Note: Descriptions are shown in the official language in which they were submitted.
CA 02380850 2002-O1-31
WO 01/09363 PCT/US00/21052
MUTANT TN5 TRANSPOSASE ENZYMES AND METHOD FOR THEIR USE
CROSS-REFERENCE TO RELATED APPLICATIONS
This application claims the benefit of US provisional patent application
number
60/146,686, filed on August 2, 1999, which is incorporated herein by reference
in its
entirety.
STATEMENT REGARDING FEDERALLY SPONSORED RESEARCH
OR DEVELOPMENT
This invention was made with United States government support awarded by the
following agency: NIH, Grant No. GM50692. The United States government has
certain rights in this invention.
BACKGROUND OF THE INVENTION
Bacterial transposons such as Tn5 evolved within the cell by maintaining a low
mobility level. While necessary for the transposon to survive, the low
mobility level has
inhibited the ability of researchers to detail the molecular transposition
process and to
exploit the transposition process for use, e.g., in the development of new
diagnostic and
therapeutic resources. Tn5 is a conservative "cut and paste" transposon of the
IS4
family (Rezsohazy,R., Hallet,B., Delcour,J., and Mahillon,J, "The IS4 family
of
insertion sequences: evidence for a conserved transposase motif," Mol
Microbiol.
9:1283-1295 (1993)) that encodes a 53kD transposase protein (Tnp) that is
responsible
for its movement. The wild-type Tn5 transposase amino acid and nucleic acid
sequences are known. Ahmed, A. and Podemski, L. The Revised Sequence of TnS.
Gene 154(1),129-130(1995), incorporated by reference as if set forth herein in
its
entirety. A nucleic acid sequence that encodes wild-type Tn5 transposase is
attached as
SEQ ID NO:1. A polypeptide sequence encoded by SEQ ID NO:1 which corresponds
to wild-type Tn5 transposase is attached as SEQ ID N0:2.
The Tnp protein facilitates movement of the entire element by binding
initially
to each of two l9bp specific binding sequences termed outside end (OE; SEQ ID
N0:3),
followed by formation of a nucleoprotein structure termed a synapse, blunt
ended
cleavage of each end, association with a target DNA, and then strand transfer
(Reznikoff,W.S., Bhasin,A., Davies,D.R., Goryshin,LY., Mahnke,L.A.,
Naumann,T.,
Rayment,L, Steiniger-White,M., and Twining,S.S., "TnS: A molecular window on
-1-
CA 02380850 2002-O1-31
WO 01/09363 PCT/US00/21052
transposition," Biochem. Biophys. Res. Commun. 266:729-34 (1999)). Tn5
transposase can also promote movement of a single insertion sequence by using
a
combination of OE and inside end (IE; SEQ ID N0:4) sequences. The IE is also
l9bp
long and is identical to OE at 12 of 19 positions (Fig. 1). In vivo, Tn5
transposase
exhibits a marked preference for OE in E. coli. Transposase recognition and
binding to
IE is inhibited in E. coli by the presence of two dam methylation sites (GATC
palindromes) which add four methyl groups per inside end sequence (IE"'ø~ also
depicted
as SEQ ID N0:4, methylation not shown) (Yin,J.C.P., Krebs,M.P., and
Reznikoff,W.S.,
"Effect of dam Methylation on Tn5 Transposition," J. Mol Biol., 199:35-45
(1988),
incorporated by reference as if set forth herein in its entirety). This
methylation reduces
transposition by reducing protein-DNA primary recognition (Jilk, R.A.,
York,D., and
Reznikoff, W.S., "The organization of the outside end of transposon TnS," J.
Bacteriol.
178:1671-1679 (1996)).
A principal roadblock to understanding how Tn5 transposes is the fact that
purified wild-type Tnp has no detectable activity in vitro. Recently, a double
mutant
hyperactive form of transposase ("Tnp EK/LP") that promotes all steps of the
transposition reaction in vitro was developed. The Tnp EK/LP protein differs
from
wild-type Tn5 Tnp at position 54 (Glu to Lys mutation) and at position 372
(Leu to Pro
mutation), in addition to a non-essential but advantageous change at position
56 that
prevents production of a so-called inhibitor protein. The modified hyperactive
Tnp
protein retains the dramatic preference for OE (or OE-like) termini of wild-
type Tn5
transposase. Tnp EK/LP has clarified many aspects of Tn5 transposition that
were not
previously adequately addressable in vivo.
In vitro polynucleotide transposition is a powerful tool for introducing
random or
targeted mutations into a genome. Useful in vitro transposition systems based
upon the
Tn5 transposon are disclosed in US Patent No. 5,925,545 and International
Publication
No. WO 00/17343, both of which are incorporated herein by reference in their
entirety
as if set forth herein.
A Tnp protein having an ability to discriminate between IE and OE and having a
preference for binding IE is desired to permit directed nucleic acid
transposition and to
facilitate more complex transposition and genetic engineering strategies of
the type
disclosed in the above-mentioned patent and application than are available
using a Tnp
having a single specificity for OE. A Tnp having an enhanced preference for
IE'"ø is
also desired because methylation of DNA in common dam+ bacterial hosts
inhibits
binding of existing Tn5 transposases and reduces the ability of existing
transposases to
facilitate movement of IE-defined transposons.
-2-
CA 02380850 2002-O1-31
WO 01/09363 PCT/US00/21052
BRIEF SUMMARY OF THE INVENTION
The present invention is summarized in that a transposase protein modified
relative to wild-type Tn5 Tnp as disclosed herein preferentially promotes
transposition
of a target sequence flanked with wild-type Tn5 transposon inside ends (IE)
rather than
outside ends (OE) without regard to whether the IE sequences are methylated.
In a related aspect, the present invention is also summarized in that a
transposase
modified relative to wild-type Tn5 Tnp as disclosed herein has a preference
for IE over
OE and is hyperactive with regard to transposition frequency.
In another related aspect, the present invention is also summarized in that a
transposase modified relative to wild-type Tn5 Tnp as disclosed herein has a
preference
for IE over OE and catalyzes transposition at a high level even when the IE
sequences
are methylated. In contrast, wild-type Tn5 transposase does not efficiently
recognize
methylated IE sequences.
In yet another related aspect, the present invention is summarized in that a
transposase according to the invention includes a mutation relative to wild
type Tn5
transposase that either ( 1 ) alters binding of the transposase to the DNA
termini or (2)
enhances transposition or (3) both. The mutation can be end-sequence-specific
(as in the
exemplified embodiments that alter DNA binding) or non-specific (as in the
exemplified
embodiment that enhances transposition.
In still another related aspect, the present invention is summarized in that a
transposase according to the invention has ( 1 ) a greater preference for IE
than OE and
(2) differs from wild-type Tnp in at least one an amino acid selected from the
group
consisting of amino acid 58, amino acid 344 and amino acid 372.
In still another related aspect, the present invention is summarized in that a
transposase according to the invention differs from wild-type Tnp in that it
contains at
least one of a mutation from glutamic acid to valine at amino acid 58, a
mutation from
glutamic acid to lysine at amino acid 344, and a mutation from leucine to
glutamic acid
at amino acid 372.
In another related aspect the invention is summarized in that the transposase
of
the invention can also exhibit a greater preference for IE by reducing the
preference for
OE. A mutation at amino acid position 8 relative to wild-type transposase can
reduce the
preference of a transposase for OE, and thereby increase the apparent
preference for IE.
A mutation from arginine to cysteine can accomplish this modification.
A transposase protein of the invention can promote more transposition of an IE-
flanked target sequence in vivo or in vitro than wild-type Tn5 transposase
does. A
suitable method for determining transposase enzyme activity in vitro is
disclosed herein
and in US Patent No. 5,925,545, incorporated herein by reference in its
entirety. A
-3-
CA 02380850 2002-O1-31
WO 01/09363 PCT/US00/21052
suitable method for determining transposase activity in vivo is disclosed
herein.
The modified Tn5 Tnp of the present invention differs from wild-type Tn5 Tnp
by virtue of at least one a change to an amino acid position, where the change
is selected
from the group consisting of (1) a change at amino acid position 58 that
reduces or
S eliminates a negative interaction between the Tnp and a methylated DNA
residue and
(2) a change at amino acid position 344 that alters DNA binding. In addition
to the
changes noted herein, the modified Tnp's of the invention can also include a
change at
position 56 (such as a Met to Ala change) that prevents production of the so-
called
inhibitor protein that interferes with transposition. Moreover, the mutant Tn5
transposase proteins can contain mutations in addition to those noted above.
Additional
mutations relative to the wild-type Tn5 Tnp are disclosed below. The effect of
each
mutation is disclosed below and it is understood that the applicants have
identified
amino acid residues of the protein that have a direct impact upon function and
that other
modifications at the same positions can have effects comparable to, greater
than or
lesser than those noted on the preference of the protein for inside ends. The
wild-type
Tn5 transposase amino acid sequence is presented in the Sequence Listing.
Exemplified
changes relative to that wild-type transposase are presented in the text of
this
application.
The invention is further summarized in that the enzymes disclosed herein
facilitate a simple, in vitro system and method for introducing any
transposable element
from a donor DNA into a target DNA when the transposable element DNA is
flanked on
either side by IE termini inverted relative to one another. Few other
requirements on
either the donor DNA or the target DNA are envisioned. It is thought that Tn5
has few,
if any, preferences for insertion sites, so it is possible to use the system
to introduce
desired sequences at random into target DNA. Therefore, it is believed that
this system
and method, employing the modified transposase described herein and a simple
donor
DNA, is broadly applicable to introduce changes into any target DNA, without
regard to
its nucleotide sequence. It will, thus, be applied to many problems of
interest to those
skilled in the art of molecular biology.
Finally, although the changes noted herein are disclosed in terms of changes
at
the protein level, it is well within the ability of a skilled artisan to
modify a
polynucleotide that encodes a Tn5 transposase protein to encode the modified
proteins
of the invention. The skilled artisan also understands the degeneracy of the
genetic code
and understands that a plurality of codons can direct the production of a
single amino
acid residue.
The invention will be more fully understood upon consideration of the
following
detailed description taken in conjunction with the accompanying drawings.
-4-
CA 02380850 2002-O1-31
WO 01/09363 PCT/US00/21052
BRIEF DESCRIPTION OF THE SEVERAL VIEWS OF THE DRAWINGS
Fig. 1 depicts the structure of Tn5 transposase and the nucleotide sequences
of
Tn5 outside ends (OE), inside ends (IE), methylated inside ends (IEi''ø), and
modified IE
(IE 12A).
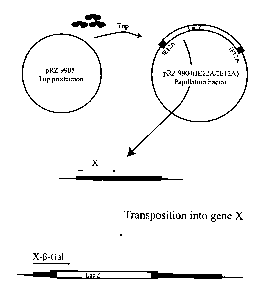
Fig. 2 is a schematic depiction of the molecular basis for a papillation assay
for
observing transposition in vivo.
Fig. 3A depicts the locations of mutations observed in four successive rounds
(A, B, C, D) of mutagenesis/recombination. Fig. 3B depicts the in vivo
transposition
profile of each of the mutants of Fig. 3A.
Fig. 4 depicts the relative preference of mutant transposases obtained in
successive rounds of mutagenesis/recombination for OE and IE in a dam- strain
of E.
coli.
Fig. 5A depicts a plasmid suitable for use in an in vitro transposition
method.
Fig. 5B depicts the transposition products obtained using a mutant transposase
of the
invention to catalyze transposition in vitro. Fig. SC further characterizes
the products of
lane 2 of Fig. 5B.
DETAILED DESCRIPTION OF THE PREFERRED EMBODIMENTS
Random mutagenesis studies performed on the Tnp gene have demonstrated that
it is possible to isolate mutations in the protein that increase the rate of
transposition
(Krebs and Reznikoff, 1988; DeLong, A., and Syvanen, M., "Trans-acting
transposase
mutant from TnS," P.N.A.S. U.S.A. 88:6072-6 (1991); Wiegand, T.W., and
Reznikoff,
W.S., "Characterization of two hypertransposing Tn5 mutants," J. Bacteriol.
174:1229-1239 (1992); Weinreich, M.D., Gasch, A., & Reznikoff, W.S., "Evidence
that
the cis preference of the Tn5 transposase is caused by nonproductive
multimerization,"
Genes. Dev. 8: 2363-2374 (1994); Zhou, M., & Reznikoff, W.S., "Tn5 mutants
that alter
DNA binding specificity," J. Mol. Biol. 271:362-73. (1997), incorporated by
reference
as if set forth herein in its entirety). This makes transposase unique in
comparison to
most enzymatic proteins that appear to have evolved for maximum in vivo
activity.
This is likely due to the fact that high rates of transposition would be
detrimental to the
survival of the transposon and hence transposase has evolved to have
an'optimaf level
that is much lower than its maximum attainable level.
Applicants have isolated a set of related mutant transposase proteins that
differ
from wild type Tn5 transposase in that the mutant proteins show a preference
in a
transposition system for inside ends (IE), and in some cases, methylated
inside ends
(IE'''ø), rather than for outside ends (OE), which are unmethylated because
they lack a
methylation site. The end preference of a mutant transposase can characterized
either
-5-
CA 02380850 2002-O1-31
WO 01/09363 PCT/US00/21052
(1) by the in vivo transposition frequency observed when it is used in a
system in which
the target polynucleotide is flanked with either OE and IE'''ø termini, or (2)
by the ratio
of the in vivo transposition frequencies observed when it is used in a pair of
systems in
which the target polynucleotide is flanked with OE and IE"'ø termini,
respectively.
Although applicants have exemplied a number of transposase proteins differing
from
wild-type Tn5 Tnp at one, four, five, and seven mutations, one can reasonably
predict
from their analysis the effects of particular individual mutations.
EXAMPLE
Overview
A number of related methods were used to obtain the family of mutants
disclosed
herein. In a first method, the applicants obtained mutants that restored in
vivo
transposition activity to a mutated end binding sequence that is not
recognized as a
substrate by wild type transposase. In a second method, the applicants
introduced
directed mutations into certain products of the first method to determine the
preferred
structure of a mutant transposase according to the invention.
In the first method, the mutant IE end binding sequence contains an adenine in
place of thymine at position 12 ("IE12A"; SEQ ID NO:S). The thymine-to-adenine
change in IE12A destroys one of the two methylation sites of wild-type IE.
Applicants
used sPCR, a combinatorial, random directed mutagenesis technique to obtain
modified
transposase proteins that can restore transposition activity to
polynucleotides flanked
with IE12A. sPCR was developed and described Stemmer, W.P., "Rapid evolution
of a
protein in vitro by DNA shuffling," Nature 370:389-391 (1994) and Stemmer,
W.P.,
"DNA shuffling by random fragmentation and reassembly: in vitro recombination
for
molecular evolution," Proc. Natl. Acad. Sci. U.S.A. 91:10747-10751 (1994),
both of
which are incorporated by reference herein as if set forth in their entirety.
Briefly, DNA
is manipulated in vitro in the sPCR method to introduce point mutations and to
allow
random recombination within a population of mutant sequences. The mutated
genes can
be cloned into plasmids which can be selected for increased activity in vivo.
Clones
having desirable phenotypes are then used as substrates for subsequent rounds
of
mutagenesis/recombination and selection for a further improved phenotype.
The sPCR method can be used in conjunction with a screen (instead of a
selection) in which a modest number of colonies 0104) are analyzed per round
(Crameri,
A., Whitehorn, E.A., and Stemmer, W.P., "Improved green fluorescent protein by
molecular evolution using DNA shuffling," Nat. Biotechnol. 14:315-319 (1996),
and
Zhang, J.H., Dawes, G., and Stemmer, W.P., "Directed evolution of a fucosidase
from a
galactosidase by DNA shuffling and screening," Proc. Natl. Acad. Sci. U.S.A.
94,
-6-
CA 02380850 2002-O1-31
WO 01/09363 PCT/US00/21052
4504-4509 (1997), both of which are incorporated by reference herein as if set
forth in
their entirety. A papillation assay described in the Example that follows was
used as a
screen for transposase mutants that restore transposition activity to
polynucleotides
flanked with IE12A. T'he papillation assay is a modification of the assay
described by
Krebs, M.P., and Reznikoff, W.S., "Use of a Tn5 derivative that creates lacZ
translational fusions to obtain a transposition mutant," Gene 63:277-85
(1988),
incorporated by reference in its entirety as if set forth herein. In the
papillation assay,
productive transposition in frame into an actively expressed gene results in
formation of
a ~-Gal fusion protein. These cells turn blue (due to presence of X-gal) and
grow at an
increased rate within the colony (lactose utilization). The rate at which
these papillae
form can be used to compare transposition rates promoted by mutated proteins.
The applicants determined the ability of mutant transposases identified using
the
first method to catalyze in vivo transposition of polynucleotides flanked
either with wild-
type OE or with wild-type IE in a dam- strain (i.e., the nucleic acid was
unmethylated).
Among the mutants identified using the method, applicants identified a mutant
transposase ("Tnp sC7") that retained near-wild-type activity with OE-flanked
polynucleotides but which had very high activity with IE-flanked
polynucleotides and
even higher activity with IE-flanked polynucleotides when tested in a dam+
strain (i.e.,
the nucleic acid was methylated, subsequently "IE'''ø"). Tnp sC7 contains
seven
mutations relative to wild-type transposase.
In the second method, it was subsequently determined that a related mutant
transposase ("Tnp sC7v2.0") having only four of the seven mutations of Tnp sC7
exhibited a still higher IE"'ø:OE activity ratio. Both Tnp sC7 and Tnp sC7v2.0
contain a
mutation that inhibits OE related activity (R8C), two mutations that
specifically increase
IE'''ø related activity (E58V, E344K), and a mutation that increases
transposition of
polynucleotides flanked by either IE"'ø or OE (L372Q).
Obtainin;~ the mutant Tnp's
The modified papillation assay used to screen for productive transposition is
shown and described in Fig. 2. In a first plasmid ("pRZ9904 (IE12A/IE12A)")
used, a
pair of inverted IE12A termini flank a polynucleotide that contains a lacZ
gene but
which lacks both a promoter and a translational start site. A second plasmid
("pRZ9905") used in the assay, encodes for transposase that can move the IacZ-
containing polynucleotide. Plasmids having these attributes can readily be
constructed
by a skilled artisan. Materials and methods are detailed below.
Five random mutants that suppressed the end sequence mutation and yielded
papillae after an initial cycle of mutagenesis/recombination. Equal amounts of
plasmid
_7_
CA 02380850 2002-O1-31
WO 01/09363 PCT/US00/21052
encoding each of these mutant transposases were then used as the initial
substrates for a
second round of mutagenesis/recombination. Following this second round, the
mutated
transposase genes were cloned into vector DNA and screened a second time for
transposition activity with IE12A-defined polynucleotides via the papillation
screen.
From this second round, a total of 6 active mutants were isolated. A third
round of
mutagenesis/recombination was then performed followed by screening for
activity with
the mutant end sequence. This time hundreds of colonies were positive for
papillation.
Of these, 7 were clearly more active than the others and were isolated to
serve as a
template for a fourth round. In the fourth round of screening there were
hundreds of
transposing colonies visualized during the screening process. None of these,
however,
were as active as the most active mutant from the third round of
mutagenesislrecombination (Tnp sC6).
The most active isolate from each round was sequenced and tested for
transposition activity with IE12A defined transposons in a quantitative
papillation assay
(see Figs. 3A and 3B). Fig. 3A depicts the mutations relative to wild-type Tnp
of Tnp
sA5 (best first round papillator), sB2 (best second round papillator), sC6
(best third
round papillator), and sD5 (best fourth round papillator). Fig. 3B depicts the
transposition activities of the four isolates in vivo in the papillation
assay. Tnp WT was
also tested but failed to promote a single detectable transposition event. The
mutation
Q81H is the only mutation that distinguishes the most active mutant, sC6,
isolated in the
third round, from the noticeably less active mutant sB2, isolated in the
second round. A
second isolate from the third round, Tnp sC7, is similar to the fourth round
isolate sDS,
except that it has two additional mutations (D217A and E344K). Tnp sC7's
activity with
IE12A defined transposons is similar to that of the fourth round isolate sD5
(data not
shown).
The mutant Tnp's can promote transposition of OE- and IE-defined transposons
The mutant Tnp's described above were of initial interest because they could
restore transposition activity to transposons flanked with IE12A termini that
are inactive
in the presence of wild-type Tnp. Although the transposase mutants functioned
increasingly well with these transposons, the transposition rate did not
approach the level
of activity that is required in vitro. In fact, the in vivo activity of Tnp
sC6 with IE12A
ends only restored activity to a level similar to that of Tnp WT with
transposons defined
by OE (data not shown).
Interestingly, however, most of the isolated mutants were hyperactive with
transposons defined by at least one of the native end sequences (IE or OE).
These
transposition preferences were determined in a dam- (DNA unmethylated) strain
using
_g_
CA 02380850 2002-O1-31
WO 01/09363 PCT/US00/21052
the mating out assay of Goryshin,LY., Kil, Y.V., and Reznikoff, W.S., "DNA
length,
and twisting constraints on IS50 transposition," Proc Natl Acad Sci U S A
91:10834-10838 (1994), incorporated by reference as if set forth herein in its
entirety.
Fig. 4 depicts the in vivo transposition activity of 25 mutant Tnps with IE
and OE
defined transposons in the mating-out assay in a dam- strain, normalized to
the activity
of wild-type Tnp (6.5X10-5 normalized to 1 ). In this assay in a dam-
environment, Tnp
WT shows generally equivalent activity levels whether the substrate
polynucleotide is
flanked with IE or with OE. On the other hand, many of the mutants exhibited
higher
activity with IE than with OE. This is not surprising, since the mutants were
obtained in
a screen using IE12A, which differs from IE at only 1 nucleotide. In contrast,
OE differs
from IE12A at 6 nucleotides. One mutant in particular, Tnp sC7, displayed a
very
interesting phenotype. It is markedly hyperactive with IE transposons while
exhibiting
little change in the frequency of OE transposon movement. The ability to
discriminate
between IE and OE is important because it facilitates mufti-part
transpositions that
separately employ IE and OE ends, where a reaction can be directed one way or
another
by providing a transposase that prefers either IE or OE. A preference for IE
over OE of
greater than about 5-fold may be suitable, though a preference of greater than
about 10-
fold for IE is more preferred. A preference of greater than about 20-fold is
still more
preferred. Fig. 4 demonstrates that a skilled artisan can obtain such mutant
transposases
using the methods disclosed herein. In particular, mutants sBl, sC6, sC7, sDl
and sD3
are examples of such mutants.
Even more significantly, however, Tnp sC7 is not only not inhibited for
transposition activity by methylated IE (which reduces Tnp WT levels by 102)
but
actually prefers IE'"ø transposons to those flanked by IE, as is shown in the
mating out
results of Table 1 which indicate that transposition frequency with OE is
reduced in a
dam+ strain for both Tnp sC7 and Tnp WT. Since binding of Tnp to OE is not
affected
by dam methylation, this difference merely reflects the difference in
transposition
activity between dam+ strains and dam- strains. Despite this reduction, the
rate of IE
defined transposition facilitated by Tnp sC7 is even higher in the dam+ strain
due to the
presence of methylation in the ends. In contrast, methylation of the end
sequence
inhibits recognition by wild-type transposase. On the basis of its ability to
discriminate
between IE and OE, and because of its insensitivity to methylation of IE,
subsequent
attention was directed to Tnp sC7.
-9-
CA 02380850 2002-O1-31
WO 01/09363 PCT/US00/21052
Table 1. In vivo transposition rate of Tnp WT and Tnp sC7.
JCM 1 O 1 (dam-) RZ212 (dam+)
IE freq. OE freq. IE/OE IEl''ø freq. OE freq. IE'''ø/OE
Tnp WT 6.5X10-5 6.5X10'5 1.0 1.0X10-8 3.1X10-6 3.2X10-3
Tnp sC7 1.8X10-3 3.7X10-5 50 2.6X10-3 3.3X10-6 794
The role of the individual Tnp sC7 mutations in tran~osition activity
To understand how individual mutations affect the activity of the protein and
in
an effort to maximize the activity of the protein and its ability to
discriminate between
IE"'ø and OE, the applicants strategically prepared more variant transposases
using
information obtained from sC7, which as noted above, contained seven mutations
relative to wild type transposase. Since a comprehensive trial of all possible
combinations of these seven mutations was cumbersome (128 possible
combinations)
two classes of seven mutants each were engineered. In each mutant of the first
class, a
unique mutation from sC7 was engineered to revert to wild-type at that
position. This
1 S set of so-called "minus one" mutant transposases included all possible
mutants having 6
of the 7 mutations. In each mutant of the second class, wild-type transposase
was
engineered to contain exactly one of the 7 mutations from sC7. The set of so-
called
"plus one" mutant transposases included all possible mutants having only 1 of
the 7
mutations. These mutant proteins were all assayed for in vivo transposition
activity with
IEr''ø and OE by mating out assays. The results of the analysis are shown in
Table 2.
-10-
CA 02380850 2002-O1-31
WO 01/09363 PCT/US00/21052
Table 2. Results of 'minus ones/plus ones' analysis.
'minus ' IE"''minus 'plus IE'''~'plus OE
one one' one' one'
OE
freq. norm. freq. norm.freq. norm.freq. norm.
Wild-type 1.0X 1.0 3.1 1.0
10-8 X 10-6
sC7 2.6X10-31.0 3.3X10-51.0
RCBC 3.1X10-31.2 6.0X10-61.8 ------------ 4.0X10-'0.1
E58V 1.8X10-66.9X101.1X10-60.3 4.0X10-44X1041.4X10-60.5
A157T 3.1X10-31.2 4X10-6 1.2 ------------ 6.6X10-'0.2
T171S 2.8X10-31.1 2.6X10-60.8 5.6X10-90.6 1.8X10-60.6
D217A 3.7X103 1.3 4.5X10-61.4 ------------ 3.4X10-61.1
E344K S.SX10~ 0.2 1.7X10-55.2 4.3X108 4.3 6.8X10-'0.2
L372Q 2.6X10-40.1 2.8X10-'0.1 8.9X10-88.9 1.1X10-53.5
a. 'Minus ones' contain all mutations present in Tnp sC7 except at indicated
position. e.8. R8C 'minus one' contains the wild-type argenine at position 8.
b. 'Plus ones' are Tnp WT except that the indicated amino acid is mutated to
the
residue present in Tnp sC7. E.8. R8C 'plus one' contains a cysteine at amino
acid 8.
Sequence-specific mutations. The mutation E58V has the most profound effect
of all the mutations on the activity of Tnp sC7. This mutation in the wild-
type
background (E58V 'plus one') increases IE"'ø related transposition by 40,000
fold while
removal of the mutation from Tnp sC7 (E58V 'minus one') drops the total
activity by
more than 1,000 fold. The mutation has comparatively little effect on OE
related
activity.
The mutation E344K exhibits a similar, though much weaker, sequence specific
effect on activity. When removed from Tnp sC7 (E344K'minus one'), IE"'ø
related
activity decreases by 5 fold while OE related activity is stimulated about 5
fold. This
result is mirrored in the 'plus ones' data as E344K in the wild type
background stimulates
IEi''ø related activity 4 fold and decreases OE related activity about 5 fold.
Non-sequence-specific mutations. The mutation L372Q strongly stimulates Tnp
sC7 activity with IE"'ø. When removed from sC7 both IE"'~ and OE related
activity are
reduced to the same degree. When added to wild-type transposase, its 'plus
one'
phenotype stimulates activity with both substrates.
Other mutations. The mutation R8C was the most interesting of the remaining
four mutations. When the mutation is added to wild-type transposase, OE
related
-11-
CA 02380850 2002-O1-31
WO 01/09363 PCT/US00/21052
transposition was reduced nearly 10 fold. When the mutation was removed from
sC7 OE
related activity increased approximately 2 fold. In a methylating host, its
removal from
sC7 had little effect on IE"'ø related activity while by itself it decreased
IE'''ø related
activity to below detectable levels. None of the remaining three (A157T, T171
S,
D217A) mutations has much effect on either IE"'ø or OE related activity when
removed
from sC7. To determine whether any combination of these mutations would lead
to an
increase in overall IE"'ø related activity without sacrificing its specificity
for IE'''ø over
OE, the applicants engineered pairwise reversions of sC7 at these positions as
well as a
triple-reversion mutant. The construct with all three of these mutations
reverted had the
second highest IE"'ø related activity and the best ability to discriminate
between IE'''ø and
OE (Table 3). This four-mutant construct, Tnp (RBC, E58V, E344K, L372Q), was
renamed Tnp sC7 v2Ø
Table 3. In vivo frequency of Tnp sC7 with indicated mutations reverted to
wild type.
Numbers in parenthesis indicate positions changed to wild-type.
IE"' OE IE'''~/OE
freq. norm. freq. norm.
Tnp sC7 2.6X103 1.0 3.3X10-61.0 794
Tnp sC7(157,171) 6.5X10-32.5 7.0X10-62.1 933
Tnp sC7(157,217) 8.4X10'33.3 2.25X10-56.8 373
Tnp sC7(171,217) 5.8X10-32.3 1.18X10-53.6 502
Tnp sC7(157,171,217)a7.4X103 2.8 7.1X10-62.1 1042
a Re-named sC7v2Ø
Abbreviations: OE = outside end
IE = inside end
IE'"ø = dam methylated inside end
Tnp sC7 is less than the sum of its individual mutations. The Tnp E58V (E58V
'plus one') mutant has an activity increase of 4X104 over Tnp WT alone. The
composite
mutant with E58V removed (E58V 'minus one') has an increase of 1.8X102 over
Tnp
WT. Additivity would then predict that Tnp sC7 would have an activity increase
of:
(4X104)(1.8X102) = 7.2X106
-12-
CA 02380850 2002-O1-31
WO 01/09363 PCT/US00/21052
However the stimulation of the composite mutation is actually much lower
(2X105). In
other words the increase obtained by adding E58V to the other 6 mutations is
1.1X103
fold, and not the 4X104 fold stimulation seen by adding E58V into the wild-
type
background. This may be because that E58V and E344K are both stimulating the
same
step (primary DNA binding) and are less than additive when combined.
Tnp sC7v2.0 effectively transposes IE"'ø defined transposons in vitro
The ability of Tnp sC7v2.0 to promote transposition of IE"'ø defined
transposons
in vitro was tested under the same conditions developed for movement of OE
defined
transposons by Tnp EK/LP (Goryshin, LY., and Reznikoff, W.S., "Tn5 in vitro
transposition," J. Biol. Chem. 273:7367-7374 (1998), incorporated by reference
as if set
forth herein in its entirety). Substrate plasmid pGT4, a high-copy number pUC
19-based
vector in which inverted IE'"ø end sequences flank a kanamycin resistance
gene, was
purified as a supercoiled monomer (see Materials and Methods). This plasmid
was
constructed so that digestion with PvuII restriction endonuclease causes
release of the
1 S transposon from the donor backbone DNA (see Fig. 5A). Tnp sC7v2.0 was
produced
and purified by cloning the nucleotide sequence in an expression vector,
expressing the
protein in a host cell and isolating the protein from an extract from the host
cell, all using
standard methods known to a skilled artisan.
Incubation with Tnp sC7v2.0 results in conversion of 66% of supercoiled
plasmid into transposition products and intermediates after a three hour
incubation. In
Fig. 5B, lane 1 is unreacted substrate pGT4. The transposition activity
promoted by Tnp
sC7v2.0 on IE"'ø transposons is shown in lane 2 Lane 3 is the result of pGT4
digestion
with PvuII restriction endonuclease.
Though transposition reactions performed in this fashion can lead to many
different DNA products, the reaction can be interpreted by analyzing certain
diagnostic
fragments as defined previously by Goryshin and Reznikoff (1998), supra. Fig.
SC is a
reproduction of lane 2 of Fig. 5B. Band 1 is the excised transposon. It is an
intermediate
that has undergone double ended break from the plasmid but has not undergone
strand
transfer. Band 2 is the donor backbone DNA that is released upon double-ended
excision of the transposon. These bands have the same molecular weight as the
products
of the PvuII digest shown in lane 3 of Fig. 5B. Band 3 represents substrate
plasmid that
has undergone cleavage at one transposon end. It migrates at the same position
as
linearized plasmid (data not shown). Bands 4 and 5 are two different types of
strand
transfer products. Band 4 is the result of a transposon inserting
intermolecularly into an
unreacted plasmid. This results in a relaxed circular DNA that is longer than
the original
substrate plasmid by the length of the inserted transposon. The bands denoted
as 5 are
-13-
CA 02380850 2002-O1-31
WO 01/09363 PCT/US00/21052
the result of intramolecular inversion events. These transposition products
are the size of
the transposon but circularized. These circularized transposition products can
contain
differing numbers of nodes and hence migrate in different positions on the
gel.
Structure/function analysis
The hyperactive Tn5 transposase mutant Tnp sC7v2.0, which increases
transposition of IE"'ø flanked transposons by 7.4X105 times, includes
mutations at amino
acids 8, 58, 344, and 372. Analysis of Tnp sC7 revealed that the hyperactivity
conferred
by both the E58V and E344K mutations depends upon the transposon termini
sequence.
This phenotype occurring for E58V was not surprising, as previous random
mutagenesis
screens performed with OE flanked transposons resulted in the isolation of
proteins with
mutations at either amino acid 47 or amino acid 54 which had sequence specific
activity
(Zhou and Reznikoff, 1997; Zhou,M., Bhasin,A., and Reznikoff,W.S., "Molecular
genetic analysis of transposase-end DNA sequence recognition: cooperativity of
three
adjacent base-pairs in specific interaction with a mutant Tn5 transposase," J.
Mol. Biol.
276:913-925 ( 1997)). The sequence specific activity of a mutation at amino
acid 344
that suggests that the residue also interacts with transposon end DNA,
however, was
indeed very surprising.
Recently a protein-DNA co-crystal representing Tnp EK/LP complexed with
pre-cut (no donor backbone) OE DNA has been solved. In this complex amino acid
58
is shown to interact specifically with OE at position 10. This places the
mutant residue
in the vicinity of position 12, the nucleotide mutation that it was initially
chosen to
repress. Positions 10, 11, and 12 are all different between IE and OE and
furthermore it
is near one of two areas that contain major groove modification by dam
methylase
(position 11 of top strand and position 12 of bottom strand).
The structure further shows that amino acid 344 interacts with DNA at position
7
of OE. While position 7 is not one of the seven that distinguishes IE from OE,
it is
between both position 4 and the region of nucleotides 10, 11, and 12 that
differentiate the
two. It is therefore plausible that the activity difference of Tnp E344K with
IE and OE
can be attributed to context effects of these two regions. We therefore
propose that both
mutations E58V and E344K are interacting with end sequence DNA and alter
transposase function at the level of primary sequence recognition.
The leucine to glutamine mutation (L372Q), which was present in all sequenced
mutants (sAS, sB2, sC6, sC7, and sD5), was both familiar and surprising. It
was familiar
because a hyperactive mutation at this position was previously isolated. It
was
surprising because the previously isolated mutation causes a leucine to
proline (L372P)
substitution at this position (Weinreich et al., 1994). The L372P mutation
results in two
-14-
CA 02380850 2002-O1-31
WO 01/09363 PCT/US00/21052
consecutive proline residues in an a helix. In the co-crystal this
destabilizes amino acids
372-390. It is proposed that the mutation changes the conformation of the
catalytic
domain in relation to the C-terminal dimerization domain. This alteration can
improve
activity by increasing the distance between the N and C termini. The close
proximity of
these termini in Tnp WT is thought to decrease the rate of transposition of
Tnp WT
(Reznikoff et al., 1999). The wild-type leucine residue, visible in an earlier
partial Tnp
structure (Davies,D.R., Braam,L.M., Reznikoff,W.S. and Rayment,L, "The
three-dimensional structure of a Tn5 transposase-related protein determined to
2.9-A
resolution," J Biol Chem. 274: 11904-11913 (1999)), is buried in a hydrophobic
pocket.
It is likely that substitution with a glutamine destabilizes this hydrophobic
packaging.
Given that two different hyperactive mutations have been isolated by PCR-base
random
mutagenesis techniques that only result in single nucleotide changes in the
codon and
can only make five different amino acid substitutions at this residue
(methionine, valine,
arginine, proline, or glutamine), it is likely that a more direct mutagenesis
approach such
as codon randomization could reveal other interesting mutants.
The mutation RBC, which reduces OE related transposition almost 10-fold in the
wild-type background (R8C 'plus one') and increases it approximately 2-fold
when
removed from the sC7 background (R8C 'minus one') is less easy to interpret.
In the
co-crystallographic structure the arginine residue is not in an area of DNA
contact.
However this structure only represents a still picture of the complex after
cleavage has
occurred and it is possible that this region contacts DNA in the initial
synapse.
IE methylation
The inside end of the Tn5 transposon contains two GATC signal sequences that
add four methyl groups into the major groove of each end. In this study, we
were able to
isolate a single mutation, E58V, which not only overcomes this binding
inhibition but
also appears to preferentially function on transposons in which the methyl
groups are
present (presumably due to increased binding affinity). Furthermore, the co-
crystal
structure of Tnp EK/LP complexed with pre-cleaved DNA shows that glutamate 58
interacts directly with position 10 of OE in the major groove. This region is
in the
vicinity of the methyl group that is present on the adenine of the non-
transferred strand
of IE'''ø. The fact that a single amino acid change can result in this extreme
change in
phenotype leads us to propose that this methyl group alone is responsible for
the
inhibition of binding of transposase to IE"'ø. The inhibition of binding by
Tnp WT to
IEr"ø is likely caused by an interaction between this methyl group and the
negatively
charged side chain of glutamate 58. Replacing this residue with a valine can
not only
remove this unfavorable interaction but also lead to an increase in binding
affinity due to
-15-
CA 02380850 2002-O1-31
WO 01/09363 PCT/US00/21052
hydrophobic packaging between the side chain of the valine residue and this
methyl
group.
General Materials and Methods
Media and Rea~~ents
Papillation assays were performed using glucose minimal Miller media (Miller,
J., Experiments in Molecular Genetics, Cold Spring Harbor Laboratory Press,
Cold
Spring Harbor, NY. (1992)) supplemented with ampicillin, chloramphenicol,
S-bromo-4-chloro-3-indolyl-(3- D-galactoside, and phenyl-~-D-galactoside (Trp--
XG-PG
plates) as described previously by Zhou, M., & Reznikoff, W.S., supra. After
transformation during site directed mutagenesis, the cells were outgrown in
SOC media
as indicated in the altered sites protocol (Promega). All other bacterial
growth was
performed in Luria Broth (Sambrook, J., Fritsch, E.F. & Maniatis, T. (1989).
Molecular
Cloning: A Laboratory Manual, Cold Spring Harbor Laboratory Press, Cold Spring
Harbor, NY.). When necessary, antibiotics (Sigma) were added at the following
concentrations: ampicillin 100p,g/ml; chloramphenicol 20~g/ml; nalidixic acid
20~g/ml;
gentamycin Spg/ml; tetracycline l5p.g/ml. Taq DNA Polymerase, T4 polymerase,
T4
ligase, dNTP's, and all components of the Altered Sites Mutagenesis kit were
purchased
from Promega. Restriction enzymes were purchased either from Promega or New
England Biolabs. Oligonucleotides used in site directed mutagenesis, sPCR, and
sequencing were purchased from Research Genetics. Radionucleotides used in
sequencing were from Amersham.
Construction of plasmids
Plasmid pGT4 was constructed as a high copy number plasmid containing a
kanamycin resistance gene flanked by two inside ends. It is designed so that
digestion
with PvuII releases the transposon from its pUC vector backbone.
The fourteen pRZ9905 (sC7) derivatives and pRZ9905 (sC7 version 2.0) were
constructed by swapping restriction fragments between pRZ9905 and pRZ9905
(sC7).
Bacterial Strains
Cloning of plasmids and the directed evolution process were performed in JM109
(Promega). The site-directed mutagenesis protocol utilized strain ES1301
(Promega)
and JM109. In the mating out assay, transposase plasmids were transformed into
strain
RZ212 [0(lac-proA,B), ara, str, recA56, srl, thi/pOX38-Gen], followed by
conjugation
into 148525[F-nalr].
-16-
CA 02380850 2002-O1-31
WO 01/09363 PCT/US00/21052
Directed Evolution Process
Directed evolution of Tn5 transposase was carried out by sPCR, basically as
described previously by Stemmer (1994). Forty micro liters (~4~,g) of a
pRZ9905
plasmid mini prep (Wizard SV preps, Promega) were partially digested in a SOpI
volume
containing 100mM Tris-HCl (ph = 7.0), SnM MgCI, and 90ng of DNase I. After a
7-minute incubation at room temperature, the reactions were stopped by adding
EDTA to
lOmM. Following the addition of loading dye (Sambrook et. al, 1989) the
digested
DNA was electrophoresed in a 2% NuSieve gel (FMC BioProducts) next to pGEM DNA
markers (Promega). A gel slice containing DNA fragments of 200-600bp in size
as well
as a second slice containing DNA fragments from 600-1000bp in size were
excised from
the gel. The DNA from these two slices was purified separately by phenol
chloroform
extraction (Sambrook et. al, 1989). After ethanol precipitation, the DNA
pellets were
dried and resuspended directly into 50 p.1 of an assembly reaction mix. The
assembly
mix contained 0.2mM dNTP's; 2.OmM MgCI; SOmM KCI; lOmM Tris-HCl (pH9.0 at
25C); and 0.1% Triton X-100. After addition of 0.5 units of Taq DNA
Polymerase, the
DNA was reassembled by the following thermo cycling program: 94C for
30seconds; SO
cycles of 94C for 20 seconds, 65C for 1 minute, and 72C for 2 minutes; and
cooling to
4C. A standard PCR amplification reaction using 5 ~1 of the assembly reaction
product
as a DNA template was performed for each sample (200-600bp and 600-I OOObp) to
amplify the transposase gene. This transposase-encoding fragment was digested
to
completion with AflII and BgIII and ligated into purified AflIIBgIII digested
vector
DNA from pRZ9905.
Ligation products were transformed into electrocompetent JM109 cells that
contained plasmid pRZ9904 (IE12A/IE12A). After outgrowth the cells were plated
on
2S Trp--XG-PG plates with chloramphenicol and ampicillin selection. The plates
were
incubated at 32C for 14 days. At this time pRZ9905 plasmid DNA from all
colonies that
exhibited at least one papillae were isolated and re-transformed into the
papillation assay
to confirm their papillation plus phenotype. A total of 5 pRZ9905 derivatives
(out of
20,000 original colonies screened) were confirmed to be papillation plus. An
equal
amount of all five plasmids was then used as the substrate for a second round
of
mutagenesis and screening. This process was repeated for a total of four
rounds of
screening (~ 20,000 colonies / round)
(quantitative Papillation Assay
The IE12A in vivo transposition activity of Tnp WT, Tnp sAS, Tnp sB2, Tnp
sC6, and Tnp sD5 were compared by a quantitative papillation assay. Competent
cells
of strain JM109 harboring plasmid pRZ9904 (IE12A/IE12A) were transformed with
the
-17-
CA 02380850 2002-O1-31
WO 01/09363 PCT/US00/21052
appropriate transposase-encoding version of pRZ9905. After outgrowth,
transformed
cells were plated on Trp--XG-PG plates with chloramphenicol and ampicillin.
The
plates were grown at 32C until colonies began to appear (~18 hours).
Individual
colonies were then picked with sterile sticks and spotted onto a fresh plate
in a 4X4 grid
pattern to evenly space all colonies. One plate of 16 colonies was spotted for
each
protein. Plates were incubated at 32C and quantified for transposition by
observing the
appearance of papillae at 24-hour intervals. Data are expressed as the average
number of
papillae present per colony.
Mating out assays
Mating out assays were performed as described previously (Yin et al., 1988;
Goryshin et al., 1994). Bacterial cells with the transposon containing
plasmids
pFMA52-187 (with either two OEs or two IEs) and the F factor pox-Gen were
transformed with the appropriate transposase encoding plasmid pRZ9905. The
donor
used for the library screening was the strain JCM101 [OIacZX74, raps, dam-3].
All other
mating out was performed in E. coli strain RZ212 [~(lac-proA,B), ara, str,
recA56, srl,
thi]. The recipient strain used was 148525[F-nalr]. A total of three assays
were
performed for each combination of transposase and end sequence. The values
reported
are the average of these three data points.
In vitro transposition assays
Substrate plasmid pGT4 was isolated from DHSa cells using a qiafilter plasmid
mega kit (Qiagen). Supercoiled monomer plasmid was isolated from a 1 % agarose
gel
by use of the qiaquick gel purification kit (Qiagen). Reactions were performed
at 37C
under conditions determined by Reznikoff and Goryshin ( 1998). The
concentration of
pGT4 was 35.SnM. Tnp sC7v2.0 was added to a concentration of 280nM.
The foregoing is not intended to limit the scope of the invention. Rather the
invention is understood to encompass all the variations and modifications that
come
within the scope of the appended claims.
-18-
CA 02380850 2002-O1-31
WO 01/09363 PCT/LJS00/21052
SEQUENCE LISTING
<110> Reznikoff, William S
Naumann, Todd A
<120> Transposase Enzyme and Method for Use
<130> 960296.96471
<140>
<141>
<150> 60/146686
<151> 1999-08-02
<160> 5
<170> PatentIn Ver. 2.1
<210> 1
<211> 1431
<212> DNA
1$ <213> Transposon Tn5
<220>
<221> CDS
<222> (1)..(1428)
<400> 1
atg ata act tct get ctt cat cgt gcg gcc gac tgg get aaa tct gtg 48
Met Ile Thr Ser Ala Leu His Arg Ala Ala Asp Trp Ala Lys Ser Val
1 5 10 15
ttc tct tcg gcg gcg ctg ggt gat cct cgc cgt act gcc cgc ttg gtt 96
Phe Ser Ser Ala Ala Leu Gly Asp Pro Arg Arg Thr Ala Arg Leu Val
2,5 20 25 30
aac gtc gcc gcc caa ttg gca aaa tat tct ggt aaa tca ata acc atc 144
Asn Val Ala Ala Gln Leu Ala Lys Tyr Ser Gly Lys Ser Ile Thr Ile
35 40 45
tca tca gag ggt agt gaa gcc atg cag gaa ggc get tac cga ttt atc 192
Ser Ser Glu Gly Ser Glu Ala Met Gln Glu Gly Ala Tyr Arg Phe Ile
50 55 60
-1-
CA 02380850 2002-O1-31
WO 01/09363 PCT/US00/21052
cgc aat ccc aac gtt tct gcc gag gcg atc aga aag get ggc gcc atg 240
Arg Asn Pro Asn Val Ser Ala Glu Ala Ile Arg Lys Ala Gly Ala Met
65 70 75 80
caa aca gtc aag ttg get cag gag ttt ccc gaa ctg ctg gcc att gag 288
$ Gln Thr Val Lys Leu Ala Gln Glu Phe Pro Glu Leu Leu Ala Ile Glu
85 90 95
gac acc acc tct ttg agt tat cgc cac cag gtc gcc gaa gag ctt ggc 336
Asp Thr Thr Ser Leu Ser Tyr Arg His Gln Val Ala Glu Glu Leu Gly
100 105 110
aag ctg ggc tct att cag gat aaa tcc cgc gga tgg tgg gtt cac tcc 384
Lys Leu Gly Ser Ile Gln Asp Lys Ser Arg Gly Trp Trp Val His Ser
115 120 125
gtt ctc ttg ctc gag gcc acc aca ttc cgc acc gta gga tta ctg cat 432
Val Leu Leu Leu Glu Ala Thr Thr Phe Arg Thr Val Gly Leu Leu His
IS 130 135 140
cag gag tgg tgg atg cgc ccg gat gac cct gcc gat gcg gat gaa aag 480
Gln Glu Trp Trp Met Arg Pro Asp Asp Pro Ala Asp Ala Asp Glu Lys
145 150 155 160
gag agt ggc aaa tgg ctg gca gcg gcc gca act agc cgg tta cgc atg 528
Glu Ser Gly Lys Trp Leu Ala Ala Ala Ala Thr Ser Arg Leu Arg Met
165 170 175
ggc agc atg atg agc aac gtg att gcg gtc tgt gac cgc gaa gcc gat 576
Gly Ser Met Met Ser Asn Val Ile Ala Val Cys Asp Arg Glu Ala Asp
180 185 190
25 att cat get tat ctg cag gac aaa ctg gcg cat aac gag cgc ttc gtg 624
Ile His Ala Tyr Leu Gln Asp Lys Leu Ala His Asn Glu Arg Phe Val
195 200 205
gtg cgc tcc aag cac cca cgc aag gac gta gag tct ggg ttg tat ctg 672
Val Arg Ser Lys His Pro Arg Lys Asp Val Glu Ser Gly Leu Tyr Leu
210 215 220
tac gac cat ctg aag aac caa ccg gag ttg ggt ggc tat cag atc agc 720
Tyr Asp His Leu Lys Asn Gln Pro Glu Leu Gly Gly Tyr Gln Ile Ser
225 230 235 240
-2-
CA 02380850 2002-O1-31
WO 01/09363 PCT/US00/21052
att ccg caa aag ggc gtg gtg gat aaa cgc ggt aaa cgt aaa aat cga 768
Ile Pro Gln Lys Gly Val Val Asp Lys Arg Gly Lys Arg Lys Asn Arg
245 250 255
cca gcc cgc aag gcg agc ttg agc ctg cgc agt ggg cgc atc acg cta 816
$ Pro Ala Arg Lys Ala Ser Leu Ser Leu Arg Ser Gly Arg Ile Thr Leu
260 265 270
aaa cag ggg aat atc acg ctc aac gcg gtg ctg gcc gag gag att aac 864
Lys Gln Gly Asn Ile Thr Leu Asn Ala Val Leu Ala Glu Glu Ile Asn
275 280 285
1~ ccg ccc aag ggt gag acc ccg ttg aaa tgg ttg ttg ctg acc agc gaa 912
Pro Pro Lys Gly Glu Thr Pro Leu Lys Trp Leu Leu Leu Thr Ser Glu
290 295 300
ccg gtc gag tcg cta gcc caa gcc ttg cgc gtc atc gac att tat acc 960
Pro Val Glu Ser Leu Ala Gln Ala Leu Arg Val Ile Asp Ile Tyr Thr
1$ 305 310 315 320
cat cgc tgg cgg atc gag gag ttc cat aag gca tgg aaa acc gga gca 1008
His Arg Trp Arg Ile Glu Glu Phe His Lys Ala Trp Lys Thr Gly Ala
325 330 335
gga gcc gag agg caa cgc atg gag gag ccg gat aat ctg gag cgg atg 1056
Gly Ala Glu Arg Gln Arg Met Glu Glu Pro Asp Asn Leu Glu Arg Met
340 345 350
gtc tcg atc ctc tcg ttt gtt gcg gtc agg ctg tta cag ctc aga gaa 1104
Val Ser Ile Leu Ser Phe Val Ala Val Arg Leu Leu Gln Leu Arg Glu
355 360 365
2$ agc ttc acg ctg ccg caa gca ctc agg gcg caa ggg ctg cta aag gaa 1152
Ser Phe Thr Leu Pro Gln Ala Leu Arg Ala Gln Gly Leu Leu Lys Glu
370 375 380
gcg gaa cac gta gaa agc cag tcc gca gaa acg gtg ctg acc ccg gat 1200
Ala Glu His Val Glu Ser Gln Ser Ala Glu Thr Val Leu Thr Pro Asp
385 390 395 400
gaa tgt cag cta ctg ggc tat ctg gac aag gga aaa cgc aag cgc aaa 1248
Glu Cys Gln Leu Leu Gly Tyr Leu Asp Lys Gly Lys Arg Lys Arg Lys
405 410 415
-3-
CA 02380850 2002-O1-31
WO 01/09363 PCT/US00/21052
gag aaa gca ggt agc ttg cag tgg get tac atg gcg ata get aga ctg 1296
Glu Lys Ala Gly Ser Leu Gln Trp Ala Tyr Met Ala Ile Ala Arg Leu
420 425 430
ggc ggt ttt atg gac agc aag cga acc gga att gcc agc tgg ggc gcc 1344
$ Gly Gly Phe Met Asp Ser Lys Arg Thr Gly Ile Ala Ser Trp Gly Ala
435 440 445
ctc tgg gaa ggt tgg gaa gcc ctg caa agt aaa ctg gat ggc ttt ctt 1392
Leu Trp Glu Gly Trp Glu Ala Leu Gln Ser Lys Leu Asp Gly Phe Leu
450 455 460
gcc gcc aag gat ctg atg gcg cag ggg atc aag atc tga 1431
Ala Ala Lys Asp Leu Met Ala Gln Gly Ile Lys Ile
465 470 475
<210> 2
<211> 476
<212> PRT
<213> Transposon Tn5
<400> 2
Met Ile Thr Ser Ala Leu His Arg Ala Ala Asp Trp Ala Lys Ser Val
1 5 10 15
Phe Ser Ser Ala Ala Leu Gly Asp Pro Arg Arg Thr Ala Arg Leu Val
25 30
Asn Val Ala Ala Gln Leu Ala Lys Tyr Ser Gly Lys Ser Ile Thr Ile
35 40 45
Ser Ser Glu Gly Ser Glu Ala Met Gln Glu Gly Ala Tyr Arg Phe Ile
50 55 60
Arg Asn Pro Asn Val Ser Ala Glu Ala Ile Arg Lys Ala Gly Ala Met
65 70 75 80
Gln Thr Val Lys Leu Ala Gln Glu Phe Pro Glu Leu Leu Ala Ile Glu
85 90 95
Asp Thr Thr Ser Leu Ser Tyr Arg His Gln Val Ala Glu Glu Leu Gly
100 105 110
-4-
CA 02380850 2002-O1-31
WO 01/09363 PCT/US00/21052
Lys Leu Gly Ser Ile Gln Asp Lys Ser Arg Gly Trp Trp Val His Ser
115 120 125
Val Leu Leu Leu Glu Ala Thr Thr Phe Arg Thr Val Gly Leu Leu His
130 135 140
$ Gln Glu Trp Trp Met Arg Pro Asp Asp Pro Ala Asp Ala Asp Glu Lys
145 150 155 160
Glu Ser Gly Lys Trp Leu Ala Ala Ala Ala Thr Ser Arg Leu Arg Met
165 170 175
Gly Ser Met Met Ser Asn Val Ile Ala Val Cys Asp Arg Glu Ala Asp
180 185 190
Ile His Ala Tyr Leu Gln Asp Lys Leu Ala His Asn Glu Arg Phe Val
195 200 205
Val Arg Ser Lys His Pro Arg Lys Asp Val Glu Ser Gly Leu Tyr Leu
210 215 220
1$ Tyr Asp His Leu Lys Asn Gln Pro Glu Leu Gly Gly Tyr Gln Ile Ser
225 230 235 240
Ile Pro Gln Lys Gly Val Val Asp Lys Arg Gly Lys Arg Lys Asn Arg
245 250 255
Pro Ala Arg Lys Ala Ser Leu Ser Leu Arg Ser Gly Arg Ile Thr Leu
260 265 270
Lys Gln Gly Asn Ile Thr Leu Asn Ala Val Leu Ala Glu Glu Ile Asn
275 280 285
Pro Pro Lys Gly Glu Thr Pro Leu Lys Trp Leu Leu Leu Thr Ser Glu
290 295 300
2$ Pro Val Glu Ser Leu Ala Gln Ala Leu Arg Val Ile Asp Ile Tyr Thr
305 310 315 320
His Arg Trp Arg Ile Glu Glu Phe His Lys Ala Trp Lys Thr Gly Ala
325 330 335
Gly Ala Glu Arg Gln Arg Met Glu Glu Pro Asp Asn Leu Glu Arg Met
340 345 350
-5-
CA 02380850 2002-O1-31
WO 01/09363 PCT/US00/21052
Val Ser Ile Leu Ser Phe Val Ala Val Arg Leu Leu Gln Leu Arg Glu
355 360 365
Ser Phe Thr Leu Pro Gln Ala Leu Arg Ala Gln Gly Leu Leu Lys Glu
370 375 380
Ala Glu His Val Glu Ser Gln Ser Ala Glu Thr Val Leu Thr Pro Asp
385 390 395 400
Glu Cys Gln Leu Leu Gly Tyr Leu Asp Lys Gly Lys Arg Lys Arg Lys
405 410 415
Glu Lys Ala Gly Ser Leu Gln Trp Ala Tyr Met Ala Ile Ala Arg Leu
1~ 420 425 430
Gly Gly Phe Met Asp Ser Lys Arg Thr Gly Ile Ala Ser Trp Gly Ala
435 440 445
Leu Trp Glu Gly Trp Glu Ala Leu Gln Ser Lys Leu Asp Gly Phe Leu
450 455 460
1$ Ala Ala Lys Asp Leu Met Ala Gln Gly Ile Lys Ile
465 470 475
<210> 3
<211> 19
<212> DNA
2~ <213> Transposon Tn5
<400> 3
ctgactctta tacacaagt 19
<210> 4
<211> 19
25 <212> DNA
<213> Transposon Tn5
<400> 4
ctgtctcttg atcagatct 19
-6-
CA 02380850 2002-O1-31
WO 01/09363 PCT/US00/21052
<210>5
<211>19
<212>DNA
<213>Transposon
Tn5
<400> 5
ctgtctcttg aacagatct 19