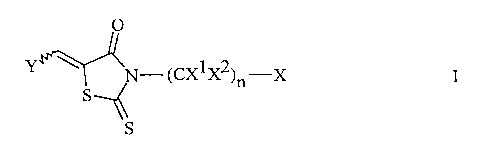Note: Descriptions are shown in the official language in which they were submitted.
CA 02370316 2001-10-15
WO 00/76988 PCT/US00/15072
-1-
RHODANINE DERIVATIVES AND THEIR USE IN INI~BITING AND IMAGING AMYLOIDS
FIELD OF THE INVENTION
S This invention relates to a method of inhibiting amyloid protein
aggregation and imaging amyloid deposits. More particularly, this invention
relates to a method of inhibiting amyloid protein aggregation in order to
treat
amyloid aggregation disorders such as Alzheimer's disease using substituted
rhodanine derivatives.
BACKGROUND OF THE INVENTION
Amyloidosis is a condition characterized by the accumulation of various
insoluble, fibrillar proteins in the tissues of a patient. The fibrillar
proteins that
comprise the accumulations or deposits are called amyloid proteins. While the
particular proteins or peptides found in the deposits vary, the presence of
fibrillar
morphology and a large amount of [3-sheet secondary structure is common to
many types of amyloids. An amyloid deposit is formed by the aggregation of
amyloid proteins, followed by the further combination of aggregates and/or
amyloid proteins.
The presence of amyloid deposits has been shown in various diseases,
each with its particular associated protein, such as Mediterranean fever,
Muckle-
Wells syndrome, idiopathetic myeloma, amyloid polyneuropathy, amyloid
cardiomyopathy, systemic senile amyloidosis, amyloid polyneuropathy,
hereditary cerebral hemorrhage with amyloidosis, Alzheimer's disease, Down's
syndrome, Scrapie, Creutzfeldt-Jacob disease, Kuru, Gerstmann-Straussler-
Scheinker syndrome, medullary carcinoma of the thyroid, isolated atrial
amyloid,
~i2-microglobulin amyloid in dialysis patients, inclusion body myositis,
CA 02370316 2001-10-15
WO 00/76988 PCT/US00/15072
-2-
(32-amyloid deposits in muscle wasting disease, sickle cell anemia,
Parkinson's
disease, and Islets of Langerhans diabetes type 2 insulinoma.
Alzheimer's disease is a degenerative brain disorder characterized
clinically by progressive loss of memory, cognition, reasoning, judgement, and
emotional stability that gradually leads to mental deterioration and
ultimately
death. Because Alzheimer's disease and related degenerative brain disorders
are a
major medical issue for an increasingly aging population, the need for new
treatments and methods for diagnosing the disorders are needed.
A simple, noninvasive method for detecting and quantitating amyloid
deposits in a patient has been eagerly sought. Presently, detection of amyloid
deposits involves histological analysis of biopsy or autopsy materials. Both
methods have major drawbacks. For example, an autopsy can only be used for a
postmortem diagnosis.
The direct imaging of amyloid deposits in vivo is difficult, as the deposits
have many of the same physical properties (ie, density and water content) as
normal tissues. Attempts to image amyloid deposits directly using magnetic
resonance imaging (MRI) and computer-assisted tomography (CAT) have been
disappointing and have detected amyloid deposits only under certain favorable
conditions. In addition, efforts to label amyloid deposits with antibodies,
serum
amyloid P protein, or other probe molecules has provided some selectivity on
the
periphery of tissues, but has provided for poor imaging of tissue interiors.
Thus, it would be useful to have a noninvasive technique for imaging and
quantitating amyloid deposits in a patient. In addition, it would be useful to
have
compounds that inhibit the aggregation of amyloid proteins to form amyloid
deposits. United States Patent No. 5,523,314 describes certain rhodanines said
to
be useful for treating Alzheimer's disease. The present compounds differ in
structure and are surprisingly potent inhibitors of amyloid aggregation.
SUMMARY OF THE INVENTION
The present invention provides compounds having the Formula I:
CA 02370316 2001-10-15
WO 00/76988 PCT/US00/15072
-3-
O
Y \~ ~N-(CX1X2)n-X
S
\\S
or a pharmaceutically acceptable salts thereof,
wherein:
RS
R1 / R4
I \
R2-N RS R1 \ /
Y is , or \ , or ( 0 or / ;
/I
\ R2 R3
each n is independently 1 to 3 inclusive;
X1 and X2 are independently hydrogen or C1-Cg alkyl, or -(CH2)y Z;
y is 0 to 4 inclusive;
Z is hydrogen, C1-Cg alkyl, C3-Cg cycloalkyl, C1-Cg perfluoroalkyl, C2-Cg
alkenyl, phenyl, substituted phenyl, naphthyl, substituted naphthyl, -OH,
-OC1-Cg alkyl, -SC1-Cg alkyl,-S03H, -C02H, -C02C1-Cg alkyl,
O O O
-CHN2, -CNH(C1-Cgalkyl), -CN(C1-Cgalkyl)2, -NH2, -NH(C1-Cgalkyl),
O
-N(C1-Cgalkyl)2, -NCC1-Cg alkyl, guanidinyl, thienyl, imidazolyl,
thiazolyl, or indolyl;
R1 and R2 are independently C1-Cgalkyl or -(CH2)n C3-C6cycloalkyl,
-(CH2)n-phenyl, or R1 and R2 taken together with the nitrogen atom to
which they are attached form a cyclic structure selected from
CA 02370316 2001-10-15
WO 00/76988 PCT/US00/15072
_4_
R3
-
N N- R , N (CH2)m ,
R R4
R3 R4 R3 R4
~N
or N
where R3 and R4 independently are hydrogen, C1-Cg alkyl, -(CH2)n-phenyl, or
-(CH2)n cycloalkyl;
RS is hydrogen, C1-Cg alkyl, halogen, or -CF3;
each m is 2 to 8 inclusive;
O O O O
X is -S-OH, -S-NR3R4, -SNHC(C1-C6perfluoroalkyl), tetrazolyl,
O O O
O O O
-SNHC-phenyl, -SNH-phenyl,
O O
~O~
O
O
O
N
I
~OH
N-
O O O O O
-SNHC(C1-C6alkyl), -CNR3R4, -CNHSC1-C6alkyl,
O O
O
-CNH-phenyl,
CA 02370316 2001-10-15
WO 00/76988 PCT/US00/15072
-5-
O O O O O
-CNHSC1-C6perfluoroalkyl, -CNHS-phenyl, -NS-C1-C6alkyl,
H ~~
O O O
O O O
-N-SOH, -NSC1-C6perfluoroalkyl, -NHS-phenyl,
H II H II II
O O O
O
-NH-C-phenyl,
O O
-NHCC1-C6perfluoroalkyl; or NHCC1-C6alkyl;
wherein phenyl includes substituted phenyl.
In a preferred embodiment of the compounds of Formula I,
R1 is methyl and R2 is pentyl or hexyl.
Also preferred are compounds of Formula I wherein X1 and X2 both are
hydrogen.
In another preferred embodiment of the compounds of Formula I,
R~
the ~ group is located at the para position on the aryl ring, for example
R2
4-aminophenyl.
Also preferred are compounds of Formula I where Y has the Z geometry at
the double bond.
In another preferred embodiment, R1 and R2 are taken together with the
nitrogen to which they are attached to form a cyclic structure.
Also preferred are compounds wherein R2 is -(CH2)n-C3-C6 cycloalkyl or
-(CH2)n-phenyl when R1 is C1-Cg alkyl.
Especially preferred compounds are benzylidenes of Formula II
CA 02370316 2001-10-15
WO 00/76988 PCT/US00/15072
-6-
O
1
R N \ \ N-(CX1X2)n-X II
2~ i / S \\
R S
wherein n, X1, X2, and X are as defined above.
Further preferred compounds are naphthalenylmethylene derivatives of
Formula III
O
\~ \N-(CX1X2)n-X III
S
R1~N~ R2
S
wherein R1, R2, X1, X2, n, and X are as defined above.
Still other preferred compounds are quinolinylmethylene derivatives of
Formula IV
R4
I O
N / \ \ 1 2 IV
N- (CX X )n -X
R3 S \\
S
wherein R3, R4, X1, X2, n, and X are as defined above.
The most preferred invention compounds have Formula V
O
R' \~ ~r1- (CH2)n-X . V
S
R2 S
In a more preferred embodiment, the present invention provides the
compounds:
(Z) 2-{5-[4-(Hexyl-methyl-amino)-benzylidene]-4-oxo-2-thioxo-
thiazolidin-3-yl}-ethanesulfonic acid;
CA 02370316 2001-10-15
WO 00/76988 PCT/US00/15072
(Z) 2-{5-[4-(Hexyl-methyl-amino)-benzylidene]-4-oxo-2-thioxo-
thiazolidin-3-yl}-ethanesulfonic acid methylamide;
(Z) 2-{5-[4-(Hexyl-methyl-amino)-benzylidene]-4-oxo-2-thioxo-
thiazolidin-3-yl}-ethanesulfonic acid trifluoroacetyl-amide;
(Z) 2-{5-[4-(Hexyl-methyl-amino)-benzylidene]-4-oxo-2-thioxo-
thiazolidin-3-yl}-N- methyl-acetamide;
(Z) N-({5-[4-(Hexyl-methyl-amino)-benzylidene]-4-oxo-2-thioxo-
thiazolidin-3-yl }-acetyl)-methanesulfonamide;
(Z) N-{5-[4-(Dipentylamino-benzylidene)-4-oxo-2-thioxo-thiazolidin-3-
yl]-acetyl}-methanesulfonamide;
(Z) C,C,C-Trifluoro-N-({5-[4-(hexyl-methyl-amino)-benzylidene]-4-oxo-
2-thioxo-thiazolidin-3-yl}-acetyl)-methanesulfonamide;
(Z) N-{5-[4-(Dipentylamino-benzylidene)-4-oxo-2-thioxo-thiazolidin-3-
yl]-acetyl}-C,C,C-trifluoro-methanesulfonamide;
(Z) N-({5-[4-(Hexyl-methyl-amino)-benzylidene]-4-oxo-2-thioxo-
thiazolidin-3-yl}-acetyl)-benzenesulfonamide;
(Z) N-(2-{5-[4-(Hexyl-methyl-amino)-benzylidene]-4-oxo-2-thioxo-
thiazolin-3-yl}-ethyl)-methanesulfonamide;
(Z) N-(2-{5-[4-(Hexyl-methyl-amino)-benzylidene]-4-oxo-2-thioxo-
thiazolidin-3-yl}-ethyl)-benzenesulfonamide;
(Z) C,C,C-Trifluoro-N-(2-{5-[4-(hexyl-methyl-amino)-benzylidene]-4-
oxo-2-thioxo-thiazolidin-3-yl }-ethyl)-methanesulfonamide;
(Z) 2,2,2-Trifluoro-N-(2-{5-[4-(hexyl-methyl-amino)-benzylidene]-4-oxo-
2-thioxo-thiazolidin-3-yl}-ethyl)-acetamide;
(Z) N-(2-{5-[4-Hexyl-methyl-amino)-benzylidene]-4-oxo-2-thioxo-
thiazolidin-3-yl } ethyl)-acetamide;
(Z) {5-[4-(Hexyl-methyl-amino)-benzylidene]-4-oxo-2-thioxo-thiazolidin-
3-yl}-methanesulfonic acid;
(Z) 5-[4-(Hexyl-methyl-amino)-benzylidene]-3-(1H-tetrazol-5-ylmethyl)-
2-thioxo-thiazolidin-4-one;
(Z) 5-(4-Dipentylamino-benzylidene)-3-( 1 H-tetrazol-5-ylmethyl)-2-
thioxo-thiazolidin-4-one;
CA 02370316 2001-10-15
WO 00/76988 PCT/US00/15072
_g_
(Z) N-{[S-(4-Dibutylamino-benzylidene)-4-oxo-2-thioxo-thiazolidin-3-yl]-
acetyl}-C,C,C-trifluoro-methanesulfonamide;
(Z) N-{[5-(4-Dibutylamino-benzylidene)-4-oxo-2-thioxo-thiazolidin-3-yl]-
acetyl }-benzenesulfonamide;
(Z) 5-(4-Dibutylamino-benzylidene)-3-(1H-tetrazol-5-ylmethyl)-2-thioxo-
thiazolidin-4-one;
(Z) N-{2-[5-(4-Dibutylamino-benzylidene)-4-oxo-2-thioxo-thiazolidin-3-
yl]-acetyl}-methanesulfonamide;
(Z) N-{2-[5-(4-Dipentylamino-benzylidene)-4-oxo-2-thioxo-thiazolidin-3-
yl]-acetyl}-benzenesulfonamide;
(Z) 5-[(4aS,8aR)-4-(Octahydro-isoquinolin-2-yl)-benzylidene]-3-(1H-
tetrazol-5-ylmethyl)-2-thioxo-thiazolidin-4-one;
(Z) N-(2-{5-[(4aS,8aR)-4-(Octahydro-isoquinolin-2-yl)-benzylidene]-4-
oxo-2-thioxo-thiazolidin-3-yl}-acetyl)-benzenesulfonamide;
(Z) N-{2-[5-(4-Dibutylamino-benzylidene)-4-oxo-2-thioxo-thiazolidin-3-
yl]-acetyl}-4-fluoro-benzenesulfonamide;
(Z) 2-[5-(4-Dibutylamino-benzylidene)-4-oxo-2-thioxo-thiazolidin-3-yl]-
ethanesulfonic acid 4-fluoro-benzoylamide;
(Z) N-{2-[5-(4-Dipentylamino-benzylidene)-4-oxo-2-thioxo-thiazolidin-3-
yl]-acetyl}-4-fluoro-benzenesulfonamide;
(Z) 2-[5-(4-Dibutylamino-benzylidene)-4-oxo-2-thioxo-thiazolidin-3-yl]-
ethanesulfonic acid benzoylamide;
(Z) 2-{S-[4-(Octahydro-isoquinolin-2-yl)-benzylidene]-4-oxo-2-thioxo-
thiazolidin-3-yl}-ethanesulfonic acid benzoylamide;
(Z) 2-{5-[4-(Octahydro-isoquinolin-2-yl)-benzylidene]-4-oxo-2-thioxo-
thiazolidin-3-yl}-ethanesulfonic acid 4-fluoro-benzoylamide;
(Z) 2-[5-(4-Dipentylamino-benzylidene)-4-oxo-2-thioxo-thiazolidin-3-yl]-
ethanesulfonic acid 4-fluoro-benzoylamide;
(Z) 3-(5-Hydroxy-4-oxo-4H-pyran-2-ylmethyl)-5-[4-(octahydro-
isoquinolin-2-yl)-benzylidene]-2-thioxo-thiazolidin-4-one;
(Z) 5-(4-Dibutylamino-benzylidene)-3-(5-hydroxy-4-oxo-4H-pyran-2-
ylmethyl)-2-thioxo-thiazolidin-4-one;
CA 02370316 2001-10-15
WO 00/76988 PCT/US00/15072
-9-
(Z) 3-(5-Hydroxy-4-oxo-4H-pyran-2-ylmethyl)-5-[4-(4-propyl-piperidin-
1-yl)-benzylidene]-2-thioxo-thiazolidin-4-one;
(Z) 5-[4[(4-Propyl-piperidin-1-yl)-benzylidene]-3-(1H-tetrazol-5-
ylmethyl)-2-thioxo-thiazolidin-4-one;
(Z) N-(2-{4-Oxo-5-[4-(4-propyl-piperidin-1-yl)-benzylidene]-2-thioxo-
thiazolidin-3-yl}-acetyl)-benzenesulfonamide;
(Z) N-(2-{4-Oxo-5-[4-(4-propyl-piperidin-1-yl)-benzylidene]-2-thioxo-
thiazolidin-3-yl}-acetyl)-methanesulfonamide;
(Z) 4-Fluoro-N-(2-{5-[(4aS,8aR)-4-(octahydro-isoquinolin-2-yl)-
benzylidene]-4-oxo-2-thioxo-thiazolidin-3-yl}-acetyl)-benzenesulfonamide;
(Z) 4-Fluoro-N-(2-{4-oxo-5-[4-(4-propyl-piperidin-1-yl)-benzylidene]-2-
thioxo-thiazolidin-3-yl }-acetyl)-benzenesulfonamide;
(Z) 2-[5-(4-Hexyl-methyl-amino-benzylidene)-4-oxo-2-thioxo-thiazolidin-
3-yl]-ethanesulfonic acid 4-fluoro-benzoylamide;
(Z) N-({5-[4[(Octahydro-isoquinolin-2-yl)-benzylidene]-4-oxo-2-thioxo-
thiazolidin-3-yl}-acetyl)-methanesulfonamide;
(Z) N-({S-[4[(Octahydro-isoquinolin-2-yl)-benzylidene]-4-oxo-2-thioxo-
thiazolidin-3-yl}-acetyl)-C,C,C-trifluoro-methanesulfonamide;
(Z) N-(2-{4-Oxo-5-[4-(4-propyl-piperidin-1-yl)-benzylidene]-2-thioxo-
thiazolidin-3-yl}-acetyl)-C,C,C-trifluoro-methanesulfonamide;
(Z) 2-[5-(4-Dibutylamino-benzylidene)-4-oxo-2-thioxo-thiazolidin-3-yl]-
ethanesulfonic acid methylamide;
(Z) 2-[5-(4-Dipentylamino-benzylidene)-4-oxo-2-thioxo-thiazolidin-3-yl]-
ethanesulfonic acid methylamide;
(Z) 2-[5-(4-Hexyl-methyl-amino-benzylidene)-4-oxo-2-thioxo-thiazolidin-
3-yl]-ethanesulfonic acid methylamide;
(Z) N-2-{4-Oxo-5-[4-(4-propyl-piperidin-1-yl)-benzylidene]-2-thioxo-
thiazolidin-3-yl}-ethanesulfonic acid methylamide;
(Z) 2-{5-[4-(octahydro-isoquinolin-2-yl)-benzylidene]-4-oxo-2-thioxo-
thiazolidin-3-yl}S-ethanesulfonic acid methylamide;
(Z) 2-{5-[4-(Octahydro-isoquinolin-2-yl)-benzylidene]-4-oxo-2-thioxo-
thiazolidin-3-yl}S-ethanesulfonic acid trifluoroacetylamide;
CA 02370316 2001-10-15 .
WO 00/76988 PCT/US00/15072
-10-
(Z) N-2-{4-Oxo-5-[4-(4-propyl-piperidin-1-yl)-benzylidene]-2-thioxo-
thiazolidin-3-yl}-ethanesulfonic acid trifluoroacetylamide;
(Z) 2-[5-(4-Dipentylamino-benzylidene)-4-oxo-2-thioxo-thiazolidin-3-yl]-
ethanesulfonic acid trifluoroacetylamide;
(Z) 2-[5-(4-Dibutylamino-benzylidene)-4-oxo-2-thioxo-thiazolidin-3-yl]-
ethanesulfonic acid trifluoroacetylamide;
(Z) 2-[5-(4-Dipentylamino-benzylidene)-4-oxo-2-thioxo-thiazolidin-3-yl]-
ethanesulfonic acid benzoylamide;
(Z) 2-[5-(4-Hexyl-methyl-benzylidene)-4-oxo-2-thioxo-thiazolidin-3-yl]-
ethanesulfonic acid benzoylamide;
(Z) N-2-{4-Oxo-5-[4-(4-propyl-piperidin-1-yl)-benzylidene]-2-thioxo-
thiazolidin-3-yl}-ethanesulfonic acid benzoylamide;
(Z) N-2-{4-Oxo-5-[4-(4-propyl-piperidin-1-yl)-benzylidene]-2-thioxo-
thiazolidin-3-yl}-ethanesulfonic acid 4-fluoro-benzoylamide;
(Z) 2-[5-(4-Hexyl-methyl-benzylidene)-4-oxo-2-thioxo-thiazolidin-3-yl]-
ethanesulfonic acid 4-fluoro-benzoylamide;
(Z) [5-(4-Hexyl-methyl-amino)-benzylidene]-3-(5-oxo-4,5-dihydro-
[ 1,2,4] oxadiazol-3-ylmethyl)-2-thioxo-thiazolidin-4one;
(Z) [5-(4-Propyl-piperidin-1-yl)-benzylidene]-3-(5-oxo-4,5-dihydro-
[1,2,4]oxadiazol-3-ylmethyl)-2-thioxo-thiazolidin-4one;
(Z) [5-(4-Octahydro-isoquinolin-2-yl)-benzylidene]-3-(5-oxo-4,5-dihydro-
[ 1,2,4] oxadiazol-3-ylmethyl)-2-thioxo-thiazolidin-4one;
(Z) 5-(4-Dipentylamino-benzylidene)-3-(5-oxo-4,5-dihydro-
[1,2,4]oxadiazol-3-ylmethyl)-2-thioxo-thiazolidin-4-one; or
(Z) 5-(4-Dibutylamino-benzylidene)-3-(5-oxo-4,5-dihydro-
[ 1,2,4]oxadiazol-3-ylmethyl)-2-thioxo-thiazolidin-4-one.
Also provided is a pharmaceutical composition comprising a compound of
Formula I together with a pharmaceutically acceptable diluent, excipient, or
carrier therefor.
Also provided is a method of treating Alzheimer's disease, the method
comprising administering to a patient having Alzheimer's disease a
therapeutically effective amount of a compound of Formula I.
CA 02370316 2001-10-15
WO 00/76988 PCT/US00/15072
-11-
Also provided is a method of inhibiting the aggregation of amyloid
proteins to form amyloid deposits, the method comprising administering to a
patient in need of inhibition of the aggregation of amyloid proteins an
amyloid
protein aggregation inhibiting amount of a compound of Formula I.
Also provided is a method of imaging amyloid deposits, the method
comprising the steps o~
a. introducing into a patient a detectable quantity of a labeled
compound of Formula I;
b. allowing sufficient time for the labeled compound to become
associated with amyloid deposits; and
detecting the labeled compound associated with the amyloid
deposits.
In a preferred embodiment of the method of imaging, the patient has or is
suspected to have Alzheimer's disease.
In another preferred embodiment of the method of imaging, the labeled
compound is a radiolabeled compound.
In another preferred embodiment of the method of imaging, the labeled
compound is detected using MRI.
DETAILED DESCRIPTION OF THE INVENTION
The term "alkyl" means a straight or branched chain hydrocarbon.
Representative examples of alkyl groups are methyl, ethyl, propyl, isopropyl,
isobutyl, butyl, tert-butyl, sec-butyl, pentyl, and hexyl.
Preferred alkyl groups are C1-Cg alkyl.
The term "alkoxy" means an alkyl group attached to an oxygen atom.
Representative examples of alkoxy groups include methoxy, ethoxy, tent-butoxy,
propoxy, and isobutoxy.
The term "halogen" includes chlorine, fluorine, bromine, and iodine.
The term "substituted" means that one or more hydrogen atom in a
molecule has been replaced with another atom or group of atoms. For example,
substituents include halogen, -OH, -CF3, -N02, -NH2, -NH(C1-C6alkyl),
CA 02370316 2001-10-15
WO 00/76988 PCT/US00/15072
-12-
-N(C1-C6alkyl)2, C1-C6 alkyl, -OC1-C6 alkyl, -CN, -CF3, -C02H, and
-C02C1-C6 alkyl.
The term "substituted phenyl" means a phenyl ring in which from 1 to
4 hydrogen atoms have been independently replaced with a substituent,
preferably
one selected from the list above. Typical "substituted phenyl" groups include
4-fluorophenyl, 3-chlorophenyl, 3-methyoxyphenyl, 4-trifluoromethylphenyl,
4-dimethylamino phenyl, and 2,6-difluorophenyl.
The symbol "-" means a covalent bond.
The term pharmaceutically acceptable salt, ester, amide, and prodrug as
used herein refers to those carboxylate salts, amino acid addition salts,
esters,
amides, and prodrugs of the compounds of the present invention which are,
within
the scope of sound medical judgement, suitable for use in contact with the
tissues
of patients without undue toxicity, irritation, allergic response, and the
like,
commensurate with a reasonable benefitlrisk ratio, and effective for their
intended
use, as well as the zwitterionic forms, where possible, of the compounds of
the
invention. The term "salts" refers to the relatively nontoxic, inorganic and
organic
acid addition salts of compounds of the present invention. These salts can be
prepared in situ during the final isolation and purification of the compounds
or by
separately reacting the purified compound in its free base form with a
suitable
organic or inorganic acid and isolating the salt thus formed. Representative
salts
include the hydrobromide, hydrochloride, sulfate, bisulfate, nitrate, acetate,
oxalate, valerate, oleate, palmitate, stearate, laurate, borate, benzoate,
lactate,
phosphate, tosylate, citrate, maleate, fumarate, succinate, tartrate,
naphthylate
mesylate, glucoheptonate, lactiobionate and laurylsulphonate salts, and the
like.
These may include cations based on the alkali and alkaline earth metals, such
as
sodium, lithium, potassium, calcium, magnesium, and the like, as well as,
nontoxic ammonium, quaternary ammonium and amine cations including, but not
limited to ammonium, tetramethylammonium, tetraethylammonium, methylamine,
dimethylamine, trimethylamine, triethylamine, ethylamine, and the like. (See,
for
example, Berge S.M., et al., Pharmaceutical Salts, J. Pharm. Sci., 1977;66:1-
19
which is incorporated herein by reference.)
CA 02370316 2001-10-15
WO 00/76988 PCT/US00/15072
-13-
Examples of pharmaceutically acceptable, nontoxic esters of the
compounds of this invention include C1-C6 alkyl esters wherein the alkyl group
is
a straight or branched chain. Acceptable esters also include C5-C7 cycloalkyl
esters as well as arylalkyl esters such as, but not limited to benzyl. C1-C4
alkyl
esters are preferred. Esters of the compounds of the present invention may be
prepared according to conventional methods.
Examples of pharmaceutically acceptable, nontoxic amides of the
compounds of this invention include amides derived from ammonia, primary
C1-C6 alkyl amines and secondary C1-C6 dialkyl amines wherein the alkyl
groups are straight or branched chain. In the case of secondary amines, the
amine
may also be in the form of a 5- or 6-membered heterocycle containing one
nitrogen atom. Amides derived from ammonia, C1-C3 alkyl primary amides and
C1-C2 dialkyl secondary amides are preferred. Amides of the compounds of the
invention may be prepared according to conventional methods.
The term "prodrug" refers to compounds that are rapidly transformed
in vivo to yield the parent compound of the above formulas, for example, by
hydrolysis in blood. A thorough discussion is provided in T. Higuchi and
V. Stella, Pro-drugs as Novel Delivery Systems, Vol. 14 of the A.C.S.
Symposium
Series, and in Bioreversible Carriers in Drug Design, ed. Edward B. Roche,
American Pharmaceutical Association and Pergamon Press, 1987, both of which
are incorporated herein by reference.
In addition, the compounds of the present invention can exist in unsolvated
as well as solvated forms with pharmaceutically acceptable solvents such as
water,
ethanol, and the like. In general, the solvated forms are considered
equivalent to
the unsolvated forms for the purposes of the present invention.
The compounds of the present invention can exist in different
stereoisometric forms by virtue of the presence of asymmetric centers in the
compounds. It is contemplated that all stereoisometric forms of the compounds,
as
well as mixture thereof, including racemic mixtures, form part of this
invention.
In the first step of the present method of imaging, a labeled compound of
Formula I is introduced into a tissue or a patient in a detectable quantity.
The
CA 02370316 2001-10-15
WO 00/76988 PCT/US00/15072
-14-
compound is typically part of a pharmaceutical composition and is administered
to
the tissue or the patient by methods well-known to those skilled in the art.
In the methods of the present invention, a compound can be administered
either orally, rectally, parenterally (intravenous, by intramuscularly or
subcutaneously), intracisternally, intravaginally, intraperitoneally,
intravesically,
locally (powders, ointments or drops), or as a buccal or nasal spray.
Compositions suitable for parenteral injection may comprise
physiologically acceptable sterile aqueous or nonaqueous solutions,
dispersions,
suspensions or emulsions, and sterile powders for reconstitution into sterile
injectable solutions or dispersions. Examples of suitable aqueous and
nonaqueous
carriers, diluents, solvents, or vehicles include water, ethanol, polyols
(propyleneglycol, polyethyleneglycol, glycerol, and the like), suitable
mixtures
thereof, vegetable oils (such as olive oil), and injectable organic esters
such as
ethyl oleate. Proper fluidity can be maintained, for example, by the use of a
coating such as lecithin, by the maintenance of the required particle size in
the
case of dispersions and by the use of surfactants.
These compositions may also contain adjuvants such as preserving,
wetting, emulsifying, and dispensing agents. Prevention of the action of
microorganisms can be ensured by various antibacterial and antifungal agents,
for
example, parabens, chlorobutanol, phenol, sorbic acid, and the like. It may
also be
desirable to include isotonic agents, for example sugars, sodium chloride, and
the
like. Prolonged absorption of the injectable pharmaceutical form can be
brought
about by the use of agents delaying absorption, for example, aluminum
monostearate and gelatin.
Solid dosage forms for oral administration include capsules, tablets, pills,
powders, and granules. In such solid dosage forms, the active compound is
admixed with at least one inert customary excipient (or carrier) such as
sodium
citrate or dicalcium phosphate or (a) fillers or extenders, as for example,
starches,
lactose, sucrose, glucose, mannitol, and silicic acid; (b) binders, as for
example,
carboxymethylcellulose, alignates, gelatin, polyvinylpyrrolidone, sucrose, and
acacia; (c) humectants, as for example, glycerol; (d) disintegrating agents,
as for
example, agar-agar, calcium carbonate, potato or tapioca starch, alginic acid,
certain complex silicates and sodium carbonate; (e) solution retarders, as for
CA 02370316 2001-10-15
WO 00/76988 PCT/US00/15072
-15-
example paraffin; (f) absorption accelerators, as for example, quaternary
ammonium compounds; (g) wetting agents, as for example, cetyl alcohol and
glycerol monostearate; (h) adsorbents, as for example, kaolin and bentonite;
and
(i) lubricants, as for example, talc, calcium stearate, magnesium stearate,
solid
polyethylene glycols, sodium lauryl sulfate, or mixtures thereof. In the case
of
capsules, tablets, and pills, the dosage forms may also comprise buffering
agents.
Solid compositions of a similar type may also be employed as fillers in
soft- and hard-filled gelatin capsules using such excipients as lactose or
milk
sugar, as well as high molecular weight polyethyleneglycols, and the like.
Solid dosage forms such as tablets, dragees, capsules, pills, and granules
can be prepared with coatings and shells, such as enteric coatings and others
well
known in the art. They may contain opacifying agents, and can also be of such
composition that they release the active compound or compounds in a certain
part
of the intestinal tract in a delayed manner. Examples of embedding
compositions
which can be used are polymeric substances and waxes. The active compounds
can also be in microencapsulated form, if appropriate, with one or more of the
above-mentioned excipients.
Liquid dosage forms for oral administration include pharmaceutically
acceptable emulsions, solutions, suspensions, syrups, and elixirs. In addition
to the
active compounds, the liquid dosage forms may contain inert diluents commonly
used in the art, such as water or other solvents, solubilizing agents and
emulsifiers,
as for example, ethyl alcohol, isopropyl alcohol, ethyl carbonate, ethyl
acetate,
benzyl alcohol, benzyl benzoate, propyleneglycol, 1,3-butyleneglycol,
dimethylformamide, oils, in particular, cottonseed oil, groundnut oil, corn
germ
oil, olive oil, castor oil, and sesame oil, glycerol, tetrahydrofurfuryl
alcohol,
polyethyleneglycols, and fatty acid esters of sorbitan or mixtures of these
substances, and the like.
Besides such inert diluents, the composition can also include adjuvants,
such as wetting agents, emulsifying and suspending agents, sweetening,
flavoring,
and perfuming agents.
Suspensions, in addition to the active compounds, may contain suspending
agents, as for example, ethoxylated isostearyl alcohols, polyoxyethylene
sorbitol
CA 02370316 2001-10-15
WO 00/76988 PCT/US00/15072
-16-
and sorbitan esters, microcrystalline cellulose, aluminum metahydroxide,
bentonite, agar-agar and tragacanth, or mixtures of these substances, and the
like.
Compositions for rectal administrations are preferably suppositories which
can be prepared by mixing the compounds of the present invention with suitable
nonirritating excipients or carriers such as cocoa butter, polyethyleneglycol
or a
suppository wax, which are solid at ordinary temperatures but liquid at body
temperature and therefore, melt in the rectum or vaginal cavity and release
the
active component.
Dosage forms for topical administration of a compound of this invention
include ointments, powders, sprays, and inhalants. The active component is
admixed under sterile conditions with a physiologically acceptable carrier and
any
preservatives, buffers or propellants as may be required. Ophthalmic
formulations,
eye ointments, powders, and solutions are also contemplated as being within
the
scope of this invention.
In a preferred embodiment of the invention, the labeled compound is
introduced into a patient in a detectable quantity and after sufficient time
has
passed for the compound to become associated with amyloid deposits, the
labeled
compound is detected noninvasively inside the patient. In another embodiment
of
the invention, a labeled compound of Formula I is introduced into a patient,
sufficient time is allowed for the compound to become associated with amyloid
deposits, and then a sample of tissue from the patient is removed and the
labeled
compound in the tissue is detected apart from the patient. In a third
embodiment
of the invention, a tissue sample is removed from a patient and a labeled
compound of Formula I is introduced into the tissue sample. After a sufficient
amount of time for the compound to become bound to amyloid deposits, the
compound is detected.
The administration of the labeled compound to a patient can be by a
general or local administration route. For example, the labeled compound may
be
administered to the patient such that it is delivered throughout the body.
Alternatively, the labeled compound can be administered to a specific organ or
tissue of interest. For example, it is desirable to locate and quantitate
amyloid
deposits in the brain in order to diagnose or track the progress of
Alzheimer's
disease in a patient.
CA 02370316 2001-10-15
WO 00/76988 PCT/US00/15072
-17-
The term "tissue" means a part of a patient's body. Examples of tissues
include the brain, heart, liver, blood vessels, and arteries. A detectable
quantity is
a quantity of labeled compound necessary to be detected by the detection
method
chosen. The amount of a labeled compound to be introduced into a patient in
order
to provide for detection can readily be determined by those skilled in the
art. For
example, increasing amounts of the labeled compound can be given to a patient
until the compound is detected by the detection method of choice. A label is
introduced into the compounds to provide for detection of the compounds.
The term "patient" means humans and other animals. Those skilled in the
art are also familiar with determining the amount of time sufficient for a
compound to become associated with amyloid deposits. The amount of time
necessary can easily be determined by introducing a detectable amount of a
labeled compound of Formula I into a patient and then detecting the labeled
compound at various times after administration.
The term "associated" means a chemical interaction between the labeled
compound and the amyloid deposit. Examples of associations include covalent
bonds, ionic bonds, hydrophilic-hydrophilic interactions, hydrophobic-
hydrophobic interactions, and complexes.
Those skilled in the art are familiar with the various ways to detect labeled
compounds. For example, magnetic resonance imaging (MRI), positron emission
tomography (PET), or single photon emission computed tomography (SPELT)
can be used to detect radiolabeled compounds. The label that is introduced
into the
compound will depend on the detection method desired. For example, if PET is
selected as a detection method, the compound must possess a positron-emitting
atom, such as 11 C or 1 gF.
Another example of a suitable label in a compound of Formula I is an atom
such as 13C, 15N, or 19F which can be detected using magnetic resonance
imaging (MRI) which is also sometimes called nuclear magnetic resonance
(NMR). In addition, the labeled compounds of Formula I may also be detected by
MRI using paramagnetic contrast agents.
CA 02370316 2001-10-15
WO 00/76988 PCT/US00/15072
-18-
Another example of detection is electron paramagnetic resonance (EPR).
In this case, EPR probes which are well-known in the art, such as nitroxides,
can
be used.
The imaging of amyloid deposits can also be carried out quantitatively so
that the amount of amyloid deposits can be determined.
The present invention also provides a method of inhibiting the aggregation
of amyloid proteins to form amyloid deposits, by administering to a patient in
need of inhibition of the aggregation of amyloid protein an amyloid protein
inhibiting amount of a compound of Formula I. Those skilled in the art are
readily
able to determine an amyloid inhibiting amount by simply administering a
compound of Formula I to a patient in increasing amounts until the growth of
amyloid deposits is decreased or stopped. The rate of growth can be assessed
using imaging or by taking a tissue sample from a patient and observing the
amyloid deposits therein.
A patient in need of inhibition of the aggregation of amyloid proteins is a
patient having a disease or condition in which amyloid proteins aggregate.
Examples of such diseases and conditions include Mediterranean fever, Muckle-
Wells syndrome, idiopathetic myeloma, amyloid polyneuropathy, amyloid
cardiomyopathy, systemic senile amyloidosis, amyloid polyneuropathy,
hereditary
cerebral hemorrhage with amyloidosis, Alzheimer's disease, Down's syndrome,
Scrapie, Creutzfeldt-Jacob disease, Kuru, Gerstmann-Straussler-Scheinker
syndrome, medullary carcinoma of the thyroid, isolated atrial amyloid,
(32-microglobulin amyloid in dialysis patients, inclusion body myositis,
~i2-amyloid deposits in muscle wasting disease, and Islets of Langerhans
diabetes
Type II insulinoma.
Also provided by the present invention are compounds of Formula I
wherein one or more atom in the compound has been replaced with a
radioisotope.
The radioisotope can be any radioisotope. However,3H, 123h 125h 131h 13C~
and 18F are preferred. Those skilled in the art are familiar with the
procedure used
to introduce a radioisotope into a compound. For example, compounds of
Formula I wherein one carbon atom is 13C are readily prepared by standard
method in organic chemistry.
CA 02370316 2001-10-15
WO 00/76988 PCT/US00/15072
-19-
T'he compounds of the present invention can be administered to a patient at
dosage levels in the range of about 0.1 to about 1,000 mg per day. For a
normal
human adult having a body weight of about 70 kg, a dosage in the range of
about
0.01 to about 100 mg per kilogram of body weight per day is sufficient. The
S specific dosage used, however, can vary. For example, the dosage can depend
on a
number of factors including the requirements of the patient, the severity of
the
condition being treated, and the pharmacological activity of the compound
being
used. The determination of optimum dosages for a particular patient is well-
known to those skilled in the art.
The examples presented below are intended to illustrate particular
embodiments of the invention and are not intended to limit the scope of the
specification, including the claims, in any manner.
EXAMPLES
The compounds of the present invention can be generally prepared as
illustrated in Scheme 1 below. With regard to Scheme 1, appropriately
substituted
amino benzaldehydes are commercially available or are prepared by reacting
4-fluorobenzaldehyde with an amine in the presence of a base such as potassium
carbonate in a solvent such as dimethylacetamide or dimethylformamide.
N-substituted rhodanines that are not commercially available are prepared by
condensing carbon disulfide and chloroacetic acid with the appropriate amine.
The
compounds of the present invention can be prepared by condensation of an
appropriately N-substituted rhodanine with an appropriately substituted
aromatic
aldehyde in refluxing glacial acetic acid in the presence of sodium acetate.
Other
methods of preparing invention compounds will be readily available to those
skilled in organic chemistry.
CA 02370316 2001-10-15
WO 00/76988 PCT/US00/15072
-20-
Scheme 1
Rl K2C03 ~ CHO
CHO
I + NH _ 1
/ R2~ dimethylacetamide RAN /
95°C I
R2
O X1 X2
CS2 S C1CH2C02H
H N(CX1X2) X ~ ~ N n
2 n H NS' 'N CX1X2 X X
NH40H 4 ( )n H O S'
2 S
CHO O Xl X2
NaOAc, HOAc
Rv I ~ + N\J
S ~ X reflux
R2 S
O X1 X2
N\
S X
R~ /
N S
I 2
R
CA 02370316 2001-10-15
WO 00/76988 PCT/US00/15072
-21
Example 1
(Z) 2-{5-[4-(Hexyl-methyl-amino)-benzylidene]-4-oxo-2-thioxo-thiazolidin-3-
yl}- ethanesulfonic acid
Step A:
Hexylmethyl amine (10 g, 86.8 mmol), 4-fluorobenzaldehyde (8.0 mL,
75.0 mmol) and potassium carbonate ( 12.4 g, 90.0 mmol) in dimethylacetamide
(15 mL) are heated to 95°C for 3 days with vigorous stirring. The
reaction mixture
is cooled, diluted with water (200 mL) and extracted with diethyl ether. The
organic extract is dried (magnesium sulfate) and concentrated in vacuo. The
resulting oil is purified by medium pressure liquid chromatography (MPLC) on
silica gel eluting with 5% ethyl acetate/hexane to give 14.9 g of 4-(n-
hexylmethylamino)benzaldehyde as a yellow oil.
Step B:
To a mixture of carbon disulfide (5.5 mL, 90 mmol) and ammonium
hydroxide (20 mL) at 0°C is added 2-aminoethane sulfonic acid (9.0 g,
72 mmol).
The reaction mixture is warmed to room temperature and stirred for 18 hours
then
concentrated to dryness. This dithiocarbamate is added slowly to a cold
(0°C)
solution of sodium chloroacetate (8.5 g, 75 mmol) in water (25 mL) made basic
with sodium carbonate. The reaction mixture is warmed to room temperature and
poured into a warm (70°C) HCl solution (160 mL, 5 M) and heated to
90°C for
1 hour. The reaction mixture is cooled, the product collected on a filter,
washed
with water, and dried to give 10.6 g of rhodanine-3-ethane sulfonic acid.
Step C:
4-(n-Hexylmethylamino)benzaldehyde (0.91 g, 4.14 mmol), rhodanine-3-
ethyl sulfonic acid (1.00 g, 4.14 mmol), and sodium acetate acetate (0.42 g,
4.97 mmol) in acetic acid (15 mL) are heated to reflux for 15 hours with
stirring.
The reaction mixture is cooled, diluted with water, and the precipitated
solids are
collected by filtration, washed with water, and dried under vacuum to provide
0.85 g of the title compound as the sodium salt; melting point (mp)
>250°C.
Elemental analysis calculated for C19H25N2~4S3Na~(2.01 mol) H20:
Calculated: C, 45.57; H, 5.84; N, 5.59. Found: C, 45.57; H, 5.62; N, 5.41.
CA 02370316 2001-10-15
WO 00/76988 PCT/US00/15072
-22
Example 2
(Z) 2-{5-[4-(Hexyl-methyl-amino)-benzylidene]-4-oxo-2-thioxo-thiazolidin-3-
yl}-ethanesulfonic acid methylamide
(Z) 2-{5-[4-(Hexyl-methyl-amino)-benzylidene]-4-oxo-2-thioxo-
thiazolidin-3-yl}-ethanesulfonic acid (0.50 g, 1.13 mmol) is suspended in
anhydrous (anh.) dichloromethane (25 mL) under N2. To this mixture is added
anh. dimethylformamide (6 mL) followed by oxalyl chloride (0.11 mL,
1.24 mmol). The reaction mixture is stirred for 3.5 hours at room temperature.
Methyl amine (2.0 M in tetrahydrofuran (THF), 1.7 mL, 3.39 mmol) is added, and
stirring is continued overnight. The reaction mixture is concentrated in vacuo
and
purified by MPLC (1% to 10% methanol (MeOH) in CH2C12) to give 0.106 g of
the title compound, mp 147-149°C. Elemental analysis calculated for
C20H29N303s3~ Calculated: C, 52.72; H, 6.42; N, 9.22. Found: C, 52.72; H,
6.38; N, 9.11.
Example 3
(Z) 2-{5-[4-(Hexyl-methyl-amino)-benzylidene]-4-oxo-2-thioxo-thiazolidin-3-
yl}-ethanesulfonic acid trifluoroacetyl-amide
(Z) 2-{5-[4-(Hexyl-methyl-amino)-benzylidene]-4-oxo-2-thioxo-
thiazolidin-3-yl}-ethanesulfonic acid (0.70 g, 1.58 mmol) is suspended in
(anh.) dichloromethane (35 mL) under N2. To this mixture was added anh.
dimethylformamide (10 mL) followed by oxalyl chloride (3.6 mL, 7.27 mmol).
The reaction mixture is stirred 15 hours at room temperature. In a separate
flask,
sodium hydride (60% dispersion on mineral oil, 0.76 g, 19.0 mmol) is suspended
in anh. dimethylformamide (10 mL) under N2. Triflouromethyl acetamide (2.15 g,
19.0 mmol) is slowly added. This mixture is stirred 20 minutes, added to the
sulfonyl chloride solution, and stirred an additional 2 hours at room
temperature.
The reaction mixture is concentrated in vacuo and purified by MPLC (5% MeOH
in CH2Cl2) to give 0.176 g of the title compound as an orange solid,
mp 158-162°C. Elemental analysis calculated for C21H26F3N304S3~
Calculated:
C, 46.91; H, 4.87; N, 7.82. Found: C, 44.40; H, 4.59; N, 7.44.
CA 02370316 2001-10-15
WO 00/76988 PCT/US00/15072
-23
Example 4
(Z) 2-{5-[4-(Hexyl-methyl-amino)-benzylidene]-4-oxo-2-thioxo-thiazolidin-3-
yl}-N-methyl-acetamide
Ethyl chloroformate (0.23 mL, 2.42 mmol) is dissolved in anh.
tetrahydrofuran (10 mL) and cooled to 0°C under N2. A solution of {5-[4-
(hexyl-
methyl-amino)-benzylidene]-4-oxo-2-thioxo-thiazolidin-3-yl}-acetic acid (0.50
g,
1.27 mmol) and triethylamine (0.32 mL, 2.29 mmol) in anh. tetrahydrofuran
(30 mL) is added dropwise. Stirred 2 hours at 0°C, then allowed to warm
to room
temperature. Methyl amine (2.0 M in THF, 1.91 mL, 3.81 mmol) is added and
stirred at room temperature under N2 for 15 hours. The reaction mixture is
concentrated in vacuo and purified by MPLC (3% MeOH/CH2C12) to give
0.254 g of the title compound as an orange solid, mp 228-230°C.
Elemental
analysis calculated for C2pH27N302S2: Calculated: C, 59.23; H, 6.71; N, 10.36.
Found: C, 58.82; H, 6.61; N, 10.00.
Example 5
(Z) N-({5-[4-(Hexyl-methyl-amino)-benzylidene]-4-oxo-2-thioxo-thiazolidin-3-
yl}-acetyl)-methanesulfonamide
A solution of {5-[4-(hexyl-methyl-amino)-benzylidene]-4-oxo-2-thioxo-
thiazolidin-3-yl}-acetic acid (0.50 g, 1.27 mmol) in anh. dimethylformamide
(12 mL) is treated with 1,1'-carbonyldiimidazole (0.62 g, 3.82 mmol) and
stirred
at 60°C for 4 hours under N2. A solution of methylsulfonamide (0.36 g,
3.77 mmol) in anh. dimethylformamide (10 mL) is treated with sodium hydride
(60% dispersion on mineral oil, 0.16 g, 4.04 mmol), stirred 4 hours under N2,
added to the reaction mixture and stirred for 15 hours at room temperature.
The
reaction mixture is poured into 1N HCl (100 mL) at 0°C. The resulting
suspension
is collected, washed with water and purified by MPLC (3% MeOH/CH2Cl2) to
give 0.375 g of the title compound as an orange solid, mp 227-230°C.
Elemental
analysis calculated for C2pH27N304S3x0.28H20: Calculated: C, 50.61; H, 5.85;
N, 8.85. Found: C, 50.37; H, 5.60; N, 8.62.
CA 02370316 2001-10-15
WO 00/76988 PCT/US00/15072
-24-
Example 6
(Z) N-{5-[4-(Dipentylamino-benzylidene)-4-oxo-2-thioxo-thiazolidin-3-yl]-
acetyl}-methanesulfonamide
Example 6 was prepared according to Example 5, except that [5-(4-
S dipentylamino-benzylidene)-4-oxo-2-thioxo-thiazolidin-3-yl]-acetic acid is
substituted for {5-[4-(hexyl-methyl-amino)-benzylidene]-4-oxo-2-thioxo-
thiazolidin-3-yl}-acetic acid, mp 134-138°C. Elemental analysis
calculated for
C23H33N304S3X2.OCH30H: Calculated: C, 52.15; H, 7.18; N, 7.30. Found: C,
52.36; H, 7.05; N, 6.97.
Example 7
(Z) C,C,C-Trifluoro-N-({5-[4-(hexyl-methyl-amino)-benzylidene]-4-oxo-2-
thioxo-thiazolidin-3-yl}-acetyl)-methanesulfonamide
Example 7 was prepared according to Example 5, except that
trifluoromethanesuflonamide is substituted for methanesulfonamide, mp 94-
97°C.
Elemental analysis calculated for C2pH24F3N3O4S3x1.OC6H15N: Calculated: C,
49.98; H, 6.29; N, 8.97. Found: C, 50.01; H, 6.20; N, 8.77.
Example 8
(Z) N-{5-[4-(Dipentylamino-benzylidene)-4-oxo-2-thioxo-thiazolidin-3-yl]-
acetyl}-C,C,C-trifluoro-methanesulfonamide
Example 8 was prepared according to Example 6, except that
trifluoromethanesuflonamide is substituted methanesulfonamide, mp 286-
288°C.
Elemental analysis calculated for C23H30F3N304S3 ~ Calculated: C, 48.83; H,
5.35; N, 7.43. Found: C, 47.03; H, 4.90; N, 7.12.
Example 9
(Z) N-({5-[4-(Hexyl-methyl-amino)-benzylidene]-4-oxo-2-thioxo-thiazolidin-3-
yl}-acetyl)-benzenesulfonamide
Example 9 was prepared according to Example 5, except
benzenesulfonamide is substituted for methanesulfonamide, mp 173-177°C.
CA 02370316 2001-10-15
WO 00/76988 PCT/US00/15072
-25-
Elemental analysis calculated for C25H29N3~4S3X0.33H20: Calculated: C,
55.85; H, 5.56; N, 7.82. Found: C, 55.81; H, 5.48; N, 7.59.
Example 10
(Z) N-(2-{5-[4-(Hexyl-methyl-amino)-benzylidene]-4-oxo-2-thioxo-thiazolin-3-
yl}-ethyl)-methanesulfonamide
Step A:
(Z) N-(2-{5-[4-(Hexyl-methyl-amino)-benzylidene]-4-oxo-2-thioxo-
thiazolidin-3-yl}-ethyl)-acetamide (0.50 g, 1.19 mmol) is suspended in 2N HC1
( 100 mL) and heated at reflux for 6 hours. The reaction mixture is cooled to
room
temperature, concentrated and purified by MPLC (5% MeOH/CH2C12) to give
0.380 g of (Z) N-(2-{5-[4-(hexyl-methyl-amino)-benzylidene]-4-oxo-2-thioxo-
thiazolidin-3-yl}-ethyl)-amine.
Step B:
Triethylamine (0.12 mL, 0.83 mmol) is added to (Z) N-(2-{5-[4-(hexyl-
methyl-amino)-benzylidene]-4-oxo-2-thioxo-thiazolidin-3-yl}-ethyl)-amine
(0.175 g, 0.46 mmol) in anh. dimethylformamide (20 mL), followed by
methanesulfonyl chloride (0.07 mL, 0.93 mmol). The reaction is stirred
overnight
at room temperature under N2, concentrated under vacuum, and purified by
MPLC (1% MeOH/CH2C12) to give 50 mg of the title compound, mp 150-
153°C.
Elemental analysis calculated for C2pH29N3~3S3~ Calculated: C, 52.72; H, 6.42;
N, 9.22. Found: C, 52.84; H, 6.39; N, 9.14.
Example 11
(Z) N-(2-{5-[4-(Hexyl-methyl-amino)-benzylidene]-4-oxo-2-thioxo-thiazolidin-
3-yl}-ethyl)-benzenesulfonamide
Example 11 was prepared according to Example 10, except
benzenesulfonyl chloride is substituted for methanesulfonyl chloride,
mp 152-155°C. Elemental analysis calculated for C25H31N303S3~
Calculated: C,
58.00; H, 6.04; N, 8.12. Found: C, 58.28; H, 6.07; N, 8.05.
CA 02370316 2001-10-15
WO 00/76988 PCT/US00/15072
-26-
Example 12
(Z) C,C,C-Trifluoro-N-(2-{5-[4-(hexyl-methyl-amino)-benzylidene]-4-oxo-2-
thioxo-thiazolidin-3-yl}-ethyl)-methanesulfonamide
Example 12 was prepared according to Example 10, except
trifluoromethanesulfonyl chloride is substituted for methanesulfonyl chloride,
mp 170-173°C. Elemental analysis calculated for C2pH26F3N3~3S3~
Calculated:
C, 47.14; H, 5.14; N, 8.25. Found: C, 47.43; H, 5.06; N, 8.16.
Example 13
(Z) 2,2,2-Trifluoro-N-(2-{5-[4-(hexyl-methyl-amino)-benzylidene]-4-oxo-2-
thioxo-thiazolidin-3-yl}-ethyl)-acetamide
Triflouroacetic anhydride (0.76 mL, 5.38 mmol) is added to (Z) N-(2-{5-
[4-(hexyl-methyl-amino)-benzylidene]-4-oxo-2-thioxo-thiazolidin-3-yl}-ethyl)-
amine (0.48 g, 1.27 mmol) and sodium carbonate (0.57 g, 5.38 mmol) in anh.
acetonitrile (30 mL). The reaction mixture is stirred overnight at room
temperature
under N2, concentrated in vacuo, and purified by MPLC (5% MeOH/CH2Cl2) to
give 0.100 g of the title compound. mp 136-139°C. Elemental analysis
calculated
for C21H26F3N3~2S2~ Calculated: C, 53.26; H, 5.53; N, 8.87. Found: C, 53.28;
H, 5.43; N, 8.79.
Example 14
(Z) N-(2-{5-[4-(Hexyl-methyl-amino)-benzylidene]-4-oxo-2-thioxo-thiazolidin-
3-yl}-ethyl)-acetamide
Step A:
Rhodanine-3-ethyl acetamide was prepared according to Example 1,
Step B, except acetyl ethylenediamine is substituted for 2-aminoethane
sulfonic
acid.
Step B:
Example 14 was prepared according to Example 1, Step C, except
rhodanine-3-ethyl acetamide is substituted for rhodanine-3-ethyl sulfonic
acid,
CA 02370316 2001-10-15
WO 00/76988 PCT/US00/15072
-27-
mp 137-140°C. Elemental analysis calculated for C21H29N~2S2=
Calculated: C,
60.11; H, 6.97; N, 10.01. Found: C, 60.3 8; H, 7.06; N, 10.01.
Example 15
(Z) {5-[4-(n-Hexylmethylamine)-benzylidene]-4-oxo-2-thioxo-thiazolidin-3-
yl}-methanesulfonic acid
By following the general procedure of Example 1, 4-(n-
hexylmethylamino)benzaldehyde was reacted with rhodanine-3-methane sulfonic
acid to give (Z) {5-[4-(n-hexylmethylamine)-benzylidene]-4-oxo-2-thioxo-
thiazolidin-3-yl}-methanesulfonic acid.
MS 429 (M+).
Example 16
(Z) 5-(4-(Hexyl-methyl-amino)-benzylidene]-3-(1H-tetrazol-5-ylmethyl)-2-
thioxo-thiazolidin-4-one
Step A:
Phthalimide (19.5 g, 0.132 mol) is suspended in anhydrous DMF (80 mL)
under N2. Potassium t-butoxide (17.8 g, 0.159 mol) is added slowly and the
suspension stirred at room temperature for 10 minutes. Chloracetonitrile
(10.1 mL, 0.159 mol) is then added and the mixture stirred overnight. Methanol
(50 mL) is added and the mixture is concentrated in vacuo. The phthalimide
acetonitrile is purified by MPLC (100% CH2C12) to give 16.0 g of white
crystalline solid.
Step B:
Sodium azide (5.81 g, 89.4 mmol) and ammonium chloride (4.6 g,
85.9 mmol) are added to the phthalimide acetonitrile (16.0 g, 85.9 mmol) in
DMF
(100 mL) and heated to 100°C for 6 hours. The solids are filtered and
the filtrate
concentrated in vacuo to give 17.25 g of the phthalimide methyl tetrazole as a
white crystalline solid, mp 164-167°C.
CA 02370316 2001-10-15
WO 00/76988 PCT/US00/15072
-28-
Step C:
Hydrazine monohydrate (2.7 mL, 56.0 mmol) is added to the phthalimide
methyl tetrazole (12.6 g, 55.0 mmol) suspended in EtOH (400 mL) and heated to
reflux for 3 hours. The mixture is concentrated in vacuo and resuspended in 3N
HCl (500 mL). The solids are filtered, washed with water, and dried to give
7.8 g
of the tetrazole methyl amine as a white solid hydrochloride salt, mp 148-
153°C.
Step D:
1H-tetrazol-5-ylmethyl-2-thioxo-thiazolidin-4-one was prepared from the
tetrazole methyl amine as previously described in Example 1, Step B.
Step E:
5-[4-(Hexyl-methyl-amino)-benzylidene]-3-( 1 H-tetrazol-5-ylmethyl)-2-
thioxo-thiazolidin-4-one was prepared as previously described in Example 1,
Step C, mp 198-200°C.
MS 417 (M+).
0 0 0
\ t-BuOK \ NaN , DMF \
I NH + C1~CN I N~ ~ I N
/ DMF / ~ NH4CI / NH
O O O NN~
H2NNH2 x H20
EtOH
O H 1.) CS2, NH40H HCIx H2N
N N~ 2.) C1CH2C02Na, H20 ~NH
/ 1
~ 3.) HC1 N~
S'\S N~N N~
\ CHO
I NaOAc
HOAc
O
\ \~ \N
/ S~ N/ NN
S ~%
CA 02370316 2001-10-15
WO 00/76988 PCT/US00/15072
-29-
The following invention compounds were prepared by following the
general procedures of the foregoing examples.
Example 17
(Z) 5-(4-Dipentylamino-benzylidene)-3-(1H-tetrazol-5-ylmethyl)-2-thioxo-
thiazolidin-4-one. mp 208-210°C.
MS 459 (M+).
Example 18
(Z) N-{[5-(4-Dibutylamino-benzylidene)-4-oxo-2-thioxo-thiazolidin-3-yl]-
acetyl}-C,C,C-trifluoro-methanesulfonamide. mp 228-230°C.
MS 538 (M+)
Example 19
(Z) N-{[5-(4-Dibutylamino-benzylidene)-4-oxo-2-thioxo-thiazolidin-3-yl]-
acetyl}-benzenesulfonamide. mp 112-115°C.
MS 545 (M+)
Example 20
(Z) 5-(4-Dibutylamino-benzylidene)-3-(1H-tetrazol-5-ylmethyl)-2-thioxo-
thiazolidin-4-one. mp 243-246°C.
MS 431 (M+).
Example 21
(Z) N-{2-[5-(4-Dibutylamino-benzylidene)-4-oxo-2-thioxo-thiazolidin-3-yl)-
acetyl}-methanesulfonamide. mp 219-222°C.
MS 484 (M+).
Example 22
(Z) N-{2-[5-(4-Dipentylamino-benzylidene)-4-oxo-2-thioxo-thiazolidin-3-yl]-
acetyl}-benzenesulfonamide. mp 121-123°C.
MS 574 (M+).
CA 02370316 2001-10-15
WO 00/76988 PCT/US00/15072
-30-
Example 23
(Z) 5-[(4aS,8aR)-4-(Octahydro-isoquinolin-2-yl)-benzylidene]-3-(1H-tetrazol-
5-ylmethyl)-2-thioxo-thiazolidin-4-one. mp 247°C.
MS 441 (M+).
Example 24
(Z) N-(2-{5-[(4aS,8aR)-4-(Octahydro-isoquinolin-2-yl)-benzylidene]-4-oxo-2-
thioxo-thiazolidin-3-yl}-acetyl)-benzenesulfonamide. mp 222°C.
MS 556 (M+).
Example 25
(Z) N-{2-[5-(4-Dibutylamino-benzylidene)-4-oxo-2-thioxo-thiazolidin-3-yl]-
acetyl}-4-fluoro-benzenesulfonamide. mp 125°C.
MS 564 (M+)
Example 26
(Z) 2-[5-(4-Dibutylamino-benzylidene)-4-oxo-2-thioxo-thiazolidin-3-yl]-
ethanesulfonic acid 4-fluoro-benzoylamide. mp 191-192°C.
MS 578 (M+).
Example 27
(Z) N-{2-[5-(4-Dipentylamino-benzylidene)-4-oxo-2-thioxo-thiazolidin-3-yl]-
acetyl}-4-fluoro-benzenesulfonamide. mp 130-132°C.
MS 592 (M+).
Example 28
(Z) 2-[5-(4-Dibutylamino-benzylidene)-4-oxo-2-thioxo-thiazolidin-3-yl]-
ethanesulfonic acid benzoylamide. mp 183°C.
MS 560 (M+).
CA 02370316 2001-10-15
WO 00/76988 PCT/US00/15072
-31-
Example 29
(Z) 2-{5-[4-(Octahydro-isoquinolin-2-yl)-benzylidene]-4-oxo-2-thioxo-
thiazolidin-3-yl}-ethanesulfonic acid benzoylamide. mp 213°C.
MS 570 (M+).
Example 30
(Z) 2-{5-[4-(Octahydro-isoquinolin-2-yl)-benzylidene]-4-oxo-2-thioxo-
thiazolidin-3-yl}-ethanesulfonic acid 4-fluoro-benzoylamide. mp 248°C.
MS 588 (M+).
Example 31
(Z) 2-[5-(4-Dipentylamino-benzylidene)-4-oxo-2-thioxo-thiazolidin-3-yl]-
ethanesulfonic acid 4-fluoro-benzoylamide. mp 195-196°C.
MS 604 (M-)
Example 32
(Z) 3-(5-Hydroxy-4-oxo-4H-pyran-2-ylmethyl)-5-[4-(octahydro-isoquinolin-
2-yl)-benzylidene]-2-thioxo-thiazolidin-4-one
Step A:
Chlorokojic acid (5.0 g, 31.1 mmol) and sodium azide (2.06 g, 31.8 mmol)
are stirred in DMF (20 mL) overnight at room temperature. The reaction mixture
is diluted with water (120 mL) and the white precipitation is collected,
washed
with water, and dried to give 3.72 g of the azide as a white solid.
Step B:
Triphenylphosphine (3.53 g, 13.45 mmol) is slowly added to the kojic acid
azide (1.5 g, 8.97 mmol) in THF (20 mL). The evolution of gas is immediate.
Water (0.8 mL) is added and the reaction heated to 55°C for 18
hours. The
reaction mixture is cooled and solids are collected and washed with diethyl
ether.
The kojic acid amine (0.74 g) is obtained as a light tan solid.
Step C:
The 2-thioxo-thiazolidin-4-one of kojic acid is prepared as previously
described in Example 1.
CA 02370316 2001-10-15
WO 00/76988 PCT/US00/15072
-32-
Step D:
3-(5-Hydroxy-4-oxo-4H-pyran-2-ylmethyl)-5-[4-(octahydro-isoquinolin-
2-yl)-benzylidene]-2-thioxo-thiazolidin-4-one is prepared as previously
described
in Example 1, Step C, mp 238.
MS 483 (M+)
0 0 0
OH OH PPh3 OH
Cl I O J NaN3, DMF N I OJ THF, HZO 2 ( OJ
3 H N
1.) CSZ, NH40H
2.) CICH2C02NA, HZO
O 3.) HCI
O OH
\ \ I I O
H I '~ '-O
N / S_ % O NaOAc, HOAc S'/~IN I I OH
~S \ CHO
H I / S
~N
H
The following compounds of Formula I (Examples 33-62) were prepared
according to the general procedures described above.
Example 33
(Z) 5-(4-Dibutylamino-benzylidene)-3-(5-hydroxy-4-oxo-4H-pyran-2-
ylmethyl)-2-thioxo-thiazolidin-4-one. mp 226-227°C.
MS 473 (M+).
Example 34
(Z) 3-(5-Hydroxy-4-oxo-4H-pyran-2-ylmethyl)-5-[4-(4-propyl-piperidin-1-yl)-
benzylidene]-2-thioxo-thiazolidin-4-one. mp 253°C.
MS 471 (M+).
CA 02370316 2001-10-15
WO 00/76988 PCT/US00/15072
-33-
Example 35
(Z) 5-[4[(4-Propyl-piperidin-1-yl)-benzylidene]-3-(1H-tetrazol-5-ylmethyl)-2-
thioxo-thiazolidin-4-one. mp 263°C.
MS 429 (M+)
Example 36
(Z) N-(2-{4-Oxo-5-[4-(4-propyl-piperidin-1-yl)-benzylidene]-2-thioxo-
thiazolidin-3-yl}-acetyl)-benzenesulfonamide. mp 201 °C.
MS 544 (M+)
Example 37
(Z) N-(2-{4-Oxo-5-[4-(4-propyl-piperidin-1-yl)-benzylidene]-2-thioxo-
thiazolidin-3-yl}-acetyl)-methanesulfonamide. mp 254°C.
MS 482 (M+)
Example 38
(Z) 4-Fluoro-N-(2-{5-[(4aS,8aR)-4-(octahydro-isoquinolin-2-yl)-benzylidene]-
4-oxo-2-thioxo-thiazolidin-3-yl}-acetyl)-benzenesulfonamide. mp 220-221
°C.
MS 574 (M+)
Example 39
(Z) 4-Fluoro-N-(2-{4-oxo-5-[4-(4-propyl-piperidin-1-yl)-benzylidene]-2-
thioxo-thiazolidin-3-yl}-acetyl)-benzenesulfonamide. mp 197°C.
MS 562 (M+).
Example 40
(Z) 2-[5-(4-Hexyl-methyl-amino-benzylidene)-4-oxo-2-thioxo-thiazolidin-3-
yl]-ethanesulfonic acid 4-fluoro-benzoylamide.
MS 550 (M+).
CA 02370316 2001-10-15
WO 00/76988 PCT/US00/15072
-34-
Example 41
(Z) N-({5-[4[(Octahydro-isoquinolin-2-yl)-benzylidene]-4-oxo-2-thioxo-
thiazolidin-3-yl}-acetyl)-methanesulfonamide
Example 42
(Z) N-({5-[4[(Octahydro-isoquinolin-2-yl)-benzylidene]-4-oxo-2-thioxo-
thiazolidin-3-yl}-acetyl)-C,C,C-trifluoro-methanesulfonamide
Example 43
(Z) N-(2-{4-Oxo-5-[4-(4-propyl-piperidin-1-yl)-benzylidene]-2-thioxo-
thiazolidin-3-yl}-acetyl)-C,C,C-trifluoro-methanesulfonamide
Example 44
(Z) 2-[5-(4-Dibutylamino-benzylidene)-4-oxo-2-thioxo-thiazolidin-3-yl]-
ethanesulfonic acid methylamide
Example 45
(Z) 2-[5-(4-Dipentylamino-benzylidene)-4-oxo-2-thioxo-thiazolidin-3-yl]-
ethanesulfonic acid methylamide
Example 46
(Z) 2-[5-(4-Hexyl-methyl-amino-benzylidene)-4-oxo-2-thioxo-thiazolidin-3-
yl]-ethanesulfonic acid methylamide
Example 47
(Z) N-2-{4-Oxo-5-[4-(4-propyl-piperidin-1-yl)-benzylidene]-2-thioxo-
thiazolidin-3-yl}-ethanesulfonic acid methylamide
Example 48
(Z) 2-{5-[4-(Octahydro-isoquinolin-2-yl)-benzylidene]-4-oxo-2-thioxo-
thiazolidin-3-yl}S-ethanesulfonic acid methylamide
CA 02370316 2001-10-15
WO 00/76988 PCT/US00/15072
-3 5-
Example 49
(Z) 2-{5-[4-(Octahydro-isoquinolin-2-yl)-benzylidene]-4-oxo-2-thioxo-
thiazolidin-3-yl}S-ethanesulfonic acid trifluoroacetylamide
Example 50
(Z) N-2-{4-Oxo-5-[4-(4-propyl-piperidin-1-yl)-benzylidene]-2-thioxo-
thiazolidin-3-yl}-ethanesulfonic acid trifluoroacetylamide
Example 51
(Z) 2-[5-(4-Dipentylamino-benzylidene)-4-oxo-2-thioxo-thiazolidin-3-y1J-
ethanesulfonic acid trifluoroacetylamide
Example 52
(Z) 2-[5-(4-Dibutylamino-benzylidene)-4-oxo-2-thioxo-thiazolidin-3-yl]-
ethanesulfonic acid trifluoroacetylamide
Example 53
(Z) 2-[5-(4-Dipentylamino-benzylidene)-4-oxo-2-thioxo-thiazolidin-3-yl]-
ethanesulfonic acid benzoylamide
Example 54
(Z) 2-[5-(4-Hexyl-methyl-benzylidene)-4-oxo-2-thioxo-thiazolidin-3-yl]-
ethanesulfonic acid benzoylamide
Example 55
(Z) N-2-{4-Oxo-5-[4-(4-propyl-piperidin-1-yl)-benzylidene]-2-thioxo-
thiazolidin-3-yl}-ethanesulfonic acid benzoylamide
Example 56
(Z) N-2-{4-Oxo-5-[4-(4-propyl-piperidin-1-yl)-benzylideneJ-2-thioxo-
thiazolidin-3-yl}-ethanesulfonic acid 4-fluoro-benzoylamide
CA 02370316 2001-10-15
WO 00/76988 PCT/US00/15072
-3 6-
Example 57
(Z) 2-[5-(4-Hexyl-methyl-benzylidene)-4-oxo-2-thioxo-thiazolidin-3-y1J-
ethanesulfonic acid 4-fluoro-benzoylamide
Example 58
(Z) [5-(4-Hexyl-methyl-amino)-benzylidene]-3-(5-oxo-4,5-dihydro-
[1,2,4] oxadiazol-3-ylmethyl)-2-thioxo-thiazolidin-4one
Example 59
(Z) [5-(4-Propyl-piperidin-1-yl)-benzylideneJ-3-(5-oxo-4,5-dihydro-
[1,2,4]oxadiazol-3-ylmethyl)-2-thioxo-thiazolidin-4one
Example 60
(Z) [5-(4-Octahydro-isoquinolin-2-yl)-benzylidene]-3-(5-oxo-4,5-dihydro-
[1,2,4] oxadiazol-3-ylmethyl)-2-thioxo-thiazolidin-4one
Example 61
(Z) 5-(4-Dipentylamino-benzylidene)-3-(5-oxo-4,5-dihydro-[1,2,4]oxadiazol-3-
ylmethyl)-2-thioxo-thiazolidin-4-one
Example 62
(Z) 5-(4-Dibutylamino-benzylidene)-3-(5-oxo-4,5-dihydro-[1,2,4]oxadiazol-3-
ylmethyl)-2-thioxo-thiazolidin-4-one
Other typical invention compounds that can be prepared by following the
foregoing general methods include:
(Z) N-(2-{5-[(4-Hexylmethylamino)-naphthalan-1-ylmethylene]-4-oxo-2-
thioxo-thiazolidin-3-yl}-acetyl)-benzenesulfonamide;
(Z) 2-[5-(1-Ethyl-2,3-dihydro-1H-indol-5-ylmethylene)-4-oxo-2-thioxo-
thiazolidin-3-yl]-ethanesulfonic acid;
(Z) 3-[S-(1-Isopropyl-1,2,3,4-tetrahydro-quinolin-6-yl-methylene)-4-oxo-
2-thioxo-thiazolidin-3-yl]-propanesulfonic acid; and
CA 02370316 2001-10-15
WO 00/76988 PCT/US00/15072
-3 7-
(Z) 1-[5-(1-tert.Butyl-1,2,3,4-tetrahydro-quinolin-5-yl-methylene)-4-oxo-
2-thioxo-thiazolidin-3-yl]-1-methylethanesulfonic acid.
BIOLOGICAL EXAMPLES
Representative compounds of Formula I have been evaluated in the
following standard in vitro and in vivo assays which are commonly used to
indicate clinical utility in inhibiting amyloid formation and to treat
diseases
associated with amyloid, such as Alzheimer's disease.
AMYLOID ASSAYS
BASSR (Beta-Amyloid Self Seeding Radioassayl
An assay for inhibitors of self seeded amyloid fibril growth
Materials:
Stock Solutions:
Assay Buffer - 50 mM sodium phosphate, pH 7.5, 100 mM NaCI, 0.02% NaN3,
1 M urea (filter and store at 4°C)
Soluble Af.3(1-40) peptide (Bachem, Torrance, CA) - 2.2 mg/mL in deionized H20
(stored in aliquots at -20°C, keep on ice when thawed) will self seed
after 1 week
storage. Typically, the solution should be stored until no lag phase is seen
in the
assay.
1251 labeled A/.i (1-40) - 150K-350K cpm/~.L in 100% acetonitrile -0.1%
trifluoroacetic acid (TFA) -1% ~3-mercaptoethanol (aliquots stored at -
20°C).
125I_labeled A~3(1-40) can be made in accordance with the procedure set forth
by
H. Levine, III in Neurobiol. Aging, 16:755 (1995), which is hereby
incorporated
by reference, or this reagent may be purchased from Amersham, Arlington
Heights, Illinois.
CA 02370316 2001-10-15
WO 00/76988 PCT/US00/15072
-3 8-
Final assay conditions: 30 ~,M soluble A/3(1-40) in deionized water in assay
buffer + 20-SOK cpm 1251-labeled A/.3 (1-40) per assay. Compound to be tested
is
dissolved in dimethylsulfoxide (DMSO), typically 5-50 mM stock, such that the
final concentration of DMSO is <1% v/v in the assay.
Assay: Reaction mixture for 50 assays (on ice) is comprised of 0.1-0.2 ~tL of
1251_labeled A1251 labeled A/3 (1-40) + 1 ~.L of soluble A~i (1-40) + 13.5 ~L
assay buffer per assay. The following are the amounts of the components of the
reaction mixture sufficient for 50 assay wells.
5-10 ~L 125I_labeledA~3(1-40) dried down
675 ~.L assay buffer
50 ~.L soluble A/3 (1-40)
Assay Method
1 ) Prepare reaction mixture above by mixing components and storing on ice.
2) Pipet 14.5 ~.L of reaction mixture into each of 50 wells on a polypropylene
U-bottom 96-well microtiter plate on ice (Costar 3794).
3) Add 1.7 pL of diluted compound to be tested to each well in a column of
eight, including solvent control. Serial 3-fold dilutions from 1 mM (100 ~M
final) in assay buffer -urea = 7 dilutions + zero. Each 96-well plate can
therefore accommodate 11 samples + 1 Congo Red control (0.039-5 ~,M final
in 2-fold steps).
4) Seal the plate with aluminum film (Beckman 538619) and incubate for
10 minutes on ice.
5) Raise the temperature to 37°C and incubate for 3 to 5 hours
(depending on
the lot of the peptide).
6) Remove the aluminum film and add 200 ~,Llwell of ice cold assay buffer with
urea, collecting the radiolabeled fibrils by vacuum filtration through 0.2 ~,m
pore size GVWP filters in 96-well plates (Millipore MAGV N22, Bedford,
MA). Determine the radioactivity of the filters using standard methods well-
known to those skilled in the art.
CA 02370316 2001-10-15
WO 00/76988 PCT/US00/15072
-39-
BASST (Beta-Amyloid Self seeding, ThioflavinT)
An assay for inhibitors of self seeded amyloid fibril growth
METHODS:
Materials
Stock Solutions:
Assay Buffer - 50 mM sodium phosphate, pH 7.5, 100 mM NaCI, 0.02% NaN3,
1 M urea (filter and store at 4°C).
Soluble A~3 (1-40) - 2.2 mg/mL in deionized H20 (store in aliquots at -
20°C, keep
on ice when thawed) will self seed after 1 week storage. Typically, the
solution
should be stored until no lag phase is seen in the assay.
Final assay conditions: 30 ~M soluble A~3(1-40) in deionized water in assay
buffer. Compound to be tested is dissolved in DMSO, typically 5-50 mM stock,
such that the final concentration of DMSO is <1 % v/v in the assay.
Assay: Reaction mixture for 50 assays (on ice) comprised of 1 ~,L of soluble
A/j (1-40) + 13.5 ~.L assay buffer per assay. The following are the amounts of
the
components of the reaction mixture that result in each of the 50 assay wells.
50 ~L soluble A/3 (1-40)
675 ~,L assay buffer
Assay Method
1) Prepare the reaction mix above by mixing the components and storing on ice.
2) Pipet 14.5 ~.L of reaction mixture into each of 50 wells of a polystyrene
U-bottom 96-well microtiter plate (Corning 25881-96) on ice.
3) Add 1.7 ~.L of diluted compound to be tested to each well in a column of
eight, including solvent control. Serial 3-fold dilutions from 1 mM (100 ~,M
final) in assay buffer -urea = 7 dilutions + zero. Each 96-well plate can
therefore accommodate 11 samples + 1 Congo Red control (0.039-5 ~M final
in 2-fold steps).
CA 02370316 2001-10-15
WO 00/76988 PCT/US00/15072
-40-
4) Seal the plate with aluminum film and incubate for 10 minutes on ice.
5) Raise the temperature to 37°C and incubate for 3 to 5 hours (depends
on the
lot of the peptide).
6) Remove the aluminum film and add 250 ~.L/well of 5 ~.M thioflavin T (ThT)
[T-3516, Sigma-Aldrich] in 50 mM glycine-NaOH, pH 8.5. Read fluorescence
on a plate reader (ex = 440 nm/20 nm; em = 485 nm/20 nm) within 5 minutes.
BAPA (Beta-Amyloid Peptide A~~re~ation)
This assay is used to provide a measure of inhibition by a compound
against the aggregation behavior of the beta amyloid peptide.
The purpose of this assay is to provide a higher volume method of
assaying the amount of beta amyloid aggregation using an endpoint assay based
on filtration. In this assay, hexafluoroisopropanol (HFIP) is used to break
down
the initial amyloid peptide to a monomer state and use a concentration of 33
pM
which is high enough so that aggregation will occur at pH 6.0 in several
hours.
METHODS:
(3-Amyloid Peptide A~~re atg ion, pH 6.0 (BAPA)
In a 96-well plate (Costar 3794), we add 25 pL 50 mM Phosphate Buffer,
pH 6.0, 10 ~,L 0.5 mg/mL A(3 (1-40) peptide in 20% HFIP + 0.1 ~,L/assay
radioiodinated 1251 A~i (1-40) [1251 A[3(1-40)], and 1 ~.L of the compound to
be
tested starting at 50 mM with a concentration of DMSO <1%. Then, we incubate
for 2 to 4 hours at room temperature. We stop the reaction with 200 ~L of 50
mM
phosphate buffer, pH 6.0, and filter it through a 0.2 ~m 96-well filter plate
(Millipore MAGU N22). We wash the filter plate with 100 ~.L of the same
phosphate buffer. Aggregation was detected on a Microbeta counter after
impregnating the filters with Meltilex (1450-441) and is corrected for
background.
CA 02370316 2001-10-15
WO 00/76988 PCT/US00/15072
-41-
BATYM ASSAY
METHODS:
Required A(3 (1-42)(California Peptide) was dried from its
hexafluoroisopropanol (HFIP) stock solution. The A(3 (1-42) was dissolved in
dimethylsulfoxide (DMSO) and then mixed with phosphate buffered saline (PBS)
(pH 7.4). The mixed A(3 (1-42) solution was filtered with a 0.2 ~,m Omnipore
membrane syringe filter (Millipore, Bedford, MA). The compound to be tested in
DMSO (50 times concentrate) was put into each well (0.5 ~.L/well) of a 96-well
plate. The A(3 (1-42) solution was added into each well (24.5 ~.L/well). The
plate
was centrifuged at 1,000 g for 5 minutes and incubated at 37°C for 1
day
(A(3 1-42; final concentration 100 p,M).
After incubation Thioflavin T (ThT) (30 ~.M) solution in glycine-NaOH
buffer (pH 8.5, 50 mM) was added into each well (250 ~.L/well), fluorescence
was
measured (ex. 440/20 nm; em 485/20 nm) using a fluorescence plate reader. The
inhibitory activity was calculated as the reduction of fluorescence with the
following formula:
Inhibition (%) _ {(F(A(3)-F(A(3+compound)}/{F(A(3)-F (solvent + compound)} x
100
The ICSps were calculated by a curve fitting program using the following
equation. The data were obtained from two different experiments in triplicate.
Inhibition(x) = 100-100/{ 1 + (x/IC50)n},
x = concentration of tested compound (M),
IC50 = (M),
n = Hill coefficient.
Representative compounds of Formula 1 have exhibited inhibitory
activities (IC50) ranging from about 0.1 ~m to greater than 100 ~,m in the
foregoing assays. The results of these assays for several specific compounds
of the
present invention are shown in Table 1 below.
CA 02370316 2001-10-15
WO 00/76988 PCT/US00/15072
-42
TABLE 1. Amyloid Inhibitory Activity
Example BASSR BASST BATYM BAPA
No. IC M IC M IC M IC M
50 ~ 50 I~ 50 N~ 50 I~
1 >100 1 1.72 23
2 >100 0.8 2.32 7
3 8 0.3 2.24 >100
4 >100 3 8.26 6
9 0.8 1.83 >10
6 >100 28.5 2.92 3
7 1 0.4 1.7 70
8 >100 1 1.96 2.5
9 > 100 1.75 2.15 4
>100 1.1 2.51 8
11 > 100 9 2.19 25
12 > 100 1 1.64 26
13 >100 10 3.5 28
14 >100 0.22 >100 30
>100 >100 3.39 8
16 7 3 1.94 38
17 10 ,5 1, 1.5 1.87 2
18 5 0.6 1.88 122
19 5 1.1 1.77 129
1, 8.5 2 2.04 >100
21 >100 2.91 >100
22 3 1 1.99 >60
23 >100 1, 0.5 3.33 30
24 6.5, 10, 1, 0.8 3.84 60
6
6 0.6, 0.5 2.38 100
26 6.1, 10 0.5, 10 2.14
27 7 2 2.02 93
28 21 0.3 2.19 69
CA 02370316 2001-10-15
WO 00/76988 PCT/US00/15072
-43
TABLE 1. Amyloid Inhibitory Activity (cont'd)
Example BASSR BASST BATYM BAPA
No. IC M IC M IC M IC M
50 !~ 50 !~ 50 I~ 50 ~
29 11 0.6 3.42 > 100
30 >100 0.3 2.26 >60, >60
31 9 0.3 3.11 >100
32 >100 1 2.55 46
33 >100 2 3.03 >60
34 > 100 2 2.3 8 >60
35 15 0.4 3.31 >60
36 >100 1 3.3 >60
37 50 0.4 3.83 >60
3 8 10, 5 0.7, 0.1 2.54 66
39 >100, 21 l, 0.1 2.86 37
40 2.84
Activity of the invention compounds is also evaluated in standard in vivo
assays commonly used to evaluate agents to treat diseases related to
aggregation
of amyloid proteins, especially Alzheimer's disease. Such assays are described
by
Axelrad et al., Lab. Invest., 1982;47(2):139-146; and by Stenstad et al., J.
Biochem., 1994;303 (Pt 2):663-670. In one assay, amyloid protein is induced
into
the spleen of mice by subcutaneous injections of silver nitrate, Freund's
complete
adjuvant, and an intravenous injection of amyloid enhancing factor. Silver
nitrate
is administered each day through Day 11. Test compounds are administered to
the
mice daily starting on Day 1 through Day 11. On Day 12, the animals are
sacrificed, and the spleens are removed, histologically prepared, stained with
Congo red, and the percent area of the spleen occupied by birefringent, Congo
red-stained amyloid is quantitated microscopically.
Another in vivo assay in which the invention compounds are evaluated
uses transgenic mice. The mice bear a human (3-amyloid precursor protein
transgene with a prior promoter and are described by Hsiao et al.,
"Correlative
Memory Deficits, A(3 Elevation, and Amyloid Plaques in Transgenic Mice,''
CA 02370316 2001-10-15
WO 00/76988 PCT/US00/15072
-44-
Science 1996;274:99-102. These transgenic mice develop ~3-amyloid deposits at
about 9 months of age. By 15 months, diffuse and compact senile plaques are
abundant, primarily in the neocortex, olfactory bulb, and hippocampus.
Invention
compounds are administered orally to the mice beginning at the age of 8 months
(just prior to the onset of amyloid deposits) and continuing for several
months (up
to about age 14-18 months). The animals are then sacrificed, and the brains
are
removed. The amount of amyloid in the brain is quantitated both histologically
and biochemically.
The above data establishes that representative invention compounds are
active in standard assays used to measure inhibition of protein aggregation.
The
compounds are thus useful to clinically inhibit amyloid protein aggregation
and to
image amyloid deposits for diagnostic use. The compounds will be used in the
form of pharmaceutical formulations, and the following examples illustrate
typical
compositions.
Example 63
Tablet Formulation
Ingredient Amount
Compound of Example 1 50 mg
Lactose 80 mg
Cornstarch (for mix) 10 mg
Cornstarch (for paste) 8 mg
Magnesium Stearate (1%) 2 mg
150 mg
The compound of Example 1 is mixed with the lactose and cornstarch (for
mix) and blended to uniformity to a powder. The cornstarch (for paste) is
suspended in 6 mL of water and heated with stirring to form a paste. The paste
is
added to the mixed powder, and the mixture is granulated. The wet granules are
passed through a No. 8 hard screen and dried at 50°C. The mixture is
lubricated
with 1 % magnesium sterate and compressed into a tablet. The tablets are
administered to a patient at the rate of 1 to 4 each day for prevention of
amyloid
protein aggregation and treatment of Alzheimer's disease.
CA 02370316 2001-10-15
WO 00/76988 PCT/US00/15072
-45-
EXAMPLE 64
Parenteral Solution
In a solution of 700 mL of propylene glycol and 200 mL of water for
injection is added 20.0 g of Compound No. 26) (from Example 26). The mixture
is stirred and the pH is adjusted to 5.5 with hydrochloric acid. The volume is
adjusted to 1000 mL with water for injection. The solution is sterilized,
filled into
5.0 mL ampoules, each containing 2.0 mL (40 mg of Compound No. 26), and
sealed under nitrogen. The solution is administered by injection to a patient
suffering from medullary carcinoma of the thyroid and in need of treatment.
EXAMPLE 65
Patch Formulation
Ten milligrams of 5-[4-(4-propyl-piperidin-1-yl)-benzylidene]-3-(1H-
tetrazol-5-yl-methyl)-2-thioxo-thiazolidin-4-one is mixed with 1 mL of
propylene
glycol and 2 mg of acrylic-based polymer adhesive containing a resinous cross-
linking agent. The mixture is applied to an impermeable backing (30 cm2) and
applied to the upper back of a patient for sustained release treatment of
amyloid
polyneuropathy.
The invention and the manner and process of making and using it, are now
described in such full, clear, concise, and exact terms as to enable any
person
skilled in the art to which it pertains, to make and use the same. It is to be
understood that the foregoing describes preferred embodiments of the present
invention and that modifications may be made therein without departing from
the
spirit or scope of the present invention as set forth in the claims. To
particularly
point out and distinctly claim the subject matter regarded as invention, the
following claims conclude this specification.