Note: Descriptions are shown in the official language in which they were submitted.
J'~-~ -- ---t 3:35 PM FR MO-FOM SRN DIEG0858 720 51.25 TO 854A#t99999t#9~~"n ~
"°
06-06-2001 , ~ US 00000991 ~
300622003340
- -1-
MACROLIDE A,N~mYFEC'IfYE AGENTS
Technical icld
The invention is dizectod to antibacterial compounds that expand the
repertoire of
eiytlwomycin-Like antibiotics. More particularly, the invention concerns
macrolide antibiotics
con~tainin an a
g rythronolide nucleus modified at least at the substituent at C-13.
i
Back, and Art
1'he increasing nunnber of microbial strains that have acqerired resistance to
the
currently available known anfs'biotic compounds is recognized as a dangerous
threat to public
health. As the use of such compounds has p i liferated, so too has the need
for eatpanding the
options available to treat ~a wide variety of microbial based conditions. The
need for a larger
choice of antimicrobial compounds extends ~eyond treatment of human infection
and to a
need to preserve food and other perishable commodities. New antibiotics can
also be
essential for resistant plants and animals as well as to provide resistance to
materials that
otherwise are subject to microbially caused corrosion.
Thus, there is a clear need for an expanded armament of compounds which can
provide a multifaceted defense against unwanted microbial activity.
WO 98109978 published 12 March 1998 aid incorporated herein by reference
2Q discloses modified forms of erythxomycin which lack a cladinose residue at
the 3 position and
which are derivatized ><n various ways in positions 9-12 of the macrolide
ring" Similarly, U.S.
Patent No. 5,750,5I0, issued 12 May 1998 aErd incorporated herein by
reference, discloses
i
modified erythromycin derivatives. '
Empfangsteit 7.Juni 0:37
AMENDED SHEET
CA 02369886 2001-10-03
"~~"' ,s:,so ran rr~ I'IV-f Vfl JHI~I L~GVVO1.~P ILYJ J1LJ 1V UJ-
tVfIJJvJVVIIVIV~-~~-~~ ~ -~~~~
06-06-2001 j US 00000991 E
300622003340
' '~
I
The naturally occurring erythromycirj s have the structure
~vthramycin $' $"
A -OH -CH3
B ' ~i -CH3
C -OH -H
D -H -H
wherein R' can be H or OH and lZ" can be H ox GFi3.
All of the compounds disclosed in the above-referenced patent documents
contain a~n
ethyl group ax pasitivn 13 of the macrolide nii~g. The present inventors have
found that
alterations in the substituent at position 13 results in a largo number of
compounds with
excelle~ntt antibacterial activity. .
Disclosure of the yention ,
The invention is directed to erythronolide derivatives that contain
modifications firm
l0 the native structure. All of the compounds of the invention are modified at
least at position
13.
AMENDED SHEET
EmpfanBsZeit 7.Juni 0:37
CA 02369886 2001-10-03
3 : 36 PM FR MO-FOM SHN U 1 t~,ud~tt ~ce~ m c~ ~ V OJ~IYJtISJvJJJV tIJVVW ~ ~
m
06-06-2001 ' US 00000991:
i
300622003340 '
3.
Thus, in one aspect, the invention is directed to compounds of the formula
or.
or the 10,11-anhydro fornas thereof; ,
wherein
Rn is H or OH, preferably OH;
Rb is H or halogen;
Rr is H or a protecting group;
Ra is methyl; unsubstituted alkyl (3-10C); substituted alkyl (1-lOC);
substituted or
unsubstitutcd alkenyl (2-IflC); substituted ar unsubstituted alkynyl (2-l OC);
substituted or
unsubstituted aryl (4-14C); substituted or unsubstituted arylalkyl (5-20C);
substituted or
EmDfan6steit 7.Juni 0:37 AMENDED SHEET
CA 02369886 2001-10-03
' ."~-' wi:v30 fl'1 f rt 1'IV-rVnl amv mcuwu,~u mu ..rw,.r ..,.
...,.~~..,.r......_.....,.-._--_ . .._
06-06-2001 , US 00000991 ~
3006220Q3340
unsubsfiituted arylalkenyI (5-ZOC); substituted or unsubstituted arylalkynyl
(5-20C);
.
substituted or unsubstituted amidoarylalkyl (5-20C); substituted or
unsubstituted
amidoaryialkenyl (5-24C); or substituted or unsubstitutcd amidoarylalkynyl (5-
20C);
R~ is H or a protecting group or is mono- or disubstituted amino carbonyl;
i
S Rr is H; substituted or unsubstituted alkyl ( I-l OC); substituted or
unsubstituted
!.
atkenyl (1-I OC); substituted or unsubstituted all<ynyl (1-lOC); substituted
or unsubstituted
aryl (4-14C); substituted or unsubstituted arylalkyl (5-20C); or ORr may be
replaced by H;
one of Z and Y is H and the other is OH or protected OH, or is amino, mono- or
dialkyl-amino, pmtectcd amino, or an aminoheterocycle or
Z and Y togethex are ~, =NOH or aj derivatizcd oxime;
i
including any pharmaceutically acceptable salts thereof and any stereoisomenic
forms
and mixtures of stereoisomeric forms thereof
In another aspect, the invention is directed to pharmaceutical or preservative
compositions containing the compounds offormulas (1)-(3) and to methodsto
treat infectious
diseases by administering these compounds or to preserve materials by
providing them.
The compounds of the invention have antibiotic acfiivity, but preferably are
useful as
semi-synthetic intermediates for forming 10, i 1 I aahydro forms of the
compounds that arc
further converted to compounds having an erythronolide nucleus and having a
ring between
the C10 and CI 1 positions of the eryrhronolide nucleus as descn'bed in PCT
Publication No.
WO 00/63224 which claims priority to U.S. provisional patent application
Serial Al'os.
60II40,I75 filed I8 3une 1999 and 60/172,159 filed 17 December 1999 and U.S.
utilitypatent
application Serial No. 091550,045 filed 14 April 2000 entitled "Macmiide
,A,ntiinfectives",
which are incorporated by reference.
AsBrief Descrintioa o_f the Drawines
Figure 1 shows a schematic of the synthesis of the compounds of the invention.
Figure 2 shows the post PKS biosynthesis of mycins. This pathway is
employed in the present invention, as shown in Figure 1.
Figure 3 shows the syntlxesis of compounds of formula (3) wherein Rr is
methyl.
Figure 4 shows the synthesis of compounds of formula (1) and their
con~esponding
10,11-anhydro forms.
AMENDED SHEET
Empfangsteit 7.Juni 0:37
CA 02369886 2001-10-03
_. .._._1 ,j:,,ip rn rK rW-rUl'I 5H(V iJltUUi3Jti 1610 51G' lu t5J410R~~~~bpy
.._._ .. ._..
06-Ofi-2001 ~ US 00000991 E
300622003340
-5-
Figure 5 shows the synthesis of compounds of formula (3) wherein QRf is
replaced by
H.
Figure 6 illustrates the conversion of 15-azidoerythmmycin A into 15-
amidoerythmmycins.
Modes of Carrvint Out the invention
The compounds of the invention are conveniently synthesized by combining
synthetic
chemical techniques with,microbiological processes involving genetically
engineered
microorganisms. Briefly, in a preferred mode of carrying out the invention, a
microbial host,
preferably a host which does not itself pmduce a macrolide antibiotic, is
provided with a
recombinant expression system for the production of modified 6-
deoxyerythronolide B
(6-dE~), which expression system in some instances will have been altered by a
disruption in
the catalytic domain of the ketvsyathase moiety in the first module. For
substituents is which
Rd is methyl, host cells arc used which do not have a disrupted domain of the
ketosynthase
moiety. This alteration in the d-dEB pol3tketide synthase (PKS) results in the
inability of this
PKS to utilize its native starter unit, and thus;permits inclusion of a
synthetic diketide
thiocster far its initial condensation pmduct in the sequence of reactions
leading to modified
6-dTrB without competition from the diketide; that would otherwise, natively,
have been
produced. Thus, the recombinant host can be provided a synthetic diketide
thioester for
incorporation into the resulting polyketide. The incorporation of this
diketide into the
resulting polyketide results in a polyketide with a substituent at position 13
that may be
selected as desired. Preferred methods for preparing the synthetic polyketide
thioesters are set
forti~ in PCT Publicafion No. WQ 00/44717 which claims priority to copending
application
1J'.S. Serial No. 60/117,384 filed 27 January 1999 and 09/492,733 filed on 27
January 2000,
which are incorporated herein by reference. ,
Recombinant forms of the 6-dEB PKS containing inactivated ketosynthase (KS)
domains in the first module {KS1) and appropriate organisms modified to
contain an
expression system far this PKS are described~in PCT applications WO 97!02358,
published
28 3anuary 1997 and WO 99/03986, published 28 January 1999, incorporated
herein by
reference.
Additional manipulations which provide alternative substituents on the
macrolide ring
are disclosed in PCT Publication No. WO 98/49315 which claims priority to U.S.
Sezial No.
AMENDED SHEET
EmvfangsZeit l.Juni 0:31
CA 02369886 2001-10-03
r.... v... . .~..~ ~ V ~ vl 1 I 11 l~~ 1\ I IV-f V11 JI'11'1 L~ G~:1VGJ0 ( LYJ
J 1 GJ 1 V OJ~~i1J1~1.7J.7.7CJhh~~~u"~ .- . r.
06-06-2001 i US 000009915
E
~oos2~oo3340
-6-
09!073,538 filed 6 May 1998, PCT Publication No. WO 00163361 which claims
priority to
U.S. Serial No. 601129,731 filed 16 April 1999, and PCT Publication No. WO
00124907
which claims priority to U.S. Serial No. 091x29,349 filed 28 October 1999 and
are
r
irrcarporated herein in their entirety by reference.
The polyketide resulting from expres lion of the modified PKS is then isolated
and
purified, if desired, from the recombinantly ~odificd organism and fed to
Saccharopolyspora
erythraea, which contains the functionality for postpolyketide modifications,
including
glycosylation. Other modifications include hydroxylation at positions 6 and/or
12. The
resultin modified
g erythromycin is then isolated and chemically modified to obtain the
compounds of the invention. Synthetic methods for providing these
modifications are
1
described in WO 98109978 and U.S. Patent No. 5,750,510, referenced
hereinabove.
The general method for synthesizing compounds of the invention is shown in
Figure
I.
The resulting antiinfective compound is active in vitro and in vivo for
activity against
a panel of representative microorganisms. The compounds of the invention thus
exhibit a
sufficient diversity in specificity to cover the' spectrum of antibiotic
activities desired,
For use in treating infectious disease, the compounds of the invention are
formulated
1
into suitable compositions which will include typical excipients,
pharmaceutically acceptable
counterions if the compound is a salt, fiuthez~ additives as desired, such as
antioxidants,
buff=ers, and the lOce, and administered to aninnals or humans. The types of
formulations that
are appropriate for these compounds are similar to those for the macrolide
antibiotics in
general. Formulations may be foand, for example, in min en's Pharmaceutical
Sciences
Mack Publishing Co., latest edition. The compounds can be administered by any
desired
mute, including injection, oral administration, tisnsdermal administration,
transmucosal
adminustration, or any combination. The compounds of the invention can also be
administered with additional active iugredieitts if desired.
The compounds of the invention are of fornaulas (1~(3) as set forth above, as
well as
any stereoisomeric forms of these compounds as shown. The particular
stereoisomers
depicted are those resulting from the preferred method of synthesis set forth
above and
exemplified herein; however, by modifying the expression system for the PK.S,
or by altering
r
the chirality of the diketide, or by synthetic chemical conversion, other
stereoisomers may
also be prepared. Additional chiral centers ti2ay be present in the
substituents, such as Rd and
AMENDED SHEET
Emafanasteit l.Juni 0:31
CA 02369886 2001-10-03
... ...... i ;a:;~ r rr~ rK r~u-rur~ 5HN uttuu~5~s rye 5m5 i a
~s54ea~a~aetts~~,--~. .. . .
Ofi-06-2001 i US 000009915
.i
i
30062003340
i
Rt: The stereoisomers may be administered as mixtures, or individual
stereoisomers may be
separated and utilized as is known in the art.
The properties of the compounds of formulas (1 ~(3) are defined by the
substituents
Ra-R~, Y and Z. Preferred embodiments of these substituents are set forth
hereinbeiow. They
contain moieties which are defined as follows: '
"Halogen" includes ffuoro, chloro, b imo and iodo, aad most preferably fluoro.
"Alkyl" refers to a saturated straight-chain, branched chain or cyclic
hydrocarbyl
moiety containing a specified number of carbons and that may contain one or
more suitable
heteroatoms; similarly, alkenyl aad aIkynyl refer to straight or branched
chain or cyclic
hydrocarbon substituents containing one or more double bonds or one or more
triple bonds,
respectively, and containing one or more suitable heteroatoms.
"Aryl" refers to an aromatic substituerit that may contain one or more
suitable
i
heteroatoms such as phenyl, naphthyl, quiaolyl, or phenanthryl.
"ArylaIkyl," "atylalkenyl," or "arylalkynyl" refer to substituents wherein an
aryl group
is linked to the substituted moiety through an alkyl, alkenyl or alkynyl
linkage, respectively.
Again, the number of carbons in the arylalkyl; arylalkenyl or arylalkynyl
groups will be
specified.
"Amidoarylalkyl," "amidoarylalkenyl,i or "anudoarylalkynyl" refer to
substituents
wherein an aryl group is linked to the substituted moiety through an amido and
an alkyl,
alkenyl or alkynyl linkage, respectively. Again, the number of carbons in the
amidoarylalkyl,
amidaarylalkenyl or amidoarylalkynyl groups~will be speeded.
Thus, included among the defined substituents herein are "hcteroalkyl,"
"heteroalkenyl," "heteroalkynyl;' "heteroatyl," "heteroarylallCyl," and the
like. Suitable
heteroatoms include N, O, and S.
All of the foregoing substituents may be unsubstituted ox may be further
substituted.
Typical substituents include R, -OR, -SR, -NR,a, -COR, -COOR, -CONR~, -QOCR,
NRCOit,
-OCONRz, -CN, -CF3, -NOi, -SOR, -S02R, Halogen, wherein each R is
independently H or is
alkyl, allcenyl, allcynyl, aryl, arylalkyl, or the Iietero forms of these as
definal above. In
addition -al l, alken 1 and
ky y alkynyl may be substituted by aryl or heteroaryl, which may,
themselves, be further substituted. Aryl and lieteroaryl may also be
substituted by alkyl,
allcenyl or alkynyl, or by additional aryl or hetGroaryl moieties.
EmpfangsZeit 7.Jvni 0:37
AMENDED SHEET
CA 02369886 2001-10-03
... ... . .... _ ~ ~ : ~~ t..~.l t-K 1''IV-hUrl SHIV U 1 tlaUttSii ~Gl~ 51 G5
I U t35410R~~~~ldfi'J~ .. ._.. , .
06-06-2001 , s US 00000991 E
30U6zz003340
.g-
"A derivatized oxime" is of the formula N-O-R, wherein R is other than H and
is
otherwise defined as above.
A "protecting group" far a hydroxy includes aryl groups, silyl groups, and
tire like.
Suitable protecting groups are described by Cneene, T.W., et al., in
Protecting Grouvs in
Organic S~~nthesis, 2"d Ed., John Wiley & Sons, Inc. (1991), incorporated
herein byreference.
The invention includes more preferred embodiments of the compound defined
above.
Ra is preferably butyl, pentyl, methoxyethoxyinethyl, isobutyl,
methylcyclo>~exyl, phenyl,
benzyl, ethylphenyl, 3-{benzyIoxy)propyl, z-(pyrimidin-2-ylthio)ethyl, propyl,
tluoroethyl,
chloroethyl, vinyl, 3-buteriyl, ar azidaethyl and more preferably propyl,
fluoroethyl,
chloroethyl, vinyl, 3-butenyl, or azidoethyl. PCT Publication No. WO 00/44717
which
. claims priority to U.S_ Serial No. 40/117,384 filed 27 January 1999 and U.S.
Serial
No. 09/492,733 filed 27 January 2000 all of v~hich are incorporated herein by
reference
describe various oligoketide thioesters, preferably diketide thio~esters, that
can be incorporated
at the C-13 position. Such diketide thioesters! as described therein are
incorporated into the
compounds of the invention and thus determine preferred Ra groups at the C-13
position.
In another preferred embodiment, Rr is H or lower C1-C3 alkyl, and more
preferably
methyl. Rf is also preferably arylalkenyI or arylalkynyl such as 3-arylprop-2-
enyl or 3-
arylprop-2-ynyl. Preferably the aryl group in the preferred arylalkenyl or
arylalkynyl
embodiments are 3-quinolyl, 4-quinolyl, 5-quinolyl, phenyl, 4-fluorophenyl, 4-
chlorophenyl,
4-methoxyphenyl, 6-quinolyl, 6-quinoxalyl, ~6~ amino-3-quinolyl, or
4~isoquinolyl.
Synthesis of the Invention Compounds
A.s dcscn'bed above, the antibiotic starting matezia,is for any further
chemical synthesis
to obtain the compounds of the invention are prepared, preferably, by feeding
a suitable
diketide to a microorganism modified to contain an expression system for the
6~EB PKS
containing a KS 1 knockout, or by a host cell that provides a methyl at the 13-
position,
followed by feeding the resulting polyketide to a recombinant strain of
Saccharopolyspora
erythraea that has been altered to eliminate pioduetion of 6-dEB. A strain can
be prepared
that is able to hydroxylate either both the 6- and 12-positions or the 12-
position only. In this
case, -ORf is replaced by -bI: Alternatively, a atrain can be prepared that
hydroxylates only
the 6-position. The recombinant S erythraea atrain, K40-57, is obtained by
transforming an
S. erythraea slain that produces high levels of erythromycin A with a plasnud
comprising a
AMENDED SHEET
Emafangsteit 7.Juni 0:37
CA 02369886 2001-10-03
" ! ;~ : ;~d NI'1 h-ht MU-f-OM SRN D 1 EG0858 720 S 1 25 TO 8540it9999et#9~"""
" ' "
06-06-2001 I US 00000991 ~
r
340622003340
i9_
mutated eryAl sequence encoding an inactivated KS I domain. By homologous
recombination, the resulting transformants no~cv are unable to produce b-dEB
as a competitor
to the fed polykebde and, instead, hydroxylate the 6-position and 12 position
and glycosylate
the 3-position and 5-position of the modified polyketide that has been made in
Streptomyces
or other polyketide-producing transformant If a macrolide having only the 12-
position, and
not the 6-position hydroxylated is desired (ORr is replaced by H), an S
erythraea strain is
constructed by disrupting the tryF hydroxylase gene in strain K40-6?.
Alternatively, the eryK
gene can be disabled, wherein embodiments of compo~mds (1)-(3) wherein R" is H
ntay
readily be produced.
14 The glycosylation reactions far the production of the erythromycins result
in the
diglycosylated forms analogous to the naturally occumng erythromycins. If the
compounds
r
of formula (3) are to be prepared from the initial product, the hydroxyl gmup
of the cladinose
ring (attached to position 3) may then need to~be protected for subsequent
modification of the
macrolide substituents.
The modified erythromycins of the invention, in addition to modification at C-
13, xnay
contain an -OH group at position 6, unless ORf is replaced by H as desedbed
above. To
construct the compounds of formulas (1), (2) and (3) where position b is ORr,
the compound.
of formula (3) is provided with protecting groups which form one embodiment of
R~ and Rt.
Such otection is effected usin suitable mtec
Pr g p ~ ting reagents such as acetic at~ydride,
benzoic anhydride, benzochloro formate, hexamethyldisilazane, or a
trialkylsilyl chloride in
an aprotic solvent. Aprotic solvents include, for example, dichloromethane,
chloroform,
tetrahydrofl~ran, N methyl pymolidone, dimethyl sulfoxide (DMSO), dimethyl
formamide
(pMF) and the like. Mixtures may also be used. Protection of both sugar
hydroxyls in
formula (3) may be done simultaneously or sequentially. .
In addition to protecting the 2' and 4" hydroxyl groups of the two glycose
residues, the
keto ou at ~ '
gr p position 9 of the macrolide ring:must also be protected. Typically, this
is
effected by converting the keto group to a derxvatized oxime. Particularly
preferred
embodiments for R in the formula NOR include unsubstituted or substituted
alkyl (I -12C),
substituted or unsubstitutcd aryl (6-lOC), alkyl (1-t2C), substituted or
unsubstituted
heteroaryl (6-lOC), alkyl (1-I2C), and heteraa'lkyl (such as substitucuts
ofthe formula. '
CR'20R wherein each R', in addition to being independently embodied as R as
set forth
Emafan8sieit l.Juni 0:37
AMENDED SHEET
CA 02369886 2001-10-03
.... v.uu n m n w n . . v.. ........ ... ......._ . ~._ -__- . _ ._ _
06-06-2001 f US 00000991
30o6Z2003340
10-
above, rnay, together with the other, form s cycloalkyl ring (3-12C)). A
pref~rod derivatizcd
i
crime is of the formula NOR wherein R is isopropoxycyclohexyI.
With the 9-kcto group and the 2' and 4, " hydroxyls protected, it is then
possible to
alkylate the fi-hydroxy group in the precursor to the compound of formula (3)
by reaction with
S an alkylating agent in the presence of base. ~ilkylating agents include
alkyl halides and
sulfonates. For example, the alkylating agents may include methyl tosylate, 2-
fluoroethyl
I
bromide, cinnamyl bromide, crotonyl bromide , allyl bromide, propargyl
bromide, and the
like. The alkylation is coimducted in the presence of base, such as potassium
hydroxide,
sodium hydride, potassium isopropoxide, potassium t-butoxide, and an aprotic
solvcat.
Especially preferred for R.f are methyl; alIyl and ethyl.
Once the alkylation of the 6-hydroxxliis completed, the sugar residues and the
macrolidc ring may be dcpmtectcd. Dcprotection of the glycoside moieties is
conducted as
described by Green, T.W., et al., in Protective Groups in Organic S n~, fnfra.
Similar
conditions result in converting the derivati2ed crime to NOH. Zf formation of
the
1S undcrivatized crime is rat concumxent with deprotection, the conversion to
the crime is
conducted separately.
The crime can then be removed and converted to a keto group by standard
methods
known in the art. Deoximating agents include inorganic sulfur oxide compounds
such as
sodium hydrogen sulfite, 'sodium pyrosulfate,~ sodium thiosulfxte, and the
fke. In this case,
I
profit solvents arc used, such as water, methanol, ethanol, isopropanol,
trimethyl silanol and
mixtures of these. In general, the deoximation reaction is conducted in the
presence of an
organic acid. ~ .
At this point in the process, or later, after the compound of formula (3) has
been
converted to the compounds of formulas (1) or (2} as further described below,
the group
introduced at the 6 hydroxyl can further be manipulated. Conveniently, the
initial
substitution may provide a 6-O-allyl -- i.e., O' CHzCH=CH2 -- which can
further be
derivatized by reduction to give the 6-0 propyl compound, or be treated with
osmium
tetroxide to provide the 2;3-dihydroxypropyl compound, which can further be
esterified at
tech oxygen atom. The O-allyl derivative can also be oxidized with m-
chloroperoxxbenzoic
acid in am aprotic solvent to provide the epoxy compound which can be opened
with amines
or N-containing heteroaryl compounds to provide compounds with N-containing
sid0-chains,
or can be oxidized vender blacker conditions to provide time substituent O-CH2-
C(O)-CH3, or
Emafangszeit 7.Juni 0:37
AMENDED SHEET
CA 02369886 2001-10-03
_. ..... 1 ;S : ;,i~ rn rtC hIU-rur~ 5f-IfV ~ 1 tCaUli5li r~l~ 51 G5 I U
ti54l~ii~~~~idii~' ...... ..
06-06-2001 ; US 000009915
300622003340
-~11-
can be ozonized to provide the aldehyde. The aldehyde can then be converted to
the oxime or
reacted with a suitable amine and reduced in the presence of a borohydride
reducing agent to
i
provide an amine. The oxime can also be corivertod to a nitrite by reaction
with a dehydration
agent in an aprotic solvent. The O-allyl derivative can also be reacted with
an aryl halide
under Heck conditions (Pd(II) or Pd(O), phosphine and amine or inorganic base}
tv provide a
3-aryl prop-2-enyl derivative. This derivative can then be reduced with
hydxogen and
palladium on carbon to provide a 3-arylpropyl derivative. If the initial
su6stituent Rf is a
2-propyae, similar reactions can be employod to provide alterations in the
side-chain,
including arylation. '
In order to convert the compound of formula (3) iato the compound of formula
(1), by
first removing the cladinose moiety, the compound of formula (3) is treated
with mild
aqueous acid or with a deglycosylating enzyzr'te. Suitable acids include
hydrochloric, sulfuric,
chloroacetic, trifluoroacetic and the like, in ttie presence of alcohol.
Reaction times are
typically 0.5-24 hours at a temperature of -10:35°C. During this
reaction, the 2' group of the
I 5 remaining sugar is protected as set forth above and deprotected subsequent
to the
decladinizing reaction. The resulting hydroxyl group at the 3-position of the
macmlide ring is
then oxidized to the ketone using a modified Swern oxidation procedure. In
this procedure,
an oxidizing agent such as N-chlorosuccinimide-dimethyl sulfide or a
carbodiamide-
dimethylsulfoxide is used: Typically, a compound of formula (3) is added to
pre-fonxted
N-chlorosuceinimide and'dimethyl sulfide complex in a chlorinated solvent such
as
methylene chloride at -I O-25°C. After being stilted for 0.5-4 hours, a
tertiary amine such as
triethylamine is added to produce the corresponding ketone and the 2'
protecting group is then
removed. .
In order to halogenate the macrolide at position 2 (converting Rb is H to
halogens the
i
compound of formula (1) is treated with a base and an electrophilic
halogenating reagent such
as pyridinium perbromide or N fluorobetazene sulfonic acid. The position 2 can
be
halogenated at any time after the 3 keto compound is prepared.
The appropriate substitucnt such as viayl, ethenyl, butenyl or azida at the C-
13
position can be further maruipulatod. Por example, are amidoacetate salt of
the compound of
the invention can be derivatized using an arylacetyl chloride to yield az~
arylamino alkyl group
on the C-I3 position. Preferably the C13 derivatives of an azido group take
place before the
AMENDED SHEET
EmufangsZeit 7.Juni 0:37
CA 02369886 2001-10-03
J.06=06-2001 t 3 : 39 PM FR MO-FOM SAN D I EG0858 720 5 125 TO 8540#99990#9 US
00000991 ~
300622003340
-;12 -
kctolide is formed. Derivations of an ethenyligroup can take place either
before or after the
ketolide is formed.
In order to obtain the compounds of formula (Z), the compound resulting from
the
deglycosylation reaction of formula ( 1 ) is treated with a dehydrating agent
such as carbonyl
diimidazole and base.
In order to prepare compounds of forri~ulas (1 )-(3) wherein one of Z and Y is
H and
i
the other OH or protected OH or is an amino derivative as described above,
either the
i
carbonyl or, oximo or derivatized oxime is reduced using a suitable reducing
agent, such as
i
sodium horohydzide, Raney nickel/H2 or reductive aniiaation with the use of
sodium
IO cyanoborohydride and an amine. Substitutedjamines can also be obtained by
alkylation.
Novel methods of synthesis of the compounds of the invention are also
providod.
Exemplary Embodiments
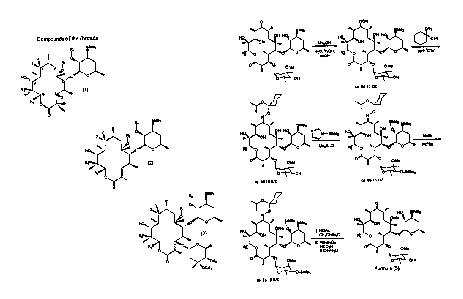
The compounds of formulas (1), (2) and (3) are defined by their various
substituents.
Table 1 illustrates compounds within the scope of the present invention which
are:
of formula (I) wherein R, is H or OHRb is H, Cl, or F, and R~ is H;
of formula (2) wherein R, is H or OH~and R~ is H; and
of formula (3) wherein R" is H or OH; R~ is H, and R~ is H, or a radical a, b,
c, oar d:
Vin).
v
F F
F
-~N ~ (c): ~ ~..~ ~ (d).
Y
v
Table 1
Rd . Rf Y Z
-CH3 -CHICHI =O
-~CH=CH2 -CHZCH=CH-~ ~ -_O
-CHZCH2CH3-CHZCti2NHCH3 =NOH
-CHa -CH2CHOHCH~ ' =NOCHzCH3
-CH(CH3}2 -CHz~ ~ H OH
-CHs -CH2-CH=CHZ . =O
-CH3 -CHa-CH=CH-(3-qeiinolyl)
-CH3 -CHZ-CHa-CHZ-(3-QuitiOlyl) =O
Emafangsteit l.Juni 0:37
AMENDED SHEET
CA 02369886 2001-10-03
J"" "" ""'"1 3:40 PM FR MO-FOM SRN DIEGOB58 720 5125 TO 8540#99990#°--
"- - '-
06-06-2001 i US 00000991 ~
3'00622003340 '
-~ 13 -
Table 1
Rd fRf Y Z
-CH9 =CHI-CH~H-(2-methi',yl-6-quinolyl) =O
-CH, -CHz-CH=CH-(5-isoquinalyl) =O
-CHs -CHrCH=CH-(3-bronio-6-quinofyl) =O
~
-CH3 -CHrC=CH-(6-meiho~2-naphthyl) =O
-CH3 -CHTC ~C-(2-phenylethenyt) =O
-CH$ -CH~-C eC-(3-quinolyl) O
-CH3 -CHrC eC-naphthyt =O
;
-CHI -CHI-C=C-(6-methyl.2-naphthyl) =O
-CH3 -CHrC~Cr(3-(2-furanyl)-6-quinolyl) =O
-CH=CHZ -CH3 , =O
-CHzOH -CHZ-C=CH-(4-fluorophenyl) =O
'
-CHZOH -CHI-C=CH-(3-quinolyl) =O
-CH20H -CH2-C=CH-(6-quinolyl)
-CH20CH9 -CHz-C=CH-(3-pyridylt) =O
-CHZCHzGH3-CHI-C=CH-(3-quinoljrl) =O
-CH=CHZCH3-CHI-C=CH-(6-chloro~3-qulnolyl) -_O
-CH2CH2CH3-CHrC=CH-(4-quinolyl) =O
-CHzCHzCH~-CHrC~H-(6-chtoro&quinolyt) =O
-CH2CH2CH3-CHI-C=CH-(8-hycbroXyr,3-quinolyl) =O
-CHZCHZCHa-CHz-C=CH-(fi-methoxy =O
3-quinolyl)
-CHzCHZCH3-CHx-CCH-(G-aminocarbonyl-3-quinolyl) =O
-CH2CHZCH~-CHZ-C=CH-(3-(2-thiophenyl)-6-quinolyl) =Q
-CHxCHZCH~-CH2-C=CH-(6-hyclrouy =p
2-naphthyl)
-CHZCHzCHa-CH~-C ~-(3-quinolyl) _O
-CHzCHZCH3-CHrC~-(6-chlora-2;
naphthyl)
-CHZCHzCH~-CHZ-C=C-(6-quinoly!)
=O
-CHzCH2CH3-CH2CH2NHGHZCHr(2-chlorophenyl) -_O
-CHS -CHZCH=NH2
'
-CH3 OR, replaced
by H -NHs H
-CH3 -CH3 ~ -NHi H
-CH3 OR, replaced ~~ H
by H '
-CH3 . ~~ ~ H
-CH3 ' ~~, ~ H
0
AMENDED SHEET
Empfansszeit 7.Juni 0:37
CA 02369886 2001-10-03
.. .._.. ~ ;,f : 410 t'rl 1-K I'IU-1-UI''I 5f-lfV 1J 1 tlaUtiStf f~19 51 ~5 I
U t354bii~J'JJIJix~.,"",' ,-' . '.
06-06-2001 ~ , ~ US 000009915
I
304622003340 '
-14_
Table
1
Rd Rf Y Z
-CH3 -CH2CHCIChl3 ~ H
v
-CH3 ~ "; H
-CH3 j H -
-CHs -CHs ~ -H~., H
-CHZCH2CH3ORr replaced by H ~ H _,~NH
-CHzCHiCH'~~ -NHZ H
-CHzCHZChf-CHCH(OCH~)CH~ ~ .~~", H
-CH2CH2CH$-CNa H _,,
v
-CH2CH2CH3-CH2CH2CH3 : ~"~ H
-CH2CHZCH3-CHZCHBrCH3 . H
-CHs -CHZCHOHCHs ~ =NOCHCH$
-CH2CHzCHs-CHiCH2CH3 ~ -NHz H
x ' les
i
The following examples are intended to illustrate but not to limit the
invention.
Compound cumbers and designations fare found in TIlustrative Scheme 1
In these examples,, in the first general 'step of the method, a 6-
deoxyerythronolide B
(6-dEB) derivative compound is prepared by ferrreentation of a recombinant
Streptomyces
host cell.
The fermentation to produce 15-methyl-ti-deoxyerythronolide B and 14,15-
dehydro-d-
deoxyerythronolide B requires a synthetic diketide intermediate to be fed to
the fermenting
1 D cells. The preparation of these s~thetic diketides is described in Example
1. These synthetic
diketides are substrates for a 6-deoxyerythronolide B synthase (DEBS) that is
unable to act on
its natural substrate (propionyl CoA) due to a ,mutation in the ketosynthase
domain of module
1 of DEBS. This recombipant DEBS is provided by plasmid pJRJ2 in
Streptorriyce~r
coelicolor CH999_ S. coelicolor CH999 is described in U.S. Patent No.
5,672,491,
incorporated herein by reference. A derivative of S. coelicolor CH999, S.
coelicolor K39-02,
AMENDED SHEET
Emvfangszeit 7.Juni 0:31 .
CA 02369886 2001-10-03
J"" "'' '""'l 3:40 PM FR MO-FOM SRN DIEG0858 720 5125 TO 8540#99990#9'"""" "
'"
06-06-2001 ; US 00000991 ~
300622003340 ,
- X15 -
that has been genetically modified to include aptpA gene, is described in U.S.
patent
application Serial No. 09!181,833, incorporated herein by reference can also
be employed for
this purpose.
Plasmid 3RJ2 encodes the
p cryAr, eryATX, and eryABI genes; the eryAl gene contained
in the plasmid contains the KS 1 null mutation. The KS 1 null mutation
prevents formation of
the 6-deoxyerythronolide B produced by the wild-type gene unless exogenous
substrate is
provided. Plasmid pJRJ2 and a process.for using the plasmid to prepare novel
13-substituted
erythromycins are descn'bed in PCT Publication Nos. WO 99!03986 and WO
97102358 and in
PCT Publication No. WO 97/02358 which claims priority to U.S. patent
application Serial
Nos. 081675,817, filed 5 July 1996; 08/896,323, filed 17 July 1997; and
091311,756, bled 14
May 1999, each of which is incorporated herein by reference. The exogenous
substrates
provided can be prepared by the methods and~include the compounds described in
PCT
Publication No. WO 00!44717 which claims priority to U.S. patent application
Serial No.
09/492,733, both filed 27 Jan. 2000, by inventors G. Ashley et al., and both
of which claim
priority to U.S. patent application Serial No. 60!117,384, filed 27 Jan.1999,
each of which is
incorporated herein by reference. PK"S genes wither than the ery genes can
also be employed;
suitable genes include the KS 1 null mutation containing oleaadoIide and
megalomicin PKS
genes described in PCT Publication No. WO 01/27284 which claims priority to
U.S. patent
application Serial Nos. 60/158,305, filed 8 Oc~~t.1999 and 091428,517, filed
28 Oct.1999, and
PCT Publication No. WO ~ 00/26349, filed 22 Oct.1999, each of which is
incorporated herein
by reference. .
The fermentation to produce x4-nor-6deoxyeTythronolida B does not require
diketide
feeding, bocause the desired compound is produced by the recombinant host cell
Streptomyces coelicolor CH9991pQ~7. Plasatid pCK? is described in U.S. Patent
No.
5,672,491 and comprises the DEBS genes. A~derivative of plasmid pCK?, pROS011-
26, can
also be used. The host cell comprising pI~OS01 I-26 and a recombinant ptpA
gene is
S coelicolor 27-261pKOS01 I-26. These hosti cells produce both b-
deoxyerftt~onolide B and
I4-nor-6-deoxyerythronolide, due to the incorporation of propionyl CoA and
acetyl CoA,
both of wb,ich serve as substrates for DEBS.
The fermentation of Streptomyces coelicolor CH999lpJRJ2 and S. coeltcolor
C~999IpCK7 is described in Example 2_ The isolation of the 6-
deoxyerythranolide products
resulting from this fermentation can be achieved by separation,.
AMENDED SHEET
Emafaossteit 7.Juni~ 0:37
CA 02369886 2001-10-03
J"" '" """ 1 :,i : 4 1 rri rK MV-r VI''I 5HN 1J 1 t(~Ul35ti lib 51 ~5 TO
854f~ti99990fi~9"'""" " "-
06-06-2001 ~ US 000009915
300b22003340
-;lb-
The isolated products are then added to the fermentation broth of
Saccharopodyspora
eryrhraea strains to make,other useful intermediate compounds of the
invention. The
S. erythraea strains catalyze the biosynthesis and attachment of sugar
residues to the 3 and 5
positions of the b-dEB derivative compouads~ These strains also comprise a
functional eryK
gene product and so hydroxylate the 6-dEB derivative compounds at the 12
position. The
strains differ in regard to whether a functional eryF gene product is
produced. If so, then the
compounds produced are hydroxylated at the 6 position as well. If not, then a
6-
dcoxyerythromycin A derivative is produced ~ These S erythraea fermentations
are described
in Example 3, together with the isolation of the erythromycin A derivative
compounds from
the fermentation broth. '
The isolated products are then used ash intermediates in the chemical
synthesis of other
intermediate compounds of the inve~ntaon. Por erythromycin A derivative
intermediates that
comprise a b-hydroxyl, Eacamples 4-6 descn-be the process for alkylatiung the
compounds to
make the 6-O-alkyl intermediates of the invention and Example 11 describes the
process for
allylation to make 6-O-allyl intermediates which can be further derivatized as
shown is
Example 15 upon protection of the 2' u~d 4" Hydroxyl groups and protection of
the 9-position
as shown in Example 14. , The schematic for these reactions is shown in Figure
3.
i
Examples ?-9 describe the conversions of the above-described compounds of
formula
(3) to compounds of formula {1), and corresponding compounds that are the
10,11-anhydro
fozxns. 'This is shown schematically in Figure 4.
Example 10 also sets forth the process for making the 10,1 I-anhydro compounds
of
formula (3), but wherein ORf is replaced by H. The reaction scheme far these
conversions is
shown in Figure 5.
The compoc~nds in Example 11 can be converted to compounds of formula (1) or
(2)
as shown in Examples 12 and 13, respecdvcly.
Example 16 illustrates the halogenation of the 2 position.
Example 17 illustrates the conversion ~of 15-azidoerythromycin A into 15-
anudoerythromycins, as shown in Figure 6.
AMENDED SHEET
Emufangszeit 7.Juni 0:37
CA 02369886 2001-10-03
J"" '"" '""" 1 3 : 41 PM FR MO-FOM SRN D I EG0858 720 5125 TO
8540#99990#9"'""" " "'
06-06-2001 ' US 00000991 ~
300622003340
-.17 -
Example 1
P~e~aratian of Diketide Thioesters
The processes used to prepare the N-acetylcysteaminethiotsters (NAcS) used to
feed
. the recombinant Str~eptomyces host cells to make the 15-methyl and 14,15-
dehydr~o-6-
I
deoxyerythronolide B intermediate compounds are described in this Example The
synthesis
i
protocols d~cn'bed below are also described in PCT Publication No. W0 00/44717
which
claims priority to U.S. provisional patent application Serial No. 501117,384,
filed 27 Jan.
1999 and U.S. utility patent application Serial No. 091492,733, filed 27 Jan.
2000, both of
Which are incorporated herein by reference. ,
Thus, (2S,3R)-2-methyl-3-hydroxyhe~anoate NAcS (Preparation E), which is used
to
prepare the 15-methyl-6-deoxyerythronalide B intermediate, is prepared from
reacting (4S)-
N-[(2S,3R)-Z-methyl-3 hydraxyhexauoyl]-4-benzyl 2-oxa2olidinone (Preparation
D) with N-
acetylcysteamine (Preparation B). N acetylcysteamine is, in taro, prepared
from N,S-
diacetylcysteamine (Preparation A). (4S)-N-[(2S,3R~Z-methyl-3 hydroxyhexanoyl]-
benzyl-2-oxazolidinone (Preparation D) is prepared from (4S~N-Propionyl-4-
benzyl-Z-
oxazolidinone (Prapionyl-NOx; Preparation
In similar fashion,. (2S,3R}-2-methyl-3-hydroxy-4-pentenoate NAcS (Prcgaration
G),
which isused to prepare the 14,15-dchydro-6; deoxyerythtonolide B
intermediate, is prepared
from reacting (4S) N [(2S,3R~2-methyl-3-hydroxy-4-pentenoyl]-4-benzyl-2-
oxazolidinone
(Preparation F) with N-acetylcysteamine (preparation B). (4S)-N [(2S,3R~2-
methyl-3-
hydroxy~4-pentenoyl]-4-beazyl-Z-oxazoiidinone (Preparation F) is prepared from
(4S~N
Propionyl-4-benzyl-2-oxazolidinone (Propionyl-NOx; Preparation C).
A. 1~T.S-Diacetvlcvsteamine: G~sfeamine hydrochloride (50.0 g) is added to a 1
L
3-neck round bottom flask fitted with a magnetic stir bar, 2 addition funnels,
and a pH
electrode. Water (300 mL) is added, and the stirred solution is cooled on ice.
The pH is
adjusted to 8.0 by addition of 8 N KOH. Acetic anhydride {125 mL) is placed in
one addition
funnel, and 8N KO~I (350 mL) is placed in tl ie other addition ftmnel. The
acetic anhydride is
added dropwise to the cysteamine solution, with 8 N KOH being added sv as to
keep the
reaction pH at 8 +i- l . After addition of acetic anhydride is complete, the
pH was adjusted to
7.0 using 1 N HCl and the mixture is allowed;to stir for 75 min. on ice. Solid
NaCI is added
to saxuration, and'the solution is extracted 4 tinnes using 400 mL portions of
CHiCl2. The
organic extracts are combined, diied over lVlgSOa, filtered, and concentrated
under reduced
AMENDED SHEET
EmPfanasZeit 7.Ju~i 0:37
CA 02369886 2001-10-03
JI06-06-2001 ~ 3 ~ 42 PM FR MO-FOM SRN D I EG0858 720 5 I 25 TO 8548#99990#9
US 00000991
3oosz2oo~3ao
-iI8 :.
pressure to yield 68.9 g (97% yield) of a pale yellow oil, which crystallizes
upon standing at
4°C. . i
B. ~j-,~9;ce3ylc3rsteamine: N,S-diacetylcysteamine (42.64 g3 is placed in a 2
L
round bottom Mask fitted with a magnetic s 'tuier, and dissolved in 1400 mL of
water. The
flask is purged with N2, and the nuxture is chilled in an ice bath. Potassium
hydroxide (49.42
g) is added, and the mixture is stirred for 2 hr! on ice under inert
atmosphere. The pH is
adjusted to 7 using 6 N HCI, and solid NaCI '~s added to saturation. The
mixture is extracted
7 times with 500 mL pvrtians of GHZCIZ. Thi organic extracts are combined,
dried over
MgSOa, filtered, and concentrated under reduced pressure to yield 30.2 g (96%
y~eld~ of
product. This material is distilled immediately prior to use, by 138-
140°C/7 mmHg.
C. (4S)-N-Pro~ionvl-4-benzvl-2-oxazolidinone (Prooionvl-NOx): A dry, 1 L
three-necked round bottomed flask equipped with a 500 mL addition funnel and a
stir bar was
charged with 20 g of (4S)-4-benzyl-2-oxazolidinone, capped with septa and
flushed with
nitrogen. Anhydrous THF (300 mL) was add~~ed by cannula and the resulting
solution was
cooled with a -78°C bath of dry i~sopropani 1. The addition futmel was
charged with 78
mL of n-butyllithivart (1.6 M in hexane) by eannula, which was added in a stow
stream to the
reaction. Distilled propionyl chloride (bp 77 ~79°C), 8.0 mL, was added
rapidly via syringe.
The reaction was allowed to stir for 30 min. is the dry icelisopropanol bath.
The reaction was removed from the cold bath, allowed to warm to
>0°C, and
quenched with 50 mL of saturated aqueous I~iIi~Cl. The mixture was
concentrated to a slurry
on a rotary evaporator. The slurry was extracted three times with 250 rnL
portions of ethyl
ether. The organic extracts were combined and washed with 50 mL each of
saturated aqueous
NaHC03 and brine, dried with MgSp4, filtered, and concentrated to give a
yellow oil. The
material
crystallized upon sitting. The crystals were triturated once with cold (-
20°C) hexanes
to give 2l.Q~g (80% yield) ofwhite crystalline material, m.p. 41-43°C.
a ,
APCI-MS: m/z = 234 (Ngi+),178,117. 1H-NMR (360 MHz, CDC13): 87.2-7.4
(SH,m); 4.67 (IH,m,H4); ,4.14-4.22 (2H,m,HS); 3.30 (lH,dd,J=3,13 Hz,benzylic);
2.89-3.03
(2H,m,H2'; 2.77 (lH,dd,J=9,13,benzylic); 1.20 (3H,t,J=7 Hz,H2~.
D. (4S1-N f(2S 381-2 methyl-3 hvdroxvhexanovll 4 benzYl 2-oxazolidinone: A
dry, 2 i, three-necked round bottomed flask equipped with a 500 mI, addition
funnel, a low-
temperature thermometer,'. and a stir bar was charged with 19.84 g of N
propionyl-
oxazolidinone, capped with septa and flushed~with nitrogen. Anhydrous
dichloromethane
Empfao~szeit 7.Juni 0:31
AMENDED SHEET
CA 02369886 2001-10-03
w w ~i ~i:4C h~t~l rK I~IU-rui~i 5HN UltUUtiSa 720 5125 TO 8540tt99990i~9""""
06-06-2001 ~ : US 00000991:
340622003340
-~19-
(i00 mL) was added by cannula, and the resulting solution was cooled to -
65°C in a bath of
dry icelisopropanol. The addition funnel was charged by cannula with 100 mL of
dibutylboron triflate (1.0 M in dichlomtnethaiie), which was added in a slow
stream to the
reaction. Triethylamine (15.6 mf.) was added dropwise by syringe, keeping the
reaction
temperature below -If°C. The reaction was then transferred to an ice
bath and allowed to stir
i
at 0°C for 30 min. A.fteic that period, the reac i on was placed. back
into the dry icelisopropanol
bath and altowed to cool to -65°C. Butyraldehyde (8.6 mL) was added
rapidly by syringe, aad
the reaction was allowed to stir for 30 mia.
The reaction was transferred to an icelbath and the addition funnel was
charged with
I O I00 mL of a I M aqueous phosphate solution; pH ?.0 (the phosphate solution
is comprised of
equal molar amounts of mono- and dibasic potassiuan phosphate). The phosphate
solution
was added as quickly as possible while keeping the reaction temperature below
10°G. The
addition funnel was then charged with 300 mL methanol which was added as
quickly as
possible while keeping the reaction temperature below 10°C. Finally,
the addition funnel was
charged with 300 mL of 2:1 methano1:30% hydrogea peroxide. This was added
dropwise to
ensure that the temperature was kept below 10°C. The reaction was
stirred for one hx. after
completion of addition. The solvent was then removed on a rotary evaporator
until a slurry
remained. The slurry was extracted 4 times with 500 rnL portions of ethyl
ether. The
combined organic extracts were washed with;250 mL each of saturated aqueous
sodium
bicarbonate and brine. The extract was then dried with MgS04, filtered, and
concentrated to
give a slightly yellow oil. , T'he nnaterial was then chromatographed on SiOz
using 2:1
hexanes:ethyl acetate (product Rf= 0.4) resulting in 2z.0 g (8S% yield) of
title compound as a
colorless oil. i
APCI-MS: mile 306 (MH+); !~ ~ (360 MHz, CDC13): 87.2-7.4 (SH,rn, Phenyl);
4.71 (lH,tn,H4); 4.17-4.25 (2H,m,HS); 3.96 (~IH,m,H3~; 3.77 (lH,dq,J~.S,? Hz,
H2'; 3.26
(iH,dd,J=4,13 Hzbenzylic); 2.79 (lH,dd,r 9;13 Hz,benzylic);1.5-1.6
(2H,m,H4';1.3-1.5
(2H,m,HS~j;1.27 (3H,d,J=7 Hz,2: Me); 0.94 (3H,t,J=7 Hz,H6'~. .
E. (2S.3R)-2-nnethvl-3-hydrox hexanoate Iwacetvlcvsteamir~e thioeater: N
acetylcysteamine was distilled at 130°Cl7 mm Hg to give a colorless
liquid at room
temperature. A dry, 1 ~ t~_necked round bottomed flask equipped with a 500 mh
addition
funnel and a star bar was capped with septa and flushed with nitrogen. The
flask was then
charged with 10.7 mL of N-acetylcysteamine by syringe aad with 400 mL of
anhydrous THF
AMENDED SHEET
EmpfangsZeit 7.Ju~i 0:37
CA 02369886 2001-10-03
J"" '"" ~"" l 3 : 42 PM FR MO-FOM SRN D I EG0858 720 5125 TO 8540t399990tt9--"-
- - '
06-06-2001 , ~ US 000009915
t
300622003340 '
-20 -
by cannula. The mixture was cooled with a lv~fIeOHlice bath. Butyllithium (64
mL of 1.6 M
in hexanes) was added dtopwise by syringe, resulting in formation of a white
precipitate.
After stirring for 30 min., arimethylaluminum~ (51 mL of 2.0 M in hexanes) was
added
dropwise by syringe. The reaction became clear after addition of
trimethylaluminum and was
allowed to stir an additional 30 min. During this period, 20.5 g (0.068 mot)
of (4S~N-
[(2S,3R}-2-methyl-3-hydroxylhexanoylJ-4-beilzyl-2-oxazolidinone was put under
a blanket of
nitrogen and dissvived in 100 mL of anhydrous THF; this solution was then
transferred in a
.
slow stream by camiula into the reaction. The resulting reaction mixture
turned a yellow-
I
green color and was allowed to stir for 1 hr. The reaction was finished whey
the starting
material could no longer be seen by thin-layer chromatographic analysis (ca. 1
hr.).
The reaction was treated with enough saturated oxalic acid to give a neutral
reaction
with pH paper (approximately 90 mL). The solvents were then removed on a
rotary
evaporator to give a white slurry. The slurry Gras extracted six times with
250 mL portions of
ethyl ether. The organic extracts were combined and washed with brine, dried
with MgS04,
filtered, and concentrated to give a slightly yellow oil. The thioester
product was purified by
flash chromatography on SiOZ using 1:1 hexai~es:EtOAc until the elution of 4-
benzyl-2-
i
oxazolidinone. At that point, the solvent system was switched to 100% EtOAc to
give pure
fractions of dl'ketide thioester. The product fractions were combined and
concentrated to give
14.9 g {89% yield) of title compound. Tbis compound is referred to as the
propyl diketide
thioester in Example 2.
i
APCI MS: m/z 248 (N1~I+);1H-NMR (360 Nffiz, CDCi~): a5.8 (br s,lH); 3.94
(dt,ll~, 3.46 (m,2~, 3.03 (dt,2I~, 2.71 (dq,l!1~, 1.97 (s,3Ii}, 1.50
(m,2I~,1.37 (m,2I~,1.21
(d,3H}, 0.94 (t,3HJ. ' t
F. (4S)-N f (ZS.3Rl-2-methyl-3-hvdroxv-4-pentenoxl,)-4-Yl 2-oxazolidinone:
t
A dry, 2 L three-necked round bottomed flask equipped with a 500 mL addition
funnel, a
low-temperature thermometer, and a stir bar v~ras charged with 20.0 g of
propionyl
oxazolidinone A, capped with septa and flushed with nitrogen. Anhydrous
dichloromethane
(100 ml) was added and the resulting solution was cooled to -15°C in a
bath of methanollice.
Dibutylboron triflate (100 mL of i.0 M in dichlorometi~ane) was added in a
slow stream via
the addition funnel at such a rate as to keep the reaction temperature below
3°C.
Diisopropylethylamine (17.9 mL) was added dropwise by syringe, again keeping
the internal
temperature below 3°C. The reaction was then cooled to -65°C
using a dry ice/isopropanol
AMENDED SHEET
Empfangszeit 7.Juni 0:37
CA 02369886 2001-10-03
.. ~ : 4:3 h'I") rht nlu-rlJ~'~ ShIIV L 1 CtWIts~ts r eYJ ~ 1 e~ i v o:W
r~rro~~~ri"oo_-,»,~, ,
06-06-2001 , ~ US 00000991 ~
300622003340
-! Z1-
bath. Acrolein was added over S min. by syringe. The reaction was allowod to
stir for 30
min. after completion of addition.
The reaction was then transferred to an ice bath and the addition funnel was
charged
with I20 mL (0.1 mol) of a 1 M aqueous phosphate solution, pH 7.0 (the
phosphate solution
is comprised of equal molar amounts of mono- and dibasic phosphate). The
phosphate
solution was added as quickly as possible while keeping the reaction
temperature belaw 10°C.
The addition funnel was then charged with 400 mL ofmethanol that wen added as
quickly as
. i
possible while keeping tha reaction temporatye below 10°C. Finally, the
addition funnel was
charged with 400 mL of 2,:1 methano1:30% hydrogen peroxide by initial drapwise
addition to
i
keep the temperature below 10°C. The reaction was stirred for one hour.
The solvent was
removed using a rotary evaporator, leaving a slurry. The slurry was extracted
4 tunes with
500 mL poxtions of ethyl ether. The organic extracts were combined and washed
with 250
mL each of saturated sodium bicarbonate andjbrine, then dried with MgS04,
fltered, and
concentrated to give a slightly yellow oil. Trituration with hexane induced
crystallization.
RecrystalIization from ether by addition of he~cane resulted in 13.67 g (55%
yield) of product.
IH NMR (360 MHz, CDC13): a7.2-?4 (m,51~; 5.86 (ddd,II~, 5.35 (dt,lI~, 5.22
(dt,lI~, 4.71 {m,lT~, 4.51 (m,II~, 4.21 (rn,2H), 3.89 (dq,lH), 3.26 (dd,lI~,
2.80 (dd,lH),
1.25 (d,3IT).
;
G. (2S.3R) 2-methyl-3-hy~,y-4! ventenoate N acet~rlc3rsteamine thioester: N
acetylcysteamine was distilled at 130°CJ7 mtri Hg to give a colorless
liquid at room
temperature. A dry, 1 L thrx-necked round bottomed flask equipped with a 500
mL addition
funnel and a stir bar was capped with septa anld flushed with nitrogen. The
flask was then
charged with 7.5 mL of N-aeetyleystearnine by syringe and with 500 mL of
anhydrous THF
by cannula. The reaction was then cooled with a MaOH/ice bath. 8utyllithium
(44 mL of
1.6 M in hexane) was added dropwise by sy~ge. A white precipitate formed as
the n BnLi
was added. After stirring for 30 min., 35.5 mL (0.071 moi) of
trim;ethylaluminum (2.0 M in
I
hexane) were added drop-wise by syringe. The reaction became clear after
addition of
trimethylaluminum and was allowed to stir an'jadditional 30 min. (4S)-N
[(25,31't)-2-methyl-
3-hydroxy 4-pentenoylJ-øbenzyl-2-oxazolidinone front Preparation F (13.6 g)
yeas put under
a'blanket of nitrogen, dissolved in 50 mL of anhydrous TTY, and this solution
was then
transferred in a slow stream by cannnla into the reaction. The resulting
reaction mixture
fumed a yellow-green color and was allowed to stir for 1 hr. The reaction was
judged to be
Emvfanssieit 7.Juni 0:31 AMENDED SHEET
CA 02369886 2001-10-03
J"" '"'" ""1 3:43 PM FR MO-FOM SRN DIEG0858 720 5125 TO 8540#99990#9--"- - -'
06-06-2001 . ; US 000009915
(00622003340
_;
finished when starting material could no longer be seen by thin-layer
chromatography (ea. 30
min.).
Enough saturated oxalic acid was adds to give a neutral reaction with pH paper
(approximately b0 mL). The solvents were then removed by rotary evaporator to
give a white
slurry. The slurry was extt'aeted six times with 250 mL portions of ethyl
ether. The organic
extracts were combined, washed with brine, dried with MgS04, filtered, and
concentrated to
give a slightly yellow oil.. The thioestes was then purified by flash
chromatography on SiOZ.
The column was run with 1:1 hexanes:ethyl acetate until the elution of
oxazolidinone. At that
point, the eluent was switched to 100% ethyl acetate to give pure fractions of
product. ?he
fractions wero combined and concentrated to give 7.7 g (71 % yield) of Title
compound
product. This product is referred to as the vinyl diketide thioester in
Example 2.
1H-NMR (360 MHz, CDCl3): 85.82 (ddd,lH), 5.78 (br s, lf~, 5.32 (dt,lH?, 5.21
(dt,lH), 4.47 tm,lI~, 3.45 (m,2I~, 3.04 (m,2I~, 2.81 (dq,lFi), 1.96 (s,3H),
1.22 (d,3~.
. atn a
I
' Preparation of lrrvthronolides
A. ~ 5-methyl-6-deoxvervthronolide B (Compound P, R, H, Rd~ro 1
Py )~
Streptomyces coelicolor CI3999/pJRl2 is described in PCT Publication No. WO
97/02358 which claims priority to U.S. patent application Serial Nos.
08/896,323, filed 17
fuly 1997, and 081675,817, filed 5 ~ulx 1996,1 each of which is incorporated
herein by
reference. Plasmid p11ZT2 encodes a mutated~forrn ofAEBS in which the
ketosynthase
domain of module I (KS 1 ) has been inactivated via mutagenesis (KS I
°). S. coelicolor strains
comprising this plasmid that are fed (2S, 3R)'2-meth 1-3-h o exanoate-N
i y Y~ ~
acetylcysteamine (Preparation E, propyl diketide) of Example 1 produce 15-
methyl-5-
i
deoxyerythronolide B.
A 1 mI, vial of the CH9991pJRJ2 wor~la'ng cell bank is~thawed and the contents
of the
vial are added to 50 mL of Inoculum Medium 1 in a 250 mL baffled flask. The
flask is
placed in art incubatorlshaker maintained at 30t1°C and 1755 RPM for
48~10 hours. The
I
50 mL culture is then added to a 2.8 L baffled flask containing 500. mi, of
lnoculum Medium
1. This flask is incubated'in an incubator/shaker at 3011°C and 175~25
RPM for 48~10
hours. The 500.mL culture is divided equallyi among ten 2.8 L baffled flasks
each containing
540 rrd:, of Inoculmn Medium 1. All flasks are then incubated as described
previously.
AMENDED SHEET
Emvfan8szeit 7.Juni 0:37
CA 02369886 2001-10-03
J"" "~ ~"" 1 3 : 44 PM FR MO-FOM SAN D I EG0858 720 51 25 TO
8540ii99990tt9"""" '" ",
06-06-2001 , US 00000991 E
300622003340
-'23
A 150 L fertnenter is prepared by sten'lizing ~I00 L of Production Medium 1 at
121°C
for 45 minutes. After incubation, all 10 flasks are combined in a 5 L sterile
inoculation bottle
i
and aseptically added to a. I50 L fermenter. The fcrmenter is controlled at
30°C, pH 6.5 by
addition of 2.5 N HIS04 and 2.5 N NaOH, 'dissolved oxygen >_ 80% air
saturation by ag'station
i
rate (500-700 RPM), air flow rate (10-50 LPM), andlor back pressure control
(0.1-0.4 bar).
Foam is controlled by the~intcrmittent atiditioin of a 50% solution of
Antifoam B.
At 2415 hours (2S, 3R)-2-methyl-3-hydroxyhexanoyl-N aceiylcysteaminc (prapyl
diketide, Preparation E in Example 1) is add i to a final concentration of 1
g/L. Propyl
diketide is prepared by solubilizing in dimethyl sulfoxido at a ratio of 1:4
(diketide~to DMSO)
i
and then filter sterili2ed (0.2 ~tm, nylon filter). Production of 15-methyl-G-
deoxyerythronolide B (15-methyl-6dEB) ceases on day 7 and the fermenter is
harvested. The
a
fermentation broth is centrifuged at 20,500 g ~ n an Algha Laval AS-26
centrifuge. The
product is predominantly in the centrate; the centrifuged cell mass is
discarded.
This process has also been completed~in a 1000 L fermenter (700 L working
volume).
The inoculum process is identical to the above process except that the 150 L
fermenter is
charged with Inoculum Medium 1 and the 1000 L fermenter is charged with
Production
Mediuan 1. The fermenter is controlled at 30°C, pH 6.5 by addition of
2.5-5 N HzS04 aad
2.5-5 N NaOH, dissolved oxygen Z70% au saturation by agitation rate (140-205
RPNn, air
flow rate (100-Z00 LP11~, andlor back press i 'e control (0.2-0.5 bar). Foam
is controlled by
the addition of a 50% solution of Antifoam B as needed. At 24~5 hours racemic
2-methyl-3-
hydroxyhexanoyl-N propionylcysteamine (300 grams) is added to the 1000 L
fermenter. The
femnenter is harvested at 4.6 days by centrifugation as desen'bed above.
Media used in this process include the following:
Inoculum Medium 1
Component ' C
oncentradon
KN03 _
i 2 g/L
'Yeast extract ; 2p
H case SF ; 20
Feso4-7H~o . as
NaCl I2.5% stock ; ~ ~,
M 04 I2.5% stock '
MaS04-HZO 0.5% stock ~ 1 ~
ZnSOa-7H20 1.0% stock ; 1 mLJL
CaCIZ-2H20 2.0% stock ~ 1 ~
AMENDED SHEET
Emafanssteit 7.Juni 0:37
CA 02369886 2001-10-03
v 06-06-2001 3 ~ 44 PM FR MO-FOM SRN D I EGOB58 72A 5125 TO B540tt9999ett~w "'
US 000009915
i
300b22003340
' ~-24-
Sterilized by autoclaving for 60 minutes at r21 °C.
Post-sterile additions: I
1) 1 mL/L of 50 mglml Thiostrcpton in 100~o DMSO, sterile filtered.
2) 1 mT-!L 100% Antifoatn B silicon emulsion (J.T. Baker), autoclaved.
3) 40 mL of 500 g/L glucose, sterile filtered.
Production Medium 1
Com onent i
Corn Starch I 45
Corn st li uor ~ 10
Dried, inactivated brewers ; 10
east
CaC03 1
Sterilixed in fermenter for 45 minutes at 121°C.
Post-sterile additions for J'roduction Mcdiuui 1
I
1) 1 mIJf, of SO mg/mI Thiostrept~on in 100% DMSO, sterile filtered.
2) 1 mIJL of I00% Antifoam B (J.T. Baker), autoclaved.
After centrifugation, the centrate is filtered. The filtrate (approximately
700 L) are
passed through an Amicon Moduline column (20 x 350 cm) containing 20 L of I-
B'20 resin
(Mitsubishi). The flow rate during loading i5 4 Lminute with a presswre drop
below 8 psi.
After loading the resin is iwashed with 20 L of watcr and then 40 L of 30%
methanol. 15-
tnethyl-6dEB is eluted using 100% methanol: Four 12 L fractions were collected
with
i
fractions 2, 3 and 4 containing all of the detectable 15-methltl~dEB. The 15-
methyl-6dEB
product pool is diluted with 36.7 L of water giving 7S L of a clear solution.
This solution is
loaded dir~tly onto a 5 L Amicon Vantage Column containing HP20SS resin
(Mitsubislui}.
Column loading is carried out at I LJminute. The column is eluted with 20 L of
65%
methanol, 20 L of 70% uiethanol, 20 L of $0% methanol, and finally 20 L of
100% methanol
A total of I6 x 5 L fractious were collected. The 80% fractions along with the
last 70%
fraction were combined (25 L) and evaporated to dryness. The resulting residue
is dissolved
in 1 L of 100% methanol, filtered, evaporated; and dried iri a vacuum oven at
40oC. This
process resulted in 33 g of a solid product containing 93% 15 methyl-6dEB.
B. ~4.I 5-dehvdro-6-deoxvervthronolide B (Compound P --H Rd-alIvl):
S. coelicolor strains comprising this plasmid that are fed (2S,3Rr2-methyl-3-
hydroxy-
4-pentenoate NAc Cysteamine thioester (Preparation G) of Example 1 produce
14,15-
Empfangsieit 7.Juni 0:3~AMENDEDSHEET
CA 02369886 2001-10-03
x.06-06-2001 3 ~ 44 PM I-K InU-1-UhI 'JHIV L I Cl7VCI~CS f GYJ J 1 GJ 1 V
OJ'nYJfTi7JJJCJfIJO:JHeJ n . wr
US 00000991
r
300622003340
25-
dehydro-6-deoxyerythroztolide B when prepared in accordance with the process
described in
Preparation A about to produce 15-methyl-6-deoxyerythronolide B.
C. 14-nor-6-iieoxvtrYthronolide B tCom~ P Ra,~I;I R.~amethvll;
i
Similarly, 14-not 6-denxyerythmnolide B is produced u~g S. coelicolor
't
CH999/pCK7 host, without using a diketide ithioester., when prepared in
accordance with the
process descn'bed in Example 2A.
xam le 3
. l~~r~aration of Enr~yg
IO The 6-dE8 dezivative compounds produced in Example 2, Preparations ArC are
converted to erythromycin derivatives using a recombinant strain of
Saccharopolyspora
erythraea. For production of erythromycins having both the 6 and 12 hydroxyl
groups, the
S. erythraea strain used was K40-67 or K39-14V. This strain was created by
transforming sn
S. erythraea strain capable of producing highs levels of arythromyein A with a
pVVHIYI3-
derived plasmid comprising a mutated eryAl sequence encoding an inactivated KS
1 domain.
By homologous recombination, the resulting transformants were rendered
incapable of
producing 6-dcoxyerythrnnolide B. Thus the~dE$ analog fed is not subject to
competition for
hydroxylation at the b position. For production of erythromycin derivatives
having only the
12-hydroxyl group, the S.. erythraea strain used was K39.07. This strain was
constructed
i
from strain K40-67 by disruption of the eryF hydroxylase gene; this destroys
ability to
hydroxylate the analog at the 6-position. Both strains were fermented under
substantially
similar conditions, as described below.
15-sncthyl-e~cin A: 15-methyl-orytbromycin A is produced according to tha
following protocol: A I mL vial of the K39-~4V working cell bank is thawed and
the
contents of the vial arc added to 50 mL of Inoculum Medium 2 in a 250 mL
baffled flask. The
flask is placed in an incubator/shaker maintains at 3411°C and 175125
RPM for 48110
hours. The 50 nnh culture is then added to a 2.'8 L baffled flask containing
504 mL of
xnoculuna Medium 2. The flask is incubated hi an incubator/s~haker at 34~1
°C and 175125
RPM for 48110 hours. The 500 mL culture is divided equally among ten 2.$ L
bafllcd flasks
i
each containing 500 mL of Iraoculum Medium~2. All flasks are then incubated as
described
previously.
AMENDED SHEET
Empfaogsreit 7.Ju~oi 0:37
CA 02369886 2001-10-03
Un, ,~,~ ~.,...~. J,aJ ry ry ..v ~ v.. .rv", .,.... ~ ....... ,__ ___._ _ __
06-06-2001 , US 000009915
300622003340
26 -
i
I
A 150 L fermenter is prepared by steFilizing 100 L of Production Mcdiutn 2 at
12I °C
for 45 minutes. After incubation, all 10 flask's are combined in a 5 L sterile
inoculation bottle
and aseptically added tv a I 50 L fermenter. The fermenter is controlled at
34°C, pH 7.0 by
addition of 2.5 N H2SOa and 2.5 N NaOH, dissolved oxygen >_ 80% air saturation
by agitation
rate (500-700 RPM), air flow rate (15-50 LPlvn, andlor back pressure control
(O.I-0.4 bar).
Foam is controlled by the addition of a 50% solution of Antifoam B.
At 24~5 hours a 58-60 mZJhour 15%idextrin (w/v) feed is initiated. The dextrin
solution is continuously mixed during the fc I period. At 24~S hours 25 grams
of 15-methyl-
bdEB (Preparation A in Example 2) are added to the fermenter. The 15-methyl-
6dEB is
prepa3red by solubilizing 25 grams of 15-met$yl-6dFB in 400-600 mL of I00%
ethanol and
filtering (0.2 Etm, nylon filter). Conversion of 15 methyl-6dEB to 15-methyl-
erythromycin A
ceases after 60110 hours and the fermenter is harvested. The fermentation
broth is centrifuged
at 20,500 g in an Alpha Laval AS-26 ccntrifiige. The product is predominantly
in the centrate;
the centrifuged cell mass is discarded. , '
Media used in this process include the following:
Inoculum Medium 2
Component ~ g/1,
Corn Starch i 16.0
Corn dextrin ~ 10.0
Sa Meal Flour ' 15.0
CaCO~ ' 4.0
Corn st li uor i 5,0
So Bean Oil i 6.0
NaCI 2.5
~)aSOa j Z.O
Sterilized by autoclaving for 60 minutes at 121°C.
Post-sterile addition:
1 mL/L 100% Antifoam B (J.?. Baker), autoclaved.
Production Medium 2 '
Com onent
Corn Starch , - ~ 17.5
Com dextrin T a 3 I6.0
So Meal Flog 16.5
CaC03 ' ' 4.0
Corn ste li uor . g.0
So Bean OiI . 3.0
AMENDED SHEET
EmvfanasZeit l.Juni 0:37
CA 02369886 2001-10-03
X06-06-2001 3:45 PM FR MO-FOM SRN DIEG0858 720 5125 TO 8540~99990tt969"'
US 000009915
300522003340
._ z~ _
i
NaCI ~ 3.5
2Sd4 1.~
Sterilized in fermenter for 45 minutes at 121 °C.
Centrifuged fermentation broth (127 L) containing 34 g of the target molecule
is
passed through 18.3 L of HP20 sorbant packed into an Amicon P350 Moduline 2
chromatography column. At 4 Llmin loading, backpressure is found to be less
than 5 psi.
Following loading, the resin is washed with 20 L deionized water and then 40 L
of 30%
methanol. I S-Methyl-Erythromycin A is eluted using 54 L of 100% methanol. The
product
pool is evaporated using a Buchi mtary evaporator (R-152). The solids were
dissolved in a
nunimal amount of 100% methanol, filtered and the filtrate evaporated to
dryness. This
resulted in 123 g of material containigg 30% i I5-Methyl-Faythromycin A by
weight. 80 grams
. of the 30% material is extracted twice with 1;L of 40°C acetone. The
acetone extract is
i
filtered, and the filtrate is dried on the inside surface of a 20 L rotary
evaporation flask. The
r
solids were extracted with 9:1 hexane to acetone three times at 40°C.
The organic extracts
were pooled and evaporated to dryness ,giving 32 g of solids enriched (68%) in
I5-Methyl
Erythromycin A. The product pool from the acetonelhexane extraction is
dissolved in 1 L of
methanol to which an equal amount of water is added. The methanol solution is
loaded onto a
Hp20SS chromato~raphy~calumn (Kontes) pr~feviously washed arid equilibrated
with 50%
methanol. Column dimensions were'4.8 x 115 em. Column loading with respect to
I5-
I
Methyl-Erythromycin A is I 1 g/L. The colump is washed with 50% (0.8 L) and
50% (8 L)
methanol in water. Elution of the target molecule is carried out using
70°!0 (8L), 80% (16 L)
and 85% (8 L) methanol iit water. 1 L fractions were collected. Fractions 11-
29 were
combined, evaporated and dried in a vacuum oven giving 23 g of product with
93°!o purity.
This material served as starting material for the chemical derivatization
procedures
descn'bed in the following.examples. The following compounds are also produced
by this
methodology: 14-norerythromycin A (Re=Met; 14,15-dehydro-erythromycin A
(Ra=allyl);
la-nor-6-deoxy-czythromycin A;14,15-dehyclro-6-deoxy erythromycin A; and t5
methyl-6-
deoxy erytluomycin A. When used to make 3descladinose-3-oxo-derivatives, the
erythromycin A derivatives were not separated, from the orythrvmxein C
derivatives; instead,
mixtures of the erytbmmycin A and erythromycin C compounds were used as
starting
materials for chemical derivatization.
These products were extracted and puri'~~fied as follows:
Emvfan8steit 7.Juni 0:3AMENDEDSHEET
CA 02369886 2001-10-03
~~06-06-2001 3' 45 PM FK ~tU-t-Ut~ 5Hrv m Cuvo~~ r c~ ,.~. ~~ ~ .,
~~..~.,~.,.,.,~".....,~.. . . __
i US 00000991 ~
300622003340 ~ ' .
=28-
In general, fermentation troths are brought to phi 8.0 by addition of NaOH and
ethanol is added (0.1 l:ll: broth). The broth is clarified by centrifugation
and loaded onto an
XAD-16 resin (Rohm and Haas) column (1 kg XAD/1 g erythromycin analogs) at a
flow, rate
of 2-4 mLlcmz-min. The loaded resin is washod with 2 column volumes of 20%
(vlv) ethanol
in water and the erythromycin analogs are eluted from the resin with acetone
and collected in
l l2 column volume fractions. The fractions containing erythromycin analogs
are identified
by thin-layer chromatography (ethyl acetate:hexanes I:1) and HPLC/MS.
The acetone fractions containing erythromycin analogs are pooled and the
volatiles are
removed under reduced pressure. The resulting aqueous mixture is extracted
with ethyl
acetate. The ethyl acetate extract is washed with saturated Nal~sC03 and brine
solutions,
dried over sodium or magnesium sulfate, ~tltered, and concentrated to dryness
under reduced
pressure. Crude material is dissolved in dich~loromethane and loaded onto a
pad of silica gel
and washed with dichloromethane:methanol (96:4 vlv) until the eluent is no
longer yellow.
The desired material is with dichloromethaneunethanolariethylamine (94:4:2
v/v) and
IS collected in fractions. Fractions containing ei y0uomycin are identified
bythin-layer
chromatography, collected and concentrated under xeduced pressure. This
material is
recrystallized from dichloromethanelhexanes~
This general procedure is illustrated a's follows:
(i) 14-nor
~rt~c~nvcins: 1 liter of ethanol was added to each of 10 liters of
fermentation broth. The broth was centrifu ed and the su
8 : pernatant was passed through 0.6
liters of XA.D (column dimensions 17 cm x 6~ 5 cm) at a flow rate of 100
mLniin. ARer
I
loading, the column was washed with 1.5 liters of 20% (vlv) ethanol in water.
The desired
I
material was then eluted with acetone. The fiactions containing this material
were
concentrated under reduced pressure until thej volatiles were removed and the
aqueous
remainder was extracted with ethyl acetate. The ethyl acetate layers were
washed with
.
saturated sodium bicarbonate solution, brine, dried with mtagaesium sulfate
and concentrated
under reduced pressure to give the crude extract.
Crude material (0.6 g) was dissolved in dichloromethane and gravity filtered
through a
3 cm pad of silica gel in a'6 cm diameter fritEed funnel. The material was
eluted with 400 mL
of dichloromethane fnilovved by 400 mL
dichloro~methane:methanolariethylatxtine (90,10:2
v/v) and collected in 40 mL fractions. Fractions containing eryth~coutycin
were identified by
thin-layer claromato h ~ ether:methanol.
~P Y ( -NH~OH 90:8:2 v/v, Rf.- 0,35 and
AMENDED SHEET
Emvfa~sszeit l.Juni 0;37
CA 02369886 2001-10-03
~ 06-06-2001 3 ~ 46 PM FR MO-FOM SRN U 1 E~UtsSts red ~ t ~c~ 1 V
0.7~fYJN.7JJJ'UffJO-~~~~ r . ..,..
US 000009915
300622003340
29 -
dichloramethane:methanol 95:5 v/v, Rf~ Q)~and concentrated under reduced
pressure. This
material was recrystallized from dichloromethane/hcxanes.
i
(ii) 1 S-methyl-ervri~romvr 'ns: 8 liters of ethanol was added to
approximately 80
liters of fermentation broth. The broth was centrifuged and the supernatant
was passod
through 2.5 liters of SAD at a flow rate of 230 mlJmin. After loading the
column was
washed with 1 Liter of water and 5 liters of 20% (vlv) ethanol in water. The
desired material
was theft eluted with acetone. The fiactions containing this material were
concentrated under
reduced pressure until the volatiles were rea loved and the aqueous remainder
was extracted
with ethyl acebte. The ethyl acetate layers were washed with saturatod sodium
bicarbonate
solution, brine, dried with magnesium sulfate and concentrated under reduced
pressure to
give the crude extract
Exude material (8.3 g) was dissolved in dichloromethane and gravity filtered
through a
3 cm pad of silica gel in a 9 cm diameter ftitt~ed funnel. The material was
eluted with 200 mL
of dichloromethane followed by 600 mL of dichloromethane: methanol (96:4 v/v)
followed
by 900 ml. dichloromethane:methanolariethylamine (89:9:2 v/v) and collected in
40 mL
fractions. Fractions containing erythromycini were identified by thin-layer
chromatography
(ether:methanol:NH40H 90:8:2 v/v, R f ~ 0.4 ;and dichloromethane:methanol
95:5, R,f ~ 0.05)
and concentrated under reduced pressure. This material was re-subjected to the
above
procedure before it was sui#able for recrystalloiZation.
(iii) 14 nor-6-deoxv exvthsom~; 1 liter of ethanol was added to each of 210
liter fermenting. The broths were centrifuged and the supernatants were
combined for a total
of approximately 22 liters. The combined broths were then passed through 1
liter of XE~,D
(column dimensions 23.5 cm x 6.5 cm (i.d.) a't a flow rate of I70 mL/min.
After loading the
column was washed with 2 liars of 20% (vlvj ethanol in water. The desired
material was
then eluted with acetone. The fractions containing this material were
concentrated under
reduced presswc until thc;volatiles were removed and the aqueous ranainder was
extracted
with ethyl acetate. The ethyl acetate layers were washed with saturated sodium
bicarbonate
soIutio brine dried with
'magnesium sulfate and concentrated under reduced pressure to
give the crude extract_ ' '
(iv) 15-methyl-6-dcoxv-a ~rnmvr~ine~ 1 liter of ethanol was added to each of 3
ferrnentors containing 10 liters of broth. The broths were centrifuged and the
supernatant was
passed over I.25 liters of ~A,D (column dime~isions 40 cm x 6.5 cm) at a flow
rate of 130
' AMENDED SHEET
Emufaasszeit 7.Juni 0:37
CA 02369886 2001-10-03
~ 06-06-2001 3 ~ 46 PM FR MO-FOM SHN 1J 1 ttauti't~ r ~e~ ~ i e:.r ~ ~
o.r..,~".r,~....,~.,,.,~,. ._.. . . _ .
US 00000991
300622003340
~. 30 _
mIJmin. The column was then washed with 3 liters of 20% (v/v) ethaaol in
water. T'he
desired material was they eluted with acctoi je. The fractions containing this
material we1'e
concentrated under reduced pressure until the volatiles were removed and the
aqueous
remainder was extracted with ethyl acetate. iThe ethyl acetate layers were
washed with
saturated sodium bicarbonate solution, brine dried with magnesium sulfate and
concezttratcd
under reduced pressure to give the crude extiact.
Crude material (2.8 g) was dissolved) in dichlorotnethane and gravity filtered
through a
3 cm pad of silica gel in a 6 cm diameter frilled funnel. The material was
eluted with 400.mL
of dichloromethane:methanol (96:4 v/v) followed by 400 mL
dichloromethane :methanolariethylatnine (8 x:9:2 v/v) and collected in 40 mL
fractions.
Fractions eoatainmg eryrhromycm were identified by thin-layer chromatography
(ether:methaaol:NH40H.90:8:2 v/v and dieh~oromethane~ethanol 95:5) and
concentrated
under reduced pressure. This material required further purification by silica
gel
chromatography.
IS
RQ=OH. R, Mc. R~Me ,RS=H, Rg=H Z ~'~
A. I4-N~ 'rthrom~vcin A g_Oxizne: A soh~tion of 14-noceryt~omycin A (4.621
g, 80% pure), hydroxytamine (0.5 ml of 50%j aqueous solution) and acetic acid
(0.2 ml) in
isopropanol (2 mi) was kept at 50°C for Z2 hours. It was extracted with
chloroform/ethanol
(3I2), washed with sodium bicarbonate, brine, and dried over MgS04. Filtration
and
evaporation in vacuo yielded a crude product~(0.65 g) as a white solid which
was used
directly for next transforaration.
B. 14-Noretvtin~omvcin A-9-f0-.~~ -isooropox~cvclohe;ryl)lox~: To a solution
of above crude I4-noreythiomycin p~, goxime (O.fS g) and 1,1-diisopropoxy
cyelohexanone
(0.95 ml) in mcthylene chloride (2 ml) was added pyridiruiump-toluenesnlfonate
(PATS)
0.333 y ~ ( )
( g) in meth lone chloride 2 ml . After~stirring overnight, the mixture was
extracted
a
(chloroform/ethanol 3:2), washed (NaF3C03 HzO, brine), and dried (lvlgsp4).
After. filtration
and evaporation in vacuo, ahe crude product was repeatedly driven with toluene
and
isopropanol to yield 0.74 g of product, which was used directly for next
reaction.
AMENDED SHEET
Emafan~sieit 7.Juni 0:37
CA 02369886 2001-10-03
~.~~ ~~ ~~ IIU ~UI~I ~JH~ LICUVCS.7O IGrJ J1GJ I V VJ'lV.Iv.IVJW JVff.~VvJrtV
n ..r v..
06-06-2001 , i US 00000991;
300622003340
-3I-
C. ~ 4"-bis-0-trimethylsilvl-14-norervthromycin A-9-f O-f 1
~ycvclohexy oxime: To a solutia i of 14-aore~hmm in A 9
Yc -gyp-(1_
isopropoxycyclohexyl)]oxime (0.74 g) is methylene chloride (6 ml) was added a
solution of
tnimethylsilyl imidazole (0.33 ml) and trimethylsilyl chloride {0.18 ml) in
methylene chloride
(2 mI) at 0°C. After 5 minute stirring, ethyl acetate was added, washed
(NaHC03-HzO,
brine), and dried (MgS04). Flash chromatography on silica gel (10:1
hexanes:acetone, l
triethylamine) afforded port product as a white solid (0.50 g). Mass
spectrometry reveals
(M+H+] ~ 1020.
D. 6-O-Methyl-2' 4"- a il 1-14-no in A- - O- 1-
isonropoxycvclohexyl)loXime: A solution of 2',4"-bis-Qtrimethylsilyl-!4-
norerytluomycin A
9-[Q(1-isopropoxycyclohexyl)]oxime (0.3 g0.29. mmol) in 1:1
mcthylsulfoxide/tetrahydrofiuan (I?NISp!>"T~ (1.4 ml) was treated with 0.3 ml
of a 2114
solution of methyl bmmide in ether arnd cooled to 10°C. A mixture of 1
M solution of
potassium tent-butoxide in THF (0.6 ml ) and DMSO (0.6 mI) was added over 6
hours using a
syringe pump. The reaction was then diluted) with ethyl acetate, washed with
saturated
NaHC03, brine, and dried over MgS04. Filttation and evaporation in vacuo
yielded a crude
product (0.29 g) as a white solid. Mass speetiometry reveals [M+I~j = I 034.
1
E. 6-D-Meth ~ 1-I4- orcr hxom~ ' in A -oxime: A mixture of 6-O-methyl-2',4"-
bis-O-trimethylsilyl-14-norerythromycin A 9 ~(O-(1-isopropoxycyclohexyl)]oxime
(0.29 g),
acetic acid (3.6 ml), acctonitrile (6 ml) and water (3 ml) was stirred at
ambient temperature
for 4.5 hours. The mixture was driven to dryness using toluene to give a crude
product as
white solid (0.24 g), which was used directly for next step without further
purification.
F. b-Q Mcthlrl- 4-note m tin ~ A mixture of 6-Q-methyl-14.
norerythromycin A 9-oxime (0.24 g), sodium~hydrosulfite (0.45 g, 85% pure),
water (3 mI),
ethanol (3 ml) and formic acid (0.07 ml) was kept at 85°C for 8 hours.
The reaction was
brought to pH 8 with 1 N NaOH and extracted with ethyl acetate. The organic
extract was
washed with brim dried over MgSOa, filtered, and concentrated to yield a made
preduct as a
white solid (0.2 g). Mass Spectrometry revesis [M+H~'J = 735.
AMENDED SHEET
Emafangszeit l.Juni 0:37
CA 02369886 2001-10-03
L.... ...... _......, u,,,m v m ~ m my m,.. r.... ... ...... .__ __._ _ _.
06-06-2001 ~ US 00000991
300522003340
-32-
~' le
S a ' of eth 1- 41 -de
i. orm w ere
~~.-.Rs~
A. 14 15-deh er throw in A, 9-0 ' a
A suspension of 14,15-dehydroersrth ' myein A (L984 g, 47% purity,1.2 mmol) in
6
mL of 2-propanol was treated with 1.97 mL ~ f 50% aqueous hydroxylamine and
stirred until
dissolved. Acetic acid (0.62 mL} was added~and the mixture was stirred for 25
hours at 50°C.
Upon cooling to ambient temperature, saturated NaHC03 was added and the
mixture was
concentrated en vacuo to, remove isopropano'l. The resulting aqueous mixture
was extracted
I
three times with 250-mI, portions of CHC13. i The organic extracts were
combined, washed
with saturated NaHC03, water, and brine, then dried over MgSO4, filtered, and
concentrated
to yield 0.92 g of product.
B. I4 1 -do ' m in A 9- 1-iso o loh 1 oxime
The oxime from (A) (0.92 gJ was dissolved in 6.2 mL of CH~C1Z and treated with
1,1
diisopropoxycyclohexane (1.Z3 g) and pyridinium p-toluenesulfonate (0.464 gar)
for 15 hours
at ambient temperature. The mixture was diluted with 160 mI, of CH2Clz, then
washed
sequentially with saturated NaHC03, water, and brine. The organic phase was
dried with
MgS04, filtered, and evaporated to yield a brown syrup. Chromatography on
silica gel
(gradient from toluene to~l:l toluene/acetone~+ 1°/a Et~ 3tielded 0.99$
g ofproduct.
C. ~',4"-bis(O-trimeth silv1~,14! 15-dehydr~, hromvcin A 9 (,O (I
iso 0 ox c rcIohex 1 oxime
A solution of 14,15-dehydroeryttrCOmjre~ A 9_[~(1-isopropoxycyclohexyl)Ioxime
(998 mg, 9.96) in 1 i.25 mL of CHZCIZ was cooled on iCe under inert atmosphere
arid treated
with a solution of chlorotrimethylsilane (0.24~mL) and 1-
trimcthylsilylimidazole (0.44 mL ).
After 30 minutes, the reaction was diluted with 250. mI. of ethyl acetate and
washed
sequentially with saturated NaHC03, water, aad brine. The organic phase was
dried with
MgS04, filtered, aad evaporated to yield 1.002 g ofproduct.
D. ,~' 4"-bis O- eth 1 ' -6-O-meth 1- 4 15-deh oe m cin A 9- O-
1-iso ro oxycvclohcx~_1 oxiime I
A solution of2',4"-bis-O-trimothylsilyl-14,15-dehydroerythromycin A 9-(O-(1-
isopropoxycyclohexyl)]oxime ( 1.00 g, 20.7 mmol) in 9.69 mL of 1:1
t
tetrahydrofuranlmethylsulfoxide was cooled to 10°C and treated with
0.97 mL of 2.0 M
AMENDED SHEET
Empfangsteit 7.Juni 0:37
CA 02369886 2001-10-03
L,06-06-2001 x:41 r'I'1 1'tC wV-rWn Jnlv u~wlvu.mr ,~c, ...~.. ...
....._.._____ ___
U S 00000991:
. a
~oo6z2oo3340
33 -
methyl bromide in ether under inert atmosphere. A mixture of methylsulfaxide
(1.94 rnL}
and 1.0 M potassium tern-butoxide in tetrah idrofutan (1.94 mL) was added
slowly. The
reactioa was monitored by thin-layer chromatography (silica gel, 10:1
toluenelacetone), and
was j udged complete after addition of 1 _6 molar equivalents of base. The
reaction was
diluted with 200 mL of ethyl acetate and 70 inL of satiuated NaHC03. The
mixture was
transferred to a separatory funnel, diluted with 850 mL of ethyl acetate and
280 nzL of
saturated NaHCO3, then washed soquentially with water and brine. The organic
phase was
dried with MgSO4, f Itered thmugh Celitc, aid evaporated to yield 21.2 g of
cmde 6-O-
methyl 2',4"-bis-4-tcirnethylsilyl-14,15-dehydroezythromy~cin A 9-[O-(1- .
IO isopmpoxycyclohexyl))oxime. This was carded on without furtherpurifeation.
E. 6-O-me , v - 4 -dehvdroenit~mm~n a g~xitn~e
.,
A solution of 6-O-methyl-2',4" bis-p-trimethylsilyl-14,15-dehydroerythromycin
A 9-
[O-{1-isopropoxycyclohexyl)joxime (1.0 g) in 9.8 mL of 2:1 acetonitrilelwater
was treated
with 5.3 mL of acetic acid, and stirred for 8 Hours at ambient temperature.
The mixture was
I
concentrated en vacuo, then repeatedlit concentxated after addition of tolu~e
to yield 0.797 g
of crude 6-O-methyl-14,15-dehydroerytiuomycin A 9-oxirne.
.
F. 6-O-methyl-14.I5-dehyd-rocr rfthro~mvcin A
A solution of 6-O-methyl-14,15-dehydroerythromycin A 9-oxime {0.797 g) and
sodium hydrosuIfite (85%, 1.02 g~ in 7.5 mL'of 1:1 ethanol/water was placed
under inert
atmosphere. Formic acid (0.186 mL) was added dt~opwise, and the mixture was
stirred at
80°C for 3 hours. After cooling to ambient temperature, the reaction
wa,s adjusted to p~I 10
with 6 N NaOI~ and extracted three times with I50-mL portions of ethyl
acetate. The organic
extracts wear combined and washed sequentially with saturated NaHC03, water,
acrd brine.
The organic phase was dried with MgS04, filtered, and evaporated to yield 0.68
g of 6-0-
meth I-14 15-deh
Y ~ Yaroe~rthrOmycin A suitable for furrhez conversion.
Exainple_6,
Synthesis of 6-O-methyt_ I 5-methylervt_h~mycin A i a Formula t,3~ w a R~ OH.
R~~roDVL R~Me
A. t5-Meth r~"erythromvcin A 9-Ozime: A suspension of IS-methyierythromycin
A (20.0 g, 85% puzity, 22.6 mmol) in 40 m1~ of 2-pmpanol was treatod with 20.5
mL of 50"/0
ueous h '
aQ ydroxylaniine and stirred until dissolved. Acetic acid (6.41, mL) was ceded
~d the
AMENDED SHEET
EmafanasZeit I.Juni 0:37
CA 02369886 2001-10-03
~ 06-06-2001 3 ~ 48 PM FR MO-FOM SRN D I EG0858 720 51 25 TO
854ett~~~~eu~b'~~~ r .ara
US 00000991
, ,
30o62Z003340
-34-
mixture was stirred for 15 hours at SQ°C. Upon cooling to ambient
temperature, saturated
NaHC03 was added and,the mixture was concentrated en vacuo to remove
isopropanol. The
resulting aqueous mixture was extracted threie times with 250-mL portions of
CHCI3. The
organic extracts were combined, washed with saturated NaHCOa, water, and
brine, then dried
over MgSOo, filtered, arid concentrated to yield 20.5 g of crude product.
Analysis by LC/MS
revealed a 94:6 mixture of E and Z oximes, [M+N]+ = 764,
B. 15-Methvlervthromyc'~A 9-jOJl-isoptopol,;)r~yclohexvl)loxime: The cmde
oxime from above (20.5 g) was dissolved in 55 mL of CHZC12 and treated with
1,1-
diisopropoxycyclohexane (27.3 mL) and pyridinium p-toluenesulfonate (9.8 gm)
for 15 hours
at ambient temperature. The mixtnre was diluted with 160 mL of CH2C11, then
washed
sequentially with saturated NaHC03, water, and brume. The organic phase was
dried with
MgS04, fxltcred, and ev ~ orated to eld a brown s
ap Yi ~ yrup. Chromatography on silica gel
(gradient from 2:I to 3:2 hexanes/acetone + 1% Et3l~ yielded 18.0 g ofproduct
C. 2'. 4"-bis-~-trimethvlsitvi-1 S ii,pthvirr~rt",r~r"".~.:~, a o_rn_n _
isoflropoxvcvclohexvl)loicime: A solution of 15-Methyter~rthromycin A g-~O-(1-
isopropoxycyclohexyl)Joacime (9.00 g, 9.96 trimol) in 25 mL of CHzCIz was
cooled an ice
under inert atmosphere and treated with a solution of chlorotrimethylsilatte
(1.89 mL) aad 1-
trimethyls~ylimidazoIc (3.65 mL ) in 8 mL of C1i2C1Z. After 30 minutes, the
reaction was
diluted with 250 mL of ethyl acetate and washed sequentially with saturated
NaI~C03, water,
and brine. The organic phase was dried with iVIgSO4, filtered, and evaporated.
The crude
product was purified by silica gel chromatography (gradient from hexanes to
10:1
hexaneslacetone + 1 % Et3I~, yielding 7.8 g of product.
D. 6-O-Mcth 1-2' 4"-bis-O- eth lsil - -meth m i 9- O-
iso ~ c ohe oxi e: A solution of 2',4' =bis-O-trimethylsilyl-15-
,ZS methylerythramycin A 9-[O-(1-isopropoxycyclohexyl)Joxime (21.7 g, 20.7
mmol) in 41.4 mi,
of tetrahydrofuran wa$ cooled to I 0°C and treafed with 41.4 mL of
methylsulfoxide and 20.?
mL of 2.0 M methyl bromide in ether under inert atmosphere. A mixture of
methylsulfoxide
(41.4 mL) and 1.0 M potassium tent butoxide in tetrahydrofuran (41.4 mL) was
added at a
rate of ca 20 mL pes hour.' The reaction was ryonitored by thin-layer
chromatography (silica
gel, 1 O:I tolucnetacetone),, and was judged complete after addition of 1.6
molar equivalents of
base. The reaction was diluted with 200 mL of ethyl acetate and 70 mL of
saturated
Na1-TCO3. The mixture was transferred to a separatory funnel, diluted with 850
mL of ethyl
AMENDED SHEET
Em~faneszeit 7.Juni 0:37
CA 02369886 2001-10-03
~ 06-06-2001 3: 48 PM FR MO-FOM SRN D I EG0858 720 5125 TO
8540tt99~5ertab;~~'~
. US 000009915
300622003340
35 -
acetate and 280 mL of saturated NaHC03, then washed sequentially with water
and br~me.
The organic phase was dried with MgS04, filtered through Celite, and
evaporated to yield
21.2 g of crude 6-O-methyl-2',4"-bis-O-trimethylsilyI-I S-methylerythromycin A
9-[0-(I-
isopropoxycyclohexyl)]oarime. This was cairied on without further purif
ration.
E. 6-0-Methvl-I5-methylerythnomvcin A 9-oxime: A solution of 6-0-methyl-
2',4"-bis-O-trimeth lsil 1-15-meth 1
y y y erythromycin A 9-[O-(1-isopropoxycyclohexyl)Joxime
(21.2 g) in 110 mL of acetonitzile was treated with 55 mI. of water and 67 mL
of acetic said,
and stirred for 8 hours at,ambient tempcratuTe. The mixture was concentrated
en vacuo, then
repeatedly concentrated after addition of toluene to yield 14.7 g of 5-O-
methyl-1S-
methylerythromycin A 9-oxime.
F. b-O-~ethyl-15-methy a omycin A: A solution of 6-O-methyl-15-
methylerythromycin A 9-oxime (19.7 g) and sodium hydrosulfite (85%, 23.1 g~ in
280 mL of
I :1 ethanol/water was placed under inert atm' sphere. Formic acid (3.75 rn~,)
was added
dropwise, acrd the mixtwe was stirred at 80°C for 4.5 hours. After
cooling to ambient
temperature, the reaction was treated with saturated NaHC03 and extracted
three tirries with
400-mL portions of ethyl acetate. The orgaruic extracts were combined atld
washod
sequentially with saturated NaHC03, water, and brine. The organic phase was
dried with
MgS 04, filtered, and evaporated to yield 15.1! g of 6-O-methyl-1 S-
methylerythmmycin A,
suitable for further coavcrsion.
Exam a 7
Svnthcsis of 5-O-!2'-A,ectvldesosaminYl)~b 1 I-anh~eaxy~ -O methltl 14
ore onolide A fo o:f Formula =OH =Me Rp ~Ac
i
A.. S-O-Desosaminyl-6-O-methy -I4-norervthronolide A: A mixture of 6-Q
methyl-I 4-norerythromyein A (77 rng), 0.073 ~ml of 12 N HCl and water (2 ml)
was stirred at
ambient temperature for 3~hours. The mixture was brought to pH 8 with 8 N KOH,
and
r
extracted with ethyl acetate. The organic extract was washed with brine, dried
with Mgs04,
filtered, and evaporated. The residue was chromatographed on silica gel
(3:IJhexanes:aCetone, l % triethylamine) to gibe pure product as a white solid
(42 mg). Mass
spectrometry reveals [M+H~') = 576.
B. 5-O-f2'-Acetvldesosaminyll-6-O-methvt-14-norervthronolide A: A mixtuxe of
5-p desosamin 1-6-O-meth 1-14-pore
Y y rythronolide A (73 mg), potassium carbonate(20 mg), ,
-AMENDED SHEET
Empfangszeit 7.Juni 0.3.
CA 02369886 2001-10-03
~u06-06-2001. 3~49 PM FR MO-FUh1 bHN mtuub~ts r~r~ ,m~~ ~.. ~~-.~.,...-...-
.~.,~~.."_ _ _
US 00000991 ~
300622003340
-3b-
acetic anhydride (1411,1) and acetone (I ml) was shard at ambient temperature
for 18 boars.
Ethyl acetate was added, washed with wateriaad brine, dried over MgSO4,
fittcred, and
evaporated. The residue; was chromatograplied on silica gel
(3:llhexanes:acetone, l%
triethylamine) to yield the pare product (71 x~ng~ as a white solid. Mass
spectrometry reveals
s [M+xr~ = 61 s.
C. 5-O-f2 =AoeriIdesosaminyl)-3-deoxy 3-oxo-6-O-meth 1-14 noretvthronolide
._
A (Formula (1 ) R~~H -Me, Rr=Me ~=H ~Acl: A solution of 5-D-(2'-
acetyidcsosaminyl~6-t~rncthyl-14-norerythronolide A (99 mg) and 1-(3-
dimethylatninopropyl)-3-ethylcarbodiidmide,' (EDC) hydrochloride (206 mph in
dichloromethane (2 ml} was treated with DMSO (O.Z1 ml} and cooled to
5°C. A solution of
i
pyridinium trifluoroacetate (208 mg) in dichloromethane (2 ml) was added viu a
syringe
pump in 4 hours. Ethyl acetate was then added, washed with saturated NaHC03,
water, brine,
and dried over M,gS04, filtered, and evaporated. The residue was
chromatographed on silica
gel (3:1/hexaries:acetone, '1% triethyIamine) to yield the pure product (94
mg) as a whito
i 5 solid. Mass spectrometry reveals [M+I~ = 616.
D. 5- 2'
meth ~~1-14-nor'eiythronolide A: To a solution of 5-0-(2'-acetyldesosaminyl)-3-
deoxy-3-oxo-
6-D loath 1-I4-note
Y rythronolidc A (93 mg) m dry pyridine (I ml) was added methanesulfonyl
chloride (0.057 ml) at 5°C. A#ler 3 hours at 5°C, the reaction
was warmed to ambient
temperature and kept for~an additional 15 hours. The mixture was diluted with
ethyl acetate,
washed with saturated NaHC03(Zx}, water (~x}, b~nine, and dried over MgS04,
filtered, and
evaporated. The residue was chromatographed on silica gel
(2:1/hexanes:acetone, 1
triethylamine) to yield the port product (72 trig) as a white solid. Mass
spectrometry reveals
[M+H~] = 695. , '
E. 5-O-t2'- ce , desosamimvl>-10.11-anhyd~o-3-deoxv-3-oxo-6 D methyl 14-
z~or~rvthronolide A: A solution of 5-O (2'-acetyldesosaminyl)-3-deoxy-3-oxo-11-
O
methanesulfonyl-b-t~methyl-14-norerythronolidc A (73 mg) in acetone {l m1) was
treated
with diazabic loundecene 32
Yo , { ~I) at ambient temperature for 18 hours. The mixture was
diluted with ethyl acetate, washed with saturated NapiC03, water, brine, and
dried over
MgS04, filtered, and evaporated. The residue;. was chromatographed on silica
gel
(2:1/hexanes:acetone, 1 % triethylamine) to yield the pure product (50 mg) as
a white solid.
Mass spectrometry reveals [M+1-1~ = 5gg. ' 3C NM~ (~C13, 100 MHz): 8 207.02,
204.50,
EmPfangsZeit 7.Juni 0;31 AMENDED SHEET
CA 02369886 2001-10-03
J,OG-06-2001 1 3 : 49 PM FR MO-FOM SRN D I EG0858 720 5125 TO 8540ii99990#9""'
' ' '
US 000009915
300b22003340
-37-
169.b3,168.72,142.52,139.40,101.87, 80.6 i, 80.02, 77,14, ?2.66, 71.48, 69.09,
63.56,
51.35, 50.56, 47.12, 40.61, 39.73, 37.36, 30.36, 21.32, 21.06, 20.96,
20.67,18.45,14.34,
13.89, 13.55,13.45.
$&-Benzovl)
A. ~'-O-Ben~ovl-6-O-methyl-14.15-dehyrthlom~c'n A
A solution of G-O-methyl-14,15-deh~ecythromycin A (668 m~, benzoic anhydride
(385 rnp~, and triethylamine (0.25 mL) in 3.6 .ImI. of CHZCIz was stirred for
2 days. After
I
addition of saturated NaHCQ3, the mixture was extracted three times with
CH2Cl2. The
i
organic extracts were combined and evaporated to dryness, and the product was
purified by
silica chromatography (90:9:1 toluene/acetone/Et~ to give 477 mg of product;
LC-MS
shows [M+Fi]+ = 850.6.
B. ~'~-Henzoyl-6-O-methyl-4":11 bis(O-methanesulfonvl)-1415
dehvdroervthromvcin A ,
A solution of2'-O-benzoyl-6-O-methyl-14,15-dehydrocxythromycin A (549 mg) and
methanesulfonyl chloride (0.50 mL) in 2.39 ~rtiL of pyridine was stirred for
24 hours, then
diluted with CI3zC12 and saturated NaHC03. The mixture was extracted three
times with
CHZClz. The organic extracts were combined and evaporated to dryness, and the
product was
purified by silica chmmato~raphy (90:9:1 toluenelacetoneJEt~ to give 530 mg of
product;
LC-MS shows [M+ITJ* =1006.5.
C. __2'-O-l3enzovl-6-O-methyl,4"-0-methanesulfon7rl-1011-anhvdro-I41~
dehvdroervthromvein A ~ '
I
A mixture of 2'-O ; benzoyl-b-Q-methyl-4",11-bis(O-methaaesulfonyl) 14,15-
dehydroerythromycin A (59 mg) and diazabicyclotmdecene. (0.018 mL) in 0.195 mL
of
acetone was stirred for 24~hours, then dried ini vacuo. The pmduct was
purifxad by silica
~ P Y~
chromato a h 90:9:1 toluene/acetonelEt~ to give 50 mg of product; LC-MS shows
.jM+Hj* = 910.5. ,
AMENDED SHEET
~Empfansszeit 7.Juni 0:37
CA 02369886 2001-10-03
a : 4~ ri~i rK nu-ruri bNIV U 1 thuliJti (C1~ ~ 1 C' I a C'41~~i~~~~IOFIW "'
06-06-2001 ~ US 00000991
300622003340 ~ ,
- 38 -
I
D. 2'-O-Benzoyl-6-O-methyl-3-descladinosyl-l0~ydro-14,1 S-
dehvdroervthiomv~cin A
A mixture of 2'-O-benzoyl-f>--O-methyl-4"-O-methu~esulfonyl-10, I 1-anhydro-
I4, I S
dehydroerythromycin A (337 mg),1.5 mL of acetonitrile, and G.9 mL of 3 N HC!
was stirred
for 22 hours. The acetonitrile was removed iri vacuo, the pH of the aqueous
residue was
adjusted to 12 by addition of NaOH, and the product was extracted using 4
portions of
CHZCIz. The combined extracts were dried aid evaporated. The product was
purified by
i
silica chromatography (gradient from 96:4 C~i2Cl2lMeOH to 95:4:1
CHZCl2/McOI3lEt3N) to
give 197 mg, [M+H)+ = 674.4. .
IO E. 2_'-O-Benzovl-6-O-methyl-3-descladinosyl-3-oxo-10 lI-aa~hydro 14,~~5
dehydroeivrhromvcin A ~ ~ ,
,
A suspension of 2'-O-be~zoyl-6-O-methyl-3-desoladinosyl-10,11-anhydro-I4,15-
dehydroerythromycin A (22G mg) and the Dess-Martin periodiuaue (427 mg) in
14.6 mL of
CHiCI2 ( 14.6 mL) was stirred for 1 hour. The mixture was diluted with C1i2C1Z
aad saturated
NaHCO3. The product was extracted using 3',,po~t~ions of CHzCIZ, and the
extracts were
combined, driod, and evaporated. Silica gel chromatography (90:9:1
tolueaelacetone/Et~
yielded the product, 168 mg. [M+I~~* = 672.4. i3C-NMR (CDC13,100 IvlHz): S
206.78, 203
(br), 158.19, 165.08,141.36, 139.58, 132.74, .131.51,130.46, 129.79, 128.25,
120.18, 102.09,
80.79, 80.40, 78.70, 72.52, 71.91, 69.19, 63.76, 51.10, 50.54, 47.08, 40.73,
39.87, 37.77,
31.23, 22.13, 20.98,18.52,14.28,14.15,13.5. .
Ex~mle 9
~ Ac
A. 6O- methyl-3-descladinosvl-I S-methyler~~
A mixture of 6-O-meth 1-15-meth 1
Y Y ~Y~'omycin A (15.1 g) and 280 mL of 0.5 N
HCl was stirred at ambient temperature for 3 fours. The pH was adjusted to 9
by addition of
6 N NaOH, and the resulting precipitate was collected by vacuum filtration,
washed with .
water, and dried. The filtrate was extracted thiee times with 400-mL portions
of ethyl acetate.
The orga'rtic extracts were combined, washed s
equentially with saturated NaHC03, water, and
brine, then dried over MgSOd, filtered, and evaporated to provide. further
product. .The
AMENDED SHEET
Emvfangszeit 7.Juni 0:31
CA 02369886 2001-10-03
J.06-06-2001 1 3 : 50 PM FR MO-FOM SRN D I EG0858 720 5125 TO
8540tt99999#39~~"~ ~ '~
US 00000991
300622003340 ,
' -'39 -
combined crude products were chromatographed on silica gel to yield 9.35 g of
pure 6-O- .
methyl-3-descladinosyl-I S-methylerythromycin A. ES-LClMS shows [M+HJ+ = 605.
B. ~'-O-Acetyl-6-O-meth~rl-3-descladino~y -15-methyl-~e ~rcin A
A solution of acetic anhydride (2.92 m~ L) in 35 mL of ethyl acetate was added
dropwise to a solution of 6-O-methyl-3-descli~adinosyl-15-methylerythromycin A
(9.35 g) in
40 tnL of ethyl acetate. 'fhe mixture was st'~ed for 30 minutes after
completion of addition,
then concentrated. Chromatography on silicai gel (2:1 hexanes/acetone) gave
8.35 g of 2'-0-
acetyl-6-O-methyl-3-descladinosyl-15 methylerythromycin A. ES-LCIMS shows
[M+FI]'
647. '
C. 2'-O-Acctyl-6-O-methyl-3-descladinosvl-3-oxo-15-methvlcrvthmmvcin A
A solution of 2'-O=acetyl-6-O-methyl ~3-descladinosyl-15-methylerytbromycita A
(8.3
g) and I-ethyl-3-(dimcthylamino 1 carbo,~diimide b~ drochloride 16.51
PmPY ) ~ Y ( g) in 64 nlL, of
dichloromethane and 15.47 mL of rnethylsulfoxide was placed under inert
atmosphere and
cooled on ice. A solution ofpyridinium trifluoroacetate (I6.63 g) in 64 mL of
dichloromethane was added at a rate such that addition would be complete in 4
hours, and the
reaction was monitored by thin-layer chromatography. Complete reaction was
observed after
addition of 73% of the solution, and so the reaction was then quenched by
addition of b00 mL
of ethyl acetate and 200 mL of saturated NaHC03. The organic layer was
collected and
washed sequentially with saturated NaHC03, iwater, and brine, then dried over
MgS04,
filtered, and evaporated to yield 8.4 g of crude product. Chromatography on
silica gel (3: I
hexaneslacetone) gave 6.75 g of 2'-O-ace~rl-6-p-methyl-3-descladinosyl-3-oxo-
15-
methylerythromycin A. ES-Y.CIMS shows [M+H]' = 645.
b. 2'-O Acetyl-6-O-methyl-3-descla inocvl oxo-l I-d methanesulfonvl 15
methvlervthrom cin A ;
. Methanesulfonylchloride (5.68 mL) was added dropwise to a solution of 2'-O-
acetyl-
6-O-methyl-3-descladinosyl-3-oxo-15-methylerythromycin A (6.73 g) in 35 rnL of
pyridine at
0°C. The mixture was brought to ambient teriiperature and quenched by
addition of 700 mL
of ethyl acetate and 200 niL of saturated NaHCO3. The organuc layer was
collected and
washed sequentially with 'saturated NaIiC03,'water, and brine, then dried over
MgS04,
3a filtered, and eva orated to
P yield 8.2 g of crude product. Chromatography on silica. gel (5:2
hexaaeslacetone
gave 5_04 g of 2'-O-acetyl-6-O-methyl-3-descladinosyl-3-oxo-l 1-O-
methanesulfonyl-IS-methylerythromycin A. ES-LCIMS shows [M+H~' _ X23.
AMENDED SHEET
EmPfansszeit 7.Juni 0;31
CA 02369886 2001-10-03
J.06-06-2001 I 3 : 50 PM FR MO-FOM SRN D I EG085B 720 51 25 TO
B540t399990tt9~~"~ ~ ' '
US 00000991
300622003340
-40_
E. _2'-O-Acetyl-6-O-methyl-3-descladinosyt-3-oxo-10,11-~hydro-15
methvlervthromvcin A
l,&Diazabicyclo(5.4.0]under 7-ere (5.22 raL) was added dropwise to a solution
of 2'-
O-acetyl-6-O-methyl-3-descladinosyl-3-oxo-11-O-mcthanesulfonyl-15-
methyierythromycin
A (5_03 g) in 23 mL of ace~tane. The solution~was concentrated after 4.5
hours, and the
residue was chromatographed on silica gel (S:I2 hexanes/acetone) to give 3.?2
g of 2'-O-
acetyl-d-O-methyl-3-descladinosyl-3-oxo-10,1 I-anhydro-IS-methylerythromycin
A. ES-
LC/MS shows (M+H)' = 627.
! ~ Facatiiple 10
Synthesis of 5-O-(Z'-acetvldesosaminvl)-10 11-anhydro-3 6-dideoxv 3-0~ xo 15
meth le nolide A o a ~ dro fo
- ro 1 OR re 1 ed
by H,1t~=H, R~ Acl
To a solution of 6-deoxy 15-methyl ezythromycin C (220 mg, 0.307 mmol) in .
dichloromethane {5 mL) were given potassium carbonate (50 mph and acetic
anhydride (100
L, 0.9 mmol), and the reaction was stirred at room temperature for 16 hours.
The solution
was filtered, sodium hydroxide (1N, 25 mL) and brine (25 mL) added and the
aqueous Iayer
was extracted with ethyl acetate 6 times. Thel combined organic layers were
dried with
sodium sulfate, filtered, and the solvent removed in vacuo. The crude product
the 2'
24 acetylated form of the starting material was carried on to the next step.
The chide product;was dissolved in pyridine (5 tnL) and mesyl chloride (70 L,
0.9
mmoi) was added. The reaction was stirred at 20°C for 2' days, poured
on sodium hydroxide
(1 N, 2S mL) and brine (25 mL) and the ueous 1
aq ~ ayer was extracted with ethyl acetaxe 6
times. Tlae combined organic Layers were dried with sodium sulfate, filtered,
and the solvent
removed in vaeuo. The residue was purifaed liy chromatography on silica ,gel
(toluenelacetone = 3:1,1% ammonium hydroatide) to yield 11,4"-dimesylated form
(190 mg,
68% over two steps). '
The 11, 4"-dimesylated form (190 mg; 0.21 mmol) was dissolved in acetone {7
mL)
and DBU (63 L, 0.42 mmol) was added, and the reaction was stirred at room
temperature
. over night. The muixture was poured an sodium hydroxide (1 N, 2~ ~,) ~d
brine (25 mL)
. and the aqueous layer was; extracted with ethyl acetate 6 times. The
combined organic layers
were dried with sodium sulfate, filtered, and the solvent removed in vacuo.
The crude
.
AMENDED SHEET
Emvfangszeit 7.Ju~i 0;37
CA 02369886 2001-10-03
' """ ;5:~1~ t'1'1 f K I'IV-1'VI'1 JHIV LlGl7VO.J0 ~ LY., .J~c_.J ~ v ..J-
.un....,....v."....v~~ ~ . ,_
06-06-2001 ; US 000009915
300622003340
i -41 -
product, tire 10,11-d~hydro form of G-deoxy li5-methyl erythromycin was
carried on to the.
next step. '
To the crude product from the above step was added hydrochloric acid (30 ntL,
3 I~
and ethanol (2 mL) and the mixture was stirred vigorously for 6 hours. Sodium
hydroxide (S
mL,10 N) was added and 'the aqueous layer v ias extracted with ethyl acetate b
times. The
combined organic layers were dried with sodium sulfate, filtered, and the
solvent removed in
vacuo_ The crude product; the anhydro form of formula (1) (but with aH at
position 3) where
1~~OH, Rd=prapyl, ORf is replaced by H, Rb~R~~H, was carried on to the next
step.
To the crude product from the above step in dichloromethane (5 mL) was added
acetic
I O anhydride (50 L, 0.45 mmol) and potassium carbonate (100 mg) and the
mixture was stored
vigorously for 9 hours. The reaction was filtered, sodium hydroxide (20 mL, l
I~ and brine
(ZS mL) were added and the aqueous layer was extracted with ethyl acetate 6
times. The
combined organic layers were dried with sodium sulfate, filtered, and the
solvent removed in
vacuo. The residue was ptuified by chromatography on silica gel
(toluene/acetone 3 3:1, 1%
ammonium hydroxide) to yield the 2' acctylatcd form of the starting material
(110 mg, $9%
over three steps).
The product of the, above step (110 mg, 0.184 mmol) was dissolved in
dichlommethanc (10 mL) and Dess-Martin reagent (220 mg, 0.53 mmol) was added.
The
reaction was stirred at room temperature for 45 min. The reaction was quenched
with
Sodium hydroxide (20 mL,11~ and brine (25, mL) and the aqueous layer was
extracted with
ethyl acetate 6 times. The~combined organic layers were dried with sodium
sulfate, filtered,
and the solvent removed in vacuo. The residue was purified by chromatography
on silica gel
(tolueneJacetone; gradient ,= 6:1-3:1, 1 % ammonium hydroxide) to yield the
compound of
formula (1), anhydro form, where R,~H, R~ropyl, OR,f is replaced by H, Rb H,
R~Ac
(94 m,g, 86%).
Exaranle 11
I. Cvmnound of Formula ~): Rg=OH, R~~ropyl R~allvl
i
Step I . AIlyIation of Tntermediate Antibiotic at : OH: A solution of 2',4"-
bis-Q
trimethylsilyl-i5 methylerythromycin A 9- O- 1-iso ro ox clohex 1 oxime
formula
I C P P YcY Y )1 (
{R, is OH, Rd is propyl, protected at 2' and 4" yrith. trimethylsiIyl and at
C9~ by the .
isoproxycyclohexyl oxime)) (7.8 g, ?.44 mmol) it 30 mL of tetrahydro~uran was
cooled on
AMENDED SHEET
Empfangsteit 7.Juni 0:37
CA 02369886 2001-10-03
J"" "- ---1 3:51 PM FR MO-FOM SRN DIEG0858 720 5125 TO 8540#99990#9"-"- ' "'
06-06-2001 . ! US 00000991 E
300622003340
-42-
ice and treated with 30 mL of methylsulfoxid~ and 2.58 mL of freshly distilled
allyl bromide
under inert atmosphere. A mixture of methylsulfoxide (29.8 mL) and 1.0 M
potassium tert-
butoxide in tetrahydrofuran (29.8 mL) was added at a rate of 1.33 molar
equivalents of base
per hour. The reaction was monitored by think layer chromatography (silica
gel,10:1
toluenelacetone), and was~judged complete after addition of 3.6 molar
equivalents of base.
The reaction was diluted with 700 mL of ethyl acetate and washed sequentially
with saturated
NaHC03, water, and brine. The organic phase was dried with MgS04, filtered,
and
evaporated to yield 8.08 g; of crude 6-O-allyl-2',4' ~bis-O-trimethylsilyl-1 S-
methylerythromycin A 9-[O-(1-isopropoxycyclohexyl)Joxime. This was carried on
without
further purification.
Step 2: A solution of 6-O-allyi-2',4"~iis-O-trimethylsilyl-15-
methylerythromycin A 9-
[O-(1-isopropoxycycloheXyl)joxime {8.08 g) in 42 mL of acetonitrile was
treated with 21 mL
of water and 24 mL of acetic acid, and stined jfor 18 hours at ambient
temperature. The
mixture was concentrated after addition of 2 propanol, then repeatedly after
addition of
toluene to yield 7.7 g of crude product. Chromatography on silica gel
(gradient ii'om 2:1 to
1: x hexanes/acetone + 1 % Et3I~ gave 3.75 g of 6-O-allyl-15-
methylerythroznyoin A 9-oxime.
Step 3: A solution of b-O-allyt-15-methylerythromycin A 9-oxime (3.75 g) and
sodium hydrosulfite (85%, 5.37 g) in 65 rnL o'f 1:1 ethanollwater was placad
under inert
atmosphere. Formic acid (0.845 mL) was added dropwise, and the mixture was
stiaed at
r
80°C for 3.5 hours. After cooling to ambient temperature, the reaction
was adjusted to pH 1 p
with 6 N NaOH and extracted three times with 150-mL portions of ethyl acetate.
The organic
extracts were combined and washed sequentially with saturated NaHC03, water,
and brine.
The organic phase was dried with MgSOs, filtered, and evaporated to yield 3.42
g~of 6-O-
allyl-1 S-methylerythrotxlycin A suitable for ~u~cther conversion.
ZS
II. Compound of Formula l3)
' R~=O~ Me R~allvl
Step 1: Alllrlation'of Intermediate Antibiotic at 5-OH: A solution of 2',4"-
bis-O-
trimtethylsilyl-I4-norerythromycin A 9-[O-(1-~sopropoxycyclohexyl)]oxime,
Formula ()7, (Ra
is OH, Rd is methyl, protected at 2' and 4" with trimethylsilyI and at C9=O by
the
isoproxycyclohexyl oxime) (202 mg) in tetrahydrofuran (0.4 mL), DMSO (0.4 mL),
and ether
(0.04 mL) was cooled to 10°C and treated with 0.035 mL of freshly
distilled allyl bromide
r
under inert atmosphere. A mixture of methylsWfoxide (0.4 mL) and 1.0 M
potassiuru iert-
' AMENDED SHEET
Emvfanssteit 7.Joni 0:31
CA 02369886 2001-10-03
J"~~ -- ---t 3:51 PM FR MO-FOM SRN DIEG0858 720 5125 TO 8540#99990#9"'""'" "
"'
06=06-2001 , US 00000991 E
300622003340
- 43 -
butoxide in tetrahydrofuran {0.4 mL) was add! at a rate 0.22 mLlhour. The
reaction was
monitored by thin-layer chromatography (silica gel, 5:1 toluenelacetone. The
reaction was
i
diluted with ethyl acetate and washed scquent~ally with saturated NaHC03,
water, and brine.
The organic phase was dried with MgS04, filtered, and evaporated to yield 222
mg of crude
fi-0-allyl-2',4°-bis-0-trimethylsilyl-14-norerythromycin A 9-(O-(1-
isopropoxycyclohexyl)]oxime. This was carried on without further purification.
a
Step 2: A solution of 6-0-allyl-2',4"-firs-O-trimcthylsilyl-14-norerythmmycin
A 9-[O-
(1-isopropoxycyclohexyl)]oxime (222 mg) in~4 mL'of acetonitrile was treated
with 2 mL of
water and 2.4 mL of acetic acid, and stirred f i r I8 hours at ambient
temperature. Tha mixture
was concentrated after addition of 2-propanol; then repeatedly after addition
of toluene to
yield 220 mg of crude 6-0-allyl-14-norerythromycin A 9-oxime.
Step 3: A solution of 6-O-allyl-14-noierythromycin A 9-oxime (220 mg) and
sodium
hydrosulfite (8S%, 322 mg) in 4 mL of 1:1 ethanol/water was placed under inert
atmosphere.
Formic acid (0.050 mL) was added dropwise,j and the mixture was stirred at
80°C for 15
hours. After cooling to ambient temperature, jthe reaction was adjusted to pH
10 with 6 N
NaOII and extracted three times with 1 SO-mL portions of ethyl acetate. The
organic extr8cts
were combined and washed sequentially with] saturated NaHC03, water, and
brine. The
organic phase was dried with MgS04, filtered, and evaporated to yield 15d mg
of 6-O-allyl-
14-norerythromycin A suitable for fiuther eoaversion.
Other embodiments: In a similar manner, compounds of formula (3) wherein Y aad
Z
are, together, =O, Ra is OH, Rf is allyl, is prepared from an intermediate
where R~ is butyl,
benzyl, vinyl, or 3-hydroXybutyl.
Ex ~ 12
' Conversion;to Formula (1)
Step 1: A mixture of the compound prepared in Example 11, II (77 mg, crude),
0.073
mI of l2 N HCl and water (2 mi) was stirred at ambient temperature for 3
hours. The mixture
was brought to pH 8 with :8 N KOTd, and extracted with ethyl acetate. The
organic extract
was washed with brine, deed with MgS04, filtered, and evaporated. The residue
was
chromatographed on silica gel (3:1/hexanes:acetone, I% triethyIamine) to give
pure product
as a white solid (42 mg).
AMENDED SHEET
Fmofan~s~ait 7..luni 0:37
CA 02369886 2001-10-03
_ __ -___ _ _. . .. , ,. . , . ..... ,r..., ~yVVJU IGVJ J1GJ IV
CY~4YJH~~~~~j~"'-~~».. .. ....
06-06-2001 ; US 00000991
300622003340 , ,
-44-
Step 2: To protect rthe 2' OH, a mixture the above compound (73 mg), potassium
i
carbonate(20 mg), acetic anhydride (14w1) and acetone (1 ml) was stirred at
ambient
temperature for I 8 hours. Ethyl acetate was added, washed with water and
brine, dried over
MgSO~, filtered, and evaporated. The residuelwas chromatographed on silica gel
(3:Ilhexanes:acetone,1 % ~iethylamine) to yield the pure product (7I mg) as a
white solid.
Step 3: A solution of the compound resulting from step 2 (99 mg) and 1-(3-
dimethylaminopropyl)-3-ethylcarbodiid~ide(EDC) hydrochloride (206 mg) in
dichloromcthane (2 ml) was treated with AMSO (0.21 ml) and cooled to
5°C. A solution of
pyridinium trifluoroacetate (208 mg) in dichloromethane (2 ml) was added via a
syringe
pump in 4 hours. Ethyl acetate was then added, washed with saturated NaHC03,
water, brine,
and dried over MgSOa, filtered, and evaporated. Tlie residue was
chmmatographed on silica
gel (3:llhexanes:acetone, I% triethylanzinc) to yield the pure compound of
formula (1) (94
r
mg, R~ is OH, R~ is acetate, Re is CHs, and Rfis allyl).
Step 4: To deprotect 2' OH, a solutioti of the compound resulting 6om step 3
(94
mg) in 5 mL methanol was stirred at room Eemperature for 24 hours. The solvent
was
removed in vacuo to give~the desired compound of formula (1) (R' is OH, R~ is
H, Ita is CH3,
and Rf is allyl).
Other embodiments: In a similar ma inner, compounds of formula (1 ) wherein RQ
is
OH, R~ is H, R~ is allyl, and Rd is propyl, butxl, benzyl, vinyl, or 3-
hyd~roxybutyl is prepared.
Prepaxatxon of Compounds of Formula l2)
The compound of formula (3), prepared as the 6-aliyl derivative in Example 11,
is
protected at the 2' position, treated with acid and dehydrated, then
deprotected to obtain the
compound of formula (2), as shown in Figure 1, wherein R, is OH, R~ is H, and
Rf is allyl.
Similarly, compounds of'~ormula (1) wherein Rd is propyl, butyl, benzyl,
vinyl, or
3-h drox a 1 are
Y Yb tY , prepared as descn~bed above using as starting material the compounds
of
formula (n wherein Rd is as set forth above.
Emufansszeit 7.Juni 0:37 AMENDED SHEET
CA 02369886 2001-10-03
J um uu ~rm.~ 1 J ~ ,.rte r m r r~ ~~m-r vrm Jr»v L 1 CqVi3~t5 fGVJ ~ J. C~ 1
V tf~41011~~~~19ii~'~..r.""
06-06-2001 . ; US 000009915
300622003340
- 45
i
x le 1
Conversion of ~ at Position 9 to NOH
According to the procedure of Example 6A, the carbonyl at position 9 of
eryrhromycins are converted to the corresponding oximes.
S
Fxaniple 15
Conversions at -OR
A. Aryl --° Propyl: A solution of any of the compounds prepared
above (0.2
i
mmol) in ethanol is flushed with nitrogen and~IO% palladium on carbon (20 mg)
added. The
mixture is then flushed with hydrogen and theireaction mixture stimd overnight
under
positive hydrogen pressure. The reaction mixture is filtered and concentrated
in vacuo to give
.
a glass. Chromatography on silica gel (95:5:0( 5 dichloromethane-methanol-
ammonia) gives
the propyl compounds as white solids.
B. A.llvl --y -CH3CH0: Ozone is passed through a -?8°C solution is
dichloromethane (I00 mL) of any of the compounds resulting above (4.0 mmol)
for 45
minutes. The reaction mixture is then flushed with nitrogen for 10 minutes.
Dimethyl sulfide
(1.46 mL, 20 mmol) is ad~.ed at -78°C and the reaction mixture stirred
for 30 minutes at 0°C.
The reaction mixture is concentrated in vacuo to give a white foam which is
used without
further purification by heating a solution of the compound in THF (40 mL, 4.0
mmol) and
triphenylphosphine (2.62 g, 10.0 mmoi) at 55~ C for 2.5 hours. The reaction
mixture is
concentrated in vacuo to give a white foam. Chromatography on silica gel (1:1
acetone-
hexane, then ?5:25:0.5 acetone-hexane-tniethylamane) gives !he desired
compound as a white
solid. , ;
C. AIlyl ~ -CH,~CH-NOH: To a solution in methanol (5 mL) of the compound
prepared in B wherein Rf is -CHzCHO, (0.08 ~mmol) is added triethylamine (31
p.L, 0.225
mmol) and hydroxylamine hydrochloride (?.7, mg, 0.1 I2 mmol) and the reaction
mixtwe
stirred for 6 hours at ambient temperature. The xeaction mixture is taken up
in ethyl acetate
,
and washed with aqueous. 5% sodium bicarbonate and brine, dried over sodium
sulfate, and
concentrated in vacuo to give a clear glass. Chromatography on silica gel
(g5:5 :0.5
dichloronaethane-methanol-ammonia) gives the compound as a white solid.
D. -__CH~CH NOH i -CHzCN: ~'o a solution under nitrogen of the compound
prepared in C {0.267 mmol) in T'1~ {5 mL) is added diisopropylcarbodiimide (83
uL, 0.534
AMENDED SHEET
Emofanosteit 7.J.uni 0:31
CA 02369886 2001-10-03
""' "'" '""~1 3:53 FM FR MO-FOM SRN DIEG0858 720 5125 TO 8540#99990#~'-'
06-06-2001 ' ~ US 00000991 ~
~30062Z003340 ,
- 46 -
tnmol) and CuCI (2.7 mg, 0.027 mmol) and the reaction mixture is stirred
overnight at
ambient tomperature. The reaction mixture is taken up in ethyl acetate and
washed witth
aqueous 5% sodium bicailionate and brine, dried over sodium sulfate, and
concentratod in
' I
vacuo to give a clear glass: Chromatography on silica gel (95:5:0.5
dichloromethane-
methanol-ammonia) gives the desired compound as a white solid.
E. -CH~CHO --f -CH~CH21~'i~: To a solution in methanol (10 mL) of the
compound prepared in B (0.276 mmol) is added ammonium acetate (212 mg, 2.76
mmol) and
I
the mixture is cooled to 0°C. Sodium cyanoborohydride (34 mg, 0.553
mmol) is added and
the reaction mixtuie stirred for 30 hours at 0°C. The reaction mixture
is taken up in ethyl
' ~ I
acetate and washed with aqueous 5% sodium carbonate, aqueous 2%
tris(hydroxymethyl)aminomethane, and brines dried over sodium sulfate,
filtered, and
I
concentrated in vacuo. Chromatography on silica gel (90:10:0.5 dichloromethane-
methanol
i
ammonia) gives the desired compound as a white solid.
p'. -CH"~,CHO'-~ -CH~~~iCH~-Phenol: To a 0°C solution in methanol (10
mL of the co
) mpound prepared in B (0.200 mmol) is added acetic acid (114 ~tL, 2.00 ramol)
and benzylamine (2I 8 ~r,Y., 2.00 moron and the mixture is stirred for IO
minutes. Sodium
cyanobomhydride (24.8 mg, 0.400 mmol) is added and the reaction mixture
stirred for lb
hours. Additional sodium cyanoborohydride x(24.8 mg, 0.400 mmol) is then added
and
stirring continued for 5 hours. The reaction mixture is taken up in ethyl
acetate and washed
with aqueous 5% sodium.carbonate, aqueous~2% tris(hydroxymethyl)aminolmethane,
and
brine, dried over sodium sulfate, filtered, and concentrated in vacuo.
Chromatographx on
silica gel (95:5:0.5 dichloromethane-methanol-ammonia) followed by a second
chromatography (50:50:0.5 acetone-hexanes !triethylamiae) gives the desired
compound as a
white foam. ~ ~ '
i
G. -CH~CHO ~ -CH_CHZNHC1:I2CHa Phenyl: To a 0°C solution in
methanol
(10 mT.) of the compound prepared in B (0.200 mmol) is added acetic acid (114
p,L,, 2.00
nlmol) and phenethylamine (218 ~,, 2.00 minol) and the mixture stirred for 10
minutes.
Sodium cyanoborohydride (24.8 mg, 0.400 ~mol) is added and the reaction
mixture stirred
. for 16 hours. The reaction mixhire is taken up in ethyl acetate and washed
with aqueous 5%
sodium carbonate, aqueous 2% tris(hydroxymethyl)aminornethaue, and brine,
dried over
sodium sulfate, filtered, and concentrated in vacuo. Chromatography on silica
gel (90:10:0.5
dicbloxoznethane-methanol-ammonia) gives the desired compound.
Emvfa~ssteit 7.Juni 0:37 AMENDED SHEET
CA 02369886 2001-10-03
w" ,,:~,, rm rK r~u-r~r~ SHrv ~ttuua~a rib 515 rU 8540~99990a~'US~OOOOO9915
06-06-2001 '
300622003340 f
-47-
i
H. -G'~H~S~ O i -CH~CH~NHyC42C,.~~1(.~'H~ Phenyl: To a 0°C solution
in
i
methanol (10 mL) of the compound prepared i~ B (0.200 mmol) is added L-
phenylalanine
methyl ester hydrochloride (129 mg, 0.604 znrirol) and the mixture stirred for
10 minutes.
Sodium cyanoborohydride 924.8 mg, 0.400 minol) is added and the reaction
mixture stin~ed
for 22 hours. The ruction mixture is taken up in ethyl acetate and washed with
aqueous 5%
sodium carbonate, aqueous 2% tcis(hydroxymethyl)aminomethane, and brine, dried
over
sodium sulfate, filtered, arid concentrated in vacuo. Chromatography on silica
gel (95:5:0.5
dichloromethane-methanol-ammonia) gives the desired compound.
i
r. -CHaCHO -: -C 2CHZNHCHZ-(4 p~ri~l): The desired compound is
prepared according to the inethod in G, except substituting 4-
aminomethylpyridiune for
phenethylamine. ;
J. -CH2C~NHg -~ -CH,2CH~NH~ 'nolxl~: To a solution of the
compound prepared in E (0.15 mmol) in methanol (2 mT.) is added
4-quinolinecarboxaldehyde (23 mg, O.I S mmol), acetic acid (8.6 ~cL, 0.15
mmol), and sodium
a
cyanoborohydride (9.4 mg, 0.15 mmol) and the reaction mixture is stirred for
i5 hours. The
reaction mixtuxe is taken up in ethyl acetate and washed with aqueous 5%
sodium carbonate,,
aqueous 2% tris(hydroxymethyl)aminomcthane, and brine, dried over sodium
sulfate, filtered,
I
and concentrated in vacua. Chromatographyion silica gel (95;10:0.5
dichloromethane-
methanol-ammonia) gives the desired compound.
ZO K. All~rl -~ -CHaCH=CH-Phenyl: To a solution under nitrogen of the 2'
protected
compound prepared in Example 10 (1.00 miriol), palladium(II)acetate (22 mg,
0.100 nrmol),
and triphenylphosphine (52 mg, 0.200 mmol) in acetonitrile (5 mL) was added
iodobenzene
(220 uL, 2.00 mmol) and triethylamine (280 ~L, 2.00 mmol) and the mixture is
cooled to
-78°C, degassed, and sealed. The reaction mixture is then warmed to
60°C for 0.5 hours and
stirred at 80°C far 12 hours, taken up in ethyl acetate and washed
twice with aqueous 5%
sodium bicarbonate, once with aqueous 2% tiffs(hydroxymethyl}aminomethane, and
once with
brine, dried over sodium sulfate, filtered, and concentrated in vacuo.
Chromatographx on
silica gel (95:5:0.5 dichlommethano-methanol-ammonia) gives the desired
compound.
Deproteetion is accomplished by heating in methanol
Other embodiments of formulas (1)-(.3} where Rb is H, Its is H, R,, is OH, Y
and Z are
together -0 and Rd is propyl, butyl, bent 1, vinyl, or 3 h
Y , - ydroxybutyl are those wherein Rris:
-CHzCHzCHz phenyl;
AMENDED SHEET
Emvfansszeit 7.Juni 0:37
CA 02369886 2001-10-03
J'"' -- 3:54 PM FR MO-FOM SRN DIEG085B
---t 720 5125 TO 8540~19~~~dtt9''""~
06-06-2001 i US 00000991 ~
l
3oos22oo~ao
, i .
- 4 8 -
-CH2CH~CH (4-methoxyphenyl);
-CHZCH~CH-(4-cb~lorophenyl);
-CHZCH~CH-(3-quinolyl);
,
-~z~z~zOH; ,
-CHzC(O)OH;
-CHZCHzNHCH3; ~ I
-CHzCHzNHCH20H; '
_~z~zN(Cg3~y
-CHyCH2( 1 3riOrph011Ilyj);
1 ~ -CH2C(O)NH2;
-CHzNHC(O)NHz;
-CH~VHC(O)CH3; . ;
-CHIF;
-CHzCHzOCH3;
-CHz~3; ~ .
-~Z~=C~(CH3)2i .
-CHzCHzCH(CH3)CH3;
-CH2CHZOCHZC~izOCH3;
-CHZSCH3; ~ i
-cyclopropyl;
-CHzOCHs;
-CHzCHzF; ~ i
_CH2-cyclopropyl; .
-CH2CH2CH0; ;
-C(O)CHzCHZCH3;
-CHZ-(4-~itrophenyl);
-CHz-(4-chiorophenyl);
-CHz-(4-methoxyphenyl);
-CHZ-(4-c o hen 1
Y~ p Y )~
-CHZCH=CHC(O)OCH3;
i
-CH2CH=CHC(O)OCHzCH3;
-CHZCH=CHCH3; .
AMENDED SHEET
EmPfangszeit 7.Juni 0:37
CA 02369886 2001-10-03
.. v' m-.~. .-~.v ~ a . ,-,~ n v' ' m ' m ' w' Jim a a LuVU.~O i LYJ J 1 GJ I
V OJai'UH055JU' ~1;'~~'~aw ~~~ w<
06-06-2001 ; ~ US 00000991 ~
300622003340
I
-49-
-CH2CHaCHCHzCH3; ;
-CHzCH~HCH2CHaCFi3; i
-CHiCH=CHSOz-phcayl;
-CHiC ~Si(CH3~
-CHIC ~CH2CH2CH2CHZCHZCH3; I
-CH2C ~CH3;
-~2-(2-P~dyl)~
-CHr{3-PYridYI)i
-CHz-(4-PYndY1)~
-CHz-{4-quinolyl);
I I
-CH2N0z;
-CH2C{O)OCH3; ; .
-CHZC(U)-phenyl; ~ .
-CH2C(O)CHiCH3;
-CH3C1; , ;
-cHZs(o)2 phe~yi;
-cHZcH~z~r ~ -;
-CHZCH=CFi-(4-quinolyl); j
-CHaCHzCHZ-{4-quinolyl);
-CHzCH=CH-(5-quinolyl);
-CH2CHZCH2-(5-quinolyl);
i
-CH2GH~H-(4-tienzoxazolyl); or
-CH2CH=CH-(7-benzimidazolyl).
Any of the foregoing compounds can~be converted to the correspoztding
derivatives
wherein Y and Z are together NOH in the manner descn'bed in Example 34 above.
i
am le 16
Fluorinatioa of C2 Position
Synthesis of 2'-O benzovl-6-O-provar~~rl-3-descladinosStl-3-oxo 10 i 1 anhydro
2
i
fluoro-15-meths tomycin A
34 A solution of 2'-O-benzoyl -6-O-propargyl-3-descladinosyl-3-oxo-10,11-
anhydro-! 5-
methyl-erythromycin A in tet<ahydrofuran, under inert atmosphere is cooled to -
7$°C and
treated with 1.0 M potassium tern-butoxide iit tetrahydz~ofuran. The mixture
is stirred for 5
'
. i
'
AMENDED SHEET
Empfangsteit l.Juni 0:31 .
CA 02369886 2001-10-03
J"~'~ "~~ ~~""W ~J'~f rll 1I< IIV-rVl1 Jf71'1 L1L17VIJJU 1LCJ JiLJ W
OJ'lCJff.JJJJYJr1.70JY1YJ f .J'I
06-06-2001 ~ US 00000991 E
i
300622003340
i
' - 50 -
minutes, and a solution of N-ffuorobenzenesulfonimide in tetrahydrofuran is
added in three
portions over 2 hours. After addition, the reaction is allowed to warm to
ambient temperature
and kept for an additional 5 hours. Aqueous KiC03 is added, and the mixture is
extracted
With CHZC12. The organic extracts are combined, drib over MgS04, filtered, and
evaporated.
1
Chromatography on silica gel gives the prod ~ t.
c ! of
berivatization ~of C-13 Position
. .~
Starting Material. 15-Aminoerythromyeiti A diacetate salt
apHOAc
H3
A solution of 15-azidoerythromycin A (7.75 g, 10 mmol) in 50 mL of methanol is
treated with acetic acid (2.0 mL) and 10% palladium on carbon (0.1 g) and
stirred under 1
i
atm of hydrogen gas until, thin-layer chromatographic analysis reveals
complete reduction of
the starting material. The suspension is littered through Celite to remove the
catalyst, then
evaporated to dryness to yield the product, which is used as a starting
material for the
following derivatizations.
A. Synthesis of 15-lquinol-4-ylacetatnido~er~rthromycm A
~Me2
A solution of 15-aminoerythromycin A diacetate salt (1.0 g) in 10 mL of
dichloromethaxxe is treated sequentially with qninol-4-ylacetyl chloride (350
mg) and
1
AMENDED SHEET
Emvfangszeit 7.Juni 0:31
CA 02369886 2001-10-03
06-06-2001 , ~ . _ .. _ _ _ _ , ~ -US~ ~~0000991
i
300622003340 ~' i
. _51 _
triethylamine (0.5 mL) at 0°C. . After 3 hours, tie reaction is diluted
with dichloromethane
and washed three times with saturated aqueous NaHC03. The organic phase is
dried ovar
MgS04, filtered, and evaporated to yield the ciude groduct. Purification by
silica gel
chmmatographyyields the.pure product. !
;
B. Synthesis of 1 S-(3dquinol-4.. llarovio,~amidol~pr~yein A
i
H: H
\.
"' ~ ~OH "" M
\ N~,,. ~~, ~ H~7~T~~~a2
~O
H . y,
O~ ~ ~ Me
O fiI..OH
A solution of 15-aminoerythromycin A diacetate salt (1.0 g) in 10 mL of
dichloroFnGthane is treated sequentially with 3-(quinol-4-yl)propionyl
chloride (400 mph and
triethylamine (0.5 mL) at 0°C. After 3 hours, ;the reaction is diluted
with dachloromethane
and washed three times with saturated aqueous NaHC03. The organic phase is
dried over
MgS04, filtered, and evaporated to yield the crude product. Purification by
silica. gel
chromatography yields the pure product. i
i
C. S~mthesis of 15-rsgquinol-4-vlacetaniidol~~,~.A
.;
~I H
OH "~ e2
V,, ~4, ., HO
i
A solution of 15-amino
erythromycin A diacetate salt (1.0 g) in 10 mL of
dichlommethane is treated s
equentially with isoquinol-4-ylacetyl chloride (350 mg) and
tricthylanunc (0.5 mL) at!0°C. After 3 hours ~ the reaction is diluted
with dichlorrnnethane
and washed three times. with saturated aqueous NaHC03. The organic phase is
dried over
i
' AMENDED SHEET
Emvfangszeit 7.Juni 0:37
CA 02369886 2001-10-03
J' -- ---1 3:55 PM FR MO-FOM SRN D1EG0858 728 5125 TO 854C~~t9S5~4~tt9''~""
06=06-2001 ~ US 00000991 E
300622003340
-5,2-
MgS4a~ fiitcred, and evaporated to yield tire ar~udc product. PuriEcation by
silica gel
chromatography yields the;pure product.
D. ~ynthcsis of 15-(3-(isoquinol-4-yl)nrooio~lamidolen~thromvcin A
.
A solution of 15-aminoersrthromycin A diacetate salt (1.0 g) in 10 mL of
dichioromethane is treated sequentially with 3~ (isoqwinol-4-yl)propionyl
chloride (400 mg)
1
and triethylamine {0.5 mL) at 0°C. After 3 hoe~rs, the reaction is
diluted with
dichlorornethaae and washed three times withlsaturated aqueous NaHCOa. The
organic phase
TO is dried over MgSOa, filtered, and evaporated xo yield the crude product.
Purification by
silica gel chromatography;yields the pure product.
j
E. Synthesis of 15-((quinol-5-ylamino)acetamido?erythsom~rcin A
p ,
N I ~. N
i
A solution of 15-amino
erythromycin A diacetate salt (1.0 g) in 10 mL of
dichloromethane is treated sequentially with (quinol-5 ylamino)acetic acid
(0.30 g},
' dicyclahexyicarbodiimide:(0.4 g}, l~ydroxybenzotriazole (0.25 g}, and
triethylamine (0.5
mL) at 0°C. After 3 hours, the reaction is diluted with dichlommethane
and washed three
times with saturated a evus NaHC . The or
qu 03 , ganic phase is dried over MgSOa, filtered, and
evaporated to yield the. crude produet_ Purification by silica gel
chromatography yields the
pure product. ~ j
'
Fmofanx~~ait 7.,Ivni 0;37
AMENDED SHEET
CA 02369886 2001-10-03
06-06-2001 ; ~ US 00000991 ~
300622003340
i
F. Svnthesis of 15-(fauinol-6-vlamino)acetamido)ervthromvcin A
I
N~
A solution of 15-aiminoerythromycin ~r diacetate salt (1.0 g) in 10 mL of
dichloromethane is treated sequentially with (quinol-6-ylamino)acetic acid
(0.30 g),
i
dicyclohexylcarbodiinude (0.4 g), I-hydroxybenzotriazole (0.25 g), and
triethylamine (0.5
mL) at 4°C. After 3 hours, the reactuon is diluted with dichloromethane
and washed three
times with saturated aqueous NaHC03. The organic phase is dried over MgS04,
filtered, and
I 4 evaporated to yield the crude product. Purification by silica gel
chromatography yields the
pure product. ' I
G.
N~ j
I S A solution of I S-atninoerythromycin A diacetate salt (I .0 g) in 10 mI,
of
dichloromethane is treated sequentially with quinoline,-q..methoxycarbonyl
chloride (400 tng)
and triethylamine (0.5 znL) at 0°C. After 3 hours, the reaction is
diluted with
dichloromethane and washed three times with saturated aqueous NaHC03. The
organic phase
is dried over MgSp4, filtered, and, evaporated to yield the crude product.
Purification by
20 silica gel chromatography yields the pure product.
Emvfangszeit 7.Jiuni 0:31 AMENDED SHEET
CA 02369886 2001-10-03