Note: Descriptions are shown in the official language in which they were submitted.
CA 02363695 2001-08-15
WO 00/49000 PCT/GB00/00570
Dopamine D1 Receptor Agonist Compounds
The present invention relates to Dopamine D1 receptor agonist compounds, to
methods for
preparing such compounds and to their use.
GB 1 599 705 discloses 1-thienyl and 1-furyl-2,3,4,5-tetrahydro-1H-3-
benzazepines having
utility as cardiovascular agents. Some benzazepines as Dopamine D1 receptor
agonists have
been described. For example, 1-phenyl-3-benzazepines are disclosed in EP 0 230
755-A and
carbamates of 6-chloro-7,8-dihydroxy-1 (4'-hydroxyphenyl)-2,3,4,5-tetrahydro-
1H-3-
benzazepine are disclosed in
EP 0 380 355-A.
The present invention provides compounds which are potent and selective
ligands for the
Dopamine Dl receptor. Such compounds can be used in the treatment of
neurodegenerative
diseases especially, but not limited to, Parkinson's disease. Parkinson's
disease is a progressive
neurodegenerative disorder characterized by the progressive death of
presynaptic dopamine
nuerones in the substantia nigra that innervate postsynaptic striatal neurones
and the resultant loss
of striatal dopamine. The primary therapy for Parkinson's disease focuses upon
compensation
for this loss of dopamine in the striatum. The current main-stay for this
replacement in the
administrattion of the metabolic precursor of dopamine, namely, L-DOPA which
is converted
into dopmaine in the central nervous system. However, L-DOPA can cause severe
adverse effects
such as nausea, vomiting, cardiac arrythmias and hypotension. Additionally,
long-term use of L-
DOPA is associated with the development of abnormal involuntary movements
(dyskinesias) and
psychosis. Furthermore, the positive benefits associated with chronic L-DOPA
therapy
experienced by suffers is lessened, typically several years after treatment
was first initiated.
Therapeutic agents that selectively interact positively with postsynaptic
dopamine D 1 receptors in
the striatum, directly or in-directly (hereafter termed dopamine Dl agonists)
are particularly
valuable as anti-Parkinsonian agents.
CA 02363695 2001-08-15
WO 00/49000 PCT/GB00/00570
2
HO
-RZ
HO
3
4
(I)
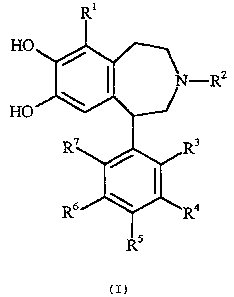
According to the present invention there are provided 2,3,4,5-tetrahydro-1H-3-
benzazepines of
the general formula I
wherein: RI is hydrogen, halogen, C1-C4 alkyl, or CF3;
RZ is hydrogen, methyl, or lower alkenyl of 3-5 carbon atoms;
R3 and R4 together form a furan, dihydrofuran, thiophene, dihydrothiophene,
cyclopentane or
cyclohexane ring and RS is hydrogen or R4 and R5 together form a furan,
dihydrofuran,
thiophene, dihydrothiophene, cyclopentane or cyclohexane ring and R3 is
hydrogen;
R6 is hydrogen, halogen, CF3, CN, NOZ or NH2;
R' is hydrogen, halogen, CF3, CN, NOZ or NH2.
The specific combination of substituents: Rl = H, R2 = H and R4 and RS
together forming a
cyclohexane ring is excluded, namely 1-(5,6,7,8-tetrahydronaphthalen-2-yl)-
2,3,4,5-tetrahydro-
1H-benzo[d]azepine-7,8-diol.
04-04-2001 CA 02363695 2001-08-15
- 2a -
K. S. Sugamori, et al., (Journal of Neurochemistry. 1998. 71,4,1685-93)
describes
the functional differentiation of multiple Dopamine D1 like receptors by
compound
NNC O1-0012. The compound disclosed which is closest to that of the invention
(Compound NNC OI-0127), differs from that of the invention in that it has a
hydrogen at the R' position.
US 4,265,889 discloses 6-lower alkyl-7,8-dihydroxy-1-phenyl 1,2,3,4,5-
tetrahydro-
lh-3-benzazepines. The compounds disclosed in this patent differ from that of
the
invention in that the substituents on the phenyl group at position 1, do not
have a
furan, dihydrofuran, thiopene, dihydrothiopene, cyclopentane, or cyclohexane
ring.
1 o DE 2629887 discloses medicaments with peripheral Dopamine receptor
stimulation
for kidney disorders, diuretics and anti-Parkinson syndrome defects. Preferred
compounds have a hydrogen at the R' position and do not have a furan,
dihydrofuran, thiopene, dihydrothiopene, cyclopentane, or cyclohexane ring
structure on the phenyl group.
i5 GB 1 599 705 discloses 1-thienyl and 1-furyl-2,3,4,5-tetrahydro,lH-3-
benzazepines
being medicinally active compounds especially as cardiovascular agents due to
their
peripheral Dopaminergic activity. These compounds do not have a phenyl group
at
position 1 on the benzazepine ring.
EP 0 380 355 discloses carbamates of 6-chloro-7,-dihydroxy-1-(4'-
Hydroxyphenyl)-
20 2,3,4,5-tetra-hydro-IH-3-benzazepines as prodrugs. The compounds disclosed
do not
have hydroxyl substituents at positions 7 and 8 on the benzazepine ring, and
they do
not have a ring substituent on the phenyl group at position 1.
EP 0 230 755 discloses 1-phenyl-3-benzazepines. These compounds have a
hydrogen
residue at position RI and do not a furan, dihydrofuran, thiopene,
dihydrothiopene,
z s cyclopentane, or cyclohexane ring structure on the phenyl group.
AMENDED SHEET
CA 02363695 2001-08-15
WO 00/49000 PCT/GB00/00570
3
The compounds of formula I may be presented as a mixture of enantiomers, which
may be
resolved into the individual pure enantiomers. This resolution may
conveniently be performed by
fractional crystallisation, from various solvents, of the salts of compounds
of the formula I with
optically active acids or by other methods known from the literature e.g.
chiral column
chromatography. Therefore, this invention includes all isomers, whether
resolved or mixtures
thereof.
Particularly valuable embodiments of this invention are non-toxic,
pharmaceutically acceptable
acid addition salts of benzazepines of formula I. Such salts include those
derived from inorganic
and organic acids such as hydrochloric, hydrobromic, sulphuric, phosphoric,
methanesulfonic,
acetic, lactic, malefic, phthalic and tartaric acids.
The compounds of the invention are useful because of their pharmacological
activity. In
particular, the compounds of the invention are potent (high aW nity) and
selective ligands for the
central dopamine D 1 receptor (Table 1 ) as measured by competitive radio-
ligand displacement
assays using rat striatal tissue homogenates as per the method described in
Psychopharmacology
117:275-286(1995).
Test Compound Ki (nM) Ki (nM)
Dopamine D 1 receptorDopamine D2 receptor
affinity affinity
Example 1 31 790
Example 4 6.7 2500
Table 1
The benzazepine compounds of Formula I possess anti-Parkinsonian activity due
to central
dopaminergic activity as demonstrated by employing the standard
pharmacological test procedure
as reported by Ungerstedt et al., in Brain Research 24:485-493 (1970). This
procedure is based
on the drug-induced rotation (circling) of rats having extensive unilateral
dopaminergic lesions of
CA 02363695 2001-08-15
WO 00/49000 PCT/GB00/00570
4
the substantia nigra. Briefly, the test comprises the quantitative recording
of rotational behaviour
in rats in which 6-hydroxydopamine lesions of the nigrostriatal dopamine
system have been
produced. Unilateral brain lesioning of the substantia nigra in one hemisphere
results in the
dopamine receptor system in that region to become hypersensitive following the
degeneration of
the nigral cell bodies. Activation of these super-sensitive dopamine receptors
by drugs induce
asymmetrical movement of the animal, contralateral rotation (with respect to
the lesioned side of
the brain). The rate and duration of contralateral rotation induced upon drug
administration is an
index of central dopaminergic activity of the agent. Compounds which are known
to be clinically
effective in controlling Parkinsonism, e.g. L-DOPA and apomorphine, are also
effective in this
rat circling model. By way of example the compound 1-indan-5-yl-6-chloro-3-
methyl-2,3,4,5-
tetrahydro-1H-3-benzazepine-7,8-diol produces robust circling in the
unilateral lesioned 6-
hydroxydopamine rat model in a dose-related fashion from 0.438 to 5.79
micromoles/kg when
administered by subcutaneous injection. Cumulative rotations over a set time-
period (190mins)
were as follows: 0.438 micromoles/kg = 23, 0.965 micromole/kg = 397 rotations,
1.93
micromoles/kg = 867 rotations, 3.86 micromoles/kg = 1078 rotations, 5.79
micromoles/kg =
1388 rotations.
The invention is further described by way of example only.
Examule 1
a) 1-(Benzofuran-7 yl)-2-~2-(2-chloro-3,4-dimethoxyphenyl)ethylaminoJethanol
A solution of 2-chloro-3,4-dimethoxyphenylethylamine (6.35g, 0.0296mo1) and (7-
benzofuranyl)oxirane (4.298, 0.0268mo1) in 15m1 acetonitrile was refluxed for
16 hours. The
reaction mixture was cooled to 0°C (ice-bath), filtered and the crude
product re-crystallised from
hot acetonitrile to afford the title compound (3.52g, 35%) as a white
crystalline solid. 1H-NMR in
CDC13 [8, ppm]: 2.86-3.16 (m, 6H); 3.85 (s, 6H); 5.26 (dd, 1H); 6.73-6.77 (m,
2H); 6.91 (d, 1H);
7.21-7.26 (m, 1H); 7.39 (d, 1H),;7.51 (d, 1H); 7.61 (d, 1H).
CA 02363695 2001-08-15
WO 00/49000 PCT/GB00/00570
b) 1-(BenZofuran-7 yl)-6-chloro-7,8-dimethoxy-2,3,4,5-tetrahydro-1H 3-
benZaZepine
1-(Benzofuran-7-yl)-2-[2-(2-chloro-3,4-dimethoxyphenyl)ethylamino]ethanol
(2.20g, 5.85mmo1)
in 70m1 trifluoroacetic acid was treated with concentrated sulphuric acid
(0.71m1, 0.0135mo1) and
stirred at ambient temperature for 90 minutes. The solution was evaporated in
vacuo and the
residue dissolved in 30m1 4M sodium hydroxide and extracted with
dichloromethane (4 x 50m1).
The organic fractions are combined, dried, filtered and evaporated to afford
the crude product as
a yellow/green glass. Subsequent purification by column chromatography on
silica with
dichloromethane/methanol (9:1 ) as eluant afforded the title compound as a
sticky white solid
(1.42g, 68%).
iH-NMR in CDC13 [8, ppm]: 2.95-3.80 (m, 6H); 3.57 (s, 3H); 3.85 (s, 3H); 4.76
(dd, 1H); 6.30 (s,
1H); 6.80 (d, 1H); 6.95 (d, 1H); 7.21 (t, 1H); 7.54 (d, 1H); 7.62 (d, 1H).
c) 1-(Benzofuran-7 yl)-6-chloro-7,8 hydroxy-2,3,4,5-tetrahydro-IH 3-
benzazepine
hydrobromide
1-(Benzofuran-7-yl)-6-chloro-7,8-dimethoxy-2,3,4,5-tetrahydro-1H-3-benzazepine
(1.19g,
3.32mmo1) dissolved in dry dichloromethane (20m1). The solution was cooled to -
78°C and
boron tribromide (133m1, 133mmol) added slowly via syringe. The reaction
mixture was
maintained at -78°C for 30 minutes, allowed to warm to 0°C and
stirred for 2 hours. The reaction
mixture was subsequently cooled to -78°C, methanol (25m1) added slowly
and stirred for 30
minutes. After refluxing the reaction mixture for 1 hour the solvents were
removed in vacuo to
afford the crude product. Trituration with diethyl ether afforded the title
compound as a off white
solid (1.26g, 92%).
1H-NMR in CD30D [~, ppm]: 3.15 (m, 1H); 3.30-3.99 (m, 5H); 5.00 (m, 1H); 6.10
(s, 1H); 6.93
(d, 1H); 7.07 (d, 1H); 7.30 (t, 1H); 7.64 (m, 1H); 7.79 (d, 1H).
Example 2
a) 1-(Benzo~bJthiophen-7 yl)-2-~2-(2-chloro-3,4
dimethoxyphenyl)ethylaminoJethanol
CA 02363695 2001-08-15
WO 00/49000 PCT/GB00/00570
6
A solution of 2-chloro-3,4-dimethoxyphenylethylamine (7.OOg, 32.5mmo1) and 7-
benzo[b]thiophenyl oxirane (5.30g, 30.lmmol) in 20m1 acetonitrile was refluxed
for 72 hours.
The reaction mixture was cooled to 0°C (ice-bath), filtered and the
crude product re-crystallised
from hot acetonitrile to word the title compound (5.57g, 47%) as a white
crystalline solid. 1H-
NMR in CDCl3 [8, ppm]: 2.83-3.08 (m, 6H); 3.84 (s, 3H); 3.85 (s, 3H); 5.06 (m,
1H); 6.73 (d,
1H); 6.88 (d, 1H); 7.35 (m, 3H); 7.43 (d, 1H); 7.73 (m, 1H).
b) 1-(Benzo~bJthiophen-7 yl)-6-chloro-7,&dirnethoxy-2,3,4,5-tetrahydro-IH 3-
benzazepine
1-(Benzo[b]thiophen-7-yl)-2-[2-(2-chloro-3,4-
dimethoxyphenyl)ethylamino]ethanol (3.90g,
lOmmol) in 30m1 trifluoroacetic acid was treated with methane sulphonic acid
(0.7m1,
10.7mmo1),under a nitrogen atmosphere, and the solution heated under reflux
for 18 hours. The
solution was evaporated in vacuo and the residue dissolved in dichloromethane
(100m1) and the
solution washed with concentrated aqueous ammonia (2x50m1, 0.880),water
(100m1) and
saturated aqueous sodium chloride solution (100m1), dried, filtered and
evaporated to afford the
crude product as a yellow/green glass. Subsequent purification by column
chromatography on
silica with dichloromethane/methanol (9:1) as eluant afforded the title
compound as a pale brown
gum (3 .01 g, 81 %).
1H-NMR in CDC13 [8, ppm]: 2.82-2.92 (m, 2H); 3.13-3.23 (m, ZH); 3.41-3.55 (m,
2H); 3.49 (s
3H); 3.83 (s, 3H); 4.66 (d, 1H); 6.21 (s, 1H); 7.11 (d, 1H); 7.37-7.41 (m,
3H); 7.76 (d, 1H).
c) 1-(Benzo~bJthiophen-7 yl)-6 chloro-7,8 hydroxy-2,3,4,5-tetrahydro-IH 3-
benzazepine
1-(B enzo [b]thiophen-7-yl)-6-chloro-7, 8-dimethoxy-2, 3,4, 5-tetrahydro-1 H-3
-benzazepine
(1.31g, 3.5mmo1) dissolved in dry dichloromethane (ZOmI). The solution was
cooled to -78°C
and boron tribromide (14m1, l4mmol) added slowly via syringe. The reaction
mixture was
maintained at -78°C for 30 minutes, allowed to warm to 0°C and
stirred for 2 hours. The reaction
mixture was subsequently cooled to -78°C, methanol (lOml) added slowly
and stirred for 30
minutes. After refluxing the reaction mixture for 1 hour the solvents were
removed in vacuo to
afford the crude product. Purification by recrystallisation from methanol
afforded the title
compound as an off white solid (0.68g, 45%). Mpt 185-188°C. Anal.
(Calc.)
CA 02363695 2001-08-15
WO 00/49000 PCT/GB00/00570
7
C18H16C1NOZS.HBr.H20 C 48.61 (48.61), H 4.27 (4.30), N 2.98 (3.15). 1H-NMR in
CD30D [~,
ppm]: 3.02 (t, 1H); 3.30-3.38 (m, 1H); 3.60-3.74 (m, 3H); 3.89 (d, 1H); 4.91
(d, 1*H); 5.97 (s,
1H); 7.23 (d, 1H); 7.41 (d, 1H); 7.45-7.52 (m, 2H); 7.84 (d, 1H).
Examule 3
a) 2-(Z-Chloro-3,ødimethoxyphenyl)ethylaminoJ 1-indan-5 yl ethanol
To the solution of 2-indan-5-yl oxirane (3.38 g, 21.1 mmol) in anhydrous
acetonitrile (20 ml)
was added 2-(2-chloro-3,4-dimethoxyphenyl)ethylamine (2.0 g, 23.2 mmol) and
the solution
refluxed for 20h. Upon cooling a white precipitate formed and was collected by
filtration and
washed with diethyl ether, giving the title compound as a white solid (2.85 g,
36%). 1H NMR
(400 MHz, DMSO-d6): b (ppm) 1.96-2.03 (2H, m, CH2-CH2-CH2), 2.50-2.84 (lOH, m,
5xCH2),
3.72 (3H, s, CH30), 3.80 (3H, s, CH30), 4.56 (1H, t, J 6.04, H 1), 5.14 (1H,
broad, NH) and
6.94-7.14 (5H, m, Ar-H). This material was used for the next step without
further purification.
b) 1-~ndan-S yl ~rchloro-7,8 dimethoxy-2,3,4,5-tetrahydro-IH 3-benzazepi~e
2-(2-Chloro-3,4-dimethoxyphenyl)ethylamino]-1-indan-5-yl-ethanol (2.7 g, 7.18
mmol) was
dissolved in trifluoroacetic acid (50 ml), to which was added methane sulfonic
acid (0.76 g, 7.90
mmol). The reaction mixture was stirred under reflux for 20 h, and was then
allowed cooling to
rt. Removal of the solvent afforded an oily residue, which was dissolved in
dichloromethane (200
ml) and washed with ammonia solution (0.88 M, 150 ml), water (2x 150 ml),
brine (100 ml) and
dried. Removal of the solvent gave the crude product as a white solid. 'H NMR
(400 MHz,
CDC13): 8 (ppm) 2.04-3.52 (12H, m, 6xCH2), 2.55 (1H, broad, NH), 3.70 (3H, s,
CH30), 3.86
(3H, s, CH30), 4.27 (1H, d, H 1), 6.43 (1H, s, H 9), 6.88-7.20 (3H, m, other
Ar-H). This material
was used for the next step without further purification.
CA 02363695 2001-08-15
WO 00/49000 PCT/GB00/00570
g
c) 1-Indan-S yl 6-chloro-7,8-dimetho~y-3-methyl 2,3,4,5-tetrahydro-IH 3-
benzazepine
1-Indan-5-yl-6-chloro-7,8-dimethoxy-2,3,4,5-tetrahydro-1H-3-benzazepine (1.3
g, 3.63
mmol) was dissolved in methanol (20 ml), to which was added dropwise
formaldehyde solution
(37%, 1.87 ml, 23.2 mmol). White precipitate formed with the addition. Sodium
cyanoborohydride (97%, 0.92 g, 14.9 mmol) was added, bringing most of the
solid into solution.
The reaction mixture was then stirred at r.t for 3 h. Removal of the solvent
gave a residue
containing a colorless oil and a white solid (3.8 g). This residue was
purified by column
chromatography (Petroleum etherlethyl acetate, 1:1, Rf 0.25), giving the
desired product as
colourless oil (1.16 g, 86%). 1H NMR (400 MHz, CDC13): 8 (ppm) 2.08 (2H, t, J
7.5, lxCH2),
2.37 (3H, s, N CH3), 2.86-3.54 (lOH, m, other 5xCH2), 3.61 (3H, s, OCH3), 3.83
(3H, s, OCH3),
4.32 (1H, d, 1-H), 6.26 (1H, s, 9-H), 6.92-7.22 (3H, m, other Ar-H).
d) 1-Indan-S yl ~chloro-3-methyl 2,3,4,5-tetrahydro-1H 3-benzazepine-7,8-diol
1-Indan-5-yl-6-chloro-7,8-dimethoxy-3-methyl-2,3,4,5-tetrahydro-1H-3-
benzazepine (1.03 g,
2.77 mmol) was dissolved in anhydrous dichloromethane (15 ml), which was
cooled to -78°C. To
this solution was added dropwise BBr3 (1.0 M solution in dichloromethane, 13.8
ml, 13.8 mmol)
over 25 min. The reaction mixture was stirred at -78°C for 1 h, at
0°C for 3 h and at r.t for further
1 h. The reaction mixture was cooled to -78°C again and treated with
methanol (20 ml) and was
then stirred at r.t overnight. Removal of the solvent afforded a brown
residue. Methanol (10 ml)
was added and removed under reduced pressure. This process was repeated four
times, giving the
crude product as a brown residue, which was recrystallised from methanol/ether
to give a pale
solid (0.87 g, 74%). The material was recrystallised again from methanol/ether
to give the title
compound as a pale solid (0.53 g, 45%), mp. 255-257°C (decomp.); Found:
%C, 56.42; %H,
5.58; %N, 3.14. CZOHz3BrC1N02 requires %C, 56.55; %H, 5.46; %N, 3.30. Mass 354
(m-81). iH
NMR (400 MHz, DMSO-d6): 8 (ppm) 2.05-3.79 (m, 6xCH2), 4.60 (1H, d, H 1), 6.16
(1H, broad,
H 9), 6.97-7.28 (3H, m, other Ar-H).
CA 02363695 2001-08-15
WO 00/49000 PCT/GB00/00570
9
Example 4
a) 1-(Benzo~bJthiophen-S yl)-2-~2-(2-chloro-3,4
dimetho~.yphenyl)ethylaminoJethanol
A solution of 2-chloro-3,4-dimethoxyphenylethylamine (7.OOg, 32.Smmol) and (5-
benzo[b]thiophenyl oxirane (5.30g, 30mmo1) in 30m1 acetonitrile was refluxed
for 48 hours. The
reaction mixture was cooled to 0°C (ice-bath), filtered and the crude
product re-crystallised from
hot acetonitrile to afford the title compound (4.408, 37%) as a white
crystalline solid. Mpt 137-9
°C. 1H-NMR in CDC13 [8, ppm]: 2.75 (m, 6H); 3.72 (s, 3H); 3.79 (s, 3H);
4.76 (m, 1H); 6.90 (d,
1H); 6.99 (d, 1H); 7.35 (d, 1H); 7.43 (d, 1H); 7.73 (d, 1H); 7.83 (s, 1H);
7.91 (d, 1H).
b) 1-(Benzo~bJthiophen-S yl)- 3- methyl 6-chloro-7,&dimetho.~y-2,3,4,5-
tetrahydro-lH 3-
benzazepine
1-(Benzo[b]thiophen-5-yl)-2-[2-(2-chloro-3,4-
dimethoxyphenyl)ethylamino]ethanol (2.OOg,
S.lmmol) in 40m1 trifluoroacetic acid was treated with methane sulphonic acid
(0.36m1,
S.Smmol), under a nitrogen atmosphere, and heated under reflux for 18 hours.
The solution was
evaporated in vacuo and the residue taken up in dichloromethane (100m1) and
washed with
concentrated aqueous ammonia (100m1,0.880), water (2x 100m1) and saturated
aqueous sodium
chloride solution (100m1), dried, filtered and evaporated to afford the crude
product as a
yellow/green glass. The crude amine was taken up in methanol (40m1) and
aqueous formaldehyde
(2.8m1, 37% wt, 37 mmol) was added followed by sodium cyanoborohydride (1.35g,
Zlmmol)
and the resulting solution stirred for 18 hours. The solvents were removed in
vacuo, and the
residue taken up in hydrochloric acid (100m1, 1M). The solution was washed
with diethyl ether
(2x 100m1) and basified with concentrated aqueous ammonia (100m1, 0.880), the
mixture was
extracted with dichloromethane (2x 100m1). The combined extracts were washed
with water (2x
100m1) and saturated aqueous sodium chloride solution (150m1), dried, filtered
and concentrated
in vacuo. Subsequent purification by column chromatography on silica with
diethyl ether as
eluant afforded the title compound as a white solid (640mg, 34%).
CA 02363695 2001-08-15
WO 00/49000 PCT/GB00/00570
1H-NMR in CDC13 [8, ppm]: 2.35 (m, 1H); 2.39 (s, 3H) ;2.85-2.98 (m, 2H); 3.13
(m, 2H); 3.31
(m, 1H); 3.56 (s 3H); 3.83 (s, 3H); 4.47 (d, 1H); 6.25 (s, 1H); 7.11 (d, 1H);
7.37-7.41 (m, 3H);
7.76 (d, 1H).
c) 1-(Benzo~bJthiophen-S yl)-3-methyl 6-chloro-7,8-dihydroxy-2,3,4,5-
tetrahydro-IH 3-
benzazepine
1-(Benzo[b]thiophen-5-yl)-3-methyl-6-chloro-7,8-dimethoxy-2,3,4,5-tetrahydro-
1H-3-
benzazepine
(470mg, l.2mmol) dissolved in dry dichloromethane (15m1). The solution was
cooled to -78°C
and boron tribromide (6m1, 6mmol) added slowly via syringe. The reaction
mixture was
maintained at -78°C for 60 minutes, allowed to warm to 0°C and
stirred for 2 hours. The reaction
mixture was subsequently cooled to -78°C, methanol (40m1) added slowly
and stirred for
30 minutes. After the solvents were removed in vacuo purification by column
chromatography on
silica using methanol/dichloromethane (1:9) as eluant afforded the title
compound as a yellow
solid (127mg, 30%). 1H-NMR in (CD3)2S0 [8, ppm]: 2.29 (s, 3H); 2.4 (m, 1H);
2.95-3.12 (m,
4H); 3.30-3.4 (m, 3H); 3.89 (d, 1H); 4.38 (d, 1*H); 6.09 (s, 1H); 7.21 (dd,
1H); 7.44 (d, 1H); 7.68
(s, 1H); 7.74 (d, 1H); 7.96 (d, 1H).
d) 1-(Benzo~bJthiophen-S yl)-3-methyl 6-chloro-7,&dihydroxy-2,3,4,5-tetrahydro-
IH 3-
benzazepine. monohydrochloride
1-(Benzo[b]thiophen-5-yl)-3-methyl-6-chloro-7,8-dihydroxy-2,3,4,5-tetrahydro-
1H-3-
benzazepine
(lllmg, 0.31mmol) was dissolved in a mixture of dry diethylether (30m1) and
dry chloroform
(6m1). The solution was treated with 2N hydrochloric acid in dry diethylether
( l2ml, 24mm1) and
stirred for 5 hours. The reaction mixture was filtered and the crude product
re-crystallised from
methanol/diethylether to afford the title compound as a pale yellow solid
(95mg, 78%). Mpt
>220C (decomp), 1H-NMR in (CD3)ZSO [8, ppm]: 2.82 (s, 3H); 2.9-3.0 (m, 2H);
3.5-3.6 (m, 2H);
3.7 (m, 1H); 3.84 (d, 1H); 4.87 (d, 1H); 5.89 (s, 1H); 7.23 (dd, 1H); 7.52 (d,
1H); 7.77 (s, 1H);
7.84 (d, 1H); 8.10 (d, 1H); 9.04 (s, OH); 9.40 (s, OH); 11.15 (broad s, HCl).
CA 02363695 2001-08-15
WO 00/49000 PCT/GB00/00570
11
Calculated for C19H18N02C1S.HC1: C, 57.71; H, 4.85; N, 3.54; Cl, 17.70. Found:
C, 55.51;1-1,
5.18; N, 3.08; Cl, 17.66.
Example 5
a) 1-(Benzo~bJfuran-7 yl)-3-methyl 6-chloro-7,8-dimethoxy-2,3,Q,5 tetrahydro-
1H 3-
benzazepine
1-(Benzo[b]furan-7-yl)-6-chloro-7,8-dimethoxy-2,3,4,5-tetrahydro-1H-3-
benzazepine (0.96g,
2.7mmol) was taken up in methanol (25m1) and aqueous formaldehyde (l.6ml,
37%wt, 21mmo1)
was added, followed by sodium cyanoborohydride (0.75g, l2mmol) and the
resulting solution
stirred for 24 hours. The solution was concentrated in vacuo and the residue
was taken up in
dichloromethane (100m1), the solution was washed with water (2x 100m1) and
saturated sodium
chloride solution (100m1), dried, filtered and concentrated in vacuo. After
purification by column
chromatography on silica using dichloromethane/ methanol (9:1) as eluant the
title compound
was obtained as a pale orange gum (0.88g, 88%).'H-NMR in CDC13 [8, ppm]: 2.34
(m, 1H); 2.37
(s, 3H); 2.96 (m, 1H); 3.07 (m, 1H); 3.18 (m, 1H); 3.45 (s, 3H); 3.81 (s, 3H);
4.84 (d, 1*H); 6.10
(s, 1H); 6.78 (d, 1H); 7.06 (d, 1H); 7.23 (m, 1H); 7.53 (dd, 1H) 7.58 (d, 1H).
b) 1-(Benzo~bJfuran-7 yl)-3-methyl fi-chloro-7,8-hydroxy-2,3,4,5-tetrahydro~IH
3-
benzazepine
1-(B enzo [b] furan-7-yl)-3-methyl-6-chloro-7, 8-dimethoxy-2,3,4, 5-tetrahydro-
1 H-3 -benzazepine
(0.52g, l.4mmo1) dissolved in dry dichloromethane (l5ml). The solution was
cooled to -78°C
and boron tribromide (6m1, 6mmo1) added slowly via syringe. The reaction
mixture was
maintained at -78°C for 1 hour, allowed to warm to 0°C and
stirred for 2 hours. The reaction
mixture was subsequently cooled to -78°C, methanol (lOml) added slowly
and stirred for 1 hour,
and for 18 hours at ambient temperature. The solvents were removed in vacuo to
afford the crude
product. Purification by column chromatography on silica using
dichloromethane/ methanol (9:1)
as eluant and re-crystallisation from propan-2-ol/ diethyl ether afforded the
title compound as a
CA 02363695 2001-08-15
WO 00/49000 PCT/GB00/00570
12
buff solid (100mg, 17%).Anal. (Calc.) C19H1gC1N03.HBr.l.5H20 C 50.58 (50.51) H
4.78 (4.90)
N 2.78 (3.10). iH-NMR in (CD3)ZSO [8, ppm]: 2.51(s, 3H) 3.02 (t, 1IT); 3.30-
3.38 (m, 1H); 3.60-
3.74 (m, 3H); 3.89 (d, 1H); 5.03 (d, 1*H); 5.82 (s, 1H); 7.05 (d, 1H); 7.20
(d, 1H); 7.36 (m, 1H);
7.72 (m, 1H); 8.00 (d, 1H).
Example 6
a) 1-(Benzo~bJthiophen-7 yl)-3-methyl ~rchloro-7,&dihydroxy-2,3,4,5-tetrahydro-
IH 3-
benzazepine
1-(B enzo [b]thiophen-7-yl)-6-chloro-7, 8-dihydroxy-2, 3,4, 5-tetrahydro-1 H-3
-b enzazepine
hydrobromide. (180mg, 0.4mmol) was suspended in dry methanol (5ml) and aqueous
formaldehyde (0.2m1, 37% wt., 2.7 mmol) was added followed by sodium
cyanoborohydride
(O.lOg, 1.6 mmol) to give a clear colourless solution. The solution was
stirred for 18 hours to
give a white suspension. The suspension was cooled to 0°C and
hydrobromic acid (lml, 48% wt)
was added to give a clear solution stirred for 90 minutes. The solution was
evaporated in vacuo
and the residue purified by column chromatography on silica with chloroform/
methanol (9/1) as
eluant gave the title compound as a yellow solid (170mg, 94%). 1H-NMR in (CD3
)2 SO [8, ppm]:
2.11 (t, 1H); 2.29 (s, 3H); 2.80 (dd, 1H); 2.95 (m, 2H); 3.18 (d, 1H); 3.35
(m, 3H): 4.53 (d, 1*H);
5.88 (s, 1H); 7.23 (d, 1H); 7.46 (m, 2H); 7.66 (d, 2H); 7.83 (d, 1H).