Note: Descriptions are shown in the official language in which they were submitted.
*
2 ' CA 02364862 2001-08-24
Wo 00/51961 - 1 - PCT/EPOO/01162
75961pct
Pro q.q for i re ap ring deri vat i ves of lhi_nhenyl -.-rarboxvl i c-
acid
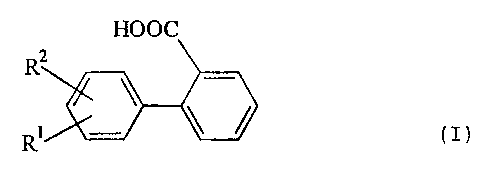
The invention relates to a process which can be used on an
industrial scale for preparing biphenyl-2-carboxylic acid
derivatives (I)
HOOC
R2
R (I),
wherein R1 and R2 may have the meanings given in the
specification and claims.
Background of the i nv _n .i_on
Biphenyl-2-carboxylic acids of formula (I) are very
important as intermediate products in the production of
pharmaceutically valuable active substances, particularly
in the production of pharmaceutical substances which may
be used as angiotensin-II-antagonists.
Processes for preparing biphenyl-2-carboxylic acid and the
derivatives (I) thereof are known from the prior art. One
method essential to the background of the invention is the
coupling of aromatic Grignard compounds (II) with
optionally substituted (2-methoxyphenyl)-2-oxazolines
(III) according to Diagram 1, as described by Meyers et
al. (e.g. Tetrahedron (1985) Vol. 41, 837-860), in which
the corresponding (2-oxazolinyl)-2-biphenyl derivatives
(IV) are obtained to begin with.
CA 02364862 2001-08-24
WO 00/51961 - 2 - PCT/EPOO/01162
RZ R2
Br [(J_MBr]
R~ R2 R x R20_ OH
R
R "
CH30 / \ (IV) (~)
(III)
Di agram ~
The group R i denotes an optionally substituted oxazolin-
2-yl group. The definition of the groups Rland RZ can be
found in the later part of the specification and in the
claims. Conversion into the corresponding carboxylic acids
of formula (I) is effected by saponification of the
oxazolines (IV). This saponification of (IV) can be
carried out by two different reaction methods, from a
formal point of view. In Diagram 2 these reaction methods
are illustrated by way of example with reference to the
preparation of biphenyl-2-carboxylic acid, starting from
biphenyl-oxazoline unsubstituted at the oxazoline group
(i.e. R' and R2 = hydrogen; R " = oxazolin-2-yl).
CA 02364862 2001-08-24
WO 00/51961 - 3 - PCT/EPOO/01162
~OH
O
N
H
rO (Va)
~
N HOOC
6-:b
(IV) H2N O (~)
O ~
~
Diagram 2 : (Vb)
The saponification of the oxazoline under reaction
conditions known in the art leads in the first step to the
formation of the aminoester (Vb) (Meyers et al. J. Org.
Chem. (1974) Vol. 39, 2787-2793). The aminoester (Vb) can
then be saponified to the carboxylic acid (I) in a second
reaction step, e.g. by boiling for several hours in 10 -
25 o sodium hydroxide solution.
For a large-scale manufacturing process, however, it is
desirable to carry out the saponification process as a
one-pot process.
The acid saponification by a one-pot process (e.g.
according to EP59983) carried out using the methods known
from the prior art, however, led to unsatisfactory results
when done on a large scale.
It was observed that because of the low solubility in the
solvents which are used according to the prior art (e.g.
aqueous hydrochloric acid according to EP 59983) aniino-
ester (Vb) is partially precipitated after being formed. A
precipitate of (Vb) is formed on the stirring mechanism
CA 02364862 2007-06-04
, . , .
27400-212
- 4 -
and on the walls of the reaction vessel. This causes
aminoester (Vb) to be removed successively from the reaction
solution and, because of the poor solubility, it is then
virtually no longer available for further reaction to form
the desired end compound (I). A further reduction in yield
occurs as a result of the inclusion of product (I) in the
crystallised aminoester (Vb) which is clumping normally.
The abovementioned disadvantages lead to an
increased cost in the large-scale production of (I), since
within the scope of the working up and processing of the end
product, on the one hand, the aminoester (Vb) has to be
separated off and, on the other hand, a separate synthesis
step has to take place for reacting the precipitated
aminoester (Vb) to form the end product.
The aim and objective of the present invention is
therefore to provide a process for preparing
derivatives/homologues of biphenyl-2-carboxylic acid on a
large scale which overcomes the disadvantages of the
processes known from the prior art.
According to one aspect of the present invention,
there is provided a process for preparing a biphenyl-2-
carboxylic acid derivative of general formula (I)
HOOC
R2 b
~
RI - (I),
wherein
R1 and R 2 independently denote hydrogen;
C1-C6-alkyl, which is unsubstituted or substituted by
CA 02364862 2007-06-04
27400-212
- 4a -
halogen; C1-C6-alkoxy; C1-C6-acyl; C1-C6-alkoxycarbonyl; COOH;
phenyl; benzyl; halogen; hydroxy; nitro or amino, or
wherein
Ri and R2 together with adjacent carbon atoms of
the phenyl ring form a saturated or unsaturated 5- or
6-membered carbocyclic group which is unsubstituted or
substituted by C1-C4-alkyl, halogen, COOH, phenyl or hydroxy;
wherein a(2-oxazolinyl)-2-biphenyl derivative of general
formula (IV)
RoX
2
Rt
(IV),
wherein
R1 and R2 are as hereinbefore defined and
R " denotes an oxazolin-2-yl group, which is
unsubstituted or mono-, di-, tri- or tetra-substituted by
one or more of the groups C1-C6-alkyl, which is unsubstituted
or substituted by halogen, hydroxy or C1-C4-alkoxy;
C1-C6-alkoxy; phenyl; which is unsubstituted or substituted
by C1-C4-alkyl, C1-C4-alkoxy, hydroxy, nitro or amino; benzyl;
pyridyl or C1-C6-alkoxycarbonyl,
is saponified with hydrochloric acid at a temperature of 120
to 160 C and under a pressure of 3 to 6 baro, in the
presence of an inert organic solvent which is immiscible
with water.
CA 02364862 2007-06-04
27400-212
- 4b -
Detailed description of the invention
Surprisingly, it has been found that the
disadvantages encountered in the methods of preparing
biphenyl-2-carboxylic acid derivatives known from the prior
art can be avoided if the saponification of the
oxazoline (IV) is carried out with hydrochloric acid at
elevated temperature under pressure, in the presence of an
inert organic solvent which is immiscible with water.
The present invention is consequently directed to
a large-scale process for preparing biphenyl-2-carboxylic
acid derivatives of general formula (I)
CA 02364862 2001-08-24
WO 00/51961 - 5 - _ PCT/EP00/01162
HOOC
RZ -
~ ~
R1
(I) ,
wherein
R1 and R2 which may be identical or different denote
hydrogen, C1-C6-alkyl, which may optionally be substituted
by halogen, C1-C6-alkoxy, Cl-C6-acyl, Cl-C6-alkoxycarbonyl,
COOH, phenyl, benzyl, halogen, hydroxy, nitro or amino, or
wherein
R1 and R2 together with adjacent carbon atoms of the
phenyl ring form a saturated or unsaturated 5- or 6-
membered carbocyclic group which may optionally be
substituted by C1-C4-alkyl, halogen, COOH, phenyl or-
hydroxy;
characterised in that a (2-oxazolinyl)-2-biphenyl
derivative of general formula (IV)
:0x\_/
~
(IV)
,
wherein
R' and Rz are as hereinbefore defined and
R i denotes an oxazolin-2-yl group, which may optionally
be mono-, di-, tri- or tetra-substituted by one or more of
the groups C1-C6-alkyl, which may optionally be
substituted by halogen, hydroxy or C1-C4-alkoxy, Cl-C6-
alkoxy, phenyl, which may optionally be substituted by C1-
C4-alkyl, C1-C4-alkoxy, hydroxy, nitro or amino, benzyl,
pyridyl or C1-C6-alkoxycarbonyl,
is saponified with hydrochloric acid at elevated
temperature under pressure, in the presence of an inert
organic solvent which is immiscible with water.
CA 02364862 2001-08-24
WO 00/51961 - 6 - PCT/EP00/01162
A preferred process according to the invention is a
process for preparing biphenyl-2-carboxylic acid
derivatives of general formula (I) wherein
R' and R2, which may be identical or different, denote
hydrogen, C1-C4-alkyl, which may optionally be substituted
by fluorine, chlorine or bromine, C1-C4-alkoxy, Cl-C4-acyl,
C1-C4-alkoxycarbonyl, COOH, phenyl, benzyl, fluorine,
chlorine, bromine, hydroxy, nitro or amino, or wherei.n
R1 and R 2 together with adjacent carbon atoms of the
phenyl ring form an unsaturated 6-membered carbocyclic
group which may optionally be substituted by C1-C4-alkyl,
fluorine, chlorine, bromine, COOH, phenyl or hydroxy;
characterised in that a (2-oxazolinyl)-2-biphenyl
derivative of general formula (IV), wherein
R1 and R2 are as hereinbefore defined and
R " denotes an oxazolin-2-yl group, which may optionally
be mono- or disubstituted by one or more of the groups C1-
C4-alkyl, which may optionally be substituted by fluorine,
chlorine, bromine, hydroxy or Cl-C4-alkoxy, Cl-C4-alkoxy,
phenyl, which may optionally be substituted by C1-C,-
alkyl, C1-C4-alkoxy, hydroxy, nitro or amino, benzyl or C1-
C4-alkoxycarbonyl,
is saponified with hydrochloric acid at elevated
temperature under pressure, in the presence of an inert
organic solvent which is immiscible with water.
Particularly preferred is a process for preparing
biphenyl-2-carboxylic acid derivatives of general formula
(I), wherein
R' and R2 which may be identical or different, denote
hydrogen, methyl, ethyl, n-propyl, iso-propyl, n-butyl,
tert-butyl, CF3, methoxy, ethoxy, COOH, phenyl, benzyl,
fluorine, chlorine, bromine, hydroxy, nitro or amino, or
wherein
R1 and R2 together with adjacent carbon atoms of the
phenyl ring form an anellated phenyl ring which may
optionally be substituted by methyl, ethyl, n-propyl,
CA 02364862 2001-08-24
WO 00/51961 - 7 - PCT/EPOO/01162
isopropyl, tert.-butyl, fluorine, chlorine, bromine, COOH,
phenyl or hydroxy,
characterised in that a (2-oxazolinyl)-2-biphenyl
derivative of general formula (IV) wherein
R1 and R 2 are as hereinbefore defined and
R x denotes an oxazolin-2-yl group, which may optionally
be mono- or disubstituted by one or more of the groups
methyl, ethyl, n-propyl, iso-propyl, n-butyl, tert.-butyl,
methoxymethyl, hydroxymethyl, methoxy or ethoxy, or
phenyl, which may optionally be substituted by methyl,
ethyl, n-propyl, iso-propyl, n-butyl, tert.-butyl,
methoxy, ethoxy or hydroxy, or benzyl, methoxycarbonyl or
ethoxycarbonyl,
is saponified with hydrochloric acid at elevated
temperature under pressure, in the presence of an inert
organic solvent which is immiscible with water.
Also of importance according to the invention is a process
for preparing biphenyl-2-carboxylic acid derivatives of
general formula (I), wherein
R1 and R2 which may be identical or different, denote
hydrogen, methyl, CF3, COOH, phenyl, fluorine or hydroxy,
or wherein
R1 and R2 together with adjacent carbon atoms of the
phenyl ring form an anellated phenyl ring,
characterised in that a (2-oxazolinyl)-2-biphenyl
derivative of general formula (IV), wherein
R1 and R2 are as hereinbefore defined and
R x denotes an oxazolin-2-yl group, which may optionally
be mono- or disubstituted by one or more of the groups
methyl, ethyl, methoxy, ethoxy, phenyl or benzyl,
is saponified with hydrochloric acid at elevated
temperature under pressure, in the presence of an inert
organic solvent which is immiscible with water.
Of particular importance is a process for preparing
CA 02364862 2001-08-24
WO 00/51961 - 8 - PCT/EPOO/01162
biphenyl-2-carboxylic acid derivatives of general formula
(I), wherein
R1 and Rz which may be identical or different, denote
hydrogen, methyl or CF31
characterised in that a (2-oxazolinyl)-2-biphenyl
derivative of general formula (IV), wherein
R1 and R2 are as hereinbefore defined and
R x denotes an oxazolin-2-yl group optionally mono- or
disubstituted by methyl,
is saponified with hydrochloric acid at elevated
temperature under pressure, in the presence of an inert
organic solvent which is immiscible with water.
It is particularly preferable according to the invention
to operate as follows. 0.08-0.8, preferably 0.15-0.5, most
preferably about 0.2 1 water and 3.0-6.0 mol, preferably
3.5 - 5.0 mol, most preferably about 4.0 mol hydrochloric
acid per mol of oxazolin-2-yl-biphenyl (IV)are placed in a
reaction vessel of suitable size. Preferably, the
abovementioned hydrochloric acid is added in the form of
aqueous solutions, most preferably in the form of 36.5%
aqueous solution, so that a hydrochloric acid
concentration of 20 - 30 %, most preferably about 24 % is
produced.
After rendering inert with protective gas, preferably
nitrogen, the reaction vessel is evacuated (to about 50
mbar) and 0.05-0.2, preferably 0.08-0.15, most preferably
about 0.1 litre of an inert organic solvent is added per
mol of starting compound (IV) used. According to the
invention, aliphatic or aromatic hydrocarbons and aromatic
chlorohydrocarbons with 6-10 carbon atoms may be used as
inert organic solvents. Aliphatic or aromatic hydrocarbons
with 7-8 carbon atoms are preferred. The solvents which
may be used according to the invention are preferably
CA 02364862 2001-08-24
WO 00/51961 - 9 - PCT/EPOO/01162
toluene, xylene, chlorobenzene and methylcyclohexane.
Methylcyclohexane is particularly preferred.
After the addition of the inert organic solvent the
reaction solution is heated to a temperature in the range
from 120-160 C, preferably 130-150 C, most preferably 140-
145 C. At constant temperature the mixture is stiri-ed for
a further 3-10 h, preferably 4-8 hours. The apparatus is
sealed (in practice by closing the vapour seal valve), so
that the heating of the reaction solution mentioneci above
produces an internal pressure of 3-6 baro (= bars
overpressure), preferably 4-5 baro within the apparatus.
The temperature can be varied depending on the boiling
point of the solvent used, so that the abovementioried
internal pressure is built up. This results in the
additional advantage according to the invention that
conventional apparatus such as DIN enamel apparatus (to
pressure level 6 baro) can be used.
The reaction vessel is then cooled to a temperature at
which the apparatus is at maximum atmospheric pressure (20
- 50 C). Any underpressure is optionally equalised with
inert gas. For working up, the reaction mixture is
combined with a suitable solvent or mixture of solvents
which enables the aqueous hydrochloric acid phase to be
separated off without loss of product. It is preferable to
use toluene, xylene or methylcyclohexane in admixture with
tetrahydrofuran. A mixture of toluene and tetrahydrofuran
in a ratio of about 1:1 is particularly preferred. Between
0.1 and 1 litre of the above mentioned organic solvent or
mixture of solvents are used per mol of starting cc>mpound
(IV) used. Preferably, 0.2-0.5 1 of the above mentioned
organic solvent or mixture of solvents are used per mol of
oxazoline (IV) put in. Most preferably, about 0.3 to 0.35
1 of the organic solvent or mixture of solvents are: used
per mol of oxazoline (IV) put in.
CA 02364862 2001-08-24
WO 00/51961 - 10 - PCT/EPOO/01162
The aqueous lower phase is then separated off and the
remaining upper phase is extracted several times,
preferably 2-3 times, most preferably twice with water.
According to the invention, the amount of washing water
used for each extraction process is within a range of from
0.05-0.5 1 water per mol of oxazoline (IV) used.
Preferably, in each extraction step, 0.1-0.2 litres of
water are used per mol of starting compound (IV) put in.
The washed organic upper phase is then made alkaline. This
may be done according to the invention with aqueous
solutions of alkali metal or alkaline earth metal
hydroxides. Preferably, aqueous solutions of lithium,
sodium or potassium hydroxide are used. According to the
invention, aqueous sodium hydroxide solution is
particularly preferred as the base. 0.7-1 mol of base,
preferably 0.8-0.9 mol of base are used per mol of
starting compound (IV).
After the phase separation has taken place the lower phase
is decanted into another reaction vessel. The upper phase
remaining is then subjected to the above mentioned
alkalisation. According to the invention, however, only
about 10% w/w of the quantity of base used in the first
alkalisation step are added. Once the lower phase has been
separated off, the combined aqueous extracts are freed
from any entrained solvent by distillation. About 0.05-0.5
1 water, preferably between 0.07 and 0.2 1, most
preferably about 0.1 litres of water are distilled off per
mol of starting compound (IV) used. After cooling to a
temperature below 40 C, preferably to a temperature in the
range from 20-30 C, most preferably to 25 C, 0.1-0.5 1,
preferably about 0.2 1 of water are added per mol of
starting compound put in and the mixture is then made
acidic with 1-5 mol, preferably, 2-4 mol, most preferably
about 3.5 mol hydrochloric acid.
The product precipitated is centrifuged, washed with water
and dried.
CA 02364862 2001-08-24
WO 00/51961 - 11 - PCT/EP00/01162
The Examples which follow serve to illustrate some methods
of synthesising derivatives of biphenyl-2-carboxylic acid
of general formula (I) carried out by way of example,
according to the invention. They should be understood as
being purely possible procedures described by way of
example, without restricting the invention to their
contents.
Example 1
265 kg of 41-methyl-2-(4,4-dimethyloxazolin-2-yl)biphenyl,
205 1 of water and 400 kg of 36.5 o hydrochloric acid are
placed in a 1200 1 enamel stirring apparatus. After it has
been rendered inert with nitrogen it is evacuated to about
50 mbar and then 102.5 1 of methylcyclohexane are added.
After the vapour seal valve has been closed the apparatus
is heated to about 140 C within about 1 h and then stirred
for a further 4 to 8 h at 140 - 145 C. An internal
pressure of 4 - 5 baro is built up. Then the apparatus is
cooled to 20 - 30 C, adjusted to atmospheric pressure with
nitrogen and 175 1 of toluene and 150 1 of THF are added.
The aqueous lower phase is separated off and the organic
upper phase remaining is extracted with 205 1 and then
with 103 1 of water. A further 512 1 of water and 80 kg of
45 o sodium hydroxide solution are added to the upper
phase and, after settling, the lower phase is drained off
into another 1200 1 enamel stirring apparatus. This
operation is repeated with 103 litres of water and 8.9 kg
of 45 o sodium hydroxide solution. To begin with, about
103 1 are distilled off from the combined aqueous extracts
and after cooling to 25 C, 205 litres of water and then 97
kg of 36.5 a hydrochloric acid are added. The product is
centrifuged, washed with water and dried.
Yield : 190 kg of 4'-methylbiphenyl-2-carboxylic acid
(90%)
CA 02364862 2001-08-24
WO 00/51961 - 12 - PCT/EP00/01162
RxaID 1~P 2
251 kg of 2-(4,4-dimethyloxazolin-2-yl)biphenyl, 205 1 of
water and 400 kg of 36.5 o hydrochloric acid are placed in
a 1200 1 enamel stirring apparatus. After it has been
rendered inert with nitrogen it is evacuated to about 50
mbar and then 102.5 1 of methylcyclohexane are added.
After the vapour seal valve has been closed the apparatus
is heated to about 140 C within about 1 h and then stirred
for a further 4 to 8 h at 140 - 145 C. An internal.
pressure of 4 - 5 baro is built up. Then the apparatus is
cooled to 20 - 30 C, adjusted to atmospheric pressure with
nitrogen and 175 1 of toluene and 150 1 of THF are added.
The aqueous lower phase is separated off and the organic
upper phase remaining is extracted with 205 1 and then
with 103 1 of water. A further 512 1 of water and 80 kg of
45 % sodium hydroxide solution are added to the upper
phase and, after settling, the lower phase is drained off
into another 1200 1 enamel stirring apparatus. This
operation is repeated with 103 litres of water and 8.9 kg
of 45 o sodium hydroxide solution. To begin with, about
103 1 are distilled off from the combined aqueous extracts
and after cooling to 25 C, 205 litres of water and then 97
kg of 36.5 o hydrochloric acid are added. The product is
centrifuged, washed with water and dried.
Yield : 180 kg of biphenyl-2-carboxylic acid (91%)
S'_ompariso xamnle:
265 kg of 41-methyl-2-(4,4-dimethyloxazolin-2-yl)bi.phenyl,
205 1 of water and 400 kg of 36.5 % hydrochloric acid are
placed in a 1200 1 enamel stirring apparatus. After it has
been rendered inert with nitrogen, evacuated to about 50
mbar and the vapour seal valve has been closed, the
contents of the apparatus are heated to about 140 C within
about 1 h and then stirred for a further 4 to 8 h at 140 -
CA 02364862 2001-08-24
WO 00/51961 - 13 - PCT/EPOO/01162
145 C, during which time an internal pressure of 4 5
baro is built up. Then the apparatus is cooled to 20 -
30 C, adjusted to atmospheric pressure with nitrogen and
175 1 of toluene and 150 1 of THF are added. The aqueous
lower phase is added to the waste water and the organic
upper phase remaining is extracted with 205 1 and then
with 103 1 of water. A further 512 1 of water and 80 kg of
45 % sodium hydroxide solution are added to the upper
phase and, after settling, the lower phase is drained off
into another 1200 1 enamel stirring apparatus. This
operation is repeated with 103 litres of water and 8.9 kg
of 45 % sodium hydroxide solution. To begin with, about
103 1 are distilled off from the combined aqueous extracts
and after cooling to 25 C, 205 litres of water and then
97 kg of 36.5 % hydrochloric acid are added. The product
is centrifuged, washed with water and dried.
Yield : 100 kg of 4'-methylbiphenyl-2-carboxylic acid
(47%)