myc
tag and the 8F9, 3C7,
3 0 6E10, G4, B7 and 9E10 antibodies thereto [Evan et al., Molecular and
Cellular Biology, 5:3610-3616
(1985)]; and the Herpes Simplex virus glycoprotein D (gD) tag and its antibody
[Paborsky et al.,
Protein Engineering, 3(6):547-553 (1990)]. Other tag polypeptides include the
Flag-peptide [Hopp et
al., BioTechnology, 6:1204-1210 (1988)]; the KT3 epitope peptide [Martin et
al., Science, 255:192-194
CA 02362541 2001-08-10
WO 00/47612 PCT/US00/03665
(1992)]; tubulin epitope peptide (Skinner et al., J. Biol. Chem., 266:15163-
15166 (1991)]; and the T7
gene 10 protein peptide tag [Lutz-Freyermuth et al., Proc. Natl. Acad. Sci.
USA, 87:6393-6397 (1990)].
In a preferred embodiment, the analog protein is purified or isolated after
expression. Analog proteins
may be isolated or purified in a variety of ways known to those skilled in the
art depending on what
other components are present in the sample. Standard purification methods
include electrophoretic,
molecular, immunological and chromatographic techniques, including ion
exchange, hydrophobic,
affinity, and reverse-phase HPLC chromatography, and chromatofocusing. For
example, the analog
protein may be purified using a standard anti-library antibody column.
Ultrafiltration and diafiltration
techniques, in conjunction with protein concentration, are also useful. For
general guidance in suitable
purification techniques, see Scopes, R., Protein Purification, Springer-
Verlag, NY (1982). The degree
of purification necessary will vary depending on the use of the analog
protein. In some instances no
purification may be necessary. A preferred method for purification is outlined
in the examples.
Once made, the analog proteins and nucleic acids of the invention find use in
a number of
applications.
In a preferred embodiment, the receptor analogs are used in methods designed
for high throughput
screening for ligand analogs and bioactive agents.
In a preferred embodiment, the receptor analogs are used in a method of
screening for ligand analogs,
comprising adding a candidate ligand to a receptor analog or to a naturally
occurring receptor. By
"candidate ligand" or grammatical equivalents thereof, herein is meant any
molecule, e.g., protein,
2 0 small organic molecule, polysaccharide, lipid, polynucleotide, etc., or
mixtures thereof with the
capability of binding to a receptor analog or to a naturally occurring
receptor. Included within this
definition are any molecules, as defined above, that have the capability to
modulate the signaling
activity of a receptor analog or of a naturally occurring receptor.
"Modulating the signaling activity of a
receptor analog or of a naturally occurring receptor" by a candidate ligand
includes an increase (i.e.,
more efficient signaling of a receptor analog or of a naturally occurring
receptor) or a decrease (i.e.,
less efficient signaling of a receptor analog or of a naturally occurring
receptor), when compared to the
signaling activity of a receptor analog or of a naturally occurring receptor
in the absence of a candidate
ligand. Assays used to determine signaling activity of naturally occurring
cell surface receptors are
also used to determine the signaling activity of the receptor analogs of the
invention. These assays
3 0 are known in the art and some are described further below.
A candidate ligand, once shown to bind to a receptor analog or to a naturally
occurring receptor or
modulates its activity is termed a "ligand analog." Of particular interest are
candidate ligands that
46
WO 00/47612 CA 02362541 2001-08-10 pCT/US00/03665
either have a low or no toxicity for human cells. Candidate ligands may be
added as individual
ligands, as combined samples of individual ligands or as more complex
libraries as is discussed
further below. Generally a plurality of assay mixtures is run in parallel with
different candidate ligand
concentrations to obtain a differential response to the various
concentrations. Typically, one of these
concentrations serves as a negative control, i.e., at zero concentration or
below the level of detection.
Candidate ligands encompass numerous chemical classes, though typically they
are organic
molecules, preferably small organic compounds having a molecular weight of
more than 100 and less
than about 2,500 daltons. Candidate ligands comprise functional groups
necessary far structural
interaction with proteins, particularly hydrogen bonding, and typically
include at least an amine,
carbonyl, hydroxyl or carboxyl group, preferably at least two of the
functional chemical groups. The
candidate ligands often comprise cyclical carbon or heterocyclic structures
and/or aromatic or
polyaromatic structures substituted with one or more of the above functional
groups. Candidate
ligands are also found among biomolecules including peptides, saccharides,
fatty acids, steroids,
purines, pyrimidines and derivatives, structural analogs or combinations
thereof. Particularly preferred
are peptides. In fact, virtually any small organic molecule that is
potentially capable of binding to a
receptor analog or to a naturally occurring receptor of interest may find use
in the present invention
provided that it is sufficiently soluble and stable in aqueous solutions to be
tested for its ability to bind
to the receptor analog or to the naturally occurring receptor analog.
Candidate ligands are obtained from a wide variety of sources including
libraries of synthetic or natural
2 0 compounds. For example, numerous means are available for random and
directed synthesis of a
wide variety of organic compounds and biomolecules, including expression of
randomized
oligonucleotides. Alternatively, libraries of natural compounds in the form of
bacterial, fungal, plant
and animal extracts are available or readily produced. Additionally, natural
or synthetically produced
libraries and compounds are readily modified through conventional chemical,
physical and biochemical
means. Known pharmacological agents may be subjected to directed or random
chemical
modifications, such as acylation, alkylation, esterification, amidification to
produce structural analogs.
In a preferred embodiment, the candidate ligands are proteins.
In another preferred embodiment, the candidate ligands are naturally occurring
proteins, fragments of
naturally occurring proteins, ligand analogs, as described above, or fragments
of ligand analogs.
3 0 Thus, for example, cellular extracts containing proteins, or random or
directed digests of
proteinaceous cellular extracts, may be used. In this way libraries of
prokaryotic and eukaryotic
proteins may be made. Particularly preferred in this embodiment are libraries
of bacterial, fungal, viral,
47
CA 02362541 2001-08-10
WO 00/47612 PCT/US00/03665
and mammalian proteins, with the latter being preferred, and human proteins
being especially
prefer-ed.
In a preferred embodiment, the candidate ligands are peptides of from about 5
to about 30 amino
acids, with from about 5 to about 20 amino acids being preferred, and from
about 7 to about 15 being
particularly preferred. The peptides may be digests of naturally occurring
proteins as is outlined
above, random peptides, or "biased" random peptides. By "randomized" or
grammatical equivalents
herein is meant that each peptide consists of essentially random amino acids.
Since generally these
random peptides are chemically synthesized, they may incorporate any amino
acid at any position.
The synthetic process can be designed to generate randomized proteins to allow
the formation of all or
most of the possible combinations over the length of the sequence, thus
forming a library of
randomized candidate proteinaceous ligands.
In one embodiment, the library is fully randomized, with no sequence
preferences or constants at any
position. In a preferred embodiment, the library is biased. That is, some
positions within the sequence
are either held constant, or are selected from a limited number of
possibilities. For example, in a
preferred embodiment, the amino acid residues are randomized within a defined
class, for example, of
hydrophobic amino acids, hydrophilic residues, sterically biased (either small
or large) residues,
towards the creation of cysteines, for cross-linking, prolines for SH-3
domains, serines, threonines,
tyrosines or histidines for phosphorylation sites, or the like.
In one embodiment, a library of protein encoding nucleotide sequences may be
obtained from genomic
2 0 DNA, from cDNAs or from random nucleotides. Particularly preferred in this
embodiment are libraries
encoding bacterial, fungal, viral, and mammalian proteins and peptides, with
the latter being preferred,
and human encoding proteins and peptides being especially preferred. As
described above and as
known in the art the protein and peptide encoding nucleotide sequences may be
inserted into any
vector suitable for expression in mammalian cells, other eukaryotic cells,
prokaryotic cells and viruses.
2 5 In a preferred embodiment, a library of candidate ligands is generated
using protein design such as
the PDA methodology, described herein. In this embodiment, a virtual library
of candidate ligands is
first generated and evaluated for its potential to generate candidate ligands
capable of binding to a
receptor analog of the invention. Following this analysis, an experimental
random library is generated
that is only randomized at the readily changeable, non-disruptive sequence
positions of a naturally
3 0 occurring protein. Thus, by limiting the number of randomized positions
and the number of
possibilities at these positions, the probability of finding sequences with
useful properties does
increase.
48
WD 00/47612 CA 02362541 2001-08-10
PCT/US00/03665
In another preferred embodiment, the candidate ligands are obtained from
combinatorial chemical
libraries, a wide variety of which are available in the literature. By
"combinatorial chemical library"
herein is meant a collection of diverse chemical compounds generated in a
defined or random manner,
generally by chemical synthesis. Millions of chemical compounds can be
synthesized through
combinatorial mixing.
Two types of assays are generally used for high throughput screening in drug
discovery: (1) cell-free
(i.e., biochemical assays or in vitro assays), which measure the binding
affinity between a ligand and a
receptor, and (2) cell-based assays, which measure the biological response
triggered by the
interaction between a ligand and a receptor displayed on the cell surface.
Cell-free assays have
several advantages over cell-based assays: (i) A far larger library may be
screened allowing e.g., the
use of highly diverse encoded small molecule libraries or peptide libraries,
thereby greatly increasing
the likelihood of a hit; (ii) a greater sensitivity in comparison to cell-
based assays allows lower affinity
molecules to be identified. Cell-based assays (i) generally require reporter
gene expression or
downstream signals to be detected, (ii) are generally more time-consuming, and
(iii) are generally
more expensive than cell-free assays. However, the signaling activity of a
receptor or receptor analog,
usually the biological response upon binding of a cognate ligand, does involve
cellular components
and as such the biological activity of ligand analogs or bioactive agents
identified in cell-free assays, is
generally verified in cell-based assays. Thus, the present invention provides
receptor analogs useful
in methods of screening in cell-free assays and/or cell-based assays.
2 0 As outlined above, the novel receptor analogs of the present invention are
useful for high throughput
screening of ligand analogs and/or bioactive agents. The receptor analogs
employed in the screening
methods, detailed herein, are designed to maintain a stable, biologically
active structure when used in
cell-free assays or in in vitro assays.
In another preferred embodiment a library of candidate ligands is used in in
vitro binding assays to
detect binding of a candidate ligand to a receptor analog bound non-diffusably
to an insoluble support
having isolated sample receiving areas (e.g. a microtiter plate, an array,
etc.). These assays are
particularly useful for high throughput screening for ligand analogs. The
insoluble support may be
made of any composition to which the receptor analog can be bound, is readily
separated from soluble
material, and is otherwise compatible with the overall method of screening.
The surface of such a
3 0 support may be solid or porous and of any convenient shape. Examples of
suitable insoluble supports
include microtiter plates, an-ays, membranes and beads. These are typically
made of glass, plastic
(e.g., polystyrene), polysaccharides, nylon or nitrocellulose, teflonT"', etc.
Microtiter plates and arrays
are especially convenient because a large number of assays can be carried out
simultaneously, using
small amounts of reagents and samples. The particular manner of binding of the
receptor analog is
49
CA 02362541 2001-08-10
WO 00/47612 PCT/US00/03665
not crucial so long as it is compatible with the reagents and overall methods
of the invention, maintains
the characteristics of the receptor analog and is non-diffusable. The receptor
analog may be either
bound directly to the insoluble support (e.g. via cross-linking) or indirectly
(e.g., via antibody, other
protein or nucleic acid, etc.). Preferred methods of binding include the use
of antibodies (which do not
sterically block the interaction surface for the candidate ligand and
preferably are directed against a
tag polypeptide which may be incorporated into the surface receptor analog),
direct binding to "sticky"
or ionic supports, chemical crosslinking, etc. Following binding of the
receptor analog, excess
unbound material is removed by washing. The sample receiving areas may then be
blocked through
incubation with bovine serum albumin (BSA), casein or other innocuous protein.
The candidate ligand is added to the binding assay. Determination of the
binding of the candidate
ligand to the receptor analog may be done using a wide variety of assays,
including labeled in vitro
protein-protein binding assays, electrophoretic mobility shift assays (EMSA),
immunoassays for
protein binding, functional assays (phosphorylation assays, etc.) and the
like. (e.g., see, Harlow and
Lane, Antibodies: A Laboratory Manual (New York, Cold Spring Harbor Laboratory
Press, 1988) and
Ausubel et al., Short Protocols in Molecular Biology (John Wiley & Sons, Inc.,
1995).
By "labeled" herein is meant that the compound (e.g., the candidate ligand
which is tested for binding)
is either directly or indirectly labeled with a label which provides a
detectable signal, e.g. radioisotope,
fluorescers, enzyme, antibodies, particles such as magnetic particles,
chemiluminescers, or specific
binding molecules, etc. Specific binding molecules include pairs, such as
biotin and streptavidin,
2 0 digoxin and antidigoxin etc. For the specific binding members, the
complementary member would
normally be labeled with a molecule which provides for detection, in
accordance with known
procedures, as outlined above. The label can directly or indirectly provide a
detectable signal.
In some embodiments, only one of the components is labeled. For example, the
candidate ligand may
be labeled at tyrosine positions using'z51, or at methionine positions using
35S, or with fluorophores.
Alternatively, more than one component may be labeled with different labels
using '251 or 35S for one
protein, for example, and a fluorophor for a potential additional component.
In a preferred embodiment, the candidate ligand is labeled, and binding is
determined directly. For
example, this may be done by attaching all or a portion of receptor analog to
a solid support, adding a
labeled candidate ligand (for example a fluorescent label), washing off excess
reagent, and
3 0 determining whether the label is present on the solid support. Various
blocking and washing steps
may be utilized as is known in the art.
CA 02362541 2001-08-10
WO 00/47612 PCT/US00/03665
In another preferred embodiment, the candidate ligand and the receptor analog
are combined first and
after a certain incubation period, one protein, preferably the non-labeled
protein (e.g. receptor analog)
is bound either directly or indirectly to an insoluble support. The second
protein, preferably labeled
(e.g., the candidate ligand) which is bound to the receptor analog is
visualized in accordance with the
label incorporated.
Exemplified herein by the naturally occurring EPOR and/or EPOR analogs (as
described above), but
applicable to all naturally occurring receptors and receptor analogs of the
invention, the naturally
occurring EPOR and/or EPOR analogs are immobilized following standard
procedures in the literature.
Binding conditions may be further optimized for increased immobilization.
Immobilization may be
tested by determining the binding affinity of the natural ligand. The goal is
to have an active and
robust immobilized receptor analog for use in methods of screening for ligand
analogs and bioactive
agents (as described herein). Several methods, known to the skilled artisan,
are used to immobilize
the EPOR analog.
In one embodiment, the EPOR and/or EPOR analog is immobilized by attaching its
free sulfhydryl
group to Sulfolink agarose beads (Pierce Chemical Co), as described in the
literature.
In another embodiment, the EPOR andlor EPOR analog or their disulfide-linked
dimer are coated on
Maxisorp microtiter plates (NUNC, Roskilde, Denmark), following published
protocols.
In a preferred embodiment, the EPOR and/or EPOR analog containing a His-tag
fusion peptide may
be attached to a Ni-containing support, as described in the literature.
2 0 In another preferred embodiment, the EPOR andlor EPOR analog may be
immobilized to a functional
plate by cross-linking random lysines, as known in the art.
The binding assays described herein are exemplified by the natural occurring
EPOR, EPOR analogs,
natural occurring EPO, EPO analogs, and peptide mimics, however, as outlined
above apply to other
receptors and ligands, both naturally occurring and analogs thereof.
In one aspect of this embodiment, the equilibrium and kinetic constants
between EPO or its mimics
(e.g., EMP1) and immobilized receptor analogs using surface plasmon resonance
(SPR) is measured.
Following literature procedures, the naturally occurring EPOR, EPOR analogs or
their disulfide-linked
homodimers are coupled to the sensor chip by random lysines to yield two
different resonance units
(RU), and the dissociation and association rates are determined by global
fitting. The equilibrium
3 0 constant is given by Kd = Ko~/Ko~.
51
CA 02362541 2001-08-10
WO 00/47612 PCT/US00/03665
In another embodiment, the equilibrium constant is determined using
competition binding assay.
Using the EPOR attached to sulfolink agarose,'251 labeled EPO is used in
competition assays with
EPO and EPO mimetic peptides to measure the equilibrium constant between EPOR
and ligand
following standard procedures. Comparison of the equilibrium constant between
the naturally
occurring EPOR and EPO (or EPO mimetic peptides) with those from EPOR analogs
provide a
quantitative comparison on the EPOR analogs with EPO and its peptide mimetics.
Biophysical characterization is used to assess protein design and binding
studies. Increased stability
and oligomerization state both suggest a successful design. Also, receptor
robustness is tested here.
The following biochemical characterization is exemplified by the naturally
occurring EPOR and/or
EPOR analogs, but is applicable to all naturally occurring receptors and
receptor analogs of the
invention,
In a preferred embodiment the stoichiometry of EPOR analogs in complex with
EPO, EPO analogs, or
EPO mimetics is determined. In one aspect of this embodiment, size exclusion
chromatography
(SEC) is used to evaluate the receptor dimerization in the presence or absence
of EPO, EPO analogs,
or EPO mimetics, as shown in the literature. EPO, EPOR and disulfide-linked
EPOR homodimer are
used, together with protein standards, to calibrate the system. In another
aspect of this embodiment,
equilibrium sedimentation is used to confirm the result from SEC if necessary.
EPOR analogs are
expected to elute as dimers due to their stabilized complex conformation.
In another preferred embodiment, the binding constant between EPOR analogs and
EPO, EPO
2 0 analogs, or EPO mimetics is estimated using SEC at various ratios of EPOR
analog vs. EPO, EPO
analogs, or EPO mimetics. this results in useful information with respect to
the binding affinity of
EPOR analogs relative to each other.
In a preferred embodiment, the stability of-naturally occurring EPOR and EPOR
analogs is monitored
by circular dichroism (CD) and fluorescence upon thermal and/or chemical
denaturation, as is known
2 5 in the art.
In another embodiment, the conformational stability or mobility of naturally
occurring EPOR and EPOR
analogs is determined. As described in the literature, fluorescence can
monitor the interface between
two receptors by dye quenching and energy transfer.
In a preferred embodiment, the shelf life of EPOR analogs and EPO analogs is
determined at various
3 0 conditions, in particular at conditions used for screening. This can,
e.g., be performed by incubating
52
WO 00/47612 CA 02362541 2001-08-10 pCT~S00/03665
the proteins at high temperature and analyzing the protein left as a function
of incubation time by
analytical HPLC.
In a preferred embodiment, receptor analogs (i.e. structurally constrained
receptors) are tested for
their ability to screen phage displayed peptide libraries for agonists and
antagonists. Standard phage
library generation techniques are used to create libraries.
In a preferred embodiment, the present invention provides methods of screening
for ligand analogs
that are capable of binding to a receptor analog using a cell-based assay.
Whenever available and
applicable, a naturally occurring receptor and/or a naturally occurring ligand
is/are used as a control
within the assays outlined below. Briefly, a labeled candidate ligand analog
is added to a cell
comprising a receptor analog of the invention or a naturally occurring
receptor and binding of the
labeled candidate ligand analog is detected by virtue of the label as
described above.
In a preferred embodiment, a plurality of cells is screened. By a "plurality
of cells" herein is meant
roughly from about 103 cells to 108 or 109, with from 106 to 108 being
preferred. This plurality of cells
comprises a cellular library, wherein generally each cell within this cellular
library contains a member of
the molecular library, i.e. a different candidate ligand analog or a different
ligand analog encoding
nucleic acid, although as will be appreciated by those in the art, some cells
within the cellular library.
may not contain a member of the molecular library, and some may contain more
than one. Methods
such as retroviral infection, electroporation and others known in the art can
be used to introduce the
candidate analog protein into a plurality of cells; the distribution of
candidate nucleic acids within the
2 0 individual cell members of the cellular library may vary widely, depending
on the method used.
As used in this specification and the appended claims, the singular forms "a",
"an" and "the" include
plural references unless the content clearly dictates otherwise. Likewise,
plural forms, unless the
content clearly dictates otherwise, include singular references. Thus,
reference to "a monomer"
includes mixtures of monomers, reference to a "receptor analog" includes
mixtures of receptor
2 5 analogs, and the like. Likewise, reference to "cells" includes a cell, and
the like.
In a preferred embodiment, the receptor analogs of the invention are used in
cell based assays to
screen for ligand analogs that have the ability to modulate the signaling of
receptor analogs and or the
signaling of naturally occurring cell surface receptors. Receptor signaling
generally leads to an
altered phenotype of the host cell or to a change in cell physiology .
3 0 By "altered phenotype" or "changed physiology" or other grammatical
equivalents herein is meant that
the phenotype of the cell is altered in some way, preferably in some
detectable and/or measurable
way. As will be appreciated in the art, a strength of the present invention is
the wide variety of cell
53
CA 02362541 2001-08-10
WO 00/47612 PCT/US00/03665
types and potential phenotypic changes which may be tested using the present
methods. Accordingly,
any phenotypic change which may be observed, detected, or measured may be the
basis of the
screening methods herein. Suitable phenotypic changes include, but are not
limited to: gross physical
changes such as changes in cell morphology, cell growth, cell viability,
adhesion to substrates or other
cells, and cellular density; changes in the expression of one or more RNAs,
mRNAs, proteins, lipids,
hormones, cytokines, or other molecules; changes in the equilibrium state
(i.e. half life) of one or more
RNAs, mRNAs, proteins, lipids, hormones, cytokines, or other molecules;
changes in the localization
of one or more RNAs, mRNAs, proteins, lipids, hormones, cytokines, or other
molecules; changes in
the bioactivity or specific activity of one or more RNAs, mRNAs, proteins,
lipids, hormones, cytokines,
receptors, or other molecules; changes in the secretion of ions, cytokines,
hormones, growth factors,
proteins, or other molecules; alterations in cellular membrane potentials,
polarization, integrity or
transport; changes in infectivity, susceptibility, latency, adhesion, and
uptake of viruses and bacterial
pathogens; etc.
By "capable of altering the phenotype" or grammatical equivalents, herein is
meant that a candidate
ligand analog can change the phenotype of the cell in some detectable and/or
measurable way.
The altered phenotype may be detected in a wide variety of ways, as is
described more fully below and
in PCT/US97/01019, and will generally depend and correspond to the phenotype
that is being
changed. Generally, the changed phenotype is detected using, for example:
microscopic analysis of
cell morphology; standard cell viability assays, including both increased cell
death and increased cell
2 0 viability, for example, cells that are now resistant to cell death via
virus, bacteria, or bacterial or
synthetic toxins; standard labeling assays such as fluorometric indicator
assays for the presence or
level of a particular cell or molecule, including FACS or other dye staining
techniques; biochemical
detection of the expression of target compounds after killing the cells;
monitoring changes in gene
expression within a target cell, etc. In some cases, as is more fully
described herein, the altered
2 5 phenotype is detected in the cell in which the molecular library
comprising the randomized nucleic acid
or randomized proteins was introduced; in other embodiments, the altered
phenotype is detected in a
second cell which is responding to some molecular signal from the first cell.
In one aspect of this embodiment, the candidate ligand analogs, as part of a
molecular library,
generally are added to suitable host cells or are introduced into suitable
host cells to screen for ligand
3 0 analogs, capable of altering the phenotype of the host cell, harboring or
expressing a receptor analog.
If necessary, the cells are treated to conditions suitable for the expression
of genes encoding the
candidate analog proteins (for example, when inducible promoters are used), to
produce the candidate
expression products.
I n another aspect of this embodiment, the methods of the present invention
comprise introducing a
3 5 molecular library of randomized candidate nucleic acids into a plurality
of cells, generating a cellular
54
CA 02362541 2001-08-10
WO 00/47612 PCT/US00/03665
library. Each of the nucleic acids comprises a different, generally
randomized, nucleotide sequence,
encoding a different ligand analog. The plurality of cells is then screened,
as is more fully outlined
below, for a cell exhibiting an altered phenotype. The altered phenotype is
generally due to the
presence of a ligand analog.
The present invention further provides methods of screening for ligand analogs
that are capable of
modulating the signaling activity of a receptor analog.
In a preferred embodiment, the method of screening for ligand analogs that are
capable of modulating
the signaling activity of a receptor analog comprises the steps of (1)
providing a host cell comprising a
vector composition, comprising a gene encoding a receptor analog. This vector
composition may or
may not comprise retroviral vectors, and may or may not be integrated into the
genome of the host
cell. (2) The host cell is subjected to conditions under which the gene
encoding the receptor analog is
expressed to produce a receptor analog. Optionally, it is determined (3)
whether the receptor analog
is displayed on the surface of the host cell, e.g., by using
immunohistochemical and other methods as
known in the art. (4) Optionally a natural ligand known to bind and activate a
corresponding naturally
occurring cell surface receptor is added. (5) Optionally the signaling
activity of the receptor analog in
response to the natural ligand is determined. (6) Candidate ligands that are
capable of modulating the
signaling activity of the receptor analog are added. Simultaneously,
sequentially or at a later step (7)
the modulation of the signaling activity of the receptor analog in response to
the candidate ligand is
determined by screening the cell for an altered phenotype or changed
physiology e.g., by using
2 0 cytokine, cell proliferation, cell differentiation assays, and other
assays that are further described
below. Preferably, (6) the candidate ligands are identified. Candidate ligands
identified by this method
are named ligand analogs.
In another preferred embodiment, the method of screening for ligand analogs
that are capable of
modulating the signaling activity of a receptor analog comprises the steps of
(1 ) providing a host cell
comprising a vector composition, comprising vector composition comprising a
gene encoding a
receptor analog and a gene encoding a natural ligand that is capable of
binding to and activating the
receptor analog. This vector composition may or may not comprise retroviral
vectors, and may or may
not be integrated into the genome of the host cell. (2) The host cell is
subjected to conditions under
which the genes encoding the receptor analog and the natural ligand are
expressed to produce a
3 0 receptor analog and a natural ligand. Optionally, it is determined (3)
whether the receptor analog is
displayed on the surface of the host cell, e.g., by using immunohistochemical
and other methods as
known in the art. (4) Optionally the signaling activity of the receptor analog
in response to the natural
ligand is determined. (5) Candidate ligands that are capable of modulating the
signaling activity of the
receptor analog are added. Simultaneously, sequentially or at a later step (7)
the modulation of the
CA 02362541 2001-08-10
WO 00/47612 PCT/US00/03665
signaling activity of the receptor analog in response to the candidate ligand
is determined by screening
the cell for an altered phenotype or changed physiology e.g., by using
cytokine, cell proliferation, cell
differentiation assays, and other assays that are further described below.
Preferably, (6) the candidate
ligands are identified. Candidate ligands identified by this method are named
ligand analogs.
In a preferred embodiment a library of candidate ligands is added to a host
cell comprising a receptor
analog of the invention.
In another preferred embodiment a library of candidate ligands is added to a
host cell displaying a
receptor analog on its surface.
In another preferred embodiment a library of candidate ligands is added to a
virus displaying a
receptor analog on its surface.
As outlined above ligand analogs or bioactive agents (as further outlined
below) of the present
invention may modulate signaling activity of a receptor analog and as such
they may exhibit cytokine,
cell proliferation (either inducing or inhibiting), cell differentiation
(either inducing or inhibiting),
chemotactic or chemokinetic activity. The activity of the proteins of the
invention, comprising receptor
analogs, ligand analogs and bioactive agents may, among other means, be
measured using a variety
of assays.
In one embodiment, the natural ligands, known peptide agonists and known
antagonists are used to
measure the EC50 for proliferation and the level of JAK2 tyrosine kinase
phosphorylation in cytokine
receptor responsive cell lines.
2 0 In another embodiment, binding affinities of fluorescently labeled natural
ligands, known peptide
agonists and known antagonists to receptor analogs and naturally occurring
cell surface receptors are
assayed by competition assays.
In a preferred embodiment, assays for proliferation and differentiation of
hematopoietic and
lymphopoietic cells are provided that include, but are not limited to those
described in: Current
Protocols in Immunology (Ed by J.E. Coligan et al. Vol 1; Greene Publishing
Associates and Wiley-
Interscience; John Wiley and Sons, Toronto (1994)); deVries et al., J. Exp.
Med. 173:1205-1211
(1991); Moreau et al., Nature 336:690-692 (1988); Greenberger et al., Proc.
Natl. Acad. Sci. U.S.A.
83:1857-1861 (1986); incorporated as references in their entirety.
56
WO 00/47612 CA 02362541 2001-08-10
PCT/US00/03665
In another embodiment, assays for T-cell or thymocyte proliferation are
provided that include , but are
not limited to those described in: Current Protocols in Immunology, supra;
Takai et al., J. Immunol.
137:3494-3500 (1986); Bertagnolli et al., J. Immunol. 145:1706-1712 (1990);
Bertagnolli et al., Cellular
Immunology 133:327-341 (1991); Bertagnolli et al., J. Immunol. 149:3778-3783
(1992); Bowman et al.,
J. Immunol. 152:1756-1761 (1994); incorporated as references in their
entirety.
In another embodiment, assays for cytokine production andlor proliferation of
spleen cells, lymph node
cells or thymocytes are provided that include, but are not limited to those
described in: Current
Protocols in Immunology, supra.
In one embodiment, assays for T-cell clone responses to antigens are provided
that include, but are
not limited to those described in: Currenf Protocols in Immunology, supra;
Weinberger et al., Proc Natl.
Acad. Sci. U.S.A. 77:6091-6095 (1980); Weinberger et al., Eur. J. Immun.
11:405-411 (1981); Takai et
al., J. Immunol. 137:3494-3500 (1986); Takai et al., J. Immunol. 140:508-512
(1988); incorporated as
references in their entirety.
In a preferred embodiment. assays for thymocyte or splenocyte cytotoxicity are
provided that include,
but are not limited to those described in: Current Protocols in Immunology,
supra; Herrmann et al.,
Proc. Natl. Acad. Sci. U.S.A. 78:2488-2492 (1981); Herrman et al., J. Immunol.
128:1968-1974 (1982);
Handa et al., J. Immunol. 135:1564-1572 (1985); Takai et al., J. Immunol.
137:3494-3500 (1986);
Takai et al., J. Immunol. 140:508-512 (1988); Bowman et al., J. Virology
61:1992-1998; Bertagnolli et
al., Cellular Immunology 133:327-341 (1991); Brown et al., J. Immunol.
153:3079-3092 (1994);
2 0 incorporated as references in their entirety.
In one embodiment assays for T-cell-dependent immunoglobulin responses and
isotope switching are
provided that include, but are not limited to those described in: Maliszewski,
J. Immunol. 144:3028-
3033 (1990); incorporated as reference in its entirety.
In another embodiment assays for B cell function are provided that include,
but are not limited to those
2 5 described in: Current Protocols in Immunology, supra;
In another embodiment, mixed lymphocyte reaction (MLR) assays are provided
that include, but are
not limited to those described in: Current Protocols in Immunology, supra;
Takai et al., J. Immunol.
137:3494-3500 (1986); Takai et al., J. Immunol. 140:508-512 (1988);
Bertagnolli et al., J. Immunol.
149:3778-3783 (1992); incorporated as reference in their entirety.
57
CA 02362541 2001-08-10 PCT~S00/03665
WO 00/47612
In a preferred embodiment, dendritic cell-dependent assays are provided that
include, but are not
limited to those described in: Guery et al., J. Immunol. 134:536-544 (1995);
Inaba et al., J. Exp. Med.
173:549-559 (1991); Macatonia et al., J. Immunol. 154:5071-5079 (1995);
Porgador et al., J. Exp.
Med. 182:255-260 (1995); Nair et al. J. Virology 67:4062-4069 (1993); Huang et
al., Science 264:961-
965 (1994); Macatonia et al., J. Exp. Med. 169:1255-1264 (1989); Bhardwaj et
al., J. Clinic. Invest.
94:797-807 (1994); Inaba et al., J. Exp. Med. 172:631-640 (1990); incorporated
as reference in its
entirety.
In another embodiment assays for lymphocyte survival/apoptosis are provided
that include, but are not
limited to those described in: Darzynkiewicz et al., Cytometry 13:795-808
(1992); Gorczyca et al.,
Leukemia 7:659-670 (1993); Gorczyca et al., Cancer Research 53:1945-1951
(1993); Itoh et al., Cell
66:233-243 (1991); Zacharchuk, J. Immunol. 145:4037-4045 (1990); Zamai et al.,
Cytometry 14: 891-
897 (1993); Gorczyca et al., Intl. J. Oncology 1:639-648 (1992); incorporated
as references in their
entirety.
In a preferred embodiment, assays for proteins that influence early steps of T-
cell commitment and
development are provided that include, but are not limited to those described
in: Antica et al., Blood
84:111-117 (1994); Fine et al., Cell. Immunol. 155:111-122 (1994); Galy et
al., Blood 85:2770-2778
(1995); Toki et al., Proc. NatLAcad. Sci. U.S.A. 88: 7548-7551 (1991);
incorporated as references in
their entirety.
In another preferred embodiment, assays for proliferation and differentiation
of various hematopoietic
cells are provided (cited above).
In one embodiment, assays for embryonic stem cell differentiation are provided
that include, but are
not limited to those described in: Johansson et al., Cell. Biol. 15:141-151
(1995); Keller et al., Mol. Cell.
Biol. 13:473-486 (1993); McClanahan et al., Blood 81:2903-2915 (1993);
incorporated as references in
their entirety.
2 5 In a further embodiment assays for stem cell survival and differentiation
are provided that include, but
are not limited to those described in: Culture of Hemai'opoietic Cells (R.I.
Freshney et al., eds . Wiley-
Liss, Inc. New York 1994); Hirayama et al., Proc. Natl. Acad. Sci. U.S.A.
89:5907-5911 (1992);
incorporated as references in their entirety.
In a preferred embodiment, assays for chemotactic activity are provided that
include, but are not
3 0 limited to those described in: Currenf Protocols in Immunology, supra;
Taub et al., J. Clin. Invest.
95:1370-1376 (1995); Lind et al., AP/MIS 103:140-146 (1995); Muller et al.,
Eur. J. Immunol. 25:1744-
58
CA 02362541 2001-08-10 pC'j'/US00/03665
WO 00/47612
1748; Gruber et al., J. Immunol. 152:5860-5867 (1994); Johnston et al., J.
Immunol. 153:1762-1768
(1994); incorporated as reference in their entirety.
In another preferred embodiment, assays for hemostatic and thrombolytic
activity are provided that
include, but are not limited to those described in: Linet et al., J. Clin.
Pharmacol. 26:131-140 (1986);
Burdick et al., Thrombosis Res. 45:413-419 (1987); Humphrey et al.,
Fibrinolysis 5:71-79 (1991);
Schaub, Prostaglandins 35:467-474 (1988); incorporated as references in their
entirety.
In another embodiment, assays for receptor-ligand activity are included and
include, but are not limited
to those described in: Currenf Protocols in Immunology, supra; Takai et al.,
Proc. Natl. Acad. Sci.
U.S.A. 84:6864-6868 (1987); Bierer et al., J. Exp. Med. 168:1145-1156 (1988);
Rosenstein et al., J.
Exp. Med. 169:149-160 (1989); Stoltenberg et al., J. Immunol. Methods 175:59-
68 (1994); Sitt et al.,
Cell 80:661-670 (1995); incorporated as reference in their entirety.
Generally, the biological activity of a cytokine is determined by cytokine
receptor-mediated signal
transduction events. Accordingly, the biological activity of a receptor analog
can be determined by
measuring the EC50 from cell proliferation and the level of tyrosine
phosphorylation following standard
procedures. Assays for determining biological activity are exemplified for the
naturally occurring
EPOR and/or EPOR analogs, but are applicable to all naturally occurring
receptors and receptor
analogs of the invention,
In a preferred embodiment, the EC50 is determined using a cell proliferation
assay. In one aspect of
this embodiment, FD-P1 cells are transfected with the full-length human EPOR,
(which includes either
2 0 a naturally occurring EPOR or a designed EPOR analog or comprises either a
naturally occurring ECD
or a designed ECD). EPO, EPO analogs and EMP are used to determine the EC50 by
incubating
them with the transfected cell following standard procedures.
In another preferred embodiment, the level of tyrosine phosphorylation is
determined. In one aspect
of this embodiment, FD-P1 cells are transfected with the full-length human
EPOR, (which includes
either a naturally occurring EPOR or a PDA designed EPOR analog or comprises
either a naturally
occurring ECD or a PDA designed ECD). Following published procedures, these
cells are stimulated
by EPO, EPO analogs and EMP, and then processed and isolated by
immunoprecipitation and
electrophoresis. The phosphorylation level may be measured by immunoblotting
antibodies.
Yeast and mammalian protein-protein interaction cloning systems (termed two-
hybrid interaction
3 0 screening systems) are described in the art (Fields et al., Nature 340:245
(1989); Vasavada et al.,
Proc. Natl. Acad. Sci. U.S.A. 88:10686 (1991); Fearon et al., Proc. Natl.
Acad. Sci. U.S.A. 89:7958
59
CA 02362541 2001-08-10 pCT/US00/03665
WO 00/47612
(1992); Dang et al., Mol. Cell. Biol. 11:954 (1991); Chien et al., Proc. Natl.
Acad. Sci. U.S.A. 88:9578
(1991); Luo et al., Biotechniques 22:350-352 (1997); and U.S. Patent Nos.
5,283,173; 5,667,973;
5,468,614; 5,525,490; and 5,637,463). The basic system requires a protein-
protein interaction in order
to tum on transcription of a reporter gene.
In a preferred embodiment, the receptor analogs of the invention are used in
cell free assays to screen
for ligand analogs that have the ability to modulate the signaling of receptor
analogs and or the
signaling of naturally occurring cell surface receptors.
In another preferred embodiment, the invention provides a three hybrid
interaction system for the
detection of ligand-receptor interaction in vivo. Briefly, the sequence of a
receptor analog is fused to a
DNA-binding domain, including, but not limited to those derived from Gal4 and
LexA, to generate a
DNA-binding-analog receptor fusion protein ("Fusion protein I"). Another
receptor analog sequence is
fused to a transcription activation domain including, but not limited to those
derived from VP16 and
Gal4 to generate a transcription-activation-domain-receptor analog fusion
protein ("Fusion protein II).
A reporter gene construct comprising a detectable marker and the genes
encoding fusion proteins I
and II are introduced into a eukaryotic cell (e.g., yeast or any mammalian
cell) by methods known in
the art. Detectable markers include, but are not limited to luciferase, GFP,
etc . Fusion protein I has
the capability to bind to a DNA binding site in the proximity of the
transcriptional start site for the gene
encoding the detectable marker. By adding a candidate ligand capable of
binding to the analog
receptor, and due to the binding of the candidate ligand to both receptor
analogs, i.e. to those receptor
2 0 analogs comprised by fusion proteins I and II, fusion protein II is
recruited to fusion protein I that is
bound to the promoter region of the detectable marker gene. As a consequence
thereof, the
transcription activation domain is brought into the vicinity of the
transcriptional machinery and
stimulates transcription of the detectable marker gene. The expression of the
detectable marker is
detected using various methods known in the art and depends on the detectable
marker used.
In another aspect of this embodiment, a candidate bioactive agent is added and
bioactive agents are
identified that in the above described three-hybrid system, lead to an
increase or decrease of reporter
gene activity. Thereby, bioactive agents having the capability of stabilizing
and/or destabilizing the
ligand-receptor interaction are identified.
In a preferred embodiment, the invention provides methods for screening for
bioactive agents that are
3 0 capable of modulating the interaction between the receptor analog and the
ligand analog.
In a preferred embodiment, the invention provides methods for screening for
bioactive agents that are
capable of modulating the interaction between the receptor analog and the
ligand analog. "Modulating
WO 00/47612 CA 02362541 2001-08-to pCT/US00/03665
the interaction between the receptor analog and the ligand analog" includes an
increase (i.e., tighter
affinity between the receptor analog and the ligand analog), a decrease (i.e.,
lower affinity between the
receptor analog and the ligand analog), or a change in the type or kind of
this interaction. Both in vivo
and in vitro systems (ceel free systems) are used in this invention to
identify bioactive agents that are
capable of modulating the interaction between the receptor analog and the
ligand analog.
By "bioactive agent" or grammatical equivalents thereof herein is meant any
molecule, e.g., protein,
small organic molecule, polysaccharide, lipid, polynucleotide, etc., or
mixtures thereof with the
capability of modulate the interaction between the receptor analog and the
ligand analog. Various
classes and libraries of proteins, small organic molecules, polysaccharides,
lipids, polynucleotides,
etc. are described above and also apply with respect to bioactive agents.
Further included within this
definition are molecules, as defined above, with the capability of modulating
the signaling activity of the
receptor analog.
Addition of the candidate bioactive agent is performed under conditions which
allow the modulation of
the interaction between the receptor analog and the ligand analog or the
modulation of the signaling
activity of the receptor analog to occur. As will be appreciated by those in
the art, those conditions will
depend upon the nature of the interaction, the nature of the candidate
bioactive agent, and are
determined routinely and empirically, as will the concentration of the
candidate bioactive agents to be
employed. Thus, in this embodiment, the candidate bioactive agent possesses a
size or structure
2 0 which allows binding to either receptor analog or the ligand analog
(although this may not be
necessary), and modulate the interaction between them. This modulation
preferably results in a
measurable change of the signaling activity of the receptor analog.
Accordingly, in one embodiment, the method of screening for bioactive agents
that are capable of
modulating the interaction between a receptor analog and a ligand analog
comprises the steps of (1)
providing a host cell comprising a vector composition, comprising a gene
encoding a receptor analog
and a gene encoding a ligand analog. This vector composition may or may not
comprise retroviral
vectors, and may or may not be integrated into the genome of the host cell.
(2) The host cell is
subjected to conditions under which the genes encoding the receptor analog and
the ligand analog are
expressed to produce a receptor analog and a ligand analog. Optionally, it is
determined (3) whether
3 0 the receptor analog is displayed on the surface of the host cell, e.g., by
using immunohistochemical
and other methods as known in the art. (4) Optionally it is determined whether
the ligand analog is
bound to the receptor analog. (5) Candidate bioactive agents that are capable
of modulating the
interaction between the receptor analog and the ligand analog are added.
Simultaneously,
sequentially or at a later step (6) the interaction between the ligand analog
and the receptor analog is
3 5 determined, e.g., by using cytokine, cell proliferation, cell
differentiation assays, and other assays that
61
WO 00/47612 CA 02362541 2001-08-to pCT~S00/03665
are further described below. (7) The interaction between the ligand analog and
the receptor analog in
the presence of a bioactive agent is compared to the interaction between the
ligand analog and the
receptor analog in the absence of a bioactive agent. Preferably, (8) the
bioactive agents are identified.
Bioactive agents identified by the, subject methods may find use as new small
molecule drug leads,
inhibitors, activators, diagnostic reagents, and the like.
In another preferred embodiment, the method of screening for bioactive agents
that are capable of
modulating the signaling activity of a receptor analog comprises the steps of
(1 ) providing a host cell
comprising a vector composition, comprising a gene encoding a receptor analog
and a gene encoding
a ligand analog. This vector composition may or may not comprise retroviral
vectors, and may or may
not be integrated into the genome of the host cell. (2) The host cell is
subjected to conditions under
which the genes encoding the receptor analog and the ligand analog are
expressed to produce a
receptor analog and a ligand analog. Optionally, it is determined (3) whether
the receptor analog is
displayed on the surface of the host cell, e.g., by using immunohistochemical
and other methods as
known in the art. (4) Optionally it is determined whether the ligand analog is
bound to the receptor
analog. (5) Optionally the signaling activity of the receptor analog in
response to the ligand analog is
determined. (6) Candidate bioactive agents that are capable of modulating the
signaling activity of the
receptor analog are added. Simultaneously, sequentially or at a later step (7)
the modulation of
signaling activity of the receptor analog is determined, e.g., by using
cytokine, cell proliferation, cell
differentiation assays, and other assays that are further described below. (8)
The signaling activity of
2 0 the receptor analog in the presence of a bioactive agent is compared to
the signaling activity of the
receptor analog in the absence of a bioactive agent. Preferably, (9) the
bioactive agents are identified.
Bioactive agents identified by the subject methods may find use as new small
molecule drug leads,
inhibitors, activators, diagnostic reagents, and the like.
In another preferred embodiment of the above invention, instead of providing a
gene encoding a ligand
analog, the method comprises the step of providing a gene encoding a natural
ligand.
In another preferred embodiment of the above invention, instead of providing a
gene encoding a ligand
analog, the method comprises the step of providing the ligand analog as a
recombinant protein to the
host cell comprising a receptor analog.
In another preferred embodiment of the above invention, instead of providing a
gene encoding a ligand
3 0 analog, the method comprises the step of providing a natural ligand as a
recombinant protein to the
host cell comprising a receptor analog.
62
CA 02362541 2001-08-10
WO 00/47612 PCT/US00/03665
In a preferred embodiment, it is desired to screen for bioactive agents that
are antagonists, i.e., the
libraries of bioactive agents is used to identify bioactive agents that (i)
decrease the interaction
between the receptor analog and the analog ligand or natural ligand or (ii)
decrease the signaling
activity of the receptor analog.
In a preferred embodiment, it is desired to screen for bioactive agents that
are agonists, i.e., the
libraries of bioactive agents is used to identify bioactive agents that (i)
increase the interaction between
the receptor analog and the analog ligand or natural ligand or (ii) increase
the signaling activity of the
receptor analog.
In a preferred embodiment, the candidate bioactive agent or the candidate
ligand is a protein which is
encoded by a cDNA, cDNA fragment or genomic DNA fragment (for example, as part
of a cDNA or
genomic library) and is readily identified by rescuing the nucleic acid
encoding the candidate bioactive
agent. The nucleic acid sequence is determined. As known in the art, the
obtained information may
be used to isolate a full-length cDNA encoding the full-length candidate
bioactive agent and to express
the candidate bioactive agent as a recombinant protein. Preferably, the full-
length recombinant
candidate bioactive agent (either in form of a full-length cDNA or as a full-
length protein) may be
purified, labeled and used in in vivo and in in vitro binding assays (as
outlined herein) to confirm e.g.,
its modulation of the signaling activity of a receptor analog.
In a preferred embodiment, the modulation of the signaling activity of a
surface receptor analog by a
candidate bioactive agent is optimized. The identified candidate bioactive
agent is either chemically
2 0 modified or the nucleic acid encoding the candidate bioactive agent is
subjected to in vitro
mutagenesis or to the PDA methodology, as described herein. These
modifications result in the
synthesis of candidate bioactive agent variants. Preferably, these variants
are purified, labeled and
used in in vivo and in in vitro binding assays (as outlined herein) to test
their modulation of the
signaling activity of receptor analog. These variants lead either to more
potent, more tolerable or less
toxic small molecule drug leads, inhibitors, activators, diagnostic reagents,
and the like.
In a preferred embodiment, the efficacy of the candidate bioactive agent
variant (i.e., its
characteristics, its modulation of the signaling activity of the receptor
analog, its binding to the receptor
analog, etc.) is compared to the efficacy of the originally isolated candidate
bioactive variant using in
vitro binding assays and in vivo assays (as outlined herein). In this
embodiment, the in vitro binding
3 0 assays comprise at least four components: a receptor analog, a ligand
analog (or a natural ligand), an
originally identified candidate bioactive agent and a candidate bioactive
agent variant.
63
CA 02362541 2001-08-10
WO 00/47612 PCT/US00/03665
In a preferred embodiment, the invention provides in vitro (i.e. cell free)
methods for screening for
bioactive agents that are capable of modulating the interaction between a
receptor analog and a ligand
analog.
In one embodiment, a receptor analog is bound to an insoluble support and a
ligand analog (or natural
ligand), which may be labeled, is added and allowed to bind to the receptor
analog. Incubations are
performed at any temperature which facilitates optimal binding, typically
between 4°C and 40°C.
Incubation periods are selected for optimum binding, but are also optimized to
facilitate rapid high
through put screening. Typically between 0.1 and 3 hour is sufficient. Excess
of labeled ligand
analogs (or natural ligands) is generally removed or washed away. The original
candidate bioactive
agent or a variant thereof is then added, and the presence or absence of the
labeled ligand analog (or
natural ligand) in the wash solution or supernatant is followed, to indicate a
possible displacement by
the candidate bioactive agent or its variant.
In this embodiment, displacement of the ligand analog (or natural ligand) is
an indication that the
candidate bioactive agent or a variant thereof is modulating the interaction
between the receptor
analog and the ligand analog (or natural ligand) and thus functions as
antagonist. A displacement of
more ligand analog (or natural ligand) by the candidate bioactive agent
variant (i.e., when compared to
the original candidate bioactive agent) indicates that the variant is a
stronger antagonist which may be
developed as a more potent small molecule drug lead, inhibitor, activator,
diagnostic reagent, or the
like. Alternatively, a displacement of less ligand analog (or natural ligand)
by the candidate bioactive
2 0 agent variant (i.e., when compared to the original candidate bioactive
agent) which indicates that the
variant is a weaker antagonist can lead to the development of a more tolerable
or less toxic small
molecule drug lead, inhibitor, activator, diagnostic reagent, or the like.
In another embodiment, the original candidate bioactive agent or a variant
thereof is added first to the
receptor analog, which is bound to an insoluble support, with incubation and
washing, followed by the
addition of the ligand analog (or natural ligand), which may be labeled, with
incubation and washing.
Absence of binding of the ligand analog (or natural ligand) or reduced binding
thereof when compared
to a control sample may indicate that the original bioactive agent is bound to
the receptor analog with a
high affinity and may mask the interaction surface for the ligand analog (or
natural ligand).
Alternatively, the original candidate bioactive agent may have changed the
tertiary structure of the
3 0 receptor analog and thereby rendered the receptor analog unable to
functionally interact with the
ligand analog (or natural ligand). More or less binding of the ligand analog
(or natural ligand), when
used in combination with the candidate bioactive agent variant (and when
compared to the original
candidate bioactive agent) indicates a weaker or stronger binding of the
variant to the receptor analog.
The ramifications drawn, are similar to those outlined above.
64
WO 00/47612 CA 02362541 2001-08-10 pCT/US00/03665
The following examples serve to more fully describe the manner of using the
above-described
invention, as well as to set forth the best modes contemplated for carrying
out various aspects of the
invention. It is understood that these examples in no way serve to limit the
true scope of this invention,
but rather are presented for illustrative purposes.
The practice of the present invention will employ, unless otherwise indicated,
conventional methods of
chemistry, biochemistry, microbiology, molecular biology, cell biology,
recombinant DNA techniques
and computational analyses within the skill of the art. Such techniques are
explained fully in the
literature. All publications, patents and patent applications cited herein,
whether supra or infra, are
hereby incorporated by reference in their entirety.
Example 1
Designing the EPOR se4uences using PDA with multiple strategies and structure
complexes
Following the steps outlined above for designing receptor analogs using PDA,
the following sequences
using three structures of EPOR in 1 ebp, 1 eer and 1 blw (also called 1 cn4)
were designed, resulting in EPOR
analogs (see Figure 8).
~ First PDA Design: D1 and D2 domain
1. PDA design of EPO receptors based on (EPOR+EMP1)Z dimer complex 1ebp,
(EPOR)zEPO complex
1 eer, and 1 blw.
2. The core was divided into two subdomains: D1 and D2
3. Sequences with betterthan wild type energy are chosen: two for D1 and only
onefor D2 and a combination
2 0 of D1 and D2 using backbone independent rotamer libraries and one for D1
using backbone dependent
rotamer library for 1 ebp alone.
~. Second PDA design:
1. elbow PDA design based on (EPOR+EMP1)2 dimer complex 1ebp, (EPOR)ZEPO
complex leer,
2 5 and 1 blw.
2. PDA design of EPOR elbow involving the buried residues between the
interfaces of domain D1, domain
D2, the N-terminal helix H and the WSXWS-box.
3. Both the EPOR dimer and its monomers (amino acid residues 1-211 ) and
(amino acid resid ues 222-422)
were designed and listed (see Figure 8).
CA 02362541 2001-08-10
WO 00/47612 PCT/US00/03665
Example 2
Fusion of coiled coil to EPOR analogs
The designed EPOR analog is linked bya GGGGS linker sequence to a PDA designed
coiled-coil sequence
(RMEKLEQKVKELLRKNERLEEEVERLKQLVGER, based on the GCN4 structure.
For example: 1 ebp d12 GCN4 (see also Example 3) is composed of 1 ebp d12
(1ebp_d1 add 1ebp_d2
mutants together), plus GGGGS, plus designed GCN4 sequence.
Example 3
Cloning expression refolding purification and characterization of EPOR analogs
Human wild type EPOR (amino acid positions 1-225) (called EPOR), its fusion
construct linked bya GGGGS
linker sequence to a coiled-coil (called EPOR GCN4) and a mutant linked by a
GGGS linker sequence
to a coiled-coil (called 1 ebp_d12 GCN4) were cloned using standard
techniques. The DNA sequences
were synthesized using a series of overlapping oligonucleotides and amplified
by the polymerase chain
reaction (PCR), cloned into the expression vector PET21 a, and transfected
into E. coli. Some proteins
were expressed in yeast using known methods in the art. Using standard
techniques, the proteins were
refolded from inclusion bodies and purified at the expected molecular weight
using size-exclusion
chromatography calibrated by standard proteins. These purified proteins were
further purified using C4
reverse phase column chromatography to obtain the mass spectra of these
proteins at the expected
molecularweight. The proteins show cooperative thermal melting curves as
monitored by circular dichroism
(CD) (data not shown). A western blot analysis of all three proteins, EPOR and
EPOR analogs with the
2 0 EPOR antibody confirmed the expression, the size of the respective
proteins and the crossreactivity with
EPOR antibodies (data not shown).
66