Note: Descriptions are shown in the official language in which they were submitted.
CA 02372051 2001-11-15
WO 00/69860 PCT/US00/13413
IMIDAZO-CONTAINING HETEROCYCLIC COMPOUNDS,
THEIR COMPOSITIONS AND USES
FIELD OF THE INVENTION
The subject invention relates to novel imidazo-containing heterocyclic
compounds, pharmaceutical compositions containing them, and their therapeutic
or
preventative use in the areas of cardiovascular, oncology, infectious and
inflammatory
diseases.
BACKGROUND
Certain imidazo-isoquinoline compounds are disclosed in U.S. Patent No.
3,917,610 issued November 4, 1975; they are reported to have certain
cardiovascular
activities. One such compound is further reported on in Borchard, Fox, and
Greeff, "The
Positive Isotropic, Antiarrhythmic and Na+, K+-ATPase Inhibitory Effects of
the
Isoquinoline Derivative, BIIA", Achives of Pharmacology, vol. 312 (1980), pp.
187-192.
SUMMARY OF THE INVENTION
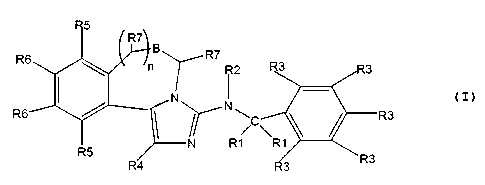
The subject invention includes compounds having the structure:
RE
R2
R6 N ~C
R1
wherein:
(a) each R1 is independently selected from alkyl, aryl, and heterocycle;
(b) R2 is selected from hydrogen, alkyl, alkylacyl, arylacyl, alkylsulfonyl
and
arylsulfonyl;
CA 02372051 2001-11-15
WO 00/69860 PCT/US00/13413
(c) each R3 is independently selected from hydrogen, halo, alkyl, aryl,
heterocycle, vitro, cyano, and unsubstituted or alkyl- or aryl- or
heterocycle-substituted hydroxy, thio, amino, amide, formyl (acyl),
carboxy, and carboxamide; two R3's on adj acent carbons may optionally
together be alkylene or heteroalkylene, thereby forming a fused ring with
the phenyl to which they are attached;
(d) R4 is selected from hydrogen, halo, alkyl, aryl, heterocycle, and carboxy
and its alkyl and aryl esters and amides;
(e) each RS is independently selected from hydrogen, alkyl, and aryl;
(f) each R6 is independently selected from hydrogen, halo, alkyl, aryl,
heterocycle, vitro, cyano, and unsubstituted or alkyl- or aryl- or
heterocycle-substituted hydroxy, thio, amino, amide, sulfonamide, formyl
(acyl), carboxy, and carboxamide; or the two R6's may optionally together
be alkylene or heteroalkylene, thereby forming a fused ring with the
phenyl to which they are attached;
(g) B is nil or -C(R7)=C(R7)-;
(h) if B is nil, n is an integer from 0 to about 3, otherwise n is from 0 to
about
1;
(j) each R7 is independently selected from hydrogen, alkyl and aryl;
and an optical isomer, diestereomer or enantiomer thereof; a pharmaceutically
acceptable
salt, hydrate, or biohydrolyzable ester, amide or imide thereof.
The subject invention also includes compositions comprising a subject compound
and a pharmaceutically-acceptable excipient; and methods for treating or
preventing
diseases or disorders by administering to a human or lower animal in need
thereof, a safe
and effective amount of a subject compound.
DETAILED DESCRIPTION OF THE INVENTION
As used herein unless specified otherwise, "alkyl" means a hydrocarbon chain
which is branched, linear or cyclic, saturated or unsaturated (but not
aromatic),
substituted or unsubstituted. The term "alkyl" may be used alone or as part of
another
word where it may be shortened to "alk" (e.g., in alkoxy, alkylacyl).
Preferred linear alkyl
2
CA 02372051 2001-11-15
WO 00/69860 PCT/US00/13413
have from one to about twenty carbon atoms, more preferably from one to about
ten
carbon atoms, more preferably still from one to about six carbon atoms, still
more
preferably from one to about four carbon atoms; most preferred are methyl or
ethyl.
Preferred cyclic and branched alkyl have from three to about twenty carbon
atoms, more
preferably from three to about ten carbon atoms, more preferably still from
three to about
seven carbon atoms, still more preferably from three to about five carbon
atoms.
Preferred cyclic alkyl have one hydrocarbon ring, but may have two, three, or
more, fused
hydrocarbon rings. Preferred alkyl are unsaturated with from one to about
three double or
triple bonds, preferably double bonds; more preferably they are mono-
unsaturated with
one double bond. Still more preferred alkyl are saturated. Saturated alkyl are
referred to
herein as "alkanyl". Alkyl unsaturated only with one or more double bonds (no
triple
bonds) are referred to herein as "alkenyl". Preferred substituents of alkyl
include halo,
alkyl, aryl, heterocycle, hydroxy, alkoxy, aryloxy, thio, alkylthio, arylthio,
amino,
alkylamino, arylamino, amide, alkylamide, arylamide, formyl, alkylacyl,
arylacyl, carboxy
and its alkyl and aryl esters and amides, nitro, and cyano. Also,
unsubstituted alkyl are
preferred.
As used herein, "heteroatom" means a nitrogen, oxygen, or sulfur atom.
As used herein, "alkylene" means an alkyl which connects two other moieties,
"heteroalkylene" means an alkylene having one or more heteroatoms in the
connecting
chain.
As used herein unless specified otherwise, "aryl" means an aromatic
hydrocarbon
ring (or fused rings) which is substituted or unsubstituted. The term "aryl"
may be used
alone or as part of another word (e.g., in aryloxy, arylacyl). Preferred aryl
have from six
to about fourteen, preferably to about ten, carbon atoms in the aromatic
ring(s), and a total
of from about six to about twenty, preferably to about twelve, carbon atoms.
Preferred
aryl is phenyl or naphthyl; most preferred is phenyl. Preferred substituents
of aryl include
halo, alkyl, aryl, heterocycle, hydroxy, alkoxy, aryloxy, thio, alkylthio,
arylthio, amino,
alkylamino, arylamino, amide, alkylamide, arylamide, formyl, alkylacyl,
arylacyl, carboxy
and its alkyl and aryl esters and amides, nitro, and cyano. Also,
unsubstituted aryl are
preferred.
3
CA 02372051 2001-11-15
WO 00/69860 PCTlUS00/13413
As used herein unless specified otherwise, "heterocycle" means a saturated,
unsaturated or aromatic cyclic hydrocarbon ring (or fused rings) with one or
more
heteroatoms in the hydrocarbon ring(s). Preferred heterocycles have from one
to about
six heteroatoms in the ring(s), more preferably one or two or three
heteroatoms in the
ring(s). Preferred heterocycles have from three to about fourteen, preferably
to about ten,
carbon plus heteroatoms in the ring(s), more preferably from three to about
seven, more
preferably still five or six, carbon plus heteroatoms in the rings(s); and a
total of from
three to about twenty carbon plus heteroatoms, more preferably from three to
about ten,
more preferably still five or six, carbon plus heteroatoms. Preferred
heterocycles have
one ring, but may have two, three, or more, fused rings. More preferred
heterocycle rings
include those which are one ring with 5 or 6 carbon plus heteroatoms in the
ring with no
more than three ring heteroatoms, no more than two of which are O and S. Still
more
preferred are such 5- or 6-ring atom heterocycles with one or two ring atoms
being O or S
and the others being C; or with one, two or three ring atoms being N and the
others being
C. Such preferred 5- or 6-ring atom heterocycles are preferably saturated,
unsaturated
with one or two double bonds, or aromatic. Such preferred 5- or 6-ring atom
heterocycles
are preferably a single ring; or fused with a 3- to 6-ring atom hydrocarbon
ring which is
saturated, unsaturated with one double bond, or aromatic (phenyl); or fused
with another
such S- or 6-ring atom heterocyclic ring. Heterocycles are unsubstituted or
substituted.
Preferred heterocycle substituents are the same as for alkyl.
Compounds of the Invention
The subj ect invention involves compounds having the following structure:
R7
R7
Rs
N
Z6 ~ N U
N F
R5
4
CA 02372051 2001-11-15
WO 00/69860 PCT/US00/13413
In structure 1, each R1 is independently selected from alkyl, aryl, and
heterocycle. Preferred Rl include linear alkanyl having from 1 to about 6
carbon atoms,
linear alkenyl having from 2 to about 6 carbon atoms, and branched and cyclic
alkanyl
and alkenyl having from 3 to about 6 carbon atoms, such alkenyl preferably
having 1
double bond. Such preferred alkanyl and alkenyl are preferably unsubstituted,
or
substituted with phenyl, heterocycle having 5 or 6 ring atoms, carboxy and its
C 1 - C6
alkyl and phenyl esters, or cyano. More preferably such alkanyl and alkenyl
have up to
about 5 carbon atoms, more preferably still up to 4 carbon atoms. More
preferred R1 are
methyl, ethyl and isopropyl. Most preferred R1 is unsubstituted methyl. Also
preferred is
both R1 being the same moiety.
In structure l, R2 is selected from hydrogen, alkyl, alkylacyl, arylacyl,
alkylsulfonyl, and arylsulfonyl. Preferred R2 is selected from hydrogen; Cl-C6
alkyl,
such alkyl being saturated or unsaturated with one double bond and
unsubstituted or
substituted with phenyl; C1-C6 alkylacyl, the alkyl being saturated or
unsaturated with
one double bond; and phenylacyl. More preferred is the alkyl portions of the
aforementioned moieties being Cl-C4 and saturated. More preferred still is R2
being
methyl. Most preferred R2 is hydrogen.
In structure 1, each R3 is each independently selected from hydrogen, halo,
alkyl,
aryl, heterocycle, nitro and cyano; also from hydroxy, thio, amino, amide,
formyl (acyl),
carboxy, and carboxamide which are unsubstituted or substituted, preferably
with alkyl or
aryl or heterocycle; or two R3 together are alkylene or heteroalkylene
attached to adjacent
carbon atoms of the phenyl ring, thereby forming a cycloalkyl or aryl or
heterocycle ring
which is fused to the phenyl ring. Preferred R3 are independently selected
from
hydrogen, halo, alkyl, aryl, heterocycle, hydroxy, alkoxy, aryloxy, thio,
alkylthio, arylthio,
amino, alkylamino, arylamino, amide, alkylamide, arylamide, formyl, alkylacyl,
arylacyl,
carboxy and its alkyl or aryl esters and amides; more preferably from
hydrogen, halo, C1-
C4 alkyl, thio, Cl-C4 alkylthio, C1-Cq. mono-or dialkylamino, and C1-C4
alkylacyl.
More preferred is for from one to three R3's being halo, the others being
hydrogen. Also
more preferred is for from one to three R3's being methyl or ethyl, the others
being
CA 02372051 2001-11-15
WO 00/69860 PCT/US00/13413
hydrogen. Also preferred is one R3 being dialkylamino, the alkyls having from
1 to about
6 carbon atoms, preferably from about 1 to about 4 carbon atoms, and the
others being
hydrogen, such R3 preferably being attached to the 4' carbon. More preferred
still is for
from one to three R3's being independently selected from F, Cl and Br, the
others being
hydrogen; still more preferred, when two or three R3's are F, C1 or Br, they
are the same.
Also more preferred is from one to three R3's being unsubstituted methyl, the
others
being hydrogen. Also more preferred is one or two R3's being trifluoromethyl,
the others
being hydrogen.
Also preferred are two R3's which are attached to adjacent carbon atoms of the
phenyl ring, together being a saturated or unsaturated alkylene or
heteroalkylene having
from 1 to about 6 carbon atoms and from 0 to about 3 heteroatoms, thus forming
a ring
fused to the phenyl, such ring having from about 5 to about 8 ring atoms. Such
ring fused
to the phenyl preferably has from about 5 to about 6 ring atoms of which from
0 to 2,
more preferably 0 or 1, are heteroatoms. Preferred fused rings (including the
phenyl to
which the R3's are attached) include naphthyl, indolyl, benzimidazoyl,
benzofuryl,
benzopyranyl. When two R3's form a ring fused with the phenyl, other R3's are
preferably H.
In structure 1, R4 is selected from hydrogen, halo, alkyl, aryl, heterocycle,
carboxy
and its alkyl esters and amides. Preferred R4 is selected from hydrogen, halo,
Cl- C4
alkyl, phenyl. More preferred R4 is selected from hydrogen and unsubstituted
and
substitituted phenyl; substituents on such phenyl are preferably selected from
hydroxy,
alkoxy, thio and alkylthio. Most preferred R4 is hydrogen.
In structure l, each RS is independently selected from hydrogen, alkyl and
aryl.
Preferred RS are selected from hydrogen and alkyl having from 1 to about 4
carbon
atoms, especially unsubstituted methyl or ethyl. Most preferred is for both RS
to all be
hydrogen.
In structure 1, each R6 is independently selected from hydrogen, halo, alkyl,
aryl,
heterocycle, cyano and nitro; also from hydroxy, thio, amino, amide, formyl
(acyl),
carboxy, carboxamide, and sulfonamide which are unsubstituted or substituted,
preferably
with alkyl or aryl or heterocycle. Preferred R6's are selected from hydrogen,
halo, alkyl,
6
CA 02372051 2001-11-15
WO 00/69860 PCT/US00/13413
aryl, heterocycle, hydroxy, alkoxy, aryloxy, thio, alkylthio, arylthio, amino,
alkylamino,
arylamino, amide, alkylamide, arylamide, sulfonamide, alkylsulfonamide,
arylsulfonamide, formyl, alkylacyl, arylacyl, carboxy and its alkyl and aryl
esters and
amides; more preferably from hydrogen, halo, hydroxy, C1-C4 alkoxy, thio, C1-
C4
alkylthio, Cl-C4 alkyl esters and amides of carboxy, and heterocycle having 5
or 6 ring
atoms. More preferably one or both R6's are selected such that at least one
heteroatom is
bonded directly to the phenyl ring. When both R6's have heteroatoms bonded
directly to
the adjacent carbons of the phenyl ring, the two R6's may together form an
alkylene
moiety connecting the two heteroatoms, the alkylene moiety preferably having
from 1 to
about 4, more preferably 1 or 2, carbon atoms. Preferred is that one or both
of the R6's
are non-hydrogen moieties; more preferred is that both of them are non-
hydrogen
moieties. More preferred still is that both R6's are alkoxy or both alkylthio;
preferably
both are the same. Preferred alkyl portions of R6's have from 1 to about 4
carbon atoms,
more preferably 1 or 2 carbon atoms; most preferred is methyl. Such alkyl
portions are
preferably unsubstituted. Such alkyl portions are preferably saturated. Most
preferred is
that both R6's are methoxy or ethoxy, especially methoxy.
Also preferred are a RS and a R6, which are attached to adjacent carbon atoms
of
the phenyl ring, together being a saturated or unsaturated alkylene or
heteroalkylene
having from 1 to about 6 carbon atoms and from 0 to about 3 heteroatoms, thus
forming a
ring fused to the phenyl, such ring having from about 5 to about 8 ring atoms.
Such ring
fused to the phenyl preferably has from about 5 to about 6 ring atoms of which
from 0 to
2, more preferably 0 or 1, are heteroatoms. Preferred rings formed by such RS
and R6
include phenyl, furyl, pyrrolyl, dioxanyl, imidazoyl, pyridinyl, pyrrolidinyl,
piperidinyl.
When such RS and R6 form a fused ring, the other RS and R6 are both preferably
hydrogen.
In structure 1, B is nil or -C(R7)=C(R7)-. When B is nil, n is an integer from
0 to
about 3, preferably 1 or 2. When B is -C(R7)=C(R7)-, n is an integer from 0 to
about 1,
preferably 0. Preferred B is nil.
In structure 1 and in B, each R7 is independently selected from hydrogen,
alkyl,
and aryl. Non-hydrogen R7 are preferably phenyl, or alkyl having from 1 to
about 4
7
CA 02372051 2001-11-15
WO 00/69860 PCT/US00/13413
carbon atoms, preferably 1 or 2 carbon atoms. Such non-hydrogen R7 are
preferably
unsubstituted. Alkyl R7 are preferably saturated. Preferably no more than one
of all the
R7's is other than hydrogen. More preferably all R7's are hydrogen.
The subject invention includes optical isomers, diastereomers, and enantiomers
of
the compounds of structure 1. The subject invention includes pharmaceutically-
acceptable salts, hydrates, and biohydrolizable esters, amides and imides of
such
compounds.
A "pharmaceutically-acceptable salt" is a cationic salt formed at any acidic
group
(e.g., carboxy group), or an anionic salt formed at any basic group (e.g.,
amino group) on
a compound of structure 1. Many pharmaceutically-acceptable salts are known.
Preferred cationic salts include the alkali metal salts, such as sodium and
potassium,
alkaline earth metal salts, such as magnesium and calcium, and organic salts,
such as
ammonium. Preferred anionic salts include halides, sulfonates, carboxolates,
phosphates,
and the like. Salts of addition may provide an optical center where once there
was none.
The compounds of the subject invention, and salts thereof, may have one or
more
chiral centers. The invention includes all optical isomers of the compounds of
structure 1
and salts thereof, including diastereomers and enanteomers The subject
invention
includes and contemplates each optical isomer, diastereomer or enanteomer
thereof, in
purified form, substantially purified form, and mixtures, including racemic
mixtures.
Preferred compounds of the subject invention include those having structure 2:
R ~ R3
2' 3, R3
x R1
N
R6 / NH~I 1' ~ ~ 4 R3
c~>
5'
N R1 R3 6' R3
In structure 2, R1's, R3's and R6's are as described hereinabove, and x is 0
or 1.
In structure 2, each R1 is preferably independently selected from linear
alkanyl
having from one to four carbon atoms, linear alkenyl having one double bond
and from
two to four carbon atoms, branched and cyclic alkanyl having from three to
five carbon
atoms, and branched and cyclic alkenyl having one double bond and from three
to five
8
CA 02372051 2001-11-15
WO 00/69860 PCT/iJS00/13413
carbon atoms. Such preferred R1 are unsubstituted or substituted with one
phenyl, more
preferably are unsubstituted. More preferred Rl are selected from methyl,
ethyl, ethenyl,
n-propyl, i-propyl, n-propenyl, i-propenyl, s-butyl, cyclopropyl, cyclobutyl,
and
cyclopentyl. More preferred still Rl are selected from methyl, ethyl, ethenyl,
i-propyl,
and n-propenyl. Most preferred R1 are methyl. Also preferred is for both R1 to
be the
same.
In structure 2, one or both of R6, preferably both, are preferably alkylthio
or more
preferably alkoxy with alkanyl having from one to four carbon atoms. If one R6
is not
alkylthio or alkoxy, it is preferably hydrogen. More preferred is both R6
being methoxy
or ethoxy; most preferred is both R6 being methoxy.
In structure 2, the R3's are preferably selected from all hydrogen; mono-, di-
, or
trihalo, preferably selected from fluoro, chloro and bromo, preferably in one
or more of
the 2', 3', 4' and 5' positions; mono- di-, and trimethyl, preferably in one
or more of the
2', 3', 4' and 6' positions; and mono- or di-trifluoromethyl, preferably in
one or both of
the 3' and 5' positions. Also preferred is one R3 being diakylamino,
preferably in the 4'
position, the two alkyls preferably being the same and preferably having from
1 to 4
carbon atoms, and the other R3's being hydrogen. More preferred combinations
of R3's
are selected from 4'-fluoro, 4'-chloro, 4'-bromo, 2',4'-difluoro, 2',4'-
dichloro, 2',4'-
dibromo, 2',4',5'-trifluoro, 2',4',5'-trichloro, 3',4'-difluoro, 3',4'-
dichloro, 3',4'-
dibromo, 4'-methyl, 2',4'-dimethyl, 2',4',6'-trimethyl, 3'-trifluoromethyl,
3',5'-di-
trifluoromethyl, and 4'-dibutylamino; in each cased all other R3's being
hydrogen. Also
preferred R3 combinations are selected from 2',4'-dihalo and 3',4'-dihalo,
where one
halo is selected from fluoro, chloro, and bromo, and the other halo is a
different one of
those three; more preferably one of such halo is fluoro, all other R3's having
hydrogen.
Most preferred R3 combinations are selected from 4'-chloro, 4'-bromo, and
2',4'-
dichloro, all other R3's being hydrogen.
Non-limiting examples of compounds of the subject invention include those of
structure 2 wherein both R6's are methoxy, and R1's and R3's are as indicated
in the
following table (R3's not specified are all hydrogen):
Example x Rl R3
9
CA 02372051 2001-11-15
WO 00/69860 PCT/US00/13413
1 0 methyl, methyl hydrogen
2 0 methyl, methyl 4'-fluoro
3 1 methyl, methyl 4'-chloro
4 0 methyl, methyl 4'-bromo
0 methyl, methyl 2',4'-difluoro
6 1 methyl, methyl 2',4'-dichloro
7 0 methyl, methyl 2'-fluoro, 4'-chloro
8 1 methyl, methyl 2'-fluoro, 4'-bromo
Example x R1 R3
9 0 methyl, methyl 2'-chloro, 4'-fluoro
1 methyl, methyl 2'-bromo, 4'-fluoro
11 1 methyl, methyl 3',4'-difluoro
12 0 methyl, methyl 3',4'-dichloro
13 0 methyl, methyl 3'-fluoro, 4'-chloro
14 1 methyl, methyl 3'-fluoro, 4'-bromo
0 methyl, methyl 3'-chloro, 4'-fluoro
16 1 methyl, methyl 3'-bromo, 4'-fluoro
17 0 methyl, methyl 2',4',5'-trifluoro
18 0 methyl, methyl 4'-methyl
19 1 methyl, methyl 2',4'-dimethyl
0 methyl, methyl 3',4'-dimethyl
21 1 methyl, methyl 2',4',6'-trimethyl
22 1 methyl, methyl 3'-trifluoromethyl
23 0 methyl, methyl 3',5'-di-trifluoromethyl
24 0 methyl, methyl 4'-fluoro, 3'-trifluoromethyl
1 ethyl, ethyl 4'-fluoro
26 0 ethyl, methyl 4'-chloro
27 0 ethyl, ethyl 4'-bromo
28 1 ethyl, methyl 2',4'-dichloro
29 1 ethyl, ethyl 3'-fluoro, 4'-chloro
CA 02372051 2001-11-15
WO 00/69860 PCT/US00/13413
30 1 ethenyl, ethenyl 4'-chloro
31 0 ethenyl, methyl 2',4'-dichloro
32 0 i-propyl, i-propyl 4'-fluoro
33 0 i-propyl, methyl 4'-chloro
34 1 i-propyl, i-propyl 4'-bromo
35 1 i-propyl, methyl 2',4'-dichloro
36 0 i-propyl, i-propyl 3'-fluoro, 4'-chloro
37 0 -CH2-CH=CH2, methyl 4'-fluoro
38 1 -CH2-CH=CH2, 4'-chloro
-CH2-CH=CH2
Example x R1 R3
39 1 -CH2-CH=CH2, ethyl 2',4'-dichloro
40 0 s-butyl, s-butyl 4'-chloro
41 1 cyclopentyl, methyl 4'-chloro
42 1 methyl, methyl 2',4',5'-trifluoro
43 0 methyl, methyl 2',3',4'-trifluoro
44 0 ethyl, ethyl 2',4',5'-trifluoro
45 1 ethyl, ethyl 2',3',4'-trifluoro
46 1 isopropyl, isopropyl 2',4',5'-trifluoro
47 0 isopropyl, isopropyl 2',3',4'-trifluoro
48 0 methyl, methyl 4'-dibutylamino
49 1 ethyl, ethyl 4'-dimethylamino
50 0 isopropyl, isopropyl 4'-diethylamino
In addition, it is istration, and
recognized the like, the
that
for
purification,
admin
salts and derivativesof the above compoundsan be used. Thus
other c a
pharmaceutically-acceptable salt, hydrate, or biohydrolizable
ester, amide or imide
thereof is ated
contempl as
part
of
the
subject
invention.
Methods
of
Making
the
Compounds
11
CA 02372051 2001-11-15
WO 00/69860 PCT/US00/13413
In making the compounds of the subject invention, the order of synthetic steps
may be varied to increase yield of desired product. The skilled artisan will
recognize that
the judicious choice of reactants, solvents, and temperatures is important in
successful
synthesis. The starting materials used in preparing the subject compounds are
known,
made by known methods, or are commercially available.
It is recognized that the skilled artisan can readily carry out standard
manipulations of organic compounds without further direction. These include,
but are not
limited to, reduction, oxidation, acylation, substitution, etherification,
esterification,
sulfonation, and the like. Examples of these manipulations are discussed in
standard
texts.
Procedures for preparing some imidazo-isoquinoline compounds are disclosed in
U.S. Patent No. 3,917,610 issued November 4, 1975, and U.S. Patent No.
4,143,143
issued on March 6, 1979, both of which are incorporated herein by reference.
The following general schemes can be used for synthesizing compounds of the
subject invention. In these schemes, R1, R2, R3, R4, RS, R6, R7, B, and n are
as defined
hereinabove. Non-conventional symbols and abbreviations for other moieties and
chemicals, which may not be clear to the skilled chemist, are defined in the
Examples
hereinbelow.
General Scheme I
H SEM
N S
N 1. NaH/SEMCI_ nBuLi/Bu3SnCl
2. nBuLi/PhSSPh
3. MCPBA/CH2CI2 N
N
12
CA 02372051 2001-11-15
WO 00/69860 PCT/~JS00/13413
R5 R7
SEM O
"
Bu3Sn N ~S/O R6 ~ n B R7
N I / R6 / Br OH
R5
Pd(PPhg)4
Dioxane
R5 R7 R7
B
R6, " ~OH
SEM O
II/O MsCI/EtgN
R6 ~ S
R5 \ CH2CI2
N
R5 R7
R6 ~ n B R7 R1 R1 R3
R3
R6 ~ ~ O O H2N
N IS/ \
R5 ~ \ R3 R3
I
N / R3
nBuLi/THF
13
CA 02372051 2001-11-15
WO 00/69860 PCT/LJS00/13413
R5 R
R7 R3
R6 \ n B/
H R3 (R2)1
N N
R6 ~ NaH/THF
R5 ~ R3
N R~ R1 R3
R5 R
R7 R3
R6 \ n B' /
/y R2 R3 / R3
/ N N
R6
R5 ~ R3
N R1 R1
R3
General Scheme II
R5 R7
R6 ~ nB R7
/ O O NBS
R6 N S~//
R5
I
N /
R5
R6 ~ n R7 ( 3)4
B Pd PPh
O Toluene
R6 / N ~I/O
S R4-SnMe3
R5 ~ ~ or R4-B(OH)2
I
N /
Br
14
CA 02372051 2001-11-15
WO 00/69860 PCT/US00/13413
R5 R~
R6 \ n B R7 R1 R1 R3
R3
R6 ~ N S~ % H2N \
R5 ~ ~ ~ \ R3 R3
N / R3
R4
nBul_i/THF
R5
R7 R3
R6 \ n B/
H R3 / R3 (R2)1
R6 N N \ ~ NaH/THF
R3
R5
N R1 \R1 R3
R6 R7 R3
\ n B/
R2 R3 / R3
N N
R6 ~ ~ \
R5 R3
N R1 ~R1
R3
The following examples provide further information regarding synthesis of
the subject invention compounds.
Precursor Example 1
H SEM
N N
SEM = ~~D~SiMeg
N ~ N
A
CA 02372051 2001-11-15
WO 00/69860 PCT/US00/13413
A suspended solution of NaH (6.5 g, 0.162 mol, washed with hexane twice) in
anhydrous
DMF (300 ml) is cooled in an ice/acetone bath (bath temp. -15 °C).
Solid imidazole (10
g, 0.145 mol) is added in small portions and the mixture is stirred at room
temperature
(r.t.) for 0.5 h; the solution becomes clear. SEM-Cl (25 g, 0.150 mol) is
added dropwise
by a syringe pump at r.t. over 1 h; NaCI precipitates during the addition. The
mixture is
stirred at r.t. for about 1 h. Progress of the reaction is monitored by TLC
(CH2Cl2/MeOH, 9:1). H20 (10 ml) is added with caution to quench the reaction.
The
solvent is evaporated in vacuo. The residue is dissolved in Et20 (200 ml) and
washed
with H20 (4 x 50m1), brine (50 ml), dried (MgS04), filtered, and evaporated in
vacuo to
give compound A as an orange liquid.
Precursor Example 2
SEM SEM
N N
\
\ N N
A
To a solution of SEM-protected imidazole A (1.48 g, 7.50 mmol) in dry THF (75
ml)
under argon at -78 °C, n-BuLi (1.6 M in hexane) (6 ml, 9.60 mmol) is
added dropwise
and the mixture is stirred at -78 °C for 30 min. Phenyl disulfide (2.1
g, 9.60 mmol) in
THF (2 ml) is then added dropwise. The dry ice/acetone bath is replaced with
an ice bath
after this addition. The mixture is stirred at 0 °C for 1 h, then at
r.t. for 1 h. Progress is
monitored by TLC (CH2Cl2/MeOH, 9:1). H20 (5 ml) is added to quench the
reaction.
The solvent is evaporated in vacuo, and the residue is dissolved in Et20,
washed with 5%
NaHC03 (3 x 20m1), brine (20 ml), dried (MgS04), evaporated in vacuo, and
purified by
chromatography (silica gel, hexane/EtOAc 3:1 ) to give B as a yellow oil.
Precursor Example 3
16
CA 02372051 2001-11-15
WO 00/69860 PCTlUS00/13413
SEM SEM
N S N SI//o
w \~- w
N ~ N
B C
3-chloroperoxybenzoic acid (MCPBA, 80-85%) (17.03 g, 78.9 mol) is added to a
solution
of SEM-protected 2-phenylsulfide imidazole B (9.71 g, 31.6 mmol) in anhydrous
CH2C12
(160mL), and the reaction is stirred under argon at room temperature for 15
hours.
Progress monitored by TLC (hexane/EtOAc, 3:1). Sodium thiosulfate (3.9 g) is
added to
remove excess MCPBA. The mixture is filtered. The filtrate is washed with 5%
Na2C03
(3 x 150 mL), brine (150 mL), dried (MgS04), filtered, evaporated in vacuo,
and purified
by chromatography (silica gel, hexaneBtOAc 3:1) to give C as a light yellow
oil.
Precusor Example 4
SEM
SEM O
N SI/ \ nBugSn ~ S~O
N
N
C D
To a solution of SEM-protected 2-phenylsulfone imidazole C (8.61 g, 25.4 mmol)
in anhydrous THF (250 mL) under argon at -78 °C, n-BuLi (1.6 M in
hexane)
(19.0 mL, 30.0 mmol) is added dropwise by a syringe pump; the solution is
stirred at -78 °C for 30 minutes. Tributyltin chloride (6.9 mL, 25.4
mmol) is
added dropwise by a syringe pump. The mixture is stirred at room temperature
for one hour. Progress is monitored by TLC (hexane/EtOAc, 9:1 ). H20 (30 mL)
is added to quench the reaction. The solvent is evaporated in vacuo. The
residue is dissolved in ether (550 mL) and washed with saturated NH4C1 (3 x
150
mL), brine (150 mL), dried (MgS04), filtered, evaporated in vacuo, and
purified
17
CA 02372051 2001-11-15
WO 00/69860 PCT/US00/13413
by chromatography (silica gel, gradient: hexane (500 mL), hexane/EtOAc, 50:1;
hexane/EtOAc, 12:1 ) to give D as a clear oil.
Precursor Example 5
SEM
O O
nBu3Sn N S~~ Me0
OH
N
Me0 Br
D E
nu
M
:M
~~O
M // S
_ N
Pd(PPh3)q. (0.0177g, 0.015 mmol) is added to a solution of stannylimidazole D
(0.51 g,
0.80 mmol), 4,5-dimethoxy-2-(2-hydroxyethyl)phenyl bromide E (0.33 g, 1.1
mmol), and
LiCI (0.087 g, 2.1 mmol) in anhydrous dioxane (4.0 mL) at room temperature. A
few
crystals (~ 2 mg) of a radical scavenger, 2,6-di-tert-butyl-4-methylphenol, is
added and
the reaction is heated to reflux under argon for 5 hours. The reaction is
cooled to room
temperature and treated with a 1:1 mixture of ether and saturated aqueous KF
solution (10
mL) for 15 hours. Progress is monitored by TLC (hexane/EtOAc, 3:1). The
mixture is
filtered through a pad of Celite with ether rinses. The filtrate is washed
with water (3 x
12 mL), brine (3 x 12 mL), dried (MgS04), filtered, evaporated in vacuo, and
purified by
chromatography (silica gel, hexane/EtOAc, 2:3) to give F as an orange oil.
Precursor Example 6
18
CA 02372051 2001-11-15
WO 00/69860 PCT/US00/13413
M
:M
O
O
M
N
F
Me0
O
Me0
N
G
To a solution of F (2.2 g, 4.24 mmol) and Et3N (886 ~L ) in dichloromethane
(200
mL) under argon atmosphere at 0 °C, methylsulfonyl chloride (MsCI, 492
~L) is
added over a peroid of 0.5 h. The reaction is then warmed to room temperature
and stirred for 1 h. TLC (EtOAc:Hexane, 1:1 ) is used to monitor the reaction;
it
indicates that Ms0-ester formation and the SEM-cleavage followed by ring
closure occur in one pot. The mixture is diluted with CH2C12 (25 mL), and
washed with cold HCI aqueous (0.5N), NaHC03 aqueous, H20, brine and dried
over MgS04. Filtration and evaporation of solvent gives a yellow solid.
Purification using preparative HPLC (EtOAc:hexane, a gradient from 1:1 to 1:0)
provides product G (1.3 g).
Invention Compound Example I
19
CA 02372051 2001-11-15
WO 00/69860 PCT/US00/13413
Me0 Me Me
° H N
~~/° z /
Me0 / S/ ~ +
N
_H
G
Me0
/
Me0
N
\\~~e
Me
J
In a 25 mL single neck round bottom flask, equipped with a magnetic stir bar,
argon inlet, and rubber septum, 1-methyl-1-phenyl-ethylamine (~ (164 mg, 1.2
mmol) in anhydrous THF under argon atmosphere is cooled to 0 °C. nBuLi
in
hexane (0.5 mL, 2.4 M) is added to the solution slowly. The reaction turns
light
yellow. After stirring for 45 minutes, compound G (150 mg, 0.405 mmol) in THF
(1 mL) is added. The solution is warmed to room temperature, and is further
heated to reflux for 12 h. TLC (CH2C12:CHgOH, 99:1 ) indicates completion of
the reaction. The solution is quenched with MeOH and evaporated to give a
residue which is redissoleved in CH2C12, washed with aqueous NaHCOg (5%),
H20, brine, and dried over Na2S04. The crude product, after filtration and
evaporation in high vacuum in order to remove any excess amine H, is purified
by chromatography (CH2C12:CHgOH, 99:1 ) to provide subject invention
compound J.
Invention Compound Example II
CA 02372051 2001-11-15
WO 00/69860 PCT/US00/13413
Me0 \
\
N
Me0
N
Me Me
J
Me0 \
Me \
N
Me0
N
Me Me
P
In a 25 mL single neck round bottom flask, compound J (181 mg, 0.5 mmol) in
anhydrous THF (10 mL) under argon atmosphere is cooled to 0 °C, then
NaH (18 mg)
pre-washed with hexane is added as a suspension in hexane to the solution.
After stirnng
for 15 minutes, methyl iodide (71 mg, 0.5 mmoll) in THF (0.5 mL) is added. The
solution is warmed to room temperature, and further heated to reflux for 2 h
to complete
the reaction. The solution is quenched with MeOH and evaporated to give a
residue
which is redissolved in CH2Cl2, washed with aqueous NaHC03 (5%), H20, brine
and
dried over Na2SOq.. The crude product is purified by chromatography
(CH2C12:CH30H
from 99:1 to 95:5) to provide subject invention compound P.
Precursor Example 7
21
CA 02372051 2001-11-15
WO 00/69860 PCT/US00/13413
Me0 \
O
S~~O
Me0 ~ \
N
G
Me0 \
O
N S~~O
Me0 ~ \
N
Br
K
N-bromosuccinimide (NBS) solid (98 mg, 0.26 mmol) is added to a solution of
compound G (0.5 mmol) in 15 mL of CCIq.. Radical initiator benzoyl peroxide (2
mol%) is subsequently added. The flask is placed into a 90 °C oil bath.
After 10
min stirring, the reaction is complete. Filtration of the mixture through a
celite
pad, and evaporation of the filtrate gives a residue. Purification by
chromatography (EtOAc:hexane, 1:3 to 1:1 ) affords compound K.
Precursor Example 8
Me0 \
S~O
Me0 ~ I/~ \
N
Br
K
Me0 \
O
S~~O
Me0 ~ \
N
L
22
WO 00/69860 CA 02372051 2001-11-15 pCT~S00/13413
To a solution of K (0.22mmol) and Pd(PPh3)4 (13mg, 0.056mmol) in 7 mL of
anhydrous toluene are added phenyltributyltin (0.26mmol) and a few crystals
(~ 2 mg) of 2,6-di-tert-butyl-4-methylphenol. The reaction mixture is allowed
to
reflux at 110 °C under nitrogen for 6 hours to complete the reaction.
The
reaction mixture is allowed to cool, and is then diluted with 1-2 mL of ethyl
acetate (EtOAc). The resultant mixture is washed with water, then brine,
extracted with CH2C12, dried over Na2SOq., and filtered. The filtrate is
treated
with 3 mL of 30% aqueous KF at room temperature for 2h. The solid is filtered
off. The filtrate is diluted with CH2C12 and washed with water, 30% aqueous
NHq.OH (3X), brine, extracted with EtOAc, dried (Na2S04), and concentrated in
vacuo to yield crude product. Chromatography purification (silica gel,
EtOAc:hexane, 1:1 to 1:0) yields compound L.
Subject Invention Example III
O Me Me
O
s/ \ + H2N
N / \
L M
H
\' N
Me Me
N
In a 25 mL single neck round bottom flask, equipped with a magnetic stir bar,
argon inlet, and rubber septum, (1-methyl-1-phenyl-ethylamine) (M) (1.2 mmol)
in
anhydrous THF under argon atmosphere is cooled to 0 °C. nBuLi in hexane
(0.5
23
CA 02372051 2001-11-15
WO 00/69860 PCT/US00/13413
mL, 2.4 M) is added slowly to the solution. The reaction turns light yellow.
After
stirring for 45 minutes, compound L (180 mg, 0.405 mmol) in THF (1 mL) is
added. The solution is warmed to room temperature, and is further heated to
reflux for 12 h to complete the reaction. The solution is quenched with MeOH
and evaporated to give a residue which is redissoleved in CH2C12, washed with
aqueous NaHC03 (5%), H20, brine and dried over Na2S04. The crude product,
after filtration and evaporation in high vacuum in order to remove excess
amine
M, is purified by chromatography to provide subject invention compound N.
Compositions of the Invention
A composition of the subject invention comprises:
a) a safe and effective amount of a compound of the invention; and
b) a pharmaceutically-acceptable excipient.
Typically, such composition comprises several excipients. It may also
optionally
comprise other active compounds which do not substantially interfere with the
activity of
the subject invention compound.
The compositions of the subject invention may be in any of a variety of forms,
suitable (for example) for oral, rectal, topical or parenteral administration.
Compositions
of the subject invention are preferably provided in unit dosage form. As used
herein, a
"unit dosage form" is a composition containing an amount of a subject compound
that is
suitable for administration to a human or lower animal subject, in a single
dose, according
to good medical practice.
As used herein, a "safe and effective amount" of a subject compound is an
amount
large enough to significantly induce a positive modification in the symptoms
and/or
condition to be treated in a host, but small enough to avoid serious adverse
side effects in
the host (such as toxicity, irntation, or allergic response), commensurate
with a
reasonable benefit/risk ratio. The safe and effective amount will vary with
such factors as
the particular condition being treated, the age and physical condition of the
patient, the
duration of treatment, the nature of concurrent therapy (if any), the specific
dosage form
to be used, and the dosage regimen employed.
24
CA 02372051 2001-11-15
WO 00/69860 PCT/US00/13413
The term "pharmaceutically-acceptable excipient", as used herein, includes
physiologically inert, pharmacologically inactive substances which are
compatible with
the physical and chemical characteristics of the subject invention compound
used, and
which are of sufficiently high purity and sufficiently low toxicity to be
suitable for
administration to a human or lower animal. The term "compatible", as used
herein,
means that the excipients of the subject composition are capable of being
commingled
with the subject invention compound, and with each other in a manner such that
there is
no interaction which would substantially reduce the pharmaceutical efficacy of
the
compound, under ordinary use situations.
Depending upon the particular route of administration desired, a variety of
pharmaceutically-acceptable excipients well-known in the art may be used.
Excipients
include, but are not limited to, polymers, resins, plasticizers, fillers,
binders, lubricants,
glidants, disintegrants, solvents, co-solvents, buffer systems, surfactants,
preservatives,
sweetening agents, flavoring agents, and dyes or pigments. The amount of
excipients
employed in conjunction with the subject compound is sufficient to provide a
practical
quantity of material for administration per unit dose of the subject compound.
Some examples of substances which can serve as pharmaceutically-acceptable
excipients are sugars, such as lactose, dextrose, glucose and sucrose;
starches, such as
cornstarch and potato starch; cellulose and its derivatives, such as
methylcellulose,
sodium carboxymethylcellulose, ethylcellulose, hydroxypropylcellulose and
cellulose
acetate; polymers, such as povidone and carbomers; powdered tragacanth; gums,
such as
xanthan, guar and acacia; malt; solid lubricants, such as stearic acid,
magnesium stearate,
and talc; inorganic fillers, such as calcium phosphates and calcium sulfate;
disintigrants,
such as sodium starch glycolate, crospovidone, croscarmelose sodium, and
microcrystalline cellulose; encapsulating and coating materials, such as
gelatins, waxes,
and cellulose derivatives; vegetable oils, such as peanut oil, cottonseed oil,
sesame oil,
olive oil, corn oil and oil of the obroma; polyols such as propylene glycol,
glycerin,
sorbitol, mannitol, and polyethylene glycol; alginic acid; surfactants such as
the
Tweens~, alkyl sulfate salts, salts of fatty acids, sucrose esters; ethyl
oleate; coloring
agents; flavoring agents; tableting agents; stabilizers; antioxidants;
preservatives;
CA 02372051 2001-11-15
WO 00/69860 PCT/US00/13413
solvents, such as ethanol, pyrogen-free water; isotonic saline; and buffer
solutions, such
as phosphoric, tartaric, citric, and acetic acids, and their sodium,
potassium, and
ammonium salts.
Preferred compositions of the subject invention are oral dosage forms. The
term
"oral dosage form", as used herein, means any pharmaceutical composition
intended to be
systemically administered to an individual by delivering the composition via
the mouth to
the gastrointestinal tract of an individual. Preferred are oral unit dosage
forms, such as
tablets, coated or non-coated, and capsules, hard or soft gel. Subject oral
unit dosage
form compositions comprise preferably at least about 4 mg, more preferably at
least about
20 mg, more preferably still at least about 100 mg, and preferably at most
about 1000 mg,
more preferably at most about 500 mg, more preferably still at most about 250
mg, of a
subject compound. Subject oral dosage form compositions comprise preferably at
least
about 1 %, more preferably at least about 10%, and preferably at most about
70%, more
preferably at most about 40%, of a subject compound; and comprise preferably
at least
about 30%, more preferably at least about 60%, and preferably at most about
99%, more
preferably at most about 90%, pharmaceutically-acceptable excipients.
Parenteral dosage forms are also preferred subject invention compositions. The
term "parenteral dosage form", as used herein, means any pharmaceutical
composition
intended to be systemically administered to a human or lower animal via
delivery of a
solution or emulsion containing the active ingredient, by puncturing the skin
of the
individual, in order to deliver the solution or emulsion to the circulatory
system of the
individual either by intravenous, intramuscular, intraperitoneal or
subcutaneous injection.
Subject parenteral unit dosage form compositions comprise preferably at least
about 1
mg, more preferably at least about 6 mg, more preferably still at least about
30 mg, and
preferably at most about 400 mg, more preferably at most about 100 mg, more
preferably
still at most about 40 mg, of a subject compound. Subject parenteral dosage
form
compositions comprise preferably at least about 1 %, more preferably at least
about 5%,
and preferably at most about 20%, more preferably at most about 10%, of a
subject
compound; and comprises preferably at least about 80%, more preferably at
least about
90%, and preferably at most about 99%, more preferably at most about 95%,
26
CA 02372051 2001-11-15
WO 00/69860 PCT/US00/13413
pharmaceutically-acceptable excipients. In addition, dosages for injection may
be
prepared in dried or lyophilized form. Such forms can be reconstituted with
water, saline
solution, or a buffer solution, depending on the preparation of the dosage
form. Such
forms may be packaged as individual dosages or multiple dosages for easier
handling.
Where lyophilized or dried dosages are used, the reconstituted dosage form is
preferably
isotonic, and at a physiologically compatible pH, and comprises the subject
compound
and excipients in the amounts and percentages indicated previously in this
paragraph.
Methods of Treatment Usin~the Compounds
Subject invention compounds have demonstrated pharmacological activity in
processes known to be associated with one or more of cardiovascular activity,
inflammatory mechanisms, oncology, and regulation of protein transport from
cells. The
subject invention includes methods of using the above compounds of the subject
invention for therapeutic or preventative treatment of one or more of the
following
diseases or disorders: congestive heart failure, arrhythmia, hypotension,
cardiac
reperfusion injury, arteriosclerosis, restenosis, vascular tone, bacterial
infection, cancer,
Kaposi's sarcoma, psoriasis, migraine, nasal congestion, allergic responses,
rheumatoid
arthritis, and osteoporosis. Such methods comprise administering to a human or
lower
animal in need of such treatment or prevention a safe and effective amount of
a subject
invention compound.
For preferred oral administration of compounds and/or compositions of the
subject invention, preferably at least about 0.1 mg/kg, more preferably at
least about 0.5
mg/kg, more preferably still at least about 2 mg/kg, and preferably at most
about 20
mg/kg, more preferably at most about 5 mg/kg, more preferably still at most
about 2
mg/kg, of a subject compound is administered to a human or lower animal,
preferably at
least about 1 time, more preferably at least about 2 times, and preferably at
most about 4
times, more preferably at most about 2 times, daily. Treatment duration using
such oral
daily dosages is dependent on the disease or disorder being treated; it is
preferably at least
about 1 day, more preferably at least about 3 days, more preferably still at
least 7 days,
and preferably at most about 5 years, more preferably at most about 60 days,
more
preferably still at most about 15 days.
27
CA 02372051 2001-11-15
WO 00/69860 PCT/US00/13413
For preferred parenteral administration of compounds and/or compositions of
the
subject invention, preferably at least about 0.04 mg/kg, more preferably at
least about 0.2
mg/kg, more preferably still at least about 1 mg/kg, and preferably at most
about 10
mg/kg, more preferably at most about 4 mg/kg, more preferably still at most
about 1
mg/kg, of a subject compound is administered to a human or lower animal,
preferably at
least about 1 time, more preferably at least about 2 times, and preferably at
most about 4
times, more preferably at most about 2 times, daily. Treatment duration using
such
parenteral daily dosages is dependent on the disease or disorder being
treated; it is
preferably at least about 1 day, more preferably at least about 3 days, more
preferably still
at least 7 days, and preferably at most about 60 days, more preferably at most
about 20
days, more preferably still at most about 5 days.
While particular embodiments of the subject invention have been described, it
will be obvious to those skilled in the arts that various changes and
modifications of the
subject invention can be made without departing from the spirit and scope of
the
invention. It is intended to cover, in the appended claims, all such
modifications that are
within the scope of this invention.
28