Note: Descriptions are shown in the official language in which they were submitted.
CA 02368793 2001-10-22
WO 00/64483 PCT/US00/10830
1
RELEASABLE LINKAGE AND COMPOSITIONS CONTAINING SAME
Field of the Invention
The present invention relates to a compound comprised of a hydrophilic
polymer,
s such as polyethyleneglycol, cleavably linked to an amine-containing ligand,
which in
preferred embodiments can be an amine-containing lipid, drug or protein. The
compounds are cleavable under mild thiolytic conditions to regenerate the
amine-
containing ligand in its original form.
1 o Background of the Invention
Hydrophilic polymers, such as polyethylene glycol (PEG), have been used for
modification of various substrates, such as polypeptides, drugs and liposomes,
in order to
reduce immunogenicity of the substrate and/or to improve its blood circulation
lifetime.
For example, parenterally administered proteins can be immunogenic and may
~s have a short pharmacological half life. Proteins can also be relatively
water insoluble.
Consequently, it can be difficult to achieve therapeutically useful blood
levels of the
proteins in patients. Conjugation of PEG to proteins has been described as an
approach to
overcoming these difficulties. Davis et al. in U. S. Pat. No. 4,179,337
disclose
conjugating PEG to proteins such as enzymes and insulin to form PEG-protein
zo conjugates having less immunogenicity yet which retain a substantial
proportion of
physiological activity. Veronese et al. (Applied Biochem. and Biotech, 11:141-
152
(1985)) disclose activating polyethylene glycols with phenyl chloroformates to
modify a
ribonuclease and a superoxide dimutase. Katre et al. in U.S. Pat. Nos.
4,766,106 and
4,917,888 disclose solubilizing proteins by polymer conjugation. PEG and other
zs polymers are conjugated to recombinant proteins to reduce immunogenicity
and increase
half life. (Nitecki et al., U.S. Pat. No. 4,902,502; Enzon, Inc.,
PCT/US90/02133).
Garman (U.S. Patent No. 4,935,465) describes proteins modified with a water
soluble
polymer joined to the protein through a reversible linking group.
However, PEG-protein conjugates described to date suffer from several
so disadvantages. For example, modification of the protein with PEG often
inactivates the
protein so that the resulting conjugate has poor biological activity.
Typically in the
CA 02368793 2001-10-22
WO 00/64483 PCT/US00/10830
2
prior art to date, it is desired to have the PEG stably linked to the protein
so that the
beneficial properties provided by PEG remain. Another problem with some
protein
PEG conjugates is that upon decomposition of the conjugate undesirable
products may
be formed.
s PEG has also been described for use in improving the blood circulation
lifetime of
liposomes (U.S. Patent No. 5,103,556). Here, the PEG is covalently attached to
the
polar head group of a lipid in order to mask or shield the liposomes from
being
recognized and removed by the reticuloendothelial system. Liposomes having
releasable PEG chains have also been described, where the PEG chain is
released from
the liposome upon exposure to a suitable stimulus, such as a change in pH
(PCT/US97/18813). However, release of the PEG chain from the liposome suffers
from the drawback that the decomposition products are chemically modified and
can
have unpredictable, potentially negative effects in vivo.
1 s Summary of the Invention
Accordingly, it is an object of the invention to provide a compound where a
ligand is covalently yet reversibly linked to a hydrophilic polymer. Upon
cleavage of
the linkage, the ligand in its native form is regenerated.
In one aspect, the invention includes a compound having the general structure:
3
1
R
R5 R2
wherein R' is a hydrophilic polymer comprising a linkage for attachment to the
dithiobenzyl moiety; RZ is selected from the group consisting of H, alkyl and
aryl; R3 is
2s selected from the group consisting of O(C=O)R4, S(C=O)R4, and O(C=S)R4; R4
comprises an amine-containing ligand; and RS is selected from the group
consisting of
H, alkyl and aryl; and where orientation of CHZ-R3 is selected from the ortho
position
and the para position.
In one embodiment, RS is H and RZ is selected from the group consisting of
CH3,
CA 02368793 2001-10-22
WO 00/64483 PCT/US00/10830
3
CZHS and C3H8. In another embodiment, RZ and RS are alkyls.
In another embodiment, the amine-containing ligand R4 is selected from the
group
consisting of a polypeptide, an amine-containing drug and an amine-containing
lipid. In
an embodiment where the amine-containing ligand R4 is an amine-containing
lipid, the
lipid includes either a single hydrocarbon tail or a double hydrocarbon tail.
In one
preferred embodiment, the lipid is a phospholipid having a double hydrocarbon
tail.
The hydrophilic polymer R' can be, in yet another embodiment, selected from
the
group consisting of polyvinylpyrrolidone, polyvinylmethylether,
polymethyloxazoline,
polyethyloxazoline, polyhydroxypropyloxazoline, polyhydroxypropyl-
methacrylamide,
i o polymethacrylamide, polydimethyl-acrylamide,
polyhydroxypropylmethacrylate,
polyhydroxyethylacrylate, hydroxymethylcellulose, hydroxyethylcellulose,
polyethyleneglycol, polyaspartamide, copolymers thereof, and polyethyleneoxide-
polypropylene oxide.
In one preferred embodiment, the hydrophilic polymer R' is polyethyleneglycol.
i s In another embodiment, when R' is polyethylene glycol, RS is H and RZ is
CH3 or CzHS.
In still another embodiment, the amine-containing ligand R4 is a polypeptide.
The
polypeptide can be, in another embodiment, a recombinant polypeptide.
Exemplary and
preferred polypeptides include cytokines, such as interferons, interleukins,
and growth
factors, and enzymes.
2o In another aspect, the invention includes a composition comprising a
conjugate
obtainable by reaction with a compound having the general structural formula:
~\ Ji
~S~~ R3
R ~~~~~
R' RZ
2s wherein R' is a hydrophilic polymer comprising a linkage for attachment to
the
dithiobenzyl moiety; RZ is selected from the group consisting of H, alkyl and
aryl; R3 is
selected from the group consisting of O(C=O)R4, S(C=O)R4, and O(C=S)R4; R4
comprises a leaving group; and RS is selected from the group consisting of H,
alkyl and
aryl; and where orientation of CHZ-R3 is selected from the ortho position and
the para
_ _ v_ ~..~,~",
T""' ~ X901 2~17PM IOTR PI LAW GROUP o ,,
N0.402
05-Ofi-2001 ~ US 00001083
CA 02368793 2001-10-22
i
~, i
position. The composition also includes a p ~ aceutically-acceptable carrier,
such as
saline, buffer or the like, ;
In one embodiment of this aspect, R2 is s ted from the group consisting of
CH3,
CHs and C.3Hs.
In another embodiment, R' is O(C ~ O)R' and R' is a hydroxy- or oxy-containing
t ~ ~ ~ leaving group. The leaving group, in another ~ odiraent, is derived
from a compound
selected from the ou c of
gr p onsistxng chloride, porn-nttropheanl, ortho-mtrophenol, N-
bydroxy-tetrahydrophthalinnide, N-hydroxysucci~de, N-hydroxy-glutarimide, N- ~
;.
hydroxynorbornene 2,3-dicarboxyimide, 1-hydr I xybenzotxiazole, 3
hydroxypyridine, 4-. ,
o hydroxypyridine, Z-hydroxypyridine, 1-hydroxy 6-
ttifluoromethylbenzotriazole,
imtnidazole, triazole, N-methyl-imidazole, pentafluorophenol, trifluorophenol
and
~~ ; trichlorophenol. ,
In another embodiment, the compound is zjeacted with an amine=containing
ligand ~ . " ~~
is that displaces Rd to form a conjugate that include s the amine~ontaining
ligand. For
. example, the amine-containing ligand can be a phospholipid.
In a preferred embodiment, tha hydrop ' . polymer Rl is polyethylcneglycol, RS
is
' i : i H and R~ is C~I3 or C2Hs.
In yet another embodimern of this aspect, a composition containing the
conjugate
zo comprises a liposome. The liposome can furthe comprise an entrapped
therapeutic
agent.
In another embodiment; the amine-contai~g ligand comprises a polypepiide.
Yn yet another aspect, the invention incl s a liposome composition comprising
..
t v ~ '~ liposomes which include a surface coating of by ophilic polymer
chains wherein at Ieast
2 s a portion of the hydrophilic polymer chains hav I' the general structure:
. _ ; . . F. . . ,
r .,i _ , . R5
' wherein Rl is a hydrophilic polymer comprising a linkage for attachment to
the ~ ; ,
3 o dithiobenzyl moiety; R2 is selected from the gro~p consisting of H, alkyl
and aryl; R3
4
' ! ,~; ~ - Em~fan8steit. 6.J~uni 0.13 AMENDED SHEET
CA 02368793 2001-10-22
WO 00/64483 PCT/US00/10830
selected from the group consisting of O(C=O)R4, S(C=O)R4, and O(C=S)R4; R4
comprises an amine-containing ligand; and RS is selected from the group
consisting of
H, alkyl and aryl; and where orientation of CHZ-R3 is selected from the ortho
position
and the para position. The liposomes have a longer blood circulation lifetime
than
s liposomes having hydrophilic polymer chains joined to the liposome via an
aliphatic
disulfide linkage.
In one embodiment, the liposome further comprises an entrapped therapeutic
agent.
In still another aspect, the invention includes a method for improving the
blood
i o circulation lifetime of liposomes having a surface coating of releasable
hydrophilic
polymer chains. The method includes preparing liposomes that have between
about 1 %
to about 20 % of a compound having the general structure:
3
i ~S~ ~~
R
R5 R2
~s wherein R', R2, R3, and RS are as described above and R4 comprises an amine-
containing lipid.
In a preferred embodiment of this aspect, RS is H and RZ is selected from the
group consisting of CH3, CZHS and C3H8.
In another embodiment, the amine-containing lipid comprises a phospholipid and
z o R' is polyethyleneglycol.
In this aspect, the liposomes can further comprise an entrapped therapeutic
agent.
These and other objects and features of the invention will be more fully
appreciated when the following detailed description of the invention is read
in
conjunction with the accompanying drawings.
2s
Brief Description of the Drawings
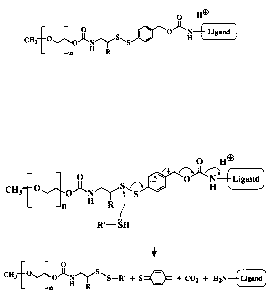
Fig. lA shows an embodiment of the invention where the dithiobenzyl (DTB)
links a methoxy-polyethyelene glycol (mPEG) moiety and the amine-containing
ligand;
Fig. 1B shows the products after thiolytic cleavage of the compound in Fig.
lA;
CA 02368793 2001-10-22
WO 00/64483 PCT/US00/10830
6
Fig. 2 illustrates a synthetic reaction scheme for synthesis of the mPEG-DTB-
amine-lipid, where the amine-ligand is the lipid
distearoylphosphatidylethanolamine
(DSPE);
Fig. 3 illustrates the thiolytic cleavage mechanism of a para-dithiobenzyl
urethane
s (DTB)-linked mPEG-DSPE conjugate;
Figs. 4A-4B show a synthetic reaction scheme for preparation of an mPEG-DTB-
DSPE compound in accord with the invention where the DTB linkage is sterically
hindered by an alkyl group;
Fig. 5 shows another synthetic reaction scheme for preparation of an mPEG-
~o DTB-ligand compound in accord with the invention;
Fig. 6A is a synthetic reaction scheme for synthesis of an mPEG-DTB-lipid
which
upon thiolytic cleavage yields a cationic lipid;
Fig. 6B shows the products after thiolytic cleavage of the compound in Fig.
6A;
Fig. 7A shows the rate of cleavage of ortho-mPEG-DTB-DSPE and para-mPEG-
~s DTB-DSPE conjugates in solution to form micelles in buffer alone (ortho-
conjugate (*);
para-conjugate (+)) and in the presence of 150 ~,M cysteine (ortho-conjugate
(open
circles); para-conjugate (open squares);
Fig. 7B shows the rate of cleavage of micellar mPEG-DTB-DSPE conjugates as
described in Fig. 7A and of ortho-mPEG-DTB-DSPE (solid circles) and para-mPEG-
zo DTB-DSPE (solid squares) conjugates formulated in liposomes and incubated
in the
presence of 150 ~,M cysteine;
Figs. 8A-8B show percentage of content release of entrapped fluorophore from
liposomes comprised of DOPE:ortho-mPEG-DTB-DSPE (Fig. 8A) or of DOPE:para-
zs mPEG-DTB-DSPE (Fig. 8B) incubated in the presence of cysteine at the
indicated
concentrations;
Fig. 9A shows normalized percent release of entrapped fluorophore as a
function of
time for liposomes comprised of DOPE and para-mPEG-DTB-DSPE. The percent
release
of entrapped fluorophore is normalized with respect to percent release of
fluorophore from
30 liposomes incubated in the absence of cysteine. The release rate from
liposomes incubated
in the presence of cysteine at concentrations of 15 ~.M (solid squares), 75
~,M (open
CA 02368793 2001-10-22
WO 00/64483 PCT/US00/10830
7
triangles), 150 wM (X symbols), 300 ~.M (open circles), 1500 ~.M (solid
circles), 3000 wM
(+ symbols), and 15000 ~,M (open diamonds) is shown;
Fig. 9B shows normalized percent release of entrapped fluorophore as a
function of
time for liposomes comprised of DOPE and para-mPEG-MeDTB-DSPE. The percent
s release of entrapped fluorophore is normalized with respect to percent
release of
fluorophore from liposomes incubated in the absence of cysteine. The release
rate for
liposomes incubated in the presence of cysteine at concentrations of 15 ~,M
(solid squares),
75 ~M (open triangles), 150 ~M (X symbols), 300 ~,M (open circles), 1500 ~.M
(solid
circles), 3000 ~,M (+ symbols), and 15000 p,M (open diamonds) is shown;
~o Fig. 9C shows normalized percent release of entrapped fluorophore as a
function of
time for liposomes comprised of DOPE and mPEG-meDTB-distearoyl-glycerol
compound
of Fig. 6A. The percent release of entrapped fluorophore is normalized with
respect to
percent release of fluorophore from liposomes incubated in the absence of
cysteine. The
release rate of dye upon cleavage of the compound from liposomes incubated in
the
~s presence of cysteine at concentrations of 15 ~,M (solid squares), 75 ~,M
(open triangles),
150 ~,M (X symbols), 300 ~M (open circles), 1500 ~M (solid circles), 3,000 ~cM
(+
symbols), and 15,000 ~M (open diamonds) is shown;
Fig. 10 is a plot showing the amount of liposomes, in counts per minute/mL of
liposomes containing entrapped In"', in blood samples taken from mice at
various times
zo after injection of liposomes comprised of PHPC:cholesterol:mPEG-DTB-DSPE
(55:40:5 molar ratio). One group of animals received a 200 wL injection of 200
mM
cysteine at time zero (solid squares). The control group was injection with
saline at
time zero (open circles);
Fig. 1 lA shows a synthetic reaction scheme for synthesis of an mPEG-DTB-
z s protein compound in accord with another embodiment of the invention;
Fig. 11B shows the decomposition products after thiolytic cleavage of the
compound in Fig. 11A;
Fig. 12 is a rendering of a photograph of an sodium-dodecyl-sulfate
polyacrylamide gel electrophoresis (SDS-PAGE) profile of lysozyme reacted for
15
3 o minutes (Lane 1 ) or for 1 hour (Lane 2) with mPEG-MeDTB-
nitrophyenylchloroformate to form a mPEG-MeDTB-lysozyme conjugate, native
CA 02368793 2001-10-22
WO 00/64483 PCT/tTS00/10830
8
lysozyme (Lane 3), lysozyme reacted for 1 hour with mPEG-
nitrophenylchloroformate
(Lane 4), molecular weight markers (Lane 5), and the samples of Lanes 1-4
treated with
2 % (3-mercaptoethanol for 10 minutes at 70°C (Lanes 6-9);
Fig. 13 shows the decomposition products after thiolytic cleavage of the an
s mPEG-DTB p-nitroanilide conjugate;
Fig. 14A shows the absorbence as a function of wavelength, in nm, of mPEG-
MeDTB-para-nitroanilide (closed diamonds) and after in vitro incubation with 5
mM
cysteine for 2 minutes (closed squares), 5 minutes (x symbols), 10 minutes
(open
squares), 20 minutes (triangles), 40 minutes (open diamonds) and 80 minutes
(closed
to circles); and
Fig. 14B shows the amount of para-nitroanilide, in mole/L, released in vitro
as a
function of time, in minutes, from mPEG-MeDTB para-nitroanilide conjugate
incubated
in the presence of 5 mM cysteine (closed circles), 1 mM cysteine (closed
squares) and
0.15 mM cysteine (closed diamonds).
is
Detailed Description of the Invention
I. Definitions
"Polypeptide" as used herein refers to a polymer of amino acids and does not
refer to a specific length of a polymer of amino acids. Thus, for example, the
terms
2o peptide, oligopeptide, protein, and enzyme are included within the
definition of
polypeptide. This term also includes post-expression modifications of the
polypeptide,
for example, glycosylations, acetylations, phosphorylations, and the like.
"Amine-containing" intends any compound having a moiety derived from
ammonia by replacing one or two of the hydrogen atoms by alkyl or aryl groups
to
as yield general structures RNHZ (primary amines) and RZNH (secondary amines),
where
R is any hydrocarbyl group.
"Hydrophilic polymer" as used herein refers to a polymer having moieties
soluble
in water, which lend to the polymer some degree of water solubility at room
temperature. Exemplary hydrophilic polymers include polyvinylpyrrolidone,
s o polyvinylmethylether, polymethyloxazoline, polyethyloxazoline,
polyhydroxypropyloxazoline, polyhydroxypropyl-methacrylamide,
polymethacrylamide,
CA 02368793 2001-10-22
WO 00/64483 PCT/US00/10830
9
polydimethyl-acrylamide, polyhydroxypropylmethacrylate,
polyhydroxyethylacrylate,
hydroxymethylcellulose, hydroxyethylcellulose, polyethyleneglycol,
polyaspartamide,
copolymers of the above-recited polymers, and polyethyleneoxide-polypropylene
oxide
copolymers. Properties and reactions with many of these polymers are described
in U.S.
s Patent Nos. 5,395,619 and 5,631,018.
"Polymer comprising a reactive functional group" or "polymer comprising a
linkage for attachment" refers to a polymer that has been modified, typically
but not
necessarily, at a terminal end moiety for reaction with another compound to
form a
covalent linkage. Reaction schemes to functionalize a polymer to have such a
reactive
i o functional group of moiety are readily determined by those of skill in the
art and/or have
been described, for example in U.S. Patent No. 5,613,018 or by Zalipsky et
al., in for
example, Eur. Polymer. J., 19(12):1177-1183 (1983); Bioconj. Chem., 4(4):296-
299
( 1993).
"Recombinant" as in "recombinant polypeptide" implies joining of amino acids
~s through laboratory manipulation into a desired sequence.
"Alkyl" as used herein intends a group derived from an alkane by removal of a
hydrogen atom from any carbon atom: "C~HZ"+,". The groups derived by removal
of a
hydrogen atom from a terminal carbon atom of unbranched alkanes form a
subclass of
normal alkyl (n-alkyl) groups: H[CHZ]n. The groups RCHZ-, RZCH- (R not equal
to H),
zo and R3C- (R not equal to H) are primary, secondary and tertiary alkyl
groups
respectively.
"Aryl" refers to a substituted or unsubstituted monovalent aromatic radical
having a
single ring (e.g., benzene) or two condensed rings (e.g., naphthyl). This term
includes
heteroaryl groups, which are aromatic ring groups having one or more nitrogen,
oxygen,
2s or sulfur atoms in the ring, such as furyl, pyrrole, pyridyl, and indole.
By "substituted" is
meant that one or more ring hydrogens in the aryl group is replaced with a
halide such as
fluorine, chlorine, or bromine; with a lower alkyl group containing one or two
carbon
atoms; vitro, amino, methylamino, dimethylamino, methoxy, halomethoxy,
halomethyl, or
haloethyl.
3o An "aliphatic disulfide" linkage intends a linkage of the form R'-S-S-R",
where
R' and R" are linear or branched alkyl chains that may be further substituted.
CA 02368793 2001-10-22
WO 00/64483 PCT/US00/10830
The following abbreviations are used herein: PEG, polyethylene glycol); mPEG,
methoxy-PEG; DTB, dithiobenzyl; MeDTB, methyl-dithiobenzyl, EtDTB, ethyl-
dithiobenzyl; DSPE, distearoyl phosphatidylethanolamine; DOPE, dioleoyl
phosphatidylethanolamine; PHPC, partially hydrogenated phosphatidylcholine;
MALDI-
s TOFMS, matrix-assisted laser desorption / ionization time-of-flight mass
spectrometry.
II. The Compound of the Invention
In one aspect, the invention comprises a compound of the form:
~S~ ~ R3
R ~~~~~
R5 Rz
io
wherein R' comprises a hydrophilic polymer including functional group suitable
for covalently attaching the polymer to the dithiobenzyl moiety. Rz and RS are
independently selected to be H, an alkyl or an aryl, and, as will be seen, can
be varied
to tailor the rate of disulfide cleavage. For example, to achieve a faster
rate of
~ s cleavage, Rz and RS are hydrogen. A slower rate of cleavage is achieved by
sterically
hindering the disulfide by selecting an alkyl or aryl for one or both of RZ
and R5. R3
comprises a linking moiety joined to R4, which comprises an amine-containing
ligand.
The linking moiety in preferred embodiments is O(C=O), S(C=O) or O(C=O). The
amine-containing ligand R4 can be a primary or a secondary amine and can be
selected
2o from any number of substrates, including, but not limited to lipids, drugs,
polypeptides,
viruses, surfaces of biomaterials and aminoglycosides. In preferred
embodiments, R4 is
a primary or secondary amine-containing lipid, drug or polypeptide. In the
compound
of the invention, the orientation of the group CHz R3 can be either ortho or
para.
Fig. lA shows the structure of an exemplary compound in accord with the
z s invention, where R' is the hydrophilic polymer methoxy-polyetheylene
glycol,
mPEG=CH30(CHZCH20)~ where n is from about 10 to about 2300, which corresponds
to
molecular weights of about 440 Daltons to about 100,000 Daltons. The molecular
weight
of the polymer depends to some extent on the selection of R3. In embodiments
where R3 is
an amine-containing lipid for use in a liposome a preferred range of PEG
molecular weight
CA 02368793 2001-10-22
WO 00/64483 PCT/US00/10830
11
is from about 750 to about 10,000 Daltons, more preferably from about 2,000 to
about
5,000 Daltons. The mPEG in this embodiment includes a urethane linking moiety.
In
embodiments where R3 is an amine-containing polypeptide a preferred range of
PEG
molecular weight is from about 2,000 to about 40,000 Daltons, more preferably
from
s about 2,000 to about 20,000 Daltons. It will be appreciated that R' can be
selected from a
variety of hydrophilic polymers, and exemplar polymers are recited above. It
will also be
appreciated that for some ligands, such as polypeptides, the molecular weight
of the
polymer may depend on the number of polymer chains attached to the ligand,
where a
larger molecular weight polymer is often selected when the number of attached
polymer
i o chains is small.
With continuing reference to Fig. la, RZ and RS in this exemplary compound are
H,
however either or both RZ and RS can also be a straight chain or branched
alkyl or an aryl
group. In a preferred embodiment, RS is H and RZ is an alkyl, and several
examples are
given below. In the compound shown in Fig. lA, R3 takes the general form of
O(C=O)-
~s (NHZ ligand), where the NHZ ligand can be any amine-containing polypeptide,
drug or
lipid, and specific examples of each embodiment are given below. R3 can also
be of the
form O(C=S)-(NHZ-ligand) or S(C=O)-(NHZ-ligand).
Fig. 1B shows the mechanism of thiolytic cleavage of the mPEG-DTB-(NHZ
ligand) compound of Fig. lA. The ortho- or para-dithiobenzyl carbamate moiety
is
zo cleavable under mild thiolytic conditions, such as in the presence of
cysteine or other
naturally-occurring reducing agents. Upon cleavage, the amine-containing
ligand is
regenerated in its natural, unmodified form. Studies in support of the
invention,
described below, show that natural, physiologic conditions in vivo are
sufficient to
initiate and achieve cleavage of the DTB linkage. It will be appreciated that
a reducing
zs agent can also be administered to artificially induce thiolytic conditions
sufficient for
cleavage and decomposition of the compound.
As noted above, R3 takes the general form of a linking moiety, such as O(C=O),
S(C=O) or O(C=S) joined to an amine-containing ligand. In preferred
embodiment, the
amine-containing ligand comprises an amine-containing polypeptide, drug or
lipid.
3o Examples of these embodiments will now be described.
CA 02368793 2001-10-22
WO 00/64483 PCT/US00/10830
12
A. Amine-Containing Lipid
In one embodiment, the amine-containing ligand is an amine-containing lipid.
Lipids as referred to herein intend water-insoluble molecules having at least
one acyl
chain containing at least about eight carbon atoms, more preferably an acyl
chain
s containing between about 8-24 carbon atoms. A preferred lipid is a lipid
having an
amine-containing polar head group and an acyl chain. Exemplary lipids are
phospholipids having a single acyl chain, such as stearoylamine, or two acyl
chains.
Preferred phospholipids with an amine-containing head group include
phosphatidylethanolamine and phosphatidylserine. The lipid tails) can have
between
about 12 to about 24 carbon atoms and can be fully saturated or unsaturated.
One
preferred lipid is distearoylphosphatidylethanolamine (DSPE), however those of
skill in
the art will appreciate the wide variety of lipids that fall within this
description. It will
also be appreciated that the lipid can naturally include an amine group or can
be
derivatized to include an amine group. Other lipid moieties that do not have
an acyl
~s tail, such as cholesterolamine, are also suitable.
The synthesis of a polymer-DTB-lipid compound is schematically depicted in
Fig.
2. mPEG derivatives (MW 2000 and 5000 Daltons) having a
methoxycarbonyldithioalkyl end group were prepared by reacting 2-
(methoxycarbonyldithio)ethaneamine with mPEG-chloroformate, which was readily
zo prepared by phosgenation of dried mPEG-OH solution (Zalipsky, 5., et al.,
Biotechnol.
Appl. Biochem. 15:100-114 (1992).). The former compound was obtained through 2-
aminoethanethiol hydrochloride reaction with an equivalent amount of
methoxycarbonylsulfenyl chloride, according to published procedures (Brois,
S.J., et
al. , J. Amer. Chem. Soc. 92: 7629-7631 ( 1970); Koneko, T . , et al. ,
Bioconjugate
zs Chem. 2:133-141 (1991)). Both the para and ortho isomers of mercaptobenzyl
alcohol
(Grice, R., et al., J. Chem. Soc. 1947-1954 (1963)) coupled cleanly with the
resulting
PEG-linked acyldisulfide, yielding mPEG bearing a dithio benzyl alcohol end
group.
Active carbonate introduction proceeded as with underivatized mPEG-OH, to give
the
para-nitrophenyl carbonate. Addition of DSPE in ethanolamine formed the
desired
3o mPEG-DTB-DSPE product. Both ortho- andpara-DTB-lipid compounds were
prepared and purified by silica gel chromatography and characterized by NMR
and
CA 02368793 2001-10-22
WO 00/64483 PCT/IJS00/10830
13
MALDI-TOFMS, the details of which are given in Example 1.
Fig. 3 shows the mechanism of thiolytic cleavage of the mPEG-DTB-DSPE
conjugate. Upon cleavage, the phosphatidylethanolamine lipid is regenerated in
its
natural, unmodified form.
s Figs. 4A-4B show a reaction scheme for synthesis of mPEG-DTB-DSPE conjugates
having an alkyl group adjacent the disulfide linkage, e.g., a more hindered
disulfide
linkage. As described more fully in Example 2A, mPEG-OH in dichloromethane was
reacted with p-nitrophenylchloroformate in the presence of triethylamine (TEA)
to form
mPEG-nitrophenyl carbonate. An amino alcohol, such as 1-amino-2-propanol or 1-
amino-
i o 2-butanol, in dimethylformamide (DMF) was reacted with the mPEG-
nitrophenyl
carbonate in the presence of TEA to form a secondary alcohol attached to PEG.
The
secondary alcohol was then converted to the desired mPEG-DTB-DSPE compound as
illustrated in Fig. 4A and detailed in Example 2A.
In this reaction scheme, mPEG-methyl-dithiobenzyl- nitrophenyl chloroformate
was
~ s reacted with DSPE to form the desired compound. The nitrophenyl
chloroformate moiety
in the mPEG-methyl-dithiobenzyl-nitrophenyl chloroformate compound acts as a
leaving
group to yield the desired product upon reaction with a selected lipid. The
invention
contemplates, in another aspect, a composition that comprises a compound
produced by
reaction with a compound such as mPEG-methyl-dithiobenzyl-R3, where R3
represents a
20 leaving group joined through a linking moiety to the benzene ring. The
leaving group is
displaced upon reaction with an amine-containing ligand, such as DSPE, a
polypeptide or
an amine-containing drug. The leaving group is selected according to the
reactivity of the
amine in the ligand, and is preferably derived from various acidic alcohols
that have a
hydroxy- or oxy-containing leaving group. These include chloride, p-
nitrophenol, o-
zs nitrophenol, N-hydroxy-tetrahydrophthalimide, N-hydroxysuccinimide, N-
hydroxy-
glutarimide, N-hydroxynorbornene-2,3-dicarboxyimide, 1-hydroxybenzotriazole, 3-
hydroxypyridine, 4- hydroxypyridine, 2-hydroxypyridine, 1-hydroxy-6-
trifluoromethylbenzotriazole, immidazole, triazole, N-methyl-imidazole,
pentafluorophenol, trifluorophenol and trichlorophenol.
3o Example 2B describes preparation of an mPEG-EtDTB-lipid conjugate where the
disulfide linkage is hindered by an ethyl moiety.
CA 02368793 2001-10-22
WO 00/64483 PCT/US00/10830
14
Fig. 5 shows another synthetic reaction scheme for preparation of an mPEG-
DTB-ligand compound in accord with the invention. The details of the reaction
procedure are given in Examples 3A-3B. Briefly, cold 1-amino-2-propanol was
reacted
with sulfuric acid to form 2-amino-1-methylethyl hydrogen sulfate. This
product was
s reacted with carbon disulfide and sodium hydroxide in aqueous ethanol to
yield 5-
methylthiazolidine-2-thione. An aqueous solution of hydrochloric acid was
added to the
5-methylthiazolidine-2-thione and heated. After refluxing for one week, the
product, 1-
mercapto(methyl)ethyl ammonium chloride, was crystallized and recovered. This
product was reacted with methoxy carbonylsulfenyl chloride to yield 2-
(methoxycarbonyldithio)ethaneamine. Reaction of the 2-
(methoxycarbonyldithio)ethaneamine with mPEG-chloroformate using the procedure
described above with respect to Fig. 2 yields the desired mPEG-DTB-nitrophenyl
compound suitable for reaction with a selected amine-containing ligand to form
a
compound in accord with the invention.
is Example 3B describes the reaction for synthesis of mPEG-(ethyl)DTB-
nitrophenyl.
Fig. 6A shows a reaction scheme for preparation of another mPEG-DTB-lipid
compound in accord with the invention. The reaction details are provided in
Example 4.
The lipid 1,2-distearoyl-sn-glycerol is activated for reaction with mPEG-DTB-
nitropheynl,
2 o prepared as described in Fig. 4A or Fig. 5. The resulting mPEG-DTB-lipid
differs from
the compounds described above in the absence of a phosphate head group. The
mPEG-
DTB-lipid of Fig. 6A is neutral prior to cleavage. As shown in Fig. 6B, upon
thiolytic
reduction of the disulfide bond, the compound decomposes to yield a cationic
lipid. The
positively-charged lipid provides for electrostatic interaction in vivo and
commensurate
zs advantages in in vivo targeting.
In the reaction schemes described above, RS of the claimed compound is H.
However, in other embodiments RS is an alkyl or an aryl moiety. In this
approach, for
example where Rz and RS are both CH3 moieties, an a, (3-unsaturated acyl
chloride
(R'R"C=CHCOCI, where R' is, for example CH3 and R" is CH3, however any alkyl
30 or aryl is contemplated) i~ reacted with an amine-terminated PEG to give
the
corresponding N-PEG-substituted a, (3-unsaturated amide. This compound is
reacted
CA 02368793 2001-10-22
WO 00/64483 PCT/US00/10830
with thiolacetic acid, giving the corresponding N-PEG-substituted (3-
(acetylthio) amide
via conjugate addition to the C=C bond. The acetylthio group (-SCOCH3) is
hydrolyzed to a thiol group (-SH), which is then reacted with methyl
(chlorosulfenyl)formate (C1SCOOCH3), generating a methoxycarbonyl diothio
group (-
s SSCOOCH3); this intermediate is then reacted with p-mercapto benzyl alcohol
to give
the N-PEG-substituted (3-(dithiobenzyl alcohol) amide (having the structure
PEG-NH-
CO-CHzCR'R"-SS p-phenyl-CHZOH). The benzyl alcohol moiety is then reacted with
nitrophenyl chloroformate to give the nitrophenyl carbonate leaving group, as
above.
i o 1. In vitro Cleavage of mPEG-DTB-DSPE Co found
The in vitro rate of cleavage of ortho-mPEG-DTB-DSPE and para-mPEG-DTB-
DSPE (prepared as described in Example 1) was studied by preparing micellar
solutions
of the compounds in a buffered aqueous solution (pH 7.2). Thiolytic cleavage
of the
compounds was monitored in the presence and absence of 150 p.M cysteine by
is analyzing for disappearance of the compounds by HPLC, as described in
Example 5.
The results are illustrated in Fig. 7A where the ortho- and para-compounds in
the
absence of cysteine (* symbols and + symbols, respectively) show no cleavage
and are
stable under these conditions in the absence of cysteine. The ortho- and para-
compounds, represented by the open circles and the open squares, respectively,
in the
a o presence of 150 wM cysteine cleave as shown in Fig. 7A. The ortho-compound
exhibited a slightly faster rate of decomposition than its para counterpart
(T1,2 X12
minutes vs. X18 minutes).
2. Liposome Compositions Comprisi~ an mPEG-DTB-lipid Compound
z s a. In vitro Characterization
In one embodiment, the mPEG-DTB-lipid compound is formulated into liposomes.
Liposomes are closed lipid vesicles used for a variety of therapeutic
purposes, and in
particular, for carrying therapeutic agents to a target region or cell by
systemic
administration of liposomes. In particular, liposomes having a surface coating
of
so hydrophilic polymer chains, such as polyethylene glycol (PEG), are
desirable as drug
carries, since these liposomes offer an extended blood circulation lifetime
over
._ _ - ~ _: _. r _ .: _., _ .. . .. . . . _ _
- T~ ~ ~ ?001 2: iBPM IOTA PI LRLJ GROUP ~ N0. 402 ~ ' ~
05-06-2001 US 00001083
' CA 02368793 2001-10-22
' liposomes lacking the polymer coating. The p~lymer chains in the polymer
coating
shield the liposomes and form a "stiff brush ~f water solvated polymer chains
about the
v ~ ~ ~ - ~ liposomes.. . Thus, the polymer acts as a barne~ to blood
proteins, preventing binding of
the protein and recognition of the liposomes for uptake and removal by
macrophages and
other cells of the reticuloendothelial system.
Typically,-liposomes having a surface cdating of polymer chains are prepared
by
including in the lipid mixture between about 1 to abort 20 mole percent of the
lipid
derivatized with the polymer. The actual amo of polymer derivatized lipid can
be
'higher or lower depending on the molecular
fight of the polymer. In the present
io inventioa, liposomes are prepared by adding bptwecn about 1 to about 20
mole percem of.
the polymer-DTB-lipid conjugate to other lipoi ome lipid bilayer components.
As will~be w
demonstrated in the studies described below, iposomes contaiaing the polymer-
DTB-
lipid conjugate of the invention have a blood circulation lifetime longer than
liposomes
containing a polymer-lipid conjugate where th polymer aad lipid are joined by
a
. w w, ; .
i5 '~ ~ cleavable aliphatic disulfide bond.
In studies performed in support of the in ention, liposomes comprised of the .
. ~ ..
vesicle-farming lipid partially hydrogenated p osphatidyl choline along with
cholesterol i
and the ortho-mPEG-DTB-DSPE or the para- ~mFEG-DTB-DSP~ compound were
prepared as descrl'bed in Example 6. Cysteine~ mediated. cleavage of the mPEG-
DTB-
2o DSPE compounds was monitored in the presence and absence of x50 uM cysteine
in an
f i.
~ ~~ aqueous buffer. The results axe shown in Fig.l 7B, which includes the
data of Fig. 7A for
comparison, In Fig. 7B, the ortho- and para mpouads in micellar form in the
absence
of cysteine (* symbols and + symbols, respec 'vely) show no cleavage, .which
indicates ' ' .
stability of the conjugate in the absence of thx . The open circles and the
open squares
25 correspond to the ortho- aadpara-compounds re
I spectively, in nucellar form in the
.~ .f , ~ , ., presence of cysteine, as discussed above with respect to Fig,
7A. The solid circles and
the solid squares correspond to the ortho- and ara-compou~sds, respectively,
in
liposomal form in the presence of cysteine. .
The data in Fig. 7B shows that both the ortho and para compoundlwere slightly
. . .
30 ~ more resistant to thiolytic cleavage when inco ~ orated into liposomes.
Examination of
the thiolysis reaction products by TLC (silica ~el G,..chloroform l methanol !
water
. .. ~ _
' 16
._
'c
Empfangsieit 6.Juni 0.13 AMENDED SHEET
i
CA 02368793 2001-10-22
WO 00/64483 PCT/US00/10830
17
90:18:2) (Dittmer, J.C., et al., J. Lipid Res. 5:126-127 (1964)) showed DSPE
as the
sole lipid component and another spot corresponding to a thiol-bearing, lipid-
free
mPEG derivative.
In another study performed in support of the invention, liposomes were
prepared
s from the lipid dioleoyl phosphatidylethanolamine (DOPE) and either the ortho-
mPEG-
DTB-DSPE or the para-mPEG-DTB-DSPE compound were prepared. DOPE is a
hexagonal phase lipid which alone does not form lipid vesicles. However,
liposomes
will form when DOPE is combined with a few mole percent of the mPEG-DTB-DSPE
compound. Cleavage of the mPEG-DTB-DSPE compound triggers decomposition of
o the liposomes and release of liposomally-entrapped contents. Thus, the
content release
characteristics of such liposomes provides for a convenient quantitative
evaluation of
cleavable PEG-bearing liposomes.
Liposomes comprised of DOPE and the ortho- or para-mPEG-DTB-DSPE
compound were prepared as described in Example 7A with entrapped fluorophores,
p-
is xylene-bis-pyridinium bromide and trisodium 8-hydroxypyrenetrisulfonate.
Release of
the fluorophores from liposomes incubated in the presence of cysteine at
various
concentrations was monitored as described in Example 7B.
Results for liposomes comprising the ortho-compound are shown in Fig. 8A,
where percentage of content release of entrapped fluorophore from liposomes
incubated
2o in the presence of cysteine at concentrations of 15 ~M (solid diamonds),
150 ~,M (solid
inverted triangles), 300 ~,M (solid triangles) and 1.5 mM (solid circles) are
shown.
Fig. 8B is a similar plot for liposomes comprising the para-compound, where
the
liposomes are incubated in cysteine at concentrations of 15 ~,M (solid
diamonds), 300
~,M (solid triangles), 1 ~,M (solid squares) and 1.5 mM (solid circles).
2s Figs. 8A-8B show that both the ortho- and para-compounds when incorporated
into liposome are cleaved, as evidenced by release of the entrapped dye, at a
rate
dependent on the concentration of cysteine. Control studies with non-cleavable
mPEG-
DSPE containing liposomes produced no content release (results not shown
here).
These results also suggest that the ortho conjugate is somewhat more
susceptible to
3o thiolytic cleavage. For example, 300 pM cysteine liberates most of the
contents of
DOPE liposomes within 20 minutes. Under the same conditions, only a fraction
of
CA 02368793 2001-10-22
WO 00/64483 PCT/US00/10830
18
liposomes having para-mPEG-DTB-DSPE decomposed. Similarly, after incubation
for
20 minutes at 150 pM cysteine, half of the entrapped contents was released for
the
ortho-containing liposomes, while only approximately 10% of the contents were
release
in liposomes containing the para-compound. Both ortho and para compounds have
s half-lives of less than 20 minutes at a cysteine level of 150 ~cM (see data
in Fig. 7B).
This suggests that more than half of the original three mole percent of the
mPEG-DTB-
lipid must be cleaved to observe content release from the liposomes.
Decomposition of the mPEG-DTB-DSPE/DOPE liposomes in 15 pM cysteine,
the average plasma concentration in both humans and rodents (Lash, L. H. , et
al. , Arch.
Biochem: Biophys. 240:583-592 (1985)), was minimal in the time frame of these
experiments (60 minutes). This suggests that the mPEG-DTB-lipid compounds
should
have sufficiently long lifetimes in plasma to allow the PEG-grafted vesicles
to distribute
systemically in vivo, or to accumulate in a specific site either passively or
through
ligand-mediated targeting. Local or short term increase in cysteine
concentration can
~ s potentially be achieved by its intravenous of into a-arterial
administration. The results
shown in Figs. 8A-8B also suggest that a prolonged exposure to the natural
plasma
cysteine concentration (~ 15 p,M) would be sufficient to decompose most of
these
compounds. These suggestions were studied in in vivo experiments, described
below.
In another study performed in support of the invention, liposomes comprised of
zo DOPE and three different mPEG-DTB-lipid compounds were prepared. The
liposomes
were prepared as described in Example 7 and included and entrapped
fluorophore. The
three mPEG-DTB-lipid compounds were mPEG-DTB-DSPE as shown in Fig. lA; mPEG-
MeDTB-DSPE as shown in Fig. 4B, where R is CH3, and mPEG-MeDTB-distearoyl-
glycerol, as shown in Fig. 6A. The liposomes were comprised of 97 mole percent
DOPE
z s and 3 mole percent of one of the mPEG-DTB-lipid compounds. Cysteine-
mediated rate of
cleavage of the compounds was determined by monitoring the release of
entrapped
fluorophore as a function of time in the presence of various cysteine
concentrations. The
results are shown in Figs. 9A-9C where the percent release of entrapped
fluorophore is
normalized for the release rate from liposomes incubated in buffer alone.
3o Fig. 9A shows the percent release of entrapped fluorophore as a function of
time for
liposomes comprised of DOPE and para-mPEG-DTB-DSPE (compound of Fig. lA). The
CA 02368793 2001-10-22
WO 00/64483 PCT/US00/10830
19
release rate from liposomes containing the conjugate and incubated in the
presence of
cysteine at concentrations of 15 ~,M (solid squares), 75 wM (open triangles),
150 ~M (X
symbols), 300 ~,M (open circles), 1500 ~cM (solid circles), 3000 wM (+
symbols), and
15000 ~.M (open diamonds) is shown.
s Fig. 9B shows the percent release of entrapped fluorophore as a function of
time for
liposomes comprised of DOPE and para mPEG-MeDTB-DSPE (compound of Fig. 4B).
The release rate of the fluorophore from liposomes incubated in the presence
of cysteine at
concentrations of 15 ~M (solid squares), 75 ~.M (open triangles), 150 ~.M (X
symbols),
300 ~,M (open circles), 1500 ~M (solid circles), 3000 ~,M (+ symbols), and
15000 ~,M
~ o (open diamonds) is shown.
Fig. 9C is a similar plot for liposomes formed with DOPE and mPEG-MeDTB-
distearoyl glycerol (compound of Fig. 6A). The release rate of dye from
liposomes
incubated in the presence of cysteine at concentrations of 15 ~,M (solid
squares), 75 ~M
(open triangles), 150 ~,M (X symbols), 300 ~,M (open circles), 1500 ~.M (solid
circles),
~s 3000 ~.M (+ symbols), and 15000 ~,M (open diamonds) is shown.
Figs. 9A-9C show that the rate of mPEG-MeDTB-lipid cleavage is cysteine-
concentration dependent, with a slow rate of cleavage, as evidenced by release
of
entrapped fluorophore, at cysteine concentrations of 15-75 ~.M. In comparing
the data in
Fig. 9A with that in Fig. 9B, it is seen that the mPEG-MeDTB-DSPE compound
(Fig. 9B)
z o cleaves approximately 10 times more slowly than the mPEG-DTB-DSPE compound
(Fig.
9A). Thus, the rate of cleavage can be tailored according to the R moiety (see
Fig. 2) in
the DTB linkage.
b. In vivo Characterization
zs The blood circulation lifetime of liposomes prepared as described in
Example 8
and that include a polymer-DTB-lipid conjugate in accord with the invention
was
determined in mice. In"' was entrapped in the liposomes and the liposomes were
administered by intravenous injection. One group of test animals additionally
received
an injection of cysteine, the control group of animals additionally received
an injection
30 of saline. Blood samples were taken at various times and analyzed for the
presence of
liposomes, as evidenced by the presence of In"'.
CA 02368793 2001-10-22
WO 00/64483 PCT/US00/10830
Fig. 10 shows the results where the counts per minute (CPM) of In'~' is shown
as a
function of time following injection of the liposomes and saline (open
circles) or 200 mM
cysteine (solid squares). As seen, the cleavage of the mPEG-DTB-DSPE occurred
upon
exposure to the naturally-occurring physiologic conditions, as evidenced by
the cleavage in
s the group of mice treated with saline after administration of the liposomes.
Administration
of an exogeneous reducing agent, cysteine, to the mice was effective to
increase the rate of
cleavage of the mPEG-DTB-lipid compound in the time frame from between about 2
hours
to about 8 hours.
Importantly, cleavage of the polymer-DTB-lipid compound of the invention
results
~ o in regeneration of the original lipid in unmodified form. This is
desirable since unnatural,
modified lipids can have undesirable in vivo effects. At the same time, the
compound is
stable when stored in the absence of reducing agents.
In other studies, not shown here, the blood circulation lifetime of liposomes
containing the mPEG-DTB-lipid were compared to liposomes containing a polymer-
lipid
~s conjugate where the polymer and lipid are joined by a cleavable aliphatic
disulfide
bond. Aliphatic disulfide linkages are readily cleaved in vivo and the blood
circulation
lifetime of liposomes having polymer chairs grafted to their surface by an
aliphatic
disulfide typically do not have the extended blood circulation lifetime
observed for
liposomes having stably linked polymer chains. The dithiolbenzyl linkage of
the
zo invention, and in particular the more hindered DTB linkages, are more
stable in vivo
and achieve a longer blood circulation lifetime than liposomes with polymer
chains
attached via an aliphatic disulfide linkage.
B. Amine-Containing Polvpentide
z s In another embodiment, the invention includes a compound as described with
respect
to Fig. lA, where the amine-containing ligand is a polypeptide. A synthetic
reaction
scheme showing preparation of a polymer-DTB-polypeptide is shown in Fig. 11A,
with
mPEG as the exemplary polymer. In general, a mPEG-DTB-leaving group compound
is
prepared according to one the synthetic routes described above in Figs. 2, 4A
and 5. The
leaving group can be nitrophenyl carbonate or any one of the others described
above. The
mPEG-DTB-nitrophenyl carbonate compound is coupled to an amine moiety in a
CA 02368793 2001-10-22
WO 00/64483 PCT/US00/10830
21
polypeptide by a urethane linkage. The R group adjacent the disulfide in the
compound
can be H, CH3, CZHS or the like and is selected according to the desired rate
of disulfide
cleavage.
Fig. 11B shows the decomposition products upon cysteine-mediated cleavage of
the
s compound. As seen the native protein with no modification to the protein
amine group is
regenerated upon cleavage.
Attachment of polymer chains, such as PEG, to a polypeptide often diminishes
the
enzymatic or other biological activity, e. g. , receptor binding, of the
polypeptide.
However, polymer modification of a polypeptide increases the blood circulation
lifetime of
the polypeptide. In the present invention, the polymer-modified polypeptide is
administered to a subject. As the polymer-modified polypeptide circulates
exposure to
physiologic reducing conditions, such as blood cysteine and other in vivo
thiols, initiates
cleavage of the polymer chains from the polypeptide. As the polymer chains are
released
from the polypeptide, the biological activity of the polypeptide is gradually
restored. In
~s this way, the polypeptide initially has a sufficient blood circulation
lifetime for
biodistribution, and over time regains its full biological activity as the
polymer chains are
cleaved.
In a study performed in support of the invention, lysozyme was used as a model
polypeptide and an mPEG-MeDTB-lysozyme conjugate was prepared by a synthetic
route
zo similar to those described above. Lysozyme was incubated with mPEG-MeDTB-
nitrophenylcarbonate in 0.1 M borate, at pH 9 at a 2:1 ratio of
nitrophenylcarbonate to
amino group of lysozyme. After reactions times of 15 minutes and 3 hours,
samples were
characterized by SDS-PAGE. A comparative compound was prepared by reacting
lysozyme under the same conditions for 60 minutes with a conjugate of mPEG-
nitrophenyl
zs carbonate, which will form a stable mPEG-lysozyme conjugate.
Fig. 12 shows a rendering of the SDS-PAGE gel. Lane 1 corresponds to the
compound formed after 15 minutes reaction of lysozyme with mPEG-MeDTB-
nitrophyenylcarbonate and Lane 2 represents the compound formed after a 1 hour
reaction time of the same compounds. Lane 3 represents native lysozyme and
Lane 4
so corresponds to lysozyme reacted for 1 hour with mPEG-nitrophenylcarbonate.
The
molecular weight markers in Lane 5 are as follows, from the top down:
CA 02368793 2001-10-22
WO 00/64483 PCT/US00/10830
22
Molecular Weight (kDaltons) Marker
1163 (3-galactosidase
97.4 phosphorylase b
66.3 bovine serum albumin
55.4 glutamic dehydrogenase
36.5 lactate dehydrogenase
31 carbonic anhydrase
21.5 trypsin inhibitor
14.4 lysozyme
Comparison of Lane 1 and Lane 2 shows that the longer reaction time results in
an increase in compound molecular weight, consistent with additional mPEG
chains
conjugated to the polypeptide at longer incubation time.
s Lanes 6-9 of the SDS-PAGE profile correspond to the samples in Lanes 1-4
after
treatment with 2 % (3-mercaptoethanol for 10 minutes at 70°C. The mPEG-
MeDTB-
lysozyme conjugate after exposure to a reducing agent decomposed to regenerate
native
lysozyme, as evidenced by the band in Lanes 6 and 7 at 14.4 kDa. In contrast,
the
stable mPEG-lysozome compound was not affected upon incubation with a reducing
agent, as evidenced by the agreement in the profile in Lane 9 and Lane 4.
Also evident from the SDS-PAGE profile is that covalent attachment of mPEG-
MeDTB to a protein forms a mixture of conjugates containing various mPEG-
protein
ratios. This ratio is dependent on the reaction time and conditions. This is
clearly seen
in viewing the bands in Lanes 1 and 2, where Lane 1 shows lysozyme derivatized
with
~s from about 1-6 PEG chains. In Lane 2, the longer reaction time yielded mPEG-
MeDTB-lysozyme conjugates with a higher mPEG-protein ratio. All cleavable
conjugates were readily cleaved to regenerate the native protein, as seen in
the bands of
Lanes 6 and 7.
It will be appreciated that any of the hydrophilic polymers described above
are
zo contemplated for use. The molecular weight of the polymer is selected
depending on the
polypeptide, the number of reactive amines on the polypeptide and the desired
size of the
polymer-modified compound.
CA 02368793 2001-10-22
WO 00/64483 PCT/US00/10830
23
Polypeptides contemplated for use are unlimited and can be naturally-occurring
or
recombinantly produced polypeptides. Small, human recombinant polypeptides are
preferred, and polypeptides in the range of 10-30 KDa are preferred. Exemplary
polypeptides include cytokines, such as tumor necrosis factor (TNF),
interleukins and
s interferons, erythropoietin (EPO), granulocyte colony stimulating factor
(GCSF),
enzymes, and the like. Viral polypeptides are also contemplated, where the
surface of a
virus is modified to include one or more polymer chain linked via a DTB
reversible
linkage. Modification of a virus containing a gene for cell transfection would
extend the
circulation time of the virus and reduce its immunogenicity, thereby improving
delivery of
io an exogeneous gene.
C. Amine-Containing Drug
In yet another embodiment of the invention, a compound of the form polymer-DTB-
amine-containing drug is contemplated. The compound is of the structure
described
~s above, and in particular with respect to Fig. lA where the amine-containing
ligand in the
figure is the amine-containing drug. Modification of therapeutic drugs with
PEG is
effective to improve the blood circulation lifetime of the drug and to reduce
any
immunogenicity .
A polymer-DTB-amine-containing drug is prepared according to any of the
reaction
2 o schemes described above, with modifications as necessary to provide for
the particular
drug. A wide variety of therapeutic drugs have a reactive amine moiety, such
as
mitomycin C, bleomycin, doxorubicin and ciprofloxacin, and the invention
contemplates
any of these drugs with no limitation. It will be appreciated that the
invention is also
useful for drugs containing an alcohol or carboxyl moiety. In the case where
the drug
z s contains a hydroxyl or carboxyl moiety suitable for reaction, the polymer-
DTB moiety can
be linked to the drug via urethane, ester, ether, thioether or thioester
linkages. In all of
these embodiments, the polymer-DTB-drug compound after administration in vivo
thiolytically decomposes to regenerate the amine-containing drug in its
native, active form,
therapeutic activity of the compound after modification and prior to
administration is not
3o necessary. Thus, in cases where modification of the drug with the DTB-
polymer causes a
reduction or loss of therapeutic activity, after administration and cleavage
of the DTB-
CA 02368793 2001-10-22
WO 00/64483 PCT/US00/10830
24
polymer from the drug, activity of the drug is regained.
In studies performed in support of the invention, the drug nitroanilide was
reacted
with mPEG-MeDTB-nitrophenylcarbonate to form an mPEG-MeDTB para-nitroanilide
compound, as shown in Fig. 13. Decomposition of the compound upon exposure to
a
s reducing agent yields the products shown in the figure, with the drug para-
nitroanilide
regenerated in an unmodified state.
The mPEG-MeDTB para-nitroanilide compound was incubated in vitro in buffer
containing 5 mM cysteine and the absorbence of samples withdrawn at various
times is
shown in Fig. 14A. Seen in the figure are samples measured at the following
time
~o points: time zero (closed diamonds), 2 minutes (closed squares), 5 minutes
(x
symbols), 10 minutes (open squares), 20 minutes (triangles), 40 minutes (open
diamonds) and 80 minutes (closed circles). The change in the UV spectra as a
function
of incubation time in cysteine is evident, showing cysteine-mediated release
of para-
nitroanilide from the mPEG-MeDTB- para-nitroanilide compound.
~s Fig. 14B shows the amount ofpara-nitroanilide, in mole/L, released in vitro
from
the mPEG-MeDTB para-nitroanilide conjugate incubated in the presence of 5 mM
cysteine (closed circles), 1 mM cysteine (closed squares) and 0.15 mM cysteine
(closed
diamonds). The rate of drug release from the conjugate was dependent on the
concentration of reducing agent present.
z o From the foregoing, it can be seen how various objects and features of the
invention
are met. The compounds of the invention comprise an amine-containing ligand
reversibly
joined to a hydrophilic polymer via an ortho or para-disulfide of a benzyl
urethane linkage.
This linkage when subjected to mild thiolytic conditions is cleaved to
regenerate the
original amine-containing ligand in its unmodified form. The rate of cleavage
can be
z s controlled by steric hinderance of the disulfide in the linkage and/or by
controlling the
thiolytic conditions in vivo. The compounds prior to cleavage of the
dithiobenzyl linkage
are provided with an increased blood circulation lifetime, improved stability
and reduced
immunogenicity.
CA 02368793 2001-10-22
WO 00/64483 PCT/US00/10830
III. Examples
The following examples further illustrate the invention described herein and
are in
no way intended to limit the scope of the invention.
s Materials
All materials were obtained from commercially suitable vendors, such as
Aldrich
Corporation.
Example 1
~ o Synthesis of mPEGDTB-DSPE
mPEG-MeDTB-nitrophenylcarbonate (300 mg, 0.12 mmol, 1.29 eq) was
dissolved in CHC1~ (3 ml). DSPE (70 mg, 0.093 mol) and TEA (58.5 p.l, 0.42
mmol,
4.5 eq) were added to PEG-solution, and was stirred at 50°C (oil bath
temp). After 15
minutes, TLC showed that the reaction didn't go to completion. Then two
portions of
is TEA (10 ~1, and 20 ~1), and few portions of mPEG-MeDTB-nitrophenylcarbonate
(50
mg, 30 mg, 10 mg) were added every after 10 minutes, until the reaction went
to
completion. Solvent was evaporated. Product mixture was dissolved in MeOH, and
1
g of C8 silica was added. Solvent was evaporated again. Product containing C8
silica
was added on the top of the column, and was eluted with MeOH: H20 gradient
zo (pressure), MeOH: H20 = 30: 70, 60 ml; MeOH: H20 = 50: 50, 60 ml; MeOH: H20
= 70: 30, 140 ml (starting material eluted); MeOH: H20 = 75: 25 = 40 ml; MeOH:
H20 = 80: 20, 80 ml (product eluted); MeOH: Hz0 = 85: 15, 40 ml; MeOH: HZO =
90: 10, 40 ml; MeOH = 40 ml; CHC13: MeOH: H20 = 90: 18: 10, 40 ml. Fractions
containing pure product were combined and evaporated to give product as
colorless
zs thick liquid. Tertiary butanol (5 ml) was added to it, lyophilized and the
dried in vacua
over P205 to give product as white fluffy solid (252 mg, 89% yield).
The ortho- and para-DTB-DSPE compounds were purified by silica gel
chromatography (methanol gradient 0-10 % in chloroform, X70 % isolated yield)
and the
structures confirmed by NMR and MALDI-TOFMS. ('H NMR for para conjugate: (d6-
so DMSO, 360 MHz) 8 0.86 (t, CH3, 6 H), 1.22 (s, CHz of lipid, 56H), 1.57 (m,
CHzCHZCOZ, 4H), 2.50 (2xt, CHZCO2, 4H), 2.82 (t, CHZS, 2H), 3.32 (s, OCH3,
3H),
CA 02368793 2001-10-22
WO 00/64483 PCT/US00/10830
26
3.51 (m, PEG, X180 H), 4.07 (t, PEG-CHZOCONH, 2H), 4.11 & 4.28 (2 x dd CHZCH
of glycerol, 2H), 4.98 (s, benzyl-CH2, 2H), 5.09 (m, CHCHZ of lipid), 7.35 &
7.53 (2
x d, aromatic, 4H) ppm. The ortho conjugate differed only in benzyl and
aromatic
signals at 5.11 (s, CHz, 2H), and 7.31 (d, 1H), 7.39 (m, 2H) 7.75(d, 1H) ppm.
s MALDI-TOFMS produced a distribution of ions spaced at equal 44 Da intervals,
corresponding to the ethylene oxide repeating units. The average molecular
weights of
the compounds was 3127 and 3139 Da for para and ortho isomers respectively
(theoretical molecular weight X3100 Da).
The reaction scheme is illustrated in Fig. 2.
Example 2
Synthesis of mPEGDTB-DSPE
A. mPEG-MeDTB-DSPE
This reaction scheme is illustrated in Figs. 4A-4B.
i s mPEG(5K)-OH (40 g, 8 mmol) was dried azeotropically with toluene (total
volume was 270 ml, 250 ml was distilled off by Dean-Stark). Dichloromethane
(100
ml) was added to mPEG-OH. P-nitrophenyl chloroformate (2.42 g, 12 mmol, 1.5
eq),
and TEA (3.3 ml, 24 mmol, 3 eq) were added to PEG solution at 4°C (ice
water), while
taking precautions against moisture. Light yellow TEA hydrochloride salt was
formed.
ao After 15 minutes cooling bath was removed, and the reaction mixture was
stirred at
room temperature overnight. TLC showed (CHC 13: MeOH: Hz0 = 90: 18: 2) that
the
reaction was complete. Solvent was evaporated. The residue was dissolved in
ethyl
acetate ('50°C). TEA hydrochloride salt was filtered off and washed
with warm ethyl
acetate. Solvent was evaporated and the product recrystallized with
isopropanol (thres
as times). Yield: 38.2 g (92%). 'H NMR (DMSO-db, 360 MHz) 8 3.55 (s, PEG,
450H);
4.37 (t, PEG-CH2, 2H); 7.55 (d, C6H5, 2H); 8.31 (d, C6FI5, 2H).
1-Amino-2-propanol (l.l ml, 14.52 mmol, 3 eq), and TEA (2.02 ml, 14.52
mmol, 3 eq) were added to mPEG (5K)-nitrophenyl carbonate (25 g, 4.84 mmol) in
DMF (60 ml) and CHZCIz (40 ml). It was a yellow clear solution. The reaction
3o mixture was stirred at room temperature for 30 minutes. TLC (CHC13: MeOH =
90:
10) showed that the reaction went to completion. Solvent (dichloromethane) was
CA 02368793 2001-10-22
WO 00/64483 PCT/US00/10830
27
evaporated. Isopropanol (250 ml) was added to the product mixture in DMF (60
ml).
Product precipitated immediately, and then recrystallized with iPrOH (three
times).
Yield: 22.12 g (90%). 'H NMR (DMSO-db, 360 MHz) 8 .98 (d, CH3CH(OH)CH2,
3H); 3.50 (s, PEG, 180H); 4.03 (t, PEG-CH2, 2H); 4.50 (d, CH3CHOH, 1H); 7.0
(t,
s mPEG-OCONH).
mPEG(SK)-urethane-2-methyl propanol (22.12 g, 4.34 mmol) was dried
azeotropically with toluene (45 ml). Dichloromethane (60 ml) was added to it.
Methane sulfonyl chloride (604.6 pl, 7.81 mmol, 1.8 eq) and TEA (3.93 ml,
28.21
mmol, 6.5 eq) were added to mPEG-solution at 0°C while maintaining
stirring and
o taking precautions against moisture. After 30 minutes, cooling bath was
removed, and
the reaction mixture was stirred at room temperature for 16 h. Solvent was
evaporated.
Ethyl acetate was added to remove TEA salts. The product was recrystallized
with
isopropanol (three times). Yield: 20.27 g (90%). 'H NMR (DMSO-d6, 360 MHz) 8
1.27 (d, CH3CHOSOZCH3, 3H); 3.162 (s, CH302SOCH, 3H); 3.50 (s, PEG, 180H);
is 4.07 (t, PEG-CH2, 2H); 4.64 (q, CH3CHOH, 1H); 7.43 (t, mPEG-OCONH).
mPEG(SK)-urethane-2methyl-methane sulfone ( 10.27 g, 1.98 mmol) was dried
azeotropically with toluene (20 ml, each time). Sodium hydride (377 mg, 9.4
mmol,
4.75 eq) was added in anhydrous toluene (60 ml) at 0°C (in ice water).
After 5
minutes, triphenylmethanethiol (3.92 g, 14.6 mmol, 7.15 eq) was added to the
solution.
2 o After 10 minutes, mPEG-urethane-2methyl-methane sulfone ( 10.27 gm, 1.98
mmol)
was added to the reaction mixture. It became a yellow solution. After 45
minutes,
TLC (CHC13: MeOH: H20 = 90: 18: 2) showed that the reaction went to
completion.
Acetic acid (445.57 pl, 7.42 mmol, 3.75 eq) was added to the reaction mixture
to
neutralize excess of sodium hydride. The solution became thick and whitish.
Solvent
zs was evaporated and the solid was recrystallized with ethyl acetate (30 ml)
and
isopropanol (70m1). The product mixture did not dissolve completely, while
precipitate
filtered off. Then the product mixture was recrystallyzed with
isopropanol/tert-butyl
alcohol (100m1/20m1). Yield: 8.87 g (84%). 1H NMR (DMSO-db, 360 MHz) 8 .74 (d,
CH3CHSC(C6H5)3, 3H), 3.50 (s, PEG, 180H), 4.0 (t, PEG-CHZ, 2H), 4.64 (q,
3o CH3CHOH, 1H); 7.49 (t, mPEG-OCONH); 7.20-7.41 (m, SC(C6H5)3, 15H).
mPEG(SK)-urethane-2methyl-triphenylmethanethiol (8.87 g, 1.65 mmol) was
CA 02368793 2001-10-22
WO 00/64483 PCT/US00/10830
28
dissolved in TFA/CHZC12 (lOml/lOml) at 0°C. Under vigorous stirring,
methoxy
carbonylsulfenyl chloride (185.5 pl, 1.99 mmol, 1.2 eq) was added to the
solution. The
reaction mixture was stirred at room temperature for 15 minutes. TLC (CHCl3:
MeOH
= 90: 10) showed that the reaction was complete. Solvents were evaporated. The
s product mixture was recrystallized with isopropanol:tert-butyl alcohol (80
ml: 20 ml)
two times. Tertiary butanol (5 ml) was added to the product, which was then
lyophilized and dried in vacuo over P205 to give product as white fluffy solid
(8.32g,
97 % yield). 'H NMR (DMSO-db, 360 MHz) 8 1.17 (d, CH3CHSSCOOCH3, 3H); 3.42
(s, PEG, 180H); 3.84 (s, CH30COSSCH, 3H); 4.05 (t, mPEG-CH2, 2H); 7.38 (t,
~o mPEG-OCONH, 1H).
mPEG(SK)-urethane ethyl(methyl)dithiocarbonyl methoxide (8.32 g, 1.6 mmol)
was dissolved in dry methanol (20 ml), and chloroform (2.5 ml). A solution of
mercapto benzyl alcohol (592 mg, 4 mmol, 2.5 eq) in dry methanol (2 ml) was
added to
the PEG-solution. The reaction mixture was stirred at room temperature for 18
h.
~s Solvent was evaporated, product mixture was recrystallized with ethyl
acetate/isopropanol, 30 ml/ 100 ml (3 times). NMR showed -16 % product was
formed.
So, another portion of mercapto benzyl alcohol (322 mg, 2.18 mmol, 1.8 eq) in
MeOH
(2m1) was added dropwise to the product mixture in MeOH/CHC13 (24 ml/1 ml) at
0°C
(ice water). After addition (' 10 minutes) completion, ice bath was removed,
and the
2 o reaction mixture was stirred at room temperature for 24 h. TLC (CHC 13:
MeOH: H20
= 90: 18: 2) showed that the reaction was complete. Solvent was evaporated,
and then
product mixture was recrystallized with ethyl acetate/isopropanol, 30 ml/100
ml.
Yield: 7.25 g, (94%). 'H NMR (DMSO-d6, 360 MHz) 8 1.56 (d,
CH3CHSSC6HSCHZOH, 3H); 3.29 (CH30-PEG, 3H); 3.50 (s, PEG, 450H); 4.03
as (t, mPEG-CH2, 2H); 4.46 (d, HOCHZC6H5, 2H); 5.16 (t, HOCHZC6H5, 1H);
7.30 (d, C6H5, 2H); 7.40 (br t, mPEG-OCONH, 1H); 7.50 (d, C~15, 2H).
mPEG(SK)-urethane-ethyl(methyl)-dithiobenzyl alcohol (6.75 g, 1.27 mmol)
was dissolved in CHC13 (30 ml), P-nitrophenyl chloroformate (513 mg, 2.54
mmol, 2
eq) was added to it at 0°C (ice water). After 5 minutes triethylamine
(531 ~1, 3.81
3o mmol, 3 eq) was added. After 30 minutes ice bath was removed, and the
reaction
mixture was stirred at room temperature overnight. Solvent was evaporated. The
CA 02368793 2001-10-22
WO 00/64483 PCT/US00/10830
29
product mixture was dissolved in ethyl acetate. TEA salt was filtered off, and
then
solvent was evaporated. Then the product mixture was recrystallized with ethyl
acetate/isopropanol, 30 ml/100 ml (three times). Yield: 6.55 g (94%). 'H NMR
(DMSO-db, 360 MHz) 8 1.17 (d, CH3CHSSC6H5, 3H); 3.24 (CH30-PEG, 3H); 3.40 (s,
s PEG, 180H); 4.03 (br t, mPEG-CHZ, 2H); 5.28 (S, C6HSCHzOCO, 2H); 7.45-8.35
(m,
C6H5)2~ 8H)
mPEG-MeDTB-nitrophenylcarbonate (766 mg, 0.14 mmol, 1.29 eq) was
dissolved in CHC13 (5 ml). DSPE (70 mg, 0.093 mol) and TEA (58.5 pl, 0.42
mmol,
4.5 eq) were added to PEG-solution, and was stirred at 50°C (oil bath
temp). After 20
o minutes, TLC showed that the reaction didn't go to completion. More mPEG-
MeDTB-
nitrophenylcarbonate (total 1239 mg, 0.23 mmol, 2.47 eq) and 1-
hydroxybenztriazole
(HOBt) (25 mg, 0.19 mmol, 2 eq) were added. After 20 minutes, TLC (CHC 13:
MeOH: H20 = 90: 18: 2, with molybdenum and ninhydrin) showed that the reaction
was complete. Solvent was evaporated. Product mixture was dissolved in warm
(42°C)
~s ethyl acetate. It was a cloudy solution (TEA salt precipitated). The
solution was
filtered, and solvent was evaporated. MeOH, and 2 g of C8 silica was added to
the
product mixture. Solvent was evaporated again. Product containing C8 silica
was
added on the top of the column, and was eluted with MeOH: H20 gradient
(pressure),
MeOH : H20 30: 70, 100 ml; MeOH Hz0 50: 50, 100 ml; MeOH HZO 70: 30, 250 ml
zo (starting material eluted); MeOH H20 75: 25 = 40 ml; MeOH Hz0 80: 20, 200
ml
(product eluted); MeOH = 100 ml; CHC 13: MeOH: H20 = 90: 18: 2, 100 ml; CHC
13:
MeOH Hz0 = 75: 36: 6, 100 ml. Fractions containing pure product were combined
and evaporated to give product as colorless thick liquid. Tertiary butanol (5
ml) was
added to it, lyophilized and then dried in vacuo over P205 to give product as
white
2s fluffy solid (467 mg, 83% yield). 'H NMR (DMSO-d6, 360 MHz) 8 0.83 (d,
2(CH3),
3H); 1.16 (d, CH3CHSSC6H5, 3H); 1.21 (s, 28(CH2, 56H); 1.47 (br m, CHZCHZCO,
4H); 2.23 (2 x t, CHZCHzCO, 4H); 3.50 (s, PEG, 180H); 4.04 (br t, mPEG-CH2,
2H);
4.05 (traps d, POQCHZCHCHZ, 1H); 4.24 (cis d, P04CHZCHCH2, 1H); 4.97 (s,
C6HSCHZOCO-DSPE, 2H); 5.03 (br s, (P04CHZCH, 1H); 7.32 (d, C6II5, 2H); 7.53
(d,
3o C6H5, 2H); 7.52 (br s, mPEG-OCONH, 1H). MALDI-TOFMS produced a bell shaped
distribution of ions spaced at equal 44 Da intervals, corresponding to the
ethylene oxide
CA 02368793 2001-10-22
WO 00/64483 PCT/US00/10830
repeating units. The average molecular mass of the conjugate and mPEG-thiol
(mostly
cleaved disulfide) is 6376 and 5368 Da (theoretical molecular mass -6053, and
5305
Daltons).
s B. mPEG-ethylDTB-DSPE
mPEG-urethane ethyl(ethyl)dithiocarbonyl methoxide (2 g, 0.90 mmol) was
dissolved in dry methanol (8 ml). At the beginning the solution was cloudy,
but after 5
minutes it became a clear solution. Mercaptobenzyl alcohol (265.2 mg, 1.79
mmol, 2
eq) was added to the PEG-solution. The reaction mixture was stirred at room
~o temperature for 30 hours. Ether (70 ml) was added to the reaction solution
to
precipitate the product, and kept at 4°C overnight. The white solid was
filtered and
recrystallized with ethyl acetate/ether, 30 ml/70 ml. Yield : 1.96 g, (94%).
'H NMR
(DMSO-d6, 360 MHz) 80.86 (d, CH3CHZCHSSC6HSCHZOH, 3H); 1.42 (p,
CH3CHZCHSSC6HSCHZOH, 1H); 1.64 (p, CH3CHZCHSSC6HSCHZOH, 1H); 3.51 (s,
is PEG, 180H); 4.03 (t, mPEG-CHz, 2H); 4.47 (d, HOCHzC6H5, 2H); 5.20 (t,
HOCHZC6H5, 1H); 7.31(d, C6H5, 2H); 7.42 (br t, mPEG-OCONH, 1H); 7.49 (d,
C6FI5,
2H) .
N-hydroxy-s-norbornene-2,3-dicarboxylic acid imide (HONB) (48 mg, 0.269
mmol) was added to DSPE (55 mg, 0.073 mmol) in CHC13 (3 ml) at 50°C
(oil bath
zo temperature). After 3-4 minutes it became a clear solution. Then mPEG-EtDTB-
nitrophenylchloroformate (334 mg, 0.134 mmol) was added, followed by
triethylamine
(TEA, 45 ~,1, 0.329 mmol). After 20 minutes TLC (CHCl3 : MeOH : Hz0 = 90 : 18
2) showed that the reaction went to completion (molybdenum and ninhydrin
sprays).
Solvent was evaporated. Product mixture was dissolved in methanol, mixed with
C8
as silica (lg) and striped of the solvent by rotary evaporation. The solid
residue was
added on the top of the C8-column, which was then eluted with MeOH : H20
gradient
(pressure), MeOH : HZO = 30 : 70, 60 ml; MeOH : H20 = 50 : 50, 60 ml; MeOH
H20 = 70 : 30, 140 ml; MeOH : H20 = 75 : 25 = 140 ml (starting material
eluted);
MeOH : H20 = 80 : 20, 80 ml; MeOH : H20 = 90 : 10, 140 ml (product eluted);
3o MeOH = 40 ml; CHC13 : MeOH : Hz0 = 90 : 18 : 10, 40 ml. Fractions
containing
pure product were combined and evaporated to give product as colorless thick
liquid.
CA 02368793 2001-10-22
WO 00/64483 PCT/US00/10830
31
Tertiary butanol (5 ml) was added, lyophilized and then dried in vacuo over
P205 to
give product as white fluffy solid (175 mg, 78% yield). 'H NMR (DMSO-d6, 360
MHz) b 0.85 (d, 2(CH3), 6H; d, CH3CHSSC6H5, 3H); 1.22 (s, 28(CHz), 56H); 1.49
(br
m, CHZCHZCO, 4H); 2.24 (2 x t, CHZCHZCO, 4H); 3.50 (s, PEG, 180H); 4.04 (br t,
s mPEG-CHz, 2H); 4.08 (trans d, P04CHZCHCHz, 1H); 4.27 (cis d, POQCHZCHCHZ,
1H); 4.98 (s, C6HSCHZOCO-DSPE, 2H); 5.06 (br s, (P04CHZCH, 1H); 7.34 (d,
C6FI5,
2H); 7.53 (d, C6H5, 2H); 7.55 (br s, mPEG-OCONH, 1H).
Example 3
o Synthesis of mPEGDTB-nitrophenylchloroformate
A. Procedures for synthesis of 1-(mercaptomethyl) ethylammonium chloride
1. 2-Amino-1-methylethyl hydrogen sulfate. 1-Amino-2-propanol (22.53g, 0.3
mol) was vigorously stirred in an ice bath. Sulfuric acid (16.10 ml, 0.3 mol)
was added
very slowly, over the course of one hour. Thick vapors and a very viscous
solution
i s were formed in the flask. After addition was complete, the reaction was
heated
between 170°C and 180°C, under reduced pressure, connected to
the house vacuum.
Upon heating, the reaction turned light brown. After all water was removed
(approximately 1 hour) it was allowed to cool to room temperature. Upon
cooling a
brown, glassy solid was formed which would crystallize when triturated with
methanol.
zo It was dissolved in water (50 ml) at 60°C. Enough warm methanol was
added to make
the solution 80 % methanol. Upon cooling, crystals formed which were then
filtered
and dried over P205. Yield: 17.17g (37 %). 'H NMR (D6-DMSO): 8 1.16 (d, CH3,
3H); 8 2.78 (dd, NH3-CH2, 1H); 8 2.97 (dd, NH3-CH2, 1H); 8 4.41 (m, CH-OS03,
1H); 8 7.69 (s, H3N, 3H). Melting point: 248°-250°C (lit:
250°C)
zs 2. 5-Methylthiazolidine-2-thione. 2-Amino-1-methylethyl hydrogen sulfate
(23.03 g, 148 mmol) and carbon disulfide (10.71 ml, 178 mmol, 1.2 eq.) were
stirred
in a 250 ml round-bottom-flask in 50% aqueous ethanol (40 ml). To this, sodium
hydroxide ( 13.06 g, 327 mmol, 2.2 eq. ) in 50 % aqueous ethanol (50 ml) was
added
drop-wise, very slowly. Upon addition of sodium hydroxide, all starting
materials
3o dissolved and the solution turned orange. The reaction was refluxed
(85°C) for 40
minutes, after which time it turned bright yellow and a thick precipitate was
formed.
CA 02368793 2001-10-22
WO 00/64483 PCT/US00/10830
32
Ethanol was evaporated and then the aqueous solution was warmed and then
filtered
through a Buchner funnel to remove all water-soluble impurities. The remaining
crystals were dissolved in warm ethanol and then warm water was added until
the
solution was 80 % water. The mixture was allowed to cool and then
refrigerated,
s yielding long, needle-like crystals. Yield: 14.64 g (75 %). 'H NMR (D6
DMSO): 8
1.33 (d, CH3, 3H); 8 3.50 (m, R3CH, 1H); b 3.95 (dd, N-CH2, 1H); 8 4.05 (m, N-
CH2, 1H); 8 10.05 (s, NH, 1H). Melting point: 92.5-93.5 (lit: 94-95).
3. 1-(mercaptomethyl)ethylammonium chloride. 5-Methylthiazolidine-2-thione
~o (6.5 g, 49 mmol) was placed in a 250 ml round-bottom-flask. A solution of
aqueous
hydrochloric acid (40 ml, 18 % in H20) was added and the flask was heated in
an oil
bath. The reaction refluxed (120°C) for one week. Three times
throughout the week 1
ml of concentrated hydrochloric acid was added. The reaction was monitored
using
TLC with ethyl acetate as eluent. They were visualized using UV, ninhydrin,
and
is iodine vapors. Through most of the week the reaction was a heterogeneous
mixture,
with the starting material as oil which was denser than water. After one week
the oil
starting material was gone, although still visible on TLC. The reaction was
removed
from heat and allowed to cool to room temperature, and then was refrigerated
to
crystallize starting material. The crystallized starting material was
filtered. Filtrate was
zo evaporated and it was dried over Pz05 and NaOH to remove all water and HCI.
The
crude product was washed with two portions of diethyl ether (50 ml each) to
remove ull
starting material. It was again dried over PZOS. Yield: 2.83 g (45%). 'H NMR
(D6
DMSO): 8 1.33 (d, CH3, 3H); b 2.92 (m, N-CH2, 2H); 8 3.12 (m, SH, 1H); b 3.18
(m, R3-CH, 1H); 8 8.23 (bs, NH3, 3H). Melting point: 80-82°C (lit: 92-
94).
2s The reaction scheme is illustrated in Fig. 5.
B. Synthesis of mPEG-ethyl-DTB-nitrophenylchloroformate
1. 2-Amino-1-ethylethyl hydro~en sulfate. 1-Amino-2-butanol (15 ml, 158
mmol) was vigorously stirred in a 100 ml round-bottom-flask in an ice bath.
Sulfuric
3o acid (8.43 ml, 158 mmol) was added very slowly, over the course of one
hour. Thick
vapors and a very viscous solution were formed in the flask. After addition
was
CA 02368793 2001-10-22
WO 00/64483 PCT/US00/10830
33
complete, the reaction was heated between 170° and 180°C, under
reduced pressure,
connected to the house vacuum. Upon heating, the reaction turned light brown.
After
all water was removed (approximately 1 hour) it was allowed to cool to room
temperature. Upon cooling a brown, glassy solid was formed. It was dissolved
in hot
s water (50 ml) and then placed in the refrigerator overnight. Upon cooling,
crystals
formed which were then filtered and dried over PZOS. Yield: 9.98 g (37%). 'H
NMR
(D6 DMSO): 8 0.87 (t, CH3, 3H); 8 1.51 (q, CH3-CH2, 2H); 8 2.82 (dd, NH3-CHz,
1H); 8 3.00 (dd, NH3-CH2, 1H); 8 4.21 (m, CH-OS03, 1H); 8 7.70 (s, H3N, 3H).
io 2. 5-Ethylthiazolidine-2-thione. 2-Amino-1-ethyl-ethyl hydrogen sulfate
(9.98
g, 59 mmol) and carbon disulfide (4.26m1, 71 mmol, 1.2 eq.) were stirred in a
100 ml
round-bottom-flask in 50% aqueous ethanol (15m1). To this, sodium hydroxide
(5.20
g, 130 mmol, 2.2 eq.) in 50% aqueous ethanol (20 ml) was added drop-wise, very
slowly.
Upon addition of sodium hydroxide, all starting materials dissolved and the
solution
~s turned orange. The reaction was refluxed (85°C) for 40 minutes,
after which time it
turned bright yellow and a thick precipitate was formed. Ethanol was
evaporated and
then the aqueous solution was warmed and then filtered through a Buchner
funnel to
remove all water-soluble impurities. The remaining crystals were dissolved in
warm
ethanol and then warm water was added until the solution was 80 % water. The
zo mixture was allowed to cool and then refrigerated, yielding needle-like
crystals.
Yield: 7.28g (86%). 'H NMR (D6 DMSO): 8 0.88 (t, CH3, 3H); 8 1.66 (in, CH3-
CH2,
2H); 8 3.58 (m, R3CH, 1H); 8 3.93 (m, N-CH2, 2H); S 10.06 (s, NH, 1H). Melting
point: 76-78° (lit: 76.6-76.9).
zs 3. 1-(mercaptoethyl)ethylammonium chloride. 5-Ethylthiazolidine-2-thione
(7.24 g, 50 mmol) was placed in a 250 ml round-bottom-flask. A solution of
aqueous
hydrochloric acid (45 ml, 18 % in H20) was added and the flask was heated in
an oil
bath. Upon heating, the starting material melted, forming, all heterogeneous
mixture.
The reaction refluxed ( 120°C) for one week. Four times throughout the
week 1 ml of
3o concentrated hydrochloric acid was added. The reaction was monitored using
TLC
with ethyl acetate as eluent. They were visualized using UV, ninhydrin, and
iodine
CA 02368793 2001-10-22
WO 00/64483 PCT/US00/10830
34
vapors. Throughout the week the reaction was a heterogeneous mixture, with the
starting material as oil which was denser than water. The reaction was removed
from
heat and allowed to cool to room temperature, and then was refrigerated to
crystallize
starting material. The crystallized starting material was filtered. Filtrate
was
s evaporated and it was dried over Pz05 and NaOH to remove all water and HCI.
The
crude product was washed with two portions of diethyl ether (50 ml each) to
remove
all starting material. It was again dried over Pz05. Yield: 3.66 g (52%). 'H
NMR
(D6 DMSO):
The reaction scheme is illustrated in Fig. 5.
~o
Example 4
Synthesis of mPEGDTB-lipid
1, 2-distereoyl-sn-glycerol (500 mg, 0.8 mmol) was dried azeotropically with
benzene (3 times). Para-nitrophenyl chloroformate (242 mg, 1.2 mmol, 1.5 eq),
is dimethylaminopyridine (DMAP) (10 mg, 0.08 mmol, 0.1 eq), and TEA (334.5
~.1, 2.4
mmol, 3 eq) were added to 1, 2-distereoyl glycerol in CHC13 (5 ml). The
reaction
mixture was stirred at room temperature for 2 h. TLC (Toluene: ethyl acetate =
7: 3)
showed that the reaction was complete. Then the product mixture was extracted
with
10% citric acid to remove dimethylaminopyridine (DMAP), washed with
acetonitrile (3
zo ml, 4 times) to remove excess ofp-nitrophenyl chloroformate. Pure product
was dried
in vacuo over P205. Yield: 557 mg(88 % ). % ). 'H NMR (CHC 13, 360 MHz) 8 0.88
(t,
end CH3, 6H); 1.25 (s, 28xCH2, 56H); 1.58 (m, CHZCHZCO, 4H); 2.34 (2xt, CHZCO,
4H); 4.22 (trans d, CHzOCOC,~H35, 1H); 4.35 (m, OCOOCHZCH, 2H); 4.51 (cis d,
CHZOCOC"H35, 1H); 5.37 (m, OCOOCHZCH, 1H); 7.39 (d, C6FI5, 2H); 8.28 (d,
zs C6H5, 2H).
Ethylene diamine (42 ~1, 0.63 mmol, 5 fold excess), and pyridine (200 ~.1,
were
added in CHC 13 ( 1 ml) . 2-disteroyl-sn-p-nitrophenyl carbonate ( 100 mg,
0.13 mmol)
was dissolved in CHC13 ( 1 ml) and added dropwise to ethylene diamine solution
with a
pastuer pipette at 0°C (ice water) and continued overnight (16 h). TLC
(CHC13:
so MeOH: HZO 90: 18: 2, and CHC13: MeOH = 90: 10) showed that the reaction was
complete. Solvent was evaporated to remove pyridine. Then the product mixture
was
CA 02368793 2001-10-22
WO 00/64483 PCT/US00/10830
dissolved in CHC 13, loaded onto the column (Aldrich, Silica gel, 60°A,
200-400 mesh),
and eluted with CHC 13: CH3COCH3, and CHC 13: MeOH gradient, CHC 13:
CH3COCH3 = 90: 10, 60 ml (upper spot eluted); CHC 13: NeOH = 90: 10, 60 ml
(product eluted). Fractions containing pure product were combined and
evaporated.
s Tert-butanol was added and dried in vacuo over P205. Yield: 64 mg (75 % ).
'H NMR
(DMSO-d6, 360 MHz) 8 .83 (t, end CH3, 6H); 1.22 (s, 28xCH2, 56H); 1.51 (m,
CHZCHZCO, 4H); 2.25 (2xt, CHZCO, 4H); 2.83 (m, HzNCH2CH2NH, 2H); 3.21 (m,
HZNCHZCHZNH, 2H); 4.10-4.14 (m & cis d, COOCHzCHCH2, 4H); 5.17 (m,
OCOOCHZCH, 1H); 7.78 (m, HzNCH2CHZNH, 2H).
io mPEG-MeDTB-nitrophenylchloroformate (400 mg, 0.162 mmol, 2.2 eq) was
dissolved in CHC13 in (2 ml). 1,2-steroyl-sn-ethylene amine (51 mg, 0.075
mmol) and
TEA (37 ~1, 0.264 mmol, 3.52 eq) were added to the solution. Then the reaction
mixture was stirred at 45°C for 20 minutes. TLC (CHC13: MeOH: H20 = 90:
18: 2,
and CHC 13: MeOH = 90: 10) showed that the reaction went to completion.
Solvent
~s was evaporated. The product mixture was dissolved in methanol. 2 g of C8
silica was
added and then solvent was evaporated. C8 silica containing product mixture
was
added on the top of the C8 column ((Supelco, Supel clean. Lot no. SP0824), and
was
eluted with MeOH: Hz0 gradient (pressure), MeOH: H20 = 60: 40, 40 ml; MeOH:
HZO = 70: 30, 80 ml (starting material eluted); MeOH: HZO = 80: 20, 40 ml;
MeOH:
a o Hz0 = 90: 10 = 20 ml; CHC 13: MeOH: H20 = 5: 80: 15, 20 mi; CHC 13: MeOH:
HZO = 90: 18: 10, 40 ml (product eluted). Fractions containing pure product
were
combined and evaporated to give product as colorless thick liquid. Tertiary
butanol (5
ml) was added and the solution was lyophilized and then dried in vacuo over
PZOS to
give product as white solid (200 mg, 89% yield). 'H NMR (DMSO-d6, 360 MHz) 8 8
2s .83 (t, end CH3, 6H); 1.22 (s, 28xCH2, 56H); 1.48 (m, CHZCHZCO, 4H); 2.25
(2 x t,
CHZCO, 4H); 3.10 (m, HNCHZCHZNH, 4H); 3.50 (s, PEG, 180H); 4.04 (t, mPEG-
CH2, 2H); 4.09 (trans d, COOCHzCHCH2, 1H); 4.25 (cis d, COOCHzCHCH2, 1H);
4.98 (s, C6HSCHzOCO, 2H); 5.23 (m, COOCHzCHCHz, 1H); 7.18 (m,
NHCHZCHzNH, 2H); 7.33 (d, C6H5, 2H); 7.38 (m, mPEG-OCONH, 1H); 7.52 (d,
3o C6H5, 2H).
CA 02368793 2001-10-22
WO 00/64483 PCT/CJS00/10830
36
The reaction scheme is illustrated in Fig. 6A.
Example 5
In vitro Cleavage of mPEG-DTB-DSPE Compound
s Ortho-mPEG-DTB-DSPE and para- mPEG-DTB-DSPE (prepared as described in
Example 1) were added to a buffered aqueous solution (pH 7.2) in the presence
and
absence of 150 q,M cysteine. Disappearance of the conjugates was monitored by
HPLC
(Phenomenex Cg Prodigy, 4.6 x 50 mm column, detection at 277 nm, mobile phase
methanol / water 95:5 with 0.1 % trifluoroacetic acid at 1 mL/min). The
results are
to illustrated in Fig. 7A where the ortho-conjugate is represented by the open
circles and
the para-conjugate by the open squares.
Example 6
In vitro Cleavage of o- and p-mPEGDTB-DSPE Compound in Liposomes
is A. Liposome Preparation
The lipids partially hydrogenated phosphatidylcholine (PHPC), cholesterol and
ortho- or para-mPEG-DTB-DSPE (prepared as described in Example 1, mPEG
MW =1980 Daltons) were dissolved in a 95:5:3 molar ratio, respectively, in a
suitable
organic solvent, typically cholorform/methanol in a l: l or 1:3 ratio. The
solvent was
ao removed by rotary evaporation to form a dried lipid film. The film was
hydrated with
aqueous buffer to from liposomes that were sized via extrusion to an average
diameter
of 120 nm.
B. In vitro Characterization
The ~liposomes were incubated in phosphate buffered saline, pH 7.2, containing
S
2s mM EDTA at 37°C in the presence of 150 ~.M cysteine. Disappearance
of the
conjugates was monitored by HPLC (Phenomenex C8 Prodigy, 4.6 x 50 mm column,
detection at 277 nm, mobile phase methanol / water 95:5 with 0.1 %
trifluoroacetic acid
at 1 mL/min). Results are shown in Fig. 7B where the liposomes comprising the
ortho-
conjugate are represented by the solid circles and liposomes comprising the
para-
so conjugate by the solid squares. The open circles and the open squares
correspond to
ortho-mPEG-DTB-DSPE and para-mPEG-DTB-DSPE in micellar form (discussed
above in Example 5, Fig. 7A).
:~y-; ,~.- .-:~ ~ ,.v."r .. ~.:>rx-:-~;~.~- ~c~.e~aerrrr~ .,e~.c:- --
;a..~.__., r. ~ ..._.p.~:.-:;a,~a,.-~~.~.,~""",~
" ''.001 2:18PM IOTR PI LHW GROUP ~ N0.402 US 00001083
o5-os-2oo1
~ ' CA 02368793 2001-10-22
' . E7CSn'lp~C 7
In vitro Cleavage of o- and p-mPEGD DSPE Compound in Liposomes
A. Liposome Preparation , _
The lipids dioleoyl phosphatidylethanol (DOPE) and ortho- or para- mPEG-
s DTI-DSPE (prepared as described in Exampl 1, mPEG MW =1980 Daltons) were
dissolved a 97:3 molar ratio in chloroform/tne of ~1:1. The solvent was
removed by
rotary,evaporation to form a dried lipid elm. a lipid film was hydrated with
an
aqueous solution conraining 30 mM each of th fluorophorcs p xylene-his-
pyridinium
bromide and trisodium 8-hydroxypyrenetrisulf hate. was hydrated with aqueous
buffer to ;. .
io , from liposomes that were sized via extrusion tq an average diameter of
100 nm.
B. ~Tn vitro ~lharacterization
~ ; , .. The liposomes.were incubated in HEPESi buffer, pH 7.2, at 37°C
in the presence of
cysteine at concentrations of IS ~M, 150 ~M, 00 ~,cM and 1.5 mM. Percent of
released
15 dye was determined as the increase fn sample luoreseenee (J~~=5x2 nm, ~=413
nni - '
pH-independent isobestic point) over that of th preincubation sample (zero
release)
,normalized to the increase in fluorescence ob d after lysis of preincubatioa
sample
with 0.2% Triton X-100 (I00% release) (Kirp tin, D. et al., F~BSLetters,
x$$:115-lI8
t y ~ ! ~ (1996)). Results at various cysteine concentra~oas for ' osome co
the ortho-
' hP
2o compound are shown in Fig. 8A and for the fo thepara-eompoutid are shown in
Fig.
8B. : '. - '.'~
Examp~e $
In vivo Characterization o Liposomes comprising
;2s, . mP'EG-DTB-DS Compownd
'~ ' ! ' ~ ~ A. Liposome Preparation
The lipids partially hydrogenated phosp tidylcholiae (PHPC), cholesterol and
para-mPEG-DTB laSPB {prepared as describ d in Example 1, mPEG MW=1980
. Daltons) were dissolved in a 55:40:5 mole p~ceat ratio res eetivel i
p y, n an organic
3o solvent. The solvent was removed by rotary evaporation to form a dried
lipid film.
The film was hydrated with aqueous buffer ontaining diethylene trianline
pentacetic
~ ' '', acid (DTPA) to form liposomes. After do izing the liposomes to an
average .
r , ..
37
Empfanosteit 6.Juni 0:13 AMENDED SHEET
i_ . E
-..:. ....- _~ . . _. _ _
~" .. ~:.."~"t,
'" r ''901 2:19PM IOTR PI Lf~.J GROUP ~' 402 [~S 00001083C
05-06-2001
CA 02368793 2001-10-22
diameter of 120 nm unentrapped DTPA was re oved and In"' was added to the
external
medium. The Iiposonzes were incubated for a time suffcieat for In"' to cross
the lipid
i ~ ; ;,~ bilayer and chelate with DTPA.
.. .
B. In viva Administration . ,.
Mice were divided into two study groups The Iiposome composition described
above was injected irno all test animals. One ~roup of the test animals also
received a
200 p.L, injection of 200 mM cysteiue at I, 3 ~ 5 hours post liposome
injection. The
y ; t ~ ~ other test group received an inEjection of saline t the same time
points. Liposome
1 o content in the blood was deterrmined by monito ' blood samples for Inl" ,
The resalLs
are shown in Fig. 10. ~ ~ ~ "
Although the invention has been describe with respect to particular
embodiments,
it will be apparent to those sidlled in the art th t various changes and
modifications can
be made without departing from the invenrion.
v:~f; - . , .. .
~'j : I, '
i .
i~ ~ ~ :. ~ , . .
,. .
..
y: v ~ . . ~~ . ,
38 i
.',~ ; ; ,
Emafangsteit 6.Juni 0.13 AMENDED SHEET
c