Note: Descriptions are shown in the official language in which they were submitted.
CA 02368431 2001-09-28
WO 00/58361 PCT/US00/07473
1
MELANOCORTIN RECEPTOR LIGANDS
FIELD OF THE INVENTION
The present invention relates to new melanocortin receptor ligands. These
ligands are
cyclic peptide analogs that preferably exhibit selectivity for the MC-4 and/or
the MC-3 receptors
io relative to the other melanocortin receptors (in particular the MC-1
receptor). The invention also
relates to pharmaceutical compositions comprising the peptide analogs, and to
their use in the
prevention or treatment of conditions affected by those receptors in animals,
including man.
BACKGROUND OF THE INVENTION
Melanocortin peptides (melanocortins) are natural peptide hormones in animals
and man
i5 that bind to and stimulate MC-receptors. Examples of melanocortins are a-
MSH (melanocyte
stimulating hormone), (3-MSH, y-MSH, ACTH (adrenocorticotropic hormone) and
their peptide
fragments. MSH is mainly Irnown for its ability to regulate peripheral
pigmentation (Eberle
1988), whereas ACTH is lrnown to induce steroidoneogenesis (Simpson and
Waterman, 1988).
The melanocortin peptides also mediate a number of other physiological
effects. They are
2o reported to affect motivation, learning, memory, behavior, inflammation,
body temperature, pain
perception, blood pressure, heart rate, vascular tone, natriuresis, brain
blood flow, ner~~e growth
and repair, placental development, aldosterone synthesis and release, thyroxin
release,
spermatogenesis, ovarian weight, prolactin and FSH secretion, uterine bleeding
in women, sebum
and pheromone secretion, sexual activity, penile erection, blood glucose
levels, intrauterine fetal
25 growth, food motivated behavior, as well as other events related to
parturition.
ACTH and the various MSH peptides share the tetrapeptide core His-Phe-Arg-Trp.
All
of the peptides are derived from the proteolytic processing of the pro-peptide
pre-
opiomelanocortin (POMC). In the past several years, five distinct melanocortin
receptor
subtypes have been identified. These MC receptors belong to the class of 7
transmembrane
3o domain G-protein coupled receptors. The five MC receptors, termed MC-l, MC-
2, MC-3, MC-4
and MC-5, all couple in a stimulatory fashion to cAMP. Of these, the MC-2
receptor is the
ACTH receptor, whereas the others constitute subtypes of MSH receptors. The MC-
1 receptor is
CA 02368431 2001-09-28
WO 00/58361 PCT/US00/07473
-2-
present on melanocytes and melanoma. The MC-2 receptor is present
predominantly in the
adrenal gland. The mRNA for the MC-3 receptor has been found in the brain, as
well as in
placental and gut tissues (Gantz et al. 1993a, Desarnaud et al. 1994, Roselli
Rehfuss et al. 1993).
The MC-4 receptor has been found primarily in the brain (Gantz et al. 1993b;
Mountjoy et al
1994). The MC-5 receptor is expressed in the brain, as well as in several
peripheral tissues
(Chhajlani et al 1993: Gantz et al 1994; Griffon et al 1994; Labbu et al.
1994; Barrett et al. 1994;
Fathi et a1.1995). More recent data from humans indicate that all of the
cloned MC-receptors
have a wider tissue distribution (Chhajlani, 1996) than originally thought.
As discussed above, the members of the melanocortin receptor family can be
~o differentiated on the basis of their tissue distribution. Both the MC-4 and
MC-3 receptors have
been localized to the hypothalamus, a region of the brain believed to be
involved in the
modulation of feeding behavior. Compounds showing selectivity for the MC-4/MC-
3 receptors
have been shown to alter food intake following intracerebroventricular and
peripheral injection in
rodents. Specifically, agonists have been shown to reduce feeding, while
antagonists have been
shown to increase feeding. See, Fan, W. et al., "Role of Melanocortinergic
Neurons in Feeding
and the Agouti Obesity Syndrome", Nature, 385(6612), pp. 165-8 (Jan. 9, 1997).
The role of the MC-4 receptor subtype has been more clearly defined in the
control of
eating and body weight regulation in mammals. See, e.g., Huszer, D. et al.,
"Targeted Disruption
of the Melanocortin-4 Receptor Results in Obesity in Mice", Cell, pp. 131-141
(1997); Klebig,
2o M.L. et al., "Ectopic Expression of the Agouti Gene in Transgenic Mice
Causes Obesity,
Features of Type II Diabetes, and Yellow Fur", Proc. Natl Acad Sci., Vol. 92,
pp. 4728-32
(1995); Karbon, W. et al., "Expression and Function of Argt, a Novel Gene
Related to Agouti",
Abstract from the Nineteenth Annual Winter Neuropeptide Conference ( 1998);
Fan, W. et al.,
"Role of Melanocortinergic Neurons in Feeding and the Agouti Obesity
Syndrome", Nature, Vol.
2s 385, pp. 165-168 (1997); Seely, R.J., "Melanocortin Receptors in Leptin
Effects", Nature, Vol.
390, p. 349 (1997); Comuzzie, A.G., "A Major Quantitative Trait Lacus
Determining Serum
Leptin Levels and Fat Mass is Located on Human Chromosome 2", Nat. Gen., Vol.
15, pp. 273-
276 (1997); Chagnon, Y.C. et al., "Linkage and Association Studies Between the
Melanocortin
Receptors 4 and 5 Genes and Obesity-Related Phenotypes in the Quebec Family
Study", Mol.
so Med., Vol 3(10), pp. 663-673 (1997); Lee, F. and Huszar, D, "Screening
Methods for Compounds
Useful in the Regulation of Body Weight", World Patent Publication WO 97/47316
(1997); and
Shutter, J.R. et al., "Hypothalamic Expression of ART, a Novel Gene Related to
Agouti, is Up-
Regulated in Obese and Diabetic Mutant Mice", Gen. & Dev. Vol. 11, pp. 593-602
(1997).
Stimulation of the MC-4 receptor by its endogenous ligand, aMSH, produces a
satiety signal and
CA 02368431 2001-09-28
WO 00/58361 PCT/US00/07473
-3-
may be the downstream mediator of the leptin satiety signal. It is believed
that by providing
potent MC-4 receptor agonists, appetite may be suppressed and weight loss
benefits may be
achieved.
Applicants have discovered a class of compounds that surprisingly have high
affinity for
the MC-4 and/or the MC-3 receptor subtypes, and that are typically selective
for these MC
receptors relative to the other melanocortin receptor subtypes, particularly
the MC-1 subtype. It
is therefore an object of this invention to provide chemical compounds that
activate or antagonize
the MC-4 and/or the MC-3 receptor subtypes. It is a further object of the
invention to provide
means for administration of said compounds to animals or man. Still other
objects of the
~o invention will be evident from the following disclosure of the invention.
DISCLOSURE OF THE INVENTION
Applicants have discovered certain structural requirements for a class of
cyclic peptide
analogs that are ligands for receptors of the MC-4 and/or the MC-3 subtype.
The structural
requirements constitute an optimal ring size of the peptide analog cycle at
the proper location in
s the analog, as is described below. Thus, the present invention relates to a
cyclic peptide analog
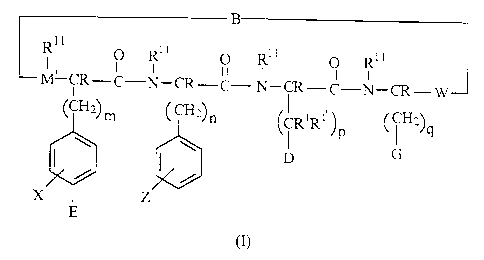
having a structure according to Formula (I):
B
I1
O R1' O R1' o
O R
M~ ~-C-N-CR-C- N-~-C-N-~- W
~~Z~m CCHZ~n CR~RI'1 ~CH2~
Jp ~ q
D G
X Z
E
(I)
wherein
zo (A) each of m, n, and q is independently selected from 0 to about 4 and p
is from 0 to about
5;
(B) X, which represents the four substituents on the phenyl ring other than E
and (CH,)m, is
independently selected from hydrogen; halo; ORB; -SRg; -NRBRg~; -N(R8)SO,RB~~;
-
SO~RB~~; -SO~-NRgRB~; -(CH,)T PO~HR'~' where r is 0 to about 10 and- R'4 is
selected from -
2s OH, hydrogen and alkyl; alkyl; alkene; alkyne; cyano; nitro; CF3; aryl;
heteroaryl;
cycloalkyl; and heterocycloalkyl; where each R8 and R8~ is independently
selected from
CA 02368431 2001-09-28
WO 00/58361 PCT/US00/07473
-4-
hydrogen, alkyl, acyl, heteroalkyl, aryl, heteroaryl, cycloalkyl, and
heterocycloalkyl and
RB~~ is selected from hydrogen, alkyl, heteroalkyl, aryl, heteroaryl,
cycloalkyl, and
heterocycloalkyl; or two X moieties can together form a fused ring with the
depicted
phenyl ring;
(C) E is selected from hydrogen; halo; -OR'3; -SR'3; -NR"R''~; -N(R'3)SO,R"~~;
-SO~R'3~~; -
SO,-NR'3R'3~; -(CH~)~ PO~HR'' where r is 0 to about 10 and R'' is selected
from -OH,
hydrogen and alkyl; alkyl; alkene; alkyne; cyano; nitro; CF3; aryl;
heteroaryl; cycloalkyl;
and heterocycloalkyl; provided that when each X is hydrogen, E is not
hydrogen; where
each R'3 and R'3~ is independently selected from hydrogen, alkyl, acyl,
heteroalkyl, aryl,
~o heteroaryl, cycloalkyl, and heterocycloalkyl and R'3~~ is selected from
hydrogen, alkyl,
heteroalkyl, aryl, heteroaryl, cycloalkyl, and heterocycloalkyl;
(D) Z is one or more substituents independently selected from hydrogen,
hydroxy, halo, thiol,
-OR9, -SR9, -NR9R9~, alkyl, acyl, alkene, alkyne, cyano, nitro, aryl,
heteroaryl, cycloalkyl,
and heterocycloalkyl; where each R9 and R9~ is independently selected from
hydrogen,
i5 alkyl, acyl, heteroalkyl, aryl, heteroaryl, cycloalkyl, and
heterocycloalkyl; or some Z
moieties can form a fused ring with the depicted phenyl ring;
(E) D is selected from -N(R'')C(=NR')NR4R', an optionally substituted imidazol
ring, and
NR'~R', wherein
( 1 ) Rr and R3 are independently selected from hydrogen, alkyl, alkene, and
alkyne;
20 or R' and R3, together with the atoms to which they are bonded, join to
form a
heterocycloalkyl or a heteroaryl; or R' and R°, together with the atoms
to which
they are bonded, join to form a heterocycloalkyl or a heteroaryl; or R' and
R'',
together with the atoms to which they are bonded, join to form a
heterocycloalkyl
or a heteroaryl; and
25 (2) R~ and RS are independently selected from hydrogen, alkyl, alkene, and
alkyne;
or R~ and R5, together with the atoms to which they are bonded, join to form a
heterocycloalkyl or a heteroaryl;
(F) each R' and R'~ is independently selected from hydrogen, alkyl, aryl and
heteroaryl; or
two R' moieties, together with the carbon atoms to which they are bonded, join
to form a
so cycloalkyl or aryl ring; or an R' and R'' (if present), together with the
atoms to which they
are bonded, join to form a heterocycloalkyl or a heteroaryl; or an R' and R3
(if present),
together with the atoms to which they are bonded, join to form a
heterocycloalkyl or a
CA 02368431 2001-09-28
WO 00/58361 PCT/US00/07473
-5-
heteroaryl; or an R' and R'' (if present), together with the atoms to which
they are
bonded, join to form a heterocycloalkyl or a heteroaryl;
(G) G is selected from an optionally substituted bicyclic aryl ring and an
optionally
substituted bicyclic heteroaryl ring;
(H) each R" is independently selected from hydrogen, alkyl, alkene, alkyne,
aryl, heteroaryl,
and cycloalkyl; and each R is independently selected from hydrogen, alkyl,
alkene,
alkyne, aryl, heteroaryl, and cycloalkyl; or an R" moiety can join with an
adjacent R
moiety to form a ring;
(I) W is selected from covalent bond, -CH,- and -C(=O)-;
io (J) M' is selected from covalent bond, -N- and -CH-; and
(K) B is an optionally substituted bridge moiety that links M' and W to form a
ring and
comprises either a covalent bond or an ionic bond, wherein when the bridge
moiety
comprises an ionic bond it is unsubstituted or is substituted with not more
than 3 amino
acid residues;
provided that when the compound comprises less than 25 ring atoms, then the
phenyl ring
substituted with Z is of the D-configuration ("D-Phe" or "f ') and further
provided that when B
comprises two or more Cys residues that form one or more disulfide bonds, said
disulfide bonds)
is not necessary for the existence of the cyclic peptide analog of Formula
(I).
The invention also relates to pharmaceutical compositions comprising the above
zo compounds, and to methods of treating disorders mediated by the MC-3 or MC-
4 receptor by
administering these compounds.
DETAILED DESCRIPTION OF THE INVENTION
I. Definitions:
"Amino acid" refers to alanine (Ala; A), arginine (Arg; R), asparagine (Asn;
N), aspartic
acid (Asp; D), cysteine (Cys; C), glutamic acid (Glu; Q), glutamine (Gln; E),
glycine (Gly; G),
histidine (His; H), isoleucine (Ile; I), leucine (Leu; L), lysine (Lys; K),
methionine (Met; M),
phenylalanine (Phe; F), proline (Pro; P), serine (Ser; S), threonine (Thr; T),
tryptophan (Trp; W),
tyrosine (Tyr; Y), and valine (Val; V). The common 3-letter and 1-letter
abbreviations are
indicated parenthetically. Modified amino acids also useful herein are the
following (the 3-letter
3o abbreviation for each moiety is noted parenthetically): p-Benzoyl-
phenylalanine (Bpa);
(3-(2-Naphtyl)-alanine (Nal); ~3-Cyclohexylalanine (Cha), 3,4-
Dichlorophenylalanine (3,4-Dcp);
4-Fluorophenylalanine (4-Fpa); 4-Nitrophenylalanine (4-Npa); 2-Thienylalanine
(Tha); 1,2,3,4-
Tetrahydroisoquinoline-3-carboxylic acid (Tic); 3-Benzothienylalaninem (3-
Bal);
CA 02368431 2001-09-28
WO 00/58361 PCT/US00/07473
-6-
4-Cyanophenylalanine (4-Ypa); 4-lodophenylalanine (4-lpa); 4-
Bromophenylalanine (4-Rpa);
4,4'-Biphenylalanine (Bip); Pentafluorophenylalanine (Pfp); and (3,(3-
Diphenylalanine (Dip).
With respect to moieties depicted on Formula (I) and Formula (A), moieties
referred to using a
single letter designation are as defined and do not refer to the single letter
amino acids
corresponding to those letters.
The letter "D" preceding the above three-letter abbreviations, e.g. as in "D-
Nal" or
"D-Phe", denotes the D-form of the amino acid. The letter "L" preceding an
amino acid three-
letter abbreviation denotes the natural L-form of the amino acid. For purposes
of this disclosure,
unless otherwise indicated, absence of a "D" or "L" designation indicates that
the abbreviation
to refers to both the D- and L-forms. Where the common ~;noiP_~PrrPT
~~,~".o..;"t:,.., .,. ......a
capitalization refers to the L-form and small letter designation refers to the
D-form, unless
otherwise indicated.
"Ac" refers to acetyl (i.e., CH3C(=O)-).
"Alkoxy" is an oxygen radical having a hydrocarbon chain substituent, where
the
i5 hydrocarbon chain is an alkyl or alkene (i.e., -O-alkyl or -O-alkene).
Preferred alkoxy groups
include (for example) methoxy (Me0), ethoxy, propoxy and allyloxy.
"Alkyl" is a saturated hydrocarbon chain having 1 to 15 carbon atoms,
preferably 1 to 10,
more preferably 1 to 4 carbon atoms. "Alkene" is a hydrocarbon chain having at
least one
(preferably only one) carbon-carbon double bond and having 2 to 1~ carbon
atoms, preferably 2
2o to 10, more preferably 2 to 4 carbon atoms. "Alkyne" is a hydrocarbon chain
having at least one
(preferably only one) carbon-carbon triple bond and having 2 to 15 carbon
atoms, preferably 2 to
10, more preferably 2 to 4 carbon atoms. Alkyl, alkene and alkyne chains
(referred to
collectively as "hydrocarbon chains") may be straight or branched and may be
unsubstituted or
substituted. Preferred branched alkyl, alkene and alkyne chains have one or
two branches,
2s preferably one branch. Preferred chains are alkyl. Alkyl, alkene and alkyne
hydrocarbon chains
each may be unsubstituted or substituted with from 1 to 4 substituents; when
substituted,
preferred chains are mono-, di-, or tri-substituted. Alkyl, alkene and alkyne
hydrocarbon chains
each may be substituted with halo, hydroxy, aryloxy (e.g., phenoxy),
heteroaryloxy, acyloxy (e.g.,
acetoxy), carboxy, aryl (e.g., phenyl), heteroaryl, cycloalkyl,
heterocycloalkyl, spirocycle, amino,
3o amido, acylamino, keto, thioketo, cyano, or any combination thereof.
Preferred hydrocarbon
groups include methyl (Me), ethyl, propyl, isopropyl, butyl, vinyl, allyl,
butenyl, and
exomethylenyl.
CA 02368431 2001-09-28
WO 00/58361 PCT/US00/07473
_7_
Also, as referred to herein, a "lower" alkyl, alkene or alkyne moiety (e.g.,
"lower
alkyl") is a chain comprised of 1 to 6, preferably from 1 to 4, carbon atoms
in the case of
alkyl and 2 to 6, preferably 2 to 4, carbon atoms in the case of alkene and
alkyne.
"Alkylthio" is a sulfur radical having a hydrocarbon chain substituent, where
the
hydrocarbon chain is an alkyl or alkene (i.e., -S-alkyl or -S-alkene).
Preferred alkylthio groups
include (for example) methylthio (MeS) and ethylthio.
"Aryl" is an aromatic hydrocarbon ring. Aryl rings are monocyclic or fused
bicyclic ring
systems. Monocyclic aryl rings contain 6 carbon atoms in the ring. Monocyclic
aryl rings are
also referred to as phenyl rings. Bicyclic aryl rings contain from about 8 to
about 17 carbon
io atoms, preferably about 9 to about 12 carbon atoms in the ring. Bicyclic
aryl rings include ring
systems wherein one ring is aryl and the other ring is aryl, cycloalkyl, or
heterocycloakyl.
Preferred bicyclic aryl rings comprise 5-, 6- or 7-membered rings fused to 5-,
6-, or 7-membered
rings. Aryl rings may be unsubstituted or substituted with from 1 to 4
substituents on the ring.
Aryl may be substituted with halo, cyano, nitro, hydroxy, carboxy, amino,
acylamino, alkyl,
i5 heteroalkyl, haloalkyl, phenyl, aryloxy, heteroaryloxy, or any combination
thereof. Preferred aryl
rings include naphthyl, tolyl, xylyl, and phenyl. The most preferred aryl ring
radical is phenyl.
"Aryloxy" is an oxygen radical having an aryl substituent (i.e., -O-aryl).
Preferred
aryloxy groups include (for example) phenoxy, napthyloxy, methoxyphenoxy, and
methylenedioxyphenoxy.
2o As used herein, "basic amino acids" refers to His, Lys, and Arg.
"Bc" refers to butyryl (i.e., CH3CH~CH~C(=O)-).
"Cycloalkyl" is a saturated or unsaturated hydrocarbon ring. Cycloalkyl rings
are not
aromatic. Cycloalkyl rings are monocyclic, or are fused, spiro, or bridged
bicyclic ring systems.
Monocyclic cycloalkyl rings contain from about 3 to about 9 carbon atoms,
preferably from 3 to 7
25 carbon atoms in the ring. Bicyclic cycloalkyl rings contain from 7 to 17
carbon atoms, preferably
from about 7 to about 12 carbon atoms in the ring. Preferred bicyclic
cycloalkyl rings comprise
4-, 5-, 6- or 7-membered rings fused to 5-, 6-, or 7-membered rings.
Cycloalkyl rings may be
unsubstituted or substituted with from 1 to 4 substituents on the ring.
Cycloalkyl may be
substituted with halo, cyano, alkyl, heteroalkyl, haloalkyl, phenyl, keto,
hydroxy, carboxy, amino,
3o acylamino, aryloxy, heteroaryloxy, or any combination thereof. Preferred
cycloalkyl rings
include cyclopropyl, cyclopentyl, and cyclohexyl.
"Fused" refers to cyclic moieties having at least two common ring atoms, the
preferred
maximum number of fused cycles being three.
"Halo" is fluoro (F), chloro (Cl), bromo (Br) or iodo (I).
CA 02368431 2001-09-28
WO 00/58361 PCT/US00/07473
_g_
"Heteroatom" is a nitrogen, sulfur, or oxygen atom, to which one or more
moieties may
be connected according to heteroatom valence; in the case of nitrogen, one
oxygen atom may be
optionally connected to it by a donor or acceptor bond, such as forming an N-
oxide. Groups
containing more than one heteroatom may contain different heteroatoms.
"Heteroalkyl" is a saturated or unsaturated chain containing carbon and at
least one
heteroatom, wherein no two heteroatoms are adjacent. Heteroalkyl chains
contain from 2 to
about 1 ~ member atoms (carbon and heteroatoms) in the chain, preferably 2 to
about 10, more
preferably 2 to about 5. For example, alkoxy (i.e., -O-alkyl or -O-
heteroalkyl) radicals are
included in heteroalkyl. Heteroalkyl chains may be straight or branched.
Preferred branched
~o heteroalkyl have one or two branches, preferably one branch. Preferred
heteroalkyl are saturated.
Unsaturated heteroalkyl have one or more double bonds (also referred to herein
as
"heteroalkenyl") and/or one or more triple bonds (also referred to herein as
"heteroalkynyl").
Preferred unsaturated heteroalkyl have one or two double bonds or one triple
bond, more
preferably one double bond. Heteroalkyl chains may be unsubstituted or
substituted with from 1
is to 4 substituents. Preferred substituted heteroalkyl are mono-, di-, or tri-
substituted. Heteroalkyl
may be substituted with lower alkyl, halo, hydroxy, aryloxy, heteroaryloxy,
acyloxy, carboxy,
monocyclic aryl, heteroaryl, cycloalkyl, heterocycloalkyl, spirocycle, amino,
acylamino, amido,
keto, thioketo, cyano, or any combination thereof.
"Heterocycloalkyl" is a saturated or unsaturated, non-aromatic ring containing
carbon
zo and from 1 to about 4 (preferably 1 to 3) heteroatoms in the ring, wherein
no two heteroatoms are
adjacent in the ring and no carbon in the ring that has a heteroatom attached
to it also has a
hydroxyl, amino, or thiol radical attached to it. Heterocycloalkyl rings are
monocyclic, or are
fused, bridged, or spiro bicyclic ring systems. Monocyclic heterocycloalkyl
rings contain from
about 4 to about 9 member atoms (carbon and heteroatoms), preferably from 5 to
7 member
2s atoms in the ring. Bicyclic heterocycloalkyl rings contain from about 7 to
about 17 atoms,
preferably from 7 to 12 atoms. Bicyclic heterocycloalkyl rings may be fused,
spiro, or bridged
ring systems. Preferred bicyclic heterocycloalkyl rings comprise 5-, 6- or 7-
membered rings
fused to 5-, 6-, or 7-membered rings. Heterocycloalkyl rings may be
unsubstituted or substituted
with from 1 to 4 substituents on the ring. Heterocycloalkyl may be substituted
with halo, cyano,
3o hydroxy, carboxy, keto, thioketo, amino, acylamino, acyl, amido, alkyl,
heteroalkyl, haloalkyl,
phenyl, phenoxy or any combination thereof. Preferred substituents on
heterocycloalkyl include
halo and haloalkvl.
"Heteroaryl" is an aromatic ring containing carbon and from 1 to about 6
heteroatoms in
the ring. Heteroaryl rings are monocyclic or fused bicyclic ring systems.
Monocyclic heteroaryl
CA 02368431 2004-04-22
-9-
rings contain from about p to about 9 member atoms (carbon and heteroatoms),
preferably 5 or 6
member atoms in the ring. Bicyclic heteroaryl rings contain from about 8 to
about 17 member
atoms, preferably about 8 to about 12 member atoms in the ring. Bicyclic
heteroaryl rings
include ring systems wherein one ring is heteroaryl and the other ring is
aryl, heteroaryl,
s cycloalkyl, or heterocycloalkyl. Preferred bicyclic heteroaryl ring systems
comprise S-, 6- or 7-
membered rings fused to ~-, 6-, or 7-membered rings. Heteroaryl rings may be
unsubstituted or
substituted with from I to 4 substituents on the ring. Heteroaryl may be
substituted with halo,
cyano, nitro, hydroxy, carboxy, amino, acylamino, alkyl, heteroalkyl,
haloalkyl, phenyl, aryloxy,
heteroaryloxy, or any combination thereof. Preferred heteroaryl rings include
thienyl, thiazolo,
~o imidazyl, purinyt, pyrimidyl, pyridyl, and furanyl.
As used herein, "MC-4 agonist" and "MC-3 agonist" refers to a compound with
affinity
for the MC-=1 receptor or MC-3 receptor, respectively, that results in
measurable biological
actiozty in cells, tissues, or organisms which contain the MC-4 or MC-3
receptor. Assays which
demonstrate MC-4lMC-3 agonistic activity of compounds are well known in the
art. One
t5 particuEarly useful assay is the BioTrak TM cAMP direct enzymeimmunoassay
(EIA) system
from Amersham Pharmacia Biotech, which quantitates the CAMP response of cells
to MC
ligands. This system allows the simple quantitation of total cellular cAMP
measurement in cells
exposed to selective ligands. Briefly summarized: HEK cells stably transfected
with the MC-1,
MC-3 or MCP receptors are plated into 96 well microtiter plates and grown
overnight. Cells are
zo dosed with the appropriate MC ligand for I hour and then lysed. A fraction
of the lysed cell
extract is transfened to the assay plate. The ELISA assay is performed
according to kit
instructions. Each plate contains a series of cAMP standards for calculating a
standard curve, as
well as a full MC agonist as a positive control for each MC receptor. cAMP
activity is calculated
as a % of the maximum cAMP activity of the full MC agonist control.
2s As used herein, "MCP antagonist" and "MC-3 antagonist" refer to compounds
with
affinity for the MC-4 receptor or MC-3 receptor, respectively, and blocks
stimulation by a known
MC agonist. Assays which demonstrate MC-4/MC-3 antagonistic activity of
compounds are well
latown in the art.
As used herein, "MC-3 receptor" and "MC-~4 receptor ' mean the known MC-3 and
MC-:l
so receptors, their splice variants, and undescribed receptors. MC-3 receptors
are described by
Gantz et al., supra (human MC-3): Desarnaud et al., supra (mouse MC-3) and L.
Reyfuss et al..
"Identification of a Receptor far Gamma Melanotropin and Other
Proopiomelanocortin Peptides
in the Hypothalamus and Limbic System., Proc. Natl. Acad. Sci. USA, vol. 90,
pp. 8856-8860
( 1993) (rat MC-3). MC-4 receptors are described by Gantz et al., supra (human
MC-4), J.D.
*trade-mark
CA 02368431 2004-04-22
-10-
Alvaro et al., "Morphine Down-Regulates Melanocortin-4 Receptor Expression in
Brain Regions
that Mediate Opiate Addiction", Mol-Pharmacol. Sep, vol. 50(3), pp. 583-91
(1996) (tat MC-4)
and Takeuchi, S. and Takahashi, S., "Melanocortin Receptor Genes in the
Chicken--Tissue
Distributions", Gen-Comp-Endocrinol., vol. 112(2), pp 220-31 (Nov, 1998)
(chicken MCli).
s As used herein, "measurable" means the biologic effect is both reproducible
and
significantly different from the baseline variability of the assay.
A "pharmaceutically-acceptable salt" is a cationic salt formed at any acidic
(carboxylic
acid) group, or an anionic salt formed at any basic (e.g., amino) group. Nfany
such salts are
known in the art, as described in World Patent Publication 87105297, Johnston
et al., published
to September 11, 1987 . Preferred cationic salts include the alkali
metal salts (such as sodium and potassium), and alkaline earth metal salts
(such as magnesium
and calcium) and organic salts. Preferred anionic salts include the halides
(such as chloride
salts), sulfonates, carboxylates, phosphates, and the like. Clearly
contemplated in such salts are
addition salts that may provide an optical center where once there is none.
For example, a chiral
za tartrate salt may be prepared from the compounds of the invention, and this
definition includes
such chiral salts.
Such salts are well understood by the skilled artisan, and the skilled artisan
is able to
prepare any number of salts given the knowledge in the art. Furthermore, it is
recognized that the
skilled artisan may prefer one salt over another for reasons of solubility,
stability, formulation
zo ease and the like. Determination and optimization of such salts is within
the purview of the
skilled artisan's practice.
As used herein, "selective" means having an activation preference for a
specific receptor
over other receptors which can be quantified based upon whole cell, tissue, or
organism assays
which demonstrate receptor activity, such as the cAMP enzyme in~ununoassay
(EIA) system
2s discussed above. A compound's selectivity is determined from a comparison
of its ECso values at
the relevant receptors being referenced. As used herein, use of the term
"selective over the other
MC receptors" means selective with respect to all of the MC-1, MC-2 and MC-~
receptors. For
example, a compound having an ECso of 8 nM at the MC-4 receptor and an ECso of
>_ 80 nM at
the MC-1, MC-2 and MC-5 receptors has a selectivity ratio for the MC-4
receptor over the other
3a MC receptors of at least 1:10. Additionally, it will be recognized that
selectivity may also refer
to one of the MC-l, MC-2 or MC-5 receptors individually. For example, a
compound having an
ECso of 8 nM at the MC-4 receptor and an EC;o of 80 nM at the MC-1 receptor
has a selectivity
ratio for the MC-4 receptor over the MC-1 receptor of 1:10. Such a compound is
selective over
the MC-1 receptor, regardless of its ECso value for MC-2 or MC-5. Selectivity
is described in
CA 02368431 2001-09-28
WO 00/58361 PCT/US00/07473
-11-
more detail below and may be determined by using, for example, the software
Prism v 2.0 which
is available from GraphPad, Inc.
"Spirocycle" is an alkyl or heteroalkyl diradical substituent of an alkyl or
heteroalkyl,
wherein said diradical substituent is attached geminally and wherein said
diradical substituent
s forms a ring, said ring containing about 4 to about 8 member atoms (carbon
or heteroatoms),
preferably 5 or 6 member atoms.
"Substituted" refers to one or several hydrogens being substituted,
independently, by
alkyl, haloginated alkyl, alkenyl, halogenated alkenyl, alkynyl, halogenated
alkynyl, cycloalkyl,
halogenated cycloalkyl, cycloheteroalkyl, halogenated cycloheteroalkyl,
cycloalkenyl,
io halogenated cycloalkenyl, cycloheteroalkenyl, halogenated
cycloheteroalkenyl, aryl, halogenated
aryl, heteroaryl, halogenated heteroaryl and/or functional group. Moreover, if
a "substituted"
structure is a cyclic structure fused with other cyclic structures) these
latter cyclic structures)
may also be substituted.
A "solvate" is a complex formed by the combination of a solute (e.g., a cyclic
MC-4/MC-
i5 3 receptor ligand of the present invention) and a solvent (e.g., water).
See J. Honig et al., The
van Nostrand Chemist's Dictionary, p. 650 (1953). Pharmaceutically-acceptable
solvents used
according to this invention include those that do not interfere with the
biological activity of the
compound (e.g., water, ethanol, acetic acid, N,N-dimethylformamide and others
known or readily
determined by the skilled artisan).
2o II. The Compounds
The compounds of the present invention are MC-4 and/or MC-3 receptor ligands
having
a structure according to Formula (I):
B
II
O Rl~ O Rll a
II I O R
M-~-C-N-CR-C- N-~-C-N-~- W
~~z~m ~CHz~n CR~RI'l ~CHz~
/p I q
D G
X Z
E
(I)
25 wherein B, X, E, Z, D, G, R, R', R'~, R", m, n, p, and q are as described
in the Disclosure of the
Invention section above.
CA 02368431 2001-09-28
WO 00/58361 PCT/US00/07473
-12-
With reference to Formula (I), it is seen that the compounds comprise an
important
backbone based on the natural amino acid sequence Tyr (or substituted Phe)-Phe-
J-M (per the
description above, when the first amino acid is Phe it should be substituted
and is preferably
Tyr), where J is an amino acid whose side chain is a nitrogen containing group
(e.g., Arg, His or
Lys) or a derivative thereof, and M is a bicyclic aromatic moiety (e.g., Trp
or napthylalanine, or a
derivative thereof). Preferred are those compounds wherein J is Arg or a
derivative of Arg and M
is Trp or a derivative of Trp. It is noted, as is depicted in Formula (I),
that substitution of the
natural amino acids is possible without losing the MC-3/ MC-4 ligand
properties. In this regard,
while reference is made herein to the Phe(Tyr)-Phe-J-M sequence, it is
understood that
o substitution is allowed per the description of Formula (I).
Applicants have found that to obtain optimum agonistic or antagonistic
activity, the ring
moiety of the compound will preferably comprise from 25 to 27 ring atoms. That
is, the ring
comprising the depicted residues and the bridging moiety, B, preferably
contains from 25 to 27
ring atoms. It will be recognized that additional amino acids or other
chemical entities may be
is included as substituents on the cyclic structure depicted in Formula (I)
without negatively
impacting interaction with MC-3/MC-4 receptor.
That the compounds of Formula (I) have affinity for the MC-4. andlor the MC-3
receptor
is surprising, given that the linear compound BIM-22015 exhibits essentially
no affinity for any
of the melanocortin receptors, including MC-3 and MC-4, while differing from
endogenous a-
2o MSH in the 6-9 region by only the substitution of Tyr for His at position 6
(using natural a-MSH
numbering). See, e.g., Schioth, H.B. et al., "Selectivity of [Phe-17], [Ala6)
and [D-
Ala4,G1n5,Tyr6] Substituted ACTH (4-10) Analogues for the Melanocortin
Receptors", Peptides,
vol. 18(5), pp. 761-3 (1997).
-Tyr-Phe-Arg -Trp- BIM-22015
6 7 8 9
- His- Phe- Arg -Trp- alpha-MSH
25 6 7 8 9
One would have also predicted that Applicants' compounds would lack affinity
for these
receptors given the common -Tyr-Phe-Arg-Trp- domain the preferred compounds
share with
BIM-22015. That is, the logical conclusion is that the His at position 6 is
critical, or at a
minimum that His at position 6 cannot be replaced with a substituted Phe or
Tyr. Surprisingly,
3o this is not the case, as Applicants compounds exhibit significant affinity
for one or both of the
MC-3 and MC-4 receptors. Without being bound by theory, Applicants believe the
surprisingly
high affinity of the present compounds is due to the shape of these residues
induced by the cyclic
CA 02368431 2001-09-28
WO 00/58361 PCT/US00/07473
-13-
nature of the molecules. That is, the cyclic aspect of the compounds provides
rigidity which
allows them to effectively interact with the relevant binding sites of the MC-
4/MC-3 receptor.
Moreover, it appears that the surprising ability to retain the preferred Tyr-
like residue provides
desired selectivity relative to the other MC receptors, particularly the MC-1
receptor.
With respect to B, this bridge can be in the form of covalent bond linkages or
alternatively can include a salt bridge resulting from the formation of ionic
bonds. The bridging
moiety can be wholly peptidic in nature (i.e., containing amino acids only),
non-peptidic (i.e.,
containing no amino acids) in nature, or it can include both peptidic and non-
peptidic moieties
introduced using well known chemistry. The bridge can comprise aliphatic
residues, aromatic
~o residues or heteroaromatic residues, or any combination thereof. The bridge
preferably
comprises at least 2 amino acids, such that the compounds of the present
invention comprise at
least 6 amino acid residues. Preferably, B will not contain 3 adjacent amino
acids that are all are
basic amino acids. In addition, when B comprises two or more Cys residues that
form one or
more disulfide bonds, said disulfide bonds) is not necessary for the existence
of the cyclic
i5 molecule of Formula (I). In other words, cleavage of such disulfide bonds)
does not result in the
loss of the ring formed by joining M' and W of Formula (I).
In one embodiment, the bridge will preferably comprise long chain omega-amino
acids in
which amino and carboxyl groups are separated by from about 4 to about 6
methylene groups or a
combination of said omega-amino acids and aminobenzoic acids.
2o In another embodiment, which is a preferred embodiment, the bridging moiety
will
contain all covalent bonds, such as an amide bond. For example, the bridge can
comprise an
amide formed through the chemical coupling of a side-chain amino group of
amino acids such as
Lys or Orn, and a side-chain carboxyl group of the amino acid residue such as
Asp or Glu.
Alternatively, the bridging moiety can comprise an amide formed between the
amino and
25 carboxylate groups attached to the a-carbon of the bridging moiety amino
acids. (Hereafter
referred to as the "a-amino" moiety of an amino acid or the "a-carboxyl"
moiety of an amino
acid.) In another alternative, the bridging moiety can comprise an amide
formed between any
combination of the side-chain amino group or side-chain carboxyl group and the
a-amino and the
a-carboxyl moieties. The bridging residues may be amine- or carboxyl-
containing structures
so other than natural amino acids, including, e.g., 6-aminohexanoic acid as an
amine-containing
residue and succinic acid as a carboxyl-containing residue. Furthermore, the
invention allows for
bridging of the Tyr-Phe-Arg-Trp core sequence using other types of chemical
functionalities. In
this case, these bridging residues may contain a variety of groups and
substituents, including
aliphatic, aromatic and heterocyclic moieties. When covalently linked, the
bridge can be
CA 02368431 2004-04-22
-14-
connected through a variety of linkages including but not limited to amide,
ester, ether, thioether,
aminoalkyl or aminoaryl bonds. When B is a covalent bond. preferred are
compounds having
from about 24 to about 30 ring atoms, more preferred are compounds having from
about 25 to
about 27 ring atoms.
The bridging moiety can alternatively be an ionic bond/association that favors
a cyclic
structure. This "ionic" bridge is comprised of salt-forming basic and acid
functionalities. For
example, the bridge can comprise an ionic bond formed between the side-chain
amino group of
amino acids such as Lys or Om, and the side-chain carboxyl group of the amino
acid residue such
as Asp or Glu. Alternatively, the bridging moiety can comprise an ionic bond
formed between
~o the amino and carboxylate groups attached to the a-carbon of the bridging
moiety amino acids.
In another alternative, the bridging moiety may comprise an amide formed
between any
combination of the side-chain amino group or side-chain carboxyl and the a-
amino and the a-
carboxyl moieties. Since an ionic bond is typically weaker than a covalent
bond, it is easier to
distort the topography of a cyclic structure based on such an ionic bond. This
distortion may
i5 occur when additional groups are attached to the bridging moiety, thereby
negatively impacting
interaction with the receptor, Thus, the bridging moiety preferably will not
be substituted with
more than 3 amino acid residues when the moiety is in the form of an ionic
bond. In one
particularly preferred aspect, when B is an ionic bond, the compounds wilt
have from about 26 to
about 29 ring atoms.
2o It will be recognized that any free peptidic a-carboxy and a-amino groups
(i.e., amino
acid a-carboxy and a-amino groups) not involved in formation of the ring can
optionally be in
the form of a carboxyamide or an acylamino moiety, respectively.
1n addition to the compounds described by Formula (I), it is envisioned that
the core
peptide residues can be pegylated to provide enhanced therapeutic benefits
such as, for example,
2s increased efficacy by extending half Life in vfvo. Peptide pegylation
methods are well (mown in
the literature. For example pegylation of peptides is described in the
following references,
Lu, Y.A. et al., "Pegylated
peptides. II. Solid-phase synthesis of amino-, carboxy- and side-chain
pegylated peptides", Inr. J.
Pept. Protein: Res., Vol. 43(2), pp. I27-38 (1994); Lu, Y.A. et al.,
"Pegylated peptides. I. Solid-
3o phase synthesis of N alpha-pegylated peptides using Fmoc strategy", Pept.
Res., Vol. 6(3), pp.
140-6 (1993); Felix, A.M. et al., "Pegylated peptides. CV. Enhanced biological
activity of site-
directed pegylated GRF analogs.", Int. J. Pept. Protein Res., Vol. 46(3-4),
pp. 253-64 (1990:
Gaertner, H.F. et al., "Site-speciftc attachment of functionalized
poly(ethyiene glycol) to the
amino terminus of proteins", Bioconjug Chem., Vol. 7(1), pp. 38-44 (1996);
Tsutsumi. Y. et al.,
CA 02368431 2001-09-28
WO 00/58361 PCT/US00/07473
-15-
"PEGylation of interleuken-6 effectively increases its thrombopoietic
potency", Thromb
Haentost, Vol. 77(1), pp. 168-73 (1997); Francis, G.E. et al., "PEGylation of
cytokines and other
therapeutic proteins and peptides: the importance of biological optimisation
of coupling
techniques", Int. J Hematol., Vol. 68(1), pp. 1-18 (1998); Roberts, M.J. et
al., "Attachment of
degradable polyethylene glycol) to proteins has the potential to increase
therapeutic efficacy", J.
Pharrn. Sci., Vol 87(11), pp. 1440-45 (1998); and Tan, Y. et al.,
"Polyethylene glycol conjugation
of recombinant methioninase for cancer therapy", Protein Expr. Purif., Vol.
12(1), pp. 45-52
( 1998). The compounds of Formula (I) can be pegylated directly, or a "linker
arm" may be added
to the compounds to facilitate pegylation.
~o With reference to Formula (I), the following is a non-limiting list of
preferred
substituents:
For m, n, and q, preferred is 1. For p, preferred is 3.
For X, preferred substituents are hydrogen, hydroxy, halo, -ORB. -NRgRB~,
alkyl, cyano,
nitro, aryl, heteroaryl, cycloalkyl, and heterocycloalkyl; more preferred is
where X is hydrogen,
5 hydroxy, halo, -ORB, -NRBRB~, alkyl, cyano or nitro; most preferred is where
X is hydrogen. For
RB and RB~ preferred are hydrogen, alkyl, acyl, aryl, and cycloalkyl; more
preferred is where RB is
hydrogen and RB~ is hydrogen, alkyl or acyl. Also preferred is where two X
moieties form a
fused ring with the depicted phenyl ring.
For E, preferred substituents are halo, especially fluoro, chloro and bromo; -
OH; -SH;
20 -OR's; -SR'3; -NHR'3, where R'' is preferably acyl; -NHSO,R'3~~; -(CH~)~
PO~HR'' where r is 0 to
about 10 and R" is selected from -OH, hydrogen and alkyl; alkyl; cyano; nitro;
and CF3. R'3~~ is
preferably selected from hydrogen and alkyl. Most preferred is -OH.
For Z, preferred substituents are hydrogen, hydroxy, halo, -OR9, -NR9R9~,
alkyl, cyano,
nitro, aryl, heteroaryl, cycloalkyl, and heterocycloalkyl; more preferred is
where Z is hydrogen,
2s hydroxy, halo, -OR9, -NR9R9~, alkyl, cyano or nitro; most preferred is
where Z is hydrogen. For
R9 and R9~ preferred are hydrogen, alkyl, acyl, aryl, and cycloalkyl; more
preferred is where R9 is
hydrogen and R9~ is hydrogen, alkyl or acyl. Also preferred is where two Z
moieties form a fused
ring with the depicted phenyl ring.
For each existence of R' and R'~, preferred are hydrogen and alkyl.
Alternative preferred
3o compounds are those where an R' and R~, together with the atoms to which
they are bonded, join
to form a heterocycloalkyl or a heteroaryl ring.
For D, preferred is -N(R')C(=NR')NR''R'.
CA 02368431 2001-09-28
WO 00/58361 PCT/US00/07473
-16-
For each of R- and R3, when present, preferred are hydrogen and alkyl, more
preferred is
hydrogen. Alternative preferred compounds are those where R' and R'', together
with the atoms
to which they are bonded, join to form a heterocycloalkyl or a heteroaryl
ring.
For each R'' and R5, when present, preferred are hydrogen and alkyl, most
preferred is
hydrogen.
For G, preferred are optionally substituted napthylene rings and optionally
substituted
indoles (i.e., the residue of Formula (I) is Trp); more preferred is an
optionally substituted indole.
For each R" preferred are hydrogen and alkyl, more preferred is hydrogen.
For each R, preferred are hydrogen, alkyl and cycloalkyl; most preferred is
hydrogen.
io For B, preferred is where B results in a compound having greater than about
25 ring
atoms. In one aspect, preferred are compounds having greater than about 25
ring atoms and B
consists of amino acid (natural or unnatural, e.g., a, (3, y, etc.) residues,
preferably 3-5 residues,
more preferably 3 or 4 residues. Preferred are compounds where B is a
covalently bonded bridge.
Most preferred is where B comprises three amino acids wherein an
intramolecular amide is
15 formed through the chemical coupling of a side-chain amino group of one of
the amino acids (e.g.
Lys or Orn), and a side-chain carboxyl group of a second amino acid residue
(e.g. Asp or Glu).
A preferred subclass of compounds of Formula (I) are compounds having a
structure of
Formula (A) as follows:
B
R"
O R" O RI ~ 11
O R
M ~ CR- C-N- CR- C- N- CR- C-~;- CR- W
CHZ CHz CCR~R'~\ 6 CHZ
\ \ N_R213 R /
I
i ~\J
/ / C=N-R3 Y
X ~ Z N Q
OH Ra ~ ~ RS
20 (A)
where B, X, Z, M', W, R, R', R'~, R', R3, R'', R' and R" are as described
above; R6 is selected
from hydrogen, alkyl, hydroxy, alkoxy, aryl, heteroaryl, halogen, and SOrR''
where x is 0, 1 or 2
and R'' is aryl; Y is selected from -NR'-, -CR'~R'~, -CR'~=CR'~, -CR'~=N- and -
N=CR'~-,
wherein R' and R'~ are independently selected from hydrogen, alkyl, aryl, and
heteroaryl, or R' or
25 R'~ is a covalent bond that links Y to the R6 or -CH,- moiety depicted in
Formula (A); and Q is
one or more substituents independently selected from hydrogen, hydroxy, halo,
thiol, -OR'°,
CA 02368431 2001-09-28
WO 00/58361 PCT/US00/07473
-17-
SR'°, -NR'°R'°~, alkyl, alkene, alkyne, cyano, vitro,
aryl, heteroaryl, cycloalkyl, and
heterocycloalkyl; where each R'° and R'°~ is independently
selected from alkyl, heteroalkyl, aryl,
heteroaryl, cycloalkyl, and heterocycloalkyl; or two Q moieties can form a
fused ring with the
depicted phenyl ring. With reference to Formula (I), in this subclass of
Formula (A), m, n and q
are all l, p is 3, D is an optionally substituted guanidino moiety and G is an
optionally substituted
11 or 12 membered bicyclic aryl or heteroaryl.
Another preferred sub-class of compounds of Formula (I) are compounds having a
structure according to Formula (B), as follows:
B
11 11
O R O Rl1 O R11 O
N-CR-C-N-CR-C- N-CR-C-N-CR-C
cH~ I ~
CHI
< <CH2 ~ CHz
NH 3 /
/ I / C = NH
I N
OH NH, H
i o where R, R" and B are as defined above.
The following is a non-limiting list of preferred substituents for the
moieties of the
Formula (A) compounds:
For Q, preferred substituents are hydrogen, hydroxy, halo, -OR'°, -
NR'°R'°~, alkyl, cyano,
vitro, aryl, heteroaryl, cycloalkyl, and heterocycloalkyl; more preferred are
hydrogen, hydroxy,
halo, -OR'°, -NR'°R'°~, alkyl, cyano, and vitro; most
preferred is where Q is hydrogen. For R'°
and R'°~, preferred are hydrogen, alkyl, acyl, heteroalkyl, aryl,
heteroaryl, cycloalkyl, and
heterocycloalkyl; more preferred is where R'° is hydrogen and
R'°~ is hydrogen, alkyl or acyl.
Also preferred is where two Q moieties form a fused ring with the depicted
bicyclic ring.
For Rb,.preferred are hydrogen and alkyl, more preferred is hydrogen.
2o For R', preferred are hydrogen, alkyl and aryl, more preferred is hydrogen.
The following is a non-limiting list of preferred cyclic compounds of the
present
invention. (The use of brackets ([ ]) denotes amino acid points of
cyclization, where possible, via
side-chain moieties. Where typical amino acid termini capping groups are
indicated, e.g Ac- or -
NH~ (carboxamide capping group), these capping groups are utilized on the
peptidic a-carboxy or
z5 a-amino groups.) In this list, "Nal" refers to napthylalanine and "Orn"
refers to ornithine.
aDYfRWK-NH~ a[DYfRWK]-NH,
CA 02368431 2001-09-28
WO 00/58361 PCT/US00/07473
-18-
a[DY(D-2-Nal)RWK]-NH, Ac-aDYfRWK-NH,
Ac-a[DYfRWK]-NHS Ac-a[DY(D-Phe(4-CI))RWK]-NH,
Ac-a[DY(D-1-Nal)RWK]-NHS Ac-[EYfRWGK]-NH,
Ac-a[EYfRWG(Orn)]-NHS Ac-SYSa[DYfRWGK]-NH,
Ac-GGGa[DYfRWGK]-NH, Ac-[DY(D-1-Nal)RWGK]-NH=
Ac-aEYfRWGK-NH, Ac-aDYfRWGK-NH,
Ac-a[DYfRWGK]-NHS Ac-a[EYfRWGK]-NH,
Ac-aDYfRWG(Orn)-NHS Ac-a[DYfRWG(Orn)]-NH,
Ac-aEYfRWG(Orn)-NHS Ac-a[EYfRWG(Orn)]-NH,
io Ac-a[EFfRWGK]-NHS Ac-a[DFfRWGK]-NH,
Ac-a[DyfRWGK]-NHS Ac-[DYfRWGK]-NH,
Ac-a[DYyRWGK]-NHS Ac-a[DY(D-Phe(4-I))RWGK]-NH,
Ac-[DY(D-Phe(4-I))RWGK]- NH, Ac-a[EY(D-Phe(4-I))RWGK]-NH,
Ac-a[DY(D-Phe(4-I))RWG(Orn)]-NHS Ac-[EY(D-Phe(4-I))RWGK]-NHS
i s Ac-[DY(D-Phe(4-I))RWG(Orn)]-NH, Ac-[EYfRWGK]-NH,
Ac-[EYfRWG(Orn)]-NHS Ac-[DYfRWG(Orn)]-NHS
[GYfRWGGG] [GGYfRWGGG]
[GGYfRWGGGG] [GGYfRWGK]-NHS
[GGYfRWGK]-OH [GGYfRWGGAG]
20 [GGYfRWGLAG] [GGYfRWGFAG]
[AGYfRWGGG] [GGYfRWAAA]
Ac-[D(Tyr(3-OH)fRWGK]-NHS Ac-[DF(D-Phe(4-I))RWGK]-NHS
Ac-DYfRWK-NHS Ac-DYfRWGK-NH~
Ac-EYfRWGOrn-NHS Ac-a[DY(D-Phe(4-I))RWGK]-NH,
2s Ac-[DY(D-Phe(4-Cl))RWK]-NHS Ac-[D(Tyr(3-OH))fRWGK]-NHS
Ac-[DY(D-Phe(4-Cl)))RWGK]-NHz Ac-a[D(Tyr(Me))fRWGK]-NHS
Ac-a[DYfR(Trp(5-Br)GK]-NHS Ac-a[DYfR(Trp(5-F)GK]-NH~
Ac-a[DYfR(Trp(5-OMe)GK]-NHS Ac-a[DYfR(Trp(S-Me)GK]-NH,
Ac-a[DYfR(Trp(6-F)GK]-NHS Ac-a[DYfR(Trp( 1-Me)GK]-NHS
3o Ac-a[DYfR(Trp(4-F)GK]-NHS Ac-a[DYfR(Trp(6-Br)GK]-NHS
Ac-a[DYfR(Trp(7-Me)GK]-NHS Ac-a[EYfR(Trp(5-OH))G(Om)]-NH,
Ac-a[EYfR(Trp(6-OH))G(Orn)]-NH, Ac-a[EYfR(Trp(6-Cl))G(Orn)]-NHS
Ac-a[E(Tyr(Me))fRWG(Orn)]-NHS Ac-a[E(Tyr(CH~Ph))fRWG(Orn)]-NH,
Ac-[E(Tyr(2-Br))fRWG(Orn)]-NHS Ac-[E(Tyr(3-F))fRWG(Orn)]-NH,
CA 02368431 2001-09-28
WO 00/58361 PCT/US00/07473
-19-
Ac-[E(Tyr(3-I))fRWG(Orn)]-NHS Ac-[E(Tyr(2,5-OH))fRWG(Orn)]-NHS
Ac-[D(Tyr(3-NO,))fRWGK]-NHS Ac-[D(Tyr(3-NH~))fRWGK]-NH,
Ac=[D(Tyr(3-Me0))fRWGK]-NHS Ac-[D(Tyr(3-Cl))fRWGK]-NHS
Ac-a[DY(D-Phe(5-Br))RWGK]-NHS Ac-a[DY(D-Phe(4-NH,))RWGK]-NHS
s Ac-a[DY(D-Phe(4-NO~))RWGK]-NHS Ac-[DY(D-Phe(4-F))RWGK]-NH~
Ac-[DY(D-Phe(2,5-OH))RWGK]-NHS Ac-[DY(D-Phe(3-F))RWGK]-NHS
Ac-[DY(D-Phe(5-F))RWGK]-NHz Ac-[EY(D-Phe(3-Me0))RWGOrn]-NH,
Ac-[EY(D-Phe(3-NHZ))RWGOrn]-NHS Ac-[EY(D-Phe(3-NO~))RWGOrn]-NHS
Ac-[EY(D-Phe(3-I))RWGOrn]-NH, Ac-[D(Phe(4-Cl))fRWGK]-NHS
io Ac-[DYfRW(Gly-(N-CH3))K]-NHS Ac-[D(Phe(4- NH~))fRWGK]-NH,
Ac-[E(Phe(4-Br))fRWGK]-NHZ Ac-a[D(Phe(4-Cl))fRWGK]-NHS
Ac-a[DYFRW(Gly-(N-CH3))K]-NHS Ac-[DYFRW(Gly-(N-CH3))K]-NHS
Ac-a[D(Phe(4-Cl))FRWGK]-NHZ Ac-[D(Phe(4-CI))FRWGK]-NHS
Ac-[D(Phe(4-Br))FRWGK]-NH, Ac-[D(Phe(4-CN))fRWGK]-NHS
is Ac-[D(Phe(4-F))fRWGK]-NHZ Ac-[E(Phe(4-F))fRWG(Orn)]-NH,
Ac-[D(Phe(4-F))fRWGK]-NHS Ac-[DYf(Arg-(N-CH3))WGK]-NHS
Ac-[D(Phe(4-NO,))fRWGK]-NHS Bc-[DYfRWGK]-NH,
Ac-[D(Phe(4-NH~))fRW(Gly-(N-CH3))K]-NHS Bc-DYfRWGK-NHS
Ac-[D(Phe(4-Cl))fRW(Gly-(N-CH3))K]-NHS Bc-[DYfRW(Gly-(N-CH3))K]-NHS Ac-
20 [D(Phe(4-NO~))fRW(Gly-(N-CH3))K)-NHS Bc-aEYfRWGK-NH,
Ac-[D(Phe(4-NHCOCH3))fRW(Gly-(N-CH3))K]-NHZ
Ac-a[E(Phe(4-Cl))fRW(Gly-(N-CH3))K]-NHS
Ac-a[E(Phe(4-Cl))fRW(Gly-(N-CH3))(Orn)]-NHS
Ac-[E(Phe(4-CI))fRW(Gly-(N-CH3))(Orn)]-NH,
2s Ac-a[D(Phe(4-F))fRW(Gly-(N-CH3))K]-NH,
Ac-a[D(Phe(4-F))FRW(Gly-(N-CH3))K]-NHS
Bc-DYfRW(Gly-(N-CH3))K-NHS
Bc-EYfRW(Gly-(N-CH3))K-NHZ
Bc-EYfRWGK-NH~
3o Bc-D(Phe(4-Cl))fRW(Gly-(N-CH3))K-NHS
Bc-D(Phe(4-Cl))fRWGK-NHS
Bc-E(Phe(4-Cl))fRW(Gly-(N-CH3))K-NH,
Bc-[D(Phe(4-Cl))fRW(Gly-(N-CH3))K]-NH,
Bc-[DY(D-2-Nal)RWGK]-NH,
CA 02368431 2001-09-28
WO 00/58361 PCT/US00/07473
-20-
Bc-[DY(L-2-Nal)RWGK]-NHS
Bc-[DY(D-1-Nal)RWGK]-NH,
Bc-[DY(Phe(4-Br))RWGK]-NH,
Ac-[DY(D-2-Nal)RWGK]-NH,
Bc-EY(D-2-Nal)RWGK-NH,
Bc-EY(D-2-Nal)RW(Gly-(N-CH3))K-NHS
Ac-[DYf(homo-Arg)WGK]-NHS
Ac-[DYyRWGK]-NHS
Ac-[DYYRWGK]-NHS
i o DYfRWGK-NH~
Ac-a[DY(D-2-Nal)RWGK]-NHS
Ac-DYfRWGK-NH,
Ac-EYfRWGK-NHZ
Ac-EY(D-2-Nal)RWGK-NHS
Ac-[EY(D-2-Nal)RWGK]-NH,
Ac-a[DY(D-2-Nal)RWK]-NH,
a[DY(D-1-Nal)RWK]-NHS
In the following specific examples of compounds of the present invention,
absence of a "D" or "L" designation refers to the L-form.
CA 02368431 2001-09-28
WO 00/58361 PCT/US00/07473
-21-
~yr-DPhe-Arg-Tr~ ~Yr-DPhe-Arg-Trp-Gly
\ O
O (CH2)6NH-CO(CH2)6N (CH2)6NH-CO(CH2)6NH
H
~yr-DPhe-Arg-Trp-jly
yr-DPhe-Arg-Tr O (CH2)5NH-CO(CH2)6NH
~
~
O
CH NH-CO CH ,Tyr-DPhe-Arg-Trp~ly
H
( 2)5 ( 2)s
o
~(CH2)4NH-CO(CHz)6N/H
~yr-DPhe-Arg-Tr
\
O (CH2)4NH-CO(CH2)6N ~Yr-DPhe-Arg-Trp-~ly
H /
O (CH2)6NH-CO(CH2)5N
H
,Tyr-DPhe-Arg-Tr
\
O (CH2)6NH-CO(CH2)5N ~Yr-DPhe-Arg-Trp-Gly
H /
O (CH2)6NH-CO(CH2)4N
H
,Tyr-DPhe-Arg-Tr
p ~ yr-DPhe-Arg-Trp-~ly
(CH2)6NH-CO(CH2)4NH
O~CH NH-CO H
( 2)5 (C 2)5NH
o~yr-DPhe-Arg-Tr~ o~yr-DPhe-Arg-Trp-~ly
(CH2)5NH-CO(CH2)5N\H (CH2)4NH-CO(CH2)5N/H
yr-DPhe-Arg-Tr~ ~Yr-DPhe-Arg-Trp-~ly
O \ O (CH2)5NH-CO(CH2)4N/H
~CH NH-CO CH NH
( 2)4 ( 2)5
yr-DPhe-Arg-Tr~ ~Yr-DPhe-Arg-Trp-~ly
O \ O (CH2)4NH-CO(CHZ)4N/H
~CH NH-CO CH NH
( 2)5 ( 2)4
~yr-D P h e-Arg-T r~ ~yr- D P h e-Arg-T rp-~ I y
O O (CH2)aNH-CO(CH2)4N~
(CH2)4NH-CO(CH2)4NH
~yr-DPhe-Arg-
O (CH2)»NH ~Yr-DPhe-Arg-Trp-Gly
O (CH2)sNH-CO(CH2)sN
o~yr-DPhe-Arg- yr-DPhe-Arg-Trp-Giy
(CH2)8NH O~CH NH-CO CH N/
( 2)5 ( 2)s w
CA 02368431 2001-09-28
WO 00/58361 PCT/US00/07473
_22_
yr-DPhe-Arg-Trp-~ly
yr-DPhe-Arg-Trp-~y o
o ~ / ~ ~ NH(CH2)5NH
NH(CH2)sNH
NOz
No2 yr-DPhe-Arg-Trp- ly
yr-DPhe-Arg-Trp- ly o
~ ~ ~ NH(CH2)4NH
NH(CH2)4NH
NHz
NO2
yr-D P h e-A rg-T rp-G l y
yr-DPhe-Arg-Trp-Gly o
/0
w ~ ~ NH(CHZ)5NH
NH(CH2)sNH
NHZ
NHZ
o yr-DPhe-Arg-Trp-~ly yr-DPhe-Arg-Trp- ly
0
NH(CH2)5NH ~ ~ NH(CH2)5N~
NHz NOz
yr-DPhe-Arg-Trp- y yr-DPhe-Arg-Trp-Gly
o ~
NH(CH2)sN ( ~ NH(CH2)5N~
NOZ NHZ
yr-DPhe-Arg-Trp-~ly
0
NH(CH2)sN~
NHz
CA 02368431 2001-09-28
WO 00/58361 PCT/US00/07473
-23-
Tyr-D Phe-Arg-Trp-G'y ~yr_D P h e-Arg-Trp-G IN
r0
(CH2)4NH (CH2)aNH / NH
NH' O Q
~yr-DPhe-Arg-Trp-Gly
Tyr-D P h e-A rg-Trp-G~y 10
(CHz)sNH NH
(CH2)sNH o
NH' O
~yr-D P he-Arg-Trp-G~y
Tyr-DPhe-Arg-Trp ly o NH
o \ (CH2)sNH
~ ~~(CH2)sNH o _
NH O
~yr-DPhe-Arg-Trp-~ly
Tyr-DPhe-Arg-Trp- ly /o
o \ o (CH2)4NH \ NH
I
N~(CH2)4NH o
H
~yr-DPhe-Arg-Trp-~ly
Tyr-DPhe-Arg-Trp-Gly o0
o (CH2)sNH ~ I NH
O I \ \
~N~--(CH2)5NH o
H
~yr-DPhe-Arg-Trp-Gly
Tyr-DPhe-Arg-Trp-Gyly °°
o ~ o (CH2)sNH ~ I NH
-(CH2)sNH
N O
H
Tyr-DPhe-Arg-Trp-G y ~yr-DPhe-Arg-Trp-GI~
° N
(CH2)sNH
(CH2)5N~
NH' O
~yr-DPhe-Arg-Trp-Gly
Tyr-DPhe-Arg-Trp-Grly °°
(CH2)sNH~N~
o ~ \ o ~ \ II
N~--(CH2)sN~ o
H
CA 02368431 2001-09-28
WO 00/58361 PCT/US00/07473
-24-
NH
O ~--NH2 N~NHZ
IIH
~N O HN ".,
O HN~ N.H
N-{3-[9-Benzyl-12-(4-hydroxy-benzyl)-3-(1 H-indol-3-
ylmethyl)-2,5,8,11-tetraoxo-1-oxa-4,7,10-triaza-
cyclononadec-6-yl]-propyl}-guanidine N-{3-[9-Benzyl-12-(4-hydroxy-benzyl)-3-(1
H-indol-3-
ylmethyl)-2,5,8,11-tetraoxo-1-oxa-4,7,10-triaza-
cyclohexadec-6-yl]-propyl}-guanidine
HzN~NH
O
\
HO
N-{3-[2-Benzyl-24-(4-hydroxy-benzyl)-8-(1 H-indol-3- N-{3-[2-Benzyl-24-(4-
hydroxy-benzyl)-10-methyl-8-
ylmethyl)-10-methyl-3,6,11,25-tetraoxo-1,4,7,10- naphthalen-1-ylmethyl-
3,6,11,25-tetraoxo-1,4,7,10-
tetraaza-cyclopentacos-5-ylj-propyl}-guanidine tetraaza-cyclopentacos-5-yl]-
propyl}-guanidine
H2N~NH HZN' /'NH
HN H'~N
O O
O
I \ ,,~ N NH \ ~ \ O N NH \
\ ,,~ O U
V HN
O N- O HN O
O
\
\
HO
HO
CA 02368431 2001-09-28
WO 00/58361 PCT/US00/07473
_2J_
N-{3-[2-Benzyl-24-(4-hydroxy-benzyl)-10-methyl-8- N-{3-[9-Benzyl-12-(4-hydroxy-
benzyl)-3-naphthalen-
naphthalen-2-ylmethyl-3,6,11,25-tetraoxo-1,4,7,10- 2-ylmethyl-2,5,8,11-
tetraoxo-1-oxa-4,7,10-triaza-
tetraaza-cyclopentacos-5-yl]-propyl}-guanidine cyclopentacos-6-yl]-propyl}-
guanidine
H NH2
N
O NH
O
H ''yN O
HN O HN H HN O
O ~NH
\ l \ / ,'/~O ( ~ i
HO HO
9-Benzyl-6-(3-guanidino-propyl)-12-(4-hydroxy- 10-Benzyl-7-(3-guanidino-
propyl)-13-(4-hydroxy-
benzyl )-5,8,11-trioxo-1-oxa-4,7,10-triaza- benzyl )-6,9,12-trioxo-1-oxa-
5,8,11-triaza-
cyclohexadecane-3-carboxylic acid naphthalen-1- cycloheptadecane-4-carboxylic
acid naphthalen-1-
ylamide ylamide
N~,( NHz N~NHZ
NH ~ / NH
OII O
',,~N O ',,~N O
HN O O HN H HN O
.,,,~ ~ NH ,~ ~ NH
W W
\ l ~ i i \ l ~ i i
HO HO
2-Benzyl-5-(3-guanidino-propyl)-13-(4-hydroxy- 2-Benzyl-5-(3-guanidino-propyl)-
12-(4-hydroxy-
benzyl)-3,6,14-trioxo-1,4,7-triaza-cyclotetradecane- benzyl)-3,6,13-trioxo-
1,4,7-triaza-cyclotridecane-8-
8-carboxylic acid naphthalen-1-ylamide carboxylic acid naphthalen-1-ylamide
CA 02368431 2001-09-28
WO 00/58361 PCT/US00/07473
-26-
N NHz N NHz
I / ~H I / ~H
OII OII
',,.~N O ',,.~N O
H~0 HN /l0 HN 0 HN
HN~~ : NH HN~ ~NH
O '~
\ 0 ~~'~ \ \
I I i i i O
I /
\ \ I
12-Benzoylamino-9-benzyl-6-(3-guanidino-propyl)- 3-Benzoylamino-6-benzyl-9-(3-
guanidino-propyl)-
5,8,11-trioxo-1-oxa-4,7,10-triaza- 4,7,10-trioxo-1-oxa-5,8,11-triaza-
cyclopentadecane-3-carboxylic acid naphthalen-1- cyclopentadecane-12-
carboxylic acid naphthalen-1-
ylamide ylamide
\ N NHZ N~NH2
I
/ NH I / NH
OI~ O
',,I N 0 ''y N O
H~O HN, //O HN O HN, //O
HN~/ :' NH HN~ : NH
O ,' '
0 I \ \ 0 ~ ~ ~ ~'O \ \
/ I i / / 0
I
\ \
12-Benzoylamino-9-benzyl-6-(3-guanidino-propyl)- 12-Benzoylamino-9-benzyl-6-(3-
guanidino-propyl)-
5,8,11-trioxo-1,14-dioxa-4,7,10-triaza- 5,8,11-trioxo-1,15-dioxa-4,7,10-triaza-
cyclooctadecane-3-carboxylic acid naphthalen-1- cyclononadecane-3-carboxylic
acid naphthalen-1-
ylamide ylamide
~( NH2 N~NH2
I \1
/ NH I / NH
0 0II
N O ''-~N O
H 0 O HN H HN~ /Ol
HN HN NH O HN~~~NH
O '
I \ \ \ \
/ CO O . ~ / / I
I -/
\ ~ \I
CA 02368431 2001-09-28
WO 00/58361 PCT/US00/07473
-2 i -
13-Benzoylamino-10-benzyl-7-(3-guanidino-propyl)- 13-Benzoylamino-2-benryl-5-
(3-guanidino-propyl)-
6,9,12-trioxo-1,16-dioxa-5,8,11-triaza- 3,6,14-trioxo-1,4,7-triaza-
cyclotetradecane-8-
cycloeicosane-4-carboxylic acid naphthalen-1- carboxylic acid naphthalen-1-
ylamide
ylamide
N~( NH2 N~NH2
/ NH ~ / NH
OII O
.,,~N O ~,,~N O
H~O HN~ H~O HN~ ,O/
H NH HN~/ ~NH
~; ~ o -..,--~:
0
o / / / ~ / /
13-Benzoylamino-10-benryl-7-(3-guanidino-propyl)- 12-Benzoylamino-2-benryl-5-
(3-guanidino-propyl)-
6,9,12-trioxo-1,15-dioxa-5,8,11-triaza- 3,6,13-trioxo-1,4,7-triaza-
cyclotridecane-8-
cyclononadecane-4-carboxylic acid naphthalen-1- carboxylic acid naphthalen-1-
ylamide
ylamide
N~NHZ
/ NH
O
~,.~N O
HN H HN, //O
~ ~O
HN--% NH
O '~~,~-;
/ / /
11-Benzoylamino-2-benryl-5-(3-guanidino-propyl)-
3,6,12-trioxo-1,4,7-triaza-cyclododecane-8-
carboxylic acid naphthalen-1-ylamide
III. Synthesis of the Compounds
The compounds of the invention can be prepared using a variety of procedures.
The
starting materials used in preparing the compounds of the invention are known,
made by known
CA 02368431 2001-09-28
WO 00/58361 PCT/US00/07473
-28-
methods, or are commercially available. A general reaction for making the
compounds is set
forth below. Representative examples for synthesizing representative compounds
of the present
invention are disclosed in Examples 1-18.
According to one general scheme, the claimed peptides are synthesized using
Fmoc (9-
Fluorenylmethoxycarbonyl as protection group for alpha NH,) chemistry followed
by
deprotection, solution phase cyclization and detailed characterization and
purification. One
general synthesis scheme for the claimed compounds is as follows:
Fmoc-N H-R1-COOH
activation
deprotection coupling
Fmoc-NH-Resin H2N-Resin ----- Fmoc-NH-R1-CO-NH-Resin
deprotection
coupling
Fmoc-N H-R2-OC-H N-R1-CO-N H-Resin H2N-R 1-CO-N H-Resin
activation
Fmoc-NH-R1-COOH
The R-group designations in the above scheme do not correspond to the R groups
used to define
the Formula (I) compounds.
Linear Peptide Synthesis: The linear version of the compounds are synthesized
with a Perkin-
Elmer Applied Biosystem Division (PE-ABD) Model 433 automated synthesizer. All
the
reagents used for peptide synthesis, Fmoc amino acids (except Fmoc-L-Arg-Pbf
is from
~5 AnaSpec) and resins can be purchased from PE-ABD. Standard 0.1 mmole
FastMoc chemistry
with single coupling is used. The general Fmoc chemistry protocol for SPPS
(solid phase peptide
synthesis) includes: 1 ) cleavage of the Fmoc protection groups with
piperidine; 2) activation of
the carboxyl group of amino acids; and 3) coupling of the activated amino
acids to the amino-
terminal of the resin bound peptide chain to form peptide bonds. FastMoc
cycles in which amino
2o acids are activated with 2-(1H-benzotriazol-1-yl)-1,1,3,3-
tetramethyluronium
hexafluorophosphate (HBTU). 1.0 mmole of dry protected amino acid in a
carnidge is dissolved
in a solution of HBTU, N,N-diisopropylethylamine (DIEA), and 1-
hydroxybenzotriazole (HOBt)
in N,N-dimethylformamide (DMF) with additional N-methylpyrrolidone (NMP)
added. The
CA 02368431 2004-04-22
-29-
activated Fmoc amino acid is formed almost instantaneously and the solution is
transferred
directly to the reaction vessel. The step of Fmoc deprotection is monitored
and controlled by
conductivity measurement. The peptide chain is built on a Rink Amide resin
since the C-terminal
amide is needed. The acetyl group is added on the N-terminal side of the
peptide after the full
s length of the peptide chain is made. It is accomplished by reaction of
acetic anhydrite (4.75%
V:V acetic anhydrite, 0.2% HOBt W:V, 2.25°% DIEA in NMP) with the a-
amino group on N-
terminal side of residue. The final synthesis product is washed extensively
with NMP and
dichloromethane (DCM).
Denrotection: The resins containing synthesized peptides are unloaded from the
synthesizer and
io briefly air-dried. Using 1.~-Z.0 ml of the cleavage cocktail (93%
trifluoroacetic acid (TFA),
2.3% ethanodithiol in water) for 1.5-3.0 hours at room temperature, the
peptides are cleaved off
the resin and at the same time, the side chain protection groups [O-t-butyl
(OtBu) for Asp, Glu,
Tyr and Ser, pentamethyldihydrobenzofuran-S-sulfonyl (Pbf) for Arg, t-
butoxycarbonyl (Boc) for
Trp, Otn, Lys] are removed under the deprotection condition. The cleavage
solution is separated
i5 from the resin by filtration. The filtrate is then diluted with 15 ml of
water. Six rounds of ether
extraction are performed to clean the peptide product. The peptide is
lyophilized and stored at -
20°C before cyclization.
Solution Phase cvclization: The peptide is characterized by reversed phase
high performance
liquid chromatography (RP-HPLC) and mass spectrometry (MS) prior to
cyclization process.
2o The lyophilized peptide is dissolved in cold DMF with addition of dibasic
potassium phosphate.
Diphenylphosphoryl azide (DPPA, from Sigma), the cyclization reagent, to
peptide molar ratio is
4:1. The reaction vessel is chilled with dry ice when the DPPA is introduced.
The overall
cyclization reaction is carried out in 4°C for 24 hours with another
DPPA addition in 4 hours
after the reaction is started. Analytical RP-HPLC and electrospray MS is used
to monitor the
25 cyclization reaction. A HP1090 HPLC system with a Vadyc C-8 column with 2.1
mm )D, 15 cm
length, 300 t~ pore size, and 10 p.m particle size. A uv detector is used for
detection of the
cyclization process. With the described cyclization protocol, the reaction is
completed within 24
hours.
Puriffcation and Characterization: The cyclized peptide product is then
lyophilized to remove
3o DMF solvent. The peptide powder along with phosphate salt, DPPA and other
by-products are
re-dissolved in 50% acetic acid solution and injected onto a Vydac 1.0 cm LD.
25 cm length C-8
column with 5 um particle size, and 300 ~ pore size for purification. A
Beckman System Gold
HPLC system with dual wavelength u.v. detector is used. Linear gradient of
acetonitrile is
*trade-mark
CA 02368431 2001-09-28
WO 00/58361 PCT/US00/07473
-30-
programmed and introduced to the column to separate the cyclic peptide product
from other
substances. The elute is collected by a Pharmacia fraction collector, and the
individual
separation fractions are subjected to both analytical HPLC and electrospray MS
for
characterization to ensure the identity and purity.
A variety of additional compounds can be generated using the guidance of the
scheme
above.
It is recognized that it is preferable to use a protecting group for any
reactive
functionality such as a carboxyl, hydroxyl and the like. This is standard
practice. well within the
normal practice of the skilled artisan.
The indicated steps may be varied to increase yield of desired product. The
skilled
artisan will recognize the judicious choice of reactants, solvents, and
temperatures is an important
component in any successful synthesis. Determination of optimal conditions,
etc. is routine.
Thus the skilled artisan can make a variety of compounds using the guidance of
the above
general description.
~5 It is recognized that the skilled artisan in the art of organic chemistry
can readily carry
out standard manipulations of organic compounds without further direction;
that is, it is well
within the scope and practice of the skilled artisan to carry out such
manipulations. These
include, but are not limited to, reduction of carbonyl compounds to their
corresponding alcohols,
oxidations of hydroxyls and the like, acylations, aromatic substitutions, both
electrophilic and
2o nucleophilic, etherifications, esterification and saponification and the
like. Examples of these
manipulations are discussed in standard texts such as March, Advanced Oreanic
Chemistry
(Wiley), Carey and Sundberg, Advanced Organic Chemistry (Vol. 2) and other art
that the skilled
artisan is aware of.
The skilled artisan will also readily appreciate that certain reactions are
best carried out
25 when potentially reactive functionalities on the molecule are masked or
protected, thus avoiding
any undesirable side reactions and/or increasing the yield of the reaction.
Often the skilled
artisan utilizes protecting groups to accomplish such increased yields or to
avoid the undesired
reactions. These reactions are found in the literature and are also well
within the scope of the
skilled artisan. Examples of many of these manipulations can be found for
example in T. Greene,
3o Protecting Groups in Organic Synthesis. Of course, amino acids with
reactive side chains used as
starting materials are preferably blocked to prevent undesired side reactions.
IV. Melanocortin Functional Activity and Selectivity
CA 02368431 2001-09-28
WO 00/58361 PCT/US00/07473
-31-
Functional activity can be evaluated using various methods lrnown in the art.
Examples
of such methods are measurement of second messenger responses, in particular
cAMP, the use of
modified cell systems yielding color reaction upon accumulation of second
messenger elements
such as cAMP, e.g. as described by Chen et al. 1995 (Anal Biochem: 1995, 226,
349-54),
Cytosensor Microphysiometer techniques (see Boyfield et al. 1996), or the
study of physiological
effects caused by the compounds of the invention may be applied by using the
compounds of the
invention alone, or in combination with natural or synthetic MSH-peptides.
The compounds of the present invention will interact preferentially (i.e.,
selectively) to
MC-4 and/or MC-3, relative to the other melanocortin receptors. Selectivity is
particularly
1o important when the compounds are administered to humans or other animals,
to minimize the
number of side effects associated with their administration. MC-3/MC-4
selectivity of a
compound is defined herein as the ratio of the EC;o of the compound for an MC-
1 receptor
("EC;o-MC-1 ") over the EC;o of the compound for the MC-3 (EC;o-MC-3) / MC-4
(EC;o-MC-4)
receptor, the EC;o values being measured as described above. The formulas are
as follows:
MC-3 selectivity= [EC;o-MC-1] / [EC;o-MC-3]
MC-4 selectivity = [EC;o-MC-1] / [EC;o-MC-4]
A compound is defined herein as being "selective for the MC-3 receptor" when
the above
mentioned ratio "MC-3-selectivity" is at least about 10, preferably at least
about 100, and more
preferably at least about 500.
2o A compound is defined herein as being "selective for the MC-4 receptor"
when the above
mentioned ratio "MC-4-selectivity" is at least about 10, preferably at least
about 100, and more
preferably at least about 500.
The following data demonstrate both the agonistic activity and selectivity of
the
representative compounds of the present invention.
Ac,a [DYfRW GI~-NH2
'' 100
~ 80
~ 60
Q 20
0 ~--1R
1x 1x10- 1x10- + 4R
10-8 6 4
3R
CA 02368431 2001-09-28
WO 00/58361 PCT/US00/07473
-32-
Ac-a [EYfRW G(Orn)]-NH2
.~ 100
' 80
40 I
Q 20
a
a 0 ~1R
1x10- 1x10- 1x10-
8 6 4
3RD
Ac-a [DyfRW GK]NH2
~~ 100
60
40 I
Q 20
0 ~~T I
1x10- 1x10- 1x1 ~1R
8 6 4 -!-4R
5
Ac-a [EYfRW GK]-NH2
.~ 100
'~ 80
40
Q 20
a 0 ~ ~ --~ 1 R
1x 1x10- 1x10- +4R
10-8 6 4
3R
Ac-a DYfRW GK-NH2
.~ 100
'~ 80
40
Q 20
0 ~ ~ t 1R
1x10- 1x10- 1x10- ~4RI
8 6 4
2 exp 3R;
CA 02368431 2001-09-28
WO 00/58361 PCT/US00/07473
-33-
Ac-a [DYfRW G(Orn)]-NH2
_~., 100
~ 60
a 40
0 -f-1 R
1x10- 1x10- 1x10- +4R1
8 6 4
3R
Ac-a DYfRW G(Orn)-NH2
_~, 100
~> 80
.~ 60
a 20
0
0
1x10- 1x10- 1x10- -~-1R
8 6 4 ~ --~- 4R
Ac-aEYfRWG(Orn)-NH2
_~, 100
.cvo 60
20
° 0
1x10- 1x10- 1x10- +1R
8 6 4
f4R
5
Ac-a DYfRW K-N H2
_~, 100
'> 80 _
.~ 60
20
0
1x10- 1x10- 1x10- t 1R
8 6 4 --~- 4R
CA 02368431 2001-09-28
WO 00/58361 PCT/US00/07473
-34-
Ac-(DYfRW GK]-NH2
_~. 100
~' 80
.~ 60
20
a 0 ~.~ ~-,I ~ 1 R
1x10- 1x10- 1x10- ~4R
8 6 4
3R
The following data demonstrate the lack of selectivity over the MC-1 receptor
when the
Tyr residue of the present compounds is replaced by His in the tetrapeptide
backbone.
Ac-SYSNIeEHfRWGKPV-NH2 (MT-I)
_a, 100
60
20
0 -~ i
° --~ 1 R
1x10- 1x10- 1x10-
11 9 ~ -f-4R
3R
5
Ac-NIeDHfRWK-NH2 (MT-II)
~ 100
.R 60
20
a
--~-1 R
1x10- 1x10- 1x10- +4R
11 9 7
3R
V. Methods of Use and Compositions:
Based on their ability to agonize or antagonize the MC-4 and/or MC-3 receptor,
the
1o present inventions also relates to the use of the ligands of the present
invention in methods for
treating obesity as well as other body weight disorders, including anorexia
and cachexia. The
invention further relates to the treatment of disorders relating .to behavior,
memory (including
learning), cardiovascular function, inflammation, sepsis and septic,
cardiogenic and hypovolemic
CA 02368431 2001-09-28
WO 00/58361 PCT/US00/07473
-3 5-
shock, sexual dysfunction, penile erection, muscle atrophy, nerve growth and
repair, intrauterine
fetal growth, and the like.
The terms treating and treatment are used herein to mean that, at a minimum,
administration of a compound of the present invention mitigates a disease
state by acting via the
MC-3 or MC-4 receptor. Thus, the terms include: preventing a disease state
from occurring in a
mammal, particularly when the mammal is predisposed to acquiring the disease,
but has not yet
been diagnosed with the disease; inhibiting progression of the disease state;
and/or alleviating or
reversing the disease state.
The invention compounds can therefore be formulated into pharmaceutical
compositions
for use in treatment or prophylaxis of these conditions. Standard
pharmaceutical formulation
techniques are used, such as those disclosed in Remington 's Pharrnaceutical
Sciences, Mack
Publishing Company. Easton, Pa., latest edition and Peptide and Protein Drug
Delivery, Marcel
Dekker, NY, 1991.
The compositions of the invention comprise:
a. a safe and effective amount of a compound of Formula (I); and
b. a pharmaceutically-acceptable excipient.
A "safe and effective amount" of a Formula (I) compound is an amount that is
effective to interact with the MC-4 and/or MC-3 receptor, in an animal,
preferably a
mammal, more preferably a human subject, without undue adverse side effects
(such as
2o toxicity, irritation, or allergic response), commensurate with a reasonable
benefit/risk ratio
when used in the manner of this invention. The specific "safe and effective
amount" will,
obviously, vary with such factors as the particular condition being treated,
the physical
condition of the patient, the duration of treatment, the nature of concurrent
therapy (if any),
the specific dosage form to be used, the excipient employed, the solubility of
the Formula (I)
compound therein, and the dosage regimen desired for the composition.
In addition to the subject compound, the compositions of the subject invention
contain
one or more pharmaceutically-acceptable excipients. The term "pharmaceutically-
acceptable
excipient", as used herein, means one or more compatible solid or liquid
ingredients which are
suitable for administration to an animal, preferably a mammal, more preferably
a human. The
3o term "compatible", as used herein, means that the components of the
composition are capable of
being commingled with the subject compound, and with each other, in a manner
such that there is
no interaction which would substantially reduce the pharmaceutical efficacy of
the composition
under ordinary use situations. Pharmaceutically-acceptable excipients must, of
course, be of
CA 02368431 2001-09-28
WO 00/58361 PCT/US00/07473
-36-
sufficiently high purity and sufficiently low toxicity to render them suitable
for administration to
the animal, preferably a mammal, more preferably a human being treated.
Some examples of substances which can serve as pharmaceutically-acceptable
excipients
or components thereof are sugars, such as lactose, glucose and sucrose;
starches, such as corn
starch and potato starch; cellulose and its derivatives, such as sodium
carboxymethyl cellulose,
ethyl cellulose, and methyl cellulose; powdered tragacanth; malt; gelatin;
talc; solid lubricants,
such as stearic acid and magnesium stearate; vegetable oils, such as peanut
oil, cottonseed oil,
sesame oil, olive oil, corn oil and oil of theobroma; polyols such as
propylene glycol, glycerin,
sorbitol, mannitol, and polyethylene glycol; agar; alginic acid; wetting
agents and lubricants, such
~o as sodium lauryl sulfate; coloring agents; flavoring agents; tableting
agents, stabilizers;
antioxidants; preservatives; pyrogen-free water; isotonic saline; and buffers,
such as phosphate,
citrate and acetate.
The choice of pharmaceutically-acceptable excipients to be used in conjunction
with the
subject compound is basically determined by the way the compound is to be
administered. If the
i5 subject compound is to be injected, the preferred pharmaceutically-
acceptable excipient is sterile
water, physiological saline, or mixtures thereof, the pH of which has
preferably been adjusted to
about 4-10 with a pharmaceutical buffer; a compatible suspending agent may
also be desirable.
In particular, pharmaceutically-acceptable excipients for systemic
administration
include sugars, starches, cellulose and its derivatives, malt, gelatin, talc,
calcium sulfate,
20 lactose, vegetable oils, synthetic oils, polyols, alginic acid, phosphate,
acetate and citrate
buffer solutions, emulsifiers, isotonic saline, and pyrogen-free water.
Preferred excipients
for parenteral administration include propylene glycol, ethyl oleate,
pyrrolidone, ethanol,
and sesame oil. Preferably, the pharmaceutically-acceptable excipient, in
compositions for
parenteral administration, comprises at least about 90% by weight of the total
composition.
2s The compositions of this invention are preferably provided in unit dosage
form. As
used herein, a "unit dosage form" is a composition of this invention
containing an amount of
a Formula (I) compound that is suitable for administration to an animal,
preferably a
mammal, more preferably a human subject, in a single dose, according to good
medical prac-
tice. These compositions preferably contain from about 1 mg to about 750 mg,
more
3o preferably from about 3 mg to about 500 mg, still more preferably from
about ~ mg to about
300 mg, of a Formula (I) compound.
The compositions of this invention may be in any of a variety of forms,
suitable (for
example) for oral, rectal, topical, nasal, ocular, transdermal, pulmonary or
parenteral
administration. Depending upon the particular route of administration desired,
a variety of
CA 02368431 2004-04-22
-37-
pharmaceutically-acceptable excipients well-known in the art may be used.
These include
solid or liquid fillers, diluents, hydrotropes, surface-active agents, and
encapsulating
substances. Optional pharmaceutically-active materials may be included, which
do not
substantially interfere with the inhibitory activity of the Formula (I)
compound. The amount
of excipient employed in conjunction with the Formula (I) compound is
sufficient to provide
a practical quantity of material for administration per unit dose of the
compound.
Techniques and compositions for making dosage forms useful in the methods of
this
invention are described in the following references
Modern Pharmaceutics, Chapters 9 and 10 (Banker & Rhodes, editors. 1979);
Lieberman et
to al., Pharmaceutical Dosage Forms: Tablets (1981); and Ansel, Introduction
to
Pharmaceurical Dosage Forms 2d Edition ( 1976).
Various oral dosage forms can be used, including such solid forms as tablets.
capsules, granules and bulk powders. These oral forms comprise a safe and
effective
amount, usually at least about 5%, and preferably from about 25% to about 50%,
of the
~s Formula (I) compound. Tablets can be compressed, tablet triturates, enteric-
coated, sugar-
coated, film-coated, or multiple-compressed, containing suitable binders,
lubricants,
diluents, disintegrating agents, coloring agents, flavoring agents, flow-
inducing agents, and
melting agents. Liquid oral dosage forms include aqueous solutions, emulsions,
suspensions, solutions andlor suspensions reconstituted from non-effervescent
granules, and
2o effervescent preparations reconstituted from effervescent granules,
containing suitable
solvents, preservatives, emulsifying agents, suspending agents, diluents,
sweeteners, melting
agents, coloring agents and flavoring agents.
The pharmaceutically-acceptable excipient suitable for the preparation of unit
dosage
forms for peroral administration are well-known in the art. Tablets typically
comprise
zs conventional pharmaceutically-compatible adjuvants as inert diluents, such
as calcium carbonate,
sodium carbonate, mannitol, lactose and cellulose; binders such as starch,
gelatin.
polyvinylpyrrolidone and sucrose; disintegrants such as starch, alginic acid
and croscarmelose;
lubricants such as magnesium stearate, stearic acid and talc. Glidants such as
silicon dioxide can
be used to improve flow characteristics of the powder mixture. Coloring
agents, such as the
so FD&C dyes, can be added for appearance. Sweeteners and flavoring agents,
such as aspartame.
saccharin, menthol, peppermint, and fruit flavors, are useful adjuvants for
chewable tablets.
Capsules typically comprise one or more solid diluents disclosed above. The
selection of
excipient components depends on secondary considerations like taste, cost, and
shelf stability,
CA 02368431 2001-09-28
WO 00/58361 PCT/US00/07473
-3 8-
which are not critical for the purposes of the subject invention, and can be
readily made by a
person skilled in the art.
Peroral compositions also include liquid solutions, emulsions, suspensions,
and the like.
The pharmaceutically-acceptable excipients suitable for preparation of such
compositions are
s well known in the art. Typical components of excipients for syrups, elixirs,
emulsions and
suspensions include ethanol, glycerol, propylene glycol, polyethylene glycol,
liquid sucrose,
sorbitol and water. For a suspension, typical suspending agents include methyl
cellulose, sodium
carboxymethyl cellulose, Avicel~ RC-591, tragacanth and sodium alginate;
typical wetting
agents include lecithin and polysorbate 80; and typical preservatives include
methyl paraben,
~o propyl paraben and sodium benzoate. Peroral liquid compositions may also
contain one or more
components such as sweeteners, flavoring agents and colorants disclosed above.
Such compositions may also be coated by conventional methods, typically with
pH or
time-dependent coatings, such that the subject compound is released in the
gastrointestinal tract
in the vicinity of the desired topical application, or at various times to
extend the desired action.
~s Such dosage forms typically include, but are not limited to, one or more of
cellulose acetate
phthalate, polyvinylacetate phthalate, hydroxypropyl methyl cellulose
phthalate, ethyl cellulose,
Eudragit~ coatings, waxes and shellac.
Because the compounds of the present invention are peptidic in nature, a
preferred mode
of administration is parenteral (more preferably intravenous injection) or
nasal administration, in
2o the form of a unit dose form. Preferred unit dose forms include suspensions
and solutions,
comprising a safe and effective amount of a Formula I compound. When
administered
parenterally, the unit dose form will most preferably comprise from about 3 to
about 300 mg of
the Formula (I) compound.
Compositions of the subject invention may optionally include other drug
actives.
25 Other compositions useful for attaining systemic delivery of the subject
compounds
include sublingual, buccal and nasal dosage forms. Such compositions typically
comprise one or
more of soluble filler substances such as sucrose, sorbitol and mannitol; and
binders such as
acacia, microcrystalline cellulose, carboxymethyl cellulose and hydroxypropyl
methyl cellulose.
Glidants, lubricants, sweeteners, colorants, antioxidants and flavoring agents
disclosed above
3o may also be included.
VI. Methods of Administration
As indicated, compositions of this invention can be administered topically or
systemically. Systemic application includes any method of introducing a
Formula (I)
CA 02368431 2001-09-28
WO 00/58361 PCT/US00/07473
-39-
compound into the tissues of the body, e.g., intra-articular, intrathecal,
epidural,
intramuscular, transdermal, intravenous, intraperitoneal, subcutaneous,
sublingual, rectal,
nasal, pulmonary, and oral administration. The Formula (I) compounds of the
present
invention are preferably administered systemically, more preferably
parenterally and most
preferably via intravenous injection.
The specific dosage of compound to be administered, as well as the duration of
treatment, and whether the treatment is topical or systemic are
interdependent. The dosage
and treatment regimen will also depend upon such factors as the specific
Formula (I)
compound used, the treatment indication, the ability of the Formula (I)
compound to reach
minimum inhibitory concentrations at the site of the metalloprotease to be
inhibited, the
personal attributes of the subject (such as weight), compliance with the
treatment regimen,
and the presence and severity of any side effects of the treatment.
Typically, for a human adult (weighing approximately 70 kilograms), from about
0.003 mg to about 300 mg, more preferably from about 0.03 mg to about 100 mg,
of Formula
i5 (I) compound are administered per day for systemic administration. It is
understood that
these dosage ranges are by way of example only, and that daily administration
can be
adjusted depending on the factors listed above.
As is known and practiced in the art, all formulations for parenteral
administration
must be sterile. For mammals, especially humans, (assuming an approximate body
weight of
20 70 kilograms) individual doses of from about 0.001 mg to about 100 mg are
preferred.
A preferred method of systemic administration is intravenous delivery.
Individual
doses of from about 0.01 mg to about 100 mg, preferably from about 0.1 mg to
about 100 mg are
preferred when using this mode of delivery.
In all of the foregoing, of course, the compounds of the invention can be
administered
25 alone or as mixtures, and the compositions may further include additional
drugs or excipients as
appropriate for the indication.
The compound of the invention can be delivered to the preferred site in the
body by using
a suitable drug delivery system. Drug delivery systems are well known in the
art. For example, a
drug delivery technique useful for the compounds of the present invention is
the conjugation of
3o the compound to an active molecule capable of being transported through a
biological barrier (see
e.g. Zlokovic, B.V., Pharmaceutical Research, Vol. 12, pp. 1395-1406 (1995)).
A specific
example constitutes the coupling of the compound of the invention to fragments
of insulin to
achieve transport across the blood brain barrier (Fukuta, M., et al.
Pharmaceutical Res., Vol. 11,
pp. 1681-1688 (1994)). For general reviews of technologies for drug delivery
suitable for the
CA 02368431 2001-09-28
WO 00/58361 PCT/US00/07473
-40-
compounds of the invention see Zlokovic, B.V., Pharmaceutical Res., Vol. 12,
pp. 139-1406
(1995) and Pardridge, WM, Pharmacol. Toxicol., Vol. 71, pp. 3-10 (1992).
VII. Representative Examples
In the following examples the invention will be described in greater detail by
reference to
a number of preferred embodiments which are only given for purposes of
illustration and should
not be considered to limit the invention in any way.
The following abbreviations are used in the Examples:
OtBu: tert-butoxy [-O-C(CH3)3] tBu: tent-butyl [-C(CH3)s]
Pbf: penta fluorophenyl Boc: tert-butyloxycarbonyl
~o TFA: trifluoroacetic acid DMF: N,N-Dimethylformamide
Fmoc: 9-Fluorenylmethoxycarbonyl DPPA: Diphenylphosphoryl azide
HOAt: 1-hydroxy-7-azabenzotriazole HOBt: N-hydroxybenzotriazole
EDCI: 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride
HATU: O-(7-azabenzotiazol-1-yl)-N,N,N',N'-tetramethyluronium
hexafluorophosphate
Pbf: 2,2,4,6,7-pentamethyl-dihydrobenzofuran-5-sulfonyl-
Pmc: 2,2,5,7,8-pentamethyl-chroman-6-sulfonyl
Trt: trityl
A. Synthetic Examples
2o Example 1
Synthesis of Ac-a(DYfRWGK]-NHZ [M+H]': 1065.2
Based on the 0.55 mmole/g substitution rate for the Rink Amide resin, 0.182g
of the resin
is weighted out for 0.1 mmole scale synthesis. The performance of the PE-ABD
433 peptide
synthesizer is checked before the run with various flow tests to ensure the
accurate reagent
2s delivery. Fmoc amino acids: Asp-OtBu, Tyr-OtBu, Arg-Pbf, Trp-Boc, Lys-Boc,
and Gly are
purchased commercially in 1 mmole cartridges. Fmoc-ala (311 mg, 1 mmole) and
Fmoc-phe (387
mg, 1 mmole) is measured and added in the synthesis cartridges, respectively.
The freshly made
acetic anhydride solution is loaded on the instrument at 4 bottle position.
Other synthesis
reagents and solvents are purchased commercially and loaded on the instrument
according to the
3o instrument's instruction. A chemistry nroQram called NAc.-(l 1 mmnlc
MnnPraPlr ;c "~P(1 F'."-
synthesizing this peptide. The Fmoc deprotection is monitored and controlled
by conductivity
measurement with set criteria of 5% or less conductivity comparing to previous
deprotection
CA 02368431 2001-09-28
WO 00/58361 PCT/US00/07473
-41-
cycle. The overall synthesis yield for this series of peptides (linear) is
better than 90%,
determined by analytical HPLC.
The resin is air-dried and transferred into a glass vial and a freshly
prepared cleavage
reagent (2 ml) is added. The deprotection reaction is carried out for 2 hours
at room temperature
s with constant stirring. The supernatant is then separated from the resin by
filtration. The resin is
sequentially washed with TFA (2x2 ml) and double distilled water. The combined
wash
solutions are extracted with ether (3x20 ml). The upper (ether) layer is
discarded after each
extraction. The peptide solution is freeze-dried overnight. The identity of
the linear peptide is
determined by both MS and HPLC. Expected peptide molecular weight is detected.
Analytical
HPLC profile indicates that the peptide purity is over 90%.
The peptide powder is dissolved in DMF (15 ml) and KZHPO,~ (60 mg) is added.
The
reaction mixture is cooled in dry-ice and treated with DPPA (70 ~1), agitated
for 20 min., and
warmed up to 4°C. An additional, equal portion of DPPA is added after 2
hours. The total
cyclization reaction proceeds for 24 hours. DMF is then removed by vacuum.
Cyclic peptide is
is re-dissolved in 50% acetic acid and purified by a C8 reverse phase HPLC
using a linear gradient
of 0-70% solvent B with solvent A in 70 min at a flow rate 3 ml/min. The
composition of
solvents A and B are as follows: A: 0.1% TFA, 2% acetonitrile in water; B:
0.1% TFA in 95%
acetonitrile. The fractions are collected at every 0.5 min. Aliquots of each
fraction are analyzed
by both MS and analytical RP-HPLC. The fractions that contain a single u.v.
220 nm absorbance
2o peak with expected mass unit for the cyclized peptide ([M+H]~: 1066.2) are
combined and
lyophilized. The final purity (95%) of the peptide is determined by an
analytical RP-HPLC of the
combined fractions.
The cyclic peptides listed below are readily synthesized according to the same
protocol
as Example l, but with certain modifications as noted.
2s Example 2
Synthesis of Ac-[DYfRWGK]-NHZ, [M+H]+: 995.1
Prepared according to Example 1, with the exception that Fmoc-D-Ala is not
used.
Example 3
Synthesis of Ac-a[DyfRWGK]-NHZ, [M+H]+: 1066.2
3o Prepared according to Example l, with the exception that Fmoc-D-Tyr-OtBu is
used
instead of Fmoc-L-Tyr-OtBu.
Example 4
CA 02368431 2001-09-28
WO 00/58361 PCT/US00/07473
-42-
Synthesis of Ac-a[EYfRWG(Orn)]-NHZ [M+H]': 1052.1
Prepared according to Example 1, except Fmoc-L-Glu-OtBu and Fmoc-L-Orn-Boc are
used instead of Fmoc-Asp-OtBu and Fmoc-Lys-Boc, respectively.
Example 5
Synthesis of Ac-a[EYfRWGK]-NHZ [M+H]+; 1080.2
Prepared according to Example 1, except Fmoc-L-Glu-OtBu is used instead of
Fmoc-L-
Asp-OtBu.
Example 6
Synthesis of Ac-a[DYyRWGK)-NHZ[M+H]-: 1082.2
Prepared according to Example 1, except Fmoc-D-Tyr-OtBu is used instead of
Fmoc-D-
Phe.
Example 7
Synthesis of Ac-a[DY(D-Phe(pCl))RWK]-NHZ [M+H]+; 1061.7
Prepared according to Example 1, except Fmoc-D-Phe(pCl) is used instead of
Fmoc-D-
i 5 Phe.
Example 8
Synthesis of Ac-SYSa[DYfRWGK]-NHZ [M+H]': 1403.5
Prepared according to Example 1, except three additional amino acid (Fmoc-L-
Ser-OtBu,
Fmoc-L-Tyr-OtBu and Fmoc-L-Ser-OtBu) are used at the N-terminus of the peptide
of Example
20 1.
Example 9
Synthesis of Ac-[EYfRWGK]-NHZ, [M+H]+: 1009.2
Prepared according to Example 1, except Fmoc-L-Glu-OtBu is used instead of
Fmoc-L-
Asp-OtBu and Fmoc-D-Ala is not used.
25 Example 10
Synthesis of Ac-a(DY(D-Nal)RWK]-NHZ, [M+H]+: 1060.2
Prepared according to Example 1, except Fmoc-D-Nal is used instead of Fmoc-D-
Phe
and Fmoc-L-Gly is not used.
Example 11
so Synthesis of Ac-a(DYfRWK]-NHZ, [M+H]+: 1009.1
Prepared according to Example 1, except Fmoc-L-Gly is not used.
CA 02368431 2001-09-28
WO 00/58361 PCT/US00/07473
-43-
Example 12
Synthesis of Ac-GGGa[DYfRWGK]-NHz, [M+H]+; 1237.3
Prepared according to Example 1, except three additional amino acid (Fmoc-L-
Gly,
Fmoc-L-Gly and Fmoc-L-Gly) are used at the N-terminus of the peptide of
Example 1.
Example 13
Synthesis of Ac-[DY(D-Nal)RWGK]-NHz, [M+H]+; 1045.1
Prepared according to Example 1, except Fmoc-D-Nal is used instead of Fmoc-D-
Phe
and Fmoc-D-Ala is not used.
io The linear peptides described in Examples 14-17 below are synthesized
according to the
same protocol as Example l, except without the cyclization steps.
Example 14
Synthesis of Ac-aDYfRWK-NHZ, [M+H]+: 1010.0
Prepared according to Example 1, except Fmoc-L-Gly is not used.
Example 15
Synthesis of Ac-aEYfRWGK-NH2, [M+H]+: 1098.2
Prepared according to Example 1, except Fmoc-L-Glu-OtBu is used instead of
Fmoc-L-
Asp-OtBu.
Example 16
2o Synthesis of Ac-aDYfRWGK-NHZ, [M+H]+: 1084.2
Compound is the same as example 1 without cyclization.
Example 17
Synthesis of Ac-aDYfRWG(Orn)-NH2, [M+H]+: 1070.1
Prepared according to Example 1, except Fmoc-L-Orn-Boc is used instead of Fmoc-
Lys-
BOC.
Example 18
Synthesis of [(5-Ava)YfRW(5-Ava)], [M+H] ':
YfRW WNH
so NH(CH2)5CONH(CH2)5
CA 02368431 2001-09-28
WO 00/58361 PCT/US00/07473
-44-
Prepared according to Example 1. The following Fmoc - protected amino acids
were
used: Y, f, R, W, 5-Ave; Fmoc - protected amino acids D, a, G, K were not
used.
Example 19
Synthesis of [(5-Ava)YfRW], [M+H]T:
YfRW ~NH
~o CH2)s
Prepared according to Example 1. The following Fmoc - protected amino acids
were
used: Y, f, R, W, 5-Ave; Fmoc - protected amino acids D, a, G, K were not
used.
Example 20
Synthesis of [(6-Ahx)YfRW], [M+H]~:
YfRW ~NH
CH2
Prepared according to Example 1. The following Fmoc - protected amino acids
were
used: Y, f, R, W, 6-Ahx. Fmoc - protected amino acids D, a, G, K were not
used.
Example 21
2s Synthesis of Ac-[DYfRWG(Orn)]-NHZ [M+H]T: 980.2
Prepared according to Example 1, except Fmoc-L-Orn-Boc is used instead of Fmoc-
Lys-
Boc and Fmoc-D-Ala is not used.
Example 22
Synthesis of Ac-(DYf(homoArg)WGK]-NHZ [M+H]': 1008.6
CA 02368431 2001-09-28
WO 00/58361 PCT/US00/07473
-45-
Prepared according to Example l, except Fmoc- homoArg-(Pmc) is used instead of
Fmoc-L-Arg-Pbf and Fmoc-D-Ala is not used.
Example 23
Synthesis of [GGYfRWGGG] [M+H]': 938.5
Prepared according to Example 1, except Fmoc-D-Ala, Fmoc-Asp-OtBu, Fmoc-Lys-
Boc
are not used, and two additional amino acids (Fmoc-Gly, Fmoc-Gly) are used at
the N-terminus
and C-terminus of the peptide, respectively.
Example 24
Synthesis of Ac-[DYfHWGK]-NHZ [M+H]+: 975.5
Prepared according to Example l, except Fmoc-L-His-Trt is used instead of Fmoc-
L-Arg-
Pbf and Fmoc-D-Ala is not used
Example 25
Synthesis of Ac-[DYfR(D-Nal)GK]-NHZ[M+H]': 1043.4
Prepared according to Example 1, except Fmoc-D-Nal is used instead of Fmoc-L-
Trp and
i s Fmoc-D-Ala is not used.
Example 26
Synthesis of Ac-Nle[DYfRWGK]-NHZ [M+H]T: 1191.7
Prepared according to Example 1, except Fmoc-L-Nle is used instead of Fmoc-D-
Ala.
2o Example 27
Synthesis of Ac-a[DHfRWGK]-NHZ, [M+H]+; 1039.4
Prepared according to Example 1, except Fmoc-L-His-Trt is used instead of Fmoc-
L-Tyr-
OtBu.
Example 28
25 Synthesis of Ac-a[D(homoTyr)fRWGK]-NHZ, [M+H]+; 1079.2
Prepared according to Example l, except Fmoc-L-homoTyr-OtBu is used instead of
Fmoc-L-Tyr-OtBu.
Example 29
Synthesis of Ac-a(DFfRWGK]-NHZ, [M+H]+; 1049.5
3o Prepared according to Example 1, except Fmoc-L-Phe is used instead of Fmoc-
L-Tyr-
OtBu.
CA 02368431 2001-09-28
WO 00/58361 PCT/US00/07473
-46-
Example 30
Synthesis of Ac-Nle(DHyRWK]-NHZ, [M+H]+: 1040.6
Prepared according to Example l, except Fmoc-Nle is used instead of Fmoc-D-
Ala,
Fmoc-His-Trt is used instead of Fmoc-L-Tyr-OtBu, and Fmoc-L-Gly is not used.
Example 31
Synthesis of Ac-a[DYfRWG(Orn)J-NHZ, [M+H~+; 1051.5
Prepared according to Example 1, except Fmoc-L-Orn-Boc is used instead of Fmoc-
L-
Lys-Boc.
Many compounds of this invention, including those described in the specific
examples
above, can be made on a solid support with the aid of an automated peptide
synthesizer such as
PE-ABD 433. However, other structures may require methodology that cannot be
easily
accommodated by the synthesizer. For the latter products, the solution phase
synthesis is more
~ 5 appropriate. The synthetic example described below illustrates application
of both solid
supported automated synthesis and preparation in solution.
Example 32
Synthesis of N-{3-[9-Benzyl-12-(4-hydroxy-benzyl)-3-(1H-indol-3-ylmethyl)-
2,5,8,11,14-
2o pentaoxo-1,4,7,10,13pentaaza-cyclopentacos-6-yl]-propyl}-guanidine
(a). Step 1:
02N ~ NH2 H \ ~ / O N
2~
NH H N N ~ NH2
(2)
O O OMe
°~ O
\ I ,~N HOBt, EDCI, NMM ~ , O N
O \
HN~HO OH ~ \ I ~~.~N
/' ~HN HN
O (1 ) DMF ~H
O
O O
OMe
(3)
A mixture of dipeptide (1) (0.233 g, 0.5 mmol), L-tryptophan methyl ester (2)
(0.127 g,
0.5 mmol), HOBt (O.I35 g, 1.0 mmol), and N-methylmorpholine (0.17 ml, 1.5
mmol) in
CA 02368431 2001-09-28
WO 00/58361 PCT/US00/07473
-47-
dimethylformamide ( 1 ml) is treated with EDCI (0.115 g, 0.6 mmol) and stirred
overnight at
room temperature. A treatment of the reaction mixture with water (30 ml)
results in the
precipitation of a crude product which is separated by decanting the liquid.
The crude product is
mixed again with water, filtered, washed with water and purified on a silica
column with a 20:1
solution of dichloromethane and methanol as the eluant. The yield of
tripeptide (3) is 0.26 g.
(b). Step 2:
OzN OzN
N\'NHZ N\'NHZ
~N H ~N H
O H TFA p H
~( O N ~ p-Ts H ~ , O N
~~~~ ~N H N ~ I ~ ~ I + ~~ ~N
HN~H ~ H3N H HN U
O
O O p-Ts0- O
(4)
OMe OMe
(3)
Tripeptide (3) (0.26 g) is treated with 25% (v/v) solution of trifluoroacetic
acid in
io dichloromethane (2 ml) for 2 hr. at room temperature. The reaction mixture
is diluted with 1,2-
dichloroethane ( 10 ml), treated with p-toluenesulfonic acid hydrate (95 mg,
0.5 mmol) and
evaporated under reduced pressure to give 0.34 g of the product (4).
(c). Step 3:
0 OH
OzN
i
NH N~NHz
OyN ~ '(NH
N~NHz \ ~ 0 (CHz)~~lHBoc
'(NH O N
O (5) ~ O
0 H i ~ O /'N~H HN
~ 0 N \ I HOBt, EDCI, NMM
~,','~N ~ ~ \ O
\ +H3N H HN ~ ~NH OMe
p-Ts0- 0 (4) DMF ~ 0
OMe \ ~ ~(CHz)~lNHBoc
O (6)
i5 The procedure analogous to that applied for the preparation of (3) is
applied for the
reaction of (4) (0.344 g 0.46 mmol) with (5) (0.262 g, 0.46 mmol). The crude
product is purified
CA 02368431 2001-09-28
WO 00/58361 PCT/US00/07473
-48-
on a silica column using a 9:1 solution of ethyl acetate and methanol as the
eluant to give 0.21 g
of (6).
(d). Step 4:
OzN
NYNHZ O2N pzN
NYNHz NYNHz
NH
NH NH
O H
O N ~ 0 H
N ~ I i '' O N
NH H HN
O NaOH TFA ~ I /' ~N
0 NH H HN U
0
NH OMe
NH OH
O~(OHZO~H8oc O
i
I O~(CFizW ~'~FIz
O (6)
0 (e)
,,
d
Tetrapeptide (6) (0.233 g, 0.21 mmol) in tetrahydrofuran (5 ml) is stirred
with 1 N NaOH
(0.5 ml) at room temperature for 3 hr. The reaction mixture is acidified with
1 N HCl to pH 2
and partitioned between water and ethyl acetate. The aqueous layer is
repeatedly extracted with
fresh ethyl acetate, combined organic extracts are washed with 0.1 N HCI,
dried with anhydrous
magnesium sulfate and filtered. Concentration under reduced pressure produced
0.226 g of the
io product (7).
The crude residue of (7) is stirred with 25% (v/v) solution of trifluoroacetic
acid in
dichloromethane (2 ml) for 2 hr. at room temperature. The reaction mixture is
diluted with 1,2-
dichloroethane (10 ml) and evaporated under reduced pressure. The residue is
purified on HPLC
C4 reverse phase column using a linear gradient of 0.1% aqueous
trifluoroacetic acid and
~5 acetonitrile to give 0.155 g of (8).
(e). Step 5:
CA 02368431 2001-09-28
WO 00/58361 PCT/US00/07473
-49-
OZtJ
N~NHz
NOz
NH
-NHz
O H HATU ~ ~NH
O N HOAt HN w
//,N~H HN ~ I \ DIPEA
O
DMF t
~NH O OH
n%\ ~H
($)
\ I ~NHz
Tetrapeptide (8) ( 100 mg, 0.1 mmol), HATU (46 mg, 0.12 mmol) and HOAt ( 14
mg, 0.1
mmol) are placed in a dry flask under argon atmosphere. Dimethylformamide (
100 ml) is added
and the flask is cooled to about 0°C in an ice bath. DIPEA (0.053 ml,
0.3 mmol) is added and the
reaction mixture is stirred in the ice bath for 3 hr. After solvent
evaporation under reduced
pressure, the crude material is separated on a silica column using a 15:1
solution of
dichloromethane and methanol as the eluant to give 90 mg of the macrocyclic
product (9).
(f). Step 6:
NOz
_ N~NH HN
z ~--NHz
NH NH
0 HN w \ ~ = IOI N HN w
HN ~~ I / Pd(OH)z O HN~'- 0
I \ NH IH1 \ NH
HN 0 ~ HO ~ / HN
0 (9) NH O (gyp) 0
NH
The macrocylic compound (9) (0.09 g) is hydrogenated in ethanol (20 ml) with
20%
Pd(OH)~ at 45 psi. for 48 hr. The catalyst is removed by filtration through
Celite. After
evaporation of the filtrate, the crude product is purified on HPLC using a C4
reverse phase
column and a linear gradient of 0.1% aqueous trifluoroacetic acid and
acetonitrile to give final
product N-{3-[9-Benzyl-12-(4-hydroxy-benzyl)-3-(1H-indol-3-ylmethyl)-
2,5,8,11,14-pentaoxo
1,4,7,10,13pentaaza-cyclopentacos-6-yl]-propyl}-guanidine (10).
Example 33
CA 02368431 2001-09-28
WO 00/58361 PCT/US00/07473
-50-
Synthesis of 12-Benzoylamino-9-benzyl-6-(3-guanidino-propyl)-5,8,11-trioxo-
1,14-dioxa
4,7,10-triaza-cyclooctadecane-3-carboxylic acid naphthalen-1-ylamide
(a) Step 1:
Boc O Boc O
H-N~ 1 ) NaH/DMF H-N O
y OH 2) allyl bromide ~O ~H2N~
OH 3) dicyclohexyl amine
1 / 2
A solution of Boc-Ser-OH (1, 2.05 g, 10 mmol) in DMF (30 ml) is added to a
stirred
suspension of sodium hydride (60 wt% in mineral oil, 880 mg, 22 mmol) in DMF
(30 ml) at 0°C.
After the evolution of hydrogen gas ceased, allyl bromide (0.95 ml, 11 mmol)
is added to the
milk-colored solution. The resulting mixture is stirred at room temperature
for 5 h to give a clear
solution. The solvent is removed in vacuo, water (50 ml) is added, and the
aqueous solution is
~o extracted with ether (2 x 20 ml.) The aqueous solution is then acidified to
pH 3.0 with 1.0 N HCl
and extracted further with ethyl acetate (5 x 20 ml.) The combined ethyl
acetate extracts are
washed with water (20 ml), brine (20 ml), dried over anhydrous magnesium
sulfate, and
concentrated to give crude Boc-Ser(allyl)-OH ( 1.96 g) as a light yellow oil.
A solution of this oil
in ether (30 ml) is treated with dicyclohexylamine (1.594 ml, 8.0 mmol), the
solvent removed,
i 5 and the residue triturated with 1:9 ethyl acetate/hexane to yield 2 (2.26
g) as a colorless solid. An
additional amount of 2 (0.58 g) is isolated from the trituration solution also
as a colorless solid.
(b) Step 2:
H NHz
N N-NOZ
O N NH2
H ~H O ( ~ O ~-NOz
Boc O s + OMe
_ p-TSA- 3 ~~-.~ O
H N~O ~H ON~ HOBt, EDCI, NMM B°c O~1~.NH H
,N~ OMe
O DMF H
~O
2 ~ 4
A mixture of dipeptide (3, 1.382 g, 2.5 mmol), Boc-Ser(allyl)-OH ~ DCA salt
(2, 1.067 g,
20 2.5 mmol), HOBt (0.338 g, 2.5 mmol), and N methylmorpholine (0.6 ml, 5.5
mmol) in DMF (40
ml) is treated with EDCI (0.527 g, 2.75 mmol) and stirred at room temperature
16 h. The
reaction mixture is diluted with ethyl acetate (300 ml) and washed
successively with water (50
CA 02368431 2001-09-28
WO 00/58361 PCT/US00/07473
-51-
ml), 5°io aqueous citric acid (2 x 25 ml), 5% aqueous sodium
bicarbonate (2 x 25 ml), and brine
(25 ml.) The ethyl acetate solution is dried over a combination of anhydrous
sodium and
magnesium sulfates and is concentrated to yield 4 (1.519 g) as a colorless
syrup.
(c ) Step 3:
N NH2 H NH2
\ \ N
O N NO2 ~ / ~-NO2
O
O ~~ N O 1 ) DCM-TFA/H20 O ~~' ~ N O
Boc~ ,.~H TS - + H
,N~NH OMe 2) P-TSA '°- A H ~~-NH OMe
H
WO WO
4 5
Trifluoroacetic acid (6.0 ml) is added at room temperature,to a well-stirred
mixture of
Boc-Ser(allyl)-D-Phe-Arg(NO~)-OMe (4, 1.519 g, 2.5 mmol), dichloromethane (30
ml) and water
(0.6 ml.) After stirring at room temperature for 3 h, p-toluenesulfonic acid
hydrate (0.380 g, 2.00
mmol) is added and the volatiles are removed in vacuo. Trituration of the
residue with ether (50
i o ml) afforded 5 ( 1.46 g) as an off white solid.
(d) Step 4:
N NHz H NH2
-NOZ O
O I ~ pH ~ , O N-NO2
O
O ~N O '~-.~N O
p-TSA- O+ N'H H HOBt, EDCI, NMM / H ' H
H3N~ OMe DMF ~ ~ N~NH OMe
O O ~O
5 ~ 6
A mixture of tripeptide (5, 1.46 g, 2.15 mmol), benzoic acid (0.263 g, 2.15
mmol), HOBt
(0.291 g, 2.15 mmol), and N methylmorpholine (0.52 ml, 4.7 mmol) in DMF (22
ml) is treated
~5 with EDCI (0.454 g, 2.37 mmol) and stirred at room temperature 16 h. The
reaction mixture is
diluted with ethyl acetate (220 ml) and washed successively with water (50
ml), 1 N HCl (2 x 25
ml), 1 N aqueous sodium bicarbonate (2 x 25 ml), and brine (25 ml.) The ethyl
acetate solution is
dried over a combination of anhydrous sodium and magnesium sulfates and is
concentrated to
yield 6 ( 1.10 g) as a colorless solid.
20 (e) Step 5:
CA 02368431 2001-09-28
WO 00/58361 PCT/US00/07473
-52-
N NHz H NHz
\ ~ \ N
/ O N-NOZ I / ~-NOZ
O
O ~ ~N O ~ ) 1 N NaOH ~ O
H 'N'H H 2) 1 N HCI ~ H O /'NIH H
N OMe \ ~ N~ OH
O O O ~O
6 ~ 7
Aqueous sodium hydroxide ( 1.0 N, 3.0 ml, 3.0 mmol) is added to a well-stirred
solution
of the benzoyl-tripeptide ester (6, 1.10 g, 1.80 mmol) in methanol ( 18 ml) at
room temperature.
The resulting mixture is stirred at room temperature for 2.5 h and the
volatiles are removed by
rotary evaporation. The residue is dissolved in water (20 ml), the solution
acidified to pH 3 with
1 N HCl (3.2-3.5 ml), and the solution extracted with ethyl acetate (1 x 70
ml, 2 x 25 ml.) The
combined ethyl acetate extracts are dried over anhydrous sodium sulfate and
concentrated in
vacuo. Trituration of the residue with ether gives 7 (0.884 g) as a colorless
solid.
(f) Step 6:
NH2
Boc O $ Boc
EDCI, NMM, HOBt
O H2N'~ NH
DMF
O
9
N methylmorpholine (0.33m1, 3.0 mmol) is added dropwise to a well-stirred
mixture of
Boc-Ser(allyl)-OH ~ DCA salt (2, 1.067 g, 2.5 mmol), 1-napthylamine (8, 0.358
g, 2.5 mmol),
HOBt (0.338 g, 2.5 mmol), and EDCI (0.527 g, 2.75 mmol) in DMF (28 ml) at
0°C. The resulting
mixture is stirred at 0°C for 0.5 h and then at room temperature for 16
h. The reaction mixture is
then diluted with ethyl acetate (200 ml) and washed successively with water
(50 ml), 1 N HCl (2
x 25 ml), 1 N aqueous sodium bicarbonate (2 x 25 ml), and brine (20 ml.) The
ethyl acetate
solution is dried over anhydrous sodium sulfate and is concentrated to a brown
oil which is
shown by HPLC/MS analysis to consist of a mixture of 8 and 9. This crude
product is purified
on an HPLC C4 reverse-phase column using a linear gradient of 0.1% aqueous
trifluoroacetic
2o acid and acetonitrile to give 0.26 g of 9.
CA 02368431 2001-09-28
WO 00/58361 PCT/US00/07473
-53-
(g) Step 7:
Boc ~ p-TSA- OO
HJN~ 1) DCM-TFA/H20 HsN
NH 2) p-TSA NH
O \ \ p \ \
/ / I / /
Trifluoroacetic acid (1.0 ml) is added at room temperature to a well-stirred
mixture of
5 Boc-Ser(allyl)-1-napthamide (9, 0.26 g, 0.702 mmol), dichloromethane (5.0
ml), and water (0.1
ml.) After stirring at room temperature for 88 h, p-toluenesulfonic acid
hydrate (0.133 g, 0.7
mmol) is added and the volatiles are removed in vacuo. Trituration of the
residue with
ether/hexane (1:1, 25 ml) afforded 10 (0.311 g) as a brown solid.
(h) Step 8:
p-TSA' O O
H3N. I/
NHy ;~NH N NHz
I / ~-NOZ O I w W I / O -NOz
O
O ,, N ~ ~,..~ O
O 10 / O H
I N~NH H OH HOBt, EDCI, NMM \ I N _ NH HN O
~NH
O ~O DMF O
O I
i i
10 ~ 11
lV-methylmorpholine (0.18 ml, 1.61 mmol) is added dropwise to a well-stirred
mixture of
the benzoyl-tripeptide acid (7, 0.436 g, 0.73 mmol), the p-TSA salt of H-
Ser(allyl)-1-napthamide
(10, 0.322 g, 0.73 mmol), HOBt (0.099 g, 0.73 mmol), and EDCI (0.154 g, 0.803
mmol) in DMF
(6 ml) at 0°C. The resulting mixture is stirred at 0°C for 0.5 h
and then at room temperature for 16
h. The reaction mixture is then diluted with ethyl acetate (100 ml) and washed
successively with
water ( 15 ml), 1 N HCl (2 x 10 ml), water ( 15 ml), 1 N aqueous sodium
bicarbonate (2 x 10 ml),
and brine (2 x 10 ml.) The ethyl acetate solution is dried over a combination
of anhydrous sodium
and magnesium sulfates, is concentrated in vacuo, and then the residue
triturated with ether (50
ml) to yield 11 (0.531 g) as a light brown solid.
(i) Step 9:
CA 02368431 2001-09-28
WO 00/58361 PCT/US00/07473
-54-
H NHz CI,,PCY~Ph N NHz
\ N 1\ CI~Ru \
/ N-NOz PCy3 I / O N-NOz
O 12 O
/ O ~'yH O CHC13 N O
O H
N~NH HN. //p (RCM) / NH HN~ lI
~NH ~ HN~ ~NH
O O \ ,
p \ \ O O~O I \ \
11 13
A degassed solution of Grubb's catalyst (12, 0.0504 g, 0.061 mmol) in
chloroform (5.0
ml) is added to a well-stirred and degassed solution of the dime (11, 0.260 g,
0.306 mmol) in
chloroform (20 ml) at room temperature. The resulting purple solution is
stirred at room
temperature under argon for 22 h. More catalyst (0.0504 g) in chloroform (5.0
ml) is added, the
mixture is stirred at room temperature for 5 h, and more catalyst (0.0504 g)
in chloroform (5.0
ml) is again added. After stirring 60 h at room temperature, triethylamine
(3.0 ml) is added and
the solution is concentrated by rotary evaporation. The residue is
chromatographed on silica gel
using ethyl acetate and 1:9 ethyl acetate/methanol as eluants to afford a
crude product (0.125 g)
~o which is purified further on an HPLC C4 reverse-phase column using a linear
gradient of 0.1%
aqueous trifluoracetic acid and acetonitrile to yield 13 (8 mg) as a 1:1
mixture of E and Z
isomers.
(j) Step 10:
H~/ NHz H NHz
I \ 1 N1~
/ N N-NOz ~ / NH
O O
O H O Hz/Pd/BaS04 O H O
O \\ _
~NH HN~ MeOH ~NH HN
HN ; NH ~ ~ HN ~NH
O O~O \\ O p, \\
O ~--~\i I
13 14
A solution of the macrocycle (13, 8 mg, 0.097 mmol) in methanol (10 ml) is
treated
with 10% palladium on barium sulfate (unreduced, 1 mg) and shaken under a
hydrogen
atmosphere (40 psi) for 48 h. The catalyst is removed by filtration through
Celite and the solvent
is removed in vacuo to give 14 as a colorless glass. Treatment of this glass
in methanol with
trifluoroacetic acid followed by removal of the volatiles and lyophilization
of the residue from a
20 5% acetonitrile/water mixture gives the TFA salt of 14 (8 mg) as an off
white powder.
CA 02368431 2001-09-28
WO 00/58361 PCT/US00/07473
-55-
B. Composition and Method Examples
Example A
An obese human female subject weighing 130 kg is treated by this method to
incur
weight loss. Specifically, once each day for a period of 6 months, the subject
is administered, via
intravenous injection, 15 ml of an aqueous solution comprising the following:
Component Concentration (mg/ml)
Compound of Ex. 1
Sodium bisulfate 1
~o Sodium chloride 7
Chlorobutanol
Citric acid 10
Sterile water qs to 1 mL,
Sodium Hydroxide adjust to pH 5
At the end of the treatment period, the patient exhibits measurable weight
loss.