Note: Descriptions are shown in the official language in which they were submitted.
CA 02374926 2001-12-06
WO 00/71549 PCT/EP00/04279
1
THIAZOLODERIVATIVES AND PHARMACEUTICAL COMPOSTTIONS CONTAINING THEM
The present invention relates to certain novel substituted dihydroimidazo[2,1-
b]thiazole and dihydro-5H-thiazolo[3,2-a]pyrimidine compounds which have
affinity
for 5-HT1A receptors and which inhibit neuronal reuptake of 5-
hydroxytryptamine
and/or noradrenaline, to processes for their preparation, to pharmaceutical
compositions containing them and to their use in the treatment of depression,
anxiety, psychoses (for example schizophrenia), tardive dyskinesia; obesity,
drug
addiction, drug abuse, cognitive disorders, Alzheimer's disease, obsessive-
compulsive behaviour, panic attacks, social phobias, eating disorders such as
bulimia, anorexia, snacking and binge eating, non-insulin dependent diabetes
mellitus, hyperglycaemia, hyperlipidaemia, stress, as an aid to smoking
cessation
and in the treatment and/or prophylaxis of seizures, neurological disorders
such as
epilepsy and/or conditions in which there is neurological damage such as
stroke,
brain trauma, cerebral ischaemia, head injuries and haemorrhage.
WO 98/41528 discloses that compounds of formula A
R3
S
Ar
N A
N
4
R Rs R~ R2
including pharmaceutically acceptable salts thereof in the form of individual
enantiomers, racemates, or other mixtures of enantiomers,
in which:
Ar is phenyl, naphthyl or benzo[b]thiophenyl, each of which may be optionally
substituted by one or more substituents selected from a) halo, b) an alkyl
group
containing 1 to 3 carbon atoms optionally substituted by one or more halo, c)
an
alkoxy group containing 1 to 3 carbon atoms optionally substituted by one or
more
halo, d) an alkylthio group containing 1 to 3 carbon atoms optionally
substituted by
one or more halo, e) a phenoxy group optionally substituted by one or more
halo or f)
phenyl optionally substituted by one or more halo;
R~ and R2 , which may be the same or different, independently are a) H, b) an
alkyl
group containing 1 to 6 carbon atoms, c) an alkenyl group containing 3 to 6
carbon
CA 02374926 2001-12-06
WO 00/71549 PCT/EP00/04279
2
atoms, d) a cycloalkyl group containing 3 to 7 carbon atoms, e) a
cycloalkylmethyl
group in which the ring contains 3 to 7 carbon atoms, f) an aryl or heteroaryl
group
optionally substituted by one or more substituents selected from i) halo, ii)
an alkyl
group containing 1 to 3 carbon atoms optionally substituted by one or more
halo, iii)
an alkoxy group containing 1 to 3 carbon atoms optionally substituted by one
or
more halo, iv) an alkylthio group containing 1 to 3 carbon atoms optionally
substituted by one or more halo, g) an arylalkyl or heteroarylalkyl group in
which the
alkyl chain contains 1 to 3 carbon atoms and in which the aryl or heteroaryl
group
may optionally be substituted by one or more substituents selected from i)
halo, ii) an
alkyl group containing 1 to 3 carbon atoms optionally substituted by one or
more
halo, iii) an alkoxy group containing 1 to 3 carbon atoms optionally
substituted by
one or more halo, iv) an alkylthio group containing 1 to 3 carbon atoms
optionally
substituted by one or more halo; or R, and RZ form an alkylene chain
optionally
substituted by one or more alkyl groups each containing 1 to 3 carbon atoms,
such
that, together with the atoms to which they are attached, they form a 5 or 6
membered ring,
R3 is a) H, b) an aryl or heteroaryl group optionally substituted by one or
more
substituents selected from i) halo, ii) an alkyl group containing 1 to 3
carbon atoms
optionally substituted by one or more halo, iii) an alkoxy group containing 1
to 3
carbon atoms optionally substituted by one or more halo, iv) an alkylthio
group
containing 1 to 3 carbon atoms optionally substituted by one or more halo, c)
an
arylmethyl group in which the aryl is optionally substituted by one or more
substituents selected from i) halo, ii) an alkyl group containing 1 to 3
carbon atoms
optionally substituted by one or more halo, iii) an alkoxy group containing 1
to 3
carbon atoms optionally substituted by one or more halo, iv) an alkylthio
group
containing 1 to 3 carbon atoms optionally substituted by one or more halo; or
d) an
alkoxyalkyl group containing 3 to 6 carbon atoms; and
R4 and R5 , which may be the same or different, independently are an alkyl
group
containing 1 to 3 carbon atoms, or R4 and RS together with the atom to which
they
are attached form a cycloalkyl ring containing 3 to 6 carbon atoms;
are useful in the treatment of depression, anxiety, Parkinson's disease,
obesity,
cognitive disorders, seizures, neurological disorders such as epilepsy, and as
neuroprotective agents to protect against conditions such as stroke. The
compounds of the present invention are riot disclosed or suggested in this
document.
CA 02374926 2001-12-06
WO 00/71549 PCT/EP00/04279
3
Sharpe C.J and Shadbolt R.S. (Journal of Medicinal Chemistry, 1971, Vol 14
No.10, p977-982) disclose certain dihydroimidazo[2,1-b]thiazole compounds
having
antidepressant activity. However, the document also states that these
compounds
were generally less active and more toxic than the imidazolines also disclosed
in the
document. The compounds of the present invention are not disclosed or
suggested
in this document.
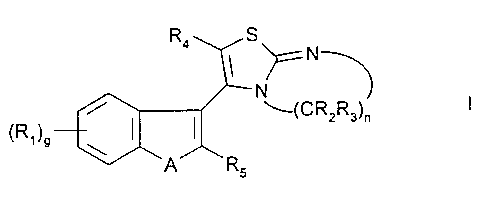
WO 97/02269 discloses that compounds of formula B
R4 S
~ ~ N~
N g
/ ~(CR2Rs)n
(R')9 \
A RS
including pharmaceutically acceptable salts thereof
in which
A is S(O)P or O;
p is 0, 1 or 2;
g is 0, 1, 2, 3, or 4;
nis2or3;
R~ is a) halo, b) an alkyl group containing 1 to 3 carbon atoms optionally
substituted
by one or more halo, c) an alkoxy group containing 1 to 3 carbon atoms
optionally
substituted by one or more halo, d) an alkylthio group, an alkylsulphinyl
group or an
alkylsulphonyl group each containing 1 to 3 carbon atoms optionally
substituted by
one or more halo, e) hydroxy, f) an acyloxy group containing 1 to 3 carbon
atoms, g)
a hydroxyalkyl group containing 1 to 3 carbon atoms, h) cyano, i) an alkanoyl
group
containing 1 to 6 carbon atoms, j) an alkoxycarbonyl group containing 2 to 6
carbon
atoms, k) a carbamoyl group or a carbamoylmethyl group each optionally
N-substituted by one or two alkyl groups each containing 1 to 3 carbon atoms,
I) a
sulphamoyl or sulphamoylmethyl group each optionally N-substituted by one or
two
alkyl groups each containing 1 to 3 carbon atoms, or m) an amino group
optionally
substituted by one or two alkyl groups each containing 1 to 3 carbon atoms; R~
being
the same or different when g is 2, 3 or 4;
CA 02374926 2001-12-06
WO 00/71549 PCT/EP00/04279
4
R2, R3 and R4 independently are H or an alkyl group containing 1 to 3 carbon
atoms
optionally substituted by one or more halo; and
R5 is a) halo, b) an alkyl group containing 1 to 3 carbon atoms optionally
substituted
by one or more halo, c) an alkoxy group containing 1 to 3 carbon atoms
optionally
substituted by one or more halo, d) an alkylthio group, an alkylsulphinyl
group or an
alkylsulphonyl group each containing 1 to 3 carbon atoms optionally
substituted by
one or more halo, e) hydroxy, f) an acyloxy group containing 1 to 3 carbon
atoms, g)
a hydroxyalkyl group containing 1 to 3 carbon atoms, h) cyano, i) an alkanoyl
group
containing 1 to 6 carbon atoms, j) an alkoxycarbonyl group containing 2 to 6
carbon
atoms, k) a carbamoyl group or a carbamoylmethyl group each optionally
N-substituted by one or two alkyl groups each containing 1 to 3 carbon atoms,
I) a
sulphamoyl or sulphamoylmethyl group each optionally N-substituted by one or
two
alkyl groups each containing 1 to 3 carbon atoms, m) an amino group optionally
substituted by one or two alkyl groups each containing 1 to 3 carbon atoms, or
n) H ;
have affinity for 5-HT~A receptors and inhibit neuronal reuptake of
5-hydroxytryptamine and/or noradrenaline. These compounds are stated to be
useful in the treatment of CNS disorders. However, these compounds exhibit
activity
as monoamine oxidase inhibitors and/or have affinity for other receptors, for
example
muscarinic receptors, and are therefore likely to cause undesired side
effects.
Surprisingly the present invention provides compounds with unexpectedly
superior
selectivity and efficacy. The compounds of the present invention are not
disclosed
or suggested in WO 97/02269 .
US4,160,768 discloses that 3-(2-benzofuranyl)-5,6-dihydroimidazo(2,1-
b]thiazole is useful as an anti-inflammatory agent. This document does not
disclose
or suggest the compounds of the present invention.
The present invention provides compounds of Formula I
R4 S
~N~
N
(R~)9 \ ~ ~ ~(CRZR3)~
_A, ~Rs
CA 02374926 2001-12-06
WO 00/71549 PCT/EP00/04279
including pharmaceutically acceptable salts thereof in which
AisSorO;
5
g is 0, 1, 2, 3 or 4;
n is 2 or 3;
R, is a) halo, b) an alkyl group containing 1 to 3 carbon atoms optionally
substituted
by one or more halo, c) an alkoxy group containing 1 to 3 carbon atoms
optionally
substituted by one or more halo, d) an alkylthio group, an alkylsulphinyl
group or an
alkylsulphonyl group each containing 1 to 3 carbon atoms optionally
substituted by
one or more halo, e) hydroxy, f) an acyloxy group containing 1 to 3 carbon
atoms, g)
a hydroxyalkyl group containing 1 to 3 carbon atoms, h) cyano, i) an alkanoyl
group
containing 1 to 6 carbon atoms, j) an alkoxycarbonyl group containing 2 to 6
carbon
atoms, k) a carbamoyl group or a carbamoylmethyl group each optionally
N-substituted by one or two alkyl groups each containing 1 to 3 carbon atoms,
I) a
sulphamoyl or sulphamoylmethyl group each optionally N-substituted by one or
two
alkyl groups each containing 1 to 3 carbon atoms, or m) an amino group
optionally
substituted by one or two alkyl groups each containing 1 to 3 carbon atoms; R~
being
the same or different when g is 2, 3 or 4;
R2 and R3 are each H ;
R4 represents a hydroxyalkyl group containing 1 to 6 carbon atoms, an a-
hydroxy(2-
C~_3alkoxyphenyl)methyl group, a hydroxyalkenyl group containing 3 to 6 carbon
atoms in which hydroxy is not attached directly to either carbon of the double
bond, a
hydroxyalkynyl group containing 3 to 6 carbon atoms in which hydroxy is not
attached directly to either carbon of the triple bond, a hydroxycycloalkyl
group
containing 3 to 6 carbon atoms, an alkenyl group containing 2 to 8 carbon
atoms, an
arylalkenyl group containing 8 to 10 carbon atoms, a cycloalkyl group
containing 3 to
6 carbon atoms, a C3_~alkynyfalkoxyC,_3alkyl group, a
C4_~cycloalkylalkoxyC,_3alkyl
group, a C,_3alkoxyC,_3alkyl group, a C,_3alkylthioC,_3alkyt group, a
C,_3alkoxy group,
a C,_3alkylthio group, an arylthio group, a C,_6 alkanoyl group, a C3_s
CA 02374926 2001-12-06
WO 00/71549 PCT/EP00/04279
6
alkoxycarbonylalkyl group, cyano, halo, a C,~alkyliminomethyl group, a C~_
4alkylaminoalkyl group or a hydroxyiminomethyl group ;
RS is H or halo.
In a preferred aspect the present invention provides compounds of Formula I
R4 S
I ~N~
N I
(R~)9 ~ ~ ~ ~(CRZR3)~
_A_ -Rs
including pharmaceutically acceptable salts thereof in which
AisSorO;
g is 0, 1, 2, 3, or 4;
n is 2 or 3;
R, is a) halo, b) an alkyl group containing 1 to 3 carbon atoms optionally
substituted
by one or more halo, c) an alkoxy group containing 1 to 3 carbon atoms
optionally
substituted by one or more halo, d) an alkylthio group, an alkylsulphinyl
group or an
alkylsulphonyl group each containing 1 to 3 carbon atoms optionally
substituted by
one or more halo, e) hydroxy, f) an acyloxy group containing 1 to 3 carbon
atoms, g)
a hydroxyalkyl group containing 1 to 3 carbon atoms, h) cyano, i) an alkanoyl
group
containing 1 to 6 carbon atoms, j) an alkoxycarbonyl group containing 2 to 6
carbon
atoms, k) a carbamoyl group or a carbamoylmethyl group each optionally
N-substituted by one or two alkyl groups each containing 1 to 3 carbon atoms,
I) a
sulphamoyl or sulphamoylmethyl group each optionally N-substituted by one or
two
alkyl groups each containing 1 to 3 carbon atoms, or m) an amino group
optionally
substituted by one or two alkyl groups each containing 1 to 3 carbon atoms; R~
being
the same or different when g is 2, 3 or 4;
RZ and R3 are each H ;
CA 02374926 2001-12-06
WO 00/71549 PCT/EP00/04279
7
R4 represents a hydroxyalkyl group containing 1 to 6 carbon atoms, a
hydroxyalkenyl
group containing 3 to 6 carbon atoms in which hydroxy is not attached directly
to
either carbon of the double bond, a hydroxyalkynyl group containing 3 to 6
carbon
atoms in which hydroxy is not attached directly to either carbon of the triple
bond, a
hydroxycycloalkyl group containing 3 to 6 carbon atoms, an alkenyl group
containing
2 to 8 carbon atoms, a cycloalkyl group containing 3 to 6 carbon atoms, a
C,_3alkoxyC,_3alkyl group, a C,_3alkylthioC,_3alkyl group, a C,_3alkoxy group,
a
C,_3alkylthio group, a C,_6 alkanoyl group, halo; a C,~alkyliminomethyl group
or a
hydroxyiminomethyl group ; and
R5 is H or halo.
It will be understood that the term halo, when used herein, includes fluoro,
chloro, bromo and iodo. It will be understood that in alkyl groups, alkenyl
groups,
alkynyl groups, alkylthio groups and alkoxy groups containing more than two
carbon
atoms the alkyl group may be straight or branched. Aryl is used to indicate
phenyl
optionally substituted by one or more of the following:a C~_3alkyl group, a
C1_3 alkoxy
group or halo.
In a first group of preferred compounds of the present invention A is S.
In a second group of preferred compounds of the present invention A is O.
Preferably g is 0 or 1 and R, is a) halo, b) an alkyl group containing 1 to 3
carbon atoms optionally substituted by one or more halo, or c) an alkoxy group
containing 1 to 3 carbon atoms optionally substituted by one or more halo.
Preferably R, is in the 5-position of the benzo(b]thiophene ring. More
preferably g is
0 or 1 and R, is halo or an alkoxy group containing 1 to 3 carbon atoms. Most
preferably g is 0 or 1 and R, is chloro or methoxy.
Preferably n is 2.
Preferably Rz and R3 are each H.
Preferably R4 represents a hydroxyalkyl group containing 1 to 6 carbon
atoms, a hydroxyalkenyl group containing 3 to 6 carbon atoms in which hydroxy
is
CA 02374926 2001-12-06
WO 00/71549 PCT/EP00/04279
8
not attached directly to either carbon of the double bond, a hydroxyalkynyl
group
containing 3 to 6 carbon atoms in which hydroxy is not attached directly to
either
carbon of the triple bond, an alkenyl group containing 2 carbon atoms
optionally
substituted by one or more C~_2alkyl groups , a C,~ alkyliminomethyl group or
a
hydroxyiminomethyl group. More preferably R4 represents a hydroxyalkyl group
containing 1 to 5 carbon atoms, a hydroxyalkenyl group containing 3 to 5
carbon
atoms in which hydroxy is not attached directly to either carbon of the double
bond, a
hydroxyalkynyl group containing 3 to 4 carbon atoms in which hydroxy is not
attached directly to either carbon of the triple bond or an alkenyl group
containing 2
carbon atoms optionally substituted by one or more methyl groups. Most
preferably
R4 represents hydroxymethyl or vinyl. Hydroxymethyl is especially preferred
for R4.
Preferred values of R4 are hydroxymethyl, 1-hydroxyethyl, 1-hydroxy-1-
methylethyl; 1-hydroxypropyl, 1-hydroxy-2-methylpropyl, 1-hydroxybutyl, 1-
hydroxy-
3-methylbutyl, 1-hydroxypentyl, 1-hydroxypropenyl, 1-hydroxybut-3-enyl, 1-
hydroxy-
2-methylpropenyl, 1-hydroxy-2-methylbut-3-enyl, 1-hydroxypent-4-enyl, 1-
hydroxypropynyl, 1-hydroxybut-2-ynyl, methoxymethyl, ethoxymethyl, methylthio,
bromo, chloro, vinyl, allyl, 1-methylvinyl, formyl, acetyl,N-(1-
methylethyl)iminomethyl,
and hydroxyiminomethyl. More preferably R4 is hydroxymethyl, 1-hydroxyethyl, 1-
hydroxy-1-methylethyl; 1-hydroxypropyl, 1-hydroxy-2-methylpropyl, 1-
hydroxybutyl,
1-hydroxy-3-methylbutyl, 1-hydroxypentyl, 1-hydroxypropenyl, 1-hydroxybut-3-
enyl,
1-hydroxy-2-methylpropenyl, 1-hydroxy-2-methylbut-3-enyl, 1-hydroxypent-4-
enyl, 1
hydroxypropynyl, 1-hydroxybut-2-ynyl, vinyl, 1-methylvinyl, acetyl, N-(1
methylethyl)iminomethyl, and hydroxyiminomethyl. Most preferably R4 is
hydroxymethyl or vinyl. Hydroxymethyl is especially preferred.
Preferably R5 is H.
Preferably n is 2.
In a preferred group of compounds of Formula I, A is S, g is 0 or 1; n is 2;
R~
is halo or an alkoxy group containing 1 to 3 carbon atoms; Rz and R3 are each
H;
R4 represents a hydroxyalkyl group containing 1 to 5 carbon atoms, a
hydroxyalkenyl
group containing 3 to 5 carbon atoms in which hydroxy is not attached directly
to
either carbon of the double bond, a hydroxyalkynyl group containing 3 to 4
carbon
atoms in which hydroxy is not attached directly to either carbon of the triple
bond or
CA 02374926 2001-12-06
WO 00/71549 PCT/EP00/04279
9
an alkenyl group containing 2 carbon atoms optionally substituted by one or
more
methyl groups; and R5 is H.
In a preferred group of compounds of Formula I, preferably g is 0 or 1 and R,
is chloro or methoxy. More preferably R, is in the 5-position of the
benzo[b]thiophene ring.
In a further preferred aspect the present invention provides compounds of
Formula la
Ka ~~N
N (CH2)n
Ia
H
including pharmaceutically acceptabae salts thereof in which
AisSorO;
g is 0, 1, 2, 3 or 4;
n is 2 or 3;
R, is a) halo, b) an alkyl group containing 1 to 3 carbon atoms optionally
substituted
by one or more halo, c) an alkoxy group containing 1 to 3 carbon atoms
optionally
substituted by one or more halo, d) an alkylthio group, an alkylsulphinyl
group or an
alkylsulphonyl group each containing 1 to 3 carbon atoms optionally
substituted by
one or more halo, e) hydroxy, f) an acyloxy group containing 1 to 3 carbon
atoms, g)
a hydroxyalkyl group containing 1 to 3 carbon atoms, h) cyano, i) an alkanoyl
group
containing 1 to 6 carbon atoms, j) an alkoxycarbonyl group containing 2 to 6
carbon
atoms, k) a carbamoyl group or a carbamoylmethyl group each optionally
N-substituted by one or two alkyl groups each containing 1 to 3 carbon atoms,
I) a
sulphamoyl or sulphamoylmethyl group each optionally N-substituted by one or
two
alkyl groups each containing 1 to 3 carbon atoms, or m) an amino group
optionally
substituted by one or two alkyl groups each containing 1 to 3 carbon atoms; R~
being
the same or different when g is 2, 3 or 4; and
CA 02374926 2001-12-06
WO 00/71549 PCT/EP00/04279
R4 represents a hydroxyalkyl group containing 1 to 6 carbon atoms, an a-
hydroxy(2-
C~_3 alkoxyphenyl)methyl group, a hydroxyalkenyl group containing 3 to 6
carbon
atoms in which hydroxy is not attached directly to either carbon of the double
bond, a
5 hydroxyalkynyl group containing 3 to 6 carbon atoms in which hydroxy is not
attached directly to either carbon of the triple bond, an alkenyl group
containing 2 to
8 carbon atoms, an arylalkenyl group containing 8 to 10 carbon atoms, a
cycloalkyl
group containing 3 to 6 carbon atoms, a C3_,alkynylalkoxyC,_3alkyl group, a
C4_
~cycloalkylalkoxyC,_3alkyl group, a C,_3alkoxyC,_3alkyl group, a
C,_3alkylthioC,_3alkyl
10 group, a C,_3alkoxy group, a C,_3alkylthio group, an arylthio group, a
C,_salkanoyl
group, a C3_6 alkoxycarbonylalkyl group, cyano, halo, a C,~alkyliminomethyl
group, a
C~_4alkylaminomethyl group or a hydroxyiminomethyl group.
In a first group of preferred compounds of the present invention A is S.
Preferably n is 2 in this group of compounds. Preferably g is 0 or 1 in this
group of
compounds. Preferably R~ is halo, an alkoxy group containing 1 to 3 carbon
atoms,
or an alkylthio group containing 1 to 3 carbon atoms.
In a second group of preferred compounds of the present invention A is O.
Preferably n is 2 in this group of compounds. Preferably g is 0 or 1 in this
group of
compounds. Preferably R~ is halo, an alkoxy group containing 1 to 3 carbon
atoms,
or an alkylthio group containing 1 to 3 carbon atoms.
In compounds of Formula I and Formula la preferred values of Ri are methyl,
ethyl, propyl, isopropyl, cyclopropyl, methoxy, ethoxy, isopropoxy, bromo,
chloro,
fluoro, iodo, trifluoromethyl, trifluoromethoxy, methylthio, methylsulphinyl,
methylsulphonyl, hydroxy, formyloxy, acetoxy, hydroxymethyl, 1-hydroxyethyl, 1-
hydroxy-1-methylethyl, 1-hydroxypropyl, cyano, formyl, acetyl,
methoxycarbonyl,
ethoxycarbonyl, carbamoyl, carbamoylmethyl, sulphamoyl, sulphamoylmethyl,
amino, methylamino, dimethylamino, ethylamino or diethylamino. More preferably
R1 is methoxy, chloro or methylthio.
In compounds of Formula I and Formula la preferred values of R4 are
cyclopropyl, methoxy, ethoxy, bromo, chloro, fluoro, iodo, trifluoromethyl,
trifluoromethoxy, hydroxymethyl, 1-hydroxyethyl, 1-hydroxy-1-methylethyl; 1-
CA 02374926 2001-12-06
WO 00/71549 PCT/EP00/04279
11
hydroxypropyl, 1-hydroxy-2-methylpropyl, 1-hydroxybutyl, 1-hydroxy-3-
methylbutyl,
1-hydroxypentyl, 1-hydroxypropenyl, 1-hydroxybut-3-enyl, 1-hydroxy-2-
methylpropenyl, 1-hydroxy-2-methylbut-3-enyl, 1-hydroxypent-4-enyl, 3-
hydroxybut-
1-enyl, 1-hydroxypropynyl, 1-hydroxybut-2-ynyl, a-hydroxy-2-methoxybenzyl,
methoxymethyl, ethoxymethyl, isopropoxymethyl, cyclopropylmethoxymethyl,
cyclobutylmethoxymethyl, prop-2-ynyloxymethyl, methylthio, phenylthio, vinyl,
allyl,
prop-1-enyl, 2-methylprop-2-enyl, 1-methylvinyl, styryl, formyl, acetyl,
cyano,
ethoxycarbonylmethyl, N-(1-methylethyl)iminomethyl, N-methylaminomethyl and
hydroxyiminomethyl. More preferably R4 is hydroxymethyl, 1-hydroxyethyl or
vinyl.
Most preferably R4 is hydroxymethyl or vinyl.
In especially preferred compounds of Formula la, A is S or O; g is 0 or 1, n
is
2; R~ represents halo, an alkoxy group containing 1 to 3 carbon atoms or an
alkylthio
group containing 1 to 3 carbon atoms; and R4 represents a hydroxyalkyl group
containing 1 to 4 carbon atoms, an a-hydroxy(2-C~_3alkoxyphenyl)methyl group,
a
hydroxyalkenyl group containing 3 to 4 carbon atoms in which hydroxy is not
attached directly to either carbon of the double bond, a hydroxyalkynyl group
containing 3 to 4 carbon atoms in which hydroxy is not attached directly to
either
carbon of the triple bond, an alkenyl group containing 2 to 3 carbon atoms, a
C,_3alkylthio group, a C,_2 alkanoyl group or a hydroxyiminomethyl group.
In the remainder of this description the term "compounds of Formula I" means
compounds of Formula I or compounds of Formula la. Similarly the term "a
compound of Formula I" means a compound of Formula I or a compound of Formula
la.
Compounds of Formula I may exist as salts with pharmaceutically acceptable
acids. The present invention includes all such salts. Examples of such salts
include
hydrochlorides, hydrobromides, sulphates, methanesulphonates, nitrates,
maleates,
formates, acetates, citrates, fumarates, tartrates [eg (+)-tartrates, (-)-
tartrates or
mixtures thereof including racemic mixtures], succinates, oxalates, benzoates
and
salts with amino acids such as glutamic acid. Such salts are prepared by
methods
known to those skilled in the art as illustrated in the Examples.
CA 02374926 2001-12-06
WO 00/71549 PCT/EP00/04279
12
Certain compounds of Formula I may exist in different tautomeric forms or as
different geometric isomers, and the present invention includes each tautomer
and/or
geometric isomer of compounds of Formula I and mixtures thereof.
Certain compounds of Formula I may exist in different stable conformational
forms which may be separable. For example, if a bulky group is present there
may
be restricted rotation about one or more single bond or bonds due to steric
hindrance. Torsional asymmetry due to restricted rotation about an asymmetric
single bond, for example because of steric hindrance or ring strain, may
permit
separation of different conformers. The present invention includes each
conformational isomer of compounds of Formula I and mixtures thereof.
Certain compounds of Formula I and their salts may exist in more than one
crystal form and the present invention includes each crystal form and mixtures
thereof. Certain compounds of Formula I and their salts may also exist in the
form of
solvates, for example hydrates, and the present invention includes each
solvate and
mixtures thereof.
Certain compounds of Formula I contain one or more chiral centres, and exist
in different optically active forms. When compounds of Formula I contain one
chiral
centre, the compounds exist in two enantiomeric forms and the present
invention
includes both enantiomers and mixtures of enantiomers. The enantiomers may be
resolved by methods known to those skilled in the art, for example by
formation of
diastereoisomeric salts which may be separated, for example, by
crystallisation;
formation of diastereoisomeric derivatives or complexes which may be
separated, for
example, by crystallisation, gas-liquid or liquid chromatography; or gas-
liquid or
liquid chromatography in a chiral environment, for example on a chiral support
for
example silica with a bound chiral ligand or in the presence of a chiral
solvent. It will
be appreciated that where the desired enantiomer is converted into another
chemical
entity by one of the separation procedures described above, a further step is
required to liberate the desired enantiomeric form. Alternatively, specific
enantiomers may be synthesised by asymmetric synthesis using optically active
reagents, substrates, catalysts or solvents, or by converting one enantiomer
into the
other by asymmetric transformation.
CA 02374926 2001-12-06
WO 00/71549 PCT/EP00/04279
13
When a compound of Formula I contains more than one chiral centre it may
exist in diastereoisomeric forms. The diastereoisomeric pairs may be separated
by
methods known to those skilled in the art, for example chromatography or
crystallisation and the individual enantiomers within each pair may be
separated as
described above. The present invention includes each diastereoisomer of
compounds of Formula I and mixtures thereof.
15
Specific compounds of Formula I are:-
3-(benzo[b]thiophen-3-yl)-5,6-dihydroimidazo[2,1-b]thiazole-2-carboxaldehyde;
[3-(benzo[b]thiophen-3-yl)-5,6-dihydroimidazo[2,1-b]thiazol-2-yl]methanol;
3-(benzo[b]thiophen-3-yl)-5,6-dihydroimidazo[2,1-b]thiazole-2-carboxaldehyde
oxime;
1-[3-(benzo[b]thiophen-3-yl)-5,6-dihydroimidazo[2,1-b]thiazol-2-yl]ethanol;
3-(benzo[b]thiophen-3-yl)-2-bromo-5,6-dihydroimidazo(2,1-b]thiazole;
3-(benzo[b]thiophen-3-yl)-2-bromo-6,7-dihydro-5H-thiazolo[3,2-a]pyrimidine;
1-[3-(benzo[b]thiophen-3-yl)-5, 6-dihydroimidazo[2,1-b]thiazol-2-yl]-1-
methylethanol;
1-[3-(benzo[b]thiophen-3-yl)-5,6-dihydroimidazo[2,1-b]thiazol-2-yl]propan-1-
ol;
2-bromo-3-(5-methoxybenzo[b]thiophen-3-yl)-5,6-dihydroimidazo[2,1-b]thiazole;
2-bromo-3-(5-chlorobenzo[b]thiophen-3-yl)-5,6-dihydroimidazo[2,1-b]thiazole;
3-(benzo[b]thiophen-3-yl)-2-ethoxymethyl-5,6-dihydroimidazo[2,1-b]thiazole;
1-[3-(benzo[b]thiophen-3-yl)-5,6-dihydroimidazo[2,1-b]thiazol-2-yl]prop-2-en-1-
ol;
1-[3-(benzo[b]thiophen-3-yl)-5, 6-dihydroimidazo[2,1-b]thiazol-2-yl]but-2-yn-1-
ol;
3-(benzo[b]thiophen-3-yl)-2-vinyl-5,6-dihydroimidazo[2,1-b]thiazole;
2-allyl-3-(benzo[b]thiophen-3-yl)-5,6-dihydroimidazo[2,1-b]thiazole;
[3-(benzo[b]furan-3-yl)-5,6-dihydroimidazo[2,1-b]thiazol-2-yl]methanol;
N-[3-(benzo[b]thiophen-3-yl)-5,6-dihydroimidazo[2,1-b]thiazol-2-ylmethylidene]-
1-
methylethylamine;
3-(benzo[b]thiophen-3-yl)-2-chloro-5,6-dihydroimidazo[2,1-b]thiazole;
2-acetyl-3-(benzo[b]thiophen-3-yl)-5,6-dihydroimidazo[2,1-b]thiazole;
3-(benzo[b]thiophen-3-yl)-2-(methoxymethyl)-5,6-dihydroimidazo[2,1-b]thiazole;
CA 02374926 2001-12-06
WO 00/71549 PCT/EP00/04279
14
3-(benzo[b]thiophen-3-yl)-2-(methylfhio)-5,6-dihydroimidazo[2,1-b]thiazole;
3-(benzo[b]thiophen-3-yl)-2-(1-methylvinyl)-5,6-dihydroimidazo[2,1-b]thiazole;
1-[3-(benzo[b]thiophen-3-yl)-5,6-dihydroimidazo[2,1-b]thiazol-2-yl]-2-
methylpropan-1-
ol ;
1-[3-(benzo[b]thiophen-3-yl)-5,6-dihydroimidazo[2,1-b)thiazol-2-yl]butan-1-of
;
1-[3-(benzo[b]thiophen-3-yl)-5,6-dihydroimidazo[2,1-b]thiazol-2-yl]-2-
methylbut-3-en-
1-0l ;
1-[3-(benzo[b]thiophen-3-yl)-5,6-dihydroimidazo[2,1-b]thiazol-2-yl]-3-
methylbutan-1-
ol;
1-[3-(benzo[b]thiophen-3-yl)-5,6-dihydroimidazo[2,1-b)thiazol-2-yl]pentan-1-
ol;
1-[3-(benzo[b]thiophen-3-yl)-5,6-dihydroimidazo[2,1-b]thiazol-2-yl]prop-2-yn-1-
of ;
1- [3-(benzo[b]thiophen-3-yl)-5,6-dihydroimidazo[2,1-b]thiazol-2-yl]but-3-en-1-
of ;
1-[3-(benzo[b)thiophen-3-yl)-5,6-dihydroim idazo[2,1-b]thiazol-2-yl]-2-
methylprop-2-
en-1-ol;
1-[3-(benzo[b]thiophen-3-yl)-5, 6-dihydroimidazo[2,1-b]thiazol-2-yl]pent-4-en-
1-ol;
[3-(benzo[b]thiophen-3-yl)-5,6-dihydroimidazo[2,1-b]thiazol-2-yl](2-
methoxyphenyl)methanol;
3-(benzo[b]thiophen-3-yl)-2-prop-1-enyl-5,6-dihydroimidazo[2,1-b]thiazole;
[3-(benzo[b]thiophen-3-yl)-6,7-dihydro-5H-thiazolo[3,2-a]pyrimidin-2-
yl]methanol;
3-(benzo[b]thiophen-3-yl)-5,6-dihydroimidazo[2,1-b]thiazole-2-carbonitrile;
40
3-(benzo[b]thiophen-3-yl)-2-styryl-5,6-dihydroimidazo[2,1-b]thiazole;
3-(5-chlorobenzo[b]thiophen-3-yl)-5,6-dihydroimidazo[2,1-b]thiazole-2-
carboxaldehyde;
[3-(5-chlorobenzo[b]thiophen-3-yl)-5,6-dihydroimidazo[2,1-b]thiazol-2-
yl]methanol;
3-(benzo[b]thiophen-3-yl)-2-(phenylthio)-5,6-dihydroimidazo[2,1-b]thiazole;
[3-(benzo[b]thiophen-3-yl)-5,6-dihydroimidazo[2,1-b]thiazol-2-yl]-N-
methylmethylamine;
3-(benzo[b]thiophen-3-yl)-2-cyclopropyl-5,6-dihydroimidazo[2,1-b]thiazole;
3-(benzo[b]thiophen-3-yl)-2-iodo-5,6-dihydroimidazo[2,1-b]thiazo1e;
4-[3-(benzo[b]thiophen-3-yl)-5,6-dihydroimidazo[2,1-b]thiazol-2-yl]but-3-en-2-
ol;
3-(benzo[b]thiophen-3-yl)-2-(2-methylprop-2-enyl)-5,6-dihydroimidazo[2,1-
b]thiazole;
CA 02374926 2001-12-06
WO 00/71549 PCT/EP00/04279
5
ethyl [3-(benzo[b]thiophen-3-yl)-5,6-dihydroimidazo[2,1-b]thiazol-2-
yl]acetate;
2-bromo-3-[5-(methylthio)benzo[b]thiophen-3-yl]-5,6-dihydroimidazo[2,1-
b)thiazole;
f 3-[5-(methylthio)benzo[b]thiophen-3-yl]-5,6-dihydroimidazo[2,1-b]thiazol-2-
yl}methanol;
3-(benzo[b]thiophen-3-yl)-2-cyclopropylmethoxymethyl-5,6-dihydroimidazo[2,1-
10 b]thiazole;
20
3-(benzo[b]thiophen-3-yl)-2-prop-2-ynyloxymethyl-5,6-dihydroimidazo[2,1-
b]thiazole;
2-bromo-3-(7-methoxybenzo[b]thiophen-3-yl)-5,6-dihydroimidazo[2,1-b]thiazole;
3-(benzo[b]thiophen-3-yl)-2-isopropoxymethyl-5,6-dihydroimidazo[2,1-
b]thiazole; and
3-(benzo[b]thiophen-3-yl)-2-cyclobutylmethoxymethyl-5,6-dihydroimidazo[2,1-
b]thiazole
including pharmaceutically acceptable salts thereof and individual
enantiomers,
racemates or other mixtures of enantiomers.
The present invention also includes pharmaceutical compositions comprising
a therapeutically effective amount of a compound of Formula I or a salt
thereof
together with a pharmaceutically acceptable diluent or carrier.
As used hereinafter, the term "active compound" denotes a compound of
Formula I or a salt thereof. In therapeutic use, the active compound may be
administered orally, rectally, parenterally or topically, preferably orally.
Thus the
therapeutic compositions of the present invention may take the form of any of
the
known pharmaceutical compositions for oral, rectal, parenteral or topical
administration. Pharmaceutically acceptable carriers suitable for use in such
compositions are well known in the art of pharmacy. The compositions of the
invention may contain 0.1-99% by weight of active compound. The compositions
of
the invention are generally prepared in unit dosage form. Preferably the unit
dosage
of active ingredient is 1-500 mg. The excipients used in the preparation of
these
compositions are the excipients known in the pharmacist's art.
Compositions for oral administration are the preferred compositions of the
invention and these are the known pharmaceutical forms for such
administration, for
example tablets, capsules, syrups and aqueous or oil suspensions. The
excipients
CA 02374926 2001-12-06
WO 00/71549 PCT/EP00/04279
16
used in the preparation of these compositions are the excipients known in the
pharmacist's art. Tablets may be prepared by mixing the active compound with
an
inert diluent such as calcium phosphate in the presence of disintegrating
agents, for
example maize starch, and lubricating agents, for example magnesium stearate,
and
tableting the mixture by known methods. The tablets may be formulated in a
manner
known to those skilled in the art so as to give a sustained release of the
compounds
of the present invention. Such tablets may, if desired, be provided with
enteric
coatings by known methods, for example by the use of cellulose acetate
phthalate.
Similarly, capsules, for example hard or soft gelatin capsules, containing the
active
compound with or without added excipients, may be prepared by conventional
means and, if desired, provided with enteric coatings in a known manner. The
tablets and capsules may conveniently each contain 1 to 500 mg of the active
compound. Other compositions for oral administration include, for example,
aqueous suspensions containing the active compound in an aqueous medium in the
presence of a non-toxic suspending agent such as sodium
carboxymethylcellulose,
and oily suspensions containing a compound of the present invention in a
suitable
vegetable oil, for example arachis oil.
Preferably the compositions of the invention are administered orally in the
known pharmaceutical forms for such administration. Dosage forms suitable for
oral
administration may comprise tablets, pills, capsules, caplets,
multiparticulates
including: granules, beads, pellets and micro-encapsulated particles; powders,
elixirs, syrups, suspensions and solutions.
Solid oral dosage forms, for example tablets, may be prepared by mixing the
pharmaceutical composition of the present invention with one or more of the
following ingredients or mixtures thereof:
inert diluents, for example calcium carbonate, calcium sulphate, compressible
sugar,
confectioner's sugar, dextrates, dextrin, dextrose, dibasic calcium phosphate
dihydrate, glyceryl palmitostearate, hydrogenated vegetable oil, kaolin,
lactose,
magnesium carbonate, magnesium oxide, maltodextrin, mannitol, microcrystalline
cellulose, polymethacrylates, potassium chloride, powdered cellulose,
pregelatinized
starch, sodium chloride, sorbitol, starch, sucrose, sugar spheres, talc and
tribasic
calcium phosphate;
disintegrating agents, for example alginic acid, carboxymethylcellulose
calcium,
carboxymethylcellulose sodium, colloidal silicon dioxide, croscarmellose
sodium,
CA 02374926 2001-12-06
WO 00/71549 PCT/EP00/04279
17
crospovidone, guar gum, magnesium aluminium silicate, methylcellulose,
microcrystalline cellulose, polacrilin potassium, powdered cellulose,
pregelatinized
starch, sodium alginate, sodium starch glycolate, starch including maize
starch and
agar;
lubricating agents, for example calcium stearate, glyceryl monostearate,
glyceryl
palmitostearate, hydrogenated castor oil, hydrogenated vegetable oil, light
mineral
oil, magnesium stearate, mineral oil, polyethylene glycol, sodium benzoate,
sodium
lauryl sulphate, sodium stearyl fumarate, stearic acid, talc and zinc
stearate;
binders, for example acacia, alginic acid, carbomer, carboxymethylcellulose
sodium,
dextrin, ethylcellulose, gelatin, guar gum, hydrogenated vegetable oil,
hydroxyethyl
cellulose, hydroxypropyl cellulose, hydroxypropyl methylcellulose, liquid
glucose,
magnesium aluminium silicate, maltodextrin, methylcellulose,
polymethacrylates,
povidone, pregelatinized starch, sodium alginate, starch including maize
starch, zein,
sugars (such as sucrose, molasses and lactose), and natural and synthetic gums
(such as extract of Irish moss, polyethylene glycol, waxes, microcrystalline
cellulose
and polyvinylpyrrolidone);
colouring agents, for example conventional pharmaceutically acceptable dyes;
sweetening and flavouring agents;
preservatives;
one or more pharmaceutically acceptable couple or couples (such as those
comprising an acid and a carbonate or bicarbonate salt), which effervesces to
aid
dissolution when the solid dosage form is added to water; and
other optional ingredients known in the art to permit production of oral
dosage forms
by known methods such as tabletting.
Solid oral dosage forms may be formulated in a manner known to those
skilled in the art so as to give a sustained release of the active compound.
Film
coated, solid oral dosage forms comprising compositions of the present
invention
may be advantageous, depending on the nature of the active compound. Various
materials, for example shellac and/or sugar, may be present as coatings, or to
otherwise modify the physical form of the oral dosage form. For example
tablets or
pills may, if desired, be provided with enteric coatings by known methods, for
example by the use of cellulose acetate phthalate and/or hydroxy propyl
methylcellulose phthalate.
CA 02374926 2001-12-06
WO 00/71549 PCT/EP00/04279
18
Capsules and/or caplets (for example hard or soft gelatin capsules)
comprising the active compound (with or without added excipients such as a
fatty
oil), may be prepared by conventional means and, if desired, provided with
enteric
coatings in a known manner. The contents of the capsule and/or caplet may be
formulated using known methods to give sustained release of the active
compound.
Liquid oral dosage forms comprising compositions of the present invention
may be an elixir, suspension andlor syrup (for example, aqueous suspensions
containing the active compound in an aqueous medium in the presence of a non-
toxic suspending agent [such as sodium carboxymethylcellulose] and/or oily
suspensions containing the active compound in a suitable vegetable oil [such
as
arachis oil and/or sunflower oil]). Liquid oral dosage forms may also comprise
one or
more sweetening agent, flavouring agent, preservatives and/or mixtures
thereof.
The active compound may be formulated into granules with or without
additional excipients. The granules may be ingested directly by the patient or
they
may be added to a suitable liquid carrier (for example water) before
ingestion. The
granules may contain disintegrants (for example a pharmaceutically acceptable
effervescent couple formed from an acid and a carbonate or bicarbonate salt)
to
facilitate dispersion in the liquid medium.
Preferably each of the above oral dosage forms may contain from about 1 mg
to about 1000 mg, more preferably from about 5 mg to about 500 mg (for example
10 mg, 50 mg, 100 mg, 200 mg, or 400 mg) of the active compound.
Compositions of the invention suitable for rectal administration are the known
pharmaceutical forms for such administration, for example, suppositories with
hard
fat, semi-synthetic glyceride, cocoa butter and/or polyethylene glycol bases.
Pharmaceutical compositions may also be administered parenterally (for
example subcutaneously, intramuscularly, intradermally and/or intravenously
[such
as by injection and/or infusion] in the known pharmaceutical dosage forms for
parenteral administration (for example sterile suspensions in aqueous and/or
oily
media and/or sterile solutions in suitable solvents, preferably isotonic with
the blood
of the intended patient). Parenteral dosage forms may be sterilised (for
example by
micro-filtration and/or using suitable sterilising agents [such as ethylene
oxide]).
CA 02374926 2001-12-06
WO 00/71549 PCT/EP00/04279
19
Optionally one or more of the following pharmaceutically acceptable adjuvants
suitable for parenteral administration may be added to parenteral dosage
forms:
local anaesthethetics, preservatives, buffering agents and/or mixtures
thereof.
Parenteral dosage forms may be stored in suitable sterile sealed containers
(for
example ampoules and/or vials) until use. To enhance stability during storage
the
parenteral dosage form may be frozen after filling the container and fluid
(for
example water) may be removed under reduced pressure.
Pharmaceutical compositions may be administered nasally in known
pharmaceutical forms for such administration (for example sprays, aerosols,
nebulised solutions and/or powders). Metered dose systems known to those
skilled
in the art (for example aerosols and/or inhalers) may be used.
Pharmaceutical compositions may be administered to the buccal cavity (for
example sub-lingually) in known pharmaceutical forms for such administration
(for
example slow dissolving tablets, chewing gums, troches, lozenges, pastilles,
gels,
pastes, mouthwashes, rinses and/or powders).
Compositions for topical administration may comprise a matrix in which the
pharmacologically active compounds of the present invention are dispersed so
that
the compounds are held in contact with the skin in order to administer the
compounds transdermally. A suitable transdermal composition may be prepared by
mixing the pharmaceutically active compound with a topical vehicle, such as a
mineral oil, petrolatum and/or a wax, for example paraffin wax or beeswax,
together
with a potential transdermal accelerant such as dimethyl sulphoxide or
propylene
glycol. Alternatively the active compounds may be dispersed in a
pharmaceutically
acceptable cream or ointment base. The amount of active compound contained in
a
topical formulation should be such that a therapeutically effective amount of
the
compound is delivered during the period of time for which the topical
formulation is
intended to be on the skin.
The compounds of the present invention may also be administered by
continuous infusion either from an external source, for example by intravenous
infusion or from a source of the compound placed within the body. Internal
sources
include implanted reservoirs containing the compound to be infused which is
continuously released for example by osmosis and implants which may be (a)
liquid
CA 02374926 2001-12-06
WO 00/71549 PCT/EP00/04279
such as a suspension or solution in a pharmaceutically acceptable oil of the
compound to be infused for example in the form of a very sparingly water-
soluble
derivative such as a dodecanoate salt or (b) solid in the form of an implanted
support, for example of a synthetic resin or waxy material, for the compound
to be
5 infused. The support may be a single body containing all the compound or a
series
of several bodies each containing part of the compound to be delivered. The
amount
of active compound present in an internal source should be such that a
therapeutically effective amount of the compound is delivered over a long
period of
time.
In some formulations it may be beneficial to use the compounds of the
present invention in the form of particles of very small size, for example as
obtained
by fluid energy milling.
In the compositions of the present invention the active compound may, if
desired, be associated with other compatible pharmacologically active
ingredients.
The present invention also comprises a compound of Formula I for use as a
medicament.
The pharmaceutical compositions containing a therapeutically effective
amount of a compound of Formula I may be used to treat depression, anxiety,
psychoses (for example schizophrenia), tardive dyskinesia, obesity, drug
addiction,
drug abuse, cognitive disorders, Alzheimer's disease, obsessive-compulsive
behaviour, panic attacks, social phobias, eating disorders such as bulimia,
anorexia,
snacking and binge eating, non-insulin dependent diabetes mellitus,
hyperglycaemia,
hyperlipidaemia, stress in mammals particularly humans, and as an aid to
smoking
cessation in human beings. In addition such compositions may be used in the
treatment andlor prophylaxis of seizures, neurological disorders such as
epilepsy
and/or conditions in which there is neurological damage such as stroke, brain
trauma, cerebral ischaemia, head injuries and haemorrhage. Whilst the precise
amount of active compound administered in such treatment will depend on a
number
of factors, for example the age of the patient, the severity of the condition
and the
past medical history, and always lies within the sound discretion of the
administering
physician, the amount of active compound administered per day is in the range
1 to
CA 02374926 2001-12-06
WO 00/71549 PCT/EP00/04279
21
1000 mg preferably 5 to 500 mg given in single or divided doses at one or more
times during the day.
In yet another aspect, the present invention provides the use of a compound
of Formula I in the manufacture of a medicament for use in the treatment of
depression, anxiety, psychoses (for example schizophrenia), tardive
dyskinesia,
obesity, drug addiction, drug abuse, cognitive disorders, Alzheimer's disease,
obsessive-compulsive behaviour, panic attacks, social phobias, eating
disorders
such as bulimia, anorexia, snacking and binge eating, non-insulin dependent
diabetes mellitus, hyperglycaemia, hyperlipidaemia, stress, as an aid to
smoking
cessation and in the treatment and/or prophylaxis of seizures, neurological
disorders
such as epilepsy and/or conditions in which there is neurological damage such
as
stroke, brain trauma, cerebral ischaemia, head injuries and haemorrhage.
The present invention also provides a method of treating depression, anxiety,
psychoses (for example schizophrenia), tardive dyskinesia, obesity, drug
addiction,
drug abuse, cognitive disorders, Alzheimer's disease, obsessive-compulsive
behaviour, panic attacks, social phobias, eating disorders such as bulimia,
anorexia,
snacking and binge eating, non-insulin dependent diabetes mellitus,
hyperglycaemia,
hyperlipidaemia, stress and seizures, neurological disorders such as epilepsy
and/or
conditions in which there is neurological damage such as stroke, brain trauma,
cerebral ischaemia, head injuries and haemorrhage which comprises the
administration of a therapeutically effective amount of a compound of Formula
I to a
patient in need thereof.
The present invention also provides a method of reducing the craving to
smoke in human beings which comprises the administration of a therapeutically
effective amount of a compound of Formula I to a patient in need thereof. The
present invention also provides a method of reducing weight gain after smoking
cessation in human beings which comprises the administration of a
therapeutically
effective amount of a compound of Formula I to a patient in need thereof.
In addition the compounds of the present invention may be useful in the
treatment or prevention of metabolic diseases and conditions arising
therefrom, for
example non exercise activity thermogenesis and increased metabolic rate,
sexual
dysfunction, sleep apnoea, premenstrual syndrome, urinary incontinence,
CA 02374926 2001-12-06
WO 00/71549 PCT/EP00/04279
22
hyperactivity disorders, hiatial hernia and reflux esophagitis, pain,
especially
neuropathic pain, weight gain associated with drug treatment, chronic fatigue
syndrome, osteoarthritis and gout, cancers associated with weight gain,
menstrual
dysfunction, gallstones, orthostatic hypotension and pulmonary hypertension.
The compounds of the present invention may be useful in preventing cardio-
vascular disease, and in reducing platelet adhesiveness, in aiding weight loss
after
pregnancy and in aiding weight loss after smoking cessation.
The compounds of the present invention are particularly useful in treating
obesity and related co-morbid conditions, for example, diabetes,
hyperglycaemia and
hyperlipidaemia. It is known that monoamine reuptake inhibitors which are used
to
treat obesity are often associated with cardiovascular side effects, for
example,
increased heart rate and increased blood pressure. The compounds of the
present
invention reduce the cardiovascular side effects which might be expected to
occur
from the administration of a monoamine reuptake inhibitor particularly a
noradrenaline reuptake inhibitor. Whilst not wishing to be bound by theory it
is likely
that the combination of 5-HT~A agonism in the compounds of the present
invention
reduces the cardiovascular side effects which might have arisen from their
monoamine reuptake inhibition particularly their noradrenaline reuptake
inhibition.
In another aspect the present invention provides a method of reducing the
cardiovascular side effects of an anti-obesity drug comprising incorporating
into the
compound 5-HT~A agonism.
In another aspect the present invention provides the use of a compound
which is a 5-HT~A agonist and which is a monoamine reuptake inhibitor
particularly a
noradrenaline reuptake inhibitor in the treatment of obesity and related co-
morbid
conditions without causing cardiovascular side effects.
The beneficial properties of especially preferred compounds of the present
invention in reducing cardiovascular side-effects may be demonstrated in rat
telemetry studies in which heart rate, blood pressure, body temperature and
locomotor activity are recorded continuously over time. Suitable methods are
described in:
CA 02374926 2001-12-06
WO 00/71549 PCT/EP00/04279
23
Brockway, BP, Mills, PA & Azar, SH (1991 ) A new method for continuous chronic
measurement of blood pressure, heart rate and activity in the rat via radio-
telemetry.
Clinical and Experimental Hypertension - Theory and Practice A13(5), 885-895
and
Guiol, C, Ledoussal, C & Surge, J-M (1992) A radiotelemetry system for chronic
measurement of blood pressure and heart rate in the unrestrained rat.
Validation of
method. Journal of Pharmacological and Toxicological Methods 28, 99-105.
The 5-HT~A agonism of especially preferred compounds of the present
invention may be determined by electrophysiology by methods known to those
skilled in the art.
Processes for the preparation of compounds of Formula I will now be
described. The processes may be performed on an individual basis, or by
multiple
parallel synthesis, also known as High Speed Analoguing. The processes are
preferably carried out at atmospheric pressure.
Compounds of Formula I may be prepared by methods disclosed in WO
97/02269. Additionally compounds of Formula I may be prepared by methods
described below.
Compounds of Formula I may be prepared by dehydrating a compound of
Formula II
R4 S
N
H JJO
N I I
(R~)9 \ ~ ~ ~(CRzR3)~
_A_ ~Rs
in which A, R~, R2, R3, R4, R5, g and n are as hereinbefore defined,
optionally in the
presence of an acid, for example acetic or sulphuric acid, at a temperature in
the
range 0-200°C; preferably in the range 20-150°C.
Compounds of Formula II may be prepared by reacting a compound of
Formula III
CA 02374926 2001-12-06
WO 00/71549 PCT/EP00/04279
24
H
N~
(CR2R3)~ III
N -~
i
H
in which R2,R3 and n are as hereinbefore defined, with a compound of Formula
IV
CO.CHR4.Z
(R')9 \ I _ IV
A Rs
in which Z is a leaving group, for example a halo such as bromo, and A, R~,
R4, R5
and g are as hereinbefore defined, at a temperature in the range 0-
200°C, in the
presence of a solvent, for example ethanol and optionally in the presence of
an acid,
for example acetic acid; preferably by heating at a temperature in the range
20°C to
the boiling point of the solvent used.
Compounds of Formula I may also be prepared directly by reacting a
compound of Formula III with a compound of Formula IV at a temperature in the
range of 0-200°C, optionally in the presence of an acid, for example
acetic acid, and
optionally in the presence of a solvent, for example ethanol, without
isolation of the
intermediate of Formula II; preferably by heating at a temperature in the
range
20-150°C.
Compounds of Formula I in which RQ represents halo may be prepared by
reacting a compound of Formula V
S
I ~ N
/ ~(CRZR3)~
(R~)9 \
A Rs
in which A, R~, Rz, R3, R5, n and g are as hereinbefore defined, with a
halogenating
agent for example bromine, phenyltrimethylammonium tribromide, iodine
rnonochloride or benzyltrimethylammonium tetrachloroiodate at a temperature in
the
CA 02374926 2001-12-06
WO 00/71549 PCT/EP00/04279
range -50-200°C optionally in the presence of a solvent, for example
dichloromethane, tetrahydrofuran or acetone.
Compounds of Formula I in which R4 represents a group of Formula
5 -CH(OH)RX in which RX is a C,_5 alkyl group, an alkenyl group containing 2-5
carbon
atoms, an alkynyl group containing 2-5 carbon atoms or (2-C~_3alkoxyphenyl)
may
be prepared by reacting a compound of Formula VI
O
S
r
R I ~ N~ VI
19
A ~ Rs
10 in which A, R1, R2, R3, R5, n and g are as hereinbefore defined and Ry is H
with an
organometallic reagent, for example a compound of formula RXMgX or RXLi in
which
Rx is as hereinbefore defined and X is halo, for example bromo, in the
presence of a
solvent, for example tetrahydrofuran or ether, at a temperature in the range
of -50°C
to the boiling point of the solvent used.
Compounds of Formula I in which R4 represents a group of Formula
-CH(OH)RY in which Ry is a C,_5 alkyl group, an alkenyl group containing 2-5
carbon
atoms, an alkynyl group containing 2-5 carbon atoms or (2-C~_3alkoxyphenyl)
may be
prepared by reacting a compound of Formula VI in which A, R~, Rz, R3, F~, g
and n
are as hereinbefore defined and Ry is a C,_5 alkyl group, an alkenyl group
containing
2-5 carbon atoms, an alkynyl group containing 2-5 carbon atoms or (2-Ci_
3alkoxyphenyl) with a reducing agent, for example sodium borohydride, in the
presence of a solvent, for example ethanol, at a temperature in the range of
0°C to
the boiling point of the solvent used.
Compounds of Formula I in which R4 is hydroxymethyl may be prepared by
reacting a compound of Formula VI in which A, R~, F~, R3, R4, R5, g and are as
hereinbefore defined and Ry is H with a reducing agent, for example sodium
borohydride, in a solvent, for example methanol, at a temperature in the range
of
-50°C to the boiling point of the solvent°used.
CA 02374926 2001-12-06
WO 00/71549 PCT/EP00/04279
26
Compounds of Formula I in which R4 is hydroxyiminomethyl may be prepared
by reacting a compound of Formula VI in which A, R~, RZ, R3, R5, g and n are
as
hereinbefore defined and Ry is H with hydroxylamine or a salt thereof
optionally in
the presence of a solvent, for example an alcohol, eg ethanol, at a
temperature in
the range of 0-250°C.
Compounds of Formula I in which R4 is cyano may be prepared by reacting a
compound of Formula VI in which A, R~, RZ, R3, R5, n and g are as hereinbefore
defined and Ry is H with hydroxylamine or a salt thereof in the presence of
formic
acid at a temperature in the range of 0-250°C.
Compounds of Formula I in which R4 represents a C»alkyliminomethylene
group may be prepared by reacting a compound of Formula VI in which A, R~, R2,
R3, R5, n and g are as hereinbefore defined and Ry represents H with an amine
of
Formula RaNHz wherein Ra represents a C~.~ alkyl group optionally in the
presence
of a solvent, for example ethanol, optionally in the presence of an acid
catalyst, for
example acetic acid, at a temperature in the range 0-250°C.
Compounds of Formula I in which R4 represents a C~.~alkylaminomethylene
group may be prepared by reacting a compound of Formula I in which R4
represents
a C»alkyliminomethylene group, and A, R~, Rz, R3, R5, n and g are as
hereinbefore
defined, with a reducing agent, for example sodium borohydride, in the
presence of a
solvent, for example an alcohol e.g. ethanol, at a temperature in the range
0°C to the
boiling point of the solvent used.
Compounds of Formula I in which R4 represents a C»alkylaminomethylene
group may be prepared directly from a compound of Formula VI in which A, Ri,
Rz,
R3, R5 , n and g are as hereinbefore defined and Ry represents hydrogen by
reaction
with an amine of formula RaNH2 wherein Ra represents a C~~ alkyl group and a
reducing agent, for example sodium triacetoxyborohydride, in the presence of a
solvent, for example tetrahydrofuran, at a temperature in the range 0°C
to the boiling
point of the solvent used.
Compounds of Formula I in which R4 represents a group of formula
-C(OH)RxRY in which RX and Ry are each independently a C,.S alkyl group may be
CA 02374926 2001-12-06
WO 00/71549 PCT/EP00/04279
27
prepared by reacting a compound of Formula VI in which RY is a C,_5 alkyl
group, and
A, R~, R2, R3, R5, n and g are as previously defined, with an organometallic
reagent,
for example a compound of formula RxMgX or RXLi in which RX is as hereinbefore
defined and X is halo, for example bromo, in the presence of a solvent, for
example
tetrahydrofuran or ether, at a temperature in the range of -50°C to the
boiling point of
the solvent used.
Compounds of Formula I in which R4 represents a group of formula
-C(OH)RXRy in which RX and Ry are the same C,_z alkyl group may be prepared by
reacting a compound of Formula VI, as hereinbefore defined except that Ry is
ORZ in
which RZ is a C,_salkyl group, with an organometallic reagent, for example a
compound of formula RXMgX or RXLi in which RX is as hereinbefore defined and X
is
halo, for example bromo, in the presence of a solvent, for example
tetrahydrofuran or
ether, at a temperature in the range of -50°C to the boiling point of
the solvent used.
Compounds of Formula I in which R4 represents a C2_6 alkenyl group in which
the double bond is attached to the carbon alpha to the thiazole ring or a
styryl group
may be prepared by reacting compounds of Formula VI, in which RY represents
hydrogen or a C» alkyl group, and A, R~, R2, R3, R5, n and g are as previously
defined, with a phosphonium salt of formula RZPh3P+Br- in which RZ represents
a
C~_5 alkyl group or a benzyl group in the presence of a base for example n-
butyllithium, in a solvent for example, an ether, e.g. tetrahydrofuran, at a
temperature
in the range -78°C to the boiling point of the solvent used.
Compounds of Formula I in which R4 represents a C2_6 alkanoyl group may
be prepared by reacting a compound of Formula I in which R4 represents halo,
for
example bromo or chloro, and A, R~, R2, R3, R5, n and g are as previously
defined,
or a compound of Formula V with a compound of formula RbMgX or RbLi in which
Rb
is a C~_6 alkyl group and X is halo, for example bromo or chloro, in the
presence of a
solvent for example an ether, eg diethyl ether or tetrahydrofuran, at a
temperature in
the range -78°C to the boiling point of the solvent used, and then
reacting the
product obtained with an acylating agent for example a compound of Formula
R~CON(CH3)OCH3 in which R~ represents a C~_5 alkyl group in a solvent, for
example an ether e.g. tetrahydrofuran, at a temperature in the range
0°C to the
CA 02374926 2001-12-06
WO 00/71549 PCT/EP00/04279
28
boiling point of the solvent used. Compounds of Formula VI may be prepared in
a
similar manner.
Compounds of Formula I in which R4 represents a C~_3 alkoxyC~_3 alkyl group
may be prepared by reacting a compound of Formula I in which R4 represents a
hydroxyC~_3 alkyl group, and A, R~, R2, R3, R5, n and g are as previously
defined,
with a C~_3 alkylating agent, for example a C~_3 alkyl halide e.g. a C~_3
alkyl iodide in
the presence of a base, for example sodium hydride, in a solvent, for
exampIeN,N-
dimethylformamide, at a temperature in the range of -50 to 150°C.
Compounds of Formula I in which R4 represents a C4_
,cycloalkylalkoxyC,_3alkyl group may be prepared by reacting a compound of
Formula I in which R4 represents a hydroxyC~_3 alkyl group, and A, R~, R2, R3,
R5, n
and g are as previously defined, with a C4_~cycloalkylalkylating agent, for
example a
C4_~ cycloalkylalkyl halide e.g. a C4_~ cycloalkylalkyl iodide in the presence
of a base,
for example sodium hydride, in a solvent, for example N,N-dimethylformamide,
at a
temperature in the range of -50 to 150°C.
Compounds of Formula I in which R4 represents a C3_~alkynylalkoxyC,_3alkyl
group may be prepared by reacting a compound of Formula I in which R4
represents
a hydroxyC~_3 alkyl group, and A, R~, R2, R3, R5, n and g are as previously
defined,
with a C3_~ alkynylalkylating agent, for example a C3_, alkynylalkyl halide
e.g. a C3_~
alkynylalkyl iodide in the presence of a base, for example sodium hydride, in
a
solvent, for example N,N-dimethylformamide, at a temperature in the range of -
50 to
150°C.
Compounds of Formula I in which R4 represents a C~_3 alkylthioC~_3 alkyl
group may be prepared by reacting a compound of Formula I in which R4
represents
a mercaptoC~_3 alkyl group, and A, R~, R2, R3, R5, n and g are as previously
defined,
with a Ci_3 alkylating agent, for example a C~_3 alkyl halide e.g. a C~_3
alkyl iodide in
the presence of a base, for example sodium hydride or sodium hydroxide, in a
solvent, for example N,N-dimethylformamide, at a temperature in the range of -
50 to
150°C.
CA 02374926 2001-12-06
WO 00/71549 PCT/EP00/04279
29
Compounds of Formula I in which R4 represents a C~_3 alkylthio group or an
arylthio group and A, R~, R2, R3, R5, n and g are as previously defined may be
prepared by reacting a compound of Formula I in which R4 represents halo or a
compound of Formula V with a metallating agent, for example a compound of
formula RMgX or RLi in which R is a C~_6 alkyl group and X is halo, for
example
chloro, bromo or iodo, in a solvent, for example an ether or a mixture of
ethers, eg
tetrahydrofuran, or diethyl ether, at a temperature in the range of -
100°C to the
boiling point of the solvent used to give an intermediate complex, which is
reacted
with a disulphide of formula RdS-SRd in which Rd is a C~_3 alkyl group or an
aryl
group, at a temperature in the range of -100°C to the boiling point of
the solvent
used.
Compounds of Formula I in which R4 represents a C~_3 alkoxy group and A,
R~, R2, R3, R5, n and g are as previously defined may be prepared by reacting
a
compound of Formula I in which R4 represents halo, for example bromo or iodo,
with
an C~_3 alkoxide salt, for example a sodium or potassium salt, optionally in
the
presence of a solvent, for example a C~_3 alcohol or dimethylformamide,
optionally in
the presence of a catalyst, for example a copper (I) salt, at a temperature in
the
range of 0-350°C.
Compounds of Formula I in which R4 represents a C3_6 alkenyl group in which
the double bond is not attached to the carbon alpha to the thiazole ring may
be
prepared by reacting a compound of Formula I in which R4 represents halo, for
example bromo or chloro, and A, R~, R2, R3, R5, n and g are as previously
defined,
or a compound of Formula V with a compound of formula RbMgX or RbLi in which
Rb
is a C~_6 alkyl group and X is halo, for example bromo or chloro, in the
presence of a
solvent for example an ether, eg diethyl ether or tetrahydrofuran, at a
temperature in
the range -78°C to the boiling point of the solvent used, and then
reacting the
product obtained with an alkenylating agent, for example a C3_salkenylmethyl
halide
e.g. a C3_salkenylmethyl iodide, in a solvent, for example an ether e.g.
tetrahydrofuran, at a temperature in the range 0°C to the boiling point
of the solvent
used.
CA 02374926 2001-12-06
WO 00/71549 PCT/EP00/04279
Compounds of Formula I in which R4 is a C3_salkoxycarbonylalkyl group may
be prepared by reacting compounds of Formula VI, in which Ry represents
hydrogen
and A, R~, R2, R3, R5, n and g are as previously defined, with a phosphonate
of
formula Me2NCH[PO(ORZ)2]2 in which RZ represents a C~.~ alkyl group in the
5 presence of a base, for example sodium hydride, in a solvent for example an
ether,
e.g. 1,4-dioxane, at a temperature in the range -78°C to the boiling
point of the
solvent used, then subjecting the resulting intermediate product to partial
hydrolysis
in the presence of an acid, for example hydrochloric acid.
10 Compounds of Formula I in which R4 is a C4_shydroxyalkenyl group in which
the double bond is attached to the carbon alpha to the thiazole ring may be
prepared
by reacting compounds of Formula VI, in which Ry represents hydrogen and A,
R~,
R2, R3, R5, n and g are as previously defined, with a compound of Formula
(RZO)ZPOCH2COR~ in which RZ represents a Ci_2 alkyl group and R~ represents a
15 C~_3 alkyl group in the presence of a base, for example sodium hydride, in
a solvent
for example an ether, e.g. tetrahydrofuran, at a temperature in the range -
78°C to
the boiling point of the solvent used, then subjecting the resulting
intermediate
product to reaction with a reducing agent, for example sodium borohydride, in
a
solvent, for example ethanol, at a temperature in the range -20°C to
the boiling point
20 of the solvent used.
Compounds of Formula III are commercially available or may be prepared by
methods known to those skilled in the art.Compounds of Formula IV may be
prepared by methods known to those skilled in the art as specified in the
individual
25 Examples described herein.
The ability of compounds of Formula I to interact with 5-hydroxytryptamine
(5-HT) receptors has been demonstrated for the products of Examples 1 to 56 by
the
following test which determines the ability of the compounds to inhibit
tritiated ligand
30 binding to 5-HT receptors in vitro and in particular to 5-HT1A receptors.
Hippocampal tissue from the brains of male Sprague-Dawley rats (Charles
River; weight range 150-250 g) was homogenised in ice-cold 50 mM Tris-HCI
buffer
(pH 7.7 when measured at 25°C, 1:40 w/v) and centrifuged at 40,000 g at
4°C for 10
minutes. The pellet was rehomogenised in the same buffer, incubated at
37°C for 10
CA 02374926 2001-12-06
WO 00/71549 PCT/EP00/04279
31
minutes and centrifuged at 40,000 g at 4°C for 10 minutes. The final
pellet was
resuspended in 50 mM Tris-HCI buffer (pH 7.7) containing 4 mM CaCl2, 0.1
L-ascorbic acid and 10 uM pargyline hydrochloride (equivalent to 6.25 mg wet
weight
of tissue/ml) and used immediately in the binding assay.
Membranes (400 ~I; equivalent to 2.5 mg wet weight of tissue/tube) were
incubated with 50 ~I of [3H]8-hydroxy-2-(dipropylamino)tetralin ([3H]8-OH-
DPAT) at a
single concentration of 1 nM and 50 ~I of distilled water (total binding) or
50 p.1 of test
compound (at a single concentration of 10-6 M or at 10 concentrations ranging
from
10-"-10-3 M) or 50 p.1 of 5-HT (10 ~.M, non-specific binding) at 25°C
for 30 minutes.
The incubation was terminated by rapid filtration under vacuum through Skatron
11734 filters using a Skatron Cell Harvester. Filters were washed with ice-
cold 50
mM Tris-HCI buffer, pH 7.7 (at 25°C, wash setting 9,9,0). The scored
filter paper
discs were punched out into vials, scintillation fluid added and radioactivity
determined by liquid scintillation counting.
The ability of compounds of Formula I to interact with 5-hydroxytryptamine
(5-HT) reuptake sites has been demonstrated for the products of Examples 1 to
56
by the following test which determines the ability of compounds to displace
the
standard ligand, [3H]citalopram, from 5-HT reuptake sites in vitro.
Frontal cortical tissue from the brains of male Charles River rats weighing
150-250 g was homogenised in ice-cold 50 mM Tris-HCI, pH 7.4 (when measured at
25°C) containing 120 mM sodium chloride and 5 mM potassium chloride
(Tris buffer;
1:30 w/v) and centrifuged at 40,000 g for 10 minutes. The supernatant was
discarded and the pellet rehomogenised in Tris buffer, 1:60 w/v, and
centrifuged at
40,000 g for 10 minutes. This step was repeated a further time. The final
pellet was
resuspended in 50 mM Tris-HCI, pH 7.4 containing 120 mM sodium chloride and 5
mM potassium chloride (equivalent to 3.125 mg wet weight of tissue/ml) and
used
immediately in the binding assay. All centrifugations were performed at
4°C.
Membranes (400 ~I; equivalent to 1.25 mg wet weight of tissue/tube) were
incubated with 50 ~I [3H]citalopram at a single concentration of 1.3 nM and 50
~I of
distilled water (total binding) or 50 ~I of test compound (at a single
concentration of
10-6 M or at 10 concentrations ranging from 10-"-10-3 M) or 50 ~.I of
paroxetine
CA 02374926 2001-12-06
WO 00/71549 PCT/EP00/04279
32
(0.5 p.M; non-specific binding) for 1 h at 27°C. Membrane bound
radioactivity was
recovered by filtration under vacuum through Skatron 11734 filters presoaked
in
0.5% PEI using a Skatron Cell Harvester. Filters were then washed in ice-cold
50 mM Tris-HCI buffer, pH 7.4 (at 25°C, wash setting 9,9,0). The scored
filter paper
discs were punched out into vials, scintillation fluid added and radioactivity
determined by liquid scintillation counting.
The ability of compounds of Formula I to interact with noradrenaline (NA)
reuptake sites has been demonstrated for the products of Examples 1 to 33 by
the
following test which determines the ability of compounds to displace the
standard
ligand, [3H]nisoxetine, from noradrenaline reuptake sites in vitro.
Frontal cortical tissue from the brains of male Charles River rats weighing
150-250 g was homogenised in ice-cold 50 mM Tris-HCI, pH 7.4 (at 25°C)
containing
120 mM sodium chloride and 5 mM potassium chloride (Tris buffer; 1:60 w/v)
using a
Kinematic polytron (speed setting 6 for 10 seconds) and centrifuged at 40,000
g for
10 minutes. The supernatant was discarded and the pellet rehomogenised in Tris
buffer, 1:60 w/v, and centrifuged at 40,000 g for 10 minutes. This step was
repeated
twice more so that, in total, the brain tissue was homogenised and centrifuged
four
times. The final pellet was resuspended in 50 mM Tris-HCI, pH 7.4 containing
300 mM sodium chloride and 5 mM potassium chloride (equivalent to 18.75 mg wet
weight of tissue/ml) and used immediately in the binding assay. All
centrifugations
were performed at 4°C.
Membranes (400 ~I; equivalent to 7.5 mg wet weight of tissue/tube) were
incubated with 50 ~I [3H)nisoxetine at a single concentration of 0.6 nM and 50
p1 of
distilled water (total binding) or 50 w1 of test compound (at a single
concentration
10'6 M or at 10 concentrations ranging from 10-"-10-3 M) or 50 ~.I of mazindol
(1 wM;
non-specific binding) for 4 h at 4°C. Membrane bound radioactivity was
recovered
by filtration under vacuum through Skatron 11734 filters using a Skatron cell
harvester. Filters were rapidly washed with ice-cold 50 mM Tris-HCI, pH 7.4
containing 120 mM sodium chloride and 5 mM potassium chloride (wash setting
9,9,0). The scored filter paper discs were punched out into vials,
scintillation fluid
added and radioactivity determined by liquid scintillation counting.
CA 02374926 2001-12-06
WO 00/71549 PCT/EP00/04279
33
The ability of compounds of Formula I to interact with muscarinic receptors
has been demonstrated for the products of Examples 1-56 by the following test
which determines the ability of compounds to displace the standard ligand,
[3H]N-methylscopolamine, from muscarinic receptors in vitro.
Frontal cortical tissue from the brains of male Charles River rats weighing
150-250 g was homogenised in ice-cold 20 mM HEPES buffer, pH 7.5 (measured at
25°C) containing 100 mM sodium chloride and 10 mM magnesium chloride
(1:10
w/v) using a Polytron PT3100 (speed setting 21,700rpm, 3 x 5 seconds) and
centrifuged at 49,500 g for 30 minutes at 4°C. The supernatant was
discarded and
the pellet rehomogenised in 20 mM HEPES buffer, pH 7.5 containing 100 mM
sodium chloride and 10 mM magnesium chloride (equivalent to 12.5 mg wet weight
of tissue/ml). Membranes were stored at -80°C until required.
Membranes were thawed, diluted 1:10 in ice-cold 20 mM HEPES buffer, pH
7.5 containing 100 mM sodium chloride and 10 mM magnesium chloride and
homogenised using a Polytron PT3100 as above. Diluted membranes (200 NI;
equivalent to 0.25 mg wet weight of tissue/tube) were incubated with 200 NI of
mM HEPES buffer, pH 7.5 containing 100 mM sodium chloride and 10 mM
20 magnesium chloride and 50 NI of [3H]N-methylscopolamine at a single
concentration
of 0.15 nM and 50 NI of distilled water (total binding) or 50 NI of test
compound (at a
single concentration of 10-6 M or at 10 concentrations ranging from 10-" - 10-
3 M) or
50 NI of atropine sulphate (1 NM; non-specific binding) for 30 min at
30°C.
Membrane bound radioactivity was recovered by filtration under vacuum through
Skatron 11734 filters using a Skatron cell harvester. Filters were rapidly
washed
with ice-cold 20 mM HEPES buffer, pH 7.5 (wash 1,2 at setting 5,5). The scored
fitter paper discs were punched out into vials, scintillation fluid added and
radioactivity determined by liquid scintillation counting.
For each of these tests measuring the ability of compounds of Formula I to
displace standard ligands from 5-HT,A receptors and 5-hydroxytryptamine (5-HT)
and noradrenaline (NA) reuptake sites and muscarinic receptors in vitro, the
percentage displacement of specific binding of tritiated ligand by 106 M test
compound was calculated in the following way.
CA 02374926 2001-12-06
WO 00/71549 PCT/EP00/04279
34
Firstly, specific binding of tritiated ligand in the absence (A) and presence
(B)
of test compound was determined:
In the absence of compound:
A (dpm) = Total binding (dpm) - Non-specific binding (dpm)
In the presence of compound (10~ M):
B (dpm) = Binding at 10-6M (dpm) - Non-specific binding (dpm)
The specific binding of tritiated ligand in the presence (B) of compound was
then converted to a percentage of specific binding of tritiated ligand in the
absence
(A) of compound:
Specific binding at 10-6 M = B (dpm) / A (dpm) x 100
The percentage displacement of specific binding of tritiated ligand by the
test
compound (10'6 M) was then obtained by subtraction of the percentage specific
binding in the presence of compound from the percentage specific binding in
the
absence of compound, which is taken as the maximum binding and so equals 100%:
Displacement at 10-6 M = 100 - % Specific binding at 10-6 M.
In some cases, displacement curves were then produced for compounds which
displaced >_50% of specific binding of the tritiated ligand at 10-6 M using a
range of
concentrations of the compound. The Ki was then calculated by fitting the
following
simultaneous equations (which are derived from the Feldman equations) by
robust
non-linear regression to data from three experiments simultaneously:
CA 02374926 2001-12-06
WO 00/71549 PCT/EP00/04279
F~ = LL~r'» - B
F
K; ' = K; 1 + K
a
ab = Ck r, - L + K;'
2
FZ =-ab+ abz +K;'L
Nk F'~ Non - specific data
B = C K r, F, + Nk F, Otherwise
K~,+F,+K''FZ
K;
where B is the concentration of bound ligand-receptor complex. This is
calculated
for each observation as:
DPM
5 B=
Specific activity x Volume of incubation
L is the concentration of compound
[L],°, is the concentration of the tritiated ligand used, calculated
as:
_ Mean DPM for Total DPM added samples x Dilution
LL~r°' Specific activity x Volume of incubation
Kd is the equilibrium dissociation constant for the ligand.
10 F, and F2 are the concentrations of free ligand and free compound
respectively.
r, is the total concentration of the receptor in the first experiment. This
must be
multiplied by CK for subsequent experiments (C~=1 ).
NK is the non-specific binding constant.
The results obtained in the above tests for 5-HT1A binding and 5-HT and NA
uptake, and muscarinic binding for the final products of Examples 1 - 56
hereinafter
are given in Table 1 below. K;s are in nM and are means of three independent
determinations. % Figures are for % displacement at 10-6 M for a single
determination.
CA 02374926 2001-12-06
WO 00/71549 PCT/EP00/04279
36
TABLE 1
Example 5_HT1A 5-HT NA uptake Muscarinic
No. uptake
1 51 % 1 % 58% O%
2 13 398 3.7 19%
3 201 200 12 28%
4 26 72% 6.2 0%
11 162 3.1 16%
6 7.7 378 3.5 9%
7 18 492 8.9 338
8 79% 48% 85% 87%
9 114 119 17 17%
73% 19% 94% 16%
11 97% 8% 61 % 39%
12 93% 18% 87% 54%
13 101 89 2.0 15
14 82% 39% 3.1 % 16%
60 269 8.1 4%
16 27 510 3.8 418
17 16 162 2.7 10.4
18 2.0 14% 46 11%
19 59% 8% 57% 19%
5.0 623 9.3 420
21 62% 13% 93% 22%
22 93 283 2.2 49
23 ~ 27 ~ 71 ~ 1.1 ~ 63
CA 02374926 2001-12-06
WO 00/71549 PCT/EP00/04279
37
Example 5-HT1A 5-HT NA uptake Muscarinic
No. uptake
24 98 245 5.6 353
25 74% 34% 99% 51
26 61 % 57% 99% 33%
27 64% 43% 99% 49%
28 73% 48% 97% 48%
29 55% 42% 99% 35%
30 58% 42% 99% -5%
31 53% 62% 101 % 28%
32 69% 54% 100% 36%
33 55% 48% 101 % 28%
34 76 234 61 23%
35 36.5 347 2.2 245
36 62% 68% 104% 95%
37 189 30% 146 20%
38 50% 5% 84% 16%
39 73% 64% 64% 64%
40 75% 62% 44% 16%
41 2.9 110 7.5 28%
42 100% 65% 101 % 31
43 57% 60% 102% 99%
44 53% 4% 86% 32%
45 8.8 28 1.6 7.9
46 94% 92% 100% 88%
47 53% 65% 97% 39%
48 23 24.3 4.1 3.9
CA 02374926 2001-12-06
WO 00/71549 PCT/EP00/04279
38
Example 5-HT1A 5-HT NA uptake Muscarinic
No. uptake
49 77% 61 % 102% 73%
50 98% 46% 82% 81
51 2.1 251 17 39%
52 66% 86% 103% 97%
53 64% 71 % 101 % 100%
54 54% 97% 101 % 2%
55 60% 85% 101 % NT
56 61% 81% 101% 96%
Ki values are n=1, mean of n=2 or mean of n=3.
NT = Not tested.
The ability of compounds of the invention to inhibit monoamine oxidase A
activity is demonstrable by the following test.
The assay was performed using the following general procedure in which the
tissue
source was human placenta:
Enzyme Substrate IncubationReaction Method of detection
product
MAO-A(h) Kynuramine30min/30C 4-OHquinolineSpectrophotometry
(0.15mM)
The compounds were tested at 1 and 10micromolar in duplicate.
Ref: Weyler, W. and Salach, J.I. (1985) Purification and properties of
mitochondrial
monoamine oxidase type A from human placenta. J. Biol. Chem., 260: 13199-
13207.
The combination of inhibition of monoamine oxidase activity and 5-HT
reuptake inhibition may cause serotonin syndrome (Sternbach, H. Serotonin
syndrome. Am. J. Psychiatry 148, 705-713, 1991 ) which is highly undesirable.
CA 02374926 2001-12-06
WO 00/71549 PCT/EP00/04279
39
Acute feeding studies
Animals and environment
Experiments were performed on male Sprague-Dawley rats (300-450 g at the
start of the experiment) which were obtained from Charles River (Margate).
Animals
were individually-housed in polypropylene cages with metal grid floors at a
temperature of 2111 °C and 55% humidity. Polypropylene trays were
placed below
each cage. Animals were maintained on a reverse phase light-dark cycle. Lights
were off from 09.30 h to 17.30 h during which time the room was illuminated by
red
light. Animals had free access to a powdered rat diet and tap water at all
times. The
diet was contained in glass feeding jars (10 cm diameter; 8 cm deep) with
aluminium
lids. Each lid had a hole (3 cm diameter) cut in it to allow access to the
food.
Animals were accustomed to these conditions for at least two weeks before
experimentation.
Test procedure
On the day prior to testing, the animals were randomly allocated to treatment
groups containing 6-8 rats, weighed and their food intakes over a 6 h period
were
measured. These baseline readings were taken to ensure that the body weights
and
food intakes of the different groups of rats were not significantly different
before drug
treatment. On the test day, animals were given vehicle or one of three doses
of the
test drug. All drugs were dosed orally at the onset of the dark phase since
rats
consume most of their food during this period. Feeding jars were weighed (to
the
nearest 0.1 g) at the time of drug administration and 1, 2, 4, 6 and 24 h
after dosing.
At each reading, the trays below the cages were examined for spilt food which
was
then returned to the feeding jar. However, spillage of food from the feeding
jars was
generally negligible.
All drug doses are expressed as the free base. Drugs were dissolved in
deionised water or suspended in 0.4% cellosize using a sonic bath.
CA 02374926 2001-12-06
WO 00/71549 PCT/EP00/04279
Data analysis
Variations in body weight were accounted for by expressing the results as
g/kg rat weight (treatment group means t s.e.mean). EDso values (the dose of a
5 drug required to reduce food intake to 50% of the control values) were
calculated
from a logistic sigmoid curve using a dedicated computer program. Statistical
comparisons between mean group intakes were made using analysis of variance
and Dunnett's test (two-tailed).
10 Especially preferred compounds of Formula la have surprisingly lower
affinity
for muscarinic receptors compared to the Examples of W097/02269 and/or have
significantly reduced MAOA inhibitory activity compared to compounds
exemplified in
W097/02269. For example, Example 1 of W097/02269 has a muscarinic receptor
binding Ki of 130 nM. Muscarinic affinity may cause undesired side-effects,
for
15 example dry mouth, blurred vision, sweating, palpitations, constipation and
aggravation of narrow angle glaucoma (Blackwell, B. Adverse effects of
antidepressant drugs. Part 1 Monoamine oxidase inhibitors and tricyclics.
Drugs 21,
202-219, 1981 ). Obviously it is desirable for compounds to have minimal
affinity for
muscarinic receptors.
Particularly preferred compounds of the present invention have superior
activity in acute feeding studies compared to the compounds exemplified in WO
97/02269.
The invention is illustrated by the following Examples which are given by way
of example only. The final product of each of these Examples was characterised
by
one or more of the following procedures: high performance liquid
chromatography;
elemental analysis, nuclear magnetic resonance spectroscopy, mass spectroscopy
and infrared spectroscopy.
Exam les
Example 1
Triethylamine (75 ml) was added dropwise at < 10 °C to a stirred
suspension
of 3-(benzo[b]thiophen-3-yl)-5,6-dihydroimidazo[2,1-b]thiazole hydrobromide
(50 g;
CA 02374926 2001-12-06
WO 00/71549 PCT/EP00/04279
41
prepared in a manner similar to that described in WO 97/02269) in
dichloromethane
(400 ml), then the mixture was stirred at ambient temperature for 1 hour.
Water (300
ml) was added, then the organic phase was separated, washed with water (2 x
100
ml) and saturated aqueous sodium chloride solution (100 ml), dried (Na2S04),
and
the solvent removed in vacuo to leave 3-(benzo[b]thiophen-3-yl)-5,6-
dihydroimidazo[2,1-b]thiazole as a pale yellow solid (34 g) which was used
without
further purification.
n-Butyllithium (2.5 M solution in hexanes; 17.5 ml) was added dropwise under
nitrogen at -70 °C to a stirred solution of 3-(benzo[b]thiophen-3-yl)-
5,6-dihydro-
imidazo[2,1-b]thiazole (10.14 g) in tetrahydrofuran (260 ml), then the mixture
was
stirred at -70 °C for 20 minutes, allowed to warm to 0 °C, and
stirred at 0 °C for 30
minutes. Dimethylformamide (2.86 ml) was added, and the mixture was stirred at
ambient temperature for 20 minutes. Saturated aqueous sodium chloride solution
(200 ml) and ether (400 ml) were added, then the organic phase was separated,
washed with water (100 ml) and saturated aqueous sodium chloride solution (100
ml), dried (Na2S04), and the solvents were removed in vacuo. The residue was
purified by flash chromatography over silica using a 10:1 mixture of
dichloromethane
and methanol as eluant. Appropriate fractions were combined and the solvents
removed in vacuo. The residue was heated under reflux for 5 minutes with
propan-
2-0l (100 ml), then the mixture was filtered while hot and allowed to cool to
ambient
temperature. The resulting solid was collected by filtration and dried in
vacuo at 60
°C to give 3-(benzo[b]thiophen-3-yl)-5,6-dihydroimidazo[2,1-b]thiazole-
2-carbox-
aldehyde as a yellow solid (1.4 g), m.p. 206 °C.
Example 2
A mixture of 3-(benzo[b]thiophen-3-yl)-5,6-dihydroimidazo[2,1-b]thiazole-2-
carboxaldehyde (0.198 g), sodium borohydride (0.026 g) and methanol (20 ml)
was
stirred at ambient temperature under nitrogen for 1 hour, then water (2 ml)
was
added and the mixture was concentrated in vacuo to remove methanol. The
residue
was partitioned between water (30 ml) and ethyl acetate (50 ml), then the
organic
phase was separated, washed with saturated aqueous sodium chloride solution
(20
ml), dried (NazS04), and the solvent was removed in vacuo to leave [3-
CA 02374926 2001-12-06
WO 00/71549 PCT/EP00/04279
42
(benzo[b]thiophen-3-yl)-5,6-dihydroimidazo[2,1-b]thiazol-2-yl]methanol as a
yellow
solid (0.077 g), m.p. 168 - 170 °C.
Example 3
A mixture of 3-(benzo[b]thiophen-3-yl)-5,6-dihydroimidazo[2,1-b]thiazole-2-
carboxaldehyde (0.44 g), hydroxylamine hydrochloride (0.118 g) and ethanol (30
ml)
was heated under reflux for 2.75 hours, then cooled to ambient temperature.
The
resulting solid was collected by filtration and suspended in dichloromethane
(50 ml).
Triethylamine (3 ml) was added, then the resulting solution was washed with
water
(20 ml) and saturated aqueous sodium chloride solution (20 ml), dried (MgS04),
and
the solvent was removed in vacuo to leave 3-(benzo[b]thiophen-3-yl)-5,6-
dihydro-
imidazo[2,1-b]thiazole-2-carboxaldehyde oxime as a white solid (0.07 g),
m.p.226-228°C.
Example 4
Methylmagnesium bromide (3 M solution in ether; 1.2 ml) was added
dropwise at 0°C under nitrogen to a stirred solution of 3-
(benzo[b]thiophen-3-yl)-5,6-
dihydroimidazo[2,1-b]thiazole-2-carboxaldehyde (0.286 g) in tetrahydrofuran
(30 ml),
then the mixture was stirred at ambient temperature for 15 minutes. Water (5
ml)
and ethyl acetate (70 ml) were added, then the organic phase was separated,
washed with water (20 ml) and saturated aqueous sodium chloride solution (20
ml),
dried (Na2S04), and the solvents were removed in vacuo. The residue was
triturated
with ether (30 ml) and the resulting solid was collected by filtration and
driedin vacuo
at 100°C to give 1-[3-(benzo[b]thiophen-3-yl)-5,6-dihydroimidazo[2,1-
b]thiazol-2-y1]-
ethanol as an off-white solid (0.213 g), m.p. 174-176°C.
Example 5
A suspension of [3-(benzo[b]thiophen-3-yl)-5,6-dihydroimidazo[2,1-b]thiazol-
2-yl]methanol (0.5 g) in ethanol (25 ml) was heated under reflux until all of
the solid
had dissolved. Ethereal hydrogen chloride solution (1 M; 2 ml) was added, then
the
mixture was heated under reflux for 3 minutes and allowed to cool to ambient
CA 02374926 2001-12-06
WO 00/71549 PCT/EP00/04279
43
temperature. The resulting solia was collected by filtration, washed with
ether
(20 ml) and dried in vacuo at 60°C to give 3-(benzo[b]thiophen-3-yl)-
5,6-dihydro-
imidazo[2,1-b]thiazole-2-methanol hydrochloride as a white solid (0.32 g),
m.p.
240-250°C (decomposes). Ethereal hydrogen chloride solution (1 M; 2 ml)
was
added to the filtrate remaining from isolation of the above solid, and the
mixture was
stirred at ambient temperature for 18 hours. The resulting solid was collected
by
filtration, washed with ether (20 ml) and dried in vacuo at 60°C to
give a second crop
of [3-(benzo[b]thiophen-3-yl)-5,6-dihydroimidazo[2,1-b]thiazol-2-yl]methanol
hydro-
chloride as a white solid (0.1 g), m.p. 240-250°C (decomposes).
Example 6
A mixture of 3-(benzo[b]thiophen-3-yl)-5,6-dihydroimidazo[2,1-b]thiazole
hydrobromide (200 g; prepared in a manner similar to that described in
WO 97/02269), saturated aqueous sodium carbonate solution (1000 ml) and
dichloromethane (2000 ml) was stirred vigorously at ambient temperature for
1.5
hours, then the organic layer was separated, washed with water (500 ml), dried
(MgS04) and the solvent was removed in vacuo. The process was repeated on the
same scale, and the two products were combined to give 3-(benzo[b]thiophen-3-
yl)-
5,6-dihydroimidazo[2,1-b]thiazole as a pale yellow solid (264.3 g), which was
used
without further purification.
Bromine (55.5 ml) was added dropwise at 0-5 °C over 1.75 hours to a
stirred
solution of 3-(benzo[b]thiophen-3-yl)-5,6-dihydroimidazo[2,1-b]thiazole (264.3
g) in
dichloromethane, then the mixture was stirred at 0°C for 30 minutes and
at ambient
temperature for 1 hour. The resulting solid was collected by filtration,
washed with
dichloromethane (300 ml) and dried in vacuo at 70°C to give 3-
(benzo[b]thiophen-3-
yl)-2-bromo-5,6-dihydroimidazo[2,1-b]thiazole hydrobromide as a pale yellow
solid
(431 g) which was used without further purification.
3-(Benzo[b]thiophen-3-yl)-2-bromo-5,6-dihydroimidazo[2,1-b]thiazole hydro-
bromide (170 g) was added in portions under nitrogen at 0-8°C over 1
hour to a
stirred solution of ethylmagnesium chloride [2.0 M solution in ether (620 ml)]
in
tetrahydrofuran (1700 ml), then the mixture was stirred at 3°C for 1.5
hours.
Dimethylformamide (136 ml) was added at 3-8 °C over 30 minutes, then
the mixture
CA 02374926 2001-12-06
WO 00/71549 PCT/EP00/04279
44
was stirred at ambient temperature for 2 hours, cooled to 8°C and
quenched by the
cautious addition of saturated aqueous ammonium chloride solution (600 ml) and
water (350 ml). Ethyl acetate (1500 ml) was added, the mixture was stirred at
ambient temperature for 18 hours, and the resulting solid (Fraction 1 ) was
collected
by filtration. The organic layer of the filtrate was separated, washed with
saturated
aqueous sodium chloride solution (500 ml), dried (MgS04), and the solvents
were
removed in vacuo. The residue was dissolved in hot propan-2-of (1000 ml) and
the
solution was filtered while hot then allowed to stand at ambient temperature
for 20
hours. The resulting solid was collected by filtration, washed with propan-2-
of
(100 ml) and dried in vacuo at 70°C to give 3-(benzo[b]thiophen-3-yl)-
5,6-dihydro-
imidazo[2,1-b]thiazole-2-carboxaldehyde as a yellow solid (19.4 g), m.p.
206°C. A
mixture of the solid Fraction 1, dichloromethane (2100 ml), 2 M hydrochloric
acid
(250 ml) and water (1000 ml) was stirred at ambient temperature for 15
minutes,
then triethylamine (80 ml) was added. The dichloromethane layer was separated,
and further product was isolated from the aqueous layer by extraction into
dichloromethane (500 ml). The combined dichloromethane solutions were dried
(MgS04), and the solvent was removed in vacuo to give further 3-
(benzo[b]thiophen-
3-yl)-5,6-dihydroimidazo[2,1-b]thiazole-2-carboxaldehyde as a yellow solid
(63.0 g),
m.p. 208°C.
Sodium borohydride (12.5 g) was added in portions over 10 minutes to an
ice-cold stirred suspension of 3-(benzo[bJthiophen-3-yl)-5,6-
dihydroimidazo[2,1-b]-
thiazole-2-carboxaldehyde (63 g) in methanol (1200 ml), then the mixture was
stirred
at 5°C for 30 minutes, at ambient temperature for 4 hours, and at
reflux temperature
for 20 minutes. The mixture was cooled to ambient temperature over 1 hour then
water (200 ml) was added and stirring at ambient temperature was continued for
1
hour. The resulting solid was collected by filtration, washed with water (200
ml),
ethanol (200 ml) and ether (200 ml), then dried in vacuo at 60°C to
give
[3-(benzo[b]thiophen-3-yl)-5,6-dihydroimidazo[2,1-b]thiazol-2-yl]methanol as
an off-
white solid (52.1 g) which was used without purification.
A stirred suspension of [3-(benzo[b]thiophen-3-yl)-5,6-dihydroimidazo[2,1-
b]thiazol-2-yl]methanol (52.1 g) in methanol (2200 ml) was heated under reflux
until
virtually all of the solid had dissolved. The heat source was removed, and a
solution
of fumaric acid (21 g) in methanol (250 ml) was added over 1 minute. The
mixture
CA 02374926 2001-12-06
WO 00/71549 PCT/EP00/04279
was stirred for 10 minutes and allowed to stand at ambient temperature for 1
hour,
then it was cooled in ice for 3 hours. The resulting solid was collected by
filtration,
washed with ice-cold methanol (200 ml), and dried in vacuo at 60°C for
3 hours and
at 80°C for 2 hours to give a white solid which was shown by nmr
spectroscopy to be
5 solvated by 1 equivalent of methanol. This solid was combined with a second
sample of product (5.2 g; also solvated by methanol) prepared in a manner
similar to
that described above, and the combined material was ground using a pestle and
mortar, then dried in vacuo at 90°C and 133Pa for 15 hours to give
[3-(benzo[b]thiophen-3-yl)-5,6-dihydroimidazo[2,1-b]thiazol-2-yl]methanol
fumarate
10 (solvated by 0.05 equivalent of methanol) as a white solid (53.4 g), m.p.
258-262°C
(decomposes).
Example 7
15 Phenyltrimethylammonium tribromide (3.0 g) was added in portions under
nitrogen at 0°C to a stirred suspension of 3-(benzo[b]thiophen-3-yl)-
5,6-dihydro-
imidazo[2,1-b]thiazole (2.0 g) in tetrahydrofuran (50 ml), then the mixture
was stirred
at 0°C for 1 hour and at ambient temperature for 18 hours. Water (50
ml) and
triethylamine (50 ml) were added, and the organic phase was separated, washed
20 with saturated aqueous sodium chloride solution (50 ml), dried (MgS04), and
the
solvents were removed in vacuo. The residue was purified by flash
chromatography
over silica using a 9:1 mixture of ethyl acetate and methanol as eluant.
Appropriate
fractions were combined and the solvents were removed in vacuo to leave
3-(benzo[b)thiophen-3-yl)-2-bromo-5,6-dihydroimidazo[2,1-b]thiazole as a
yellow
25 solid (1.0 g), m.p. 196-200°C.
Example 8
Phenyltrimethylammonium tribromide (0.75 g) was added in portions under
30 nitrogen at 0°C to a stirred suspension of 3-(benzo[b]thiophen-3-yl)-
6,7-dihydro-5H-
thiazolo[3,2-a]pyrimidine (0.5 g; prepared in a manner similar to that
described in
WO 97/02269)) in tetrahydrofuran (15 ml), then the mixture was stirred at
0°C for 4
hours and allowed to warm to ambient temperature. Water (50 ml) and
triethylamine
(50 ml) were added, and the organic phase was separated, washed with saturated
35 aqueous sodium chloride solution (50 ml), dried (MgS04), and the solvents
were
CA 02374926 2001-12-06
WO 00/71549 PCT/EP00/04279
46
removed in vacuo. The residue was purified by flash chromatography over silica
using a 9:1 mixture of ethyl acetate and methanol as eluant. Appropriate
fractions
were combined and the solvents were removed in vacuo to leave 3
(benzo[b]thiophen-3-yl)-2-bromo-6,7-dihydro-5H-thiazolo[3,2-a]pyrimidine as a
pale
yellow solid (0.2 g), m.p. 200-202°C.
Example 9
Sodium hydride (60% dispersion in mineral oil; 1.35 g) was added in portions
at ambient temperature under nitrogen to a stirred solution of 3-acetyl-
benzo[b]thiophen (2.9 g) in diethyl carbonate (50 ml), then the mixture was
stirred at
80°C for 1.5 hours and poured onto a mixture of water (300 ml) and
acetic acid
(5 ml). The product was extracted into ether (3 x 150 ml), then the combined
extracts were washed with water (2 x 50 ml) and saturated aqueous sodium
chloride
solution (50 ml), dried (MgS04), and the solvent was removed in vacuo. The
residue
was purified by flash chromatography over silica in Biotage Flash 40i ~
equipment
using a 9:1 mixture of hexane and ethyl acetate as eluant. Appropriate
fractions
were combined and the solvents were removed in vacuo to leave ethyl 3
(benzo[b]thiophen-3-yl)-3-oxopropanoate as a brown oil (1.5 g) which was used
without further purification.
Phenyltrimethylammonium tribromide (2.15 g) was added in portions at
0°C
under nitrogen to a stirred solution of ethyl 3-(benzo[b]thiophen-3-yl)-3-oxo-
propanoate (1.5 g) in tetrahydrofuran (30 ml), then the mixture was stirred at
0°C for
30 minutes and at ambient temperature for 1.5 hours. The resulting solid was
removed by filtration and washed with tetrahydrofuran (30 ml). The filtrate
and
washings were combined and the solvent was removed in vacuo. The residue was
purified by flash chromatography over silica in Biotage Flash 40i ~equipment
using
a 9:1 mixture of petroleum ether (b.p. 60-80 °C) and ethyl acetate as
eluant.
Appropriate fractions were combined and the solvents were removed in vacuo to
leave ethyl 3-(benzo[b]-thiophen-3-yl)-2-bromo-3-oxopropanoate as a brown
solid
(1.7 g), m.p. 81-83°C.
A mixture of ethyl 3-(benzo[b]thiophen-3-yl)-2-bromo-3-oxopropanoate
(1.7 g), 2-imidazolidinethione (0.53 g) and ethanol (30 ml) was heated under
reflux
for 10 minutes, then acetic acid (15 ml) was added and the mixture was heated
CA 02374926 2001-12-06
WO 00/71549 PCT/EP00/04279
47
under reflux for 18 hours. The solvents were removed in vacuo and the residue
was
triturated with ethanol (20 ml). The resulting solid was collected by
filtration, washed
with ethanol (10 ml) and ether (20 ml), then dried in vacuo at 60°C to
give ethyl 3
(benzo[b]thiophen-3-yl)-5,6-dihydroimidazo[2,1-b]thiazole-2-carboxylate as an
off
white solid (1.15 g), m.p. 209-211°C.
A mixture of ethyl 3-(benzo[b]thiophen-3-yl)-5,6-dihydroimidazo[2,1-b]-
thiazole-2-carboxylate (0.4 g), triethylamine (2 ml) and dichloromethane (15
ml) was
stirred at ambient temperature for 20 minutes, then it was diluted with
dichloromethane (40 ml), washed with water (2 x 20 ml) and saturated aqueous
sodium chloride solution (20 ml), dried (Na2S04), and the solvents were
removed in
vacuo. The residue (0.22 g) was dissolved in tetrahydrofuran (7 ml), and
methylmagnesium bromide (3 M solution in ether; 0.66 ml) was added under
nitrogen. The mixture was stirred at ambient temperature for 2 hours, then
further
methylmagnesium bromide (3 M solution in ether; 0.42 ml) and toluene (5 ml)
were
added. The mixture was stirred at ambient temperature for 5 minutes and at
90-95°C for 5 hours, then it was cooled to ambient temperature and
diluted with
water (30 ml). The product was extracted into ethyl acetate (2 x 30 ml), and
the
combined extracts were washed with water (30 ml) and saturated aqueous sodium
chloride solution (30 ml), dried (MgS04), and the solvents were removed in
vacuo.
A mixture of the residue, fumaric acid (0.037 g) and ethanol (5 ml) was heated
under
reflux for 5 minutes, then the hot solution was decanted from a small trace of
undissolved solid and allowed to cool to ambient temperature. The resulting
solid
was collected by filtration, washed with ether (10 ml) and dried in vacuo at
60°C to
give 1-[3-(benzo[b]thiophen-3-yl)-5,6-dihydroimidazo[2,1-b]thiazol-2-yl]-1-
methyl-
ethanol fumarate as an off-white solid (0.057 g), m.p. 180-182°C.
Example 10
Ethylmagnesium chloride (2M solution in ether; 1.4 ml) was added dropwise
at ambient temperature to a stirred solution of 3-(benzo[b]thiophen-3-yl)-5,6-
dihydro-
imidazo[2,1-b]-thiazole-2-carboxaldehyde (0.52 g) in tetrahydrofuran (30 ml),
then
the mixture was stirred at ambient temperature for 30 minutes. Further
ethylmagnesium chloride (2M solution in ether; 0.5 ml) was added, the mixture
was
stirred at ambient temperature for 1 hour, then it was quenched by the
addition of
CA 02374926 2001-12-06
WO 00/71549 PCT/EP00/04279
48
water (30 ml). The product was extracted into ether (50 ml) followed by ethyl
acetate
(2 x 50 ml), then the combined extracts were washed with saturated aqueous
sodium
chloride solution (2 x 30 ml), dried (Na2S04), and the solvents were removed
in
vacuo. The residue was purified by flash chromatography over silica in Biotage
Flash 40i ~ equipment using a 99:1 mixture of dichloromethane and methanol as
eluant. Appropriate fractions were combined and the solvents removed in vacuo
to
leave 1-[3-(benzo[b]thiophen-3-yl)-5,6-dihydroimidazo[2,1-b)thiazol-2-
yl]propan-1-of
as an orange solid (0.24 g), m.p. 92-94°C.
Example 11
Triethylamine (50 ml) was added dropwise at ambient temperature to a
stirred suspension of 3-(5-methoxybenzo[b]thiophen-3-yl)-5,6-
dihydroimidazo[2,1-b]-
thiazole hydrobromide (1 g; prepared in a manner similar to that described in
WO 97/02269) in dichloromethane (25 ml), then the mixture was stirred at
ambient
temperature for 10 minutes. Water (25 ml) was added, then the organic phase
was
separated, washed with water (25 ml), dried (Na2S04), and the solvents were
removed in vacuo to leave 3-(5-methoxybenzo[b)thiophen-3-yl)-5,6-dihydro
imidazo[2,1-b]thiazole as a brown solid (0.65 g) which was used without
further
purification.
Phenyltrimethylammonium tribromide (1.5 g) was added in portions at
0°C
under nitrogen to a stirred solution of 3-(5-methoxybenzo[b]thiophen-3-yl)-5,6-
dihydroimidazo[2,1-b]thiazole (0.65 g) in tetrahydrofuran (25 ml), the mixture
was
stirred at 0°C for 16 hours, then it was allowed to warm to ambient
temperature.
Triethylamine (50 ml) and water (50 ml) were added, then the organic phase was
separated, dried (Na2S04), and the solvents were removed in vacuo. The residue
was purified by flash chromatography over silica using a 99:1:0.1 mixture of
ethyl
acetate, methanol and triethylamine as eluant. Appropriate fractions were
combined
and the solvents were removed in vacuo. The residue was crystallised from
methanol and the resulting solid was collected by filtration and dried in
vacuo at
ambient temperature to give 2-bromo-3-(5-methoxybenzo[b)thiophen-3-yl)-5,6-
dihydroimidazo[2,1-b]thiazole as an off-white solid (0.05 g), m.p. 210'C
(decomposes).
CA 02374926 2001-12-06
WO 00/71549 PCT/EP00/04279
49
Example 12
Phenyltrimethylammonium tribromide (1.0 g) was added in portions at 0 -
5°C under nitrogen over 15 minutes to a stirred solution of 3-(5-
chlorobenzo[b]thiophen-3-yl)-5,6-dihydroimidazo[2,1-b]thiazole (0.8 g;
prepared in a
manner similar to that described in WO 97/02269) in tetrahydrofuran (20 ml),
then
the mixture was stirred at ambient temperature for 18 hours. Water (30 ml) and
triethylamine (5 ml) were added, then the product was extracted into
dichloromethane (2 x 20 ml), and the combined extracts were washed with water
(4 x 20 ml), dried (MgS04), and the solvents were removed in vacuo to leave 2-
bromo-3-(5-chlorobenzo[b]thiophen-3-yl)-5,6-dihydroimidazo[2,1-b]thiazole as a
yellow solid (0.72 g), m.p. 205 - 208°C (decomposes).
Example 13
A mixture of 3-acetylbenzo(b]thiophene (25 g), dimethylamine hydrochloride
(15.05 g), paraformaldehyde (5.7 g), concentrated hydrochloric acid (1 ml) and
ethanol (75 ml) was heated under reflux for 18 hours then allowed to cool to
ambient
temperature. The resulting solid was collected by filtration and dried in
vacuo at
ambient temperature to give 1-(benzo[b]thiophen-3-yl)-3-(dimethylamino)propan-
1-
one hydrochloride as a pink solid (15.7 g), m.p. 169 - 171°C. The
solvent was
removed in vacuo from the filtrate, and the residue was triturated with ether
(100 ml).
The resulting solid was collected by filtration and dried in vacuo at ambient
temperature to give a second crop of 1-(benzo[b]thiophen-3-yl)-3-
(dimethylamino)-
propan-1-one hydrochloride as a pink solid (13.8 g).
A mixture of 1-(benzo[b]thiophen-3-yl)-3-(dimethylamino)propan-1-one
hydrochloride (29.4 g) and water (600 ml) was basified to pH 9.0 by the
addition of
saturated aqueous sodium carbonate solution, the mixture was stirred at
ambient
temperature for 1 hour, then the free base was extracted into ether (3 x 100
ml). The
combined extracts were dried (MgS04) and the solvent was removed in vacuo. The
residue was dissolved in methanol (50 ml), then the solution was cooled in
ice, and
iodomethane (15.7 ml) was added dropwise. The mixture was stirred at ambient
temperature for 1 hour, then the resulting solid was collected by filtration,
washed
well with ether, and dried in vacuo at ambient temperature to give [3-
(benzo[b]-
CA 02374926 2001-12-06
WO 00/71549 PCT/EP00/04279
thiophen-3-yl)-3-oxopropyl]trimethylammonium iodide as a pink solid (28.7 g),
m.p. 165 - 167°C.
A mixture of [3-(benzo[b]thiophen-3-yl)-3-oxopropyl]trimethylammonium
5 iodide (5.0 g), sodium hydrogencarbonate (5.0 g), ether (150 ml) and water
(130 ml)
was stirred at ambient temperature for 4 hours, then the product was extracted
into
ether (3 x 150 ml). The combined extracts were dried (MgS04) and the solvent
was
removed in vacuo to leave 1-(benzo[b]thiophen-3-yl)propenone as a pink solid
(2.1 g) which was used without further purification.
A solution of 1-(benzo[b]thiophen-3-yl)propenone (0.75 g) and benzyl alcohol
(0.41 ml) in dichloromethane (2 ml) was cooled to 0°C, and concentrated
sulphuric
acid (2 drops) was added. The mixture was stirred at 0°C for 3 hours,
then allowed
to stand at 4°C for 18 hours. Further benzyl alcohol (0.82 ml) was
added, the
mixture was stirred at 0°C for 7 hours, then it was diluted with
dichloromethane
(20 ml), washed with saturated aqueous sodium hydrogencarbonate solution
(2 x 10 ml) and water (10 ml), and dried (MgS04). The solvent was removed in
vacuo, and the residue was purified by preparative-scale thin layer
chromatography
on a silica-coated glass plate using dichloromethane as eluant. Appropriate
sections
of silica were removed from the developed plate, and the product was extracted
by
trituration with dichloromethane (30 ml). The extract was filtered, and the
solvent
removed in vacuo to leave 1-(benzo[b]thiophen-3-yl)-3-benzyloxypropan-1-one as
an
orange oil (0.49 g) which was used without further purification.
Phenyltrimethylammonium tribromide (0.4 g) was added in portions under
nitrogen to a stirred solution of 1-(benzo[b]thiophen-3-yl)-3-benzyloxypropan-
1-one
(0.43 g) in tetrahydrofuran (5 ml), the mixture was stirred at ambient
temperature for
18 hours, then it was filtered and the solvent was removed in vacuo to leave
1-(benzo[b]thiophen-3-yl)-3-benzyloxy-2-bromopropan-1-one as a yellow oil
(0.56 g)
which was used without further purification.
A mixture of 1-(benzo[b]thiophen-3-yl)-3-benzyloxy-2-bromopropan-1-one
(0.54 g), 2-imidazolidinethione (0.15 g), ethanol (3 ml) and acetic acid (1
ml) was
heated under reflux under nitrogen for 18 hours, then the solvents were
removed in
vacuo. The residue was triturated with ice-cold ethanol (5 ml) and the
resulting solid
CA 02374926 2001-12-06
WO 00/71549 PCT/EP00/04279
51
was collected by filtration, washed with ethanol (5 ml), and dried in vacuo at
60°C to
give 3-(benzo[b]thiophen-3-yl)-2-ethoxymethyl-5,6-dihydroimidazo[2,1-
b]thiazole
hydrobromide as a cream solid (0.19 g), m.p. > 250°C.
Example 14
Vinylmagnesium chloride (1 M solution in tetrahydrofuran; 6.7 ml) was added
dropwise at 0°C under nitrogen to a stirred suspension of 3-
(benzo[b]thiophen-3-yl)-
5,6-dihydroimidazo[2,1-b]thiazole-2-carboxaldehyde (0.64 g) in tetrahydrofuran
(30 ml), then the mixture was stirred at 0°C for 10 minutes and at
ambient
temperature for 30 minutes. Water (50 ml) was added, and the mixture was
concentrated in vacuo to remove tetrahydrofuran, then the product was
extracted
into ethyl acetate (3 x 30 ml). The combined extracts were washed with
saturated
aqueous sodium chloride solution (2 x 25 ml), dried (MgS04), and the solvents
were
removed in vacuo. The residue was purified by flash chromatography over silica
in
Biotage Flash 40i ~ equipment using 1 - 5% mixtures of methanol in
dichloromethane as eluant. Appropriate fractions were combined and the
solvents
were removed in vacuo to leave 1-[3-(benzo[b]thiophen-3-yl)-5,6-
dihydroimidazo[2,1-
b]thiazol-2-yl]prop-2-en-1-of as a pale yellow foam (0.12 g), m.p. 60-
65°C.
Example 15
1-Propynylmagnesium bromide (0.5 M solution in ether; 10.5 ml) was added
dropwise at 0°C under nitrogen to a stirred suspension of 3-
(benzo[b]thiophen-3-yl)-
5,6-dihydroimidazo[2,1-b]thiazole-2-carboxaldehyde (0.5 g) in tetrahydrofuran
(30 ml), then the mixture was stirred at 0°C for 10 minutes and at
ambient
temperature for 30 minutes. Water (60 ml) and ethyl acetate (100 ml) were
added,
then the organic phase was separated, washed with saturated aqueous sodium
chloride solution (2 x 25 ml), dried (MgS04), and the solvents were removed in
vacuo. The residue was purified by flash chromatography over silica in Biotage
Flash 40i ~ equipment using 1 - 5% mixtures of methanol in dichloromethane as
eluant. Appropriate fractions were combined and the solvents were removed in
vacuo to leave 1-[3-(benzo[b]thiophen-3-yl)-5,6-dihydroimidazo[2,1-b]thiazol-2-
yl]but-
2-yn-1-of as a white solid (0.11 g), m.p. 190 - 200°C (decomposes).
CA 02374926 2001-12-06
WO 00/71549 PCT/EP00/04279
52
Example 16
n-Butyllithium (2.5 M solution in hexanes; 4.7 ml) was added dropwise at
0°C
under nitrogen to a stirred solution of methyltriphenylphosphonium bromide
(4.2 g) in
tetrahydrofuran (30 ml), then the mixture was stirred at 0°C for 5
minutes and at
ambient temperature for 30 minutes. 3-(Benzo[b]thiophen-3-yl)-5,6-
dihydroimidazo-
[2,1-b]thiazole-2-carboxaldehyde (3.05 g) was added in portions, then the
mixture
was heated under reflux for 3 hours and allowed to cool to ambient
temperature.
Ethyl acetate (75 ml) and water (50 ml) were added, then the organic phase was
separated, washed with saturated aqueous sodium chloride solution (50 ml),
dried
(MgS04), and the solvents were removed in vacuo. The residue was diluted with
2.5 M hydrochloric acid (75 ml), the mixture was stirred at ambient
temperature for 3
hours, then it was filtered to remove a gummy semisolid. The filtrate was
shaken
with ethyl acetate (25 ml), then the organic phase was separated, dried
(MgS04),
and the solvent was removed in vacuo to leave slightly impure 3-
(benzo[b]thiophen
3-yl)-2-vinyl-5,6-dihydroimidazo[2,1-b]thiazole hydrochloride as a white solid
(0.85 g). Further product was isolated by extraction with dichloromethane
(2 x 50 ml), drying (MgS04), and removal of the solvent in vacuo to leave 3
(benzo[b]thiophen-3-yl)-2-vinyl-5,6-dihydroimidazo[2,1-b]thiazole
hydrochloride as a
white solid (0.49 g), m.p. 221 - 223°C.
Example 17
3-(Benzo[b]thiophen-3-yl)-2-bromo-5,6-dihydroimidazo[2,1-b]thiazole hydro-
bromide (2 g) was added in portions under nitrogen at 0 - 5°C over 10
minutes to a
stirred solution of ethylmagnesium chloride (2 M solution in ether; 7.2 ml) in
tetrahydrofuran (20 ml), then the mixture was stirred at 0 - 5°C for 30
minutes. Allyl
bromide (0.87 ml) was added dropwise, the mixture was stirred at ambient
temperature for 18 hours, then it was quenched by the addition of saturated
aqueous
ammonium chloride solution (15 ml) followed by water (10 ml). The product was
extracted into ethyl acetate (50 ml), then the extract was washed with water
(25 ml)
and saturated aqueous sodium chloride solution (25 ml), dried (MgS04), and the
solvent was removed in vacuo. The residue was purified by flash chromatography
over silica using 5 - 7% mixtures of methanol in dichloromethane as eluant.
Appropriate fractions were combined and the solvents removed in vacuo to give
a
CA 02374926 2001-12-06
WO 00/71549 PCT/EP00/04279
53
brown gum (0.36 g). The gum was dissolved in ethanol (2 ml), and a solution of
fumaric acid (0.13 g) in ethanol (2 ml) was added. The resulting solid was
collected
by filtration, washed with ethanol (10 ml) and dried in vacuo at 75°C
to give 2-allyl-3
(benzo[b]thiophen-3-yl)-5,6-dihydroimidazo[2,1-b]thiazole fumarate as an off-
white
solid (0.22 g), m.p. 163 - 164°C.
Example 18
n-Butyllithium (2.5 M solution in hexanes; 1 ml) was added dropwise under
nitrogen at -70°C over 10 minutes to a stirred solution of 3-
(benzo(b)furan-3-yl)-5,6-
dihydroimidazo(2,1-b]thiazole (0.5 g; prepared in a manner similar to that
described
in WO 97/02269) in tetrahydrofuran (6 ml), then the mixture was stirred at -
70°C for
30 minutes. Dimethylformamide (0.2 ml) was added, the mixture was stirred at
-70°C for 5 minutes, then it was allowed to warm to ambient
temperature. Saturated
aqueous ammonium chloride solution (30 ml) was added, and the product was
extracted into dichloromethane (3 x 30 ml). The combined extracts were washed
with water (30 ml), dried (Na2S04), and the solvents were removed in vacuo.
The
residue was purified by flash chromatography over silica using a 9:1 mixture
of
dichloromethane and methanol as eluant. Appropriate fractions were combined,
and
the solvents were removed in vacuo to leave 3-(benzo[b]furan-3-yl)-5,6-
dihydroimidazo[2,1-b]thiazole-2-carboxaldehyde as a brown solid (0.24 g),
m.p. 192 - 195°C.
3-(Benzo[b]furan-3-yl)-5,6-dihydroimidazo[2,1-b]thiazole-2-carboxaldehyde
(0.17 g) was dissolved in methanol (6 ml) by gentle warming, then sodium
borohydride (0.035 g) was added and the mixture was stirred at ambient
temperature
for 30 minutes. Water (50 ml) was added, the mixture was stirred at ambient
temperature for 1 hour, then the resulting solid was collected by filtration,
washed
with water (10 ml) and dried in vacuo at 60°C to give [3-(benzo[b)furan-
3-yl)-5,6-
dihydroimidazo[2,1-b]thiazol-2-yl]methanol as a white solid (0.08 g),
m.p. 184 - 187°C.
CA 02374926 2001-12-06
WO 00/71549 PCT/EP00/04279
54
Example 19
A stirred mixture of 3-(benzo[b]thiophen-3-yl)-5,6-dihydroimidazo[2,1-b]-
thiazole-2-carboxaldehyde (0.5 g), isopropylamine (1 ml), ethanol (50 ml) and
acetic
acid (1 drop) was heated under reflux for 4 hours, then the solvent was
removed in
vacuo. The residue was triturated with ether (30 ml), and the resulting solid
was
collected by filtration, washed with ether (10 ml) and dried in vacuo at
ambient
temperature to give N-[3-(benzo[b)thiophen-3-yl)-5,6-dihydroimidazo[2,1-
b]thiazol-2-
ylmethylidene]-1-methylethylamine as a pale brown solid (0.38 g), m.p. 180-
182°C.
Example 20
Benzyltrimethylammonium chloride (8 g) was added in portions at ambient
temperature to a stirred solution of iodine trichloride (10 g) in
dichloromethane
(120 ml), then the mixture was stirred at ambient temperature for 2.5 hours.
The
resulting solid was collected by filtration and dried in vacuo at ambient
temperature
to give benzyltrimethylammonium tetrachloroiodate as a yellow solid (16.2 g)
which
was used without further purification.
Benzyltrimethylammonium tetrachloroiodate (5 g) was added in portions at
0°C over 10 minutes to a stirred solution of 3-(benzo[b]thiophen-3-yl)-
5,6-dihydro-
imidazo[2,1-b)thiazole (3 g) in acetone (125 ml), then the mixture was stirred
at 0°C
for 1 hour. The resulting solid was collected by filtration, triturated with
hot propan-2-
ol (150 ml) and crystallised from ethanol (150 ml) to give an off-white solid
(0.74 g).
Concentration of the mother liquor to 75 ml gave a second crop of solid (0.41
g).
The two crops of solid were combined, triturated with hot ethanol (40 ml), and
the
resulting solid was collected by filtration, washed with ethanol (10 ml) and
dried in
vacuo at 60°C to give 3-(benzo[b]thiophen-3-yl)-2-chloro-5,6-
dihydroimidazo[2,1-
b]thiazole hydrochloride 0.5 hydrate as an off-white solid (0.91 g), m.p. 255-
257°C.
Example 21
3-(Benzo[b]thiophen-3-yl)-2-bromo-5,6-dihydroimidazo[2,1-b]thiazole hydro-
bromide (6 g) was added in portions under nitrogen at 0- 5°C over 10
minutes to a
stirred solution of ethylmagnesium chloride (2 M solution in ether; 21.6 ml)
in
tetrahydrofuran (100 ml), then the mixture was stirred at 0- 5°C for 1
hour. This
CA 02374926 2001-12-06
WO 00/71549 PCT/EP00/04279
mixture was added over 10 minutes at 50 - 70°C to a stirred solution
ofN-methoxy-
N-methylacetamide (5 g) in tetrahydrofuran (50 ml), then stirring at
70°C was
continued for a further 2.5 hours. The mixture was cooled in ice, then
saturated
aqueous ammonium chloride solution (100 ml), water (100 ml) and ethyl acetate
5 (150 ml) were added. The organic layer was separated, washed with a mixture
of
saturated aqueous sodium chloride solution (100 ml) and water (100 ml), dried
(MgS04), and the solvents were removed in vacuo. The residue was triturated
with
ether (3 x 50 ml), and the resulting solid was collected by filtration, washed
with ether
(30 ml) and dried in vacuo to give a yellow solid (1.84 g). A sample (0.25 g)
of the
10 solid was crystallised from ethanol (3.5 ml), and the resulting solid was
collected by
filtration, washed with ethanol (5 ml), and dried in vacuo at 75°C to
give 2-acetyl-3-
(benzo[b]thiophen-3-yl)-5,6-dihydroimidazo[2,1-b]thiazole as a yellow solid
(0.043 g),
m.p. 203 - 205°C.
15 Example 22
Sodium hydride (60% dispersion in mineral oil; 0.15 g) was added in portions
at ambient temperature over 10 minutes to a stirred suspension of [3-(benzo[b]-
thiophen-3-yl)-5,6-dihydroimidazo[2,1-b]thiazol-2-yl]methanol (1 g) in
dimethylform-
20 amide (20 ml), then the mixture was stirred at ambient temperature for 45
minutes.
lodomethane (240 ~I) was added, and stirring at ambient temperature was
continued
for a further 2 hours. Water (25 ml) and ethyl acetate (50 ml) were added,
then the
organic phase was separated, washed with water (4 x 25 ml) and saturated
aqueous
sodium chloride solution (25 ml), dried (MgS04), and the solvents were removed
in
25 vacuo. The residue was purified by flash chromatography over silica using 5-
8%
mixtures of methanol in dichloromethane as eluant. Appropriate fractions were
combined and the solvents were removed in vacuo. The residue was dissolved in
warm ethanol (3 ml), added to a solution of fumaric acid (0.085 g) in warm
ethanol
(2 ml), and the mixture was allowed to cool to ambient temperature. The
resulting
30 solid was collected by filtration, washed with ethanol (3 ml), and dried in
vacuo at
75°C to give 3-(benzo[b]thiophen-3-yl)-2-(methoxymethyl)-5,6-
dihydroimidazo[2,1-
b]thiazole fumarate as a white solid (0.23 g), m.p. 175- 176°C.
CA 02374926 2001-12-06
WO 00/71549 PCT/EP00/04279
56
Example 23
3-(Benzo[b]thiophen-3-yl)-2-bromo-5,6-dihydroimidazo[2,1-b]thiazole hydro-
bromide (5 g) was added in portions under nitrogen at 0-5°C over 30
minutes to a
stirred solution of ethylmagnesium chloride [2.0 M solution in ether (15 ml)]
in
tetrahydrofuran (75 ml), then the mixture was stirred at ambient temperature
for 1
hour. The mixture was cooled to 0 °C, dimethyl disulphide (1.8 ml) was
added , then
the mixture was stirred at ambient temperature for 24 hours, and quenched by
the
cautious addition of saturated aqueous ammonium chloride solution (50 ml). The
product was extracted into ethyl acetate (150 ml), then the extract was washed
with
water (50 ml) and saturated aqueous sodium chloride solution (50m1), dried
(Na2S04), and the solvents were removed in vacuo. The residue was purified by
flash chromatography over silica in Biotage Flash 40i ~ equipment using a 19:1
mixture of ethyl acetate and methanol as eluant. Appropriate fractions were
combined and the solvents were removed in vacuo. The residue was triturated
with
ether (20 ml) and the resulting solid was collected by filtration and dried in
vacuo to
leave 3-(benzo[b]thiophen-3-yl)-2-(methylthio)-5,6-dihydroimidazo[2,1-
b]thiazole
(1.9g) as an off-white solid, m.p. 129 - 131 °C.
Example 24
n-Butyllithium (2.5 M solution in hexanes; 1.7 ml) was added under nitrogen
to an ice-cold, stirred suspension of methyltriphenylphosphonium bromide (1.5
g) in
tetrahydrofuran (25 ml), then the mixture was stirred at ambient temperature
for 30
minutes. A solution of 2-acetyl-3-(benzo[b]thiophen-3-yl)-5,6-
dihydroimidazo[2,1-
b]thiazole (1.6 g) in tetrahydrofuran (15 ml) was added, the mixture was
heated
under reflux for 4h, allowed to stand at ambient temperature for 18 hours,
then it was
quenched by the addition of water (50 ml). The product was extracted into
ethyl
acetate (50 ml), then the extract was washed with saturated aqueous sodium
chloride solution (50 ml), dried (MgS04), and the solvents were removed in
vacuo.
The residue was purified by flash chromatography over silica using 5- 6%
mixtures
of methanol in dichloromethane as eluant. Appropriate fractions were combined
and
the solvents were removed in vacuo to leave 3-(benzo[b]thiophen-3-yl)-2-(1-
methylvinyl)-5,6-dihydroimidazo[2,1-b]thiazole (0.14 g) as a brown solid, m.p.
76 °C.
CA 02374926 2001-12-06
WO 00/71549 PCT/EP00/04279
57
Examples 25-33
Examples 25 - 33 were prepared as part of a High Speed Analogue library
using the following general method
The appropriate commercially-available Grignard reagent (3 molar
equivalents) was added to a solution of 3-(benzo[b]thiophen-3-yl)-5,6-
dihydroimidazo[2,1-b]thiazole-2-carboxaldehyde (approximately 50 mg) in
tetrahydrofuran (4 ml), the mixture was stirred at ambient temperature for 1
hour,
water (1 ml) was added, then the mixture was left exposed to the atmosphere
for 18
hours to allow the solvent to evaporate. Dichloromethane (4 ml) was added and
the
solution was pipetted onto a ChemElute (CE 1103; pH9) cartridge, allowed to
stand
for 15 minutes, then the product was eluted from the cartridge with
dichloromethane
(3 x 4 ml). The solvent was removed in vacuo to leave the required product
which
was analysed by high performance liquid chromatography on a Hypersil BDS C18
column (100 x 4.6mm) using gradient elution with mixtures of acetonitrile and
0.1 M
ammonium acetate buffer according to the following time schedule:
Ammonium
Time (minutes)Acetonitrile
(%) acetate (%)
0 10 90
8-9 100 0
11 10 90
All of the products of the following Examples gave satisfactory mass spectra
Hplc retention time and % purity are reported for each Example.
Example 25
1-[3-(Benzo[b]thiophen-3-yl)-5,6-dihydroimidazo[2,1-b]thiazol-2-yl]-2-
methylpropan-
1-0l (Retention time: 3.28 minutes - Purity: 80%).
CA 02374926 2001-12-06
WO 00/71549 PCT/EP00/04279
58
Example 26
1-[3-(Benzo[b]thiophen-3-yl)-5,6-dihydroimidazo[2,1-b]thiazol-2-yl]butan-1-of
(Retention time: 3.29 minutes - Purity: 100%).
Example 27
1-[3-(Benzo[b]thiophen-3-yl)-5,6-dihydroimidazo[2,1-b]thiazol-2-yl]-2-
methylbut-3-en-
1-0l (Retention time: 3.42 minutes - Purity: 100%).
Example 28
1-[3-(Benzo[b]thiophen-3-yl)-5,6-dihydroimidazo[2,1-b]thiazol-2-yl]-3-
methylbutan-1-
ol (Retention time: 3.61 minutes - Purity: 90%).
Example 29
1-[3-(Benzo[b]thiophen-3-yl)-5,6-dihydroimidazo(2,1-b]thiazol-2-yl]pentan-1-of
(Retention time: 3.68 minutes - Purity: 100%).
Example 30
1-[3-(Benzo[b]thiophen-3-yl)-5, 6-dihydroimidazo[2,1-b]thiazol-2-yl]prop-2-yn-
1-of
(Retention time: 2.71 minutes - Purity: 100%).
Example 31
1-[3-(Benzo[b]thiophen-3-yl)-5,6-dihydroimidazo[2,1-b]thiazol-2-yl]but-3-en-1-
of
(Retention time: 3.12 minutes - Purity: 96%).
Example 32
1-[3-(Benzo[b]thiophen-3-yl)-5,6-dihydroimidazo[2,1-b]thiazol-2-yl]-2-
methylprop-2-
en-1-of (Retention time: 3.13 minutes - Purity: 97%).
CA 02374926 2001-12-06
WO 00/71549 PCT/EP00/04279
59
Example 33
1-[3-(Benzo[b]thiophen-3-yl)-5,6-dihydroimidazo[2,1-b]thiazol-2-yl]pent-4-en-1-
of
(Retention time: 3.44 minutes - Purity: 85%).
Example 34
In a manner similar to that described in Example 4, 3-(benzo[b]thiophen-3-yl)-
5,6-dihydroimidazo[2,1-b]thiazole-2-carboxaldehyde (prepared via the
methodology
described in Example 6) was reacted with 2-methoxyphenylmagnesium bromide and
the product was recrystallised from a mixture of methanol and propan-2-of to
give
[3-(benzo[b]thiophen-3-yl)-5,6-dihydroimidazo[2,1-b]thiazol-2-yl](2-
methoxyphenyl)-
methanol as a white solid, m.p. 195 - 197 °C.
Example 35
n-Butyllithium (2.5M solution in hexanes; 73.8 ml) was added dropwise over
45 minutes at 0 - 4 °C under nitrogen to a stirred mixture of
methyltriphenylphosphonium bromide (65.7 g) and tetrahydrofuran (680 ml), then
the
mixture was stirred at 4 °C for 10 minutes and at ambient temperature
for 30
minutes. 3-(Benzo[b]thiophen-3-yl)-5,6-dihydroimidazo[2,1-b]thiazole-2-
carboxaldehyde (48 g; prepared in a manner similar to that described in
Example 6)
was added in portions at ambient temperature, then the mixture was heated
under
reflux for 3 hours, cooled to ambient temperature, and added to water (500
ml). The
product was extracted into ethyl acetate (3 x 400 ml), then the combined
extracts
were washed with water (400 ml), dried (MgS04), and the solvents were removed
in
vacuo. The residue was triturated with ethyl acetate (300 ml) and the
resulting solid
was collected by filtration and recrystallised from ethyl acetate to give an
off-white
solid. The liquors from the trituration and recrystallisation were combined
and
concentrated to give a further crop of solid. This was repeated until no
further solid
was isolable. All of the crops of solid were combined and repeatedly
recrystallised
from ethyl acetate until >99% pure (by hplc) to give 3-(benzo[b]thiophen-3-yl)-
2-vinyf-
5,6-dihydroimidazo[2,1-b]thiazole as an off-white solid (43.5 g). The majority
of the
solid (41 g) was dissolved in warm methanol (500 ml) and added to a saturated
solution of fumaric acid (16.7 g) in methanol, then the solvent was removedin
vacuo.
The residue was stirred with ether (500 ml) for 3 hours and the resulting
solid was
CA 02374926 2001-12-06
WO 00/71549 PCT/EP00/04279
collected by filtration and dried in vacuo at ambient temperature for 24 hours
to give
3-(benzo[b]thiophen-3-yl)-2-vinyl-5,6-dihydroimidazo[2,1-b]thiazole fumarate
(56.8 g)
as a white solid, m.p. 161 - 162 °C.
5 Example 36
In a manner similar to that described in Example 35, 3-(benzo[b]thiophen-3-
yl)-5,6-dihydroimidazo[2,1-b]thiazole-2-carboxaldehyde was reacted with
ethyltriphenylphosphonium bromide and n-butyllithium to give crude product
which
was purified by flash chromatography over silica using a 19:1 mixture of
10 dichloromethane and methanol as eluant. Appropriate fractions were combined
and
the solvents removed in vacuo to give a 3.7:1 mixture of E- and Z-3-
(benzo[b]thiophen-3-yl)-2-prop-1-enyl-5,6-dihydroimidazo[2,1-b]thiazole as a
yellow
solid, m.p. 62 - 68 C.
15 Example 37
In a manner similar to that described in Example 6, 3-(benzo[b]thiophen-3-yl)-
6,7-dihydro-5H-thiazolo[3,2-a]pyrimidine (prepared in a manner similar to that
described in WO 97/02269) was brominated, then reacted with ethylmagnesium
chloride followed by dimethylformamide to give 3-(benzo[b]thiophen-3-yl)-6,7-
20 dihydro-5H-thiazolo[3,2-a]pyrimidine-2-carboxaldehyde. This was reduced
with
sodium borohydride in a manner similar to that described in Example 2 to give
[3-
(benzo[b]thiophen-3-yl)-6,7-dihydro-5H-thiazolo[3,2-a]pyrimidin-2-yl]methanol
as an
off-white solid, m.p. 174 - 176 °C.
25 Example 38
A mixture of 3-(benzo[b]thiophen-3-yl)-5,6-dihydroimidazo[2,1-b]thiazole-2-
carboxaldehyde (0.5 g; prepared in a manner similar to that described in
Example 6),
hydroxylamine hydrochloride (0.16 g) and formic acid (1.3 ml) was heated at 90
- 95
°C for 25 hours, then diluted with ether (50 ml). The resulting solid
was collected by
30 filtration, washed with ether (30 ml) and purified by flash chromatography
over silica
CA 02374926 2001-12-06
WO 00/71549 PCT/EP00/04279
61
using 95:5 followed by 85:15 mixtures of dichloromethane and methanol as
eluants.
Appropriate fractions were combined, and the solvents were removed in vacuo to
leave 3-(benzo[b]thiophen-3-yl)-5,6-dihydroimidazo[2,1-b]thiazole-2-
carbonitrile 0.3
formate (0.12 g) as a white solid, m.p. 195 - 196 °C.
Example 39
A solution of diethyl benzylphosphonate (1.5 ml) in tetrahydrofuran (10 ml)
was added dropwise at ambient temperature under nitrogen to a stirred
suspension
of sodium hydride (60% dispersal in mineral oil; 0.26 g) in tetrahydrofuran
(15 ml),
then the mixture was stirred at ambient temperature for 20 minutes.
3-(Benzo[b]thiophen-3-yl)-5,6-dihydroimidazo[2,1-b]thiazole-2-carboxaldehyde
(0.83
g; prepared in a manner similar to that described in Example 6) was added in
one
portion, the mixture was stirred at ambient temperature for 72 hours, then it
was
quenched by the addition of water (30 ml). The product was extracted into
dichloromethane (3 x 30 ml), the combined extracts were dried (MgS04), and the
solvents were removed in vacuo. The residue was triturated with ether (20 ml)
and
the resulting solid was collected by filtration. The ethereal liquors were
concentrated
in vacuo and the residue was triturated with ether (10 ml) to give a second
crop of
solid. The combined solids were dried in vacuo to give 3-(benzo[b]thiophen-3-
yl)-2-
styryl-5,6-dihydroimidazo[2,1-b]thiazole (0.49 g) as a yellow solid, m.p. 153 -
155 °C.
Example 40
In a manner similar to that described in Example 6, 3-(5-chloro-
benzo[b]thiophen-3-yl)-5,6-dihydroimidazo[2,1-b]thiazole (prepared by
basification of
the hydrobromide salt obtained in a manner similar to that described in
WO 97/02269) was brominated, then reacted with ethylmagnesium chloride
followed
by dimethylformamide to give 3-(5-chlorobenzo[b]thiophen-3-yl)-5,6-dihydro-
imidazo[2,1-b]thiazole-2-carboxaldehyde as a yellow solid, m.p. 258 - 260
°C,
CA 02374926 2001-12-06
WO 00/71549 PCT/EP00/04279
62
Example 41
Sodium borohydride (0.06 g) was added to a stirred suspension of 3-(5-
chlorobenzo[b]thiophen-3-yl)-5,6-dihydroimidazo[2,1-b]thiazole-2-
carboxaldehyde
(0.31 g) in ethanol (15 ml), the mixture was stirred at ambient temperature
for 4
hours, then water (15 ml) was added. The resulting solid was collected by
filtration,
washed with ether (15 ml) and dried in vacuo at 60 °C to give [3-(5-
chlorobenzo[b]thiophen-3-yl)-5,6-dihydroimidazo[2,1-b]thiazol-2-yl]methanol
(0.14 g)
as a white solid, m.p. 204 - 206 °C.
Example 42
[3-(5-Chlorobenzo[b]thiophen-3-yl)-5,6-dihydroimidazo[2,1-b]thiazol-2-yl]-
methanol (0.6 g) was prepared in a manner similar to that described in
Examples 40
and 41. However, on this occasion the product was impure. Consequently it was
purified by preparative-scale hplc using mixtures of acetonitrile and aqueous
triethylammonium formate buffer as eluant. Appropriate fractions were combined
and the solvents were removed in vacuo. The residue was triturated with water
(2 x
3 ml) and the resulting solid was collected by filtration and dried in vacuo
at 60 °C to
give [3-(5-chlorobenzo[b]thiophen-3-yl)-5,6-dihydroimidazo[2,1-bJthiazol-2-ylJ-
methanol formate (0.19 g) as an off-white solid, m.p. 171 - 173 °C.
Example 43
In a manner similar to that described in Example 23, 3-(benzo[b]thiophen-3-
yl)-2-bromo-5,6-dihydroimidazo[2,1-b]thiazole hydrobromide (5 g) was reacted
with
ethylmagnesium chloride followed by Biphenyl disulphide to give
3-(benzo[b]thiophen-3-yl)-2-(phenylthio)-5,6-dihydroimidazo[2,1-b]thiazole
(0.6 g) as
a yellow solid, m.p. 123 - 125 °C.
CA 02374926 2001-12-06
WO 00/71549 PCT/EP00/04279
63
Example 44
Methylamine (2M solution in tetrahydrofuran; 8.7 ml) and sodium
triacetoxyborohydride (0.56 g) were added under nitrogen to a stirred solution
of
3-(benzo[b]thiophen-3-yl)-5,6-dihydroimidazo[2,1-b]thiazole-2-carboxaldehyde
(0.5
g; prepared in a manner similar to that described in Example 6) in
tetrahydrofuran
(20 ml), then the mixture was stirred at ambient temperature for 72 hours.
Further
methylamine solution (4.3 ml) was added, the mixture was stirred at ambient
temperature for 48 hours, then it was quenched by the addition of saturated
aqueous
sodium hydrogencarbonate solution (50 ml). The product was extracted into
ethyl
acetate (3 x 30 ml), the combined extracts were washed with water (30 ml) and
saturated aqueous sodium chloride solution (30 ml), then they were dried
(MgS04)
and the solvents were removed in vacuo. The residue was purified by flash
chromatography over silica using a 9:1 mixture of dichloromethane and methanol
as
eluant. Appropriate fractions were combined and the solvents were removed in
vacuo to give [3-(benzo[b]thiophen-3-yl)-5,6-dihydroimidazo[2,1-b]thiazol-2-
yl]-N-
methylmethylamine (0.19 g) as a waxy solid, m.p. 102 - 104 °C.
Example 45
A solution of oxalyl chloride (13 ml) in dichloromethane (20 ml) was added
dropwise at -10 - 0 °C under nitrogen to a stirred solution of
cyclopropylacetic acid
(5 g) and dimethylformamide (2 drops) in dichloromethane (20 ml), then the
mixture
was stirred at ambient temperature for 24 hours and the solvent was removed in
vacuo to leave cyclopropylacetyl chloride as a brown oil which was used
without
purification.
Potassium carbonate (9.2 g) was added in portions at 0 - 5 °C over
10
minutes to a stirred solution of N, O-dimethylhydroxylamine hydrochloride (5.1
g) in
the minimum volume of water, then dichloromethane (30 ml) was added. The above
cyclopropylacetyl chloride was dissolved in dichloromethane (20 ml) and the
solution
was added dropwise at -5 - 0 °C to the dimethylhydroxylamine solution.
The
mixture was stirred at 0 °C for 30 minutes and at ambient temperature
for 2 hours,
then the product was extracted into dichloromethane (3 x 50 ml). The combined
extracts were dried (NazS04) and the solvent was removed in vacuo to give
CA 02374926 2001-12-06
WO 00/71549 PCT/EP00/04279
64
cyclopropyl-N-methoxy-N-methylacetamide (6.2 g) as a pale brown oil which was
used without purification.
A few drops of a solution of 3-bromobenzo[b]thiophene (8.7 g) in
tetrahydrofuran (35 ml) were added under nitrogen to a mixture of magnesium
turnings (1.05 g), 2 crystals of iodine and tetrahydrofuran (5 ml), and the
mixture was
warmed gently to initiate the reaction. The remainder of the solution was then
added
at a rate sufficient to maintain gentle relux. When the addition was complete,
the
mixture was stirred at reflux temperature for 1.5 hours then allowed to cool
to
ambient temperature. A solution of cyclopropyl-N-methoxy-N-methylacetamide (6
g)
in tetrahydrofuran (35 ml) was added at ambient temperature, the mixture was
stirred
at reflux temperature for 5 hours, and allowed to stand at ambient temperature
for 18
hours, then it was quenched by the addition of 2M hydrochloric acid (50 ml)
and
stirred at ambient temperature for 1 hour. The product was extracted into
ethyl
acetate (2 x 100 ml), the combined extracts were washed with water (2 x 30 ml)
and
saturated aqueous sodium chloride solution (2 x 30 ml), then they were dried
(MgS04) and the solvents were removed in vacuo. The residue was purified by
flash
chromatography over silica using a 1:3 mixture of dichloromethane and
petroleum
ether (b.p. 60 - 80 °C) as eluant. Appropriate fractions were combined
and the
solvents were removed in vacuo to give 1-(benzo[b]thiophen-3-yl)-2-cyclopropyl-
ethan-1-one (4.15 g) as an orange oil which was used without further
purification.
Phenyltrimethylammonium tribromide (1.74 g) was added under nitrogen to a
stirred solution of 1-(benzo[b]thiophen-3-yl)-2-cyclopropylethan-1-one (1 g)
in
tetrahydrofuran (15 ml), the mixture was stirred at ambient temperature for 18
hours,
then it was filtered and the solvent was removed in vacuo. The residue was
dissolved in ethanol (12 ml), 2-imidazolidinethione (0.47 g) and acetic acid
(4 ml)
were added, the mixture was heated under reflux under nitrogen for 18 hours,
then
the solvents were removed in vacuo. The residue was purified by flash
chromatography over silica using a 1:3 mixture of dichloromethane and ether as
eluant. Appropriate fractions were combined and the solvents were removed in
vacuo to leave 3-(benzo[b]thiophen-3-yl)-2-cyclopropyl-5,6-dihydroimidazo[2,1-
b]-
thiazole hydrobromide (0.72 g) as an off-white solid, m.p. 181 - 183
°C.
CA 02374926 2001-12-06
WO 00/71549 PCT/EP00/04279
Example 46
A solution of iodine monochloride (1.83 g) in dichloromethane (5 ml) was
added dropwise at ambient temperature to a stirred solution of 3-
(benzo[b]thiophen-
3-yl)-5,6-dihydroimidazo[2,1-b]thiazole (3.8 g; prepared in a manner similar
to that
5 described in Example 6) in dichloromethane (200 ml) and the mixture was
stirred at
ambient temperature for 15 minutes. The resulting solid was colected by
filtration,
washed with dichloromethane (50 ml) and dried in air to give 3-
(benzo[b]thiophen-3-
yl)-2-iodo-5,6-dihydroimidazo[2,1-b]thiazo1e hydrochloride (1.3 g) as a pale
yellow
solid, m.p. 194.7 - 195.2 °C.
Example 47
A solution of dimethyl 2-oxopropylphosphonate (1.28 g) in tetrahydrofuran (10
ml) was added dropwise at ambient temperature under nitrogen to a stirred
suspension of sodium hydride (60% dispersion in mineral oil; 0.46 g) in
tetrahydrofuran (15 ml), then the mixture was stirred at ambient temperature
for 20
minutes. 3-(Benzo[b]thiophen-3-yl)-5,6-dihydroimidazo[2,1-b]thiazole-2-carbox-
aldehyde (2 g; prepared in a manner similar to that described in Example 6)
was
added in portions, the mixture was stirred at ambient temperature for 18 hours
and at
reflux temperature for 7 hours, then it was allowed to stand at ambient
temperature
for 18 hours. The solvent was removed in vacuo, the residue was diluted with
water
(200 ml), and the product was extracted into dichloromethane (200 ml). The
extract
was dried (Na2S04), the solvent was removed in vacuo, then the residue was
purified by flash chromatography over silica using a 19:1 mixture of
dichloromethane
and methanol as eluant. Appropriate fractions were combined and the solvents
were
removed in vacuo to give 4-[3-(benzo[b]thiophen-3-yl)-5,6-dihydroimidazo[2,1-
b]thiazol-2-yl]but-3-ene-2-one (1.15 g) as a yellow solid, m.p. 164 - 166
°C.
Sodium borohydride (36 mg) was added to a stirred solution of
4-[3-(benzo[b]thiophen-3-yl)-5,6-dihydroimidazo[2,1-b]thiazol-2-yl]but-3-ene-2-
one
(0.28 g) in ethanol (10 ml), the mixture was stirred at ambient temperature
for 3
hours, then the solvent was removed in vacuo. The residue was diluted with
water
(100 ml) and the product was extracted into dichloromethane (100 ml), then the
extract was dried (NazS04) and the solvent was removed in vacuo. The residue
was
triturated with ether (20 ml) and the resulting solid was collected by
filtration and
CA 02374926 2001-12-06
WO 00/71549 PCT/EP00/04279
66
dried in vacuo to give 4-[3-(benzo[b]thiophen-3-yl)-5,6-dihydroimidazo[2,1-
b]thiazol-
2-yl]but-3-en-2-of (0.16 g) as an off-white solid, m.p. 161 - 162 °C.
Example 48
In a manner similar to that described in Example 17, 3-(benzo[b]thiophen-3-
yl)-2-bromo-5,6-dihydroimidazo[2,1-b]thiazole hydrobromide was reacted with
ethylmagnesium chloride followed by 3-bromo-2-methylpropene and fumaric acid
to
give 3-(benzo[b)thiophen-3-yl)-2-(2-methylprop-2-enyl)-5,6-dihydroimidazo[2,1-
b]-
thiazole fumarate as an off-white solid, m.p. 54 - 64 °C.
Example 49
A solution of tetraethyl (dimethylamino)methylenediphosphonate (2.32 g) in
1,4-dioxane (5 ml) was added dropwise at ambient temperature under nitrogen to
a
stirred suspension of sodium hydride (60% dispersion in mineral oil; 0.28 g)
in 1,4-
dioxane (5 ml), and the mixture was stirred at ambient temperature until the
evolution
of hydrogen ceased. 3-(Benzo[b]thiophen-3-yl)-5,6-dihydroimidazo[2,1-
b]thiazole-2-
carboxaldehyde (2 g; prepared in a manner similar to that described in Example
6)
was added, the mixture was stirred at 60 °C for 1.3 hours and at
ambient
temperature for 18 hours, then it was poured into water (50 ml). The product
was
extracted into ethyl acetate (3 x 30 ml), the combined extracts were dried
(MgS04),
and the solvents were removed in vacuo. The residue was purified by flash
chromatography over silica using a 93:7 mixture of dichloromethane and
methanol
as eluant. Appropriate fractions were combined and the solvents were removed
in
vacuo to give diethyl 2-[3-(benzo[b]thiophen-3-yl)-5,6-dihydroimidazo[2,1-
b]thiazol-2-
yl]-1-(dimethylamino)ethenylphosphonate (1.78 g) as a red oil which was used
without further purification.
A mixture of 2-[3-(benzo[b]thiophen-3-yl)-5,6-dihydroimidazo[2,1-b]thiazol-2-
yl]-1-(dimethylamino)ethenylphosphonate (1.7 g) and concentrated hydrochloric
acid
(12 ml) was heated under reflux for 1 hour then cooled in ice. The product was
extracted into dichloromethane (3 x 30 ml), the combined extracts were dried
(MgS04) and the solvent was removed in vacuo. The residue was dissolved in
water
CA 02374926 2001-12-06
WO 00/71549 PCT/EP00/04279
67
(30 ml), the solution was neutralised by the addition of an excess of
saturated
aqueous sodium hydrogencarbonate solution, and the product was extracted into
dichloromethane (3 x 30 ml). The combined extracts were dried (MgS04) and the
solvent was removed in vacuo. The residue was dissolved in ethanol (2 ml),
oxalic
acid (15 mg) was added, and the solvent was removed in vacuo. The residue was
triturated with ether (10 ml) and the resulting solid was collected by
filtration and
dried in vacuo to give ethyl [3-(benzo[b]thiophen-3-yl)-5,6-dihydroimidazo[2,1-
b]thiazol-2-yl]acetate oxalate (40 mg) as an off-white solid, m.p.102 - 104
°C.
Example 50
sec-Butyllithium (1.25M solution in a 92:8 mixture of cyclohexane and
hexane; 100 ml) was added dropwise at -70 °C under nitrogen to a
stirred solution of
4-fluorophenyl methyl sulphide (22.0 g) in tetrahydrofuran, then the mixture
was
stirred at -70 °C for 65 minutes. Dimethylformamide (13.2 ml) was added
dropwise
at -68 °C to -70 °C, the stirred mixture was allowed to warm to
ambient temperature
slowly over 20 hours, then it was added to a solution of acetic acid (10 ml)
in water
(500 ml). The product was extracted into ether (3 x 150 ml), then the combined
extracts were washed with 2M hydrochloric acid (200 ml) and saturated aqueous
sodium chloride solution (200 ml), dried (NaZS04), and the solvents were
removed in
vacuo. The residue (25 g), which was used without further purification, was
estimated to consist predominantly of a 2:3 mixture of 4-fluorophenyl methyl
sulphide
and 2-fluoro-5-(methylthio)benzaldehyde respectively by nuclear magnetic
resonance spectroscopy.
A stirred mixture of methyl thioglycolate (8.9 ml), crude 2-fluoro-5-
(methylthio)benzaldehyde (25 g), dimethyl acetamide (250 ml) and N,N-
diisopropyl-
ethylamine (42 ml) was heated at 140 - 150 °C under nitrogen for 3.5
hours, then
the solvent was removed in vacuo. Water (650 ml) was added to the residue,
then
the product was extracted into dichloromethane (3 x 150 ml). The combined
extracts
were washed with water (3 x 150 ml), dried (Na2S04), and the solvent removed
in
vacuo. The residue was purified by trituration with ether (250 ml), then the
resulting
solid was collected by filtration, washed with ether (2 x 50 ml), and dried in
vacuo to
give methyl 5-(methylthio)benzo[b]thiophene-2-carboxylate (13.3 g) as a pale
yellow
solid, m.p. 89.7 - 90.4 °C.
CA 02374926 2001-12-06
WO 00/71549 PCT/EP00/04279
68
Sodium hydroxide (8 g) was dissolved in water (100 ml) then the solution was
added to a stirred solution of methyl 5-(methylthio)benzo[b)thiophene-2-
carboxylate
(23.8 g; prepared in a manner similar to that described above) in methanol
(300 ml).
The mixture was heated under reflux for 10 minutes then allowedto stand at
ambient
temperature for 3 days. The suspension was concentrated to a total volume of
approximately 250 ml by heating, then water (100 ml) was added and the mixture
hot
filtered to remove insoluble material. A solution of concentrated hydrochloric
acid
(40 ml) in water (20 ml) was added to the filtrate and the resulting solid was
collected
by filtration and washed with water, then dried in vacuo at 80 °C to
give
5-(methylthio)benzo[b]thiophene-2-carboxylic acid (21 g) as a pale yellow
solid, m.p.
186 - 186.5 °C.
A mixture of copper powder (5.3 g), 5-(methylthio)benzo[b]thiophene-2-
carboxylic acid (20 g) and quinoline (100 ml) was stirred and heated under
reflux
under nitrogen for 30 minutes, then hot filtered. The filtrate was added to a
mixture of
concentrated hydrochloric acid (100 ml), ice (500 g) and ether (200 ml), then
the
resulting solid was collected by filtration and washed with ether (200 ml).
The
aqueous phase was separated and further product extracted from it into ether
(2 x
150 ml). The combined ethereal solutions were washed with 2M hydrochloric acid
(200 ml) and water (200 ml), dried (Na2S04), and the solvent removed in vacuo
to
give 5-(methylthio)benzo[b]thiophene (14.7 g) as a light brown solid which was
used
without further purification.
A solution of 5-(methylthio)benzo[b]thiophene (14.7 g) in dichloromethane
(180 ml) was added at <0 °C under nitrogen to a stirred mixture of
aluminium
bromide (26.2 g), bromoacetyl bromide (7.12 ml) and dichloromethane (120 ml),
the
resulting dark red-brown solution was stirred at <0 °C for 30 minutes
and at ambient
temperature for 20 hours, then it was added to a mixture of ice (600 g) and
concentrated hydrochloric acid (50 ml). Dichloromethane (300 ml) was added and
insoluble materials were removed by filtration (Celite ~). The aqueous phase
was
separated and further product extracted from it into dichloromethane (300 ml),
then
the combined dichloromethane solutions were dried (MgS04), and the solvent was
removed in vacuo. The residue was dissolved in a mixture of acetic acid (200
ml)
and dichloromethane (200 ml), a solution of 2-imidazolidinethione (5.29 g) in
acetic
acid (200 ml) was added, then the dichloromethane was removed by distillation.
The
CA 02374926 2001-12-06
WO 00/71549 PCT/EP00/04279
69
remaining mixture was heated under reflux for two hours, then the total volume
was
reduced to approximately 200 ml by heating in vacuo. The hot solution was
removed
from insoluble material by decantation, then allowed to cool. Further solid
precipitated and was removed as before, then the solvents were removed in
vacuo
at 50 °C. The residue was mixed with water (300 ml) and 5M aqueous
sodium
hydroxide solution (300 ml) then the product was extracted into
dichloromethane
(300 ml). The extract was dried (Na2S04), and the solvent was removed in
vacuo.
The residue was partially purified by flash chromatography over silica using
ethyl
acetate then an 8:1:1 mixture of ethyl acetate, methanol and triethylamine as
eluants. Appropriate fractions were combined and the solvents removed in vacuo
to
leave 3-[5-(methylthio)benzo[b]thiophen-3-yl]-5,6-dihydroimidazo[2,1-
b]thiazole as a
yellow-brown gum (3.6 g), which was used without further purification.
A solution of bromine (1.87 g) in dichloromethane (10 ml) was added
dropwise at 15 - 20 °C to a stirred solution of 3-[5-
(methylthio)benzo[b]thiophen-3-
yl]-5,6-dihydroimidazo[2,1-b]thiazole (3.55 g) in dichloromethane (80 ml), the
mixture
was stirred at ambient temperature for 10 minutes, then the dichloromethane
solution was separated by decantation from insoluble materials and
concentrated in
vacuo. The residue was triturated with dichloromethane (10 ml) and the
resulting
solid was collected by filtration and dried in vacuo to give 2-bromo-3-[5-
(methylthio)-
benzo[b]thiophen-3-yl]-5,6-dihydroimidazo[2,1-b]thiazole hydrobromide (2.97 g)
as a
yellow-brown solid, m.p. 260 - 265 °C.
Example 51
Ethylmagnesium chloride (2.8M solution in tetrahydrofuran; 8.76 ml) was
added at -10 °C under nitrogen to a stirred suspension of 2-bromo-3-[5-
(methylthio)-
benzo[b]thiophen-3-yl]-5,6-dihydroimidazo[2,1-b]thiazole hydrobromide (2.9 g)
in
tetrahydrofuran (40 ml), then the mixture was stirred at ambient temperature
for 90
minutes and cooled to -5 °C. Dimethylformamide (5 ml) was added, the
resulting
suspension was stirred for 2 hours at ambient temperature, then ethyl acetate
(200
ml) and saturated aqueous ammonium chloride solution (200 ml) were added. The
ethyl acetate layer was separated, dried (Na2S04), and the solvents removed in
vacuo to give 3-[5-(methylthio)benzo[b]thiophen-3-yl]-5,6-dihydroimidazo[2,1-
b]-
thiazole-2-carboxaldehyde (1.4 g) as a solid, m.p. 157-158.5 °C, which
was used
without further purification.
CA 02374926 2001-12-06
WO 00/71549 PCT/EP00/04279
Sodium borohydride (0.114 g) was added to a stirred solution of
3-[5-(methylthio)benzo(b]thiophen-3-yl]-5,6-dihydroimidazo(2,1-b]thiazole-2-
carbox-
aldehyde (0.664 g) in ethanol (40 ml), then the mixture was stirred at ambient
5 temperature for 24 hours and quenched by the addition of 5M hydrochloric
acid (10
ml). The mixture was basified by the addition of an excess of 1 M aqueous
sodium
hydroxide solution and concentrated in vacuo to remove ethanol, then the
product
was extracted into dichloromethane (3 x 30 ml). The combined extracts were
dried
(MgS04), the solvent was removed in vacuo, and the residue was purified by
Biotage
10 flash chromatography over silica using a 34:3:3 mixture of ethyl acetate,
industrial
methylated spirit and triethylamine as eluant. Appropriate fractions were
combined
and the solvents were removed in vacuo, then the residue was triturated with
ether
(10 ml). The resulting solid was collected by filtration and dried in air to
give
{3-[5-(methylthio)benzo[b]thiophen-3-yl]-5,6-dihydroimidazo[2,1-b]thiazol-2-
yl}-
15 methanol (0.13 g) as a beige solid, m.p. 164 - 167 °C.
Example 52
In a manner similar to that described in Example 22, [3-(benzo[b]thiophen-3-
20 yl)-5,6-dihydroimidazo[2,1-b]thiazol-2-yl]methanol (0.5 g) was reacted with
sodium
hydride and bromomethylcyclopropane followed by fumaric acid. In this case the
salt
formation was carried out in methanol solution and no solid product
precipitated.
Consequently, the solvent was removed in vacuo and the residue was triturated
with
ether (10 ml). The resulting solid was collected by filtration and dried in
vacuo to
25 give 3-(benzo[b]thiophen-3-yl)-2-cyclopropylmethoxymethyl-5,6-
dihydroimidazo-
[2,1-b]thiazole fumarate (40 mg) as a pale brown solid, m.p. 82 - 98
°C.
Example 53
30 Sodium hydride (60% dispersion in mineral oil; 0.153 g) was added in
portions at ambient temperature over 10 minutes to a stirred suspension of
[3-(benzo[b]thiophen-3-yl)-5,6-dihydroimidazo[2,1-b]thiazol-2-yl]methanol (1
g) in
dimethylformamide (50 ml), then the mixture was stirred at ambient temperature
for 2
hours. 1-Chloroprop-2-yne (276 ~I) was added, and stirring at ambient
temperature
35 was continued for a further 2 hours. Further 1-chloroprop-2-yne (27.1) was
added
and the mixture was stirred at ambient temperature for 18 hours. Water (50 ml)
was
CA 02374926 2001-12-06
WO 00/71549 PCT/EP00/04279
71
added, the product was extracted into ethyl acetate (3 x 50 ml), then the
combined
extracts were washed with water (4 x 25 ml) and saturated aqueous sodium
chloride
solution (25 ml), dried (MgS04), and the solvents were removed in vacuo. The
residue was purified by flash chromatography over silica using 5 - 8% mixtures
of
methanol in dichloromethane as eluant. Appropriate fractions were combined and
the solvents were removed in vacuo. The residue was dissolved in methanol (15
ml), fumaric acid (0.185 g) was added, the mixture was stirred at ambient
temperature for 5 hours, then it was warmed to dissolve the product and
filtered to
remove a small amount of insoluble material. The solvent was removed in vacuo
to
give 3-(benzo[b]thiophen-3-yl)-2-prop-2-ynyloxymethyl-5,6-dihydroimidazo[2,1-
b]
thiazole fumarate (0.67 g) as a brown solid, m.p. 120 °C (softens 80 -
90 °C).
Example 54
A solution of potassium hydroxide (4.87 g) in a mixture of ethanol (140 ml)
and water (35 ml) was added in one portion at ambient temperature under
nitrogen
to a stirred solution of 2-methoxybenzenethiol (10.6 ml) in a mixture of
ethanol (125
ml) and water (7.5 ml), then the mixture was stirred at ambient temperature
for 3.5
hours. A solution of 1-chloro-4-phenoxybut-2-yne (15.7 g) in a mixture of
ethanol
(125 ml) and water (7.5 ml) was added dropwise over 1.5 hours, then the
mixture
was stirred at ambient temperature for 18 hours, and the solvents were removed
in
vacuo. The residue was diluted with water (75 ml), the product was extracted
into
ethyl acetate (2 x 115 ml), then the combined extracts were washed with water
(50
ml) and saturated aqueous sodium chloride solution (50 ml), dried (MgS04), and
the
solvent was removed in vacuo to leave 1-(2-methoxyphenylthio)-4-phenoxybut-2-
yne
(24.7 g) as a yellow oil which was used without further purification.
A solution of 3-chloroperoxybenzoic acid (70% purity; 9.1 g) in chloroform
(210 ml) was added dropwise at 0 - 5 °C over 1.5 hours to a stirred
solution of
1-(2-methoxyphenylthio)-4-phenoxybut-2-yne (10.5 g) in chloroform (95 ml), the
mixture was stirred at ambient temperature for 18 hours, then it was washed
with 5%
aqueous sodium carbonate solution (3 x 120 ml) and water (3 x 80 ml) and dried
(MgS04). The chloroform solution was stirred and heated under reflux for 7
hours
and allowed to stand at ambient temperature for 18 hours, then it was washed
with
5M aqueous sodium hydroxide solution (115 ml), water (4 x 110 ml) and
saturated
aqueous sodium chloride solution (100 ml) and dried (MgS04). The solvent was
CA 02374926 2001-12-06
WO 00/71549 PCT/EP00/04279
72
removed in vacuo to leave 1-(7-methoxy-2,3-dihydrobenzo[b]thiophen-3-yl)-2-
phenoxyethan-1-one (10.7 g) as a brown gum which was used without further
purification.
A mixture of 1-(7-methoxy-2,3-dihydrobenzo[b]thiophen-3-yl)-2-phenoxy-
ethan-1-one (10.7 g), acetic acid (100 ml) and concentrated sulphuric acid (12
drops)
was heated at 90 - 95 °C for 3 hours then cooled to ambient temperature
and
poured onto water (800 ml). The product was extracted into dichloromethane (2
x
250 ml), the combined extracts were washed with 2M aqueous sodium hydroxide
solution (2 x 200 ml) and water (3 x 200 ml), then they were dried (MgS04) and
the
solvents were removed in vacuo. The residue was purified by flash
chromatography
over silica using a 1:1 mixture of dichloromethane and petroleum ether (b.p.
60 - 80
°C) as eluant. Appropriate fractions were combined and the solvents
were removed
in vacuo to give 1-(7-methoxybenzo[b]thiophen-3-yl)ethan-1-one (2 g) as a
brown oil
which was used without further purification.
Phenyltrimethylammonium tribromide (2.55 g) was added in portions at
ambient temperature under nitrogen over 20 minutes to a stirred solution of
1-(7-methoxybenzo[b]thiophen-3-yl)ethan-1-one (1.4 g) in tetrahydrofuran (40
ml),
the mixture was stirred at ambient temperature for 1 hour, then it was
filtered and the
solvent was removed in vacuo. The residue was dissolved in ethanol (30 ml),
2-imidazolidinethione (0.69 g) and acetic acid (20 ml) were added, the mixture
was
heated under reflex for 18 hours, then it was cooled to ambient temperature.
The
resulting solid was collected by filtration, washed with ether (20 ml) and
dried in
vacuo at 60 °C to give 3-(7-methoxybenzo[b]thiophen-3-yl)-5,6-
dihydroimidazo[2,1-
b]thiazole hydrobromide (1.48 g) as a grey solid, m.p. 277 - 279 °C.
3-(7-Methoxybenzo[b]thiophen-3-yl)-5,6-dihydroimidazo[2,1-b]thiazole
hydrobromide (1.48 g) was basified by the addition of 2M aqueous sodium
hydroxide
solution (110 ml) and the free base was extracted into dichloromethane (150
ml).
The extract was washed with water (2 x 70 ml), dried (MgS04), and the solvent
was
removed in vacuo. The residue was dissolved in dichloromethane (23 ml), then
the
stirred solution was cooled to 0 - 5°C and a solution of bromine (0.68
g) in
dichloromethane (4.7 ml) was added dropwise over 45 minutes. The mixture was
stirred at 0 - 5 °C for 30 minutes and allowed to stand at ambient
temperature for 18
CA 02374926 2001-12-06
WO 00/71549 PCT/EP00/04279
73
hours, then the resulting solid was collected by filtration, washed with
dichloromethane (20 ml) and dried in vacuo to give 2-bromo-3-(7-methoxy-
benzo[b]thiophen-3-yl)-5,6-dihydroimidazo[2,1-b]thiazole hydrobromide (1.22g)
as a
white solid, m.p. 240 - 242 °C.
Example 55
Sodium hydride (60% dispersion in mineral oil; 0.38 g) was added in portions
at ambient temperature over 10 minutes to a stirred suspension of [3-(benzo[b]-
thiophen-3-yl)-5,6-dihydroimidazo[2,1-b]thiazol-2-yl]methanol (2.5 g) in
dimethyl-
formamide (50 ml), then the mixture was stirred at ambient temperature for 2
hours.
2-Bromopropane (0.9 ml) was added and stirring at ambient temperature was
continued for 2 hours. Further 2-bromopropane (0.4 ml) was added and the
mixture
was stirred at ambient temperature for 72 hours. Sodium hydride (60%
dispersion in
mineral oil; 0.07 g) was added, the mixture was stirred for 10 minutes, then
2-bromopropane (0.16 ml) was added and the mixture was stirred at ambient
temperature for 18 hours. Water (100 ml) was added, the product was extracted
into
ethyl acetate (100 + 2 x 75 ml), then the combined extracts were washed with
water
(3 x 100 ml) and saturated aqueous sodium chloride solution (100 ml), dried
(MgS04), and the solvents were removed in vacuo. The residue was purified by
flash chromatography over silica using a 1:19 mixture of methanol and
dichloromethane as eluant. Appropriate fractions were combined and the
solvents
were removed in vacuo. The residue was purified by flash chromatography over
silica using a 9:1 mixture of ethyl acetate and methanol as eluant.
Appropriate
fractions were combined and the solvents were removed in vacuo to give
3-(benzo[b)thiophen-3-yl)-2-isopropoxymethyl-5,6-dihydroimidazo[2,1-b]thiazole
(0.12 g) as a yellow solid, m.p. 64 - 66 °C.
Example 56
Sodium hydride (60% dispersion in mineral oil; 0.46 g) was added in portions
at ambient temperature over 10 minutes to a stirred suspension of [3-(benzo[b]-
thiophen-3-yl)-5,6-dihydroimidazo[2,1-b)thiazol-2-yl]methanol (3 g) in
dimethyl-
formamide (80 ml), then the mixture was stirred at ambient temperature for 2
hours.
Bromomethylcyclobutane (1.7 g) was added dropwise and the mixture was stirred
at
ambient temperature for 2.5 hours. Further bromomethylcyclobutane (0.16 g) was
CA 02374926 2001-12-06
WO 00/71549 PCT/EP00/04279
74
added dropwise and the mixture was stirred at ambient temperature for 1 hour.
Further bromomethylcyclobutane (0.16 g) was added dropwise and the mixture was
stirred at ambient temperature for 18 hours. Tlc showed starting material
still
remained, so bromomethylcyclobutane (0.32 g) was added and the mixture was
stirred at ambient temperature for 2 hours and at 55 °C for 1 hour.
Further sodium
hydride (0.042 g) and bromomethylcyclobutane (0.8 g) were added and the
mixture
was stirred at 55 °C for 1 hour and at ambient temperature for 18
hours. Water
(100 ml) was added, the product was extracted into ethyl acetate (100 + 3 x 75
ml),
then the combined extracts were washed with water (4 x 75 ml) and saturated
aqueous sodium chloride solution (75 ml), dried (MgS04), and the solvents were
removed in vacuo. The residue was purified by flash chromatography over silica
using a 1:19 mixture of methanol and dichloromethane as eluant. Appropriate
fractions were combined and the solvents were removed in vacuo. The residue
was
purified by flash chromatography over silica using a 3:1 mixture of petroleum
ether
(b.p. 60 - 80 °C) and ethyl acetate followed by a 19:1 mixture of
dichloromethane
and methanol as eluants. Appropriate fractions were combined and the solvents
were removed in vacuo. A sample (30 mg from 70mg) of the residue was further
purified by preparative scale hplc on a C8 symmetry shield column using a 2:3
mixture of acetonitrile and triethylammonium formate buffer as eluant.
Appropriate
fractions were combined and the solvents removed in vacuo to give
3-(benzo[b]thiophen-3-yl)-2-cyclobutylmethoxymethyl-5,6-dihydroimidazo[2,1-b]-
thiazole (12 mg) as a brown gum, ~H-nmr (DMSO-ds): 8H 1.61 - 1.84 (6H, m, 3 x
cyclobutane CH2), 2.36 - 2.41 (1 H, m, cyclobutane CH), 3.22 (2H, d, CH20),
3.49 -
3.61 (2H, m, CH2N), 3.98 - 4.05 (2H, m, CH20), 4.11 - 4.15 (2H, m, CH2N), 7.45
-
7.49 (2H, m, 2 x ArH), 7.77 - 7.81 (1 H, m, ArH), 8.01 (1 H, s, ArH), 8.08 -
8.11 (1 H,
m, ArH).
Example A
The use of compounds of the present invention in the manufacture of
pharmaceutical compositions is illustrated by the following description. In
this
description the term "active compound" denotes any compound of the invention
but
particularly any compound which is the final product of one of the preceding
Examples.
CA 02374926 2001-12-06
WO 00/71549 PCT/EP00/04279
a) Capsules
In the preparation of capsules, 10 parts by weight of active compound and
240 parts by weight of lactose are de-aggregated and blended. The mixture is
filled
5 into hard gelatin capsules, each capsule containing a unit dose or part of a
unit dose
of active compound.
b) Tablets
10 Tablets are prepared from the following ingredients.
Parts by weight
Active compound 10
Lactose 190
Maize starch 22
15 Polyvinylpyrrolidone 10
Magnesium stearate 3
The active compound, the lactose and some of the starch are de-aggregated,
blended and the resulting mixture is granulated with a solution of the
polyvinyl-
20 pyrrolidone in ethanol. The dry granulate is blended with the magnesium
stearate
and the rest of the starch. The mixture is then compressed in a tabletting
machine to
give tablets each containing a unit dose or a part of a unit dose of active
compound.
c) Enteric coated tablets
Tablets are prepared by the method described in (b) above. The tablets are
enteric coated in a conventional manner using a solution of 20% cellulose
acetate
phthalate and 3% diethyl phthalate in ethanol:dichloromethane (1:1 ).
d) Suppositories
In the preparation of suppositories, 100 parts by weight of active compound is
incorporated in 1300 parts by weight of triglyceride suppository base and the
mixture
formed into suppositories each containing a therapeutically effective amount
of
active ingredient.