Note: Descriptions are shown in the official language in which they were submitted.
CA 02365776 2001-09-21
WO 00/66113 PCT/EP00/04262
METHOD FOR TREATING ABNORMAL
CELL PROLIFERATION IN THE BRAIN
The present invention relates to a method for treating abnormal cell
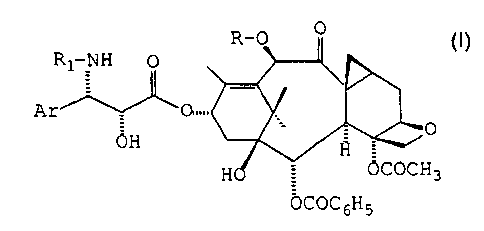
proliferation in the brain by administering a compound of general formula (I)
s or a pharmaceutically acceptable salt or hydrate thereof
0
OCOCH3
OCOC6H5
The compounds of general formula (I) manifest a significant inhibitory
activity with respect to abnormal cell proliferation of malignant and
nonmalignant cells of the brain. The compounds of formula (I) also possess
io therapeutic properties permitting the treatment of pathological conditions
associated with abnormal cell proliferation. The compounds of formula (I)
exhibit these properties in particular in the brain due to their longer
residence
time in the brain as compared to other tissues and/or organs. The term
«treatment,» as used in the present application refers both to decreasing cell
Is proliferation and to preventing cell proliferation.
The term « abnormal cell proliferation of the brain » as used in the
present application refers to brain tumor and brain metastasis, which may
occur in the development of cancers of brain or other tissues.
In general formula (I), Ar represents an aryl radical, R represents a
2o hydrogen atom or an acetyl, alkoxyacetyl or alkyl radical, R, represents a
benzoyl radical or a radical R2-O-CO- in which R2 represents
- a straight or branched alkyl radical containing 1 to 8 carbon atoms,
CA 02365776 2001-09-21
WO 00/66113 PCT/EP00/04262
2
an alkenyl radical containing 2 to 8 carbon atoms, an alkynyl radical
containing 3 to 8 carbon atoms, a cycloalkyl radical containing 3 to 6 carbon
atoms; a cycloalkenyl radical containing 4 to 6 carbon atoms or a bicycloalkyl
radical containing 7 to 11 carbon atoms, these radicals being optionally
s substituted by one or more substituents chosen from halogen atoms and
hydroxy radicals, alkyloxy radicals containing 1 to 4 carbon atoms,
dialkylamino radicals in which each alkyl portion contains 1 to 4 carbon
atoms, piperidino radicals, morpholino radicals, 1-piperazinyl radicals
(optionally substituted at position 4 by an alkyl radical containing 1' to 4
io carbon atoms or by a phenylalkyl radical whose alkyl portion contains 1 to
4
carbon atoms), cycloalkyl radicals containing 3 to 6 carbon atoms,
cycloalkenyl radicals containing 4 to 6 carbon atoms, phenyl radicals, cyano
radicals, carboxy radicals or alkyloxycarbonyl radicals whose alkyl portion
contains 1 to 4 carbon atoms,
is - or a phenyl radical optionally substituted by one or more atoms or
radicals chosen from halogen atoms and alkyl radicals containing 1 to 4
carbon atoms or alkyloxy radicals containing 1 to 4 carbon atoms,
or a saturated or unsaturated 4- to 6-membered nitrogen-containing
heterocyclyl radical optionally substituted by one or more alkyl radicals
2o containing 1 to 4 carbon atoms,
it being understood that the cycloalkyl, cycloalkenyl or bicycloalkyl radicals
may be optionally substituted by one or more alkyl radicals containing 1 to 4
carbon atoms.
Preferably, Ar represents a phenyl or a- or ~-naphthyl radical
2s optionally substituted by one or more atoms or radicals chosen from halogen
atoms (fluorine, chlorine, bromine, or iodine) and alkyl, alkenyl, alkynyl,
aryl,
arylalkyl, alkoxy, alkylthio, aryloxy, arylthio, hydroxy, hydroxyalkyl,
mercapto,
formyl, acyl, acylamino, aroylamino, alkoxycarbonylamino, amino, alkylamino,
CA 02365776 2001-09-21
WO 00/66113 PCT/EP00/04262
3
dialkylamino, carboxy, alkoxycarbonyl, carbamoyl, dialkylcarbamoyl, cyano,
nitro and trifluoromethyl radicals, it being understood that the alkyl
radicals
and the alkyl portions of the other radicals contain 1 to 4 carbon atoms, that
the alkenyl and alkynyl radicals contain 2 to 8 carbon atoms and that the aryl
s radicals are phenyl or a- or ~3-naphthyl radicals or alternatively Ar
represents
a 5-membered aromatic heterocyclic radical containing one or more atoms,
which are identical or different, chosen from nitrogen, oxygen or sulphur
atoms, optionally substituted by one or more substituents, which are identical
or different, chosen from halogen atoms (fluorine, chlorine, bromine or
io iodine) and alkyl radicals containing 1 to 4 carbon atoms, aryl radicals
containing 6 to 10 carbon atoms, alkoxy radicals containing 1 to 4 carbon
atoms, aryloxy radicals containing 6 to 10 carbon atoms, amino radicals,
alkylamino radicals containing 1 to 4 carbon atoms, dialkylamino radicals in
which each alkyl portion contains 1 to 4 carbon atoms, acylamino radicals in
is which the acyl portion contains 1 to 4 carbon atoms, alkoxycarbonylamino
radicals containing 1 to 4 carbon atoms, acyl radicals containing 1 to 4
carbon atoms, arylcarbonyl radicals in which the aryl portion contains 6 to 10
carbon atoms, cyano radicals, carboxy radicals, carbamoyl radicals,
alkylcarbamoyl radicals in which the alkyl portion contains 1 to 4 carbon
2o atoms, dialkylcarbamoyl radicals in which each alkyl portion contains 1 to
4
carbon atoms or alkoxycarbonyl radicals in which the alkoxy portion contains
1 to 4 carbon atoms.
More particularly, Ar represents a phenyl, 2- or 3-thienyl or 2- or 3-
furyl radical optionally substituted by one or more atoms or radicals, which
2s are identical or different, chosen from halogen atoms and alkyl, alkoxy,
amino, alkylamino, dialkylamino, acylamino, alkoxycarbonylamino and
trifluoromethyl radicals.
Still more particularly, Ar represents a phenyl radical optionally
CA 02365776 2001-09-21
WO 00/66113 PCT/EP00/04262
4
substituted by a chlorine or fluorine atom or by an alkyl (methyl), alkoxy
(methoxy), dialkylamino (diethylamino), acylamino (acetylamino) or
alkoxycarbonylamino (tert-butoxycarbonylamino) or 2- or 3-thienyl or 2- or
3-furyl radical.
s Of even more special interest are the products of general formula (I) in
which Ar represents a phenyl radical and R, represents a benzoyl or tert-
butoxycarbonyl radical.
According to the present invention, the taxoids of general formula (I)
can be obtained from a product of general formula
0
~i-~ II
0
.~
0....
R1_ 0 H O
R H bCOCH3
~COC6H5
in which Ar and R, are defined as above and R3 and R4, which are identical
or different represent a hydrogen atom or an alkyl radical containing 1 to 4
carbon atoms, or an aralkyl radical whose alkyl portion contains 1 to 4 carbon
atoms and the aryl portion preferably represents a phenyl radical optionally
is substituted by one or more alkoxy radicals containing 1 to 4 carbon atoms,
or
an aryl radical preferably representing a phenyl radical optionally
substituted
by one or more alkoxy radicals containing 1 to 4 carbon atoms, or
alternatively R3 represents an alkoxy radical containing 1 to 4 carbon atoms
or a trihalomethyl radical such as trichloromethyl or a phenyl radical
2o substituted by a trihalomethyl radical such as trichloromethyl and R4
represents a hydrogen atom, or alternatively R3 and R4 form, together with
the carbon atom to which they are attached, a 4- to 7-membered ring, and G,
represents a hydrogen atom or an acetyl, alkoxyacetyl or alkyl radical or a
CA 02365776 2001-09-21
WO 00/66113 PCT/EP00/04262
hydroxy-protecting group, the procedure being carried out, according to the
meanings of R3 and R4, in the following manner
1 ) when R3 represents a hydrogen atom or an alkoxy radical
containing 1 to 4 carbon atoms or an optionally substituted aryl radical and
s R4 represents a hydrogen atom, the product of general formula (II) is
treated
in acidic medium in order to obtain a product of general formula:
Gl-O 0
O
il
OCOCH3
OCOC6H5
in which Ar, R, and G, are defined as above, whose G, radical is, if
necessary, replaced by a hydrogen atom.
to The deprotection of the side chain of the product of general formula
(II) can also be carried out in the presence of an inorganic acid
(hydrochloric
acid or sulphuric acid) or an organic acid (acetic acid, methanesulphonic
acid, trifluoromethanesulphonic acid or p-toluenesulphonic acid), used alone
or in the form of a mixture, the procedure being carried out in an organic
is solvent chosen from alcohols (methanol, ethanol or isopropanol), ethers
(tetrahydrofuran, diisopropyl ether or methyl t-butyl ether), esters (ethyl
acetate, isopropyl acetate or n-butyl acetate), aliphatic hydrocarbons
(pentane, hexane or heptane), halogenated aliphatic hydrocarbons
(dichloromethane or 1,2-dichloroethane), aromatic hydrocarbons (benzene,
2o toluene or xylenes) and nitrites (acetonitrile) at a temperature of between
-10
and 60°C, preferably between 15 and 30°C. The acid may be used
in a
catalytic or stoichiometric quantity or in excess.
The deprotection can also be carried out under oxidizing conditions,
CA 02365776 2001-09-21
WO 00/66113 PCT/EP00/04262
6
using for example ammonium cerium (IV) nitrate in an acetonitrile-water
mixture or 2,3-dichloro-5,6-dicyano-1,4-benzoquinone in water.
The deprotection can also be carried out under reducing conditions,
for example by hydrogenolysis in the presence of a catalyst.
s When G, represents a protecting group, it is preferably a 2,2,2-
trichloroethoxycarbonyl or 2-(2-trichloromethylpropoxy)carbonyl radical
whose replacement by a hydrogen atom is carried out using zinc, optionally
combined with copper, in the presence of acetic acid, at a temperature of
between 20 and 60°C or by means of an inorganic or organic acid such as
io hydrochloric acid or acetic acid in a solution in an aliphatic alcohol
containing
1 to 3 carbon atoms or in an aliphatic ester such as ethyl acetate, isopropyl
acetate or n-butyl acetate in the presence of zinc optionally combined with
copper, or alternatively, when G, represents an alkoxycarbonyl radical, its
optional replacement by a hydrogen atom is carried out by treatment in
is alkaline medium or by the action of a zinc halide under conditions which do
not affect the rest of the molecule. Generally, the alkaline treatment is
carried
out by the action of ammonia in aqueous-alcoholic medium, at a temperature
close to 20°C. Generally, the treatment with a zinc halide, and
preferably zinc
iodide, is carried out in methanol at a temperature close to 20°C.
20 2) when R3 and R4, which are identical or different, represent an
alkyl radical containing 1 to 4 carbon atoms, or an aralkyl radical whose
alkyl
portion contains 1 to 4 carbon atoms and the aryl portion is preferably an
optionally substituted phenyl radical, or alternatively R3 represents a
trihalomethyl radical or a phenyl radical substituted by a trihalomethyl
radical
2s and R4 represents a hydrogen atom, or alternatively R3 and R4 form,
together
with the carbon atom to which they are attached, a 4- to 7-membered ring,
the product of general formula (II) is converted to the product of general
formula
CA 02365776 2001-09-21
WO 00/66113 PCT/EP00/04262
7
G1-0
0
Ar
Q.... -
H2N -OH H 0
HO v bCOCH3
~COC6H5
in which Ar and G, are defined as above, which is acylated by means of
benzoyl chloride or a reactive derivative of general formula
R2-O-CO-X (V)
s in which R2 is defined as above and X represents a halogen atom (fluorine or
chlorine) or a residue -O-R2 or -O-CO-O-R2, to give a product of general
formula (III) in which Ar, R, and G, are defined as above, whose G, radical
is,
if necessary, replaced by a hydrogen atom.
The products of general formula (IV) can be obtained by treating a
to product of general formula (II), in which Ar, R, and G, are defined as
above,
R3 and R4, which are identical or different, represent an alkyl, aralkyl or
aryl
radical, or alternatively R3 and R4 form together with the carbon atom to
which they are attached a 4- to 7-membered ring, with an inorganic acid
(hydrochloric acid or sulphuric acid) or an organic acid (formic acid)
Is optionally in an alcohol containing 1 to 3 carbon atoms (methanol, ethanol
or
isopropanol) at a temperature of between 0 and 50°C. Preferably, formic
acid
is used at a temperature close to 20°C.
The acylation of the product of general formula (IV) by means of
benzoyl chloride or a reactive derivative of general formula (V) is carried
out
2o in an inert organic solvent chosen from esters such as ethyl acetate,
isopropyl acetate or n-butyl acetate and halogenated aliphatic hydrocarbons
such as dichloromethane or 1,2-dichloroethane in the presence of an
inorganic base such as sodium bicarbonate or an organic base such as
CA 02365776 2001-09-21
WO 00/66113 PCT/EP00/04262
8
triethylamine. The reaction is carried out at a temperature of between 0 and
50°C, preferably close to 20°C.
When the radical G, represents a protecting group, its replacement by
a hydrogen atom is carried out under the conditions described above.
s The products of general formula (II) can be obtained according to one
of the following methods:
1 ) by esterification of the product of general formula:
HO ~~~~~
0
in which G, is defined as above, by means of an acid of general formula:
Ar ' ',.OOOH
R 1-N/~\O
to R3~R4
in which Ar, R,, R3 and R4 are defined as above, or of a derivative of this
acid.
The esterification by means of an acid of general formula (VII) Can be
carried out in the presence of a condensing agent (carbodiimide, reactive
is carbonate) and an activating agent (aminopyridine) in an organic solvent
(ether, ester, ketones, nitrites, aliphatic hydrocarbons, halogenated
aliphatic
hydrocarbons or aromatic hydrocarbons) at a temperature of between -10
and 90°C.
The esterification may also be performed using the acid of general
2o formula (VII) in anhydride form, the procedure being carried out in the
HO "''"''~i3
bCOC6H5
CA 02365776 2001-09-21
WO 00/66113 PCT/EP00/04262
9
presence of an activating agent (aminopyridine) in an organic solvent
(ethers, esters, ketones, nitrites, aliphatic hydrocarbons, halogenated
aliphatic hydrocarbons or aromatic hydrocarbons) at a temperature of
between 0 and 90°C.
s The esterification can also be performed using the acid of general
formula (VII) in halide form or in anhydride form with an aliphatic or
aromatic
acid, optionally prepared in situ, in the presence of a base (tertiary
aliphatic
amine), the procedure being carried out in an organic solvent (ethers, esters,
ketones, nitrites, aliphatic hydrocarbons, halogenated aliphatic hydrocarbons
io or aromatic hydrocarbons) at a temperature of between 0 and 80°C.
The acid of general formula (VII) can be obtained by saponification of
an ester of general formula:
Ar~COORS
R1-N O
R3~R4 (VIII)
in which Ar, R,, R3 and R4 are defined as above and R5 represents an alkyl
is radical containing 1 to 4 carbon atoms optionally substituted by a phenyl
radical.
Generally, the saponification is carried out by means of an inorganic
base (alkali metal hydroxide, carbonate or bicarbonate) in aqueous-alcoholic
medium (methanol-water) at a temperature of between 10 and 40°C.
2o The ester of general formula (VIII) can be obtained by the action of a
product of general formula
R3
- 0
in which R3 and R4 are defined as above in the form of a dialkylacetal or an
CA 02365776 2001-09-21
WO 00/66113 PCT/EP00/04262
enol alkyl ether, on an ester of general formula:
R1-NH
~COOR5
Ar
OH
in which Ar, R, and R5 are defined as above, the procedure being carried out
in an inert organic solvent (aromatic hydrocarbon) in the presence of a strong
s inorganic acid (sulphuric acid) or organic acid (p-toluenesulphonic acid
optionally in the form of a pyridinium salt) at a temperature of between
0°C
and the boiling temperature of the reaction mixture.
The ester of general formula (X) can be obtained by the action of a
product of general formula (V) on an ester of general formula:
H2N~ COORS
,;
~~°''OH
10 Ar
in which Ar and R5 are defined as above, the procedure being carried out in
an organic solvent (ester, halogenated aliphatic hydrocarbon) in the
presence of an inorganic or organic base at a temperature of between 0 and
50°C.
is The product of general formula (XI) can be obtained by reduction of an
azide of general formula
N3
~COOR5
Ar
OH
in which Ar and R5 are defined as above, by means of hydrogen in the
presence of a catalyst such as palladium on carbon, the procedure being
2o carried out in an organic solvent (ester).
CA 02365776 2001-09-21
WO 00/66113 PCT/EP00/04262
11
The product of general formula (XII) can be obtained by the action of
an azide such as trimethylsilyl azide in the presence of zinc chloride or an
alkali metal (sodium, potassium or lithium) azide in aqueous-organic medium
(water-tetrahydrofuran) at a temperature of between 20°C and the
boiling
s temperature of the reaction mixture, on an epoxide of general formula
0
Ar COOR5 (X111)
in which Ar and R5 are defined as above, optionally prepared in situ.
The epoxide of general formula (X111) can be obtained, optionally in
situ, by dehydrohalogenation of a product of general formula
OH O O
N' -O
Ar
Hal
R 6,,''' ''',R 7
in which Ar is defined as above, Hal represents a halogen atom, preferably a
bromine atom, and R6 and R,, which are identical or different, represent a
hydrogen atom or an alkyl radical containing 1 to 4 carbon atoms or a phenyl
radical, at least one being an alkyl radical or a phenyl radical, by means of
a
is alkali-metal alcoholate, optionally prepared in situ, in an inert organic
solvent
such as tetrahydrofuran at a temperature of between -80°C and
25°C.
The product of general formula (XIV) can be obtained by the action of
an aldehyde of general formula
Ar-CHO (XV)
2o in which Ar is defined as above, on a halide of general formula
CA 02365776 2001-09-21
WO 00/66113 PCT/EP00/04262
12
0 O
Hale
0
RG,,,,. ,,,,R7
in which Hal, R6 and R, are defined as above, anionized beforehand.
Generally, the procedure is carried out in an inert organic solvent
chosen from ethers (ethyl ether) and halogenated aliphatic hydrocarbons
s (methylene chloride) at a temperature of between -80 and 25°C, in the
presence of a tertiary amine (triethylamine) and an enolysing agent (di-n-
butylboron triflate).
The product of general formula (XVI) can be obtained by the action of
a halide of a haloacetic acid, preferably bromoacetic acid bromide, on the
io corresponding oxazolidinone.
The product of general formula (XI) can be obtained by hydrogenolysis
of a product of general formula:
CH3
Ph NH
COORS
Ar
OH
in which Ar and R5 are defined as above and Ph represents an optionally
Is substituted phenyl radical.
Generally, the hydrogenolysis is carried out by means of hydrogen in
the presence of a catalyst. More particularly, palladium on carbon containing
1 to 10% by weight of palladium or palladium dihydroxide containing 20 % by
weight of palladium is used as catalyst.
2o The hydrogenolysis is carried out in an organic solvent or in a mixture
of organic solvents. It is advantageous to carry out the procedure in acetic
CA 02365776 2001-09-21
WO 00/66113 PCT/EP00/04262
13
acid optionally combined with an aliphatic alcohol containing 1 to 4 carbon
atoms such as a mixture of acetic acid-methanol at a temperature of between
20 and 80°C.
The hydrogen necessary for the hydrogenolysis can also be provided
s by a compound which liberates hydrogen by chemical reaction or by thermal
decomposition (ammonium formate). It is advantageous to carry out the
procedure at a hydrogen pressure of between 1 and 50 bar.
The product of general formula (XVII) can be obtained by hydrolysis or
alcoholysis of a product of general formula
HO, Ar
N~ Ph
CH3
in which Ar and Ph are defined as above.
It is particularly advantageous to carry out an alcoholysis by means of
an alcohol of formula R5-OH in which R5 is defined as above, the procedure
being carried out in acidic medium.
is Preferably, the alcoholysis is carried out by means of methanol in the
presence of a strong inorganic acid such as hydrochloric acid at a
temperature close to the reflux temperature of the reaction mixture.
The product of general formula (XVIII) can be obtained by
saponification of an ester of general formula
CA 02365776 2001-09-21
WO 00/66113 PCT/EP00/04262
14
Re-CO-0 Ar
0 ~ Ph
CH3
in which Ar and Ph are defined as above and R$ represents an alkyl,
phenylalkyl or phenyl radical, followed by separation of the 3R, 4S
diastereoisomer of general formula (XVII) from the other diastereoisomers.
s Generally, the saponification is carried out by means of an inorganic
or organic base such as ammonium hydroxide, lithium hydroxide, sodium
hydroxide or potassium hydroxide in a suitable solvent such as a methanol-
water or tetrahydrofuran-water mixture at a temperature of between -
10°C
and 20°C.
io The separation of the 3R, 4S diastereoisomer can be carried out by
selective crystallization from a suitable organic solvent such as ethyl
acetate.
The product of general formula (XIX) can be obtained by cycloaddition
of an imine of general formula
Ar
N~Ph
CH3
is in which Ar and Ph are defined as above, onto an acid halide of general
formula
R8 -CO-
_Y
in which R$ is defined as above and Y represents a halogen atom such as a
CA 02365776 2001-09-21
WO 00/66113 PCT/EP00/04262
bromine or chlorine atom.
Generally, the reaction is carried out at a temperature of between 0
and 50°C in the presence of a base chosen from aliphatic tertiary
amines
(triethylamine) or pyridine in an organic solvent chosen from optionally
s halogenated aliphatic hydrocarbons (methylene chloride or chloroform) and
aromatic hydrocarbons (benzene, toluene or xylenes).
The product of general formula (XX) can be obtained under conditions
analogous to those described by M. Furukawa et al., Chem. Phar. Bull., 25
(1 ), 181-184 (1977).
io The product of general formula (VI) can be obtained by the action of
an alkali metal halide (sodium iodide or potassium fluoride) or an alkali
metal
azide (sodium azide) or a quaternary ammonium salt or an alkali metal
phosphate, on a baccatin III or 10-deacetylbaccatin III derivative of general
formula:
0 0 _ S02 _ CF3
HO""
0
y OCOCH3
HO
15 OCOC6H5
in which G, is defined as above.
Generally, the reaction is carried out in an organic solvent chosen
from ethers (tetrahydrofuran, diisopropyl ether, methyl t-butyl ether) and
nitrites (acetonitrile), alone or in the form of a mixture, at a temperature
of
2o between 20°C and the boiling temperature of the reaction mixture.
The product of formula (XXII) in which G, represents a hydrogen atom
or an acetyl, alkoxyacetyl or alkyl radical can be obtained by the action of a
trifluoromethanesulphonic acid derivative such as the anhydride or
CA 02365776 2001-09-21
WO 00/66113 PCT/EP00/04262
16
N-phenyltrifluoromethanesulphonimide, on baccatin III or 10-
deacetylbaccatin III, which can be extracted according to known methods
from yew leaves (Taxus baccata), optionally followed by protection in position
10, it being understood that in order to obtain a product of general formula
s (XXII) in which G, represents an alkoxyacetyl or alkyl radical, it is
necessary
to treat beforehand the 10-deacetylbaccatin III protected in position 7,
preferably with a silylated radical, with an alkoxy acetic acid halide or with
an
alkyl halide.
Generally, the reaction of a trifluoromethanesulphonic acid derivative
io is carried out in an inert organic solvent (optionally halogenated
aliphatic
hydrocarbons, or aromatic hydrocarbons) in the presence of an organic base
such as an aliphatic tertiary amine (triethylamine) or pyridine, at a
temperature in between -50 and +20°C.
Generally, the introduction of an alkoxyacetyl group is carried out by
Is treating the protected 10-deacetylbaccatin III with an alkoxyacetic acid
halide, the procedure being carried out in a basic organic solvent such as
pyridine at a temperature close to 20°C.
Generally, the introduction of an alkyl radical is carried out by treating
the 10-deacetylbaccatin III, protected and metallized ~ in position 10, by
2o means, for example, of an alkali metal hydride (sodium hydride) or a
metallic
alkylide (butyllithium), with an alkyl halide.
2) by the action of an alkali metal halide (sodium iodide or potassium
fluoride) or an alkali metal azide (sodium azide) or a quaternary ammonium
salt or an alkali metal phosphate on a product of general formula
CA 02365776 2001-09-21
WO 00/66113 PCT/EP00/04262
17
Gl_O O O_S02_CF3
0
Ar
O
R1_~ _ H
R3~ H OCOCH3
6COC6H5
in which Ar, R,, R3, R4 and G, are defined as above.
Generally, the reaction is carried out in an organic solvent chosen
from ethers (tetrahydrofuran, diisopropyl ether or methyl t-butyl ether) and
s nitrites (acetonitrile), alone or in the form of a mixture, at a temperature
of
between 20°C and the boiling temperature of the reaction mixture.
The product of general formula (XXIII) can be obtained by the action of
a trifluoromethanesulphonic acid derivative such as the anhydride or
N-phenyltrifluoromethanesulphonimide on a taxoid of general formula:
0
II OH
y _ 0
TT
.~
H~ OCOCH3
OCOC6H5
in which Ar, R,, R3, R4 and G, are defined as above.
Generally, the reaction is carried out in an inert organic solvent
(optionally halogenated aliphatic hydrocarbons, or aromatic hydrocarbons) in
the presence of an organic base such as an aliphatic tertiary amine
is (triethylamine) or pyridine, at a temperature of between -50 and
+20°C.
The taxoid of general formula (XXIV), in which G, represents a
hydrogen atom or an acetyl radical, can be obtained from a product of
general formula
CA 02365776 2001-09-21
WO 00/66113 PCT/EP00/04262
18
G~2 O O O-Gy
O
Ar ,,~~~O"",
_~ - 0
H
R3 H bCOCH3
~COC6H5
in which Ar, R,, R3, R4 are defined as above, G', represents a hydroxy-
protecting group and G'2 represents an acetyl, alkoxyacetyl or alkyl radical
or
a hydroxy-protecting group, by replacement of the protecting groups G', and
s optionally G'2 by hydrogen atoms.
The radicals G', and G'2, when they represent a hydroxy-protecting
group, are preferably 2,2,2-trichloroethoxycarbonyl or 2-(2-trichloromethyl-
propoxy)carbonyl radicals or trialkylsilyl, dialkylarylsilyl, alkyldiarylsilyl
or
triarylsilyl radicals in which the alkyl portions contain 1 to 4 carbon atoms
and
io the aryl portions are preferably phenyl radicals, it being possible, in
addition,
for G'2 to represent an alkoxyacetyl radical.
When G', and G'2 represent a 2,2,2-trichloroethoxycarbonyl or 2-(2-
trichloromethylpropoxy)carbonyl radical, the replacement of the protecting
groups by hydrogen atoms is carried out using zinc, optionally combined with
is copper, in the presence of acetic acid at a temperature of between 20 and
60°C or by means of an inorganic or organic acid such as hydrochloric
acid
or acetic acid in solution in an aliphatic alcohol containing 1 to 3 carbon
atoms or an aliphatic ester such as ethyl acetate, isopropyl acetate or n-
butyl
acetate in the presence of zinc optionally combined with copper.
2o When G', represents a silylated radical and G'2 represents an acetyl,
alkoxyacetyl or alkyl radical, the replacement of the protecting group G', by
a
hydrogen atom can be carried out by means of, for example, gaseous
hydrochloric acid in ethanolic solution at a temperature close to 0°C,
under
CA 02365776 2001-09-21
WO 00/66113 PCT/EP00/04262
19
conditions which are without effect on the rest of the molecule.
When G'2 represents an alkoxyacetyl radical, its optional replacement
by a hydrogen atom is carried out by treatment in alkaline medium or by the
action of a zinc halide under conditions which do not affect the rest of the
s molecule. Generally, the alkaline treatment is carried out by the action of
ammonia in aqueous-alcoholic medium, at a temperature close to 20°C.
Generally, the treatment with a zinc halide, preferably zinc iodide, is
carried
out in methanol at a temperature close to 20°C.
The product of general formula (XXV) can be obtained under the
to conditions described in international patent application PCT/WO 9209589.
The derivatives of general formula (I) can also be obtained by
esterification of a product of general formula (VI) by means of an acid of
general formula
R1- NH COOH
~'O-G
Ar
is in which Ar and R, are defined as above and G3 represents a hydroxy-
protecting group chosen from methoxymethyl, 1-ethoxyethyl,
benzyloxymethyl, (~-trimethylsilyloxy)methyl, tetrahydropyranyl, 2,2,2-
trichloroethoxymethyl, 2,2,2-trichloroethoxycarbonyl or 2-(2-trichloromethyl-
propoxy)carbonyl radicals or CH2-Ph radicals in which Ph represents a
2o phenyl radical optionally substituted by one or more atoms or radicals,
which
are identical or different, chosen from halogen atoms and alkyl radicals
containing 1 to 4 carbon atoms or alkoxy radicals containing 1 to 4 carbon
atoms, or an activated derivative of this acid, to give a product of general
formula
CA 02365776 2001-09-21
WO 00/66113 PCT/EP00/04262
G1-O
R1-NH 0
Ar 0~~~
- - 0
~_G3 H
v
H OCOCH3
OCOC6H5
in which Ar, R,, G,, and G3 are defined as above, followed by the
replacement of the protecting groups G,, G2 and G3 by hydrogen atoms to
give a product of general formula (I).
s The esterification can be performed under the conditions described
above for the esterification of the product of general formula (VI) by means
of
an acid of general formula (VII).
The replacement of the protecting groups G, and G3 of the product of
general formula (XXVII) by a hydrogen atom is carried out by treatment with
Io zinc, optionally combined with copper, in the presence of acetic acid at a
temperature of between 30 and 60°C or by means of an inorganic or
organic
acid such as hydrochloric acid or acetic acid in solution in an aliphatic
alcohol containing 1 to 3 carbon atoms or an aliphatic ester such as ethyl
acetate, isopropyl acetate or n-butyl acetate in the presence of zinc
is optionally combined with copper, when G, and G3 represent a 2,2,2-
trichloroethoxycarbonyl or 2-(2-trichloromethylpropoxy)carbonyl radical. The
replacement of the protecting group G3, when it represents a silylated radical
or an acetal residue, can be carried out by treatment in acidic medium such
as for example hydrochloric acid in solution in an aliphatic alcohol
containing
20 1 to 3 carbon atoms (methanol, ethanol, propanol or isopropanol) or aqueous
hydrofluoric acid at a temperature of between 0 and 40°C, when it
represents
an acetal residue, the replacement of the protecting group G, then being
carried out under the conditions described above. When G3 represents a
group -CH2-Ph, the replacement of this protecting group with a hydrogen
CA 02365776 2001-09-21
WO 00/66113 PCT/EP00/04262
21
atom can be carried out by hydrogenolysis in the presence of a catalyst.
The acid of general formula (XXVI) can be obtained by saponification
of an ester of general formula
Rl ~ COOR5
,
,,,,,, , ,,,,,0 _ G3
Ar
s in which Ar, R,, R5 and G3 are defined as above.
Generally, the saponification is carried out by means of an inorganic
base (alkali metal hydroxide, carbonate or bicarbonate) in aqueous-alcoholic
medium (methanol-water) at a temperature of between 10 and 40°C.
The ester of general formula (XXVIII) can be obtained according to the
io usual methods for the preparation of ethers, and more particularly
according
to the procedures described by J-N. DENIS et al., J. Org. Chem., 51, 46-50
(1986), from a product of general formula (XI).
The products of general formula (I) obtained using the procedures
according to the invention can be purified according to known methods such
is as crystallization or chromatography.
The products of general formula (I) have remarkable biological
properties.
In vitro, measurement of the biological activity is carried out on tubulin
extracted from pig brain by the method of M.L. Shelanski et al., Proc. Natl.
2o Acad. Sci. USA, 70, 765-768 (1973). The study of the depolymerization of
the microtubules into tubulin is carried out according to the method of
G. Chauviere et al., C.R. Acad. Sci., 293, serie II, 501-503 (1981). In this
study, the products of general formula (I) proved at least as active as
Taxol°
and Taxotere~.
2s In vivo, the products of general formula (I) proved active in mice
CA 02365776 2001-09-21
WO 00/66113 PCT/EP00/04262
22
grafted with the B16 melanoma at doses of between 1 and 10 mg/kg
intraperitoneally, as well as on other liquid or solid tumours.
The compounds have anti-tumor properties, more particularly, activity
against tumors which are resistant to Taxol° and Taxotere~. Such tumors
s include, for example, colon tumors which have an elevated expression of mdr
1 gene (multi-drug resistant gene). Multi-drug resistance is the usual term
relating to the resistance by a tumor against various compounds having
differing structures and mechanisms of action. Taxoids are generally known
to be highly recognized by experimental tumors such as P388/DOX, a P388
io murine leukemia cell line selected for doxorubicin (DOX) resistance, which
express mdr 1. The compounds according to the present invention are less
recognized by P388/DOX. More particularly, the compounds are less
recognized than Taxotere~ by mdr 1.
In particular, it has been found that the compounds of the present
is invention including the compounds of example 1, example 2 and example 3
have better multi-drug resistance properties than Taxol~ and Taxotere~.
Additionally, it has surprisingly been found that the compound of example 3
has substantially better multi-drug resistance properties than the compounds
of example 1 and example 2.
2o The products of general formula (I) manifest a significant inhibitory
activity with respect to abnormal cell proliferation and possess therapeutic
properties which permit the treatment of patients having pathological
conditions associated with abnormal cell proliferation. The pathological
conditions include the abnormal cell proliferation of malignant and
2s nonmalignant cells of various tissues and/or organs, comprising, with no
limitation being implied, muscle, bone or connective tissues, the skin, brain,
lungs, bladder, sex organs, the lymphatic or renal systems, mammary or
blood cells, liver, the digestive tract, pancreas and thyroid or adrenal
glands.
These pathological conditions can also include psoriasis, solid tumours,
CA 02365776 2001-09-21
WO 00/66113 PCT/EP00/04262
23
cancers of the lungs, bladder, ovary, breast, brain, prostate, colon, stomach,
kidney or testicles, Kaposi's sarcoma, cholangioma, chorioma,
neuroblastoma, Wilms' tumour, Hodgkin's disease, melanomas, multiple
myelomas, lymphatic leukaemias and acute or chronic granulocytic
s lymphomas. The products according to the invention are particularly useful
for the treatment of cancer of the lungs, or the bladder ; more especially of
the lungs. The products according to the invention can be used to prevent or
retard the appearance or reappearance of the pathological conditions or to
treat these pathological conditions.
io In particular, according to the present invention, the compounds of
general formula (I), especially that of example 3 are useful for the treatment
of abnormal cell proliferation of the brain, such as brain cancer or brain
metastasis, which may occur in the development of cancers of brain or other
tissues, especially lung cancer, and more especially non-small-cell lung
is cancer.
It has been found according to the invention that the compounds of
general formula (I), especially that of example 3 are useful for the treatment
of non-small-cell lung cancer with brain metastasis.
According to preferred object of the invention, it has been found that
2o the compounds of general formula (I), especially that of example 3 are
useful
for the treatment of non-small-cell lung cancer with brain metastasis, as 1-
hour IV-infusion every 3 weeks.
According to a still more preferred object of the invention, the
2s compounds of general formula (I), especially that of example 3 are useful
for
the treatment of non-small-cell lung cancer with brain metastasis, as 1-hour
IV-infusion every 3 weeks at doses from 75 mg/m2 to 90 mg/m2 ; still more
specifically, suitable doses may be 75 mg/m2 for patients with previous
CA 02365776 2001-09-21
WO 00/66113 PCT/EP00/04262
24
radiotherapy and 90 mg/m2 for patients without previous radiotherapy.
This is because they manifest a significant inhibitory activity with
respect to abnormal cell proliferation of malignant and nonmalignant cells of
s the brain. The compounds of formula (I) exhibit these properties in
particular
in the brain due to their longer residence time in the brain as compared to
other tissues and/or organs.
The products according to the invention can be administered to a
patient in various forms adapted to the chosen route of administration which
to is preferably the parenteral or oral route. Parenteral administration
comprises intravenous, intraperitoneal, intramuscular or subcutaneous
administrations. Intraperitoneal, intravenous, or oral administration is more
particularly preferred.
The present invention also comprises pharmaceutical compositions
Is containing at least one compound of general formula (I) in a sufficient
quantity adapted to use in human or veterinary therapy. The compositions
can be prepared according to the customary methods, using one or more
pharmaceutically acceptable adjuvants, carriers or excipients. Suitable
carriers include diluents, sterile aqueous media and various nontoxic
2o solvents. Preferably, the compositions are provided in the form of aqueous
solutions or suspensions, of injectable solutions which may contain
emulsifying agents, colorants, preservatives or stabilizers.
The choice of adjuvants or excipients may be determined by the
solubility and the chemical properties of the product, the particular mode of
2~ administration and good pharmaceutical practice, all known to the skilled
artisan.
For parenteral administration, aqueous or nonaqueous sterile
solutions or suspensions are used. For the preparation of nonaqueous
CA 02365776 2001-09-21
WO 00/66113 PCT/EP00/04262
solutions or suspensions, natural vegetable oils such as olive oil, sesame oil
or paraffin oil or injectable organic esters such as ethyl oleate can be used.
The aqueous sterile solutions may consist of a solution of a pharmaceutically
acceptable salt in solution in water. The aqueous solutions are suitable for
s intravenous administration insofar as the pH is appropriately adjusted and
isotonicity is achieved, for example, with a sufficient quantity of sodium
chloride or glucose. The sterilization can be performed by heating or by any
other means which does not adversely affect the composition.
It is clearly understood that all the products entering into the
io compositions according to the invention should be pure and nontoxic for the
quantities used.
The compositions may contain at least 0.01 °/a of therapeutically
active
product. The quantity of active product in a composition is such that a
suitable dosage can be prescribed. Preferably, the compositions are
is prepared such that a single dose contains about 0.01 to 1000 mg of active
product for parenteral administration.
The therapeutic treatment can be performed concurrently with other
therapeutic treatments including antineoplastic drugs, monoclonal antibodies,
immunotherapies or radiotherapies or biological response modifiers. The
2o response modifiers include, with no limitation being implied, lymphokines
and
cytokines such as interleukins, interferons (a, ~ or 8) and TNF. Other
chemotherapeutic agents which are useful in the treatment of disorders
caused by abnormal proliferation of cells include, with no limitation being
implied, alkylating agents like nitrogen mustards such as mechloretamine,
2s cyclophosphamide, melphalan and chlorambucil, alkyl sulphonates such as
busulfan, nitrosoureas such as carmustine, lomustine, semustine and
streptozocin, triazenes such as dacarbazine, antimetabolites such as folic
acid analogues like methotrexate, pyrimidine analogues such as fluorouracil
and cytarabine, purine analogues such as mercaptopurine and thioguanine,
CA 02365776 2001-09-21
WO 00/66113 26 PCT/EP00/04262
natural products like vinca alkaloids such as vinblastine, vincristine and
vindesine, epipodophyllotoxins such as etoposide and teniposide, antibiotics
such as dactinomycin, daunorubicin, doxorubicin, bleomycin, plicamycin and
mitomycin, enzymes such as L-asparaginase, various agents such as
s coordination complexes of platinum like cisplatin, substituted ureas like
hydroxyurea, methylhydrazine derivatives such as procarbazine,
adrenocortical suppressants such as mitotane and aminoglutethymide,
hormones and antagonists such as adrenocorticosteroids such as
prednisone, progestins such as hydroxyprogesterone caproate,
io methoxyprogesterone acetate and megestrol acetate, oestrogens such as
diethylstilbestrol and ethynylestradiol, antioestrogens such as tamoxifen, and
androgens such as testosterone propionate and fluoroxymesterone.
The doses used for carrying out the methods according to the
invention are those which permit a prophylactic treatment or a maximum
is therapeutic response. The doses vary according to the form of
administration, the particular product selected and the characteristics
specific
to the subject to be treated. In general, the doses are those which are
therapeutically effective for the treatment of disorders caused by abnormal
cell proliferation. The products according to the invention can be
2o administered as often as necessary to obtain the desired therapeutic
effect.
Some patients may respond rapidly to relatively high or low doses, and then
require low or zero maintenance doses. Generally, low doses will be used at
the beginning of the treatment and, if necessary, increasingly higher doses
will be administered until an optimum effect is obtained. For other patients,
it
2s may be necessary to administer maintenance doses 1 to 8 times per day,
preferably 1 to 4 times according to the physiological needs of the patient
considered. It is also possible that for certain patients it may be necessary
to
use only one to two daily administrations.
According to an object of the present invention, the compounds of
CA 02365776 2001-09-21
WO 00/66113 PCT/EP00/04262
27
general formula (I) and especially that of example (3) may preferably be
admistered as 1- or 3-hour IV-infusion every 3 weeks ;according to a
preferred object, the compounds of general formula (I) and especially that of
example (3) may be admistered as 1-hour IV-infusion every 3 weeks ;
s according to another object, the compounds may suitably be administered
weekly.
It is understood that, in order to choose the most appropriate dosage,
account should be taken of the route of administration, the patient's weight,
his general state of health, his age and all factors which may influence the
io efficacy of the treatment.
According to the present invention, the doses of the compounds
generally range from 50 to 150 mg/m2 for parenteral administration ;
preferably, from 75 mg/m2 to 90 mg/m2. Still more specifically, suitable doses
may be 75 mg/m2 for patients with previous radiotherapy and 90 mg/m2 for
is patients without previous radiotherapy.
The following examples illustrate, however, do not limit the present
invention.
EXAMPLE 1
A solution of 2.01 g of 4-acetoxy-2a-benzoyloxy-5,20-epoxy-1 (i,10~
2o dihydroxy-7~,8a-methylene-9-oxo-19-nor-11-taxen-13a-yl (4S,5R)-3-tert
butoxycarbonyl-2,2-dimethyl-4-phenyl-5-oxazolidinecarboxylate in 20 cm3 of
formic acid was stirred for 4 hours at a temperature close to 20°C and
then
concentrated to dryness under reduced pressure (0.27 kPa) at 40°C. The
foam obtained was dissolved in 100 cm3 of dichloromethane and the solution
2s obtained was supplemented with 20 cm3 of a saturated aqueous sodium
hydrogen carbonate solution. The aqueous phase was separated after
settling had taken place and extracted with 20 cm3 of dichloromethane. The
organic phases were pooled, dried over magnesium sulphate, filtered and
CA 02365776 2001-09-21
WO 00/66113 PCT/EP00/04262
28
then concentrated to dryness under reduced pressure (2.7 kPa) at 40°C.
1.95
g of a white foam were obtained which were purified by chromatography on
200 g of silica (0.063-0.2 mm) contained in a column 7 cm in diameter, eluted
with a dichloromethane-methanol mixture (98-2 by volume) and collected in
s 30 cm3 fractions. The fractions containing only the desired product were
pooled and concentrated to dryness under reduced pressure (0.27 kPa) at
40°C for 2 hours. 1.57 g of 4-acetoxy-2a-benzoyloxy-53,20-epoxy-1
(i,10(i-
dihydroxy-7a,8(3-methylene-9-oxo-19-nor-11-taxen-13a-yl (2R,3S)-3-amino-
2-hydroxy-3-phenylpropionate were obtained in the form of a white foam.
io To a solution of 400 mg of 4-acetoxy-2a-benzoyloxy-5(i,20-epoxy-
1(3,10(i-dihydroxy-7(i,8(i-methylene-9-oxo-19-nor-11-taxen-13a-yl (2R,3S)-3-
amino-2-hydroxy-3-phenylpropionate in 1 cm3 of dichloromethane, kept under
an argon atmosphere, were added 60 mg of sodium hydrogen carbonate and
then, dropwise, at a temperature close to 20°C, a solution of 0.16 g of
di-tert-
is butyl dicarbonate in 1 cm3 of dichloromethane. The solution obtained was
stirred for 64 hours at a temperature close to 20°C and then
supplemented
with a mixture of 5 cm3 of distilled water and 10 cm3 of dichloromethane. The
organic phase was washed three times with 2 cm3 of distilled water. The
organic phase was dried over magnesium sulphate, filtered and then
2o concentrated to dryness under reduced pressure (2.7 kPa) at 40°C.
317 mg
of a white foam were thus obtained which were purified by chromatography
on 30 g of silica (0.063-0.2 mm) contained in a column 3 cm in diameter,
eluted with a dichloromethane-methanol mixture (95-5 by volume) and
collected in 5 cm3 fractions. The fractions containing only the desired
2s product were pooled and concentrated to dryness under reduced pressure
(0.27 kPa) at 40°C for 2 hours. 161 mg of 4-acetoxy-2a-benzoyloxy-
5~3,20-
epoxy-1 ~,10(i-dihydroxy-7~,8~-methylene-9-oxo-19-nor-11-taxen-13a-y1
(2R, 3S)-3-tert-butoxycarbonylamino-2-hydroxy-3-phenylpropionate were
thus obtained in the form of a white foam whose characteristics were the
CA 02365776 2001-09-21
WO 00/66113 PCT/EP00/04262
29
following
- specific rotation: [a]D2° - -17° (c = 0.482; methanol)
- proton NMR spectrum: (400 MHz; CDC13; temperature of 323 K; b in ppm;
coupling constants J in Hz):1.21 (s, 3H:-CH3 16 or 17); 1.28 (s, 3H:-CH3 16 or
s 17); 1.34 [s, 9H:-C(CH3)3]; from 1.30 to 1.50 (mt, 1 H:-H7); 1.80 and 2.36
(2mt,
1 H each:-CHI- of cyclopropane); 1.88 (s, 3H:-CH3 18); 2.13 [mt, 1 H:-(CH)-H
6]; 2.26 [dd, 1 H, J = 15 to 8.5:-(CH)-H 14]; 2.35 (s, 3H:-COCH~); from 2.35
to
2.50 [mt, 2H:-(CH) -H 14 and -(CH)-H 6]; 3.21 (d, 1 H, J = 4:-OH 2'); 4.08 [d,
1 H, J = 8:-(CH)-H 20]; 4.16 (d, 1 H, J = 7: -H 3); 4.18 (s, 1 H, -OH 10);
4.31 [d,
io 1 H, J = 8:-(CH)-H 20]:4.61 (dd, 1 H, J = 4 and 2:-H 2'); 4.74 (d, 1 H, J =
4:-H
5); 5.00 (s, 1 H:-H 10); 5.26 (dd, 1 H, J = 9 and 2:-H 3'); 5.33 (d, 1 H, J =
9:-NH
3'); 5.69 (d, 1 H, J = 7:-H 2); 6.29 (d, 1 H, J = 8.5:-H 13); from 7.30 to
7.50 [mt,
5H:-C6H5 in 3' (-H 2 to -H 6); 7.51 [t, 2H, J = 7.5:-OCOCsHS (-H 3 to H 5)];
7.60 [t, 1 H, J = 7.5:-OCOCsH5 (-H 4)]; 8.14 [d, 2H, J = 7.5:-OCOC6H5 (-H 2
is and H 6)].
The 4-acetoxy-2a-benzoyloxy-5~i,20-epoxy-1 ~,10~-dihydroxy-7[i,8~-
methylene-9-oxo-19-nor-11-taxen-13a-yl (4S, 5R)-3-tert-butoxycarbonyl-2,2-
dimethyl-4-phenyl-5-oxazolidinecarboxylate was prepared in the following
manner
2o To a solution of 2.5 g of 4-acetoxy-2a-benzoyloxy-5~, 20-epoxy-
1~,10[i-dihydroxy-9-oxo-7(3-trifluoromethanesulphonate-11-taxen-13a-yl (4S,
5R)-3-tert-butoxycarbonyl-2,2-dimethyl-4-phenyl-5-oxazolidinecarboxylate in
25 cm3 of anhydrous acetonitrile and 3 cm3 of anhydrous tetrahydrofuran,
kept under an argon atmosphere, were added 2.5 g of sodium azide. The
2s reaction mixture was heated for 2 hours, with stirring and under an argon
atmosphere at a temperature close to 80°C, then cooled to a temperature
close to 20°C and supplemented with 30 cm3 of distilled water. The
aqueous
phase was separated by decantation and then extracted with 20 cm3 of
dichloromethane. The combined organic phases were dried over magnesium
CA 02365776 2001-09-21
WO 00/66113 PCT/EP00/04262
sulphate, filtered and then concentrated to dryness under reduced pressure
(2.7 kPa) at 40°C. 2.44 g of a yellow foam were thus obtained which
were
purified by chromatography on 300 g of silica (0.063-0.2 mm) contained in a
column 8 cm in diameter, eluted with a dichloromethane-ethyl acetate mixture
s (90-10 by volume) and collected in 60 cm3 fractions. Fractions 47 to 70 were
pooled and concentrated to dryness under reduced pressure (0.27 kPa) at
40°C for 2 hours. 2.01 g of 4-acetoxy-2a-benzoyloxy-5(i,20-epoxy-1 Vii,
10(3
dihydroxy-7(i,8(3-methylene-9-oxo-19-nor-11-taxen-13a-yl (4S, 5R)-3-tert
butoxycarbonyl-2,2-dimethyl-4-phenyl-5-oxazolidinecarboxylate were thus
to obtained in the form of a white foam.
The 4-acetoxy-2a-benzoyloxy-5(3,20-epoxy-1 ~i,10~3-dihydroxy-9-oxo-
7~-trifluoromethanesulphonate-11-taxen-13a-yl (4S,5R)-3-tert-butoxy-
carbonyl-2,2-dimethyl-4-phenyl-5-oxazolidinecarboxylate was prepared in the
following manner
is To a solution of 2.86 g of 4-acetoxy-2a-benzoyloxy-5(i,20-epoxy-
1 (3,7(i,10~-trihydroxy-9-oxo-11-taxen-13a-yl (4S,5R)-3-tert-butoxycarbonyl-
2,2-dimethyl-4-phenyl-5-oxazolidinecarboxylate in 29 cm3 of anhydrous
dichloromethane, kept under an argon atmosphere, were added 0.955 cm3 of
pyridine and 50 mg of powdered activated 4A molecular sieve. The reaction
2o mixture was cooled to a temperature close to -35°C, slowly
supplemented
with 0.85 cm3 of trifluoromethanesulphonic anhydride, stirred at a
temperature close to -5°C for 15 minutes and supplemented with 10 cm3
of
distilled water. After filtration on sintered glass provided with celite and
rinsing off the sintered glass 3 times with 10 cm3 of a methanol-
2s dichloromethane mixture (10-90 by volume), the aqueous phase was
separated after settling had taken place and extracted twice with 10 cm3 of
dichloromethane. The organic phases were pooled, dried over magnesium
sulphate, filtered and then concentrated to dryness under reduced pressure
(2.7 kPa) at 40°C. 3.87 g of a white foam were obtained which were
purified
CA 02365776 2001-09-21
WO 00/66113 PCT/EP00/04262
31
by chromatography on 400 g of silica (0.063-0.2 mm) contained in a column
cm in diameter, eluted with a dichloromethane-ethyl acetate gradient (from
97.5-2.5 to 90-10 by volume) and collected in 80 cm3 fractions. The fractions
containing only the desired product were pooled and concentrated to dryness
s under reduced pressure (0.27 kPa) at 40°C for 2 hours. 3.0 g of 4-
acetoxy-
2a-benzoyloxy-5~i,20-epoxy-1 [3,10~-dihydroxy-9-oxo-7(3-trifluoromethane-
sulphonate-11-taxen-13a-yl(4S,5R)-3-tert-butoxycarbonyl-2,2-dimethyl-4-
phenyl-5-oxazolidinecarboxylate were thus obtained in the form of a white
foam.
to The 4-acetoxy-2a-benzoyloxy-5[i,20-epoxy-1 [3,7[i,l0a-trihydroxy-9-
oxo-11-taxen-13a-yl (4S, 5R)-3-tert-butoxycarbonyl-2,2-dimethyl-4-phenyl-5-
oxazolidinecarboxylate were prepared in the following manner
A solution of 24.35 g of 4-acetoxy-2a-benzoyloxy-5(3,20-epoxy-9-oxo-
7[i,10~-[bis(2,2,2-trichloroethoxy)carbonyloxy]-1 [3-hydroxy-11-taxen-13a-
is yl(4S,5R)-3-tert-butoxycarbonyl-2,2-dimethyl-4-phenyl-5-oxazolidine-
carboxylate in a mixture of 130 cm3 of ethyl acetate and 46.5 cm3 of acetic
acid was heated, with stirring and under an argon atmosphere up to a
temperature close to 60°C and then supplemented with 40 g of zinc
powder.
The reaction mixture was then stirred for 30 minutes at 60°C and then
cooled
?o to a temperature close to 20°C and filtered on sintered glass
provided with
celite. The sintered glass was washed with 100 cm3 of a methanol-
dichloromethane mixture (20-80 by volume); the filtrates were pooled and
then concentrated to dryness under reduced pressure (0.27 kPa) at a
temperature close to 40°C.
2s The residue was supplemented with 500 cm3 of dichloromethane. The
organic phase was washed with twice 50 cm3 of a saturated aqueous sodium
hydrogen carbonate solution and then with 50 cm3 of distilled water. The
aqueous phases obtained after settling was taken place and pooled were
CA 02365776 2001-09-21
WO 00/66113 PCT/EP00/04262
32
extracted twice with 30 cm3 of dichloromethane. The organic phases were
pooled, dried over magnesium sulphate, filtered and then concentrated to
dryness under reduced pressure (2.7 kPa) at 40°C. 19.7 g of a white
foam
were obtained which were purified by chromatography on 800 g of silica
s (0.063-0.2 mm) contained in a column 10 cm in diameter, eluted with a
dichloromethane-methanol gradient (from 100-0 to 97-3 by volume) and
collected in 80 cm3 fractions. The fractions containing only the desired
product were pooled and concentrated to dryness under reduced pressure
(0.27 kPa) at 40°C for 2 hours. 16.53 g of 4-acetoxy-2a-benzoyloxy-
5[i,20-
to epoxy-1[3,7~,10~i-trihydroxy-9-oxo-11-taxen-13a-yl (4S,5R)-3-tert-
butoxycarbonyl-2,2-dimethyl-4-phenyl-5-oxazolidinecarboxylate in the form of
a white foam was formed.
The 4-acetoxy-2a-benzoyloxy-5~i,20-epoxy-9-oxo-7~3,10~i-[bis(2,2,2-
trichloroethoxy)carbonyloxyJ-1 [i-hydroxy-11-taxen-13a-yl (4S,5R)-3-tert-
Is butoxycarbonyl-2,2-dimethyl-4-phenyl-5-oxazolidinecarboxylate was
prepared according to the method described in international application PCT
WO 9209589, the disclosure of which is incorporated by reference herein.
EXAMPLE 2
To a solution of 550 mg of 4a,10~-diacetoxy-2a-benzoyloxy-5~,20-
2o epoxy-1 ~-hydroxy-7~i,8~-methylene-9-oxo-19-nor-11-taxen-13a-yl (2R,3S)-3-
amino-2-hydroxy-3-phenylpropionate were added 45 cm3 of distilled water,
45 cm3 of a saturated aqueous sodium hydrogen carbonate solution and
then, dropwise, at a temperature close to 20°C, 0.096 cm3 of benzoyl
chloride. The mixture obtained was stirred for 10 minutes at a temperature
2s close to 20°C. After settling had taken place, the aqueous phase was
extracted with twice 30 cm3 of ethyl acetate. The combined organic phases
were dried over magnesium sulphate, filtered and then concentrated to
dryness under reduced pressure (2.7 kPa) at 40°C. 670 mg of a white
foam
were thus obtained which were purified by chromatography at atmospheric
CA 02365776 2001-09-21
WO 00/66113 PCT/EP00/04262
33
pressure on 50 g of silica (0.063-0.2 mm) contained in a column 2.5 cm in
diameter, eluted with a methanol-dichloromethane mixture (1-99 then 2.5-
97.5 by volume) and collected in 10 cm3 fractions. The fractions containing
only the desired product were pooled and concentrated to dryness under
s reduced pressure (2.7 kPa) at 40°C. 610 mg of a white foam were thus
obtained. A sample of 300 mg was purified by preparative chromatography
on 12 thin-layer silica plates (Kieselgel 60F254, Merck; thickness 0.25 mm),
eluted with a methanol-dichloromethane mixture (3-97 by volume). After
elution of the zone corresponding to the main product with a methanol-
io dichloromethane mixture (10-90 by volume) and then evaporation of the
solvents under reduced pressure (0.27 kPa) at a temperature close to
40°C,
155.2 mg of 4a,10[3-diacetoxy-2a-benzoyloxy-5[i,20-epoxy-1 [i-hydroxy
7(3,8~-methylene-9-oxo-19-nor-11-taxen-13a-yl (2R,3S)-3-benzoylamino-2
hydroxy-3-phenyl-propionate are obtained in the form of a white foam whose
is characteristics were the following
- specific rotation: [a]D2° - -30.5° (c = 0.491; methanol)
- proton NMR spectrum: (300 MHz; CDC13; 8 in ppm; coupling constants J in
Hz):1.27 (s, 3H: -CH3 16 or 17); 1.30 (s, 3H: -CH3 16 or 17); 1.40 (mt, 1 H: -
H7); 1.62 and 2.25 (q and m, 1 H each: CH,2- of cyclopropane); 1.85 (s, 3H: -
2o CH3 18); 1.96 (s, 1 H: -OH in 1 ); 2.05 and 2.48 (d and m, 1 H each: -Cue-
in 6);
2.24 (s, 3H: -COCH~ in 10); 2.28 and 2.50 (m, 1 H each: -CHI in 14); 2.45 (s,
3H: -COCH~ in 4); 3.52 (d, 1 H: -OH in 2'); 4.10 and 4.35 (d, 1 H each: -CHI
in
20); 4.11 (d, 1 H: -H3); 4.77 (broad d, 1 H: -H5); 4.82 (dd, 1 H: -H2'); 5.70
(d,
1 H: -H in 2); 5.84 (dd, 1 H: -H3'); 6.30 (broad t, 1 H: -H13); 6.36 (s, 1 H: -
H10);
2s 7.00 (d, 1 H: -CONH-); from 7.35 to 8.30 (m, 15H: -C6H5 in 3', -OCOCsHS and
NHCOC6H~).
The 4a,10[i-diacetoxy-2a-benzoyloxy-5[i,20-epoxy-1[i-hydroxy-7[i,8[3-
methylene-9-oxo-19-nor-11-taxen-13a-yl (2R,3S)-3-amino-2-hydroxy-3-
phenylpropionate was prepared by carrying out the procedure under the
CA 02365776 2001-09-21
WO 00/66113 PCT/EP00/04262
34
conditions described in Example 1 for the preparation of 4-acetoxy-2a-
benzoyloxy-53,20-epoxy-1 (3,10(i-dihydroxy-7(3,8-methylene-9-oxo-19-nor-
11-taxen-13a-yl (2R,3S)-3-amino-2-hydroxy-3-phenylpropionate. Thus,
starting with 1.6 g of 4a,10(i-diacetoxy-2a-benzoyloxy-5(i,20-epoxy-1 (i-
s hydroxy-7(3,8(3-methylene-9-oxo-19-nor-11-taxen-13a-yl (4S,5R)-3-tert-
butoxycarbonyl-2,2-dimethyl-4-phenyl-5-oxazolidincarboxylate, 1.14 g of
4a,10(3-diacetoxy-2a-benzoyloxy-5(i,20-epoxy-1 (i-hydroxy-7(i,8(i-methylene-
9-oxo-19-nor-11-taxen-13a-yl were obtained in the form of a white foam.
The 4a,10(3-diacetoxy-2a-benzoyloxy-5(i,20-epoxy-1 (i-hydroxy-7~3,8~i-
io methylene-9-oxo-19-nor-11-taxen-13a-yl (4S,5R)-3-tert-butoxycarbonyl-2,2-
dimethyl-4-phenyl-5-oxazolidincarboxylate was prepared under the
conditions described in Example 1 for the preparation of 4a-acetoxy-2a-
benzoyloxy-5(i,20-epoxy-1 (3,10(i-dihydroxy-7(i,8(3-methylene-9-oxo-19-nor-
11-taxen-13a-yl (4S,5R)-3-tert-butoxycarbonyl-2,2-dimethyl-4-phenyl-5-
is oxazolidine-carboxylate. Thus, starting with 2.2 g of 4a,10(i-diacetoxy-2a-
benzoyloxy-5~i,20-epoxy-1 (i-hydroxy-9-oxo-7~-trifluoromethanesulphonate-
11-taxen-13a-yl (4S,5R)-3-tert-butoxycarbonyl-2,2-dimethyl-4-phenyl-5-
oxazolidine-carboxylate, 1.62 g of 4a,10(3-diacetoxy-2a-benzoyloxy-5(i,20-
epoxy-1 (3-hydroxy-7~,8~-methylene-9-oxo-19-nor-11-taxen-13a-yl (4S,5R)-3-
2o tert-butoxycarbonyl-2,2-dimethyl-4-phenyl-5-oxazolidincarboxylate were
obtained in the form of a white foam.
The 4a,10(3-diacetoxy-2a-benzoyloxy-5(i,20-epoxy-1 ~-hydroxy-9-oxo-
7(i-trifluoromethanesulphonate-11-taxen-13a-yl (4S,5R)-3-tert-butoxy-
carbonyl-2,2-dimethyl-4-phenyl-5-oxazolidincarboxylate was prepared under
2s the conditions described in Example 1 for the preparation of 4a-acetoxy-2a-
benzoyloxy-5(3,20-epoxy-1 ~,10(i-dihydroxy-9-oxo-7(i-trifluoromethane-
sulphonate-19-nor-11-taxen-13a-yl (4S,5R)-3-tert-butoxycarbonyl-2,2-
dimethyl-4-phenyl-5-oxazolidincarboxylate. Thus, starting with 2.4 g of
CA 02365776 2001-09-21
WO 00/66113 PCT/EP00/04262
4a,10~i-diacetoxy-2a-benzoyloxy-5~i,20-epoxy-1 ~i,7~-dihydroxy-9-oxo-11-
taxen-13a-yl (4S,5R)-3-tert-butoxycarbonyl-2,2-dimethyl-4-phenyl-5-
oxazolidinecarboxylate, 2.46 g of 4a,10~i-diacetoxy-2a-benzoyloxy-5(i,20-
epoxy-1 ~3-hydroxy-9-oxo-7~3-trifluoromethanesulphonate-11-taxen-13a-yl
s (4S,5R)-3-tert-butoxycarbonyl-2,2-dimethyl-4-phenyl-5-oxazolidincarboxylate
were obtained in the form of a white foam.
The 4a,10~i-diacetoxy-2a-benzoyloxy-5~i,20-epoxy-1(i,7~i-dihydroxy-9-
oxo-11-taxen-13a-yl (4S,5R)-3-tert-butoxycarbonyl-2,2-dimethyl-4-phenyl-5-
oxazolidincarboxylate was prepared under the conditions described in
io International Application PCT WO 9209589 the disclosure of which is
incorporated by reference herein.
EXAMPLE 3
To a solution of 550 mg of 4a,10~i-diacetoxy-2a-benzoyloxy-5~3,20-
epoxy-1 ~i-hydroxy-7~3,8~3-methylene-9-oxo-19-nor-11-taxen-13a-yl (2R,3S)-3-
Is amino-2-hydroxy-3-phenylpropionate in 1 cm3 of dichloromethane, kept under
an argon atmosphere, was added 76 mg of sodium hydrogen carbonate and
then, dropwise, at a temperature close to 20°C, a solution of 197 mg of
di-
tert-butyl dicarbonate in 1 cm3 of dichloromethane. The solution obtained
was stirred for 15 hours at a temperature close to 20°C and then
2o supplemented with a mixture of 5 cm3 of distilled water and 10 cm3 of
dichloromethane. The aqueous phase was extracted with 5 cm3 of
dichloromethane. The combined organic phases were dried over magnesium
sulphate, filtered and then concentrated to dryness under reduced pressure
(2.7 kPa) at 40°C. 780 mg of a white foam were thus obtained which were
2s purified by chromatography at atmospheric pressure on 50 g of silica (0.063-
0.2 mm) contained in a column 2.5 cm in diameter, eluted with a methanol-
dichloromethane mixture (1-99 then 2.5-97.5 by volume) and collected in 10
cm3 fractions. The fractions containing only the desired product were pooled
CA 02365776 2001-09-21
WO 00/66113 PCT/EP00/04262
36
and concentrated to dryness under reduced pressure (2.7 kPa) at 40°C.
660
mg of a white foam were thus obtained. A sample of 300 mg was purified by
preparative chromatography on 12 thin-layer silica plates (Kieselgel 60F254,
Merck; thickness 0.25 mm), eluted with a methanol-dichloromethane mixture
s (4-96 by volume). After elution of the zone corresponding to the main
product with a methanol-dichloromethane mixture (10-90 by volume) and
then evaporation of the solvents under reduced pressure (0.27 kPa) at a
temperature close to 40°C, 159.7 mg of 4a,10[3-diacetoxy-2a-benzoyloxy-
5[i,20-epoxy-1 (i-hydroxy-7[3,8[i-methylene-9-oxo-19-nor-11-taxen-13a-yl
io (2R,3S)-3-tert-butoxycarbonylamino-2-hydroxy-3-phenylpropionate were
obtained in the form of a white foam whose characteristics were the following:
- specific rotation: [a]D2° - -34° (c = 0.564; methanol)
- proton NMR spectrum: (400 MHz; CDC13; 8 in ppm; coupling constants J in
Hz): 1.28 (s, 3H: -CH3 16 or 17); 1.30 [s, 9H: -C(CH,~)3]; 1.38 (mt, 1 H: -
H7);
is 1.60 (s, 3H; -CH3 16 or 17); 1.68 and 2.25 (t and m, 1 H each; CH2- of
cyclopropane); 1.85 (s, 3H: -CHI 18); 2.10 and 2.45 (d and td, 1 H each: -
CHz- in 6); 2.23 (s, 3H: -COCH,~ in 10); 2.22 and 2.40 (m, 1 H each: -Cue- in
14); 2.40 (s, 3H: -COCH~ in 4); 3.28 (d, 1 H: -OH in 2'); 4.05 and 4.22 (d, 1
H
each: -CHI- in 20); 4.10 (d, 1 H: -H3); 4.62 (broad s, 1 H: -H2'); 4.73 (d, 1
H: -
2o H5); 5.29 (broad d, 1 H: -H3'); 5.37 (d, 1 H: -CONH-); 5.67 (d, 1 H: -H in
2);
6.28 (broad t, 1 H: -H13); 6.33 (s, 1 H: -H10); from 7.30 to 7.45 (mt, 5H: -
CsHS
in 3'); 7.51 [t, 2H: -OCOCsHS (-H3 and -H5)]; 7.61 [t, 1 H: -OCOC6H5 (-H4)];
8.17 [d, 2H: -OCOC6H5 (-H2 and -H6)].
EXAMPLE 4
2s To a solution of 100 mg of 10-deacetylbaccatin III in a mixture of 3 cm3
of tetrahydrofuran and 0.05 cm3 of pyridine cooled to a temperature close to
-78°C and kept under an argon atmosphere, was added, dropwise, 0.09 cm3
of trifluoro-methanesulphonic anhydride. The temperature was allowed to
rise slowly to a temperature close to 0°C over approximately one hour,
then
CA 02365776 2001-09-21
WO 00/66113 PCT/EP00/04262
37
up to a temperature close to 20°C over approximately one hour. After 2
hours at a temperature close to 20°C, 200 mg of tetrabutylammonium
iodide
were added, then the solution was heated at the boiling temperature of the
solvent for 15 hours. After cooling to a temperature close to 20°C, 10
cm3 of
s ethyl acetate and then 1 cm3 of distilled water were added. After separation
after settling had taken place, the organic phase was dried over magnesium
sulphate, filtered and concentrated to dryness under reduced pressure (2.7
kPa) at 40°C. 116 mg of a yellow oil were thus obtained which were
purified
by chromatography at atmospheric pressure on 30 g of silica (0.063-0.2 mm)
io contained in a column 2.5 cm in diameter, eluted with an ethyl acetate-
dichloromethane mixture, with an elution gradient from 0-100 to 80-20 by
volume. The fractions containing the desired product were pooled and
concentrated to dryness under reduced pressure (0.27 kPa) at 40°C. 10.3
mg of 10-deacetyl-7(3,8(i-methylene-19-norbaccatin III were thus obtained in
is the form of a white foam whose characteristics were the following:
- proton NMR spectrum: (400 MHz; CDC13; 8 in ppm; coupling constants J in
Hz): 1.14 (s, 3H: -CH3 in 16 or 17); 1.42 (mt, 1 H: -H in 7); 1.76 and 2.31 (t
and m, 1 H each; C~ of cyclopropane); 2.07 (s, 3H; -CH3 in 18); 2.15 and
2.50 (broad d and td, 1 H each: CHI- in 6); 2.30 (s, 3H: -COCH,~ in 4); 2.28
2o and 2.35 (m, 1 H each: -CH2 in 14); 4.11 and 4.37 (d, 1 H each: -CH2 in
20);
4.28 (d, 1 H: -H3 in 3); 4.79 (d, 1 H: -H in 5); 4.88 (broad t, 1 H: -H in
13); 5.09
(s, 1 H: -H in 10); 5.66 (d, 1 H: -H in 2); 7.51 [t, 2H: -OCOC6H5 (-H in 3 and
5)];
7.61 [t, 1 H: -OCOC6H5 (-H in 4)]; 8.17 [d, 2H: -OCOC6H5 (-H in 2 and 6)].
'3C NMR spectrum: (100 MHz; CDC13; 8 in ppm; uncoupled; s = singlet, d
2s =doublet; t = triplet; q = quadruplet): 15 (q, C18); 16.5 (t, C19); 20 and
27 (q,
C16 and C17); 22.5 (q, -COCH3); 26.5 (t, C6); 33 (d, C7); 35 (s, C8); 39 (d,
C3); 39.5 (t, C14); 43 (s, C15); 68 (d, C13); 76 (t, C20); 76.2 (d, C10); 79.5
(s, C1 ); 80 (s, C4); 81 (d, C2); 85 (d, C5); 129 (d, C2: -OCOC6H5); 130 (s,
CI
of -OCOC6H5); 130.5 (d, C3 of -OCOC6H5); 134 (d, C4 of
CA 02365776 2001-09-21
WO 00/66113 PCT/EP00/04262
38
-OCOC6H5); 136 (s, C11 ); 143 (s, C12); 168 (s, -OCOC6H5); 171 (s, -COCH3);
210 (s, C9).
EXAMPLE 5
Mammary C/16 adenocarcinomas were implanted in C3H/HeN female
s mice. At least 7 days later, 4a,10f3-diacetoxy-2a-benzoyloxy-5f3,20-epoxy
1 f3-hydroxy-7f3,8f3-methylene-9-oxo-19-nor-11-taxen-13a-yl (2R,3S)-3-tert
butoxycarbonylamino-2-hydroxy-3-phenylpropionate («Compound A») was
administered to groups of female mice at doses of 9.5, 15.4, 24.8, 40 or 60.5
mg/kg by intravenous injection under 0.4 or 0.5 ml/injection over 15 seconds.
Io The body weight and date of implantation were recorded for each mouse.
Blood and tissue samples were taken 2, 5, 15, 30 and 45 minutes and 1, 4,
14, 24 and 48 hours, after administration. The plasma was separated from
the blood samples at 4°C and frozen immediately. The tissue and tumors
were weighed and frozen immediately (-20°C).
is The plasma and tissue samples were then analyzed for the
concentration of Compound A. The plasma was assayed by high pressure
liquid chromatography with spectrophotometric detection. For the tissue,
liver, heart, kidney, lung, spleen and tumor tissue, samples were assayed by
a first method, while a second method was used for the brain tissue
2o analyzed. In the first method, 0.2 g of tissue was combined with 2 ml of
sodium phosphate buffer 0.1 M (pH:7) and homogenized. The homogenate
was centrifuged and 0.5 ml of supernatant was injected into the
chromatographic column. In the second method, 10 ~I of standard solution
was combined with 0.2 g of brain tissue. 2 ml of methanol/5 % perchloric
2s acid (50/50) were added and the mixture was homogenized. The
homogenate was centrifuged and 100 ~I of supernatant was injected into the
chromatographic column.
The kinetics of Compound A in plasma and tumor tissue were linear
CA 02365776 2001-09-21
WO 00/66113 PCT/EP00/04262
39
over the range studied (9.5 to 60.5 mg/kg). The observed maximum plasma
concentrations (at time 2 minutes) were linear with respect to dose and equal
to 14 ~ 1, 21 ~ 2, 35 ~ 2, 51 ~ 1 and 133 ~ 30 ~g/ml, respectively. The
pharmacokinetics of Compound A in mice with mammary tumors were
s similarly linear at the range of doses administered. This pharmacokinetic
linearity and the comparable kinetic profile in mice with and without tumors
enabled Compound A distribution in tissues (liver, heart, kidney, lung,
spleen, brain, tumor), to be studied at a single, intermediate dose of 40
mg/kg, which was the highest non toxic dose (HNTD) observed in the mouse.
io After administration of the 40 mg/kg intermediate dose to mice with
tumors (mammary carcinoma), the kinetic profiles of Compound A exhibited
fast uptake of the drug. The drug concentrations were then decreased in two
phases. A rapid phase occurred first during which 70 % of the drug that was
present at 0.033 h after administration was cleared in one hour, except from
is the tumor and brain tissue. A slower phase followed, characterized by an
elimination half-life ranging from 1.6 to 7.5 hours for the tissues tested,
with
the exception of the tumor and the brain tissue. This decrease in
concentration is illustrated in Table 1, which reports the areas under tissue
concentration (AUC) curves and observed elimination half-life. While brain
2o and plasma AUCs in mice were similar at 4 hours, the 0 to infinity AUC for
Compound A in mouse brain tissue was ten times that observed for plasma.
These results are consistent with the long half-life of Compound A in brain
tissue (47.6 h), in comparison with the half-life observed in the other
tissues
(1.6- 9.3 h).
CA 02365776 2001-09-21
WO 00/66113 PCT/EP00/04262
TABLE 1
Pharmacokinetic
parameters in
tissues (dose
= 40 mg/kg)
Tissue AUC (~g/ml.h) t'/2 (h)
or (~g/g.h)
0-4 Hours 0 to Infinity
Plasma 35.0 58.6 4.4
Liver 70.8 93 3.1
Heart 24.8 28 1.6
Kidney 74.4 121 2.7
Lung 34.3 52 2.9
Spleen 34.6 80 7.5
Brain 36.3 627 47.6
Tumor 35.6 107 9.3
The concentration of Compound A (~g/g) in the tissues and plasma is
illustrated in the Figures. Tissue and plasma concentration ratios at each
point in time after the 40 mg/kg dose was administered are shown in Table 3
s (liver, heart, kidney, lung, spleen and tumor), Tables 4 and 5 (brain) and
Table 6 (tumor - all doses). These data show that Compound A was quickly
distributed in all tissues sampled. The half-life of the distribution phase in
each of the tissue samples was determined to be 0.29 h in liver, 0.05 h in
kidney, 0.34 h in lung, 0.23 h in spleen, 0.07 h in brain and 0.23 + 0.20 h in
to tumor tissue. For heart tissue, the half-life of the distribution phase was
not
calculated because of a bad fitting.
The maximum concentration of Compound A in the sampled tissues
occurred at 0.033 h (heart and kidney), at 0.083 h (liver, lung, spleen) and
at
0.25 h (tumor and brain). The percentages of the dose recovered in the
CA 02365776 2001-09-21
WO 00/66113 PCT/EP00/04262
41
tissues sampled are reported in Table 2:
TABLE 2
Percentage
of Compund
A in
tissues
(dose
= 40
mg/kg)
Time 0.033 1 h 4 h 24 h 48 h
h
Liver 9.73 2.10 0.47 - -
Heart 0.46 0.08 0.03 - -
Kidney 2.06 0.56 0.23 - -
Lung 0.67 0.18 0.08 - -
Spleen 0.27 0.10 0.05 - -
Tumor 1.07 0.94 0.37 0.09 -
Brain 0.19 0.41 0.39 0.33 0.19
Total 14.45 4.37 1.62 0.42 0.19
- not detected
The percentage (concentration) of Compound A in the sampled tissue
s fell off rapidly over the first few hours following drug administration,
except in
the tumor and brain tissue. In terms of kinetics, after rapid tissue uptake of
Compound A, concentrations declined according to a two-phase process: a
rapid rate of decrease and then a gradual decline. See the Figures.
EXAMPLE 6
to The safety and the efficacy of the treatment according to the present
invention may be implemented on patients according to the following
protocol
Inclusion criteria:
is
Signed informed consent prior to beginning protocol specific procedures -
CA 02365776 2001-09-21
WO 00/66113 PCT/EP00/04262
42
Histologically (adenocarcinoma, large cell, or squamous carcinoma) proven
metastatic non small cell lung cancer - Objective progressive disease - Brain
metastase demonstrated by CT/MR scans - 18 < Age < 70 years - Life
expectancy of at least 12 weeks - ECOG performance status of 0 to 1
s Adequate organ function including: neutrophils > 2 x 109/L; platelets > 100
x
109/L; creatinine within upper normal limit. If borderline values, the
creatinine
clearance should be > 60 ml/mn; total bilirubin within upper normal limit and
ALAT/ASAT/AP < 2.5 times the upper normal limits of the institutional norms.
- Patients registered for this trial must be treated and followed at the
to participating center - Prior therapy: _< 1 line of chemotherapy for
advanced
disease; no prior radiotherapy on brain; prior radiotherapy is allowed on
other sites (mediastinum, bone, lymphnode...) but not extensive (s20% of
bone marrow area). - Off previous anti-cancer (radio- or chemo-) therapy or
surgery for at least 4 weeks and 6 weeks if prior nitrosoureas, mitomycin C. -
Is Recovery from toxic effects of prior treatment (except alopecia any grade,
peripheral neuropathy and neuro hearing grade 1 according to the NCIC
CTC). - Previous or concomitant corticosteroids should be administered to
the patients for symptom control but stable doses should be given for at least
2 weeks before inclusion in the trial. - Presence of at least one extracranial
2o bidimensionally measurable lesion not previously irradiated. - Patients
should
receive the study drug within 5 working days of patient registration -
Complete initial work up within 3 weeks prior to patient registration.
Non-inclusion criteria:
Pregnant or lactating women or women of childbearing potential (e.g. not
using adequate contraception). - History of prior malignancies other than
previously excised or curatively irradiated basal cell skin cancer or in situ
cervical cancer. - Symptomatic brain metastasis except seizures not
3o controlled by adequate anticonvulsiant therapy - Motor deficit - Peripheral
CA 02365776 2001-09-21
WO 00/66113 PCT/EP00/04262
43
neuropathy > grade 1 or neuro hearing > grade 1 according to the NCIC
CTC. - More than one chemotherapy line for advanced disease. - Pulmonary
fibrosis - Other serious illness or medical conditions: congestive heart
failure
or angina pectoris even if medically controlled, previous history of
myocardial
s infarction within 1 year from study entry, uncontrolled hypertension or
arrhythmias; history of significant neurologic or psychiatric disorders
impairing the ability to obtain consent; active infection; peptic ulcer,
unstable
diabetes mellitus or other contra-indications for the use of corticosteroids;
superior vena cava syndrom. - Concurrent treatment with other experimental
to drugs. - Participation in another clinical trial with any investigational
drug
within 30 days prior to patient registration - Concurrent treatment with any
other anticancer therapy - Concomitant radiotherapy - Pleural or pericardial
effusion requiring intervention (drainage, tapping). - Previous treatment by
taxanes (paclitaxel, docetaxel).
~ Compound of Example 3 is supplied as a single-dose vial containing a
total of 94.4 mg of Compound of Example 3 in 2.36 ml of polysorbate 80
at the concentration of 40 mg/ml of Compound of Example 3. The solvent
for Compound of Example 3, ethyl alcohol 95%/water (13:87 m/m), is
2o supplied as a single dose in a 15 ml glass vial.
~ Premedication consists of Dexamethasone administered as a 8 mg PO or
equivalent dose of other corticosteroids every 12 hours starting on Day - 1
(-25 and -13 hours before IV-infusion of Compound of Example 3) and one
hour before IV-infusion of Compound of Example 3 on Day 1.
~ For patients with previous extracranial radiotherapy : compound of
example 3 is given at dose of 75 mg/m2 as a one-hour IV infusion every 21
days.
For patients without previous extracranial radiotherapy, compound of
CA 02365776 2001-09-21
WO 00/66113 PCT/EP00/04262
44
Example 3 is given at dose of 90 mg/m2 as a one-hour IV infusion every 21
days.
~ Patients will be treated until there is evidence of disease progression in
s extracranial lesions or unacceptable toxicity or patient refusal. In case of
progression in brain metastasis associated to an objective response in
other extracranial sites, brain radiotherapy will be done and the treatment
with compound of Example 3 will be continued.
io ~ If G-CSF (Granulocyte Colony Stimulating Factor) is required as a
secondary prophylaxis for patients, C-CSF will be administered at the dose
and schedule recommended by the manufacturer as a subcutaneous
injection starting on Day 2-5 of the treatment of the cycle.
i s ~ Criteria for evaluation
Efficacy: Eligible patients will be evaluable for response if they.receive a
minimum of 2 cycles of treatment with at least one follow-up tumor
assessment unless « early progression » occurs in which case they will be
considered evaluable. Brain antitumor activity will be assessed using 2
2o parameters: CT/MR brain scans and clinical neurologic assessment.All
CT/MR scans will be reviewed by an external panel of one oncologist and
one neuroradiologist. Extracranial activity will be evaluated with adequate
exams (see section...)
Pharmacokinetics: by reversed phase HPLC method.
2s Safety: All patients who started at least one infusion of compound of
Example
3 will be analyzed for safety. Adverse events (toxicities), vital signs,
physical
examinations (including ECOG performance status and neurological
examination), hematology and serum chemistry laboratory findings will be
analyzed.
~ For patients wit
CA 02365776 2001-09-21
WO 00/66113 PCT/EP00/04262
Response rate, time to progression and duration of response are the efficacy
endpoints. Quality of life is described.
Response criteria are
s ~ complete response : disappearance of all radiographically and/or visually
apparent tumour for a minimum period of 4 weeks ;
~ partial response : a reduction of at least 50 % in the sum of the products
of
the perpendicular diameters of all measurable lesions for a minimum
period of 4 weeks ;
io ~ minor response : a reduction of 25 % to 49 % in the sum of the products
of
the perpendicular diameters of all measurable lesions ;
~ stable disease no change of greater than 25 % in the size of measurable
lesions ;
t progressive disease : objective evidence of an increase of 25 % or more in
is any measurable lesion ;
or else : time to disease progression, progression-free, time to treatment
failure and survival.
Although the invention has been described in conjunction with specific
embodiments, it is evident that many alternatives and variations will be
2o apparent to those skilled in the art in light of the foregoing description.
Accordingly, the invention is intended to embrace all of the alternatives and
variations that fall within the spirit and scope of the appended claims.