Note: Descriptions are shown in the official language in which they were submitted.
CA 02381297 2002-02-06
WO 01/27114 PCT/C1S00/22674
PYRROLO[2,3-d]PYRIMIDINE NUCLEOSIDE ANALOGS
Field of The Invention
The field of the invention is nucleoside analogs.
Background of The Invention
Nucleoside analogs have long been used as antimetabolites for treatment of
cancers and
viral infections. After entry into the cell, nucleoside analogs are frequently
phosphorylated by
nucleoside salvage pathways, in which the analogs are typically phosphorylated
to the correspon-
ding mono-, di-, and triphosphates. Among other intracellular destinations,
triphosphorylated
nucleoside analogs often are used as substrate for DNA or RNA polymerases and
consequently
incorporated into DNA or RNA. Where triphosphorylated nucleoside analogs are
strong
polymerase inhibitors, they may induce premature termination of a nascent
nucleic acid
molecule. Where triphosphorylated nucleoside analogs are incorporated into
nucleic acid
replicates or transcripts, gene expression or disruption of function may
result.
On a more cellular level, the nucleoside analogs can also interfere with the
cell cycle, and
especially desirable effects of nucleoside analogs include induction of
apoptosis of cancer cells.
Furthermore, nucleoside analogs are also known to modulate certain immune
responses.
Various nucleoside analogs with relatively potent anti-cancer activity are
known in the
art. For example, known drugs include thymidylate synthase inhibitors such as
5-fluorouridine,
adenosine deaminase inhibitors, including 2-chloroadenosine, and neplanocin A,
which is an
inhibitor of S-adenosylhomocysteine hydrolase. However, all or almost all of
the known
nucleoside analogs also imply a threat to normal mammalian cells, primarily
because these
nucleoside analog's lack adequate selectivity between normal cells and tumor
cells.
Unfortunately, lack of adequate selectivity is frequently associated with
severe side effects, and
therefore often limits the potential of such analog therapeutics.
Although there are various nucleoside analogs known in the art, all or almost
all of them,
suffer from one or more disadvantage. Therefore, there is still a need to
provide improved
methods and compositions for nucleoside analogs.
CA 02381297 2002-02-06
Attorney Reference No.: 330.48-PCT _ _ IPEA/US ~r ~ MllR
Summary of the InventionSummarv of the Invention
The present invention is directed to nucleoside analogs with modifications on
the sugar
moieties of the pyrrolo[2,3-d]pyrimidine nucleoside analogs, which can
significantly reduce the
toxicity of the nucleoside analogs to the mammalian cells while they also
provide significant
cytotoxicity to cancer cells. These modifications include but are not limited
to substitutions at
the C4' and CS' positions of ribofuranose moieties. The present invention also
demonstrates that
certain pyrrolo[2,3-d]pyrimidine nucleoside analogs have desired
immunomodulation effects,
including enhancement of Type 1 cytokines such as IL-2 and suppression of Type
2 cytokines
such as IL-4. These immunomodulation properties can be useful in anticancer,
antiviral and
autoimmune diseases, treating inflammation and preventing graft rejection.
In one aspect of the inventive subject matter, the nucleoside analog is a
pyrrolo[2,3)pyrimidine nucleoside having a structure according to the formula
(I):
x z
N~
(I)
Y N
RS
Rs~ A
~ R ~R2
f-.~. wherein A is O, S, or CH2; X is H, NHZ or OH; Y is H, halogen or NHZ; ;Z
is selected from the
;~;.
group consisting of H, halogen, R, OH, OR, SH, SR, NHz, NHR, NRZ, CN, C(O)NH2,
COOH,
COOR, CHzNH2, C(=NOH)NH2, and C(=NH)NHZ, where R is alkyl, alkenyl, alkynyl,
or aralkyl;
RZ and R3 are independently selected from the group consisting of H, F, and
OH; R4 is selected
from the group consisting of a hydrogen, an alkyl, an alkenyl, an alkynyl, and
an aralkyl, wherein
R4 optionally has at least one of a heteroatom and a functional group; RS is
H, OH, OP(O)(OH)2,
P(O)(OH)Z, OP(O)(OR')2, or P(O)(OR')2, wherein R' is a masking group; and R5~
is selected from
the group consisting of an alkyl, an alkenyl, an alkynyl, and an aralkyl,
wherein R5. has at least
two carbon atoms, and optionally has at least one of a heteroatom and a
functional group.
In another aspect of the inventive subject matter, the nucleoside analog is a
pyrrolo[2,3d]pyrimidine nucleoside having a structure according to the formula
(II):
2
AMENDED SHEET
CA 02381297 2002-02-06
Attorney Reference No.: 330.48-PCT
w o MAR 2001
O z
N N (II)
RS' O
Rt R3/ 'R2
wherein Z is CN, C(O)NH2, C(=NH)NH2, or C(=NOH)NH2 and R4 and Rs~ are
independently
selected from the group consisting of a hydrogen, an alkyl, an alkenyl, an
alkynyl, and an aralkyl,
wherein R4 and R5. independently and optionally contain at least one of a
heteroatom and a
functional group; with the proviso that R4 and R5~ are not together hydrogen.
In a further aspect of the inventive subject matter, contemplated compounds
are utilized
~:.,;y to inhibit tumor growth or to modulate Type l and Type 2 cytokine, and
chemokine production.
Various objects, features, aspects and advantages of the present invention
will become
more apparent from the following detailed description of preferred embodiments
of the
invention, along with the accompanying drawings.
Brief Description of The Drawing
Fig. 1 is a first exemplary synthetic scheme of reactions included in the
production of
compounds according to the inventive subject matter.
°~ 15 Fig. 2 is a second exemplary synthetic scheme of reactions
included in the production of
compounds according to the inventive subject matter.
Fig.3 is a third exemplary synthetic scheme of reactions included in the
production of
compounds according to the inventive subject matter.
Fig. 4 is a fourth exemplary synthetic scheme of reactions included in the
production of
compounds according to the inventive subject matter.
Fig. 5 is a fifth exemplary synthetic scheme of reactions included in the
production of
compounds according to the inventive subject matter.
3
AMENDED SHfE'T
CA 02381297 2002-02-06 ~ ~ ~ 7 4
Attorney Reference No.: 330.48-PCT
6 MA R 2001
Fig. 6 is a sixth exemplary synthetic scheme of reactions included in the
production of
compounds according to the inventive subject matter.
Fig. 7 depicts exemplary compounds according to the inventive subject matter.
Fig. 8A and 8B are graphs representing the effect of compounds according to
the
inventive subject matter on the expression of Type 1 and Type 2 cytokines,
respectively.
Fig. 9 is a table indicating cytotoxicity of various compounds according to
the inventive
subject matter.
Fig. 10 is a table indicating rates of DNA synthesis in cells treated with of
various
compounds according to the inventive subject matter.
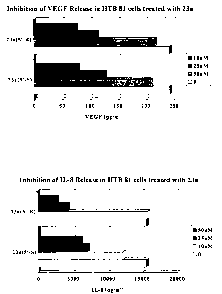
Kt,_ 10 Fig. 11 is a graph depicting the inhibition of VEGF release from human
prostate cancer
cells upon treatment with compounds according to the inventive subject matter.
Fig. 12 is a graph depicting the inhibition of IL-8 release from human
prostate cancer
cells upon treatment with compounds according to the inventive subject matter.
Detailed DescrintionDetailed Description
Pyrrolo[2,3-d]pyrimidine nucleoside analogs according to the general formulae
(I) and
(II) were found to have various biological effects on normal and
hyperproliferative cells.
'""' X
z
N
Y N (I)
Rs
Rs~ A
Ra R3~R~
4
AtuIIENDED SHEET
CA 02381297 2002-02-06
Attorney Reference No.: 330.48-PCT - ~S ~ G MAR
ZOOt
wherein A is O, S, or CH2; X is H, NHZ or OH; Y is H, halogen or NHZ; Z is
selected from the
group consisting of H, halogen, R, OH, OR, SH, SR, NHz, NHR, NR2, CN, C(O)NHz,
COOH,
COOR, CHZNH2, C(=NOH)NHZ, and C(=NH)NHZ, where R is alkyl, alkenyl, alkynyl,
or aralkyl;
R2 and R3 are independently selected from the group consisting of H, F, and
OH; R4 is selected
from the group consisting of a hydrogen, an alkyl, an alkenyl, an alkynyl, and
an aralkyl, wherein
R4 optionally has at least one of a heteroatom and a functional group; RS is
H, OH, OP(O)(OH)2,
P(O)(OH)z, OP(O)(OR')Z, or P(O)(OR')2, wherein R' is a masking group; and R5.
is selected from
the group consisting of an alkyl, an alkenyl, an alkynyl, and an aralkyl,
wherein R5~ has at least
two carbon atoms, and optionally has at least one of a heteroatom and a
fimctional group.
It should be especially appreciated that the terms "alkyl", "alkenyl",
"alkynyl", and
"aralkyl" as used herein refer to both linear and branched species. With
respect to the
substituents RZ and R3, it should be appreciated that both RZ and R3 may be
independently
~.'.r~l,
°"'~ directed to the a- or [3-face. Furthermore, where the substituents
on C5~ are non-identical,
substitution on CS may result in an R- or S-chiral center. The term
"heteroatom", as used herein,
refers to non-carbon atoms in an organic molecule, and particularly
contemplated heteroatoms
include halogens, nitrogen, oxygen, and sulfur. The term "functional group" as
used herein refers
to a reactive bond (e.g., double or triple bond) or reactive group (e.g., -OH,
-SH, -NH2, -N3, -CN,
COOH, -CHO, -CONH2, etc.).
Particularly contemplated pyrrolo[2,3-d]pyrimidine nucleoside analogs are
those
according to formula (I) wherein Z is CN, C(O)NH2, or C(=NH)NHZ , and wherein
R5, has at
least two carbon atoms and is selected from the group consisting of an alkyl,
an alkenyl, an
alkynyl, and an aralkyl.
The pyrrolo[2,3-d]pyrimidine nucleoside analog according to formula (II) has
the
following structure:
o z
R wN N (II)
s
Rs~ O
R4
R~R,
5
AMENDED SHEEN
CA 02381297 2002-02-06
WO 01/27114 PCT/US00/22674
wherein Z is CN, C(O)NH2, C(=NH)NH2, or C(=NOH)NHz and R~ and R;. are
independently
selected from the group consisting of a hydrogen, an alkyl, an alkenyl, an
alkynyl, and an aralkyl,
wherein R4 and R5- independently and optionally contain at least one of a
heteroatom and a
functional group; with the proviso that R.~ and R5~ are not together hydrogen;
and wherein the
remaining substituents are as defined in formula (I).
It should be appreciated, however, that compounds according to the inventive
subject
matter may also be in forms and formulations other than previously described,
and especially
contemplated forms include prodrug forms or otherwise modified forms in which
contemplated
molecules are chemically and/or enzymatically modified to improve
pharmacological and/or
pharmacodynamical properties, including higher specificity towards target
organs, cells or
subcellular compartments and increased half life time in the organism.
For example. cholesterol adducts may be formed to increase target specificity
towards the
liver, or apolipoprotein adducts may be formed to enhance penetration of the
modified drug
across the blood brain barrier to the brain. In another example, receptor
ligand complexes may
be synthesized to target the modified drug to a particular cell expressing a
receptor specific for
the ligand. Alternatively, antibody or antibody icagment complexes may be
formed to increase
selective delivery of the modified drug to a subcellular location. There are
many prodrug and
modified forms known in the art, and particularly contemplated prodrug forms
include prodrugs
described in U.S. Provisional Application 60/216418, filed 04/17/00, and
incorporated herein by
?0 reference. In still further examples, charged or uncharged groups,
lipophilic or polar groups may
be added to contemplated molecules to increase the half life time in serum or
other target organs
and/or cells. In further examples, it should be appreciated that the
contemplated compounds,
where phosphorylated at the C5 atom, may also be di-, or tri-phosphorylated,
or incorporate a
thiophosphate.
While it is generally preferred that contemplated compounds have a sugar
moiety in the
D-configuration, it is also contemplated that the compounds may have a sugar
moiety in the
L-configuration. Further stereochemical aspects especially include R and S
configurations at the
CS atom where appropriate, and it should be appreciated that the substituents
in contemplated
compounds may be directed to a or ~i phase.
6
CA 02381297 2002-02-06
WO 01/27114 PCT/US00/22674
It should also be appreciated that contemplated compounds can be formulated in
various
formulations, including liquid, syrup or gel forms (e.g., for injection,
ingestion, or topical
administration) and solid forms (e.g., for ingestion, injection, or deposition
in a body cavity). For
example, where it is contemplated that compounds according to the inventive
subject matter are
instable in the gastric environment, injection of a preferably isotonic
solution is particularly
contemplated. Alternatively, intranasal application or inhalation of a liquid
form may be
appropriate to circumvent acid degradation. On the other hand, where
contemplated compounds
are known to be resistant to digestive degradation, contemplated forms may be
administered in
form of a syrup or tablet. Depending on the particular use, contemplated
compounds may also be
formulated for topical or transdermal applications. There are many more
formulations known in
the art, all of which are also contemplated suitable in conjunction with the
inventive subject
matter presented herein, and particularly contemplated formulations are
described in "Drug
Products for Clinical Trials: An Intl. Guide to Formulation, Production,
Quality Control" by
Donald C. Monkhouse and Christopher T. Rhodes (Editors); ISBN:082479852X.
1 S It should still further be appreciated that contemplated compounds and
formulations may
include functional and non-functional additives. For example, where
transcutaneous drug
delivery is desired, skin penetration enhancers may be added. Alternatively,
pharmaceuticals
including cytostatic, antiviral, or immunomodulatory agents, may be added to
synergistically or
additively improve the function of contemplated compounds. Examples for non-
functional
additives include fillers, antioxidants, flavor, or color agents to enhance a
particular quality of
contemplated compounds.
With respect to the concentration of contemplated compounds, it is preferred
that the
concentration is in a range of approximately 1 ~M to about 1 OO~M when
measured at the site of
action. However, and particularly where the affinity of contemplated compounds
is below 1 ~M,
appropriate concentrations may also be in the range of 999nM to l OnM, and
less. On the other
hand, where contemplated compounds exhibit relatively short half life times,
or have a high
turnover, contemplated concentrations may be 0.1 mM and 1 OOmM, and more.
Consequently, the
dosage of contemplated compounds may vary significantly, but appropriate
dosages can readily
be determined in in vitro or animal experiments.
7
CA 02381297 2002-02-0 ~~~~~~ ~ ~ ~ t... L b ~ 1,~,
Attorney Reference No.: 330.48-PCT ~S ~ 6 MAR 2001
Among the various biological effects of both 5'- and 4'-modified
pyrrolopyrimidine
nucleoside analogs, particularly significant biological effects include
modulation of the Type 1
and Type 2 cytokine production, control of neoplastic conditions (i.e.,
reduction of DNA synthe-
sis or reduction in cell growth), and reduction of chemokine and growth factor
release as
described below.
Consequently, a contemplated method of changing secretion of a cytokine from a
cell
may comprise a step in which a compound according to formula (I) is provided
and has a further
step in which the cell is presented with the compound according to formula (I)
at a concentration
''y effective to change the secretion of the cytokine. While all possible
combinations of substituents
in formula (I) are generally contemplated, particularly contemplated compounds
are compounds
according to formula (I) wherein R4 and R5. are independently selected from
the group consisting
of a hydrogen, an alkyl, an alkenyl, an alkynyl, and an aralkyl, and wherein
R4 and R5,
independently and optionally contain at least one of a heteroatom and a
functional group, with
the remaining substituents as defined above in formula (I). In an alternative
aspect, the
compound employed to change the secretion of a cytokine from a cell may also
be a compound
according to the following structure:
,.,4
z
N N
RS O
HO OH
wherein Z is CN, C(O)NH2, C(=NH)NHZ, C(=NNHZ)NH2, or -C(=NOH)NH2, and wherein
RS is
H, OH, OP(O)(OH)2, P(O)(OH)Z, OP(O)(OR')2, or P(O)(OR')z, with R' being a
masking group.
Contemplated cytokines particularly include Type 1 (e. g., IFNy) and Type 2
(e.g., IL-4)
cytokines. With respect to the cells, it is contemplated that all cells known
to produce and/or
secrete cytokines are appropriate, however, especially contemplated cells
include lymphocytes
and cancer cells (e.g., prostate cancer cells, infra).
AMENDED SNFEt
CA 02381297 2002-02-06
WO 01/27114 PCT/US00/22674
In a further aspect of the inventive subject matter. a method of reducing
growth of a
hyperproliferative cell may comprise a step in which a compound according to
formula (I) is
provided, and another step in which the hyperproliferative cell is presented
with the compound at
a concentration effective to reduce the growth of the hyperproliferative cell.
Particularly
preferred compounds include compounds according to formula (I) wherein R~ is
selected from
the group consisting of a hydrogen, an alkyl, an alkenyl, an alkynyl, and an
aralkyl, wherein R.~
optionally contains at least one of a heteroatom and a functional group, and
wherein RS- is
selected from the group consisting of a hydrogen, an alkyl, an alkenyl. an
alkynyl. and an aralkyl,
wherein R;- has at least two carbon atoms, and optionally contains at least
one of a heteroatom
and a functional group, with the proviso that R.~ and R5~ are not together
hydrogen, and with the
remaining substituents as defined above in formula (I).
Particularly contemplated hyperproliferative cells include cancer cells, and
an especially
contemplated cancer cell is a prostate cancer cell. While not whishing to be
bound to a particular
theory, it is contemplated that the reduction of growth comprises reduction of
DNA synthesis.
In a still further aspect of the inventive subject matter, it is contemplated
that a method of
reducing a release of a growth factor from a cell has a step in which a
compound according to
formula (I) is provided, and another step in which the cell is presented with
the compound at a
concentration effective to reduce the release of the growth factor. It is
contemplated that the
release of various growth factors may be reduced by the method presented
herein, however
?0 reduction of VEGF release is especially contemplated. Similarly, while all
cells known to secrete
growth factors are contemplated in conjunction with the method presented
herein, particularly
contemplated cells include cancer cells, and especially prostate cancer cells.
With respect to the synthesis of contemplated compounds, it should be
appreciated that
pyrrolo[2,3-d]pyrimidine nucleoside analogs according to the inventive subject
matter can be
synthesized via various synthetic routes, and the following procedures are
provided by way of
example only.
9
CA 02381297 2002-02-06
WO 01/27114 PCT/US00/22674
Synthesis of CS'-modified pyrrolo~2, 3-dJpyrimidine nucleoside analogs
The 5'-substituted nucleoside analogs are prepared from the condensation of
the
pyrrolo[2,3-d]pyrimidine bases and the properly protected. 5'-substituted
ribofuranoses. As
shown in Figure 1, Compound 1, prepared according to a published procedure
(Jones et al.
Methods in carbohydrate Chemistry (edited by Whistler and Moffat), vol. VI,
pp315-322,
Academic Press, New York, (1972)), was treated with a variety of nucleophiles
such as Grignard
reagents to give compound 2, which was benzoylated and subsequently treated
with
trifluoroacetic acid to give compound 4. Benzoylation and the following
treatment with acetic
anhydride/acetic acid in the presence of sulfuric acid to give compound 6,
which was used for
condensation with pyrrolo[2,3-d]pyrimidine bases.
Compound 7 (Jones et al. Methods in carbohydrate Chemistry (edited by Whistler
and
Moffat), vol. VI. pp315-322, Academic Press, New York, (1972)), prepared
according to a
published procedure, was converted to a tosylate derivative, which was reduced
with lithium
aluminum hydride to give compound 8. By similar procedures shown in Figure 1,
compound 8
was converted to compound 9. The condensation of 9 and the pyrrolo[2,3-
d]pyrimidine 19 and
the subsequent transformations as shown in scheme 2 gave compounds 10-15 as
illustrated in
Figure 2.
As shown in Figure 3, Compound 2 was converted to the sulfonate 16, which was
subjected to nucleophilic replacement to give the configurationally inverted
compound 17.
Deprotection of the isopropylidene and the subsequent acetylation gave the
tetraacetate 18.
Condensation of the 5-C-substituted, protected ribofuranoses with nucleoside
bases is depicted
in Figure 4. 5-Cyanopyrrolo[2,3-d]pyrimidine 19, prepared according to a
published procedure
(Tolman et al. J. Org. Chem. 1969, 91, 2102-2108), was converted to the
trimethylsilyl
derivative and then condensed with compound 6 in the presence of
trimethylsilyl triflate by a
similar procedure described for toyocamycin (Sharma et al. Nucleosides
Nucleotides 1993, 12,
643-648). The resulting coupling product was subjected to debromination
through hydrogenation
to give compound 20. Treatment of 20 with ammonia in anhydrous methanol gave
compounds
21 and 23. Compound 21 was oxidized to give compound 22. Compound 23 was
converted to
the carboxamide derivative 24. Compound 23 and 24 were oxidized to give
compound 25.
Treatment of compound 25 with hydroxyamine yielded compound 26, which was
hydrogenated
CA 02381297 2002-02-06
WO 01/27114 PCT/US00/22674
over Raney Nickel to give 27. Alternatively, compound 27 was also prepared by
heating
compound 25 with ammonia in a pressured bomb.
Synthesis ofC=I'-modified pyrrolo~2,3-dJpyrimidine nucleoside analogs
In Figure 5, compound 1 was treated with formaldehyde in aqueous sodium
hydroxide to
give 4'-hydroxymethyl derivative 28, which was selectively protected to afford
compound 29.
The subsequent protection with DMT and removal of TBS gave compound 31, which
can be
converted to a variety of substituents. The 4-C-substituted derivatives
subjected to similar
transformations as 5-C-substituted ribofuranoses (scheme 1 ) can be converted
to compound 35,
which is used for condensation with nucleoside bases.
Similar to the CS'-substituted pyrrolopyrimidine nucleoiside analogs, the 4'-
substituted
analogs 36 can be obtained by condensation of compound 35 with compound 19 as
depicted in
Figure 6. The subsequent transformations can give the 4'-C-substituted
pyrrolopyrimidine
nucleoside 37-42.
Synthesis of 2'-modified, and other pyrrolo~2,3-dJpyrimidine nucleoside
analogs
The following pyrrolopyrimidine nucleoside analogs were prepared for
biological testing,
some of which were published (indicated as known compounds), and are shown in
Figure 7.
The known compounds 43, 44, 52-55, and 57 were prepared according to a
published procedure
(Hinshaw et al. J. Org. Chem. 1970, 92, 236-241 ). Compound 56 was prepared by
hydrogenation
of compound 52. The known compound 49 (Krawczyk et al. Nucleosides Nucleotides
1989, 8,
97-115) was treated with sodium nitrite to give compound 50. The known
compounds 45 and 48
were prepared according to a published procedure (Ramasamy et al. J.
Heterocyclic Chem. 1988,
25, 1043-1046). Compound 45 was treated with ammonia-methanol to give compound
46 and
hydrogenated to give compound 47. Compounds 58-63 were prepared from compound
45 by
similar procedures used for compounds 52-57. The known compound 64 (Krawczyk
et al.
Nucleosides Nucleotides 1989, 8, 97-115) was converted to compounds 65-67. The
known
compound 68 (Ramasamy et al. Tetrahedron 1986, -I2, 5869-5878) was converted
to compounds
69 and 70.
CA 02381297 2002-02-06
WO 01/27114 PCT/US00/22674
Preparation of human T cells and activation in vitro
Peripheral blood mononuclear cells were isolated from healthy donors by
density
gradient centrifugation followed by T cell enrichment using Lymphokwik (One
Lambda, Canoga
Park CA). Contaminating monocytes were removed by adherence to plastic.
Purified T cells
were > 99% CD2+ , <1% HLA-DR+ and < 5% CD25+ and were maintained in RPMI-AP5
(RPMI1640 medium containing 5% autologous plasma, 1 % L-glutamine, 1
penicillin/streptomycin and 0.05% 2-mercaptoethanol). For determination of
cytokine protein
levels, T-cells (0.2 x 106 cells in a volume of 0.2 ml) were activated by the
addition of 2 ng
phorbol myristate acetate plus 0.1 mg ionomycin (PMA-ION, both from
Calbiochem, San Diego,
I0 CA) and incubated in 96 well plates in the presence of 0 or 10 pM of
various guanosine
nucleosides for 48 h at 37°C. Following activation, supernatants were
analyzed for cell-derived
cytokine production.
Extracellular cytokine analyses.
Human cytokine levels were determined in cell supernatants, following
appropriate
dilution, using ELISA kits specific for IFNy and IL-4 (Biosource, Camarillo,
CA). All ELISA
results were expressed as pg/ml.
Effect ofpyrrolo-~2,3,dJpyrimidine nucleoside analogs on extracellular
cytokine levels in
activated human T cells.
The effect of pyrrolo-[2,3-d]pyrimidine nucleoside analogs at 0 and 10 ~M, on
PMA/ionomycin stimulated T cell expression of the Type 1 cytokine, IFNy, and
the Type 2
cytokine, IL-4, is shown in Figures 8A and 8B for 5 individual human donors.
Cytokine levels
were determined in cell free supernatants by ELISA. The most potent effect was
observed with
7-b-D-ribofuransyl-4-oxopyrrolo-[2,3-d] pyrimidine-5-carboxamidine. This
compound enhanced
activated IL-4 production by 498% t 83 and suppressed IFNy by 43% t 4 of the
activated
control levels of each cytokine. Data are shown as percentage of activated
control calculated as
the ratio of activated T cell cytokine level in the presence of test
nucleosides over the cytokine
level of untreated activated T cells x 100 %. Zero effect on cytokine levels
by test nucleosides
would give a percentage of activated control value of 100 %. The absolute
level (pg/ml t
standard deviation) of PMA-ION-induced cytokine secretion was for IFNy, 22954
f 3391; and
for IL-4, 162 ~ 40. Resting levels were < 30 pg/ml for all cytokines tested.
12
CA 02381297 2002-02-06
WO 01/27114 PCT/US00/22674
Cytotoxicity of the pyrrolo(2,3-dJpyrimidine nucleoside analogs in vitro
The pyrrolo[2,3-d]pyrimidine nucleoside analogs of the present invention are
bioactive
since they indicate some level of cytotoxicity in vitro. In these studies, the
compounds tested
were applied to cell culture of normal human fibroblasts, human Prostate
cancer cells 81, human
Melanoma cancer cells 140, Human Lung Cancer cells 177, and human Ovarian
Cancer cells R
and NR (all available from ATCC). In these experiments cells were plated at
density of 2000
cells per 2001 of medium per well (96-well plate). The compounds tested were
applied to the
wells once, at concentration range 0.78-100pM, just after plating of cells.
The colorimetric
cytotoxicity assay MTS was performed after 72 hrs of treatment. EC50 was
calculated based on
readings collected and they are presented in Figure 9. Several compounds
indicate lack of
cytotoxicity in concentration below 1 OOpM. In such cases EC50 is marked as >
100. In other
cases, EC50 indicates the concentration of the compound tested needed to
damage 50% of cell
population.
The pryrrolo(2,3-dJpyrimidine nucleoside analogs inhibit DNA synthesis in
cells cultured in
vitro in a dose-dependent manner
Analogs of pyrrolo[2,3-d]pyrimidine nucleoside inhibit growth of human cells
cultured in
vitro as measured by the level of DNA. The experimental setup was the same as
described
above. The compounds were given once and DNA level was measured after 72 hrs.
At that time,
half of the medium was removed from culture wells and replaced by pure water.
After that, the
cells were transferred to -70°C for at least 12 hrs. In the next step,
cells were transferred back
from -70°C to room temperature and 1 ~M of Hoechst 33342 was given to
each well. After 2 hrs
of incubation, the fluorescence signal (360-530nm) was measured. According to
this method.
intensity of florescence is proportional to amount of DNA due to presence of
DNA-Hoechst
33342 complex formed. The results are presented in Figure 10. The numbers
express fold of
DNA amount increase compared to amount of DNA at the beginning of experiment
(2 hrs after
plating of cells). In the untreated prostate cancer cells and normal cells the
DNA level increased
5.78 and 4.47 times, respectively, during 72 hrs of culture.
13
CA 02381297 2002-02-06
WO 01/27114 PCT/US00/22674
The compounds 23a(5'-R) and 23a(5'-S) inhibit secretion of VEGFfrom Human
Prostate
Cancer in vitro.
The compounds 23a(5'-R) and 23a(5'-S) are potent in inhibition of secretion of
Vascular
Endothelial Growth Factor (VEGF) from Human Prostate cancer cells, HTB81. VEGF
is
recognized as angiogenesis marker since this molecule is crucial for migration
and growth of
endothelial cells and microvessel formation in vivo. In order to prove this,
0.5x105 of the cells
were plated in 5 ml of culture medium into a l Ocm diameter petri dish. The
compounds were
applied just after plating for 72 hrs. After that, the medium was collected
and the level of VEGF
was measured using VEGF Elisa Assay (R&D Systems) and expressed as pg of VEGF
per ml of
the medium. The results are presented in Figure 11. According to these
results, both compounds
inhibit secretion of VEGF in a dose-dependent manner.
The compounds 23a(5'-R) and 23a(5'-S) inhibit release oflL-8,fi-om human
prostate cancer
cells carltured in vitro.
The compound 23a(5'-R) and 23a(5'-S) indicate an inhibitory effect on
secretion of
Interleukin-8 (IL-8) from human prostate cancer cells, HTB81. IL-8 belongs to
the class of
chemo-attractant chemokines (type alpha), which are involved in inflammation
processes due to
attraction of neutrophils. Chemokines, in general, are known to be produced by
various types of
cancer. It is proven in several studies that inhibition of chemokine
production by cancer cells is
beneficial for the host. In order to prove the potency of these two compounds
to inhibit IL-8
secretion from prostate cancer cells, prostate cancer cells HTB 81 were
treated in vitro with
compounds 23a in both the 5'-R and 5'-S configuration at concentrations
indicated in the graph.
The medium collected from the culture was analyzed for IL-8 level using IL-8
Elisa Assay from
R&D Systems. According to the results collected, both compounds are able to
inhibit secretion
of IL-8 in a dose-dependent manner, as depicted in Figure 12.
It should be appreciated, however, that the biological effects of contemplated
compounds
need not be limited to the particular effects as described above. In
particular, it is contemplated
that the compounds according to the inventive subject matter generally exhibit
cytostatic effect
in various hyperproliferative disorders, including localized and/or metastatic
cancers (e.g"
lymphomas and carcinomas), benign prostate hyperplasia, and keratoses. While
the inventors
found substantial biological effects on IL-4 (a Type 2 cytokine) and IFNy (a
Type 1 cytokine), it
is generally contemplated that the compounds according to the inventive
subject matter are
biologically active in modulation of cytokines other than IL-4 and IFN-y. It
is especially
14
CA 02381297 2002-02-06
WO 01/27114 PCT/US00/22674
contemplated that the compounds may increase or decrease the
expressionlsecretion of a
particular cytokine or set of cytokines. Therefore, it is contemplated that
compounds according
to the inventive subject matter may modulate the immune system of an organism
such that a
more pronounced Type 1 or Type 2 response may be achieved. Consequently, it is
contemplated
that the compounds according to the inventive subject matter may be effective
to reduce the titer
of a virus in a living system by either direct action as inhibitor of a viral
polymerase and/or
indirectly by activating the immune system to a particular humoral or cellular
response. It is
further contemplated that compounds according to the inventive subject matter
may also be
useful in reducing a response of an immune system towards an alto- or
xenograft by reducing the
severity of the cellular reponse towards the allo- or xenograft.
Examples
The following protocols describe an exemplary synthesis of various compounds
according to the inventive subject matter and are intended only to illustrate
but not to limit the
inventive concept presented herein.
Preparation ofmethyl2,3-O-isopropylidene-.5(R,S)-C-ethynyl-~ribofuranoside
(26)
To a stirred solution of methyl 4-C,5-O-didehydro-2,3-O-isopropylidene-~i-D-
ribo-
furanoside (Jones et al. Methods in Carbohydrate Chemistry Vol 1, pp315-322
(1972), 4.00 g,
19.78 mmol) in anhydrous THF (20 mL) at -42 °C under argon was added
dropwise
ethynylmagnesium bromide (0.5 M in THF, 80 mL, 40 mmol). Upon addition, the
resulting
mixture was slowly warmed up to 0 °C (~90 min.). The reaction was
quenched by adding ice (50
g)/water (50 mL) and the mixture was stirred for 30 min. After neutralization
with 10 % aq.
acetic acid, the mixture was extracted with ethyl acetate twice. The combined
organic layer was
dried (NazS04) and concentrated. Chromatography on silica (ethyl acetate-
hexanes 1:4) gave
3.48 g of the titled compound (R/S ratio 1:1 ) as a white solid. The following
compounds were
prepared in a similar fashion: Methyl 2,3-O-isopropylidene-5(R)-C-methyl-~i-D-
ribofuranoside
(2a) was prepared from methyl 4-C,5-O-dihehydro-2,3-O-isopropylidene-~i-D-
ribofuranoside
and ethylmagnesium bromide. Methyl 2,3-O-isopropylidene-5(R)-C-vinyl-(3-D-
ribofuranoside
(2c) was prepared from methyl 4-C,5-O-dihehydro-2,3-O-isopropylidene-(3-D-
ribofuranoside
and vinylmagnesium bromide. Methyl 5(R)-C-allyl-2,3-O-isopropylidene-(3-D-
ribofuranoside
(2d) was prepared from methyl 4-C,5-O-dihehydro-2,3-O-isopropylidene-(3-D-
ribofuranoside
and allylmagnesium bromide.
CA 02381297 2002-02-06
WO 01/27114 PCT/US00/22674
Preparation ofmethyl2,3-O-isoproplidene-.S-O-methanesulfonyl-5(R)-C-methyl-~-D
ribofuranoside (16)
To a stirred solution of methyl 2,3-O-isopropylidene-5(R)-C-methyl-(3-D-
ribofuranoside
(2a, 7.24 g, 33.17 mmol) in anhydrous pyridine (50 mL) at 0 °C was
added methanesulfonyl
chloride (3.1 mL, 39.92 mmol). The resulting mixture was stirred at room
temperature for 1 h,
cooled to 0 °C, quenched by adding water ( 1.0 mL), and stirred at room
temperature for 30 min.
The solvent was evaporated and the residue was dissolved in ethyl acetate,
washed with brine
three times, dried (NazS04) and concentrated. Chromatography on silica (30%
EtOAc in
hexanes) gave 8.62 g of the titled compound 16 as a colorless syrup.
Preparation of methyl 2,3-O-isopropylidene-S-O-acetyl-5(S)-C-methyl-~D
ribofuranoside (17)
A stirred suspension of methyl 2,3-O-isopropylidene-5-O-methanesulfonyl-5(R)-C-
methyl-~3-D-ribofuranoside (16, 8.62 g, 29.1 mmol) and NaOAc (anhydrous, 3.5
g, 42.5 mmol)
I S in anhydrous DMF (350 mL) was heated at 125 °C under argon for 4
days. The solvent was
evaporated and the residue chromatographed on silica (25% EtOAc in hexanes) to
give 4.0 g of
the titled compound 17 as a white solid.
Preparation ofmethyl2,3-O-isopropylidene--I-C-hydroxymethyl-~3-D-
ribofuranoside (28)
To a stirred solution of methyl 4-C,5-O-didehydro-2,3-O-isopropylidene-(3-D-
ribofuranoside 1 (20.22 g, 100 mmol) in dioxane (380 mL) at 0 °C was
added dropwise
formaldehyde (37 % solution, 76 mL) and then 2 M NaOH (188 mL). The resulting
reaction
mixture was stirred at room temperature for 20 h, cooled to 0 °C,
neutralized (10% acetic acid),
concentrated (~50 %), and extracted with methylene chloride twice. The
combined organic layer
was dried (NazS04) and concentrated to dryness. Chromatography on silica (4 %
methanol in
chloroform) gave 20.2 g of the titled compound 28 as a white solid.
Preparation of methyl 2, 3-O-isopropylidene-5-deoxy-~3-D-ribofuranoside (8)
To a stirred solution of methyl 2,3-O-isopropylidene-(3-D-ribofuranoside (14.2
g, 70.0
mmol) in anhydrous pyridine (250 mL) at 10 °C was added in portions
(over 30 min) p-
toluenesulfonyl chloride ( I 9. I g, 100 mmol). The resulting mixture was
stirred at room
temperature for 18 h, cooled to 0°C, quenched by adding water (5.0 mL),
and stirred at room
temperature for 30 min. The solvent was evaporated. The residue was dissolved
in ethyl acetate,
16
CA 02381297 2002-02-06
WO 01/27114 PCT/US00/22674
washed with brine three times, dried (Na~S04) and concentrated to dryness.
Chromatography on
silica (ethyl acetate-hexanes 1:3) gave 24.1 g of the titled compound as a
white solid.
To a stirred suspension of LiAlH4 (4.58 g, 120.5 mmol) in anhydrous diethyl
ether ( 120
mL) was added methyl 2,3-O-isopropylidene-5-O-p-toluenesulfonyl-(3-D-
ribofuranoside ( 13.1 g,
36.55 mmol) in diethyl ether-toluene (2.5:1, 140 mL). The resulting mixture
was refluxed for 22
h, cooled to room temperature, diluted with ethyl acetate (25 mL) quenched by
adding water (5.0
mL). The solvent was evaporated. The residue was dissolved in ethyl acetate,
washed with brine
three times, dried (NaZS04) and concentrated to dryness. Chromatography on
silica (ethyl acetate
hexanes 1:3) gave 3.58 g of the titled compound as a colorless liquid.
Preparation of~methyl5(R)-C-allvl-5-O-benzoyl-2.3-O-isopropylidene-~3-D-
ribofuranoside (3d)
To a stirred solution of methyl 5(R)-C-allyl-2,3-O-isoproplidene-(3-D-
ribofuranoside
(4.49 g, 18.38 mmol) in anhydrous pyridine (40 mL) at 0 °C was added
benzoyl chloride (2.7
mL, 23.0 mmol). The resulting mixture was stirred at room temperature for 18
h, cooled with
ice, quenched by adding water ( 1 mL), and stirred at room temperature for 30
min. The solvent
was evaporated and the residue was dissolved in ethyl acetate, washed with
brine three times,
dried (Na~S04) and concentrated. Chromatography on silica ( 12% ethyl acetate
in hexanes) gave
6.26 g of the titled compound 3d as a colorless syrup. The following compounds
were prepared
in a similar fashion: Methyl 5-O-benzoyl-5(R,S)-C-ethynyl-2,3-O-isoproplidene-
(3-D-ribofurano-
side (3b, R/S ratio: 1:1 ) from methyl 5(R,S)-C-ethynyl-2,3-O-isoproplidene-~3-
D-ribofuranoside
(2b). Methyl 4-C-benzoyloxymethyl-5-O-benzoyl-2,3-O-isoproplidene-(3-D-
ribofuranoside from
methyl 2,3-O-isoproplidene-4-C-hydroxymethyl-(3-D-ribofuranoside.
Preparation of methyl S(R)-C-ullyl-5-O-benzoyl-~-D-ribofuranoside (4d)
A solution of methyl 5(R)-C-allyl-5-O-benzoyl-2,3-O-isopropylidene-(3-D-
ribofuranoside
(3d, 6.2 g, 17.8 mmol) in TFA-Hz0 mixture (9: I ) was stirred at 0 °C
for 90 min and
concentrated to dryness at 0°C. The residue was dissolved in methanol-
toluene mixture (20 mL,
1:1 ) and concentrated to dryness. Chromatography on silica (ethyl acetate -
hexanes 1:1 ) gave
3.70 g of the titled compound 4d as a white solid. The following compounds
were prepared in a
similar fashion: Methyl 5-O-benzoyl-5(R,S)-C-ethynyl-(3-D-ribofuranoside (4b,
R/S ratio: 1:1 )
from methyl 5-O-benzoyl-5(R,S)-C-ethynyl-2,3-O-isopropylidene-(3-D-
ribofuranoside (3b).
17
CA 02381297 2002-02-06
WO 01/27114 PCT/US00/22674
Methyl 5-O-benzoyl-4-C-benzoyloxymethyl-(3-D-ribofuranoside from methyl 5-O-
benzoyl-4-C-
benzoyloxymethyl-2,3-O-isopropylidene-(3-D-ribofuranoside.
Preparation of methyl S(R)-C-allyl-2, 3, S-tri-O-benzoyl-~3-D-ribofuranoside
(5d)
To a stirred solution of methyl 5(R)-C-allyl-5-O-benzoyl-(3-D-ribofuranoside
(4d,
3.60mg, 11.68 mmol) in anhydrous pyridine (80 mL) at 0 °C was added
benzoyl chloride (3.0
mL, 25.84 mmol). The resulting mixture was stirred at room temperature for 18
h, cooled with
ice, quenched by adding water (1 mL), then stirred at room temperature for 30
min. The mixture
was concentrated, diluted with ethyl acetate, washed with brine three times,
dried (Na2S04) and
concentrated to dryness. Chromatography on silica ( 15% ethyl acetate in
hexanes) gave 5.3 g of
the titled compound Sd as a colorless syrup. The following compounds were
prepared in a
similar fashion: Methyl 5(R,S)-C-ethynyl-2,3,5-tri-O-benzoyl-(3-D-
ribofuranoside (5b, R/S ratio:
1:1 ) from methyl 5-O-benzolyl-5(R,S)-C-ethynyl-(3-D-ribofuranoside (4b).
Methyl 4-C-benzoyl-
oxomethyl-2,3,5-tri-O-benzoyl-~3-D-ribofuranoside from methyl 4-C-
benzoyloxymethyl-5-O-
benzoyl-(3-D-ribofuranoside.
Preparation of I -O-methyl-2, 3, 5-tri-O-benzoyl-5(R)-C-vinyl-,Q-D-
ribofuranose (5c)
A solution of methyl 2,3-O-isopropylidene-5(R)-C-vinyl-[3-D-ribofuranosise
(2c, 1.0 g,
4.3 mmol) in a mixture of trifluoroacetic acid and water (9:1, v/v, 11 mL) was
strirred at 0 °C for
30 min and concentrated to dryness. The residue was dissolved in methanol and
concentrated to
dryness (3 times), then dissolved in pyridine and evaporated, and finally was
dissolved in
anhydrous pyridine ( 11 mL). To this solution was added benzoyl chloride ( 1.9
mL, 16 mmol).
The reaction mixture was stirred at 25 °C for 16 h and poured into ice
water (20 mL). The
mixture was extracted with dichloromethane (20 mL) and the organic layer was
dried over
sodium sulfate, and concentrated to dryness. The residue was chromatographed
on silica (0-5%
ethyl acetate in dichloromethane) to give 1.0 g of the titled compound Sc as a
syrup.
Preparation of 1-O-acetyl-2, 3, 5-tri-O-benzoyl-5(R)-C-allyl-D-ribofuranose
(6d)
To a stirred solution of methyl 5(R)-C-allyl-2,3,5-tri-O-benzolyl-(3-D-
ribofuranoside (5d,
4.0 g, 7.74 mmol) in acetic acid (14 mL) and acetic anhydride (1.75 mL, 18.36
mmol) at 0 °C
18
CA 02381297 2002-02-06
WO 01/27114 PCT/US00/22674
was added concentrated sulfuric acid (200 ~L, 3.79 mmol in 4.0 mL of acetic
acid). The
resulting mixture was stirred at room temperature for 20 h, cooled to 0
°C, diluted with cold
ethyl acetate, washed with water, 5% aq. NaHC03 and then brine, dried
(NaZS04), and
concentrated. Chromatography on silica (ethyl acetate-hexanes 1:4) gave 2.82 g
of the titled
compound 6d (a/(3 ratio: I :2) as a colorless foam. The following compounds
were prepared in a
similar fashion: 1-O-Acetyl-5(R,S)-C-ethynyl-2,3,5-tri-O-benzolyl-(3-D-
ribofuranose (6b, R/S
ratio: 1:1 and a/(3 ratio: 1:2) from methyl 5(R,S)-C-ethynyl-2,3,5-tri-O-
benzolyl-(3-D-
ribofuranoside (5b). 1-O-Acethyl-4-C-benzoyloxymethyl-2,3,5-tri-O-benzoyl-D-
ribofuranose (a/
(3 ratio: 1:3) from methyl 4-C-benzoyloxymethyl-2,3,5-tri-O-benzoyl-(3-D-
ribofuranoside. 5(R)-
C-Methyl-1,2,3,5-tetra-O-acetyl-(3-D-ribofuranose from methyl 2,3-O-
isopropylidene-5(R)-C-
methyl-[i-D-ribofuranoside.
1,2,3,5-Tetra-O-acetyl-S(S)-C-methyl-D-ribofuranose 6a from methyl S-O-acetyl-
2,3-O-
isopropylidene-5(R)-C-methyl-(3-D-ribofuranoside. 5-deoxy-1,2,3-tri-O-acetyl-D-
ribofuranose 9
from methyl 5-O-acetyl-2,3-O-isopropylidene-(3-D-ribofuranoside. 1-O-Acetyl-
2,3,5-tri-O-
benzoyl-5(R)-C-vinyl-(3-D-ribofuranose 6c from methyl 2,3,5-tri-O-benzoyl-5(R)-
C-vinyl-[i-D-
ribofuranoside.
Preparation of =l-amino-6-bromo-~-cyano-7-(2, 3, 5-tri-O-benzoyl-5(R)-C-allyl-
~D
ribofuranosyl)pyrrolo~2, 3-dJpyrimidine
A suspension of 4-amino-6-bromo-5-cyanopyrrolo[2,3-d]pyrimidine (Tolman et al.
J.
Org. Chem. 1969, 91, 2102-2108, 1.05 g, 4.41 mmol) and ammonium sulfate (50
mg) in HMDS
(75 mL) and anhydrous m-xylene (25 mL) was refluxed under argon for 18 h.
Solvents were
evaporated and the residue was dried under vacuum, The residue was dissolved
in anhydrous
1,2-dichloroethane (80 mL) and mixed with 1-O-acetyl-2,3,5-tri-O-benzoyl-5(R)-
C-allyl-D-
ribofuranose (2.00 g, 3.67 mmol). Under cooling with ice, TMSOTf (1.3 mL, 7.30
mmol in 5
mL of anhydrous 1,2-dichloroethane) was added. The mixture under argon was
stirred at room
temperature for 30 min, then refluxed for 90 h, quenched by pouring it (cold)
onto ice/NaHC03
(50 mL), and filtered. The organic layer was separated, dried (NazS04), and
concentrated.
Chromatography on silica (EtOAc-hexanes 2:3) gave 1.81 g of the titled
compound as a
colorless solid. The following compounds were prepared in a similar fashion: 4-
Amino-6-
bromo-5-cyano-7-(2,3,5-tri-O-benzoyl-5(R,S)-C-ethynyl-[3-D-
ribofuranosyl)pyrrolo[2,3-
d]pyrimidine (R/S ratio: I:1) was prepared froml-O-acethyl-2,3,5-tri-O-
benzolyl-S(R,S)-C-
19
CA 02381297 2002-02-06
WO 01/27114 PCT/US00/22674
ethynyl-D-ribofuranose and 4-amino-6-bromo-5-cyanopyrrolo[2,3-dJpyrimidine. 4-
amino-6-
bromo-5-cyano-7-(4-enzoyloxomethyl-2,3,5-tri-O-benzoyl-[3-D-
ribofuranosyl)pyrrolo[2,3-
d]pyrimidine was prepared from 1-O-acetyl-4-benzoyloxymethyl-2,3,5-tri-O-
benzoyl-D-
ribofuranose and 4-amino-6-bromo-S-cyanopyrrolo[2,3-d]pyrimidine. 4-Amino-6-
bromo-5-
cyano-7-(1,2,3,5-tetra-O-acethyl-5(R)-C-methyl-[i-D-ribofuranosyl)pyrrolo[2,3-
d]pyrimidine
was prepared from 1,2,3,5-tetra-O-acethyl-5(R)-C-methyl-D-ribofuranose and 4-
amino-6-
bromo-5-cyanopyrrolo[2,3-d]pyrimidine. 4-Amino-6-bromo-5-cyano-7-( 1,2,3,5-
tetra-O-
acetyl-5(S)-C-methyl-[i-D-ibofuranosyl)pyrrolo[2,3-d]pyrimidine was prepared
from 1,2,3,5-
tetra-O-acethyl-5(S)-C-methyl-D-ribofuranose and 4-amino-6-bromo-5-
cyanopyrrolo[2,3-
d]pyrimidine.4-Amino-6-bromo-5-cyano-7-(2,3-di-O-acetyl-S-deoxy-(3-D-
ribofuranosyl)pyrrolo[2,3-d]pyrimidine was prepared from 1,2,3-tri-O-acetyl-5-
deoxy-D-
ribofuranose and 4-amino-6-bromo-S-cyanopyrrolo[2,3-d]pyrimidine. 4-Amino-6-
bromo-5-
cyano-7-(2,3,5-tri-O-benzoyl-5(R)-C-vinyl-(3-D-ribofuranosyl)pyrrolo[2,3-
d]pyrimidine was
prepared from 1-O-acetyl-2,3,5-tri-O-benzoyl-5(R)-C-vinyl-(3-D-ribofuranose
and 4-amino-6-
bromo-5-cyanopyrrolo[2,3-d]pyrimidine.
Preparation of ~-amino-5-cyano-7-(2, 3, 5-tri-O-benzoyl-5(R)-C-allyl-~3 D
ribofuranosyl)pyrrolo~2,3-dJpyrimidine (20e)
To a solution of 4-amino-6-bromo-5-cyano-7-(2,3,5-tri-O-benzoyl-5(R)-C-allyl-
(3-D-
ribofuranosyl)pyrrolo[2,3-dJpyrimidine (738 mg 1.0 mmol) in acetic acid (25
mL) was added
zinc dust (1.04 g, 16.0 mmol) in two portions (one hour apart). The reaction
mixture was stirred
at room temperature for 20 h and filtered. The filtrate was evaporated to
dryness and the residue
chromatographed on silica (ethyl acetate-hexanes 1:1 ) to give 450 mg of
titled compound 20e as
a colorless foam. The following compounds were prepared in a similar fashion:
4-Amino-5-
cyano-7-(2,3,5-tri-O-benzoyl-5(R,S)-C-ethynyl-(3-D-ribofuranosyl)pyrrolo[2,3-
d]pyrimidine
(R/S ratio: 1:1) 20b from 4-amino-6-bromo-5-cyano-7-(2,3,5-tri-O-benzoyl-
S(R,S)-C-ethynyl-[3-
D-ribofuranosyl)pyrrolo[2,3-dJpyrimidine. 4-Amino-5-cyano-7-(2,3,5-tri-O-
benzoyl-S(R)-C-
vinyl-(3-D-ribofuranosyl)pyrrolo[2,3-d]pyrimidine 20c from 4-amino-6-bromo-5-
cyano-7-(2,3,5-
tri-O-benzoyl-5(R)-C-vinyl-(3-D-ribofuranosyl)pyrrolo[2,3-d]pyrimidine.
Preparation of 4-amino-5-cyano-7-(2,3,5-tri-O-benzoyl-5(R)-C propyl-~i-D
ribofuranosyl)pyrrolo~2,3-dJpyrimidine (20,fi
CA 02381297 2002-02-06
WO 01/27114 PCT/US00/22674
A suspension of 4-amino-6-bromo-5-cyano-7-(2,3,5-tri-O-benzoyl-5(R)-C-allyl-[i-
D-
ribofuranosyl)pyrrolo[2,3-d]pyrimidine (400 mg, 0.54 mmol) and 10% Pd/C (100
mg, ~50%
water) in dioxane (50 mL) and triethylamine (0.5 mL) was shaken in
hydrogenation apparatus
(H2, 20 psi) for 4 h. The catalyst was filtered and washed (dioxane). The
combined filtrate was
concentrated and the residue chromatographed on silica (ethyl acetate-hexanes
1:1 ) to give 340
mg of the titled compound 20f as a colorless foam. The following compounds
were prepared in a
similar fashion: 4-Amino-5-cyano-7-(2,3,5-tri-O-benzoyl-5(R,S)-C-ethyl-(3-D-
ribofuranosyl)pyrrolo[2,3-d]pyrimidine (R/S ratio: 1:1 ) 20d from 4-amino-6-
bromo-5-cyano-7-
(2,3,5-tri-O-benzoyl-5(R,S)-C-ethynyl-(3-D-ribofuranosyl)pyrrolo[2,3-
d]pyrimidine. 4-Amino-5-
cyano-7-(4-benzoyloxomethyl-2,3,5-tri-O-benzoyl-[3-D-ribofuranosyl)pyrrolo[2,3-
d]pyrimidine
from 4-amino-6-bromo-5-cyano-7-(4-benzoyloxomethyl-2,3,5-tri-O-benzoyl-(3-D-
ribofuranosyl)pyrrolo[2,3-d]pyrimidine. 4-Amino-5-cyano-7-( 1,2,3,5-tetra-O-
acethyl-5(R)-C-
methyl-(3-D-ribofuranosyl)pyrrolo[2,3-cfJpyrimidine 20a from 4-Amino-6-bromo-5-
cyano-7-
( 1,2,3,5-tetra-O-acethyl-5(R)-C-methyl-[3-D-ribofuranosyl)pyrrolo[2.3-
cf]pyrimidine. 4-Amino-
5-cyano-7-(1,2,3,5-tetra-O-acetyl-5(S)-C-methyl-(3-D-ribofuranosyl)pyrrolo[2,3-
d]pyrimidine
20a tcom 4-Amino-6-bromo-5-cyano-7-(1,2,3,5-tetra-O-acethyl-5(S)-C-methyl-[3-D-
ribofuranosyl)pyrrolo[2,3-cf]pyrimidine. 4-Amino-5-cyano-7-( 1,2,3-tri-O-
acethyl-5-deoxy-(3-D-
ribofuranosyl)pyrrolo[2,3-cl]pyrimidine from 4-amino-6-bromo-5-cyano-7-(1,2,3-
tri-O-acethyl-
5-deoxi-[3-D-ribofuranosyl)pyrrolo[2,3-cl]pyrimidine. 4-Amino-5-cyano-7-(2,3-
dideoxy-(3-D-
glicero-pentofuranosyl)pyrrolo[2,3-c~]pyrimidine was prepared from 4-amino-5-
cyano-7-(2,3-
dideoxy-(3-D-pent-2-enofuranosyl)pyrrolo[2,3-d]pyrimidine.
Preparation of -l-amino-3-cyano-7-(5(R)-C-ullyl-~3 D-ribofuranosyl)pyrrolo~2,
3-dJpyrimidine
(23e)
A solution of 4-amino-5-cyano-7-(2,3,5-tri-O-benzoyl-5(R)-C-allyl-(3-D-
ribofuranosyl)-
pyrrolo[2,3-d]pyrimidine (300 mg, 0.454 mmol) in methanol (40 mL) at 0
°C was saturated with
ammonia. The solution stood at room temperature for 2 days. Solvent was
evaporated and the
residue together with NaOAc (anhydrous, 20 mg) was suspended in DMF (20 mL).
The mixture
was stirred under argon at 120 °C for 5 h. Solvent was evaporated. The
residue was adsorbed
onto silica gel and eluted from silica gel column (methanol-ethyl acetate
1:25) to give 145 mg of
the titled compound as a colorless solid.
21
CA 02381297 2002-02-06
WO 01/27114 PCT/US00/22674
Before heating in DMF, the product contained two major compounds 21 and 23.
which
coulde be separated by chromatography on silica gel. Compounds 21 were
prepared through this
procedure. The following compounds were prepared in a similar fashion: 4-Amino-
5-cyano-7-
(5(R)-C-propyl-(3-D-ribofuranosyl)pyrrolo[2,3-d]pyrimidine 23f from 4-amino-5-
cyano-7-(2,3,5-
tri-O-benzoyl-5(R)-C-propyl-(3-D-ribofuranosyl)-pyrrolo[2,3-d]pyrimidine.4-
Amino-5-cyano-7-
(5(R,S)-C-ethynyl-(3-D-ribofuranosyl)pyrrolo[2,3-d]pyrimidine (R-S ratio: 1:1
) 23b from
4-amino-5-cyano-7-(2,3,5-tri-O-benzoyl-5(R,S)-C-ethynyl-(3-D-
ribofuranosyl)pyrrolo[2,3-d]pyr-
imidine. 4-Amino-5-cyano-7-(5(R,S)-C-ethyl-(3-D-ribofuranosyl)pyrrolo[2,3-
d]pyrimidine (R-S
ratio: 1:1 ) 23d from 4-amino-5-cyano-7-(2,3,5-tri-O-benzoyl-5(R,S)-C-ethyl-(3-
D-ribofuranos-
yl)pyrrolo[2,3-dJpyrimidine. 4-Amino-5-cyano-7-(4-hydroxymethyl-(3-D-
ribofuranosyl)pyrrolo-
[2,3-d]pyrimidine 33d from 4-Amino-5-cyano-7-(4-benzoyloxomethyl-2,3,5-tri-O-
benzoyl-(3-D-
ribofuranosyl)pyrrolo[2,3-d]pyrimidine. 4-Amino-5-cyano-7-(5(R)-C-methyl-(3-D-
ribofuranos-
yl)pyrrolo[2,3-dJpyrimidine 23a(5'-R) from 4-amino-5-cyano-7-(1,2,3,5-tetra-O-
acethyl-5(R)-C-
methyl-(3-D-ribofuranosyl)pyrrolo[2,3-d]pyrimidine. 4-Amino-5-cyano-7-(5(S)-C-
methyl-(3-D-
ribofuranosyl)pyrrolo[2,3-d]pyrimidine 23a(5'-S) from 4-amino-5-cyano-7-
(1,2,3,5-tetra-O-
acethyl-5(S)-C-methyl-(3-D-ribofuranosyl)pyrrolo[2,3-cf]pyrimidine. 4-Amino-5-
cyano-7-(5-
deoxy-(3-D-ribofuranosyl)pyrrolo[2,3-d]pyrimidine 10 from 4-amino-5-cyano-7-(
1,2,3-tri-O-
acethyl-5-deoxi-[i-D-ribofuranosyl)pyrrolo[2,3-d]pyrimidine. 4-Amino-5-cyano-7-
(5(R)-C-
vinyl-(3-D-ribofuranosyl)pyrrolo[2,3-d]pyrimidine 23c from 4-amino-5-cyano-7-
(2,3,5-tri-O-
benzoyl-5(R)-C-vinyl-[3-D-ribofuranosyl)pyrrolo-[2,3-u~]pyrimidine.
Preparation of-l-amino-~-cyano-7-(2,3-di-O-methanesulfonyl-~-O-tert-
butyldiphenylsilyl-/~D
ribofuranosyl)pyrrolo~2, 3-dJpyrimidine
To a stirred solution of toyocamicin 43 (5.83 g, 20.0 mmol) in anhydrous
pyridine ( 100
mL) at 0°C was added tert-butylchlorodiphenylsilane (6.2 mL, 24.0
mmol). The resulting
mixture was stirred at room temperature for 18 h and then cooled to
0°C, and methanesulfonyl
chloride (3.4 mL, 44.0 mmol) was added. The resulting mixture was stirred at
room temperature
for 2 h, cooled with ice, quenched by adding water (2 mL), and stirred at room
temperature for
min. The solvent was evaporated. The residue was dissolved in ethyl acetate,
washed with
30 brine three times, dried (NaZS04) and concentrated. Chromatography on
silica (ethyl
acetatehexanes 3:2) gave 8.41 g of the titled compound as a colorless solid.
22
CA 02381297 2002-02-06
WO 01/27114 PCT/US00/22674
Preparation of ~-amino-3-cyano-7-(5-O-tert-butvldiphenylsilyl-2.3-didehydro-
2.3-dideoxy-~3-D
ribofuranosyl)pyrrolo~2. 3-dJpyrimidine
Tellurium powder (200 mesh, 640 mg S.0 mmol) under argon was sealed, mixed
with
lithium triethylborohydrate ( 1.0 M in THF, 11.25 mL, 11.25 mmol). The mixture
was stirred at
room temperature for 6 h and then cooled to S °C, and 4-amino-S-cyano-7-
(2,3-di-O-
methanesulfonyl-5-O-tert-butyldiphenylsilyl-[i-D-ribofuranosyl)pyrrolo[2,3-
d]pyrimidine (1.40
g, 2.09 mmol) in THF ( 12 mL) was added. The resulting mixture was stirred at
room
temperature for 18 h, cooled with ice, quenched by adding water (0°C, 5
mL), and stirred at
room temperature for 30 min. Solvent was evaporated and the residue extracted
with ethyl
acetate. The extracts were concentrated and the residue chromatographed on
silica ( 1 S% ethyl
acetate in hexanes) to give 640 mg of the titled compound as a colorless foam.
Preparation of-l-amino-~-cyano-7-(2,3-didehvdro-2,3-dideoxy-/j D-
riboftrrano.syl)pyrrolo~2,3
dJpyrimidine (49)
1 S To a stirred solution of 4-amino-S-cyano-7-(S-O-tert-butyldiphenylsilyl-
2,3-didehydro-
2,3-dideoxy-(3-D-ribofuranosyl)pyrrolo[2,3-d]pyrimidine (2.5S g, 5.32 mmol) in
anhydrous THF
( 100 mL) at S °C was added terabutylammonium fluoride ( 1.0 M in THF,
6.6 mL). The resulting
mixture was stirred at room temperature for 3 h and concentrated.
Chromatography on silica (6%
methanol in ethyl acetate) gave 1.09 g of the titled compound 49 as a
colorless solid.
Preparation of3-cvano-7-(5(R)-C,-methyl-~-D-ribofuranosyl)pyrrolo(2,3-dJ--l-
pvrimidone (25a)
To a stirred solution of 4-amino-S-cyano-7-(S(R)-C-methyl-(3-D-ribofuranosyl)-
pyrrolo-
[2,3-d]pyrimidine (306 mg, 1.0 mmol) in water (30 mL) and acetic acid (2.0 mL)
at 55 °C was
added in portions sodium nitrite (590 mg, 8.SS mmol). The resulting mixture
was stirred at 70 °
C for 3 h and more sodium nitrite (300 mg, 4.30 mmol) was added. The mixture
was stirred at
same temperature for additional 18 h. Solvent was evaporated and the residue
chromatographed
on silica ( 12 % methanol in methylene chloride) to give 210 mg of the titled
compound
25a(5'-R) as a colorless solid. Similarly, the following compounds were
prepared: 4-Amino-S-
cyano-7-(S(S)-C-methyl-[3-D-ribofuranosyl)pyrrolo[2,3-d]-4-pyrimidone 25a(5'-
S) from
4-amino-S-cyano-7-(S(S)-C-methyl-[3-D-ribofuranosyl)-pyrrolo[2,3-
d]pyrimidine.4-Amino-5-
cyano-7-([i-D-arabinofuranosyl)pyrrolo[2,3-d]-4-pyrimidone 58 from 4-amino-S-
cyano-7-
(S-deoxi-(3-D-arabinofuranosy)pyrrolo[2,3-d]pyrimidine. 4-Amino-S-cyano-7-(5-
deoxy-(3-D-
ribofuranosyl)pyrrolo[2,3-dJ-4-pyrimidone 11 from 4-amino-S-cyano-7-(S-deoxy-
[3-D-
23
CA 02381297 2002-02-06
WO 01/27114 PCT/LTS00/22674
ribofuranosyl)pyrrolo[2,3-d]pyrimidine. 4-Amino-5-cyano-7-(2,3-dideoxy-2,3-
didehydro-(3-D-
glycero-pento-furanosyl) pyrrolo[2,3-d]-4-pyrimidone 50 from 4-amino-5-cyano-7-
(2,3-dideoxy-
(3-D-pent-2-enofuranosyl)pyrrolo-[2,3-d]pyrimidine. 4-Amino-5-cyano-7-(2,3-
dideoxy-(3-D-
glycero-pentofuranosyl)pyrrolo[2,3-d]-4-pyrimidone 65 from 4-amino-5-cyano-7-
(2,3-dideoxy-(3
-D-glycero-pentofuranosyl)pyrrolo[2,3-d]pyrimidine. 4-Amino-5-cyano-7-(2-deoxy-
/3-D-
furanosyl)pyrrolo[2,3-d)-4-pyrimidone 69 from 4-amino-5-cyano-7-(2-deoxy-[i-D-
eritropentofuranosyl)pyrrolo [2,3-d]pyrimidine.
Preparation of 7-(5(R)-C-methyl-~-D-ribofuranosyl)pyrrolo~2, 3-dJ-=1
pyrimidone-3-
carboxamidoxime (24a)
A stirred suspension of 5-cyano-7-(5(R)-C-methyl-[3-D-
ribofuranosyl)pyrrolo[2,3-d]-4-
pyrimidone (240 mg, 0.784 mmol), hydroxylamine hydrochloride (163 mg, 2.352
mmol), and
potassim carbonate ( 162 mg, 1.176 mmol) in ethanol (50 mL) was refluxed under
argon for 18 h.
Preciptate was filtered and washed with warm ethanol. The filtrate was
concentrated and the
residue chromatographed on silica (20 % methanol in methylene chloride) to
give 170 mg of the
titled compound 26a(5'-R) as a colorless solid. Similarly, the following
compounds were
prepared: 4-Amino-5-cyano-7-([i-D-arabinofuranosyl)pyrrolo[2,3-d]-4-pyrimidone-
5-
carboxamidoxime 60 from 4-amino-5-cyano-7-(5-deoxi-[3-D-arabinofuranosy)-
pyrrolo[2,3-d]-4-
pyrimidone.
4-Amino-5-cyano-7-(5-deoxy-[3-D-ribofuranosyl)pyrrolo[2,3-d]-4-pyrimidone-5-
carbox-
amidoxime 13 from 4-amino-5-cyano-7-(5-deoxy-(3-D-ribofuranosyl)pyrrolo[2,3-dJ-
4-
pyrimidone. 4-Amino-5-cyano-7-(2,3-didehydro-2,3-dideoxy-(3-D-
ribofuranosyl)pyrrolo[2,3-d]-
4-pyrimidone-5-carboxamidoxime 51 from 4-amino-5-cyano-7-(2,3-didehydro-2,3-
dideoxy-[i-D-
ribofuranosyl)pyrrolo[2,3-d]-4-pyrimidone. 4-Amino-5-cyano-7-(2-deoxy-(3-D-
ribofuranosyl)pyrrolo[2,3-d)-4-pyrimidone-S-carboxamidoxime from 4-amino-5-
cyano-7-(2-
deoxy-(3-D-ribofuranosyl)pyrrolo[2,3-d]-4-pyrimidone.
Preparation of 7-(S(R)-C-methyl-~i-D-ribofuranosyl)pyrrolo~2,3-dJ-4 pyrimidone-
3-
carboxamidine hydrochloride (27a)
A suspension of 7-(5(R)-C-methyl-[3-D-ribofuranosyl)pyrrolo[2,3-d]-4-
pyrimidone-5-
carboxamidoxime (110 mg, 0.324 mmol), ammonium chloride (20 mg, 0.374 mmol),
and Raney
nickel (50% slurry in water, 200 mg) in water (75 mL) was shaken in a
hydrogenation apparatus
(H2, 50 psi) at room temperature for 18 h. The catalyst was filtered and
washed (warm water).
24
CA 02381297 2002-02-06
WO 01/27114 PCT/US00/22674
The combined filtrate was concentrated and the product was recrystallized from
methanol to give
100 mg of the titled compound 27a(5'-R) as a colorless solid. The following
compounds were
prepared in similar fashion: 4-Amino-5-cyano-7-((3-D-
arabinofuranosyl)pyrrolo[2,3-c~-4-
pyrimidone-5-carboxamidine hydrochloride 63 from 4-amino-5-cyano-7-(5-deoxi-(3-
D-
arabinofuranosy)pyrrolo[2,3-d]-4-pyrimidone-5-carboxamidoxime. 4-Amino-5-cyano-
7-(5-
deoxy-(3-D-ribofuranosyl)pyrrolo[2,3-a']-4-pyrimidone-~-carboxamidine
hydrochloride 15 from
4-amino-5-cyano-7-(5-deoxi-(3-D-ribofuranosyl)pyrrolo[2,3-cl]-4-pyrimidone-~-
carboxamidoxime. 4-Amino-5-cyano-7-(2-deoxy-(3-D-ribofuranosyl)pyrrolo[2,3-d]-
4-
pyrimidone-5-carboxamidine hydrochloride 70 from 4-amino-5-cyano-7-(2-deoxy-(3-
D-
ribofuranosyl)pyrrolo[2,3-d]-4-pyrimidone-5-carboxamidoxime.
Thus, specific embodiments and applications of pyrrolo[2,3-d]pyrimidine
nucleoside
analogs have been disclosed. It should be apparent, however, to those skilled
in the art that
many more modifications besides those already described are possible without
departing from
the inventive concepts herein. The inventive subject matter, therefore, is not
to be restricted
except in the spirit of the appended claims. Moreover, in interpreting both
the specification and
the claims, all terms should be interpreted in the broadest possible manner
consistent with the
context. In particular, the terms "comprises" and "comprising" should be
interpreted as referring
to elements, components, or steps in a non-exclusive manner, indicating that
the referenced
elements, components, or steps may be present, or utilized, or combined with
other elements,
components, or steps that are not expressly referenced.