Note: Descriptions are shown in the official language in which they were submitted.
CA 02365460 2001-09-25
WO 00/59963 PCT/EP00/02733
1
FLUORINATED COPOLYMERS FOR COATING BIOMEDICAL DEVICES AND
A PROCESS FOR THEIR MANUFACTURE.
The present invention relates to a new class of fluorinated copolymers
which, due to their good combination of low or negligible cytotoxicity,
biocompatibility (especially blood compatibility), infection and encrustation
resistance and due to their interaction with tissue cells leading to
integration, are
useful for biomedical applications. The present invention also relates to a
process
for manufacturing such fluorinated copolymers. Furthermore, this invention
also
relates to implantable and invasive biomedical devices such as implants,
stents,
catheters, staples and the like which are coated with a layer of such a
fluorinated
copolymer. Another use of such polymers is as a substrate for seeding
endothelial
cells. Finally, the present invention relates to a method for treating
atherosclerosis
by making use of a coronary stent coated with a layer of such a fluorinated
copolymer, the said layer being optionally loaded with biologically-active
ingredients. Thus the invention is in the field of biomaterials, more
particularly for
stenting of coronary arteries and for other implant therapies.
BACKGROUND OF THE INVENTION
For the treatment of atherosclerosis, it is now common to use a coronary
stent made from a metal tubing which is brought to the place of obstruction,
i.e.
inserted into a vessel, for instance via percutanic transluminal coronary
angioplasty
and then dilatated, e.g. by a balloon catheter, in order to prevent closure of
the said
vessel due to a stricture or other external cause of compression. Stents
provide a
permanent support of the vessel wall and ensure an efficient flow
therethrough.
However, acute occlusion due to thrombogenecity of the metal and restenosis
due
to neointimal proliferation of smooth muscle cells tend to impede the success
of the
presently existing stents. In particular, their thrombotic character induces
the need
for a simultaneous anti-coagulation therapy which adds to the discomfort of
the
treatment.
CA 02365460 2001-09-25
WO 00/59963 PCT/EP00/02733
2
Restenosis of blood vessels occurs after narrowed or occluded arteries are
forcibly dilated by balloon catheters, drills, lasers and the like in an
angioplastic
procedure. Such forcible dilation is required in order to reopen arteries
which have
been narrowed or occluded by atherosclerosis. However, nearly half of all
arteries
which have been treated by angioplasty return to their narrowed state through
the
process of restenosis. Restenosis is caused both by recoil of the vessel wall
towards its original dimensions and by neointimal hyperplasia induced by
trauma to
the vessel wall. Restenosis can therefore significantly reduce the efficacy of
angioplasty and as such is a major barrier to the effective treatment of
narrowed
arteries.
Attempts to reduce or eliminate restenosis have generally focused on the
design of biomedical devices, such as stents, within the treated artery.
Stents are
expected to reduce restenosis by preventing recoil of the treated blood vessel
to its
original dimensions. Various stents are known in the art, including those
which are
expandable by balloon catheters, heat expandable or self-expandable.
Unfortunately, stents alone cannot prevent restenosis caused by neointimal
hyperplasia of the tissues of the vessel wall. In fact, the stent material
itself may
accelerate such hyperplasia, since it is foreign to the body tissues.
Therefore there
is a need in the art for coating stents with a material which is not
recognized as a
foreign body. Further, there is also a need for a stent coating material which
can be
applied onto the stent body as a flat thin layer, because during stenting,
expansion
usually leads to a rough surface which will cause more tissue damage, e.g.
more
restenosis. Solving these problems would open the way to a number of further
medical applications since stents are not only used for coronary arteries, but
also
in the oesophagus for strictures or cancer, the urether for maintaining
drainage
from the kidneys, or the bile duct for pancreatic cancer or
cholangiocarcinoma.
Another requirement of biomedical devices is the resistance to bacterial,
viral, fungal and other undesirable infections. This is especially true of
some
devices which become unworkable after a short period of time and must be
replaced from time to time. For example frequent replacement of urinary
catheters
CA 02365460 2001-09-25
WO 00/59963 PCT/EP00/02733
3
may cause excessive discomfort to the patient and prolonged hospitalization.
Infections in the case of intravenous catheters used for critical care
patients can
prove to be life threatening. Therefore there is a need in the art for devices
of all
kinds exhibiting an improved infection resistance, in particular bacterial
resistance.
SUMMARY OF THE INVENTION
The present invention provides a novel class of copolymers comprising
moieties derived from at least one first fluorinated comonomer having the
formula
CH2 = C - COOCH2 Y(CF2)~ CF2Z (I)
r
x
wherein
- X is hydrogen or methyl,
- Y is a single bond or a radical of the formula CH2NRS02 , R being a C~_6
alkyl group,
- Z is hydrogen or fluorine, and
- nisOto12
and moieties derived from at least one second non-fluorinated comonomer, the
glass transition temperature of the homopolymer of said second comonomer
being lower than the glass transition temperature of the homopolymer of said
first comonomer.
The present invention further provides terpolymers and tetrapolymers
additionally comprising moieties derived from at least one hydrophilic, e.g.
cationic,
third comonomer and/or from at least one non-hydrophilic fourth comonomer
copolymerizable with the first comonomer and the second comonomer. In view of
their uses for medical applications, the present invention provides such
copolymers, terpolymers and tetrapolymers in the form of a layer preferably
having
a thickness in the range of about 0.1 to 15 pm and optionally further
comprising a
biologically active ingredient. More specifically, the present invention
provides
biomedical devices, e.g. implantable and invasive biomedical devices such as
implants, catheters (both cardiovascular and urinary), artificial veins and
arteries,
valves, stents (especially coronary stents comprising a stent body having a
metal
CA 02365460 2001-09-25
WO 00/59963 PCT/EP00/02733
4
surface) and the like coated with at least one such layer and optionally
coated with
at least one barrier layer. When used in conjunction with a biologically
active
ingredient for the coating of coronary stents, the copolymers, terpolymers and
tetrapolymers of the invention are capable of providing a sustained or
controlled
release of the biologically active ingredient over an extended period of time.
Moreover, the present invention provides use of such a copolymer, terpolymer
or
tetrapolymer as a substrate for seeding of endothelial cells.
Finally, the present invention further provides a method for treating
atherosclerosis by making use of a coronary stent coated with at least one
layer of
such a copolymer, terpolymer or tetrapolymer.
BRIEF DESCRIPTION OF THE DRAWINGS
Figure 1 shows the rate of conversion and the proportions of comonomers,
versus time, incorporated into a copolymer of the invention.
Figure 2 shows the sensorgrams of human serum albumin-antibody
responses in a surface plasmon resonance study of a copolymer of the
invention.
Figure 3 shows antibody binding of various proteins in a surface plasmon
resonance study of a copolymer of the invention.
Figure 4 shows radioactive counts for various copolymers and terpolymers
of the invention in an in vitro bacterial adhesion study via radiolabelling.
Figure 5 shows the detection of adenosine triphosphate for various
copolymers and terpolymers of the invention in a bacterial resistance study.
DETAILED DESCRIPTION OF THE INVENTION
The present invention is based on the unexpected discovery that the polymeric
combination of a fluorinated monomer and a non-fluorinated monomer able to
decrease the glass transition temperature of the polymer sequence derived from
the fluorinated monomer results in useful biomaterials. More specifically, it
was
observed that this polymeric combination, in the form of a copolymer
optionally
comprising additional hydrophilic and non-hydrophilic comonomers, provides
some
generally useful biocompatibility properties. The term "biocompatibility" as
used
herein comprises in particular a low or negligible cytotoxicity, as well as
anti-
CA 02365460 2001-09-25
WO 00/59963 PCT/EP00/02733
S
thrombogenic, anti-inflammatory and anti-immunological properties (i.e.
through
low interaction with activating blood proteins on cell types and attraction of
passivating blood components), bacterial and encrustation resistance. This
promising balance of properties may be tailored to specific needs thanks to
the
wide range of comonomers available within the frame of the invention as well
as
the various manufacturing possibilities for such polymeric combinations. This
may
be achieved in particular by designing a copolymer with a specified glass
transition
temperature (preferably within a range of about -20°C to 20°C)
and/or a specified
number average molecular weight (preferably within a range of about 25,000 to
200,000) and/or a specified molecular weight polydispersity (preferably within
a
range of about 1.3 to 5.5).
The copolymers of the present invention first comprise moieties derived
from at least one first fluorinated comonomer having the formula
CH2= C - COOCH2 Y(CF2)~ CF2Z (I)
1
X
wherein
- X is hydrogen or methyl,
- Y is a single bond or a radical of the formula CH2NRS02 or NRS02, R being
a C~_6 alkyl group,
- Z is hydrogen or fluorine, and
nisOto12.
Thus, depending on the meaning of X, the comonomer of formula (I) is a
fluorinated acrylate or methacrylate optionally (i.e. when Y is not a single
bond)
bearing a sulfonamido group. As can be seen from the meaning of Z, the
comonomer of formula (I) is not necessarily a perfluoro acrylate or
methacrylate. The term « C~_6 alkyl » as used herein, unless otherwise stated,
means a linear or branched alkyl group having from 1 to 6 carbon atoms such
as methyl, ethyl, n-propyl, isopropyl, n-butyl, sec-butyl, tert-butyl, n-
pentyl, n-
hexyl and the like. Non limiting examples of comonomers of formula (I) include
trifluoroethyl methacryfate, octafluoropentyl methacrylate, dodecafluoroheptyl
CA 02365460 2001-09-25
WO 00/59963 PCT/EP00/02733
6
methacrylate, pentadecafluorooctyl methacrylate, heptadecafluorooctyl
ethylsulfonamidoethyl acrylate and heptadecafluorooctyl butylsulfonamidoethyl
acrylate.
Since the homopolymer of the fluorinated monomer of formula (I) usually (i.e.
for a few possible exceptions) usually exhibits a relatively high glass
transition
temperature (Tg), i.e. a T9 of at least about 25°C, it is generally not
entirely suitable
for biomedical applications, most of which require some degree of flexibility
and
elastomeric character which is able to be maintained after sterilization.
Therefore it
is an essential feature of the present invention that the said first
fluorinated
monomer of formula (I) be admixed in a polymeric composition together with at
least one second non-fluorinated comonomer, the glass transition temperature
of
the homopolymer of said second non-fluorinated comonomer being lower than the
glass transition temperature of the homopolymer of said first fluorinated
comonomer. Preferably, the second non-fluorinated comonomer must be
copolymerizable in some way with the first fluorinated monomer of formula (I),
so
that a copolymer comprising moieties from each comonomer species can be
obtained. For reasons of biocompatibility, it is preferred that the second non-
fluorinated comonomer be an acrylic monomer. The term « acrylic » as used
herein, unless otherwise stated, means a monomer bearing either an acrylic or
a
methacrylic function. In view to achieve a desirable degree of flexibility and
elastomeric character for the copolymer of the present invention, it is
usually
preferred to select the second non-fluorinated comonomer in such a way that
the
glass transition temperature of the homopolymer of said second comonomer is
not
above 5°C. A non limiting example of such a non-fluorinated commercial
comonomer is 2-ethylhexyl acrylate. Keeping in mind the desirable Tg of the
homopolymer, those skilled in the art will readily be able to access
additional
suitable comonomers, in particular acrylic comonomers.
In view of the biomedical applications for which the copolymers of the present
invention have been designed, it is preferred that such copolymers exhibit a
glass
transition temperature in the range of about -20°C to 20°C. As
already mentioned
CA 02365460 2001-09-25
WO 00/59963 PCT/EP00/02733
7
before, copolymers with a T9 above about 20°C will generally exhibit
some kind of
brittleness which will make them inappropriate for most biomedical uses,
namely
for use in implants such as stents and catheters. At the opposite, copolymers
with
a T9 below about -20°C will generally exhibit some substantial tendency
to
stickiness which will make them uneasy to handle in manufacturing processes
and/or in application of biomedical devices. Tailoring the glass transition
temperature of the copolymer of the invention in the range of about -
20°C to 20°C
is easily available to those skilled in the art and can be achieved by a
number of
different ways such as, for instance
- selecting the first fluorinated comonomer within the frame of formula (I),
- selecting the second non-fluorinated comonomer according to the Tg of its
homopolymer,
- selecting the respective proportions of said first and second comonomers,
namely by making use of Fox's equation as mentioned by L.M.Sperling in
« Introduction to physical polymer science » page 357 (John Wiley and
Sons, New-York, 1992).
Depending on the choice made for each of the first and second comonomers,
the average composition of the copolymers of the present invention may thus be
from about 1 to 99 mole %, preferably from about 25 to 75 mole %, of the first
fluorinated comonomer and from about 99 to 1 mole %, preferably from about 75
to
mole. %, of the second non-fluorinated comonomer. As further indicated below,
this average composition does not exclude the presence in the copolymer of
some
fragments or sequences or blocks exclusively constituted from either comonomer
moieties.
25 The detailed structure of the copolymers of the present invention may in
some
respect play a role in their suitability for biomedical applications and was
therefore
investigated. This detailed structure, as is well known to those skilled in
the art,
mostly depends on the process of manufacture of the relevant copolymers.
Therefore, the following explanations should be understood as being linked to
their
obtention via a free radical polymerization process. Within such a process, it
was
CA 02365460 2001-09-25
WO 00/59963 PCT/EP00/02733
8
observed that the copolymers of the present invention most often exhibit a
multi-
sequence structure with variable proportions of the first comonomer and of the
second comonomer in each sequence, i.e. the comonomer proportions are not
homogeneous within all macromolecular chains. In some cases, as will be
further
demonstrated in the following examples, the copolymers of the present
invention
include at least one block derived from the homopolymer of the first
fluorinated
comonomer and/or at least one block derived from the homopolymer of the second
non-fluorinated comonomer, i.e. they may include a sequence from either or
both
homopolymers of the building comonomers. This may be derived from the fact
that
one of the comonomers, usually the fluorinated comonomer of the formula (I),
is
substantially consumed at the very beginning of the copolymerization reaction
(i.e.
is incorporated faster in the macromolecular chains) whereas the othe
comonomer,
usually the non-fluorinated comonomer, is substantially consumed at the very
end
of the copolymerization reaction. This heterogeneity of at least some of the
copolymers of the present invention is also confirmed by the presence of
multimodal peaks in their gas permeation chromatograms.
Depending on the type of polymerization process used, it is also possible to
obtain the copolymers of the present invention with a branched, grafted, star-
shaped or comb-shaped structure.
As already suggested hereinabove, the copolymers of the present invention
may conveniently be obtained via a free radical polymerization process.
Therefore
another object of the present invention is a process for making the said
copolymers, comprising providing a mixture comprising
- at least one first fluorinated comonomer having the formula (I) as stated
hereinabove,
- at least one second non-fluorinated comonomer, the glass transition
temperature of the homopolymer of said second comonomer being lower
than the glass transition temperature of the homopolymer of said first
comonomer,
CA 02365460 2001-09-25
WO 00/59963 PCT/EP00/02733
9
- at least one solvent for the said first fluorinated comonomer and the said
second non-fluorinated comonomer, and
- at least one free radical initiator
to a reactor and submitting the said reactor to temperature and pressure
conditions
under which the said first fluorinated comonomer and the said second non
fluorinated comonomer are able to copolymerize. As a solvent for the said
first
fluorinated comonomer and the said second non-fluorinated comonomer there can
be used a halogenated aliphatic or aromatic hydrocarbon or a mixture thereof.
For
instance, the said solvent may suitably be selected from 1,1,1-
trichloroethane,
trichlorotoluene, trifluorotoluene and their mixtures. Preferably, the
temperature
conditions used in performing the process of the invention include a
temperature
not exceeding the boiling point of the solvent. If it is desired, the pressure
in the
reactor may be either raised above or reduced below the normal pressure,
although this is in general not necessary. The free radical initiator to be
used in the
said process may be selected from azoic compounds such as 2,2'-
azobisisobutyronitrile peroxides (e.g. benzoyl peroxide) or conventional
alternatives) and redox initiators such as benzoyl peroxide combined with N,N-
dimethyltoluidine. Selection of an appropriate free-radical initiator as a
function of
the selected copolymerization temperature and of the effective amount of such
initiator for performing the process of the invention is within the general
knowledge
of those, skilled in the art of polymer synthesis.
An alternative method for preparing the copolymers of the present invention
involves anionically copolymerizing a mixture comprising
- at least one first fluorinated comonomer having the formula (I) as stated
hereinabove, and
- at least one second non-fluorinated comonomer, the glass transition
temperature of the homopolymer of said second comonomer being lower
than the glass transition temperature of the homopolymer of said first
comonomer,
CA 02365460 2001-09-25
WO 00/59963 PCT/EP00/02733
at a temperature below about 30°C in the presence of an aprotic
solvent, for
instance tetrahydrofuran or the like, and further in the presence of at least
an
anionic polymerization catalyst such as an alcaline metal alkoxide of a
tertiary
alcohol (e.g. potassium or cesium tertiary butoxide) and/or a crown-ether such
as
5 1,4,7,10,13,16-hexaoxocyclooctane (18-crown-6). As is well known to those
skilled
in the art, such anionic polymerization can take place at very low
temperatures,
down to -78°C. Selection of an appropriate anionic polymerization
catalyst and of
its effective amount for performing such a process is within the knowledge of
those
skilled in the art of polymer synthesis.
10 For the purpose of some specific medical applications, it has been found
useful to incorporate into the copolymers of the invention, besides the
required
moieties derived from both the first fluorinated comonomer and the second non-
fluorinated comonomer, additional moieties derived from other comonomers, in
particular from comonomers which are able to impart some specific physical and
/or chemical properties to the resulting copolymers. For instance, in order to
impart
some hydrophilicity to the copolymers of the invention, the latter may further
comprising moieties derived from at least one hydrophilic third comonomer
copolymerizable with the first comonomer and the second comonomer. The ability
of the third comonomer to copolymerize with the other :omonomers is easily
determinable by those skilled in the art. For practical reasons, namely for
ensuring
a good biocompatibility of the resulting terpolymer, it is usually preferred
that the
the said hydrophilic third comonomer is an acrylic monomer. Many such acrylic
monomers may be contemplated for this purpose. For instance, the hydrophilic
comonomer may be selected from acrylic acid, methacrylic acid and their alcali
or
alcaline-earth salts, N,N-dialkylaminoalkyl acrylates and methacrylates (e.g.
N,N-
dimethylaminoethylmethacrylate), quaternized salts of N,N-dialkylaminoalkyl
acrylates and methacrylates (e.g. N,N-dimethylaminoethylmethacrylate
benzalkonium chloride useful for its aseptic properties) and
polyalkyleneglycol-
containing monomers wherein the polyalkyleneglycol sequence (e.g.
polyethyleneglycol or polypropyleneglycol) preferably has a molecular weight
in
CA 02365460 2001-09-25
WO 00/59963 PCT/EP00/02733
11
the range of about 400 to 10,000. The latter monomers include, among others,
monoacrylates and monomethacrylates as well as monomers derived from
polyalkyleneglycol-containing macro-initiators.
Incorporation of such a hydrophilic third comonomer results in the formation
of a terpolymer with hydrophilic properties. It may be achieved, when the said
hydrophilic third comonomer is not a quaternized N,N-dialkylaminoalkyl
acrylate or
methacrylate, directly copolymerizing the said hydrophilic third comonomer in
a
reactor together with the first fluorinated comonomer and the second non
fluorinated comonomer under the conditions stated hereinabove. When the
hydrophilic third comonomer is a quaternized N,N-dialkylaminoalkyl acrylate or
methacrylate, it is generally preferable to proceed in two steps, by first
copolymerizing a N,N-dialkylaminoalkyl acrylate or methacrylate in a reactor
together with the first fluorinated comonomer and the second non-fluorinated
comonomer under the conditions stated hereinabove (i.e. using a free-radical
initiator and a solvent), and then further reacting the terpolymer obtained
with at
least one halogenated hydrocarbon, preferably a chlorinated aromatic
hydrocarbon
such as benzyl chloride, in a solvent such as methylene chloride. In this way,
hydrocarbon groups such as arylalkyl groups may be covalently linked to the
main
terpolymer chain via the nitrogen atom of the N,N-dialkylaminoalkyl acrylate
or
methacrylate.
When the hydrophilic third comonomer is acrylic acid or methacrylic acid or
their salts and/or a quaternized N,N-dialkylaminoalkyl acrylate or
methacrylate,
copolymerization may result in an ionic terpolymer, i.e. a so-called ionomer,
obtainable in the form of an aqueous solution following procedures well known
to
those skilled in the art. When the hydrophilic third comonomer is a
polyalkyleneglycol-containing monomer, free-radical copolymerization results
in an
amphiphilic terpolymer which is especially useful for its protein- and blood
platelet-
repelling properties. Finally, when the polyalkyleneglycol-containing monomer
derives from a polyalkyleneglycol-containing macro-initiator, the latter may
be
prepared in a two-steps reaction involving, in a first step, reacting an azoic
CA 02365460 2001-09-25
WO 00/59963 PCT/EP00/02733
12
compound having terminal carboxylic acid groups, such as 4,4-
azobiscyanopentanoic acid, with for instance a N-hydroxyimide in a solvent
(such
as tetrahydrofuran) and further in the presence of a coupling agent such as
cyclohexylcarbodiimide, and in a second step reacting the resulting azoic
compound having terminal imido-ester groups with an a-amino-~-methoxy-
polyalkyleneglycol, the polyalkyleneglycol sequence (e.g. polyethyleneglycol
or
polypropyleneglycol) preferably having a molecular weight in the range of
about
400 to 10,000. Such a polyalkyleneglycol-containing macro-initiator can then
be
used as an initiator-comonomer in a free-radical copolymerization process
together
with the first fluorinated and second non-fluorinated comonomers, thus leading
to a
resulting terpolymer with a terminal moiety having e.g. the formula
O CH3
n
CH30 - (PEG) - NH - C - (CH2) 2 - C - (II)
CN
wherein PEG stands for polyethyleneglycol.
The amount or proportion of hydrophilic third comonomer incorporated into
the terpolymers is not critical to the present invention. It usually needs not
to be a
high proportion in order to impart useful hydrophilic properties to the
resulting
terpolymer. A proportion of the hydrophilic third comonomer of up to about 40
mole
%, preferably from about 1 to 30 mole %, with respect to the combined amounts
of
the first.comonomer and the second comonomer is usually a sufficient amount
for
the purpose of the present invention. This proportion will also depend upon
the
nature of the hydrophilic third comonomer ; for instance a proportion of about
1 to
10 mole % should be sufficient when acrylic acid or methacrylic acid
constitutes the
said hydrophilic third comonomer.
The copolymers of the present invention may further comprise moieties
derived from at least one additional non-hydrophilic comonomer (hereinafter
the
« fourth comonomer ») copolymerizable with the first fluorinated comonomer and
the second non-fluorinated comonomer. Various types of comonomers may fall
within this category, for instance the said non-hydrophilic fourth comonomer
may
CA 02365460 2001-09-25
WO 00/59963 PCT/EP00/02733
13
be selected for its ability to modify the copolymer surface properties by
bearing at
least one functional group which is able to react with the copolymer's
environment
in medical applications, such as the amino groups present in protein. Again
for
practical reasons of biocompatibility, the said non-hydrophilic fourth
comonomer
should preferably be an acrylic monomer. A non limiting example of such a
comonomer is succinimidyl methacrylate. Incorporation of such a non-
hydrophilic
fourth comonomer results in the formation of a ter- or tetrapolymer with
additional
specific physical or chemical properties, including a suitable reactivity with
amino
groups. For instance, as demonstrated in the following examples, the
succinimidyl
methacrylate moieties are able to react with an a-amino-~-methoxy
polyethyleneglycol in order to introduce polyethyleneglycol methacrylamido
groups
into the polymeric chain. The amount or proportion of the non-hydrophilic
fourth
comonomer is not critical to the present invention. It usually needs not to be
a high
proportion in order to impart additional useful properties to the resulting
ter- or
tetrapolymer. A proportion of the non-hydrophilic fourth comonomer of up to
about
15 mole %, preferably from about 1 to 10 mole %, with respect to the combined
amounts of the first and second comonomers - and optionally third comonomer -
is
usually a sufficient amount for the purpose of the present invention.
Although this is not a limiting factor of the present invention, it was
observed
that the copolymers and terpolymers of this invention most often exhibit a
number
average, molecular weight in the range of about 25,000 to 200,000, more
preferably
about 30,000 to 130,000 and/or a molecular weight polydispersity in the range
of
about 1.3 to 5.5, more preferably about 1.5 to 3.5, i.e. a relatively narrow
distribution of molecular weights.
In view of biomedical applications, it is generally desirable that the
copolymers of this invention may be sterilized. This may conveniently be
effected
by means of irradiation, for instance via electron-beam or X-rays. As is well
known
to those skilled in the art, the choice between electron-beam and X-ray
technologies will mostly depend on the size and thickness of the material to
be
irradiated. For the complete destruction of all living micro-organisms usually
CA 02365460 2001-09-25
WO 00/59963 PCT/EP00/02733
14
involved with infection of biomedical equipment, irradiation doses within a
range of
about 0.5 to 100 kGy, preferably 10 to 50 kGy are recommended.
In view of the main biomedical applications of this invention, it is generally
desirable, and this has proven to be easily feasible, that the copolymers of
the
present invention may be processed in the form of flat thin layers,. The term
« thin
layer » as used herein, unless otherwise stated, means a layer that can
obtained
by either of the conventional dip coating and spray coating techniques. This
usually
corresponds to a thickness in the range of about 0.1 to 15 pm. As already
explained hereinabove, the need for a flat layer is especially crucial for the
coating
of stents. Hence stents coated with a copolymer layer according to the
invention
will help reducing restenosis and therefore improve the success of
angioplasty.
For many biomedical applications of the copolymers of the present
invention, it is generally useful that such copolymer layers may further
comprise a
biologically effective amount of at least one biologically active ingredient
such as a
therapeutic, diagnostic or prophylactic agent. The said therapeutic agent can
be
selected for its specific properties such as for instance its anti-thrombotic,
anti-
inflammatory, anti-proliferative or anti-microbial efficiency. The latter
include for
instance anti-microbial agents such as broad spectrum antibiotics for
combating
clinical and sub-clinical infections. Other therapeutic r.~gents which can be
considered for incorporation into the copolymer layers of this invention may
be
naturally occurring or synthetic organic or inorganic compounds, including
proteins
and peptides (produced either by isolation from natural sources or
recombinantly),
hormones, carbohydrates, antineoplastic agents or anti-proliferative agents,
anti-
inflammatory agents, antiangiogenic agents, vasoactive agents, anticoagulants,
immunomodulators, cytotoxic agents, antiviral agents, antibodies,
neurotransmitters, oligonucleotides, lipids, plasmids, DNA and the like.
Therapeutically active proteins which can additionally be present in the
formulations of this invention include fibroblast growth factors, epidermal
growth
factors, platelet-derived growth factors, macrophage-derived growth factors
such
as granulocyte macrophage colony stimulating factors, ciliary neurotrophic
factors,
CA 02365460 2001-09-25
WO 00/59963 PCT/EP00/02733
cystic fibrosis regulator genes, tissue plasminogen activator, B cell
stimulating
factors, cartilage induction factor, differentiating factors, growth hormone
releasing
factors, human growth hormone, hepatocyte growth factors, immunoglobulins,
insulin-like growth factors, interleukins, cytokines, interferons, tumor
necrosis
5 factors, nerve growth factors, endothelial growth factors, non-steroidal
anti-
inflammatory drugs, osteogenic factor extract, T cell growth factors, tumor
growth
inhibitors, enzymes and the like, as well as fragments thereof. Diagnostic
agents
which can be present in the copolymer layers of this invention include
conventional
imaging agents (for instance as used in tomography, fluoroscopy, magnetic
10 resonance imaging and the like) such as transition metal chelates. Such
agents
should be incorporated into the layers of the invention in an effective amount
for
performing the relevant diagnostic. As is well known to those skilled in the
art of
pharmacology, the at least one biologically active ingredient may be
formulated
together with additional pharmaceutically acceptable excipients and carriers,
as
15 well as together with a biological delivery system able to perform a
specific
pharmacological function, such as for instance a composition for providing a
controlled or sustained release of the said biologically active ingredient
into the
body of a mammal, in particular of a human patient in need of a specific
therapy.
As will readily be understood by those skilled in the art of such biomedical
applications, the nature and amount of the at least one biologically active
ingredient incorporated in the copolymer layers of the present invention
depends
on the specific biological action to be performed when implanting a device
coated
with such a layer into the body of a patient in need of a specific therapy.
Another embodiment of the present invention consists of a biomedical
device, for instance an implantable or invasive biomedical device, coated with
at
least one layer comprising a copolymer as hereinabove described (i.e.
including
ter- and tetrapolymers possibly incorporating a third and/or fourth comonomer)
and
possibly further comprising a biologically effective amount of at least one
biologically active ingredient. The form, shape and size of the biomedical
device is
not critical for the present invention. Non limiting examples of implantable
and
CA 02365460 2001-09-25
WO 00/59963 PCT/EP00/02733
16
invasive biomedical devices which fall within the framework of this invention
include, for instance, catheters, stents, staples, threads, needles,
pacemakers,
valves, artificial veins and arteries, electronic pumps and sensors, tubings,
prostheses, i.e. implants and materials of all kinds designed for use in the
body of
a mammal, and more specifically for the human body. Such implantable and
invasive biomedical devices are well known to those skilled in the art of
surgery
making use of implants. If necessary for some end uses, they may additionally
be
coated with at least one barrier layer. Preferably, the biomedical devices of
this
invention have a bio-inert surface, for instance a hydrophobic surface with
low
surface energy and possibly with amphiphilic heterogeneous microdomains. The
biomedical device of this invention may also usefully exhibit antagonist
properties
namely against protein activation, cell activation (e.g. anti-inflammatory),
bacteria,
and/or anti-thrombotic properties, encrustation repellency and the like.
In particular, the biomedical device of this invention may be a stent, for
example a coronary stent, comprising an expandable stent body having a plastic
or
metal surface and wherein the copolymer layer is coated onto at least a part
of the
said surface, for instance onto at least one side thereof. Such a stent is
particularly
useful for the treatment of atherosclerosis, consequently the present
invention also
provides a method for such treatment by making use of, i.e. by implanting into
the
body of a patient in need of such treatment, a coronary stent coated with a
copolymer layer (such as disclosed hereinbefore) on at least a part of its
surface.
Moreover, another embodiment of the present invention consists of a process
for
making a coated stent, comprising coating a stent body having a non-
biocompatible surface, for instance a metal or plastic surface, with at least
one
polymer, the said polymer comprising a copolymer as hereinabove described
(i.e.
including ter- and tetrapolymers possibly incorporating a third and/or fourth
comonomer) and possibly further comprising a biologically effective amount of
at
least one biologically active ingredient. Coating may be efFected by any of
the
conventional dip coating or spray coating techniques and is preferably
effected in a
manner such as to provide~a thin copolymer layer such as hereinabove
described.
CA 02365460 2001-09-25
WO 00/59963 PCT/EP00/02733
17
As is readily understandable by those skilled in the art, the use of the
copolymer layer of this invention in biomedical applications, especially in
implantable and invasive biomedical devices, is among others based on a
satisfactory adhesion to the copolymer of those proteins which are abundantly
present in plasma and which fulfill transport and passivating functions in the
last
step of the coagulation cascade in immune reactions such as albumin,
fibrinogen
and immunoglobulin G. Adherence of such proteins is believed to facilitate
endothelial cell adherence. The formation of a thin natural endothelial layer,
possibly obtained via tissue engineering techniques such as cell seeding, will
substantially prevent the surface, for instance the metal surface, of the said
implantable or invasive biomedical device to be recognized as a foreign body
by
the cells of the patient in which the device is implanted. Tissue reactions
such as
inflammation, severe immunological reactions, thrombus formation, accute
occlusion or restenosis are thereby minimized or suppressed. A steady or
controlled release of restenosis-suppressing agents over a period of about 10
to 30
days would also allow to inhibit the start of the restenosis process. Protein
adhesion was found to be especially satisfactory for those terpolymers of the
invention which include moieties derived from a cationic third comonomer, such
as
a quaternized N,N-dialkylaminoalkyl acrylate or methacrylate or a
(meth)acrylic
acid or acid salt.
The present invention will now be explained in further details by reference to
the following exampleswhich are provided for illustrative purposes only and
without
any limiting intention.
EXAMPLE 1 - preparation of octafluoropentylmethacr~rlate
100 g (0.43 mole) octafluoropentanol from Acros Chemical was dissolved in
100 ml methylene chloride. After addition of 60 ml triethylamine (0.43 mole),
the
solution was cooled down to 0°C. Next, 65 ml methacrylic acid anhydride
(0.43
mole) was added dropwise. After 4 hours reaction at 40°C, the reaction
mixture
was further stirred overnight at room temperature. In order to neutralize
traces of
unreacted methacrylic acid anhydride, the reaction mixture was reacted during
20
CA 02365460 2001-09-25
WO 00/59963 PCT/EP00/02733
18
minutes with 5.2 ml (0.086 mole) aminoethanol and then diluted with 100 ml
CH2C12 and 100 ml water and additionally stirred for 1 hour. Extraction of the
organic phase was then effected respectively by means of a 3M HCI solution (3
times), a saturated NaCI solution (3 times) and a NaHC03 solution (3 times).
The
resulting CH2C12 phase was dried with MgS04 and evaporated, leaving, after
vacuum distillation, octafluoropentylmethacrylate with a yield of 83 %.
Monomer characterization was effected by means of'H-NMR in deuterated
acetone and provided the following spectrum: 8(CH3) 1.92 ppm (s), 8(OCH2) 4.82
ppm (t), 8(CH2=C) 5.78 ppm (s) and 6.17 ppm (s), 8(CH2F) 6.78 ppm (tt).
EXAMPLE 2 - preparation of succinimidyl methacr I
In a two-neck reactor vessel, 4.3 g of N-hydroxysuccinimide from Acros
Chemical (0.043 mole) and 30 ml dichloromethane (CH2C12) were added. Only
after addition of 4.4 g (0.043 mole) of triethylamine, a clear mixture was
obtained.
After cooling down to 0°C, 3 g (0.029 mole) methacrylic acid chloride
was added.
After 5 hours reaction at 0°C, the reaction mixture was poured into 40
ml of a
saturated NaHC03 solution. After 2 times extraction with 40 ml of a saturated
NaHC03 solution, and 2 times extraction with 40 ml water, the organic phase
was
dried with MgS04 , filtered and evaporated. The solid succinimidyl
methacrylate
resulting monomer was vacuum dried and characterize; by 'H-NMR in CDC13,
providing the following spectrum: 8(CH3) 2.05 ppm (s), 8(CH2) 2.85 ppm (s),
8(CH2=C) 5.87 ppm (s) and 6.42 ppm (s).
EXAMPLE 3 -preparation of a poly(octafluoropentylmethacrylate-co-2-
ethylhexylacrylatel
10g octafluoropentylmethacrylate prepared according to example 1 (0.333
mole), 2.05 g of 2-ethylhexyl acrylate (0.111 mole) were brought to a
polymerization tube and dissolved in 50 ml CH3CC13 (i.e. 25% weight/volume)
together with 0.23 mmole 2,2'-azobisisobutyronitrile. The polymerization
mixture
was frozen in liquid nitrogen and degassed under vacuum. This was repeated
trice,
after which the polymerization tube was sealed under vacuum. After 24 hours
heating at 65°C, the resulting copolymer obtained with a yield of 93%
was
CA 02365460 2001-09-25
WO 00/59963 PCT/EP00/02733
19
precipitated twice in pentane and vacuum dried. Follow-up of the amounts of
incorporated monomer and unreacted monomer via gas chromatography (as
further detailed in example 9) and via 'H-NMR performed on a Brucker WH 360
MHz apparatus (namely the peak areas for the OCH2 groups of the fluorinated
comonomer at b = 4.55 ppm and of the non-fluorinated comonomer at s = 3.9
ppm) shows that the fluorinated comonomer was incorporated faster, i.e. in the
starting phase preferentially the fluorinated comonomer is incorporated, next
a
random copolymer is formed and finally the remaining non-fluorinated comonomer
reacts. This heterogeneity explains the existence of multimodal peaks for the
copolymer in its gel permeation chromatogram.
The glass transition temperature of this copolymer (measured by means of a
thermal analysis differential scanning calorimeter DSC 2920, at heating and
cooling speeds of 10°C/min.) is 14°C. Its average number
molecular weight
(determined by gel permeation chromatography while using a Styragel mixed B
column, 10p, 2 x 30 cm from Polymer Laboratories on N-methylpyrrolidone and
using polystyrene as a standard) is 89,000 and its molecular weight
polydispersity
MW/M" is 1.7.
EXAMPLE 4 -preparation of a pol~~pentadecafluorooctylmethacr)date-co-2-
ethylhex, I~r)rlatel
The procedure of example 3 is repeated, except that 0.65 mole of
pentadecafluorooctylmethacrylate, either prepared in accordance with a
procedure
similar to example 1 or originating from Polyscience (Germany), is
copolymerized
with 0.35 mole 2-ethylhexyl acrylate while using trichlorotrifluoroethane or
trifluorotoluene as the reaction solvent. The copolymer is obtained with a
yield of
87%. The glass transition temperature (measured as in example 3) of this
copolymer is 13°C. Its average number molecular weight (determined as
in eample
3) is 123,000 and its molecular weight polydispersity MW/M" is 1.8.
EXAMPLE 5 - areaaration of a aolv(trifluoroethvlmethacrvlate-co-2-
eth, I~ylacrylate
CA 02365460 2001-09-25
WO 00/59963 PCT/EP00/02733
The procedure of example 3 is repeated, except that 0.5 mole of
trifluoroethylmethacrylate, either prepared in accordance with a procedure
similar
to example 1 or originating from Polyscience (Germany), is copolymerized with
0.5
mole 2-ethylhexyl acrylate. The copolymer was obtained with a yield of 95%.
The
5 glass transition temperature of this copolymer (measured as in example 3) is
14°C.
Its average number molecular weight (determined as in example 3) is 40,000 and
its molecular weight polydispersity MW/M~ is 3Ø
EXAMPLE 6 - preparation of a poly~octafluoropentylmethacrylate-co-2-
ethylhexylacrylate-co-dimethylaminoethylmethacrylatel
10 Terpolymerization is effected according to a procedure similar to that of
example 3, except that 0.37 g dimethylaminoethylmethacrylate is added into the
polymerization tube. A yield of 80% of terpolymer was obtained.
Characterization
of the said terpolymer was effected by 'H-NMR in CDC13 providing the following
spectrum (for the identification of each signal,
dimethylaminoethylmethacrylate was
15 abbreviated as DM, 2-ethylhexylacrylate as EHA and
octafluoropentylmethacrylate
as FMP): 8(NCH3_oM) 2.25 ppm, 8(NCH2_oM) 2.55 ppm, 8(OCH2_pM) 4.05 ppm,
8(OCH2_EHa) 3.85 ppm, 8(OCH2_FMP) 4.55 ppm, 8(CF2H) 5.85-6.35 ppm (t).
EXAMPLE 7 - preparation of a quaternized polX(octafluoropentylmethacr~-
late-co-2-eth, I~ylacrylate-co-dimethylaminoethylmethac , late
20 To 12.5g of the terpolymer obtained in example 6 (comprising 0.0238 mole
dimethylaminoethylmethacrylate) in solution in CH2C12 (30% (weight/volume) was
added 3.02g of benzylchloride (0.238 mole). After 48 refluxes, a quaternized
terpolymer was obtained with a yield of 91 % via precipitation in pentane and
was
characterized by 'H-NMR in CDC13, providing the following spectrum (monomer
abbreviations are as in example 5): 8(NCH3_pM) 3.45 ppm, 8(NCH2_pM + OCH2_EHA)
3.65-4.00 ppm, 8(OCH2_pM + OCH2_FMP) 4.40ppm, 8(NCH2_pM, a of aromatic ring)
5.13 ppm, 8(CF2H) 5.85-6.35 ppm (t), 8(aromatic H) 7.3-7.8 ppm.
EXAMPLE 8 - preparation of a poly~octafluoropentXlmethacrylate-co-2-
eth~lhexylacrylate-co-succinimidyl methacr~rlatel
CA 02365460 2001-09-25
WO 00/59963 PCT/EP00/02733
21
A procedure similar to that of example 3 is used, except that 0.7125
mole of octafluoropenttylmethacrylate prepared according to example 1 is
copolymerized with 0.2375 mole of 2-ethylhexylacrylate and 0.05 mole of
succinimidyl methacrylate prepared according to example 2, while using
methylene
chloride as the reaction solvent. The copolymer was obtained with a yield of
90%.
EXAMPLE 9 - preparation of a poly(octafluoropentvlmethacr)date-co-2-
ethylhexylacrylate-co-polyeth I~ene~glycolmethacr)rlamidel
1 g of the terpolymer prepared in example 8 was dissolved in 2 ml of
CH2C12, then 0.22 g of an a-amino-c.~-methoxy polyethyleneglycol (also
dissolved in
CH2C12) obtained from Fluka AG and having a molecular weight of 750 (i.e. 17
ethyleneglycol units) was added thereto. The mixture was refluxed during 48
hours
at 60°C. The resulting viscous solution was diluted with an additional
4 ml of
CH2C12 and subsequently extracted with a saturated solution of NaHC03 (twice
10
ml) and then with water (twice 10 ml). The dichloromethane phase was dried by
means of MgS04 and evaporated and the resulting terpolymer, bearing
polyethyleneglycol methacrylamido groups, was further vacuum dried and
characterized by 'H-NMR in CDC13 providing the following spectrum (monomer
abbreviations are as in example 6, with PEG designating polyethyleneglycol):
cS(OCH3_EHA) 3.38 ppm (s), S(OCH2_PEG) 3.65 ppm, 8(OCH2_EHa) 3.90 ppm, 8(OCH2_
FMP) 4.45 ppm, 8(CF2H) 5.85-6.35 ppm (t). Infrared spectroscopy (by means of
Perkin-Elmer 1600 FTIR) additionally shows the appearance of polyether groups
at
1272 and 1259 cm-' and therefor confirms the resulting structure. Similar
syntheses were performed with a-amino-c~-methoxy polyethyleneglycols having
molecular weights from 400 to 4,000 and the resulting terpolymers were used
for
protein adhesion tests via surface plasmon resonance in example 14 and for
sterilization tests in example 17 below.
EXAMPLE 10 - Determination of the degree of conversion in the radical
copolymerisation of octafluoro~ent~rlmethacrvlate and 2-ethylhex I~rylate by
means of cias chromatography
CA 02365460 2001-09-25
WO 00/59963 PCT/EP00/02733
22
0.01 mole of a monomer mixture of octafluoropentylmethacrylate and 2-
ethylhexylacrylate-was weighed in a 10 ml flask. 8.2 mg 2,2'-
azobisisobutyronitrile
(free radical initiator) and 0.67 g n-decane (internal standard) were added.
The
mixture was further diluted with CHC13 to a total volume of 10 ml and brought
to a
two-neck flask, which was sealed with a septum and a valve. Degassing was done
by bringing the reaction mixture several times briefly under vacuum. During
copolymerization, effected at 65°C like in example 3, an inert nitrogen
atmosphere
was maintained. During 28 hours, at regular time intervals, a 50 pl sample was
taken and diluted chloroform to 300 pl. 0.5 pl was injected on a
polydiphenyldimethylsiloxane column (RSL-200 bonded FSOT, 30 m x 0.25 mm,
split injection). After having maintained during 4 minutes a temperature of
35°C,
the oven was heated to 250°C at a rate of 10°C/min. The
retention times for
CH3CC13, octafluoropentylmethacrylate (FMP), n-decane and 2-ethylhexylacrylate
(EHA) were respectively 1'4, 8'0, 8'6 and 12'7. A calibration curve for
concentrations of FMP and EHA between 5 and 20 ng/ml was set in advance. The
peak area of each comonomer, divided by that of n-decane, at initial time
(t=0) was
set to 100% of the corresponding monomer. From the percentages of comonomers
present in the reaction mixture, the percentage of comonomer incorporated in
the
copolymer structure and the conversion rate of the reaction ~ ~Nere deduced.
Curve
fitting was performed using the program "Graphpath Prism" (non-linear
regression).
Figure 1 shows results of this experiment for the copolymer obtained from a
mixture of 50 mole% FMP and 50 mole% EHA. As an additional information, it may
be noted that the glass transition temperature of this copolymer is
2°C, its average
number molecular weight (determined by gel permeation chromatography) is
57,000 and its molecular weight polydispersity MW/M" is 1.8.
EXAMPLE 11 - determination of copolymer compositions by'H-NMR.
The composition of the copolymers of examples 3-5 and 10 was
determined, after recording their 'H-NMR spectra in a deuterated
acetone/chloroform (1/1) mixture, on the basis of the OCH2 peak integrations
for
CA 02365460 2001-09-25
WO 00/59963 PCT/EP00/02733
23
the fluorinated comonomer (e.g. 8 - 4.45 ppm for FMP) and for 2-
ethylhexylacrylate (8 = 3.90 ppm). Results are as follows
Copolymer of example 3 contains 81 mole% octafluoropentylmethacrylate
and 19 mole% 2-ethylhexylacrylate. Copolymer of example 4 contains 67 mole%
pentadecafluorooctylmethacrylate and 33 mole% 2-ethylhexylacrylate. Copolymer
of example 5 contains 51 mole% trifluoroethylmethacrylate and 49 mole% 2-
ethylhexylacrylate. Copolymer of example 10 contains 63 mole% octafluoro-
pentylmethacrylate and 37 mole% 2-ethylhexylacrylate.
EXAMPLE 12 - preparation of polyethyleneglycol-containing macro-
initiators.
A polyethyleneglycol macro-initiator was prepared following a procedure
similar to that described by A.Ueda et al. in J.Polym.Sci., Part A,
Polym.Chem.(1986) 24:405. 1 g of 4,4-azo-bis (4-cyanopentanoic acid) and 0.83g
N-hydroxysuccinimide were dissolved in 10 ml tetrahydrofurane. 1.48 g
dicyclohexylcarbodiimide in 5 ml tetrahydrofurane was slowly added to the
reaction
medium at 0°C. After 24 hours stirring, dicyclohexylurea formed by the
reaction
was removed by filtering. The solvent was evaporated and the azoic compound
with terminal ester groups produced was dissolved in ethyleneglycol
dimethylether.
Next 7.12 mmole of an a-amino-~-methoxy-polyethyleneglycol with a molecular
weight of 750 to 5,000 from Fluka AG (dried via azeotropic distillation with
toluene)
in 20 ml dry CH2C12 was added to the azoic compound with terminal ester
groups.
After 24 hours reaction at room temperature, the polyethyleneglycol-containing
macro-initiator formed was precipitated in pentane, vacuum dried and further
characterized by infrared spectroscopy (linking amide at 1716 cm-',
polyethyleneglycol ether at 1113 cm-', methylene at 2885 cm-') and 'H-NMR in
CDC13: 8(CH3) 1.7 ppm, 8(OCH2) 2.4 ppm and 2.5 ppm, 8(OCH3) 3.4 ppm, 8(OCH~)
3.65 ppm.
EXAMPLE 13 - preparation of polar(octafluoropentylmethacrylate-co-2-
ethylhexylacr~late-co-pol~reth I~eneglycol derived monomer).
CA 02365460 2001-09-25
WO 00/59963 PCT/EP00/02733
24
A polyethyleneglycol macro-initiator according to example 12 was brought,
in a proportion (from 0.5 to 2% by mole of the total monomers) specified in
the
following table 1, to a polymerization tube, then 1 g
octafluoropentylmethacrylate
(3.33 mmole) and 0.205 g of 2-ethylhexylacrylate (1.11 mmole) were added and
toluene was used as the polymerization solvent. The polymerization mixture was
frozen in liquid nitrogen and degassed under vacuum. This was repeated trice,
after which the polymerization tube was sealed. After 24 hours of
polymerization in
a thermostatic bath at 65 °C, the resulting terpolymer was precipitated
twice in
pentane and vacuum dried. A thin film was prepared from the said terpolymer
via
solvent casting from a CH2C12/acetone (80/20) solution and was extracted
during
24 hours at 55 °C with different portions of water, the water fraction
was freeze
dried and the isolated product identified by means of 'H-NMR-spectroscopy in
CDC13 as follows: c~(OCH3_pEG) 3.4 ppm (s), 8(OCH2_pEG) 3.6 ppm, 8(OCH2_EHA)
4.0
ppm, 8(OCH2_FnnP) 4.7 ppm, 8(CF2H) 5.3-6.3 ppm.
Using this procedure, terpolymers with variable proportions of
polyethyleneglycol sequences having various molecular weights from 750 to
5,000
were prepared and used for protein adhesion tests via surface plasmon
resonance
in example 15 and for sterilization tests in example 18 below. Their physico-
chemical properties are shown in table 1 below.
Table 1
Terpolymer M~ (PEG) % mole PEG Tg (C) Cristallinity
reference (%)
13 A 750 0.5 - 7 0.1
13 B 750 2.0 - 20 1.1 i
13 C 2,000 1.0 - 10 1.2
13 D 5,000 0.5 3 5.1
13 E 5,000 1.0 10 11.7
Cristallinity was determined by X-ray measurements.
CA 02365460 2001-09-25
WO 00/59963 PCT/EP00/02733
EXAMPLE 14 - preparation of a poly(octafluoropentylmethacr)date-co-2-
ethylhexylacrylate-co-methacnrlic acidl.
10 g octafluoropentylmethacrylate, 2 g 2-ethylhexylmethacrylate and 0.2 g
methacrylic acid were dissolved in chloroform, then 0.5 mole % 2,2'
5 azobisisobutyronitrile was added. The reaction vessel was degassed three
times
and then sealed. Polymerization was allowed to proceed at 65°C for 24
hours. The
resulting terpolymer, obtained with a yield of 87 %, was precipitated in
pentane,
filtered and dried.
EXAMPLE 15 - protein adhesion to copolymer surfaces measured throuah
10 surface plasmon resonance.
Surface plasmon resonance (SPR) is an optical technique via which
adsorption to a material can be followed in situ. Laser light incides at the
underside
of a prism optically coupled to a microscopy glass covered by a thin gold or
silver
layer. Total internal reflection takes place if light goes to a medium with
smaller
15 refractive index and if the angle of incidence is bigger than the critical
angle. Under
a well defined angle, the resonance angle BSP~, resonance takes place between
the
extinguishing wave of the photon and the free oscillating surface electrons
from the
metal layer (plasmons). Hereby, a minimal intensity of the reflected light
beam
arises and is registered by a photodiode array detector. The angle under which
this
20 occurs depends on the refractive index and on the thickness of the absorbed
layer.
A sensogram is obtained by recording the shift in resonance angle (00Sp~)
versus
time, the slope of the curve being a measure for the amount of adhered
material.
Protein adsorption takes place immediately after introduction of a foreign
body into the body of a mammal and this initially adsorbed protein layer
further
25 influences physiological and cellular processes such as blood coagulation,
fibrinolysis, thrombogenesis, leukocyte activation, immunological and
inflammatory
responses. The driving interaction forces, such as H-bridges, electrostatic
and
hydrophobic interactions, for protein adhesion are non-covalent. Protein
adsorption
is a complex, competitive process whereby some proteins can desorb again and
be replaced by other proteins. Many factors, such as affinity of the protein
for the
CA 02365460 2001-09-25
WO 00/59963 PCT/EP00/02733
26
surface, contact time, concentration and flow speed of the protein play a role
in this
substitution process. When a protein has a high affinity for the surface, it
will
undergo conformational changes so that multiple bonds with the surface are
possible. The longer a protein remains attached, the more it will spread and
the
stronger the bond is. An irreversible protein monolayer is then formed.
Affinity
itself is determined in part by the protein, in part by the material surface
properties.
The Vroman effect is based on the substitution of proteins and suggests that
proteins present in high concentrations bind first and are later replaced by
other
proteins present in smaller amounts, although with high affinity for the
surface.
Plasma proteins such as albumin, immunoglobulin G, fibrinogen, fibronectin,
Hageman factor, high molecular weight kininogen and high density lipoprotein
(HDL) will adhere sequentially.
Material surfaces entail a differential humoral and cellular activity,
depending on the protein composition and conformation in the adsorbed layer. A
change in conformation can entail activation but also inhibition of the
biological
function of the protein. Generally speaking, extremely hydrophilic materials
impede
protein adsorption whereas extremely hydrophobic materials adhere a strongly
attached monolayer. Micro-heterogeneous materials usually adsorb proteins in
an
ordered way, whereby changes in protein structure and cell activation are
avoided.
In the following experiment, it will be shown to which extent human serum
albumin, (HSA) and fibrinogen (HFB) adhere to the copolymer surface and to
which
extent binding is irreversible. 0.5% copolymer solutions and 0.05
(weight/volume) protein solutions were used in this study. Copolymer solutions
were spin-coated onto silver coated glass plates and the shifts in resonance
angle
(06SP~) expressed in millidegrees of angle (mdA) obtained for both HSA and
HFB,
as measured by a surface plasmon resonance instrument (WB1006, Johnson &
Johnson Clinical Diagnostics Ltd., England) are provided in the following
table 2.
CA 02365460 2001-09-25
WO 00/59963 PCT/EP00/02733
27
Table 2
Copolymer of example oesp~ (HAS) oesp~ (HFB)
No
3 65 438
4 61 394
115 526
115 594
7 114 529
9 (PEG 400) 120 439
9 (PEG 4,000) 99 411
13 B 82 345
13 E 65 242
14 134 469
The steep slopes of the SPR curves prove the fast adsorption of proteins, a
maximal adhesion being observed already after 40 seconds. Upon overflowing a
second time, no extra adhesion was observed, indicating the formation of a
protein
monolayer. All copolymers, except for that of example 13, adhered a monolayer
of
fibrinogen. Important variations of ~85p~ (HFB) within a range of about 350 to
600
mdA are believed to be due to the different ways according to which fibrinogen
can
adhere to the copolymer surface: in particuler, the high values of 06Sp~ (HFB)
for the
copolymers of example 6 and 7 are deemed to be due to the presence of a
positively charged nitrogen group strongly interacting with the negatively
charged
fibrinogen (pl = 5.5) at physiological conditions (pH = 7.4).
EXAMPLE 16 =protein adhesion and substitution to a coaolvmer surface
measured through surface plasmon resonance.
The copolymer of example 3 was tested as follows for protein adsorption
and substitution upon subsequent contact with single component solutions of
albumin, fibrinogen and immunoglobulin G (available from Sigma
Immunochemicals).
CA 02365460 2001-09-25
WO 00/59963 PCT/EP00/02733
28
In a first experiment the 00Sp~ for maximal amounts of adhered protein were
determined. Next, the protein layer was reacted with its corresponding
monoclonal
antibody. The second shift in resonance angle, due to the binding of the
antibody,
is also a measure for the amount of protein adhered.
Substitution experiments were then performed, in which the surface
composition, protein concentration and flow speed were kept constant. From a
0.05% weight/volume HSA solution, in the first channel a maximal amount of HSA
adhered and was quantified via binding with the HSA-antibody. The shift in
resonance angle (06SP~~) was related to maximal HSA-binding (100% HSA
present). Next, in the second channel, after an albumin solution, a fibrinogen
solution was pumped over the surface. The second angle shift (06Sp~2) was due
to
the substitution of albumin by fibrinogen molecules. Using the HSA-antibody,
the
percentage HSA still present in the adhered protein layer was determined by
dividing the antibody response by the antibody response for maximal HSA-
adsorption. In the third channel, the HSA solution was followed by a human
immunoglobulin-G (HigG) solution. The sensorgram of the various HSA-antibody
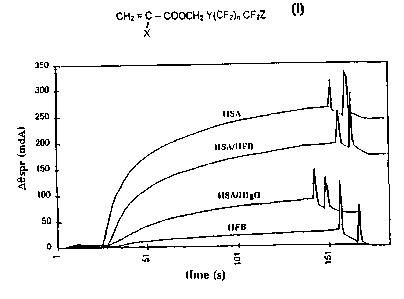
responses is provided in figure 2 and the data from the substitution
experiments
(06SP~ being expressed in mdA) are presented in the following table 3.
Table 3
Protein layer~0SPr1 09Sp~2 HSA antibody%HSA present
(mdA) (mdA) response
HAS 66 234 100
HSA/HFB 64 58 177 76
HSA/HigG 69 217 68 29
HFB 314 0 0
CA 02365460 2001-09-25
WO 00/59963 PCT/EP00/02733
29
Table 3 ~ollowina)
Protein layer08Sp~~ ~8SP~2 HFB antibody %HFB present
(mdA) (mdA) response
HFB 322 113 100
HFB/HAS 320 - 43 131 100
HFB/HigG 344 104 99 88
HAS 314 0 0
Protein layer~BSP~~ ~8SP~2 HigG antibody%IgG present
(mdA) (mdA) response
HigG 378 344 100
HIgG/HSA 367 - 5 344 100
HigG/HFB 352 88 224 65
HAS 72 41 12
From the above results, it may be concluded that HFB and HIgG possess a high
affinity for the hydrophobic fluorinated copolymer surface and that they
replace
HSA reversibly. Upon substitution, an equilibrium with both HFB and HIgG
present
at the surface is obtained. The substituted protein fraction depends on the
extent to
which the first adhered protein is able to relax and bind irreversibly.
EXAMPLE 17 - competitive adsorption of plasma proteins to a copolymer
surface measured throuah surface plasmon resonance.
The competitive adsorption of albumin, fibrinogen and immunoglobulin G
from diluted plasma to the surface of the copolymer of example 3 was followed
via
SPR versus time. Diluted human plasma was pumped over the copolymer surface
at various time intervals (5 s to 500 s) with a flow speed of 2 pl/s. For each
time
interval, the concentration of each protein was determined via binding to the
corresponding antibody. In order to obtain the percentage of protein coverage
after
a certain time period, 08SP~ was divided by the antibody response for maximal
CA 02365460 2001-09-25
WO 00/59963 PCT/EP00/02733
protein adsorption. Results are provided in the following table 4 and
graphically
shown in figure 3.
Table 4
Time HSA HSA HFB HFB HigG HIgG
(seconds)response response response response response response
mdA % MdA % MdA
Maximal
Adsorpt. 234 100 113 100 344 100
5 161 68 46 41 107 31
15 140 60 39 35 116 33
30 132 56 32 28 147 43
60 120 51 33 29 157 45
100 127 54 27 24 172 50
200 98 41 25 22 188 55
300 76 32 18 16 185 54
400 71 30 20 18 145 42
500 80 34 20 18 140 40
5 From the above results, it can be seen that no Vroman effect was observed.
The proteins adhered very fast to the surface. From the SPR-sensogram it can
be
deduced that within the first 100 seconds a stable plasma protein layer is
built and
substitution of proteins is still possible.
EXAMPLE 18 - Effect of irradiation sterilization on the macromolecular
10 characteristics of terpolymers.
Terpolymers of the previous examples were submitted to gamma
sterilization via 6°Co at various irradiation doses. Number average
molecular
weights M" and polydispersity index d (defined as MW/M~) of the copolymers
were
determined by gel permeation chromatography as indicated hereinbefore and are
15 provided in table 5 below.
CA 02365460 2001-09-25
WO 00/59963 PCT/EP00/02733
31
Table 5
Copolymer 0 kGy 0 kGy 10 kGy 10 kGy
of M~ d M~ d
example No.
3 89,000 1.7 88,200 1.9
4 123,000 1.8 126,000 1.8
40,000 3.0 39,400 3.0
9 (PEG 400) 40,600 2.4 44,600 3.2
9 (PEG 4,000)80,800 2.0 100,000 3.0
13 B 29,400 1.9 31,600 2.1
13 C 55,500 1.8 54,500 2.3
13 E 50,100 1.5 54,000 1.7
14 46,600 2.4 49,000 2.8
Copolymer 25 kGy 25 kGy 50 kGy 50 kGy
of M" d M~ d
example No.
3 91,300 2.0 87,600 2.4
4 122,000 2.0 116,400 2.2
5 43,100 3.8 52,100 4.3
9 (PEG 400) 49,500 3.7 39,900 4.7
9 (PEG 4,000)93.600 3.3 61,500 5.5
13 B 31,200 2.3 34,500 2.7
13 C 69,500 2.1 54,100 3.6
13 E 50,100 1.9 48,200 2.6
14 53,800 2.7 55,600 3.2
From the above data it can be concluded that although some increase in the
5 molecular weight and some broadening of the molecular weight range occurs
with
increasing irradiation doses, these modifications in the macromolecular
structure
CA 02365460 2001-09-25
WO 00/59963 PCT/EP00/02733
32
are not susceptible to significantly alter the mechanical properties, in
particular the
elasticity of these terpolymers as required in biomedical applications.
EXAMPLE 19 - in vitro endothelization of an ionic terpolymer.
Microscopy glasses (2.7 x 7.6 cm x 0.1 cm) were silanated and dipcoated
with a 2 % weight/volume solution of the ionic terpolymer of example 7. After
ethylene oxide sterilization, their surfaces were seeded with human umbelial
vein
endothelial cells (HUVEC) at a concentration of 36,000 cellsicm2. After 24
hours
incubation, the coated glasses were placed under sterile conditions into a
flow
chamber where they were brought into contact with a phosphate buffer saline
(PBS) flow. Culture medium was pumped over the surface, first under a venous
pressure (0.1 Pa, 10 minutes) and later under arterial pressure (0.74 Pa, 20
minutes). Using a phase contrast video camera focused and fixed on a selected
region in the middle of the chamber, a picture of the endothelial cell layer
was
recorded each 5 minutes (magnification:200 x). After 30 minutes of flow, the
chamber was rinsed with phosphate buffered saline. The set of pictures proved
that the ionic terpolymer adheres well to endothelial cells under static
conditions.
This observation is in contrast with the teachings of the literature, namely
P.Van
Wachem in Biomaterials (1985) 6:403, reporting a poor cell adhesion to
hydrophobic fluorinated surfaces.
EXAMPLE 20 - study of in vitro bacterial adhesion via radiolabellina.
The presence of Staphylococcus epidermis, the most frequent pathogen of
infested implants, was studied via radiolabelling (e.g. 3H-adenin). The
"bacterial
adhesion to hydrocarbons" (BATH) test was used for this purpose since it is
widely
recognized for its accuracy to test the relative hydrophobicity of micro-
organisms,
based on the adhesion to n-decane.
For quantification of the adhered micro-organisms by means of radio labelling,
the samples were prepared by coating silanated scintillation bottles with a 2%
(weight/volume) copolymer solution. As reference materials, polymer plates
(2.5 x
3 cm) made of polyurethane (Chronoflex-80A, Cardio.Tech. Int., Italy), low
density
polyethylene LDPE (Quantum Chemical Corporation, USA) and polystyrene (PS)
CA 02365460 2001-09-25
WO 00/59963 PCT/EP00/02733
33
were prepared by means of a mechanical press (10 kN) at 200 °C. Glass
and
stainless steel (SS, Inox 64 K°, Metaplast) plates were also used as
reference
materials.
Staphylococcus epidermis (LMG 10474, ATCC 11775) was obtained from the
Culture Collection of the University of Gent and cultured on a nutrient agar
(pH 7.4)
supplemented with 1 % glucose. Growth was followed by means of absorbency
measurements at 550 nm (Vitalab 10 photometer, Vital Scientific). Only
Staphylococcus epidermis in the stationary growth phase (after more than 10
hours
of incubation) was used for the experiments. The hydrophobicity of the
bacterial
surface was determined following the method of Rosenberg. Several volumes of n-
octane (0.1 -1 ml) were added to 3 ml of the bacterial suspension in phosphate
buffer (9.5 x 10$ cells). After two minutes shaking, two separated phases were
obtained again after 15 minutes, then the aqueous phase was removed. The
difference in absorbency between the original and the final suspension is a
measure for the bacterial hydrophobicity.
Staphylococcus epidermis was radioactively labelled with (2,8-3H)-adenine
(Dupont, Belgium, specific activity 288,000 Ci/mol, conc. 10,000 Ci/ml). 100
pl (100
pCi) of 3H-adenine was carefully smeared with a sterile trihalsky over a
nutrient
agar plate recently inoculated with Staphylococcus epidermis. After 16 hours
of
incubation at 37°C, the micro-organisms were harvested (1 M KH2P04,
Na2HP04,
phosphate buffer pH= 7.4, 15m1/plate) and concentrated via centrifugation
(Serval
Superspeed RC-2, 2500 g, 10 min). The bacterial pellet was slowly brought into
suspension again with the phosphate buffer, in order to reach a final
concentration
of 1.8 x 109 cellsiml (Abs= 1.5). The activity of this suspension was measured
and
the specific activity determined (2.7 x 10-4 countsimicro-organism). Next, the
radioactive suspension was brought during one hour into contact with the
polymer
coatings and the reference plates. The scintillation bottles were mounted in a
rotating disc and rotated at 60 rpm through a water bath at 37°C. A
dynamic
bacterial flow was secured and the contact with the materials was favoured.
The
materials were washed twice with phosphate buffer in order to remove
reversibly
CA 02365460 2001-09-25
WO 00/59963 PCT/EP00/02733
34
adhered bacteria (3 minutes at 60 rpm). After 3 minutes sonication of the
materials
in contact with a third volume of phosphate buffer, a sample (20 ~I) was taken
and
scintillation liquid (10 ml, Ultima Gold LSC Cocktail, Hewlett Packard) added.
The
radioactivity was measured via a liquid scintillation spectrometer (Beckman LS-
100, Analis, 20 minutes). Results are shown on figure 4 for the copolymers and
terpolymers of examples 3, 4, 7, 9 (PEG 4,000), 13 E and 14. These data
demonstrate that the strongly fluorinated copolymer of example 4 and, to a
lower
extent, the ionic terpolymer of example 14, adhere extremely high
concentrations
of Staphylococcus epidermis. On the other hand, the copolymer of example 3 and
the polyethyleneglycol-containing terpolymers of examples 9 and 13 E exhibit
the
lowest adhesion.and therefore are promising materials to reduce bacterial
adhesion.
EXAMPLE 21 - determination of the bacterial resistance of a
copo~mer via adenosine triphosphate (,ATP) intracellular concentration.
For quantification of the adhered micro-organisms by means of ATP
determination, the samples were prepared by coating Eppendorf tubes with a 2%
(weight/volume) copolymer solution. Polypropylene (PP) was used as the
reference material. The micro-organism (Staphylococcus epidermis) was selected
and prepared in the same way as in example 20. ThE ATP-kit (Bio-orbit Oy,
Finland) is based on the ATP-dependent behaviour of luciferase. Staphylococcus
epidermis was cultured on nutrient agar plates overnight at 37°C,
harvested,
centrifuged (Sorvall RT 6000B, 20 min, 3500 rpm) and washed twice. The pellet
was resuspended in phosphate buffer until a dense suspension of 1.8 x 109
cellsiml was obtained (Spectrophotometer Pharmacia LKB, Novaspec II, 550 nm).
The coated Eppendorf tubes were brought into contact with the bacterial
suspension for 1 hour at 37°C (Innova incubator 4230, 150 rpm) then
washed five
times by adding 1 ml of phosphate buffer and suctioning this again. The
adhering
micro-organisms were lysed by 5 to 10 minutes incubation with 1 ml of a
trichloroacetic acid (TCA) solution (TCA 1 %, EDTA 2mM, xylenol blue 0.002% in
distilled water). A 20 NI sample was pipetted in a 96-well microtiter plate
(Dynatech
CA 02365460 2001-09-25
WO 00/59963 PCT/EP00/02733
Microlite 1 ). The pink lytic solution was neutralized with 180 pl tris
acetate buffer,
as a consequence of which a change in colour (pink to yellow) took place. The
microtiter plate was kept on ice. Measurements were effected using an Amerlite
illuminometer (Amersham) at a maximal emission wavelength of 562 nm. First the
5 background noise of 40 NI of luciferase was measured (background value),
next
150 pl of the bacterial sample was pipetted over and the emitted signal
registered
once more (sample value). Finally, 50 NI of an internal ATP- standard solution
was
added to each well (standard value). The ATP-level per sample was calculated
as
follows: [(sample value - background value)/(standard value - sample value)]x
N
10 mole added standard. Results are shown on figure 5 for the copolymers and
terpolymers of examples 3, 4, 7, 9 (PEG 4,000), 13 E and 14. These data
confirm
the results obtained by the adenosine triphosphate concentration method of
example 21, i.e. the copolymer of example 3 and the polyethyleneglycol-
containing
terpolymers of examples 9 and 13 E are amenable to reduce bacterial adhesion.
15 EXAMPLE 22 - in vivo encrustation resistance of a poly(octafluoropentyl-
methacrxlate-co-2-ethylhexylacr)rlatel
Eight discs (3 mm diameter, 1 mm thick) of the copolymer of example 3 were
punched out of a pressed polymer plate. Discs made from materials used in
commercial catheters like silicon (Bard, urinary catheter) and polyurethane
20 (Chronoflex, Cardio.Tech.lnt., Italy) were used as reference materials.
After X-ray
sterilization, the discs were implanted into the bladder of adult male Wistar
rats (~
200 g) under sterile conditions, full narcosis (2 to 3% halothane in oxygen)
and
administration of a single dose of antibiotics (Clamoxil~, 15 mg/kg). An
incision
through the subcutanic tissue and the peritoneum led to the bladder. Urine pH
was
25 measured and uropathogenic micro-organisms determined. A polymer disc was
sutured with vicryl suture thread into the mucus wall, in order to impair
obstruction
of the passage to the urether. After suture, the wound was rinsed thoroughly
with
an isobetadin solution. A long-acting analgesic (brupenorphin, 0.3 mg/kg) was
administered subcutanously. After 9 weeks, the rats were killed via an
overdose
30 C02. Again, urine pH was determined and microbiological analyses performed.
CA 02365460 2001-09-25
WO 00/59963 PCT/EP00/02733
36
The polymeric discs were collected and the deposited calcium salts dissolved
with
ml of an acidic lanthanum chloride solution. The amounts of calcium were
determined via atomic absorption spectroscopy for each type of disc and
expressed in p mole. Results were as follows:
5 2.51 p mole for the copolymer of example 3
3.17 N mole for silicon
1.83 p mole for polyurethane
These data indicate that the copolymer of example 3 provides a low
encrustation value under the conditions of this experiment. This finding,
combined
10 with its low bacterial adhesiveness demonstrated in examples 20 and 21,
makes
this polymer a good candidate for coating urological catheters and stents.
EXAMPLE 23 - drug-loaded copolymer coatings obtained bhp-coatina.
Methylprednisolon (MP) (Sigma Chemicals) and valsartan (VAL) (Ciba
Geigy AG, Basel, Switzerland) were incorporated into a coating of the
copolymer of
example 3. Using the dip coating technique, stainless steal plates (2x1x0.1
cm3)
were immersed trice in a 2% (weight/volume) solution of drug and polymer, a
CHCl3/acetone (85/15) mixture being used as the solvent. Respectively 5 and
10%
by weight drug relative to the copolymer was incorporated. Scanning electron
microscopy by means of an SEM 505 apparatus (Philips, 30 kV, WDX-24 electron
accelerator) showed the presence of a 225 nm thick copolymer layer with a
predominance of fluorinated units at its surface. The inclusion of drugs did
not
result in topographic changes.
EXAMPLE 24 - drug-loaded copolymer coatings obtained by spray-coatina.
Solutions (100 mg/10 ml) of the copolymer of example 3, in which
respectively 9, 33 and 50% by weight of a drug (MP or VAL) was dissolved or
suspended, were used for the spray coating of stents. The stents were mounted
on
a rotary platform and spray-dusted via capillary atomisation under air
pressure. A
barrier coating was applied by, after 2 hours drying of the first polymer
layer,
atomizing for a second time 10 ml of the 1 % (weight/volume) solution of the
copolymer over the stents. Scanning electron microscopy by means of an SEM
CA 02365460 2001-09-25
WO 00/59963 PCT/EP00/02733
37
505 apparatus showed the presence of a 5 pm thick copolymer layer. A rough
topography and/or drug spots were observed at the surface for coatings loaded
with 50% by weight of a drug. 9 % by weight loded spay coatings had a flat
surface.
EXAMPLE 25 - in vivo biocompatibilitv of polXmer coated stents
Balloon expandable, stainless steal stents, 16 mm long and with specific
zigzag shape were folded out of a 0.18 mm diameter thread. The stents were
provided with a coating of the copolymer of example 3 by dipping or spraying
according to the methods of examples 23 and 24. Dust-free drying was effected
at
room temperature. The stents were sterilized by X-ray irradiation at 25kGy.
Cross-bred pigs (20-25 kg) were used as laboratory animals, a standard
grain diet without any fat or cholesterol supplements being fed to them. The
animals were treated according to the standards of the 'National Institute of
Health
Guide for the care and use of laboratory animals'. Under full narcosis, the
stents
were implanted in the crown/coronary artery of the animals. Heparin and
acetylsalicylic acid were administered during the operation. Via the thigh
artery, per
pig, one (coated or non-coated) stent with a conventional 3.0 mm coronary
angioplastic balloon catheter was placed in the right coronary and expanded
under
a pressure of 8 atm. during 60 seconds. By means of coronary angiography the
procedure was being followed. During the follow-up, anti-blood platelet agents
or
anticoagulants were no longer administered. After 6 weeks, a control
angiography
was performed on the stented coronary artery. An angiographic analysis of the
lumen diameter, before, just after and 6 weeks after implantation, was
performed
with a Polytron 1000~-apparatus (Siemens AG, Germany). lohexol (Omnipaque,
Nycomed, Oslo) was used as contrast fluid. The calibration wad one by means of
a
metal bar. The pigs were killed by intravenous administering of 10 ml of a
saturated
NaCI solution. The right coronary was fixed under pressure (80 mm Hg) with a
10%
formalin solution. The stented segment (from 1 cm proximal to 1 cm distal from
the
stent) was carefully dissected and fixed in a 2% formaiin solution. The stent
thread
was removed with a stereomicroscope, avoiding thereby deformation of or damage
CA 02365460 2001-09-25
WO 00/59963 PCT/EP00/02733
38
to the artery. The segments were imbedded in a cold-polymerising holder
(Technovit 710-Heraeus Kulzer GmbH, Germany) and 5 Nm thick sections were
prepared with a microtome (HM 360, Microm, Germany). The microscopic sections
were stained with haemalaun-eosin, Masson's trichome, elastics von Gieson and
phophotungstic acid heamatoxylin (PTAH). The tissue sections/samples were
studied with a light microscope by an experienced pathologist, who was unaware
of the type of coating. Morphometric analyses were performed via a computer
steered program (Leitz CBA 8000). For the statistical processing of the
angiographic measurements, a paired t-test was used. For comparison of two
different groups (coated-not coated, with-without drug), the non-paired t-test
was
used. The data were presented as mean values ~ standard deviations.
Measurements with a p-value below 0.05 were considered to differ
significantly.
20
30