Note: Descriptions are shown in the official language in which they were submitted.
CA 02363503 2008-10-17
Diagnostics and Therapeutics for Drusen Associated Ocular Disorders
1. Background of the Invention
Macular degeneration is a clinical term that is used to describe a variety of
diseases that are all characterized by a progressive loss of central vision
associated with
abnormalities of Bruch's membrane, the neural retina and the retinal pigment
epithelium.
These disorders include very common conditions that affect older patients (age-
related
macular degeneration or AMD) as well as rarer, earlier-onset dystrophies that
in some cases
can be detected in the fnst decade of life (Best F. Z, Augenheilkd., 13:199-
212, 1905; Sorsby,
A., et al., Br J. Opthalmol. 33:67-97, 1949; Stargardt, K., Albrecht Von
Graefes Arch Klin
Exp Opthalmol. 71: 534-550, 1909; Ferrell, R. E., et al., Am J. Hum
Genet.35:78-84, 1983;
Jacobson, D. M., et al., Ophthalmology, 96:885-895, 1989; Small, K. W., et al.
Genomics
13:681-685, 1992; Stone, E. M., et al., Nature Genet. 1:246-250, 1992;
Forsman, K., et al.
Clin Genet. 42:156-159, 1992; Kaplan, J. S., et al. Nature Genet. 5:308-311,
1993; Stone, E.
M., et al. Arch Opthalmol. 112:763-772,1994; Zhang, K., et al. Arch Opthalmol.
112:759-
764, 1994; Evans, K., et al. Nature Genet. 6:210-213, 1994; Kremer, H., et al.
Hum Mol
Genet. 3:299-302, 1994; Kelsell, R E., et al. Hum Mol Genet. 4:1653-1656,
1995; Nathans, J.,
et al. Science 245:831-838, 1989; Wells, J., et al. Nature Genet. 3:213-218,
1993; Nichols, B.
E., et al. Nature Genet.3:202-207, 1993a; Weber, B. H. F. , et al. Nature
Genet. 8:352-355,
1994). Macular degeneration
diseases include, for example, age- related macular degeneration, North
Carolina macular
dystrophy, Sorsby's fundus dystrophy, Stargardt's disease, pattern dystrophy,
Best disease,
malattia leventinese, Doyne's honeycomb choroiditis, dominant drusen and
radial drusen.
A number of gene loci have been reported as indicating a predisposition to
macular degeneration: l p21-q i 3, for recessive Stargardt's disease or fundus
flavi maculatus
(Allikmets, R. et al. Science 277:1805-1807,1997; Anderson, K. L. et al., Am.
J. Hum. Genet.
55:1477, 1994; Cremers, F. P. M. et al., Hum. Mol. Genet. 7:355-362, 1998;
Gerber, S. et al.,
Am. J. Hum. Genet. 56:396-399, 1995; Gerber, S. et al., Genomics 48:139-142,
1998; Kaplan,
J. et al., Nat. Genet. 5:308-311, 1993; Kaplan; J. et a1., Am. J. Hum. Genet.
55:190, 1994;
Martinez-Mir, A. et al., Genomics 40:142-146, 1997; Nasonkin, I. et al., Hum.
Genet. 102:21-
26, 1998; Stone, E. M. et al., Nat. Genet. 20:328-329, 1998); 1q25-q31, for
recessive age-
related macular degeneration (Klein, M. L. et al., Arch. Ophthalmol. 116:1082-
1088, 1988);
1
CA 02363503 2001-08-24
WO 00/52479 PCTIUSOO/05858
2p 16, for dominant radial macular drusen, dominant Doyne honeycomb retinal
degeneration or
Malattia Leventinese (Edwards, A. O. et al., Am. J. Ophthalmol. 126:417-424,
1998; Heon, E.
et al., Arch. Ophthalmol. 114:193-198, 1996; Heon, E. et al.,. Invest.
Ophthalmol Vis. Sci.
37:1124, 1996; Gregory, C. Y. et al., Hum. Mol. Genet. 7:1055-1059, 1996);
6p21.2-cen, for
dominant macular degeneration, adult vitelloform (Felbor, U. et al. Hum.
Mutat. 10:301-309,
1997); 6p21.1 for dominant cone dystrophy (Payne, A.. M. et al. Am. J. Hum.
Genet. 61:A290,
1997; Payne, A.. M. et al., Hum. Mol. Genet. 7:273-277, 1998; Sokol, I. et
al., Mol. Cell.
2:129-133, 1998); 6q, for dominant cone-rod dystrophy (Kelsell, R. E. et al.
Am. J Hum.
Genet. 63:274-279, 1998); 6q11-q15, for dominant macular degeneration,
Stargardt's-like
(Griesinger, I. B. et al., Am. J. Hum. Genet. 63:A30, 1998; Stone, E.M. et
al., Arch.
Ophthalmol. 112:765-772, 1994); 6q14-q16.2, for dominant macular degeneration,
North
Carolina Type (Kelsell, R. E. et al., Hum. Mol. Genet. 4:653-656, 1995; Robb,
M. F. et al.,
Am. J. Ophthalmol. 125:502-508, 1998; Sauer, C. G. et al., J. Med. Genet.
34:961-966, 1997;
Small, K. W. et al., Genomics 13:681-685, 1992; Small, K. W. et al., Mol. Vis.
3:1, 1997);
6q25-q26, dominant retinal cone dystrophy 1(Online Mendelian Inheritance in
Man (TM).
Center for Medical Genetics, Johns Hopkins University, and National Center for
Biotechnology Information, National Library of Medicine
(http://www3.ncbi.nlm.nih.gov/omim, (1998)); 7p21-p15, for dominant cystoid
macular
degeneration (Inglehearn, C. F. et al., Am. J. Hum. Genet. 55:581-582, 1994;
Kremer, H. et al.,
Hum. Mol. Genet. 3:299-302, 1994); 7q31.3-32, for dominant tritanopia,
protein: blue cone
opsin (Fitzgibbon, J. et al., Hum. Genet. 93:79-80, 1994; Nathans, J. et al.,
Science 193:193-
232, 1986; Nathans, J. et al., Ann. Rev. Genet.26:403-424, 1992; Nathans, J.
et al., Am. J.
Hum. Genet. 53:987-1000, 1993; Weitz, C. J. et al., Am. J. Hum. Genet. 50:498-
507, 1992;
Weitz, C. J. et al., Am. J. Hum. Genet. 51:444-446, 1992); not 8q24, for
dominant macular
degeneration, atypical vitelliform (Daiger, S. P. et al., In `Degenerative
Retinal Diseases',
LaVail, et al., eds. Plenum Press, 1997; Ferrell, R. E. et al., Am. J. Hum.
Genet. 35:78-84,
1983; Leach, R. J. et al., Cytogenet. Cell Genet. 75:71-84, 1996; Sohocki, M.
M. et al., Am. J.
Hum. Genet. 61:239-241, 1997); 11p12-q13, for dominant macular degeneration,
Best type
(bestrophin) (Forsman, K. et al., Clin. Genet. 42:156-159, 1992; Graff, C. et
al., Genomics,
24:425-434, 1994; Petrukhin, K. et al., Nat. Genet. 19:241-247, 1998;
Marquardt, A. et al.,
Hum. Mol. Genet. 7:1517-1525, 1998; Nichols, B. E. et al., Am. J Hum. Genet.
54:95-103,
1994; Stone, E. M. et al., Nat. Genet. 1:246-250, 1992; Wadeilus, C. et al.,
Am. J. Hum. Genet.
53:1718, 1993; Weber, B. et al., Am. J. Hum. Genet. 53:1099, 1993; Weber, B.
et al., Am. J
2
CA 02363503 2008-10-17
Hum. Genet. 55:1182-1187, 1994; Weber, B. H., Genomics 20: 267-274, 1994;
Zhaung, Z. et
al., Am. J Hum. Genet. 53:1112, 1993); 13q34, for dominant macular
degeneration, Stargardt
type (Zhang, F. et al., Arch. Ophthalmol. 112:759-764, 1994); 16p12.1, for
recessive Batten
disease (ceroid-lipofuscinosis, neuronal 3), juvenile; protein:Batten disease
protein (Batten
Disease Consortium, Cell 82:949-957, 1995; Eiberg, H. et al., Clin. Genet.
36:217-218, 1989;
Gardiner, M. et al., Genomics 8:387-390, 1990; Mitchison, H. M. et al., Am. J.
Hum. Genet.
57:312-315, 1995, Mitchison, H. M. et al., Am. J. Hum. Genet. 56:654-662,
1995; Mitchison,
H. M. et al., Genomics 40:346-350, 1997; Munroe, P. B. et al., Am. J. Hum.
Genet. 61:310-
316, 1997; 17p, for dominant areolar choroidal dystrophy (Lotery, A. J. et
al., Ophthalmol.
Vis. Sci.37:1124, 1996); 17p13-p12, for dominant cone dystrophy, progressive
(Balciuniene, J.
et al., Genomics 30:281-286, 1995; Small, K. W. et al., Am. J. Hum. Genet.
57:A203, 1995;
Small, K. W. et al., Am. J. Ophthalmol. 121:13-18, 1996); 17q, for cone rod
dystrophy
(Klystra, J. A. et al., Can. J. Ophthalmol. 28:79-80, 1993); 18q21.1-q21.3,
for cone-rod
dystrophy, de Grouchy syndrome (Manhant, S. et al., Am. J. Hum. Genet. 57:A96,
1995;
Warburg, M. et al., Am. J. Med. Genet. 39:288-293, 1991); 19q13.3, for
dominant cone-rod
dystrophy; recessive, dominant and 'de novo' Leber congenital amaurosis;
dominant RP;
protein: cone-rod otx-like photoreceptor homeobox transcription factor
(Bellingham, J. et al.,
In `Degenerative Retinal Diseases', LaVail, et al., eds. Plenum Press, 1997;
Evans, K. et al.,
Nat. Genet. 6:210-213, 1994; Evans, K. et al., Arch. Ophthalmol. 113:195-201,
1995; Freund,
C. L. et al., Ce1191:543-553, 1997; Freund, C. L. et al., Nat. Genet. 18:311-
312, 1998;
Gregory, C. Y. et al., Am. J. Hum. Genet. 55:1061-1063, 1994; Li, X. et al.,
Proc. Natl. Acad.
Sci USA 95:1876-1881, 1998; Sohocki, M. M. et al., Am. J. Hum. Genet. 63:1307-
1315, 1998;
Swain, P. K. et al., Neuron 19:1329-1336, 1987; Swaroop, A. et al., Hum. Mol.
Genet. In
press, 1999); 22q 12.1-q 13.2, for dominant Sorsby's fundus dystrophy, tissue
inhibitors of
metalloproteases-3 (TIMP3) (Felbor, U. et al., Hum. Mol. Genet. 4:2415-2416,
1995; Felbor,
U. et al., Am. J Hum. Genet. 60:57-62, 1997; Jacobson, S. E. et al., Nat.
Genet. 11:27-32,
1995; Peters, A. et al., Retina 15:480-485, 1995; St6hr, H. et al., Genome
Res. 5:483-487,
1995; Weber, B. H. F. et al., Nat. Genet. 8:352-355, 1994; Weber, B. H. F. et
al., Nat. Genet.
7:158-161, 1994; Wijesvriya, S. D. et al., Genome Res. 6:92-101, 1996); and
Xp11.4, for X-
linked cone dystrophy (Bartley, J. et al., Cytogenet. Cell. Genet. 51:959,
1989; Bergen, A. A.
B. et al., Genomics 18:463-464, 1993; Dash-Modi, A. et al., Invest.
Ophthalmol. Vis. Sci.
37:998, 1996; Hong, H.-K., Am. J. Hum. Genet 55:1173-1181, 1994; Meire, F. M.
et al., Br. J.
Ophthalmol. 78:103-108, 1994; Seymour, A. B. et al., Am. J. Hum. Genet. 62:122-
129, 1998).
3
CA 02363503 2008-10-17
In addition, the world wide web
site http://VWVW.SPH.UTH.TMC.EDU /RETNET/disease.htm lists genetic
polymorphisms
for macular degenerations and for additional retinal degenerations that also
may be associated
with macular degeneration. However, none of the above genes or polymorphisms
has been
found to be responsible for a significant fraction of typical late-onset age-
related macular
degeneration. Although a recent report suggested that mutations in the
photoreceptor ABCR
rim protein cause up to 15% of AMD cases in the United States (Allikmets, et
al., 1997),
conflicting results have been obtained by different investigators (De La Paz,
et al., 1998; Stone
et al., 1998).
Age-related macular degeneration (AMD), the most prevalent macular
degeneration is associated with progressive diminution of visual acuity in the
central portion
of the visual field, changes in color vision, and abnormal dark adaptation and
sensitivity
(Steinmetz, et al., 1993; Brown & Lovie-Kitchin, 1983; Brown, et al., 1986;
Sunness, et al.,
1985; Sunness, et al., 1988; Sunness, et al., 1989; Eisner, et al., 1987;
Massof, et al., 1989;
Chen, et al., 1992).
AMD is the leading cause of legal blindness in North America and Western
Europe (Hyman, 1992) and has become a significant health problem as the
percentage of
individuals above the age of 50 increases. In the Beaver Dam, Wisconsin
population, the
incidence of AMD was estimated to be 9.2% for persons over the age of 40
(Klein, et al.,
1995). The Framingham Eye Study found the overall incidence of AlVID to be
8.8%, with a
27.9% incidence in the 75-85 year old population (Kahn, et al., 1977;
Leibowitz, et al., 1980).
In an Australian study, 18.5% of those over age 85 were estimated to be
afflicted with AMD
(O'Shea, 1996). Variations in estimated incidence are likely a result of the
use of different
criteria for a diagnosis of AMD in different studies, or they may result from
different risk
factors among the various populations studied.
Two principal clinical manifestations of AMD have been described, both of
which can occur in the same patient (Green and Key, 1977). They are referred
to as the dry, or
atrophic, form, and the wet, or exudative, form (Sarks and Sarks, 1989; Elman
and Fine, 1989;
Kincaid, 1992). The most significant risk factor for the development of both
forms are age
and the deposition of drusen, abnormal extracellular deposits, behind the
retinal pigment
epithelium (RPE). In the dry form of AMD, the RPE and retina degenerate
without coincident
neovascularization. The region of atrophy that results is referred to as
geographic atrophy.
While atrophic AMD is typically considered less severe than the exudative form
because its
4
CA 02363503 2001-08-24
WO 00/52479 PCTIUSOO/05858
onset is less sudden, no treatment is effective at halting or slowing its
progression. In the less
common, but more devastating, exudative form, neovascular "membranes" derived
from the
choroidal vasculature invade Bruch's membrane, leak, and often cause
detachments of the RPE
and/or the neural retina (Elman and Fine, 1989). This event can occur over a
short period of
time and can lead to rapid and permanent loss of central vision. If one eye is
affected, there is
a high degree of probability that the second eye will develop a choroidal
neovascular
membrane within five years of the initial event (Macular Photocoagulation
Study, 1977).
Important clinical signs of neovascular AMD include gray-green neovascular
membranes,
dome-shaped RPE detachments, and disciform scars (caused by proliferation of
fibroblasts and
retinal glial cells) which are best visualized by their hyperfluorescence on
fluorescein
angiography (Elman and Fine, 1989). Killingsworth et al. (1990) suggested that
macrophages
may participate in the breakdown of Bruch's membrane in the neovascular stage
of AMD and
in drusen regression, and show one electron micrograph depicting structures
resembling
drusen cores. Duvall and Tso (1985) showed choroidal macrophages in the region
of the
Bruch's membrane are involved in the removal of drusen in monkey eyes,
following laser
photocoagulation. Penfold and others (Penfold et al., 1985; Penfold et al.,
1986; Oppenheim
and Leonard, 1989) provided "circumstantial evidence ... for the involvement
of (choroidal)
leukocytes, in the promotion of neovascular proliferation." However, these
data were
restricted to morphological observations only and only suggest that
macrophages only
participate in the neovascularization stage of drusen formation.
A number of population-based studies indicate that AMD has a genetic
component, based upon the examination of the rates of AMD in different racial
groups and the
degree of familial aggregation of AMD (Hyman, et al., 1983). For example,
Caucasians
appear to be at greater risk than individuals of Hispanic origin
(Cruickshanks, et al., 1997). In
addition, a black population on Barbados had a lower incidence of advanced AMD
than the
local Caucasian population (Schachat, et al., 1995). Studies involving twins
and other siblings
have demonstrated that, the more related two individuals are, the more likely
they are to be at
the same risk of developing AMD (Heiba, et al., 1994; Klein, et al., 1994;
Meyers and
Zacchary, 1988; Meyers, 1994; Meyers, et al., 1995; Piguet, et al., 1993;
Seddon, et al., 1997;
Silvestri, et al., 1994). These findings suggest that heredity contributes
significantly to an
individual's risk of developing AMD, but the gene(s) responsible have not been
identified.
Other maculopathies, typically with an earlier onset of symptoms than AMD,
have been described. These include North Carolina macular dystrophy (Small, et
al., 1993),
5
CA 02363503 2001-08-24
WO 00/52479 PCTIUSOO/05858
Sorsby's fundus dystrophy (Capon, et al., 1989), Stargardt's disease (Parodi,
1994), pattern
dystrophy (Marmor and Byers, 1977), Best disease (Stone, et al., 1992),
dominant drusen
(Deutman and Jansen, 1970), and radial drusen ("malattia leventinese") (Heon,
et al., 1996).
Several of these inherited disorders, including those that map to distinct
chromosomal loci or
for which the genes have been identified, are characterized by the presence of
drusen (or other
extracellular deposits in the subRPE space). Based on this information, it is
likely that: (1)
AMD is not a single, genetic disease, since different diseases with distinct
chromosomal loci
share morphologic differences (Holz, et al., 1995a; Mansergh et al., 1995; and
(2) that drusen
may develop as a result of a biological pathway induced by a variety of
different insults,
genetic or otherwise. Determining whether AMD is a genetic or an acquired
disorder is
problematic, since AMD may actually be several diseases, and thus defy simple
categorization; indeed, both genetic and environmental factors appear to play
some role in its
development.
"Environmental" conditions may modulate the rate at which an individual
develops AMD or the severity of the disease. Light exposure has been proposed
as a possible
risk factor, since AMD most severely affects the macula, where light exposure
is high.
(Young, 1988; Taylor, et al., 1990; Schalch, 1992). The amount of time spent
outdoors is
associated with increased risk of choroidal neovascularization in men, and
wearing hats and/or
sunglasses is associated with a decreased incidence of soft drusen
(Cruickshanks, et al., 1993).
Accidental exposure to microwave irradiation has also been shown to be
associated with the
development of numerous drusen (Lim, et al., 1993). Cataract removal and light
iris
pigmentation has also been reported as a risk factor in some studies
(Sandberg, et al., 1994).
This suggests that: 1) eyes prone to cataracts may be more likely to develop
AMD; 2) the
surgical stress of cataract removal may result in increased risk of AMD, due
to inflammation
or other surgically-induced factors; or 3) cataracts prevent excessive light
exposure from
falling on the macula, and are in some way prophylactic for AMD. While it is
possible that
dark iris pigmentation may protect the macula from light damage, it is
difficult to distinguish
between iris pigmentation alone and other, cosegregating genetic factors which
may be actual
risk factors.
Dietary factors may also influence an individual's risk of developing AMD.
Anecdotal evidence from Japan suggests that the incidence of AMD, while very
low 20 years
ago, has increased as urban Japanese acquired a more Western diet and
lifestyle (Bird, 1997).
Chemical exposure (Hyman, et al., 1983), smoking (Vingerling, et al., 1996),
cardiovascular
6
CA 02363503 2001-08-24
WO 00/52479 PCT/US00/05858
disease/atherosclerosis (Hyman, et al., 1983; Vingerling, et al., 1995;
Blumenkranz, et al.,
1986), hypertension (Christen, et al., 1997), dermal elastotic changes in non-
sun exposed skin
(Blumenkranz, et al., 1986), dietary fat intake (Mares-Perlman, et al.,
1995b), low
concentrations of serum lycopene (Mares-Perlman, et al., 1995a), and alcohol
consumption
(Ritter, et al., 1995) have been identified, in some studies, as additional
risk factors for the
development of wet and/or dry AMD. One recent prospective dietary study found
that it is
often possible to increase macular pigment density and/or serum concentrations
of lutein and
zeaxanthin by dietary intake (Hammond, et al., 1997), although the
significance of this
alteration in modulating macular disease remains to be determined. Thus,
dietary consumption
of some vegetables, (e.g., spinach, collard greens, kale) may be inversely
associated with the
risk of developing AMD (Seddon, et al., 1994), an effect which is presumably
due to their
lutein and zeaxanthin content.
Histopathologic studies have documented significant and widespread
abnormalities in the extracellular matrices associated with the RPE, choroid,
and
photoreceptors of aged individuals and of those with clinically-diagnosed AMD
(Sarks, 1976;
Sarks, et al., 1988; Bird, 1992a; van der Schaft, et al., 1992; Green and
Enger, 1993; Feeney-
Burns and Ellersieck, 1985; Young, 1987; Kincaid, 1992). The most prominent
extracellular
matrix (ECM) abnormality is drusen, deposits that accumulate between the RPE
basal lamina
and the inner collagenous layer of Bruch's membrane (Figure 1). Drusen appear
to affect
vision prior to the loss of visual acuity; changes in color contrast
sensitivity (Frennesson, et
al., 1995; Holz, et al., 1995b; Midena, et al., 1994; Stangos, et al., 1995;
Tolentino, et al.,
1994), macular recovery function, central visual field sensitivity, and
spatiotemporal contrast
sensitivity (Midena, et al., 1997) have been reported.
A number of studies have demonstrated that the presence of macular drusen is a
strong risk factor for the development of both atrophic and neovascular AMD
(Gass, 1973;
Lovie-Kitchin and Bowman, 1985; Lewis, et al., 1986; Sarks, 1980; Sarks, 1982;
Small, et al.,
1976; Sarks, et al., 1985; Vinding, 1990; Bressler, et al., 1994; Bressler, et
al., 1990; Macular
Photocoagulation Study). Pauleikhoff, et al. (1990) demonstrated that the
size, number,
density and extent of confluency of drusen are important determinants of the
risk of AMD.
The risk of developing neovascular complications in patients with bilateral
drusen has been
estimated at 3-4% per year (Mimoun, et al., 1990). A recent report from the
Macular
Photocoagulation Study Group shows a relative risk of 2.1 for developing
choroidal
neovascularization in eyes possessing 5 or more drusen, and a risk of 1.5 in
eyes with one or
7
CA 02363503 2001-08-24
WO 00/52479 PCTIUSOO/05858
more large drusen (Macular Photocoagulation Study, 1997). The correlation
between drusen
and AMD is significant enough that many investigators and clinicians refer to
the presence of
soft drusen in the macula, in the absence of vision loss, as "early AMD"
(Midena, et al., 1997;
Tolentino, et al., 1994), or "early age-related maculopathy" (Bird, et al.,
1995). In addition to
macular drusen, Lewis et al. (1986) found that the degree of extramacular
drusen is also a
significant risk factor for the development of AMD. A few clinical studies
have shown that
drusen regress and that visual acuity improves in some cases, following laser
photocoagulation
(Sigelman, 1991; Little, et al., 1997; Figueroa, et al., 1994; Frenneson and
Nilsson, 1996).
While prophylactic laser treatment may be helpful for some patients (Little,
et al., 1997), it
appears that other patients react adversely to laser treatment of the macula
(Hyver, et al.,
1997). In addition, while there may be long term benefits for the patient
following
photocoagulation, these may not be worth the loss of vision frequently
associated with this
procedure.
Drusen accumulate between the RPE basal lamina and the inner collagenous
layer of Bruch's membrane. They cause a lateral stretching of the RPE
monolayer and
physical displacement of the RPE from its immediate vascular supply, the
choriocapillaris.
This displacement creates a physical barrier that may impede normal metabolite
and waste
diffusion between the choriocapillaris and the retina. It is likely that
wastes may be
concentrated near the RPE and that the diffusion of oxygen, glucose, and other
nutritive or
regulatory serum-associated molecules required to maintain the health of the
retina and RPE
are inhibited. It has also been suggested that drusen perturb photoreceptor
cell function by
placing pressure on rods and cones (Rones, 1937) and/or by distorting
photoreceptor cell
alignment (Kincaid, 1992).
The terminology most commonly used to distinguish drusen phenotypes is hard
and soft (see, for example, Eagle, 1984; Lewis, et al., 1986; Yanoff and Fine,
1992; Newsome,
et al., 1987; Mimoun, et al., 1990; van der Schaft, et al., 1992; Spraul and
Grossniklaus, 1997),
although numerous drusen phenotypes exist (Mullins & Hageman, 1999, Mol.
Vision). Hard
drusen are typically defined as small distinct deposits comprised of
homogeneous eosinophilic
material. Histologically, they are round or hemispherical, without sloped
borders. Soft drusen
are larger and have sloped, indistinct borders. Unlike hard drusen, soft
drusen are not usually
homogeneous, and typically contain inclusions and spherical profiles. An eye
with many
large/soft drusen is at a significantly higher risk of developing
complications of AMD than is
an eye with no drusen or a few, small drusen. The term "diffuse drusen," or
"basal linear
8
CA 02363503 2001-08-24
WO 00/52479 PCT/US00/05858
deposit," is used to describe the amorphous material which forms a layer
between the inner
collagenous layer of Bruch's membrane and the RPE. This material can appear
similar to soft
drusen histologically, with the exception that it is not mounded.
Knowledge of drusen composition, especially as it relates to phenotype, is
scant. Wolter and Falls (1962) observed that drusen stain with oil red 0,
indicating the
presence of neutral lipids in at least some drusen. Pauleikhoff, et al. (1992)
used lipid-based
histochemical staining approaches to show that different phenotypes of drusen
contain either
phospholipids or neutral lipids. These "hydrophilic" drusen were also bound by
an anti-
fibronectin antibody. Pauleikhoff et al. (1992) concluded that phospholipid-
containing, but
not neutral lipid-containing, drusen were anti-fibronectin antibody-reactive.
Other
investigators have not been able to reproduce the observation of an
association of fibronectin
with drusen (van der Schaft, et al., 1993; Mullins et al., 1999). These data
suggest that drusen
are either hydrophobic or hydrophilic, and that different drusen classes may
indicate
significantly different pathologies, suggesting the existence of different
compositional classes
of drusen, not solely based on morphology (i.e., hard and soft).
Farkas, et al. (1971b) analyzed drusen composition by enzymatic digestion,
organic extraction, and histochemical staining methods for carbohydrates and
other molecules.
They concluded that drusen are comprised of sialomucins (glycoproteins with 0-
glycosidically-linked oligosaccharides) and cerebrosides and/or gangliosides.
Newsome et al. (1987) described labeling of soft drusen with antibodies
directed against fibronectin, and to hard and soft drusen with antibodies
directed against IgG
and IgM. In addition, weak labeling of drusen with antibodies directed against
beta amyloid
(Loeffler, et al., 1995) and complement factors (Clq, C3c, C3d, and C4) (van
der Schaft, et al.,
1993), and more intense labeling with antibodies directed against ubiquitin
(Loeffler and
Mangini, 1997) and TIMP-3 (Fariss, et al., 1997), has been reported.
Antibodies to other
ECM molecules, including collagen types I, III, IV, and V, laminin, and
heparan sulfate
proteoglycan, have also been reported as being components of drusen in
"diffuse, mottled or
superficial laminar" patterns (Newsome, et al., 1987).
Discrepancies between the results of the immunohistochemical studies
described above are likely due to disagreement upon a universal classification
system for
drusen, the use of dehydrated, paraffin-embedded tissues (which potentially
resulting in the
extraction of some drusen constituents) as opposed to frozen sections, and the
use of
antibodies directed against different epitopes of the same protein.
Additionally, the use of
9
CA 02363503 2001-08-24
WO 00/52479 PCT/US00/05858
tissues that are fixed or frozen within a short period after death reduces
false negatives (due to
post-mortem autolysis and loss of antigenicity) and false positives (due to
post-mortem
diffusion and loss of physiologic barriers).
Though the literature contains anecdotal reports about drusen composition, a
comprehensive understanding of drusen biogenesis is lacking. At least twelve
pathways for
drusen genesis have been suggested in the literature (Duke-Elder and Dobree,
1967; Wolter
and Falls, 1962; Ishibashi, et al., 1986a). These fall into two general
categories based on
whether drusen are derived from the RPE or the choroid. Theories related to
the derivation of
drusen from RPE cells include the concepts that: drusen result from secretion
of abnormal
material derived from RPE or photoreceptors ("deposition theories"--Muller,
1856; Ishibashi,
et al., 1986; Young, 1987); transformation of degenerating RPE cells into
drusen
("transformation theories"--Donders, 1854; Rones, 1937; Fine, 1981; El Baba,
et al., 1986) or
some combination of these pathways. Specifically, some investigators have
concluded, based
on ultrastructural data, that drusen are formed when the RPE expels its basal
cytoplasm into
Bruch's membrane (Ishibashi, et al., 1986a), possibly as a mechanism for
removing damaged
cytosol (Burns and Feeney Bums, 1980). However, very few convincing images of
this
process have been demonstrated. Others have postulated that drusen are formed
by autolysis
of the RPE, due to aberrant lysosomal enzyme activity (Farkas, et al., 1971
a), although more
recent enzyme histochemical studies have failed to demonstrate the presence of
lysosomal
enzymes in drusen (Feeney-Burns, et al., 1987). Other mechanisms, including
lipoidal
degeneration of the RPE (Fine, 1981) and a derivation from vascular sources
(Friedman, et al.,
1963) have also been postulated (summarized in Duke-Elder and Dobree, 1967).
Farkas et al.
(1971 a) described the presence of numerous degenerating organelles in drusen,
including what
appeared to be lysosomes. Based on the observation that similar material was
present on the
RPE side of Bruch's membrane prior to drusen formation, they suggested that
drusen
constituents were derived from the RPE. However, lysosomal enzyme activity
within drusen
has not been verified (Feeney-Bums, et al., 1987). Burns and Feeney-Burns
(1980) described
the presence of "cytoplasmic debris" in small drusen, which they inferred was
derived from
the RPE. Feeney-Bums and Ellersieck (1985) later described a paucity of debris
in Bruch's
membrane directly beneath drusen, and suggested that drusen may result from an
inability of
the choroid to clear debris from sites of drusen deposition.
Ishibashi et al. (1986) observed cellular extensions of the RPE that protruded
through the RPE basal lamina and into Bruch's membrane in eyes that were
surgically
CA 02363503 2001-08-24
WO 00/52479 PCTIUSOO/05858
enucleated for melanoma, suggesting that drusen possess, and may be derived
from, RPE cell
constituents. However, it should be noted that changes in RPE cytoskeletal
organization and
cell shape have been described in eyes with choroidal melanoma (Wallow an Tso,
1972;
Fuchs, et al., 1991), making it difficult to draw conclusions about the
derivation of drusen
during normal senescence from these studies. Duvall et al. (1985) suggested a
role for
choroidal pericytes in keeping Bruch's membrane clear of debris. They
suggested that
dysfunction of pericytes leads to the formation of drusen, either by the
accumulation of
material from the choroid or by the failure to remove material deposited by
the RPE. Penfold
et al. (1986) have suggested a role for giant cells and mononuclear phagocytes
in the
pathology of the atrophic form of senile macular degeneration (see also
Dastgheib and Green,
1994).
Burns and Feeney-Burns (1980) suggested that apoptosis, resulting in basal
shedding of RPE cytosol, gives rise to drusen. Drusen-associated membranous
profiles were
inferred to be derived from the RPE, due to their localization between the RPE
basal lamina
and the inner collagenous zone of Bruch's membrane. While a number of
investigators cite
ultrastructural evidence for the derivation of drusen from RPE, the presence
of melanin,
lipofuscin or other RPE-derived organelles in drusen has not been reported.
It is clear that new diagnostics and therapeutics for drusen associated ocular
diseases are needed. For example, there is currently no reliable means for
diagnosing AMD.
In addition, there is no available therapy that significantly slows the
degenerative progression
of AMD for the majority of patients. Current AMD treatment is limited to laser
photocoagulation of the subretinal neovascular membranes that occur in 10-15%
of affected
patients. The latter may halt the progression of the disease but does not
reverse the
dysfunction, repair the damage, or improve vision.
2. Summary of the Invention
Based on the elucidation of the role of dendritic cells in drusen biogenesis
and
a greater understanding of the pathology of drusen associated ocular
disorders, the invention
features novel diagnostics, therapeutics, treatment modalities and drug
screening assays for
drusen associated ocular disorders.
In one aspect, the invention provides methods for diagnosing a subject for the
presence of or predisposition for developing a drusen-associated ocular
disorders. In a
preferred embodiment, the method comprises detecting the presence, activity or
level of at
11
CA 02363503 2001-08-24
WO 00/52479 PCTIUSOO/05858
least one "drusen associated marker" (i.e. a phenotypic or genotypic marker
that is involved
with the development of drusen and ultimately the etiology of a drusen-
associated ocular
disorder). Examples include markers involved in: RPE cell dysfunction and
death, immune
mediated events, drusen biogenesis, dendritic cell activation, cellular
migration and
differentiation e.g. in the choroid or sub RPE space, the presence of
geographic atrophy or
disciform scars, the presence of choroidal neovascularization and/or choroidal
fibrosis (e.g.
spiral collagens, elastin fibrils and microfilaments).
For example, genes expressed by dysfunctional or dying RPE cells include:
HLA-DR, CD68, vitronectin, apolipoprotein E, clusterin and S-100. Genes
expressed by
chorodial and RPE cell in AMD include heat shock protein 70, death protein,
proteasome,
Cu/Zn superoxide dismutase, cathepsins, and death adaptor protein RAIDD.
Markers involved
in immune mediated events associated with drusen formation include:
autoantibodies (e.g.
directed against drusen, RPE and/or retina components), leukocytes, dendritic
cells,
myofibroblasts, type VI collagen, and a cadre of chemokines and cytokines.
Molecules
associated with drusen include: immunoglobulins, amyloid A, amyloid P
component, HLA-
DR, fibrinogen, Factor X, prothrombin, complements 3, 5, 9, and 5b-9, C-
reactive protein
(CRP) apolipoprotein A, apolipoprotein E, antichymotrypsin, 02 microglobulin,
thrombospondin, and vitronectin. Markers of drusen associated dendritic cells
include: CD 1 a,
CD4, CD 14, CD68, CD83, CD86, and CD45, S 100, PECAM, MMP 14, ubiquitin, and
FGF.
Important dendritic cell-associated accessory molecules that participate in T
cell recognition
include ICAM-1, LFA1, LFA3, and B7, IL-1, IL-6, IL-12, TNF-alpha, GM-CSF and
heat
shock proteins. Markers associated with dendritic cell expression include:
colony stimulating
factor, TNFa, and Il-1. Markers associated with dendritic cell proliferation
include: GM-CSF,
IL-4, I1-3, SCF, FLT-3 and TNFa. Markers associated with dendritic cell
differentiation
include IL-10, M-CSF, IL-6 and IL-4. In a preferred embodiment, a genetic
fingerprint of the
subject is analyzed to determine whether the subject has or is predisposed to
developing a
drusen associated ocular disorder.
In another aspect, the invention provides therapeutic compositions and methods
for treating or preventing the development of a drusen-associated ocular
disease, comprising
providing to the subject an effective amount of an agent which inhibits DC
migration,
proliferation or differentiation, prevents RPE cell dysfunction and death,
prevents choroidal
fibrosis, or otherwise inhibits drusen formation or enhances drusen
resolution. In a preferred
embodiment, the agent is selected from the group consisting of cytokines,
chemokines and
12
CA 02363503 2008-10-17
agonists and antagonists thereof. Preferred agents for reducing or inhibiting
DC migration
include the DCRMs granulocyte macrophage colony stimulating factor (GM-CSF),
tumor
necrosis factor a (TNFa) or interleukin-1 (IL-1) and agonists thereof.
Preferably, the agent or
method reduces or inhibits dendritic cell migration into the subRPE space. In
a most preferred
embodiment, the agent or method provide a means for inhibiting the protrusion
of a cellular
process, such as a dendritic cell process, into the subRPE space. Preferred
agents for reducing
or inhibiting DC proliferation include DCRMs that are antagonists for GM-CSF,
IL-4, IL-3,
SCF, FLT-3 or TNFa. Preferred agents for reducing or inhibiting DC
differentiation include
the DCRMs IL-10, macrophage colony stimulating factor (M-CSF), IL-6 and IL-4
and
agonists thereof. Further preferred agents for reducing or inhibiting DC
differentiation include
DCRMs that are antagonists for LPS, TNFa, IL-1, IL-4, IL-13 or GM-CSF.
Preferred agents
that prevent or inhibit choroidal fibrosis include anti-angiogenic factors,
collagenases and
elastases.
In another aspect, the invention provides therapeutic compositions and methods
for treating or preventing drusen-associated disease, comprising providing to
the subject an
effective amount of an agent which reduces or inhibits the gene expression or
activity of one
or more drusen-associated molecules (DRAM). In a preferred embodiment, the
DRAM is an
amyloid A protein, amyloid P component, antichymotrypsin, apolipoprotein E, b2
microglobulin, complement 3, complement C5, complement C5b-9 terminal
complexes, factor
X, fibrinogen, immunoglobulins (kappa and lambda), prothrombin, thrombospondin
or
vitronectin. In a preferred embodiment, DR.AM gene regulation or activity is
reduced or
inhibited by one or more of a specific antisense nucleic acid, a ribozyme, a
peptide, an
antibody, or an enzyme. In a preferred embodiment, the DRAM antibody is
conjugated to a
reactive group. In another preferred embodiment, the reactive group is a
photoreactive dye or
a toxin.
In yet another aspect, the invention features in vitro and in vivo assays for
identifying therapeutics for drusen associated ocular disorders.
13
CA 02363503 2008-10-17
Various embodiments of this invention provide a method for diagnosing or
identifying a predisposition to the development of a drusen associated ocular
disorder in a
subject, comprising detecting the presence, activity or expression level of a
drusen
associated marker, wherein the drusen associated marker is a protein or a gene
or mRNA
transcript encoding the protein, wherein the protein is selected from the
group consisting of
complement 3(C3), complement 5 (C5), and complement reactive protein (CRP).
The
method may comprise detecting a specific allele at a polymorphic genetic locus
of the
drusen associated marker.
Other features and advantages of the invention will be apparent from the
following Detailed Description and Claims.
3. Brief Description of the Drawing
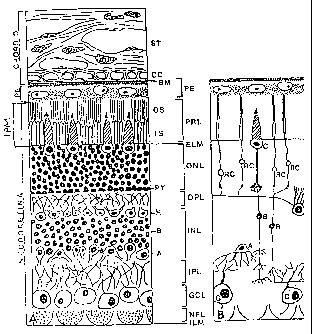
The Figure is a schematic representation of the retina and choroid, as seen in
13a
CA 02363503 2001-08-24
WO 00/52479 PCTIUSOO/05858
(A) histological section, and (B) retinal neurons shown diagrammatically. A,
amacrine cells;
B, bipolar cells; BM, Bruch's membrane; C, cone cells; CC, choriocapillaris;
ELM, external
limiting membrane; G, ganglion cells; GCL, ganglion cell layer; H, horizontal
cells; ILM,
inner limiting membrane; INL, internal nuclear layer; IPM, interphotoreceptor
matrix; IS,
inner segments of rods and cones; IPL, internal plexiform layer; NFL, nerve
fiber layer; ONL,
outer nuclear layer; OPL, outer plexiform layer; OS, outer segments of rods
and cones; PE,
pigment epithelium; PRL, photoreceptor layer; PT, photorecptor cell terminals;
R, rod cells;
ST, stroma vascularis of choroid.
4. Detailed Description of the Invention
4.1 Definitions
The meaning of certain terms and phrases as used in the following detailed
description and claims are defined as follows:
The term "agonist", as used herein, is meant to refer to an agent that
enhances
or upregulates (e.g., potentiates or supplements) the production or activity
of a gene product.
An agonist can also be a compound which increases the interaction of a gene
product,
molecule or cell with another gene product, molecule or cell, e.g., of a gene
product with
another homologous or heterologous gene product, or of a gene product with its
receptor. A
preferred agonist is a compound which enhances or increases binding or
activation of a
transcription factor to an upstream region of a gene and thereby activates the
gene. Any agent
that activates gene expression, e.g., by increasing RNA or protein synthesis
or decreasing
RNA or protein turnover, or gene product activity may be an agonist whether
the agent acts
directly on the gene or gene product or acts indirectly, e.g., upstream in the
gene regulation
pathway. Agonists may be RNAs, peptides, antibodies and small molecules, or a
combination
thereof.
The term "animal model", as used herein, includes transgenic animals,
naturally
occurring animals with genetic mutations and non-transgenic animals that have
been treated
with one or more agents, or combinations thereof (e.g., a skid mouse), any of
which may serve
as experimental models for a disease, e.g., macular degeneration. For example,
a transgenic
mouse may be a mouse in which a gene is knocked out or in which a gene is
overexpressed.
The term "antagonist" as used herein is meant to refer to an agent that
14
CA 02363503 2001-08-24
WO 00/52479 PCTIUSOO/05858
downregulates (e.g., suppresses or inhibits) the production or activity of a
gene product. Such
an antagonist can be an agent which inhibits or decreases the interaction
between a gene
product, molecule or cell and another gene product, molecule or cell. A
preferred antagonist is
a compound which inhibits or decreases binding or activation of a
transcription factor to an
upstream region of a gene and thereby blocks activation of the gene. Any agent
that inhibits
gene expression or gene product activity may be an antagonist whether the
agent acts directly
on the gene or gene product or acts indirectly, e.g., upstream in the gene
regulation pathway.
An antagonist can also be a compound that downregulates expression of a gene
or which
reduces the amount of gene product present, e.g., by decreasing RNA or protein
synthesis or
increasing RNA or protein turnover. Antagonists may be RNAs, peptides,
antibodies and
small molecules, or a combination thereof.
The term "associate" or "interact" as used herein is meant to include
detectable
relationships or associations (e.g., biochemical interactions) between
molecules, such as
interaction between protein-protein, protein-nucleic acid, nucleic acid-
nucleic acid, protein-
carbohydrate, carbohydrate-carbohydrate, protein-lipid, lipid-lipid, etc., and
protein-small
molecule or nucleic acid-small molecule in nature.
"Bruch's Membrane" is a trilaminar extracellular matrix complex that lies
between the retinal RPE and the primary capillary bed of the choroid, the
choriocapillaris.
Bruch's membrane is comprised of two collagen layers, referred to as the inner
and outer
collagenous layers, that flank a central domain comprised largely of elastin.
The strategic
location of Bruch's membrane between the retina and its primary source of
nutrition, the
choroidal vasculature, is essential for normal retinal function (Marshall et
al., 1998; Guymer
and Bird, 1998). Immunohistochemical studies have documented the presence of
collagen
types I, III, IV, V and VI within Bruch's membrane proper (Das et al., 1990;
Marshall et al.,
1992). Type VI is associated specifically with the elastic lamina, types IV
and V with the
basal laminae of the choriocapillaris and RPE, and types I and III with the
inner and outer
collagenous layers. The presence of collagen types I, III, IV and V in these
tissues has also
been confirmed biochemically.
The term "choroid" refers to the highly vascularized tissue lying between the
sclera and retinal pigment epithelium of the eye. This tissue is comprised of
numerous
pericytes, melanocytes, fibroblasts, myofibroblasts and transitional
leukocytes. "Bruch's
membrane, a trilamellar extracellular matrix comprised of inner and outer
collagenous layers
and an elastic lamina, is a component of the choroid. It is positioned between
the basal lamina
of the RPE and the choriocapillaris. The remaining extracellular matrix of the
choroid is
CA 02363503 2001-08-24
WO 00/52479 PCTIUSOO/05858
comprised of a variety of extracellular matrix constituents that are loosely
organized.
The term "dendritic cell" or "DC" as used herein refers to hematopoietic cells
characterized by their unusual dendritic morphology, their potent antigen-
presenting capability
and their lack of lineage-specific markers such as CD3, CD 19, CD 16, CD 14,
which
distinguishes them respectively from T cells, B cells, NK cells, and
monocytes. Currently
there are at least two ontogenic pathways for dendritic cell development:
those that derive
from myeloid-committed hematopoietic precursors and those that derive from
lymphoid-
committed hematopoietic precursors. Myeloid-committed precursors which give
rise to
granulocytes and monocytes can also differentiate into Langerhans cells of the
skin and
myeloid related dendritic cells in the secondary lymphoid tissue. There may
also be a class of
lymphoid-derived dendritic cells (See Lotze, M.T. and Thomson, A.W. (Eds.)
(1999)
"Dendritic Cells", Academic Press, San Diego, CA, for a number of reviews on
dendritic cells,
the teachings of which are incorporated herein by reference).
The term "dendritic cell precursor" or "DC precursor" as used herein refers to
cell types from which a dendritic cell is derived upon differentiation and
maturation. A
dendritic cell precursor may be a bone marrow stem cell, a lymphiod cell
lineage-committed
cell or a myeloid cell lineage-committed cell from which a dendritic cell may
develop after
exposure to certain DCRMs. For example, DC precursors of the myeloid lineage
can be
induced to differentiate into DCs by treatment with GM-CSF.
The term "dendritic cell process" refers to a cellular portion of a dendritic
cell
which projects or extends away from the center of the dendritic cell.
The term "drusen" as used herein encompasses a number of phenotypes, all of
which develop, between the inner collageous layer of Bruch's membrane and the
RPE basal
lamina. Hard drusen are small distinct deposits comprised of homogeneous
eosinophilic
material and are usually round or hemispherical, without sloped borders. Soft
drusen are
larger, usually not homogeneous, and typically contain inclusions and
spherical profiles.
Some drusen may be calcified. The term "diffuse drusen," or "basal linear
deposit," is used to
describe amorphous material which forms a layer between the inner collagenous
layer of
Bruch's membrane and the retinal pigment epithelium (RPE). This material can
appear similar
to soft drusen histologically, with the exception that it is not mounded.
The term "drusen-associated marker" refers to a phenotype or genotype that is
involved or associated with the development of drusen formation and ultimately
the
development of a drusen associated ocular disease or disorder. Examples of
phenotypic
16
CA 02363503 2001-08-24
WO 00/52479 PCTIUSOO/05858
markers include: RPE dysfuncation and/or death, immune mediated events,
dendritic cell
activation, migration, differentiation and extrusion of the DC process into
the sub RPE space
(e.g. by detecting the presence or level of a dendritic cell marker such as
CD68, CD1a and
S 100), the presence of geographic atrophy or disciform scars, the presence of
choroidal
neovascularization and/or choroidal fibrosis, especially in the macula.
Examples of genotypic
markers include mutant genes and/or a distinct pattern of differential gene
expression (Drusen
Development Pathway"), including genes that are upregulated or downregulated
in drusen
forming ocular tissue associated with drusen biogenesis. For example genes
expressed by
dysfunctional and/or dying RPE cells include: HLA-DR, CD68, vitronectin,
apolipoprotein E,
clusterin and S-100. Genes expressed by choroidal and RPE cells in AMD inlcude
heat shock
protein 70, death protein, proteasome, Cu/Zn superoxide dismutase, cathepsins,
and death
adaptor protein RAIDD. Markers involved in immune mediated events associated
with drusen
formation include: autoantibodies (e.g. directed against drusen, RPE and/or
retina
components), leukocytes, dendritic cells, myofibroblasts, type VI collagen,
and a cadre of
chemokines and cytokines. Molecules associated with drusen include:
immunoglobulins,
amyloid A, amyloid P component, HLA-DR, fibrinogen, Factor X, prothrombin,
complements
3, 5, 9, and 5b-9, C- reactive protein (CRP) apolipoprotein A, apolipoprotein
E,
antichymotrypsin, P2 microglobulin, thrombospondin, and vitronectin. Markers
of drusen
associated dendritic cells include: CD1a, CD4, CD14, CD31 (PECAM-1), CD45,
CD64/1
(FcR), CD68, CD83, CD86 and HLA-DR, particular preferred dendritic cell
markers include
CD1a, CD14, CD45, CD68, CD83 and HLA-DR. Important dendritic cell-associated
accessory molecules that participate in T cell recognition include ICAM-1,
LFA1, LFA3, and
B7, IL-1, IL-6, IL-12, TNF-alpha, GM-CSF and heat shock proteins. Markers
associated with
dendritic cell expression include: colony stimulating factor, TNFa, and I1-1.
Markers
associated with dendritic cell proliferation include: GM-CSF, IL-4, 11-3, SCF,
FLT-3 and
TNFa. Markers associated with dendritic cell differentiation include IL-10, M-
CSF, IL-6 and
IL-4. Markers of fibrosis include: a decrease in BIG H3, increase in (31-
integrin, increase in
various growth factors (e.g. fibroblast growth factors (FGF), chemokines and
cytokines,
increase in collagen (e.g. collagen 6 a2 and collagen 6 a3) or procollagen
e.g. I and III and
peptides thereof, increase in elastin or elastin peptides, and increase in FSP-
1 and an increase
in human metalloelastase (HME). Molecules that are known or suspected to
participate in
systemic fibrosis and are therefore potential candidates for choroidal
fibrosis include, but are
not limited to: BIGh3, calpain, cathepsin D, collagens (I, III, IV, VI, VII),
CTGF, desmosine,
17
CA 02363503 2001-08-24
WO 00/52479 PCTIUSOO/05858
elastin, emilin, endothelin, bFGF, fibrillins 1-2, fibroblasst specific
proteins (FSP-1),
fibronectin, fibrosin, fibulins 1-5, ficolin, GM-CSF, 4-hydroxy-nonenal, HLA
antigens, HME,
IFG-1, IFN-y, IL-2, 11-4, IL-6, IL-8, IL-10, integrins al(31 and a2(31,
laminins Cl-3, laminin
receptors, LOXL, LTBP 1-4, MCP-1, MFAP 1-4, MMPs, oncostatin M, osteopontin,
PAF,
PDGF, plasmin, protease inhibitors 1-3, PLOD 1-2, various proteoglycans,
RANTES, relaxin,
tenascin, TGF-(3, thromboplastin, thrombospondin, TIMPs, TNFa , transcription
factors (NF-
xB; HP-1) and VEGF.
The term "drusen-associated ocular disorder" as used herein refers to any
disease or disorder which involves drusen formation. For example, in macular
degenerations,
the accumulation of drusen creates a physical barrier that appears to impede
normal metabolite
and waste diffusion between the choriocapillaris and the retina. As a result,
the diffusion of
oxygen, glucose, and other nutritive or regulatory serum-associated molecules
required to
maintain the health of the retina and RPE are inhibited.
A "drusen-associated molecule" or "DRAM" as used herein refers to any
protein, carbohydrate, glycoconjugate (e.g., glycoprotein or glycolipid),
other lipid, nucleic
acid or other molecule which is found in association with, or interacting
with, a drusen deposit.
DRAMS may include cellular fractions or organelles that are not normally found
deposited in,
or in association with, a tissue unless it is affected by drusen or which is
not present in drusen-
affected and normal tissue in equivalent amounts.
The term "extracellular matrix" ("ECM") refers to, e.g., the collagens,
proteoglycans, non-collagenous glycoproteins and elastins that surround cells
and provide
structural and functional support for cells as well as maintain various
functions of cells, such
as cell adhesion, proliferation, differentiation and protein synthesis. A
skilled artisan will
appreciate that the precise composition and physical properties of ECM, as
well as its function,
vary between various cell types, between various tissues, and between various
organs.
"Fibrosis" as used herein refers to a disease process, typically observed in
chronic diseases, characterized by progressive accumulation and/or deposition
of extracellular
matrix proteins (e.g. collagens) and activation, differentiation and/or
transformation of various
interstitial cell types (e.g. fibroblasts).
The term "inhibit" as used herein means to prevent or prohibit and is intended
to include total inhibition, partial inhibition, reduction or decrease.
The term "macular degeneration" refers to any of a number of conditions in
which the retinal macula degenerates or becomes dysfunctional, e.g., as a
consequence of
18
CA 02363503 2001-08-24
WO 00/52479 PCT/US00/05858
decreased growth of cells of the macula, increased death or rearrangement of
the cells of the
macula (e.g., RPE cells), loss of normal biological function, or a combination
of these events.
Macular degeneration results in the loss of integrity of the histoarchitecture
of the cells of the
normal macula and/or the loss of function of the cells of the macula. The term
also
encompasses extramacular changes that occur prior to, or following dysfunction
and/or
degeneration of the macula. Any condition which alters or damages the
integrity or function
of the macula (e.g., damage to the RPE or Bruch's membrane) may be considered
to fall
within the definition of macular degeneration. Other examples of diseases in
which cellular
degeneration has been implicated include retinal detachment, chorioretinal
degenerations,
retinal degenerations, photoreceptor degenerations, RPE degenerations,
mucopolysaccharidoses, rod-cone dystrophies, cone-rod dystrophies and cone
degenerations.
The terms "modulation", "alteration", "modulate ", or "alter " are used
interchangeably herein to refer to both upregulation (i.e., activation or
stimulation (e.g., by
agonizing or potentiating) and downregulation (i.e., inhibition or suppression
(e.g., by
antagonizing, decreasing or inhibiting)) of an activity. For example, the
activity that is
modulated may be gene expression or may be the growth, proliferation,
migration or
differentiation of dendritic cells. "Modulates" or "alters" is intended to
describe both the
upregulation or downregulation of a process, since, as is well known to a
skilled artisan, a
process which is upregulated by a certain stimulant may be inhibited by an
antagonist to that
stimulant. Conversely, a process that is downregulated by a certain stimulant
may be inhibited
by an antagonist to that stimulant. Thus, e.g., the identification of an agent
that induces a
cellular response modulates or alters cellular behavior in an inductive manner
and it is
inherently understood that the response may be modulated in an inhibitory
manner by an
inhibitor of that agent (e.g., by an antibody or antisense RNA, as is well
understood and
described in the art).
The term "nucleic acid" as used herein refers to polynucleotides or
oligonucleotides such as deoxyribonucleic acid (DNA), and, where appropriate,
ribonucleic
acid (RNA). The term should also be understood to include, as equivalents,
analogs of either
RNA or DNA made from nucleotide analogs and as applicable to the embodiment
being
described, single (sense or antisense) and double-stranded polynucleotides.
The term "polymorphism" refers to the coexistence of more than one form of a
gene or portion (e.g., allelic variant) thereof. A portion of a gene of which
there are at least
two different forms, i.e., two different nucleotide sequences, is referred to
as a "polymorphic
19
CA 02363503 2001-08-24
WO 00/52479 PCT/USOO/05858
region of a gene". A polymorphic region can be a single nucleotide, the
identity of which
differs in different alleles. A polymorphic region can also be several
nucleotides long. A
"polymorphic gene" refers to a gene having at least one polymorphic region.
The terms "protein", "polypeptide" and "peptide" are used interchangeably
herein when referring to a gene product comprising amino acids. The term
"recombinant
protein" refers to a polypeptide of the present invention which is produced by
recombinant
DNA techniques, wherein generally DNA encoding a polypeptide is inserted into
a suitable
expression vector which is in turn used to transform a host cell to produce
the heterologous
protein. Likewise the term "recombinant nucleic acid" or "recombinant DNA"
refers to a
nucleic acid or DNA of the present invention which is produced by recombinant
DNA
techniques, wherein generally DNA encoding a polypeptide is inserted into a
suitable
expression vector which is in turn used to transform a host cell to produce
the heterologous
protein. Moreover, the phrase "derived from", with respect to a recombinant
gene, is meant to
include within the meaning of "recombinant protein" those proteins having an
amino acid
sequence of a native polypeptide, or an amino acid sequence similar thereto
which is generated
by mutations including substitutions and deletions (including truncation) of a
naturally
occurring form of the polypeptide.
"Retinal Pigment Epithelium" or "RPE" is defined as the cuboidal epithelial
monolayer that is situated between the neural retina and choroid. The RPE
derives
developmentally from, and is indeed contiguous with, the same neuroectodermal
layer as the
neural retina. The RPE possesses numerous large pigment granules (melanosomes)
which
participate in the prevention of light scattering. In addition, the RPE plays
a critical role in the
maintenance of photoreceptor cell viability and function by the phagocytosis
and removal of
photoreceptor outer segment disks, the processing and secretion of various
molecules
necessary for photoreceptor function and viability (such as vitamin A
derivatives and growth
factors), the regulation of macromolecular traffic between the retina and
choroid, and the
mediation of retinal adhesion.
"Small molecule" as used herein, is meant to refer to a composition which has
a
molecular weight of less than about 5 kD and most preferably less than about 4
kD. Small
molecules can be nucleic acids, peptides, polypeptides, peptidomimetics,
carbohydrates, lipids
(e.g., glycolipids and pig-tail lipids) or other organic (carbon containing)
or inorganic
molecules. Many pharmaceutical companies have extensive libraries of chemical
and/or
biological mixtures, often fungal, bacterial, or algal extracts, which can be
screened with any
of the assays of the invention to identify therapeutic compounds.
A "therapeutic" as used herein refers to an agonist or antagonist of the
CA 02363503 2001-08-24
WO 00/52479 - PCTIUSOO/05858
bioactivity of a drusen associated marker. Preferred therapeutics reduce or
inhibit RPE cell
death, factors involved in the inflammatory response, factors involved in
fibroblast
proliferation and migration resulting in fibrosis and/or dendritic cell
activation, migration or
differentiation into drusen. Examples of modulators of fibrosis include, but
are not limited to:
L-tryptophan dimer, superoxide, nitric oxide, corticosteroid, retinoid,
halofuinone, Tranilast,
CTGF, interferons, relaxin, TGFP3, HGF, prolyl hydroxylase, C-proteinase,
lysyl oxidase, and
antisense oligonucleotides. Other preferred therapeutics include agents that
have shown some
efficacy in treating or preventing aortic diseases (e.g. AAA), including:
antiinflammatory
agents (e.g. anti CD-18 antibody), protease inhibitors, inhibitors of
elastolytic MMPs (e.g. the
hydroxamate based RS312908, batimastat, antibiotics (e.g. doxycycline),
tetracycline),
inhibitors of prostaglandin synthesis and beta-blockers (e.g. propanalol).
The term "transcriptional regulatory sequence" is a generic term used
throughout the specification to refer to DNA sequences, such as initiation
signals, enhancers,
and promoters, which induce or control transcription of protein coding
sequences with which
they are operably linked.
As used herein, the term "transfection" means the introduction of a nucleic
acid,
e.g., via an expression vector, into a recipient cell by nucleic acid-mediated
gene transfer.
"Transformation", as used herein, refers to a process in which a cell's
genotype is changed as a
result of the cellular uptake of exogenous DNA or RNA.
As used herein, the term "transgene" means a nucleic acid sequence (encoding,
e.g., one of the polypeptides of the invention, or an antisense transcript
thereto) which has
been introduced into a cell. A transgene could be partly or entirely
heterologous, i.e., foreign,
to the transgenic animal or cell into which it is introduced, or can be
homologous to an
endogenous gene of the transgenic animal or cell into which it is introduced,
but which is
designed to be inserted, or is inserted, into the animal's genome in such a
way as to alter the
genome of the cell into which it is inserted (e.g., it is inserted at a
location which differs from
that of the natural gene or its insertion results in a knockout or may result
in over expression).
A transgene can also be present in a cell in the form of an episome. A
transgene can include
one or more transcriptional regulatory sequences and any other nucleic acid,
such as 5' UTR
sequences, 3' UTR sequences, or introns, that may be necessary for optimal
expression of a
selected nucleic acid.
A "transgenic animal" refers to any animal, preferably a non-human mammal,
bird or an amphibian, in which one or more of the cells of the animal contain
heterologous
nucleic acid introduced by way of human intervention, such as by transgenic
techniques well
21
CA 02363503 2001-08-24
WO 00/52479 - PCTIUSOO/05858
known in the art. The nucleic acid is introduced into the cell, directly or
indirectly by
introduction into a precursor of the cell, by way of deliberate genetic
manipulation, such as by
microinjection or by infection with a recombinant virus. The term genetic
manipulation does
not include classical cross-breeding, or in vitro fertilization, but rather is
directed to the
introduction of a recombinant DNA molecule. This molecule may be integrated
within a
chromosome, or it may be extrachromosomally replicating DNA. In the typical
transgenic
animals described herein, the transgene causes cells to fail to express a
specific normal gene
product, to express a recombinant form of one or more DRAM polypeptides, e.g.,
either
agonistic or antagonistic forrns, or molecules that regulate the biosynthesis,
accumulation or
resorption of DRAMs or dendritic cells. Transgenic knockouts may, for example,
be produced
which cause alterations in dendritic cell behavior (e.g., cell growth,
proliferation, migration,
differentiation or gene expression). For example, mice whose Rel-B,
transforming growth
factor bl (TGF-bl) or Ikaros genes are disrupted lack dendritic cells from
various cell lineages
(see Caux, C. et al., 1999). However, transgenic animals in which the
recombinant DCRM or
DRAM gene is silent are also contemplated, as for example, the FLP or CRE
recombinase
dependent constructs. Moreover, "transgenic animal" also includes those
recombinant animals
in which gene disruption is caused by human intervention, including both
recombination and
antisense techniques.
The term "treating" as used herein is intended to encompass curing as well as
ameliorating at least one symptom of the condition or disease.
The terms "vector," "cloning vector," or "replicative cloning vector," are
interchangeable as used herein, and refer to a nucleic acid molecule, which is
capable of
transporting another nucleic acid to which it has been linked. One type of
preferred vector is
an episome, i.e., a nucleic acid capable of extra-chromosomal replication.
Preferred vectors
are those capable of autonomous replication and/or expression of nucleic acids
to which they
are linked. Vectors capable of directing the expression of genes to which they
are operatively
linked are referred to herein as "expression vectors." The term "expression
system" as used
herein refers to an expression vector under conditions whereby an mRNA may be
transcribed
and/or an mRNA may be translated into protein. The expression system may be an
in vitro
expression system, which is commercially available or readily made according
to art known
techniques, or may be an in vivo expression system, such as a eukaryotic or
prokaryotic cell
containing the expression vector. In general, expression vectors of utility in
recombinant
DNA techniques are often in the form of "plasmids" which refer generally to
circular double
22
CA 02363503 2001-08-24
WO 00/52479 PCTIUSOO/05858
stranded DNA loops which, in their vector form are not bound to the
chromosome. In the
present specification, "plasmid" and "vector" are used interchangeably as a
plasmid is the most
commonly used form of vector. However, the invention is intended to include
such other
forms of expression vectors which serve equivalent functions and which become
known in the
art subsequently hereto.
The term "wild-type allele" refers to an allele of a gene which, when present
in
two copies in a subject results in a wild-type phenotype. There can be several
different wild-
type alleles of a specific gene, since certain nucleotide changes in a gene
may not affect the
phenotype of a subject having two copies of the gene with the nucleotide
changes.
4.2 General
The invention is based, at least in part, on the elucidation of the etiology
of
AMD and other drusen-associated ocular disorders, essentially as described
below.
4.2.4 Uni ing HXpothesis ofDrusen Biogenesis:
Proposed herein is a unifying theory of drusen biogenesis. This theory is put
forth with the acknowledgment that numerous AMD genotypes may exist. Thus,
only some
aspects of the proposed hypothesis may be involved in any given AMD genotype.
Importantly, the theory is based upon novel data generated by the inventors
and disclosed
herein documenting that dendritic cells are associated with drusen. This
observation invokes,
for the first time, the potential for a direct and integral role of cell-
mediated processes in
drusen biogenesis.
The presence of dendritic cells in inflammatory lesions is well-recognized. It
is
clear that dendritic cells must be recruited, activated, and migrate to, sites
of inflammation,
rather than passively migrating to these sites. Dendritic cells are typically
recruited to sites of
tissue damage by various chemoattractants, heat shock proteins, DNA fragments,
and others.
Choroidal dendritic cell processes are associated with the smallest of drusen,
and are often
observed in the sub-RPE space in association with whole, or portions of, RPE
cells that have
been shunted into Bruch's membrane, prior to the time that drusen, per se, are
detectable.
Based on these observations, proposed herein is a mechanism in which choroidal
dendritic
cells are activated and recruited by locally damaged and/or sublethally
injured RPE cells. This
idea is consistent with recent data showing that dendritic cells, and thus the
innate immune
system, can be activated by microenvironmental tissue damage. In this state,
these cells
23
CA 02363503 2001-08-24
WO 00/52479 PCTIUSOO/05858
extend a cellular process through Bruch's membrane in order to gain access to
the site of tissue
damage. In this role, choroidal dendritic cells may thus serve as sentinel
receptors with the
capacity to respond to local cell injury, and ultimately provide for the
overall integration of
immune-mediated processes that determine the outcome of the overall response.
In this model, the injured RPE itself (by whatever mechanism this occurs) may
serve as a source of soluble cytokines or other stimulatory factors that
initiate dendritic cell
recruitment and activation. The data presented herein clearly supports
accelerated RPE cell
death in eyes derived from donors with AMD, as compared to age-matched
controls. Based on
available information from other systems, and upon previous suggestions
pertaining to the
etiology of AMD, RPE cell death might occur by several mechanisms, including
ischemia,
necrosis, gene-mediated injury, Bruch's membrane-induced dysfunction,
oxidative injury from
light or systemic factors (e.g. smoking-generated compounds), lipofuscin
accumulation, or
autoimmune phenomena, to list a few. Based on data disclosed herein, it is
likely that RPE
cell death would most likely have to be due to necrosis, rather than to
apoptosis, since cells
undergoing apoptotic cell death are not known to be capable of recruiting
dendritic cells.
Indeed, the data provides compelling evidence for an absence of apoptotic RPE
cell death in
human donor eyes.
Several known pathways can initiate receptor-ligand interactions between
dendritic cell precursors and injured tissue. These include cytokines such as
IL-1, IL-6, IL-12,
TNF-alpha, and GM-CSF, heat shock proteins, altered expression of cell surface
proteins and
DNA in the presence of free radicals. The novel observation of clonal
expression of HLA-DR,
CD68, vitronectin, S-100, clusterin, and apolipoprotein E by RPE cells in eyes
from donors
with drusen may be particularly significant in this respect. Furthermore, up-
regulation of
various cell death- and immune-associated molecules by the RPE/choroid in eyes
with
developing drusen and AMD have been identified using differential display and
gene array
analyses. In addition, there is evidence that free radicals, which are known
to be present in
high concentrations at the RPE-retina-choroid interface, might be
immunostimulatory. There
is also data suggesting that ceroid (a potential component of lipofuscin)
derived from necrotic
cells may serve as an antigen in the generation of certain autoimmune
diseases. This could
explain the general contention that oxidative stress and/or lipofuscin may
lead to RPE
dysfunction and the development of AMD (Mainster, M.A., Light and macular
degeneration:
a biophysical and clinical perspective. Eye,
1987. 1(Pt 2): p. 304-10).
24
CA 02363503 2001-08-24
WO 00/52479 - PCT/US00/05858
Once inside the pre-lesion or lesion (a.k.a. the drusen, or drusen precursor
site),
dendritic cells might then contribute to the chronicity (induced chronic
inflammatory lesions)
of AMD by any number of mechanisms, including immune complex formation,
complement
activation, and/or in situ activation of choroidal T-cells, other phagocytic
cells, and matrix
proteolysis. The presence of numerous immune-associated constituents in
drusen, including
immunoglobulins, complement proteins, and some acute phase proteins, could be
explained by
such an event. One might predict that the dendritic cell response would be
down-regulated
once the local tissue damage has been repaired, thus restoring tolerance. This
type of self-
limiting control is typically accomplished in other systems via turnover of
dendritic cells; the
influx of new dendritic cell precursors and the concomitant reduction in the
influx of mature
dendritic cells into the lymph nodes is typically sufficient to shift the
balance back to
tolerance. In other cases, natural killer cells recognize mature dendritic
cells as targets,
providing a negative feedback effect on antigen presentation, forcing the
system into tolerance.
However, in the case of AMD, we suggest that a state of chronic inflammation
persist for
many years. In this scenario, cyclical events of RPE cell death may occur over
a period of
many years that do not allow the system to return to tolerance. In one
example, this might
occur as a result of genetic preprogramming, as in the case of a RPE gene
mutation. In
another example, local activation of complement and HLA-DR expression by RPE
cells,
initiated by dendritic cells recruited to the sub-RPE region, might lead to
clonal RPE cell
death, thereby maintaining a state of chronic inflammation. A negative outcome
of this entire
process may be that Bruch's membrane and the surrounding extracellular matrix
may be
degraded, angiogenic factors may be generated, resulting in opportunistic
neovascularization
of the sub-RPE and subretinal spaces. Although there is little information in
the literature
concerning matrix-degrading enzyme expression by dendritic cells, MT-1-MMP
expression
within drusen cores has been observed, suggesting a possible mechanism for DC-
mediated
matrix breakdown..
The notion that dendritic cells may be activated by local tissue injury might
also initiate an autoimmune response to retinal and/or RPE antigens that are
uncovered during
tissue damage. The availability and amount of RPE debris/antigen will most
likely determine
which ensuing pathway is involved. Such autoimmune responses have been
documented as a
consequence of ischemia or injury to the heart. The inventors have recently
identified
autoantibodies in the sera of individuals with AMD that are directed against
retinal and RPE
proteins of 35kDa and 53kDa. This might occur as a consequence of aberrant
delayed-type
CA 02363503 2001-08-24
WO 00/52479 - PCT/US00/05858
hypersensitivity responses, perhaps explaining the presence of serum
autoantibodies in at least
some AMD patients. It is also conceivable that the groundwork for this
autoimmune process
is primed earlier in life by necrosis of RPE cells. Indeed, this would explain
the inventor's
observation of a wave of peripheral RPE cell dropout in the second and third
decades of life.
In the model presented herein, the RPE injury and dendritic cell events are
followed by the continued deposition of drusen-associated constituents. Early
DRAM-matrix
complexes, such as immune complexes, or other local ligands might serve as
"nucleation sites"
for the deposition of additional self-aggregating proteins and/or lipids.
These constituents
could be derived from either the plasma and/or local cellular sources. Based
on the knowledge
that many DRAMs are circulating plasma proteins, it is plausible that some
DRAMs pass out
of choroidal vessels and into the extracellular space adjacent to the RPE
where they bind to
one or more ligands associated with Bruch's membrane in the aging eye. These
ligands could
be basement membrane components, plasma membrane receptors, secretory products
derived
from RPE or choroidal cells, or byproducts of cellular autolysis. As reported
herein, a number
of drusen-associated molecules, including apolipoprotein E, vitronectin,
fibrinogen, C reactive
protein, and transthyretin, have been synthesized by the RPE and/or retina.
Although
unexpected, these data support the concept that some DRAMs may be synthesized
and
secreted locally. It remains to be determined conclusively whether up- or down-
regulation of
DRAM synthesis by local cells correlates with drusen deposition and/or AMD,
although gene
array analyses provide support for upregulated synthesis of a number of DRAMs,
including
immunoglobins, by the RPE and choroid of AMD donors. As these abnormal drusen
deposits
increase in size they displace the RPE monolayer and are recognized clinically
as drusen.
This model also predicts an imbalance in extracellular matrix synthesis,
degradation, and/or turnover, which would thereby lead to events such as
choroidal
neovascularization, a hallmark characteristic of some forms of AMD, cellular
proliferation,
cellular differentiation, and interstitial fibrosis. In many organs, fibrosis
and fibrogenesis is a
common complication of tissue injury, independent of the initial site of said
injury. The
recruitment of immune cells, and their activation and/or modulation by
resident cells,
represents a key step in the cascade of events that ultimately lead to
fibrosis. More recent
studies suggest that distinct functional fibroblast phenotypes may play a
central role in early
fibrosis, including the initial recruitment of immune cells.
4.2.2 Involvement o Fibrosis:
26
CA 02363503 2001-08-24
WO 00/52479 - PCT/US00/05858
The inventors have documented "choroidal fibrosis" in a subset of donor eyes.
There is a significant correlation between choroidal fibrosis and age.
Furthermore,
preliminary data suggest that there is a strong correlation between choroidal
fibrosis, drusen,
AMD, aortic aneurysms, aortic stenosis, and possibly COPD. These fibrotic
choroids are
characterized ultrastructurally by massive accumulations of newly synthesized
collagen and
elastin fibrils, as well as filamentous collagens and microfilaments, that
fill the normally
loosely packed choroidal stromas. The major collagen fibrils average 0.042-
0.063 m in
diameter as compared to the fibrillar collagen in the sclera, which averages
0.211-0.253 m in
diameter. Furthermore, the collagen fibrils in these donors exhibit a classic
spiraled
morphology in longitudinal and cross sections. It is thought that spiraled
collagen results from
disaggregation of fibrils and/or to incorporation of uncleaved procollagen
molecules. This
collagen phenotype is observed in a few heritable connective tissue diseases
(Ehler's-Danlos;
PXE; dermatoparaxis), as well as in other nongenetic conditions
(collagenofibrotic
glomerulopathy, scleroderma, atherosclerosis, amyloid, emphysema, atheromatous
plaques).
The deposition of a distinct banded material is also present in donor eyes
exhibiting choroidal
fibrosis. Clear indications of active elastin synthesis (including dilated
RER, pockets of
microfilaments, and elastin exhibiting the morphological characteristics of
newly synthesized
protein) are also observed along attenuated fibroblast cell processes and
interspersed amongst
the collagen fibrils. Genes that are and are not differentially expressed in
choroidal fibrosis
are shown in Table 1, below.
TABLE 1
Molecule Expression in Choroidal Fibrosis vs Controls
BIG H3 Decreased
P1-integrin Increased
Collagen 3 al Unchanged
Collagen 1 al Unchanged
Collagen 1 a2 Unchanged
Collagen 6 al Unchanged
Collagen 6 a2 Increased
Collagen 6 0 Increased
27
CA 02363503 2001-08-24
WO 00/52479 - PCT/US00/05858
Elastin Increased
Fibulin- 1 Unchanged
Fibulin-2 Unchanged
Fibulin-3 Unchanged
Fibulin-4 Unchanged
Fibulin-5 Unchanged
Fibrillin-2 Unchanged
HLA-DR b Unchanged
HME Increased
IgK Unchanged
Laminin Receptor Unchanged
Laminin C2 Unchanged
4.2.3 Role of RPE in Drusen Biogenesis
As described herein, Applicants have discovered that retinal pigment
epithelial
cell (RPE) dysfunction and death is certainly associated with the development
of drusen and,
by extension, in the etiology of drusen-associated ocular diseases.
First, morphometric analyses of a Comprehensive Donor Database repository
comprised of 168 donors, aged between 0 and 101, with and without a clinically
documented
history of drusen and AMD, provide strong evidence that the rate of RPE cell
death in
individuals with drusen and AMD is significantly higher than in age-matched
controls. RPE
cell loss in normal individuals occurs at a rate of between 10% and 15% over
nine decades, in
contrast to a rate between 30% and 40% in individuals with AMD and drusen.
Significantly, it
appears that the majority of RPE cell death likely occurs by a process of
necrosis, rather than
apoptosis. These observations are based on employment of the TUNEL assay, an
absence of
apoptosis-associated gene expression in gene array analyses and electron
microscopic
observation.
Second, fragments of RPE cells (identified on the basis of morphologically
detectable lipofuscin and pigment granules), can be detected within drusen at
both the light
and electron microscopic levels of resolution, demonstrating that they
contribute to drusen
volume and formation.
Third, drusen-associated dendritic cell processes (as described in detail
28
CA 02363503 2001-08-24
WO 00/52479 PCTIUSOO/05858
elsewhere herein) are often observed in association with these early stages of
RPE
fragmentation and "blebbing", suggesting that the stimulus for dendritic cell
recruitment lies at
the level of RPE cells.
Fourth, RPE cells associated with the smallest of drusen (and regions presumed
to be drusen precursors) are often characterized by focal expression of
molecules not normally
associated with these cells. These molecules include HLA-DR, CD68,
vitronectin,
apolipoprotein E, and perhaps clusterin and S-100. Although it is highly
unusual for non-
immunocompetent cells to express HLA-DR, this protein is typically expressed
by cells early
in immune reactions. Indeed, its expression by RPE cells may be a marker of
RPE cell
dysfunction and is likely to be involved in recognition of dysfunction and/or
damaged RPE by
other cells. Alternatively, the expression of HLA-DR might be a secondary
phenomenon
related to the presence of dendritic cells.
Fifth, gene array analyses of RPE/choroid preparations from AMD and control
donors indicate upregulation of a number of cell death associated molecules in
AMD. These
include, but are not limited to, death protein, heat shock protein 70,
proteasome, Cu/Zn
superoxide dismutase, cathepsins and death adaptor protein RAIDD.
It is unclear if drusen (or other abnormal changes in the extracellular
environment that is Bruch's membrane) are a cause, or a consequence of RPE
dysfunction. An
accumulation of drusen could cause local interference with the exchange of
metabolites and
waste products between the choriocapillaris and an otherwise normal RPE,
leading to RPE
dysfunction and death. On the other hand, drusen may be a consequence of
aberrant RPE gene
expression, although the precise biological events that ultimately lead to RPE
dysfunction are
equally unclear. Suggestions range from gene mutations to oxidative insults to
lipofuscin
accumulation, to programmed cell death. Whatever the progression of
pathological events,
localized RPE degeneration leads to a concomitant degeneration of the
underlying
photoreceptor cells, which in turn, result in the formation of numerous
scotomas
corresponding in size and in number to the distribution of macular drusen.
4.2.4 Immune-Mediated Processes and Drusen Biogenesis
Data from a variety of studies collectively suggest that immune-mediated
events may participate in the development and/or progression of AMD.
Autoantibodies have
been detected in the sera of AMD patients (Guerne, D., et al., Antiretinal
antibodies in serum
ofpatients with age-related macular degeneration. Ophthalmology, 1991. 98: p.
602-7;
29
CA 02363503 2001-08-24
WO 00/52479 PCT/US00/05858
Penfold, P., et al., Autoantibodies to retinal astrocytes associated with age-
related macular
degeneration. Graefe's Arch. Clin. Exp. Ophthalmol., 1990. 228: p. 270-4.).
Some of these are
directed against drusen, RPE and retina components based on
immunohistochemical and
Western analyses. Accumulations of giant multinucleated cells (Penfold, P., M.
Killingsworth, and S. Sarks, Senile macular degeneration. The involvement
ofgiant cells in
atrophy of the retinal pigment epithelium. Investigative Ophthalmology &
Visual Science,
1986. 27: p. 364-71; Dastgheib, K. and W. Green, Granulomatous reaction to
Bruch's
membrane in age-related macular degeneration. Archives of Ophthalmology, 1994.
112: p.
813-818); Penfold, P.L., et al., Modulation of major histocompatibility
complex class
II expression in retinas with age-related macular degeneration. Investigative
Ophthalmology
& Visual Science, 1997. 38(10): p. 2125-33.) and other leukocytes (Penfold,
P., M.
Killingsworth, and S. Sarks, Senile macular degeneration: the involvement of
immunocompetent cells. Graefe's Archives for Clinical and Experimental
Ophthalmology,
1985. 223:p.69-76);Killingsworth, M., J. Sarks, and S. Sarks, Macrophages
related to Bruch's
membrane in age-related macular degeneration. Eye, 1990. 4: p. 613-621) in the
choroid of
donors with AMD have been described and HLA-DR immunoreactivity of retinal
microglia
increases in AMD. It is also interesting that the synthesis of type VI
collagen, a putative
component of the basal laminar deposits that are prevalent in eyes of donors
with AMD,
increases in association with inflammatory processes leading to fibrotic
remodeling in diseases
such as lung fibrosis, scleroderma, and eosinophilic myalgia syndrome.
Exhaustive immunohistochemical analyses of drusen composition have
revealed a distinct array of molecules (including immunoglobulins, amyloid A,
amyloid P
component, C5 and C5b-9 terminal complexes, HLA-DR, fibrinogen, Factor X, and
prothrombin) that are common to all phenotypes of hard and soft drusen.
Surprisingly,
additional studies have documented that a number of these constituents (many
of which have
been thought to be synthesized primarily in the liver) are synthesized locally
by RPE, retinal,
and/or choroidal cells. These include complements 3, 5 and 9, complement
reactive protein
(CRP), immunoglobulin lambda and kappa light chains, Factor X, HLA-DR,
apolipoprotein A,
apolipoprotein E, amyloid A, vitronectin and others.
Interestingly, a number of these drusen-associated constituents (DRAMs) are
participants in humoral and cellular immune processes. Moreover, it is indeed
difficult to
ignore the presence of some of these molecules, including terminal complement
complex,
immunoglobulin, and MHC class II antigens, in drusen. For example, C5b-9
complex is
CA 02363503 2001-08-24
WO 00/52479 PCTIUSOO/05858
associated with specific immune processes, often involving cell death. Thus,
the presence of
C5b-9 in drusen and the expression of complement receptor genes by RPE and
choroidal cells,
including HCR1, HCR2, clusterin, vitronectin, and gp330/megalin brings to
question the role
of complement-mediated RPE cell death in drusen biogenesis and the etiology
drusen-
associated ocular disorders. Data from differential gene expression analyses
indicate a
significant up-regulation of a number of immune system-associated molecules
(including Ig
mu, lambda, J, and kappa chains) in the RPE/choroid of AMD donors, as compared
to age-
matched controls. Taken together, these data suggest that immune-related
processes may be
important in drusen development and the etiology AMD.
4.2.5 Dendritic Cells and Drusen Biogenesis:
Dendritic cells are found in primary lymphoid organs and most non-lymphoid
tissues and organs (Ibrahim, M., B. Chain, and D. Katz, The injured cell: the
role of the
dendritic cell system as a sentinel receptor pathway. Immunology Today, 1995.
16: p. 181-6;
Matyszak, M. and V. Perry, The potential role of dendritic cells in immune-
mediated
inflammatory diseases in the central nervous system. Neuroscience, 1996. 74:
p. 599-608;
Matyszak, M. and V. Perry, Dendritic cells in inflammatory responses in the
CNS, in
Dendritic cells in fundamental and clinical immunology, Ricciardi-Castagnoli,
Editor. 1997,
Plenum Press: New York), with the possible exception of the central nervous
system.
Precursor dendritic cells reside within non-lymphoid tissues. Dendritic cells
are powerful
antigen-presenting cells that contribute to the pathogenesis of immune-
mediated responses in a
number of ways, including the primary activation of T lymphocytes, various
secondary
responses, and the induction of autoimmune responses. Antigen presentation is
important in
the induction of conventional immune responses, as well as in the induction
and maintenance
of tolerance. It has been proposed that dendritic cells may provide an
essential link between
the innate and adaptive immune systems, actively participating in determining
the outcome of
the immune response. For example, data from recent investigations suggest that
dendritic
cells, and hence the innate immune system, can be activated by local,
microenvironmental
tissue damage. In this role, dendritic cells provide a sentinel receptor
system that responds to
local tissue injury and provides an integrative mechanism that determines the
outcome of the
immune response.
After acquiring an antigen, dendritic cells typically (but not always) migrate
out
31
CA 02363503 2001-08-24
WO 00/52479 PCT/USOO/05858
of the tissue, into the blood, through the afferent lymphatics, and into the T
cell-rich regions of
the local lymphoid organs. Important dendritic cell-associated accessory
molecules that
participate in T cell recognition include ICAM-1, LFA1, LFA3, and B7, whereas
T cell
counter receptors include LFA1, CD2, and CD28. Binding of the B7 ligand to its
counter
receptor CD28 is especially important in stimulating the synthesis and
secretion of IL-2 by T
cells.
The results of studies described herein provide additional strong support for
the
involvement of immune-related processes in drusen biogenesis. Most notably, a
novel and
specific association has been noted between a subpopulation of choroidal cells
and drusen.
Ultrastructurally, processes of morphologically distinct choroidal cells are
observed to breach
Bruch's membrane and to terminate as bulbous, vesicle-filled cores within the
centers of
drusen. An association of specific cluster differentiation (CD) antigen and
MHC class II
markers indicates that these cells are certainly of monocytic origin, and are
most likely
dendritic cells. Specific marker molecules, including CDIa, CD4, CD14, CD68,
CD83,
CD86, and CD45, react with drusen-associated dendritic cells, suggesting that
these cells
belong to the DCl lineage believed to participate in the induction of
immunity. Additional
immunocytochemical analyses document an intimate association of PECAM, MMP14,
ubiquitin, and possibly FGF and HLA with drusen-associated dendritic cell
cores.
Ongoing morphometric studies suggest that 40% of drusen in any given eye
contain these structures and that at least 70% of donors with drusen possess
at least one drusen
core. Similar numbers have been obtained using different markers. Drusen cores
are observed
in all drusen phenotypes and are present in both macular and extramacular
drusen. They may
be more prevalent in drusen possessing a height-width ratio of less than 0.5.
4.2.6 Similar etiology between drusen-associated ocular disorders and other
age-related diseases
Since drusen share a number of molecular constituents in common with
abnormal deposits associated with a variety of other age-related diseases,
drusen may represent
an ocular manifestation of amyloidosis, elastosis, dense deposit disease,
and/or atherosclerosis.
Although modulated by different genes and/or environmental influences, all
these diseases
give rise to similar, yet distinguishable, pathological phenotypes by
triggering a similar set of
32
CA 02363503 2001-08-24
WO 00/52479 PCTIUSOO/05858
biological responses that include inflammation, coagulation, and activation of
the immune
system. Thus, the invention provides a valuable recognition of these
similarities but also
provides a method for diagnosing and treating drusen specifically, as compared
to other age-
related diseases which manifest themselves in deposits or plaques.
Table 2: Compositional Comparison of Extracellular Disease Plaques
Dense Drusen
Elastosis Amyloidosis Atherosclerosis Deposits
Vn + + + + +
SAP + + + + +
Apo E ? 0 + -/? +
Complement + ? + + +
Elastin + ? + -/? ?
Lipids -* ?/- + + +
Ca2+ ?** ? 0 ? +
Macrophages ? + + ? +/?
* Sudanophilia has been described with actinic elastosis.
** Calcification of elastic fibers occurs in pseudoxanthoma elasticum.
References for Table 2: Aisen, 1996; Babaev, et al., 1990; Bobryshev, et al.,
1995; Castano, et
al., 1995; Dahlback, et al., 1988; Dahlback, et al., 1989; Dahlback, et al.,
1990; Guyton and
Klemp, 1996; Hoque, et al., 1993; Jang, et al., 1993; Jansen, et al., 1993;
Li, et al., 1995;
Muda, et al., 1988; Namba, et al., 1991; Niculescu, et al., 1987; Niculescu,
et al., 1989; Pepys,
et al., 1994; Sarks and Sarks, 1989; Stary, et al., 1995; Tarnawski, et al.,
1995; Wolter and
Falls, 1962.
4.3. Diagnostic Assays
In one aspect, the invention provides a method for diagnosing, or determining
a
predisposition to developing a drusen associated disease by detecting one or
more markers
which are associated with drusen development. Examples of phenotypic markers
include:
RPE dysfuncation and/or death, immune mediated events, dendritic cell
activation, migration
and differentiation, extrusion of the dendritic cell process into the sub RPE
space (e.g. by
33
CA 02363503 2001-08-24
WO 00/52479 PCTIUSOO/05858
detecting the presence or level of a dendritic cell marker such as CD68, CD 1
a and S 100), the
presence of geographic atrophy or disciform scars, the presence of choroidal
neovascularization and/or choroidal fibrosis, especially in the macula.
Examples of genotypic
markers include mutant genes and/or a distinct pattern of differential gene
expression (Drusen
Development Pathway"), including genes that are upregulated or downregulated
in drusen
forming ocular tissue associated with drusen biogenesis. For example genes
expressed by
dysfunctional and/or dying RPE cells include: HLA-DR, CD68, vitronectin,
apolipoprotein E,
clusterin and S-100. Genes expressed by choroidal and RPE cells in AMD inlcude
heat shock
protein 70, death protein, proteasome, Cu/Zn superoxide dismutase, cathepsins,
and death
adaptor protein RAIDD. Markers involved in immune mediated events associated
with drusen
formation include: autoantibodies (e.g. directed against drusen, RPE and/or
retina
components), leukocytes, dendritic cells, myofibroblasts, type VI collagen,
and a cadre of
chemokines and cytokines. Molecules associated with drusen include:
immunoglobulins,
amyloid A, amyloid P component, HLA-DR, fibrinogen, Factor X, prothrombin,
complements
3, 5, 9, and 5b-9, c reactive protein (CRP) apolipoprotein A, apolipoprotein
E,
antichymotrypsin, 02 microglobulin, thrombospondin, and vitronectin. Markers
of drusen
associated dendritic cells include: CDIa, CD4, CD14, CD68, CD83, CD86, and
CD45,
PECAM, MMP14, ubiquitin, and FGF. Important dendritic cell-associated
accessory
molecules that participate in T cell recognition include ICAM-1, LFA1, LFA3,
and B7, IL-l,
IL-6, IL-12, TNF-alpha, GM-CSF and heat shock proteins. Markers associated
with dendritic
cell expression include: colony stimulating factor, TNFa, and II-l. Markers
associated with
dendritic cell proliferation include: GM-CSF, IL-4,11-3, SCF, FLT-3 and TNFa.
Markers
associated with dendritic cell differentiation include IL-10, M-CSF, IL-6 and
IL-4. Markers of
fibrosis include: a decrease in BIG H3, increase in 0 1- integrin, increase in
collagen (e.g.
collagen 6 a2 and collagen 6 a3), increase in elastin, and an increase in
human metallo elastase
(HME).
Some drusen-associated markers may be detected by one or more
ophthalmological procedures, such as fundus fluorescein angiography (FFA),
fundus
ophthalmoscopy or photography (FP), electroretinogram (ERG), electrooculogram
(EOG),
visual fields, scanning laser ophthalmoscopy (SLO), visual acuity
measurements, dark
adaptation measurements or other standard method.
Other drusen-associated markers can be detected on the molecular level, e.g.
by
34
CA 02363503 2001-08-24
WO 00/52479 PCTIUSOO/05858
detecting the identity, level and/or activity of the gene, mRNA transcript or
encoded protein.
For example, drusen may be detected by determining the presence of any of the
following:
amyloid A protein, amyloid P component, antichymotrypsin, apolipoprotein E,
fl2
microglobulin, complement 3, complement C5, complement C5b-9 terminal
complexes, factor
X, fibrinogen, immunoglobulins (kappa and lambda), prothrombin and
thrombospondin. In
another embodiment, the drusen-associated marker is a molecule whose
production is altered
in a drusen-associated molecular pathological process. For example, one
pathological process
associated with drusen biogenesis is cell death and/or dysfunction of the
retinal pigment
epithelium (RPE). A number of molecular markers have been associated with such
dysfunctional RPE cells including: HLA-DR, CD68, vitronectin, apolipoprotein
E, clusterin
and S-100. HLA-DR expression is particularly unique for non-immunocompetent
cells
(although it is frequently expressed by cells early in an immune reaction).
Still other
molecular markers associated with dysfunctional choroid and RPE cells of AMD-
affected eyes
include gene products associated with cell death such as: death protein, heat
shock protein 70,
proteasome, Cu/Zn superoxide dismutase, cathepsins, and death adaptor protein
RAIDD.
Furthermore, drusen biogenesis is facilitated by dendritic cells and various
immune-mediated
events such as the production of autoantibodies in the sera of AMD patients.
These
autoantibodies are directed against drusen, the RPE and other retinal
components.
Accordingly, the invention provides for diagnostic assays designed to detect
the presence and
antigen specificity of such autoantibodies by methods known in the art,
including standard
immunohistochemical and Western blot techniques. Furthermore a number of
immune
system-associated molecules, including Ig mu, lambda, J, and kappa chains and
various
cytokines are up-regulated in the RPE/choroid in conjunction with the
formation of drusen.
Accordingly, these immune-associated molecules provide another target for
protein-based (e.g.
antibody-based detection methods) and nucleic acid-based (e.g. Northern, and
RT-PCR
methods) diagnostic assays. Still other drusen-associated molecular markers
are those found
in conjunction with subpopulation of choroidal cells that possess cellular
processes which
breach Bruch's membrane and ternminate as bulbous, vesicle-filled "cores"
within the centers of
drusen. Specific marker molecules associated with these dendritic cells
include: HLA-DR,
CDIa, CD4, CD14, CD68, CD83, CD86 and CD45. Other molecular markers appear to
be
associated with drusen-associated dendritic cell cores include: PECAM, MMP14,
ubiquitin,
FGF and HLA. In yet another aspect of the invention, the drusen-associated
marker may be a
CA 02363503 2001-08-24
WO 00/52479 PCT/USOO/05858
cytokine which facilitates the development of drusen via a receptor-ligand
interaction between
a dendritic cell precursor and an injured tissue. Such cytokines include: IL-
1, IL-6, IL-12,
TNF-alpha, and GM-CSF. Other molecules involved in drusen development include
heat
shock proteins, DNA fragments, elastolytic peptides, angiogenic agents and
factors up
regulated, such as 0 integrin, collagen 6a2, collagen 6 a3, elastin, HME, or
down regulated
(e.g. BIGH3) in fibrosis.
A variety of means are currently available for detecting aberrant levels or
activities of genes and gene products. For example, many methods are available
for detecting
specific alleles at human polymorphic loci. The preferred method for detecting
a specific
polymorphic allele will depend, in part, upon the molecular nature of the
polymorphism. For
example, the various allelic forms of the polymorphic locus may differ by a
single base-pair of
the DNA. Such single nucleotide polymorphisms (or SNPs) are major contributors
to genetic
variation, comprising some 80% of all known polymorphisms, and their density
in the human
genome is estimated to be on average 1 per 1,000 base pairs. SNPs are most
frequently
biallelic- occurring in only two different forms (although up to four
different forms of an SNP,
corresponding to the four different nucleotide bases occurring in DNA, are
theoretically
possible). Nevertheless, SNPs are mutationally more stable than other
polymorphisms,
making them suitable for association studies in which linkage disequilibrium
between markers
and an unknown variant is used to map disease-causing mutations. In addition,
because SNPs
typically have only two alleles, they can be genotyped by a simple plus/minus
assay rather
than a length measurement, making them more amenable to automation.
A variety of methods are available for detecting the presence of a particular
single nucleotide polymorphic allele in an individual. Advancements in this
field have
provided accurate, easy, and inexpensive large-scale SNP genotyping. Most
recently, for
example, several new techniques have been described including dynamic allele-
specific
hybridization (DASH), microplate array diagonal gel electrophoresis (MADGE),
pyrosequencing, oligonucleotide-specific ligation, the TaqMan system as well
as various DNA
"chip" technologies such as the Affymetrix SNP chips. These methods require
amplification
of the target genetic region, typically by PCR. Still other newly developed
methods, based on
the generation of small signal molecules by invasive cleavage followed by mass
spectrometry
or immobilized padlock probes and rolling-circle amplification, might
eventually eliminate the
need for PCR. Several of the methods known in the art for detecting specific
single nucleotide
polymorphisms are summarized below. The method of the present invention is
understood to
include all available methods.
Several methods have been developed to facilitate analysis of single
nucleotide
36
CA 02363503 2008-10-17
polymorphisms. In one embodiment, the single base polymorphism can be detected
by using
a specialized exonuclease-resistant nucleotide, as disclosed, e.g., in Mundy,
C. R. (U.S. Pat.
No.4,656,127). According to the method, a primer complementary to the allelic
sequence
immediately 3' to the polymorphic site is permitted to hybridize to a target
molecule obtained
from a particular animal or human. If the polymorphic site on the target
molecule contains a
nucleotide that is complementary to the particular exonuclease-resistant
nucleotide derivative
present, then that derivative will be incorporated onto the end of the
hybridized primer. Such
incorporation renders the primer resistant to exonuclease, and thereby permits
its detection.
Since the identity of the exonuclease-resistant derivative of the sample is
Irnown, a finding that
the primer has become resistant to exonucleases reveals that the nucleotide
present in the
polymorphic site of the target molecule was complementary to that of the
nucleotide derivative
used in the reaction. This method has the advantage that it does not require
the determination
of large amounts of extraneous sequence data.
In another embodiment of the invention, a solution-based method is used for
determining the identity of the nucleotide of a polymorphic site. Cohen, D. et
al. (French
Patent 2,650,840; PCT Appin. No. W091/02087). As in the Mundy method of U.S.
Pat. No.
4,656,127, a primer is employed that is complementary to allelic sequences
immediately 3' to a
polymorphic site. The method determines the identity of the nucleotide of that
site using
labeled dideoxynucleotide derivatives, which, if complementary to the
nucleotide of the
polymorphic site will become incorporated onto the terminus of the primer.
An alternative method, known as Genetic Bit Analysis or GBA TM is described
by Goelet, P. et al. (WO 92/15712). The method of Goelet, P. et al. uses
mixtures
of labeled terminators and a primer that is complementary to the sequence 3'
to a polymorphic
site. The labeled terminator that is incorporated is thus determined by, and
complementary to,
the nucleotide present in the polymorphic site of the target molecule being
evaluated. In
contrast to the method of Cohen et al. (French Patent 2,650,840; PCT Appln.
No.
W091/02087) the method of Goelet, P. et al. is preferably a heterogeneous
phase assay, in
which the primer or the target molecule is immobilized to a solid phase.
Recently, several primer-guided nucleotide incorporation procedures for
assaying polymorphic sites in DNA have been described (Komher, J. S. et al.,
Nucl. Acids.
Res. 17:7779-7784 (1989); Sokolov, B. P., Nucl. Acids Res. 18:3671 (1990);
Syvanen, A. -C.,
et al., Genomics 8:684-692 (1990); Kuppuswamy, M. N. et al., Proc. Natl. Acad.
Sci. (U.S.A.)
88:1143-1147 (1991); Prezant, T. R. et al., Hum. Mutat. 1:159-164 (1992);
Ugozzoli, L. et al.,
GATA 9:107-112 (1992); Nyren, P. et al., Anal. Biochem. 208:171-175 (1993)).
These
methods differ from GBA TM in that they all rely on the incorporation of
labeled
deoxynucleotides to discriminate between bases at a polymorphic site. In such
a format, since
37
CA 02363503 2001-08-24
WO 00/52479 PCT/US00/05858
the signal is proportional to the number of deoxynucleotides incorporated,
polymorphisms that
occur in runs of the same nucleotide can result in signals that are
proportional to the length of
the run (Syvanen, A. -C., et al., Amer. J. Hum. Genet. 52:46-59 (1993)).
For mutations that produce premature termination of protein translation, the
protein truncation test (PTT) offers an efficient diagnostic approach (Roest,
et. al., (1993)
Hum. Mol. Genet. 2:1719-21; van der Luijt, et. al., (1994) Genomics 20:1-4).
For PTT, RNA
is initially isolated from available tissue and reverse-transcribed, and the
segment of interest is
amplified by PCR. The products of reverse transcription PCR are then used as a
template for
nested PCR amplification with a primer that contains an RNA polymerase
promoter and a
sequence for initiating eukaryotic translation. After amplification of the
region of interest, the
unique motifs incorporated into the primer permit sequential in vitro
transcription and
translation of the PCR products. Upon sodium dodecyl sulfate-polyacrylamide
gel
electrophoresis of translation products, the appearance of truncated
polypeptides signals the
presence of a mutation that causes premature termination of translation. In a
variation of this
technique, DNA (as opposed to RNA) is used as a PCR template when the target
region of
interest is derived from a single exon.
Any cell type or tissue may be utilized to obtain nucleic acid samples for use
in
the diagnostics described herein. In a preferred embodiment, the DNA sample is
obtained
from a bodily fluid, e.g, blood, obtained by known techniques (e.g.
venipuncture) or saliva.
Alternatively, nucleic acid tests can be performed on dry samples (e.g. hair
or skin).
Diagnostic procedures may also be performed in situ directly upon tissue
sections (fixed and/or frozen) of patient tissue obtained from biopsies or
resections, such that
no nucleic acid purification is necessary. Nucleic acid reagents may be used
as probes and/or
primers for such in situ procedures (see, for example, Nuovo, G.J., 1992, PCR
in situ
hybridization: protocols and applications, Raven Press, NY).
In addition to methods which focus primarily on the detection of one nucleic
acid sequence, profiles may also be assessed in such detection schemes.
Fingerprint profiles
may be generated, for example, by utilizing a differential display procedure,
Northern analysis
and/or RT-PCR.
A preferred detection method is allele specific hybridization using probes
overlapping a region of at least one allele of a drusen associated marker,
which has at least
about 5, 10, 20, 25, or 30 nucleotides around the mutation or polymorphic
region. In a
preferred embodiment of the invention, several probes capable of hybridizing
specifically to
other allelic variants involved in glaucoma are attached to a solid phase
support, e.g., a "chip"
(which can hold up to about 250,000 oligonucleotides). Oligonucleotides can be
bound to a
solid support by a variety of processes, including lithography. Mutation
detection analysis
38
CA 02363503 2001-08-24
WO 00/52479 PCT/LJS00/05858
using these chips comprising oligonucleotides, also termed "DNA probe arrays"
is described
e.g., in Cronin et al. (1996) Human Mutation 7:244. In one embodiment, a chip
comprises all
the allelic variants of at least one polymorphic region of a gene. The solid
phase support is
then contacted with a test nucleic acid and hybridization to the specific
probes is detected.
Accordingly, the identity of numerous allelic variants of one or more genes
can be identified in
a simple hybridization experiment.
These techniques may also comprise the step of amplifying the nucleic acid
before analysis. Amplification techniques are known to those of skill in the
art and include,
but are not limited to cloning, polymerase chain reaction (PCR), polymerase
chain reaction of
specific alleles (ASA), ligase chain reaction (LCR), nested polymerase chain
reaction, self
sustained sequence replication (Guatelli, J.C. et al., 1990, Proc. Natl. Acad.
Sci. USA 87:1874-
1878), transcriptional amplification system (Kwoh, D.Y. et al., 1989, Proc.
Natl. Acad. Sci.
USA 86:1173-1177), and Q- Beta Replicase (Lizardi, P.M. et al., 1988,
Bio/Technology
6:1197).
Amplification products may be assayed in a variety of ways, including size
analysis, restriction digestion followed by size analysis, detecting specific
tagged
oligonucleotide primers in the reaction products, allele-specific
oligonucleotide (ASO)
hybridization, allele specific 5' exonuclease detection, sequencing,
hybridization, and the like.
PCR based detection means can include multiplex amplification of a plurality
of markers simultaneously. For example, it is well known in the art to select
PCR primers to
generate PCR products that do not overlap in size and can be analyzed
simultaneously.
Alternatively, it is possible to amplify different markers with primers that
are differentially
labeled and thus can each be differentially detected. Of course, hybridization
based detection
means allow the differential detection of multiple PCR products in a sample.
Other techniques
are known in the art to allow multiplex analyses of a plurality of markers.
In a merely illustrative embodiment, the method includes the steps of (i)
collecting a sample of cells from a patient, (ii) isolating nucleic acid
(e.g., genomic, mRNA or
both) from the cells of the sample, (iii) contacting the nucleic acid sample
with one or more
primers which specifically hybridize 5' and 3' to at least one allele of a
drusen-associated
marker under conditions such that hybridization and amplification of the
allele occurs, and (iv)
detecting the amplification product. These detection schemes are especially
useful for the
detection of nucleic acid molecules if such molecules are present in very low
numbers.
In a preferred embodiment of the subject assay, aberrant levels or activities
of
drusen-asssociated markers are identified by alterations in restriction enzyme
cleavage
patterns. For example, sample and control DNA is isolated, amplified
(optionally), digested
with one or more restriction endonucleases, and fragment length sizes are
determined by gel
39
CA 02363503 2001-08-24
WO 00/52479 PCTIUSOO/05858
electrophoresis.
In yet another embodiment, any of a variety of sequencing reactions known in
the art can be used to directly sequence the allele. Exemplary sequencing
reactions include
those based on techniques developed by Maxim and Gilbert ((1977) Proc. Natl
Acad Sci USA
74:560) or Sanger (Sanger et al (1977) Proc. Nat. Acad. Sci USA 74:5463). It
is also
contemplated that any of a variety of automated sequencing procedures may be
utilized when
performing the subject assays (see, for example Biotechniques (1995) 19:448),
including
sequencing by mass spectrometry (see, for example PCT publication WO 94/16101;
Cohen et
al. (1996) Adv Chromatogr 36:127-162; and Griffin et al. (1993) Appl Biochem
Biotechnol
38:147-159). It will be evident to one of skill in the art that, for certain
embodiments, the
occurrence of only one, two or three of the nucleic acid bases need be
determined in the
sequencing reaction. For instance, A-track or the like, e.g., where only one
nucleic acid is
detected, can be carried out.
In a further embodiment, protection from cleavage agents (such as a nuclease,
hydroxylamine or osmium tetraoxide and with piperidine) can be used to detect
mismatched
bases in RNA/RNA or RNA/DNA or DNA/DNA heteroduplexes (Myers, et al. (1985)
Science
230:1242). In general, the art technique of "mismatch cleavage" starts by
providing
heteroduplexes formed by hybridizing (labeled) RNA or DNA containing the wild-
type allele
with the sample. The double-stranded duplexes are treated with an agent which
cleaves single-
stranded regions of the duplex such as which will exist due to base pair
mismatches between
the control and sample strands. For instance, RNA/DNA duplexes can be treated
with RNase
and DNA/DNA hybrids treated with S 1 nuclease to enzymatically digest the
mismatched
regions. In other embodiments, either DNA/DNA or RNA/DNA duplexes can be
treated with
hydroxylamine or osmium tetroxide and with piperidine in order to digest
mismatched regions.
After digestion of the mismatched regions, the resulting material is then
separated by size on
denaturing polyacrylamide gels to determine the site of mutation. See, for
example, Cotton et
al (1988) Proc. Natl Acad Sci USA 85:4397; and Saleeba et al (1992) Methods
Enzymol.
217:286-295. In a preferred embodiment, the control DNA or RNA can be labeled
for
detection.
In still another embodiment, the mismatch cleavage reaction employs one or
more proteins that recognize mismatched base pairs in double-stranded DNA (so
called "DNA
mismatch repair" enzymes). For example, the mutY enzyme of E. coli cleaves A
at G/A
mismatches and the thymidine DNA glycosylase from HeLa cells cleaves T at G/T
mismatches (Hsu et al. (1994) Carcinogenesis 15:1657-1662). According to an
exemplary
embodiment, an appropriate probe is hybridized to a cDNA or other DNA product
from a test
cell(s). The duplex is treated with a DNA mismatch repair enzyme, and the
cleavage products,
CA 02363503 2001-08-24
WO 00/52479 PCTIUSOO/05858
if any, can be detected from electrophoresis protocols or the like. See, for
example, U.S.
Patent No. 5,459,039.
In other embodiments, alterations in electrophoretic mobility will be used to
identify aberrant levels or activities of drusen-associated markers. For
example, single strand
conformation polymorphism (SSCP) may be used to detect differences in
electrophoretic
mobility between mutant and wild type nucleic acids (Orita et al. (1989) Proc
Natl. Acad. Sci
USA 86:2766, see also Cotton (1993) Mutat Res 285:125-144; and Hayashi (1992)
Genet
Anal Tech App19:73-79). Single-stranded DNA fragments of sample and control
locus
alleles are denatured and allowed to renature. The secondary structure of
single-stranded
nucleic acids varies according to sequence, the resulting alteration in
electrophoretic mobility
enables the detection of even a single base change. The DNA fragments may be
labeled or
detected with labeled probes. The sensitivity of the assay may be enhanced by
using RNA
(rather than DNA), in which the secondary structure is more sensitive to a
change in sequence.
In a preferred embodiment, the subject method utilizes heteroduplex analysis
to separate
double stranded heteroduplex molecules on the basis of changes in
electrophoretic mobility
(Keen et al. (1991) Trends Genet 7:5).
In yet another embodiment, the movement of alleles in polyacrylamide gels
containing a gradient of denaturant is assayed using denaturing gradient gel
electrophoresis
(DGGE) (Myers et al. (1985) Nature 313:495). When DGGE is used as the method
of
analysis, DNA will be modified to insure that it does not completely denature,
for example by
adding a GC clamp of approximately 40 bp of high-melting GC-rich DNA by PCR.
In a
further embodiment, a temperature gradient is used in place of a denaturing
agent gradient to
identify differences in the mobility of control and sample DNA (Rosenbaum and
Reissner
(1987) Biophys Chem 265:12753).
Examples of other techniques for detecting alleles include, but are not
limited
to, selective oligonucleotide hybridization, selective amplification, or
selective primer
extension. For example, oligonucleotide primers may be prepared in which the
known
mutation or nucleotide difference (e.g., in allelic variants) is placed
centrally and then
hybridized to target DNA under conditions which permit hybridization only if a
perfect match
is found (Saiki et al. (1986) Nature 324:163); Saiki et al (1989) Proc. Natl
Acad. Sci USA
86:6230). Such allele specific oligonucleotide hybridization techniques may be
used to test
one mutation or polymorphic region per reaction when oligonucleotides are
hybridized to PCR
amplified target DNA or a number of different mutations or polymorphic regions
when the
oligonucleotides are attached to the hybridizing membrane and hybridized with
labeled target
DNA.
Alternatively, allele specific amplification technology which depends on
41
CA 02363503 2001-08-24
WO 00/52479 PCTIUSOO/05858
selective PCR amplification may be used in conjunction with the instant
invention.
Oligonucleotides used as primers for specific amplification may carry the
mutation or
polymorphic region of interest in the center of the molecule (so that
amplification depends on
differential hybridization) (Gibbs et al (1989) Nucleic Acids Res. 17:2437-
2448) or at the
extreme 3' end of one primer where, under appropriate conditions, mismatch can
prevent, or
reduce polymerase extension (Prossner (1993) Tibtech 11:238. In addition it
may be desirable
to introduce a novel restriction site in the region of the mutation to create
cleavage-based
detection (Gasparini et al (1992) Mol. Cell Probes 6:1). It is anticipated
that in certain
embodiments amplification may also be performed using Taq ligase for
amplification (Barany
(1991) Proc. Natl. Acad. Sci USA 88:189). In such cases, ligation will occur
only if there is a
perfect match at the 3' end of the 5' sequence making it possible to detect
the presence of a
known mutation at a specific site by looking for the presence or absence of
amplification.
In another embodiment, identification of an allelic variant is carried out
using
an oligonucleotide ligation assay (OLA), as described, e.g., in U.S. Pat. No.
4,998,617 and in
Landegren, U. et al. ((1988) Science 241:1077-1080). The OLA protocol uses two
oligonucleotides which are designed to be capable of hybridizing to abutting
sequences of a
single strand of a target. One of the oligonucleotides is linked to a
separation marker, e.g,.
biotinylated, and the other is detectably labeled. If the precise
complementary sequence is
found in a target molecule, the oligonucleotides will hybridize such that
their termini abut, and
create a ligation substrate. Ligation then permits the labeled oligonucleotide
to be recovered
using avidin, or another biotin ligand. Nickerson, D. A. et al. have described
a nucleic acid
detection assay that combines attributes of PCR and OLA (Nickerson, D. A. et
al. (1990) Proc.
Natl. Acad. Sci. USA 87:8923-27). In this method, PCR is used to achieve the
exponential
amplification of target DNA, which is then detected using OLA.
Several techniques based on this OLA method have been developed and can be
used to detect aberrant levels or activities of drusen-associated markers. For
example, U.S.
Patent No. 5,593,826 discloses an OLA using an oligonucleotide having 3'-amino
group and a
5'-phosphorylated oligonucleotide to form a conjugate having a phosphoramidate
linkage. In
another variation of OLA described in Tobe et al. ((1996) Nucleic Acids Res
24: 3728), OLA
combined with PCR permits typing of two alleles in a single microliter well.
By marking each
of the allele-specific primers with a unique hapten, i.e. digoxigenin and
fluorescein, each OLA
reaction can be detected by using hapten specific antibodies that are labeled
with different
enzyme reporters, alkaline phosphatase or horseradish peroxidase. This system
permits the
detection of the two alleles using a high throughput format that leads to the
production of two
different colors.
Another embodiment of the invention is directed to kits for detecting a
42
CA 02363503 2001-08-24
WO 00/52479 PCTIUSOO/05858
predisposition for developing a drusen-associated ocular disorder. This kit
may contain one or
more oligonucleotides, including 5' and 3' oligonucleotides that hybridize 5'
and 3' to at least
one drusen-associated marker. PCR amplification oligonucleotides should
hybridize between
25 and 2500 base pairs apart, preferably between about 100 and about 500 bases
apart, in order
to produce a PCR product of convenient size for subsequent analysis.
For use in a kit, oligonucleotides may be any of a variety of natural and/or
synthetic compositions such as synthetic oligonucleotides, restriction
fragments, cDNAs,
synthetic peptide nucleic acids (PNAs), and the like. The assay kit and method
may also
employ labeled oligonucleotides to allow ease of identification in the assays.
Examples of
labels which may be employed include radio-labels, enzymes, fluorescent
compounds,
streptavidin, avidin, biotin, magnetic moieties, metal binding moieties,
antigen or antibody
moieties, and the like.
The kit may, optionally, also include DNA sampling means. DNA sampling
means are well known to one of skill in the art and can include, but not be
limited to
substrates, such as filter papers, and the like; DNA purification reagents
such as NucleonTM
kits, lysis buffers, proteinase solutions and the like; PCR reagents, such as
l Ox reaction
buffers, thermostable polymerase, dNTPs, and the like; and allele detection
means such as
restriction enzyme, allele specific oligonucleotides, degenerate
oligonucleotide primers for
nested PCR from dried blood.
4.4. Predictive Medicine
Information obtained using the diagnostic assays described herein (alone or in
conjunction with additional genetic or environmental information, which
contributes to the
drusen associated ocular disorder) may be useful for diagnosing or confirming
that a
symptomatic subject (e.g. a subject symptomatic for AMD), has a genetic defect
(e.g. in an
AMD-associated gene or in a gene that regulates the expression of a drusen-
associated marker
gene), which causes or contributes to the particular disease or disorder.
Alternatively, the
information can be used prognostically. Based on the prognostic information, a
doctor can
recommend a regimen (e.g. diet or exercise) or therapeutic protocol, useful
for preventing or
prolonging onset of the particular disease or condition in the individual.
In addition, knowledge of the particular alteration or alterations, resulting
in
defective or deficient genes or proteins in an individual (the genetic
profile), alone or in
conjunction with information on other genetic defects contributing to the same
disease (the
genetic profile of a drusen associated disease) allows customization of
therapy for the
43
CA 02363503 2001-08-24
WO 00/52479 PCTIUSOO/05858
particular disease to the individual's genetic profile, the goal of
"pharmacogenomics". For
example, an individual's genetic profile or the genetic profile of a disease
or condition, to
which genetic alterations cause or contribute, can enable a doctor to 1) more
effectively
prescribe a drug that will address the molecular basis of the disease or
condition; and 2) better
determine the appropriate dosage of a particular drug. For example, the
expression level of
drusen-associated molecular marker proteins, alone or in conjunction with the
expression level
of other genes, known to contribute to the same disease, can be measured in
many patients at
various stages of the disease to generate a transcriptional or expression
profile of the disease.
Expression patterns of individual patients can then be compared to the
expression profile of
the disease to determine the appropriate drug and dose to administer to the
patient.
The ability to target populations expected to show the highest clinical
benefit,
based on the genetic profile, can enable: 1) the repositioning of marketed
drugs with
disappointing market results; 2) the rescue of drug candidates whose clinical
development has
been discontinued as a result of safety or efficacy limitations, which are
patient subgroup-
specific; and 3) an accelerated and less costly development for drug
candidates and more
optimal drug labeling (e.g. since the use of a drusen-associated molecular
markers can be
useful for optimizing effective dose).
4.5 Screening Assays for Therapeutics for Drusen Related Ocular Disorders
4.5.1. Cell-free assa,L
Cell-free assays can be used to identify compounds which are capable of
interacting with a drusen-associated marker or binding partners thereto, to
thereby modify
their activity and/or interaction. Such a compound can, e.g., modify the
structure of a drusen-
associated marker or binding partner thereto and thereby effect its activity.
Accordingly, one exemplary screening assay of the present invention includes
the steps of contacting a drusen-associated marker or functional fragment
thereof or a binding
partner thereto with a test compound or library of test compounds and
detecting the presence
or absence of complex formation. For detection purposes, the molecule can be
labeled with a
specific marker and the test compound or library of test compounds labeled
with a different
marker. Interaction of a test compound with a drusen-associated marker,
fragment thereof or a
binding partner thereto can then be detected by determining the level of the
two labels after an
incubation step and a washing step. The presence of two labels after the
washing step is
44
CA 02363503 2001-08-24
WO 00/52479 PCTIUSOO/05858
indicative of an interaction.
An interaction between molecules can also be identified by using real-time BIA
(Biomolecular Interaction Analysis, Pharmacia Biosensor AB) which detects
surface plasmon
resonance (SPR), an optical phenomenon. Detection depends on changes in the
mass
concentration of macromolecules at the biospecific interface, and does not
require any labeling
of interactants. In one embodiment, a library of test compounds can be
immobilized on a
sensor surface, e.g., which forms one wall of a micro-flow cell. A solution
containing the
drusen-associated marker, functional fragment thereof or binding partner
thereto is then flown
continuously over the sensor surface. A change in the resonance angle as shown
on a signal
recording, indicates that an interaction has occurred. This technique is
further described, e.g.,
in BlAtechnology Handbook by Pharmacia.
Another exemplary screening assay of the present invention includes the steps
of (a) forming a reaction mixture including: (i) a drusen-associated marker,
(ii) a binding
partner, and (iii) a test compound; and (b) detecting interaction of the
drusen-associated
marker and binding partner. The drusen-associated marker and binding partner
can be
produced recombinantly, purified from a source, e.g., plasma, or chemically
synthesized, as
described herein. A statistically significant change (potentiation or
inhibition) in the
interaction of the drusen-associated marker and the binding protein in the
presence of the test
compound, relative to the interaction in the absence of the test compound,
indicates a potential
agonist (mimetic or potentiator) or antagonist (inhibitor) of drusen-
associated bioactivity for
the test compound. The compounds of this assay can be contacted
simultaneously.
Alternatively, a drusen-associated marker can first be contacted with a test
compound for an
appropriate amount of time, following which the binding partner is added to
the reaction
mixture. The efficacy of the compound can be assessed by generating dose
response curves
from data obtained using various concentrations of the test compound.
Moreover, a control
assay can also be performed to provide a baseline for comparison.
Complex formation between a drusen-associated marker and a binding partner
may be detected by a variety of techniques. Modulation of the formation of
complexes can be
quantitated using, for example, detectably labeled proteins such as
radiolabeled, fluorescently
labeled, or enzymatically labeled drusen-associated markers or binding
partners, by
immunoassay, or by chromatographic detection.
Typically, it will be desirable to immobilize either the drusen-associated
CA 02363503 2001-08-24
WO 00/52479 PCT/USOO/05858
marker or its binding partner to facilitate separation of complexes from
uncomplexed forms of
one or both of the proteins, as well as to accommodate automation of the
assay. Binding of
drusen-associated marker to a binding partner, can be accomplished in any
vessel suitable for
containing the reactants. Examples include microtitre plates, test tubes, and
micro-centrifuge
tubes. In one embodiment, a fusion protein can be provided which adds a domain
that allows
the protein to be bound to a matrix. For example, glutathione-S-transferase
(GST) fusion
proteins can be adsorbed onto glutathione sepharose beads (Sigma Chemical, St.
Louis, MO)
or glutathione derivatized microtitre plates, which are then combined with the
drusen-
associated marker gene product binding partner, e.g. an 35S-labeled drusen-
associated marker
gene product binding partner, and the test compound, and the mixture incubated
under
conditions conducive to complex formation, e.g. at physiological conditions
for salt and pH,
though slightly more stringent conditions may be desired. Following
incubation, the beads are
washed to remove any unbound label, and the matrix immobilized and radiolabel
determined
directly (e.g. beads placed in scintillant), or in the supernatant after the
complexes are
subsequently dissociated. Alternatively, the complexes can be dissociated from
the matrix,
separated by SDS-PAGE, and the level of drusen-associated marker gene product
protein or
associated binding partner found in the bead fraction quantitated from the gel
using standard
electrophoretic techniques such as described in the appended examples.
Other techniques for immobilizing proteins on matrices are also available for
use in the subject assay. For instance, either a drusen-associated marker or
its cognate binding
partner can be immobilized utilizing conjugation of biotin and streptavidin.
For instance,
biotinylated drusen-associated marker molecules can be prepared from biotin-
NHS (N-
hydroxy-succinimide) using techniques well known in the art (e.g.,
biotinylation kit, Pierce
Chemicals, Rockford, IL), and immobilized in the wells of streptavidin-coated
96 well plates
(Pierce Chemical). Alternatively, antibodies reactive with drusen-associated
marker can be
derivatized to the wells of the plate, and the drusen associated marker
trapped in the wells by
antibody conjugation. As above, preparations of a drusen-associated marker, a
binding partner
and a test compound are incubated in the presenting wells of the plate, and
the amount of
complex trapped in the well can be quantitated. Exemplary methods for
detecting such
complexes, in addition to those described above for the GST-immobilized
complexes, include
immunodetection of complexes using antibodies reactive with the drusen-
associated marker or
binding partner, or which are reactive with the drusen-associated marker and
compete with the
46
CA 02363503 2001-08-24
WO 00/52479 PCTIUSOO/05858
binding partner; as well as enzyme-linked assays which rely on detecting an
enzymatic activity
associated with the binding partner, either intrinsic or extrinsic activity.
In the instance of the
latter, the enzyme can be chemically conjugated or provided as a fusion
protein with the
drusen-associated marker or binding partner. To illustrate, the drusen-
associated marker or
binding partner can be chemically cross-linked or genetically fused with
horseradish
peroxidase, and the amount of polypeptide trapped in the complex can be
assessed with a
chromogenic substrate of the enzyme, e.g. 3,3'-diamino-benzadine
terahydrochloride or 4-
chloro-l-napthol. Likewise, a fusion protein comprising the polypeptide and
glutathione-S-
transferase can be provided, and complex formation quantitated by detecting
the GST activity
using 1-chloro-2,4-dinitrobenzene (Habig et al (1974) J Biol Chem 249:7130).
For processes which rely on immunodetection for quantitating one of the
proteins trapped in the complex, antibodies against the protein, such as anti-
drusen-associated
marker antibodies, can be used. Alternatively, the protein to be detected in
the complex can be
"epitope tagged" in the form of a fusion protein which includes, in addition
to the drusen-
associated marker, a second polypeptide for which antibodies are readily
available (e.g. from
commercial sources). For instance, the GST fusion proteins described above can
also be used
for quantification of binding using antibodies against the GST moiety. Other
useful epitope
tags include myc-epitopes (e.g., see Ellison et al. (1991) J Biol Chem
266:21150-21157)
which includes a 10-residue sequence from c-myc, as well as the pFLAG system
(International
Biotechnologies, Inc.) or the pEZZ-protein A system (Pharmacia, NJ).
Cell-free assays can also be used to identify compounds which modulate an
activity of an drusen-associated marker. Accordingly, in one embodiment, a
drusen-associated
marker is contacted with a test compound and the catalytic activity of the
drusen-associated
marker is monitored. In one embodiment, the ability of a drusen-associated
marker to bind a
target molecule is determined. The binding affinity of a drusen-associated
marker to a target
molecule can be determined according to methods known in the art.
Determination of the
enzymatic activity of a drusen-associated marker can be performed with the aid
of the
substrate furanacryloyl-L-phenylalanyl-glycyl-glycine (FAPGG) under conditions
described in
Holmquist et al. (1979) Anal. Biochem. 95:540 and in U.S. Patent No.
5,259,045.
4.5.2. Cell-based assavs
In addition to cell-free assays, such as described above, drusen-associated
47
CA 02363503 2001-08-24
WO 00/52479 PCT/US00/05858
markers provided by the present invention facilitate the generation of cell-
based assays, e.g.,
for identifying small molecule agonists or antagonists. In one embodiment, a
cell expressing a
drusen-associated marker on the outer surface of its cellular membrane is
incubated in the
presence of a test compound alone or in the presence of a test compound and a
drusen-
associated marker and the interaction between the test compound and the drusen-
associated
marker or between the drusen-associated marker and the drusen-associated
marker binding
partner is detected, e.g., by using a microphysiometer (McConnell et al.
(1992) Science
257:1906). An interaction between the drusen-associated marker and either the
test compound
or the binding partner is detected by the microphysiometer as a change in the
acidification of
the medium. This assay system thus provides a means of identifying molecular
antagonists
which, for example, function by interfering with drusen-associated marker -
ligand (e.g.
receptor) interactions, as well as molecular agonist which, for example,
function by activating
a drusen-associated marker.
Cell based assays can also be used to identify compounds which modulate
expression of a drusen-associated marker gene, modulate translation of a
drusen-associated
marker mRNA, or which modulate the stability of a drusen-associated marker
mRNA or
protein. Accordingly, in one embodiment, a cell which is capable of expressing
a drusen-
associated marker, e.g., a retinal epithelial cell, is incubated with a test
compound and the
amount of drusen-associated marker produced in the cell medium is measured and
compared
to that produced from a cell which has not been contacted with the test
compound. The
specificity of the compound vis a vis a drusen-associated marker can be
confirmed by various
control analysis, e.g., measuring the expression of one or more control genes.
Compounds
which can be tested include small molecules, proteins, and nucleic acids. In
particular, this
assay can be used to determine the efficacy of antisense or ribozymes to
drusen-associated
marker genes.
In another embodiment, the effect of a test compound on transcription of a
drusen-associated marker gene is determined by transfection experiments using
a reporter gene
operatively linked to at least a portion of the promoter of a drusen-
associated marker gene. A
promoter region of a gene can be isolated, e.g., from a genomic library
according to methods
known in the art. The reporter gene can be any gene encoding a protein which
is readily
quantifiable, e.g, the luciferase or CAT gene. Such reporter gene are well
known in the art.
This invention further pertains to novel agents identified by the above-
48
CA 02363503 2001-08-24
WO 00/52479 PCTIUSOO/05858
described screening assays and uses thereof for treatments as described
herein.
4.5.3 Animal Models
The invention further provides for animal models, including transgenic
animals, which can be used for a variety of purposes, e.g., to identify
genetic loci involved in
the common etiology of drusen associated diseases, and, further, to create
animal models for
the treatment of drusen associated diseases.
The transgenic animals can contain a transgene, such as reporter gene, under
the control of a drusen-associated marker gene promoter or fragment thereof.
These animals
are useful, e.g., for identifying drugs that modulate production of the drusen-
associated
molecular marker, such as by modulating Factor X, HLA-DR, IL-6 or elastin gene
expression.
A target gene promoter can be isolated, e.g., by screening of a genomic
library with an
appropriate cDNA fragment and characterized according to methods known in the
art. In a
preferred embodiment of the present invention, the transgenic animal
containing a reporter
gene is used to screen a class of bioactive molecules for their ability to
modulate expression of
a drusen-associated molecular marker such as a DRAM. Yet other non-human
animals within
the scope of the invention include those in which the expression of the
endogenous target gene
has been mutated or "knocked out". A "knock out" animal is one carrying a
homozygous or
heterozygous deletion of a particular gene or genes. These animals could be
useful to
determine whether the absence of the target will result in a specific
phenotype, in particular
whether these mice have or are likely to develop a drusen associated disease.
Furthermore
these animals are useful in screens for drugs which alleviate or attenuate the
disease condition
resulting from the mutation of drusen associated markers. These animals are
also useful for
determining the effect of a specific amino acid difference, or allelic
variation, in a target gene.
That is, the target knock out animals can be crossed with transgenic animals
expressing, e.g., a
mutated form or allelic variant of the target gene containing a drusen
associated marker,
thereby resulting in an animal which expresses only the mutated protein and
not the wild-type
target gene product.
Methods for obtaining transgenic and knockout non-human animals are well
known in the art. Knock out mice are generated by homologous integration of a
"knock out"
construct into a mouse embryonic stem cell chromosome which encodes the gene
to be
knocked out. In one embodiment, gene targeting, which is a method of using
homologous
49
CA 02363503 2001-08-24
WO 00/52479 PCT/USOO/05858
recombination to modify an animal's genome, can be used to introduce changes
into cultured
embryonic stem cells. By targeting a specific gene of interest in ES cells,
these changes can
be introduced into the germlines of animals to generate chimeras. The gene
targeting
procedure is accomplished by introducing into tissue culture cells a DNA
targeting construct
that includes a segment homologous to a target locus, and which also includes
an intended
sequence modification to the genomic sequence (e.g., insertion, deletion,
point mutation). The
treated cells are then screened for accurate targeting to identify and isolate
those which have
been properly targeted.
Gene targeting in embryonic stem cells is in fact a scheme contemplated by the
present invention as a means for disrupting a target gene function through the
use of a
targeting transgene construct designed to undergo homologous recombination
with one or
more target genomic sequences. The targeting construct can be arranged so
that, upon
recombination with an element of a target gene, a positive selection marker is
inserted into (or
replaces) coding sequences of the gene. The inserted sequence functionally
disrupts the target
gene, while also providing a positive selection trait. Exemplary targeting
constructs are
described in more detail below.
Generally, the embryonic stem cells (ES cells ) used to produce the knockout
animals will be of the same species as the knockout animal to be generated.
Thus for example,
mouse embryonic stem cells will usually be used for generation of knockout
mice.
Embryonic stem cells are generated and maintained using methods well known
to the skilled artisan such as those described by Doetschman et al. (1985) J.
Embryol. Exp.
Mol Biol. 87:27-45). Any line of ES cells can be used, however, the line
chosen is typically
selected for the ability of the cells to integrate into and become part of the
germ line of a
developing embryo so as to create germ line transmission of the knockout
construct. Thus, any
ES cell line that is believed to have this capability is suitable for use
herein. One mouse strain
that is typically used for production of ES cells, is the 129J strain. Another
ES cell line is
murine cell line D3 (American Type Culture Collection, catalog no. CKL 1934)
Still another
preferred ES cell line is the WW6 cell line (loffe et al. (1995) PNAS 92:7357-
7361). The cells
are cultured and prepared for knockout construct insertion using methods well
known to the
skilled artisan, such as those set forth by Robertson in: Teratocarcinomas and
Embryonic Stem
Cells: A Practical Approach, E.J. Robertson, ed. IRL Press, Washington, D.C.
[1987]); by
Bradley et al. (1986) Current Topics in Devel. Biol. 20:357-371); and by Hogan
et al.
CA 02363503 2001-08-24
WO 00/52479 PCTIUSOO/05858
(Manipulating the Mouse Embryo: A Laboratory Manual, Cold Spring Harbor
Laboratory
Press, Cold Spring Harbor, NY [1986].
A knock out construct refers to a uniquely configured fragment of nucleic acid
which is introduced into a stem cell line and allowed to recombine with the
genome at the
chromosomal locus of the gene of interest to be mutated. Thus a given knock
out construct is
specific for a given gene to be targeted for disruption. Nonetheless, many
common elements
exist among these constructs and these elements are well known in the art. A
typical knock
out construct contains nucleic acid fragments of not less than about 0.5 kb
nor more than about
10.0 kb from both the 5' and the 3' ends of the genomic locus which encodes
the gene to be
mutated. These two fragments are separated by an intervening fragment of
nucleic acid which
encodes a positive selectable marker, such as the neomycin resistance gene
(neoR). The
resulting nucleic acid fragment, consisting of a nucleic acid from the extreme
5' end of the
genomic locus linked to a nucleic acid encoding a positive selectable marker
which is in turn
linked to a nucleic acid from the extreme 3' end of the genomic locus of
interest, omits most of
the coding sequence for the gene of interest to be knocked out. When the
resulting construct
recombines homologously with the chromosome at this locus, it results in the
loss of the
omitted coding sequence, otherwise known as the structural gene, from the
genomic locus. A
stem cell in which such a rare homologous recombination event has taken place
can be
selected for by virtue of the stable integration into the genome of the
nucleic acid of the gene
encoding the positive selectable marker and subsequent selection for cells
expressing this
marker gene in the presence of an appropriate drug (neomycin in this example).
Variations on this basic technique also exist and are well known in the art.
For
example, a "knock-in" construct refers to the same basic arrangement of a
nucleic acid
encoding a 5' genomic locus fragment linked to nucleic acid encoding a
positive selectable
marker which in turn is linked to a nucleic acid encoding a 3' genomic locus
fragment, but
which differs in that none of the coding sequence is omitted and thus the 5'
and the 3' genomic
fragments used were initially contiguous before being disrupted by the
introduction of the
nucleic acid encoding the positive selectable marker gene. This "knock-in"type
of construct
is thus very useful for the construction of mutant transgenic animals when
only a limited
region of the genomic locus of the gene to be mutated, such as a single exon,
is available for
cloning and genetic manipulation. Alternatively, the "knock-in" construct can
be used to
specifically eliminate a single functional domain of the targetted gene,
resulting in a transgenic
51
CA 02363503 2001-08-24
WO 00/52479 PCT/US00/05858
animal which expresses a polypeptide of the targetted gene which is defective
in one function,
while retaining the function of other domains of the encoded polypeptide. This
type of
"knock-in" mutant frequently has the characteristic of a so-called "dominant
negative" mutant
because, especially in the case of proteins which homomultimerize, it can
specifically block
the action of (or "poison") the polypeptide product of the wild-type gene from
which it was
derived. In a variation of the knock-in technique, a marker gene is integrated
at the genomic
locus of interest such that expression of the marker gene comes under the
control of the
transcriptional regulatory elements of the targeted gene. A marker gene is one
that encodes an
enzyme whose activity can be detected (e.g., b-galactosidase), the enzyme
substrate can be
added to the cells under suitable conditions, and the enzymatic activity can
be analyzed. One
skilled in the art will be familiar with other useful markers and the means
for detecting their
presence in a given cell. All such markers are contemplated as being included
within the
scope of the teaching of this invention.
As mentioned above, the homologous recombination of the above described
"knock out" and "knock in" constructs is very rare and frequently such a
construct inserts
nonhomologously into a random region of the genome where it has no effect on
the gene
which has been targeted for deletion, and where it can potentially recombine
so as to disrupt
another gene which was otherwise not intended to be altered. Such
nonhomologous
recombination events can be selected against by modifying the abovementioned
knock out and
knock in constructs so that they are flanked by negative selectable markers at
either end
(particularly through the use of two allelic variants of the thymidine kinase
gene, the
polypeptide product of which can be selected against in expressing cell lines
in an appropriate
tissue culture medium well known in the art - i.e. one containing a drug such
as 5-
bromodeoxyuridine). Thus a preferred embodiment of such a knock out or knock
in construct
of the invention consist of a nucleic acid encoding a negative selectable
marker linked to a
nucleic acid encoding a 5' end of a genomic locus linked to a nucleic acid of
a positive
selectable marker which in turn is linked to a nucleic acid encoding a 3' end
of the same
genomic locus which in turn is linked to a second nucleic acid encoding a
negative selectable
marker Nonhomologous recombination between the resulting knock out construct
and the
genome will usually result in the stable integration of one or both of these
negative selectable
marker genes and hence cells which have undergone nonhomologous recombination
can be
selected against by growth in the appropriate selective media (e.g. media
containing a drug
52
CA 02363503 2001-08-24
WO 00/52479 PCT/US00/05858
such as 5-bromodeoxyuridine for example). Simultaneous selection for the
positive selectable
marker and against the negative selectable marker will result in a vast
enrichment for clones in
which the knock out construct has recombined homologously at the locus of the
gene intended
to be mutated. The presence of the predicted chromosomal alteration at the
targeted gene
locus in the resulting knock out stem cell line can be confirmed by means of
Southern blot
analytical techniques which are well known to those familiar in the art.
Alternatively, PCR
can be used.
Each knockout construct to be inserted into the cell must first be in the
linear
form. Therefore, if the knockout construct has been inserted into a vector
(described infra),
linearization is accomplished by digesting the DNA with a suitable restriction
endonuclease
selected to cut only within the vector sequence and not within the knockout
construct
sequence.
For insertion, the knockout construct is added to the ES cells under
appropriate
conditions for the insertion method chosen, as is known to the skilled
artisan. For example, if
the ES cells are to be electroporated, the ES cells and knockout construct DNA
are exposed to
an electric pulse using an electroporation machine and following the
manufacturer's guidelines
for use. After electroporation, the ES cells are typically allowed to recover
under suitable
incubation conditions. The cells are then screened for the presence of the
knock out construct
as explained above. Where more than one construct is to be introduced into the
ES cell, each
knockout construct can be introduced simultaneously or one at a time.
After suitable ES cells containing the knockout construct in the proper
location
have been identified by the selection techniques outlined above, the cells can
be inserted into
an embryo. Insertion may be accomplished in a variety of ways known to the
skilled artisan,
however a preferred method is by microinjection. For microinjection, about 10-
30 cells are
collected into a micropipet and injected into embryos that are at the proper
stage of
development to permit integration of the foreign ES cell containing the
knockout construct
into the developing embryo. For instance, the transformed ES cells can be
microinjected into
blastocytes. The suitable stage of development for the embryo used for
insertion of ES cells is
very species dependent, however for mice it is about 3.5 days. The embryos are
obtained by
perfusing the uterus of pregnant females. Suitable methods for accomplishing
this are known
to the skilled artisan, and are set forth by, e.g., Bradley et al. (supra).
While any embryo of the right stage of development is suitable for use,
53
CA 02363503 2001-08-24
WO 00/52479 PCT/USOO/05858
preferred embryos are male. In mice, the preferred embryos also have genes
coding for a coat
color that is different from the coat color encoded by the ES cell genes. In
this way, the
offspring can be screened easily for the presence of the knockout construct by
looking for
mosaic coat color (indicating that the ES cell was incorporated into the
developing embryo).
Thus, for example, if the ES cell line carries the genes for white fur, the
embryo selected will
carry genes for black or brown fur.
After the ES cell has been introduced into the embryo, the embryo may be
implanted into the uterus of a pseudopregnant foster mother for gestation.
While any foster
mother may be used, the foster mother is typically selected for her ability to
breed and
reproduce well, and for her ability to care for the young. Such foster mothers
are typically
prepared by mating with vasectomized males of the same species. The stage of
the
pseudopregnant foster mother is important for successful implantation, and it
is species
dependent. For mice, this stage is about 2-3 days pseudopregnant.
Offspring that are born to the foster mother may be screened initially for
mosaic coat color where the coat color selection strategy (as described above,
and in the
appended examples) has been employed. In addition, or as an alternative, DNA
from tail tissue
of the offspring may be screened for the presence of the knockout construct
using Southern
blots and/or PCR as described above. Offspring that appear to be mosaics may
then be crossed
to each other, if they are believed to carry the knockout construct in their
germ line, in order to
generate homozygous knockout animals. Homozygotes may be identified by
Southern
blotting of equivalent amounts of genomic DNA from mice that are the product
of this cross,
as well as mice that are known heterozygotes and wild type mice.
Other means of identifying and characterizing the knockout offspring are
available. For example, Northern blots can be used to probe the mRNA for the
presence or
absence of transcripts encoding either the gene knocked out, the marker gene,
or both. In
addition, Western blots can be used to assess the level of expression of the
Target gene
knocked out in various tissues of the offspring by probing the Western blot
with an antibody
against the particular target protein, or an antibody against the marker gene
product, where this
gene is expressed. Finally, in situ analysis (such as fixing the cells and
labeling with antibody)
and/or FACS (fluorescence activated cell sorting) analysis of various cells
from the offspring
can be conducted using suitable antibodies to look for the presence or absence
of the knockout
construct gene product.
54
CA 02363503 2001-08-24
WO 00/52479 PCT/US00/05858
Yet other methods of making knock-out or disruption transgenic animals are
also generally known. See, for example, Manipulating the Mouse Embryo, (Cold
Spring
Harbor Laboratory Press, Cold Spring Harbor, N.Y., 1986). Recombinase
dependent
knockouts can also be generated, e.g. by homologous recombination to insert
target sequences,
such that tissue specific and/or temporal control of inactivation of a target-
gene can be
controlled by recombinase sequences (described infra).
Animals containing more than one knockout construct and/or more than one
transgene expression construct are prepared in any of several ways. The
preferred manner of
preparation is to generate a series of mammals, each containing one of the
desired transgenic
phenotypes. Such animals are bred together through a series of crosses,
backcrosses and
selections, to ultimately generate a single animal containing all desired
knockout constructs
and/or expression constructs, where the animal is otherwise congenic
(genetically identical) to
the wild type except for the presence of the knockout construct(s) and/or
transgene(s) .
A target transgene can encode the wild-type form of the protein, or can encode
homologs thereof, including both agonists and antagonists, as well as
antisense constructs. In
preferred embodiments, the expression of the transgene is restricted to
specific subsets of cells,
tissues or developmental stages utilizing, for example, cis-acting sequences
that control
expression in the desired pattern. In the present invention, such mosaic
expression of a target
protein can be essential for many forms of lineage analysis and can
additionally provide a
means to assess the effects of, for example, lack of target expression which
might grossly alter
development in small patches of tissue within an otherwise normal embryo.
Toward this end,
tissue-specific regulatory sequences and conditional regulatory sequences can
be used to
control expression of the transgene in certain spatial patterns. Moreover,
temporal patterns of
expression can be provided by, for example, conditional recombination systems
or prokaryotic
transcriptional regulatory sequences.
Genetic techniques, which allow for the expression of transgenes can be
regulated via site-specific genetic manipulation in vivo, are known to those
skilled in the art.
For instance, genetic systems are available which allow for the regulated
expression of a
recombinase that catalyzes the genetic recombination of a target sequence. As
used herein, the
phrase "target sequence" refers to a nucleotide sequence that is genetically
recombined by a
recombinase. The target sequence is flanked by recombinase recognition
sequences and is
generally either excised or inverted in cells expressing recombinase activity.
Recombinase
CA 02363503 2001-08-24
WO 00/52479 PCTIUSOO/05858
catalyzed recombination events can be designed such that recombination of the
target
sequence results in either the activation or repression of expression of one
of the subject target
proteins. For example, excision of a target sequence which interferes with the
expression of a
recombinant target gene, such as one which encodes an antagonistic homolog or
an antisense
transcript, can be designed to activate expression of that gene. This
interference with
expression of the protein can result from a variety of mechanisms, such as
spatial separation of
the target gene from the promoter element or an internal stop codon. Moreover,
the transgene
can be made wherein the coding sequence of the gene is flanked by recombinase
recognition
sequences and is initially transfected into cells in a 3' to 5' orientation
with respect to the
promoter element. In such an instance, inversion of the target sequence will
reorient the
subject gene by placing the 5' end of the coding sequence in an orientation
with respect to the
promoter element which allow for promoter driven transcriptional activation.
The transgenic animals of the present invention all include within a plurality
of
their cells a transgene of the present invention, which transgene alters the
phenotype of the
"host cell" with respect to regulation of cell growth, death and/or
differentiation. Since it is
possible to produce transgenic organisms of the invention utilizing one or
more of the
transgene constructs described herein, a general description will be given of
the production of
transgenic organisms by referring generally to exogenous genetic material.
This general
description can be adapted by those skilled in the art in order to incorporate
specific transgene
sequences into organisms utilizing the methods and materials described below.
In an illustrative embodiment, either the cre/loxP recombinase system of
bacteriophage P1 (Lakso et al. (1992) PNAS 89:6232-6236; Orban et al. (1992)
PNAS
89:6861-6865) or the FLP recombinase system of Saccharomyces cerevisiae
(O'Gorman et al.
(1991) Science 251:1351-1355; PCT publication WO 92/15694) can be used to
generate in
vivo site-specific genetic recombination systems. Cre recombinase catalyzes
the site-specific
recombination of an intervening target sequence located between loxP
sequences. loxP
sequences are 34 base pair nucleotide repeat sequences to which the Cre
recombinase binds
and are required for Cre recombinase mediated genetic recombination. The
orientation of loxP
sequences determines whether the intervening target sequence is excised or
inverted when Cre
recombinase is present (Abremski et al. (1984) J. Biol. Chem. 259:1509-1514);
catalyzing the
excision of the target sequence when the loxP sequences are oriented as direct
repeats and
catalyzes inversion of the target sequence when loxP sequences are oriented as
inverted
56
CA 02363503 2001-08-24
WO 00/52479 PCTIUSOO/05858
repeats.
Accordingly, genetic recombination of the target sequence is dependent on
expression of the Cre recombinase. Expression of the recombinase can be
regulated by
promoter elements which are subject to regulatory control, e.g., tissue-
specific, developmental
stage-specific, inducible or repressible by externally added agents. This
regulated control will
result in genetic recombination of the target sequence only in cells where
recombinase
expression is mediated by the promoter element. Thus, the activation
expression of a
recombinant target protein can be regulated via control of recombinase
expression.
Use of the crelloxP recombinase system to regulate expression of a
recombinant target protein requires the construction of a transgenic animal
containing
transgenes encoding both the Cre recombinase and the subject protein. Animals
containing
both the Cre recombinase and a recombinant target gene can be provided through
the
construction of "double" transgenic animals. A convenient method for providing
such animals
is to mate two transgenic animals each containing a transgene, e.g., a target
gene and
recombinase gene.
One advantage derived from initially constructing transgenic animals
containing a target transgene in a recombinase-mediated expressible format
derives from the
likelihood that the subject protein, whether agonistic or antagonistic, can be
deleterious upon
expression in the transgenic animal. In such an instance, a founder
population, in which the
subject transgene is silent in all tissues, can be propagated and maintained.
Individuals of this
founder population can be crossed with animals expressing the recombinase in,
for example,
one or more tissues and/or a desired temporal pattern. Thus, the creation of a
founder
population in which, for example, an antagonistic target transgene is silent
will allow the study
of progeny from that founder in which disruption of target mediated induction
in a particular
tissue or at certain developmental stages would result in, for example, a
lethal phenotype.
Similar conditional transgenes can be provided using prokaryotic promoter
sequences which require prokaryotic proteins to be simultaneous expressed in
order to
facilitate expression of the target transgene. Exemplary promoters and the
corresponding
trans-activating prokaryotic proteins are given in U.S. Patent No. 4,833,080.
Moreover, expression of the conditional transgenes can be induced by gene
therapy-like methods wherein a gene encoding the trans-activating protein,
e.g. a recombinase
or a prokaryotic protein, is delivered to the tissue and caused to be
expressed, such as in a cell-
57
CA 02363503 2001-08-24
WO 00/52479 PCTIUSOO/05858
type specific manner. By this method, a target transgene could remain silent
into adulthood
until "turned on" by the introduction of the trans-activator.
In an exemplary embodiment, the "transgenic non-human animals" of the
invention are produced by introducing transgenes into the germline of the non-
human animal.
Embryonal target cells at various developmental stages can be used to
introduce transgenes.
Different methods are used depending on the stage of development of the
embryonal target
cell. The specific line(s) of any animal used to practice this invention are
selected for general
good health, good embryo yields, good pronuclear visibility in the embryo, and
good
reproductive fitness. In addition, the haplotype is a significant factor. For
example, when
transgenic mice are to be produced, strains such as C57BL/6 or FVB lines are
often used
(Jackson Laboratory, Bar Harbor, ME). Preferred strains are those with H-2b, H-
2d or H-2q
haplotypes such as C57BL/6 or DBA/1. The line(s) used to practice this
invention may
themselves be transgenics, and/or may be knockouts (i.e., obtained from
animals which have
one or more genes partially or completely suppressed) .
In one embodiment, the transgene construct is introduced into a single stage
embryo. The zygote is the best target for micro-injection. In the mouse, the
male pronucleus
reaches the size of approximately 20 micrometers in diameter which allows
reproducible
injection of 1-2pl of DNA solution. The use of zygotes as a target for gene
transfer has a major
advantage in that in most cases the injected DNA will be incorporated into the
host gene
before the first cleavage (Brinster et al. (1985) PNAS 82:4438-4442). As a
consequence, all
cells of the transgenic animal will carry the incorporated transgene. This
will in general also
be reflected in the efficient transmission of the transgene to offspring of
the founder since 50%
of the germ cells will harbor the transgene.
Normally, fertilized embryos are incubated in suitable media until the
pronuclei
appear. At about this time, the nucleotide sequence comprising the transgene
is introduced into
the female or male pronucleus as described below. In some species such as
mice, the male
pronucleus is preferred. It is most preferred that the exogenous genetic
material be added to
the male DNA complement of the zygote prior to its being processed by the ovum
nucleus or
the zygote female pronucleus. It is thought that the ovum nucleus or female
pronucleus release
molecules which affect the male DNA complement, perhaps by replacing the
protamines of
the male DNA with histones, thereby facilitating the combination of the female
and male
DNA complements to form the diploid zygote.
58
CA 02363503 2001-08-24
WO 00/52479 PCTIUSOO/05858
Thus, it is preferred that the exogenous genetic material be added to the male
complement of DNA or any other complement of DNA prior to its being affected
by the
female pronucleus. For example, the exogenous genetic material is added to the
early male
pronucleus, as soon as possible after the formation of the male pronucleus,
which is when the
male and female pronuclei are well separated and both are located close to the
cell membrane.
Alternatively, the exogenous genetic material could be added to the nucleus of
the sperm after
it has been induced to undergo decondensation. Sperm containing the exogenous
genetic
material can then be added to the ovum or the decondensed sperm could be added
to the ovum
with the transgene constructs being added as soon as possible thereafter.
Introduction of the transgene nucleotide sequence into the embryo may be
accomplished by any means known in the art such as, for example,
microinjection,
electroporation, or lipofection. Following introduction of the transgene
nucleotide sequence
into the embryo, the embryo may be incubated in vitro for varying amounts of
time, or
reimplanted into the surrogate host, or both. In vitro incubation to maturity
is within the scope
of this invention. One common method in to incubate the embryos in vitro for
about 1-7 days,
depending on the species, and then reimplant them into the surrogate host.
For the purposes of this invention a zygote is essentially the formation of a
diploid cell which is capable of developing into a complete organism.
Generally, the zygote
will be comprised of an egg containing a nucleus formed, either naturally or
artificially, by the
fusion of two haploid nuclei from a gamete or gametes. Thus, the gamete nuclei
must be ones
which are naturally compatible, i.e., ones which result in a viable zygote
capable of
undergoing differentiation and developing into a functioning organism.
Generally, a euploid
zygote is preferred. If an aneuploid zygote is obtained, then the number of
chromosomes
should not vary by more than one with respect to the euploid number of the
organism from
which either gamete originated.
In addition to similar biological considerations, physical ones also govern
the
amount (e.g., volume) of exogenous genetic material which can be added to the
nucleus of the
zygote or to the genetic material which forms a part of the zygote nucleus. If
no genetic
material is removed, then the amount of exogenous genetic material which can
be added is
limited by the amount which will be absorbed without being physically
disruptive. Generally,
the volume of exogenous genetic material inserted will not exceed about 10
picoliters. The
physical effects of addition must not be so great as to physically destroy the
viability of the
59
CA 02363503 2001-08-24
WO 00/52479 PCTIUSOO/05858
zygote. The biological limit of the number and variety of DNA sequences will
vary depending
upon the particular zygote and functions of the exogenous genetic material and
will be readily
apparent to one skilled in the art, because the genetic material, including
the exogenous
genetic material, of the resulting zygote must be biologically capable of
initiating and
maintaining the differentiation and development of the zygote into a
functional organism.
The number of copies of the transgene constructs which are added to the zygote
is dependent upon the total amount of exogenous genetic material added and
will be the
amount which enables the genetic transformation to occur. Theoretically only
one copy is
required; however, generally, numerous copies are utilized, for example, 1,000-
20,000 copies
of the transgene construct, in order to insure that one copy is functional. As
regards the present
invention, there will often be an advantage to having more than one
functioning copy of each
of the inserted exogenous DNA sequences to enhance the phenotypic expression
of the
exogenous DNA sequences.
Any technique which allows for the addition of the exogenous genetic material
into nucleic genetic material can be utilized so long as it is not destructive
to the cell, nuclear
membrane or other existing cellular or genetic structures. The exogenous
genetic material is
preferentially inserted into the nucleic genetic material by microinjection.
Microinjection of
cells and cellular structures is known and is used in the art.
Reimplantation is accomplished using standard methods. Usually, the surrogate
host is anesthetized, and the embryos are inserted into the oviduct. The
number of embryos
implanted into a particular host will vary by species, but will usually be
comparable to the
number of off spring the species naturally produces.
Transgenic offspring of the surrogate host may be screened for the presence
and/or expression of the transgene by any suitable method. Screening is often
accomplished by
Southern blot or Northern blot analysis, using a probe that is complementary
to at least a
portion of the transgene. Western blot analysis using an antibody against the
protein encoded
by the transgene may be employed as an alternative or additional method for
screening for the
presence of the transgene product. Typically, DNA is prepared from tail tissue
and analyzed
by Southern analysis or PCR for the transgene. Alternatively, the tissues or
cells believed to
express the transgene at the highest levels are tested for the presence and
expression of the
transgene using Southern analysis or PCR, although any tissues or cell types
may be used for
this analysis.
CA 02363503 2001-08-24
WO 00/52479 PCT/US00/05858
Alternative or additional methods for evaluating the presence of the transgene
include, without limitation, suitable biochemical assays such as enzyme and/or
immunological
assays, histological stains for particular marker or enzyme activities, flow
cytometric analysis,
and the like. Analysis of the blood may also be useful to detect the presence
of the transgene
product in the blood, as well as to evaluate the effect of the transgene on
the levels of various
types of blood cells and other blood constituents.
Progeny of the transgenic animals may be obtained by mating the transgenic
animal with a suitable partner, or by in vitro fertilization of eggs and/or
sperm obtained from
the transgenic animal. Where mating with a partner is to be performed, the
partner may or may
not be transgenic and/or a knockout; where it is transgenic, it may contain
the same or a
different transgene, or both. Alternatively, the partner may be a parental
line. Where in vitro
fertilization is used, the fertilized embryo may be implanted into a surrogate
host or incubated
in vitro, or both. Using either method, the progeny may be evaluated for the
presence of the
transgene using methods described above, or other appropriate methods.
The transgenic animals produced in accordance with the present invention will
include exogenous genetic material. As set out above, the exogenous genetic
material will, in
certain embodiments, be a DNA sequence which results in the production of a
Target protein
(either agonistic or antagonistic), and antisense transcript, or a Target
mutant. Further, in such
embodiments the sequence will be attached to a transcriptional control
element, e.g., a
promoter, which preferably allows the expression of the transgene product in a
specific type of
cell.
Retroviral infection can also be used to introduce transgene into a non-human
animal. The developing non-human embryo can be cultured in vitro to the
blastocyst stage.
During this time, the blastomeres can be targets for retroviral infection
(Jaenich, R. (1976)
PNAS 73:1260-1264). Efficient infection of the blastomeres is obtained by
enzymatic
treatment to remove the zona pellucida (Manipulating the Mouse Embryo, Hogan
eds. (Cold
Spring Harbor Laboratory Press, Cold Spring Harbor, 1986). The viral vector
system used to
introduce the transgene is typically a replication-defective retrovirus
carrying the transgene
(Jahner et al. (1985) PNAS 82:6927-693 1; Van der Putten et al. (1985) PNAS
82:6148-6152).
Transfection is easily and efficiently obtained by culturing the blastomeres
on a monolayer of
virus-producing cells (Van der Putten, supra; Stewart et al. (1987) EMBO J.
6:383-388).
Alternatively, infection can be performed at a later stage. Virus or virus-
producing cells can be
61
CA 02363503 2001-08-24
WO 00/52479 PCT/USOO/05858
injected into the blastocoele (Jahner et al. (1982) Nature 298:623-628). Most
of the founders
will be mosaic for the transgene since incorporation occurs only in a subset
of the cells which
formed the transgenic non-human animal. Further, the founder may contain
various retroviral
insertions of the transgene at different positions in the genome which
generally will segregate
in the offspring. In addition, it is also possible to introduce transgenes
into the germ line by
intrauterine retroviral infection of the midgestation embryo (Jahner et al.
(1982) supra).
A third type of target cell for transgene introduction is the embryonal stem
cell
(ES). ES cells are obtained from pre-implantation embryos cultured in vitro
and fused with
embryos (Evans et al. (1981) Nature 292:154-156; Bradley et al. (1984) Nature
309:255-258;
Gossler et al. (1986) PNAS 83: 9065-9069; and Robertson et al. (1986) Nature
322:445-448).
Transgenes can be efficiently introduced into the ES cells by DNA transfection
or by
retrovirus-mediated transduction. Such transformed ES cells can thereafter be
combined with
blastocysts from a non-human animal. The ES cells thereafter colonize the
embryo and
contribute to the germ line of the resulting chimeric animal. For review see
Jaenisch, R. (1988)
Science 240:1468-1474.
4.6. Therapeutics
In another aspect, the invention provides compositions and methods for
treating
or preventing the development of drusen associated ocular disorders.
Appropriate therapeutics can include any molecule or compound that slows or
prevents any of the processes involved in drusen biogenesis, including
dendritic cell activation
and recruitment, immune mediated events, choroidal fibrosis and
neovascularization,
extracellular matrix disequilibrium, etc. For example, an appropriate
therapeutic may be an anti-inflammatory agent, such as an antagonist of TNF-a,
IL-1, GM-
CSF, IL-4 or IL-13. The therapeutic may also be IL-10, M-CSF, IL-6 and IL-4 or
an agonist
thereof. Any therapeutic that helps to decrease drusen formation or DS/CNV may
be used. In
a preferred embodiment, the agent is selected from the group consisting of
cytokines,
chemokines and agonists and antagonists thereof. Useful therapeutics include
agents that
inhibit inflammation.
In another embodiment, the macular degeneration therapeutic is an inhibitor of
the expression of one or more DRAMs, such as, for example, amyloid A protein,
amyloid P
component, al-antichymotrypsin, apolipoprotein E, 02 microglobulin, complement
3,
62
CA 02363503 2001-08-24
WO 00/52479 PCT/US00/05858
complement C5, complement C5b-9 terminal complexes, factor X, fibrinogen,
immunoglobulins (kappa and lambda), prothrombin, thrombospondin or
vitronectin. In an
another embodiment, the invention provides method for treating a drusen
associated disease
by modulating the production of DRAMs, e.g., inhibiting or antagonizing their
gene
expression or activity. The accumulation of amyloid P and al-antichymotrypsin
(an inhibitor
of serine proteases) in drusen may act to counterbalance attempts by RPE or
choroidal cells to
clear drusen proteolytically. For example, amyloid P is also found in non-
amyloid deposits
associated with atherosclerosis (Niculescu, et al., 1987), keratin
intermediate filament
aggregates (Hintner, et al., 1988), and dense deposits associated with
glomerulonephropathy
(Yang, et al., 1992). It associates with elastic fibers and may function as an
protease inhibitor
in vivo (Li and McAdam, 1984; Vachino, et al., 1988). It is also a normal
component of
Bruch's membrane, where it might protect the elastic lamina against enzymatic
degradation
(Kivela, et al., 1994). The downregulation of the biosynthesis of these
proteins is therefore
important for inhibiting drusen formation or facilitating drusen clearance or
resolution.
Inhibiting of drusen formation or facilitating drusen clearance or resolution
may be
accomplished by a number of regimes, such as (1) inhibition of RNA synthesis
for one or
more DRAMs, (2) enhancement of RNA turnover or degradation of one or more
DRAMs, (3)
inhibition of translation of RNA for one or more DRAMs into protein, (4)
inhibition of protein
processing or transport of one or more DRAMs; (5) inhibition of drusen
formation by blocking
particular protein binding sites on one or more factors which participate in
inter- and intra-
molecular binding necessary for the association of DRAMs which results in a
drusen deposit;
(6) digestion or perturbation of protein deposits (e.g., using enzymes); (7)
targeting and
destroying DRAMs in situ (e.g., using enzyme-antibody techniques). DRAMs may
be targeted
by using photoreactive laser therapy, for example, or other means for
targeting and destroying
a protein in situ which are well known in the art. Such means may include
antibodies
conjugated to a reactive group such as a protease or chemical substance which,
when
activated, cleaves or denatures the individual components or interferes with
the interaction of
two or more components.
In another embodiment, therapeutics for drusen-associated diseases include
agents which alter the gene expression of factors that regulate the expression
of one or more
DRAMs and all other drusen biogenesis associated proteins. Such agents may be
"antagonists" which inhibit, either directly or indirectly, DRAM biosynthesis.
The agent may
63
CA 02363503 2001-08-24
WO 00/52479 PCTIUSOO/05858
specifically inhibit the transcription or translation of a DRAM, for example.
Alternatively, it
may be preferable to upregulate either directly or indirectly a gene or genes
which will
increase the synthesis of a naturally occurring therapeutic agent. For
example, the increased
gene expression of a proteolytic enzyme that degrades one or more DRAMS or a
cytokine or
drug that modulates immune responses may be desired.
The invention is therefore also useful for monitoring the efficacy of a drusen
therapeutic or preventative treatment, the absence of drusen core formation,
the disappearance
of drusen or of a drusen core providing evidence of efficacy of the
therapeutic or treatment.
In one aspect, the therapeutics of the invention relate to antisense therapy.
As
used herein, "antisense" therapy refers to administration or in situ
generation of
oligonucleotide molecules or their derivatives which specifically hybridize
(e.g., bind) under
cellular conditions, with the cellular mRNA and/or genomic DNA encoding one or
more
DRAMs so as to inhibit expression of that protein, e.g., by inhibiting
transcription and/or
translation. The binding may be by conventional base pair complementarity, or,
for example,
in the case of binding to DNA duplexes, through specific interactions in the
major groove of
the double helix. In general, "antisense" therapy refers to the range of
techniques generally
employed in the art, and includes any therapy which relies on specific binding
to
oligonucleotide sequences.
An antisense construct of the present invention can be delivered, for example,
as an expression plasmid which, when transcribed in the cell, produces RNA
which is
complementary to at least a unique portion of the cellular mRNA which encodes
a DRAM
protein. Alternatively, the antisense construct can be an oligonucleotide
probe which is
generated ex vivo and which, when introduced into the cell causes inhibition
of expression by
hybridizing with the mRNA and/or genomic sequences of a DRAM gene. Such
oligonucleotide probes are preferably modified oligonucleotides which are
resistant to
endogenous nucleases, e.g., exonucleases and/or endonucleases, and are
therefore stable in
vivo. Exemplary nucleic acid molecules for use as antisense oligonucleotides
are
phosphoramidate, phosphothioate and methylphosphonate analogs of DNA (see also
U.S.
Patent Nos. 5,176,996, 5,264,564 and 5,256,775). Approaches to constructing
oligomers
useful in antisense therapy are well known in the art. With respect to
antisense DNA,
oligodeoxyribonucleotides derived from the translation initiation site, e.g.,
between the -10
and +10 regions of the drusen-associated component nucleotide sequence of
interest, are
64
CA 02363503 2001-08-24
WO 00/52479 PCTIUSOO/05858
preferred.
Antisense approaches involve the design of oligonucleotides (either DNA or
RNA) that are complementary to a DRAM mRNA, or their agonists or antagonists.
The
antisense oligonucleotides bind to the subject mRNA transcripts and prevent
translation or
promote degradation of the transcript. Absolute complementarity, although
preferred, is not
required. In the case of double-stranded antisense nucleic acids, a single
strand of the duplex
DNA may thus be tested, or triplex formation may be assayed. The ability to
hybridize
depends on both the degree of complementarity and the length of the antisense
nucleic acid.
Generally, the longer the hybridizing nucleic acid, the more base mismatches
with an RNA it
may contain and still form a stable duplex (or triplex, as the case may be).
One skilled in the
art can ascertain a tolerable degree of mismatch by use of standard procedures
to determine the
melting point of the hybridized complex. Other features, strategies and
methods of preparing
and using antisense or ribozymes are found in U.S.S.N. 09/183,972, the
teachings of which are
incorporated herein
4.6.1 Formulation and Use
Pharmaceutical compositions for use in accordance with the present invention
may be formulated in a conventional manner using one or more physiologically
acceptable
carriers or excipients. Thus, the compounds and their physiologically
acceptable salts and
solvates may be formulated for administration by, for example, eye drops,
injection, inhalation
or insufflation (either through the mouth or the nose) or oral, buccal,
parenteral or rectal
administration.
For such therapy, the compounds of the invention can be formulated for a
variety of modes of administration, including systemic and topical or
localized administration.
Techniques and formulations generally may be found in Remmington's
Pharmaceutical
Sciences, Meade Publishing Co., Easton, PA. A preferred method of
administration is an eye
drop. For systemic administration, injection is preferred, including
intramuscular,
intravenous, intraperitoneal, and subcutaneous. For injection, the compounds
of the invention
can be formulated in liquid solutions, preferably in physiologically
compatible buffers such as
Hank's solution or Ringer's solution. In addition, the compounds may be
formulated in solid
form and redissolved or suspended immediately prior to use. Lyophilized forms
are also
included.
CA 02363503 2001-08-24
WO 00/52479 PCT/USOO/05858
Other preferred methods of administration include choroidal injection,
transscleral injection or placing a scleral patch, and selective arterial
catheterization. Other
preferred deliveries are intraocular, including transretinal, subconjunctival
bulbar, scleral
pocket and scleral cutdown injections. The agent can be alternatively
administered
intravascularly, such as intravenously (N) or intraarterially.
Techniques for choroidal injection and scleral patching are similar. The
clinician uses a local approach to the eye after initiation of appropriate
anesthesia, including
painkillers and ophthalmoplegics. A needle containing the therapeutic compound
is directed
into the patient's choroid or sclera and inserted under sterile conditions.
When the needle is
properly positioned the compound is injected into either or both of the
choroid or sclera.
When using either of these methods, the clinician may choose a sustained
release or longer
acting formulation. Thus, the procedure may need repetition only every several
months or
several years, depending on the patient's tolerance of the treatment and
response.
For oral administration, the pharmaceutical compositions may take the form of,
for example, tablets or capsules prepared by conventional means with
pharmaceutically
acceptable excipients such as binding agents (e.g., pregelatinised maize
starch,
polyvinylpyrrolidone or hydroxypropyl methylcellulose); fillers (e.g.,
lactose, microcrystalline
cellulose or calcium hydrogen phosphate); lubricants (e.g., magnesium
stearate, talc or silica);
disintegrants (e.g., potato starch or sodium starch glycolate); or wetting
agents (e.g., sodium
lauryl sulfate). The tablets may be coated by methods well known in the art.
Liquid
preparations for oral administration may take the form of, for example,
solutions, syrups or
suspensions, or they may be presented as a dry product for constitution with
water or other
suitable vehicle before use. Such liquid preparations may be prepared by
conventional means
with pharmaceutically acceptable additives such as suspending agents (e.g.,
sorbitol syrup,
cellulose derivatives or hydrogenated edible fats); emulsifying agents (e.g.,
lecithin or acacia);
non-aqueous vehicles (e.g., ationd oil, oily esters, ethyl alcohol or
fractionated vegetable oils);
and preservatives (e.g., methyl or propyl-p-hydroxybenzoates or sorbic acid).
The
preparations may also contain buffer salts, flavoring, coloring and sweetening
agents as
appropriate.
The therapeutic may be administered alone or in combination with other
molecules known to have a beneficial effect on retinal attachment or damaged
retinal tissue,
including molecules capable of tissue repair and regeneration and/or
inhibiting inflammation.
66
CA 02363503 2001-08-24
WO 00/52479 PCTIUSOO/05858
Examples of useful cofactors include basic fibroblast growth factor (bFGF),
LaVail et al.
(1998), Invest. Ophthalmol. Vis. Sci. 39:592-602, ciliary neurotrophic factor
(CNTF), LaVail
et al. (1998), Invest. Ophthalmol. Vis. Sci. 39:592-602, axokine (a mutein of
CNTF), LaVail et
al. (1998), Invest. Ophthalmol. Vis. Sci. 39:592-602, leukemia inhibitory
factor (LIF), LaVail
et al. (1998), Invest. Ophthalmol. Vis. Sci. 39:592-602. neutrotrophin 3 (NT-
3), LaVail et al.
(1998), Invest. Ophthalmol. Vis. Sci. 39:592-602, neurotrophin-4 (NT-4),
LaVail et al. (1998),
Invest. Ophthalmol. Vis. Sci. 39:592-602, nerve growth factor (NGF), LaVail et
al. (1998),
Invest. Ophthalmol. Vis. Sci. 39:592-602, insulin-like growth factor II,
LaVail et al. (1998),
Invest. Ophthalmol. Vis. Sci. 39:592-602, prostaglandin E2, La Vail et al.
(1998), Invest.
Ophthalrnol. Vis. Sci. 39:581-591, 30kD survival factor, taurine, and vitamin
A. Other useful
cofactors include symptom-alleviating cofactors, including antiseptics,
antibiotics, antiviral
and antifungal agents and analgesics and anesthetics.
A therapeutic also may be associated with means for targeting the therapeutics
to a desired tissue. Alternatively, an antibody or other binding protein that
interacts
specifically with a surface molecule on the desired target tissue cells also
may be used. Such
targeting molecules further may be covalently associated to a therapeutic,
e.g., by chemical
crosslinking, or by using standard genetic engineering means to create, for
example, an acid
labile bond such as an Asp-Pro linkage. Useful targeting molecules may be
designed, for
example, using the simple chain binding site technology disclosed, for
example, in U.S. Patent
No.5,091,513.
Preparations for oral administration may be suitably formulated to give
controlled release of the active compound. For buccal administration, the
compositions may
take the form of tablets or lozenges formulated in conventional manner. For
administration by
inhalation, the compounds for use according to the present invention are
conveniently
delivered in the form of an aerosol spray presentation from pressurized packs
or a nebuliser,
with the use of a suitable propellant, e.g., dichlorodifluoromethane,
trichlorofluoromethane,
dichlorotetrafluoroethane, carbon dioxide or other suitable gas. In the case
of a pressurized
aerosol the dosage unit may be determined by providing a valve to deliver a
metered amount.
Capsules and cartridges of e.g., gelatin for use in an inhaler or insufflator
may be formulated
containing a powder mix of the compound and a suitable powder base such as
lactose or
starch.
The compounds may be formulated for parenteral administration by injection,
67
CA 02363503 2001-08-24
WO 00/52479 PCTIUSOO/05858
e.g., by bolus injection or continuous infusion. Formulations for injection
may be presented in
unit dosage form, e.g., in ampoules or in multi-dose containers, with an added
preservative.
The compositions may take such forms as suspensions, solutions or emulsions in
oily or
aqueous vehicles, and may contain formulatory agents such as suspending,
stabilizing and/or
dispersing agents. Alternatively, the active ingredient may be in powder form
for constitution
with a suitable vehicle, e.g., sterile pyrogen-free water, before use.
The compounds may also be formulated in rectal compositions such as
suppositories or retention enemas, e.g., containing conventional suppository
bases such as
cocoa butter or other glycerides.
In addition to the formulations described previously, the compounds may also
be formulated as a depot preparation. Such long acting formulations may be
administered by
implantation (for example subcutaneously or intramuscularly) or by
intramuscular injection.
Thus, for example, the compounds may be formulated with suitable polymeric or
hydrophobic
materials (e.g., as an emulsion in an acceptable oil) or ion exchange resins,
or as sparingly
soluble derivatives, for example, as a sparingly soluble salt. Other suitable
delivery systems
include microspheres which offer the possibility of local noninvasive delivery
of drugs over an
extended period of time. This technology utilizes microspheres of precapillary
size which can
be injected via a coronary catheter into any selected part of the body, e.g.,
the eye, or other
organs without causing inflammation or ischemia. The administered therapeutic
is slowly
released from these microspheres and taken up by surrounding tissue cells
(e.g., endothelial
cells).
Systemic administration can also be by transmucosal or transdermal means.
For transmucosal or transdermal administration, penetrants appropriate to the
barrier to be
permeated are used in the formulation. Such penetrants are generally known in
the art, and
include, for example, for transmucosal administration bile salts and fusidic
acid derivatives.
In addition, detergents may be used to facilitate permeation. Transmucosal
administration
may be through nasal sprays or using suppositories. For topical
administration, the oligomers
of the invention are formulated into ointments, salves, gels, or creams as
generally known in
the art. A wash solution can be used locally to treat an injury or
inflammation to accelerate
healing.
In clinical settings, a gene delivery system for a gene therapeutic can be
introduced into a patient by any of a number of methods, each of which is
familiar in the art.
68
CA 02363503 2001-08-24
WO 00/52479 PCTIUSOO/05858
For instance, a pharmaceutical preparation of the gene delivery system can be
introduced
systemically, e.g., by intravenous injection, and specific transduction of the
protein in the
target cells occurs predominantly from specificity of transfection provided by
the gene
delivery vehicle, cell-type or tissue-type expression due to the
transcriptional regulatory
sequences controlling expression of the receptor gene, or a combination
thereof. In other
embodiments, initial delivery of the recombinant gene is more limited with
introduction into
the animal being quite localized. For example, the gene delivery vehicle can
be introduced by
catheter, See U.S. Patent 5,328,470, or by stereotactic injection, Chen et al.
(1994), Proc. Natl.
Acad. Sci., USA 91: 3054-3057. A a sequence homologous thereto can be
delivered in a gene
therapy construct by electroporation using techniques described, Dev et al.
(1994), Cancer
Treat. Rev. 20:105-115.
The pharmaceutical preparation of the gene therapy construct or compound of
the invention can consist essentially of the gene delivery system in an
acceptable diluent, or
can comprise a slow release matrix in which the gene delivery vehicle or
compound is
imbedded. Alternatively, where the complete gene delivery system can be
produced intact
from recombinant cells, e.g., retroviral vectors, the pharmaceutical
preparation can comprise
one or more cells which produce the gene delivery system.
The compositions may, if desired, be presented in a pack or dispenser device
which may contain one or more unit dosage forms containing the active
ingredient. The pack
may for example comprise metal or plastic foil, such as a blister pack. The
pack or dispenser
device may be accompanied by instructions for administration.
4.a.2 Effective Dose
Toxicity and therapeutic efficacy of such compounds can be determined by
standard pharmaceutical procedures in cell cultures or experimental animals,
e.g., for
determining the Ld50 (the dose lethal to 50% of the population) and the Edso
(the dose
therapeutically effective in 50% of the population). The dose ratio between
toxic and
therapeutic effects is the therapeutic index and it can be expressed as the
ratio LD50/ED50.
Compounds which exhibit large therapeutic indices are preferred. While
compounds that
3o exhibit toxic side effects may be used, care should be taken to design a
delivery system that
targets such compounds to the site of affected tissue in order to minimize
potential damage to
uninfected cells and, thereby, reduce side effects.
69
CA 02363503 2008-10-17
The data obtained from the cell culture assays and animal studies can be used
in
formulating a range of dosage for use in humans. The dosage of such compounds
lies
preferably within a range of circulating concentrations that include the ED50
with little or no
toxicity. The dosage may vary within this range depending upon the dosage form
employed
and the route of administration utilized. For any compound used in the method
of the
invention, the therapeutically effective dose can be estimated initially from
cell culture assays.
A dose may be formulated in animal models to achieve a circulating plasma
concentration
range that includes the IC50 (i.e., the concentration of the test compound
which achieves a half-
maximal inhibition of symptoms) as determined in cell culture. Such
information can be used
to more accurately determine useful doses in humans. Levels in plasma may be
measured, for
example, by high performance liquid chromatography.
The practice of the present invention can employ, unless otherwise indicated,
conventional techniques of cell biology, cell culture, molecular biology,
transgenic biology,
microbiology, recombinant DNA, and immunology, which are within the skill of
the art. Such
techniques are explained fully in the literature. Molecular Cloning A
Laboratory Manual
(1989), 2"' Ed., ed. by Sambrook, Fritsch and Maniatis, eds., Cold Spring
Harbor Laboratory
Press, Chapters 16 and 17; Hogan et al. (Manipulating the Mouse Embryo: A
Laboratory
Manual (1986), Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY;
See U.S.
Patent No. 4,683,195; DNA Cloning, Volumes I and II, Glover, ed., 1985;
Oligonucleotide
Synthesis, M. J. Gait, ed., 1984; Nucleic Acid Hybridization, D. Hames & S. J.
Higgins, eds.,
1984; Transcription and Translation, B. D. Hames & S. J. Higgins, eds., 1984;
Culture Of
Animal Cells, R. I. Freshney, Alan R. Liss, Inc., 1987; Immobilized Cells And
Enzymes, IRL
Press, 1986; Perbal (1984), A Practical Guide To Molecular Cloning; See
Methods In
Enzymology (Academic Press, Inc., N.Y.); Gene Transfer Vectors For Mammalian
Cells, J. H.
Miller and M. P. Calos, eds., Cold Spring Harbor Laboratory, 1987; Methods In
Enzymology,
Vols. 154 and 155, Wu et al., eds., Academic Press Inc., N.Y.; Immunochemical
Methods In
Cell And Molecular Biology (Mayer and Walker, eds., Academic Press, London,
1987;
Handbook Of Experimental Immunology, Volumes I-IV, D. M. Weir and C. C.
Blackwell,
eds., 1986.
CA 02363503 2008-10-17
The present invention is further illustrated by the following examples which
should not be construed as limiting in any way.
71
CA 02363503 2001-08-24
WO 00/52479 PCT/US00/05858
BIBLIOGRAPHY
Abreniski et al. (1984), J. Biol. Chem.. 259, 1509-1514.
Abravaya et al. (1995), Nuc. Acid. Res... 23, 675-682.
Akiyama. H., et al. (1991), J. Neuroimmunol., 32, 19-28.
Alliknlets, R., et al. (1997), cience, 277, 1805-7.
Alt, A. (1 S77), Arch. Ophthalmol. and Otolarvngol., 6, 304-323.
Babaev, V., et al. (1990), Atherosclerosis, 85, 239-247.
Badimon, J., et al. (1993), Circulation, 87 (3 Suppl), II3-116.
Barany (1991), Proc. Natl. Acad. Sci. USA, SS, 189.
Been and Cech (1986), Cell, 47, 207-216.
Bernoist and Chambon (19S 1), Nature, 290, 304-310.
Bird, A. (1992). Pathophysiology of AMD. In G. Hampton and N. PT (Eds.), Age-
related macular
degeneration: principles and practice (pp. chap. 3.). New York: Raven Press.
Bird, A. (1992a). Br. J. Ophthalmol., 763, 166-168.
Bird, A. (1992b). Pathophysiology of AN1D. In G. Hampton and N. PT (Eds.), Age-
related macular
dc,,eneration_princiPles anci practice (pp. chap. 3.). New York: Raven Press.
Bird, A. (1996). Br. J. Ophthalniol., S9, 2-3.
Bird, A. (1997), The Wolfe Lecture= Genot~~pe'nhenotype correlations in
hereditarv macular
ciystrophies No. The University of Io%va.
Bird, A., et al. (1995), Sur<,ev of Ophthal., 39(5), 367-74.
Bittorf, S. V., et al. (1993), J. Biol. Cheni., 268, 24838-24846.
BlLunenkranz, M., et al. (1986), Ophthalmol., 93, 552-558.
Bobryshev, Y., et al. (1995), Atherosclerosis, 11 S, 9-21.
Bobryshev, Y.V., and Lord, R.S. (1995), Cardiovas. Res. 29, 689-696.
Bocan, T., et al. (1958), Arteriosclerosis, 8, 499-508.
Bradley et al. (19S4), Natttre, 309,255-258.
72
CA 02363503 2001-08-24
WO 00/52479 PCTIUSOO/05858
Bradley et al. (1986), Current Topics Devel. Biol., 20, 357-371.
Bressler, N., et al. (1994), Retina, 14, 130-142.
Bressler. S., et al. (1990), Arch. of Ophthalmol., 108, 1442-1447.
Brinster et al. (1982), Nature, 296, 39-42.
Brinster et al. (19S5), Proc. Natl. Acad. Sci. USA, 82, 4438-4442.
Brown, B., aild Lovie-Kitchin, J. (1983), Amer. J. Optom. Physiol. Opt., 6-0,
645-650.
Brown, B., et al. (19S6), Ophthalmic Phvsiol. Opt., 6, 81-84.
Bunchereau, J., and Steinman, R.M. (1998), Nature 393, 245-252.
Burtls, R., and Feeney-Bums, L. (1980), Transactions of the American
Ophthalmologic Societv, 78,
206-225.
Btttler (1981), \ieth. Enzvmol., 73, 482-523.
Capon. M., et al. (19S9), Ophthalmol., 96, 1769-1777.
Caux et al. (1999) "Developmental Pathways in Httnian Myeloid Dendritic
Cells', in Dendritic Cells,
Lotze, M.T. and Thomson, A.%V. (Eds.) Academic Press, pp. 63-92.
Chan,,. B., et al. (1996), Invest. Opllthalnlol. Vis. Sci. (Sunnl), 37. S305.
Chen, J., et al. (1992), Invest Ophthalnlol. Vis. Sci., 73, 334-340.
Chc:n et al. (1994), Proc. \`atl. Acad. Sci., USA, 91, 3054-3057.
Chir-.;win, J.. et al. (1979), Biocllenl., 18, 5294-5299.
Christen, W., et al. (1997), Invest. Ophtllalnlol. Vis. Sci. (St(ppL), 38,
S472.
Cohen et al. (1996), Adv. Chromatogr.,. 36, 127-162.
Cole et al. (19S5), Monoclonal Antibodies and Cancer Theral2,Y, Alan R. Liss,
Inc. pp. 77-96.
Collier. L., et al. (1986), Invest. Opllthalmol. Vis. Sci., 27, 702-707.
Coria, F., et al. (1988), Lab. Invest., 58, 454-458.
Cotton et al (1988), Proc. Natl. Acad. Sci. USA, 8-5, 4397.
Cotton (1993), Mtttat. Res., 285, 125-144.
Cronin et al. (1996), Hunlan Mutation, 7, 244.
73
CA 02363503 2001-08-24
WO 00/52479 PCTIUSOO/05858
Cruickshanks, K., et al. (1997), Arch. Ophthalmol., 115, 242-250.
Cntickshanks, K., et al. (1993), Arch. Ophthalmol., 111, 514-518.
Curcio, C., et al. (1996), Invest. Ophthalmol. Vis. Sci., 37, 1236-49.
Ctircio, C., et al. (1993), Invest. Ophthalniol Vis. Sci., 34, 327S-3296.
Cutler, J., and AH, P. (19S1), Infection and Immunity, 31(3), 1110-1116.
Dahlback, K., et al. (1990), J. Invest. Dermatol., 94, 284-291.
Dahlback, K., et al. (1988), Acta Derm. Venereol., 68, 107-115.
Dahiback, K., et al. (1989), J. Invest. Dermatol., 92, 727-733.
Dahlback, K., et al. (1993), J. Invest. Dermatol., 100, 166-170.
Dastgheib, K. and Green, R. (1994) Arch. Opthalniol; 112, 813-818
De La Paz, N1., et al. (1998), Invest. Ophthalmol. Vis. Sci. (Supp1), 39,
S915.
Deutman, A., and Jansen, L. (1970), Br. .1. Ophthalmol., 54, 373-382.
Dcv et al. (1994), Cancer Treat. Rev., 20, 105-115.
Doetschman et al. (1985),.1. Embryol. Exp. Mol. Biol., 87, 27-45.
Donders, F. (1854), Graefe's Arch. Clin. E\h. Ophthalmol., l, 106-118.
Dorey, C., et al. (19S9), Invest. Oplithalmol. Vis. Sci., 30, 1691-1699.
Duke-Elder, S., and Dobree, J. (1967), Diseases of the Retina. St. Louis: CV
Mosby.
Duvall, J., and Tso, M. (19S5), Arch. Ophthalmol., 103, 694-703.
Duvall-Yoiui-, J., et al. (19S9), Br. J. Ophthalmol., 73, 297-302.
Eagle, R. (1984), Ophthalmol., 91, 613-25.
Eisner, A., et al. (1987), Invest. Oplitlialniol. Vis. Sci., 28, 1832-1837.
El Baba, F., et al. (1986a), Amer. J. Ophthalmol., 101, 576-83.
El Baba, F., et al. (1986b), Onhthalniol.. 93, 1581-1592.
Eglom et al. (1993), Nature, 365, 566.
Ellison et al. (1991), J. Biol. Chenl.. 206. 21150-21157.
74
CA 02363503 2001-08-24
WO 00/52479 PCTIUSOO/05858
Elman, M., and Fine, S. (1989), In S. Ryan (Ed.), The Retina (pp. 175-200). St
Louis: CV Mosby.
Elner, S. G., et al. (1992), Exp. Eye Res., 55(1), 21-8.
Engel, H., et al. (19SS), Ophthalmoloa.,iea, 196, 143-150.
Ermilov, V., and Vodovozov, A. (1995), Vestnik Oftalmoiogii, 111(4), 24-7.
Evans et al. (19S 1), \ ature. 292, 154-156.
Fariss, R. N., et al. (1997), Amer. J. Path., 150(1), 323-8.
Farkas, T., et al. (1971 a), Amer. J. Ophthalmol., 71, 1196-1205.
Farkas, T., et al. (1971 b), Amer. J. Ophthalniol., 71, 1206-1215.
Feeney-Burns, L., and Ellersieck, M. (1985), Amer. J. Ophthalmol., 100, 686-
697.
Feeney-Burns, L., et al. (19SS), Invest. Ophthalmol. Vis. Sci., 29, 708-719.
Feeney-Bu--ns, et al. (1987), Invest. Ophthalmol. Vis. Sci., 23, 1138-1147.
Fi(Yueroa, IN4., et al. (1994), Retina, 14, 391-396.
Finc, B. (19S 1), Anier. .1. Ophtlialniol., 91, 469-47 3.
Fite, K., Benuston, C., and Cousins, F. (1994). Exper. Eve Res., 59, 417-424.
Fox, P., ct al. (1995). Life Sci., 56, 1749-5S.
Frenneson, I.. and \ilsson, S. (1996), Eur. J. Ophthalmol., 6, 307-314.
Frennesson, C.. et al. (1995), Docunlenta Ophthalmoloaica, 90, 377-86.
Frennesson, I., and Nilsson, S. (1995), Br. J. Ophthalniol., 79, 905-9.
Freshney, R.I. (1987), Cultttre Of Animal Cells, Alcui R. Liss, Inc.
Friednlan, E., et al. (1963), Arch. Ophthalmol., 69, 220-230.
Fuchs, U., et al. (1991), Invest. Ophthalmol. Vis. Sci., 32(13), 3178-86.
Gait, M. J. (ed.) (1984) Oligonucleotide Synthesis.
Gasparini et al. (1992), Mol. Cell Probes, 6, 1.
Gass, J. (1973), Arch. Ophthalmol., 90, 206-217.
Gass, J. (1987), Stereoscopic Atlas of Macular Diseases: Diagnosis and
Treatment (3rd ed.). St
CA 02363503 2001-08-24
WO 00/52479 PCTIUSOO/05858
Louis: CV i`tosby.
Galy, A. (1999), "Hemaopoietic Cell Fate and the Development of Dendritic
Cells" in Dendritic
Cells, Lotze, M.T. and Thonison, A.W. (Eds.) Academic Press, pp. 3-14
Gautier et al. (1987), Nucl. Acids Res., 15, 6625-6641.
Gen~e, B., et al. (1991), J. Biol. Chenl., 266, 10678-10685.
Gliadially, F. (19SS), Ultrastnictural Pathologv of the Cell and Matrix (3rd
ed.). New York:
Buttenvorth.
Gibbs et al. (1989), Nucl. Acids Res., 17, 2437-2448.
Glover, (ed.) (1985) DNA Clonincr, Volumes I and II.
Gossler et al. (1986), Proc. Natl. Acad. Sci USA 83, 9065-9069.
Green, W., and Enger, C. (1993), Ophthalmol., 100, 1519-1535.
Green, W., and Key. S. (1977), Transactions Anier. Oplithalmol. Soc., 75, 180-
254.
Green, W., et al. (19S5), Ophthalmol., 92, 615-27.
Griffin et al. (1993), APpi. Biochem. Biotechnol., 38, 147-159.
Guatelli et al. (1990), Proc. Natl. Acad. Sci. USA, 87, 1 S74-1 S7S.
Guerne, D., et al. (1991), Ophthalniol., 9S, 602-7.
Gu'Aon, J., atiLi hlemp, K. (1996), Arterioscler., Thromb. an(i Vas. Biol.,
16, =1-1 1.
HabiLy et al (1974), J. Biol. Clieni., 249, 7130.
Hale, 1., and iMatsunioto, B. (1993), iV-Iethods in Cell Biol., 3S, 289-32=1.
Hanies, B.D. and S. J. Higgins, (eds.) (1984) Nucleic Acid Hybridization.
Hanles, B.D. and S. J. Higgins, (eds.) (1984), Transcription and Translation.
Hamniond, B. J., et al. (1997), Invest. Ophthalmol. Vis. Sci., 38, 1795-801.
Harlow and Lane (eds.) (1988) Antibodies: A Laboratory Manual, Cold Spi-iiig
Harbor Press.
Haseloff and Gerlach (1988), Nature, 334, 585-591.
Hashimoto, K., and DiBella, R. (1967), J. Invest. Dermatol., 48, 405-422.
Hayashi (1992), Genet. Anal. Tech. A12pl., 9, 73-79
76
CA 02363503 2001-08-24
WO 00/52479 PCTIUSOO/05858
Hayman, E., et al. (19S3), Proc. Nat. Acad. Sci. USA, 80, 4003-7.
Heiba, I., Elston, et al (1994), Genet. Epideniiol., 11, 51-67.
Helene (1991), Anticancer Druiz Des., 6, 569-584.
Helene et al. (1992), Ann. N.Y. Acad. Sci., 660, 27-36.
Heon, E., et al. (1996), Arch. Ophthalmol., 114, 193-8.
Herkner, K., et al. (1992), Clin. Chem., 3 8, 548-50.
Hewitt, A., and Newsome, D. (1985), Cur. Eye Res., 4, 169-173.
Hintner, H., et al. (1988), J. Invest. Derm., 91, 22-28.
Hirata, A., and Feeney-Burns, L. (1992), Invest. Ophthalmol. Vis. Sci., 33,
2079-2090.
Hogan, M. (1972), Transactions Amer. Acad. Ophthalmol. and Otola , n;ol , 76,
64-80.
Hogan et al. (1986), Manipulating the Mouse Enlbrvo= A Laboratorv Manual,
Colcl
Srriirg /larbor Laborcrtoil= Press, Cold Spring Harbor, NY.
Hogasen, K., et al. (1992), J. Biol. C'heni., 267, 2 3076-230S2.
Holniquist et al. (1979), Anal. Biochenl., 95, 540.
Holz, F., et zl. (1995u), Arch. Ophthalnlol.. 113(2), 17S-S4.
Holz, F., et al. (1995), C;ernlan J. Ophthalnlol., 4(6), 336-41.
I-lolz, F., et al. (1994), ACc}l. Opllthalillol., 112, 402-406.
Hope, G., et al. (1992), Br. J. Oplithalmol., 76, 11-16.
Hsu et al. (1994), Carcinogenesis, 15, 1657-1662.
Hussain, A., et al. (1997), Invest. Ophthalmol. Vis. Sci., 33, S353.
Hynian, L. (1992), EpidemioloQv of AMD. In G. Hampton and P. Nelsen (Eds.),
Age-related
Macular Degeneration= Principles and Practice (pp. 1-36). New York: Raven
Press.
Hynian, L., et al. (19S3), Amer. J . Epideniiol., I S, 213-227.
Inoue et al. (1987), Nucl. Acids Res., 15, 6131-6148.
lnoue et al. (1987), FEB Lett,. 215, 327-330.
77
CA 02363503 2001-08-24
WO 00/52479 PCT/US00/05858
loffe et al. (1995), Proc. Natl. Acad. Sci.. USA, 22, 7357-7361.
Ishibashi, T., et al. (1986a), Amer. J. Ophthalmol., 101, 342-353.
Ishibashi, T., et al. (19S6b), Invest. Ophthalmol. Vis. Sci., 27, 184-193.
Ishikawa, et al. (eds.) (1981), Enzvme Immunoassav, Kgaku Slroi,i, Tokyo.
Jacobson, S., et al. (1995), Nature Genet., 11, 27-32.
Jaenich (1976), Proc. Natl. Acad. Sci. USA. 73, 1260-1264.
Jaenisch (1988), Science. 240, 1468-1474.
Jang, I., et al. (1993), Eur. Heart J., 14(Supplement K), 2-6.
Jaliner et al. (19S2), Nature. 295, 623-628.
Jahner et al. (1985), Proc. Natl. Acad. Sci. USA. 82, 6927-693 1.
Jansen, J., et al. (1993), Amer. J. Path., 143, 1356-65.
JaNvorowski, A., et al. (1995), Biociiini. Biophys. Acta, 1245(l), 121-9.
Joh, K., et al. (1993), Acta Patlioloizica Japonica, 43, 552-565.
Jolinson, L., and Blanks. J. (1954), Cur. Eve Res., 3, 969-974.
.Iolinson, L., and Hageman, G. (1987), Exper. Eve Res., 44, 553-565.
Knlui, I L. ct al. (1977), :1nier. .1. Epidem., 106, 17-32.
Keen et al. (1991), Trends Genet., 7, 5.
Kida, E., et al. (1994), Neur. Lett., 167, 73-76.
Killings-,vorth, M., et al. (1990), Eye, 4, 613-621.
Kincaid, M. (1992), Pathology of AMD. In G. Hampton and P. Nelsen (Eds.), Age-
related nlacular
deuneration: principles and practice (pp. chap. 2). New York: Raven Press.
Kishinioto, T., et al. (1997), Leukocvte Typing VI. New York: Garland
Publishing.
Kivela, T. (1990), Cur. Eye Rcs., 9, 1195-1209.
Kivela, T., et al. (1994), Invest. Ophthalmol. Vis. Sci., 35, 3759-3769.
Klein, M. (1991), JAMA, 266, 2279.
78
CA 02363503 2001-08-24
WO 00/52479 - PCT/US00/05858
Klein, M., et al. (1994), Arch. Ophthalmol., 112(7), 932-7.
Klein, R., et al. (1991), Ophthalniol., 98, 1128-1134.
Klein, R., et al. (1995), Ophthalmol., 102(3), 371-81.
Kliffen, M., et al. (1995), Lab. Invest., 73(2), 267-72.
Kliffen, M., et al. (1994), Invest. Ophthalmol. Vis. Sci,, 35, 2901-2905.
Kliffen, M., et al. (1996), Arch. Ophthalmol., 114, 1009-14.
Kliffen, M., et al. (1997), Microscopy Res. Tech., 36(2), 106-22.
Kligman, A. (1969), JAMA, 210, 2377-2380.
Kohler and Milstein (1975), Nattire. 256, 495-497.
Koniher et al. (19S9), Nucl. Acids. Res., 17, 7779-7784.
Kozbar et al. (19S3), Inimunol. Todav, 4, 72.
Krol et al. (19SS), BioTeclinicttes, 6, 958-976.
Kuppuswamy et al. (1991), Proc. Natl. Acad. Sci. USA. SS, 11=I 3-1 1-17.
Kwoh et al. (19S9), Pi-oc. Natl. Acad. Sci. l_1SA, 86, 1 173-1 177.
Lacmmli, U. (1970), Nature, 227, 6S0-685.
Lakso et al. (1992;), Pi-oc. \'atl. Acad. Sci. 'SA. 89, 6232-6236.
Lamand, A., and Mann, M. (1997), Trencls in Cell Biol., 7, 139-142.
Landegran et al. (1988), Science, 241, 1077-IOSO.
LaVail et al. (1995), Invest. Onhthalmol. Vis. Sci., 39, 592-602.
La Vail et al. (1998), Invest. Ophthalmol. Vis. Sci., 39, 581-591.
Lazarus, H., et al. (1993), Exp. Eye Res., 56, 531-541.
Leibovvitz, H., et al. (1980), Survev of Ophthalmol., 24, 428-457.
Lemaitre et al. (1987), Proc. Natl. Acad. Sci. USA, 84, 648-652.
Letsinger et al. (1989), Proc. Natl. Acad. Sci. USA. 86,6553-6556.
Lewis, H., et al. (19S6), Qphthalmol., 93, 1098-1112.
79
CA 02363503 2001-08-24
WO 00/52479 PCT/USOO/05858
Leys, A., et al. (1990), Graefe's Arch. Clin. Exp. Ophthalmol., 228, 499-504.
Leys, A., et al. (1991), Graefe's Arch. Clin. Exp. Ophthalmol., 2-79, 406-10.
Li, J., and McAdam, '"'. (1984), Scand. J. of Imnlunol., 20, 219-226.
Li, X., et al. (1995), Arterioscler.. Thromb., and Vasc. Bio., 15, 252-257.
Lieni, A., et al. (1992), Anler. J. Ophthalnioi., 114, 149-157.
Lim, J., et al. (1993), Retina, 13, 230-3.
Lizardi et al. (1988), Bio/Technologv. ~, 1197.
Loeffler, K., and Mangini, M. (1997), Graefe's Arch. Clin. Exp. Ophthalniol.,
235, 248-54.
Loeffler, K., et al.(1995), Invest. Ophthalmol. Vis. Sci., G, 24-31.
Lovie-Kitchin, J., and Bowman, K. (1985), Senile %1,tacular Deaeneration.
Boston: Buttenvorth.
Luna, L. (Ed.). (196S), Manual of Histolo(gical Staining \lethods of the Armed
Forces Institute of
Patliolo-v (3rd ed.). New York: McGraw-Hill.
Macular Photocoa('ulation Study. (1997), Arch. Ophthalmol., 115(6), 741-7.
Ma~~io (ecl.) (1950), Enzyme Immunoassay, CRC Pt=ess, Boca Raton,
Maher. (1992), 13ioassavs. 14, 807-S 15.
INIanser-ll, F., et al. (1995), .1. %i-ied. Gcnet., 32, 855-S.
Mares-Perlnian, J., et al. (1995a), Arcli. Ophthalnlol., 113. 1515-23.
Mares-Perlnian, J., et al. (1995b), Arch. Ophthalmol., 1 13, 743-5.
Marnior, M., and Byers, B. (1977), Anier. J, Ophthalmol.. 84, 32-44.
Massof, R., et al. (19S9), Clin. Vision Sci., 4, 221-227.
Masini and Gilbert (1977), Proc. Natl. Acad. Sci. USA. 74, 560.
Mayer and Walker, (eds.) (1987) lnimunochemical Methods In Cell And Molecular
Biology,
Acadeiiiic Press, London.
Mehregan, A., and Hashimoto, K. (1991), Pinkus' Guide to Dermatohistonathologv
(pp. 369-382).
Englewood Cliffs, NJ: Prentice Hall.
Meyers, S. (1994), Transact. Amer. Ophthalmol. Soc., 92, 775-843.
CA 02363503 2001-08-24
WO 00/52479 PCT/US00/05858
Meyers, S., aild Zacchary, A. (19S8), Arch. Ophthalmol., 106, 651-653.
Meyers, S., et al. (1995), Amer. J. Ophthalmol., 120, 757-66.
Midena, E., et al. (1997), Invest. Ophthalmol. Vis. Sci., 38, 469-477.
Midena, E., et al. (1994), Docunlenta Opllthalmoloaiea, 88, 179-85.
J. H. Miller and M. P. Calos, (eds.) (1987) Gene Transfer Vectors For
Mammalian
Cel Cold Spring Hctrbor Laboraronv.
Mimoun, G., et al. (1990), ,LFr. Ophtalmol., 13(10), 511-30.
Monaco, W., and 'ormin,ton, C. (1990), Qptonlet. Vis. Sci., 67, 532-537.
Moore, D., et al. (1995), Invest. Ophtlialmol. Vis. Sci., 36, 1290-1297.
Muda, A., et al. (19SS), Virchows Archiv - A. Path. Anat. HistoD!th., 413, 529-
37.
Muller, H. (1S56), Albrecht von Graefe Archiv ftirOplithalmologie, 2(2), 1-69.
Mullins, R., and Hag, enian, G. (1997), Histochenlical conlparison of ocular
"dnisen" in monkey and
human. In 1%1. LaVail, J. Hollyfield. and R. Anderson (Eds.), Degenerative
RetinalDiseases
(pp. 1-10). New York: Plenum Press.
Mullins, R., et al. (1997a), Ophthalmol., 104, 288-294.
Mullins, R. et al. (1999), FA EB .1. 13:\-\.
Myers. et al. (19S5), Science. 210, 1242.
i\,Iycrs ct al. (19S5). Nature 313, 495.
Nalcaza%va et al. (1994), Proc. Nati. Acad. Sci. L'SA. 91, 360-364.
Newsonle, D. (1988), Angiold streaks and Bruch's membrane degenerations. In D.
Newsome (Ed.).
Retinal Dvstrophies and Degenerations (pp. 271-283). New York: Raven Press.
Newsonie, D., et al. (1987), Anier. J. Ophthalniol., 104, 373-38 1.
Newsonie, D., et al. (1987b), Curr. Eve Res., 6, 1211-1221.
Newsome, D., et al. (1986), Invest. Opllthalmol. Vis. Sci., 27, 1675-80.
Nickerson et al. (1990), Proc. Natl. Acad. Sci. USA. 87, 8923-8927.
Niculescu, F., et al. (1987), Atherosclerosis, 65, 1-11.
Niculescu, F., et al. (19S9), Atherosclerosis, 78, 197-203.
81
CA 02363503 2001-08-24
WO 00/52479 PCT/US00/05858
Nyren et al. (1993), Anal. Biochem., 20$, 171-175.
O'Gorman et al. (1991), cie ce 251, 1351-1355.
O'Shea, J. G. (1996), i\-led. J. Austr., 165(10), 561-4.
Olin, K., et al. (1995), Proc. Soc. Exp. Biol. Med., ?QQ, 370-377.
Oppenlleim, J., and Leonard, E. (1989), Introduction to human monocytes. In M.
Zembala and G.
Asherson (Eds.), Hunian Monocytes San Diego, CA: Academic Press.
Orban et al. (1992), Proc. Nati. Acad. Sci. USA. 89, 6861-6865.
Orita et al. (1989), Proc. Natl. Acad. Sci. USA. 86, 2766.
Osusky, R., et al. (1997), Recep. Sign. Trans., 5, 43-50.
Parodi, M. (1994), Acta Ophthalmologica, 72, 260-4.
Pauleikhoff, D., et al. (1990), Anler. J. Ophthalmol., 109, 38-43.
Pauleikhoff, D., et al. (1990b), Ophthalniol., 97, 171-178.
Pauleikhoff, D., et al. (1994)[German], Ophthalnlolo,e, 91(6), 730-4.
Pauleikhoff, D., et al. (1992), Ophthalmol., 99, 1548-1553.
Penfold, P., et al. (19S5), Graefe's Arch. Clin. Exp. Ophthalniol., 223, 69-
76.
Penfolcl, P., ct al. (19S6), lnvest. Ophthalmol. Vis. Sci., 27, 364-71.
Penfold, P., et al. (1990). Gracfc's Arch. Clin. Exl). Opltthalmol., 22s, 270-
4.
Perbal (19S4), A Practical Guide To .10olecular Cloning Metliods In Enzymoloav
(:Icaclciuic P,rss, Inc., N.Y.).
Perry-O'Keefe et al. (1996), Proc. 1\atl. Acad. Sci. USA. 93, 14670-14675.
Piguet, B., et al. (1995), Eve, 9, 34-41.
Piguet, B., et al. (1996), Oplitlialniic Gen., 17(4), 175-86.
Piguet, B., et al. (1993), Br. J. Ophthalnlol., 77, 400-403.
Preissner, K. (1991), Ann. Rev. Cell. Biol., 7, 275-310.
Prezant et al. (1992), Hum. Mutat., 1, 159-164.
Prossner (1993), Tibtech. 11,238.
82
CA 02363503 2001-08-24
WO 00/52479 PCTIUSOO/05858
Puchtler, H., et al. (1962), J. Histocheni. Cytochem., 10, 355-364.
Reblin, T., et al. (1995), Atlleroscierosis, 113, 179-88.
Rekhter, M., et al. (1993), Anier. J. Pathol., 143, 1634-1638.
Ritter, L., et al. (1995), Anier. J. Ophthalmol., 120(2), 190-6.
Robertson et al. (1986), Nature. 3?2, 445-448.
Robertson (19S7) in: Teratocarcinonlas and Embrvonic Stem Cells: A Practical
Approach, E.J.
Robertson, ed., IRL Press, Washington, D.C.
Roest, et. al. (1993), Hum. Mol. Genet., 2, 1719-21.
Roitt, I., et al. (1985), Inimunol.. St. Louis: C.V. Mosby.
*Rones, B. (1937), Formation of dnisen of the lamina vitrea.
Rosenbaum and Reissner (1987), Bionhys. Chem., 215, 12753.
Roseiifeld, ,ti1., et al. (1993), Arterioscl. Thronib., 13, 1382-9.
Rudncx\', A. (1 S71), Archiv fur Pathologische Anatoniie und Ph s~olovie und
fur Itilinische Medicin,
53, 455-465.
Rus, H., et al. (19S9), Imniunol. Lctt., 20, 305-10.
Saiki et al. (19S6), Nature. 324, 163.
Saiki et al (19S9), Proc. Natl. Acad. Sci. USA, S6, 6230.
Saitta, NI., et al. (1992). Clin. Cheni., 38, 2454-7.
Salib, E., and Hillier, V. (1997), Internat. J. Ger. Psych., 12(3), 363-8.
Saleeba et al. (1992), Meth. Enzvmol., 217, 286-295.
Sambrook, Fritsch and Maniatis, (eds.), (1989) Molecular Cloning A Laboratory
Manual, 2"`' Ed.,
ed. by Colcl Spring Ha,-bor Laboratorv Press, Chapters 16 and 17.
Sandber-, M., et al. (1994), Invest. Ophthalniol. Vis. Sci., 35(6), 2734-40.
Sanger et al (1977), Proc. Nat. Acad. Sci. USA 74, 5463
Sarin et al. (19S8), Proc. Nati. Acad. Sci. USA 85, 7448-7451.
Sarks, J., et al. (19S8), Eve, 2, 552-577.
83
CA 02363503 2001-08-24
WO 00/52479 PCT/US00/05858
Sarks, J., et al.(1994), Eye, $, 269-283.
Sarks, S. (1976), Br. J. Ophthalmol., 60, 324-341.
Sarks, S. (1980), Aust. J. Ophthalmol., $, 117-30.
Sarks, S. (19S2), Attstr. J. Ophthalmol., 10, 91-97.
Sarks, S., and Sarks, J. (1989), A~e-related macular degeneration: atrophic
form. In S. Ryan (Ed.),
The Retina St. Louis: CV Mosby.
Sarks. S., et al. (1985), Patterns in macular de~eneration. In S. Ryan, A.
Dawson, and H. Little
(Eds.), Retinal Diseases (pp. 87-93). Orlando: Grune and Stratton.
Sarks, S., et al. (1980), Trans. Ophthalmol. Soc. U.K., 100, 414-422.
Sarver et al. (1990), . cierrce, 247, 1222-1225.
Schacliat, A., et al. (1995), Arch. Oplithalniol., 113(6), 728-35.
*Schalch. W. (1992), E\s, 62, 280-98.
Seddon. J., et al. (1997), Amer. J. Ophthalnlol., 123, 199-206.
Seddon, J.. et al. (1994), AMA, 272(18), 1413-20.
Seiffert. D., et al. (1994), .1 Biol Cheni, 269, 19836-19842.
Sheraidah. G.. et al. (1993), Ophthalmol., 100, 47-51.
Siuelman. J. (1991), Ol)llthalmol., 9S, 1379-1353.
Silvestri. G., et al. (1994), Eve, S, 564-S.
Sniall, K., et al. (1993), Ophthal. Paediat. Genet., 14(4), 143-150.
Snial l, M., et al. (1976), Arch. Ophthalmol., 94, 601-607.
Sniithies et al. (1985), Nature 317, 230-234.
Sokolov (1990), Nucl. Acids Res., 18, 3671.
Solem, N-I., et al. (1991), Mol. Cell. Biochem., 100, 141-9.
Spraul, C., and Grossitiklaus, H. (1997), Arch. Ophthalmol., 115, 267-73.
Stafford, T., et al. (19S4), Ophthalmol., 9-1, 513-521.
Stanoos, N., et al. (1995), Ophthalmologica, 209, 194-8.
84
CA 02363503 2001-08-24
WO 00/52479 PCTIUSOO/05858
Starita, C., et al. (1996), Exp. Eve Res., 62, 565-572.
Stary, H., et al. (1995), Circtilation, 92, 1355-1374.
Steinmetz, R., et al. (1993), Br. J. Ophthalmol., 77, 549-554.
Stevvart et al. (1987), EMBO J., 6, 383-388.
Stockmann, A., et al. (1993), J. Biol. Chem., 268, 22874-82.
Stone, E. et al. (1998) Nature Med. December
Stone, E., and Sheffteld, V. (1993), Molecular Genetics of Inherited Eve
Disorders. Chur,
Switzerland: Academic Publishers.
Stone, E., et al. (1992), Nature Genet., 1, 246-250.
Stone, K., et al. (1990), Meth. Enzyniol., 193, 389-412.
Strittmatter, W. J., and Roses, A. D. (1995), Proc. Nat. Acad. Sci. USA,
92(11), 4725-7.
Suntiess, J., et al. (19S8), Arch. Ophthalniol., 106, 1081-1084.
Sunness, J.. et al. (19S9), Ophthalmol., 96, 375-381.
Sunness, J., et al. (1985), Arch. Ophthalmol., 103, 811-816.
Syvanen, et al. (1990), enomics. S. 684-692.
S}'-vanen et al. (1993), Amer. .i. Hum. Genet., 52, 46-59.
Tabatav. C., et al. (19S7), Arch. Ophthaimol., 105, 826-830.
Tan, S.. and Pepys, NI. (1994), Histopathol., 25, 403-414.
Tanzi, R. E., et al. (1996), Neurobiol. Dis., 3(3), 159-68.
Taylor, H. R., et al. (1990), Trans. Anier. Ophthalmol. Soc., sS, 163-73.
Tetrito, M. C., and Cline, M. J. (1975), Clin. Haematol., 4(3), 685-703.
Thontas & Capecchi (1987), ell, 51, 503-512.
Thonipson et al. (1989), Cel! 5, 313-32.
Thonler, P., and Baumal, R. (1982), Arch. Path. Lab. Med., 106, 628-3 1.
Tobe et al. (1996), Nucleic Acids Res., 24, 3728.
CA 02363503 2001-08-24
WO 00/52479 PCTIUSOO/05858
Tolentino, M., et al. (1994), Vis. Res., 24(3), 409-13.
Tomasini, B., and Mosher, D. (1991), Prog. Hemost. and Thromb., 10, 269-305.
Ugozzoli et al. (1992), GATA, 9, 107-112.
Ulshafer, R., et al. (1987), Retina, 7, 198-203.
Umemoto, J., et al. (1977), J. Biol. Chem., 252, 8609-14.
Vachino, G., et al. (1988), J. Leuk. Biol., 44, 529-34.
van der Luijt, et. al. (1994), enomics 20, 1-4.
Van der Putten et al. (1985), Proc. Natl. Acad. Sci. U A. 82, 6148-6152.
van der Schaft, T., et al. (1993), Br. J. Ophthalmol., 77, 657-661.
van der Schaft, T., et al. (1994), Graefe's Arch. Clin. Exp. Ophthalnlol., 40-
46.
van der Schaft, T., et al. (1992), Ophthainiol., 99, 278-86.
Vinding, T. (1990), Acta Opthalmlo.=ica, 6S, 410-414.
Vinyerling, J., et al. (1995), Anier. J. Epidem., 142, 404-9.
Vin~,erling, J., et al. (1996), Arcli. Ophtlialmol., 114, 1193-1196.
Volker, W., et al. (1993), J. Histocheni. Cvtocheni., 41, 1823-1832.
Voller (1978), Dia,_,nostic Horizons. 2. 1-7.
Voller et al. (1978), J. Clin. Pathol., 3, 1, 507-520.
Wagner et al. (1981), Proc. Nati. Acad. Sci. tJSA, 78, 1441-1445.
Wallow, I., and Tso, M. (1972), Anier. J. Ophthalnlol., 73, 914-926.
Weintraub, B., (1986) Principles of Radioimnuinoassays. Seventh Training,
Course on Radioligand
Assav Techniques, The Etirloct=iire Societti=.
Weir, D.M. and Blackwell, C.C. (eds.) (1986), Handbook Of Erperiniental
Immunolo~v, Volumes
I-IV
Wetzig, P. (1988), Trans. Am. Ophthalniol. oc., 86, 276-290.
Willianls, K., et al. (1996), Matrix assisted-laser desorption ionization mass
spectrometry as a
complenient to intemal aniino acid sequencing. In J. Walker (Ed.), The protein
protocols
handbook (pp. 541-555). Totowa: Huniana Press.
3S-1175.1
86
CA 02363503 2001-08-24
WO 00/52479 PCTIUSOO/05858
Wolter, J., and Falls, H. (1962), Arch. Ophthalmol., 68, 219-226.
Wu et al., (eds.) Methods In Enzymolosv, Vols. 154 and 155, Academic Press
ILic., N.Y.
Yamamoto et al. (1980), .el , 32, 787-797.
Yang, G., et al. (1992), Am. J. Path., 141, 409-419.
*Yanoff, M., and Fine, B. (Date), Ocular Patholoiay. A Color Atlas. New York:
Gower Medical
Publishing.
Yasunlitsu, H., et al. (1993), In vitro Cell. Devel. Biol., 29A, 403-407.
Young, R. (1987), Surv. Ophthalmol., 31, 291-306.
Young, R. W. (1988), Surv. Ophthalmol., 32(4), 252-69.
Zahedi, K. and %Vhitehead, A. S. (1989), J. immunol., 143,2880-2886.
Zaug et al. (1984), cience 224, 574-578.
Zaua and Cech (1986), cience, 231, 470-475.
Zauu et al. (19S6), Nature. 324, 429-433.
Zeller. C., and N-larchase, R. (1992), Ani. J. Phvsiol., 262, C1341-C1355.
Zon (19SS), Pharm. Res., 5, 539-549.
87
CA 02363503 2001-08-24
WO 00/52479 PCT/US00/05858
EXAMPLES
Example 1: Human Donor Eye Repository and "Comprehensive Donor Database" (CDD)
Tissues from the a unique human donor eye repository and comprehensive donor
database
(CDD) have been employed for the experiments described in the Examples that
follow.
This research employs a human donor eye repository that has been developed
over the past
eight years. The repository contains over 2,000 pairs of eyes. Staff are on 24
hour call to
retrieve and process donated tissue. Eyes are accepted only if they can be
processed within four
hours of death. A database of clinical, statistical and scientific information
for each donor eye
entered into the repository has been developed and will continue to be
maintained. Sera, blood
(DNA), ophthalmologic and medical histories, and family interviews are
collected for as many
donors as possible; these data have been collected for over 90% of the donors
entered into the
repository in the past two years. Over 25% of our donors in the last two years
have a clinically
documented history of AMD.
A standard procedure for processing eyes has been developed; this procedure is
modified,
when required, to meet the needs of specific studies. All eyes are
photographed immediately.
Every eye is processed similarly such that reproducible regions are available
for comparative
biochemical, molecular and morphological analyses. Briefly, the posterior pole
is pinned to a
wax plate following the placement of four incisions directed towards, but not
passing through,
the macula. Grossly, eyes (or gross photographs) are examined under a
dissecting microscope at
6-IOX. Subjective grading of drusen size, density and class is recorded.
Macular drusen are
classified into the following categories: rare (<5 drusen), few (6-50 drusen),
moderate (51-200
drusen), and numerous (>200-300); and sizes: small (<50 m), moderate (50-500
m), and large
(>500 m). Additional features of age-related macular degeneration, such as
macular increased
RPE pigmentation, macula RPE pigment clumping, RPE atrophy, subretinal or sub
RPE
hemorrhage, or subretinal fibrosis are also noted. In general, "early" AMD is
defined as 1) the
presence of indistinct ("soft") or reticular drusen, or 2) presence of any
drusen type with
associated visual loss, RPE degeneration, and/or abnormal retinal pigment in
the macular area.
"Late" AMD is defined as the presence of exudative AMD (RPE detachment,
detachment of the
retina, subretinal or subRPE hemorrhage, or subretinal fibrous scars) or
geographic atrophy. At
this stage, eyes are graded based upon an adaptation of a classification
system developed by The
International ARM Epidemiological Study Group; this information is entered
into a database
containing all information available for each donor.
Following gross examination, the vitreous is removed and various regions
excised with
trephine punches; these are frozen immediately in liquid nitrogen or fixed as
per the protocol.
The neural retina is separated from the RPE/choroid in regions that are
punched. Portions of
every eye are processed for light and electron microscopic analyses. Wedges
composed of
equatorial/peripheral retina are removed with forceps and frozen similarly.
Sections made from
all eyes are stained with hematoxylin and eosin, Mallory trichrome, PAS, oil
red 0, and Sudan
olack B. Histopathologic and electron microscopic examination of all donor
eyes, that includes
portions of the maculas from most eyes, is performed. Based on these analyses,
drusen are
classified nto distinct morphologic phenotypes. These categories resemble most
closely the
classification scheme proposed by Sarks. It is from these morphological
analyses that eyes are
divided into experimental groups for the proposed biochemical and molecular
studies.
88
CA 02363503 2001-08-24
WO 00/52479 PCT/US00/05858
We have also developed the "Comprehensive Donor Database" (CDD), a rigorously
characterized group of donors of all ages, with and without AMD. To date, we
have placed 155
donors from our repository into the CDD, ranging in age from I day to 101
years. 53 of these
donors had a clinically documented history AMD. The CDD will be comprised of
10 donors per
decade up to 50 years. Decades above the age of 50 years will ultimately be
comprised of 15
donors each with clinically documented AMD (5 each with macular drusen,
geographic atrophy,
and choroidal neovascular membranes or disciform scars) and 10 age-matched
controls. The
expansion of the CDD to include additional AMD donors with distinct phenotypes
will be
necessary.
Fixed (4% paraformaldehyde; Karnovsky) and frozen tissue is available for all
donors (see
Human Subjects section). In addition, histologic sections of all the eyes that
have been entered
to date have been made. Sections stained with H&E, PAS, Oil Red 0, and Sudan
black B, and
prepared for the examination of autofluorescence, are available for every eye
entered into the
CDD. Approximately 20% of the eyes thus far have been examined by electron
microscopy.
Micrographs from both the maculas and peripheral regions have been recorded at
standardized
magnifications. Baseline morphometric data from each eye are being obtained.
These include
measurements of drusen size, number, and phenotype; BLD density and
distribution; RPE and
photoreceptor cell densities; Bruch's membrane thickness and degree of debris
accumulation;
choriocapillaris density; and choroidal thickness and density of choroidal
fibrils. Other
parameters will be added as required.
Example 2: Identification of Distinct Core Subdomains within Drusen
Reagents: Fluorescein isothiocyanate- (FITC-) and rhodamine-conjugated lectins
derived from
Limax favus (LFA), Triticum vulgaris (WGA), Arachea hypogea (PNA), and Ricinis
communis (RCA-I) were obtained from EY Laboratories, Inc. (San Mateo, CA) and
Vector
Laboratories (Burlingame, CA). Neuraminidase (isolated fromClostridium
perfringens) was
obtained from Boehringer-Mannheim (Indianapolis, IN) and 0-glycanase (endo-a-N-
acetylgalactosaminidase) was purchased from Genzyme (Cambridge, MA) and
Boehringer-
Mannheim (Indianapolis, IN). Sudan black B solution was obtained from Poly
Scientific (Bay
Shore, NY), and PNGase F and globulin-free bovine serum albumin (BSA) were
purchased
from Sigma Chemical Company (St. Louis, MO). Immumount was purchased from
Shandon
(Pittsburgh, PA). Acrylamide and other reagents used for embedding were
purchased from
Bethesda Research Laboratories (Bethesda, MD). Optimal Cutting Temperature
compound
(OCT) was obtained from Miles Inc. (Elkhart, NY). Superfrost Plus slides were
obtained from
Fisher (Pittsburgh, PA). Materials for transmission electron microscopy were
obtained from
Fluka Chemika-BioChemika (Ronkonkoma, NY).
Human Donor Eyes: Eyes from 42 human donors, ranging from 35 to 101 years of
age, were
obtained from MidAmerica Transplant Services (St. Louis, MO) and the Iowa
Lions Eye Bank
(Iowa City, IA). Eyecups were preserved in 4% paraformaldehyde in 100mM sodium
89
CA 02363503 2001-08-24
WO 00/52479 PCT/USOO/05858
cacodylate, pH 7.4, or embedded directly in Optimal Cutting Temperature
compound (OCT)
and frozen in liquid nitrogen, without fixation, within six hours post-mortem.
Fixation and Embeddintz: After 2-4 hrs in fixative, eyecups were placed into
buffer and
embedded, as described in Example 4.
Glycosidase Treatments: Sections of fixed and unfixed tissues were incubated
with lU/mL
neuraminidase in 30mM sodium acetate buffer, pH 5, at 37oC, overnight in a
humidified
chamber. Sections from 41 eyes -- 33 fixed and acrylamide-embedded and 9
unfixed and
OCT-embedded -- were treated with neuraminidase and subsequently labeled with
FITC- or
rhodamine-PNA (below). Adjacent, control sections were incubated with buffer
alone. Serial
sections from two eyes (one fixed and one unfixed) were treated with
neuraminidase and
labeled with LFA, RCA-I, ConA, and WGA, to determine the effects of
neuraminidase on
labeling of drusen with other lectins. Tissue sections from two additional
donors were
pretreated with neuraminidase and subsequently treated with 0-glycanase (IU/mL
in 15mM
sodium cacodylate, pH 6) or PNGase F(lU/mL in PBS), for 72 hours, at 37oC.
Control
sections were treated with buffer alone.
Lectin Histochemistry: For lectin labeling, 6-8mm thick cryostat sections were
cut, mounted on
Superfrost Plus slides, and labeled with PNA, WGA, or LFA as described in
Example 4.
Unlabeled, adjacent control sections were used to distinguish between lectin
binding and
drusen autofluorescence.
In order to compare labeling of enzyme- and buffer-treated sections, intensity
and binding
patterns on serial sections containing the same drusen were compared.
Identical exposure
times for experimental pairs were used during photomicrography and during
photographic
processing.
Transmission Electron Microscony: Drusen-containing tissues were obtained as
above, and
were fixed within 4 hours of death in one half-strength Kamovsky's fixative
and processed as
described below. Reagents employed in embedding tissues for transmission
electron
microscopy were obtained from Fluka Chemical (Milwaukee, WI). All other
reagents were
obtained from Electron Microscopy Sciences (Fort Wayne, PA). Tissues were
fixed for at least
two hours in one-half strength Karnovsky fixative (1/2K; 2% formaldehyde and
2.5%
glutaraldehyde in 100mM cacodylate buffer, pH 7.4, containing 0.025% CaC12)
prior to
washing 3x10 min. in 100mM cacodylate buffer. Pellets or wedges were then post
fixed with
2% osmium tetraoxide in cacodylate buffer for 2 hours, and were then rewashed
3x10 min.
prior to dehydration through a series of graded ethanol solutions (50% ethanol
10 min. each in
70% ethanol, and 95% ethanol, followed by 2x10 min. in 100% ethanol. Tissues
were then
CA 02363503 2001-08-24
WO 00/52479 PCTIUSOO/05858
dehydrated 2x10 min. in propylene oxide, and were infiltrated overnight in a
1:1 mixture of
propylene oxide:Epon 812 solution (containing 51% Epon 812, 27% dodecenyl
succinic
anhydride, and 22% nadic methyl anhydride, with 1.5% DMP-30 added to the
solution as an
accelerator). The following day the Epon solution was changed 3 times
throughout the day,
and the samples were cured at 40%C overnight and then at 65%C for 2 days.
Thin sections (60-75nm) were taken from Epon-embedded tissues, on a Reichert-
Jung
Ultracut ultramicrotome. Sections were collected on nickel grids and were
stained with 2%
aqueous uranyl acetate and Reynold's lead citrate.
RESULTS
Neuraminidase Treatment: Incubation of drusen-containing tissue sections with
neuraminidase
completely eliminated LFA labeling of drusen and other structures in the
chorioretinal
complex, as compared to controls. Labeling of nuclei in the choroid and neural
retina persisted
after neuraminidase treatment, however. This loss of labeling was used
throughout the study to
control for enzyme efficacy, as were changes in labeling of the
interphotoreceptor matrix, as
described previously (Johnson and Hageman, 1987; Kivela, 1990).
Labeling of drusen with WGA, RCA-I, and Con A is not significantly diminished
following neuraminidase treatment, demonstrating that the binding of these
lectins to drusen-
associated glycoconjugates is not primarily due to sialic acid. In some eyes,
the intensity of
WGA labeling of the choroidal stroma decreases after neuraminidase treatment,
without a
concomitant loss of WGA binding to drusen in the same section.
PNA does not normally bind to drusen or any other structure within the RPE-
choroid
complex. Following pre-exposure to neuraminidase, the endothelial cells and/or
endothelial
cell basal laminae of the choriocapillaris and other choroidal vessels, as
well as the basal
lamina of the RPE, bound PNA intensely.
Some drusen were labeled by PNA following exposure to neuraminidase. This
intense
labeling was typically restricted to subdomains, or "cores", within drusen.
These cores were
not observed in adjacent sections treated with buffer alone. Drusen cores were
typically
spherical, centrally located within the druse, and juxtaposed against the
inner collagenous layer
of Bruch's membrane. Only one core was generally observed within any given
solitary druse,
whereas confluent or fused drusen may possess several cores. These cores
ranged from 5 to
30&m diameter, with a mean diameter of 14&m.
91
CA 02363503 2001-08-24
WO 00/52479 PCT/US00/05858
Drusen cores were observed frequently; they were present in 32 of the 42 eyes
examined in
this study. No differences in the appearance or frequency of these deposits
was noted with
respect to fixation and embedding conditions. Intense labeling of drusen cores
was observed
using both FITC-PNA and rhodamine-conjugated PNA, demonstrating that this
observation is
not due to interaction with the fluorophore. In addition, both hard and soft
drusen possessed
cores, although large, soft drusen typically had PNA-binding cores which are
larger and less
centrally localized than those of hard drusen.
Enzymatic Characterization of Drusen Cores: Following incubation with 0-
glycanase, PNA
labeling of the choroid and interphotoreceptor matrix was nearly or completely
abrogated.
Labeling of drusen cores with PNA was significantly reduced following 0-
glycanase treatment.
0-glycanase treatment had little effect on labeling of rod outer segments with
ConA,
suggesting that contaminating N-glycosidase activity was not present. In
contrast to 0-
glycanase treatment, PNGase F pretreatment did not change the intensity of PNA
labeling of
drusen cores. ConA labeling of rod outer segments was completely abrogated
following
incubation with PNGase F, demonstrating that the enzyme efficiently removed N-
linked
glycans.
When PNA-labeled or unlabeled tissue sections were stained with Sudan black B,
core-like
regions were not stained. Serial sections, stained alternatively with PNA
(following
neuraminidase pretreatment) and Sudan black B revealed that the PNA-positive
cores and
Sudan black-negative cores colocalize.
Transmission Electron Microscopy: Transmission electron microscopy revealed a
variety of
possible "core-like" structures within drusen. These include regions which
exhibited subtle
differences in contrast due to differences in quantity and/or electron density
of particles, when
compared to the rest of the drusen, as well as domains which were very dense
and osmiophilic.
In addition, drusen occasionally possessed regions that are electron lucent,
presumably due to
extraction of lipids during processing.
Example 3: Dendritic Cells and Proteins Involved in Immune-Mediated
Processes are Associated with Drusen
Introduction: Drusen are a significant risk factor for the development of age-
related macular
degeneration (AMD). Relatively little is known, however, about their
origin(s). We recently
described the presence of centralized domains comprised of distinct
saccharides within drusen (J
Histochem Cytochem 47;1533-9, 1999). Electron microscopic analyses have
revealed that cell
processes, derived from choroidal cells, breach Bruch's membrane and terminate
in bulbous
cores within drusen.
92
CA 02363503 2001-08-24
WO 00/52479 PCTIUSOO/05858
Reagents: All supplies for electron microscopy were obtained from Fluka
Chemical
(Milwaukee, WI). Antibodies to CD3, CD 15, CD45, and anti-mouse secondary
antibodies
conjugated to indocarbocyanine-3 (Cy3) were purchased from Chemicon
International
(Temecula, CA); monoclonal antibodies to CD1a, CD14, CD31, CD45, CD68, S100
and HLA-
DR were purchased from Dako (Carpenteria, CA). Fluorescein-conjugated
secondary
antibodies were obtained from Jackson Immunoresearch Laboratories (West Grove,
PA). For
some experiments, the Elite Staining Kit was employed and labeling was
visualized with the
Vector VIP substrate (Vector, Burlingame, CA). Neuraminidase (Clostridium
perfringens) was
obtained from Boehringer-Mannheim (Indianapolis, IN). Fluorescein-conjugated
peanut
agglutinin was purchased from EY Laboratories, Inc. (San Mateo, CA). Other
reagents used
for tissue fixation and embedment were obtained from Sigma Chemical Company
(St. Louis,
MO), unless otherwise noted. Studies were conducted to immunophenotype the
choroidal cells
from which these core terminations arise and to evaluate their potential
relationship to drusen
biogenesis.
Tissues: Human donor eyes employed in this study were obtained from The
University of Iowa
Lions Eye Bank (Iowa City, IA) within four hours of death. Institutional
Review Board
committee approval for the use of human donor tissues was obtained from the
Human Subjects
Committee at The University of Iowa. Posterior poles, or wedges of posterior
poles spanning
between the ora serrata and macula, were processed from 30 donors, embedded in
OCT, snap
frozen in liquid nitrogen, and stored at -80 C. Tissues were sectioned to a
thickness of 6-8um on
a cryostat. Confocal laser scanning microscopy and immunohistochemistry were
employed to
examine drusen-associated cores in human donor eyes. Immunolabeling of
sections was
performed using a battery of antibodies directed against various cell
populations including
endothelial cells, lymphocytes, granulocytes, monocytes/macrophages and
dendritic cells.
Transmission Electron Microscopy: For transmission electron microscopy,
posterior poles
were fixed in one-half strength Karnovsky's fixative (see Example 1) within 4
hours of death.
Macular punches and saggittal wedges from over 200 eyes obtained by our
laboratory were
fixed and processed for electron microscopy, as described previously (Lazarus,
et al., 1993),
and used for these analyses.
Immunohistochemistry: Eyes employed for immunohistochemistry were dissected
and
embedded into Optimal Cutting Temperature compound (Miles Inc.; Elkhart, NY)
without
prior fixation. Cryostat sections were cut at a thickness of 6-8m and were
collected on
Superfrost plus slides (Fisher; Pittsburgh, PA). Neuraminidase treatment was
performed in
some cases; sections were digested overnight with lU/mL neuraminidase in 30mM
sodium
acetate buffer, pH 5.0, overnight at 37 C. For fluorescence microscopy,
antibody and lectin
labeling was performed as described previously in Example 1. For some
experiments, the
Vector Elite kit for horseradish peroxidase staining was used, according to
the manufacturer's
instructions. In order to determine whether leukocyte antigens and PNA-binding
cores
93
CA 02363503 2001-08-24
WO 00/52479 PCT/US00/05858
colocalize, serial sections were incubated alternately with PNA (neuraminidase
pretreated) and
anti-CD antibodies. Control sections were treated with secondary antibodies
alone. Positive
controls for CD antibodies were based on their reactivity with leukocytes in
the choroidal
vasculature and stroma. Reactivity of drusen with antibodies to HLA-DR and
CD68 was
quantitated in unfixed sections by calculating the percentage of labeled
drusen. The diameters
of drusen and of drusen cores were measured with an eyepiece reticle
calibrated to a stage
micrometer.
Double Labeling/Confocal Microscopy: Cryostat sections were treated overnight
with
neuraminidase (above) followed by immunolabeling with a monoclonal antibodies
directed
against CD68 HLA-DR, and CD1a. Alexa 488-or Cy-3-conjugated secondary
antibodies
(Chemicon; Temecula, CA: Molecular Probes; Eugene, OR) were used to visualize
CD68
immunoreactivity. Sections were washed extensively and incubated with PNA
conjugated to
fluorescein. Confocal images of both probes were collected simultaneously
using a confocal
microscope (BioRad; Hercules, CA). Positive controls included choroidal and
scleral
leucocytes.
Results: Significantly, cellular processes, derived from cells in the
choroidal stroma, were
observed that breach Bruch's membrane and terminate within drusen. Extensive
serial
sectioning through five drusen revealed a single process, derived from a
choroidal cell, passing
through Bruch's membrane and terminating as a bulbous process, occupying the
same location as
drusen cores. The choroidal cells from which these processes emanate exhibited
a large degree
of rough endoplasmic reticulum, had nuclei that are lobed, and were dendritic
in shape. No large
granules or lysosomes were apparent in their cytoplasm. These processes were
also observed
lying adjacent to whole, or portions of, RPE cells, in regions without
significant numbers of
drusen.
In order to examine the association of these choroidal cell-derived processes
to drusen
cores, cryostat sections were incubated with antibodies to CD45 (leukocyte
common antigen),
while alternate serial sections were digested with neuraminidase and labeled
with PNA. A
subset of the same cores which bound PNA are also labeled with CD45
antibodies. Anti-CD45
antibodies colocalize with PNA-binding cores in smaller drusen.
Drusen cores, and the cells from which they are derived, are also strongly
reactive with
CDla, CD4, CD14, CD45, CD68, CD83, CD86, and HLA class II (CR3/43 and TAL.1B5)
antibodies. The CR3/43 antibody reacts with MHC class II antigens including
HLA-DP, HLA-
DQ, and HLA-DR, whereas the TAL.1B5 antibody is specific to HLA-DR alpha
chains. Both
antibodies react with drusen cores, although the DR-specific clone (TAL.1B5)
may react with
more restricted, cell-associated domains whereas the pan-MHC-II clone (CR3/43)
may label
more voluminous domains within drusen. This may imply that HLA-DR is largely
confined to
drusen-associated dendritic cells, in contrast to HLA-DP and HLA-DQ, which may
be
derivatives of membrane blebs from these cells, or exosomes. Ongoing studies
are be directed
94
CA 02363503 2001-08-24
WO 00/52479 PCTIUSOO/05858
toward determining whether there is a difference in the distribution of the
various MHC class-Il
antigens in drusen cores, and whether other exosomal proteins are present in
drusen.
To gain further insight into the relationship between the distribution of
leukocyte processes
and drusen, double-labeled tissue sections were examined using scanning laser
confocal
microscopy. Two relevant observations were made. In some drusen, there was a
direct
colocalization of PNA and anti-CD68 antibody to the drusen cores. However,
CD68, but not
PNA, labeled the body of the choroidal cell associated with the core. These
observations
suggest that the core-associated PNA-binding material was restricted to the
bulbous cell
process or that it was secreted by these processes into drusen. In other
cases, a small CD68-
immunoreactive core was observed that is surrounded by a larger, PNA-binding
cuff, consistent
with the proposition that this material was synthesized and secreted by core-
associated
macrophages/dendritic cells, and/or that the bulbous processes modify the
surrounding drusen-
associated matrix such that it bound PNA.
Quantitative studies indicate that these drusen-associated cores are present
in
approximately 40% of drusen. Drusen cores appear to be more prevalent in
smaller drusen,
and are also detected as putative drusen precursors, solitary cores within
Bruch's membrane
that are not surrounded by additional drusenoid accretions.
The number of HLA-DR and CD68 immunoreactive drusen were determined in unfixed
cryostat sections. Eighty-eight percent of all drusen were HLA-DR
immunoreactive; binding
was restricted to cores in some drusen, whereas in others it was observed
throughout.
The mean size of HLA-DR reactive drusen was 26um+9um. The mean size for HLA-DR
negative drusen was 22.8um+4.8um. Thus, there was no significant difference in
size between
HLA-DR positive and negative drusen (Student's t-test). In contrast,
approximately twenty
percent of all drusen in any given eye possessed anti-CD68 antibody
immunoreactive cores.
The diameters of cores that reacted with antibodies to CDla, CD45 and CD68
were
measured with an eyepiece reticle. These cores measured 10.4m + 4.4m in
diameter. This
was somewhat smaller than the average size of PNA-binding cores (14m ), and
may suggest
that the PNA-binding material in drusen surrounds the leukocyte process. This
result is
consistent with results from double labeling confocal microscopy experiments
(above).
Conclusions: The immunophenotyping data, when combined with ultrastructural
analyses,
provide strong evidence that drusen cores are derived from choroidal dendritic
cells. The
identification of dendritic cell-derived cores in smaller drusen and putative
drusen precursors,
when combined with our studies that demonstrate the presence of HLA-DR,
immunoglobulin
light chains, vitronectin, and terminal complement complexes in all drusen
phenotypes (see
Example 5), suggest a role for dendritic cells and immune-mediated processes
in drusen
biogenesis and early AMD.
CA 02363503 2001-08-24
WO 00/52479 PCT/US00/05858
Example 4: Further Characterization of Drusen-Associated Molecules: The
Development
of Procedures for the Enrichment of Drusen from Human Eyes
Reagents: Polyclonal antisera directed against vitronectin (VN) and laminin
(LN) were
obtained from Telios (San Diego, CA); antibodies to collagen type IV were
obtained from
Chemicon (Temecula, CA). Wheat germ agglutinin (WGA) and Limaxflavus
agglutinin (LFA)
were purchased from Vector (Burlingame, CA) and EY Laboratories (San Mateo,
CA),
respectively. Reagents employed in embedding tissues for immunofluorescence
were obtained
from Bethesda Research Laboratories (Bethesda, MD) and Sigma Chemical (St.
Louis, MO).
Materials employed in the preparation of tissue for transmission electron
microscopy were
obtained from Fluka Chemical (Milwaukee, WI). Sudan black B was purchased from
Poly
Scientific (Bay Shore, NY). Reagents used for hematoxylin and eosin staining
were purchased
from Richard-Allan Medical (Richland, MI). Round-tipped surgical blades
(Beaver Mini
Blade ES, #69) were obtained from Becton Dickinson (Franklin Lakes, NJ).
Human Donor Eyes: Human tissues were obtained from MidAmerica Transplant
Services (St.
Louis, MO) and the Iowa Lions Eye Bank (Iowa City, IA) within 5 hours of
death. Following
removal of the corneas, donor eyes were cut into quadrants. An inferior
saggittal wedge from
the ciliary body to the macula was removed from each eye and fixed in either
4%
paraformaldehyde or one-half strength Karnovsky's fixative (1/2K) for 2 hours,
to assess the
presence and morphology of drusen in these tissues. The neural retina was
removed from each
eye, and individual quadrants were pinned to wax-coated Petri dishes, scieral
side down.
Microdissection: Attempts were made to microdissect large drusen using number
five forceps
or narrow gauge (26G) syringe needles. Drusen were gently separated from the
choroid and
were washed with lOmM phosphate buffered saline (PBS; pH 7.4) and placed into
a Petri dish
for photomicrography or into an Eppendorf tube for transmission electron
microscopical or
biochemical analyses.
Scraped Drusen Preparations: In other experiments, the RPE aspect of the
pinned quadrants
was gently scraped with a Beaver #69, round-tipped blade to debride Bruch's
membrane in
areas with large numbers of drusen. Care was taken not to slice through the
elastic lamina, by
holding the blade at a slight angle and scraping perpendicular to the axis of
the blade. Both
RPE and drusen were harvested from these regions. Other eyes without
macroscopically-
visible drusen were also scraped. These preparations, which contain RPE but no
drusen, served
as controls. The debrided material was collected on the surface of the blade,
and was then
rinsed off with PBS containing protease inhibitors. The RPE/drusen
preparations were spun for
96
CA 02363503 2001-08-24
WO 00/52479 PCTIUSOO/05858
3 min in an Eppendorf microfuge prior to fixation or freezing of the pellet in
liquid nitrogen for
subsequent biochemical analyses.
For some experiments, enriched drusen preparations were incubated in ice cold
distilled
water in order to lyse RPE cells. These preparations were then either frozen
for electrophoresis
or were fixed and processed for immunohistochemistry, as above. In other
experiments, the
RPE was removed with a stream of buffer (using a 30 gauge needle mounted to a
10cc syringe)
and the Beaver #69 blade was used to debride Bruch's membrane of the remaining
drusen.
Tissue Processiniz: In order to determine the efficacy of the scraping
technique in removing
RPE/drusen from Bruch's membrane, portions of the scraped material and the
remaining
Bruch's membrane/choroid were fixed in 4% paraformaldehyde and prepared as
described in
Example 4. Cryostat sections of the enriched material and the remaining
choroid were
collected and employed in histochemical analyses.
Sections of drusen-enriched pellets were stained with 1% Sudan black B, WGA,
LFA, and
antibodies to vitronectin (VN), laminin (LN), complement C5, and collagen type
IV. Lectin
and antibody staining was performed as described in Examples 1 and 2.
Portions of enriched drusen/RPE specimens and post-scraped choroid (without
drusen or
RPE) from the same eyes, were also preserved in 1/2K fixative and prepared for
transmission
electron microscopy as described in Example I. Thin sections were taken from
blocks of
enriched drusen to examine the ultrastructure of these preparations, and
sections from post-
scraped choroids were prepared to examine the integrity of the Bruch's
membrane/choroid
complex.
One enriched drusen preparation was fixed in 1/2K as above, rinsed in
cacodylate, and
dried down on a polylysine coated surface for subsequent examination by
scanning electron
microscopy. The tissue was dehydrated by critical-point drying, and was
sputter coated, as
described previously.
Laser Capture Microdissection (LCM): As an additional method for the isolation
of drusen and
other ocular age-related deposits, we have employed laser capture
microdissection (LCM) on
frozen sections derived from human donor tissues. This technique allows for
the precise
identification and isolation of drusen from routinely prepared tissue
sections. CapSureTM
transfer film is placed on the tissue section surface, the structures of
interest are identified, and a
low power infrared laser is used to bond the structure of interest onto the
film, while the
remainder of the tissue section remains adhered to the slide. The instrument
delivers precise
laser pulses to cells or tissues of interest, trapping them in a polymer film
and separating them
from the remainder of the tissue components. Laser spot sizes of 7.5, 15, or
30 m may be
selected.
97
CA 02363503 2001-08-24
WO 00/52479 PCT/US00/05858
Protein Separation and Analysis: Preparation of enriched drusen proteins for
electrophoretic
separations involved sonicating total RPE/choroid tissues or enriched
RPE/drusen pellets
briefly on ice, followed by boiling of samples in sample buffer, as described
below. Following
removal of corneas, donor eyes were cut into quadrants. Neural retinae were
removed in order
to reveal more precisely the extent of RPE pathology, including geographic
atrophy, choroidal
neovascularization, pigment clumping, and/or drusen. Tissues with advanced
degeneration of
the RPE were excluded from this study. The presence or absence, and extent, of
drusen was
determined initially under a dissecting microscope. Eyes with large numbers of
drusen or no
visible drusen ("controls") were collected separately. In some cases,
different regions of the
RPE/choroid complex possessing or lacking drusen were collected from the same
eye, to
control for donor-to-donor variation in protein levels and mobility. In
addition, inferior sagittal
wedges from the ciliary body to the macula were removed from each eye,
bissected
meridionally, and fixed in either 4% paraformaldehyde or 1/2K (see Tissue
Processing:
Electron Microscopy) for two hours. These tissues were used to determine the
extent and
phenotype(s) of drusen, by routine histological techniques. For some
experiments, the entire
RPE/choroid was used. Following assessment of drusen status (outlined above),
whole
RPE/choroids were peeled from the sclera and immediately frozen in liquid
nitrogen.
When needed, these tissues were thawed, sonicated in a minimal volume of
isotonic buffer
(PBS, pH 7.4) containing protease inhibitors (see Appendix) for 20 bursts, and
then centrifuged
(11,000xg, 5 min.). Protein concentrations of the supernatant fractions were
determined using
the Micro BCA method (Pierce, Rockford, IL). Equivalent amounts of protein
from donors
with or without drusen (10-50 g of total protein per sample per lane) were
separated by SDS-
PAGE as described previously (Laemmli, 1970). For most experiments, extracts
from at least
four drusen-containing eyes and four age-matched donors without drusen were
run
simultaneously. For some experiments, samples from young donors without drusen
were also
included in order to control for age-related changes.
Enriched drusen preparations were compared to whole RPE/choroid preparations
on silver
stained gels and Western blots. In one experiment, the high molecular weight
aggregates at the
interface of the stacking gel-separating gel, characteristic of drusen-
containing preparations,
were excised and analyzed by matrix assisted laser desorption ionization mass
spectrometry
(MALDI-MS).
Amino acid sequencing was performed at the W.M. Keck Foundation's
Biotechnology
Resource Foundation (New Haven, CT) as described (Stone, et al., 1990). In
some cases, in-
gel trypsin digestion of Coomassie-stained gel bands was performed (Stone, et
al., 1990), and
98
CA 02363503 2001-08-24
WO 00/52479 PCT/US00/05858
peptides were identified and matched to their respective proteins based on
their molecular mass
using matrix assisted laser desorption ionization mass spectrometry (MALDI-
MS), as
described (Williams et al., 1996). The European Molecular Biology Laboratory
and OWL
databases were then searched for masses of tryptic peptides of known, as well
as conceptually-
translated, proteins (Lamand and Mann, 1997). Only cases in which at least
five peptides and
up to 20% of the protein mass were matched to predicted tryptic fragments were
the matches
considered significant.
MS/MS of Enriched Drusen: As an additional approach to identify the molecular
constituents of
drusen and enriched RPE/drusen preparations were collected and digested with
trypsin, followed
by identification of resultant peptides by mass spectrometry (LC/MS/MS). In
other studies,
these preparations were separated by two-dimensional SDS-PAGE, individual
spots were
collected, and analyzed, as above, employing MS/MS.
RESULTS
RPE/Choroid Biochemistry: Prior to the development of enrichment techniques
for examining
drusen constituents biochemically, proteins from the RPE/choroid complex from
donors
assessed to have drusen by gross and histological examination were separated
and compared to
those of donors without drusen. As a function of drusen status, variations in
the pattern of
proteins were observed with silver staining. RPE/choroid extracts from donors
with drusen
typically possess a doublet of approximately 35/36kDa, whereas homogenates
from age-
matched controls exhibit only a single band at this molecular weight. Of
donors with drusen,
75% were found to possess the 35/36kDa doublet, whereas none of the donors
without drusen
exhibited this alteration. A second pattern variation coinciding with the
presence of drusen is a
120kDa band which is absent in drusen-containing tissues, but is always
present in age-
matched controls. These bands were excised from preparative gradient gels, and
their
constituent proteins were identified by MALDI-MS. These analyses identified
interphotoreceptor retinoid binding protein as being present in the 120kDa
band associated with
donors without drusen. The corresponding region of the gel from drusen-
containing donors
contained ceruloplasmin, which was not identified in the control donor band,
but did not
contain IRBP. Cellular retinaldehyde binding protein (CRALBP) and annexin II
were found in
samples derived from donors with and without drusen, whereas the 26S protease
regulatory
subunit (involved in ubiquitin-mediated proteolysis) was found only in this
band from donors
with drusen.
Isolated Solitary Drusen:
99
CA 02363503 2001-08-24
WO 00/52479 PCT/US00/05858
Morphology: Large, individual drusen, relatively free of contaminants, were
isolated using
the techniques described. Isolated drusen were examined using bright field
micrography. They
were typically spherical or hemispherical, contained vesicular profiles, and
were often
associated with a few RPE cells or pigment. Ultrastructurally, this material
was comprised of
membranous debris and other structural elements characteristic of drusen in
situ and
fragmented RPE cells.
SDS-PAGE: Individual drusen were dissociated in sample buffer and separated by
SDS-
PAGE, followed by silver staining or Western blotting. Typically, we were only
able to collect
5-20 drusen per eye using this approach. Although too little protein was
obtained from each
isolated drusen sample to run preparative gels for amino acid sequencing,
sufficient material
was present for analysis by silver staining and lectin labeling of Western
blots. Silver stained
drusen preparations typically yielded 6-7 discrete bands ranging in molecular
weight from 20
to 65kDa; these preparations invariably contained a prominent band with a
molecular weight of
approximately 35kDa. Lectin labeling of Western blots indicated that isolated
drusen
contained one major WGA-binding band of approximately 65 kDa, as well as India
ink-binding
bands of 78 and 62kDa. Interestingly, vitronectin migrated at 65kDa under
reducing
conditions. The 65 kDa WGA-binding band migrated at the same apparent
molecular weight
as serum albumin. However, the drusen-associated band was bound by silver
stain, in contrast
to albumin, which was visualized as an unstained band against the background.
Enriched Drusen Preparations:
Histology and Histochemistry: Drusen in situ are typically eosinophilic when
stained with
hematoxylin/eosin. Small, hard ("hyaline") drusen stain more intensely and
uniformly than
large, soft drusen, which tend to be more heterogenous. Drusen in enriched
preparations were
stained similarly. In eyes in which differences in staining between hard and
soft drusen was
apparent in situ, this same pattern was also noted in preparations of enriched
drusen collected
from the same eye. Spherical hard drusen could be discriminated from large,
amorphous soft
drusen in these preparations. Layers of RPE cells were also readily apparent
in these
preparations.
The RPE/drusen-debrided choroid, enriched drusen preparations, and intact,
control regions
from the same eye were examined using immunohistochemistry and lectin
histochemistry. The
intact basal lamina of the choriocapillaris was observed in the debrided
choroid, as was the
autofluorescent elastic lamina of Bruch's membrane, providing evidence that
Bruch's
membrane was not breached during the enrichment procedure.
100
CA 02363503 2001-08-24
WO 00/52479 PCTIUSOO/05858
Antisera directed against VN and C5 and drusen-binding lectins were used as
markers to
follow drusen through the enrichment process. In the intact RPE/choroid
complex, these
markers labeled drusen intensely. The globular drusen within the enriched
drusen preparations
exhibited intense labeling with these probes, indicating that drusen retained
VN, C5, and
drusen-associated glyconjugate molecules after enrichment. Similarly, drusen
within enriched
pellets bound Sudan black B and oil red 0 in the same manner as was seen in
situ.
Exposure of RPE/drusen preparations to water reduced the amount of the RPE
cell material
in these preparations; only the highly insoluble melanosomes/residual bodies
remained in the
drusen rich pellet. Drusen remained highly immunoreactive to C5 antibodies
after this
treatment.
Electron Microscopy: Ultrastructural observations of enriched drusen
preparations
demonstrated that they contain RPE cells, the RPE basal lamina, free
melanosomes from the
RPE, and drusen that were morphologically identical to those observed in situ
in the same eye.
No contamination of the pellets with choroidal material was observed. Basal
laminar deposits,
that typically lie between the RPE and its basal lamina, were also present in
these preparations.
The drusen-debrided choroids possessed an intact Bruch's membrane. In eyes
without drusen,
the RPE monolayer was completely removed and much of the RPE basal lamina
typically
remained adherent to Bruch's membrane. The elastic lamina and inner
collagenous zone of
Bruch's membrane were intact and undamaged.
By scanning electron microscopy, enriched drusen preparations were visualized
as highly
heterogeneous mounds of vesicular profiles resembling drusen; RPE cell debris
and
melanosomes were also apparent.
SDS-PAGE: In preliminary attempts to determine whether enriched RPE/drusen
preparations were useful for analyzing drusen constituents, protein profiles
of enriched
RPE/drusen preparations were compared to total RPE/choroid protein profiles
following SDS-
PAGE. These experiments revealed a significant reduction in the total number
of bands in the
enriched preparations. Particularly notable was a reduction in major choroidal
constituents
such as serum albumin. Enriched drusen preparations possessed immunoreactive
vitronectin
and apolipoprotein E at the appropriate molecular weights, confirming that
known drusen-
associated molecules segregated with the drusen-enriched pellet. Western blots
of scraped
RPE/drusen exhibited bands of relatively high molecular weight, ranging from
22-150 kDa.
As described above, treatment of enriched drusen preparations with water did
not result in
a significant loss of labeling of drusen-associated molecules. Water-treated
RPE/drusen
preparations were compared to whole RPE/choroid and non-water-treated
RPE/drusen from the
101
CA 02363503 2001-08-24
WO 00/52479 PCTIUSOO/05858
same eye. A further reduction in the total number of bands was observed
secondary to lysis in
the hypotonic water solution.
As part of an initial study to characterize differences between enriched RPE
and enriched
RPE/drusen preparations, proteins were separated by SDS-PAGE. In previous
experiments, we
found that drusen-containing preparations contained significantly more high
molecular weight
protein at the gel interface than did non-drusen preparations. For this
reason, stacking gel
interfaces were excised and protein constituents of samples with and without
drusen were
identified by MALDI-MS. The matching putative proteins included myosin,
desmoplakin I/II
for the RPE preparation and Myosin, beta-spectrin, alpha-spectrin, and N-
acetylglucosamine
(G1cNAc) transferase for the RPE/drusen preparations.
Laser Capture Microdissection (LCM): We have tested this system for its
ability to collect
drusen from a complex tissue section, and have found that the Pix Ce11TM LCM
system can
efficiently and rapidly isolate drusen for further analysis.
MS/MS of Enriched Drusen: A set of molecular candidates for drusen-associated
molecules/molecules increased in the RPE-Bruch's membrane in association with
drusen have
been identified using MS/MS. Differentially-expressed proteins included an
upregulation of a
neutral pI, -30kDa and a basic, 20kDa spot in the drusen-containing sample. A
number of spots
additionally appeared to be downregulated in the drusen-containing sample,
ranging from basic
to acidic and from -15 to 80kDa. To date, these studies have conclusively
identified tissue
inhibitor of metalloproteases-3 (TIMP3) and vitronectin in the drusen-enriched
sample(s).
Collectively, these data demonstrate that a combination of novel drusen
isolation
techniques and mass spectrometry are a useful tool for the confirmation of
histochemically-
identified, and for the identification of previously uncharacterized, DRAMs.
Example 5: Charactrization of Drusen-Associated Molecules (DRAMs)
Tissues: Eyes from the human donor repository and CDD, ranging in age between
45 and 101 years,
were processed within four hours of death. Many of these donors had a
documented clinical diagnosis of
AMD (including donors with geographic atrophy, choroidal neovascularization,
and disciform scars in at
least one eye) and one donor was diagnosed with cuticular drusen. Human liver
was obtained within 2
hours of biopsy. RPE cells were isolated with 2% dispase within 5 hours of
death and were grown in
Coon's F-12 media with 10% fetal bovine serum.
Immunohistochemistry: Tissues were fixed and prepared as described in other
Examples. Slides were
blocked for 15 min. in 0.01 M sodium phosphate (pH 7.4) containing 0.85% NaCi,
1 mM calcium chloride,
1 mM magnesium chloride (PBS/MIC), and 1 mglml globulin-free bovine serum
albumin (PBSIM/CIBSA).
102
CA 02363503 2001-08-24
WO 00/52479 PCTIUSOO/05858
Sections were then rinsed for 10 min. in PBS/M/C, incubated in primary
antibody (see Table X) diluted in
PBS/M/C/BSA, for one hr., at room temperature. In some cases, sections were
pretreated, prior to
blocking, with 0.5% trypsin (Sigma, St. Louis, MO) for 10 min. as specified by
the supplier. Following
exposure to primary antibody, sections were rinsed (2x10 min.) in PBS/M/C,
incubated in the appropriate
fluorescein-conjugated secondary antibody (often adsorbed against human serum)
diluted in
PBS/M/C/BSA (30 min., room temperature), rinsed (2x10 min.) in PBS/M/C, and
mounted in Immumount
(Shandon, Pittsburgh, PA). Adjacent sections were reacted with secondary
antibody alone, as negative
controls. Some sections were pre-treated for 10 min with 0.5% trypsin (Sigma;
St. Louis, MO), or 0.2-
0.02 UImI chondroitinase ABC (Seikagaku; Rockville, MD), for use in
conjunction with antibodies for
collagen type IV or various chondroitin sulfate proteoglycans, respectively.
Drusen-containing tissues
from a minimum of five donor eyes were examined for each antibody.
For negative controls, sections were exposed to PBS/M/C/BSA containing: a) no
primary antibody; b)
1% (vol/vol) normal serum; and/or c) antibodies to irrelevant proteins. In
some cases, an additional
control included adsorption of primary antibody to purified antigen. Positive
controls included reaction of
antibodies with the extracellular matrices of sciera, choroid, and vitreous;
retinal and choroidal basal
laminae; retinal interphotoreceptor matrix; and liver. In order to determine
the "specificity" of serum
protein accumulation in drusen, drusen-containing sections were reacted with
antibodies to human
albumin (Cappel; Malvern, PA) and haptoglobin (Dako; Carpenteria, CA).
RESULTS
Reactivities of antibodies with drusen are listed in Table 2 below. In
general, all positive antibodies
bound to all drusen phenotypes. Controls confirm all antibody reactivities to
be specific. In addition, the
majority of the antibodies utilized bound to the expected regions of sclera,
choroid, RPE, retina, vitreous,
and/or other "control" tissues.
Table 2: DRAMs
ANTIGEN SOURCE DRUSEN
a1 antichymotrypsin Dako +
a1 antitrypsin Dako -/+
a2 macroglobin Biodesign -
aFGF -
AKS -
Albumin Cappel -
Amyloid A Dako +
Amyloid (i Dako - to +l-
Amyloid P Dako +
Amyloid Prec Prot B-M -
103
CA 02363503 2001-08-24
WO 00/52479 PCTIUSOO/05858
Antithrombin III Calb +l-
Apo Al Calb -
Apo E Calb +
ASPG-1 -
Atrial Natriuretic Factor Chemicon -
(32 microglobin B-M +l-
bFGF
Basement Membrane Chemicon -
Bovine nas. cart. p. ICN -
CD1 a Dako +
CD3 Pharm -l+
Dako -
CD4 Pharm +l-
CD8 Pharm -
CD14 Dako +
CD15 Chemicon -
CD31 Dako +/-
CD44 Various -
CD45 Dako +
CD68 Dako +
CD83 +
CD86 Dako +
C-Reactive Protein Dako - to +/-
Calcitonin Dako -
Carbonic Anhydrase -
Carc Assoc Ag -
cfms/CSF-1 receptor -
Chondroitin sulfate -
Chondroitin 0 sulfate -
Chondroitin 4 sulfate +
Chondroitin 6 sulfate +
Chondroitin sulfate PG Chemicon -
Collagen I Southern Biotech -
Collagen II SB -
Collagen III SB -
Collagen IV SB,Chemicon -
Collagen V SB -
Collagen VI -
Collagen VII -
Collagen IX -
Collagenases
Clq Calb -l+
Complement 3 - to +
C5 +
C5-C9 complex Calb +l-
COS -
104
CA 02363503 2001-08-24
WO 00/52479 PCTIUSOO/05858
CRALBP -
Cystatin C -
Decorin Chemicon -
Elastin Sigma -
Entactin -
Factor X Dako +
Fibrin -
Fibnnogen Dako - to +l-
Fibronectin -
Fibulin 3 Timpl -1?
Fibulin 4 Timpl -/?
FnR -
a fodrin -
(i Fodrin -/+
Gangliosides Dev Hyb -
Gelsolin -
GFAP -
Glucose Transporters 1,3,4 -
Glycolipid Dev Hyb -
Glycophorin A, C -
Haptoglobin Dako +l- (variable)
Heckenlively serum Ag +/-
Heparan sulfate (MAB) +l-
(MAC)
Kimata
Hermes -
HLA ABC -/?
HLA DR Various +
HNK-1
Heat Shock Prot 70 -
HSPG -
Human IgA -
Human IgG +/-
Hyaluronic Acid -
Ig Kappa chain - to +/-
Ig Lambda chain Dako +/- to +
Integrin a2 -
Integrin 0 -
Integrin a4 -
Integnn a5 -
Integrin a6 -
Integrin (31 -
Integrin (i2 -
Integrin P4 -
Intermediate Filaments -
Interphotoreceptor Matrix -
105
CA 02363503 2001-08-24
WO 00/52479 PCT/US00/05858
IRBP -
Keratan sulfate -
Keratin -
Laminin -
LAMP-1 Dev Hyb -
LAMP-2 Dev Hyb -
Link Protein Dev Hyb -
Lipoprotein - to +/-
Melanoma Assoc Ag -
Milk mucin core Ag -
MMPs -
Mitochindrial Ag -
N.S. Enolase -
Nerve Growth Factor -
NGFR -
Neuroflbrillary tangles -
PG40 (Decorin) -
Phospholipase A2 -
Plasminogen# Dako +
Plasminogen Act. Inhib.-1 -
Platelet Derived GF -
Prealbumin# B-M - to +
Prothrombin# +l-
S-100 (Bovine) -I?
Sialo Cell Surface Ag -
Tau -
Tenascin -
TGFb -
Thrombin Sera +/-
Thrombospondin (Gib/AMAC) - to +l-
TIMP1 -
TIMP2 -
TIMP3 +
TIMP4 +/-
Tubulin -
Ubiquitin - to +
UPAR Anderson -
Vimentin -
Vitronectin Various +
VnR -
von W Factor -
B-M=Boehringer-Mannheim
Calb=Calbiochem
Gib=GibcolBRL
Pharm=Pharmingen
106
CA 02363503 2001-08-24
WO 00/52479 PCT/US00/05858
Sera=Sera Labs
Tel=Telios
Example 6: Drusen Associated with Aging and Age-Related Macular Degeneration
Contain Proteins
Common to Extracellular Deposits Associated with Atherosclerosis, Elastosis,
Amyloidosis, and
Dense Deposit Disease
Recent studies in this laboratory revealed that vitronectin is a major
component of drusen.
Because vitronectin is also a constituent of abnormal deposits associated with
a variety of diseases, drusen
from human donor eyes were examined for compositional similarities with other
extracellular disease
deposits. The sixty-three human donor eyes employed in this study were
obtained from The Human Donor
Repository and the CDD. All eyes were collected and processed within four
hours of death; donor ages
ranged from 45 to 96 years. Drusen were categorized as hard or soft. Tissues
from a minimum of five
donors were assayed with each antibody employed, at least two of whom had
clinically-documented AMD,
and each drusen phenotype was examined in at least two donors. Institutional
Review Board committee
approval for the use of human donor tissues was obtained from the Human
Subjects Committee at The
University of Iowa.
Thirty-four antibodies to twenty-nine different proteins or protein complexes
were tested for
immunoreactivity with hard and soft drusen phenotypes. These analyses provide
a partial profile of the
molecular composition of drusen (see Table 3 below). Serum amyloid P
component, apolipoprotein E,
immunoglobulin light chains, Factor X, and complement proteins (C5 and C5b-9
complex) were identified in
all drusen phenotypes. No reaction of antibodies to the primary amyloid
proteins keratin, apolipoprotein A-
I, gelsolin, calcitonin, atrial natriuretic factor, tau, or amyloid precursor
protein was observed. Antibodies
against human serum albumin and haptoglobin bound strongly to the choroidal
stroma, but not to hard or
soft drusen. Immunoreactivity of some drusen-associated proteins was
frequently observed in distinct,
heterogeneous patterns. For example, drusen binding by prothrombin and amyloid
A antibodies, was often
localized to spherical profiles within drusen. Drusen were occasionally
labeled by anti-fibrinogen
antibodies; this binding was generally confined to peripheral regions and/or
concentric bands within drusen.
The compositional similarity between drusen and other disease deposits may be
significant in view
of the correlation between AMD and various systemic disorders, including
atherosclerosis. These data
suggest that similar pathways may be involved in the etiologies of AMD and
other systemic disorders.
Table 3: Immunoreactivity of Drusen
Antigen Su plier Cone. No. Drusen
Albumin Accurate 1:50 5 -
Amyloid A Dako 1:50 8 vesicles
Am loid Dako 1:10 7 -
Amyloid Precursor Protein Boehringer 1:20 5 -
Mannheim
Amyloid P component Dako 1:50 6 ++
Calbiochem 1:50 5 ++
al-antichymotrypsin Dako 1:50 6 +/- (var.)
Calbiochem 1:50 5 +/- (var.)
107
CA 02363503 2001-08-24
WO 00/52479 PCT/US00/05858
al anti-trypsin ICN 1:50 5 -, rare +/-
A oli o rotein Al Calbiochem 1:50 6 -
Apolipoprotein B Chemicon 1:20 6 -
Dako 1:50 5 - to +/-
A oli o rotein E Calbiochem 1:50 9 +
Atrial natriuretic factor Chemicon 1:50 5 -
C-reactive protein Dako 1:50 5 - to (var.)
Calcitonin Dako 1:50 5 -
Complement C1 Calbiochem 1:50 5 -
Complement C3 Dako 1:50 5 - to +, (var.)
Complement C5 Dako 1:50 5 ++
Complement C5b-9 Dako 1:50 5 ++
Cystatin C Accurate 1:50 5 -, (var.)
Factor X Dako 1:50 9 +
Fibrinogen Dako 1:50 5 - to +/-, (var.)
Gelsolin Chemicon 1:50 5 -
HLA-DR Accurate 1:25 10 +
Dako 1:200 10 +
Immunoglobulin kappa Boehringer 1:50 8 - to +/-
Mannheim
Immunoglobulin lambda Dako 1:50- 9 +/- to +
1:2000
R2 microglobulin Boehringer 1:50 5 - to +/-
Mannheim
Prothrombin Dako 1:50 5 + (vesicles)
Tau Dako 1:50 5 -
Transthyretin Boehringer 1:50 9 +/-
Mannheim (var.)
Ubiquitin Chemicon 1:50 5 -
StressGen 1:100 5 -, rare +/-
Key: ++ = intense, invariant labeling; + = strong labeling in most donors; +/-
= weak labeling; - no labeling
detected; (var.)= donor to donor or drusen to drusen variation; vesicles =
labeling of spherical profiles within
drusen
Example 7: Local Sources of DRAMs Common to Extracellular Deposits Associated
with
Atherosclerosis, Elastosis, Amyloidosis, and Dense Deposit Disease
Studies were conducted to determine whether any of the DRAMs that were
identified as being common
to extracellular deposits associated with atherosclerosis, elastosis,
amyloidosis, or dense deposit disease
were produced locally in the eye by RPE, retinal, andlor choroidal cells.
RNA Isolation: Total RNA was isolated from adult human liver, RPE/choroid,
retina, and enriched RPE as
described by Chirgwin et. al. (1979), except that cesium trifluoroacetate was
used instead of cesium
chloride in the density gradient ultracentrifugation step. The resulting
pellet was stored at -80oC. The
108
CA 02363503 2001-08-24
WO 00/52479 PCT/US00/05858
quality/integrity of RNA obtained was assessed on both agarose gels and
Northem blots. Total protein
was determined from identically sized punches of the ocular tissue(s) from
which the RNA was collected
and employed as an intemal reference.
RT-PCR Analyses: Total RNA was extracted from the specified tissues and cDNA
was synthesized with
reverse transcriptase using oligo(dT)16 as a primer. Reverse transcriptase was
omitted from some
reactions. cDNA was amplified using molecule-specific primer pairs. PCR
amplification products were
separated electrophore6cally on a 1.8% agarose gel.
Results: Transcripts encoding a number of DRAMs common to extracellular
deposits associated with
atherosclerosis, elastosis, amyloidosis, or dense deposit disease were found
to be synthesized by the
retina, retinal pigmented epithelium and/or choroid (see Table 4 below).
Table 4: RT-PCR results from retina, RPE/choroid, and liver.
Gene Name Primer Sequence Ret R/Ch RPE Gen Liver
Albumin SN 5' GTCGAGATGCACACAAGAGTG 3' + + + - +
AS 5' TCCTTCAGTTTACTGGAGATCG 3'
Amyloid P SN 5' GCCAGGAATATGAACAAGCCG 3' - - - -* +
AS 5' CAAATCCCCAATCTCTCCCAC3'
Apo B SN 5' TGAACACCAACTTCTTCCACG 3' + + - - +
AS 5' GGCGACCTCAGTAATTTTCTTG 3'
Apo E SN 5' GGTCGCTTITGGGATTACC3' + + + - +
AS 5' CTCCAGTTCCGATTTGTAGGC 3'
Complement SN 5' GTTCAAGTCAGAAAAGGGGC 3' + + + - +
3 AS 5' GTGTCTTGGTGAAGTGGATCTG 3'
Complement SN 5' ATGGTATGTGGACGATCAAGGC 3' + + + - +
AS 5' TATTGCTCGGTAACCTI'CCCTG 3'
Complement SN 5' AATGAGCCCCTGGAGTGAATG 3' + + - - +
9 AS 5' ATGTCAGAGTGTTTCCATCCCG 3'
Factor X SN 5' GAGCGAGTTCTACATCCTAACG 3' + + - - +
AS 5' CACGAAGTAGGTGTCCTTGAAG 3'
Fibrinogen SN 5' AGACTGGAACTACAAATGCCC 3' - + - - +
AS 5' AGATTCAGAGTGCCATTGTCC 3'
Ig kappa SN 5' ACGTTTGATITCCASYTTGGTCCC 3' - + - - +
AS 5' GAMATYSWGIATGACICAGTCTCC 3'
Ig lambda SN 5' ACCTARACGGTSASCTKGGTCCC 3' + + - - +
AS 5' TCY'I'MTGWGCTGACTCAGSMCC 3'
Prothrombin SN 5' GGGCTGGATGAGGACTCAG 3' - - - - +
AS 5' AAGGCAACAGGCTTCTTCAG 3'
Ret = retina; R/Ch = RPE/choroid; Gen = amplification of genomic DNA by the
primer pair;
higher molecular weight genomic band detected with primer pair.
Example 8: Local Sources of Additional DRAMs and Other Choroidal Fibrosis-
Associated
Molecules
109
CA 02363503 2001-08-24
WO 00/52479 PCTIUSOO/05858
Extensive studies have been conducted to determine whether AMD-, choroidal
fibrosis-, and/or
drusen-associated molecules are synthesized by local ocular sources. RT-PCR
was performed as
descnbed previously.
RNA Isolation: Total RNA was isolated from adult human liver, RPE/choroid,
enriched RPE, retina, fetal
human eye, various fetal human organs, and primary cultures of human RPE cells
using the RNeasy
system (Qiagen; Valencia, CA). Liver and peripheral blood leukocyte RNA, as
well as genomic DNA,
served as positive and negative controls, respectively. The resulting pellets
were stored at -80oC. The
quality/integrity of RNA obtained was assessed on both agarose gels and
Northem blots. Total protein
was determined from identically sized punches of the ocular tissue(s) from
which the RNA was collected
and employed as an intemal reference.
RT-PCR Analyses: Primers to nucleotide sequences were employed to amplify cDNA
molecules from
these tissue sources. Reaction mix without template and/or ommission of
reverse transcriptase during
the RT reaction were used as negative controls. PCR amplification products
were separated by agarose
gel electrophoresis and stained with ethidium bromide for visualization. cDNA
was synthesized with
reverse transcriptase using oligo(dT)16 as a primer. Reverse transcriptase was
omitted from some
reactions. cDNA was amplified using molecule-specific primer pairs. PCR
amplification products were
separated electrophoretically on a 1.8% agarose gel.
Results: The expression of various AMD-, choroidal fibrosis-, and/or drusen-
associated molecules by
local ocular sources is shown in Table 1 above.
Example 9: Identification of Fibrils in the Choroids of a Subset of Human
Donors
In a series of 91 donors from the Human Donor Repository and 160 donors from
the CDD, we
noted the appearance of a network of vitronectin-positive "fibriis" in the
choroidal stroma of donors with a
history of drusen and AMD. Statistically, there was a significant correlation
between drusen grade and the
presence of these fibrils.
Ultrastructurally, fibrils exhibiting the hallmark features of newly
synthesized elastin and coliagen fibriis
are observed in the choroids of many of these same donors, as well as in
additional donors. Although most
elastin synthesis occurs during gestation, elastin gene expression can be
reactivated postnatally during
wound healing, inflammation, and other pathological processes modulated by
TGF, ILGF-1, and/or hypoxia.
These studies document the association of choroidal fibrils with drusen, aging
and/or AMD and suggest
that choroidal elastin and/or collagen gene expression is reactivated de novo
in individuals with these
conditions. Knowledge that this process is occurring in AMD should provide
information about biological
processes involved in the development of AMD.
110
CA 02363503 2001-08-24
WO 00/52479 PCT/US00/05858
Example 10: Identification and Morphological Characterization of
"Choroidal Fibrosis"
Human donor eyes - with and without clinically-documented AMD and/or arterial
wall disruptive
disorders (AAA, TAA, aortic stenosis, and atheroscleosis) and with distinct
drusen morphologies - were
employed for simultaneous transmission electron microscopical and
immunohistochemical observation.
Eyes used in this study were selected from a repository of over 2,000 pairs of
human donor eyes
(between 0 and 102 years of age) obtained from MidAmerica Transplant Services
(St. Louis, MO), the
Iowa Lions Eye Bank (Iowa City, IA), the Heartland Eye Bank (Columbia, MO) and
the Virginia Eye Bank
(Norfolk, VA) and were processed within four hours of death. The gross
pathologic features of all eyes,
as well as the corresponding ophthalmic histories, fundus photographs and
angiograms, when available,
were read by a retina surgeon. Approximately 18% of the donors had some form
of clinically diagnosed
AMD; these included eyes with macular pigment changes, macular drusen,
geographic atrophy, choroidal
neovascularization, and/or discifomi scars. Eyes with and without clinically
documented AMD, were
employed in this study.
The RPE-choroid-sciera complex from 151 of these donors were processed for
transmission electron
microscopical examination. Tissues were fixed in one-half strength Kamovsky's
fixative within four hours
of death for a minimum of 24 hours, and transferred to 100mM sodium cacodylate
buffer, pH 7.4, prior to
dehydration, embedding, sectioning, and photomicrography.
Tissues from the same eyes processed for electron microscopy were processed
for light histological
(Elastachrome stain; H&E) and immunohistochemical studies. Anti-vitronectin
antibody was obtained
from Telios (San Diego, CA); collagens I, III, V, and VI from Chemicon and
Southern Biotech; elastin from
Elastin Products; fibriilin-1 from Chemicon; and fibulins 3 and 4 from Rupert
Timpl. Selected specimens
of human donor RPE-choroid were fixed by immersion in 4% (para)formaldehyde in
0.1 M sodium
cacodylate buffer and processed for laser scanning confocal microscopy. Images
were captured and
displayed using a BioRad 1024 laser scanning confocal microscope equipped with
a Nikon inverted
microscope.
The choroidal stromas of at least 30 of these individuals are filled with
newly synthesized collagen,
elastin, elastin-associated microfilaments, and other distinct structural
proteins and fibrils as viewed by
electron microscopy. Based on preliminary immunohistochemical analyses, the
collagen associated with
this condition appears to be largely type III and VI and typically exhibits
a"spiraled", or "frayed"
morphology that is often associated with specific hereditary and acquired
diseases. This previously
undescribed phenomenon, referred to as "choroidal fibrosis", shares many
pathological features that are
common in arterial wall disruptive disorders.
111
CA 02363503 2001-08-24
WO 00/52479 PCTIUSOO/05858
Example 11: Gene Expression of Fibrotic Molecules in the RPEJChoroid Complex
of Control and
AMD Donors
Total RNA was isolated from adult human liver and the RPE/choroid complexes
from five control
human donors (aged 18 to 58 years), one AMD/AAA donor, one AMD/aortic stenosis
donor, and one
AMD donor with a family history of AMD. The resulting pellets was stored at -
80oC. The qualitylintegrity
of RNA obtained was assessed on both agarose gels and Northem blots. cDNA was
synthesized with
reverse transcriptase using oligo(dT)16 as a primer. The enzyme was omitted
from control reactions.
RT-PCR analyses of RPE-choroid complexes derived from this series of control
(non-diseased) and
affected (AMD/AAA, AMD, AMD/aortic stenosis; all with drusen) donors with
distinct choroidal fibrosis
reveal distinct pattems of up- and down-regulated gene expression between the
two groups (see Table X
below). These include "upregulation" of b1 integrin, elastin, collagen VIa2,
collagen a3, PI-1 (antitrypsin),
PI-2, human metalloelastase (and perhaps fibrillin-2) and "downregulation" of
BigH3. No detectable
differences in expression levels of collagen IIla1, coliagen la2, collagen
6a1, fibulins-1, 2, 3, 4, and 5,
HLA-DR, Ig kappa, laminin receptor, or laminin C2 were observed. Because of
the limitations of RT-PCR,
additional real time quantitative RT-PCR studies are being conducted to assess
the precise levels of
these genes in the two groups.
Table 5: Gene Expression in Donors with Choroidal Fibrosis
Molecule Expression in Choroidal Fibrosis vs
Controls
BIG H3 Decreased
b 1-inte 'n Increased
Collagen 3 al Unchanged
Collagen lal Unchanged
Collagen la2 Unchanged
Collagen 6 al Unchanged
Collagen 6 a2 Increased
Collagen 6 0 Increased
Elastin Increased
Emilin
Fibulin-1 Unchanged
Fibulin-2 Unchanged
Fibulin-3 Unchanged
Fibulin-4 Unchanged
Fibulin-5 Unchanged
Fibrillin-1 ?
Fibrillin-2 ?
Ficolin ?
112
CA 02363503 2001-08-24
WO 00/52479 PCT/US00/05858
HLA-DR b Unchanged
HME Increased
IgK Unchanged
Laminin Receptor Unchanged
Laminin C 1
Laminin C2 Unchanged
Laminin C3 ?
L02 Unchanged
L04 Unchanged
LTBP-1 ?
LTBP-3 ?
LTBP-4 Decreased
MFAP-1 Decreased
MFAP-2 Decreased
MFAP-3 Unchanged
MFAP-4 Unchanged
MMP-2 Unchanged
MMP-7 ?
MMP-9 ?
MMP-12 Unchanged
PI-1 Decreased
PI-2 Decreased
PI-3 ?
PLOD2 Unchanged
PM5 Unchanged
RPE-65 Unchanged
TIMP-1 Unchanged
TIlVIP-2 Unchanged
TIlVIP-3 Unchanged
Vitronectin Increased?
Example 12: Choroidal Fibrosis - Characterization of Metalloproteinases/TIMPs
in
Aging and AMD Donors with Choroidal FibrosislExtracellular Matrix
Disequilibrium
Extracellular matrix tumover is initiated, at least in part, by the regulated
secretion of members of a
family of matrix metalloproteinases (MMPs) and their inhibitors, the tissue
inhibitors of metalloproteinases
(TIMPs). Leukocytes, including dendritic cells and macrophages, are major
sources of MMP production.
MMP action permits leukocyte immigration into tissues, causes tissue damage,
and generates
immunogenic fragments of normal proteins that may escalate autoimmune diseases
[Opdenakker, 1992
#681]. The MMP family of enzymes contributes to both normal and pathological
tissue remodeling.
Although the link between single MMPs and individual substrates is not as
direct as once thought, it is clear
that the MMPs are capable of breaking down most ECM components. Most MMPs,
with the exception of
the 72kDa gelatinase and the MT-MMPs, are not constituitively expressed in
normal tissues. Inflammatory
113
CA 02363503 2001-08-24
WO 00/52479 PCT/US00/05858
cytokines (IL-1 and TNF) and growth factors (TGFb) are typically required to
initiate transcription. MMPs
are expressed as inactive zymogens, which are activated extracellularly by the
action of enzymes such as
plasmin and other MMPs. Once activated, MMPs are subject to inactivation by
TIMPs and by binding to
plasma proteins such as a2-macroglobulin. This balance of expression and
activation, and the levels of
TIMPs, govem the level of destruction mediated by MMPs. Excessive or
inappropriate expression of
MMPs may contribute to the pathogenesis of many tissue destructive processes,
including diseases such
as arthritis, multiple sclerosis, atherosclerosis, and COPD.
In order to assess the notion that an imbalance of the
metailoproteinase/inhibitor system in AMD may
lead to degradation of Bruch's membrane in the macula, RPE/choroidal tissues
from 20 donors with and
without AMD were examined using zymography. Four proteases with approximate
molecular weights of 65
(MMP-2), 95 (MMP-9), 120, and 250 kDa were present in macular and peripheral
tissues. No differences in
the pattem of MMP bands were detected as a function of age or drusen
phenotype. Aprotinin and
leupeptin had no effect on proteolytic degradation of gelatin, whereas EDTA (5
mM) completely inhibited
enzyme activity in these bands, indicating that all four bands are likely
metalloproteinases. These activities
were also resistant to boiling, but extremely sensitive to reducing agents.
In order to establish the relationship of lysed bands on zymography with known
metalloproteases,
antisera to a number of known MMPs, as well as to all known TIMPs, were
employed to screen Westem
blots of RPE/choroid proteins. MMP-1, -2, and -9, but not MMP-3 or -8, were
identified in RPE/choroid
extracts and did not show changes with respect to drusen status, AMD, and/or
age. TIMP-4 antibody
bound to a band of 28 kDa in all samples, including a 2-month-old donor. The
higher molecular weight
bands may be due to smaller MMPs that have polymerized (99) or may represent
novel proteases, such as
the 300 kDa elastase identified in lung by broncheoalveolar lavage (100).
Antibodies directed against
TIMP-3 reacted with hard and soft drusen, whereas anti-TIMP-4 antibodies
reacted with drusen cores.
The development of a comprehensive picture of MMP involvement will require the
use of several
methods. Ongoing studies are being focused on furhter characterization of MMPs
2, 7, 9, 12, and 14,
members of the MMP family that exhibit elastolytic properties, using
immunohistochemistry, zymography,
ELISA, and QRT-PCR.
Example 13: Autoantibodies in the Sera of Donors with AMD and /or Drusen
In order to address the role of autoantibodies in drusen biogenesis and AMD,
we performed a series
of preliminary experiments using enriched drusen preparations in order to
identify anti-drusen/Bruch's
membranelRPE autoantibodies that might be present in the sera of donors with
AMD and/or drusen.
Protein extracts from an enriched drusen preparation (DR+) obtained by
debridement of Bruch's
membrane with a #69 Beaver blade and from a control (DR-) preparation were
prepared using PBS with
proteinase inhibitor cocktail and mild detergent. Proteins were separated by
molecular weight using 10-
20% gradient mini SDS gels (Amresco) and transferred to PVDF membranes for
Westem blot analysis.
PVDF strips with human retinal proteins from 50 normal human retinas were also
used for detection of
any anti-retinal autoantibodies in the donor sera.
114
CA 02363503 2001-08-24
WO 00/52479 PCT/US00/05858
Sera from the same eight donors described above were screened. Serum from one
AMD donor
(#90-98) positively labeled a band in the RPE (both DR+ and DR-) and
RPElchoroid preparations of
approximately 35kDa. A second band of approximately 60kDa was labeled weakly
only in the DR+
protein extract. Sera from an AAA donor (#189-97) reacted with a protein(s) of
approximately 53kDa.
This band labeled in all three protein extracts. There was one band of
approximately 64kDa that this
serum sample labeled only in the DR+ sample.
The presence of serum anti-drusen/RPE autoantibodies in donors with AMD and/or
drusen further
suggests a possible role for shared immune-mediated processes in these
conditions.
Example 14: Analyses of Autoantibodies in the Sera of Living AMD Patients
In order to determine whether the sera of AMD patients possesses
autoantibodies or alterations in
the abundance and/or mobility of serum proteins, plasma was collected from 20
patients with clinically-
diagnosed AMD and from 20 unaffected patients to serve as controls.
For some experiments, sera were separated by SDS-PAGE and proteins were
visualized with
either silver stain or Coomassie blue, or (for preparative purposes) proteins
were transferred to PVDF
membranes for amino acid sequencing. Abnormalities of serum proteins were
detected in a subset of AMD
donors. These differences included the presence of "additional" bands in the
sera of some AMD patients
(molecular weights of -25, 29, 30 and 8OkDa) that were not present in control
donors. Amino acid
sequencing of these molecules revealed N-terminal sequences consistent with
haptoglobin (25kDa) and
immunoglobulin kappa (29kDa), lambda (30kDa), and gamma (80kDa) chains.
In a second set of experiments, sera from AMD and control donors was screened
for the presence
of auto-antibodies. As an extension of experiments in which weak-moderate
immunoreactivity of drusen in
tissue sections was previously observed, purified vitronectin was
electrophoretically separated and blotted
onto PDVF. Because vitronectin had previously been identified as a DRAM (as
detailed in Example XX),
the sera from AMD patients was then evaluated for the presence of anti-
vitronectin immunoreactivity.
Strong labeling of both the 65kDa and 75kDa vitronectin species was identified
in these sera, indicating that
AMD sera contain autoantibodies directed against at least some DRAMs and/or
Bruch's membrane
constituents.
As an additional. approach toward the identification of AMD autoantibodies and
their targets in
ocular tissues, RPE-choroidal proteins from one donor with large numbers of
drusen and a nine month old
donor were separated electrophoretically according to molecular weight and
transferred to nitrocellulose.
Proteins were then immunolabeled with either sera from 3 AMD donors or
polyclonal antiserum directed
against vitronectin. The AMD sera reacted with bands of roughly 65, 150 and
200kDa only in the sample
from the donor with numerous drusen. These results are suggestive that age
and/or the presence of
drusen leads to an increase in AMD autoantigen.
Example 15: Additional Assessment of Additional Serum Markers in Drusen
Biogenesis,
Choroidal Fibrosis, and AMD
115
CA 02363503 2001-08-24
WO 00/52479 PCTIUSOO/05858
Study Design: Visual acuity measurements, stereo macula photos, and peripheral
photos will be
taken at the beginning of the study and every six months thereafter. Blood and
sera will be drawn when
subjects enter the study and every 6-12 months thereafter. DNA will be
prepared from a portion of each
blood sample for future genetic studies. The presence of serum autoantibodies
and immune complexes
will be determined using standard protocols. In addition, sera will be reacted
with tissue sections derived
from donors with and without AMD, followed by a secondary antibody that has
been adsorbed against
human immunoglobulins. Westem blots of retina/RPE/choroid from AMD and non-AMD
donors will also
be incubated with serum samples to identify specific bands against which
autoantibodies react.
In addition, levels of the following proteins, additional indicators of
autoantibody responses, chronic
inflammation and/or acute phase responses, will be assayed by a clinical
diagnostic laboratory. These
will include Bence Jones protein, serum amyloid A, M components, C-reactive
protein, mannan binding
protein, serum amyloid A, C3a, C5a, other complement proteins, coagulation
proteins, fibrinogen,
vitronectin, CD25, interleukin 1, interieukin 6, and apolipoprotein E. Serum
protein electrophoresis,
lymphocyte transformation, sedimentation rate, and spontaneous, whole blood,
white cell count will also
be measured.
The presence of antibodies directed against the following proteins (many
observed in other age-
related conditions and/or MPGN) will also be determined: type IV collagen,
glomerular basement
membrane, neutrophils, cytoplasm (c-ANCA, p-ANCA), C3 convertase (C3 nephritic
factor), alpha-1 anti-
trypsin levels (decreased in MPGN), epsilon 4 allele, apolipoprotien E, GFAP,
ANA, serum senescent cell
antigen, S-100, type 2 plasminogen activator, alpha-l-antichymotrypsin, SP-
40,40, endothelial cell,
parietal cell, mitochondria, Jo-1, islet cell, inner ear antigen,
epidermolysis Bullosa Acquista, endomysial
IgA, cancer antigen 15-3, phospholipid, neuronal nucleus, cardiolipin, and
ganglioside.
Other markers that could be present in the serum of patients having a drusen
associated ocular
disorder are listed in the following Table.
Table 6: Serological Tests for Immune-Mediated Processes, Including Autoimmune
Disease and Chronic
Inflammation
Cells:
Whole blood cell count, hemogram plus differential
CBC, hemogram.
Immunoalobulins:
lmunoglobulin A,G,M,D,E quatification
116
CA 02363503 2001-08-24
WO 00/52479 PCTIUSOO/05858
IgG subclass quantification
Kappa/lambda light chains- quantification and ratios
Miscellaneous Proteins:
Serum protein electrophoresis
Complement, total classical and altemative
Compement: C3, C4, C5 quantitative
Bence Jones proteins
M component
C reactive protein
Serum amyloid A
Coagulation proteins
Fibrinogen (and/or ESR)
Elastase inhibitors
Elastin and collagen peptide fragments
Serum beta-2-microglobulin
Serum carotine
Creatine kinase
Rheumatoid factor
C-reactive protein
Immunocompetent Cells:
Lymphocyte immunophenotyping and absolute CD4 cell count.
Anti-0KT3, IgG antibodies.
117
CA 02363503 2001-08-24
WO 00/52479 PCT/US00/05858
CD34 Stem cell count.
CD3 cell count.
CD4 cell count.
Lymphocyte mitogen and antigen profile screen (LPA).
Lymphocyte antibody screen???
NK cells.
T and B-cell markers. (which ones they screen?).
CD4ICD8 - absolute count and ratio.
HLA phenotyping, both class I and II. HLAB-27.
Cytokines:
Interleukins
Fibroblast growth factor
Vasoactive intestinal peptide (VIP)
Autoantibodies:
Anti-nuclear antibody (ANA)
Anti-neutrophil cytoplasmic antibody (ANCA)
Double stranded DNA antibody
Anti-ribonuclear protein antibody
Scl-70 antibody
SM antibody
SS-A antibody (anti-RO) and SS-B (anti-LA) antibody
Anti-neuronal nuclear antibodies
118
CA 02363503 2001-08-24
WO 00/52479 PCT/US00/05858
Antineuronal nuclear antibody (Purkinje cells).
Jo-1 antibody
Paraneoplasctic antibody A
Anti-cardiolipin antibody
Anti-glomerular basement membrane antibodies
Mitochondrial antibody
Anti-ganglioside assay
Anti-Streptolysin-0 screen
Anti-sulfatide antibody
Anti-Thyrocellular antibody
Antibody to inner ear antigen
Bullos pemphigoid antibodies
PM-1 antibody
Adrenal cortical antibody.
Liver-kidney microsomal antibody
Mitochondrial antibody
Parathyroid antibody
Parietal cell antibody
Pemphigus antibodies
Smooth muscle antibodies and striated muscle antibodies.
Islet cell antibodies
Lupus anticoagulant
119
CA 02363503 2001-08-24
WO 00/52479 PCT/US00/05858
Anti-Viral and Anti-Bacterial Antibodies:
CMV antibody
Group B strep antigen
Hepatitis B, E, C, A antibodies
Helicobacter Pylori antibodies
Antibodies to CMV, EB virus, Herpes Simplex, Measles, mycoplasma, Rubella,
Varicelia-Zoster
Others:
Cancer antigen 125
Cancer antigen 15-3
Carcinoembrionic antigen
Small fiber axonal profile
CNS serology battery
Sensorimotor neuropathy profile
Example 16: Differential Gene Expression Analyses in the RPE/Choroid Compiex
of Donors with
AMD and Choroidal Fibrosis: Toward the Development of a Diagnostic "Gene
Expression
Fingerprint" for Drusen Biogenesis, AMD, and/or Choroidal Fibrosis
One prevailing concept pertaining to the etiology of AMD is that a threshold
event occurs at some
point during the aging process that distinguishes AMD from normal aging.
Provided that AMD is heritable
in the majority of affected individuals, then the gene(s) responsible likely
initiate this threshold event. Our
working hypothesis suggests that cellular dysfunction within the RPE-choroid-
retina complex is involved in
the earliest stages of AMD, since most of the initial clinical and
histopathological signs (e.g. RPE cell death,
Bruch's membrane degradation, and choroidal fibrosis) are associated with the
RPE, Bruch's membrane,
and the choroid. However, little is known about the pattems of gene expression
in normal RPE and
choroidal cells and nothing is known about gene expression in RPE, choroidal,
or retinal cells from
individuals with AMD, drusen formation, and/or choroidal fibrosis. This is
especially surprising in view of
the strategic location of the RPE, the fact that its health appears crucial
for the maintenance of the retina-
choroid interface, and its apparent involvement in AMD. Because of our access
to a large repository of
carefully documented human donor eyes, we are in a unique position to
determine unique pattems of RPE,
120
CA 02363503 2001-08-24
WO 00/52479 PCTIUSOO/05858
choroidal, and retinal cell gene expression (AMD and drusen "gene expression
fingerprints") in defined
AMD phenotypes that are distinct from those of age-matched and younger donors
without AMD.
Differential gene expression of RPE/choroid complexes derived from four paired
donors of selected
AMD and AAA phenotypes and age-matched controls has been analyzed using gene
array analysis. The
arrays utilized in this study contained 18,380 non-redundant cDNAs derived
from the I.M.A.G.E.
consortium. Each cDNA clone was robotically spotted, in duplicate, onto a
nylon membrane in a precise
pattem, allowing easy identification. These analyses are typically performed
using first strand cDNA
which has been radiolabeled during reverse transcription of the probe mRNA.
However, due to the small
amounts of mRNA that can be isolated from the RPE layer of individual human
donor eyes, we have
modified this standard protocol. The cDNAs were radiolabeled with 33-P in a
random-primed reaction,
purified, and hybridized to the gene arrays. The arrays were phosphoimaged,
the signals were
normalized, and the data analyzed using the Genome Discovery Software package
(Genome Systems).
Analysis of the data reveals distinct pattems of clones that are significantly
up- and/or down-
regulated in the RPE/choroid of individuals with specific AMD and AMD/AAA
phenotypes as compared to
controls. At this point, these differentially-expressed mRNAs can be grouped
into three distinct
"pathways": extracellular matrix-, membrane transport-, and gene regulation-
associated pathways. In
addition, a significant number of uncharacterized expressed sequence tags
(ESTs) are differentially
expressed in the RPE-choroid of donors with specific AMD and AAA phenotypes as
compared to the
RPE from donors without the disease.
It is anticipated that large scale analyses of gene and protein expression
profiles ("fingerprints") in
tissues from donors with drusen deposits, as well as those from various "AMD
phenotypes", will provide
significant new insight into the molecular pathology of cell dysfunction
associated with the development of
AMD. These "gene expression fingerprints" will also provide powerful
diagnostic capabilities for detecting
individuals at risk for developing drusen and/or macular degenerations,
including AMD.
Example 17: Choroidal Fibrosis - Analyses of Elastin and Collagen Gene
Expression
Our observation of the de novo synthesis of elastin and coliagen fibriis in
the choroidal stroma in
donors with AMD suggests that elastin gene expression may be upregulated in
these eyes. Reactivation of
elastin gene expression typically occurs only in physiological wound healing,
where it is associated with
inflammation and tissue repair, hypoxia, or in various diseases.
Because elastin gene expression is not typically detected after birth, studies
were initiated to assess
whether elastin and various collagen genes are expressed in the choroid and/or
RPE using RT-PCR from
an array of CDD donors with and without choroidal fibrosis. We have employed
un-nested and nested
primer pair combinations to detect the presence or absence of elastin mRNA. 5'-
AGGGGTTGTGTCACCAGAAG-3' (exon 17) and 5'-AGACAATCCGAAGCCAGGTC-3' (exon 2) has
been
used as the outer pair, and 5'-GAGTTGGAGGCATTCCTAC-3' (exon 16) and 5'-
121
CA 02363503 2001-08-24
WO 00/52479 PCTIUSOO/05858
CCATATTTGGCTGCTTTAGC-3' (exon 9) as the inner pair. Significantly, elastin
mRNA upregulation has
been detected in AMD donor eyes that possess choroidal fibriis and exhibit
choroidal fibrosis.
Studies were performed to screen for abnormal exons in the elastin gene in
three donors with
choroidal fibrosis and three unaffected age-matched control donors using
primers for exons 1-34 of the
elastin gene. Genomic DNA was isolated from pelleted white blood cells that
had been subject to a
hypotonic lysis solution. Subsequent PCR analyses of elastin exons adhered to
the following reaction
conditions: IOU Taq DNA Polymerase from Promega, 1 X Taq Polymerase buffer
containing MgCI2 at a
final concentration of 2.5mM, 25 ng of both forward and reverse primers, dNTP
mixture at a final
concentration of 2.5mM,10ng of genomic template DNA. Sterile, deionized water
was added to give a final
reaction volume of 2541. Cycling was performed for 35 cycles in a Hybaid
PCRExpress thermal cycler
using the following parameters: Initial denaturing soak for 3 minutes at 94
degrees, denature at 94 degrees
for 30 seconds, anneal at each primer's respective TM for 45 seconds, extend
at 72 degrees for 45
seconds, final extension soak at 72 degrees for 3 minutes. Genomic primers for
exons 1-34 of elastin were
made according to the primer sequences given by (Reference to Tassabehji, et
al.).
Additional studies were initiated to screen for abnormal exons in the elastin
gene in 96 control
donors and 96 age-matched AMD donors. Primers for exons 2, 19, 23, 24, 26, and
33 have been
screened. These exons were selected because of their respective susceptibility
to altemative splicing. To
date, no abnormal PCR products have been identified that would suggest gene
mutations in any of the
exons screened in donors with AMD and/or choroidal fibrosis. Additional exons
are being screened in a
similar fashion.
Disequilibrium analyses of intron 18 of elastin have been performed on 96
control donors and 96
age-matched AMD donors. Primers for intron 18 were designed as follows (intron
18F: 5'-
ATGAGACGTGGTCAAGGGTAT-3'; intron 18R: 5'-GGGATCCCAGGTGCTGCGGTT-3'). The
annealing
temperature for this primer pair is 60 degrees. Additional exons will be
examined in a similar fashion.
Based on the observation of spiraled collagen in many, if not all, of the
choroidal fibrosis donors,
the above examples can be modified to include various other genes that are
identified on the basis of
pathology. These would include, but not be limited to, collagen genes
(especially collagen types I, III, and
VI).
Example 18: Choroidal Fibrosis - Bruch's Membrane Elastin Distribution
In diseases such as emphysema, atherosclerosis and arthritis,
metalloproteinases are secreted at sites
of inflammation and fibrosis where they can cause elastin destruction. These
studies were conducted in
order to determine whether choroidal fibrosis and/or the presence of drusen-
associated dendritic cells leads
to events that result in degradation of Bruch's membrane, and ultimately
choroidal neovascularization.
The reactivity of rabbit polyclonal anti-aortic elastin antibodies with the
elastic layer of Bruch's
membrane were analyzed in a small series of young (<5 years), middle-aged (20-
40 years), and AMD (>50
years) donors. The sixty-three human donor eyes employed in this study were
obtained from The University
of Iowa Lions Eye Bank (Iowa City, IA) within four hours of death.
Institutional Review Board committee
approval for the use of human donor tissues was obtained from the Human
Subjects Committee at The
122
CA 02363503 2001-08-24
WO 00/52479 PCT/US00/05858
University of Iowa. Posterior poles, or wedges of posterior poles spanning
between the ora serrata and
macula, were fixed in 4% (para)formaidehyde in 100mM sodium cacodylate, pH
7.4. After 2-4 hours of
fixation, eyes were transferred to 100mM sodium cacodylate and were rinsed
(3x10 min), infiltrated, and
embedded in acrylamide. These tissues were subsequently embedded in OCT, snap
frozen in liquid
nitrogen, and stored at -80oC. Unfixed posterior poles, or wedges thereof,
were embedded directly in OCT,
without acrylamide infiltration or embedment. Both fixed and unfixed tissues
were sectioned to a thickness
of 6-8?m on a cryostat. The presence and type(s) of drusen were documented on
adjacent sections
stained with hematoxylin/eosin, periodic acid Schiff reagent, and Sudan Black
B (1% in 70% ethanol).
Immunolabeling was performed and adjacent sections were incubated with
secondary antibody alone, to
serve as negative controls. Some immunolabeled specimens were viewed by
confocal laser scanning
microscopy .
The elastic layer in the macula differed significantly from that in
extramacular regions in all three
groups. Immunoreactive elastin was thin and highly fragmented in the macula of
AMD donors, as
compared to the peripheral region where it was contiguous and thick.
Immunoreactive elastin was absent
in the maculas of the two young donors examined. We suggest that these
observations provide a
significant clue as to why the macula may be particularly susceptible to
degeneration in individuals afflicted
with drusen deposition and AMD. The fact that the elastic lamina is absent in
the maculas of eyes from two
donors with Sorsby's fundus dystrophy (caused by a mutation in the TIMP-3
gene), further support this
concept (Hageman, unpublished).
Example 19: RPE Cell Death as Related to Drusen Biogenesis
RPE Cell Density as a Function of Age: RPE cells are generally considered
terminally-differentiated. Thus,
there is no mechanism for the replacement of lost cells in vivo. Although the
net density of RPE cells
appears to decrease in human eyes as a function of age, the rate of loss has
not rigorously examined.
Based on data from preliminary studies, we propose to determine whether this
loss is linear with age,
varies in peripheral and macular regions, is greater in eyes from donors with
AMD, especially early AMD,
and, if so, whether this loss is associated with drusen phenotype.
In order to test the feasibility of determining the relationship between RPE
cell density, age and drusen
status, we counted RPE nuclei on DAPI-stained sagittal sections (DAPI is a
probe that binds specifically to
DNA), spanning from the.ora serrata to the macula of each quadrant, in a
series of 20 CDD donors with
and without drusen/AMD. The number of RPE nuclei, basal drusen length and
drusen area were
determined per mm of Bruch's membrane. Sections were also photographed at
500nm excitation to
assess qualitatively the degree of autoflourescence due to RPE lipofuscin.
These data suggest that both RPE cell density and thickness (volume) is
reduced in eyes with
numerous drusen/AMD, supporting our hypothesis that RPE may contribute to
drusen biogenesis. Four
additional phenomena have been observed. First, a significant decrease in RPE
density is noted in the
peripheral and equatorial retina in some donors between the ages of 10 and 40.
Second, although many
RPE cells appear to be extruded toward the subRPE space where they contribute
to drusen formation,
others are extruded into the subretinal space, often as groups of cells, and
are "phagocytosed" by the
neural retina. Third, focal groups of RPE cells that express HLA-DR were
identified. Fourth, morphological
images are suggestive that the neural retina underlying drusen, even small
drusen, exhibits thinning and
local reduction in nuclear number.
123
CA 02363503 2001-08-24
WO 00/52479 PCTIUSOO/05858
.
,
ar.....r...a.a
Table 7: Average RPE cell densities (5 donors per category) in the temporal
retina from the macula to the
ora serrata. Categories depicted are AMD (black line), age-matched controls
(blue line), and young (blue
line). RPE cell nuclei were counted in two sections for each donor.
RPE Contribution to Drusen Development: The distribution of RPE-associated
lipofuscin and/or pigment
granules, in addition to nuclei, is easily detected in DAPI-stained sections.
Examination of DAPI-stained
preparations revealed the presence of autofluorescent lipofuscin and pigment
within small drusen, as well
as dispersed profiles of DAPI-reactive material, interpreted to be RPE nuclei.
When tissues from the same
donors are examined ultrastructurally, profiles of lipofuscin and pigment were
detected in small drusen.
Sometimes these were contained within membrane bound fragments of RPE cells.
Nuclei or nuclear
material are observed within the druse less commonly. In a few eyes we have
observed the remnants of
whole RPE cells in the sub-RPE space. Significantly, drusen-associated
dendritic cell processes are often
observed in association with these profiles of RPE cells, suggesting that the
dendritic cells are initially
recruited to these sites of RPE damage.
Focal RPE "Iniury" and Drusen Development: We have observed focal groups of
RPE cells that express
HLA-DR. These cells are often associated with small drusen and exhibit unique
apical-basal polarization,
and apical displacement of the majority of cellular organelles. Additional
studies of these cells indicate that
that they react with antibodies directed against CD68, HLA-DR, vitronectin,
clusterin, and apolipoprotein E.
In diseases such as rheumatoid arthritis and giomerulonephritis,
immunoglobulin and complement
activation are associated with tissue injury and subsequent cell death.
Mode of RPE Cell Death: We have never observed an apoptotic RPE cell in
examination of over 35,000
electron micrographs. In addition, the morphological profiles of dead or dying
RPE cells that we observe
exhibit the hallmarks of necrotic, rather than apoptotic, cell death. Based on
the hypothesis that the typical
mode of RPE cell death occurs by necrosis, we initiated studies to identify
apoptotic RPE cells in sections
of 20 human donor eyes of various ages, with and without AMD, from the CDD.
All experiments were run
in duplicate or triplicate. Although TUNEL positive cells were observed
consistently in the RCS rat and
human controls, no apoptotic RPE cells were identified in human donor
sections, at any age or disease
state. These data support the hypothesis that the majority of RPE cell death
occurs via cellular necrosis,
rather than by apoptosis.
Example 20: Drusen Biogenesis in Immunosuppressed Patients
124
CA 02363503 2001-08-24
WO 00/52479 PCTIUSOO/05858
with AMD, and/or Drusen
Experimental Approach
Rationale: Based on the hypothesis that drusen formation is dependent on the
activity of choroidal
dendritic cells, we propose that drusen formation will be arrested (or that
the rate of drusen formation will
be reduced significantly) in the absence or down-regulation of these cells.
The overall goal of this subaim is
to determine the prevalence, biomicroscopic characteristics and clinical
course of preexisting drusen in
AMD patients whose immunocompetent cells have been significantly depleted.
Patient Population: Twenty AMD patient volunteers will be identified who have
undergone, or will
undergo, organ transplants. In some cases, immune compromised patients will be
identified for whom
ophthalmic medical histories, fundus photographs, and angiograms were
collected prior to the
immunosuppression. In other patients who will undergo transplantation, fundus
photographs and
angiograms will be collected before radiation treatment and/or
immunosuppression. A target study
population will be patients who are to undergo cardiac transplant, as these
patients with be likely to have a
high incidence of drusen but will have limited life expectancy. The control
population will be age-matched
patients with and without AMD.
Study Design: In all participants, visual acuity measurements, fundus
photographs, and fluorescein
angiography and a number of parameters will be measured at the time of
recruitment every six months and
compared to age-matched controls with and without AMD. Blood and sera will be
drawn when subjects
enter the study and every 6-12 months thereafter. DNA will be prepared from a
portion of each blood
sample for future genetic studies.
Fundus photographs will be graded by masked readers utilizing the intemational
Age Related Eye
Disease Study grading protocol. Measurements will include: total drusen area;
sizes of drusen; number of
macular drusen; percent of drusen possessing funduscopic "cores"; rate of
appearance of new drusen; and
rate of regression of extant drusen.
The fundus photographs from the same patient, taken before and for 3-5 years
after transplantation
(including the control patients), wiil be compared for the presence of drusen,
changes in the distribution and
number of drusen, and other drusen related pathology.
Example 21: In Vitro Model for Drusen Biogenesis, RPE-Dendritic Cell
Interactions,
and Gene Expression
Our hypothesis of drusen biogenesis predicts that the essential elements to
drusen formation-
dendritic cells and the RPE-may interact across Bruch's membrane in the aging
eye, via a set of
molecular signal molecules, to result in drusen deposition and growth. It is
anticipated that this interaction
may be reproduced in vitro, as both cell types are tractable to cell culture
methodologies. Due to the fact
that there is not currently a suitable animal model for drusen biogenesis or
AMD, the prospect of testing
potential therapeutics which interfere with this interaction in vitro is
particularly attractive. Molecules to be
tested for their ability to inhibit RPE-dendritic cell interaction may include
antibodies directed against cell
125
CA 02363503 2001-08-24
WO 00/52479 PCT/US00/05858
surface proteins, cytokines and chemokines; growth factors and growth factor
inhibitors; anti-inflammatory
agents; and inhibitors of matrix degradation by dendritic cells.
Expenmental Approach
Rationale: Based on the histochemical and ultrastructural observations that
indicate a role for
dendritic cells (DCs) in the formation of drusen, the interactions between RPE
cells and DCs will be
evaluated in vitro. These studies will aid our understanding of the putative
roles of DCs in drusen formation
by examining the effects of these ceils on stimulation or depression of the
expression of specific genes by
the RPE and, subsequently, the effects of DC contact and/or DC-secreted
molecules on RPE gene
expression. These studies will also likely reveal downstream (i.e., RPE)
molecular targets for therapeutic
intervention, particularly important in the event that generallsystemic
inhibition of DC activity is assessed to
be too global to be effective in the management or prevention of AMD.
Tissues-RPE Cell Cultures: Human donor eyes, obtained within 4 hours
postmortem, will be
employed in these studies. We have found that RPE cells can be successfully
cultured up to 12 hours after
death, making 4 hours a conservative interval for the isolation of RPE cells.
We will isolate RPE cells and
other tissues from donors with diagnosed AMD, as well as from age-matched
donors without AMD.
RPE cells will be isolated either by debridement of the choroid and collection
of RPE cells from
different defined regions or surgical removal of the sclera from the choroid
and removal of RPE cells by
incubation of the eyecup in dispase. Our laboratory has considerable
experience in the successful
application of both protocols for the isolation of RPE.
For debridement of the choroid, the eye will be quartered and photographed,
the neural retina will
be removed, and the RPE surface will be scraped gently with a Beaver #69,
round-tipped blade to debride
Bruch's membrane in areas with large numbers of drusen. Care will be taken not
to slice through the
elastic lamina, by holding the blade at a slight angle and scraping
perpendicular to the axis of the blade.
The debrided material will be collected on the surface of the blade, and then
rinsed off the blade with
Coon's F-12 culture medium supplemented with either fetal calf serum (10%) or
human serum. For
isolation of RPE cells from whole eyecups following dissection of the sciera,
we will employ the protocol
described by Pfeffer (78), except that Coon's F-12 medium will be used for all
experiments.
Tissues-Dendritic Cell Cultures: DCs will be isolated using standard
techniques. Briefly, CD14
positive cells from the buffy coat fraction of peripheral blood will be
isolated, either from eye donors or from
clinic patient volunteers. In some experiments, stimulation of these cells
with the appropriate cytokines
(GM-CSF and IL-4) will be performed prior to co-incubation with RPE cells in
order to pre-differentiate these
cells into an activated dendritic cell phenotype. Unactivated DCs will be
employed in other experiments.
We will also attempt to design a protocol to isolate DCs directly from the
choroid.
RPE and Dedritic Cell Co-Culture: RPE cells derived from at least five AMD and
five non-AMD
donors will be examined for their ability to recruit, or elicit migration of,
activated and unactivated DCs.
These experiments will be conducted using modified Ussing chambers. Changes in
the expression of
various DC markers indicative of activation, including IL-12, MHC class II
antigens, CD83, and CD14 will be
monitored immunohistochemically and using RT-PCR. These experiments will help
test our hypothesis that
RPE cells from AMD patients are sublethally "injured" and/or secrete "factors"
that result in the recruitment
and maturation of DCs, thus initiating an inflammatory response that
culminates in drusen biogenesis.
126
CA 02363503 2001-08-24
WO 00/52479 PCT/US00/05858
In a second set of experiments, RPE and dendritic cells will be plated on the
opposite side of a
porous membrane, using cell culture inserts. Coon's F-12 medium will be used
in both chambers. RPE
cells will be plated at confluency and DCs will be plated at a density of 600
cells/mm2, which reflects their in
vivo distribution. Pore sizes will be employed that will permit cell processes
to penetrate (e.g. 1 pm) or that
will permit only soluble molecules to traverse (e.g. 0.45pm). RPE cells will
be co-cultured with non-immune
cells (e.g. fibroblasts; "sham-stimulated" or with no cells as a control for
"normal" RPE gene expression.
Following various periods of time in co-culture, RPE cells and DCs will be
collected by either
scraping the membrane surfaces or by trypsinization. Cells will be pelleted
and RNA will be isolated as
described by Chirgwin. Culture supematants will be collected and frozen for
future analyses of proteins,
including various growth factors and cytokines. RNA will be isolated from 1)
AMD-derived, DC-stimulated
RPE; 2) non-AMD-derived, DC-stimulated RPE; 3) AMD-derived, unstimulated/sham-
stimulated RPE; 4)
non-AMD-derived, unstimulated/sham-stimulated RPE; 5) DCs co-cultured with AMD-
derived RPE; and 6)
DCs co-cultured with non-AMD-derived RPE. RNA will be reverse-transcribed, and
33P-labeled cDNAs will
be employed to probe gene arrays. The data collected from the gene array
analyses will be analyzed as
described in Objective 1, in order to identify pathways that are up- and/or
down-regulated in DCs co-
cultured with RPE derived from AMD and non-AMD donors.
Example 22: Primate Model of Choroidal Fibrosis, Choroidal Neovascularization,
and/or AMD
A preferred animal model is an animal with a macula, such a monkey. For
example a
cynomolgus monkey was anesthetized according to methods well known in the art.
The choroidal
circulation was blocked and a 3600 peritomy was made and traction sutures were
used to rotate the eye
as far as possible supemasally to gain access to the posterior globe. A blunt
cannula was used to
separate the choroid from the edge of the sclera and 100mt of sterile balanced
salt solution (BSS)
containing 60 units of protease-free chondroitinase ABC (American Cyanimide)
was injected into the
choroidal stroma. The scierotomy was closed with 7-0 vicryl sutures. Indirect
ophthalmoscopy
demonstrated a normal choroid and retina without hemorrhage or depigmentation.
The conjunctiva was
closed with 7-0 vicryl suture and 3mg celestone was injected
subconjunctivally. The animal was
monitored non-invasively with an opthalmoscope to monitor fundus changes,
including
neovascularization, for 7 days. The animal was then euthanized with
barbiturate overdose ("Sleepaway")
and the eyes prepared for histological observation according to art known
methods. Distinct disruptions
of Bruch's membrane were observed in the experimental eye, demonstrating that
the enzyme reached
Bruch's membrane.
The above example can be modified to inject 1-100 U/ml elastase, alpha-elastin
peptides, or
elastolytic peptides in 0.05 to 0.50 ml BSS. Altematively, the method
described above can be modified to
replace the injection of enzyme for the insertion of enzyme in the form of a
slow release pellet, such slow
release pellet technology being well known in the art.
Example 23: Mouse Model of Drusen Biogenesis, Immune-Mediated Processes and/or
AMD
127
CA 02363503 2001-08-24
WO 00/52479 PCTIUSOO/05858
The transfer of human cells into mice with SCID background has been proven to
be a useful model
for immunologic studies, but has not been employed previously as a method for
studying drusen biogenesis
and its associated immune-mediated processes.
Mice homozygous for the SCID (severe combined immunodeficiency) mutation lack
functional T-
and B-cells and macrophages, and hence fail to generate either humoral or cell-
mediated immunity. The
absence of T and B-cells which normally mediate xenograft rejection enables
SCID mice to support
variable levels of growth of human lymphohematopoetic cells (LHPC). The
Emv30nu"NOD-scid mouse
strain (The Jackson Laboratory, Bar Harbor, Main) has been demonstrated to be
an improved host for
adaptive transfer of autoimmune diabetes and growth of human LHPC. This strain
has the advantage of a
higher level of human LHPC growth than the C.B-17-scid strain in which the
scid mutation originated.
When used as a host in passive transfer experiments and for the repopulation
with human cells various
SCID mice have provided great insight to the contribution of T-celis and/or
autoantibodies in various
autoimmune diseases.
Human PBLs or enriched population of monocyte/dendritic cells will be obtained
by leukapheresis
or by gradient density centrifugation (followed by GM-CSF and/or IL-4
incubation for the purpose of
enriching the dendritic cell population) from the peripheral blood of AMD
patients and from healthy
volunteers. In some experiments, DCs will be pre-incubated with either RPE
cells (with or without
AMD/fibrosis) or isolated fractions of Bruch's membrane. For each experiment,
aliquots 2 x 107PBLs from
a single human donor will be injected into aged matched female mice, ages 6-8
weeks. Injections will be
performed either interperitoneally (i.p.) or interocularly. In experiments
with serum antibody transfer, serum
Ig will be purified using protein A or G columns and Ig in concentration of 1
mg/mI in PBS will be injected
i.p. (200mg/kg/mouse).
Four weeks after the injection, splenic leukocytes from all human PBL
recipients will be
phenotyped by FACS analysis with commercially available monoclonal antibodies
that will identify the total
numbers of human LHPC (CD45+), as well as the proportion of these cells
comprised by macrophages
(CD14+), B-cells (CD19+), T-cells (CD3+), and dendritic cells (CD83+, CD86+,
CD11a+). The relative
proportions of the total T-cell population comprising the CD4+ and CD8+
subsets will be also assessed.
The effects of PBL transfer to injected mice will be evaluated by a number of
functional, histologic,
biochemical, and immunocytochemical assays. The lymphocyte proliferation assay
(LPA) will be used to
evaluate T-cell immunoreactivity against RPE/ drusen proteins. ELISA using
goat anti-human Ig (A, G and
M) will determine levels, specificity, titer and isotype of human antibodies
in mice sera. Mouse eyes will be
examined for the presence of histopathological changes and set of anti-human
antibodies (directed against
MHC class-II antigens and various, cell-specific CD antigens) will be employed
for immunocytochemical
localization of human immunocompetent cells and Ig in mouse eye tissue.
128