Note: Descriptions are shown in the official language in which they were submitted.
CA 02364914 2001-08-21
WO 00/51983 PCT/US00/04386
1
N-SUBSTITUTED BENZOYL INDOLES AS ESTROGENIC AGENTS
The present invention relates to new N-substituted benzoyl indole compounds
which are useful as estrogenic agents, as well as pharmaceutical compositions
and
methods of treatment utilizing these compounds and processes for preparing
them.
Background of the Invention
Estrogen replacement therapy has been well established as the treatment of
choice in women for the prevention of osteoporosis. [C. Christiansen, R.
Lindsay,
Estrogen , Bone Loss and Preservation, Osteoporosis International, l, 15-21
(1990)] The downside to this therapy is that when estrogen is given alone i.e.
without
the opposing effects of progestins, proliferative effects on the uterus may
result and
thereby can put the patient at risk for endometrial cancer. Although less
clear,
hormone replacement therapy has been implicated in increasing the incidence of
breast tumor formation. Non-steroidal antiestrogen drugs such as tamoxifen
have
been used in the treatment of breast cancer. The drug also is known to
maintain bone
mass, acting as a bone-sparing estrogen agonist, however it is also an agonist
in
uterine tissue. A more recent antiestrogen drug, Lilly's raloxifene, is a non-
steroidal
antiestrogen which appears to be more tissue selective. While having the
desirable
property of sparing bone, it has been demonstrated to stimulate uterine growth
in
animal models to a lesser degree than tamoxifen. Additionally, recent clinical
data
reveal no endometrial hyperplasia. A review on the tissue selective action of
estrogen
analogs has recently appeared. (G.L. Evans and R.T. Turner, Tissue Selective
Actions
ofEstrogenAnalogs, Bone, 17, no. 4, 1815-1905 (1995)].
The use of indoles as estrogen antagonists has been reported by Von Angerer,
Chemical Abstracts, Vol. 99, No. 7 (1983), Abstract No. 53886u. Also, see, J.
Med.
Chem. 1990, 33, 2635-2640; J. Med. Chem. 1987, 30, 131-136. Also see Ger.
Offen.,
DE 3821148 A1 891228 and WO 96/03375. These prior art compounds share some
CA 02364914 2001-08-21
WO 00/51983 PCT/US00/04386
2
structural similarities with the present compounds, but are functionally
different. For
compounds containing a basic amine, there is no phenyl group to ridgidify the
side
chain. The reported data for these compounds indicates that they may have a
weaker
binding to estrogen receptor than the compounds of the present invention and
the
basic side chain containing compounds show some uterotrophic effect in the rat
uterus.
WO A 95 17383 (Kar Bio AB) describes indole antiestrogens with long
straight chains. Another related patent WO A 93 10741 describes 5-
hydroxyindole
with a generic descriptor incorporating other side chains.
U.S. Patent No. 5,496,844 (Inai, et al.) teaches substituted N-indole
compounds having potent antiestrogenic activity which are useful in the
treatment of
estrogen-dependent diseases, such as anovulatory infertility, prostatic
hypertrophy,
osteoporosis, breast cancer, endometrial cancer and melanoma.
Jones et al., in their article Antiestrogens. 2.l Structure Activity Studies
in a
Series of 3-Aroyl-2-arylbenzo~bJthiophene Derivatives Leading to ~6-Hydroxy-2-
(4-
hydroxyphenyl)benzo~bJ thien-3 ylJ(4-~2-(1 piperidinyl)ethoxyJphenylJmethanone
Hydrochloride (LY156758), a Remarkably Effective Estrogen Antagonist with Only
Minimal Intrinsic Estrogenicity, J. Med. Chem. 1984, 27, 1057-1066, disclose a
series
of 3-amyl-2-arylbenzo[b]thiophene derivatives which act as non-steroidal
antiestrogens.
The compounds described in the present invention are mixed estrogen
agonists/antagonists and have potential use in treating osteoporosis,
endometriosis,
prostatic hypertrophy, breast cancer and endometrial cancer.
CA 02364914 2001-08-21
WO 00/51983 PCT/US00/04386
3
Description of the Invention
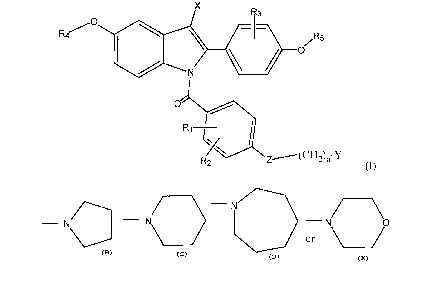
The present invention provides N-substituted indoles of Formula (I):
~ri Y
Formula (I)
wherein:
RI, RZ and R3 are independently selected from hydrogen, halogen, C1-Clz
alkoxy (straight chain or branched or cyclic), -CF3, -N02, cyano, C1-C6 alkyl
(straight
chain or branched), trifluoromethyl, -OH or the C 1-C ~ Z esters (straight
chain or
branched) thereof, or C~-C6 halogenated ethers, preferably CI-C3 halogenated
ethers,
including trifluoromethyl ether and trichloromethyl ether;
R4 and R~ are independently selected from H or benzyl, the benzyl group being
optionally substituted by C1-C6 alkyl, C1-C6 alkoxy, -CF3, or halogen;
X is H, Cl-C6 alkyl, or CF3;
ZisOorS;
nis2or3;
Y is selected from:
a) a moiety of the formula:
CA 02364914 2001-08-21
WO 00/51983 PCT/US00/04386
4
R'
-N
R'
wherein R' is C1-C6 lower alkyl the same or different; or
b) a moiety selected from the group of:
- N
- N - - N
or
or a pharmaceutically acceptable salt thereof.
Examples of R~, RZ and R3 when esters are C2-C12 alkyl esters such as
-O(C=O)(C i-Cbalkyl).
Examples of alkyl groups are methyl, ethyl, n-propyl, isopropyl and n-butyl.
Examples of alkoxy groups are methoxy, ethoxy, n-propoxy, isopropoxy and
n-butoxy.
Examples of RI and R2 are H, Cl-C6 alkyl, C1-C6 alkoxy -CF3 and NO2.
Examples of R3 and R4 are H. An example of X is methyl.
A preferred group of this invention are those compounds of Formula I wherein
Ri, RZ and R3 are independently selected from hydrogen, C1-C6 alkyl, C1-C6
alkoxy, -
CF3, or -N02; and R4, Rs, X, Z, n, and Y are as defined above, or a
pharmaceutically
acceptable salt thereof.
Another preferred group of compounds of this invention are those in which Z
is oxygen and Rl, R2, R3, and R4 are H, or a pharmaceutically acceptable salt
thereof.
CA 02364914 2001-08-21
WO 00/51983 PCT/US00/04386
Preferably RS is H. Among the most preferred compounds of these generic and
subgeneric groups are those in which Y is a piperidine ring.
This invention includes acceptable salt forms formed from the addition
5 reaction with either inorganic or organic acids. Inorganic acids such as
hydrochloric
acid, hydrobromic acid, hydroiodic acid, sulfuric acid, phosphoric acid,
nitric acid as
well as organic acids such as acetic acid, propionic acid, citric acid,
malefic acid, malic
acid, tartaric acid, phthalic acid, succinic acid, methanesulfonic acid,
toluenesulfonic
acid, naphthalenesulfonic acid, camphorsulfonic acid, benzenesulfonic acid are
useful.
Among the preferred salts of the compounds herein are the HCI, HBr, and
acetate
salts.
The compounds of the invention are partial estrogen agonists and display high
affinity for the estrogen receptor. Unlike many estrogens, however, many of
these
compounds do not cause the increases in uterine wet weight normally associated
with
natural or synthetic estrogens. These compounds are antiestrogenic in the
uterus and
antagonize the trophic effects of estrogen agonists in uterine tissue. In
addition, the
compounds may be used as estrogen agonists in bone tissue. Due to the tissue
selective nature of these compounds, they are useful in treating or preventing
in a
mammal disease states or syndromes which are caused or associated with an
estrogen
deficiency or an excess of estrogen.
The present compounds have the ability to behave like estrogen agonists by
lowering cholesterol and preventing bone loss. These compounds are useful for
treating many maladies which result from estrogen excess or deficiency
including
osteoporosis, prostatic hypertrophy, male pattern baldness, ovarian cancer,
infertility,
breast cancer, endometrial cancer, cardiovascular disease, contraception,
Alzheimer's
disease, cognitive decline and other CNS disorders, as well as certain
cancers,
including melanoma, prostrate cancer, cancers of the colon, CNS cancers, among
others. Additionally, these compounds can be used for hormone replacement
therapy
in post-menopausal women or in other estrogen deficiency states where estrogen
supplementation would be beneficial.
CA 02364914 2001-08-21
WO 00/51983 PCT/US00/04386
6
The compounds of this invention may also be used in methods of treatment for
bone loss, which may result from an imbalance in an individual's formation of
new
bone tissues and the resorption of older tissues, leading to a net loss of
bone. Such
bone depletion results in a range of individuals, particularly in post-
menopausal
women, women who have undergone hysterectomy, those receiving or who have
received extended corticosteroid therapies, those experiencing gonadal
dysgenesis,
and those suffering from Cushing's syndrome. Special needs for bone
replacement
can also be addressed using these compounds in individuals with bone
fractures,
defective bone structures, and those receiving bone-related surgeries and/or
the
implantation of prosthesis. In addition to those problems described above,
these
compounds can be used in treatments for osteoarthritis, hypocalcemia,
hypercalcemia,
Paget's disease, osteomalacia, osteohalisteresis, multiple myeloma and other
forms of
cancer having deleterious effects on bone tissues. Methods of treating the
maladies
listed herein are understood to comprise administering to an individual in
need of
such treatment a pharmaceutically effective amount of one or more of the
compounds
of this invention or a pharmaceutically acceptable salt thereof. This
invention also
includes pharmaceutical compositions utilizing one or more of the present
compounds, and/or the pharmaceutically acceptable salts thereof, along with
one or
more pharmaceutically acceptable carriers, excipients, etc.
It is understood that the dosage, regimen and mode of administration of these
compounds will vary according to the malady and the individual being treated
and
will be subject to the judgement of the medical practitioner involved. It is
preferred
that the administration of one or more of the compounds herein begin at a low
dose
and be increased until the desired effects are achieved.
Effective administration of these compounds may be given at an effective dose
of from about 0.1 mg/day to about 1,000 mg/day. Preferably, administration
will be
from about 10 mg/day to about 600 mg/day in a single dose or in two or more
divided
doses. Such doses may be administered in any manner useful in directing the
active
compounds herein to the recipient's bloodstream, including orally,
parenterally
(including intravenous, intraperitoneal and subcutaneous injections), and
transdermally. For the purposes of this disclosure, transdermal
administrations are
understood to include all administrations across the surface of the body and
the inner
linings of bodily passages including epithelial and mucosal tissues. Such
CA 02364914 2001-08-21
WO 00/51983 PCT/US00/04386
7
administrations may be carried out using the present compounds, or
pharmaceutically
acceptable salts thereof, in lotions, creams, foams, patches, suspensions,
solutions,
and suppositories (rectal and vaginal).
Oral formulations containing the active compounds of this invention may
comprise any conventionally used oral forms, including tablets, capsules,
buccal
forms, troches, lozenges and oral liquids, suspensions or solutions. Capsules
may
contain mixtures of the active compounds) with inert fillers and/or diluents
such as
the pharmaceutically acceptable starches (e.g. corn, potato or tapioca
starch), sugars,
artificial sweetening agents, powdered celluloses, such as crystalline and
microcrystalline celluloses, flours, gelatins, gums, etc. Useful tablet
formulations may
be made by conventional compression, wet granulation or dry granulation
methods
and utilize pharmaceutically acceptable diluents, binding agents, lubricants,
disintegrants, suspending or stabilizing agents, including, but not limited
to,
magnesium stearate, stearic acid, talc, sodium lauryl sulfate,
microcrystalline
cellulose, carboxymethylcellulose calcium, polyvinylpyrrolidone, gelatin,
alginic acid,
acacia gum, xanthan gum, sodium citrate, complex silicates, calcium carbonate,
glycine, dextrin, sucrose, sorbitol, dicalcium phosphate, calcium sulfate,
lactose,
kaolin, mannitol, sodium chloride, talc, dry starches and powdered sugar. Oral
formulations herein may utilize standard delay or time release formulations to
alter the
absorption of the active compound(s). Suppository formulations may be made
from
traditional materials, including cocoa butter, with or without the addition of
waxes to
alter the suppository's melting point, and glycerin. Water soluble suppository
bases,
such as polyethylene glycols of various molecular weights, may also be used.
This invention also provides processes for preparing the compounds of
formula I which processes comprises one of the following:
a) acylating a compound of formula:
CA 02364914 2001-08-21
WO 00/51983 PCT/t1S00/04386
8
X
O R3
R ~ ~ ~ _I_
4 ORs
N
H
II
wherein R3, R4, R5 and X are as defined above, with a compound of formula:
HOOC
R1
Zi(CH2)n Y
R2
(III)
or a reactive derivative thereof, e.g. acid halide wherein n, R1, R2, Z and Y
are as
defined above, to give a compound of formula I;
or
b) reacting a compound of formula
X
O R3
R
4
~R'
N
O=C
l
R ~ Z-(CH2)n -hal
(IV)
wherein X, Z, Rl, R2, R3, R4 and RS are as defined above and hal represents a
halogen
e.g chlorine or bromine, with an amine of formula:
H-Y
(V)
CA 02364914 2001-08-21
WO 00/51983 PCT/US00/04386
9
wherein Y is as defined above, to give a compound of formula I,
or
c) debenzylating a compound of formula I wherein R4 and/or RS is optionally
substituted benzyl to give a compound of formula I wherein R4 and/or RS is
hydrogen,
or
e) esterifying a compound of formula I wherein at least one of RI, R2 or R3 is
hydroxy to an ester derivative thereof.
Methods for carrying out process a) -~ e) above are known in the art and/or
are illustrated in the schemes below.
Compounds of this invention may be prepared by methods known in the art.
For instance, the starting or core indole can be prepared by the general
method of
Scheme 1, below.
Scheme No. 1
0
NH +C~
Br
(a) (b)
DMF
X
\ \ h5
0
i N \
H
(c)
The initial indole synthesis for 5-Benzyloxy-2-(4-benzyloxy-phenyl)-3-methyl-
1H-indole is accomplished by heating an appropriately substituted alpha-bromo
CA 02364914 2001-08-21
WO 00/51983 PCT/US00/04386
ketone (b) with the desired aniline (a) in DMF to form the indole (c). The
(aminoethoxy)benzoic acid side chains of the present compounds may be prepared
by
the general methods taught by Jones et al., J. Med. Chem., 1984, Vol. 27, No.
8, pp.
1057-1066 or as shown in Scheme 2 and coupled to the core indoles via the
method of
5 Scheme 3.
Scheme No. 2
O K2C03
NCI I ~ OMe pMF
HO ~ 100°C
(d) (e)
O
H CI
1. NaOH
MeOF OCI2
2. HCI ;HC13
CN
(h)
CA 02364914 2001-08-21
WO 00/51983 PCT/US00104386
11
Scheme No. 3
sodium bis(trimethyl-
silyl)amide, THF
~N'~O
(h) ~ ~ ci
O
thionyl chloride,
CHC13
~N~~
off
(9) O
HO ~ _
OH
-'O
>
THF, EtOH, O
hexadiene ~ (J)
N
Example No. 1
[5-Benzyloxy-2-(4-benzyloxy-phenyl)-3-methyl-indol-1-~l~-[4-(2-piperidin 1 yl
ethoxy)-phenyll-methanone
To a chilled (-78 °C) solution of 2.42 g (0.00577 mol) of the starting
indole
(c), (5-benzyloxy-2-(4-benzyloxy-phenyl)-3-methyl-1H-indole), in 60 mL of dry
THF,
under N2, was added 2.6 g (0.00866 mol) of the acid chloride (h), (4-(2-
piperidin-1-yl-
ethoxy)benzoyl chloride, HCl salt)), and stirred at -78 °C for 20
minutes. 22 mL
(0.0216 mol) of sodium bis(trimethylsilyl)amide (1.0 M solution in THF) was
added
CA 02364914 2001-08-21
WO 00/51983 PCT/US00/04386
12
dropwise to the reaction mixture and stirred at -78 °C for 30 minutes.
The reaction
mixture was then brought to 0 °C for 4 hours, and then to room
temperature for 1
hour. 100 mL of ethyl acetate was added to the crude reaction mixture and
washed
with aq. NaHC03 (2 X 50 mL). The organic phase was collected, washed with
water
(2 X 50 mL), saturated brine, removed, dried over magnesium sulfate, filtered
and
evaporated to dryness in a rotary evaporator. The crude product when subj
ected to
HPLC gave 1.25 g of yellow solid.
Mp = 48 - 51 °C; 1H NMR (DMSO) 7.55 - 7.30 (m, 11 H), 7.24 - 7.16 (m, 5
H), 6.94
6.88(m,SH),5.18(s,2H),S.OS(s,2H),4.09(t,2H,J=5.8Hz),2.19(t,2H,J=
5.8 Hz), 2.41 - 2.38 (m, 4 H), 2.21 (s, 3 H), 1.51 - 1.35 (m, 6 H); IR 3440;
2900, 1610
crri'; MS eI m/z 651 (M+); CHN calcd for C43H42N2O4 ~ 0.25 H20.
Example No. 2
L-Hydroxy-2-(4-hydroxy-phenyl)-3-methyl-indol-1-yll-14-(2-nineridin-1-yl-
ethoxy)-phenyll-methanone
To a solution of 0.78 g (0.00120 mol) of [5-benzyloxy-2-(4-benzyloxy-
phenyl)-3-methyl-indol-1-yl]-[4-(2-piperidin-1-yl-ethoxy)-phenyl]-methanone
(described above) in 5 mL of dry THF, and 5 mL of punctilious ethanol, under
N2,
was added 1.4 mL (0.0120 mol) of cyclohexadiene and 0.39 g (one-half the mass
of
the benzyloxy starting material) of 10% Pd/C, and stirred at room temperature
overnight. The reaction mixture was filtered and evaporated to dryness in a
rotary
evaporator. 100 mL of ethyl acetate was added to the crude product. This
organic
phase was washed with water (2 X 50 mL), saturated brine, removed, dried over
magnesium sulfate, filtered and evaporated to dryness in a rotary evaporator.
The
crude product when subjected to HPLC gave 0.30 g of pale-yellow solid.
Mp = 127 - 130°C;'H NMR (DMSO) 9.47 (s, 1 H), 9.17 (s, 1 H), 7.48 (d, 2
H, J = 8.6
Hz), 7.21 (d, 1 H, J = 8.8 Hz), 7.03 (d, 2 H, J = 8.4 Hz), 6.90 - 6.85 (m, 3
H), 6.69
6.62 (m, 3 H), 4.07 (q, 2 H, J = 5.8 Hz), 2.62 (t, 2 H, J = 5.8 Hz), 2.40 -
2.39 (m, 4
CA 02364914 2001-08-21
WO 00/51983 PCT/US00/04386
13
H), 2.15 (s, 3 H), 1.52 - 1.47 (m, 6 H); IR 3440, 2900, 1610 cm 1; MS eI m/z
471
(M+); CHN calcd for C29H3pN204 ~ 0. 5 H20.
Example No. 3
4-(2-Piperidin-1-yl-ethoxyl-benzoyl chloride Hydrochloride
The title compound was prepared as described by Jones, Charles D., Journal of
Medicinal Chemistry, 1984, Vol. 27, No. 8, pp. 1057-1066.
O SOC12 N O
CHC13 ~O ~
I
1 2
To a solution of the acid HCl salt 1 (1.0 g, 3.7 mmol) in 50 ml chloroform at
room temperature, a solution of thionyl chloride (0.3 ml, 4.4 mmol) in 10 ml
chloroform is added. The resulting solution is brought to 60 °C for 6
hours. The
reaction mixture is then allowed to cool to room temperature and diluted with
hexane.
The reaction mixture is then cooled to 0 °C and the resulting acid
chloride HCl salt, 2,
is isolated by filtration, dried and used without purification in the
acylation step.
Estrogen Receptor Bindin~/Competition Assay
Objective: To identify compounds that compete with 1713-estradiol for estrogen
receptor (ER) binding. The widely accepted mode for estrogenic action is via
its high affinity receptor protein. Compounds which demonstrate an ability to
bind to
the ER may then regulate physiological processes associated with estrogen
action.
CA 02364914 2001-08-21
WO 00/51983 PCT/US00/04386
14
Procedure: Receptor Preparation: CHO cells overexpressing the estrogen
receptor
are grown in 1 SO mm2 dishes in DMEM+ 10% dextran coated charcoal, stripped
fetal bovine serum. The plates are washed twice with PBS and once with 10 mM
Tris-HCI, pH 7.4, 1 mM EDTA. Cells are harvested by scraping the surface and
then
the cell suspension is placed on ice. Cells are disrupted with a hand-held
motorized
tissue grinder using two, 10-second bursts. The crude preparation is
centrifuged at
12,000 x g for 20 min. followed by a 60 min spin at 100,000 x g to produce a
ribosome-free cytosol. The cytosol is frozen and stored at -80 deg C. Protein
concentration of the cytosol is estimated using the BCA assay with BSA as the
reference standard protein.
Binding Assay Conditions:
The competition assay is performed in a 96-well plate (polystyrene*) which
binds <2.0% of the total input [3H]-1713-estradiol. Each data point is
gathered
in triplicate. 100 ~g/100 ql of the receptor preparation is aliquoted per
well. A
saturating dose of 2.5 nM [3H] 1713-estradiol + competitor (or buffer) in a 50
ql
volume is added in the preliminary competition when 100x and SOOx competitor
concentrations are evaluated. For an ICSO determination, where 12
concentrations of
competitor are evaluated, only 0.8 nM [3H] 17 13-estradiol is used. The plate
is
incubated at room temperature for 2.5 h. At the end of this incubation period
150 ~l
of ice-cold dextran coated charcoal (5% activated charcoal coated with 0.05%
69K
dextran) is added/well and the plate is immediately centrifuged at 900 x g for
5
minutes at 4 deg C. 200 ql of the supernatant solution is removed for
scintillation
counting. Samples are counted to 2% or 10 min, whichever occurs first.
Because polystyrene absorbs a small amount of [3H] 17 13-estradiol, wells
containing radioactivity and cytosol, but not processed with charcoal are
included to
quantitate amount of available isotope. Also, wells containing radioactivity
but no
cytosol are processed with charcoal to estimate unremovable DPM of [3H] 17 13-
CA 02364914 2001-08-21
WO 00/51983 PCT/US00/04386
estradiol. Corning #25880-96 96-well plates were used because they
demonstrated
the least binding of estradiol of those tested.
Analysis of Results:
5
Counts per minute (CPM) of radioactivity are automatically converted to
disintegrations per minute (DPM) by the Beckman LS7500 Scintillation Counter
using a set of quenched standards to generate a H# for each sample. To
calculate the
of estradiol binding in the presence of 100 or 500 fold competitor the
following
10 formula is applied:
((DPM sample-DPM not removed by charcoal/(DPM estradiol-DPM
not removed by charcoal)) x 100% _ % of estradiol binding
15 For the generation of ICSO curves, % binding is plotted vs [compound].
ICSO's
are generated for compounds that show >10% competition at up to a SOOx
competitor
concentration.
Reference Compounds:
Various reference compounds have been evaluated and their ICso
concentration determined. The concentration of these compounds required to
displace
50% of [3H] 17(3-estradiol is:
estradiol: 0.08 ~,M
tamoxifen: 4.50 ~M
raloxifene 0.04 ~M
17a-dihydroequilin 0.15 ~M
Assay Results
CA 02364914 2001-08-21
WO 00/51983 PCT/US00/04386
16
To demonstrate the utility of the compounds of this invention, the compound
of Example No. 2 was tested against the standards tamoxifen, also named (Z)-2-
[4-
(1,2-biphenyl-1-butenyl)-phenoxy]-N,N-dimethylethanamine, and raloxifene, also
named [6-Hydroxy-2-(4-hydroxy-phenyl)-benzo[b]thiophene-3-yl]-[4-(2-piperidin-
1-
yl-ethoxy)-phenyl]-methanone. It is understood that 17(3-estradiol as a
standard has
100% binding in the Receptor Binding Assay.
Receptor Binding
Compound Assay -ICso Transfection Assay
Example No. 2 2.0 x 10-~ M Concentration % Control
1 x 10-6 M 0
1x10-6M+
1 nM estradiol 13
tamoxifen 4.5 x 10-6 M Concentration % Control
1x10~6M 0
1x10-6M+
1 nM estradiol 10
raloxifene 4 x 10-8 M Concentration % Control
1x10-6M 0
1 x 10-6 M +
1 nM estradiol 0