Note: Descriptions are shown in the official language in which they were submitted.
CA 02369005 2001-09-27
WO 00/59878 PCT/US00/08840
INHIBITORS OF LFA-1 BINDING TO ICAMs
AND USES THEREOF
BACKGROUND
The leukocyte function-associated antigen (LFA-1, CDlla/CD18) is a
leukocyte-specific (3z integrin that participates in cell/cell adhesion.
Binding activity
of LFA-1 is essential to leukocyte extravasation from circulation to a site of
injury in
an inflammatory response. Three principle ligands are known to bind LFA-1,
ICAM-
1, ICAM-2, and ICAM-3, which are intercellular adhesion molecules that play an
important role in localizing leukocyte adhesion to endothelial cells at a site
of injury.
ICAM-4 and ICAM-5 have also been reported to bind LFA-1.~ Most leukocytes
constitutively express LFA-1, but ligand binding requires activation believed
to
induce a conformational change and increased avidity ligand binding. For
example,
ICAM-1 is normally expressed at low levels on the endothelium, however, injury-
1 S induced inflammatory mediators promote enhanced surface expression in
cells at the
site of the injury which, in turn, promotes localized leukocyte adhesion
through
binding with activated LFA-1.
The structure of LFA-1 includes distinct intracellular and extracellular
domains that are believed to participate and/or regulate ICAM binding. Of
particular
interest is a region in the aL chain of approximately 200 amino acids,
designated the I
domain, that is found in all ~i2 integrins, as well as many other proteins.
Evidence
suggests that the I domain is essential to LFA-1 binding to ICAM-1 and 3. For
example, anti-LFA-1 blocking monoclonal antibodies have been mapped to
epitopes
within the I domain. In addition, recombinant I domain polypeptide fragments
have
been shown to inhibit integrin-mediated adhesion and bind ICAM-1. Within the I
domain of LFA-1 (and other proteins) is a single metal ion dependent adhesion
site
(MIDAS) that preferentially binds manganese or magnesium ions. Binding of
either
cation is required for ligand interaction and is believed to induce
conformational
changes in LFA-1 necessary for binding. Cation binding may therefore be a
regulatory mechanism that responds to changes in the extracellular leukocyte
environment. This hypothesis is supported by the observation that calcium ion
CA 02369005 2001-09-27
WO 00/59878 PCT/LTS00/08840
binding actually inhibits LFA-1 interaction with ICAM-1. Indeed, it has been
proposed that an inactive LFA-1 conformation results from calcium binding, and
that
replacement of the calcium ion with a manganese or magnesium ion is a step
required
for LFA-1 activation [Griggs, et al., J. Biol. Chem. 273:22113-22119 (1998)].
Other
factors have also been shown to induce LFA-1 activation, including T cell
receptor
engagement, cytokine stimulation, and in vitro PMA stimulation.
In practical terms, the identification of LFA-1/ICAM binding sites
provides targets to modulate leukocyte inflammatory responses. Numerous
antibodies
have been isolated that are capable of inducing LFA-1 activation [see, for
example,
Landis, et al., J. Cell Biol. 120:1519-1527 (1993)] or, for example,
preventing ICAM-
1 interaction [see for example, Randi and Hogg, J. Biol. Chem. 269:12395-12398
(1994)]. The previous identification of anti-LFA-1 activating antibodies that
recognize multiple and distinct extracellular epitopes suggests the existence
of more
than one regulatory region, presumably independent of cytoplasmic signaling.
Localization of LFA-1 sites that bind ICAM-1 has been investigated through use
of
chimeric LFA-1 a subunit proteins comprising human and murine components
[Huang and Springer, J. Biol. Chem. 270:19008-19016 (1995)]. Studies have
indicated that residues that coordinate canon binding and residues proximal to
the site
are essential for binding ICAM-1 at a relatively flat interface. More precise
delineation of the extracellular regulatory regions) and the contact points
for ICAM-1
binding will permit design of efficient modulators.
Thus there exists a need in the art to precisely identify regulatory
regions for proteins that participate in inflammatory responses, and in
particular LFA-
1 and ICAMs that bind LFA-1. Determining the tertiary (or quartenary)
structure of a
protein can identify potential regulatory regions to permit the rational
design of
biologically compatible small molecules for therapeutic and prophylactic
intervention
for inflammatory disorders. There further exists a need in the art to identify
compounds that can inhibit LFA-1 binding to ICAMs that can be used in the
treatment
of inflammatory disorders.
2
CA 02369005 2001-09-27
WO 00/59878 PCT/US00/08840
SUMMARY OF THE INVENTION
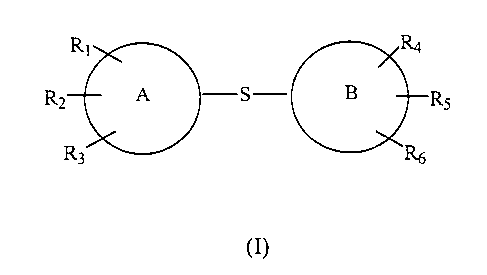
The present invention is directed to compounds that bind to a novel
regulatory site in the I domain of LFA-1, and thereby inhibit LFA-1 binding to
ICAMs
that bind LFA-1. The present invention therefore also provides methods to
regulate
S leukocyte adhesion to endothelial cells. Compounds of the invention are
useful for
the treatment of pathologies, such as those associated inflammatory diseases,
autoimmune diseases, tumor metastasis, allograft rejection and reperfusion
injury. In
particular, the present invention is directed to diaryl sulfides of general
structural
formula (I), a pharmaceutically acceptable salt, or prodrug thereof, and to
the use of
diaryl sulfides, and particularly compounds of formula (I), to inhibit LFA-1
binding to
an ICAM that binds LFA-1.
Rt Ra
RZ A S B R5
R3
(I)
wherein A and B, independently, are aryl groups selected from the group
consisting of
5- and 6-membered aromatic rings, including, but not limited to, phenyl,
thienyl,
furyl, pyrimidinyl, imidazolyl, pyrazolyl, pyridyl, pyrazinyl, pyrrolyl, and
pyridazinyl;
R,, RZ and R;, independently, are selected from the group consisting of
hydrogen,
-Ra, wherein Ra is hydrogen or an alkyl group containing one to six saturated
straight
or branched chain carbon atoms (C,_6 alkyl),
_O_Ra~
-halo, wherein halo is Cl, F, Br, or I,
-NRbR~, where Rb and R~, independently, are H, C,_6 alkyl, or -CH,-aryl,
CA 02369005 2001-09-27
WO 00/59878 PCT/US00/08840
-NO,,
-C(=O)Ra
-CN,
-perfluoroR~, such as trifluoromethyl,
-N-C(=O)Ra,
-(CHz)~-NRbR~, wherein n is an integer 1 to 6,
a 5- or 6-membered heterocyclic ring, either aliphatic or aromatic, containing
one or
more of O, N, or S, optionally substituted, such as morpholino, and
-S-aryl, wherein aryl is a S- or 6-membered aromatic ring, optionally
substituted;
and R~, RS and R6, independently, are selected from the group consisting of
hydrogen,
-O-Ra,
-halo,
_NRbR~,
-NOz
C( O)~
-CN,
-perfluoroRa,
-N-C(=O)Ra,
-(CHz)n-NRbR~, and
-a 5- or 6-membered heterocyclic ring, aliphatic or aromatic, containing one
or more
of O, N, or S, and optionally substituted,
-S-aryl, or wherein
R~ and RS are taken together to form a 5- or 6-membered aromatic ring,
optionally
containing one or more of O, N, or S in the ring, optionally substituted.
Examples of novel negative regulators of LFA-1 binding to ICAMs,
include, but are not limited to the compounds presented in Table I.
4
CA 02369005 2001-09-27
WO 00/59878 PCT/LTS00/08840
Table I
Exemplary Negative Regulators
3-Chloro-4-(2-chlorophenylsulfanyl)-phenylamine hydrochloride
4-Nitro-2-chlorophenyl-(2',3'-dichlorophenyl)-sulfide
3-Chloro-4-(2-naphthylsulfanyl)-phenylamine hydrochloride
3-Chloro-4-(2,3-dichlorophenylsulfanyl)-phenylamine hydrochloride
3-Chloro-4-(2,4,5-trichlorophenylsulfanyl)-phenylamine hydrochloride
3-Chloro-4-(2,4-dichlorophenylsulfanyl)-phenylamine hydrochloride
4-(Benzothiazol-2-ylsulfanyl)-3-chloro-phenylamine
3-Chloro-4-( 1-chloro-naphthalen-2-ylsulfanyl)-phenylamine
3-Methoxy-4-(2,3-dichlorophenylsulfanyl)-phenylamine
5-Amino-2-(2,3-dichlorophenylsulfanyl)-acetophenone hydrochloride
4-(2,3-dichlorophenylsulfanyl)-phenylamine
3-Chloro-4-(1-naphthylsulfanyl)-phenylamine hydrochloride
3-Methyl-4-(2,4-dichlorophenylsulfanyl)-phenylamine hydrochloride
1-Acetamido-3-chloro-4-(2,3-dichlorophenylsulfanyl)-benzene
4-Methylamino-2,2',4'-trichlorodiphenylsulfide
3-Bromo-4-(2,4-dichlorophenylsulfanyl)-phenylamine hydrochloride
3-Hydroxy-4-(2,3-dichlorophenylsulfanyl)-phenylamine hydrochloride
6-Chloro-5-(2,4-dichlorophenylsulfanyl)-1 H-benzimidazole
4-Amino-2-chlorophenyl-(2'4'-dimethylphenyl)-sulfide hydrochloride
2,5-Dichloro-4-(2,4-dichlorophenylsulfanyl)-phenylamine hydrochloride
4-Amino-2-chlorophenyl-(2'-methyl-4'-chlorophenyl)-sulfide
4-Amino-2-chlorophenyl-(2',4'-difluorophenyl)-sulfide hydrochloride
4-Amino-2-chlorophenyl-(2',4',6'-trichlorophenyl)-sulfide
4-Amino-2-chlorophenyl-(2'-amino-4'-chlorophenyl)-sulfide
4-Amino-2-chlorophenyl-(2'-chloro-4'-nitrophenyl)-sulfide
4-Amino-2-chlorophenyl-(2'-nitro-4'-chlorophenyl)-sulfide
4-Amino-2-chlorophenyl-(3',4'-dichlorophenyl)-sulfide
4-Amino-2-chlorophenyl-2-(3-chloro-5-trifluoromethylpyridyl)-sulfide
Bis-(4,4'-diamino-2,2'-dichlorophenyl)-sulfide
CA 02369005 2001-09-27
WO 00/59878 PCT/US00/08840
4-Amino-2-chlorophenyl-(2',4'-dichlorophenyl)-sulfide
4-Amino-2-chlorophenyl-(4'-acetamido-2'-chlorophenyl)-sulfide
4-Amino-2-chlorophenyl-6-(5-nitroquinolino)-sulfide
4-Amino-2-chlorophenyl-(4'-dimethylamino-2'-chlorophenyl)-sulfide
2-Chloro-4-amino-S-methylaminophenyl-(2',4'-dichlorophenyl)-sulfide
2-Chloro-4-amino-5-N-morpholinophenyl-(2',4'-dichlorophenyl)-sulfide
4-Amino-2-trifluoromethylphenyl-(2',4'-dichlorophenyl)-sulfide
4-Amino-2-chlorophenyl-2-(5-nitro-3-bromo)-pyridine sulfide
4-Aminomethyl-2-chlorophenyl-(2', 4'-dichlorophenyl)-sulfide
4,S-Dichloro-2-(2,4-dichlorophenylsulfanyl)-phenylamine
3,5-Dichloro-4-(2,4-dichlorophenylsulfanyl)-phenylamine
2,3-Dichloro-4-(2,4-dichlorophenylsulfanyl)-phenylamine
4-Amino-2-fluorophenyl-(2',4'-dichlorophenyl)-sulfide
5-Amino-3-chlorophenyl-(2',4'-dichlorophenyl)-sulfide
1 S 3-Chloro-4-( 1-chloro-naphthalen-2-ylsulfanyl)-phenylamine
1-(3-Nitro-4-phenylsulfanyl-phenyl)-ethanone
1-(3-Nitro-4-phenylsulfanyl-phenyl)-ethanone oxime
5-Trifluoromethyl-2-phenylsulfanyl-benzonitrile
1-(3,5-dichlorophenyl)-3-phenylsulfanyl-pyrrolidine-2,5-dione
Bis-2,4,6-Trinitrophenyl-sulfide
2-Methyl-1-(2-o-tolylsulfanyl-phenyl)-1H pyrrole
3-[2-(4-Chloro-2-nitro-phenylsulfanyl)-phenylamino-3H isobenzofuran-1-one
4-(Benzbthiazol-2-ylsulfanyl)-3-chloro-phenylamine
2-Nitro-4-chlorophenyl-(2' aminophenyl)-sulfide
6-Amino-2-chlorophenyl-(4'-methylphenyl)-sulfide
4-Nitrophenyl-(2'-chlorophenyl)-sulfide
2, 4-Dinitrophenyl-(4'-chlorophenyl)-sulfide
4-Aminophenyl-(2'-chlorophenyl)-sulfide
2, 4-Diaminophenyl-(4'-isopropylphenyl)-sulfide
4-Nitro-2-chlorophenyl-(2',3'-dichlorophenyl)-sulfide
4-Amino-2-chlorophenyl-2-(S-nitro-3-bromo)-pyridine-sulfide
6
CA 02369005 2001-09-27
WO 00/59878 PCT/US00/08840
The compounds as represented by structural formula (I) can be prepared by
synthetic methods or by metabolic processes. Preparation of the compounds by
metabolic processes include both in vivo and in vitro processes.
Pharmaceutical
compositions comprising compounds of the invention are also contemplated.
The invention also provides methods of inhibiting LFA-1 binding to
ICAMs that bind LFA-1 comprising the step of contacting LFA-1 with a diaryl
sulfide, and preferably a compound of structural formula I. Likewise, the
invention
provides methods of inhibiting leukocyte adhesion to endothelial cells
comprising the
step of contacting leukocytes expressing LFA-1 with a diaryl sulfide, and
preferably a
compound of the structural formula (I). The invention also comprehends methods
for
treating an inflammatory disorder comprising the steps of administering to a
mammal
an amount of a pharmaceutical composition of the invention sufficient to
inhibit
binding of LFA-1 to a naturlal ligand thereof that competes with ICAM-1 or
ICAM-3
for binding to LFA-1. The invention also comprehends methods for treating an
inflammatory disorder arising from LFA-1 binding to a natural ligand thereof
that
competes with ICAM-1 or ICAM-3 for binding to LFA-1, comprising administering
to a mammal in need thereof a compound that competes with 3-chloro-4-(1-chloro-
naphthalen-2-ylsulfanyl)-phenylamine for binding to LFA-1 in an amount
sufficient to
inhibit binding of the natural ligand to LFA-1. In addition, the invention
provides
methods of ameliorating a pathological condition associated with LFA-1 binding
to an
ICAM that binds LFA-1 comprising administering to an individual in need
thereof an
effective amount of a diaryl sulfide, and preferably a compound of the
structural
formula (I) to inhibit LFA-1 binding to the ICAM.
Examples of inhibitors of the present invention include, but are not
limited to, the compounds set out in Table I.
The invention also provides for use of a compound of the invention in
the production of a medicament for the treatment of pathologies associated
with LFA-
1 binding to ICAM-1.
The invention also provides methods to identify a negative regulator of
LFA-1 binding to a natural ligand thereof that competes with ICAM-1 or ICAM-3
for
binding to LFA-1 comprising the steps of: a) contacting LFA-1 with an
activator of
7
CA 02369005 2001-09-27
WO 00/59878 PCT/US00/08840
LFA-1 binding; b) measuring LFA-1 binding with the natural ligand in the
presence
and absence of a test compound; and c) identifying the test compound as an
inhibitor
when decreased LFA-1 binding to the ligand is detected in the presence of the
test
compound. In one aspect, the activator is crystal violet.
DETAILED DESCRIPTION OF THE INVENTION
An ICso value for a compound is defined as the concentration of the
compound required to produce 50% inhibition of a biological activity of
interest. As
used herein, a negative regulator is defined as a compound characterized by an
ICSO for
inhibition of LFA-1 binding to a natural ligand. Negative regulators of LFA-1
binding are defined to have an ICSO of less than about 200 ~M, less than about
100
pM, less than about 50 ~M, and preferably from about 0.05 qM to 40 ~M.
The term "pharmaceutically acceptable Garner" as used herein refers to
those prodrugs of compounds of the invention which are suitable for use in
contact
1 S with recipient animals and having undue toxicity, irritation, allergic
response
commensurate with a reasonable benefit/risk ratio, and effective for their
intended
use.
The term "prodrug" as used herein refers to compounds which are
rapidly transformed in vivo to the parent compound of the above formula, for
example, by hydrolysis. A thorough discussion is provided in Higuchi, et al.,
Prodrugs as Novel Deliver, stems, vol. 14 of the A.C.S.D. Symposium Series,
and
in Roche (ed), Bioreversible Garners in Drub Design, American Pharmaceutical
Association and Pergamon Press, 1987, both of which are incorporated herein by
reference. Prodrug design is discussed generally in Hardma, et al., (Eds),
Goodman
& Gilman's The Pharmacological Basis of Therapeutics, Ninth Edition, New York,
New York (1996), pp. 11-16. Briefly, administration of a drug is followed by
elimination from the body or some biotransformation whereby biological
activity of
the drug is reduced or eliminated. Alternatively, a biotransformation process
may
lead to a metabolic by-product which is itself more active or equally active
as
compared to the drug initially administered. Increased understanding of these
biotransformation processes permits the design of so-called "prodrugs" which,
8
CA 02369005 2001-09-27
WO 00/59878 PCT/US00/08840
following a biotransformation, become more physiologically active in an
altered state.
Prodrugs are therefore pharmacologically inactive compounds which are
converted to
biologically active metabolites. In some forms, prodrugs are rendered
pharmacologically active through hydrolysis of, for example, and ester or
amide
linkage, often times introducing or exposing a functional group on the
prodrug. The
thus modified drug may also react with an endogenous compound to form a water
soluble conjugate which further increases pharmacological properties of the
compound, for example, as a result of increased circulatory half life.
As another alternative, prodrugs can be designed to undergo covalent
modification on a functional group with, for example, glucuronic acid,
sulfate,
glutathione, amino acids, or acetate. The resulting conjugate may be
inactivated and
excreted in the urine, or rendered more potent than the parent compound. High
molecular weight conjugates may also be excreted into the bile, subjected to
enzymatic cleavage, and released back into circulation, thereby effectively
increasing
the biological half life of the originally administered compound.
Compounds of the invention may exist as stereoisomers where
asymmetric or chiral centers are present. Stereoisomers are designated by
either "S"
or "R" depending on arrangement of substituents around a chiral carbon atom.
Mixtures of stereoisomers are contemplated by the invention. Stereoisomers
include
enantiomers, diastereomers, and mixtures of the two. Individual stereoisomers
of
compounds of the invention can be prepared synthetically from commercially
available starting materials which contain asymmetric or chiral centers or by
preparation of racemic mixtures followed by separation or resolution
techniques well
known in the art. Methods of resolution include (1) attachment of a mixture of
enantiomers to a chiral auxiliary, separation of the resulting mixture by
recrystallization or chromatography, and liberation of the optically pure
product from
the auxiliary; (2) salt formation employing an optically active resolving
agent, and (3)
direct separation of the mixture of optical enantiomers on chiral
chromatographic
columns.
Compounds of the present invention include, but are not limited to
those embraced by general structural formula (I) above and the compounds set
out in
9
CA 02369005 2001-09-27
WO 00/59878 PCT/US00/08840
Table I.
The invention also provides pharmaceutical compositions comprising
one or more compounds of the invention, preferably further comprising a
pharmaceutically acceptable carrier or diluent.
The invention further provides methods for inhibiting LFA-1 binding
to an ICAM that binds LFA-1 comprising the step of contacting LFA-1, or an
ICAM-
binding fragment thereof, with a negative regulator compound; said negative
regulator
binding the LFA-1 a~ polypeptide, or a fragment thereof, at a site selected
from the
group consisting of a conformation that binds a diaryl sulfide or a binding
site defined
by Ilez59, Leuz98, Ilez3s Val's', Leu'6', and Ile3o6 of human LFA-1 aL
polypeptide and an
LFA-1 domain that binds 3-chloro-4-(1-chloro-naphthalen-2-ylsulfanyl)-
phenylamine
having the structure described above. Alternatively, the negative regulator
binding
site on LFA-1 is defined by amino acid residues Ilezs9, Leuz98, Ilez3s,
Val'S', Leu'6',
Ile3o6, Leu3°z,Tyr'S', Leu'3z, Valz33, Val'3°, and Tyr'66. In
still another alternative, the
negative regulator binding site on LFA-1 is defined by amino acid residues
LyszB',
Leuz9s, Ilezs9, Leu3oz, Ilez3s, Vallsy Tyrzsy Lys3°s~ Leu'6y Leul3z,
Valzss, Ilezss, Val'so,
T 166 Ile3°6 phe'34 phe'68 phe's3 Tyi.3o7 Val3os Ile3°9 Tlu.z31
Gluzs4~ Phezss~ Glu3°'
> > > > > > > > >
Met'S4, Ilez3', Ile'5°, and Leuz95. The LFA-1 regulatory binding site
is described in co-
pending U.S. patent application entitled "LFA-1 Regulatory Binding Site and
Uses
Thereof', filed April 2, 1999, attorney docket number 27866/35375, Serial
Number
09/285,477, incorporated herein by reference in its entirety. In one
embodiment,
methods of the invention include use of cells expressing either LFA-1 or the
ICAM.
In methods wherein one of the binding partners is expressed in a cell, the
other
binding partner is either purified and isolated, in a fluid sample (purified,
partially
purified, or crude) taken from an individual, or in a cell lysate. The
invention also
comprehends methods wherein both LFA-1 and the ICAM are expressed in cells.
The
LFA-1 and ICAM binding partners may be expressed on the same cell type or
different cell types. Preferably, the LFA-1 polypeptide is expressed on
leukocytes, i.e.
lymphocytes, monocytes, or granulocytes, and the ICAM polypeptide is expressed
on
endothelial cells.
The invention also provides methods to inhibit leukocyte adhesion to
CA 02369005 2001-09-27
WO 00/59878 PCT/LTS00/08840
endothelial cells comprising the step of contacting said leukocyte with a
negative
regulator of LFA-1 binding to an ICAM that binds LFA-1, said negative
regulator
binding an LFA-1 regulatory site selected from the group consisting of a
conformation
that binds a diaryl sulfide or a binding site defined by Ilezs~, Leu'-98,
Ile'3', Val'S',
Leu'~', and Ile3ob of human LFA-1 a~ polypeptide or an LFA-1 domain that binds
3-
chloro-4-(1-chloro-naphthalen-2-ylsulfanyl)-phenylamine. Alternatively, the
diaryl
sulfide binding conformation is defined by amino acid residues as described
above. ha
vivo and in vitro methods are contemplated.
The invention also provides methods to ameliorate a pathology arising
from LFA-1 binding to an ICAM comprising the step of administering to an
individual in need thereof a negative regulator of LFA-1 binding to the ICAM
in an
amount effective to inhibit LFA-1 binding to the ICAM, said negative regulator
binding to an LFA-1 regulatory site selected from the group consisting of a
conformation that binds a diaryl sulfide or a site defined by I1e259, Leuz98,
Ilez3s, Val'S',
1 S Leu'6', and Ile3o6 of human LFA-1 or an LFA-1 domain that binds compound 3-
chloro-4-( 1-chloro-naphthalen-2-ylsulfanyl)-phenylamine.
In a preferred embodiment, methods of the invention include use of a
diaryl sulfide compound to inhibit binding of LFA-1 to an ICAM. A preferred
method includes use of a compound of general structural formula (I), a
pharmaceutically acceptable salt, or prodrug thereof as described above.
Therapeutic Methods
To the extent that leukocyte adhesion to endothelial cells gives rise to a
pathological disorder, the invention provides methods to ameliorate
pathologies
associated with accumulation of leukocytes resulting from LFA-1 binding to an
ICAM
that binds LFA-1 comprising the step of administering to an individual in need
thereof
an amount of an inhibitor of LFA-1 binding to the ICAM effective to inhibit
LFA-1
binding to the ICAM, said inhibitor binding to LFA-1 at a site presented by
amino
acid residues Ile2s9, Leuz98, I1e235, Val'S', Leu'6' and Ile3o6. Exemplary
medical
conditions include, without limitation, inflammatory diseases, autoimmune
diseases,
reperfusion injury, myocardial infarction, stroke, hemorrhagic shock, organ
transplant,
11
CA 02369005 2001-09-27
WO 00/59878 PCT/US00/08840
and the like. Methods of the invention provide for amelioration of a variety
of
pathologies, including, for example, but not limited to adult respiratory
distress
syndrome, multiple organ injury syndrome secondary to septicemia, multiple
organ
injury secondary to trauma, reperfusion injury of tissue, acute
glomerulonephritis,
reactive arthritis, dermatosis with acute inflammatory components, stroke,
thermal
injury, Crohn's disease; necrotizing enterocolitis, granulocyte transfusion
associated
syndrome, and cytokine induced toxicity, and T cell mediated diseases.
Inflammatory cell activation and excessive or unregulated cytokine
(e:g., TNFa and IL-1 [3) production are also implicated in disorders such as
rheumatoid
arthritis, osteoarthritis, gouty arthritis, spondylitis, thyroid associated
ophthalmopathy,
Behcet disease, sepsis, septic shock, endotoxic shock, gram negative sepsis,
gram
positive sepsis, toxic shock syndrome, asthma, chronic bronchitis, allergic
respiratory
distress syndrome, chronic pulmonary inflammatory disease, such as chronic
obstructive pulmonary disease, silicosis, pulmonary sarcoidosis, reperfusion
injury of
the myocardium, brain, and extremities, fibrosis, cystic fibrosis, keloid
formation, scar
formation, atherosclerosis, transplant rejection disorders, such as graft vs.
host
reaction and allograft rejection, chronic glamerulonephritis, lupus,
inflammatory
bowel disease, such as ulcerative colitis, proliferative lymphocyte diseases,
such as
leukemia, and inflammatory dermatoses, such as atopic dermatitis, psoriasis,
urticaria,
uveitis.
Other conditions characterized by elevated cytokine levels include
brain injury due to moderate trauma (see J. Neurotrauma, 12, pp. 1035-1043
(1995);
J. Clin. Invest., '91, pp. 1421-1428 (1993)), cardiomyopathies, such as
congestive
heart failure (see Circulation, 97, pp. 1340-1341 (1998)), cachexia, cachexia
secondary to infection or malignancy, cachexia secondary to acquired immune
deficiency syndrome (AIDS), ARC (AIDS related complex), fever myalgias due to
infection, cerebral malaria, osteoporosis and bone resorption diseases, keloid
formation, scar tissue formation, and pyrexia.
The ability of the negative regulators of the invention to treat arthritis
can be demonstrated in a murine collagen-induced arthritis model [Kakimoto, et
al.
Immunol. 142:326-337 (1992)], in a rat collagen-induced arthritis model
[Knoerzer, et
12
CA 02369005 2001-09-27
WO 00/59878 PCT/US00/08840
al., Toxical Pathol. 2.5:13-19 (1997)], in a rat adjuvant arthritis model
[Halloran, et
al., As-thritis Rheum 39:810-819 (1996)], in a rat streptococcal cell wall-
induced
arthritis model [Schimmer, et al., J. Immzznol. 160:1466-1477 (1998)], or in a
SCID-
mouse human rheumatoid arthritis model [Oppenheimer-Marks, et al., J. Clin.
Invest
101:1261-1272 (1998)].
The ability of the negative regulators to treat Lyme arthritis can be
demonstrated according to the method of Gross, et al., Science, 218:703-706,
(1998).
The ability of the negative regulators to treat asthma can be
demonstrated in a murine allergic asthma model according to the method of
Wegner,
et al., Science, 247:456-459, (1990), or in a murine non-allergic asthma model
according to the method of Bloemen, et al., Am. J. Respir. Crit. Care Med.
153:521-
529 ( 1996).
The ability of the negative regulators to treat inflammatory lung injury
can be demonstrated in a murine oxygen-induced lung injury model according to
the
method of Wegner, et al., Lung, 170:267-279, (1992), in a murine immune
complex-
induced lung injury model according to the method of Mulligan, et al., J.
Immunol.,
154:1350-1363, (1995), or in a murine acid-induced lung injury model according
to
the method of Nagase, et al., Am. J. Respir. Crit. Care Med., 154:504-510,
(1996).
The ability of the negative regulators to treat inflammatory bowel
disease can be demonstrated in a murine chemical-induced colitis model
according to
the method of Bennett, et al., J. Pharmacol. Exp. Ther., 280:988-1000, (1997).
The ability of the negative regulators to treat autoimmune diabetes can
be demonstrated in an NOD mouse model according to the method of Hasagawa, et
al., Int. Immunol. 6:831-838 (1994), or in a murine streptozotocin-induced
diabetes
model according to the method of Herrold, et al., Cell Immunol. 157:489-500,
(1994).
The ability of the negative regulators to treat inflammatory liver injury
can be demonstrated in a murine liver injury model according to the method of
Tanaka, et al., J. Immunol., 151:5088-5095, (1993).
The ability of the negative regulators to treat inflammatory glomerular
injury can be demonstrated in a rat nephrotoxic serum nephritis model
according to
the method of Kawasaki, et al., J. Immunol., 150:1074-1083 (1993).
13
CA 02369005 2001-09-27
WO 00/59878 PCT/US00/08840
The ability of the negative regulators to treat radiation-induced enteritis
can be demonstrated in a rat abdominal irradiation model according to the
method of
Panes, et al., Gastroenterology, 108:1761-1769 ( 1995).
The ability of the negative regulators to treat radiation pneumonitis can
be demonstrated in a murine pulmonary irradiation model according to the
method of
Hallahan, et al., Proc. Natl. Acad. Sci (USA), 94:6432-6437 (1997).
The ability of the negative regulators to treat reperfusion injury can be
demonstrated in the isolated heart according to the method of Tamiya, et al.,
Immunopharmacology, 29:53-63 (1995), or in the anesthetized dog according to
the
model of Hartman, et al., Cardiovasc. Res. 30:47-54 (1995).
The ability of the negative regulators to treat pulmonary reperfusion
injury can be demonstrated in a rat lung allograft reperfusion injury model
according
to the method of DeMeester, et al., Transplantation, 62:1477-1485 (1996), or
in a
rabbit pulmonary edema model according to the method of Horgan, et al., Am. J.
Physiol. 261:H1578-H1584 (1991).
The ability of the negative regulators to treat stroke can be
demonstrated in a rabbit cerebral embolism stroke model according to the
method of
Bowes, et al., Exp. Neurol., 119:215-219 (1993), in a rat middle cerebral
artery
ischemia-reperfusion model according to the method of Chopp, et al., Stroke,
25:869-
875 (1994), or in a rabbit reversible spinal cord ischemia model according to
the
method of Clark et al., Neurosurg., 75:623-627 ( 1991 ). The ability of the
negative
regulators to treat cerebral vasospasm can be demonstrated in a rat
experimental
vasospasm model according to the method of Oshiro, et al., Stroke, 28:2031-
2038
( 1997).
The ability of the negative regulators to treat peripheral artery
occlusion can be demonstrated in a rat skeletal muscle ischemia/reperfusion
model
according to the method of Gute, et al., Mol. Cell Biochem., 179:169-187
(1998).
The ability of the negative regulators to treat graft rejection can be
demonstrated in a murine cardiac allograft rejection model according to the
method of
Isobe, et al., Science, 255:1125-1127 (1992), in a murine thyroid gland kidney
capsule
model according to the method of Talento, et al., Transplantation, 55:418-422
(1993),
14
CA 02369005 2001-09-27
WO 00/59878 PCT/US00/08840
in a cynomolgus monkey renal allograft model according to the method of
Cosimi, et
al., J. Irnmunol., 144:4604-4612 (1990), in a rat nerve allograft model
according to the
method of Nakao, et al., Muscle Nen~e, 18:93-102 ( 1995), in a murine skin
allograft
model according to the method of Gorczynski and Wojcik, J. Immunol. Li2:2011-
2019, ( 1994), in a murine corneal allograft model according to the method of
He, et
al., Opthalmol. Yis. Sci., 35:3218-3225 (1994), or in a xenogeneic pancreatic
islet cell
transplantation model according to the method of Zeng, et al.,
Trastsplantation,
58:681-689 (1994).
The ability of the negative regulators to treat graft-vs.-host disease
(GVHD) can be demonstrated in a murine lethal GVHD model according to the
method of Harping, et al., Transplantation, 52:842-845 (1991).
The ability of the negative regulators to treat cancers can be
demonstrated in a human lymphoma metastasis model (in mice) according to the
method of Aoudjit, et al., J. Immunol., 161:2333-2338, (1998).
Pharmaceutical Compositions
The present invention also provides pharmaceutical compositions
which comprise a diaryl sulfide formulated together with one or more
pharmaceutically-acceptable earners.
The pharmaceutical compositions of the invention can be administered
to humans and other animals by any suitable route. For example, the
compositions
can be administered orally, rectally, parenterally, intracisternally,
intravaginally,
intraperitoneally, topically (as by powders, ointments, or drops), bucally, or
nasally.
The term "parenteral" administration as used herein refers to modes of
administration
which include intravenous, intraarterial, intramuscular, intraperitoneal,
intrasternal,
intrathecal, subcutaneous and intraarticular injection and infusion.
Pharmaceutical compositions of this invention for parenteral injection
comprise pharmaceutically-acceptable sterile aqueous or nonaqueous solutions,
dispersions, suspensions or emulsions as well as sterile powders for
reconstitution
into sterile injectable solutions or dispersions just prior to use. Examples
of suitable
aqueous and nonaqueous carriers, diluents, solvents or vehicles include water,
CA 02369005 2001-09-27
WO 00/59878 PCT/US00/08840
ethanol, polyols (such as glycerol, propylene, glycol, polyethylene glycol,
and the
like), and suitable mixtures thereof, vegetable oils (such as olive oils), and
injectable
organic esters such as ethyl oleate. Proper fluidity can be maintained, for
example, by
the use of coating materials such as lecithin, by the maintenance of the
required
particle size in the case of dispersions, and by the use of surfactants.
These compositions may also contain adjuvants such as preservative,
wetting agents, emulsifying agents, and dispersing agents. Prevention of the
action of
microorganisms may be ensured by the inclusion of various antibacterial and
antifungal agents, for example, paraben, chlorobutanol, phenol sorbic acid,
and the
like. It may also be desirable to include isotonic agents such as sugars,
sodium
chloride, and the like, Prolonged absorption of the injectable pharmaceutical
form
may be brought about by the inclusion of agents which delay absorption such as
aluminum monosterate and gelatin.
In some cases, in order to prolong the effect of the drug, it is desirable
1 S to slow the absorption of the drug from subcutaneous or intramuscular inj
ection. This
may be accomplished by the use of a liquid suspension of crystalline or
amorphous
materials with poor water solubility. The rate of absorption of the drug then
depends
upon its rate of dissolution which, in turn, may depend upon crystal size and
crystalline form. Alternatively, delayed absorption of a parenterally
administered drug
from is accomplished by dissolving or suspending the drug in an oil vehicle.
Injectable depot forms are made by forming microencapsule matrices
of the drug in biodegradable polymers such a polylactide-polyglycolide.
Depending
upon the ratio of drug to polymer and the nature of the particular polymer
employed,
the rate of drug release can be controlled. Examples of other biodegradable
polymers
include poly(orthoesters) and poly(anhydrides). Depot injectable formulations
are
also prepared by entrapping the drug in liposomes or microemulsions which are
compatible with body tissue.
The injectable formulations can be sterilized, for example, by filtration
through a bacterial- or viral-retaining filter, or by incorporating
sterilizing agents in
the form of sterile solid compositions which can be dissolved or dispersed in
sterile
water or other sterile injectable medium just prior to use.
16
CA 02369005 2001-09-27
WO 00/59878 PCT/US00/08840
Solid dosage forms for oral administration include capsules, tablets,
pills, powders, and granules. In such solid dosage forms, the active compound
is
mixed with a least one inert, pharmaceutically-acceptable excipient or carrier
such as
sodium citrate or dicalcium phosphate and/or (a) fillers or extenders such as
starches,
lactose, sucrose, glucose, mannitol, and silicic acid, (b) binders such as,
for example,
carboxymethylcellulose, gums (e.g.alginates, acacia) gelatin,
polyvinylpyrrolidone,
and sucrose, (c) humectants such as glycerol, (d) disintegrating agents such
as agar-
agar, calcium carbonate, potato or tapioca starch, alginic acid, certain
silicates, and
sodium carbonate, (e) solution retarding agents such a paraffin, (f)
absorption
accelerators such as quaternary ammonium compounds, (g) wetting agents such
as, for
example, cetyl alcohol and glycerol monosterate, (h) absorbents such as kaolin
and
bentonite clay, and (I) lubricants such as talc, calcium sterate, magnesium
stearate,
solid polyethylene glycols, sodium lauryl sulfate, and mixtures thereof. In
the case of
capsules, tablets and pills, the dosage form may also comprise buffering
agents.
Solid compositions of a similar type may also be employed as fillers in
soft and hard-filled gelatin capsules using such excipients as lactose or milk
sugar as
well as high molecular weight polyethylene glycols and the like.
The solid dosage forms of tablets, dragees, capsules, pills, and granules
can be prepared with coatings and shells such as enteric coatings and other
coatings
well known in the pharmaceutical formulating art. They may optionally contain
opacifying agents and can also be of a composition that they release the
active
ingredients(s) only, or preferentially, in a part of the intestinal tract,
optionally, in a
delayed manner: Exemplary materials include polymers having pH sensitive
solubility, such as the materials available as Eudragit~. Examples of
embedding
compositions which can be used include polymeric substances and waxes.
The active compounds can also be in micro-encapsulated form if
appropriate, with one or more of the above-mentioned excipients.
Liquid dosage forms for oral administration include pharmaceutically-
acceptable emulsions, solutions, suspensions, syrups and elixirs. In addition
to the
active compounds, the liquid dosage forms may contain inert diluents commonly
used
in the art such as, for example, water or other solvents, solubilizing agents
and
17
CA 02369005 2001-09-27
WO 00/59878 PCT/US00/08840
emulsifiers such as ethyl alcohol, isopropyl alcohol, ethyl carbonate, ethyl
acetate,
benzyl alcohol, benzyl benzoate, propylene glycol, 1,3-butylene glycol,
dimethyl
formamide, oils (in particular, cottonseed, groundnut, corn, germ, olive,
castor, and
sesame oils), glycerol, tetrahydrofurfuryl alcohol, polyethylene glycols and
fatty acid
esters of sorbitan, and mixtures thereof.
Besides inert diluents, the oral compositions can also include adjuvants
such as wetting agents, emulsifying and suspending agents, sweetening,
flavoring, and
perfuming agents.
Suspensions, in addition to the active compounds, may contain
suspending agents as, for example, ethoxylated isostearyl alcohols,
polyoxyethylene
sorbitol and sorbitan esters, microcrystalline cellulose, aluminum
metahydroxide,
bentonite, agar-agar, and tragacanth, and mixtures thereof.
Compositions for rectal or vaginal administration are preferably
suppositories which can be prepared by mixing the compounds of this invention
with
suitable non-irritating excipients or carriers such as cocoa butter,
polyethylene glycol
or suppository wax which are solid at room temperature but liquid at body
temperature and therefore melt in the rectum or vaginal cavity and release the
active
compound.
Compounds of the present invention can also be administered in the
form of liposomes. As is known in the art, liposomes are generally derived
from
phospholipids or other lipid substances. Liposomes are formed by mono- or
multi-
lamellar hydrated liquid crystals that are dispersed in an aqueous medium. Any
non-
toxic, physiologically-acceptable and metabolizable lipid capable of forming
liposomes can be used. The present compositions in liposome form can contain,
in
addition to a compound of the present invention, stabilizers, preservatives,
excipients,
and the like. The preferred lipids are the phospholipids and the phosphatidyl
cholines
(lecithins), both natural and synthetic. Methods to form liposomes are known
in the
art. See, for example, Prescott, Ed., Methods in Cell Biolo~v, Volume XIV,
Academic Press, New York, N.Y. (1976), p. 33 et seq.
The compounds of the present invention may be used in the form of
pharmaceutically-acceptable salts derived from inorganic or organic acids. By
18
CA 02369005 2001-09-27
WO 00/59878 PCT/US00/08840
"pharmaceutically-acceptable salt" is meant those salts which are, within the
scope of
sound medical judgment, suitable for use in contact with the tissues of humans
and
lower animals without undue toxicity, irritation, allergic response and the
like, and are
commensurate with a reasonable benefit/risk ratio. Pharmaceutically-acceptable
salts
are well known in the art. For example, S. M. Berge, et al., describe
pharmaceutically-acceptable salts in detail in J. Pharmaceutical Sciences,
66:1
( 1977). The salts may be prepared in situ during the final isolation and
purification of
the compounds of the invention or separately by reacting a free base function
with a
suitable acid. Representative acid addition salts include, but are not limited
to acetate,
adipate, alginate, citrate, aspartate, benzoate, benzenesulfonate, bisulfate,
butyrate,
camphorate, camphorolsulfonate, digluconate, glycerophosphate, hemisulfate,
heptanoate, hexanoate, fumarate hydrochloride, hydrobromide, hydroiodide, 2-
hydroxyethanesulfonate (isothionate), lactate, maleate, methanesulfonate,
nicotinate,
2-naphthalenesulfonate, oxalate, pamoate, pectinate, persulfate, 3-
phenylpropionate,
picrate, pivalate, propionate, succinate, tartrate, thiocyanate, phosphate,
glutamate,
bicarbonate, p-toluenesulfonate and undecanoate. Examples of acids which may
be
employed to form pharmaceutically acceptable acid addition salts include
inorganic
acids as hydrochloric acid, hydrobromic acid, sulphuric acid and phosphoric
acid and
such organic acids as oxalic acid, malefic acid, succinic acid and citric
acid.
Basic nitrogen-containing groups can be quaternized with such agents
as lower alkyl halides such as methyl, ethyl, propyl, and butyl chlorides,
bromides and
iodides; dialky sulfates like dimethyl, diethyl, dibutyl and diamyl sulfates;
long chain
halides such as decyl, lauryl, myristyl and stearyl chlorides, bromides and
iodides;
arylalkyl halides like benzyl and phenethyl bromides and others. Water or oil-
soluble
or dispersible products are thereby obtained.
Basic addition salts can be prepared in situ during the final isolation
and purification of compounds of this invention by reacting a carboxylic acid-
containing moiety with a suitable base such as the hydroxide, carbonate or
bicarbonate
of a pharmaceutically acceptable metal cation or with ammonia or organic
primary,
secondary or tertiary amine. Pharmaceutically-acceptable basic addition salts
include,
but are not limited to, cations based on alkali metals or alkaline earth
metals such as
19
CA 02369005 2001-09-27
WO 00/59878 PCT/US00/08840
lithium, sodium, potassium, calcium, magnesium and aluminum salts and the like
and
nontoxic quaternary ammonia and amine canons including ammonium,
tetramethylammonium, tetraethylammonium, methylamine, dimethylamine,
trimethylamine, triethylamine, diethylamine, ethylamine and the like. Other
representative organic amines useful for the formation of base addition salts
include
ethylenediamine, ethanolamine, diethanolamine, piperidine, piperazine and the
like.
Dosage forms for topical administration of a compound of this
invention include powders, sprays, ointments and inhalants. The active
compound is
mixed under sterile conditions with a pharmaceutically-acceptable carrier and
any
needed preservatives, buffers, or propellants which may be required.
Ophthalmic
formulations, eye ointments, powders, and solutions are also contemplated as
being
within the scope of this invention.
Actual dosage levels of active ingredients in the pharmaceutical
compositions of this invention may be varied so as to obtain an amount of the
active
compounds) that is effective to achieve the desired therapeutic response for a
particular patient. compositions, and mode of administration. The selected
dosage
level will depend upon the activity of the particular compound, the route of
administration, the severity of the condition being treated, and the condition
and prior
medical history of the patient being treated. However, it is within the skill
of the art
to start doses of the compound at levels lower than required for to achieve
the desired
therapeutic effort and to gradually increase the dosage until the desired
effect is
achieved.
Generally dosage levels of about 0.1 to about 1000 mg, about 0.5 to
about S00 mg, about 1 to about 250 mg, about 1.5 to about 100, and preferably
of
about 5 to about 20 mg of active compound per kilogram of body weight per day
are
administered orally or intravenously to a mammalian patent. If desired, the
effective
daily dose may be divided into multiple doses for purposes of administration,
e.g., two
to four separate doses per day.
The present invention is illustrated by the following examples.
Example 1 described a high throughput assay to screen for negative regulators
of
CA 02369005 2001-09-27
WO 00/59878 PCT/US00/08840
LFA-1 binding to an ICAM that binds LFA-1. Example 2 relates to binding assays
to
evaluate the ability of various compounds to inhibit LFA-1 binding to an ICAM.
Example 3 describe synthesis of negative regulators. Example 4 provides
results from
cell based assays using the negative regulators.
Example 1
High Throughput Screening for LFA-1/ICAM-1 Binding Inhibitors
In an effort to identify inhibitors of LFA-1/ICAM-1 binding, a high
throughput screening (HTS) assay was designed to efficiently screen large
numbers of
chemical compounds in a proprietary library as follows.
Preliminary experiments were earned out in order to define the linear
range of LFA-1/ICAM-1 interaction. Recombinant ICAM-1/IgGI fusion protein
(comprising full length ICAM-1) was prepared as described in U.S. Patent Nos.
5,770,686, 5,837,478, and 5,869,262, each of which is incorporated herein by
reference. The fusion protein was biotinylated using a kit obtained from
Pierce
Chemical (Rockford, IL). Biotinylated protein (BioIgICAM-1) concentration was
determined by measuring absorbance at 280 nm, and serial dilutions were
prepared to
give a final concentration range of 50 ug/ml to 0.008 pg/ml. Titration of
BioIgICAM-1 was carried out with the protein first aliquoted into wells on an
assay
plate. Recombinant LFA-1 was added to each well at the same concentration and
the
experiment (as described below) was carried to completion. The amount of
binding
was determined for each well, and from a subsequent plot of the results, a
single
concentration of BioIgICAM-1 was selected for subsequent experiments. In a
similar
manner, LFA-1 was titrated using the BioIgICAM-1 concentration selected as
described above.
On day 1 of the HTS procedure, the capture antibody, i.e., a
non-blocking anti-LFA-1 monoclonal antibody (TS2/4.1; ATCC #HB244), was
diluted in plate coating buffer (50 mM sodium carbonate/bicarbonate, 0.05%
ProClin~ 300, pH 9.6) to a final concentration of 2 pg/ml. Immulon~ 4 (Dynex
Technologies, Chantilly, VA) plate wells were coated with 100 pl diluted
antibody
solution per well, and incubation was earned out overnight at 4°C. On
day 2, the
21
CA 02369005 2001-09-27
WO 00/59878 PCT/US00/08840
plates were warmed to room temperature and washed two times with wash buffer
(calcium- and magnesium-free phosphate buffered saline, CMF-PBS) with 0.05%
Tween~-20). To each well, 200 ~.1 of blocking solution (S% fish skin gelatin
in
CMF-PBS with 0.05% ProClin~ 300) was added, and the blocking incubation was
carried out at room temperature for 30 min. The blocking solution was removed
by
aspiration, and the plates were not washed. LFA-1 was diluted to a final
concentration of 1 pg/ml in assay buffer ( 1 % fish skin gelatin and 2 mM
MgCI, in
CMF-PBS), and 100 pl was added to each well. Incubation was carried out for
one
hour, and the plates were washed two times with wash buffer.
A 2X stock solution of BioIgICAM-1 was prepared containing 0.1
pg/ml BioIgICAM-l and 4 pM crystal violet (found to be an activator of
LFA-1/ICAM-1 binding) in Assay Buffer (EG&G Wallac, Gaithersburg, MD).
Aliquots (50 pl) of pooled chemicals (22 compounds/pool in 100% DMSO) from the
chemical library were added to the wells, followed by addition of 50 pl of the
2X
stock of BioIgICAM-1 to provide a final assay volume of 100 pl (containing 2%
DMSO). The plates were incubated for one hour at room temperature, and washed
once with wash buffer. Europium-labeled streptavidin (Eu-SA; #1244-360, EG&G
Wallac) was diluted 1:500 in Assay Buffer, 100 pl of the diluted Eu-SA was
added to
each well, and the plates were incubated at room temperature for one hour.
Plates were washed eight times with wash buffer, 100 pl of DELFIA~
enhancement solution (EG&G Wallac) diluted 1:2, was added to each well, and
the
plates were shaken for five minutes using a Wallac shaker at fast speed.
Plates were
read using a Wallac DELFIA~ fluorescence reader (fluorimeter). Controls
included
both positive and negative wells and 50% binding wells established using
blocking
antibodies, i.e., anti-LFA-1 monoclonal antibody (TS1/22.1, ATCC #HB202) or
anti-ICAM-1 monoclonal antibody. Chemical pools in wells showing 50% or
greater
inhibition of LFA-1 binding to ICAM-1 were identified and the experiment was
repeated using individual chemicals from those pools. Inhibitors of LFA-1/ICAM-
1
binding were identified, and a further screen was performed to determine dose
dependence of the inhibitory activity. Further study of selected compounds was
carried out using biochemical and cellular assay techniques.
22
CA 02369005 2001-09-27
WO 00/59878 PCT/US00/08840
The compounds were grouped according to common structural
features, and it was found that a subset (listed below) of the compounds
included a
characteristic diaryl sulfide structure.
3-Chloro-4-( 1-chloro-naphthalen-2-ylsulfanyl)-phenylamine
1-(3-Nitro-4-phenylsulfanyl-phenyl)-ethanone
1-(3-Nitro-4-phenylsulfanyl-phenyl)-ethanone oxime
5-Trifluoromethyl-2-phenylsulfanyl-benzonitrile
1-(3,5-dichlorophenyl)-3-phenylsulfanyl-pyrrolidine-2,5-dione
Bis-2,4,6-Trinitrophenyl-sulfide
2-Methyl-1-(2-o-tolylsulfanyl-phenyl)-1 H-pyrrole
3-[2-(4-Chloro-2-vitro-phenylsulfanyl)-phenylamino-3H isobenzofuran-1-one
4-(Benzothiazol-2-ylsulfanyl)-3-chloro-phenylamine
2-Nitro-4-chlorophenyl-(2'aminophenyl)-sulfide
6-Amino-2-chlorophenyl-(4'-methylphenyl)-sulfide
4-Nitrophenyl-(2'-chlorophenyl)-sulfide
2, 4-Dinitrophenyl-(4'-chlorophenyl)-sulfide
4-Aminophenyl-(2'-chlorophenyl)-sulfide
2, 4-Diaminophenyl-(4'-isopropylphenyl)-sulfide
In an effort to optimize the negative regulatory capacity of the
identified compounds, various diaryl sulfide derivatives were conducted as
described
below in Example 3. Additional diaryl sulfide derivatives are described in co-
pending
provisional patent application entitled "Cell Adhesion-Inhibiting
Antiinflammatory
and Immune Suppressive Compounds" filed April 2, 1999, attorney docket number
6446.US.Z3, Serial Number 09/286,645, incorporated herein by reference in its
entirety.
23
CA 02369005 2001-09-27
WO 00/59878 PCT/US00/08840
Example 2
Binding Assays
A. ICAIV1-I/LFA-1 Biochemical Interaction Assav
Compounds that antagonize the interaction between ICAM-1 and
LFA-1 can be identified, and their activities quantitated, using both
biochemical and
cell-based assays. A primary biochemical assay measures the ability of the
compound
in question to block the interaction between LFA-1 and its adhesion partner
ICAM-1,
as described below.
In this biochemical assay, 100 ~l of anti-LFA-1 antibody at a
connection of 5 ~g/ml in Dulbecco's phosphate-buffered saline (D-PBS) was used
to
coat wells of a 96-well microtiter plate overnight at 4°C. The wells
were washed
twice with wash buffer (CMF-PBS, 0.05% Tween~ 20) and blocked by addition of
200 pl of D-PBS containing 5% fish skin gelatin. Recombinant LFA-1 (100 ~l of
0.7
~g/ml) in D-PBS was added to each well. Incubation was continued for one hour
at
room temperature and the wells were washed twice with wash buffer. Serial
dilutions
of compounds being assayed as LFA-1/ICAM-1 negative regulators, prepared as 10
mM stock solutions in DMSO, were diluted in D-PBS, 2 mM MgClz, 1 % fish skin
gelatin and 500 pl of each dilution added to each well, followed by addition
of 50 ~1
of 0.8 ~g/ml BioIgICAM-1 to the wells, and the plates were incubated at room
temperature for one hour. The wells were then washed twice with wash buffer
and
100 ~l of Eu-SA (EG&G Wallac) diluted 1:100 in Delfia~ assay buffer (EG&G
Wallac) were added to the wells. Incubation was carried out for one hour at
room
temperature. The wells were washed eight times with wash buffer and 100 ~l of
enhancement solution (EG&G Wallac) was added to each well. Incubation was
continued for five minutes with constant mixing. Time-resolved fluorimetry
measurements were made using the Victor 1420 Multilabel Counter (EG&G Wallac)
and the percent inhibition of each candidate compound was calculated using the
following equation:
%inhibition = 100 X 1 _ average OD wlcompound minus background
average OD wlo compound minus background
24
CA 02369005 2001-09-27
WO 00/59878 PCT/US00/08840
where "background" refers to wells that were not coated with anti-LFA-1
antibody.
B. ICAM-I/JY-8 Cell Adhesion Assav
Biologically relevant activity of the compounds in this invention was
confirmed using a cell-based adhesion assay that measures the ability of the
compounds to block adherence of JY-8 cells (a human EBV-transformed B cell
line
expressing LFA-1 on its surface) to immobilized ICAM-1, as follows. This assay
may
be performed with or without added IL-8. For IL8 stimulation of the standard
JY-8
cells 30 ng/ml IL-8 was added in the 30 minute incubation at 37°C with
the cells.
For measurement of inhibitory activity in the cell-based adhesion
assay, 96-well microtiter plates were coated with 70 ~l of recombinant ICAM-
1/Ig at
a concentration of S ~g/ml in CMF-PBS overnight at 4°C. The wells were
washed
twice with D-PBS and blocked by addition of 200 ~l of D-PBS, 5% fish skin
gelatin
by incubation for one hour at room temperature. Fluorescent-tagged JY-8 cells
(50 ql
at 2 x 106 cells/ml in RPMI-1640/1% fetal bovine serum (FBS)) were added to
the
wells. For fluorescent labeling of JY-8 cells, 5 x 106 cells, washed once in
RPMI
1640, were resuspended in 1 ml RPMI-1640 containing 2 ~M Calcein AM (Molecular
Probes, OR), were incubated at 37°C for 30 minutes and washed once
with
RPMI-1640/1 % FBS. Dilutions of compounds to be assayed for LFA-1/ICAM-1
antagonistic activity were prepared in RPMI-1640/1 % FBS from 10 mM stock
solutions in DMSO and 50 ~1 aliquots were added to duplicate wells. Microtiter
plates were incubated for 45 min at room temperature and the wells were washed
gently once with RPMI-1640/1% FBS. Fluorescence intensity was measured in a
fluorescence plate reader with an excitation wavelength at 495 nm and an
emission
wavelength at 530 nm. The percent inhibition of a candidate compound at a
given
concentration was calculated using the following equation:
%inhibition = 100 X 1 _ average OD wlcompound
average OD w/o compound
25
CA 02369005 2001-09-27
WO 00/59878 PCT/LTS00/08840
C. ICAM-3/JY-8 Cell Adhesion Assay
Compounds of the present invention have been demonstrated to act via
interaction with the integrin LFA-1, specifically by binding to the aL I
domain which
is known to be critical for the adhesion of LFA-1 to a variety of cell
adhesion
S molecules. As such, it is expected that these compounds should block the
interaction
of LFA-1 with other CAMS, and this inhibition has been demonstrated for LFA-1
binding to ICAM-3. Compounds of the present invention were evaluated for the
ability to block the adhesion of JY-8 cells to immobilized ICAM-3, as follows.
For measurement of inhibitory activity in the cell-based adhesion
assay, 96-well microtiter plates were coated with 50 ~l of recombinant ICAM-
3/Ig at
a concentration of 10 ~g/ml in CMF-PBS overnight at 4°C. The wells were
washed
twice with D-PBS, blocked by addition of 100 ~l of D-PBS, 1% bovine serum
albumin (BSA) by incubation for one hour at room temperature, and washed once
with RPMI-1640/5% heat-inactivated FBS (adhesion buffer). Dilutions of
compounds to be assayed for LFA-1/ICAM-3 antagonistic activity were prepared
in
adhesion buffer from 10 mM stock solutions in DMSO and 100 ul aliquots were
added to duplicate wells. JY-8 cells (100 ql at 0.75 x 106 cells/ml in
adhesion buffer)
were then added to the wells (with or without 30 ng/ml IL-8). Microtiter
plates were
incubated for 30 min at room temperature, the adherent cells were fixed with
SO ~1 of
14% glutaraldehyde/D-PBS and incubation carried out for an additional 90 min.
The
wells were washed gently with dH,O and 50 ~l of dHZO was added, followed by 50
~1
of 1 % crystal violet. After five minutes, the plates were washed twice with
dHzO and
225 ~l ethanol (EtOH) was added to each well to extract the crystal violet
from the
cells. Absorbance was measured at 570 nm in an ELISA plate reader. The percent
inhibition of a candidate compound was calculated using the following
equation:
%inhibition = 100 X ~ 1- average OD wlcompound
average OD wlo compound
26
CA 02369005 2001-09-27
WO 00/59878 PCT/US00/08840
Example 3
Synthesis of Negative Regulators
Synthesis of various diaryl sulfide compounds according to the
invention is described below.
General Procedures and Starting Materials
In general, solvents were purchased anhydrous and were not dried or
purified further and starting materials were the best commercially available.
Thin
layer chromatography (TLC) was earned out using E. Merck, silica gel 60 F254,
0.25
mm, glass or aluminum coated plates, or Analtech silica gel uniplates (250
microns of
silica). Visualization was achieved by UV. Reported RCS indicate a single spot
detected. Flash chromatography was performed on E. Merck silica gel 60, 230-
400
mesh. Nuclear magnetic resonance (NMR) spectra were recorded on Bruker DPX 300
or Varian 300 Gemini 2000 spectrometers. Chemical shifts are reported in parts
per
million (ppm) downfield from tetramethylsilane (TMS). Coupling constants are
in
Hz. In other cases, NMR spectra were recorded using a Unity XL-200
spectrometer.
Mass spectra were recorded on either a VG 70 SEG instrument at the University
of
Washington, Department of Medicinal Chemistry, Mass Spectrometry Facility
(high
resolution) or a Finnigan Mat TSQ 70 spectrometer (low resolution). Only the
molecular ions are reported. Elemental analyses were performed by Quantitative
Technologies, Inc.
A. General Synthesis Method A: Aminodiarvlsulfides
1. General Description of Synthesis Method A
One molar equivalent (eq) of a desired thiolphenol and 1 molar
equivalent of the respective nitroaryl compound were placed in a dry flask
under
nitrogen and dissolved in dry acetone. One and a half molar equivalents
anhydrous
potassium carbonate (KZC03) was added and the mixture stirred vigorously
overnight.
The reaction was then diluted with ether, washed with saturated NaHC03, 3%
NaHS04, and saturated NaCI. The organics were dried over NazSO~ and filtered.
Heptane was then added and the solution was concentrated by boiling until most
of
27
CA 02369005 2001-09-27
WO 00/59878 PCT/CTS00/08840
the ether was removed. Upon cooling, the nitrodiaryl sulfide crystallized. The
product was collected by filtration, washed with pentane, and dried in vacuo.
One molar equivalent of the diaryl sulfide and S molar equivalents tin
chloride dihydrate were dissolved in ethanol (10 to 30 volumes). The mixture
was
heated to 60°C in an oil bath and concentrated HC1 was added (10 to 30
volumes).
After three hours, the reaction was allowed to cool and 20 to 60 volumes of
ice was
added. The mixture was neutralized to pH 10 - 12 by addition of 5 N NaOH. The
mixture was extracted twice with ether and the combined ether extracts were
washed
with saturated NaHC03 and saturated NaCI. The organic layer was dried over
Na,SO,,, filtered, and the solvent stripped by rotary evaporation. The
resulting solid or
oil was taken up in ether, and approximately 5 molar equivalents of 1 N HCl in
ether
was added drop-wise. The resulting solid was collected by filtration, washed
with
ether, and dried in vacuo.
The general synthesis method A is diagramed schematically below,
wherein X is a halogen, e.g., chlorine.
R R
X H
O_ I \ ~Ar K2C03 O_ I \ ~Ar
~+ R Acetone ~+ R
O O 1 ) SnC1z2H20
conc. HCI
EtOH
60 °C
2) HCI/ether
R
Ar
HC1 H2N R
28
CA 02369005 2001-09-27
WO 00/59878 PCT/US00/08840
2. Specific Example of General Synthesis Method A
General Synthesis Method A was employed as specifically described
below with the exception that tin granules were employed instead of the
preferred tin
chloride dihydrate shown in the schematic above.
S
2,4-Dichlorophenyl-(2'-chloro-4'-nitrophenyl)-sulfide
1.54 g (8.60 mmol) 2,4-dichlorothiophenol and 1.65 g (1 eq, 8.60
mmol) 3,4-dichloronitrobenzene were placed in a dry flask under an atmosphere
of
nitrogen and dissolved in 20 ml dry acetone. 1.78 g (1.5 eq, 12.9 mmol)
anhydrous
potassium carbonate (K,C03) was added and the mixture was stirred magnetically
for
hrs. The reaction was then diluted with 150 ml ether and then washed with
saturated NaHC03 (2 x 75 ml), 3% NaHSO~ (2 x 75 ml) and saturated NaCI (2 x 75
ml). The organic phase was then dried over anhydrous NazS04 and filtered. 50
ml
heptane was then added, and the solution was concentrated to approximately 50
ml by
15 boiling on a steam bath. Upon cooling high quality crystals formed. These
were
collected by filtration, rinsed with three portions of pentane, and dried in
vacuo
(70°C, 0.05 mm Hg, 2 hrs). 1.43 g (SO% yield) of yellow crystals were
obtained, mp
145-146 °C, Rf 0.37 (Ethyl acetate [EtAc]/Heptane, 1:20). 'H NMR
(CDC13) 6.71 (d,
J=8.9, 1H), 7.38 (d of d, J,=2.2, Jz=8.2, 1H), 7.59 (d, J=8.2, 1H), 7.63 (d,
J=2.2, 1H),
20 7.94 (d of d, J,=2.2, J,=8.9, 1H), 8.26 (d, J=2.5, 1H). MS (EI) m/z 333
(M+, 98).
Anal. Calcd for C,ZH6C13NOZS: C, 43.08; H, 1.81; N, 4.19. Found: C, 43.06; H,
1.77;
N, 4.02.
3-Chloro-4-(2,4-dichlorophenylsulfanyl)-phenylamine Hydrochloride
0.680 g (2.03 mmol)
2,4-Dichlorophenyl-(2'-chloro-4'-nitrophenyl)-sulfide and 0.844 g (3.5 eq,
7.11 mmol)
tin granules were slurned in 20 ml concentrated HCI. With vigorous stirring,
this was
heated to reflux for two days. The mixture was allowed to cool to room
temperature
and then was diluted with 50 ml ice. While stirnng, this mixture was
neutralized to
pH 10 by the addition of approx. 80 ml 5 N NaOH. This was then extracted with
ether (2 x 100 ml). The combined organics were washed with 100 ml saturated
29
CA 02369005 2001-09-27
WO 00/59878 PCT/US00/08840
NaHCO, and 2 x 100 ml saturated NaCI. This was dried over Na,SO~, and
filtered.
This was concentrated on a rotavapor to give a brown oil. The oil was taken up
in 50
ml ether, and 4 ml 1 N HCI in ether was then added, drop wise, producing a
white
solid. This was collected by filtration, rinsed with several portions of
ether, and dried
(90°C, 3 hrs, 0.05 mm Hg). 0.548 g (79% yield) of a white solid was
obtained, mp
183-185°C (dec), Rf0.33 (EtAc/Heptane 1:2 w/ 1% TEA). 'H NMR (DMSO-d6)
6.55 (d, J=8.6, 1H), 6.66 (d of d, J,=2.5, Jz=8.6, 1H), 6.89 (br s, 3H), 6.89
(d, J=2.5,
1H), 7.29 (d, J=2.2, 1H), 7.32 (d, J=2.2, 1H), 7.62 (d, J=2.2, 1H). MS (EI)
m/z 303
M+ ( 100]. Anal. Calcd for C,zHgCI3NS~HCI: C, 42.26; H, 2.66; N, 4.11. Found:
C,
42.39; H, 2.57; N, 4.14.
3. Additional Examples of Compounds Prepared by General Synthesis Method A
The following compounds were prepared from commercially available
reagents using General Synthesis Method A. The starting thiol and nitroaryl
compounds are provided, along with the intermediate (thiol + nitroaryl
intermediate).
3-Chloro-4-(2-chlorophenylsulfanyl)-phenylamine hydrochloride
2-chlorothiophenol + 3,4-dichloronitrobenzene ~ 4-nitro-2-chlorophenyl-(2'-
chlorophenyl)-sulfide
Grey solid, mp 197-198 ° C (dec), R f 0.16 (EtAc/Heptane 1:4 w/ 1
TEA). 'H NMR (DMSO-d6) 6.61 (d of d, J,=1.9, Jz=7.3, 1H), 6.70 (d of d,
J,=2.4,
JZ=8.4, 1 H), 6.94 (d, J=2.5, 1 H), 7.18 (m, 2H), 7.28 (d, J=8.9, 1 H), 7.45
(d of d,
J1=1.6, J2=7.6, 1H), 7.78 (br s, 3H). MS (EI) m/z 269 M+ (100]. Anal. Calcd
for
C,zH,oCI3NS~HCI: C, 47.22; H, 3.30; N, 4.59. Found: C, 47.34; H, 3.15; N,
4.45.
3-Chloro-4-(2-naphthylsulfanyl)-phenylamine hydrochloride
2-naphthalenethiol + 3,4-dichloronitrobenzene --~ 4-nitro-2-chlorophenyl-2-
naphthyl-
sulfide
Off white solid, mp 188 ° C (dec), Rf 0.16 (EtAc/Heptane 1:4 w/ 1
TEA). 'H NMR (DMSO-d6) 6.08 (br s, 3H), 6.60 (d of d, J,=2.4, JZ=8.4, 1H),
6.90
CA 02369005 2001-09-27
WO 00/59878 PCT/CTS00/08840
(d, J=2.5, 1H), 7.20 (d of d, J,=1.9, J,=8.6, 1H), 7.30 (d, J=8.6, 1H), 7.45
(m, 2H),
7.53 (s, 1H), 7.76 (m, 1H), 7.83 (m, 2H). MS (EI) m/z 285 M+ [100]. Anal.
Calcd
for C,6H,,C1NS~HC1: C, 59.64; H, 4.07; N, 4.35. Found: C, 59.59; H, 4.07; N,
4.09.
3-Chloro-4-(2,3-dichlorophenylsulfanyl)-phenylamine hydrochloride
2,3-dichlorothiophenol +3,4-dichloronitrobenzene ~ 4-nitro-chlorophenyl-(2',3'-
dichlorophenyl)-sulfide
Off white solid, mp 207-209°C (dec), Rf0.33 (EtAc/Heptane 1:2 w/
1% TEA). 'H NMR (DMSO-d6) 6.45 (d of d, J,=1.4, JZ=8.1, 1H), 6.62 (d of d,
J,=2.2, JZ=8.6, 1H), 6.79 (br s, 3H), 6.86 (d, J=2.5, 1H), 7.21 (t, J=7.9,
1H), 7.33 (d,
J=8.2, 1H), 7.38 (d of d, J,=1.4, JZ 8.1, 1H). MS (EI) m/z 303 (M+, 93). Anal.
Calcd
for C,ZH8C13NS~HC1: C, 42.26; H, 2.66; N, 4.11. Found: C, 42.49; H, 2.56; N,
4.10.
3-Chloro-4-(2,4,5-trichlorophenylsulfanyl)-phenylamine hydrochloride
2,3,4-trichlorothiophenol + 3,4-dichloronitrobenzene ~4-nitro-2-chlorophenyl-
(2',4',5'-trichlorophenyl)-sulfide
Off white solid, mp 192-194°C (dec), Rf0.33 (EtAc/Heptane 1:2 w/
1% TEA). 'H NMR (DMSO-d6) 6.46 (br s, 3H), 6.53 (s, 1H), 6.65 (d of d, J,=2.3,
JZ 8.4, 1H), 6.89 (d, J=2.2, 1H), 7.35 (d, J=8.6, 1H), 7.88 (s, 1H). MS (EI)
m/z 337
(M+, 73). Anal. Calcd for C,ZH,C14NS~HCI: C, 38.38; H, 2.15; N, 3.73. Found:
C,
38.73; H, 2.07; N, 3.60.
3-Methoxy-4-(2,3-dichlorophenylsulfanyl)-phenylamine
2,3-dichlorothiophenol + 2-bromo-5-nitroanisole ~ 4-nitro-2-methoxyphenyl-
(2',3'-
dichlorophenyl)-sulfide
This compound was isolated by crystallization from ether rather than
from the formation of the HCl salt. Off white crystals were obtained, mp 166-
167°C,
Rf 0.17 (EtAc/Heptane 1:2 w/ 1 % TEA). 'H NMR (DMSO-d6) 3.67 (s, 3H), 5.75 (br
s, 2H), 6.26 (d of d, J,=1.9, J,=8.3, 1H), 6.37 (d, J=1.9, 1H), 6.46 (d,
J=7.9, 1H), 7.13
(d, J=8.2, 1H), 7.16 (t, J=8.1, 1H), 7.31 (d, J=7.9, 1H). MS (EI) m/z 299 M+
[100].
Anal. Calcd for C,3H"C12NOS: C, 52.01; H, 3.69; N, 4.67. Found: C, 51.97; H,
3.59;
31
CA 02369005 2001-09-27
WO 00/59878 PCT/US00/08840
N, 4.67.
5-Amino-2-(2,3-dichlorophenylsulfanyl)-acetophenone hydrochloride
2,3-dichlorothiophenol + 2-chloronitroacetophenone -- 4-nitro-2-acetylphenyl-
(2',3'-
dichlorophenyl)-sulfide
Yellow solid, mp 151-153 oC (dec), Rf 0.35 (EtAc/Heptane 1:1 w/ 1% TEA).
1H NMR (DMSO-d6) 2.51 (s, 3H), 7.00-7.09 (m, 3H), 7.34 (t, J=7.9, 1H), 7.41
(br s,
1H), 7.44 (br s, 3H), 7.58 (d, J=7.9, 1H). MS (EI) m/z 311 M+ [100]. Anal.
Calcd for
C13H11C13NOS.HC1: C, 48.23; H, 3.47; N, 4.02. Found: C, 47.78; H, 3.43; N,
3.79.
4-(2,3-dichlorophenylsulfanyl)-phenylamine
2,3-dichlorothiophenol + 4-bromonitrophenol ~ 4-nitrophenyl-(2',3'-
dichlorophenyl)-
sulfide
Off white solid, mp 175 °C (dec), Rf 0.31 (EtAc/Heptane 1:2 w/ 1
TEA). 'H NMR (DMSO-d6) 7.13 (d, J=8.2, 2H), 7.26 (t, J=7.8, 1H), 7.42 (d,
J=8.6,
2H), 7.47 (d of d, J,=1.9, JZ=7.8, 1H), 7.76 (d of d, J,=1.3, J,=7.9, 1H),
8.04 (br s,
3H). MS (EI) m/z 269 M+ [100]. Anal. Calcd for C,ZH9C1ZNS~HC1: C, 47.00; H,
3.29;
N, 4.57. Found: C, 46.92; H, 3.36; N, 4.27.
3-Chloro-4-(1-naphthylsulfanyl)-phenylamine hydrochloride
1-naphthalenethiol + 3,4-dichloronitrobenzene~ 4-Nitro-2-chlorophenyl-(1-
naphthyl)-
sulfide
Off white solid, mp 192-194°C, Rf 0.37 (EtAc/Heptane 1:2 w/ 1
TEA). 'H NMR (DMSO-d6) 6.84 (m, 2H), 7.20 (d, J=1.9, 1H), 7.40 (d, J=7.0, 1H),
7.50 (t, J=7.8, 1 H), 7.56-7.61 (m, 2H), 7.94 (d, J=8.2, 1 H), 7.99 (m, 1 H),
8.12-8.15
(m, 5H). MS (EI) m/z 285 M+ [100]. Anal. Calcd for C,6H,zCINS'HCI: C, 59.64;
H,
4.07; N, 4.35. Found: C, 59.59; H, 4.19; N, 4.11.
3-Methyl-4-(2,4-dichlorophenylsulfanyl)-phenylamine hydrochloride
2,4-dichlorothiophenol + 2-bromo-5-nitrotoluene ~ 4-Nitro-2-methylphenyl-(2',
4'-
dichlorophenyl)-sulfide
32
CA 02369005 2001-09-27
WO 00/59878 PCT/US00/08840
Off white solid, mp 195-197°C, Rf0.41 (EtAc/Heptane 1:2 w/ 1%
TEA). 1H NMR (DMSO-d6) 2.22 (s, 3H), 6.62 (d, J=8.6, IH), 6.94 (d, J=8.2, 1H),
7.04 (br s, 1H), 7.29 (s, 1H), 7.32 (s, 1H), 7.66 (d, J=2.2, 1H), 7.89 (br s,
3H). MS
(EI) m/z 283 M+ [100]. Anal. Calcd for C,3H"C1,NS~HCI: C, 48.69; H, 3.77; N,
4.37.
Found: C, 48.87; H, 3.73; N, 4.34.
3-Bromo-4-(2,4-dichlorophenylsulfanyl)-phenylamine Hydrochloride
2,4-dichlorothiophenol + 3-bromo-4-chloronitrobenzene --~ 4-Nitro-2-
bromophenyl-
(2', 4'-dichlorophenyl)-sulfide
Off white solid, mp 171-173 °C (dec), Rf 0.69 (EtAc/Heptane l:l w/
1% TEA). 1H NMR (DMSO-d6) 6.66 (d, J=8.6, 1H), 6.81 (d of d, J,=2.2, JZ=8.4,
1 H), 7.19 (d, J=2.2, 1 H), 7.29 (d, J=8.2, 1 H), 7.32 (d of d, J,=2.2, Jz
8.6, 1 H), 7.64 (d,
J=2.2, 1H), 7.92 (br s, 3H). MS (EI) m/z 347 (M+, 60). Anal. Calcd for
C,ZHgBrCIzNS~HC1: C, 37.39; H, 2.35; N, 3.63. Found: C, 37.78; H, 2.28; N,
3.61.
2,5-Dichloro-4-(2,4-dichlorophenylsulfanyl)-phenylamine Hydrochloride
2,4-dichlorothiophenol + 2,4,5-trichloronitrophenol ~ 4-Nitro-2, 5-
dichlorophenyl-
(2', 4'-dichlorophenyl)-sulfide
Off white solid, mp 168-176°C, Rf 0.39 (EtAc/Heptane 1:2 w/ 1%
TEA). 1H NMR (DMSO-d6) 6.57 (d, J=8.6, 1H), 6.83 (br s, 3H), 7.06 (s, 1H),
7.30
(d of d, J,= 2.2, J,=8.6, 1H), 7.55 (s, 1H), 7.63 (d, J=2.2, 1H). MS (EI) m/z
347 (M+,
60). Anal. Calcd for C,zH,CI4NS~0.75HC1: C, 39.34; H, 2.13; N, 3.82. Found: C,
39.34;H,2.11;N,3.71.
4,5-Dichloro-2-(2,4-dichlorophenylsulfanyl)-phenylamine
2,4-dichlorothiophenol + 3,4-dichloro-2-fluoronitrobenzene ~ 4-Nitro-2-chloro-
5-
fluorophenyl-(2', 4'-dichlorophenyl)-sulfide
Starting with, 3,4-dichloro-2-fluoronitrobenzene, the fluoride is
displaced to give the 2-sulfanyl compound. This compound was purified by flash
chromatography as the free amine, an off white solid, mp 119-122°C, Rf
0.20
(EtAc/Heptane 1:20). 1H NMR (DMSO-d6) 5.93 (s, 2H), 6.61 (d, J=8.6, 1H), 7.08
(s,
33
CA 02369005 2001-09-27
WO 00/59878 PCT/US00/08840
1H), 7.31 (d of d, J,=2.1, J,=8.5, 1H), 7.55 (s, 1H), 7.65 (d, J=1.9, 1H). MS
(EI) m/z
337 (M+, 75). Anal. Calcd for C,zH~CI,NS: C, 42.51; H, 2.08; N, 4.13. Found:
C,
42.94; H, 1.97; N, 4.08.
3,5-Dichloro-4-(2,4-dichlorophenylsulfanyl)-phenylamine
2,4-dichlorothiophenol + 3,4,5-trichloronitrobenzene ~ 4-Nitro-2, 6-
dichlorophenyl-
(2', 4'-dichlorophenyl)-sulfide
This compound was recrystallized as the free amine to give a white
solid, mp 128-131 °C, Rf 0.36 (EtAc/Heptane 1:2). 'H NMR (DMSO-d6) 6.33
(br s,
2H), 6.46 (d, J=8.5, 1H), 6.83 (s, 2H), 7.32 (d of d, J,=2.1, JZ=8.5, 1H),
7.65 (d, J=2.2,
1H). MS (EI) m/z 337 (M+, 77). Anal. Calcd for C,zH~CI4NSC, 42.51; H, 2.08; N,
4.13. Found: C, 42.15; H, 2.10; N, 4.01.
2,3-Dichloro-4-(2,4-dichlorophenylsulfanyl)-phenylamine
2,4-dichlorothiophenol + 2,3,4-trinitrobenzene --~ 2,3-dichloro-4-(2,4-
dichlorophenylthio)-nitrobenzene
This compound was purified by flash chromatography as the free
amine to give a white solid, mp 116-119°C, Rf 0.33 (EtAc/Heptane 1:4 w/
1°.% TEA).
'H NMR (DMSO-d6) 6.09 (s, 2H), 6.47 (d, J=8.7, 1H), 6.86 (d, J=8.7, 1H), 7.32
(d of
d, J,=2.3, J,=8.7, 1H), 7.46 (d, J=8.7, 1H), 7.69 (d, J=2.2, 1H). MS (EI) m/z
337 (M+,
71). Anal. Calcd for C,ZH~C14NS: C, 42.51; H, 2.08; N, 4.13. Found: C, 42.83;
H,
2.02; N, 4.06.
B. General Synthesis Method B
A General Synthesis Method B is diagramed schematically below:
34
CA 02369005 2001-09-27
WO 00/59878 PCT/US00/08840
R R
X H
O_ ( \ ~Ar K?CO; O- \ ~Ar
+ --
Acetone +
R reflux l i R
O O
MeOH
aq. NH4Cl
reflux
R
Ar
H2N R
wherein X is a halogen, preferably chloro.
The following compounds were prepared using General Synthesis
Method B:
4-Nitro-2-chlorophenyl-(2',4'-dimethylphenyl)-sulfide
4-Amino-2-chlorophenyl-(2',4'-dimethylphenyl)-sulfide hydrochloride
4-Amino-2-chlorophenyl-(2'-methyl-4'-chlorophenyl)-sulfide
4-Amino-2-chlorophenyl-(2',4'-difluorophenyl)-sulfide hydrochloride
4-amino-2-chlorophenyl-(2',4',6'-trichlorophenyl)-sulfide
4-Amino-2-chlorophenyl-(2'-amino-4'-chlorophenyl)-sulfide
4-Amino-2-chlorophenyl-(3',4'-dichlorophenyl)-sulfide
4-Amino-2-chlorophenyl-2-(3-chloro-5-trifluoromethylpyridyl)-sulfide
Bis-(4,4'-diamino-2,2'-dichlorophenyl)-sulfide
4-Amino-2-chlorophenyl-(2',4'-dichlorophenyl)-sulfide
2-Chloro-4-amino-5-methylaminophenyl-(2',4'-dichlorophenyl)-sulfide
1 S 2-Chloro-4-amino-5-morpholinophenyl-(2',4'-dichlorophenyl)-sulfide
4-Amino-2-trifluoromethylphenyl-(2',4'-dichlorophenyl)-sulfide
4-Amino-2-fluorophenyl-(2',4'-dichlorophenyl)-sulfide
5-Amino-3-chlorophenyl-(2',4'-dichlorophenyl)-sulfide
CA 02369005 2001-09-27
WO 00/59878 PCT/US00/08840
The initial step in the General Synthesis Method B is illustrated by
preparation of the intermediate compound described immediately below.
4-Nitro-2-chlorophenol-(2',4'dimethylphenyl)-sulfide
2,4-Dimethylthiophenol ( 1.0 g) and 3,4-dichloronitrobenzene ( 1 eq)
were added to 100 ml acetone containing K,C03 (5 g). The mixture was refluxed
for
24 hr. After cooling to room temperature, the mixture was filtered and the
acetone
removed using a rotary vacuum. The resulting residue was dissolved in a small
volume of CHzCI, and filtered. The CH,C1, was then removed using a rotary
vacuum.
The resulting residue was treated with methanol (MeOH), which caused the
product to
precipitate. The yellow solid product was then collected by filtration and
dried at
40°C in a vacuum oven for 24 hr. The product yield was 66%, and the
melting point
was 128-130°C. Standard analytical techniques, including proton NMR
spectroscopy,
elemental analysis and thin layer chromatography, were employed to
characterize the
product.
The following compounds were prepared using an initial step generally in
accordance with the above illustrative intermediate.
4-Amino-2-chlorophenyl-(2',4'-dimethylphenyl)-sulfide Hydrochloride
2-Chloro-4-nitrophenyl-(2',4'-dimethylphenyl)-sulfide (0.85 g) and iron
powder in MeOH (300 ml) were added to 20 ml of an aqueous solution of NH4C1,
in a
molar ratio of 1:3:5 for substrate, iron powder and NH4C1, respectively. The
mixture
was stirred mechanically under reflux conditions overnight. The solvent was
removed
using a rotary vacuum. The resulting residue was dissolved in a small volume
of
chloroform and filtered. The product was obtained using preparative
chromatography
by applying the dissolved residue to a small column packed with silica gel (70-
230
mesh), followed by elution using a solvent system of chloroform:hexane (1:4).
Fractions containing the product were combined and the solvents removed using
a
rotary vacuum. The off white solid product was collected by filtration and
dried at
50°C in a vacuum oven for 24 hr. The product yield was 57%, and the
melting point
was 180-185 °C (decomposition). Standard analytical techniques,
including proton
36
CA 02369005 2001-09-27
WO 00/59878 PCT/US00/08840
NMR spectroscopy, elemental analysis and thin layer chromatography, were
employed
to characterize the product.
4-Amino-2-chlorophenyl-(2'-methyl-4'-chlorophenyl)-sulfide
4-Nitro-2-chlorophenyl-(2'-methyl-4'-chlorophenyl)-sulfide ( 1.0 g) and
iron powder in MeOH (300 ml) were added to 5 ml of an aqueous solution of
NH~C1,
in a molar ratio of 1:3:5 for substrate, iron powder and NH~C1, respectively.
The
mixture was stirred mechanically under reflux conditions overnight. The
solvent was
removed using a rotary vacuum. The resulting residue was dissolved in a small
volume of chloroform:hexanes (1:4) and filtered. The product was obtained
using
preparative chromatography by applying the dissolved residue~to a small column
packed with silica gel (70-230 mesh), followed by elution using a solvent
system of
chloroform:hexane ( 1:1 ). Fractions containing the product were combined and
the
solvents removed using a rotary vacuum. The white solid product was collected
by
filtration and dried at 60°C in a vacuum oven for 24 hr. The product
yield was 71%,
and the melting point was 91-93°C. Standard analytical techniques,
including proton
NMR spectroscopy, elemental analysis and thin layer chromatography, were
employed
to characterize the product.
4-Amino-2-chlorophenyl-(2',4'-difluorophenyl)-sulfide Hydrochloride
4-Nitro-2-chlorophenyl-(2',4'-difluorophenyl)-sulfide and iron powder
in MeOH (300 ml) were added to 20 ml of an aqueous solution of NH4Cl, in a
molar
ratio of 1:3:5 for substrate, iron powder and NH~CI, respectively. The mixture
was
stirred mechanically under reflux conditions overnight. The solvent was
removed
using a rotary vacuum. The resulting residue was dissolved in a small volume
of
chloroform and filtered. The product was obtained using preparative
chromatography
by applying the dissolved residue to a small column packed with silica gel (70-
230
mesh), followed by elution using a solvent system of chloroform:hexane (1:5).
Fractions containing the product were combined and the solvents removed using
a
rotary vacuum. The colorless solid product was collected by filtration and
dried at
SO°C in a vacuum oven for 24 hr. The product yield was 88%, and the
melting point
37
CA 02369005 2001-09-27
WO 00/59878 PCT/US00/08840
was 187-190°C. Standard analytical techniques, including proton NMR
spectroscopy,
elemental analysis and thin layer chromatography, were employed to
characterize the
product.
4-amino-2-chlorophenyl-(2',4',6'-trichlorophenyl)-sulfide
4-Nitro-2-chlorophenyl-(2',4',6;-trichlorophenyl)-sulfide (0.6 g) and
iron powder in MeOH (250 ml) were added to 32 ml of an aqueous solution of
NH~,CI,
in a molar ratio of 1:3:5 for substrate, iron powder and NH~C1, respectively.
The
mixture was stirred mechanically under reflux conditions overnight. The
solvent was
removed using a rotary vacuum. The resulting residue was dissolved in a small
volume of chloroform and filtered. The product was obtained using preparative
chromatography by applying the dissolved residue to a small column packed with
silica gel (70-230 mesh), followed by elution using a solvent system of
chloroform:hexane (1:3). Fractions containing the product were combined and
the
solvents removed using a rotary vacuum. The white solid product was collected
by
filtration and dried at 40°C in a vacuum oven for 24 hr. The product
yield was 72%,
and the melting point was 109-111 °C. Standard analytical techniques,
including
proton NMR spectroscopy, elemental analysis and thin layer chromatography,
were
employed to characterize the product.
4-Amino-2-chlorophenyl-(2'-amino-4'-chlorophenyl)-sulfide
4-Amino-2-chlorophenyl-(2'-nitro-4'-chlorophenyl)-sulfide ( 1.02 g)
and iron powder in MeOH (250 ml) were added to 60 ml of an aqueous solution of
NH~CI, in a molar ratio of 1:3:5 for substrate, iron powder and NHaCI,
respectively.
The mixture was stirred mechanically under reflux conditions overnight. The
solvent
was removed using a rotary vacuum. The resulting residue was dissolved in a
small
volume of chloroform and filtered. The product was obtained using preparative
chromatography by applying the dissolved residue to a small column packed with
silica gel (70-230 mesh), followed by elution using a solvent system of
chloroform:hexane (3:7). Fractions containing the product were combined and
the
solvents removed using a rotary vacuum. The off white solid product was
collected
38
CA 02369005 2001-09-27
WO 00/59878 PCT/US00/08840
by filtration and dried at 50°C in a vacuum oven for 24 hr. The product
yield was
59%, and the melting point was 86-88°C. Standard analytical techniques,
including
proton NMR spectroscopy, elemental analysis and thin layer chromatography,
were
employed to characterize the product.
4-Amino-2-chlorophenyl-(3',4'-dichlorophenyl)-sulfide
4-Nitro-2-chlorophenyl-(3',4'-dichlorophenyl)-sulfide (1.0 g) and iron
powder in MeOH (250 ml) were added to 40 ml of an aqueous solution of NH;CI,
in a
molar ratio of 1:3:5 for substrate, iron powder and NH~CI, respectively. The
mixture
was stirred mechanically under reflux conditions overnight. The solvent was
removed
using a rotary vacuum. The resulting residue was dissolved in a small volume
of
chloroform and filtered. The product was obtained using preparative
chromatography
by applying the dissolved residue to a small column packed with silica gel (70-
230
mesh), followed by elution using a solvent system of chloroform:hexane (3:7).
Fractions containing the product were combined and the solvents removed using
a
rotary vacuum. The colorless solid product was collected by filtration and
dried at
70 ° C in a vacuum oven for 24 hr. The product melting point was 103-
105 ° C.
Standard analytical techniques, including proton NMR spectroscopy, elemental
analysis and thin layer chromatography, were employed to characterize the
product.
4-Amino-2-chlorophenyl-2-(3-chloro-5-trifluoromethylpyridyl)-sulfide
4-Nitro-2-chlorophenyl-2-(3-chloro-5-trifluoromethylpyridyl)-sulfide
(1.5 g) and iron powder in MeOH (250 ml) were added to 80 ml of an aqueous
solution of NH4C1, in a molar ratio of 1:3:5 for substrate, iron powder and
NH4C1,
respectively. The mixture was stirred mechanically under reflux conditions
overnight.
The solvent was removed using a rotary vacuum. The resulting residue was
dissolved
in a small volume of chloroform and filtered. The product was obtained using
preparative chromatography by applying the dissolved residue to a small column
packed with silica gel (70-230 mesh), followed by elution using a solvent
system of
chloroform:hexane (3:7). Fractions containing the product were combined and
the
solvents removed using a rotary vacuum. The colorless solid product was
collected
39
CA 02369005 2001-09-27
WO 00/59878 PCT/US00/08840
by filtration and dried at 60°C in a vacuum oven for 24 hr. The product
yield was
97%, and the melting point was 96-98°C. Standard analytical techniques,
including
proton NMR spectroscopy, elemental analysis and thin layer chromatography,
were
employed to characterize the product.
Bis-(4,4'-diamino-2,2'-dichlorophenyl)-sulfide
4-Amino-2-chlorophenyl-(2'-chloro-4'-nitrophenyl)-sulfide and iron
powder in MeOH (125 ml) were added to 20 ml of an aqueous solution ofNH~CI, in
a
molar ratio of 1:3:5 for substrate, iron powder and NH~C1, respectively. The
mixture
was stirred mechanically under reflux conditions overnight. The solvent was
removed
using a rotary vacuum. The resulting residue was dissolved in a small volume
of
chloroform and filtered. The product was obtained using preparative
chromatography
by applying the dissolved residue to a small column packed with silica gel (70-
230
mesh), followed by elution using a solvent system of chloroform:hexane (3:7).
Fractions containing the product were combined and the solvents removed using
a
rotary vacuum. The off white solid product was collected by filtration and
dried at
60°C in a vacuum oven for 24 hr. The product yield was 66%, and the
melting point
was 113-115 °C. Standard analytical techniques, including proton NMR
spectroscopy,
elemental analysis and thin layer chromatography, were employed to
characterize the
product.
4-Amino-2-chlorophenyl-(2',4'-dichlorophenyl)-sulfide
2-Chloro-4-nitrophenyl-(2',4'-dichlorophenyl)-sulfide and iron powder
in MeOH (50 ml) were added to 50 ml of an aqueous solution of NH4C1, in a
molar
ratio of 1:3:5 for substrate, iron powder and NH4C1 respectively. The mixture
was
stirred mechanically under reflux conditions overnight. The solvent was
removed
using a rotary vacuum. The resulting residue was dissolved in a small volume
of
chloroform and filtered, and the solvent was then removed from the filtrate
using a
rotary evaporator. The product was obtained by stirring the residue with 75 ml
of
distilled water (dH20). The colorless solid precipitate was collected by
filtration. The
product yield was 83%, and the melting point was 105-107°C. Standard
analytical
CA 02369005 2001-09-27
WO 00/59878 PCT/US00/08840
techniques, including proton NMR spectroscopy, elemental analysis and thin
layer
chromatography, were employed to characterize the product.
4-Amino-2-chlorophenyl-(4'-acetamido-2'-chlorophenyl)-sulfide
4-Nitro-2-chlorophenyl-(4'-acetamido-2'-chlorophenyl)-sulfide and
iron powder in MeOH (250 ml) were added to 20 ml of an aqueous solution of
NH~,CI,
in a molar ratio of 1:3:5 for substrate, iron powder and NH~C1, respectively.
The
mixture was stirred mechanically under reflux conditions overnight. The
solvent was
removed using a rotary vacuum. The resulting residue was dissolved in a small
volume of chloroform and filtered. The product was obtained using preparative
chromatography by applying the dissolved residue to a small column packed with
silica gel (70-230 mesh), followed by elution using a solvent system of
chloroform-hexane (1:4). Fractions containing the product were combined and
the
solvents removed using a rotary vacuum. The off white solid product was
collected
by filtration and dried at 60°C in a vacuum oven for 24 hr. The product
yield was
76%, and the melting point was 170-175 °C. Standard analytical
techniques,
including proton NMR spectroscopy, elemental analysis and thin layer
chromatography, were employed to characterize the product.
4-Amino-2-chlorophenyl-(4'-dimethylamino-2'-chlorophenyl)-sulfide
4-Nitro-2-chlorophenyl-(4'-dimethylamino-2'-chlorophenyl)-sulfide
(0.2 g) and iron powder in MeOH (250 ml) were added to 20 ml of an aqueous
solution of NH4C1, in a molar ratio of 1:3:5 for substrate, iron powder and
NH4C1
respectively. The mixture was stirred mechanically under reflux conditions
overnight.
The solvent was removed using a rotary vacuum. The resulting residue was
dissolved
in a small volume of chloroform and filtered. The product was obtained using
preparative chromatography by applying the dissolved residue to a small column
packed with silica gel (70-230 mesh), followed by elution using a solvent
system of
chloroform:hexane (2:3). Fractions containing the product were combined and
the
solvents removed using a rotary vacuum. The pale yellow solid product was
collected
by filtration and dried at 60°C in a vacuum oven for 24 hr. The product
yield was
41
CA 02369005 2001-09-27
WO 00/59878 PCT/US00/08840
58%, and the melting point was 133-135°C. Standard analytical
techniques,
including proton NMR spectroscopy, elemental analysis and thin layer
chromatography, were employed to characterize the product.
2-Chloro-4-amino-5-methylaminophenyl-(2',4'-dichlorophenyl)-sulfide
2-Chloro-4-nitro-5-methylaminophenyl-(2',4'-dichlorophenyl)-sulfide
(0.2 g) and iron powder in methanol (250 ml) were added to 20 ml of an aqueous
solution of NH~CI, in a molar ratio of 1:3:5 for substrate, iron powder and
NH4C1
respectively. The mixture was stirred mechanically under reflux conditions
overnight.
The solvent was removed using a rotary vacuum. The resulting residue was
dissolved
in a small volume of chloroform and filtered. The product was obtained using
preparative chromatography by applying the dissolved residue to a small column
packed with silica gel (70-230 mesh), followed by elution using a solvent
system of
chloroform:hexane ( 1:4). Fractions containing the product were combined and
the
solvents removed using a rotary vacuum. The brown solid product was collected
by
filtration and dried at 50°C in a vacuum oven for 24 hr. The product
yield was 16%,
and the melting point was 65-70°C. Standard analytical techniques,
including proton
NMR spectroscopy, elemental analysis and thin layer chromatography, were
employed
to characterize the product.
2-Chloro-4-amino-5-N-morpholinophenyl-(2',4'-dichlorophenyl)-sulfide
2-Chloro-4-nitro-5-N-morpholinophenyl-(2',4'-dichlorophenyl)-sulfide
(0.9 g) and iron 'powder in MeOH (250 ml) were added to 20 ml of an aqueous
solution of NH4C1, in a molar ratio of 1:3:5 for substrate, iron powder and
NH~C1
respectively. The mixture was stirred mechanically under reflux conditions
overnight.
The solvent was removed using a rotary vacuum. The resulting residue was
dissolved
in a small volume of chloroform and filtered. The product was obtained using
preparative chromatography by applying the dissolved mixture to a small column
packed with silica gel (70-230 mesh), followed by elution using a solvent
system of
chloroform:hexane (3:2). Fractions containing the product were combined and
the
solvents removed using a rotary vacuum. The off white solid product was
collected
42
CA 02369005 2001-09-27
WO 00/59878 PCT/US00/08840
by filtration and dried at 50°C in a vacuum oven for 24 hr. The product
melting point
was 153-155 °C. Standard analytical techniques, including proton NMR
spectroscopy,
elemental analysis and thin layer chromatography, were employed to
characterize the
product.
4-Amino-2-trifluoromethylphenyl-(2',4'-dichlorophenyl)-sulfide
4-Nitro-2-trifluoromethylphenyl-(2',4'-dichlorophenyl)-sulfide (0.6 g)
and iron powder in MeOH (250 ml) were added to 20 ml of an aqueous solution of
NH4C1, in a molar ratio of 1:3:5 for substrate, iron powder and NH~CI,
respectively.
The mixture was stirred mechanically under reflux conditions overnight. The
solvent
was removed using a rotary vacuum. The resulting residue was dissolved in a
small
volume of chloroform and filtered. The product was obtained using preparative
chromatography by applying the dissolved residue to a small column packed with
silica gel (70-230 mesh), followed by elution using a solvent system of
chloroform:hexane (1:1). Fractions containing the product were combined and
the
solvents removed using a rotary vacuum. The product was collected by
filtration and
dried at 50°C in a vacuum oven for 24 hr. The product melting point was
not
determined because it was a colorless oil. Standard analytical techniques,
including
proton NMR spectroscopy, elemental analysis and thin layer chromatography,
were
employed to characterize the product.
4-Amino-2-fluorophenyl-(2',4'-dichlorophenyl)-sulfide
4-Nitro-2-fluorophenyl-(2',4'-dichlorophenyl)-sulfide and iron powder
in MeOH (250 ml) were added to 20 ml of an aqueous solution of NH~CI, in a
molar
ratio of 1:3:5 for substrate, iron powder and NH4Cl, respectively. The mixture
was
stirred mechanically under reflux conditions overnight. The solvent was
removed
using a rotary vacuum. The resulting residue was dissolved in a small volume
of
chloroform and filtered. The product was obtained using preparative
chromatography
by applying the dissolved residue to a small column packed with silica gel (70-
230
mesh), followed by elution using a solvent system of chloroform:hexane (l:l).
Fractions containing the product were combined and the solvents removed using
a
43
CA 02369005 2001-09-27
WO 00/59878 PCT/LTS00/08840
rotary vacuum. The pale yellow solid product was collected by filtration and
dried at
30°C in a vacuum oven for 24 hr. The product yield was 55%, and the
melting point
was 101-102°C. Standard analytical techniques, including proton NMR
spectroscopy,
elemental analysis and thin layer chromatography, were employed to
characterize the
product.
5-Amino-3-chlorophenyl-(2',4'-dichlorophenyl)-sulfide
3-Chloro-5-nitrophenyl-(2',4'-dichlorophenyl)-sulfide ( 1.0 g) and iron
powder in MeOH (250 ml) were added to 20 ml of an aqueous solution of NH~C1,
in a
molar ratio of 1:3:5 for substrate, iron powder and NH4C1, respectively. The
mixture
was stirred mechanically under reflux conditions overnight. The solvent was
removed
using a rotary vacuum. The resulting residue was dissolved in a small volume
of
chloroform and filtered. The product was obtained using preparative
chromatography
by applying the dissolved residue to a small column packed with silica gel (70-
230
1 S mesh), followed by elution using a solvent system of chloroform:hexane
(3:2).
Fractions containing the product were combined and the solvents removed using
a
rotary vacuum. The product was collected by filtration and dried at 25
° C in a vacuum
oven for 24 hr. The product yield was 16%, and the melting point was not
determined
because it was a brown oil. Standard analytical techniques, including proton
NMR
spectroscopy, elemental analysis and thin layer chromatography, were employed
to
characterize the product.
C. General Synthesis Method C
A General Synthesis Method C is diagramed schematically below:
Cl Cl I I
SH O~ N+.O_ K2C03 ~ ~ S~Ar N ~O_
H N ~ ~ Acetone H N
reflux
44
CA 02369005 2001-09-27
WO 00/59878 PCT/US00/08840
The following compounds were prepared by General Synthesis Method C:
4-Amino-2-chlorophenyl-(2'-chloro-4'-nitrophenyl)-sulfide
4-Amino-2-chlorophenyl-(2'-vitro-4'-chlorophenyl)-sulfide
4-Amino-2-chlorophenyl-6-(S-nitroquinolino)-sulfide
4-Amino-2-chlorophenyl-(2'-chloro-4'-nitrophenyl)-sulfide
2-Chloro-4-aminothiophenol (2.0 g) and 3,4-dichloronitrobenzene ( 1
eq.) were added to 300 ml of acetone containing K,C03 (20 g). The mixture was
refluxed for 24 hr at 60°C. After cooling to room temperature, the
mixture was
filtered and the acetone removed using a rotary vacuum. The resulting residue
was
dissolved in a small volume of chloroform and filtered. The chloroform was
then
removed using a rotary vacuum. The resulting residue was triturated with MeOH,
which caused the product to precipitate. The precipitate was collected by
filtration
and the yellow solid product dried at 80°C in a vacuum oven for 24 hr.
The product
yield was 59%, and the melting point was 135-136°C. Standard analytical
techniques, including proton NMR spectroscopy, elemental analysis and thin
layer
chromatography, were employed to characterize the product.
4-Amino-2-chlorophenyl-(2'-vitro-4'-chlorophenyl)-sulfide
2-Chloro-4-aminothiophenol (2.0 g) and 2,5-dichloronitrobenzene (1
eq.) were added to 300 ml of acetone containing K,C03 (20 g). The mixture was
refluxed for 24 hr at 60°C. After cooling to room temperature, the
mixture was
filtered and the acetone removed using a rotary vacuum. The resulting residue
was
dissolved in a small volume of CHzCI, and filtered. The CHZC12 was then
removed
using a rotary vacuum. The resulting residue was triturated with MeOH, which
caused the product to precipitate. The precipitate was collected by filtration
and the
yellow solid product dried at 60°C in a vacuum oven for 24 hr. The
product yield was
57%, and the melting point was 191-193 °C. Standard analytical
techniques,
including proton NMR spectroscopy, elemental analysis and thin layer
chromatography, were employed to characterize the product.
CA 02369005 2001-09-27
WO 00/59878 PCT/US00/08840
4-Amino-2-chlorophenyl-6-(5-nitroquinolino)-sulfide
2-Chloro-4-aminothiophenol (0.474 g) and 6-chloro-5-nitroquinoline
(1 eq.) were added to 250 ml of acetone containing K,C03 (20 g). The mixture
was
refluxed for 24 hr at 60°C. After cooling to room temperature, the
mixture was
filtered and the acetone removed using a rotary vacuum. The resulting residue
was
dissolved in a small volume of chloroform and filtered. The chloroform was
then
removed using a rotary vacuum. The resulting residue was triturated with MeOH,
which caused the product to precipitate. The product was obtained using
preparative
chromatography by applying the dissolved residue to a small column packed with
silica gel (70-230 mesh), followed by elution using a solvent system of
chloroform:hexane (3:2). Fractions containing the product were combined and
the
solvents removed using a rotary vacuum. The yellow solid product was collected
by
filtration and dried at 50°C in a vacuum oven for 24 hr. The product
yield was 62%,
and the melting point was 129-131 °C. Standard analytical techniques,
including
proton NMR spectroscopy, elemental analysis and thin layer chromatography,
were
employed to characterize the product.
D. Specific Synthesis Procedures
Full preparative methods are provided for the following compounds:
1-Acetamido-3-chloro-4-(2,3-dichlorophenylsulfanyl)-benzene
3-Hydroxy-4-(2,3-dichlorophenylsulfanyl)-phenylamine Hydrochloride
6-Chloro-5-(2,4-dichlorophenylsulfanyl)-1H-benzimidazole
1-Acetamido-3-chloro-4-(2,3-dichlorophenylsulfanyl)-benzene
0.165 g (2.1 eq, 1.35 mmol) 4-dimethylaminopyridine (4-DMAP) was
placed into a dry flask under an atmosphere of nitrogen and dissolved in 5 ml
anhydrous tetrahydrofuran (THF). 2 ml of acetic anhydride was added followed
by
0.220 g (1 eq, 0.643 mmol) 3-chloro-4-(2,3-dichlorophenylsulfanyl)-phenylamine
hydrochloride. This was stirred for 18 hrs. The mixture was then diluted with
75 ml
ether and washed with sat. NaHC03 (3 x SO ml), 0.3 N HCl (3 x 30 ml) and
saturated
NaCI (2 x 30 ml), dried over NaZSO~, filtered, and the solvent stripped on a
rotavapor.
46
CA 02369005 2001-09-27
WO 00/59878 PCT/LTS00/08840
Flash chromatography ( 1.9 x 27 cm, EtAc/Heptane ( 1:1 )) afforded 0.135 g (61
yield) of a white solid, mp 167-169°C, Rf 0.25 (EtAc/Heptane 1:1). 'H
NMR
(DMSO-d6) 2.07 (s, 3H), 6.59 (d of d, J,=1.3, J,=7.9, 1H), 7.24 (t, J=8.1,
1H), 7.46 (d
of d, J,=1.4, JZ=8.1, 1H), 7.55 (m, 2H), 8.04 (d, J=1.9, 1H), 10.35 (s, 1H).
MS (EI)
m/z 345 (M+, 82). Anal. Calcd for C,,,H,oCI3NOS: C, 48.51; H, 2.91; N, 4.04.
Found: C, 48.29; H, 2.88; N, 3.92.
3-Hydroxy-4-(2,3-dichlorophenylsulfanyl)-phenylamine Hydrochloride
0.19 g (0.67 mmol) 3-Methoxy-4-(2,3-dichlorophenylsulfanyl) phenyl-
mine was placed into a dry flask under an atmosphere of nitrogen, dissolved in
10 ml
dry CH,CI,, and cooled to -78°. 0.32 ml (5 eq., 3.3 mmol) boron
tribromide was
added drop-wise, with stirnng. The cold bath was removed and this was left to
react
for 20 hrs. The solution was again cooled to -78 °, and then 10 ml MeOH
was added
drop-wise. This was allowed to warm to room temperature and was stirred for 1
hr.
1 S The solvent was stripped on a rotavapor and the resulting oil was twice
taken up in 10
ml MeOH and again stripped. The material was again taken up in 10 ml MeOH and
then diluted with 100 ml EtAc. The white precipitate that formed was removed
by
filtration, and then the filtrate was washed with saturated NaCI (3 x 50 ml),
dried over
Na,S04, filtered, and the solvent stripped by rotary evaporation. The
resulting oil was
purified by flash chromatography (2.9 x 28 cm, EtAc/Heptane (1:3)) and was
then
dissolved in 25 ml of ether and precipitated as the HC1 salt by the addition
of 5 ml 1 N
HC1 in ether. This was collected by filtration, washed with ether, and dried
(90 oC, 3
hr, 0.3 mm Hg)'to gave 0.17 g (80% yield) of an off white solid, mp
222°C (dec), Rf
0.49 (EtAc/Heptane 1:1). 'H NMR (DMSO-d6) 6.56-6.61 (m, 2H), 6.79 (d, J=1.9,
1H), 6.90 (br hump), 7.19 (t, J=8.1, 1H), 7.27 (d, J=8.2, 1H), 7.38 (d of d,
J,=1.4,
JZ=8.1, 1H), 10.37 (br s, 1H). MS (EI) m/z 285 M+ [100]. Anal. Calcd for
C,ZH9Cl,NOS'HCI: C, 44.67; H, 3.12; N, 4.34. Found: C, 44.53; H, 2.91; N;
4.17.
6-Chloro-5-(2,4-dichlorophenylsulfanyl)-1H-benzimidazole
4-Chloro-2-nitro-S-(2,4-dichlorophenylsulfanyl)-phenylamine was
prepared by the general procedure with the exception that it was purified as
the free
47
CA 02369005 2001-09-27
WO 00/59878 PCT/LTS00/08840
amine by flash chromatography. 1.37 g (3.93 mmol) of the free amine was added
to
ml DMF and 10 ml of EtOH. 4.43 g (5 eq, 19.7 mmol) tin chloride dihydrate was
added followed by 10 ml concentrated HC1. This was heated to 60°C for
20 hrs. The
reaction was allowed to cool to ambient temperature, and was then diluted with
30 ml
5 water, and brought to pH 12 by the addition of 30 ml 5 N NaOH. This mixture
was
twice extracted with 150 ml ether. The combined organics were washed with 100
ml
sat. NaHC03 and 2 x 100 ml sat. NaCI, dried over Na,SO~, and filtered. 40 ml
heptane
was then added, and this was concentrated on a rotavapor and dried in vacuo (
100 ° C,
2 hr, 0.3 mm Hg) to yield 1.06 g (82%) of an analytically pure white solid, mp
10 206-208 ° C, Rf 0.51 (CHzCI,/MeOH (9:1 ) w/ 1 % TEA). 'H NMR (DMSO-
d6) 6.63
(d, J=8.6, 1H), 7.28 (d of d, J,=2.2, JZ=8.7, 1H), 7.69 (d, J=2.2; 1H), 7.86
(br s, 1H),
7.93 (s, 1H), 8.38 (s, 1H), 12.80 (s, 1H). MS (EI) m/z 328 (M+, 97). Anal.
Calcd for
C,3H,C13NZS: C, 47.37; H, 2.14; N, 8.50. Found: C, 47.40; H, 2.04; N, 8.32.
E. Specific Synthesis Protocols
The following compounds were prepared by methods as described below.
4-Methylamino-2,2',4'-trichlorodiphenylsulfide
4-Amino-2-chlorophenyl-(4'-acetamido-2'-chlorophenyl)-sulfide
4-Amino-2-chlorophenyl-(4'-dimethylamino-2'-chlorophenyl)-sulfide
4-Aminomethyl-2-chlorophenyl-(2',4'-dichlorophenyl)-sulfide
4-Methylamino-2,2',4'-trichlorodiphenylsulfide
4-Amino-2,2',4'-trichlorodiphenyl-sulfide (0.305 g, 1.0 mmol) was
added to 15 ml of formic acid. The mixture was stirred for 8 hr, after which
0.23 ml
of 37% formaldehyde was added and the mixture refluxed for 8 hr. The solvent
was
then removed using a rotary evaporator. The resulting residue was then applied
to a
small column containing silica gel (70-230 mesh). The product was then eluted
from
the column using chloroform. The solvent was removed from the eluate using a
rotary evaporator and the pale yellow solid product collected. The product
yield was
44%, and the melting point was 211 °C. Standard analytical techniques,
including
proton NMR spectroscopy, elemental analysis and thin layer chromatography,
were
48
CA 02369005 2001-09-27
WO 00/59878 PCT/US00/08840
employed to characterize the product.
4-Nitro-2-chlorophenyl-(4'-acetamido-2'-chlorophenyl)-sulfide
4-Amino-2,2'-dichloro-4'-nitrodiphenylsulfide (1.0 g, 3.17 mmol) was
warmed in 15 ml of acetic anhydride containing a trace ofp-toluenesulfonic
acid. The
mixture was allowed to stand for 1 hr, after which the solvent was removed
using a
rotary evaporator. The residue was dissolved in EtAc and poured over a small
column
of silica gel (70-230 mesh). The filtrate was collected, evaporated to
dryness, and
recrystallized from acetonitrile to give a yellow solid product. The product
yield was
97%, and the melting point was 163-165°C. Standard analytical
techniques,
including proton NMR spectroscopy, elemental analysis and thin layer
chromatography, were employed to characterize the product.
4-Nitro-2-chlorophenyl-(4'-dimethylamino-2'-chlorophenyl)-sulfide
4-Amino-2-chlorophenyl-(2'-chloro-4'-nitrophenyl)-sulfide ( 1.0 g, 3.17
mmol) was added to a suspension of 60% sodium hydride (1.43 g) in 250 ml of
THF
at 0°C. Iodomethane (0.2 ml, in 20 ml of THF) was then added, and the
mixture
stirred at room temperature for 48 hr. The mixture was then applied to
Analtech silica
gel plates. Each plate had a thickness of 1000 microns of silica. The two
products of
the reaction, i.e., 4-nitro-2-chlorophenyl-(4'-methylamino-2'-chlorophenyl)-
sulfide and
4-nitro-2-chlorophenyl-(4'-dimethylamino-2'-chlorophenyl)-sulfide, were eluted
using
a 1:1 mixture of CHC13 and hexane. The slower eluting band corresponded to the
former and the faster eluting band corresponded to the latter. After
collecting the
bands, the compounds were dissolved in CHCl3. The solvent was removed by
rotary
evaporation to provide a yellow solid product. The yield of the desired
product was
36%, and the melting point was 138-139°C. Standard analytical
techniques,
including proton NMR spectroscopy, elemental analysis and thin layer
chromatography, were employed to characterize the product.
4-Aminomethyl-2-chlorophenyl-(2',4'-dichlorophenyl)-sulfide
2-Chloro-4-cyanopheny-(2',4'-dichlorophenyl)-sulfide (1.0 g, 3.18
49
CA 02369005 2001-09-27
WO 00/59878 PCT/US00/08840
mmol) was added to 200 ml of THF under nitrogen at 0°C. Lithium
aluminum
hydride (0.24 g) was then added to the solution in portions. The mixture was
then
allowed to stand for 2 hr at 0°C, after which 1 ml of 20% NaOH was
added to the
mixture, followed by 1 ml of dH,O. The solution was then filtered, and
solvents were
removed using a rotary evaporator. The resulting residue was then applied to a
small
column containing silica gel (70-230 mesh). The column was washed with a 1:1
mixture of chloroform and hexane, after which the product was eluted using a
rotary
evaporator, and the product was obtained as a colorless oil. The product yield
was
40%, and the melting point was not determined. Standard analytical techniques,
including proton NMR spectroscopy, elemental analysis and thin layer
chromatography, were employed to characterize the product.
F. Other compounds
Other compounds synthesized in accordance with the general synthetic
methods include the following.
Table II
4-Nitro-2-chlorophenyl-(2',4'-dimethylphenyl)-sulfide
4-Nitro-2-chlorophenyl-(2'-methyl-4'-chlorophenyl)-sulfide
4-Nitro-2-chlorophenyl-(2', 4'-difluorophenyl)-sulfide
4-Nitro-2-chlorophenyl-(2', 4', 6'-trichlorophenyl)-sulfide
4-Nitro-2-chlorophenyl-(3', 4'-dichlorophenyl)-sulfide
4-Nitro-2-chlorophenyl-2-(3-chloro-5-trifluoromethylpyridyl)-sulfide
2-Chloro-4-nitrophenyl-(2', 4'-dichlorophenyl)-sulfide
4-Nitro-2-chlorophenyl-(4'-acetamido-2'-chlorophenyl)-sulfide
4-Nitro-2-chlorophenyl-(4'-dimethylamino-2'-chlorophenyl)-sulfide
2-Chloro-4-nitro-5-methylaminophenyl-(2', 4'-dichlorophenyl)-sulfide
2-Chloro-4-nitro-5-morpholinophenyl-(2', 4'-dichlorophenyl)-sulfide
4-Nitro-2-trifluoromethylphenyl-(2', 4'-dichlorophenyl)-sulfide
4-Nitro-2-fluorophenyl-(2', 4'-dichlorophenyl)-sulfide
3-Chloro-5-nitrophenyl-(2', 4'-dichlorophenyl)-sulfide
CA 02369005 2001-09-27
WO 00/59878 PCT/US00/08840
4-Nitro-2-chlorophenyl-( 1-naphthyl)-sulfide
4-Nitro-2-methylphenyl-(2', 4'-dichlorophenyl)-sulfide
4-Nitro-2-bromophenyl-(2', 4'-dichlorophenyl)-sulfide
4-Nitro-2, 5-dichlorophenyl-(2', 4'-dichlorophenyl)-sulfide
4-Nitro-2, 6-dichlorophenyl-(2', 4'-dichlorophenyl)-sulfide
4-Nitro-2-chloro-5-fluorophenyl-(2', 4'-dichlorophenyl)-sulfide
4-Nitro-2, 3-dichlorophenyl-(2', 4'-dichlorophenyl)-sulfide
4-Nitro-2-chlorophenyl-(4'-chloro-2'-aminophenyl)-sulfide
4-Nitro-5-acetamido-2-chlorophenyl-(2', 4'-dichlorophenyl)-sulfide
4-Nitro-2-chlorophenyl-(4'-methylamino-2'-chlorophenyl)-sulfide
4-Nitro-2-chlorophenyl-(4'-benzylamino-2'-chlorophenyl)-sulfide
4-Nitro-2-chlorophenyl-(4'-dibenzylamino-2'-chlorophenyl)-sulfide
4-Nitro-5-phenylsulfo-2-chlorophenyl-(2', 4'-dichlorophenyl)-sulfide
3-Nitro-5-chlorophenyl-(2', 4'-dichlorophenyl)-sulfide
Example 4
Cell-Based Assay Results
Compounds of the invention displayed activity in the cell-based assays
described above as set out in Table III below which provides pM ICso values
for
inhibition of LFA-1 binding to ICAM-1 and ICAM-3 where tested. Paired values
(X/Y) indicate inhibition in the absence and presence of IL-8. Multiple paired
values
(W/X; Y/Z) indicate repeated experiements. Dashes (--/X) indicate the
experiment
was not performed. "NT" indicates that the compound was not tested in a
particular
assay.
While the present invention has been described in terms of specific
embodiments, it is understood that variations and modifications will occur to
those
skilled in the art. Accordingly, only such limitations as appear in the
appended claims
should be placed on the invention.
51
CA 02369005 2001-09-27
WO 00/59878 PCT/US00/08840
0
ri
M
O ~t . M O
_ N N
U + ~-' E-~ o H cu E-- E- F- E- E-~ E-- E- F- H F- F- = F- F-y E-~ ~.» ~ n ~ E-
~ E~- = E
;zz~z~zzzzzzzzzzz~zzzzzNzzzNz
0
o O
O N
N
N
.., ~ ~ n, N .-. v~ .--~ .-, vp ~ N
w W1 O ~J W
O M ~ ~ M M
I~ O ~ ~ O ~ ~ O\ V ~ O ~ a1
.-. M . ~ M I~ V~ ~ O N N ~ O M V' 00 O O ~ V1 V~
i.r + v7 ~ ~ ~ . ., ~: ~ M N I~ ~ 'V N ~ ~ O 01
i I~ (V I~ N 01 . cV v~ /~ N .-~ /~ N ~ N .--~ N M .~ ~ .~ M M v N
I~ O .~ ~-, v'1 O O ..-. 00 ~ ~ [w w w w M w w ,_, ~ w ~ w, yp w
w w w w w w O w N 00 ~T w ~ V1 w O O W h' w ~ w O
V ~:J 00 .--~ Vwp ~ N o\o M ~ N ~ O ,~: I~ N M M ~J ~ C1 O ~ I~ M ~n Q l~
r~ r.y~ (~ ~--, I~ ~' i i i i l~ i M ~ ~ N ~ M ~-r 00 N ~-~ ~D ~-~ 01 M ~O
N
'b
U
'O a~ a~ y '~ b O a~ ~C
;'C -p b Q
U .O .-O .O O .O .O U .~ O .~ N b
b 0 ~ V .~ U
.~ o b . j .~ o N ~ "~ a~ .b x ~O .~ aW0
U ' s:. ~ >, ~ W., p 4~ ~,
~° 'd, ~ ~ x o ~ a~ ~ e.~°~. -o -o
O_~~-~Q.Ua~p~>,4:.~0..'~'y~~pp~OON~4:.~N
O. ~ t~..~ ~ cd . ~ ~ fl.. p O p" ~ .-. ..C >, .-~ U O O
chi i'~. ~ .~ ~ ~ chi ~'~~ ~ ~ ~ ~ ~ ~ V ~ ~ ~ ~ Q ~ N O N
O C w C w O >, p ~ ~ ~ C 4~ p .~. O ~ p V d' ~t O _O .b
>, .~ 4. ~ ~ ~ ~ N ~~ c~ O .C c~ ~ .w-~. U ~ ~ p y, 'O ~O ~ ,~ ~ C .O
L~.v~~~+~_,~s.UU_U
~, ~ ~Q" ~ ~ U b ~ J, can %~ ~ ~ b ~ p ~ ~ ~O .~ ~p b N
p C ~ ,~ ~ ,~ C ~ ~ p ~ p G ~ ~ ~ C1. ~ ~ ~ ~ U ~.' ~ M ~ ~t
~~ j~ ~ -O p .~ ~ .~ ~ 4~ ,~ ~j ~.~. .~ ~ ..O ~ ~ ~ ~ ~ ..Nr ~ ~ .M.r N ~ ~ yr
p p p L7.. i..~ GL ~. C1. 4~ ~ ~ ~! , U Cl. ~, t1. _i _i i _~ _i i _i _i _i U
_i _i
O ~~,_O~O.~_O_~J,OO' '_O.~_O~~-U~~.COC~pOC ~ OO
,~ U ~ U b U p ~ ~ N U "C~ U fl" cY a. ~1. ~1. L1 Q.. L1 ø. C1. N Q.. C1.
~ -p N :~ ~"~ aj p ~ O O O O O O O O ~ O O
U U C M ~ ~ N M ø' ~ ~ ' p ~ N ~ O t O O O O O O O O C O O
N N N N N ~ N ~ ~-~ N ~ ,~ ~ ~ ~ V ~ U U U U U U U U ,~ U U
'r ~ 'r ~r ~r ~' ~r O 'r ~
~ i ~ ~ i ~ b C~ ~ , ~ ~ y" ~ ~ ~ i i i ~ ~ c~ i i
d' V ~ ~ ~' X N V ~ ~ ~ ~ V ~, N N O N N N N N N N N :~ N N
O_OO_O_OOp~O~p~~~~p.OU_CCOpO~C~_~1'pp
UUUUU~d~U~d~mxUd~,dddddddd.,~dd
MMMMMMN~'MM~~MM\O~'Nd'~'d'd'd~~d'W
52
CA 02369005 2001-09-27
WO 00/59878 PCT/US00/08840
00 V~ M
~n E"' ~" E" E "' (~ ~" E'' E.., Ew' ~" p V 1 E.-. E""
NzzzzzzzzzzMOzz
M ~
M
M Vr M ~ O V~ C1 .., p p
M
t~OIMN ~V~M~N~MO\O
w w f~ l~ w p V~ w V1 w 00 O W O p
~O G1 .~ M l~ ~ M M N ct M ~ O N
N
N 'B
.~ w '"
w ~
~, >, ~ '~ ~ °'
w
,~ ~ w ~ w
o -=.
o~,.~~~~~~a~~ ~~ai
o~ ~''
~_
U ..d ~ p -C N N ~ ~ p ~ ~ N
w ~ ~ ~ ~ f~. ,~ ..C .~ ~ ~ eC
O p ~ ~ .~ p ~ Q. -t1. ~ ~ ~ ~ >, O
C C
N
U ~ ~ ~ '~ ø. t1 ~ ~~-~ 0
O O ~ O
".~O tY O _" N 7, 7, ~ "~ "~ .~ N ,.C O
i
~ M a~ b
.-C,~ ~ ~1.Q..Q..~~~cG~~
~.~ O" ~ O O O N N ~ ~ M '!
~ O O O ~ ~! ~ ~ CV
p U U U ~
N N ~ z y'
~C ~ i ~ "~ b 'C b O
S~. ~1. N v; O V ~ ~ ~ ~ ø, ~' s.. ~ t1.
O p .~ ~ ~ _N ~ ~ ~ ~ ~ N V O O
~ . .r" ~ N d' 'd' ~ ~ O ~ s..
U U cad CC w ~ p p O G".r U ~ ~ ~ U
i ~ i i i U y.., ~, ~. ~ i i U i
N N d' ~ N ~ O O p fV M ~ ~ ~ N
O O O O O O ,~ ,~ ,~ O O O O N O
.~_,G~~.C_~_U_U_U_C_~_~~0_C
:~ ~ ~~ ~1 ~ L~ ~~ ~~ °'
ddUUdd~:,~,~,dd~Uzd
~' N N ct' ~ ~ M N ~ ~ ~ M ~i'
53