Note: Descriptions are shown in the official language in which they were submitted.
CA 02378600 2002-O1-08
WO 01/04108 - 1 - PCT/US00/18935
AMINO CERAMIDE - LIKE COMPOUNDS AND
THERAPEUTIC METHODS OF USE
RELATED APPLICATIONS
The present application is a continuation-in-part of U.S. Serial No.
08/883,218,
filed June 26, 1997, which is a divisional of U.S. Serial No. 08/708,574,
filed
September 5, 1996, now U.S. Patent No. 5,916,911, which claims priority from
U.S.
Serial No. 60/004,047, filed September 20, 1995, all of which are hereby
expressly
incorporated by reference.
SPONSORSHIP
The present invention was supported by grant nos. 801 DK41487, 801
DK69255 and 80139255 from the National Institutes of Health, contract 843 CA
58159 from the National Cancer Institute, grant GM 35712 from the National
Institute
of General Medical Sciences, and by the University of Michigan Comprehensive
Cancer Center grant 2P30 CA 46592 from the National Cancer Institute, U.S.
Public
Health Service, DHHS. Grant number for Merit Award from Veteran's
Administration?
The government may have certain rights in this invention.
FIELD OF THE INVENTION
The present invention relates generally to ceramide-like compounds and, more
particularly, to ceramide-like compounds containing a tertiary amine group and
their
use in therapeutic methods.
BACKGROUND OF THE INVENTION
Hundreds of glycosphingolipids (GSLs) are derived from glucosylceramide
(GIcCer), which is enzymatically formed from ceramide and UDP-glucose. The
enzyme involved in GIcCer formation is UDP-glucose:N-acylsphingosine
glucosyltransferase (GIcCer synthase). The rate of GIcCer formation under
physiological conditions may depend on the tissue level of UDP-glucose, which
in turn
depends on the level of glucose in a particular tissue (Zador, LZ. et al., "A
Role for
Glycosphingolipid Accumulation in the Renal Hypertrophy of Streptozotocin-
Induced
Diabetes Mellitus," J. Clin. Invest. 91:797-803 (1993)). In vitro assays based
on
endogenous ceramide yield lower synthetic rates than mixtures containing added
ceramide, suggesting that tissue levels of ceramide are also normally rate-
limiting
(Brenkert, A. et al., "Synthesis of Galactosyl Ceramide and Glucosyl Ceramide
by Rat
Brain: Assay Procedures and Changes with Age," Brain Res. 36:183-193 (1972)).
It has been found that the level of GSLs controls a variety of cell functions,
such as growth, differentiation, adhesion between cells or between cells and
matrix
proteins, binding of microorganisms and vinrses to cells, and metastasis of
tumor
CA 02378600 2002-O1-08
WO 01/04108 PCT/US00/18935
-2_
cells. In addition, the GIcCer precursor, ceramide, may cause differentiation
or
inhibition of cell growth (Bielawska, A. et al., "Modulation of Cell Growth
and
Differentiation by Ceramide," FEBS Letters 307:211-214 (1992)) and be involved
in
the functioning of vitamin D3, tumor necrosis factor-a, interleukins, and
apoptosis
(programmed cell death). The sphingols (sphingoid bases), precursors of
ceramide,
and products of ceramide catabolism, have also been shown to influence many
cell
systems, possibly by inhibiting protein kinase C (PKC).
It is likely that all the GSLs undergo catabolic hydrolysis, so any blockage
in
the GIcCer synthase should ultimately lead to depletion of the GSLs and
profound
changes in the functioning of a cell or organism. An inhibitor of GIcCer
synthase,
PDMP (1 R-phenyl-2R-decanoylamino-3-morpholino-1-propanol),
previouslyidentified
as the D-threo isomer (Inokuchi, J. et al., "Preparation of the Active Isomer
of
1-Phenyl-2-Decanoylamino-3-Morpholino-1-Propanol, Inhibitor of
Glucocerebroside
Synthetase," J. Lipid Res. 28:565-571 (1987)), has been found to produce a
variety
of chemical and physiological changes in cells and animals (Radio, N.S. et
al., "Use
of 1-Phenyl-2-Decanoylamino-3-Morpholino-1-Propanol (PDMP), an Inhibitor of
Glucosylceramide Synthesis," In NeuroProtocols, A Companion to Methods in
Neurosciences, S. K. Fisher et al., Ed., (Academic Press, San Diego) 3:145-155
(1993) and Radio, N.S. et al., "Metabolic Effects of Inhibiting
Glucosylceramide
Synthesis with PDMP and Other Substances," In Advances in Lipid Research;
Sphingolipids in Signaling, Part 8., R.M. Bell et al., Ed. (Academic Press,
San Diego)
28:183-213 (1993)). Particularly interesting is the compound's ability to cure
mice of
cancer induced by Ehrlich ascites carcinoma cells (Inokuchi, J. et al.,
"Antitumor
Activity in Mice of an Inhibitor of Glycosphingolipid Biosynthesis," Cancer
Lett.
38:23-30 (1987)), to produce accumulation of sphingosine and
N,N-dimethylsphingosine (Felding-Habermann, B. et al., "A Ceramide Analog
Inhibits
T Cell Proliferative Response Through Inhibition of Glycosphingolipid
Synthesis and
Enhancement of N,N Dimethylsphingosine Synthesis," Biochemistry 29:6314-6322
(1990)), and to slow cell growth (Shayman, J.A. et al., "Modulation of Renal
Epithelial
Cell Growth by Glucosylceramide: Association with Protein Kinase C,
Sphingosine,
and Diacylglyceride," J. Biol. Chem. 266:22968-22974 (1991 )). Compounds with
longer chain fatty acyl groups have been found to be substantially more
effective
(Abe, A. et al., "Improved Inhibitors of Glucosylceramide Synthesis," J.
Biochem.
111:191-196 (1992)).
CA 02378600 2002-O1-08
WO 01/04108 PCT/US00/18935
-3-
The importance of GSL metabolism is underscored by the seriousness of
disorders resulting from defects in GSL metabolizing enzymes. For example, Tay-
Sachs, Gaucher's, and Fabry's diseases, resulting from enzymatic defects in
the GSL
degradative pathway and the accumulation of GSL in the patient, all have
severe
clinical manifestations. Another example of the importance of GSL function is
seen
in a mechanism by which blood cells, whose surfaces contain selectins, can,
under
certain conditions, bind to GSLs in the blood vessel walls and produce acute,
life-
threatening inflammation (Alon, R. et al., "Glycolipid Ligands for Selectins
Support
Leukocyte Tethering & Rolling Under Physiologic Flow Conditions." J. Immunol.,
154:5356-5366 (1995)).
At present there is only one treatment available for patients with Gaucher
disease, wherein the normal enzyme which has been isolated from normal human
tissues or cultured cells is administered to the patient. As with any drug
isolated from
human material, great care is needed to prevent contamination with a virus or
other
dangerous substances. Treatment for an individual patient is extremely
expensive,
costing hundreds of thousands, or even millions of dollars, over a patient's
lifetime.
It would thus be desirable to provide a treatment which includes
administration of a
compound that is readily available and/or producible from common materials by
simple reactions.
Possibly of even greater clinical relevance is the role of glucolipids in
cancer.
For example, it has been found that certain GSLs occur only in tumors; certain
GSLs
occur at abnormally high concentrations in tumors; certain GSLs, added to
tumor cells
in culture media, exert marked stimulatory or inhibitory actions on tumor
growth;
antibodies to certain GSLs inhibit the growth of tumors; the GSLs that are
shed by
tumors into the surrounding extracellular fluid inhibit the body's normal
immunodefense system; the composition of a tumor's GSLs changes as the tumors
become increasingly malignant; and, in certain kinds of cancer, the level of a
GSL
circulating in the blood gives useful information regarding the patient's
response to
treatment. Because of the significant impact GSLs have on several biochemical
processes, there remains a need for compounds having improved GIcCer synthase
inhibition activity.
It would thus be desirable to provide compounds which inhibit GIcCer synthase
activity. It would also be desirable to provide compounds which inhibit GIcCer
synthase activity, thereby lowering the level of GSLs and increasing GSL
precursor
levels, e.g. increasing the levels of ceramide and sphingols. It would further
be
CA 02378600 2002-O1-08
WO 01/04108 PCT/US00/18935
-4-
desirable to provide compounds which inhibit GIcCer synthase activity and
lower the
level of GSLs without also increasing ceramide levels. It would also be
desirable to
provide compounds and therapeutic methods to treat conditions and diseases
associated with altered GSL levels and/or GSL precursor levels.
SUMMARY OF THE INVENTION
Novel compounds are provided which inhibit GIcCer formation by inhibiting the
enzyme GIcCer synthase, thereby lowering the level of GSLs. The compounds of
the
present invention have improved GIcCer synthase inhibition activity and are
therefore
highly useful in therapeutic methods for treating various conditions and
diseases
associated with altered GSL levels, as well as GSL precursor levels. For
example,
the compounds of the present invention may be useful in methods involving
cancer
growth and metastasis, the growth of normal tissues, the ability of pathogenic
microorganisms to bind to normal cells, the binding between similar cells, the
binding
of toxins to human cells, and the ability of cancer cells to block the normal
process
of immunological cytotoxic attack.
Additional objects, advantages, and features of the present invention will
become apparent from the following description and appended claims, taken in
conjunction with the accompanying drawings.
BRIEF DESCRIPTION OF THE DRAWINGS
The various advantages of the present invention will become apparent to one
skilled in the art by reading the following specification and subjoined claims
and by
referencing the following drawings in which:
Figure 1 is a graph showing the growth and survival of 9L gliosarcoma cells
grown in medium containing different GIcCer synthase inhibitors;
Figure 2 is a graph showing the protein content of MDCK cells cultured for 24
hr in medium containing different concentrations of the separated eryfhro- and
fhreo-
isomers of a preferred compound of the present invention;
Figure 3 is a graph showing [3Hjthymidine incorporation into the DNA of MDCK
cells treated with a preferred compound of the present invention;
Figures 4A and 4B are graphs showing the effects of P4 and p-methoxy-P4
.~ on GIcCer synthase activity;
Figure 5 is a graph showing the linear relationship between the inhibition of
GIcCer synthase activity and electronic parameter (d) and hydrophobic
parameter (n);
Figure 6 is a graph showing the effects of dioxy P4 derivatives on GIcCer
synthase activity;
CA 02378600 2002-O1-08
WO 01/04108 PCTNS00/18935
-5-
Figure 7 is a bar graph showing the effects of D-t-3',4'-ethylenedioxy-P4 on
GIcCer synthesis and cell growth;
Figure 8 is a schematic of the synthetic pathway for 4'-hydroxy-1-phenyl-2-
palmitoylamino-3-pyrrolidino-1-propanol;
Figure 9 is an illustration of the structures of P4 and of phenyl-substituted
P4
homologues;
Figure 10 is an HPLC chromatogram showing the separation of the
enantiomers of P4 and p-methoxy-P4 by chiral chromatography;
Figure 11 is a graph showing the effects of D-threo-4'-hydroxy-P4 as
compared to D-threo-p-methoxy-P4 on GIcCer synthase activity;
Figure 12 is a graph showing the effects of D-threo enantiomers of P4, 4'-
hydroxy-P4 and 3',4'-ethylenedioxy-P4 on 1-O-acyceramide synthase activity;
Figure 13 is a graph showing the effect of D-threo-P4 on GIcCer synthesis and
cell growth;
Figure 14 is a graph showing the effect of D-threo-4'-hydroxy-P4 on GIcCer
synthesis and cell growth; and
Figure 15 is a graph showing the effect of D-threo-3',4'-ethylenedioxy-P4 on
GIcCer synthesis and cell growth.
DETAILED DESCRIPTION OF THE PREFERRED EMBODIMENTS
Novel compounds are provided which inhibit GIcCer formation by inhibiting the
enzyme GIcCer synthase, thereby lowering the level of GSLs. The compounds of
the
present invention have improved GIcCer synthase inhibitory activity and are
therefore
highly useful in therapeutic methods for treating various conditions and
diseases
associated with altered GSL levels.
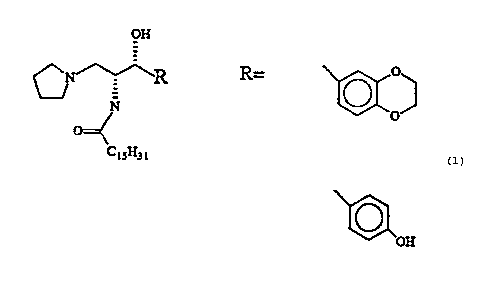
The compounds of the present invention generally have the following formula:
R~
wherein
R, is a phenyl group, preferably a substituted phenyl group such as p-methoxy,
hydroxy, dioxane substitutions such as methylenedioxy, ethylenedioxy, and
trimethylenedioxy, cyclohexyl or other acyclic group, t-butyl or other
branched aliphatic
CA 02378600 2002-O1-08
WO 01/04108 PCT/US00/18935
-6-
group, or a long alkyl or alkenyl chain, preferably 7 to 15 carbons long with
a double
bond next to the kernel of the structure. The aliphatic chain can have a
hydroxyl
group near the two asymmetric centers, corresponding to phytosphingosine.
R2 is an alkyl residue of a fatty acid, 10 to 18 carbons long. The fatty acid
can
be saturated or unsaturated, or possess a small substitution at the C-2
position (e.g.,
a hydroxyl group).
R3 is a tertiary amine, preferably a cyclic amine such as pyrrolidine,
azetidine,
morpholine or piperidine, in which the nitrogen atom is attached to the kernel
(i.e., a
tertiary amine).
All four structural isomers of the compounds are contemplated within the
present invention and may be used either singly or in combination (i.e., DL-
threo or
D L-erythro).
The preferred aliphatic compound of the present invention is D-threo-1
pyrrolidino-1-deoxyceramide, identified as IV-231 B herein and also referred
to as PD.
The preferred aromatic compound of the present invention is 1-phenyl-2
palmitoylamino-3-pyrrolidino-1-propanol, identified as BML-119 herein and also
referred to as P4. The structures of the preferred compounds are as follows:
H H
CH3(CH=)IiCH=CH- ~ - i -CHZ-N
aH NH
I
C=O
I
C15H31 ....-
PD P4
An additional preferred compound of the present invention are D-t-3',4'-
ethylenedioxy-1-phenyl-2-palmitoylamino-3-pyrrolidino-1-propanol, also
referred to
hereinasD-t 3',4'~thylenedioxy-P4, andD-t-4'-hydroxy-1-phenyl-2-palmitoylamino-
3-
pyrrolidino-1-propanol, also refer-ed to herein as D-t-4'-hydroxy-P4.
By increasing the acyl chain length of PDMP from 10 to 16 carbon atoms, the
efficacy of the compounds of the present invention as GIcCer synthase
inhibitors is
greatly enhanced. The use of a less polar cyclic amine, especially a pyn-
olidine
instead of a morpholine ring, also increases the efficacy of the compounds. In
addition, replacement of the phenyl ring by a chain corresponding to
sphingosine
yields a strongly inhibitory material. By using a chiral synthetic route, it
was
discovered that the isomers active against GIcCer synthase had the
CA 02378600 2002-O1-08
WO 01/04108 PCT/US00/18935
-7-
R,R-(D-fhreo)-configuration. However, strong inhibition of the growth of human
cancer cells in plastico was produced by both the threo and erythro racemic
compounds, showing involvement of an additional factor beyond simple depletion
of
cell glycosphingolipids by blockage of GIcCer synthesis. The growth arresting
effects
could be correlated with increases in cellular ceramide and diglyceride
levels.
Surprisingly, the aliphatic pyrrolidino compound of the present invention
(identified as IV-231 B), was strongly inhibitory toward the GIcCer synthase
and
produced almost complete depletion of glycolipids, but did not inhibit growth
or cause
an accumulation of ceramide. Attempts were made to determine if the
differences in
growth effects could be attributed to the influence of the inhibitors on
related enzymes
(ceramide and sphingomyelin synthase and ceramidase and sphingomyelinase).
While some stimulation or inhibition of enzyme activity was noted,
particularly at high
inhibitor concentrations (50 NM), these findings did not explain the differing
effects of
the different inhibitors.
By slowing the synthesis of GIcCer, the compounds of the present invention
lower the levels of all the GIcCer-derived GSLs due to the GSL hydrolases
which
normally destroy them. While the body will continue to make the more complex
GSLs
from available GIcCer, the rate of synthesis will slow down as the level of
GIcCer
diminishes. The rate of lowering depends on the normal rate of destruction of
each
GSL. These rates however, are relatively rapid in animals and cultured cells.
At higher dosages, many of the compounds of the present invention produce
an elevation in the level of ceramide. Presumably this occurs because cells
continue
to make ceramide despite their inability to utilize it for GIcCer synthesis.
Ceramide
is also normally converted to sphingomyelin, but this process does not seem to
be
able to handle the excess ceramide. It has been unexpectedly found however,
that
an additional process is also involved, since even those isomers that are
inert against
GIcCer synthase also produce an elevation in ceramide levels. Moreover, the
blockage of GIcCer synthase can occur at low inhibitor dosages, yet ceramide
accumulation is not produced. The preferred aliphatic compound of the present
invention, D-threo-1-pyrrolidino-1-deoxyceramide (PD), does not produce
ceramide
accumulation at all, despite almost complete blockage of GIcCer synthesis.
This distinction between the aromatic and the aliphatic compounds of the
present invention is important because ceramide has recently been proposed to
cause
cell death (apoptosis) by some still unknown mechanism. At lower dose levels,
the
aromatic compounds of the present invention cause GSL disappearance with only
CA 02378600 2002-O1-08
WO 01/04108 PCT/US00/18935
_g_
small accumulation of ceramide and inhibition of cell growth. Higher dosages
cause
much more ceramide deposition and very slow cell growth or cell death.
In one embodiment of the present invention, methods of treating patients
suffering from inborn genetic errors in the metabolism of GIcCer and its
normal
anabolic products (lactosylceramide and the more complex GSLs) are provided.
The
presently known disorders in this category include Gaucher, Fabry, Tay-Sachs,
Sandhoff, and GM1 gangliosidosis. The genetic errors lie in the patient's
inability to
synthesize a hydrolytic enzyme having normal efficiency. Their inefficient
hydrolase
allows the GSL to gradually accumulate to a toxic degree, debilitating or
killing the
victim. The compounds of the present invention slow the formation of GSLs,
thus
allowing the defective hydrolase to gradually "catch up" and restore the
concentrations
of GSLs to their normal levels and thus the compounds may be administered to
treat
such patients.
With respect to Gaucher disease, it has been calculated that much of the
patient's accumulated GIcCer in liver and spleen arises from the blood cells,
which
are ultimately destroyed in these organs after they have reached the end of
their life
span. The actual fraction, lipid derived from blood cells versus lipid formed
in the liver
and spleen cells, is actually quite uncertain, but the external source must be
important. Therefore it is necessary for the compounds of the present
invention to
deplete the blood cells as they are formed or (in the case of white blood
cells) while
they still circulate in the blood. Judging from toxicity tests, the white
cells continue
to function adequately despite their loss of GSLs. Although the toxicity
studies were
not of a long enough duration to produce many new red cells with low GSL
content,
it is possible that circulating red cells also undergo turnover (continual
loss plus
replacement) of GSLs.
In an alternative embodiment of the present invention, for the treatment of
disorders involving cell growth and division, high dosages of the compounds of
the
present invention are administered but only for a relatively short time. These
disorders include cancer, collagen vascular diseases, atherosclerosis, and the
renal
hypertrophy of diabetic patients. Accumulation or changes in the cellular
levels of
GSLs have been implicated in these disorders and blocking GSL biosynthesis
would
allow the normal restorative mechanisms of the body to resolve the imbalance.
With atherosclerosis, it has been shown that arterial epithelial cells grow
faster
in the presence of a GIcCer product (lactosylceramide). Oxidized serum
lipoprotein,
a material that normally circulates in the blood, stimulates the formation of
plaques
CA 02378600 2002-O1-08
WO 01/04108 PCT/US00/18935
_g_
and lactosylceramide in the inner lining of blood vessels. Treatment with the
compounds of the present invention would inhibit this mitogenic effect.
In an additional embodiment of the present invention, patients suffering from
infections may be treated with the compounds of the present invention. Many
types
of pathogenic bacteria have to bind to specific GSLs before they can induce
their toxic
effects. As shown in Svensson, M. et al., "Epithelial Glucosphingolipid
Expression as
a Determinant of Bacterial Adherence and Cytokine Production," Infect. and
Immun.
62:4404-4410 (1994), expressly incorporated by reference, PDMP treatment
reduces
the adherence of E. coli to mammalian cells. Several viruses, such as
influenza type
A, also must bind to a GSL. Several bacterial toxins, such as the verotoxins,
cannot
themselves act without first binding to a GSL. Thus, by lowering the level of
GSLs,
the degree of infection may be ameliorated. In addition, when a patient is
already
infected to a recognizable, diagnosable degree, the compounds of the present
invention may slow the further development of the infection by eliminating the
binding
sites that remain free.
It has been shown that tumors produce substances, namely gangliosides, a
family of GSLs, that prevent the host i.e., patient, from generating
antibodies against
the tumor. By blocking the tumor's ability to secrete these substances,
antibodies
against the tumor can be produced. Thus, by administering the GIcCer synthase
inhibitors of the present invention to the patient, the tumors will become
depleted of
their GSLs and the body's normal immunological defenses will come into action
and
destroy the tumor. This technique was described in Inokuchi, J. et al.,
"Antitumor
Activity in Mice of an Inhibitor of Glycosphingolipid Biosynthesis," Cancer
Left. 38:23-
30(1987), expressly incorporated by reference. The compounds of the present
invention and in particular the aliphatic compounds require much lower doses
than
those previously described. This is particularly important because the lower
dose may
reduce certain side effects. Moreover, because the aliphatic compounds of the
present invention do not produce ceramide accumulation, they are less toxic.
In
addition, 1-phenyl-2-palmitoylamino-3-pyrrolidino-1-propanol (P4), may act via
two
pathways, GSL depletion and ceramide accumulation.
In an alternative embodiment, a vaccine-like preparation is provided. Here,
cancer cells are removed from the patient (preferably as completely as
possible), and
the cells are grown in culture in order to obtain a large number of the cancer
cells.
The cells are then exposed to the inhibitor for a time sufficient to deplete
the cells of
their GLSs (generally 1 to 5 days) and are reinjected into the patient. These
CA 02378600 2002-O1-08
WO 01/04108 PCT/US00/18935
-10-
reinjected cells act like antigens and are destroyed by the patient's
immunodefense
system. The remaining cancer cells (which could not be physically removed)
will also
be attacked by the patient's antibodies. In a preferred embodiment, the
patient's
circulating gangliosides in the plasma are removed by plasmapheresis, since
the
circulating gangliosides would tend to block the immunodefense system.
It is believed that tumors are particularly dependent on GSL synthesis for
maintenance of their growth (Hakomori, S. "New Directions in Cancer Therapy
Based
on Aberrant Expression of Glycosphingolipids: Anti-adhesion and Ortho-
Signaling
Therapy," Cancer Cells 3:461-470 (1991 )). Accumulation of ceramide in treated
tumors also slows their growth or kills them. Tumors also generate large
amounts of
GSLs and secrete them into the patient's body, thereby preventing the host's
normal
response by immunoprotective cells, which should generate antibodies against
or
otherwise destroy tumor cells (e.g., tumors are weakly antigenic). It has also
been
shown that GSL depletion blocks the metastasis of tumor cells (Inokuchl, J. et
al.,
"Inhibition of Experimental Metastasis of Murine Lewis Long Carcinoma by an
Inhibitor
of Glucosylceramide Synthase and its Possible Mechanism of Action," Cancer
Res.
50:6731-6737 (1990). Tumor angiogenesis (e.g., the production of blood
capillaries)
is strongly influenced by GSLs (Ziche, M. et al., "Angiogenesis Can Be
Stimulated or
Repressed in In Vivo by a Change in GM3:GD3 Ganglioside Ratio," Lab. Invest.
67:711-715 (1992)). Depleting the tumor of its GSLs should block the tumors
from
generating the new blood vessels they need for growth.
A further important characteristic of the compounds of the present invention
is their unique ability to block the growth of multidrug resistant ("MDR")
tumor cells
even at much lower dosages. This was demonstrated with PDMP by Rosenwald,
A.G. et al., "Effects of the Glycosphingolipid Synthesis Inhibitor, PDMP, on
Lysosomes
in Cultured Cells," J. Lipid Res. 35:1232 (1994), expressly incorporated by
reference.
Tumor cells that survive an initial series of therapeutic treatments often
reappear
some years later with new properties - they are now resistant to a second
treatment
schedule, even with different drugs. This change has been attributed to the
appearance in the tumor of large amounts of a specific MDR protein (P-
glycoprotein).
It has been suggested that protein kinase C (PKC) may be involved in the
action or
formation of P-glycoprotein (Blobe, G.C. et al., "Regulation of PKC and Its
Role in
Cancer Biology," CancerMetastasis Rev. 13:411-431 (1994)). However decreases
in
PKC have other important effects, particularly slowing of growth. It is known
that
PDMP does lower the cellular content of PKC (Shayman, J.A. et al., "Modulation
of
CA 02378600 2002-O1-08
WO 01/04108 PCT/US00/18935
_11_
Renal Epithelial Cell Growth by Glucosylceramide: Association with Protein
Kinase C,
Sphingosine, and Diacylglyceride," J. BioL Chem. 266:22968-22974 (1991 )) but
it is
not clear why it so effectively blocks growth of MDR cells (Rosenwald, A.G. et
al.,
"Effects of the Glycosphingolipid Synthesis Inhibitor, PDMP, On Lysosomes in
Cultured Cells," J. Lipid Res. 35:1232 (1994)). A recent report showed that
several
lipoidai amines that block MDR action also lower the level of the enzyme acid
sphingomyelinase (Jaffrezou,J. et al., "Inhibition of Lysosorrial Acid
Sphingomyelinase
by Agents which Reverse Multidrug Resistance," 8iochim. 8iophys. Acta 1266:1-8
(1995)). One of these agents was also found to increase the cellular content
of
sphingosine 5-fold, an effect seen with PDMP as well. One agent,
chlorpromazine,
behaves like the compounds of the present invention, in its ability to lower
tissue
levels of GIcCer (Hospattankar, A.V. et al., "Changes in Liver Lipids After
Administration of 2-Decanoylamino-3-Morpholinopropiophenone and
Chlorpromazine,"
Lipids 17:538-543 (1982)).
It will be appreciated by those skilled in the art that the compounds of the
present invention can be employed in a wide variety of pharmaceutical forms;
the
compound can be employed neat or admixed with a pharmaceutically acceptable
carrier or other excipients or additives. Generally speaking, the compound
will be
administered orally or intravenously. It will be appreciated that
therapeutically
acceptable salts of the compounds of the present invention may also be
employed.
The selection of dosage, rate/frequency and means of administration is well
within the
skill of the artisan and may be left to the judgment of the treating physician
or
attending veterinarian. The method of the present invention may be employed
alone
or in conjunction with other therapeutic regimens. It will also be appreciated
that the
compounds of the present invention are also useful as a research tool e.g., to
further
investigate GSL metabolism.
The following Specific Example further describes the compounds and methods
of the present invention.
SPECIFIC EXAMPLE 1
The following formulas set forth preferred aromatic and aliphatic compounds:
FORMULA 1
7
g OH
2
R,GH=~~ CH
NH
I
C=O
R'
CA 02378600 2002-O1-08
WO 01/04108 PCT/US00/18935
-12-
identified as (1 R,2R~1-phenyl-2-acylamino-3-cyclic amino-1-propanol, and
referred
to herein as the "aromatic inhibitors," wherein
The phenyl group can be a substituted phenyl group (such as p-
methoxyphenyl).
R' is an alkyl residue of a fatty acid, 10 to 18 carbons long. The fatty acid
can
be saturated or unsaturated, or possess a small substitution at the C-2
position (e.g.,
a hydroxyl group).
R is morpholino, pyrrolidino, piperidino, azetidino (trimethyleneimino), N-
methylethanolamino, diethylamino or N-phenylpiperazino. A small substituent,
such
as a hydroxyl group, is preferably included on the cyclic amine moiety.
FORMULA II
3
t Qx
2
R,cH~~x~ x\~c~~=s
NH
C~
R'
identified as (2R,3R)-2-palmitoyl-sphingosyl amine or 1-cyclic amino-1-
deoxyceramide
or 1-cyclic amino-2-hexadecanoylamino-3-hydroxy-octadec-4,5-ene, and referred
to
herein as the "aliphatic inhibitors," wherein
R' is an alkyl residue of a fatty acid, 10 to 18 carbons long. The fatty acid
can
be saturated or unsaturated, or possess a small substitution at the C-2
position (e.g.,
a hydroxyl group).
R is morpholino, pyrrolidino, piperidino, azetidino (trimethyleneimino), N
methylethanolamino, diethylamino or N-phenylpiperazino. A small substituent,
such
as a hydroxyl group, is preferably included on the cyclic amine moiety.
The long alkyl chain shown in Formula II can be 8 to 18 carbon atoms long,
with or without a double bond near the asymmetric carbon atom (carbon 3).
Hydroxyl
groups can, with advantage, be substituted along the aliphatic chain,
particularly on
carbon 4 (as in the naturally occurring sphingol, phytosphingosine). The long
chain
can also be replaced by other aliphatic groups, such at t-butyl or
cyclopentyl.
The aromatic inhibitors (see Formula I and Table 1 ) were synthesized by the
Mannich reaction from 2-N acylaminoacetophenone, paraformaldehyde, and a
secondary amine as previously described (Inokuchi, J. et al., "Preparation of
the
CA 02378600 2002-O1-08
WO 01/04108 PCT/US00/18935
-13-
Active Isomer of 1-Phenyl-2-Decanoylamino-3-Morpholino-1-Propanol, Inhibitor
of
Glucocerebroside Synthetase," J. Lipid Res. 28:565-571 (1987) and Vunnam, R.R.
et al., "Analogs of Ceramide that Inhibit Glucocerebroside Synthetase in Mouse
Brain," Chem. Phys. Lipids 26:265-278 (1980)). For those syntheses in which
phenyl-substituted starting materials were used, the methyl group in the
acetophenone
structure was brominated and converted to the primary amine. Bromination of
p-methoxyacetophenone was performed in methanol. The acetophenones and
amines were from Aldrich Chemical Co., St. Louis, MO. Miscellaneous reagents
were
from Sigma Chemical Co. and the sphingolipids used as substrates or standards
were
prepared by methods known in the art. The reactions produce a mixture of four
isomers, due to the presence of two asymmetric centers.
The aliphatic inhibitors (See Formula II and Table 2) were synthesized from
the corresponding 3-t-butyldimethylsilyl-protected sphingols, prepared by
enantioselective aldol condensation (Evans, D.A. et al., "Stereoselective
Aldol
Condensations Via Boron Enolates," J. Am. Chem. Soc. 103:3099-3111 (1981 ) and
Abdel-Magid, A. et al., Metal-Assisted Aldol Condensation of Chiral A-
Halogenated
Imide Enolates: A Stereocontrolled Chiral Epoxide Synthesis," J. Am. Chem.
Soc.
108:4595-4602 (1986)) using a modification of the procedure of Nicolaou et al.
(Nicolaou, K.C. et al., "A Practical and Enantioselective Synthesis of
Glycosphingolipids and Related Compounds. Total Synthesis of
Globotriaosylceramide (Gb3)," J. Am. Chem. Soc. 110:7910-7912 (1988)). Each
protected sphingol was first converted to the corresponding primary triflate
ester, then
reacted with a cyclic amine. Subsequent N-acylation and desilylation led to
the final
products in good overall yield (Carson, K.G. et al., "Studies on
Morpholinosphingolipids: Potent Inhibitors of Glucosylceramide Synthase,"
Tetrahedron Lett. 35:2659-2662 (1994)). The compounds can be called
1-morpholino-(or pyrrolidino)-1-deoxyceramides.
Labeled ceramide, decanoyl sphingosine, was prepared by reaction of the acid
chloride and sphingosine (Kopaczyk, K. C. et al., "In Vivo Conversions of
Cerebroside
and Ceramide in Rat Brain," J. Lipid Res. 6:140-145 (1965)) and NBD-SM
(12-[N-methyl-N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)J-
sphingosylphosphorylcholine)
was from Molecular Probes, Inc., Eugene, OR.
Methods
TLC of the amines was carried out with HPTLC plates (E. Merck silica gel 60)
and C-M-HOAc 90:10:10 (solvent A) or 85:15:10 (solvent B) or C-M-conc.
ammonium
CA 02378600 2002-O1-08
WO 01/04108 PCT/US00/18935
-14-
hydroxide 30:10:1 (solvent C). The bands were stained with iodine or with
Coomassie
Brilliant Blue R-250 (Nakamura, K. et al., "Coomassie Brilliant Blue Staining
of Lipids
on Thin-Layer Plates," Anal. Biochem. 142:406-41 (1984)) and, in the latter
case,
quantified with a Bio-Rad Model 620 videodensitometer operated with reflected
white
light. The faster band of each PDMP analog, previously identified as the
erythro form,
corresponds to the 1 S,2R and 1 R,2S stereoisomers, and the slower band,
previously
identified as the threo form, corresponds to the 1 R,2R and 1 S,2S
stereoisomers.
TLC of the cell lipids was run with C-M-W 24:7:1 (solvent D) or 60:35:8
(solvent E).
Growth of cell lines. Comparisons of different inhibitors with regard to
suppression of human cancer cell growth were made by the University of
Michigan
Cancer Center in vitro Drug Evaluation Core Laboratory. MCF-7 breast carcinoma
cells, HT-29 colon adenocarcinoma cells, H-460 lung large cell carcinoma
cells, and
9L brain gliosarcoma cells were grown in RPMI 1640 medium with 5% fetal bovine
serum, 2 mM glutamine, 50 units/ml of penicillin, 50 mg/ml of streptomycin,
and 0.1
mg/ml of neomycin. UMSCC-10A head and neck squamous carcinoma cells were
grown in minimal essential medium with Earle salts and the same supplements.
Medium components were from Sigma Chemical Co. Cells were plated in 96-well
microtiter plates (1000 cells/well for H-460 and 9L cells, and 2000 cells/well
for the
other lines), and the test compounds were added 1 day later. The stock
inhibitor
solutions, 2 mM in 2 mM BSA, were diluted with different amounts of additional
2 mM
BSA, then each solution was diluted 500-fold with growth medium to obtain the
final
concentrations indicated in the Figures and Tables.
Five days after plating the H-460 and 9L cells, or 6 days for the other lines,
cell growth was evaluated by staining the adhering cells with sulfofiodamine B
and
measuring the absorbance at 520 nm (Skehan, P. et al., "New Colorimetric
CytotoxicityAssayforAnticancer-Drug Screening," J. Natl. Cancerlnst. 82:1107-
1112
(1990)). The absorbance of the treated cultures is reported as percent of that
of
control cultures, to provide an estimate of the fraction of the cells that
survived, or of
inhibition of growth rate.
For the experiments with labeled thymidine, each 8.5 cm dish contained
500,000 Madin-Darby canine kidney (MDCK) cells in 8 ml of Dulbecco modified
essential supplemented medium. The cells were incubated at 37°C in 5%
COZ for 24
h, then incubated another 24 h with medium containing the inhibitor-BSA
complex.
The control cells were also incubated in the presence of BSA. The cells were
washed
CA 02378600 2002-O1-08
WO 01/04108 PCT/US00/18935
-15-
with phosphate/saline and trichloroacetic acid, then scraped off the dishes,
dissolved
in alkali, and analyzed for protein and DNA incorporated tritium. [Methyl
'H]thymidine
(10 pCi) was added 4 h prior to harvesting.
Assay of sphingolipid enzymes. The inhibitors were evaluated for their
effectiveness against the GIcCer synthase of MDCK cell homogenates by
incubation
in a thermostatted ultrasonic bath (Radin N.S. et al., "Ultrasonic Baths as
Substitutes
for Shaking Incubator Baths," Enzyme 45:67-70 (1991 )) with octanoyl
sphingosine and
uridinediphospho[3H]glucose (Shukla, G.S. et al., "Glucosylceramide Synthase
of
Mouse Kidney: Further Characterization and Improved Assay Method," Arch.
Biochem. Biophys. 283:372-378 (1990)). The lipoidal substrate (85 Ng) was
added
in liposomes made from 0.57 mg dioleoylphosphatidylcholine and 0.1 mg of Na
sulfatide. Confluent cells were washed, then homogenized with a micro-tip
sonicator
at 0°C for 3 x 30 sec; -0.2 mg of protein was used in each assay tube.
In the case
of the aromatic inhibitors, the test compound was simply evaporated to dryness
from
75 solution in the incubation tube. This method of adding the inhibitor was
found to give
the same results as addition as a part of the substrate liposomes. The
aliphatic
inhibitors, which appeared to be less soluble in water, were added as part of
the
substrate liposomes.
Acid and neutral ceramidases were assayed under conditions like those above,
but the medium contained 110 NM [1-''C]decanoyl sphingosine (1 OS cpm) in 340
NM
dioleoylphosphatidylcholine liposomes and 0.34 mg of MDCK cellular protein
homogenate. The acid enzyme was incubated in 32.5 mM citrate-Na; (pH 4.5) and
the neutral enzyme buffer was 40 mM Tris-Cf (pH 7.1 at 37°C). After 60
min in the
ultrasonic bath, 3 ml of C-M 2:1, carrier decanoic acid, and 0.6 ml of 0.9%
saline were
added and the lipids in the lower layer were separated by TLC with C-HOAc 9:1.
The
liberated decanoic acid was scraped off the glass plate and counted.
Ceramide synthase was assayed with 1 NM [3 3H]sphingosine (70,000 cpm,
repurified by column chromatography), 0.2 mM stearoyl-CoA, 0.5 mM
dithiothreitol,
and -300 pg of MDCK homogenate protein in 25 mM phosphate-K' buffer, pH 7.4,
in a total volume of 0.2 ml. The incubation (for 30 min) and TLC were carried
out as
above and the ceramide band was counted.
Sphingomyelin synthase was evaluated with 44 pM ["C]decanoyl sphingosine
(105 cpm) dispersed with 136 pM dioleoyllecithin as in the ceramide synthase
assay,
and 5 mM EDTA and 50 mM Hepes-Na' pH 7.5, in a total volume of 0.5 ml. MDCK
homogenate was centrifuged at 600 X g briefly, then at 100,000 X g for 1 h,
and the
CA 02378600 2002-O1-08
WO 01/04108 PCT/US00/18935
-16-
pellet was suspended in water and sonicated with a dipping probe. A portion of
this
suspension containing 300 Ng of protein was used. Incubation was at
37°C for 30
min, after which the lipids were treated as above, using C-M-W 60:35:8 for the
isolation of the labeled decanoyl SM.
Acid and neutral SMase assays were based on the procedures of Gatt et al.
(Gaff, S. et al., "Assay of Enzymes of Lipid Metabolism With Colored and
Fluorescent
Derivatives of Natural Lipids," Meth. Enzymol. 72:351-375 (1981)), using
liposomes
containing NBD-SM dispersed like the labeled ceramide (10 NM substrate and 30
NM
lecithin). The assay medium for the neutral enzyme also contained 50 mM Tris-
Cf
(pH 7.4), 25 mM KCI, 5 mM MgCl2 and 0.29 mg of MDCK cell protein in a total
volume of 0.25 ml. Incubation was at 37°C for 30 min in the ultrasonic
bath, then the
fluorescent product, NBD-ceramide, was isolated by partitioning the assay
mixture
with 0.45 ml 2-propanol, 1.5 ml heptane, and 0.2 ml water. After
centrifugation, a
trace of contaminating NBD-SM was removed from 0.9 ml of the upper layer by
washing with 0.35 ml water. The upper layer was analyzed with a fluorometer
(460
nm excitation, 515 nm emission).
Acid SMase was assayed with the same liposomes in 0.2 ml of assay mixture
containing 125 mM NaOAc (pH 5.0) and 61 Ng of cell protein, with 60 min of
incubation at 37°C. The resultant ceramide was determined as above.
Results
Table 1 lists the aromatic compounds (see Formula I) synthesized and their
migration rates on silica gel TLC plates. Separation of the threo- and
erythro-steroisomers by TLC was generally very good, except for BML-120, -121,
and
-122 in the acidic solvent. In the basic solvent BML-119 and BML-122 yielded
poorly
resolved double bands. BML-112 was unexpectedly fast-running, especially when
compared with BML-120; both are presumably dihydrochlorides.
TABLE 1
Structures of the Aromatic Inhibitors
BML Number R Group Phenyl TLC hR,
or Name SubstituentValue'
PDMPd - morpholino 34(47)
PPMP morpholino (53)
112 N-phenylpiperazino 56
113 morpholino p-fluoro 25
CA 02378600 2002-O1-08
WO 01/04108 PCT/US00/18935
-17-
114 diethylamino 25
115 piperidino (pentamethyleneimino) 29
116 hexamethyleneimino 34
1176 morpholino p-fluoro 41
118 piperidino p-fluoro 26
119 pyrrolidino (tetramethyleneimino) 20-70(44)
120 1-methylpiperazino 7-62
121 ~ 3-dimethylaminopiperidino 1-30
122 N-methylethanolamino 6-71
123 azetidino (trimethyleneimino) 12
124 amino 15
125 morpholino p-methoxy 37
126 pyrrolidino p-methoxy (50)
a Only the relative R, value of the faster-moving band is shown. The first
value was
obtained with solvent A, the second with solvent C, and the numbers in
parentheses,
with solvent B. In the case of BML-117, -125, and -126, a 20-cm high TLC plate
was
used to improve the seperation.
b The fatty acid chain suggested by the R' group is decanoyl, not palmitoyl.
Table 2 describes four aliphatic inhibitors (see Formula II), which can be
considered to be ceramide analogs in which the C-1 hydroxyl group is replaced
by a
cyclic amine. It should be noted that the carbon frameworks of compounds in
Tables
1 and 2 are numbered differently (see Formulas I and II), thus affecting
comparisons
of stereochemical configurations. The threo- and erythro-isomers separated
very
poorly on TLC plates. Like the aromatic inhibitors, however, the morpholine
compounds ran faster than the pyrrolidine compounds. The latter are presumably
more strongly adsorbed by the silica gel because they are more basic.
CA 02378600 2002-O1-08
WO 01/04108 PCT/US00/18935
-18-
TABLE 2
Characterization of the Sphingosyl Inhibitors
Number R Group Sphingol TLC hR,
Structure Values
IV-181A morpholino 2R,3S 43
IV-206A morpholino 2R,3R 40
IV-230A pyrrolidino 2R,3S 31
IV-231 B pyrrolidino 2R,3R 31
a TLC solvent: C-M-HOAc 90:5:10. Similar but faster migrations were obtained
with
solvent A.
Structure-activity correlations. The results of testing the compounds in an
assay system for GIcCer synthase are listed in Table 3. Each inhibition
determination
(~ SD) shown in Table 3 was carried out in triplicate. Some of the inhibitors
were
tested as mixtures of DL-erythro- and DL-threo-isomers (see column 4). Only
the
D-threo enantiomer in each mixture was predicted to be the actual enzyme
inhibitor
(Inokuchi, J. et al., "Preparation of the Active Isomer of
1-Phenyl-2-Decanoylamino-3-Morpholino-1-Propanol, Inhibitor of
Glucocerebroside
Synthetase," J. Lipid Res. 28:565-571 (1987)); the content of this isomer was
calculated by measuring the proportions of the threo- and erythro- racemic
mixtures
by quantitative TLC. The DL-threo contents were found to be in the range of 40
to
72%. The comparisons, in the case of the mixtures, are therefore approximate
(most
of the samples were not purified to remove the three less-active isomers and
the
observed data were not corrected for the level of the primary enantiomers).
The
separation of the threo- and erythro- forms is most conveniently accomplished
by
crystallization, but the specific conditions vary for each substance; thus
only BML-119,
a strong inhibitor, was separated into its threo- and erythro- forms. BML-112
is not
included in Table 3 because it had no inhibitory activity against GIcCer
synthase of
rabbit liver microsomes.
CA 02378600 2002-O1-08
WO 01/04108 PCT/US00/18935
-19-
TABLE 3
Inhibition of Ceramide Glucosyltransferase of
MDCK cell Homogenates by Different Compounds
Inhibitor ~~o Inhibition at 80 Inhibition Active
Number pM at Isomer"
5 pM
i
BML-113 60 + 4.78 29
BML-114 31 + 2.98 20
BML-115 84 _+ 0.88 12.4 + 0.7' 27
82 + 0.3b _
BML-116 28 + 3.28 27
BML-117 35 + 0.6b 36
BML-118 62 + 0.4 8.3 + 1.4' 32
BML-119 94 _+ 1.4b 51 _+ 2.38 29
97+0.1' 49+0.8'
96 + 0.1 _
BML-120 11 + 3.0' 26
BML-121 11 + 0.4' 28
BML-122 58 + 1.6d 26
BML-123 86 + 0.1 d 15 + 0.8' 33
~
BML-124 -2 + 1.6d 15
BML-125 9 + 3.08 26
BML-126 60 _+ 1.88 34
54 + 0.3r
PDMP 90 + 0.88 16 + 1.8' 100
PPMP 32 _+ 1.88 100
32+0.T
IV-181 A 12 + 0.2 100
IV-206A 73 + 1.5 100
IV-230A 19 + 2.1 100
IV-231 B 87 + 0.4 100
e'° Different samples were assayed as parts of different experiments.
'' Percent of the active D-stereoisomer in the synthesized sample, estimated
by
scanning the two stained bands, assuming the slower one was the (racemic)
active
form.
Comparison of PDMP (1 R,2R-decanoate) and PPMP (1 R,2R-palmitate), when
evaluated at the same time in Expt. f, shows that an increase in the chain
length of
CA 02378600 2002-O1-08
WO 01/04108 PCT/US00/18935
-20-
the N-acyl group from 10 to 16 carbon atoms distinctly improved the inhibitory
activity
against GIcCer synthase, as noted before (Abe, A. et al., "Improved Inhibitors
of
Glucosylceramide Synthesis," J. Biochem. 111:191-196 (1992)). Accordingly,
most
of the other compounds were synthesized with the palmitoyl group for
comparison
with PPMP. The comparisons between the best inhibitors are clearer at the 5 NM
level.
Replacing the oxygen in the morpholine ring of PPMP with a methylene group
(BML-115) improved activity -1.4-fold (calculated from the inhibitions at 5 NM
in Expt.
f and relative purities, and assuming that the percent inhibition is
proportional to
concentration in this region: 12.4/27 x 100/32 = 1.4). Previous comparison
with
mouse brain, human placenta, and human Gaucher spleen glucosyltransferase also
showed that replacing the morpholino ring with the piperidino ring in a ketone
analog
of PDMP (1-phenyl-2-decanoylamino-3-piperidino-1-propanone) produced a much
more active inhibitor (Vunnam, R.R. et al., "Analogs of Ceramide that Inhibit
Glucocerebroside Synthetase in Mouse Brain," Chem. Phys. Lipids 26:265-278
(1980)).
Replacing the piperidine group with a 7-membered ring (BML-116) greatly
decreased the activity, while use of a 5-membered ring (BML-119) quadrupled
the
effectiveness (50 vs 12.4% inhibition). A 4-membered ring (BML-123) yielded a
compound about as effective as the piperidino compound. The parent amine
(BML-124), its N,N-diethyl analog (BML-114), and the sterically bulky
N-phenylpiperazine analog (BML-112) displayed little or no activity.
Replacing a hydrogen atom with a fluorine atom in the p-position of the phenyl
ring decreased the inhibitory power (BML-117 vs PDMP and BML-118 vs BML-115).
Substitution of the p-position with an electron-donating moiety, the methoxy
group,
had a similar weakening effect in the case of the morpholino compound (BML-125
vs
PPMP). Comparison of the pyrrolidino compounds, which are more basic than the
morpholino compounds, showed that the methoxy group enhanced the inhibitory
power (BML-126 vs BML-119).
Preparations of BML-119 were separated into threo and erythro racemic
mixtures by HPLC on a Waters Microbondapak C,8 column, using M-W-conc. NH40H
90:10:0.2 as the elution solvent. The material eluting earlier (but migrating
more
slowly on a TLC plate) was called BML-130; the later eluting material (faster
by TLC)
was called BML-129. Assay of GIcCer synthase with each preparation at 5 pM
showed 15% inhibition by BML-129 and 79% inhibition by BML-130. TLC analysis
of
CA 02378600 2002-O1-08
WO 01/04108 PCT/US00/18935
-21 -
the two preparations revealed incomplete separation, which could explain the
minor
inhibition by BML-129. When the two stereoisomers were separated by
preparative
TLC, the difference in effectiveness was found to be somewhat higher,
evidently due
to the better separation by this method. Thus the slower-migrating
stereoisomer
accounted for all or nearly all of the inhibitory activity, as noted with PDMP
(Inokuchi,
J. et al., "Preparation of the Active Isomer of
1-Phenyl-2-Decanoylamino-3-Morpholino-1-Propanol, Inhibitor of
Glucocerebroside
Synthetase," J. Lipid Res. 28:565-571 (1987)).
Comparison of the two pairs of aliphatic inhibitors (bottom of Table 3) showed
that the 2R,3R (D-threo) form is the primary inhibitor of glucosyltransferase.
This
finding is in agreement with previous identification of the active PDMP isomer
as
being the D-threo enantiomer. However, unlike the aromatic analog, BML-129
(2R,3S/2S,3R), there was a relatively small but significant activity in the
case of the
(erythro) 2R,3S stereoisomer. The erythro form of PDMP was found to inhibit
cell
proliferation of rabbit skin fibroblasts almost as well as R,R/S,S-PDMP but it
did not
act on the GSLs (Uemura, K. et al., "Effect of an Inhibitor of
Glucosylceramide
Synthesis on Cultured Rabbit Skin Fibroblasts," J. Biochem. (Tokyo) 108:525-
530
(1990)). As noted with the aromatic analogs, the pyrrolidine ring was more
effective
than the morpholine ring (Table 3).
Comparison of the aliphatic and corresponding aromatic inhibitors can be
made in the case of the optically active morpholine compounds PPMP and IV-
206A,
both of which have the R,R structure and the same fatty acid. Here it appears
that
the aliphatic compound is more effective (Table 3). However in a second
comparison,
at lower concentrations with the inhibitors incorporated into the substrate
liposomes,
the degree of inhibition was 77 ~ 0.9% with 3 NM IV-231 B and 89 ~ 0.6% with 6
pM
DL-threo BML-119.
Evaluations of culfured cell growth. Exposure of five different cancer cell
lines to inhibitors at different concentrations for 4 or 5 days showed that
the six BML
compounds most active against GIcCer synthase were very effective growth
inhibitors
(Table 4). The ICS° values (rounded off to one digit in the table)
ranged from 0.7 to
2.6 pM.
CA 02378600 2002-O1-08
WO 01/04108 PCT/US00/18935
_22_
TABLE 4
Inhibition of Tumor Cell Growth In Vitro by Various Inhibitors
Cell BML- BML- BML- BML- BML- BML- BML-
Type 115 118 119 123 126 129 130
MCF-7 2 2 2 2 1 3 2
H-460 2 2 1 1 1 2 3
HT-29 2 1 2 1 2 2
9L 2 2 1 2 2 2 2
UMSCC 1 1 1 1 2 2
-10A
Figure 1 shows growth and survival of 9L gliosarcoma cells grown in medium
containing different GIcCer synthase inhibitors, as described above. The BML
compounds were used as synthesized (mixtures of DL-threo and -erythro
stereoisomers) while the PDMP and PPMP were optically resolved R,R isomers.
The
concentrations shown are for the mixed racemic stereoisomers, since later work
(Table 4) showed that both forms were very similar in effectiveness. Figure 1
illustrates the relatively weak effectiveness of R,R-PPMP and even weaker
effectiveness of R,R-PDMP. The three new compounds, however, are much better
inhibitors of GIcCer synthase and growth. These differences in growth
inhibitory
power con-elate with their effectiveness in MDCK cell homogenates as GIcCer
synthase inhibitors. Some differences can be expected due to differences in
sensitivity of the synthase occurring in each cell type (the syntheses were
assayed
only in MDCK cells).
Growth inhibition by each of the most active BML compounds occurred in an
unusually small range of concentrations (e.g., the slopes of the cytotoxic
regions are
unusually steep). Similar rapid drop-offs were seen in another series of tests
with 9L
cells, in which BML-119 yielded 71 % of the control growth with 1 pM
inhibitor, but only
3% of control growth with 3 pM. Growth was 93% of control growth .with 2 pM
BML-130 but only 5% of controls with 3 NM inhibitor. While some clinically
useful
drugs also show a narrow range of effective concentrations, this is a
relatively
uncommon relationship.
When the erythro- and threo-stereoisomeric forms of BML-119 (-129 and -130)
were compared, they were found to have similar effects on tumor cell growth
(Table
4). This observation is similar to the results with PDMP isomers in
fibroblasts cited
CA 02378600 2002-O1-08
WO 01/04108 PCT/US00/18935
-23-
above (Uemura, K. et al., "Effect of an Inhibitor of Glucosylceramide
Synthesis on
Cultured Rabbit Skin Fibroblasts," J. Biochem. (Tokyo) 108:525-530 (1990)).
Since
enzymes are optically active and since stereoisomers and enantiomers of drugs
can
differ greatly in their effect on enzymes, it is likely that BML-129 and BML-
130 work
on different sites of closely related metabolic steps.
Figure 2 shows the amount of cellular protein per dish for MDCK cells cultured
for 24 h in medium containing different concentrations of the separated
erythro- and
threo- isomers of BML-119, as percent of the incorporation by cells in
standard
medium. Each point shown in Figure 2 is the average of values from three
plates,
with error bars corresponding to one standard deviation.
Figure 3 shows (3H)thymidine incorporation into DNA of MDCK cells incubated
as in Figure 2. The values in Figure 3 are normalized on the basis of the
protein
content of the incubation dishes and compared to the incorporation by cells in
standard medium.
Figures 2 and 3 thus provide comparison of the two stereoisomers with MDCK
cells. The isomers were found to inhibit growth and DNA synthesis with similar
effectiveness. Thus the MDCK cells behaved like the human tumor cells with
regard
to ICS and the narrow range of concentrations resulting in inhibition of
protein and
DNA synthesis.
Surprisingly, the aliphatic inhibitor IV-2318 exerted no inhibitory effect on
MDCK cell growth when incubated at 20 NM for 1 day or 1 pM for 3 days. Tests
with
a longer growth period, 5 days, in 5 NM inhibitor also showed no slowing of
growth.
The dishes of control cells, which contained BSA as the only additive to the
medium,
contained 3.31 ~ 0.19 mg of protein, while the IV-231 B/BSA treated cells
contained
3.30 ~ 0.04 mg.
Lipid changes induced in fhe cells. Examination by TLC of the alkali-stable
MDCK lipids after a 24 h incubation disclosed that BML-130 was more effective
than
BML-129 in lowering GIcCer levels, as expected from its greater effectiveness
in vitro
as a glucosyltransferase inhibitor. The level of GIcCer, estimated visually,
was greatly
lowered by 0.3 NM BML-130 or 0.5 pM BML-129. The levels of the other lipids
visible
on the plate (mainly sphingomyelin (SM), cholesterol, and fatty acids) were
changed
little or not at all. BML-129 and the GIcCer synthase inhibitor, BML-130, were
readily
detected by TLC at the various levels used, showing that they were taken up by
the
cells during the incubation period at dose-dependent rates. Lactosylceramide
CA 02378600 2002-O1-08
WO 01/04108 PCT/US00/18935
-24-
overlapped the inhibitor bands with solvent D but was well separated with
solvent E,
which brought the inhibitors well above lactosylceramide.
Ceramide accumulation was similar for both stereoisomers (data not shown).
An unexpected finding is that noticeable ceramide accumulation appeared only
at
inhibitor concentrations that were more than enough to bring GIcCer levels to
a very
low point (e.g., at 2 or 4 NM). The changes in ceramide concentration were
quantitated in a separate experiment by the diglyceride kinase method, which
allows
one to also determine diacylglycerol (DAG) concentration (Preiss, J.E. et al.,
"Quantitative Measurement of SN-1,2-Diacylglycerols Present in Platelets,
Hepatocytes, and Ras- and Sis-Transformed Normal Rat Kidney Cells," J. BioL
Chem.
261:8597-8600 (1986)). The results (Table 5) are similar to the visually
estimated
ones: at 0.4 NM BML-129 or -130 there was little effect on ceramide content
but at 4
NM inhibitor, a substantial increase was observed. (While the duplicate
protein
contents per incubation dish were somewhat erratic in the high-dose dishes, in
which
growth was slow, the changes were nevertheless large and clear.) Accumulation
of
ceramide had previously been observed with PDMP, at a somewhat higher level of
inhibitor in the medium (Shayman, J.A. et al., "Modulation of Renal Epithelial
Cell
Growth by Glucosylceramide: Association with Protein Kinase C, Sphingosine,
and
Diacylglyceride," J. BioL Chem. 266:22968-22974 (1991 )). From the data for
cellular
protein per incubation dish, it can be seen that there was no growth
inhibition at the
0.4 NM level with either compound but .substantial inhibition at the 4 NM
level,
especially with the glucosyltransferase inhibitor, BML-130. This finding is
similar to
the ones made in longer incubations with human cancer cells.
TABLE 5
Effects of BML-129 and -130 on MDCK Cell Growth
and the Content of Ceramide and Diacylglycerol
Growth Medium
Protein Ceramide
Diglyceride
Ngldish nmollmg
protein
Controls 490 1.04 4.52
560 0.96 5.61
0.4 Nm BML-129 500 1.29 5.51
538 0.99 5.13
0.4 Nm BML-130 544 0.94 4.73
538 0.87 5.65
4 pm BML-129 396 3.57 9.30
311 3.78 9.68
CA 02378600 2002-O1-08
WO 01/04108 PCT/US00/18935
-25-
4 Nm BML-130 160 5.41 11.9
268 3.34 8.71
In a separate study of ceramide levels in MDCK cells, BML-130 at various
concentrations was incubated with the cells for 24 h. The ceramide
concentration,
measured by TLC densitometry, was 1.0 nmol/mg protein at 0.5 NM, 1.1 at 1 pM,
1.5
at 2 pM, and 3.3 at 4 NM. The results with BML-129 were virtually identical.
It is interesting that the accumulation of ceramide paralleled an accumulation
of diacylglycerol (DAG), as observed before with PDMP (Shayman, J.A. et al.,
"Modulation of Renal Epithelial Cell Growth by Glucosylceramide: Association
with
Protein Kinase C, Sphingosine, and Diacylglyceride," J. BioL Chem. 266:22968-
22974
(1991)). DAG is ordinarily considered to be an activator of protein kinase C
and thus
a growth stimulator, but the low level of GIcCer in the inhibited cells may
counteract
the stimulatory effect. Ceramide reacts with lecithin to form SM and DAG, so
it is
possible that the increased level of the latter reflects enhanced synthesis of
the
phosphosphingolipid rather than an elevated attack on lecithin by
phospholipase D.
Arabinofuranosylcytosine (ara-C), an antitumor agent, also produces an
elevation in
the DAG and ceramide of HL-60 cells (Strum, J.C. et al.,
"1 if3-D-Arabinofuranosylcytosine Stimulates Ceramide and Diglyceride
Formation in
HL-60 Cells," J. BioL Chem. 269:15493-15497 (1994)).
TLC of MDCK cells grown in the presence of 0.02 to 1 NM IV-231 B for 3 days
showed that the inhibitor indeed penetrated the cells and that there was a
great
depletion of GIcCer, but no ceramide accumulation. The depletion of GIcCer was
evident even at the 0.1 NM level and virtually no GIcCer was visible at the 1
NM level;
however the more polar GSLs were not affected as strongly. After incubation
for 5
days in 5 ~rM inhibitor, all the GSLs were virtually undetectable. The
ceramide
concentrations in the control and depleted cells were very similar: 13.5 ~ 1.4
vs 13.9
~ 0.2 pg/mg protein.
The lack of ceramide accumulation in cells exposed to the aliphatic inhibitors
was examined further to see if it might be due to differential actions of the
different
inhibitors on additional enzymes involving ceramide metabolism. For example,
IV-231 B might block ceramide synthase and thus prevent accumulation despite
the
inability of the cells to utilize ceramide for GIcCer synthesis. However,
assay of
ceramide synthase in homogenized cells showed it was not significantly
affected by
CA 02378600 2002-O1-08
WO 01/04108 PCT/US00/18935
-26-
NM inhibitors (Table 6). There did appear to be moderate inhibition at the 50
NM
level with PDMP and the aliphatic inhibitor.
TABLE 6
Effect of Inhibitors on Acid and Neutral
5 Ceramidases and Ceramide Synthase of MDCK Cells
Enzyme Activity
(% of control)
Inhibitor Tested
Ceramidase Ceramidase
Ceramide
pH 4.5 pH 7.4 Synthase
D-threo-PDMP, 5 97 + 4 116 + 19 99 + 5
pM
D-threo-PDMP, 50 133 + 138 105 + 11 66 + g8
NM
BML-129, 5 pM 108 + 8 100 + 0 97 + 0
BML-129, 50 pM 171 + 268 99 + 2 102 + 1
BML-130, 5 ~uM 107 + 11 100 + 15 108 + 10
BML-130, 50 pM 160 + 218 100 + 15 106 + 29
IV-231 B, 5 ErM 106 + 3 116 + 20 90 + 8
IV-231 B, 50 pM 113 + 8 112 + 3 71 + 188
8 Notable differences.
Assay of the two kinds of ceramidase (Table 6) showed that there was no
effect of either the aliphatic or,aromatic inhibitors at the 5 NM level, at
which point cell
growth is completely stopped in the case of the pyrrolidino compounds. At the
50 NM
level, however, the acid enzyme was stimulated markedly by the aromatic
inhibitors,
particularly the two stereoisomeric forms of the pyrrolidino compound.
Sphingomyelin synthase was unaffected by PDMP or the aliphatic inhibitor but
BML-129 and -130 produced appreciable inhibition at 50 pM (54% and 61 %,
respectively) (Table 7).
CA 02378600 2002-O1-08
WO 01/04108 PCT/US00/18935
-27-
TABLE 7
Effect of Inhibitors on Acid and Neutral
Sphingomyeiinases and Sphingomyelin Synthase
Enzyme Activity
(% of control)
Inhibitor Tested
SPhingomyelinase
Sphingomyelinase
Sphingomyelinase
a
pH 4.5 pH 7.1
Synthase
D-threo-PDMP, 102 + 3 121 + 13
5 NM _ _
D-threo-PDMP, 100 + 3 108 + 8
50 pM - -
BML-129, 5 NM 108 + 4 105 + 11 84 + 27
~i
BML-129, 50 ErM 97 + 3 142 + 11 46 + 116
BML-130, 5 NM 109 + 1 110 + 7 87 + 14
BML-130, 50 NM 114 + 2 152 + 14 39 + 186
IV-231 B, 5 pM 101 + 7 131 + 36
IV-231 B, 50 112 + 11 120 + 3
pM
° Data for PDMP and IV-231 B are not shown here as they were tested in
other
experiments; no effect was seen.
b Notable differences.
Neutral sphingomyelinase (SMase) was distinctly stimulated by the aliphatic
inhibitor, IV-231 B, even at 5 NM (Table 7). From this one would expect that
the
inhibitor would produce accumulation of ceramide, yet it did not. The two
pyrrolidino
compounds produced appreciable stimulation at the 50 NM level. No significant
effects were obtained with acid SMase.
Discussion
The present invention shows that the nature and size of the tertiary amine on
ceramide-like compounds exerts a strong influence on GIcCer synthase
inhibition, a
5-membered ring being most active. It also shows that the phenyl ring used
previously
to simulate the traps-alkenyl chain corresponding to that of sphingosine
could, with
benefit, be replaced with the natural alkenyl chain.
Findings with the most active GIcCer synthase inhibitors in growth tests
compare favorably with evaluations of some clinically useful chemotherapeutic
agents
on three of the tumor cell lines in the same Drug Evaluation Core Laboratory.
The ICS
values were 0.2 to 6 pM for cisplatin, 0.0~ to 44 pM for carboplatin, 0.03 to
0.2 NM
for methotrexate, 0.07 to 0.2 NM for fluorouracil, and 0.1 to 1 NM for
etoposide. Unlike
CA 02378600 2002-O1-08
WO 01/04108 PCT/US00/18935
-28-
these agents, the compounds of the present invention yielded rather similar
effects
with all the cell types, including MDCK cells, and thus have wider potential
chemotherapeutic utility. This uniformity of action is consistent with the
idea that GSLs
play a wide and consistent role in cell growth and differentiation.
An important observation from the MDCK cell study is that strong inhibition of
cell growth and DNA synthesis occurred only at the same concentrations of
aromatic
inhibitor that produced marked ceramide accumulation. This observation
supports the
assertion that ceramide inhibits growth and enhances differentiation or cell
death
(Bielawska, A. et al., "Modulation of Cell Growth and Differentiation by
Ceramide,"
FEBS Letters 307:211-214 (1992)). It also agrees with previous work with
octanoyl
sphingosine, a short chain ceramide that produced greatly elevated levels of
natural
ceramide and slowed growth (Abe, A. et al., "Metabolic Effects of Short-Chain
Ceramide and Glucosylceramide on Sphingolipids and Protein Kinase C," Eur. J.
Biochem. 210:765-773 (1992)). It is also in agreement with a finding that some
synthetic, nonionic ceramide-like compounds did not inhibit GIcCer synthase
even
though they behave like ceramide in blocking growth (Bielawska, A. et al.,
"Ceramide-Mediated Biology. Determination of Structural and Stereospecific
Requirements Through the Use of N-Acyl-Phenylaminoalcohol Analogs," J. Biol.
Chem. 267:18493-18497 (1992)). Compounds tested included 20 pM
D-erythro-N-myristoyl-2-amino-1-phenyl-1-propanol, its L-enantiomer, the four
stereoisomers of N-acetylsphinganine, and N-acetylsphingosine. Furthermore,
the
lack of growth inhibition and ceramide accumulation in cells treated with the
aliphatic
inhibitor IV-231 B is also consistent with the correlation between ceramide
level and
growth rate.
The accumulation of ceramide that occurred at higher levels of GIcCer
synthase inhibitors could be attributed not only to blockage of ceramide
utilization, but
also to blockage of SM synthesis or ceramide hydrolase. This possibility is
especially
relevant to the R,S-, S,R-, and S,S-isomers, which seem to exert effects on
sphingolipids without strongly inhibiting GIcCer synthesis. The tests with
both the
DL-erythro-pyrrolidino inhibitor (BML-129) and the DL-threo-pyrrolidino
inhibitor
(BML-130), at a level producing strong growth inhibition, showed that neither
material
at a low concentration inhibited the enzymes tested in vitro (Tables 6 and 7)
but they
did cause growth inhibition as well as accumulation of ceramide. PDMP, at
relatively
high concentrations (50 pM), was found to inhibit SM synthase in growing CHO
cells
(Rosenwald, A.G. et al., "Effects of a Sphingolipid Synthesis Inhibitor on
Membrane
CA 02378600 2002-O1-08
WO 01!04108 PCT/US00/18935
_29_
Transport Through the Secretory Pathway," Biochemistry 31:3581-3590 (1992)).
In
the test with MDCK homogenates, it did not inhibit this synthase, in agreement
with
the finding that labeled palmitate incorporation into SM was stimulated by
PDMP
(Shayman, J.A. et al., "Modulation of Renal Epithelial Cell Growth by
Glucosylceramide: Association with Protein Kinase C, Sphingosine, and
Diacylglyceride," J. Biol. Chem. 266:22968-22974 (1991 )).
Retinoic acid is a growth inhibitor of interest in cancer chemotherapy and a
possible adjunct in the use of the inhibitors of the present invention. It has
been found
to elevate ceramide and DAG levels (Kalen, A. et al., "Elevated Ceramide
Levels in
GH4C1 CeIIsTreated with RetinoicAcid," Biochim. Biophys. Acta 1125:90-96
(1992))
and possibly lower lecithin content (Tang, W. et al., "Phorbol Ester Inhibits
13-Cis-Retinoic Acid-Induced Hydrolysis of Phosphatidylinositol 4,5-
Bisphosphate in
Cultured Murine Keratinocytes: a Possible Negative Feedback Via Protein Kinase
C-Activation," Cell Bioch. Funct. 9:183-191 (1991 )).
D-threo-PDMP was found to be rather active in delaying tumor cell growth or
in producing complete cures in mice (Inokuchi, J. et al., "Antitumor Activity
in Mice of
an Inhibitor of Glycosphingolipid Biosynthesis," CancerLett. 38:23-30 (1987))
but high
doses were needed. From the data in Figure 1, the inhibitors of the present
invention
are approximately 30 times as active, so the dosage levels are typical of
clinically
useful drugs. The need to use high doses with PDMP was attributed to rapid
inactivation by cytochrome P450 (Shukla, A. et al., "Metabolism of D-[3H]PDMP,
an
Inhibitor of Glucosylceramide Synthesis, and the Synergistic Action of an
Inhibitor of
Microsomal Monooxygenase," J. Lipid Res. 32:713-722 (1991 )). Cytochrome P450
can be readily blocked by various nontoxic drugs such as cimetidine, therefore
high
levels of the compounds of the present invention can be maintained.
SPECIFIC EXAMPLE 2
A series of inhibitors based on substitutions in the phenyl ring of P4 were
synthesized and studied. It was found that the potency of the inhibitors in
blocking
GIcCer synthase was mainly dependent upon hydrophobic and electronic
properties
of the substituent. Surprisingly, a linear relationship was found between log
[ICSO] and
hydrophobic parameter (rr) + electronic parameter (d). This correlation
suggested that
electron donating and hydrophilic characters of the substituent enhance the
potency
as an inhibitor. This observation resulted in the synthesis of novel compounds
that
are more active in blocking glucosylceramide formation. Two compounds, dioxy D-
t
P4 compounds, D-t-3',4'-ethylenedioxy-P4 and D-t-4'-hydroxy-P4, were observed
to
CA 02378600 2002-O1-08
WO 01/04108 PCT/US00/18935
-30-
be significantly more potent than other tested inhibitors. In particular, at
11.3 nM D-t-
3',4'-ethylenedioxy-P4, 80% of glucosylceramide in MDCK cell was depleted
without
any ceramide accumulation and cell growth inhibition. The potency of D-t-3',4'-
ethylenedioxy-P4 appears to be not only regulated by hydrophobic and
electronic
properties but also by stearic properties of the substituents on the phenyl
group.
Materials and Methods
Materials. The acetophenones and amines were from Aldrich Chemical Co.,
St. Louis, MO., Lancaster Synthesis Inc., Windham, NH. and Maybridge Chemical
Co.,
Cornwall, UK. Silica gel for column chromatography (70-230 mesh ASTM) and
Silica
gel thin layer chromatography plates were purchased from Merck Co. The
reagents
and their sources were: non-hydroxy fatty acid ceramide from bovine brain and
delipidated bovine serum albumin (BSA) from Sigma; dioleoyphosphatidylcholine
from
Avanti; DL-dithiothreitol from Calbiochem; 1-[3H]-glucose uridine diphosphate
from
NEN. Octanoylsphingosine, glucosylceramide and sodium sulfatide were prepared
as previously described. Abe, A. et al., Eur. J. Biochemistry 210:765-773
(1992).
General synthesis of inhibitors. The aromatic inhibitors were synthesized
by the Mannich reaction from 2-N-acylaminoacetophenone, paraformaldehyde, and
pyrrolidine, and then the reduction from sodium borohydride as described
before.
Inokuchi, J. et al., J. Lipid. Res. 28:565-571 (1987); Abe, A. et al., J.
Lipid. Res.
36:611-621 (1995). The reaction produces a mixture of four isomers, due to the
presence of two asymmetric centers. For these syntheses in which phenyl-
substituted
starting materials were used, the chloro, methoxy, methylenedioxy, methyl
groups in
the acetophenone structure were brominated and converted to the primary amine.
Bromation of the methoxyacetophenone, dimethyoxyacetophenone, 3',4'-
(methylenedioxy)acetophenone were performed in chloroform at room temperature
and recrystallized from ethyl acetate and hexane.
Synthesis of 1-(4-hydroxy)phenyl-2-palmitoylamino-3-pyrrolidino-1-
propanol. The synthesis of 1-(4'-hydroxy)phenyl-2-palmitoylamino-3-pyrrolidino-
1-
propanol is described in detail in Figure 8. This synthesis differs from those
of the
other compounds because of the need for the placement of a protecting group on
the
free hydroxyl (step 1 ) and its subsequent removal (step 7). All other
syntheses employ
a similar synthetic scheme (steps 2 to 6).
4 -Benzyloxyacetophenone formation (step 1): 4'-Hydroxyacetophenone
(13.62 g,100 mmol), benzylbromide (17.1 g,100 mmol), and cesium carbonate
(35.83
g, 100 mmol) were added to tetrahydrofuran at room temperature and stirred
CA 02378600 2002-O1-08
WO 01/04108 PCT/US00/18935
-31 -
overnight. The product was concentrated to dryness and recrystallized from
ether and
hexane to yield 15 g of 4'-benzyloxyacetophenone which appeared as a white
powder.
An RE of 0.42 was observed when resolved by thin layer chromatography using
methylene chloride. 'H nmr (a, ppm, CDCI3), 7.94 (2H, d, 8.8 Hz, O-Ar-C(O)),
7.42
(5H, m, Ar'CH20-), 7.01 (2H, d, 8.8 Hz, O-Ar-C(O)), 5.14 (2H, s, Ar'CH20-),
2.56 (3H,
S, CH3).
Bromination of 4'-benzyloxyacetophenone (step 2): Bromine (80 mmol)
was added dropwise over 5 min to a stirred solution of 4'-
benzyloxyacetophenone (70
mmol) in 40 ml chloroform. This mixture was stirred for an additional 5 min
and
quenched with saturated sodium bicarbonate in water until the pH reached 7.
The
organic layers were combined, dried over MgS04, and concentrated to dryness.
The
crude mixture was purified over a silica gel column and eluted with methylene
chloride to yield 2-bromo-4'-benyloxyacetophenone. An Rf of 0.62 was observed
when
resolved by thin layer chromatography using methylene chloride. 1 H nmr (d,
ppm,
CDCI3), 7.97 (2H, a, 9.2 Hz, O-Ar-C(O)), 7.43 (5H, m, Ar'CHZO-), 7.04 (2H, d,
9.0 Hz,
O-Ar-C(O)), 5.15 (2H, s, Ar'CHZO-), 4.40 (2H, s, CHZBr).
2-Amino-4'-benzyloxyacetophenone HCI formation (step 3):
Hexamethylenetetramine (methenamine, 3.8 g, 23 mmol) was added to a stirred
solution of 2-bromine-4'-benyloxyacetophenone (6.8 g, 23 mmol) in 100 ml
chloroform. After 4 h the crystalline adduct was filtered and washed with
chloroform.
The product was dried and heated with 150 ml methanol and 8 ml of concentrated
HCI in an oil bath at 85°C for 3 h. Upon cooling the precipitated
hydrochloride salt
(2.5 g) was removed by filtration. The filtrate was left at -20°C
overnight and
additional product (2.1 g) was isolated. The yield was 4.6 g (82.6%). [M;Hj':
242 for
C,SH,sN02. 'H nmr (a, ppm, CDCI3), 8.38 (2H, bs, NHZ), 7.97 (2H, d, 8.8 Hz, O-
Ar-
C(O)), 7.41 (5H, m, Ar'CH20-), 7.15 (2H, d, 8.6 Hz, O-Ar-C(O)), 5.23 (2H, s,
Ar'CHZO-
), 4.49 (2H, s, CH2NH2).
2-Palmitoylamino-4 =benyloxyacetophenone formation (step 4): Sodium
acetate (50% in water, 29 ml) was added in three portions to a stirred
solution of 2
amino-4'-benzyloxyacetophenone HCI (4.6 g,17 mmol) and tetrahydrofuran (200
ml).
Palmitoyl chloride (19 mmol) in tetrahydrofuran (25 ml) was added dropwise
over 20
min yielding a dark brown solution. The mixture was stirred overnight at room
temperature. The aqueous fraction was removed by use of a separatory funnel
and
chlorofom~/methanol (2/1, 150 ml) was added to the organic layer which was
then
washed with water (50 ml). The yellow aqueous layer was extracted once with
CA 02378600 2002-O1-08
WO 01/04108 PCT/US00/18935
-32-
chloroform (50 ml). The organic solutions were then pooled and rotoevaporated
until
near dryness. The residue was redissolved in chloroform (100 ml) and
crystallized by
the addition of hexane (400 ml). The flask was cooled to 4°C for 2 h.
The crystals
were filtered and washed with cold hexane and dried in a fume hood overnight.
The
product yield was 3.79 g (8 mmol). An Rf of 0.21 was observed when resolved by
thin
layer chromatography using methylene chloride. [M'H]': 479 for C3,H45NO3. 'H
nmr
(d, ppm, CDCI3), 7.96 (2H, d, 8.8 Hz, O-Ar-C(O)), 7.40 (5H, m, Ar'CH20-), 7.03
(2H,
a, 8.8 Hz, O-Ar-C(O)), 6.57 (1 H, bs, NH2), 5.14 (2H, s, Ar'CH20-), 4.71 (2H,
s,
C(O)CHzNHC(O)), 2.29 (2H, t, 7.4 Hz, C(O)CHZ(CHZ)~3CH3), 1.67 (2H, m,
C(O)CHz(CHZ),3CH3), 0.87 (3H, t, 6.7 Hz, C(O)CH2(CHZ),3CH3).
9-(4 -Benzyloxy)phenyl-2-palmitoylamino-3-pyrrolidino-1-propanol
formation (steps 5 and 6): 2-Palmitoylamino-4'-benyloxyacetophenone (3.79 g,
8.0
mmol), paraformaldehyde (0.25 g, 2.7 mmol), pyrrolidine (0.96 ml, 11.4 mmol)
and
ethanol (70 ml) were stirred under nitrogen. Concentrated HCI (0.26 ml) was
added
through the condensor and the mixture was heated to reflux for 16 h. The
resultant
brown solution was cooled on ice and then sodium borohydride (1.3 g, 34 mmol)
was
added in three portions. The mixture was stirred at room temperature
overnight, and
the product was dried in a solvent evaporator. The residue was redissolved in
dichloromethane (130 ml) and hydrolyzed with 3N HCI (pH-4). The aqueous layer
was
extracted twice with dichloromethane (50 ml). The organic layers were pooled
and
washed twice with water (30 ml), twice with saturated sodium chloride (30 ml),
and
dried over anhydrous magnesium sulfate. The dichloromethane solution was
rotoevaporated to a semisolid and purified by use of a silica rotor using a
solvent
consisting of 10% methanol in dichloromethane. This yielded a mixture of DL-
threo-
and DL-erythro enantiomers (2.53 g, 4.2 mmol). An Rf of 0.43 for the erythro
diastereomers and 0.36 for the threo diastereomers was observed when resolved
by
thin layer chromatography using methanol:methylene chloride (1:9). [M'H]': 565
for
C~sHssN203.
1-(4=Hydroxy)phenyl-2 palmitoylamino-3 pyrrolidino-l propano! formation
(step 7): A suspension of 20% Pd/C (40 mg) in acetic acid (15 ml) was stirred
at
room temperature under a hydrogen balloon for 15 min. 1-(4'-Benzyloxy)phenyl-2
hexadecanoylamino-3-pyrrolidino-1-propanol (420 mg, 0.74 mmol) was added and
the
solution was stirred overnight. The suspension was filtered through a glass
frit, and
the filter was rinsed with acetic acid:methylene chloride (1:1, 5 ml). The
filtrate was
concentrated in vacuo and crystallized to yield a pale yellow semisolid (190
mg, 0.4
CA 02378600 2002-O1-08
WO 01/04108 PCT/US00/18935
-33-
mmol). An Rf of 0.21 was observed when resolved by thin layer chromatography
using
methanol:methylene chloride (1:9). [M'H]+: 475 for CZ9H5°NZO3. 'H nmr
(d, ppm,
CDCI3), 7.13 (4H, m, ArCHOH-), 7.14 (1 H, a, 6.9 Hz, -NH-), 5.03 (1 H, a, 3.3
Hz,
CHOH-), 4.43 (1 H, m, c-(CH2CH2)2NCHZCH), 3.76 (2H, m, c-(CHZCHz)2N-), 3.51 (1
H,
m, c-(CHZCHZ)2NCHz ), 3.29 (1 H, m, c-(CHZCHZ)2NCHz ), 2.97 (3H, m, c-
(CHzCH2)ZN-
and ArC(OH)H-), 2.08 (6H, m, -C(O)CHZ(CHZ)13CH3 and c-(CHZCHZ)ZN-, 1.40 (2H,
m,
C(O)CHZCHZ(CHZ)12CH3), 1.25 (2H, m, -C(O)CH2CH2(CHZ)12CH3), 0.87 (3H, t, 6.7
Hz,
C(O)CH2(CH2)13CH3)'
Synthesis of D-threo-1-(3 ;4'-ethylenedioxy)phenyl-2-palmitoylamino-3-
pyrrolidino-1-propanol.
Z-Amino-3 ;4 =(ethylenedioxy)acefophenone HCI: Hexamethylenetetramine
(methenamine, 5.4 g, 0.039 mol) was added to a stirred solution of
phenacylbromide
(10.0 g, 0.039 mol) in 200 ml chloroform. After 2 h, the crystalline adduct
was filtered
and washed with chloroform. The product was then dried and heated with
methanol
(200 ml) and concentrated HCI (14 ml) in an oil bath at 85°C for 2 h.
On cooling, the
precipitated ammonium chloride was removed by filtration and the filtrate was
left in
a freezer overnight. After filtration the crystallized phenacylamine HCI was
washed
with cold isopropanol and then with ether. The yield of this product was -7.1
g (81 %).
2-Palmitoylamino-3 ;4 =(ethylenedioxy)acetophenone: Aminoacetophenone
HCI (7.1 g, 31 mmol) and tetrahydrofuran (300 ml) were placed in a 1 liter
three-neck
round bottom flask with a large stir bar. Sodium acetate (50% in water, 31 ml)
was
added in three portions to this suspension. Palmitoyl chloride (31 ml, 10 %
excess,
0.036 mol) in tetrahydrofuran (25 ml) was then added dropwise over 20 min to
yield
a dark brown solution. This mixture was then stirred for an additional 2 h at
room
temperature. The resultant mixture was poured into a separatory funnel to
remove the
aqueous solution. Chloroform/methanol (2/1, 150 ml) was then added to the
organic
layer and washed with water (50 ml). The yellow aqueous layer was extracted
once
with chloroform (50 ml). The organic solutions were pooled and rotoevaportated
until
almost dry. The residue was redissolved in chloroform (100 ml) and
crystallized by the
addition of hexane (400 ml). The flask was then cooled to 4°C for 2 h.
The crystals
were filtered and washed with cold hexane until they were almost white and
then dried
in a fume hood overnight. The yield of the product was 27 mmol (11.6 g).
D-fhreo-9-(3 ;4'-ethylenedioxy)phenyl-2 palmitoylamino-3 pyrrolidino-9-
propanol: almitoylaminoacetophenone (11.6 g, 0.027 mol), paraformaldehyde
(0.81
g, 0.009 mol), pyrrolidine (3.6 ml, 0.042 mol) and ethanol (250 ml) were added
to a
CA 02378600 2002-O1-08
WO 01/04108 PCT/US00/18935
-34-
500 ml round flask under nitrogen flow. Concentrated HCI (0.8 ml) was added to
this
mixture through the reflux condenser and the mixture was refluxed for 16 h.
The
brown solution was cooled in an ice-bath. Sodium borohydride (2.28 g, 0.06
mol) was
added in three portions. This mixture was stirred at room temperature for 3 h
and then
rotoevaporated. The residue was dissolved in 130 ml of dichloromethane and the
borate complex hydrolyzed with HCI (3N) until the pH was approximately 4. The
aqueous layer was extracted twice with 50 ml dichloromethane. The organic
layers
were pooled and washed twice with H20 (30 ml), saturated NaCI (30 ml) and
dried
over anhydrous MgS04. The dichloromethane solution was rotoevaporated to a
viscous oil which was purified by use of a Chromatotron with a solvent
consisting of
10% methanol in dichloromethane to obtain a mixture of DL-threo and erythro
enantiomers (2.24 g, 0.004 mol).
Resolufion of inhibitor enanfiomers. High performance liquid
chromatography (HPLC) resolution of the enantiomers of DL-threo and DL-erythro
are
performed using a preparative HPLC column (Chirex 3014: [(S)-val-(R)-1-(a-
naphtyl)ethylamine, 20 x 250 mm: Phenomenex], eluted with hexane-1,2-
dichloroethane-ethanol-trifluroacetic acid 64:30:5.74:0.26, at a flow rate of
8 ml/min.
The column eluent was monitored at 254 nm in both the preparative and
analytical
modes. Isolated products were reinjected until pure by analytical HPLC
analysis,
determined using an analytical Chirex 3014 column (4.6 x 250 mm) and the above
solvent mixture at flow rate of 1 ml/min.
Glycosylceramidesynthase acfivify. The enzyme activity was measured by
the method previously described in Skukla, G. et al., Biochim. Biophys. Acta
1083:101-108 (1991 ). MDCK cell homogenate (120Ng of protein) was incubated
with
uridinediphosphate [3H]glucose (100,000 cpm) and liposomes consisting of 85 Ng
octanoylsphingosine, 570Ng dioleoyphosphatidylcholine and 100Ng sodium
sulfatide
in 200 NI of reaction mixture and kept for 1 h at 37 °C. P4 and P4
derivatives
dissolved in dimethyl sulfoxide were dispersed into the reaction mixture after
adding
liposomes. The final concentration of dimethyl sulfoxide was kept 1 % under
which the
enzyme activity was not at all inhibited.
Cell culture and lipid extraction. One half million of MDCK cells were
seeded into 10 cm style dish containing 8 ml serum free DMEM supplemented
medium. Shayman, J.A. et al., J. Biol. Chem. 265:12135-12138 (1990). After 24
h
the medium was replaced with 8 ml of the medium containing 0, 11.8, 118 or
1180
nM D-t-P4, D-t 3',4'-ethylenedioxy-P4 or D-4'-hydroxy)-P4. The GIcCer synthase
CA 02378600 2002-O1-08
WO 01/04108 PCT/US00/18935
-35-
inhibitors were added into the medium as a one to one complex with delipidated
BSA.
Abe, A. et al., J. Lipid. Res. 36:611-621 (1995); Abe, A. et al., Biochim.
Biophys. Acta
1299:331-341 (1996). The cells were incubated for 24 h or 48 h with the
inhibitors.
After the incubation, the cells were washed twice with 8 ml of cold PBS and
fixed with
2 ml of cold methanol. The fixed cells were scraped and transferred to a glass
tube.
Another one ml of methanol was used to recover the remaining cells in the
dish.
Three ml of chloroform was added to the tube and briefly sonicated using a
water bath type sonicator. After centrifugation at 800g for 5 min, the
supernatant was
transferred into another glass tube. The residues were reextracted with
chloroform/methanol (1/1). After the centrifugation, the resultant supernatant
was
combined with the first one. The residues were air-dried and kept for protein
analysis.
Adding 0.9% NaCI to the supernatant combined, the ratio of
chloroform/methanol/aqueous was adjusted to 1/1/1. After centrifugation 800g
for 5
min, the upper layer was discarded. Methanol/water (1/1 ) with the same amount
of
volume of the lower layer was used to wash. The resultant lower layer was
transferred into a small glass tube and dried down under a stream of nitrogen
gas.
A part of the lipid was used for lipid phosphate determination. Ames, B.N.,
Methods
Enzymoi. 8:115-118 (1966). The remainder was analyzed using HPTLC (Merck).
Results
Synthesis of P4 and P4 derivatives. The preparation of P4 derivatives
utilized the Mannich reaction from 2-N-acylaminoacetophenone,
paraformaldehyde,
and pyrrolidine, and then the reduction of DL-pyrrodino ketone from sodium
borohydride. In most cases, no isolation of DL-pyrrodino ketones were
performed to
maintain solubility. The overall yields of the DL-fhreo and DL-erythro
syntheses were
- 10-30%. These derivatives were purified by the either silica gel column or
rotors
with solvent 5-12% methanol in dichloromethane to optimize the separation from
the
chiral column. To obtain the best separation, each injection contains no more
than
150 mg, and fractions were pooled to obtain sufficient quantity of isomer of D-
threo
for further biological characterization.
Resolution of PDMP homologues by chiral chromatography. The
structures of the parent compound, D-fhreo-P4 and the phenyl-substituted
homologues including the new dioxy-substituted and 4'-hydroxy-P4 homologues
are
shown in Figure 9. Initially the effect of each P4 isomer separated by chiral
chromatography on GIcCer synthase activity was determined (Figure 10). Four
peaks
were observed for the chiral separation of P4. Peaks 1 and 2 represented the
erythro
CA 02378600 2002-O1-08
WO 01/04108 PCT/US00/18935
-36-
diastereomers and 3 and 4 represented the threo diastereomers as determined by
a
sequential separation of the P4 mixture by reverse phase chromatography
followed
by the chiral separation. The enzyme activity was specifically inhibited by
the fourth
peak, the D-threo isomer (Figure 4A). This specificity for the D-threo
enantiomer was
consistent with the previous results observed in PDMP and PDMP homologues (2-
4).
The ICSO of D-threo-P4 was 0.5 mM for GIcCer synthase activity measured in the
MDCK cell homogenates.
Effects of P4 and P4 derivatives with a single substituenf of phenyl group
on GIcCer synthase activity. The effect of each P4 isomer on GIcCer synthase
activity was analyzed. The reaction was carried out in the absence or presence
of
0.1, 1.0 or 10 NM P4 (Figure 4A) or p-methoxy-P4 (Figure 4B). As shown in
Figure
4A, the enzyme activity was specifically inhibited by D-threo isomer. In
Figure 4A, the
symbols are denoted as follows: D-threo (o), D-erythro (o), L-threo and (~), L-
erythro
(o). This specificity is consistent with previous results observed in PDMP and
PDMP
homologs. Inokuchi, J. et al., J. Lipid. Res. 28:565-571 (1987); Abe, A. et
al., J. Lipid.
Res. 36:611-621 (1995). The ICS of D-t P4 was 500 nM.
As set forth herein, the addition of a p-methoxy group to DL-t-P4 was found
to enhance the effect of the inhibitor on the enzyme activity. Abe, A. et al.,
J. Lipid.
Res. 36:611-621 (1995). As shown in Figure 4B, it was confirmed that the
enzyme
activity was potently inhibited by D-threo-p-methoxy-P4 whose ICso was 200 nM.
In
Figure 4B, o denotes a mixture of D-erythro and L-threo isomers contaminated
with
a small amount of the D-threo isomer. Chiral chromatography of the four p-
methoxy-
P4 enantiomers failed to completely resolve to baseline each enantiomer
(Figure 10).
A slight inhibition of the enzyme activity by p-methyoxy-P4 in a combined D-
erythro
and L-threo mixture (peaks 2 and 3, Figure 10) was observed; this was due to
contamination of the D-fhreo isomer (peak 4, Figure 10) into these fractions.
A series of D-t-P4 derivatives containing a single substituent on the phenyl
group were investigated. As shown in Table 8, the potency of the derivatives
as
inhibitors were inferior to that of D-f-P4 or p-methoxy-D-f P4. In many drugs,
the
influence of an aromatic substituent on the biological activity has been known
and
predicted. Hogberg, T. et al., Theoretical and experimental methods in drug
design
applied on antipsychotic dopamine antagonists. Larsen, P.K., and Bundgaard,
H.,
'?extbook of Drug Design and Development," pp. 55-91 (1991 ). Generally ICS is
described as the following equation:
CA 02378600 2002-O1-08
WO 01/04108 PCT/US00/18935
-37-
log (1iIC5o) = a (hydrophobic parameter (n) + b (electronic parameter (Q))
+ c (stearic parameter) + d (other descriptor) + a
where a, b, c, d and a are the regression coefficients. Hogberg, T. et al.,
Theoretical
and experimental methods in drug design applied on antipsychotic dopamine
antagonists. Larsen, P.K., and Bundgaard, H., "Textbook of Drug Design and
Development," pp. 55-91 (1991 ).
The hydrophobic effect, n, is described by the equation rr = IogPx - log PH
where PX is the partition coefficient of the substituted derivative and PH is
that of the
parent compound, measured as the distribution between octanol and water.
The electronic substituent parameter, ~, was originally developed by Hammett
(Hammett, L.P., In Physical Organic Chemistry, McGraw-Hill, New York (1940))
and
is .expressed as Q = IogKx - IogKH, where Kx and KH are the ionization
constants for
a para or meta substituted derivative and benzoic acid respectively. Positive
Q values
represent electron withdrawing properties and negative ~ values represent
electron
donating properties.
The potency of D-threo-P4 and P4 derivatives as an inhibitor is mainly
dependent upon two factors, hydrophobic and electronic properties, of a
substituent
of phenyl group (Table 8). Surprisingly, a linear relationship was observed
between
log (ICSO) and n + Q (Figure 5). These findings suggest that the more negative
the
value of n + ~, the more potent is D-threo-P4 derivatives made as GIcCer
synthase
inhibitor.
The data in Table 8 indicate that the potency of D-t P4 and P4 derivatives as
an inhibitor is mainly dependent upon two properties, hydrophobic and
electronic
properties, of a substituent of the phenyl group. Surprisingly, a linear
relationship was
observed between log(ICSO) and n + ~ (Figure 5). These findings suggest that
the
more negative the value of n + o, the more potent the D-t P4 derivative as a
GIcCer
synthase inhibitor.
CA 02378600 2002-O1-08
WO 01/04108 PCT/US00/18935
-38-
Table 8
D-threo-P4 derivativeQ + ri ICSO (ErMj ~ i
p-methoxy -0.29
0.2
P-4 0.00 0. 5
m-methoxy-P4 0.10 0.6
p-methyl-P4 0.39 2.3
p-chloro-P4 0.94
7.2
These values were estimated from the Table in Hogberg, T. et al., Theoretical
and experimental methods in drug design applied on antipsychotic dopamine
antagonists. Larsen, P.K., and Bundgaard, H., 'Textbook of Drug Design and
Development," pp. 55-91 (1991 ), for methoxy, Qm = 0.12, QP = -0.27, n= -0.02;
hydro,
Q = 0, n = 0; methyl, QP = -0.17, rr = 0.56; chloro, Qp = 0.23, rr = 0.71.
~~These values were derived from Figures 4A and 4B. For other compounds
the same analytical approach as shown in Figures 4A and 4B was carried out to
obtain the ICSO.
The p-hydroxy-substituted homologue was a significantly better GIcCer
synthase inhibitor. The strong association between rr + Q and GIcCer synthase
inhibitioh suggested that a still more potent inhibitor could be produced by
increasing
the electron donating and decreasing the lipophilic properties of the phenyl
group
substituent. A predictably negative n + Q value would be observed for the p-
hydroxy
homologue. This compound was synthesized and the D-threo enantiomer isolated
by
chiral chromatography. An IC~o of 90 nM for GIcCer synthase inhibition was
observed
(Figure 11 ), suggesting that the p-hydroxy homologue was twice as active as
the p
methoxy compound. Moreover, the linear relationship between the log (1C50) and
rr
+ Q was preserved (open circle, Figure 4).
Effecfs of3;4=dioxy-D-threo-P4 derivatives on GIcCersynthase activity.
The result in Figure 5 suggested that an electron donating and hydrophilic
substituent
of phenyl group makes the GIcCer synthase inhibitor potent. To attain further
improvement of the inhibitor, another series of P4 derivatives with
methylenedioxy,
ethylenedioxy and trimethyldioxy substitutions on the phenyl group were
designed
(Figure 9).
As shown in Figure 6, the enzyme activity was markedly inhibited by D-t 3',4'-
ethylenedioxy-P4 whose IC~o was 100 nM. In Figure 6, o denotes D-t 3',4'-
methylenedioxy-P4, o denotes D-t 3',4'-ethylenedioxy-P4, a denotes D-t-3',4'-
CA 02378600 2002-O1-08
WO 01/04108 PCT/US00/18935
-39-
trimethylenedioxy-P4 and ~ denotes D-t 3',4'-dimethyoxy-P4. One the other
hand, the
ICSOs for D-t 3',4'-methylenedioxy-P4 and D-t-3',4'-trimethylenedioxy-P4 were
about
500 and 600 nM, respectively. These results suggest that the potency of D-t-
3',4'-
ethylenedioxy-P4 is not only regulated by hydrophobic and electronic
properties but
also by other factors, most likely stearic properties, induced from the dioxy
ring on the
phenyl group.
Interestingly, D-t-3',4'-dimethoxy-P4 was inferior to these dioxy derivatives,
even to D-t P4 or m- or D-t-p-methoxy-P4, as an inhibitor (Figure 6). As the
parameters, Qm, aP and n, for methoxy substituent are 0.12, -0.27 and -0.02,
respectively (Hogberg, T. et al., Theoretical and experimental methods in drug
design
applied on antipsychotic dopamine antagonists. Larsen, P.K., and Bundgaard,
H.,
"Textbook of Drug Design and Development," pp. 55-91 (1991 )), the value of n
+ o
of D-f-dimethoxy P4 is presumed to be negative. Therefore the dimethoxy-P4 is
thought to deviate quite far from the correlation as observed in Figure 5.
There may
be a repulsion between two methoxy groups in the dimethoxy-P4 molecule that
induces a stearic effect that was negligible in mono substituent D-t P4
derivatives
studied in Figure 5. GIcCer synthase is thought to possess a domain that
interacts
with D-t-PDMP and PDMP homologs and that modulates the enzyme activity.
Inokuchi, J. et al., J. Lipid. Res. 28:565-571 (1987); Abe, A. et al.,
Biochim. Biophys.
Acta 1299:331-341 (1996). The stearic effect generated by an additional
methoxy
group may affect the interaction between the enzyme and the inhibitor. As a
result,
the potency as an inhibitor is markedly changed.
Distinguishing between inhibition of GIcCer synfhase and 9-O-
acylceramide synfhase inhibition. Prior studies on PDMP and related homologues
revealed that both the threo and erythro diastereomers were capable of
increasing
cell ceramide and inhibiting cell growth in spite of the observation that only
the D-
threo enantiomers blocked GIcCer synthase. An alternative pathway for ceramide
metabolism was subsequently identified, the acylation of ceramide at the 1-
hydroxyl
position, which was blocked by both threo and erythro diastereomers of PDMP.
The
specificities of D-threo-P4, D-threo-3',4'-ethylenedioxy-P4, and D-threo-(4'-
hydroxy)-
P4 for GIcCer synthase were studied by assaying the transacylase. Although
there
was an ca. 100 fold difference in activity between D-threo-3',4'-ethylenedioxy-
P4, D-
threo-(4'-hydroxy~P4, and D-threo-P4 (ICS 0.1 mM versus 10 mM) in inhibiting
GIcCer synthase, the D-threo enantiomers of all three compounds demonstrated
comparable activity in blocking 1-O-acylceramide synthase (Figure 12).
CA 02378600 2002-O1-08
WO 01/04108 PCT/US00/18935
-40-
In order to determine whether inhibition of 1-O-acylceramide synthase was the
basis for inhibitor mediated ceramide accumulation, the ceramide and
diradylglycerol
levels of MDCK cells treated D-threo-P4, D-threo-3',4'-ethylenedioxy-P4, and D-
threo-
(4'-hydroxy)-P4 were measured (Table 9). MDCK cells (5 x 105) were seeded into
a
10 cm dish and incubated for 24 h. Following the incubation, the cells were
treated
for 24 or 48 h with or without P4 or the phenyl substitute homologues. Both
ceramide
and diradylglycerol contents were determined by the method of Preis, J. et
al., J. Biol.
Chem. 261:8597-8600 (1986). GIcCer content was measured densitometrically by a
video camera and use of NIH image 1.49. Significant increases in both ceramide
and
diradylglycerol occurred only in cells treated with inhibitor concentrations
in excess
of 1 mM. This was approximately 30-fold lower than the concentration required
for
inhibition of the 1-O-acylceramide synthase assayed in the cellular
homogenates. This
disparity in concentration effects most likely reflects the ability of the
more potent
homologues to accumulate within intact cells. Abe, A. et al., Biochim.
Biophys. Acta
1299:331-341 (1996).
Table 9
GIcCer, ceramide and diradylglycerol content of MDCK cells treated with
D-threo-P4, D-threo-3',4'-ethylenedioxy-P4, and D-threo-(4 =hydroxy)-P4
Condition Ceramide Diradylglycerol
(pmol/nmol (pmol/nmol
phospholipid) phospholipid)
II Control
24h 4.53~0.12 24.2~2.36
48h 6.68~0.49 32.3~3.11
D-threo-P4
11.3 nM
~~ 24 h 5.33 ~ 0.41 * 24.1 ~ 1.66
48 h 5.68 ~ 0.27* 29.6 ~ 0.73
113 nM
24 h 4.64 f 0.38 26.6 ~ 1.56
48 h 7.08 ~ 0.29 33.0 ~ 2.63
II 1130 nM
24 h 5.10 ~ 0.35 27.1 ~ 0.67
48 h 9.74 ~ 0.53* 38.8 ~ 1.11
CA 02378600 2002-O1-08
WO 01/04108 PCT/US00/18935
-41 -
D-threo-4'-hyd roxy-P4
11.3 nM
24h 4.290.71 30.92.01*
48 h 6.70 0.29 38.4 1.44*
113 nM
24h 5.090.95 31.53.84*
48h 7.470.29 41.50.66*
1130 nM
24h 7.380.13 38.53.84*
48 h 13.4 1.03* 47.2 2.51
D-threo-3',4'-ethylenedioxy-P4
11.3 nM
24 h 5.24 22.0
5.04 24.7
113 nM
24 h 5.21 32.5
5.21 41.6
1130 nM
24 h 9.64 32.5
13.0 41.6
*Denotes p < 0.05 by the Student t test. For the D-threo-
(ethylenedioxy)-P4 only two determinations were made.
Effects of D-threo-P4, D-threo-4 =hydroxy-P4 and D-threo-3;4 =
ethylenedioxy-P4 on GIcCer synthesis and cell growth. To confirm the cellular
specificityofD-threo-3',4'-ethylenedioxy-P4 andD-threo-(4'-hydroxy)-P4 as
compared
to D-threo-P4, MDCK cells were treated with different concentrations of the
inhibitors.
The total protein amount in each sample was determined by the BCA method. In
GIcCer analysis, lipid samples and standard lipids were applied to the same
HPTLC
plate pre-treated with borate and developed in a solvent consisting of C/M/V11
(63124/4). The level of GIcCer was estimated from a standard curve obtained
using
a computerized image scanner. The values were normalized on the basis of the
phospholipid content. The results are shown in Figure 7, wherein each bar is
the
average values from three dishes, with error bars corresponding to one
standard
CA 02378600 2002-O1-08
WO 01/04108 PCT/US00/18935
-42-
deviation. In the control, the total protein and GIcCer were 414 ~ 47.4
Ng/dish and
24.3 ~ 1.97 ng/nmol phosphate, respectively.
Approximately 66 and 78% of the GIcCer was lost from the cells treated by
11.3 nM D-threo-4'-hydroxy-P4 and D-threo-3',4'-ethylenedioxy-P4 respectively
(Figures 7, 14 and 15). By contrast, only 27 percent depletion of GIcCer
occurred in
cells exposed to D-fhreo-P4 (Figure 13). A low level of GIcCer persisted in
the cells
treated with 113 or 1130 nM of either compound. This may be due to the
contribution,
by degradation, of more highly glycosylated sphingolipids or the existence of
another
GIcCer synthase that is insensitive to the inhibitor.
On the other hand, there was little difference in the total protein content
between untreated and treated cells with 11.3 or 113 nM nM D-threo-4'-hydroxy-
P4
and D-threo-3',4'-ethylenedioxy-P4 (Figures 14 and 15). A significant decrease
in
total protein was observed in the cells treated with 1130 nM of either P4
homologue.
In addition, the level of ceramide in the cells treated with 1130 nM D-threo-
3',4'-
ethylenedioxy-P4 and D-threo-(4'-hydroxy)-P4 was two times higher than that
measured in the untreated cells (Table 9). There was no change in ceramide or
diradylglycerol levels in cells treated with 11.3 nM or 113 nM concentrations
of either
compound. Similar patterns for GIcCer levels and protein content were observed
at
48 h incubations.
The phospholipid content was unaffected at the lower concentrations of either
D-threo-3',4'-ethylenedioxy-P4 or D-threo-(4'-hydroxy)-P4. The ratios of cell
protein
to cellular phospholipid phosphate (mg protein/nmol phosphate) were 4.94 ~
0.30,
5.05 ~ 0.21, 4.84 ~ 0.90, and 3.97 ~ 0.29 for 0, 11.3, 113, and 1130 nM D-
fhreo-
3',4'-ethylenedioxy-P4 respectively, and 4.52 ~ 0.39, 4.35 ~ 0.10, and 3.68 ~
0.99 for
11.3,113, and 1130 nM D-threo-4'-hydroxy-P4 suggesting that the changes in
GIcCer
content were truly related to inhibition of GIcCer synthase activity. These
results
strongly indicate that the inhibitors D-threo-4'-hydroxy-P4 and D-threo-3',4'-
ethylenedioxy-P4, are able to potently and specifically inhibit GIcCer
synthesis in
intact cells at low nanomolar concentrations without any inhibition of cell
growth.
SPECIFIC EXAMPLE 3
Compositions within the scope of invention include those comprising a
compound of the present invention in an effective amount to achieve an
intended
purpose. Determination of an effective amount and intended purpose is within
the skill
of the art. Preferred dosages are dependent for example, on the severity of
the
disease and the individual patient's response to the treatment.
CA 02378600 2002-O1-08
WO 01/04108 PCT/US00/18935
-43-
As used herein, the term "pharmaceutically acceptable salts" is intended to
mean salts of the compounds of the present invention with pharmaceutically
acceptable acids, e.g., inorganic acids such as sulfuric, hydrochloric,
phosphoric, etc.
or organic acids such as acetic.
Pharmaceutically acceptable compositions of the present invention may also
include suitable carriers comprising excipients and auxiliaries which
facilitate
processing of the active compounds into preparations which may be used
pharmaceutically. Such preparations can be administered orally (e.g., tablets,
dragees
and capsules), rectally (e.g., suppositories), as well as administration by
injection.
The pharmaceutical preparations of the present invention are manufactured
in a manner which is itself known, e.g., using the conventional mixing,
granulating,
dragee-making, dissolving or lyophilizing processes. Thus, pharmaceutical
preparations for oral use can be obtained by combining the active compounds
with
solid excipients, optionally grinding a resulting mixture and processing the
mixture of
granules, after adding suitable auxiliaries, if desired or necessary, to
obtain tablets or
dragee cores.
Suitable excipients are, in particular, fillers such as sugars, e.g., lactose
or
sucrose, mannitol or sorbitol, cellulose preparations and/or calcium
phosphates, e.g.,
tricalcium diphosphate or calcium hydrogen phosphate, as well as binders such
as
starch paste, using, e.g., maize starch, wheat starch, rice starch, potato
starch,
gelatin, gum tragacanth, methyl cellulose and/or polyvinylpyrrolidone. If
desired,
disintegrating agents may be added such as the above-mentioned starches and
also
carboxymethyl starch, cross-linked polyvinylpyrrolidone, agar, or alginic acid
or a salt
thereof, such as sodium alginate. Auxiliaries are, above all, flow-regulating
agents and
lubricants, e.g., silica, talc, stearic acid or salts thereof, such as
magnesium stearate
or calcium stearate, and/or polyethylene glycol. Dragee cores are provided
with
suitable coatings which, if desired, are resistant to gastric juices. For this
purpose,
concentrated sugar solutions may be used, which may optionally contain gum
arabic,
talc, polyvinylpyrrolidone, polyethylene glycol and/ortitanium dioxide,
lacquersolutions
and suitable organic solvent or solvent mixtures. In order to produce coatings
resistant
to gastric juices, solutions of suitable cellulose preparations, such as
acetylcellulose
phthalate or hydroxypropylmethylcellulose phthalate, are used. Dyestuffs or
pigments
may be added to the tablets or dragee coatings, e.g., for identification or in
order to
characterize different combinations of active compound doses.
CA 02378600 2002-O1-08
WO 01/04108 PCT/US00/18935
-44-
Other pharmaceutical preparations which can be used orally include push-fit
capsules made of gelatin, as well as soft, sealed capsules made of gelatin and
a
plasticizer such as glycerol or sorbitol. The push-fit capsules may contain
the active
compounds in the form of granules which may be mixed with fillers such as
lactose,
binders such as starches, and/or lubricants such as talc or magnesium stearate
and,
optionally, stabilizers. In soft capsules, the active compounds are preferably
dissolved
or suspended in suitable liquids, such as fatty oils, liquid paraffin, or
liquid
polyethylene glycols. In addition, stabilizers may be used.
Possible pharmaceutical preparations which can be used rectally include, e.g.,
suppositories, which consist of a combination of the active compounds with a
suppository base. Suitable suppository bases are, e.g., natural or synthetic
triglycerides, paraffin hydrocarbons, polyethylene glycols or higher alkanols.
It is also
possible to use gelatin rectal capsules which consist of a combination of the
active
compounds with a base. Possible base materials include, e.g., liquid
triglycerides,
polyethylene glycols or paraffin hydrocarbons.
Suitable formulations for parenteral administration include aqueous solutions
of the active compounds in water-soluble form, e.g., water-soluble salts. In
addition,
suspension of the active compounds as appropriate oily injection suspensions
may
be administered. Suitable lipophilic solvents or vehicles include fatty oils,
such as
sesame oil, or synthetic fatty acid esters, e.g., ethyl oleate or
triglycerides. Aqueous
injection suspensions may contain substances which increase the viscosity of
the
suspension such as sodium carboxymethylcellulose, sorbitol and/or dextran.
Optionally, the suspension may also contain stabilizers.
Alternatively, the active compounds of the present invention may be
administered in the form of liposomes, pharmaceutical compositions wherein the
active compound is contained either dispersed or variously present in
corpuscles
consisting of aqueous concentrate layers adherent to hydrophobic lipidic
layer. The
active compound may be present both in the aqueous layer and in the lipidic
layer or
in the non-homogeneous system generally known as a lipophilic suspension.
The foregoing discussion discloses and describes merely exemplary
embodiments of the present invention. One skilled in the art will readily
recognize
from such discussion, and from the accompanying drawings, that various
changes,
modfications and variations can be made therein without departing from the
spirit and
scope of the invention.
All publications cited herein are expressly incorporated by reference.