Note: Descriptions are shown in the official language in which they were submitted.
CA 02376113 2001-12-03
WO 00/74657 PCT/US00/15547
.TAMES D. TALTON
FOR
METHODS FOR COATING PARTICLES AND
PARTICLES PRODUCED THEREBY
CA 02376113 2001-12-03
WO 00/74657 PCT/US00/15547
METHODS FOR COATING PARTICLES AND
PARTICLES PRODUCED THEREBY
II. BACKGROUND OF THE INVENTION
A. Related Applications
The present application claims priority under 35 U.S.C. ~ 119 to U.S.
Provisional Patent
Application Serial No. 60/137,733, filed June 7, 1999, and No. 60/138,006,
filed June 7, 1999.
The entire contents of each of the aforementioned applications is specifically
incorporated herein
by reference in its entirety.
B. Field of the Invention
The invention relates to methods of coating particles, and the particles
produced thereby.
More specifically, the invention relates to drug particles or drug delivery
particles coated with a
material, which may be biodegradable or biocompatible, such as a polymer. The
coating may
impart a number of characteristics to the particle, including altering its
surface properties, its rate
of dissolution, or its rate of diffusion and/or release of an active
component. More particularly,
the invention provides methods for preparing particulate compositions that are
coated with
ultrafine layers of coating materials, preferably organic polymeric coating
materials, applied
through a non-aqueous, non-solvent technique. A particularly preferred process
is a vapor
deposition process such as pulsed laser ablation. Among the many advantages of
the disclosed
methods are control of both the thickness and uniformity of the coating on the
surfaces of the
selected particulate drug.
2
CA 02376113 2001-12-03
WO 00/74657 PCT/US00/15547
C. Description of Related Art
Pharmaceutical formulations that provide for delivery of a drug over an
extended period
of time have revolutionized the pharmaceutical industry. Whether the delivery
is sustained,
modified, controlled, extended, or delayed, the concept is generally the same -
provide in a single
dose what previously required multiple doses. ("Sustained release" will be
used herein to
describe this generic class of release mechanisms.) The desire is to provide
an effective
concentration of the drug for an appropriate length of time.
There are several advantages to such formulations. For example, having a lower
concentration of the drug in the body for a longer period of time lowers the
incidence of toxicity
for drugs with a narrow therapeutic window. and often improves the overall
effect. Also, patient
compliance is improved when the dosing regimen is decreased, a patient is far
more likely to take
a single daily dose, than to take two, three, or even four doses daily. This
is true for drugs
delivered orally, as well as those which are injected, inhaled, or delivered
by transdermal or
transmucosal diffusion.
Traditionally, sustained release has been achieved by placing a coating
material over the
drug particles or granules. Thus, tablets, capsules, caplets, pills, and other
formulations with
coated granules have been provided. Depending on the desired drug release
properties, a drug
core may be coated with a single layer of coating, or alternating coatings may
be provided. or the
drug may actually be interdispersed within a coating material. The
possibilities are numerous,
and the particulars of the formulation are chosen based on the desired drug
release properties. A
summary of such formulations is provided in Modern Pharmaceutics, Second
Edition, edited by
CA 02376113 2001-12-03
WO 00/74657 PCT/US00/15547
Gilbert S. Banker and Christopher T Rhodes, the entire contents of which is
hereby incorporated
by reference.
Oral and other sustained release delivery systems have largely been based on
solvent-
based particulate or matrix-type systems. These systems utilize spray-coating
or mechanical
mixing of a core drug particle and/or excipient granule with a polymer, e.g.,
a cellulose.
polyacrylate, degradable polyester, etc., to control the rate of release of
the active drug substance.
In addition, traditional matrix systems may contain a gel-forming excipient,
e.g., polyvinyl
alcohol (PVA), polyethylene oxide (or polyethylene glycol, PEG), celluloses,
etc., that form a gel
layer after delivery that releases the drug over time by diffusion of the drug
through the matrix.
A limitation of these systems is that multi-stage scale-up from the laboratory
to commercial-scale
production of formulations can be lengthy and difficult, often requiring
specialized equipment
and expensive solvents. Additionally, known systems produce formulations that
have a
relatively high concentration of polymer, thick coatings, and tend not to be
reproducibly
manufactured with identical release profiles.
Therefore, what is needed are improved methods for preparing coated drug
particles that
do not suffer these limitations, and that are useful in preparing
pharmaceutical formulations with
superior drug delivery and efficacy properties.
4
CA 02376113 2001-12-03
WO 00/74657 PCT/US00/15547
III. SUMMARY OF THE INVENTION
A. Features and Advantages of the Invention
The present invention overcomes these and other inherent deficiencies in the
prior art by
providing novel coating methods for use in preparing coated particles, and in
particular, coated
drug particles for having improved pharmaceutical properties. In general, the
methods disclosed
herein provide a means for coating particulate materials with one or more
layers of discrete
coating matter or materials such that the coated matter or materials adheres
generally uniformly
to the surface of the particulate materials to form either continuous or
discontinuous coatings
depending upon the particular application of the coated particulate materials.
The invention also provides for modification of (1) the aggregation
characteristics; (2) the
flow properties; and (3) the release-rate of the drug, by applying coatings
using the methods of
the present invention to greatly enhance the pharmacokinetic profiles of
coated drugs.
Additional advantages include improved flow properties during manufacture; and
formulation stability, e.g., shelf life.
Drugs coated by the processes outlined herein have been shown to possess high
encapsulation efficiencies (>99% drug) while requiring minimal processing. The
process also
has several advantages over current coating techniques including:
1. It is a fast process with modification times (i.e.. how long it takes to
coat a particulate
from beginning to end) on the order of minutes.
2. A variety of materials can be used for producing the coatings on the
particulate
materials, thus it is possible to produce films from materials with proven
biocompatibility.
CA 02376113 2001-12-03
WO 00/74657 PCT/US00/15547
It can be a dry, solventless technique conducted under a sterile environment,
which is
an important consideration in the drug industry.
4. Particle agglomeration/adhesion can be minimized by applying coatings that
affect the
bonding nature and electrostatic charge on the surface of the particulate
materials.
5. Formation of microcapsules by depositing coatings onto the particle surface
will make
it possible to control drug release kinetics by: (a) diffusion of the drug
through the polymer; (b)
degradation of the biodegradable polymer coating off of the drug particles,
thereby releasing the
core drug material.
6. Laser ablation can be performed under normal atmospheric pressure, as
opposed to a
vacuum, thereby eliminating the need for vacuum mechanisms, including chambers
and pumps,
in the process, and allowing for a continuous production line. This advantage
significantly
improves production times, and thereby decreasing production costs and scale-
up difficulty.
B. Summary of the Invention
The present invention provides methods of coating core materials comprising:
providing
target materials and core materials; ablating the target materials to form
ablated particulate target
materials; and coating the core materials with the ablated particulate target
materials; wherein the
method occurs at a pressure of about 10 Torr or higher. The ablating may occur
at a pressure of
about 20 Torr or higher, including about 760 Torr.
The core materials may have an average diameter of about 0.5 ~cm to about 1
mm.
Coating the core materials with the ablated particulate target material may
result in a coating of
the target materials on the core materials of a thickness of less than about
1000 nm. The coating
on the core materials may have a thickness of less than about 100 nm, or less
than about 10 nm.
6
CA 02376113 2001-12-03
WO 00/74657 PCT/US00/15547
Coating the core materials with the ablated particulate target materials may
result in
coated particles having average diameters of less than about 1 mm, less than
about 100 Vim, or
less than about 10 ~cm. Preferably, the target materials include at least a
biodegradable polymer,
biocompatible polymer, polysaccharide, and/or protein.
Ablating may be achieved by the use of a high energy source, which may be a
laser.
Lasers include, but are not limited to, ion laser, diode array laser. and
pulsed excimer laser. In
preferred embodiments, the coating of the core materials with the ablated
particulate target
materials is performed by mixing the core materials with the ablated
particulate materials using
fluidization. The fluidization may be achieved by pneumatic fluidization.
The core materials may include pharmaceuticals for human or animal use,
pesticides,
herbicides, fungicides. cosmetics, paints or pigments, and/or inert particles.
Preferably, the core
materials includes at least one pharmaceutical for human or animal use. The
coating of the target
materials on the core materials may result in a continuous coating or a
discontinuous coating.
In other embodiments, the present invention includes methods of coating
particulates to a
coating thickness of less than about 100 nm, the method comprising: providing
target materials
and core materials; ablating the target materials to form ablated particulate
target materials; and
coating the core materials with the ablated particulate target materials;
wherein the core materials
are fluidized using pneumatic fluidization.
7
CA 02376113 2001-12-03
WO 00/74657 PCT/US00/15547
In other embodiments, the invention includes methods of coating core materials
comprising: providing target materials and core materials: ablating the target
materials to form
ablated particulate target materials; and coating the core materials with the
ablated particulate
target materials; wherein the method occurs at a pressure of about 760 Torr
and wherein the core
material is fluidized using pneumatic fluidization.
The present invention also provides coated particulates formed according to
these
methods.
IV. BRIEF DESCRIPTION OF THE DRAWINGS
The drawings form part of the present specification and are included to
further
demonstrate certain aspects of the present invention. The invention may be
better understood by
reference to one or more of these drawings in combination with the detailed
description of
specific embodiments presented herein.
FIGURE 1 is a diagrammatic representation of an embodiment of the invention.
FIGURE 2 is a diagrammatic representation of another embodiment of the
invention.
FIGURE 3 shows a TEM image of deposited nanoparticles film at atmospheric
pressure (scale
100,000 times).
FIGURE 4 shows another TEM image of deposited nanoparticles film at
atmospheric pressure
(scale 100,000 times).
FIGURE 5 shows a 1H-NMR spectra of A) original PLGA, B) deposited PLGA at 500
mJ/cm=
at atmospheric pressure, and C) near atmospheric pressure (10 Torr).
FIGURE 6 shows the PLGA deposition rate at different pressures.
CA 02376113 2001-12-03
WO 00/74657 PCT/US00/15547
FIGURE 7 shows a gel permeation chromatogram of A) original PLGA MW, 56,000
daltons,
and B) deposited PLGA at 500 mJ/cm' at atmospheric pressure, MW 7,000 daltons.
FIGURE 8 shows SEM micrographs of uncoated TA powder at A) 1,000, B) 5,000, C)
10,000,
and D) 20,000 times magnification.
FIGURE 9 shows SEM micrographs of PLGA-coated TA powder at A) 1,000, B) 5,000,
C)
10,000, and D) 20,000 times magnification.
FIGURE 10 shows dissolution of uncoated TA vs. PLGA-coated TA in pH 7.4 PBS
(SO mM, 1
SDS) at 37°C (n=3). Profiles are shown for uncoated TA powder (TA)~,
and coated powders
after 30 minutes at 500 mJ/cm' (PLGA30)~ at atmospheric pressure.
FIGURE 11 shows the release profile for PLGA coated BSA ~ compared to uncoated
BSA ~.
FIGURE 12 shows a 1H-NMR spectra of A) original HPMC, B) deposited HPMC at 500
mJ/cm= near atmospheric pressure ( 10 Torr).
FIGURE 13 shows the dissolution of uncoated TA vs. HPMC-coated TA in pH 7.4
PBS (50
mM, 0.5 % SDS) at 37°C (n=3). Profiles are shown for uncoated TA powder
(TA) ~, and
coated powders after 10 minutes at 500 mJ/cm'- (HPMC200) ~ and 625 mJ/cm2
(HPMC250) ~.
FIGURE 14 shows a 1H-NMR spectra of A) original Eudragit 4135, B) deposited
Eudragit at
500 mJ/cm'- near atmospheric pressure (10 Torr).
FIGURE 15 shows a 1H-NMR spectra of A) original SDS, B) deposited SDS at 500
mJ/cm'-
near atmospheric pressure ( 10 Torr).
FIGURE 16 shows an Anderson Cascade impaction profile for uncoated vs. SDS-
coated TA
powders.
9
CA 02376113 2001-12-03
WO 00/74657 PCT/US00/15547
FIGURE 17 shows the release profile for PLGA coated Griseofulvin (GRIS) ~
compared to
uncoated GRIS ~.
FIGURE 18 shows the release profile for PLGA coated Bupivacaine-HC1 (BUP) ~
compared to
uncoated BUP ~.
FIGURE 19 shows a 1 H-NMR spectra of A) original PC, B) original PEG20K, and
C)
PC+PEG20K solid target deposited at atmospheric pressure.
FIGURE 20 shows a 1H-NMR spectra of A) original PC, B) original PEG400, and C)
PC+PEG400 liquid target deposited at atmospheric pressure.
FIGURE 21 shows a 1 H-NMR spectra of A) original PC, B) original PEG20K, and
C)
PC+PEG20K gel target (heat mixed) deposited at atmospheric pressure.
FIGURE 22 shows a 1H-NMR spectra of A) original PC, B) original PEG400, and C)
PC+PEG400 frozen liquid target deposited at atmospheric pressure.
V. DETAILED DESCRIPTION OF THE INVENTION
The invention is directed to methods of coating particulate materials, and the
coated
particulate materials produced thereby. Particulates to be coated in
accordance with this
invention are those in which a thin coating is desirable. Such particulates
(cores) include, but are
not limited to, drugs or pharmaceuticals for human or animal use, cosmetics,
pesticides,
herbicides, fungicides, paints and pigments, as well as inert particles for
which a thin coating is
desirable. Of course, this invention is also applicable to the application of
thin layers of active
materials to inert particles. Examples might include nanoparticles having
biologically active
CA 02376113 2001-12-03
WO 00/74657 PCT/US00/15547
coatings, such as antigens, nucleic acids, proteins, or even pharmaceuticals.
The possibilities and
combinations are numerous.
The invention is particularly directed to particulate materials in the form of
drug or drug
delivery materials coated with a material, which may be a biodegradable or
biocompatible
matter, including biodegradable or biocompatible polymers. The coating may
impart a number
of characteristics to the particulate material, including altering its surface
properties, its rate of
dissolution, or its rate of diffusion and/or release of an active component.
More particularly, the
invention provides methods for preparing particulate material compositions
that are coated with
ultrafine layers of coating materials, preferably organic polymeric coating
materials, preferably
applied through a non-aqueous, non-solvent technique. A particularly preferred
process is a
vapor deposition process using pulsed laser ablation. Among the many
advantages of the
disclosed methods are control of both the thickness and uniformity of the
coating on the surfaces
of the selected particulate drug.
11
CA 02376113 2001-12-03
WO 00/74657 PCT/US00/15547
A. METHODS FOR PREPARING COATED DRUG PARTICLES
The method of the present invention generally involves physical vapor
deposition (PVD)
of the polymer coating onto the surface of the target particulate material.
Techniques for
achieving PVD are well-known in the art. and include such methods as thermal
evaporation,
sputtering, and laser ablation of a target material to produce a flux of
coating particulate
materials, which are then contacted with the core particulate material, and
allowed to form a
coating thereon. A most preferred method is laser ablation. Depending upon the
amount of
vapor or the length of deposition, the number of coating particles, and the
thickness of the
resulting layer of coating onto the core particulate material can be varied to
achieve the particular
objectives of a given coating process. Laser ablation for coating particles
under very low
pressure is disclosed in WO 00/28969, the entire contents of which is hereby
incorporated by
reference.
Throughout this specification, the terms "core material," "core particles,"
and "core
particulate materials" will be used interchangeably, as will the terms
"coating material," "coating
particles," and "coating particulate materials." These interchangeable terms
are intended to have
the same meanings as used herein.
In this invention, PLD or pulsed laser ablation is used in the preparation of
ultrafine, fine.
and granular drugs particles / particulate materials having atomic to
nanometric thick coatings
that impart improved pharmaceutical properties to the resulting coated drugs.
The present
coating methods are particularly desirable, since the core drug particles
themselves are not
subjected to conditions that would decompose, destroy, or alter the activity
of the drug itself.
12
CA 02376113 2001-12-03
WO 00/74657 PCT/US00/15547
The use of PLD also minimizes the thermal decomposition or denaturation of the
coating
material itself, and permits the deposition of the coating material onto core
drug particles that
may be maintained at ambient temperature and atmospheric pressure during the
deposition
process.
Through regulation of the physical parameters of the deposition process
(including
background gas and pressure and coating exposure time) the skilled artisan may
now for the first
time prepare a variety of particulate drugs that comprise ultrafine
particulate coatings. In
particular, the method affords the control of both the extent of molecular
coating, and the
thickness of the resulting coating layer on the surfaces of the drug
particles. Both relatively thick
coating layers, and relatively thin coating layers may be produced by
controlling the extent of
laser ablation process and the exposure of the coating particles to the laser
ablated coating
material.
By choosing a correct energy density, the target material for coating ablates
in a cluster-
like form that retains a majority of the characteristics of the target
material. Generally, when the
energy density (fluence) is increased, the ablation has more of an atomic
character, and is
composed of atoms that do not resemble the signature of the original material.
To provide optimum deposition of the coating onto the surface of the core
particle,
fluidization or agitation mechanisms may be employed to agitate the core
particles during the
coating process both to prevent agglomeration of the resulting coated core
particles, and also to
control the extent of coating thickness onto the core particles. Such
mechanisms may involve
subjecting the target particles to a stream of air or gas or other fluid to
agitate the particles during
the vapor deposition process, or alternatively may involve physical stirring.
Some applications
13
CA 02376113 2001-12-03
WO 00/74657 PCT/US00/15547
may require the use of both mechanical stirring and pneumatic fluidization to
achieve the
intended results. The present method provides an improvement for producing
individual coated
particles that remain essentially or substantially non-agglomerated after
coating.
Operating the coating process at approximate atmospheric pressure allows for a
continuous production process. Rather than needing to apply a vacuum on each
batch for
coating, the process of the present invention, operated at near atmospheric
pressure, allows for
continuous processing. For example, uncoated particles are transported into a
fluidized bed
coating chamber and coated using the present method, at atmospheric pressure.
The continuous
fluidizing mechanism, e.g., a gas stream, is sufficient to lift only the
uncoated particles into the
coating chamber. As the coating is applied, the particles become heavier, and
fall out of the gas
stream, to be transported out of the chamber. As an alternative, a circular
gas flow (cyclone)
may be applied to simultaneously separate and coat particles in a continuous
fashion. This
process continues as more uncoated particles are transported in, and coated
particles are
transported out. In addition, mechanical agitation may be included from the
bottom to improve
the fluidization at lower gas flow rates. A relatively inert atmosphere is
maintained by constantly
flowing a gas such as helium into the chamber. The gas may be recycled after
filtering and
scrubbing. Preferably the gas to be used is relatively light and inert.
Preferable gases helium.
argon, nitrogen, etc. Alternatively, if desired, a more reactive gas may be
included, or used
alone.
The invention is operated such that the coating chamber has a pressure of
around
atmospheric pressure, which may be a pressure as low as about 10 Torr to as
high as about 2500.
or any pressure in between. Preferably, the pressure in the coating chamber is
greater than about
14
CA 02376113 2001-12-03
WO 00/74657 PCT/US00/15547
20, or 30, or 40, or 50 Torr, more preferably greater than about 100 or X00
Torr, and most
preferably greater than about 700 Torr. Preferably, the pressure in the
coating chamber is less
than about 1000, more preferably less than about 900, and most preferably less
than about 820.
In a most preferred embodiment, the pressure in the coating chamber is about
760 Torr, or
atmospheric pressure.
The materials employed in the coating process are preferably materials such
that when
ablated by an energy source, comprise a vapor of discrete particles that are
extremely small --
typically preferred are coating particles that are sized on the order of from
about 1 to about 1000
nanometers in average diameter. It should be recognized that the particles
discussed in this
application are not necessarily spherical, but may be irregularly shaped.
Thus, reference to
diameter is meant to include an "equivalent diameter," or "geometric
equivalent diameter,"
recognizing that particles may be irregular. This measurement may be
determined by light
scattering measurements, such as by using a Coulter Counter (Beckman Coulter,
Inc., Fullerton,
CA). Techniques for measuring irregularly shaped particles are discussed in
Small Particle
Statistics, the entire contents of which is hereby incorporated by reference.
The deposition materials employed in the preparation of coated drug particles
may
comprise an inorganic or an organic material, including but not limited to,
polymers, proteins,
sugars, lipids, as well as bioactive ceramics, anionic, cationic, or
zwitterionic polymers or lipids.
and also antibodies or antigens. In preferred embodiments an organic polymer
is selected for
laser ablation and deposition onto the surface of pharmaceutical compounds.
Particularly
preferred as coating materials are organic compounds such as PLA, PGA, PLGA,
and related
biodegradable polymers, and functionalized derivatives thereof.
CA 02376113 2001-12-03
WO 00/74657 PCT/US00/15547
The materials applied as coatings may act to modify the release rate or cell
uptake of an
active compound in the particle core. Such sustained-release coatings
generally will act through
diffusion or dissolution modification mechanisms.
The coatings may also act to improve the physical stability of the drug
particle, so as to
improve, for example, its resistance to chipping or cracking. A coating may
also serve as a
moisture barrier, improving shelf life of an otherwise rapidly degrading drug.
Because of the
potential for dry coating pharmaceutical particulates, use of the present
invention is especially
advantageous for coating to improve shelf life. Thus, the present invention is
especially
applicable for coating pharmaceutical formulations which are sensitive to
moisture; or solvents
(such as proteins), and are therefore difficult to coat. This invention solves
that problem.
Moreover. the quality of the coating of the present invention, i.e., its
potential to be non-porous,
is unique and provides one more advantage for coating sensitive compounds.
A unique aspect of this invention is its ability to produce coatings that are
substantially
non-porous. Solvent-based coating techniques produce porous coatings because,
during drying,
the solvent evaporates, leaving minute pores in the coating. Because pores are
formed during the
coating, more coating is required to obtain a proper seal. Thus, a thicker
coating is required
when solvent-based techniques are used. This invention, on the other hand,
allows for extremely
thin coatings, at least in part because of their integrity -- they are almost
completely non-porous,
because applying a coating from nanometric-scale particles, the relative
thickness may still be on
the order of 10 to 50 nm.
The coating may also play a direct role in the pharmacology, or
pharmacokinetics, of the
pharmaceutical particle. For example, the coating may modify the interaction
of the particle with
16
CA 02376113 2001-12-03
WO 00/74657 PCT/US00/15547
tissues or cells. targeting specific cell or tissue types, or improving cell
uptake, or even acting to
provoke an immune response. The methods of the present invention may even be
used to coat
nucleic acids to inert particles with the purpose of particle bombardment
transfection of plants or
animals (for use in a "gene gun"). The possibilities are too numerous to list
here. In short, this
invention provides improved methods for coating particles for all known coated
particle
applications, and for applications which are disclosed herein for the first
time.
These materials may be readily deposited onto the surface of drug particles in
preferred
particle sizes and layer thicknesses using the laser ablation apparatus and
method disclosed
herein. This method may be used to deposit one or more layers of nanometric-
sized coating
(each on the order of from about 1 nm to about 1000 or so nm in thickness) on
core particles that
range on the order of from about 0.1 ,um to about 1 mm in diameter. The
average size of the
resulting coated drug particles may range from about 0.1 ,um to as high as
several millimeters or
so in diameter. Obviously, the size of the coated particle will depend on the
needs of the user,
with smaller coated particles finding application in, for example, in
molecular biology
applications, and larger coated particles finding application in, for example,
pharmaceutical
formulations.
The core particulate material to be coated in the process are preferably gas
and/or
mechanically fluidized to improve coating uniformity during deposition. By
controlling
conditions during deposition, the coating thickness, particle size, and
adhesion can be varied.
This coating method provides rapid thermal evaporation from the pulsed excimer
laser to
coat solid materials onto particles. Through this method, the coating material
is generally less
than about 1 to 5% by mass, and coating times are under one hour without the
need for drying
17
CA 02376113 2001-12-03
WO 00/74657 PCT/US00/15547
solvents. This method has a wide variety of pharmaceutical applications
ranging from coatings
to improve agglomeration and flowability, stability, cell uptake and
interactions, as well as
controlling the release rate of the drug. .
Drug particles or drug delivery particles coated with biodegradable or
biocompatible
polymer coatings with controlled thickness and controlled coating uniformity
may be produced
using the apparatus and methods as described herein. The drug particle coating
thickness can be
controlled down to nanometer thicknesses, and encapsulation can be partial or
complete.
Core particulate material, which may range in size, for example, from several
nanometers
to several millimeters in diameter, is provided with a relatively uniformly
dispersed
discontinuous or continuous coating of discrete individual coating particles
sized from atomic
scale to a few nanometers. The coating particles are created by a vapor
deposition process, and
preferably by laser ablation, where a pulsed laser beam is aimed at a target
composed of the
coating material under conditions sufficient to release individual particles
from the target in a
generally perpendicular ablation flux, e.g., a solid target material, frozen
liquid matrix target, etc.
Pulsed laser ablation is especially suited for mufti-elemental deposition in
which the
stoichiometry of the ablated species is maintained. This may be important when
organic coating
materials, such as polymers or other mesoscopic entities such as antibodies,
are employed
(Agarwal, 1998). During laser ablation, the core particulate material may be
agitated or fluidized
such that there is relatively continuous movement between all the core
particles. The degree of
coating is controlled by varying the laser parameters, energy density and
number of pulses, and
the treatment time.
18
CA 02376113 2001-12-03
WO 00/74657 PCT/US00/15547
Coated drug particles and pharmaceuticals may be prepared with a uniform
coating. Such
a coating may delay drug diffusion and dissolution until the coating degrades
or until the drug
diffuses through the coating for non-degradable coatings. The uniform coating
may also be used
to protect the drug particle from hostile environments. A coating may control
the release rate by
affecting surface area. The coating may also protect the drug particle size
during processing
steps such as compacted tablet grinding by providing a weaker interface that
separates before the
stresses fracture the drug particles themselves. The coating may also improve
flow
characteristics, which can be significant during manufacturing or in
determining the efficiency of
drug delivery mechanisms.
B. APPARATUS FOR COATING PARTICULATES
The apparatus of the invention generally includes a coating chamber in which a
target
material and particulate substrate is placed. An external evaporation or
removal source (EORS),
such as a pulsed excimer laser, enters the chamber through a window,
preferably quartz, and
interacts with the matrix target (MT). In alternative embodiments, the
evaporation or removal
source is internal, i.e., in the same chamber as the matrix and particles.
A nanometer-thin layer of target material absorbs the energy from the laser
pulse and the
surface is rapidly heated and expands from the target in the form of a plume
of ablated atomic to
micrometer sized particles. The plume of particles is then deposited onto the
fluidized core
particles.
A region of target absorbs the incident energy, for example, an excimer laser
(UV
excimer laser at 193-308 nm, solid-state Nd-Yag lasers at 255 to 1064 nm,
etc.). The absorption
19
CA 02376113 2001-12-03
WO 00/74657 PCT/US00/15547
depth of the incident laser depends on the structure of the biocompatible
target, typically the
absorption depth will range from 10-100 nanometers. This rapid (nanoseconds)
absorption and
subsequent heating of the target surface by the laser pulse provides energy
for polymer
desorption from the biocompatible target. Due to differential changes in the
heated target in a
time regime of nanoseconds, the matrix target ablates from the surface into a
dense plume of
nanometric-sized clusters, molecules, molecular chains, polymers, and/or lipid
fragments, for
example. (For a discussion of laser ablation of polymer, see Ogale, 1994,
incorporated herein by
reference.) The plume of nanometric-sized clusters, molecules, molecular
chains, polymers, and
lipid fragments, and particles are then deposited onto the fluidized core
particles. (For a
discussion of fluidization, see Kodas and Hampden-Smith. 1999, incorporated
herein by
reference. )
The MT preferably includes a matrix of biocompatible or biodegradable coating
material
and/or mesoscopic molecules that modify surface interactions. Biocompatible
coating materials
used for the MT may include polymers, proteins, sugars, lipids, and/or other
biologically active
or inactive materials. Nanofunctional molecules that modify the surface
interactions may include
bioactive ceramics, anionic or cationic polymers and lipids, antibodies, or
antigens. The MT, in
solid, liquid, or gel form, may alternatively be dispersed in a solvent that
evaporates relatively
quickly from the core particles. The core particles may be pharmaceutically
relevant particles
such as an active drug, pharmaceutically inert excipient particles, or other
preformed particulate
mixtures.
The core particles, or particulate materials, are preferably fluidized within
the coating
chamber to improve the uniformity of coating. The fluidization is preferably
achieved by air/gas
CA 02376113 2001-12-03
WO 00/74657 PCT/US00/15547
stream fluidization. That is, the core particles or core particulate materials
are placed in the path
of flowing air or gas, which fluidizes the cores, improving their mixing and
exposure to the
coating chamber atmosphere. Fluidization may also be achieved by mechanical
mixing, but
air/gas fluidization is preferable. Hybrid air/gas/mechanical fluidizing
mechanisms are also
preferable.
Having the EORS (e.g., laser) and the coating chamber separate allows great
latitude for
varying the coating structure and thickness. Also, with the proper EORS
choice, the process can
be used to create coatings on many different materials or particulates. The
composition of the
coatings is strongly dependent on the laser processing parameters such as
incident energy fluence
(J/cm'-), laser repetition frequency, fluidization gas pressure, fluidization
gas molecular weight.
target to substrate distance, and the optical absorption coefficient of the
matrix target and
components.
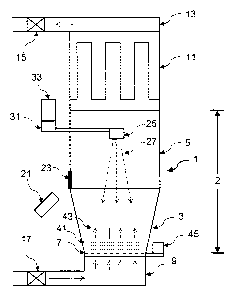
Figure 1 shows one embodiment of the present invention. The apparatus of
Figure 1 is a
top-coating apparatus 1. Top-coating apparatus 1 includes a coating chamber 2,
which is formed
from a cylindrical portion 5 connected to a conical portion 3. Although the
embodiment of
Figure 1 is shown with a cylindrical coating chamber, other shapes may be
chosen. depending
on the needs of the user or manufacturer, including, for example, square,
rectangular, or
polygonal.
Conical portion 3 is connected at its tapered end to a gas-permeable porous
plate 7, and a
gas distributor 9, adjacent to plate 7. At the opposite end of cylindrical
portion 5, a filter 11 with
cylindrical housing is mounted. An exhaust duct 13 carries gas for
recirculation back through a
filter assembly 15, through a blower (not shown), a temperature controller 17,
then back to gas
21
CA 02376113 2001-12-03
WO 00/74657 PCT/US00/15547
distributor 9 before re-entering the chamber. Recirculation, filtration, and
temperature control of
the chamber gas, are preferred aspects of the present invention.
Top-coating apparatus 1 includes an external evaporation or removal source
(EORS) 21,
which is directed upward into central chamber 2 through window 23 to the
matrix target (MT) 25
at approximately a 45 ° angle. Window 23 is formed from an optically
transparent material,
which is preferably quartz. The plume 27 leaves MT 25 downward toward the
fluidized particles
41 below MT 25. Plume 27 coats onto particles 41 which are contacted.
An external control device 31 and container 33 are used to feed or turn MT 25,
which
may involve a rotating motor control and/or feeding tube. Container 33 may
also include a
chiller to freeze material for MT 25.
Particles 41 are fluidized as a whirling layer at controlled temperature, and
solvent 43
from MT 25 is dried simultaneously during the coating process. A mechanical
vibrator 45 can
be used in conjunction with the gas fluidization to prevent particle
agglomeration and apply
fluidization at lower gas flow regimes.
Figure 2 shows another embodiment of the invention, a bottom-coating apparatus
101.
Apparatus 101 includes a coating chamber 102, which is formed from a
cylindrical portion 105
connected to a conical portion 103. Conical portion 103 is connected at its
tapered end to a gas-
permeable porous plate 107, and a gas distributor 109, adjacent to plate 107.
At the opposite end
of cylindrical portion 105, a filter 111 with cylindrical housing is mounted.
An exhaust duct 113
carries gas for recirculation back through a filter assembly 115, through a
blower (not shown), a
temperature controller 117, then back to gas distributor 109 before re-
entering the chamber.
22
CA 02376113 2001-12-03
WO 00/74657 PCT/US00/15547
External to coating chamber 102 is the EORS 121, which is directed downward
into
coating chamber 102 through window 123 to the MT 125 at approximately a 45
° angle. The
aerosol plume 127 leaves MT 125 upward toward the fluidized particles 141
above MT 125,
which attaches as a partial coating to the surfaces of exposed particles 41.
An external control device 131 and container 133 are used to feed or turn MT
125, which
may involve a rotating motor control and/or feeding tube. Container 133 may
also include a
chiller to freeze material for MT 125.
Particles 141 are fluidized as a whirling layer at controlled temperature, and
solvent 143
from MT 125 is dried simultaneously during the coating process. A mechanical
vibrator 145 can
be used in conjunction with the gas fluidization to prevent particle
agglomeration and apply
fluidization at lower gas flow regimes.
In a preferred embodiment, and as shown in Figures 1 and 2, the PVD technique
known
as laser ablation is employed in the fabrication of the coated particles. When
desirable, other
PVD techniques, such as thermal evaporation or sputtering, may be utilized to
produce a flux of
ablated species for deposition onto a host surface. A typical laser used in
the practice of the
present method is a Lambda Physik model 1248 pulsed excimer gas laser with an
operating UV
wavelength of 248 nanometers. Many other suitable lasers may be substituted
therefor, such as a
Nd:YAG laser operating at 255-1064, etc. The laser beam will produce a
particle flux generally
perpendicular to the surface of the target.
The laser wavelength is selected based on the nature of the material to be
ablated. A high
absorption coefficient and low reflectivity is a factor to consider for
efficient removal of the
material by the ablation process. The absorption coefficient is dependent on
the type of material
23
CA 02376113 2001-12-03
WO 00/74657 PCT/US00/15547
and the laser wavelength, and in some cases the intensity of the laser beam.
Typically, as the
surface temperature is increased, the absorption coefficient of the material
increases. Thus the
selection of laser wavelength is dependent on the type and characteristics of
materials ablated.
Additionally, for wavelengths in the blue and ultraviolet region of the
spectrum, the
absorption coefficient increases and the reflectivity decreases. Thus,
although any wavelength
could be used, the use of wavelengths less than 350 nm may lead to more
efficient removal of the
material.
Since the laser system and the PLD chamber are preferably separate, the
process offers
great latitude for varying experimental parameters. With the proper laser
choice this process can
be used to create coatings of many different materials on particulates. The
composition of the
coatings is dependent on the laser processing parameters, such as incident
energy fluence (J/cm=),
laser repetition frequency, target to substrate distance, and optical
absorption coefficient of the
target.
In most cases the chamber will be separate from the laser. However, if one
uses compact
lasers like a solid-state laser operating from 248 to 1056 nm, the laser can
be attached to the side
of the chamber. The specific conditions which affect the deposition of
coatings include (i)
control of the laser influence; (ii) control of the laser spot size: (iii)
control of the gas
composition and flow rate; (iv) control over the pulsation rate; and (v)
number of pulses and
wavelength of the light. By controlling each of these parameters, which are
different for
different materials, the integrity, microstructure, topology, architecture,
thickness and adhesion
of the coatings on the drug particles can be varied.
24
CA 02376113 2001-12-03
WO 00/74657 PCT/US00/15547
C. COATED PARTICLE COMPOSITIONS
The coating techniques described herein and the compositions derived therefrom
are
applicable to a wide variety of compositions, including, but not limited to,
pharmaceutical
compositions for human or veterinary use, biotechnology applications,
herbicides, or pesticides.
Pharmaceutical compositions include organic and inorganic active compounds,
including
biologically active peptides, proteins, and nucleic acids. Pharmaceutical
compositions of the
invention may be delivered by inhalation through the respiratory tract, as
well as, orally,
parenterally, or transdermally. In the embodiment of an implant, or other slow
release
formulation, such compositions may be manually placed into a body. In
addition, site specific
entities may be added to the particle surface so the drug core may be carried
to a specific tissue.
Methods of delivery of such compositions are well known in the art, and are
described, for
example, in Modern Pharmaceutics, Second Edition, edited by Gilbert S. Banker
and Christopher
T Rhodes, the entire contents of which is hereby incorporated by reference.
In one embodiment, an oral drug is formulated with a thin-film coating of the
present
invention. Exemplary pharmaceuticals that would benefit from such a coating
include drugs
used in controlled or targeted release formulation, taste-masking, or
particulate surface
modification prior to tableting or capsule filling.
In another embodiment, a pulmonary dry-powder formulation produced with a thin-
film
coating of the present invention. Exemplary pulmonary drugs that could be used
include
glucocorticoids and other localized asthma drugs, as well as drugs and
bioactive peptides and
proteins for systemic delivery, such as insulin, that have low absorption
through the oral route.
The present methods provides a high encapsulation efficiency, reduced damage
to the drug
CA 02376113 2001-12-03
WO 00/74657 PCT/US00/15547
particle during coating, and do not produce coatings of a thickness that would
reduce respiratory
fraction.
Topical drugs that could be used include localized antibiotics, antifungals,
and anti-
inflammatories. Parenteral drugs that could be used include many currently
used suspensions
and preparations for sustained or localized release, or simply to reduce
hydration and improve
shelf life of protein powders.
In illustrative embodiments, the coating material may be deposited onto the
surface of the
drug particle by a pulsed laser ablation process wherein the individual
particulate coating
materials deposited onto the core drug particles range in size from about 1 or
2 nm in average
diameter up to and including about 40 or 50 nm in diameter. More preferably
the particles that
comprise that coating may be range in size from about 3 or 4 nm in diameter up
to and including
about 20 to 30 nm in diameter. In other embodiments that particles that
comprise the coating
may range in size from about 5 or 6 nm in diameter up to and including about
10 or 15 nm in
diameter. By modifying the particular parameters of the coating process,
coatings that are
comprised of particles of slightly larger or smaller average diameter particle
sizes, may be
obtained.
Such layers do not necessarily have to be continuous in thickness over the
entire surface
of the drug particles, and in fact, in certain embodiments, it may be more
desirable to provide
substantially discontinuous deposition of the coating particles onto the
surfaces of the drug
particles to achieve coated drug particles that have particular
pharmaceutically-desirable
properties. In some cases, it may be desirable to provide coatings that are
almost entirely
discontinuous in thickness over the surfaces of the drug particles.
26
CA 02376113 2001-12-03
WO 00/74657 PCT/US00/15547
Likewise, in certain applications, it may also be desirable to coat the drug
particles with
mixtures of two or more coating materials. Such coating mixtures may be
prepared so that each
member of the plurality of coating materials may be simultaneously ablated and
applied to the
surfaces of the drug particles, or more conveniently, it may be desirable to
alternate or
sequentially apply two or more coating materials onto the surface of the drug
particles to be
coated. The ability of the method to prepare pluralities of layers of coating
materials is
particularly desirable when timed-, controlled- or sustained-release
formulations are being
prepared. Such combinations of coating materials may afford particular
pharmaceutically
desirable properties to the resulting coated drug particles. Such combinations
may include both
combinations of inert coating materials, or combinations of coating materials
and
pharmaceutically active compounds, or even multiple inert materials, and
multiple drugs, or site-
specific entities to produce targeting. The combinations are limited only by
the choice of the
user, and the compatibilities of the compounds.
The choice of core particle size, the choice of coating material(s), the size
of the coating
material particles, and the overall thickness and continuous/discontinous
nature of the coating
layers) will, of course, vary from particular application to application. The
skilled artisan will
be able to adjust such parameters to prepare coated drug particles having
particular desired
physical or pharmaceutical properties. The choice of these parameters will
often depend upon
the particular compound to be coated, and/or the particular coating to be
applied to the host
particle. Likewise, the preparation of the host particle may be varied
depending upon the
particular thickness of coating to be applied during the laser ablation
process. In some
circumstances, it may be necessary to dessicate, grind, pulverize, or
otherwise reduce the
27
CA 02376113 2001-12-03
WO 00/74657 PCT/US00/15547
particular core particulate materials to a certain uniform particle size or
consistency prior to, or
following, the deposition of the coating materials) onto the surfaces of the
host drug particles.
In addition, separation and coating may be performed in a continuous fashion
to reduce
agglomeration and remove particles once a target size is reached (cyclone). In
either
embodiment. the milling of the coated or uncoated drug particles may be
readily achieved using
methods well known to those of skill in the pharmaceutical arts. For example,
mechanical
shearing or milling may be used to reduce the particles to a particular
average particle size.
Likewise, methods such as sieving may be employed to improve the uniformity of
particle sizes
in a given sample.
When desirable, no milling or sizing may be required, and in fact, the drugs
to be coated
may be subjected to the laser ablation processes described herein in their
natural, or
commercially-available state. Moreover, in some situations, it may not even be
necessary to
assure a particular coating particle size or a coating thickness, or even to
prepare substantially
continuous layers of coating material onto the surface of the drug particle,
so long as the
resulting coated material retains all or most of its desired characteristics.
As described above, the total thickness of the coating materials) deposited
onto the
surface of the core particle may range in average thickness from about 1 nm to
about 1000
nanometers. In certain embodiments, the coating particles will form one or
more layers onto the
surface of the drug particles, each layer having a thickness of about 6, about
7, about 8, about 9.
about 10, about 11, about 12, about 13, about 14, about 15, about 16, about
17, about 18, about
19, about 20, about 21, about 22, about 23, about 24, about 25, about 26,
about 27, about 28,
about 29. or about 30 nm. In other embodiments, slightly thicker coating
layers will be desired
28
CA 02376113 2001-12-03
WO 00/74657 PCT/US00/15547
and in those instances, layers having an average thickness of about 31, about
32, about 33, about
34, about 35. about 36 about 37, about 38, about 39, about 40, about 41. about
42. about 43,
about 44, about 45, about 46, about 47, about 48, about 49, about 50, about
51, about 52, about
53, about 54, about 55, about 56, about 57, about 58, about 59, or about 60 nm
may be useful in
coating particular drug particles for use in the pharmaceutical arts.
Likewise, when slightly
thicker coating layers are required, layers having an average thickness of
about 65, about 70,
about 75, about 80, about 85, about 90, about 95, about 100, about 120, about
140, about 160,
about 180, about 200, about 225, about 250, about 275, about 300, about 400,
about 450, about
500, about 550, about 600, about 650, about 700, about 750, about 800, about
850, about 900.
about 950, or even about 1000 nm may be desired in coating particular drug
particles for use in
achieving coated drug particles having certain pharmaceutically desirable
properties. Of course.
thicker or thinner layers may be created, if desired, by modifying the
operational parameters.
As described herein, the sizes of the core drug particles to be coated may
range in average
diameter from about 0.1 ~m to about 1000 micrometers. In certain embodiments,
the host drug
particles will typically have an average size of about 0.2, about 0.3, about
0.4, about 0.5, about
0.6, about 0.7, about 0.8, about 0.9, about 1, about 2, about 3, about 4,
about 5, about 6, about 7,
about 8, about 9, about 10, about 1 l, about 12, about 13, about 14, about 15,
about 16, about 17.
about 18, about 19, or about 20 ~m in average particle diameter. For some
drugs, the average
particle diameter may be slightly larger. As such, the method may also be
employed to coat
these particles as well. In these instances, the drug particles may have an
average particle size of
about 21, about 22, about 23, about 24, about 25, about 26, about 27, about
28, about 29, about
30, about 40, about 50, about 60, about 70, about 80, about 90, about 100,
about 120, about 140,
29
CA 02376113 2001-12-03
WO 00/74657 PCT/US00/15547
about 160, about 180, about 200, about 220, about 240, about 260, about 280,
about 300, about
350, about 400, about 450, or even about 500 ,um in diameter. Intermediate
sizes in each of the
stated size ranges may be prepared using the disclosed methods, and such
intermediate sizes to
fall within the scope of the present invention.
The coated drug particles of the present invention may range in size from
about 0.1 ,um
average diameter, up to and including those coated particles that are about 2-
3 mm in average
particle size diameter. In certain embodiments, the final coated drug
particles obtained will
typically have an average particle diameter size of about 0.2, about 0.3,
about 0.4, about 0.5,
about 0.6. about 0.7, about 0.8, about 0.9, about 1, about 2, about 3, about
4, about 5, about 6,
about 7, about 8, about 9, about 10, about 1 l, about 12, about 13, about 14,
about 15, about 16,
about 17, about 18, about 19, or about 20 ~cm in average particle diameter.
For some drugs, the
average coated drug particle diameter size may be slightly larger, and may
have an average size
of about 21, about 22, about 23, about 24, about 25, about 26, about 27, about
28, about 29,
about 30, about 40, about 50, about 60, about 70, about 80, about 90, about
100, about 120, about
140, about 160, about 180, about 200, about 220, about 240, about 260, about
280, about 300,
about 350, about 400, about 450, or even about S00 ,um in average diameter,
and may be as large
as about 0.75, about 1.0, about 1.25, about 1.5, about 1.75, about 2.0, or
even about 2.5 mm
in diameter. In all cases, it is contemplated that all intermediate sizes in
each of the stated size
ranges may be prepared using the disclosed methods, and that such intermediate
sizes to fall
within the scope of the present invention.
The preferred sizes for the final coated particles will depend on the
application. Generally
preferred sizes for different applications will be described below.
CA 02376113 2001-12-03
WO 00/74657 PCT/US00/15547
D. PHARMACEUTICAL FORMULATIONS COMPRISING
COATED DRUG PARTICLES
The present invention also concerns formulations of one or more of the coated
drug
particle compositions disclosed herein in pharmaceutically acceptable
solutions for
administration to a cell or an animal, either alone, or in combination with
one or more other
drugs for the treatment of particular diseases or medical conditions.
The coated drug particle compositions disclosed herein may be administered in
combination with other agents as well, such as, e.g., proteins or polypeptides
or various
pharmaceutically-active agents. As long as the composition comprises at least
one of the coated
drug particle compositions disclosed herein, there is virtually no limit to
other components that
may also be included, given that the additional agents do not cause a
significant adverse effect
upon contact with the target cells or host tissues. The disclosed compositions
may thus be
delivered along with various other agents as required in the particular
instance. Such secondary
compositions included in the pharmaceutical formulations may be purified from
host cells or
other biological sources, or alternatively may be chemically synthesized as
described herein. The
formulations may comprise substituted or derivatized RNA, DNA, or PNA
compositions, they
may also be modified peptide or nucleic acid substituent derivatives, or other
coated or non-
coated drugs.
The formulation of pharmaceutically-acceptable excipients and carrier
solutions are well-
known to those of skill in the art, as is the development of suitable dosing
and treatment
regimens for using the particular compositions described herein in a variety
of treatment
31
CA 02376113 2001-12-03
WO 00/74657 PCT/US00/15547
regimens. including e.g., oral, parenteral, intravenous, intranasal, and
intramuscular
administration and formulation.
In general, the pharmaceutically relevant particles / particulate materials of
this invention
include particles from 0.1 pm to 2-3 mm, where oral formulations include
particles primarily
from 10 ~m to 1 or more mm, injectable powders are 80 ~m to 200 Vim, and
inhaled or nasally
delivered powders are 1 to 10 ~m (inhaled: generally 1 to 5; nasal: generally
1 to 10).
The present invention has been found to be particularly suited in the coating
of several
specific classes of drugs, including but not limited to inhaled powders, such
as glucocorticoids.
Nano-thin coatings applied to dry-powder formulations improve the flow
properties and provide
sustained-release of already established and FDA-approved formulations without
changing the
bulk product or requiring remanufacturing.
Glucocorticoids are beneficial in treating various pulmonary diseases,
including asthma,
sarcoidosis, and other conditions associated with alveolitis. Although
systemic glucocorticoid
therapy is effective in such conditions, prolonged administration carries the
risk of toxicity and
side effects (Mutschler and Derendorf, 1995). In attempts at reducing systemic
side effects,
several clinically efficacious glucocorticoids, including TA, have been
employed for delivery as
aerosols or dry powders.
In a recent study, it was shown that beneficial pulmonary effects were
achieved when
three different glucocorticoid powders and suspensions are administered
intratracheally in rats.
(Talton, 1999). In contrast, lung targeting (ratio of local to systemic
effects) was not observed
when different glucocorticoids are administered intratracheally, presumably
because of the fast
absorption of the lipophilic steroid (Hochhaus et al., 1995). This suggests
that pulmonary
32
CA 02376113 2001-12-03
WO 00/74657 PCT/US00/15547
targeting depends on slow release from the delivery that results in a
prolonged pulmonary
residence time.
The use of liposomes has been suggested to provide sustained pulmonary release
for
various drugs including glucocorticoids such as beclomethasone diproprionate
and
dexamethasone (Tremblay et al., 1993; Fielding and Abra, 1992; Vidgren et al.,
1995; Schreier et
al., 1993). However, although liposomes have a moderate loading capacity for
lipophilic
glucocorticoids (10 to 20%) such as TA under equilibrium conditions, TA is
rapidly released
under nonequilibrium conditions from the liposome matrix upon dilution or
administration
(Schreier et al.. 1994).
As the examples demonstrate, the present invention is particularly suited for
glucocorticoid formulations.
Delivery devices such as dry powder inhalers and metered dose inhalers have
been
improved in the last few years such that pulmonary deposition can range from
10% for
conventional delivery systems to up to 40% for recently developed third
generation devices
(Newman et al., 1997).
Interestingly, one of the predominant factors responsible for pulmonary
targeting, the
pulmonary mean residence time, has not been extensively evaluated. Pulmonary
residence time is
determined by the release rate of the inhaled particle from an inhaled solid
(powder) or an
alternative delivery system such as liposomes, the absorption rate of
dissolved drug across
pulmonary membranes and the mucociliary clearance which is able to remove drug
particles
from the upper portions of the lung. The absorption across membranes is a
rapid process for
lipophilic glucocorticoids (Burton and Schanker, 1974), and, consequently, the
dissolution rate of
33
CA 02376113 2001-12-03
WO 00/74657 PCT/US00/15547
a glucocorticoid powder will be the main determinant for controlling the
pulmonary residence
time. Simulations using a recently developed PD/PD model showed that for
inhalation products
with very rapid release kinetics, no targeting is observed because of the very
fast absorption from
the lung into the systemic circulation. With decreasing release rate
(dissolution rate), pulmonary
targeting is increased, as indicated by a dissociation of pulmonary and
systemic receptor
occupancies. A further decrease in release rate will consequently lead to a
decrease in pulmonary
targeting as a significant portion of the drug is removed via the mucociliary
clearance and after
swallowing is available for oral absorption. Thus, inhaled glucocorticoids
should possess certain
dissolution or release characteristics in order to show significant targeting.
However, this invention is suitable in preparing all forms of pharmaceutical
preparations,
some of which are discussed below.
1. Oral Delivery
The pharmaceutical compositions disclosed herein may be delivered by oral
administration to an animal, and as such, these compositions may be formulated
with an inert
diluent or with an assimilable edible carrier, or they may be enclosed in hard-
or soft-shell gelatin
capsule, or they may be compressed into tablets, or they may be incorporated
directly with the
food of the diet.
The coated drug particle-containing compounds may even be incorporated with
excipients and used in the form of ingestible tablets, buccal tables, troches,
capsules, elixirs,
suspensions, syrups, wafers, and the like (Mathiowitz et al., 1997; U. S.
Patent 5,641,515; U. S.
Patent 5,580,579 and U. S. Patent 5,792,451, each specifically incorporated
herein by reference
34
CA 02376113 2001-12-03
WO 00/74657 PCT/US00/15547
in its entirety). The tablets, troches, pills, capsules and the like may also
contain the following: a
binder. as gum tragacanth, acacia, cornstarch, or gelatin; excipients, such as
dicalcium phosphate;
a disintegrating agent, such as corn starch, potato starch, alginic acid and
the like; a lubricant,
such as magnesium stearate; and a sweetening agent, such as sucrose, lactose
or saccharin may
be added or a flavoring agent. such as peppermint, oil of wintergreen, or
cherry flavoring. When
the dosage unit form is a capsule, it may contain, in addition to materials of
the above type, a
liquid carrier. Various other materials may be present as coatings or to
otherwise modify the
physical form of the dosage unit. For instance, tablets, pills, or capsules
may be coated with
shellac, sugar or both. A syrup or elixir may contain the active compounds
sucrose as a
sweetening agent methyl and propylparabens as preservatives, a dye and
flavoring. such as
cherry or orange flavor. Of course, any material used in preparing any dosage
unit form should
be pharmaceutically pure and substantially non-toxic in the amounts employed.
In addition, the
active compounds may be incorporated into sustained-release preparation and
formulations.
Typically, these formulations may contain at least about 0.1 % of the active
compound or
more, although the percentage of the active ingredients) may, of course, be
varied and may
conveniently be between about 1 or 2% and about 95% or 98% or more of the
weight or volume
of the total formulation. Naturally, the amount of active compounds) in each
therapeutically
useful composition may be prepared is such a way that a suitable dosage will
be obtained in any
given unit dose of the compound. Factors such as solubility, bioavailability,
biological half life,
route of administration, product shelf life, as well as other pharmacological
considerations will
be contemplated by one skilled in the art of preparing such pharmaceutical
formulations, and as
such, a variety of dosages and treatment regimens may be desirable.
CA 02376113 2001-12-03
WO 00/74657 PCT/US00/15547
For oral administration the compositions of the present invention may
alternatively be
incorporated with one or more excipients in the form of a mouthwash,
dentifrice, buccal tablet,
oral spray, or sublingual formulation. For example, a mouthwash may be
prepared incorporating
the active ingredient in the required amount in an appropriate solvent, such
as a sodium borate
solution (Dobell's Solution). Alternatively, the active ingredient may be
incorporated into an
oral solution such as those containing sodium borate, glycerin and potassium
bicarbonate, or
dispersed in a dentifrice, including: gels, pastes, powders and slurries, or
added in a
therapeutically effective amount to a paste dentifrice that may include water.
binders. abrasives.
flavoring agents. foaming agents, and humectants, or alternatively fashioned
into a tablet or
solution form that may be placed under the tongue or otherwise dissolved in
the mouth.
2. Injectable Delivery
Alternatively, the pharmaceutical compositions disclosed herein may be
administered
parenterally, intravenously, intramuscularly, or even intraperitoneally, as
described in U.S. Patent
5,543.158, U. S. Patent 5,641,515 and U. S. Patent 5,399,363 (each
specifically incorporated
herein by reference in its entirety). Solutions of the active compounds as
free-base or
pharmacologically acceptable salts may be prepared in water suitably mixed
with a surfactant,
such as hydroxypropylcellulose. Dispersions may also be prepared in glycerol,
liquid
polyethylene glycols, and mixtures thereof and in oils. Under ordinary
conditions of storage and
use, these preparations contain a preservative to prevent the growth of
microorganisms.
The pharmaceutical forms suitable for injectable use include sterile aqueous
solutions or
dispersions and sterile powders for the extemporaneous preparation of sterile
injectable solutions
36
CA 02376113 2001-12-03
WO 00/74657 PCT/US00/15547
or dispersions (U. S. Patent 5,466,468, specifically incorporated herein by
reference in its
entirety). In all cases the form must be sterile and must be fluid to the
extent that easy
syringability exists. It must be stable under the conditions of manufacture
and storage and must
be preserved against the contaminating action of microorganisms, such as
bacteria and fungi.
The carrier can be a solvent or dispersion medium containing, for example,
water, ethanol, polyol
(e.g., glycerol, propylene glycol, and liquid polyethylene glycol, and the
like), suitable mixtures
thereof, and/or vegetable oils. Proper fluidity may be maintained, for
example, by the use of a
coating, such as lecithin, by the maintenance of the required particle size in
the case of dispersion
and by the use of surfactants. The prevention of the action of microorganisms
can be brought
about by various antibacterial and antifungal agents, for example, parabens,
chlorobutanol,
phenol, sorbic acid, thimerosal, and the like. In many cases, it will be
preferable to include
isotonic agents, for example, sugars or sodium chloride. Prolonged absorption
of the injectable
compositions can be brought about by the use in the compositions of agents
delaying absorption,
for example, aluminum monostearate and gelatin.
For parenteral administration in an aqueous solution, for example, the
solution should be
suitably buffered if necessary and the liquid diluent first rendered isotonic
with sufficient saline
or glucose. These particular aqueous solutions are especially suitable for
intravenous.
intramuscular, subcutaneous and intraperitoneal administration. In this
connection, sterile
aqueous media that can be employed will be known to those of skill in the art
in light of the
present disclosure. For example, one dosage may be dissolved in 1 ml of
isotonic NaCI solution
and either added to 1000 ml of hypodermoclysis fluid or injected at the
proposed site of infusion,
(see for example, Remin~ton's Pharmaceutical Sciences 15th Edition, pages 1035-
1038 and
37
CA 02376113 2001-12-03
WO 00/74657 PCT/US00/15547
1570-1580, which pages are hereby incorporated by reference). Some variation
in dosage will
necessarily occur depending on the condition of the subject being treated. The
person
responsible for administration will, in any event, determine the appropriate
dose for the
individual subject. Moreover, for human administration, preparations should
meet sterility,
pyrogenicity, and general safety and purity standards as required by FDA
Office of Biologics
standards.
Sterile injectable solutions are prepared by incorporating the active
compounds in the
required amount in the appropriate solvent with several of the other
ingredients enumerated
above, as required, followed by filtered sterilization. Generally, dispersions
are prepared by
incorporating the various sterilized active ingredients into a sterile vehicle
which contains the
basic dispersion medium and the required other ingredients from those
enumerated above. In the
case of sterile powders for the preparation of sterile injectable solutions,
the preferred methods of
preparation are vacuum drying and freeze-drying techniques which yield a
powder of the active
ingredient plus any additional desired ingredient from a previously sterile-
filtered solution
thereof.
The drug compositions to be coated by the methods disclosed herein may be
formulated
either in their native form, or in a salt form. Pharmaceutically-accepted
salts, include the acid
addition salts (formed with the free amino groups of the protein) and which
are formed with
inorganic acids such as, for example, hydrochloric or phosphoric acids, or
such organic acids as
acetic, oxalic, tartaric, mandelic, and the like. Salts formed with the free
carboxyl groups can
also be derived from inorganic bases such as, for example, sodium, potassium,
ammonium,
calcium, or ferric hydroxides. and such organic bases as isopropylamine,
trimethylamine,
38
CA 02376113 2001-12-03
WO 00/74657 PCT/US00/15547
histidine, procaine and the like. Upon formulation, solutions will be
administered in a manner
compatible with the dosage formulation and in such amount as is
therapeutically effective. The
formulations are easily administered in a variety of dosage forms such as
injectable solutions,
drug release capsules and the like.
As used herein, "carrier" includes any and all solvents, dispersion media,
vehicles,
coatings, diluents, antibacterial and antifungal agents, isotonic and
absorption delaying agents,
buffers, carrier solutions, suspensions, colloids, and the like. The use of
such media and agents
for pharmaceutical active substances is well known in the art. Except insofar
as any
conventional media or agent is incompatible with the active ingredient, its
use in the therapeutic
compositions is contemplated. Supplementary active ingredients can also be
incorporated into
the compositions.
The phrase "pharmaceutically-acceptable" refers to molecular entities and
compositions
that are not intended to produce an allergic or similar unexpected reaction
when administered to a
human. The preparation of an aqueous composition that contains a protein as an
active
ingredient is well understood in the art. Typically, such compositions are
prepared as injectables.
either as liquid solutions or suspensions; solid forms suitable for solution
in, or suspension in,
liquid prior to injection can also be prepared. The preparation can also be
emulsified.
Immunogenic compositions, such as vaccines, which are intended and expected to
induce an
immune response are, of course, pharmaceutically-acceptable.
39
CA 02376113 2001-12-03
WO 00/74657 PCT/US00/15547
3. Nasal Delivery
The administration of the pharmaceutical compositions by intranasal sprays;
inhalation,
and/or other aerosol delivery vehicles is also contemplated. Methods for
delivering genes,
nucleic acids. and peptide compositions directly to the lungs via nasal
aerosol sprays has been
described e.g., in U. S. Patent 5,756,353 and U. S. Patent 5,804,212 (each
specifically
incorporated herein by reference in its entirety), and delivery of drugs using
intranasal
microparticle resins (Takenaga et al., 1998) and lysophosphatidyl-glycerol
compounds (U. S.
Patent 5,725,871, specifically incorporated herein by reference in its
entirety) are also well-
known in the pharmaceutical arts. Likewise, transmucosal drug delivery in the
form of a
polytetrafluoroethylene support matrix is described in U. S. Patent 5,780,045
(specifically
incorporated herein by reference in its entirety).
The delivery of aerosol formulations of the drugs of the present invention may
be
accomplished using methods such as those described in United States Patent
5,849,265 and
United States Patent: 5,922,306 (each specifically incorporated herein by
reference in its
entirety).
Particularly preferred medicaments for administration using aerosol
formulations in
accordance with the invention include, but are not limited to, anti-allergics,
bronchodilators, and
anti-inflammatory steroids used in the treatment of respiratory disorders such
as asthma and the
like.
Medicaments which may be coated and administered in aerosol formulations
according to
the present invention include any drug useful in inhalation therapy which may
be presented in a
form which is substantially completely insoluble in the selected propellant.
Appropriate
CA 02376113 2001-12-03
WO 00/74657 PCT/US00/15547
medicaments may thus be selected from, for example, analgesics (codeine,
dihydromorphine.
ergotamine, fentanyl, morphine and the like); anginal preparations;
antiallergics (cromoglycate,
ketotifen, nedocromil and the like); anti-infectives (cephalosporins,
penicillins, rifampicin,
streptomycin, sulfonamides, macrolides, pentamidines, tetracyclines and the
like); antihistamines
(methapyrilene and the like); anti-inflammatories (flunisolide, budesonide,
tipredane,
triamcinolone acetonide, and the like); antitussives (noscapine and the like);
bronchodilators
(ephedrine, adrenaline, fenoterol, fomloterol, isoprenaline, metaproterenol,
phenylephrine,
phenylpropanolamine, pirbuterol, reproterol, rirniterol, terbutaline,
isoetharine, tulobuterol,
orciprenaline, and the like); diuretics (amiloride and the like);
anticholinergics (ipratropium,
atropine, oxitropium and the like); hormones (cortisone, hydrocortisone,
prednisolone and the
like); xanthines (including aminophylline, choline theophyllinate, lysine
theophyllinate. and
theophylline); and therapeutic proteins and peptides (e.g., insulin or
glucagons).
One of ordinary skill in the art will appreciate that in certain
circumstances, the coated
drugs particles of the present invention may be formulated in the form of
salts (such as alkali
metal or amine salts or as acid addition salts) or as esters (e.g., lower
alkyl esters) or as solvates
(e.g., hydrates) to optimize the activity and/or stability of the medicament
and/or to minimize the
solubility of the medicament in the delivery vehicle or propellant.
It will be appreciated by those skilled in the art that the aerosol
formulations according to
the invention may, if desired, contain a combination of two or more active
ingredients. Aerosol
compositions containing two active ingredients (in a conventional propellant
system) are known,
for example, for the treatment of respiratory disorders such as asthma.
Accordingly the present
invention further provides aerosol formulations that contain two or more
particulate medicaments
41
CA 02376113 2001-12-03
WO 00/74657 PCT/US00/15547
that are coated using the methods of the present invention. The medicaments
may be selected
from suitable combinations of the drugs mentioned herein, such as budesonide
(BU-D),
triamcinolone acetonide (TA), fluticasone propionate (FP), and the like, or
may even include
suitable combinations of other bronchodilatory agents (including ephedrine and
theophylline,
fenoterol, ipratropium, isoetharine, phenylephrine, and the like).
Preferred aerosol formulations in accordance with the invention comprise an
effective
amount of a polymer-coated particulate pulmonary medicament and a fluorocarbon
or hydrogen-
containing chlorofluorocarbon propellant. The final aerosol formulation may
typically contain
from about 0.005% to about 10% (wt./wt.) of the coated drug particles, more
preferably from
about 0.05% to about 5% (wt./wt.) of the coated drug particles, and more
preferably still, from
about 0.1 % to about 3.0% (wt./wt.), of the coated particles relative to the
total weight of the
formulation.
The propellants for use in the invention may be any fluorocarbon or hydrogen-
containing
chlorofluorocarbon or mixtures thereof as described in U.S. Patent 5,922,306.
4. Additional Modes of Drug Delivery
In addition to the methods of delivery described above, the following
techniques are also
contemplated as alternative methods of delivering coated drug particle
compositions.
Sonophoresis (i.e., ultrasound) has been used and described in U. S. Patent
5,656,016
(specifically incorporated herein by reference in its entirety) as a device
for enhancing the rate
and efficacy of drug permeation into and through the circulatory system. Other
drug delivery
alternatives contemplated are intraosseous injection (U. S. Patent 5,779,708),
microchip devices
42
CA 02376113 2001-12-03
WO 00/74657 PCT/US00/15547
(U. S. Patent 5,797,898), ophthalmic formulations (Bourlais et al., 1998),
transdermal matrices
(U. S. Patent 5.770,219 and U. S. Patent 5,783,208) and feed back-controlled
delivery (U. S.
Patent 5,697,899), each specifically incorporated herein by reference in its
entirety.
E. COATING COMPOSITIONS
The target materials used for the coating include most solids currently used
in the
pharmaceutical and food industries, namely any material that can be
effectively ablated by the
energy source. These materials include, but are not limited to, biodegradable
and biocompatible
polymers, polysaccharides, and proteins. Suitable biodegradable polymers
include polylactides,
polyglycolides, polycaprolactones, polydioxanones, polycarbonates,
polyhydroxybutyrates,
polyalkylene oxalates, polyanhydrides, polyamides, polyesteramides,
polyurethanes,
polyacetates, polyketals, polyorthocarbonates, polyphosphazenes,
polyhydroxyvalerates,
polyalkylene succinates, poly(malic acid), poly (amino acids),
polyvinylpyrrolidone,
polyethylene glycol, polyhydroxycellulose, polyorthoesters, and combinations
thereof, as well as
other polylactic acid polymers and copolymers, polyorthoesters, and
polycaprolactones, etc.
Suitable biocompatible polymers include polyethyleneglycols,
polyvinylpyrrolidone, and
polyvinylalcohols, etc. Suitable polysaccharides include dextrans, cellulose,
xantham, chitins
and chitosans, etc. Suitable proteins include polylysines and other
polyamines, collagen,
albumin, etc. A number of materials particularly useful as coating materials
are disclosed in U.S.
Patent No. 5,702,716.
43
CA 02376113 2001-12-03
WO 00/74657 PCT/US00/15547
F. SUBSTRATES FOR COATING
The core particles are generally large relative to the size of the coating
particles or
particulate materials, with the method proven to be very applicable to core
particles sized from
about 0.1 to about 1000 microns. It is understood that the core particulate
materials, i.e., core
particles, can be smaller, down to several nanometers in diameter, or larger,
up to several
millimeters in diameter. The core particulate materials are retained within a
processing container
that has a large enough volume to permit movement of the particles within the
container. The
top of the container is open or covered by a mesh to prevent the powder from
escaping, and the
container maintained in a vertical position during fluidization, or a portion
of the processing
container, such as a part or all of a side or bottom, is provided with
openings or apertures to
retain the core particulate materials within the processing container, if the
particle deposition is
to occur laterally or from below.
The core particulate material should be agitated or fluidized in some manner
to expose
the entire surface of each host particle to the coating particles entering the
processing container to
insure general uniformity of coating and to assist in the prevention of
agglomeration of
individual core particulate material. This fluidization may be accomplished in
a number of
equivalent manners, such as by mechanical agitation by vibration, rotation or
movement of the
processing container, by providing a stirring device within the container,
preferably by
pneumatic agitation by passing gas flow through the core particulate material.
Mechanisms for
fluidizing particles are well known in the art and examples are described in
Fluidization (Grace
and Matsen, eds., Plenum Press, NY 1980), which is hereby incorporated by
reference.
44
CA 02376113 2001-12-03
WO 00/74657 PCT/US00/15547
The percentage of deposition or coverage of the coating particles on the core
particulate
material is controlled by controlling the size of the coating particles and
the treatment time. The
longer the treatment time, the more coating particles will be adhered to the
surface of the core
particulate material, increasing both the percentage of coverage and the
thickness of the coating
layer. Surface coverage can be adjusted from below 1 percent up to 100
percent. The size of the
coating particles is controlled by the atmospheric composition. Inert gases
such as helium,
argon, or nitrogen, etc., are preferred, but reactive gases may be used.
Reactive gases such as
oxygen, ammonia or nitrous oxide produce higher concentrations of molecular,
as opposed to
atomic, species within the ablated particle flux, and are used if deposition
of oxide, nitride or
similar particles is desired.
Pressure within the system is generally around atmospheric pressure, i.e.,
about 1
atmosphere, or about 760 Torr. However, pressure may vary to some extent, and
may be as low
as about 10 Torr to as high as about 2500, or any pressure in between.
Preferably, the pressure in
the coating chamber is greater than about 20, or 30, or 40, or SO Torr, more
preferably greater
than about 100 or 500 Torr, and most preferably greater than about 700 Torr.
Preferably, the
pressure in the coating chamber is less than about 1000, more preferably less
than about 900, and
most preferably less than about 820. In a most preferred embodiment, the
pressure in the coating
chamber is about 760 Torr, or atmospheric pressure. Within these ranges, and
around these
values, the pressure may be varied.
CA 02376113 2001-12-03
WO 00/74657 PCT/US00/15547
K. MICROENCAPSULATION
The area of microencapsulation is relatively new, previously limited to
solvent
evaporation techniques (Thies, 1982; Manekar et al., 1992; Conti et al.,
1992). Currently there
are several different ways of applying coatings to particles in industry,
mainly through spray-
coating technologies (Gopferich et al., 1994). Pranlukast, a leukotriene
inhibitor, encapsulated
with hydroxypropylmethylcellulose (HPMC) nanospheres prepared by spray drying
showed an
improvement in inhalation efficiency but did not show a significant difference
in the dissolution
rate (Kawashima et al., 1998). The disadvantages of applying micron-thick
coatings for
sustained-release (10-100 microns thick) (Glatt, 1998) is that large
quantities of solvents must be
dried under strong venting and that an increase in particle size reduces the
inhalation efficiency
(Zeng et al., 1995; Talton, 1999)
L. EXAMPLES
The following examples are included to demonstrate preferred embodiments of
the
invention. It should be appreciated by those of skill in the art that the
techniques disclosed in the
examples which follow represent techniques discovered by the inventors to
function well in the
practice of the invention, and thus can be considered to constitute preferred
modes for its
practice. However, those of skill in the art should, in light of the present
disclosure, appreciate
that many changes can be made in the specific embodiments which are disclosed
and still obtain
a like or similar result without departing from the spirit and scope of the
invention.
46
CA 02376113 2001-12-03
WO 00/74657 PCT/US00/1554?
1. Matrix Target Solid Target at Room Temperature
The required biocompatible coating material (bioactive ceramics. anionic or
cationic
polymers or lipids, antibodies, or antigens, bio-polymers, drugs, proteins,
sugars, lipids,
electronic polymers, SMART polymers, functional organic molecules, metastable
compounds
and biologically inactive materials) can be combined with N number of
constituent (bioactive
ceramics, anionic or cationic polymers or lipids, antibodies, or antigens, bio-
polymers, drugs.
proteins, sugars, lipids, electronic polymers, SMART polymers, functional
organic molecules,
metastable compounds and biologically inactive materials) materials to form
solid matrix target
(SMT) for coating core particles. The overall properties of the constituent
materials must reflect
a higher absorption coefficient with respect to the EORS process, thereby
interaction with the
bio-coating material is reduced, thereby allowing transfer to the fluidized
core particles without
negative effects. Alternatively the above said constituent materials may also
be altered
chemically during interaction with the EORS process to further facilitate the
efficiency of the
core particle coating process. Depending on the composition and the removal
rate of the
constituent materials involved, removal of the constituents for toxicity
purposes may or may not
be necessary.
EXAMPLE 1
Triamcinolone acetonide (TA) was coated with a solid PLGA target for various
times
under low fluidization. Films were deposited onto glass slides before powder
runs to
characterize the deposited film material.
47
CA 02376113 2001-12-03
WO 00/74657 PCT/US00/15547
PLGA was deposited onto copper TEM grids at atmospheric pressure and a Joel
200
TEM was used to observe nanoparticle size and composition. The results are
shown in Figure 3.
which shows a transmission electron microscope (TEM) image of deposited
nanoparticles film at
atmospheric pressure (scale 100,000 times). Figure 4 shows another TEM image
of deposited
nanoparticles film at atmospheric pressure (scale 100,000 times).
Spherical PLGA nanoparticles from 20 nanometers and below are observable at
100,000
times magnification. The particles were dispersed uniformly across the
substrate after only 5
pulses from the laser at 750 mJ/cm2.
Characterization above shows the versatility of the coating process showing
characterization of original PLGA, HPMC, Eudragit 4135, and SDS.
Characterization using
NMR shows a strong correlation of deposited material characteristic peaks to
original material
(Figure 5). The deposition rate of PLGA under optimized conditions also shows
a slightly
higher deposition rate near atmospheric pressure compared to low pressures
(Figure 6). Gel
permeation chromatography (GPC) of original PLGA compared to ablated PLGA is
shown in
Figure 7. Scanning electron microscope (SEM) analysis of PLGA coatings on TA
powders
shows no increase in particle size compared to original TA powders, verifying
the relative
nanometer thin coating thicknesses obtainable by this process (Figures 8 and
9). Finally, the
sustained-release profile of PLGA coated TA is shown compared to original TA
with powders
coated for 30 minutes providing 12 to 24 hour release in vitro (Figure 10).
Other coating
materials including poly-vinyl-pyrollidone (PVP), polyethylene glycol (PEG),
amylopectin
starch, albumin protein, and chitin have also been deposited successfully.
48
CA 02376113 2001-12-03
WO 00/74657 PCT/US00/15547
EXAMPLE 2
PLGA coatings on Bovine Serum Albumin (BSA) were successful in sustaining the
release out to 2 to 3 hours. BSA powders were sieved and the 75 to 250 micron
fraction was
coated with poly(lactic-co-glycolic acid) (PLGA) for 30 minutes. Dissolution
on 20 mg coated
and uncoated powders were performed in triplicate in 40 ml isotonic saline in
centrifuge tubes on
a rotating tumbler at room temperature. Filtered samples were collected at
different time points
up to 12 hours and analyzed using the Biocinchoninic Acid (BCA) protein assay
in a 96-well
plate and plate-reader at 568 nm. The results are presented in Figure 11.
EXAMPLE 3
Another accepted material used in oral tablet dosage forms is the different
celluloses,
such as hydroxy-propyl-methyl-cellulose (HPMC). Coatings of HPMC were
deposited on flat
glass slides for characterization and then onto micronized TA powders for 30
minutes. Figure
12 shows proton NMR spectra of the original HPMC and HPMC deposited at 500
mJ/cm2 near
atmospheric pressure ( 10 Torr). For HPMC, it is believed that the 3.6 ppm
peak correlates to the
methyl protons and the multiple peaks at 6.0 ppm for the multiple ring
protons.
Figure 13 shows dissolution test results for coated and uncoated TA powders.
Coated
formulations showed 80% release after 2 to 4 hours for HPMC coatings compared
to 24 hours for
PLGA coatings, but additionally showed improved flow properties as seen by
Anderson Cascade
Impaction.
49
CA 02376113 2001-12-03
WO 00/74657 PCT/US00/15547
EXAMPLE 4
Another accepted material used in oral tablet dosage forms is the poly(acrylic
acids), such
as Eudragit (Rohm), which show specific pH sensitive release. Intact coatings
of Eudragit were
successfully deposited on flat glass slides for characterization. Proton NMR
spectra of original
Eudragit compared to deposited Eudragit are shown in Figure 14.
EXAMPLE 5
A surfactant material used in oral tablet dosage forms to increase solubility
and
flowability is sodium-dodecyl-sulfate (SDS). Intact coatings of SDS were
successfully deposited
on flat glass slides for characterization. Proton NMR spectra of original SDS
compared to
deposited SDS are shown in Figure 15.
In addition, Anderson Cascade Impaction, which measures the deposition of
powders
onto different stages based on the aerodynamic particle size, and SDS coated
TA powders, was
performed. Results, shown in Figure 16, showed a nearly double increase in
emitted powder
dose compared to uncoated powders, suggesting a higher flowability and
deposition into the
lung.
EXAMPLE 6
PLGA coatings on Griseofulvin (GRIS), an oral fungistatic, were successful in
sustaining
the release out to 12 to 24 hours. GRIS powders were coated with poly(lactic-
co-glycolic acid)
(PLGA) for 30 minutes at atmospheric pressure under helium flow and mechanical
agitation.
Dissolution of 50 mg coated and uncoated powders were performed in a USP
dissolution bath
CA 02376113 2001-12-03
WO 00/74657 PCT/US00/15547
(paddles, 50 RPM) in pH 7.4 phosphate buffer with 0.5% SDS at 37 degrees C.
Filtered samples
were collected at different time points up to 24 hours and analyzed using
HPLC. The results are
presented in Figure 17.
EXAMPLE 7
PLGA coatings on bupivacaine-HCl (BUP), a pain-blocking injectable, were
successful
in sustaining the release out to 2 to 4 hours. GRIS powders were coated with
poly(lactic-co-
glycolic acid) (PLGA) for 30 minutes at atmospheric pressure under helium flow
and mechanical
agitation. Dissolution of 4 mg coated and uncoated powders were analyzed in
triplicate in 40 ml
isotonic saline in centrifuge tubes on a rotating tumbler at room temperature.
Filtered samples
were collected at different time points up to 12 hours and analyzed at 220nm
in a Beckman UV
spectrophotometer. The results are presented in Figure 18.
EXAMPLE 8
Using a solid matrix of PEG 20,000, phosphatidylcholine (PC), a lipid present
in cell
membranes, was deposited successfully onto flat glass slides for
characterization. Proton NMR
spectra of the A) original PC, B) original PEG400, and C) deposited PEG400/PC
at 500 mJ/cm'-
for 10 minutes are shown in Figure 19.
51
CA 02376113 2001-12-03
WO 00/74657 PCT/US00/15547
2. Matrix Target Liquid at Room Temperature
The required biocompatible coating material (bioactive ceramics, anionic or-
cationic
polymers or lipids, antibodies, or antigens, bio-polymers, drugs, proteins,
sugars, lipids,
electronic polymers, SMART polymers, functional organic molecules, metastable
compounds
and biologically inactive materials) can be combined with N number of
constituent (bioactive
ceramics, anionic or cationic polymers or lipids, antibodies, or antigens, bio-
polymers, drugs,
proteins, sugars, lipids, electronic polymers. SMART polymers, functional
organic molecules,
metastable compounds and biologically inactive materials) materials to form a
liquid matrix
target (LMT) for coating core particles. The overall properties of the
constituent materials must
reflect a higher absorption coefficient with respect to the EORS process,
thereby interaction with
the bio-coating material is reduced, allowing transfer to the fluidized core
particles without
negative effects. Although the target material is a liquid, interaction with
EORS during a time
regime on the order of nano-microseconds allows the following events to occur:
1 ) Heating of laser interaction area (LIA).
2) Subsequent curing and preferential absorption with respect to the bio-
coating and constituent materials.
52
CA 02376113 2001-12-03
WO 00/74657 PCT/US00/15547
3) Evaporation of the bio-coating material and coating onto the core
particles.
Alternatively the above said constituent materials may also be altered
chemically during
interaction with the EORS process to further facilitate the efficiency of the
core particle coating
process. Depending on the composition and the removal rate of the constituent
materials
involved, removal of the constituents for toxicity purposes may or may not be
necessary.
EXAMPLE 9
Using a liquid matrix target of PEG 400, phosphatidylcholine (PC), a lipid
present in cell
membranes. was deposited successfully onto flat glass slides for
characterization. Proton NMR
spectra of the A) original PC, B) original PEG400, and C) deposited PEG400/PC
at 500 mJ/cm'-
for 10 minutes are shown in Figure 20.
3. Matrix Target Solid-Liquid Target at Room Temperature
The required biocompatible coating material (bioactive ceramics, anionic or
cationic
polymers or lipids, antibodies, or antigens, bio-polymers, drugs, proteins,
sugars, lipids,
electronic polymers, SMART polymers, functional organic molecules, metastable
compounds
and biologically inactive materials) can be combined with N number of
constituent (bioactive
ceramics, anionic or cationic polymers or lipids, antibodies, or antigens, bio-
polymers, drugs,
proteins, sugars, lipids, electronic polymers, SMART polymers, functional
organic molecules.
metastable compounds and biologically inactive materials) materials to form a
gel matrix target
53
CA 02376113 2001-12-03
WO 00/74657 PCT/US00/15547
(GMT) for coating core particles. The overall properties of the constituent
materials must reflect
a higher absorption coefficient with respect to the EORS process, thereby
interaction with the
bio-coating material is reduced, thereby allowing transfer to the fluidized
core particles without
negative effects. The difference that must be identified between cases two and
three are the
following:
1 ) The functionality is based on solid material absorption being different
than
the liquid counterpart, constituent or bio-coating material.
2) The above said solid material may precipitate out of the liquid solution
during the reaction via catalyst type reactions, constituent or bio-coating
material.
3) The constituent material will control the interaction processes associated
with the
EORS.
Although the target material may be a solid or a solid/liquid composite,
interaction with EORS
during a time regime on the order of nano-microseconds allows the following
events to occur:
1 ) Heating of laser interaction area (LIA).
2) Subsequent curing and preferential absorption with respect to the bio-
coating and constituent materials. In the case of the liquid pure liquid,
solid constituent materials may precipitate out of solution to act as
selective absorption sites, chromophores, nano-particles or entities.
3) Evaporation of the bio-coating material and coating onto the core
particles.
54
CA 02376113 2001-12-03
WO 00/74657 PCT/US00/15547
Alternatively the aforementioned constituent materials may also be altered
chemically during
interaction with the EORS process to further facilitate the efficiency of the
core particle coating
process. Depending on the composition and the removal rate of the constituent
materials
involved, removal of the constituents for toxicity purposes may or may not be
necessary.
EXAMPLE 10
Using a gel matrix of PEG 20,000, phosphatidyl choline (PC) (mixed with PEG20K
at
60°C) was deposited successfully after cooling onto flat glass slides
for characterization. Proton
NMR spectra of the A) original PC, B) original PEG20K, and C) deposited
PEG20K/PC gel at
500 mJ/cm'- for 10 minutes are shown in Figure 21.
4. Matrix Target Solid Below Room Temperature
The required biocompatible coating material (bioactive ceramics, anionic or
cationic
polymers or lipids, antibodies, or antigens, bio-polymers, drugs, proteins,
sugars, lipids,
electronic polymers, SMART polymers, functional organic molecules, metastable
compounds
and biologically inactive materials) can be combined with N number of
constituent (bioactive
ceramics, anionic or cationic polymers or lipids, antibodies, or antigens, bio-
polymers, drugs,
proteins, sugars, lipids, electronic polymers, SMART polymers, functional
organic molecules,
metastable compounds and biologically inactive materials) materials to form a
frozen matrix
target (FMT) below room temperature (<300K) for coating core particles. The
overall properties
CA 02376113 2001-12-03
WO 00/74657 PCT/US00/15547
of the constituent materials must reflect a higher absorption coefficient with
respect to the EORS
process. thereby interaction with the bio-coating material is reduced, thereby
allowing transfer to
the fluidized core particles without negative effects.
Although the target material may be a solid or a solid/liquid composite,
interaction with EORS
during a time regime on the order of nano-microseconds allows the following
events to occur:
1 ) Heating of laser interaction area (LIA).
2) Preferential absorption with respect to the bio-coating and constituent
materials.
3) Evaporation of the bio-coating material and coating onto the core
particles.
Alternatively the above said constituent materials may also be altered
chemically during
interaction with the EORS process to further facilitate the efficiency of the
core particle coating
process. Depending on the composition and the removal rate of the constituent
materials
involved, removal of the constituents for toxicity purposes may or may not be
necessary.
EXAMPLE 11
Using a frozen matrix of PEG 400, phosphatidylcholine (PC) was snap frozen in
liquid
N, and deposited successfully onto flat glass slides for characterization.
Proton NMR spectra of
the A) original PC, B) original PEG20K, and C) deposited PEG20K/PC gel at 500
mJ/cm'- for 10
minutes are shown in Figure 22.
56
CA 02376113 2001-12-03
WO 00/74657 PCT/US00/15547
VI. CITED DOCUMENTS
The following literature citations as well as those cited above are
incorporated in
pertinent part by reference herein for the reasons cited in the above text:
Agarwal and Phadke, "Laser assisted deposition of supramolecular organizates
on solid
surfaces" Mat Sci Eng C, 6:13-17, 1998.
Banker and Rhodes, Eds, Modern Pharmaceutics, Marcel Dekker, Inc., New York,
1990.
Bourlais, et al., "Ophthalmic drug delivery systems--recent advances", Prog
Retin Eye
Res, 17(1): 33-58, 1998.
Burton and Schanker, "Absorption of corticosteroids from the rat lung,"
Steroids,
23(5):617-24, 1974.
Conti, Pavanetto and Genta, "Use of polylactic acid for the preparation of
microparticulate drug delivery systems," J. Microencapsul., 9(2):153-66, 1992.
Fielding and Abra, "Factors affecting the release rate of terbutaline from
liposome
formulations after intratracheal instillation in the guinea pig," Pharm. Res.,
9(2):220-23, 1992.
Glatt, "Multi-purpose Fluid Bed Processing," Product Literature, 1998.
Gopferich, A., Alonso, M., and Langer, R., "Development and characterization
of of microencapsulated microspheres", Pharm Res, 11 ( 11 ): 1568-74, 1994.
Herdan, G., Small Particle Statistics, Second Edition, Butterworths, London,
1960.
Hochhaus, Derendorf, Mollmann and Gonzalez-Rothi,
57
CA 02376113 2001-12-03
WO 00/74657 PCT/US00/15547
"Pharmacokinetic/pharmacodynamic Aspects of Aerosol Therapy Using
Glucocorticoids as a
Model." J. Clin. Pharmacol., 37:881-92, 1997.
Hochhaus, Gonzalez-Rothi, Lukyanov, Derendorf, Schreier and Dalla Costa,
"Assessment of glucocorticoid lung targeting by ex-vivo receptor binding
studies," Pharm. Res.,
12:134-37, 1995.
Huang, Tamada, Hochhaus and Bodor, "An AM1-based model for the estimation of
the
relative binding affinity for glucocorticoids," in "1St Drug Optimization via
Retrometabolism
Conference," Amelia Island: Die Pharmazie, 1997.
Kawashima, Serigano, Hino, Yamamoto and Takeuchi, "A new powder design method
to
improve inhalation efficiency of pranlukast hydrate dry powder aerosols by
surface modification
with hydroxypropylmethylcellulose phthalate nanospheres," Pharm. Res.,
15(11):1748-52, 1998.
Kodas, T and Hampden-Smith, M., Aerosol Processing of Materials, Wiley-VCH,
New York, 1999.
Manekar, Puranik and Joshi, "Microencapsulation of propranolol hydrochloride
by the
solvent evaporation technique," J. Microencapsul., 9(1):63-66, 1992.
Mathiowitz, et. Al., "Biologically erodable microspheres as potential oral
drug delivery
systems", Nature, 386(6623): 410-4, 1997.
Mutschler and Derendorf, in "Drug Actions," CRC Press, Boca Raton, FL, pp. 286-
87,
1995.
Newman, Steed, Reader, Hooper and Zierenberg, "Efficient delivery to the lungs
of
58
CA 02376113 2001-12-03
WO 00/74657 PCT/US00/15547
flunisolide aerosol from a new portable hand-held multidose nebulizer," J.
Pharm. Sci., 85:960-
64. 1997.
Ogale, S. B., "Deposition of Polymer Thin Films by Laser Ablation," in Pulsed
Laser
Deposition of Thin Films, Chrisey, D. B. and Hubler, G.K., Eds. John Wiley &
Sons, New York.
1994, Chapter 25.
Schreier, Gonzalez-Rothi and Stecenko, J. Control Release, 24:209-23, 1993.
Schreier, Lukyanov, Hochhaus and Gonzalez-Rothi, "Thermodynamic and kinetic
aspects
of the interaction of triamcinolone acetonide with liposomes," Proceed. Inter.
Symp. Control.
Rel. Bioact. Mater., 21:228-29, 1994.
Takenaga, M., et.al., "Microparticle resins as a potential nasal drug delivery
system for
insulin", JControlled Release, 52(1-2): 81-7, 1998.
Talton, James D., Ph.D. Thesis, University of Florida. 1999.
Thies, "Microcapsules as drug delivery devices," Crit. Rev. Biomed. Eng.,
8(4):335-83,
1982.
Tremblay, Therien, Rocheleau and Cormier, Eur. J. Clin. Inv., 23:656-61, 1993.
Vidgren, Waldrep, Arppe, Black, Rodarte, Cole and Knight, "A study of
99"'technetium-
labeled beclomethasone diproprionate dilauroylphosphatidylcholine liposome
aerosol in normal
volunteers," Int. J. Pharm., 115:209-16, 1995.
Zeng, Martin and Marriott, "The Controlled Delivery of Drugs to the Lungs,"
Int. J.
Pharm. , 124:149-64, 1995.
59
CA 02376113 2001-12-03
WO 00/74657 PCT/US00/15547
All of the compositions and methods disclosed and claimed herein can be made
and
executed without undue experimentation in light of the present disclosure.
While the
compositions and methods of this invention have been described in terms of
preferred
embodiments, it will be apparent to those of skill in the art that variations
may be applied to the
composition, methods and in the steps or in the sequence of steps of the
method described herein
without departing from the concept, spirit and scope of the invention. More
specifically, it will
be apparent that certain agents which are both chemically and physiologically
related may be
substituted for the agents described herein while the same or similar results
would be achieved.
All such similar substitutes and modifications apparent to those skilled in
the art are deemed to
be within the spirit, scope and concept of the invention as defined by the
appended claims.