Note: Descriptions are shown in the official language in which they were submitted.
CA 02381771 2002-02-07
WO 01/23571 PCT/US00/27119
-1-
METHODS AND COMPOSITIONS RELATING TO SODIUM CHANNEL BETAlA SUBUNITS
RELATED APPLICATIONS
This application claims priority to U.S. Provisional Application No.
60/156,837, filed September 30, 1999, which is herein incorporated by
reference in its
entirety.
FIELD OF THE INVENTION
The present invention relates generally to sodium channel proteins and more
particularly to the (3 subunits of voltage gated sodium channel proteins, to
DNA
sequences encoding these subunits, to the polypeptide products of recombinant
expression of these DNA sequences, to peptides whose sequences are based on
amino
acid sequences deduced from these DNA sequences, and to procedures relating to
the
development of drugs that influence function of such proteins.
BACKGROUND OF THE INVENTION
Ion channels from mammalian systems are the subject of intensive scientific
investigation because of the importance and variety of their biochemical
functions.
Ion channels are now understood to be polypeptide or protein structures with
tertiary-
quaternary structures forming interior pores embedded in plasma cell
membranes, that
control the flow of ionic currents. There are many types of ion channels which
share
both similarity of function and amino acid sequence, thus defining familial
relationships between many of these channels. Current work shows there are ion
channel families comprised of voltage-gated sodium, potassium, and calcium
channels, as well as the ligand-gated acetylcholine receptors, glycine
receptors, and
gamma aminobutyric acid receptors.
Voltage-gated sodium channels have been the subject of numerous studies and
much is known about these channels and their component parts. These
transmembrane proteins are responsible for the early sodium permeability
increases
that underlie initial depolarization of the action potential in many excitable
cells such
as muscle, nerve, and cardiac cells.
CA 02381771 2002-02-07
WO 01/23571 PCT/US00/27119
-2-
More specifically, sodium channels are composed of a central pore-forming
a subunit (260 kDa) and two auxiliary subunits, (31 (36 kDa) and X32 (33kDa),
which
do not form the pore yet play critical roles in channel modulation and
expression. The
(31 subunit is of particular interest because a mutation in the (31 gene
(Scnlb) has been
implicated to play a role in fibrillar seizures and generalized epilepsy,
GEFS+
(Wallace et al., 1998).
The primary structure of the (31 subunit deduced from its cDNA sequence
predicts an integral membrane glycoprotein with type I transmembrane topology
as
well as an extracellular immunoglobulin (Ig)-fold (Isom et al., 1994; Isom and
Catterall, 1996). (31 subunits can be classified as members of the V-set of
the Ig-
superfamily which includes many cell adhesion molecules. (31 and type IIA a
subunit
co-expression has been well-characterized in Xenopus oocytes and in mammalian
cells. In oocytes, co-expression of type IIA (Scn2a) or ~I (Scn4a) a subunits
with (31
increases the proportion of sodium channels that function in a fast gating
mode,
accelerates the macroscopic rates of activation and inactivation, shifts the
voltage
dependence of inactivation in the hyperpolarizing direction, and increases the
peak
current amplitude consistent with increases in channel expression (Isom et
al., 1992;
Bennett et al., 1993; Cannon et al., 1993; Schreibmayer et al., 1994; Wallner
et al.,
1993). In Chinese hamster lung (CHL) cells, stable coexpression of [31 with
aIIA
results in increased channel expression levels at the plasma membrane as well
as
moderate hyperpolarizing shifts in the voltage dependence of channel
activation and
inactivation (Isom et al., 1995).
Northern blot analysis has shown that rat brain (31 mRNA is expressed only
after birth in the developing brain (Patton et al., 1994; Sashihara et al.,
1995).
However, previous studies showing the developmental time course of (31
expression
in rat forebrain showed a 26-kDa (31-immunoreactive protein at embryonic day
18
(McHugh-Sutkowski and Catterall 1990). This protein was also expressed in
adult
adrenal gland, heart, skeletal muscle, and PC12 cells. After birth there was a
dramatic
decrease in the level of this protein in brain, and little if any remained by
postnatal
day 14. Other excitable tissues express multiple size forms of immunoreactive
(31-
like subunits. Adult rat heart and skeletal muscle membrane preparations
exhibited
38-, and 41-kDa bands on Western blots in addition to the 26-kDa band. Day 18
WO 01/23571 cA o23amn 2002-02-0~ PCT/US00/27119
-3-
embryonic brain membranes also exhibited a low level of an immunoreactive
peptide
that migrated with an apparent molecular weight greater than 42-kDa. This
protein
was not detected in rat brain after birth. The 41-kDa immunoreactive band was
identified as the adult rat brain isoform, and was later identified as
ClAa.(31(Isom et
al., 1992). Because all of the immunoreactive peptides identified in the
McHugh-
Sutkowski and Catterall study were detected with a polyclonal antibody raised
against
purified (31 subunits, there is no clear indication of what the identity of
each of these
peptides may be.
From the discussion above it is clear that voltage-gated sodium channels are
of
great scientific and economic interest. Further, it would appear that there is
some
protein in addition to the sodium channel a-subunit and the (31 subunit that
is
involved in the regulation and/or formation of functional sodium channels. The
present invention is directed to identification and characterization of such a
protein.
BRIEF SUMMARY OF THE INVENTION
The present invention describes a novel splice variant of the sodium channel
(31 subunit and various methods and compositions for exploiting this finding.
Thus,
the present invention contemplates an isolated nucleic acid comprising a
region, or the
complement thereof, encoding a sodium channel X31 A subunit or an allelic
variant or
mutant thereof. In specific embodiments, the sodium channel (31A subunit
coding
region encodes a primate sodium channel ~i 1 A subunit. In other embodiments,
the
sodium channel /31A is a splice variant of sodium channel (31 subunit. More
particularly, the splice variant results from an intron retention of the
sodium channel
(31 subunit encoding nucleic acid. More particularly, the nucleic acid has a
sequence
of SEQ ID NO:1 (GenBankTM AF182949). In specific embodiments, the nucleic acid
is selected from the group consisting of genomic DNA, complementary DNA and
RNA. In certain embodiments, the nucleic acid is a complementary DNA and
further
comprises a promoter operably linked to said region, or the complement
thereof,
encoding said sodium channel ~31A subunit. More particularly, the nucleic acid
may
comprise a polyadenylation signal operably linked to said region encoding said
sodium channel (31 A subunit encoding region. In additional embodiments, the
nucleic
WO 01/23571 cA o23amn 2002-02-0~ pCT/L1S00/27119
-4-
acid may also comprise an origin of replication. The nucleic acid may be a
viral
vector selected from the group consisting of retrovirus, adenovirus,
herpesvirus,
vaccinia virus and adeno-associated virus. More specifically, the nucleic acid
is
packaged into a viral particle or the nucleic acid may be packaged into a
liposome or a
dendrimer formulation.
In other aspects, the present invention contemplates an isolated
oligonucleotide of between about 10 and about 50 consecutive bases of a
nucleic acid,
or complementary thereto, encoding a sodium channel (31A subunit. In
particular
embodiments, the oligonucleotide may be 17, 18, 19, 20, 21, 22, 23, 24, 25,
26, 27,
28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46,
47, 48 49, 50,
55, 60 65 consecutive bases in length or longer. In specific aspects these
consecutive
bases may be derived from the coding region of SEQ ID NO: l .
Also contemplated herein is a sodium channel (31A subunit-encoding nucleic
acid operably linked to a first promoter. Although not being limited to the
promoters
indicated herein, preferred promoters may be selected from the group
consisting of
CMV IE, SV40 IE, RSV LTR, (3-actin, tetracycline regulatable, ecdysone
regulatable,
tyrosinase, retrovirus LTR, PGK HIV-1 promoter, and HIV-2 promoter. In
addition,
the promoters may be cell specific or tissue specific for the particular cell
type being
employed to express the nucleic acid. The expression construct may be a
lentiviral,
adenoviral, adeno-associated viral, vaccinia viral, herpes viral or retroviral
expression
construct. More particularly, the (31A encoding nucleic acid sequence is as
set forth
in SEQ ID NO:1. In other embodiments, the (31A encoding expression construct
encodes a protein of SEQ ID N0:2.
Other aspects of the present invention provide an isolated polypeptide
encoding a sodium channel (31A subunit. More particularly, the sodium channel
(31A
subunit has the amino acid sequence as set forth in SEQ ID N0:2. In other
embodiments, there is provided an isolated peptide having between about 10 and
about 50 consecutive residues of a sodium channel ~31A subunit. In specific
embodiments, the peptide is conjugated to a carrier molecule. The carrier
molecule
may be any molecule commonly used to generate antibodies. For example the
carrier
molecule may be KLH and BSA. In specific embodiments, the sodium channel ~31A
subunit has the amino acid sequence as set forth in SEQ ID N0:2.
WO 01/23571 cA o23ai~~i 2002-02-0~ PCT/US00/27119
-5-
Other aspects of the invention provide a monoclonal antibody that binds
immunologically to a sodium channel (31A subunit. In particularly preferred
embodiments, the antibody does not bind immunologically to other sodium
channel
subunit polypeptides. In specific embodiments, the antibody further comprises
a
detectable label. The label may be selected from the group consisting of a
fluorescent
label, a chemiluminescent label, a radiolabel and an enzyme. Also provided by
the
present invention is a hybridoma cell that produces a monoclonal antibody that
binds
immunologically to a sodium channel (31A subunit. In preferred embodiments,
the
antibody does not bind immunologically to other sodium channel subunit
polypeptides. In addition the present invention provides a polyclonal
antisera,
antibodies of which bind immunologically to a sodium channel (31A subunit. The
antisera may be generated from any animal normally used by those of skill in
the art
to produce antisera. For example the antisera is derived from an animal
selected from
the group consisting human, mouse, horse, dog, goat, rabbit, rat, and sheep.
Additional embodiments of the present invention provide a method of
screening for a modulator of sodium channel activity comprising providing a
cell co-
expressing a sodium channel (31A subunit polypeptide with a sodium channel
a subunit; contacting said cell with a candidate modulator substance; and
determining
the effect of said candidate substance on the sodium channel function in said
cell.
In specific embodiments, the determining comprises comparing the sodium
current density of the cell in the presence of said candidate substance with
sodium
current density of said cell in the absence of said candidate substance
wherein an
alteration of said density is indicative of said candidate substance being a
modulator.
More particular embodiments describe the candidate substance as being selected
from
a small molecule library. In preferred embodiments, the candidate substance
alters
the expression of said sodium (31 A subunit. The method is such that the cell
may be
contacted in vitro or in vivo.
Other embodiments describe a modulator of sodium channel activity identified
by a method comprising providing a cell co-expressing a sodium channel (31 A
subunit
polypeptide with a sodium channel a subunit; contacting said cell with a
candidate
modulator substance; and determining the effect of said candidate substance on
the
sodium channel function in said cell. In preferred embodiments, the modulator
is an
CA 02381771 2002-02-07
WO 01/23571 PCT/US00/27119
-6-
inhibitor of sodium channel (31A subunit activity. In other embodiments, the
modulator is an activator of sodium channel (31 A subunit activity. In still
additional
embodiments, the modulator is an inhibitor of sodium channel activity. In
specific
embodiments, the candidate substance modulates the expression of sodium
channel
(31 A subunit. In certain other preferred embodiments the modulator is an
activator of
sodium channel activity. The modulator may be a naturally occurnng modulator
of
sodium channel (31A activity or is a modulator synthesized from rational drug
design.
Other aspects of the present invention contemplate a method for transforming
a cell comprising contacting the cell with a nucleic acid expression construct
(i)
encoding a sodium channel (31 A subunit and (ii) a promoter active in said
cell,
wherein said promoter is operably linked to the region encoding said sodium
channel
(31A subunit, under conditions permitting the uptake of said expression
construct by
said cell. In particular embodiments, the cell is a brain cell, lung cell,
muscle cell,
adrenal cell, fibroblast cell, or a cardiac cell. Of course these are merely
exemplary
and those of skill in the art may use any cell routinely employed in such
transformations. In preferred embodiments, the expression construct is
encapsulated
in a liposome or a dendrimer. In other embodiments, the expression construct
is a
viral vector selected from the group consisting of retrovirus, adenovirus,
adeno-
associated virus, vaccinia virus and herpesvirus. In particularly preferred
aspects the
nucleic acid is encapsulated in a viral particle.
Also provided is a method for decreasing the number of fibrillar seizures in
an
individual comprising administering to said individual a modulator of a sodium
channel (31A subunit in an amount effective to change the sodium channel
activity in
said individual. In preferred embodiments, the modulator is identified
according to a
method comprising providing a cell expressing a sodium channel (31A subunit
polypeptide; contacting said cell with a candidate substance; and determining
the
effect of said candidate substance on the activity of said sodium channel (31A
subunit.
In specific embodiments, the modulator is an anti-epileptic agent. In other
embodiments, the modulator is an inhibitor of sodium channel (31 A subunit. In
more
particular aspects of the invention the modulator affect the level of
expression of the
sodium channel (31A subunit. More specifically, the modulator decreases the
expression of sodium channel (31A subunit.
WO 01/23571 cA o23amn 2002-02-0~ pCT/US00/27119
Also provided is a method for treating fibrillar seizures in a subject
comprising
altering the activity or level of sodium channel (31A subunits of a cell in
said subject.
In preferred embodiments, the altering comprises contacting said cell with a
sodium
channel (31A subunit under conditions permitting the uptake of said sodium
channel
(31 A subunit by said cell. In specific embodiments, the contacting comprises
contacting said cell with a nucleic acid expression construct (i) encoding a
sodium
channel (31A subunit and (ii) a promoter active in said cell, wherein said
promoter is
operably linked to the region encoding said sodium channel X31 A subunit,
under
conditions permitting the uptake of said expression construct by said cell. In
preferred embodiments, the subject is a human. In other embodiments, it would
be
useful to decrease the expression of a sodium channel (31 A subunit by for
example
contacting said cell with a nucleic acid expression construct encoding sodium
channel
(31A subunit positioned antisense to a promoter active in said cell, wherein
said
promoter is operably linked to the region encoding said sodium channel ~ilA
subunit,
under conditions permitting the uptake of said expression construct by said
cell.
Another embodiment contemplates a method for decreasing neuropathic pain
in an individual comprising administering to said individual a modulator of a
sodium
channel (31A subunit in an amount effective to change the sodium channel
activity in
said individual. In specific embodiments, the modulator decreases the
expression of
sodium channel (31 A subunit of the cells of said individual. In other
embodiments,
there is provided a method for treating neuropathic pain in a subject
comprising
altering the activity or level of sodium channel ~ilA subunits of a cell in
said subject.
Similarly, sodium channel (31A is involved in certain cardiac functions
relating to the
sodium channel. As such it is contemplated that methods similar to those set
forth for
neuropathic pain also could be employed to ameliorate disorders in cardiac
function,
e.g., cardiac arrhythmia and the like. In preferred embodiments, it is
contemplated
that altering the activity or level of sodium channel ~31A subunits of a cell
in a subject
would be useful in treating cardiac arrhythmia.
Other objects, features and advantages of the present invention will become
apparent from the following detailed description. It should be understood,
however,
that the detailed description and the specific examples, while indicating
preferred
embodiments of the invention, are given by way of illustration only, since
various
WO 01/23571 cA o23amn 2002-02-0~ PCT/US00/27119
_g_
changes and modifications within the spirit and scope of the invention will
become
apparent to those skilled in the art.
BRIEF DESCRIPTION OF SEVERAL VIEWS OF THE DRAWINGS
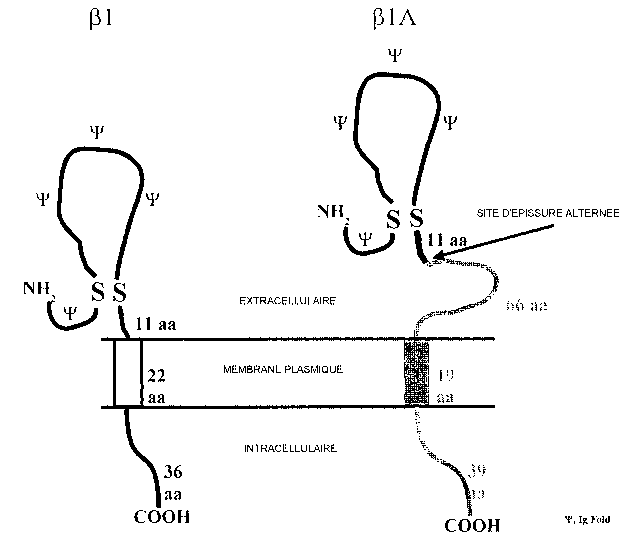
FIG. lA-FIG. 1D. Sequence analysis of (31A FIG. 1A. Deduced amino acid
sequence of (31A. Solid box: signal peptide that becomes cleaved in the mature
protein. Arrow: site of alternate splice resulting in retention of intron 3.
Asterisks: N-
linked glycosylation sites. Dotted box: transmembrane segment predicted from
Kyte-
Dolittle hydropathy profile. Solid underline: amino acid sequence used to
synthesize
(31A-MAP. FIG. 1B. Sequence analysis of (31A - Comparison of ~31A and (31
amino
acid sequences. Upper sequence: ~i 1 A. Lower sequence: (31. FIG. 1 C.
Sequence
analysis of (31A - Putative membrane topologies of (31A and (31. (31A is
predicted to
encode a type I membrane protein with similar topolgy to /31. The disulfide-
linked
immunoglobulin fold (from the indicated carboxy-terminal disulfide bond to the
amino terminus; traced by "'F") is common to both (31 and [31A. Following the
alternate splice site (arrow), (31A contains a novel 66-amino acid
juxtamembrane
region, 19-amino acid transmembrane segment, and 39-amino acid intracellular
domain. FIG. 1D. Sequence analysis of ~31A - Sequence homology to known
proteins. Results of BLAST-P search of the Swissprot database using the novel,
carboxy-terminal domain of (31A beginning at residue 130 as the query
sequence.
FIG. 2A-FIG. 2D. Effects of ~31A on the functional properties of whole
cell sodium currents. FIG. 2A. Voltage-dependent sodium currents recorded in a
SNaIIA cell (top traces) and a SNaIIA(31A cell (bottom traces). Currents were
elicited by depolarizations to -40, -30, -20, -10, 0 and +10 mV, from a
prepulse
potential of -100 mV. FIG. 2B. Mean activation (filled symbols) and
inactivation
(open symbols) curves for cell lines SNaIIA (circles), SNaIIA~ilA-7 (squares),
SNaIIA(31A-8 (triangles) and SNaIIA[31A-16 (inverted triangles). For each
cell,
activation and inactivation were analyzed as described in Methods. The symbols
show means of the activation and inactivation data for the different cell
lines. In this
and subsequent electrophysiology figures, error bars indicate standard errors
of the
means (SEM). The smooth lines were generated with the Boltzman equation (see
WO 01/23571 cA o23amn 2002-02-0~ PCT/US00/27119
-9-
Methods), using mean values of V 1,2 and k determined for each cell line, from
fits of
individual experiments. Mean values for V1,2 and k and the number of
experiments
for each cell line are as follows: activation: SNaIIA, V lie = -11.0 ~ 0.96, k
= -8.3 ~
0.28, n = 15; SNaIIA(31A-7, -8.1 ~ 2.13, -7.9 ~ 0.48, 6; SNaIIA~ilA-8, -11.6 ~
1.21,
7.4 ~ 0.31, 11; SNaIIA(31A-16, -17.0 ~ 1.41, -7.18 ~ 0.39, 1 l; inactivation:
SNaIIA,
VIiZ = -48.3 ~ 0.77, k= 6.5 ~ 0.20, n = 13; SNaIIA(31A-7, -45.2 ~ 1.25, 6.8 ~
0.22, 6,
SNaIIA(31A-8, -45.9 ~ 0.63, 6.5 ~ 0.21, n = 11; SNaIIA(31A-16, -47.2 ~ 0.50,
6.5 ~
0.15, 10. FIG. 2C. Inactivation time constants ('L;nactivation) determined
from fits of
current decay for SNaIIA (O), SNaIIA~ilA-7 (D), SNaIIA(31A-8 (O) and
SNaIIA(31A-16 (~), plotted as a function of test potential. FIG. 2D. Mean Vl,z
values for activation (filled symbols) and inactivation (open symbols) for
cell lines
SNaIIA, SNaIIA(31 A-7, SNaIIA(31 A-8, SNaIIA(31 A-16.
FIG. 3A and FIG. 3B. Effect of (31A on the level of expression of
functional sodium channels. FIG. 3A. Current densities for SNaIIA and
SNaIIA(31 A cell lines. Currents were elicited by depolarization to +10 mV
from a
prepulse potential of -100 mV. Peak current amplitude was divided by cell
capacitance to give current density. Cell capacitance was determined by
integrating
the area under transients elicited by 3 mV voltage steps applied before series
resistance compensation and capacitive transient cancellation. FIG. 3B.
Amplitude-
frequency histogram for SNaIIA (black bars) and SNaIIA(31A (white bars; data
for all
three SNaIIA(31A cell lines were combined). Currents were evoked by
depolarization
to +10 mV. The bars indicate the number of cells with peak currents that fell
within
different amplitude ranges.
DETAILED DESCRIPTION OF THE INVENTION
The present invention describes the isolation, molecular cloning and
functional
characterization of a new protein involved in voltage-gated sodium channel
formation. The data presented herein show that (31A is a splice variant of the
~i 1 gene.
(31A contains an identical amino terminal and extracellular Ig-fold region as
the (31
protein; however, this region is followed by a significantly different
extracellular
W~ 01/23$71 CA 02381771 2002-02-07
PCT/US00/27119
-10-
juxtamembrane domain, predicted transmembrane region, and predicted
intracellular
COOH-terminal domain.
The inventors show that the (31A mRNA is expressed early in embryonic brain
development and then disappears after birth. Western blot analysis of membrane
preparations using an antibody to a unique, extracellular region of (31A not
found in
~i 1 showed that (31A protein is expressed in adult rat heart, skeletal
muscle, and
adrenal gland but was not detected in adult brain or spinal cord.
Immunocytochemcial analysis of (31 A expression in adult rat tissues revealed
high expression in heart and dorsal root ganglion (DRG) and selective
expression in
some areas of the brain and spinal cord. (31A functions to increase channel
expression
at the plasma membrane when coexpressed with aIIA subunits in CHL fibroblasts.
Unlike (31, however, mean steady state inactivation curves for a(31A-
expressing cell
lines were shifted to more positive potentials than the mean inactivation
curves for
cells expressing a alone. Previous studies showed that coexpression of a and
X31 subunits in CHL cells shifted the voltage-dependence of inactivation to
more
negative potentials compared to a alone (Isom et al., 1995b). Therefore, the
novel,
carboxy-terminal domains of (31A likely is important for electrophysiological
function. It has been shown previously that the extracellular domain of (31 is
essential
for expression and function of the a(31 complex in Xenopus oocytes (Chen and
Cannon, 1995; McCormick et al., 1998). The inventors suggest that the
extracellular
Ig fold, common to (31 and (31A, is essential for the observed increases in
channel
expression levels. Thus, this report introduces a novel splice variant of (3I,
(31A, and
adds to the understanding of (31 structure/function relationships in terms of
channel
expression/stabilization and electrophysiology.
The present invention exploits these findings to provide a variety of novel
methods and compositions related to voltage-gated sodium channels. The novel
protein and DNA sequences described herein and variants thereof may be used in
the
production of antibodies, expression vectors, in methods for producing
recombinant
proteins, methods of producing recombinant cells, screening assays and
identification
of modulators of sodium channels using such assays. In addition, the sodium
channel
plays a central role in a number of biologically significant areas. For
example, the
present inventors contemplate that the sodium channel is involved in
neuropathic pain
WO 01/23571 cA o23amn 2002-02-0~ PCT/US00/27119
-11-
and as such modulators of the sodium channel (31A subunit may be identified
that
modulate neuropathic pain. Similarly these channels are important in certain
aspects
of cardiac function, and the present invention may be employed in various ways
of
altering, adjusting or otherwise treating disorders stemming form changes in
cardiac
S function. Another interesting observation is that the sodium (31 A subunit
may be
involved in altering the amount, degree, or severity of fibrillar seizures in
an
individual. The present invention describes methods and compositions that may
be
used in modifying, altering or otherwise managing these disorders as well as
providing details of the use of the sodium channel (31 A subunit related
compositions
in various research settings.
1. Voltage-Gated Sodium Channels
In order to provide a more detailed understanding of the present invention,
the
subject of voltage gated sodium channels needs some introduction. As such, the
present section provides a background discussion of sodium channels and their
function.
The localized cell surface density and the functional properties of sodium
channels are a key determinant of the threshold for action potential
generation and the
frequency of firing of neurons. Myelination of both central and peripheral
axons,
permits rapid saltatory conduction of action potentials through a cooperation
between
the axon itself and specialized glial cells which envelope the axon with
multiple
insulating layers. These cells are the oligodendrocytes in the central nervous
system
(CNS), and Schwann cells in the peripheral nervous system (PNS). Interruptions
in
the myelin sheath, known as nodes of Ranvier, contain locally high
concentrations of
voltage-gated sodium channels, the membrane proteins which are responsible for
initiation and propagation of the action potential.
A complex interplay between a number of extracellular and intracellular
signaling events is required for proper generation and propagation of an
action
potential (Salzer1997; Vabnick and Shrager 1998). It appears that there is an
unidentified factor secreted by oligodendrocytes that is necessary for the
induction of
clustering of CNS sodium channels in vitro and in vivo (Kaplan et al., 1997).
Similarly, Schwann cell contact has been shown to induce sodium channel
clustering
WO 01/23571 cA o23amn 2002-02-0~ pCT/US00/27119
-12-
in PNS axons (Dugandzija-Novakovic et al., 1995; Joe and Angelides, 1992; Joe
and
Angelides, 1993). Sodium channels colocalize with the cytoskeletal proteins
ankyrin
G spectrin at the nodes of Ranvier and there is some evidence to suggest that
sodium
channels bind ankyrin directly (Srinivasan et al., 1988).
Brain and muscle sodium channel a subunit isoforms interact with multiple
members of the syntrophin family, cytoplasmic peripheral membrane proteins
which
contain a PDZ domain (Gee et al., 1998). Cell adhesion molecules present in
the node
of Ranvier, such as neurofascin and NrCAM, have also been proposed to
participate
in linking axonal proteins with glial processes (Davis et al., 1996). Finally,
sodium
channel a subunit isoforms are differentially localized in neurons. For
example, aIIA
is located axonally and aI is located somatodendritically in hippocampal
slices
(Westenbroek et al., 1989). Thus, information regulating sodium channel
localization
may be encoded in the amino acid sequence of various subunit isoforms.
Sodium channels isolated from brain are heterotrimeric structures composed
of a central, pore-containing a-subunit and two auxiliary subunits, (31 and
(32, which
do not form the pore but play critical roles in channel gating, voltage-
dependence of
activation and inactivation, expression levels, and localization. At least 4
genes
encoding sodium channel a subunits are expressed in the CNS: aI (Noda et al.,
1986x; Noda et al., 1986b), aII/IIA (Auld et al., 1988; Noda et al., 1986x;
Noda et al.,
1986b), aIII (Kayano et al., 1988), and a6 or ScnBa (Burgess et al., 1995;
Schaller et
al., 1995). RNA encoding a subunits is sufficient to direct the synthesis of
functional
sodium channels in Xenopus oocytes (Goldin et al., 1986; Noda et al., 1986b),
but co-
injection of low molecular weight brain mRNA from brain or skeletal muscle is
required for rapid inactivation (Auld et al., 1988; Joho et al., 1990, Krafte
et al., 1988;
Ukomadu et al., 1992; Zhou et al., 1991). These results suggested a possible
role for
the low molecular weight ~3 subunits in sodium channel function. Cloning and
functional analysis of the (31 and (32 subunits of sodium channels have shown
that the
(31 subunit (Bennett et al., 1993; Cannon et al., 1993; Isom et al., 1992,
Makita et al.,
1994x, Makita et al., 1994b; Schreibmayer et al., 1994, Tong et al., 1993;
Wallner et
al., 1993) and the (32 subunit (Isom et al., 1995b) do indeed strongly
modulate sodium
channel function.
CA 02381771 2002-02-07
WO 01/23571 PCT/US00/27119
-13-
Glial-derived extracellular matrix molecules, such as tenascin-C (TN-C) and
tenascin-R (TN-R), play important roles in cell interactions in the developing
nervous
system, such as neuronal migration, neuritogenesis, and neuronal regeneration
(Bartsch, 1996; Chiquet-Ehrismann et al., 1994; Erickson, 1993; Schachner et
al.,
1994). TN-R is expressed predominantly by oligodendrocytes during the onset
and
early phases of myelin formation, and remains expressed by some
oligodendrocytes in
the adult (Bartsch et al., 1993; Fuss et al., 1991; Fuss et al., 1993; Pesheva
et al.,
1989; Wintergerst et al., 1993) as well as some neurons and interneurons in
the spinal
cord, retina, cerebellum, and hippocampus (Bartsch et al., 1993, Fuss et al.,
1993;
Wintergerst et al., 1993). Interestingly, TN-R co-localizes with other glial-
derived
molecules, such as myelin-associated glycoprotein (MAG) and a phosphacan-
related
molecule, at high density in myelinated CNS nerves (Xiao et al., 1997). TN-R
is a
multi-functional molecule that promotes neurite outgrowth when presented as a
uniform substrate, inhibits growth cone advance when offered as a sharp
substrate
boundary, and induces axonal defasciculation in vitro, resulting from
interaction with
one of its neuronal receptors, F3/contactin (Lochter and Schachner, 1993;
Lochter et
al., 1994; Pesheva et al., 1993, Taylor et al., 1993).
Cell adhesion molecules of the Ig superfamily interact homophilically and
heterophilically to transduce signals between adjacent cells or adjacent axons
where
they participate in axonal fasciculation. Cell adhesion molecules of the L1
family
have been studied extensively. For example, transfection of neuroglian, a
member of
the L1 family, into Drosophila S2 cells results in homophilic binding and
cellular
aggregation (Hortsch et al., 1998; Malhotra et al., 1998). It appears that the
/3 subunits behave as classical CAMS in addition to playing roles as channel
modulators. (3 subunits participate in modulation of the voltage-dependence of
channel activation and inactivation, channel gating mode, as well as channel
expression levels at the plasma membrane.
The ~3 subunits are members of the Ig superfamily and play roles in cellular
adhesion and repulsion. Thus, sodium channel ~3 subunits are multifunctional
proteins
with possible roles independent of the ion channel complex. The inventors
propose
that the sodium channel (3 subunits function as true CAMs, acting as bridges
between
the extra- and intracellular neuronal environments. The present invention
provides
WO 01/23571 cA o23ai~~i 2002-02-0~ pCT/US00/27119
-14-
detailed characterization, cloning and sequencing of an important and novel
sodium
channel (3 subunit.
2. ~ilA Protein
According to the present invention, there has been identified a splice variant
of
S the sodium channel (31 subunit, that results from an intron retention in the
(31 subunit
encoding gene. This splice variant is referred to herein as (31 A. Co-
expression of
(31 A with an a-subunit results in an increase in sodium current density
compared to
cells expressing a alone. This increase in current density reflected two
effects of
(31 A: 1 ) an increase in the proportion of cells expressing detectable sodium
currents,
and 2) an increase in the level of functional sodium channels in expressing
cells.
In addition to the entire (31A molecule, the present invention also relates to
fragments of the polypeptide that may or may not retain the a-subunit
regulatory (or
other) activity of [31 A. Fragments, including the N-terminus of the molecule
may be
generated by genetic engineering of translation stop sites within the coding
region
(discussed below). Alternatively, treatment of the (31A molecule with
proteolytic
enzymes, known as proteases, can produce a variety of N-terminal, C-terminal
and
internal fragments. Examples of fragments may include contiguous residues of
the
(31 A sequence given in FIG. 1 B of 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16,
17, 18, 19, 20,
21, 22, 23, 24, 25, 30, 35, 40, 45, 50, 55, 60, 65, 75, 80, 85, 90, 95, 100,
200, or more
amino acids in length. These fragments may be purified according to known
methods,
such as precipitation (e.g., ammonium sulfate), HPLC, ion exchange
chromatography,
affinity chromatography (including immunoaffinity chromatography) or various
size
separations (sedimentation, gel electrophoresis, gel filtration).
A. Structural Features
The gene for /31A encodes a 272 amino acid polypeptide. The predicted
molecular weight of this molecule is SOkDa, with a resulting pI of 6.42. Thus,
at a
minimum, this molecule may be used as a standard in assays where molecule
weight
and pI are being examined.
(31A, a splice variant of (31, is the result of the retention of intron 3
containing
an in frame stop codon. This alternate splicing event produces a novel carboxy-
WO 01/23571 cA o23amn 2002-02-0~ pCT/US00127119
-15-
terminus that includes an extracellular region, a transmembrane segment, and a
short
intracellular domain. Western blot analysis showed (31A immunoreactive
peptides of
approximately 50 kDa expressed in heart, skeletal muscle, and adrenal gland,
but not
in adult brain or spinal cord.
A number of cases of intron retention have been reported in the literature,
including alternative splicing of the genes encoding leukocyte-common antigen-
related protein tyrosine phosphatase (LAR), CD44, effector cell protease
receptor-1
(EPR-1), the microtubule-associated protein tau, thyrotropin-releasing hormone
receptor, and bovine growth hormone (Tabiti et al., 1996; Higashikawa et al.,
1996;
Zhang and Longo, 1995; Altieri, 1994; Sadot et al., 1994; de la Pene, et al.,
1992;
Hampson et al., 1989). In many cases, the retained intron contains an
alternate, in-
frame termination codon as well as a polyadenylation signal.
Alternative splicing which results in retention of the intron in the primary
transcript thus results in an isoform of the protein containing a novel
carboxy
terminus. Interestingly, this is not the first report of intron retention in
the (31 gene.
Waxman and coworkers have previously reported that intron 5 of (31 can be
retained,
creating a novel isoform which is expressed in rat brain, optic nerve, sciatic
nerve,
and skeletal muscle. This isoform contains a 86 nucleotide insert encoded by
intron 5
in the 3' untranslated region (Oh and Waxman, 1994; Dib-Hajj and Waxman,
1995).
While the intron retention reported previously does not alter the (31 coding
sequence,
the present data describe a very significant coding sequence change resulting
in a
novel carboxy terminus.
Sodium channel (3 subunits are members of the immunoglobulin (Ig)
superfamily and contain extracellular cell adhesion molecule (CAM) domains.
Because CAMs interact with extracellular protein ligands as well as with other
CAMS, the (3 subunits likely act in a similar fashion. The inventors suggest
that the
transmembrane and/or the intracellular domains of the sodium channel (3
subunits are
responsible for transducing a signal from the extracellular environment to
cytoskeletal
or signaling molecules inside the cell. Tenascin-R (TN-R), an extracellular
matrix
protein that is secreted by oligodendrocytes during formation of CNS nodes of
Ranvier, is a functional modulator of sodium channel ~3 and a subunits of TN-R
binding and can be used to study the effects of varying the subunit sequence.
WO 01/23571 cA o23amn 2002-02-0~ PCT/US00/27119
-16-
Given the disclosure of the present invention, it is now possible to construct
and express truncation mutations that eliminate the intracellular COOH-
terminal
domains of the [3 subunit, introduction of a COOH-terminal signal peptide
which
eliminates the transmembrane domain and adds a glycophosphatidyinositol (GPI)
lipid anchor to the (3 subunit, and point mutations of amino acids in (3
subunits that are
predicted to be located in the extracellular Ig fold. These mutations then are
tested in
the cell migration assay for modulation by TN-R.
The Ig loop region of the (31 A and the other (3 subunits is responsible for
mediating homophilic binding and the intracellular COOH-terminal regions of
these
molecules are then responsible for transducing a signal resulting in ankyrin
recruitment to the plasma membrane. Thus, a truncated (3 subunit lacking the
COOH-
terminal intracellular domain is predicted to display homophilic binding but
be
incapable of ankyrin recruitment. Given the disclosure presented herein it is
now
possible to construct mutants to test this Drosophila S2 cells. Further,
recombinant
fragments of TN-R can be used to inhibit homophilic binding between (3
subunits
expressed in S2 cells. This will allow determination of whether these two
binding
events are mediated by the same or different extracellular amino acid motifs
in the
(3 subunits.
Some CAMS belonging to the Ig superfamily interact heterophilically with
other CAMS to transduce signals across the plasma membrane to the cytoskeleton
and
other signaling molecules. This interaction can be intercellular or traps,
such that
different CAMs on adjacent cells interact, or it can be intracellular or cis,
such that
adjacent molecules on the same plasma membrane interact to produce a signal.
Neurofascin, ankyrin G , and sodium channels have been shown to be colocalized
at
CNS nodes of Ranvier. Thus, neurofascin and (3 subunits may interact via cis
heterophilic binding.
Using the Drosophila S2 cell model system it will be possible to test for
aggregation and ankyrin recruitment by mixing (31-, (32-, and/or neurofascin-
transfected cells, after one of the cell lines has been labeled with DiI. By
performing
coimmunoprecipitations on cells that are cotransfected with a (3 subunit plus
neurofascin it will be possible to test for cis interactions. A mutation in
the Ig loop
region of (31 has recently been implicated in fibrillar seizures and
generalized epilepsy
CA 02381771 2002-02-07
WO 01/23571 PCT/US00/27119
-17-
(Wallace et al., 1998). Mutations in L1-CAM are linked to hydrocephalus, motor
neuron defects, agenesis of the corpus callosum, and the cerebrospinal tract.
Mutations in myelin P o , a CAM with homology to (31, may be involved in
Charcot-
Marie-Tooth disease. Alterations in sodium channel clustering and localization
are
inherent in demyelinating disease such as multiple sclerosis. Given the
present
disclosure of a novel [i subunit of the sodium channel containing a COON
terminus
that is different from those described for /31 and (32, there will be an
increase the basic
understanding of the molecular and cellular biology of sodium channel (3
subunits,
especially their roles as CAMS, and lead to the future development of
therapeutic
agents for these and related diseases.
. B. Functional Aspects
The most striking functional consequence of aIIA and (31A coexpression was
a significant increase in sodium current density compared to cells expressing
aIIA
subunits alone. This increase in current density reflected two distinct
effects of (31A:
1 ) an increase in the proportion of cells expressing detectable sodium
currents, and 2)
an increase in the level of functional sodium channels in expressing cells.
Increases
in sodium channel expression with /31A are similar to previous results
obtained with
the adult (31 isoform in both mammalian cells (Isom et al., 1995) and Xenopus
oocytes
(Isom et al., 1992). These observations are consistent with the hypothesis
that [i l and
(31A subunits facilitate the expression of sodium channels, and/or stabilize
the
channels in the plasma membrane and that the molecular basis for this function
resides in the extracellular cell adhesion molecule domain common to the two
isoforms. The results of the [3H]-STX binding experiments further support this
hypothesis. It is proposed that interaction of the extracellular cell adhesion
molecule
domain (Ig fold) common to (31 and (31 A with a may be responsible for the
observed
effects on channel expression levels. Consistent with this interpretation, it
has been
shown that the extracellular domain of (31 is essential for modulation of both
brain
and skeletal muscle a subunits, whereas the intracellular carboxy-terminal
domain is
not. Truncated (31 subunits lacking the intracellular carboxy-terminus are
fully
functional in terms of kinetic modulation of brain and skeletal muscle a
subunits
expressed in Xenopus oocytes (Chen and Cannon et al., 1995; McCormick et al.,
WO 01/23571 cA o23amn 2002-02-0~ PCT/US00/27119
-18-
1998). The (31 extracellular domain, together with the residues proximal to
the
transmembrane domain, constructed in a [31/(32 subunit chimera were found to
be
sufficient to modify skeletal muscle sodium channel a subunits expressed in
oocytes
(Chen and Cannon et al., 1995; McCormick et al., 1998). Finally, residues
predicted
S to be in the Ig fold of (31 interact with type IIA a subunits (McCormick et
al., 1998).
Thus, the extracellular cell adhesion domain common to (31 and (31 A appears
to be
required for function.
Sodium currents in (31 A-expressing cell lines also exhibited subtle
functional
differences compared to the parent SNaIIA cell line. For example, inactivation
curves
in SNaIIA[31A cell lines were shifted to slightly more positive potentials
than
inactivation curves for SNaIIA cells. This finding differs from previous
results
showing that coexpression of (31 and aIIA in CHL cells shifts inactivation to
potentials approximately 10 mV more negative than for cells expressing a
alone.
However, there is a significant difference in the (31 and (31A proteins in
that the latter
contains a novel 55 residue long juxtamembrane portion which may explain this
difference in steady state inactivation.
In addition to opposite effects on steady state inactivation, the voltage-
dependence of activation and the rate of channel inactivation were also
different in
one of the three (31A-expressing cell lines, compared to the parent SNaIIA
cell line.
Thus, whole cell electrophysiological data suggest that (31 A subunits may
subtly
modulate various aspects of sodium channel function.
TTX-sensitive sodium channel a subunits expressed in brain (type I/Scala,
Smith and Goldin, 1998; type II/Scn2a, Isom et al., 1992; Isom et al., 1995;
type
III/Scn3a, Patton et al., 1994; type VI/PN4/ScnBa, Smith et al., 1998) and
skeletal
muscle (Skm 1/Scn4a Wallner et al., 1993) have been shown to be modulated by
co-
expression of (31 subunits in heterologous systems. In contrast, TTX-resistant
sodium
channel a subunits expressed in cardiac myocytes (Skm 2/H1/ScnSa; Qu et al.,
1995)
and peripheral nerve (PN3/SNS/ScnlOa; Sangemeswaran et al., 1996; Klugbauer et
al., 1995; Akopian et al., 1996) are much less sensitive or insensitive to
modulation
by (31 when co-expressed either in Xenopus oocytes or mammalian cells. It is
proposed that tissues in which TTX-resistant channels are expressed may also
express
novel auxiliary subunits, possibly X31 A. Interestingly, heart, skeletal
muscle, and
CA 02381771 2002-02-07
WO 01/23571 PCT/US00/27119
-19-
DRG express TTX-sensitive and -resistant sodium channel a subunits (Dib-Hajj
et
al., 1998). It is shown in the present study that these tissues also express
both (31 and
(31A subunits. Given the findings of the present invention, it will now be
possible to
determine whether TTX-resistant channels are modulated by ~ilA.
When the present application refers to the function of (31 A or "wild-type"
activity, it is meant that the molecule in question has the ability to
modulate the
sodium density of a cell co-expressing the sodium a-subunit. In addition the
cellular
signaling mediated by these pores is influenced by the ~i 1A. Other phenotypes
that
may be considered to be regulated by the normal (31 A gene product include
peripheral
nerve sodium channels (i.e., SCN10A, SCN11A); cardiac sodium channels (SCNSA);
(thus, neuropathic pain and cardiac arrythmias such as Long QT may be
involved);
cell adhesion/repulsion - thus including axonal fasciculation/defasciculation,
growth
cone guidance, and synaptic remodeling following epileptic seizures.
Determination
of which molecules possess this activity may be achieved using assays familiar
to
those of skill in the art. For example, transfer of genes encoding ~ilA, or
variants
thereof, into cells that do not have a functional sodium channel, and hence
exhibit
impaired anion transport, will identify, by virtue of increasing the sodium
current
density in these cells, those molecules having (31A function. In addition the
assays
using TN-R, ankyrin and/or neurofascin binding discussed herein also may be
employed to show (31A function seeing as these molecules are known to interact
with
(3 subunits. Additional studies may be performed comparing such assays from
cells
co-expressing (31A subunits and an a-subunit with cells co-expressing other (3
subunits (e.g., (31, ~i2 and the like) with an a-subunit thereby giving a
quantitative and
qualitative difference in the function/activity of a (31A as compared to e.g.,
a (31 or (32
subunit.
As stated above, (31A is belongs to the Ig superfamily of CAMS. (31A is
predicted to encode a type I membrane protein with similar topolgy to (31
(FIG. 1 C).
(31A contains a novel 66-amino acid juxtamembrane region, 19-amino acid
transmembrane segment, and 39-amino acid intracellular domain. The disulfide-
linked immunoglobulin fold is common to both X31 and ~31A. The (3 subunits are
known to regulate the function of the pore-forming a-subunit of the sodium
channel.
CA 02381771 2002-02-07 pCT/US00/27119
WO 01/23571
-20-
Given that there is a substantial difference in the COOH terminus of the ~31A
as
compared to e.g. (31, it will be desirable to determine the effect of this
novel COOH
terminus on the function in the sodium channel-regulating role of (31A. This
also may
be a fruitful approach to developing screening assays for the absence of (31A
function
or in the development of therapies, for example, in targeting the sodium
current
density and other functions of (31A, targeting the substrate upon which (31A
acts (e.g.,
a subunit, ankyrin, TN-R), and the like.
C. Variants of (31A
Amino acid sequence variants of the polypeptide can be substitution, insertion
or
deletion variants. Deletion variants lack one or more residues of the native
protein
which are not essential for function or immunogenic activity, and are
exemplified by the
variants lacking a transmembrane, COON-terminus, the Ig fold or other region
described
above. Another common type of deletion variant is one lacking secretory signal
sequences or signal sequences directing a protein to bind to a particular part
of a cell.
Insertion mutants typically involve the addition of material at a non-terminal
point in the
polypeptide. This may include the insertion of an immunoreactive epitope or
simply a
single residue. Terminal additions, called fusion proteins, are discussed
below.
Substitutional variants typically contain the exchange of one amino acid for
another at one or more sites within the protein, and may be designed to
modulate one or
more properties of the polypeptide, such as stability against proteolytic
cleavage, without
the loss of other functions or properties. Substitutions of this kind
preferably are
conservative, that is, one amino acid is replaced with one of similar shape
and charge.
Conservative substitutions are well known in the art and include, for example,
the
changes of alanine to serine; arginine to lysine; asparagine to glutamine or
histidine;
aspartate to glutamate; cysteine to serine; glutamine to asparagine; glutamate
to
aspartate; glycine to proline; histidine to asparagine or glutamine;
isoleucine to leucine
or valine; leucine to valine or isoleucine; lysine to arginine; methionine to
leucine or
isoleucine; phenylalanine to tyrosine, leucine or methionine; serine to
threonine;
threonine to serine; tryptophan to tyrosine; tyrosine to tryptophan or
phenylalanine; and
valine to isoleucine or leucine.
WO 01/23571 cA o23amn 2002-02-0~ PCT/US00/27119
-21-
The following is a discussion based upon changing of the amino acids of a
protein to create an equivalent, or even an improved, second-generation
molecule. For
example, certain amino acids may be substituted for other amino acids in a
protein
structure without appreciable loss of interactive binding capacity with
structures such as,
for example, antigen-binding regions of antibodies or binding sites on
substrate
molecules. Since it is the interactive capacity and nature of a protein that
defines that
protein's biological functional activity, certain amino acid substitutions can
be made in a
protein sequence, and its underlying DNA coding sequence, and nevertheless
obtain a
protein with like properties. It is thus contemplated by the inventors that
various changes
may be made in the DNA sequences of genes without appreciable loss of their
biological
utility or activity, as discussed below. Table 1 shows the codons that encode
particular
amino acids.
In making such changes, the hydropathic index of amino acids may be
considered. The importance of the hydropathic amino acid index in conferring
interactive biologic function on a protein is generally understood in the art
(Kyle &
Doolittle, 1982). It is accepted that the relative hydropathic character of
the amino acid
contributes to the secondary structure of the resultant protein, which in turn
defines
the interaction of the protein with other molecules, for example, enzymes,
substrates,
receptors, DNA, antibodies, antigens, and the like.
Each amino acid has been assigned a hydropathic index on the basis of their
hydrophobicity and charge characteristics (Kyle & Doolittle, 1982), these are:
isoleucine (+4.5); valine (+4.2); leucine (+3.8); phenylalanine (+2.8);
cysteine/cystine
(+2.5); methionine (+1.9); alanine (+1.8); glycine (-0.4); threonine (-0.7);
serine (-
0.8); tryptophan (-0.9); tyrosine (-1.3); proline (-1.6); histidine (-3.2);
glutamate (-
3.5); glutamine (-3.5); aspartate (-3.5); asparagine (-3.5); lysine (-3.9);
and arginine (-
4.5).
It is understood that an amino acid can be substituted for another having a
similar hydrophilicity value and still obtain a biologically equivalent and
immunologically equivalent protein. In such changes, the substitution of amino
acids
whose hydrophilicity values are within ~2 is preferred, those that are within
~l are
particularly preferred, and those within X0.5 are even more particularly
preferred.
It is also understood in the art that the substitution of like amino acids can
be
made effectively on the basis of hydrophilicity. U.S. Patent 4,554,101, states
that the
WO 01/23571 cA o23amn 2002-02-0~ PCT/US00/27119
-22-
greatest local average hydrophilicity of a protein, as governed by the
hydrophilicity of
its adjacent amino acids, correlates with a biological property of the
protein. As
detailed in U.S. Patent 4,554,101, the following hydrophilicity values have
been
assigned to amino acid residues: arginine (+3.0); lysine (+3.0); aspartate
(+3.0 ~ 1);
S glutamate (+3.0 ~ 1); serine (+0.3); asparagine (+0.2); glutamine (+0.2);
glycine (0);
threonine (-0.4); proline (-0.5 ~ 1); alanine (-0.5); histidine *-0.5);
cysteine (-1.0);
methionine (-1.3); valine (-1.5); leucine (-1.8); isoleucine (-1.8); tyrosine
(-2.3);
phenylalanine (-2.5); tryptophan (-3.4).
As outlined above, amino acid substitutions are generally based on the
relative
similarity of the amino acid side-chain substituents, for example, their
hydrophobicity, hydrophilicity, charge, size, and the like. Exemplary
substitutions
that take various of the foregoing characteristics into consideration are well
known to
those of skill in the art and include: arginine and lysine; glutamate and
aspartate;
serine and threonine; glutamine and asparagine; and valine, leucine and
isoleucine.
Another embodiment for the preparation of polypeptides according to the
invention is the use of peptide mimetics. Mimetics are peptide-containing
molecules
that mimic elements of protein secondary structure. See, for example, Johnson
et al.,
"Peptide Turn Mimetics" in BIOTECHNOLOGYAND PHARMACY, Pezzuto et al.,
Eds., Chapman and Hall, New York (1993). The underlying rationale behind the
use of
peptide mimetics is that the peptide backbone of proteins exists chiefly to
orient amino
acid side chains in such a way as to facilitate molecular interactions, such
as those of
antibody and antigen. A peptide mimetic is expected to permit molecular
interactions
similar to the natural molecule. These principles may be used, in conjunction
with the
principles outline above, to engineer second generation molecules having many
of the
natural properties of (31A, but with altered and even improved
characteristics. The
generation of specific mutants is described in the examples.
D. Domain Switching
As described in the examples, the present inventors have identified that the
(31A is a splice variant of /31 subunit of voltage-gated sodium channels. This
provides
a starting point for further mutational analysis of the molecule. One way in
which this
information can be exploited is in "domain switching."
WO 01/23571 cA o23amn 2002-02-0~ pCT/LTS00/27119
-23-
Domain switching involves the generation of chimeric molecules using
different but, in many cases, related polypeptides. By comparing the (31 and
(31 A, the
inventors have predicted the functionally significant regions of these
molecules. It is
possible, then, to switch related domains of these molecules in an effort to
determine --
S the criticality of these regions to the ~3 subunit activity and function.
These molecules
may have additional value in that these "chimeras" can be distinguished from
natural
molecules, while possibly providing the same function. Also it should be
possible to
switch the domains of (31A subunits from voltage dependant sodium channels
with
those of [3 subunits from non-voltage dependant sodium channels.
In addition to switching domains between (31 and (31 A it should also be
possible to identify the X31 A proteins, if any, from other animals such as
rat, rabbit,
monkey, gibbon, chimp, ape, baboon, cow, pig, horse, sheep and cat. Upon
identification and isolation of these homologs, variants and mutants, and in
conjunction with other analyses, certain active or functional domains can be
identified. These domains can then be switched with those already identified
herein
to yield additional information about the conservation of these domains across
species.
Based on sequence identity, at the amino acid level, of the (31A and (31
proteins, it may be inferred that even small changes in the primary sequence
of the
molecule will affect function. Further analysis of mutations and their
predicted effect
on secondary structure will add to this understanding.
Another structural aspect of /31A that provides fertile ground for domain
switching experiments is the Ig fold domain. This domain may be substituted
for Ig
folds from other members of the Ig superfamily domains in order to alter the
specificity of this function. A further investigation of the homology between
~i 1 A and
other members of the Ig superfamily is warranted by this observation.
E. Fusion Proteins
A specialized kind of insertional variant is the fusion protein. This molecule
generally has all or a substantial portion of the native molecule, linked at
the N- or C-
terminus, to all or a portion of a second polypeptide. For example, fusions
typically
employ leader sequences from other species to permit the recombinant
expression of a
CA 02381771 2002-02-07
WO 01/23571 PCT/US00/27119
-24-
protein in a heterologous host. Another useful fusion includes the addition of
a
immunologically active domain, such as an antibody epitope, to facilitate
purification of
the fusion protein. Inclusion of a cleavage site at or near the fusion
junction will
facilitate removal of the extraneous polypeptide after purification. Other
useful fusions
include linking of functional domains, such as active sites from enzymes,
glycosylation
domains, cellular targeting signals or transmembrane regions. One particular
fusion of
interest would include a deletion construct lacking the carboxy terminal site
of (31A but
containing other regions that could bind the substrate molecule. Indeed any of
the
distinct regions of the (31 A protein can be removed or replaced in order to
determine
their function in relation to the whole protein. Fusion to a polypeptide that
can be used
for purification of the substrate-(31A complex would serve to isolate the
substrate for
identification and analysis.
F. Protein Purification
It will be desirable to purify /31A or variants thereof. Protein purification
techniques are well known to those of skill in the art. These techniques
involve, at
one level, the crude fractionation of the cellular milieu to polypeptide and
non-
polypeptide fractions. Having separated the polypeptide from other proteins,
the
polypeptide of interest may be further purified using chromatographic and
electrophoretic techniques to achieve partial or complete purification (or
purification
to homogeneity). Analytical methods particularly suited to the preparation of
a pure
peptide are ion-exchange chromatography, exclusion chromatography,
polyacrylamide gel electrophoresis, and isoelectric focusing. A particularly
efficient
method of purifying peptides is fast protein liquid chromatography or even
HPLC.
Certain aspects of the present invention concern the purification, and in
particular embodiments, the substantial purification, of an encoded protein or
peptide.
The term "purified protein or peptide" as used herein, is intended to refer to
a
composition, isolatable from other components, wherein the protein or peptide
is
purified to any degree relative to its naturally-obtainable state. A purified
protein or
peptide therefore also refers to a protein or peptide, free from the
environment in
which it may naturally occur.
WO 01/23571 cA o23amn 2002-02-0~ PCT/US00/27119
-25-
Generally, "purified" will refer to a protein or peptide composition that has
been subjected to fractionation to remove various other components, and which
composition substantially retains its expressed biological activity. Where the
term
"substantially purified" is used, this designation will refer to a composition
in which
the protein or peptide forms the major component of the composition, such as
constituting about 50%, about 60%, about 70%, about 80%, about 90%, about 95%
or
more of the proteins in the composition.
Various methods for quantifying the degree of purification of the protein or
peptide will be known to those of skill in the art in light of the present
disclosure.
These include, for example, determining the specific activity of an active
fraction, or
assessing the amount of polypeptides within a fraction by SDS/PAGE analysis. A
preferred method for assessing the purity of a fraction is to calculate the
specific
activity of the fraction, to compare it to the specific activity of the
initial extract, and
to thus calculate the degree of purity, herein assessed by a "-fold
purification
number." The actual units used to represent the amount of activity will, of
course, be
dependent upon the particular assay technique chosen to follow the
purification and
whether or not the expressed protein or peptide exhibits a detectable
activity.
Various techniques suitable for use in protein purification are well known to
those of skill in the art. These include, for example, precipitation with
ammonium
sulfate, PEG, antibodies and the like or by heat denaturation, followed by
centrifugation; chromatography steps such as ion exchange, gel filtration,
reverse
phase, hydroxylapatite and affinity chromatography; isoelectric focusing; gel
electrophoresis; and combinations of such and other techniques. It is believed
that the
order of conducting the various purification steps may be changed, or that
certain
steps may be omitted, and still result in a suitable method for the
preparation of a
substantially purified protein or peptide.
There is no general requirement that the protein or peptide always be provided
in their most purified state. Indeed, it is contemplated that less
substantially purified
products will have utility in certain embodiments. Partial purification may be
accomplished by using fewer purification steps in combination, or by utilizing
different forms of the same general purification scheme. For example, it is
appreciated that a cation-exchange column chromatography performed utilizing
an
HPLC apparatus will generally result in a greater "-fold" purification than
the same
CA 02381771 2002-02-07
WO 01/23571 PCT/US00/27119
-26-
technique utilizing a low pressure chromatography system. Methods exhibiting a
lower degree of relative purification may have advantages in total recovery of
protein
product, or in maintaining the activity of an expressed protein.
It is known that the migration of a polypeptide can vary, sometimes
significantly, with different conditions of SDS/PAGE (Capaldi et al., 1977).
It will
therefore be appreciated that under differing electrophoresis conditions, the
apparent
molecular weights of purified or partially purified expression products may
vary.
High Performance Liquid Chromatography (HPLC) is characterized by a very
rapid separation with extraordinary resolution of peaks. This is achieved by
the use of
very fine particles and high pressure to maintain an adequate flow rate.
Separation
can be accomplished in a matter of minutes, or at most an hour. Moreover, only
a
very small volume of the sample is needed because the particles are so small
and
close-packed that the void volume is a very small fraction of the bed volume.
Also,
the concentration of the sample need not be very great because the bands are
so
narrow that there is very little dilution of the sample.
Gel chromatography, or molecular sieve chromatography, is a special type of
partition chromatography that is based on molecular size. The theory behind
gel
chromatography is that the column, which is prepared with tiny particles of an
inert
substance that contain small pores, separates larger molecules from smaller
molecules
as they pass through or around the pores, depending on their size. As long as
the
material of which the particles are made does not adsorb the molecules, the
sole factor
determining rate of flow is the size. Hence, molecules are eluted from the
column in
decreasing size, so long as the shape is relatively constant. Gel
chromatography is
unsurpassed for separating molecules of different size because separation is
independent of all other factors such as pH, ionic strength, temperature, etc.
There
also is virtually no adsorption, less zone spreading and the elution volume is
related in
a simple matter to molecular weight.
Affinity chromatography is a chromatographic procedure that relies on the
specific affinity between a substance to be isolated and a molecule that it
can
specifically bind. This is a receptor-ligand type interaction. The column
material is
synthesized by covalently coupling one of the binding partners to an insoluble
matrix.
The column material is then able to specifically adsorb the substance from the
CA 02381771 2002-02-07
WO 01/23571 PCT/LTS00/27119
-27-
solution. Elution occurs by changing the conditions to those in which binding
will not
occur (alter pH, ionic strength, temperature, etc.).
A particular type of affinity chromatography useful in the purification of
carbohydrate containing compounds is lectin affinity chromatography. Lectins
are a
S class of substances that bind to a variety of polysaccharides and
glycoproteins.
Lectins are usually coupled to agarose by cyanogen bromide. Conconavalin A
coupled to Sepharose was the first material of this sort to be used and has
been widely
used in the isolation of polysaccharides and glycoproteins. Other lectins that
have
been include lentil lectin, wheat germ agglutinin, which has been useful in
the
purification of N-acetyl glucosaminyl residues, and Helix pomatia lectin.
Lectins
themselves are purified using affinity chromatography with carbohydrate
ligands.
Lactose has been used to purify lectins from castor bean and peanuts; maltose
has
been useful in extracting lectins from lentils and jack bean; N-acetyl-D
galactosamine
is used for purifying lectins from soybean; N-acetyl glucosaminyl binds to
lectins
from wheat germ; D-galactosamine has been used in obtaining lectins from clams
and
L-fucose will bind to lectins from lotus.
The matrix should be a substance that itself does not adsorb molecules to any
significant extent and that has a broad range of chemical, physical and
thermal
stability. The ligand should be coupled in such a way as to not affect its
binding
properties. The ligand should also provide relatively tight binding. And it
should be
possible to elute the substance without destroying the sample or the ligand.
One of
the most common forms of affinity chromatography is immunoaffinity
chromatography. The generation of antibodies that would be suitable for use in
accord with the present invention is discussed below.
G. Synthetic Peptides
The present invention also describes smaller (31A-related peptides for use in
various embodiments of the present invention. Because of their relatively
small size,
the peptides of the invention can also be synthesized in solution or on a
solid support
in accordance with conventional techniques. Various automatic synthesizers are
commercially available and can be used in accordance with known protocols.
See, for
example, Stewart and Young, (1984); Tam et al., (1983); Merrifield, (1986);
and
Barany and Merrifield (1979). Short peptide sequences, or libraries of
overlapping
CA 02381771 2002-02-07
WO 01/23571 PCT/US00/27119
-28-
peptides, usually from about 6 up to about 35 to 50 amino acids, which
correspond to
the selected regions described herein, can be readily synthesized and then
screened in
screening assays designed to identify reactive peptides. Alternatively,
recombinant
DNA technology may be employed wherein a nucleotide sequence which encodes a
peptide of the invention is inserted into an expression vector, transformed or
transfected into an appropriate host cell and cultivated under conditions
suitable for
expression.
H. Antigen Compositions
The present invention also provides for the use of (31A proteins or peptides
as
antigens for the immunization of animals relating to the production of
antibodies. It is
envisioned that either (31A, or portions thereof, will be coupled, bonded,
bound,
conjugated or chemically-linked to one or more agents via linkers, polylinkers
or
derivatized amino acids. This may be performed such that a bispecific or
multivalent
composition or vaccine is produced. It is further envisioned that the methods
used in
the preparation of these compositions will be familiar to those of skill in
the art and
should be suitable for administration to animals, i.e., pharmaceutically
acceptable.
Preferred agents are the earners are keyhole limpet hemocyannin (KLH) or
bovine
serum albumin (BSA).
3. Nucleic Acids encoding (31A Protein
The present invention also provides, in another embodiment, nucleic acid
sequences encoding (31A. The present invention is not limited in scope to
these
genes, however, as one of ordinary skill in the art could, using these nucleic
acids,
readily identify related homologs in various other species (e.g., rat, dog,
rabbit,
monkey, gibbon, chimp, ape, baboon, cow, pig, horse, sheep, cat and other
species)
In addition, it should be clear that the present invention is not limited to
the
specific nucleic acids disclosed herein. As discussed below, a "(31A encoding
nucleic
acid" may contain a variety of different bases and yet still produce a
corresponding
polypeptide that is functionally indistinguishable, and in some cases
structurally, from
the polypeptide disclosed herein as SEQ >D N0:2.
WO 01/23571 cA o23ai~~i 2002-02-0~ pCT/US00/27119
-29-
Similarly, any reference to a nucleic acid should be read as encompassing a
host cell containing that nucleic acid and, in some cases, capable of
expressing the
product of that nucleic acid. In addition to therapeutic considerations, cells
expressing nucleic acids of the present invention may prove useful in the
context of
screening for agents that induce, repress, inhibit, augment, interfere with,
block,
abrogate, stimulate or enhance the function of (31 A or voltage gated sodium
channel
activity in general.
A. Nucleic Acids Encoding (31A
The nucleic acid given in FIG. 1 C and disclosed in SEQ ID NO:1 represent the
(31 A encoding nucleic acids of the present invention. Nucleic acids according
to the
present invention may encode an entire (31A protein as shown in SEQ ID N0:2, a
domain of (31 A that expresses a particular function attributable to (31 A, or
any other
fragment of the (31 A sequences set forth herein. The nucleic acid may be
derived from
genomic DNA, i.e., cloned directly from the genome of a particular organism.
In
preferred embodiments, however, the nucleic acid would comprise complementary
DNA
(cDNA). Also contemplated is a cDNA plus a natural intron or an intron derived
from
another gene; such engineered molecules are sometime referred to as "mini-
genes." This
is particularly of note seeing as the (31 A protein results from a splice
variant of (31 gene.
The (31A protein results from a retention of intron 3 of the (31 gene. At a
minimum, it
should be understood that these and other nucleic acids of the present
invention may be
used as molecular weight standards in, for example, gel electrophoresis.
The term "cDNA" is intended to refer to DNA prepared using messenger RNA
(mRNA) as template. The advantage of using a cDNA, as opposed to genomic DNA
or
DNA polymerized from a genomic, non- or partially-processed RNA template, is
that
the cDNA primarily contains coding sequences of the corresponding protein.
There may
be times when the full or partial genomic sequence is preferred, such as where
the non-
coding regions are required for optimal expression or where non-coding regions
such as
introns are to be targeted in an antisense strategy.
It also is contemplated that a given X31 A from a given species may be
represented
by natural variants that have slightly different nucleic acid sequences but,
nonetheless,
encode the same protein (see Table 1 below).
CA 02381771 2002-02-07
WO 01/23571 PCT/US00/27119
-30-
As used in this application, the term "a nucleic acid encoding a (31A" refers
to a
nucleic acid molecule that has been isolated free of total cellular nucleic
acid. In
preferred embodiments, the invention concerns a nucleic acid sequence
essentially as set
forth in SEQ >D NO:1. The term "as set forth in SEQ ID NO:1 " means that the
nucleic
acid sequence substantially corresponds to a portion of SEQ ID NO:1. The term
"functionally equivalent codon" is used herein to refer to codons that encode
the same
amino acid, such as the six codons for arginine or serine (Table 1, below),
and also refers
to codons that encode biologically equivalent amino acids, as discussed in the
following
pages.
TABLE 1
Amino Acids Codons
Alanine Ala A GCA GCC GCG GCU
Cysteine Cys C UGC UGU
Aspartic Asp D GAC GAU
acid
Glutamic Glu E GAA GAG
acid
PhenylalaninePhe F UUC UUU
Glycine Gly G GGA GGC GGG GGU
Histidine His H CAC CAU
Isoleucine Ile I AUA AUC AUU
Lysine Lys K AAA AAG
Leucine Leu L UUA UUG CUA CUC CUG CUU
Methionine Met M AUG
Asparagine Asn N AAC AAU
Proline Pro P CCA CCC CCG CCU
Glutamine Gln Q CAA CAG
Arginine Arg R AGA AGG CGA CGC CGG CGU
Serine Ser S AGC UCA UCC UCG UCU
AGU
Threonine Thr T ACA ACC ACG ACU
Valine Val V GUA GUC GUG GUU
Tryptophan Trp W UGG
Tyrosine Tyr Y UAC UAU
Allowing for the degeneracy of the genetic code, sequences that have at least
about 50%, usually at least about 60%, more usually about 70%, most usually
about
80%, preferably at least about 90% and most preferably about 95% of
nucleotides that
are identical to the nucleotides of SEQ ID NO:1 will be sequences that are "as
set forth
in SEQ >D NO:1." Sequences that are essentially the same as those set forth in
SEQ >D
NO:1 may also be functionally defined as sequences that are capable of
hybridizing to a
CA 02381771 2002-02-07
WO 01/23571 PCT/US00/27119
-31-
nucleic acid segment containing the complement of SEQ m NO:1 under standard
conditions.
The DNA segments of the present invention include those encoding biologically
functional equivalent (31A proteins and peptides, as described above. Such
sequences
may arise as a consequence of codon redundancy and amino acid functional
equivalency
that are known to occur naturally within nucleic acid sequences and the
proteins thus
encoded. Alternatively, functionally equivalent proteins or peptides may be
created via
the application of recombinant DNA technology, in which changes in the protein
structure may be engineered, based on considerations of the properties of the
amino
acids being exchanged. Changes designed by man may be introduced through the
application of site-directed mutagenesis techniques or may be introduced
randomly and
screened later for the desired function, as described below.
B. Oligonucleotide Probes and Primers
1 S Naturally, the present invention also encompasses DNA segments that are
complementary, or essentially complementary, to the sequence set forth in SEQ
>D
NO:l . Nucleic acid sequences that are "complementary" are those that are
capable of
base-pairing according to the standard Watson-Crick complementary rules. As
used
herein, the term "complementary sequences" means nucleic acid sequences that
are
substantially complementary, as may be assessed by the same nucleotide
comparison set
forth above, or as defined as being capable of hybridizing to the nucleic acid
segment of
SEQ >T7 NO: l under relatively stringent conditions such as those described
herein.
Complementarity between two single-stranded molecules may be "partial", in
which only some of the nucleic acids bind, or it may be complete when total
complementarity exists between the single-stranded molecules. The degree of
complementarity between nucleic acid strands has significant effects on the
efficiency
and strength of hybridization between nucleic acid strands. This is of
particular
importance in amplification reactions, which depend upon binding between
nucleic
acids strands.
The term "homology", as used herein, refers to a degree of complementarity.
There may be partial homology or complete homology (i.e., identity). A
partially
complementary sequence is one that at least partially inhibits an identical
sequence
from hybridizing to a target nucleic acid; it is referred to using the
functional term
WO 01/23571 cA o23amn 2002-02-0~ pCT/LTS00/27119
-32-
"substantially homologous." The inhibition of hybridization of the completely
complementary sequence to the target sequence may be examined using a
hybridization assay (Southern or northern blot, solution hybridization and the
like)
under conditions of low stringency. A substantially homologous sequence or
probe
S will compete for and inhibit the binding (i.e., the hybridization) of a
completely
homologous sequence or probe to the target sequence under conditions of low
stringency. This is not to say that conditions of low stringency are such that
non-
specific binding is permitted; low stringency conditions require that the
binding of
two sequences to one another be a specific (i.e., selective) interaction. The
absence of
non-specific binding may be tested by the use of a second target sequence
which lacks
even a partial degree of complementarity (e.g., less than about 30% identity);
in the
absence of non-specific binding, the probe will not hybridize to the second
non-
complementary target sequence.
Thus, the sequences of the present invention may encode the entire (31A
protein
or functional or non-functional fragments thereof. Alternatively, the
hybridizing
segments may be shorter oligonucleotides. Sequences of 17 bases long should
occur
only once in the human genome and, therefore, suffice to specify a unique
target
sequence. Although shorter oligomers are easier to make and increase in vivo
accessibility, numerous other factors are involved in determining the
specificity of
hybridization. Both binding affinity and sequence specificity of an
oligonucleotide to its
complementary target increases with increasing length. It is contemplated that
exemplary oligonucleotides of 8, 9, 10, 1 l, 12, 13, 14, 15, 16, 17, 18, 19,
20, 25, 30, 35,
40, 45, 50, 55, 60, 65, 70, 75, 80, 85, 90, 95, 100 or more base pairs will be
used,
although others are contemplated. Longer polynucleotides encoding 250, 500,
1000,
1212, 1500, 2000, 2500, 3000 or 3431 bases and longer are contemplated as
well. Such
oligonucleotides will find use, for example, as probes in Southern and
Northern blots
and as primers in amplification reactions.
The term "hybridization", as used herein, refers to any process by which a
strand of nucleic acid binds with a complementary strand through base pairing.
The
term "h 1 hybridization complex", as used herein, refers to a complex formed
between
two nucleic acid sequences by virtue of the formation of hydrogen bonds
between
complementary G and C bases and between complementary A and T bases; these
hydrogen bonds may be further stabilized by base stacking interactions. The
two
WO 01/23571 cA o23amn 2002-02-0~ pCT/US00/27119
-33-
complementary nucleic acid sequences hydrogen bond in an antiparallel
configuration. A hybridization complex may be formed in solution or between
one
nucleic acid sequence present in solution and another nucleic acid sequence
immobilized on a solid support (e.g., membranes, filters, chips, pins or glass
slides to
which cells have been fixed for in situ hybridization).
Suitable hybridization conditions will be well known to those of skill in the
art.
As known in the art, numerous conditions may be employed to comprise either
low or
high stringency conditions for hybridization. Factors such as the length and
nature
(DNA, RNA, base composition) of the sequence, nature of the target (DNA, RNA,
base composition, presence in solution or immobilization, etc.), and the
concentration
of the salts and other components (e.g., the presence or absence of formamide,
dextran sulfate and/or polyethylene glycol) are considered and the
hybridization
solution may be varied to generate conditions of either low or high stringency
different from, but equivalent to, the above listed conditions.
In certain applications, for example, substitution of amino acids by site-
directed
mutagenesis, it is appreciated that lower stringency conditions are required.
Under these
conditions, hybridization may occur even though the sequences of probe and
target
strand are not perfectly complementary, but are mismatched at one or more
positions.
Conditions may be rendered less stringent by increasing salt concentration and
decreasing temperature. For example, a medium stringency condition could be
provided
by about 0.1 to 0.25 M NaCI at temperatures of about 37°C to about
55°C, while a low
stringency condition could be provided by about 0.15 M to about 0.9 M salt, at
temperatures ranging from about 20°C to about 55°C. Thus,
hybridization conditions
can be readily manipulated, and thus will generally be a method of choice
depending on
the desired results.
In other embodiments, hybridization may be achieved under conditions of, for
example, 50 mM Tris-HCl (pH 8.3), 75 mM KCI, 3 mM MgCl2, 10 mM dithiothreitol,
at
temperatures between approximately 20°C to about 37°C. Other
hybridization
conditions utilized could include approximately 10 mM Tris-HCl (pH 8.3), 50 mM
KCI,
1.5 pM MgCl2, at temperatures ranging from approximately 40°C to about
72°C.
Formamide and SDS also may be used to alter the hybridization conditions.
WO 01/23571 cA o23ai~~i 2002-02-0~ PCT/US00/27119
-34-
One method of using probes and primers of the present invention is in the
search
for genes related to (31A or, more particularly, homologs of (31A from other
species. The
existence of a rat homolog strongly suggests that other homologs of the human
(31A will
be discovered in species more closely related, and perhaps more remote, than
mouse.
Normally, the target DNA will be a genomic or cDNA library, although screening
may
involve analysis of RNA molecules. By varying the stringency of hybridization,
and the
region of the probe, different degrees of homology may be discovered.
Another way of exploiting probes and primers of the present invention is in
site-directed, or site-specific mutagenesis. Site-specific mutagenesis is a
technique
useful in the preparation of individual peptides, or biologically functional
equivalent
proteins or peptides, through specific mutagenesis of the underlying DNA. The
technique further provides a ready ability to prepare and test sequence
variants,
incorporating one or more of the foregoing considerations, by introducing one
or
more nucleotide sequence changes into the DNA. Site-specific mutagenesis
allows the
production of mutants through the use of specific oligonucleotide sequences
which
encode the DNA sequence of the desired mutation, as well as a sufficient
number of
adjacent nucleotides, to provide a primer sequence of sufficient size and
sequence
complexity to form a stable duplex on both sides of the deletion junction
being
traversed. Typically, a primer of about 17 to 25 nucleotides in length is
preferred,
with about 5 to 10 residues on both sides of the junction of the sequence
being altered.
The technique typically employs a bacteriophage vector that exists in both a
single stranded and double stranded form. Typical vectors useful in site-
directed
mutagenesis include vectors such as the M13 phage. These phage vectors are
commercially available and their use is generally well known to those skilled
in the
art. Double stranded plasmids are also routinely employed in site directed
mutagenesis, which eliminates the step of transferring the gene of interest
from a
phage to a plasmid.
In general, site-directed mutagenesis is performed by first obtaining a single-
stranded vector, or melting of two strands of a double stranded vector which
includes
within its sequence a DNA sequence encoding the desired protein. An
oligonucleotide
primer bearing the desired mutated sequence is synthetically prepared. This
primer is
then annealed with the single-stranded DNA preparation, taking into account
the
degree of mismatch when selecting hybridization conditions, and subjected to
DNA
WO 01/23571 cA o23amn 2002-02-0~ pCT/US00/27119
-35-
polymerizing enzymes such as E. coli polymerase I Klenow fragment, in order to
complete the synthesis of the mutation-bearing strand. Thus, a heteroduplex is
formed
wherein one strand encodes the original non-mutated sequence and the second
strand
bears the desired mutation. This heteroduplex vector is then used to transform
appropriate cells, such as E. coli cells, and clones are selected that include
recombinant vectors bearing the mutated sequence arrangement.
The preparation of sequence variants of the selected gene using site-directed
mutagenesis is provided as a means of producing potentially useful species and
is not
meant to be limiting, as there are other ways in which sequence variants of
genes may
be obtained. For example, recombinant vectors encoding the desired gene may be
treated with mutagenic agents, such as hydroxylamine, to obtain sequence
variants.
C. Antisense Constructs
In some cases, mutant ~31A proteins may not be non-functional. Rather, they
1 S may have aberrant functions that cannot be overcome by replacement gene
therapy,
even where the "wild-type" molecule is expressed in amounts in excess of the
mutant
polypeptide. Antisense treatments are one way of addressing this situation.
Antisense
technology also may be used to "knock-out" function of (31 A in the
development of
cell lines or transgenic mice for research, diagnostic and screening purposes.
Antisense methodology takes advantage of the fact that nucleic acids tend to
pair with "complementary" sequences. By complementary, it is meant that
polynucleotides are those which are capable of base-pairing according to the
standard
Watson-Crick complementarity rules. That is, the larger purines will base pair
with
the smaller pyrimidines to form combinations of guanine paired with cytosine
(G:C)
and adenine paired with either thymine (A:T) in the case of DNA, or adenine
paired
with uracil (A:U) in the case of RNA. Inclusion of less common bases such as
inosine, 5-methylcytosine, 6-methyladenine, hypoxanthine and others in
hybridizing
sequences does not interfere with pairing.
Targeting double-stranded (ds) DNA with polynucleotides leads to triple-helix
formation; targeting RNA will lead to double-helix formation. Antisense
polynucleotides, when introduced into a target cell, specifically bind to
their target
polynucleotide and interfere with transcription, RNA processing, transport,
translation
and/or stability. Antisense RNA constructs, or DNA encoding such antisense
RNA's,
WO 01/23571 cA o23amn 2002-02-0~ PCT/US00/27119
-36-
may be employed to inhibit gene transcription or translation or both within a
host cell,
either in vitro or in vivo, such as within a host animal, including a human
subject.
Antisense constructs may be designed to bind to the promoter and other
control regions, exons, introns or even exon-intron boundaries of a gene. It
is
contemplated that the most effective antisense constructs will include regions
complementary to intron/exon splice junctions. Thus, it is proposed that a
preferred
embodiment includes an antisense construct with complementarity to regions
within
50-200 bases of an intron-exon splice junction. It has been observed that some
exon
sequences can be included in the construct without seriously affecting the
target
selectivity thereof. The amount of exonic material included will vary
depending on
the particular exon and intron sequences used. One can readily test whether
too much
exon DNA is included simply by testing the constructs in vitro to determine
whether
normal cellular function is affected or whether the expression of related
genes having
complementary sequences is affected.
As stated above, "complementary" or "antisense" means polynucleotide
sequences that are substantially complementary over their entire length and
have very
few base mismatches. For example, sequences of fifteen bases in length may be
termed complementary when they have complementary nucleotides at thirteen or
fourteen positions. Naturally, sequences which are completely complementary
will be
sequences which are entirely complementary throughout their entire length and
have
no base mismatches. Other sequences with lower degrees of homology also are
contemplated. For example, an antisense construct which has limited regions of
high
homology, but also contains a non-homologous region (e.g., ribozyme; see
below)
could be designed. These molecules, though having less than 50% homology,
would
bind to target sequences under appropriate conditions.
It may be advantageous to combine portions of genomic DNA with cDNA or
synthetic sequences to generate specific constructs. For example, where an
intron is
desired in the ultimate construct, a genomic clone will need to be used. The
cDNA or
a synthesized polynucleotide may provide more convenient restriction sites for
the
remaining portion of the construct and, therefore, would be used for the rest
of the
sequence.
D. Ribozymes
CA 02381771 2002-02-07
WO 01/23571 PCT/US00/27119
-37-
Another approach for addressing the "dominant negative" mutant protein is
through the use of ribozymes. Although proteins traditionally have been used
for
catalysis of nucleic acids, another class of macromolecules has emerged as
useful in
this endeavor. Ribozymes are RNA-protein complexes that cleave nucleic acids
in a
site-specific fashion. Ribozymes have specific catalytic domains that possess
endonuclease activity (Kim and Cook, 1987; Gerlach et al., 1987; Forster and
Symons, 1987). For example, a large number of ribozymes accelerate
phosphoester
transfer reactions with a high degree of specificity, often cleaving only one
of several
phosphoesters in an oligonucleotide substrate (Cook et al., 1981; Michel and
Westhof,
1990; Reinhold-Hurek and Shub, 1992). This specificity has been attributed to
the
requirement that the substrate bind via specific base-pairing interactions to
the
internal guide sequence ("IGS") of the ribozyme prior to chemical reaction.
Ribozyme catalysis has primarily been observed as part of sequence-specific
cleavage/ligation reactions involving nucleic acids (Joyce, 1989; Cook et al.,
1981).
For example, U.S. Patent No. 5,354,855 reports that certain ribozymes can act
as
endonucleases with a sequence specificity greater than that of known
ribonucleases
and approaching that of the DNA restriction enzymes. Thus, sequence-specific
ribozyme-mediated inhibition of gene expression may be particularly suited to
therapeutic applications (Scanlon et al., 1991; Sarver et al., 1990).
Recently, it was
reported that ribozymes elicited genetic changes in some cells lines to which
they
were applied; the altered genes included the oncogenes H-ras, c-fos and genes
of HIV.
Most of this work involved the modification of a target mRNA, based on a
specific
mutant codon that is cleaved by a specific ribozyme.
4. Preparation of Vectors for Cloning, Gene Transfer and Expression
Within certain embodiments expression vectors are employed to express the
(31A polypeptide product, which can then be purified and, for example, be used
to
vaccinate animals to generate antisera or monoclonal antibodies with which
further
studies may be conducted. In other embodiments, the expression vectors are
used in
gene therapy. Expression requires that appropriate signals be provided in the
vectors,
and which include various regulatory elements, such as enhancers/promoters
from
both viral and mammalian sources that drive expression of the genes of
interest in
WO 01/23571 cA o23amn 2002-02-0~ pCT/US00/27119
-3 8-
host cells. Elements designed to optimize messenger RNA stability and
translatability
in host cells also are defined. The conditions for the use of a number of
dominant
drug selection markers for establishing permanent, stable cell clones
expressing the
products are also provided, as is an element that links expression of the drug
selection
markers to expression of the polypeptide.
A. Regulatory Elements
Throughout this application, the term "expression construct" is meant to
include any type of genetic construct containing a nucleic acid coding for a
gene
product in which part or all of the nucleic acid encoding sequence is capable
of being
transcribed. The transcript may be translated into a protein; but it need not
be. In
certain embodiments, expression includes both transcription of a gene and
translation
of mRNA into a gene product. In other embodiments, expression only includes
transcription of the nucleic acid encoding a gene of interest.
In preferred embodiments, the nucleic acid encoding a gene product is under
transcriptional control of a promoter. A "promoter" refers to a DNA sequence
recognized by the synthetic machinery of the cell, or introduced synthetic
machinery,
required to initiate the specific transcription of a gene. The phrase "under
transcriptional control" means that the promoter is in the correct location
and
orientation in relation to the nucleic acid to control RNA polymerise
initiation and
expression of the gene.
The term promoter will be used here to refer to a group of transcriptional
control modules that are clustered around the initiation site for RNA
polymerise II.
Much of the thinking about how promoters are organized derives from analyses
of
several viral promoters, including those for the HSV thymidine kinase (tk) and
SV40
early transcription units. These studies, augmented by more recent work, have
shown
that promoters are composed of discrete functional modules, each consisting of
approximately 7-20 by of DNA, and containing one or more recognition sites for
transcriptional activator or repressor proteins.
At least one module in each promoter functions to position the start site for
RNA synthesis. The best known example of this is the TATA box, but in some
promoters lacking a TATA box, such as the promoter for the mammalian terminal
WO 01/23571 cA o23ai~~i 2002-02-0~ pCT/US00/27119
-39-
deoxynucleotidyl transferase gene and the promoter for the SV40 late genes, a
discrete element overlying the start site itself helps to fix the place of
initiation.
Additional promoter elements regulate the frequency of transcriptional
initiation. Typically, these are located in the region 30-110 by upstream of
the start
S site, although a number of promoters have recently been shown to contain
functional
elements downstream of the start site as well. The spacing between promoter
elements frequently is flexible, so that promoter function is preserved when
elements
are inverted or moved relative to one another. In the tk promoter, the spacing
between promoter elements can be increased to 50 by apart before activity
begins to
decline. Depending on the promoter, it appears that individual elements can
function
either co-operatively or independently to activate transcription.
The particular promoter employed to control the expression of a nucleic acid
sequence of interest is not believed to be important, so long as it is capable
of
direction the expression of the nucleic acid in the targeted cell. Thus, where
a human
cell is targeted, it is preferable to position the nucleic acid coding region
adjacent to
and under the control of a promoter that is capable of being expressed in a
human cell.
Generally speaking, such a promoter might include either a human or viral
promoter.
In various embodiments, the human cytomegalovirus (CMV) immediate early
gene promoter, the SV40 early promoter, the Rous sarcoma virus long terminal
repeat, rat insulin promoter and glyceraldehyde-3-phosphate dehydrogenase can
be
used to obtain high-level expression of the coding sequence of interest. The
use of
other viral or mammalian cellular or bacterial phage promoters which are well-
known
in the art to achieve expression of a coding sequence of interest is
contemplated as
well, provided that the levels of expression are sufficient for a given
purpose.
By employing a promoter with well-known properties, the level and pattern of
expression of the protein of interest following transfection or transformation
can be
optimized. Further, selection of a promoter that is regulated in response to
specific
physiologic signals can permit inducible expression of the gene product.
Promoters that may be useful include insulin, elastin, amylase, pdr-l, pdx-1
and glucokinase, MMTV, MT-l, ecdysone and RuBisco, c-fos, TNF-alpha, C-
reactive
protein (Arcone et al., 1988), haptoglobin (Oliviero et al., 1987), serum
amyloid A2,
C/EBP alpha, IL-1, IL-6 (Poli and Cortese, 1989), Complement C3 (Wilson et
al.,
1990), IL-8, alpha-1 acid glycoprotein (Prowse and Baumann, 1988), alpha-1
WO 01/23571 cA o23amn 2002-02-0~ pCT/US00/27119
-40-
antitypsin, lipoprotein lipase (Zechner et al., 1988), angiotensinogen (Ron et
al.,
1991 ), fibrinogen, c jun (inducible by phorbol esters, TNF-alpha, UV
radiation,
retinoic acid, and hydrogen peroxide), collagenase (induced by phorbol esters
and
retinoic acid), metallothionein (heavy metal and glucocorticoid inducible),
Stromelysin (inducible by phorbol ester, interleukin-1 and EGF), alpha-2
macroglobulin and alpha-1 antichymotrypsin. Other promoters that could be used
according to the present invention include Lac-regulatable, chemotherapy
inducible
(e.g. MDR), and heat (hyperthermia) inducible promoters, Radiation-inducible
(e.g.,
EGR (Joki et al., 1995)), Alpha-inhibin, RNA pol III tRNA met and other amino
acid
promoters, Ul snRNA (Bartlett et al., 1996), MC-l, PGK, -actin and alpha-
globin.
Many other promoters that may be useful are listed in Walther and Stein
(1996). This
list is not intended to be exhaustive of all the possible elements involved in
the
promotion of gene expression but, merely, to be exemplary thereof.
Enhancers are genetic elements that increase transcription from a promoter
located at a distant position on the same molecule of DNA. Emhancers are
organized
much like promoters. That is, they are composed of many individual elements,
each
of which binds to one or more transcriptional proteins.
The basic distinction between enhancers and promoters is operational. An
enhancer region as a whole must be able to stimulate transcription at a
distance; this
need not be true of a promoter region or its component elements. On the other
hand, a
promoter must have one or more elements that direct initiation of RNA
synthesis at a
particular site and in a particular orientation, whereas enhancers lack these
specificities. Promoters and enhancers are often overlapping and contiguous,
often
seeming to have a very similar modular organization.
The list of viral promoters, cellular promoters/enhancers and inducible
promoters/enhancers that could be used in combination with the nucleic acid
encoding
a gene of interest in an expression construct is extensive and well known to
those in
the art. Any promoter/enhancer combination (as per the Eukaryotic Promoter
Data
Base EPDB) could also be used to drive expression of the gene. Eukaryotic
cells can
support cytoplasmic transcription from certain bacterial promoters if the
appropriate
bacterial polymerase is provided, either as part of the delivery complex or as
an
additional genetic expression construct.
CA 02381771 2002-02-07
WO 01/23571 PCT/US00/27119
-41-
Where a cDNA insert is employed, one will typically desire to include a
polyadenylation signal to effect proper polyadenylation of the gene
transcript. The
nature of the polyadenylation signal is not believed to be crucial to the
successful
practice of the invention, and any such sequence may be employed such as human
growth hormone and SV40 polyadenylation signals. Also contemplated as an
element
of the expression cassette is a terminator. These elements can serve to
enhance
message levels and to minimize read through from the cassette into other
sequences.
B. Selectable Markers
In certain embodiments of the invention, the cells contain nucleic acid
constructs of the present invention, a cell may be identified iii vitro or in
vivo by
including a marker in the expression construct. Such markers would confer an
identifiable change to the cell permitting easy identification of cells
containing the
expression construct. Usually the inclusion of a drug selection marker aids in
cloning
1 S and in the selection of transformants, for example, genes that confer
resistance to
neomycin, puromycin, hygromycin, DHFR, GPT, zeocin and histidinol are useful
selectable markers. Alternatively, enzymes such as herpes simplex virus
thymidine
kinase (tk) or chloramphenicol acetyltransferase (CAT) may be employed.
Immunologic markers also can be employed. The selectable marker employed is
not
believed to be important, so long as it is capable of being expressed
simultaneously
with the nucleic acid encoding a gene product. Further examples of selectable
markers are well known to one of skill in the art.
C. Multigene Constructs and IRES
In certain embodiments of the invention, the use of internal ribosome binding
sites (IRES) elements are used to create multigene, or polycistronic,
messages. IRES
elements are able to bypass the ribosome scanning model of 5' methylated Cap
dependent translation and begin translation at internal sites (Pelletier and
Sonenberg,
1988). IKES elements from two members of the picanovirus family (polio and
encephalomyocarditis) have been described (Pelletier and Sonenberg, 1988), as
well
as an IItES from a mammalian message (Macejak and Sarnow, 1991). IRES elements
can be linked to heterologous open reading frames. Multiple open reading
frames can
be transcribed together, each separated by an IRES, creating polycistronic
messages.
CA 02381771 2002-02-07
WO 01/23571 PCT/US00/27119
-42-
By virtue of the IRES element, each open reading frame is accessible to
ribosomes for
efficient translation. Multiple genes can be efficiently expressed using a
single
promoter/enhancer to transcribe a single message.
Any heterologous open reading frame can be linked to IRES elements. This
includes genes for secreted proteins, multi-subunit proteins, encoded by
independent
genes, intracellular or membrane-bound proteins and selectable markers. In
this way,
expression of several proteins can be simultaneously engineered into a cell
with a
single construct and a single selectable marker.
5. Delivery of Expression Vectors
In order to effect expression of sense or antisense nucleic acid constructs,
the
expression construct must be delivered into a cell. This delivery may be
accomplished in vitro, as in laboratory procedures for transforming cells
lines, or in
vivo or ex vivo, as in the treatment of certain disease states. One mechanism
for
delivery is via viral infection where the expression construct is encapsidated
in an
infectious viral particle.
Thus, in certain embodiments of the invention, the expression construct
comprises a virus or engineered construct derived from a viral genome. The
ability of
certain viruses to enter cells via receptor-mediated endocytosis, to integrate
into host
cell genome and express viral genes stably and efficiently have made them
attractive
candidates for the transfer of foreign genes into mammalian cells (Ridgeway,
1988;
Nicolas and Rubenstein, 1988; Baichwal and Sugden, 1986; Temin, 1986). The
first
viruses used as gene vectors were DNA viruses including the papovaviruses
(simian
virus 40, bovine papilloma virus, and polyoma) (Ridgeway, 1988; Baichwal and
Sugden, 1986) and adenoviruses (Ridgeway, 1988; Baichwal and Sugden, 1986).
These have a relatively low capacity for foreign DNA sequences and have a
restricted
host spectrum. Furthermore, their oncogenic potential and cytopathic effects
in
permissive cells raise safety concerns. They can accommodate only up to 8 kb
of
foreign genetic material but can be readily introduced in a variety of cell
lines and
laboratory animals (Nicolas and Rubenstein, 1988; Temin, 1986).
The viral vectors used herein may be adenoviral, such as described in U.S.
Patent No. 5,824,544; U.S. Patent No. 5,707,618; U.S. Patent No. 5,693,509;
U.S.
CA 02381771 2002-02-07
WO 01/23571 PCT/US00/27119
-43-
Patent No. 5,670,488; U.S. Patent No. 5,585,362 retroviral, such as described
inU.S.
Patent No. 5,888,502; U.S. Patent No. 5,830,725; U.S. Patent No. 5,770,414;
U.S.
Patent No. 5,686,278; U.S. Patent No. 4,861,719; an adeno-associated viral,
such as
described inU.S. Patent No. 5,474,935; U.S. Patent No. 5,139,941; U.S. Patent
No.
5,622,856; U.S. Patent No. 5,658,776; U.S. Patent No. 5,773,289; U.S. Patent
No.
5,789,390; U.S. Patent No. 5,834,441; U.S. Patent No. 5,863,541; U.S. Patent
No.
5,851,521; U.S. Patent No. 5,252,479; an adenoviral-adenoassociated viral
hybrid,
such as described in U.S. Patent No. 5,856,152; and a vaccinia viral or a
herpesviral,
such as described in U.S. Patent No. 5,879,934; U.S. Patent No. 5,849,571;
U.S.
Patent No. 5,830,727; U.S. Patent No. 5,661,033; U.S. Patent No. 5,328,688
vector.
Delivery of the expression constructs through non-viral vectors also is
contemplated. Such delivery may employ microinjection (U.5. Patent No.
5,612,205), electroporation (U.S. Patent No. 5,507,724; U.S. Patent No.
5,869,326;
U.S. Patent No. 5,824,547; U.S. Patent No. 5,789,213; U.S. Patent No.
5,749,847;
U.S. Patent No. 5,019,034; Tur-Kaspa et al., 1986; Potter et al., 1984),
calcium
phosphate coprecipitation (Graham and Van Der Eb, 1973; Chen and Okayama,
1987;
Rippe et al., 1990), DEAF dextran introduction (Gopal, 1985), receptor
mediated
introduction (Wu and Wu, 1987; Wu and Wu, 1988), liposome mediated
introduction
(U.5. Patent No. 5,631,018; U.S. Patent No. 5,620,689; U.S. Patent No.
5,861,314;
U.S. Patent No. 5,855,910; U.S. Patent No. 5,851,818; U.S. Patent No.
5,827,703,
U.S. Patent No. 5,785,987; Nicolau and Sene, 1982; Fraley et al., 1979),
dendrimer
technology (U.S. Patent 5,795,581; U.S. Patent 5,714,166; U.S. Patent
5,661,025),
naked DNA injection (Harland and Weintraub, 1985) and particle bombardment
(U.S.
Patent No. 5,836,905; U.S. Patent No. 5,120,657; Yang et al., 1990).
Once the expression construct has been delivered into the cell the nucleic
acid
encoding the gene of interest may be positioned and expressed at different
sites. In
certain embodiments, the nucleic acid encoding the gene may be stably
integrated into
the genome of the cell. This integration may be in the cognate location and
orientation via homologous recombination (gene replacement) or it may be
integrated
in a random, non-specific location (gene augmentation). In yet further
embodiments,
the nucleic acid may be stably maintained in the cell as a separate, episomal
segment
of DNA. Such nucleic acid segments or "episomes" encode sequences sufficient
to
permit maintenance and replication independent of or in synchronization with
the host
CA 02381771 2002-02-07
WO 01/23571 PCT/US00/27119
-44-
cell cycle. How the expression construct is delivered to a cell and where in
the cell
the nucleic acid remains is dependent on the type of expression construct
employed.
6. Cell Culture and Propagation
Primary mammalian cell cultures may be prepared in various ways. In order
for the cells to be kept viable while in vitro and in contact with the
expression
construct, it is necessary to ensure that the cells maintain contact with the
correct ratio
of oxygen and carbon dioxide and nutrients but are protected from microbial
contamination. Cell culture techniques are well documented and are disclosed
herein
by reference (Freshner, 1992).
One embodiment of the foregoing involves the use of gene transfer to
immortalized cells for the production of proteins. The gene for the protein of
interest
may be transferred as described above into appropriate host cells followed by
culture
of cells under the appropriate conditions. The gene for virtually any
polypeptide may
be employed in this manner. The generation of recombinant expression vectors,
and
the elements included therein, are discussed above. Alternatively, the protein
to be
produced may be an endogenous protein normally synthesized by the cell in
question.
Examples of useful mammalian host cell lines are Vero and HeLa cells and
cell lines of Chinese hamster ovary, W138, BHK, COS-7, 293, HepG2, NIH3T3, RIN
and MDCK cells. In addition, a host cell strain may be chosen that modulates
the
expression of the inserted sequences, or modifies and process the gene product
in the
manner desired. Such modifications (e.g., glycosylation) and processing (e.g.,
cleavage) of protein products may be important for the function of the
protein.
Different host cells have characteristic and specific mechanisms for the post-
translational processing and modification of proteins. Appropriate cell lines
or host
systems can be chosen to insure the correct modification and processing of the
foreign
protein expressed.
A number of selection systems may be used including, but not limited to, HSV
thymidine kinase, hypoxanthine-guanine phosphoribosyltransferase and adenine
phosphoribosyltransferase genes, in tk-, hgprt- or apf-t- cells, respectively.
Also, anti-
metabolite resistance can be used as the basis of selection for dhfr, that
confers
resistance to methotrexate; gpt, that confers resistance to mycophenolic acid;
neo, that
CA 02381771 2002-02-07
WO 01/23571 PCT/US00/27119
-45-
confers resistance to the aminoglycoside 6418; and hygr-o, that confers
resistance to
hygromycin.
Animal cells can be propagated in vitro in two modes: as non-anchorage
dependent cells growing in suspension throughout the bulk of the culture or as
anchorage-dependent cells requiring attachment to a solid substrate for their
propagation (i.e., a monolayer type of cell growth).
Non-anchorage dependent or suspension cultures from continuous established
cell lines are the most widely used means of large scale production of cells
and cell
products. However, suspension cultured cells have limitations, such as
tumorigenic
potential and lower protein production than adherent cells.
Large scale suspension culture of mammalian cells in stirred tanks is a
common method for production of recombinant proteins. Two suspension culture
reactor designs are in wide use - the stirred reactor and the airlift reactor.
The stirred
design has successfully been used on an 8000 liter capacity for the production
of
interferon. Cells are grown in a stainless steel tank with a height-to-
diameter ratio of
1:1 to 3:1. The culture is usually mixed with one or more agitators, based on
bladed
disks or marine propeller patterns. Agitator systems offering less shear
forces than
blades have been described. Agitation may be driven either directly or
indirectly by
magnetically coupled drives. Indirect drives reduce the risk of microbial
contamination through seals on stirrer shafts.
The airlift reactor, also initially described for microbial fermentation and
later
adapted for mammalian culture, relies on a gas stream to both mix and
oxygenate the
culture. The gas stream enters a riser section of the reactor and drives
circulation.
Gas disengages at the culture surface, causing denser liquid free of gas
bubbles to
travel downward in the downcomer section of the reactor. The main advantage of
this
design is the simplicity and lack of need for mechanical mixing. Typically,
the
height-to-diameter ratio is 10:1. The airlift reactor scales up relatively
easily, has
good mass transfer of gases and generates relatively low shear forces.
The antibodies of the present invention are particularly useful for the
isolation
of antigens by immunoprecipitation. Immunoprecipitation involves the
separation of
the target antigen component from a complex mixture, and is used to
discriminate or
isolate minute amounts of protein. For the isolation of membrane proteins
cells must
be solubilized into detergent micelles. Nonionic salts are preferred, since
other agents
CA 02381771 2002-02-07
WO 01/23571 PCT/US00/27119
-46-
such as bile salts, precipitate at acid pH or in the presence of bivalent
cations.
Antibodies and their uses are discussed further, below.
7. Generating Antibodies Reactive With ~31A
S In another aspect, the present invention contemplates an antibody that is
immunoreactive with a (31 A molecule of the present invention, or any portion
thereof.
An antibody can be a polyclonal or a monoclonal antibody. In a preferred
embodiment, an antibody is a monoclonal antibody. Means for preparing and
characterizing antibodies are well known in the art (see, e.g., Harlow and
Lane,
1988).
Briefly, a polyclonal antibody is prepared by immunizing an animal with an
immunogen comprising a polypeptide of the present invention and collecting
antisera
from that immunized animal. A wide range of animal species can be used for the
production of antisera. Typically an animal used for production of antisera is
a non-
human animal including rabbits, mice, rats, hamsters, pigs or horses. Because
of the
relatively large blood volume of rabbits, a rabbit is a preferred choice for
production
of polyclonal antibodies.
Antibodies, both polyclonal and monoclonal, specific for isoforms of antigen
may be prepared using conventional immunization techniques, as will be
generally
known to those of skill in the art. A composition containing antigenic
epitopes of the
compounds of the present invention can be used to immunize one or more
experimental animals, such as a rabbit or mouse, which will then proceed to
produce
specific antibodies against the compounds of the present invention. Polyclonal
antisera may be obtained, after allowing time for antibody generation, simply
by
bleeding the animal and preparing serum samples from the whole blood.
It is proposed that the monoclonal antibodies of the present invention will
find
useful application in standard immunochemical procedures, such as ELISA and
Western blot methods and in immunohistochemical procedures such as tissue
staining, as well as in other procedures which may utilize antibodies specific
to (31A-
related antigen epitopes. Additionally, it is proposed that monoclonal
antibodies
specific to the particular (31A of different species may be utilized in other
useful
applications
CA 02381771 2002-02-07
WO 01/23571 PCT/US00/27119
-47-
In general, both polyclonal and monoclonal antibodies against (31 A may be
used in a variety of embodiments. For example, they may be employed in
antibody-
based cloning protocols to obtain cDNAs or genes encoding other (31 A They may
also be used in inhibition studies to analyze the effects of /31A related
peptides in cells
or animals. Anti-(31A antibodies will also be useful in immunolocalization
studies to
analyze the distribution of (31A during various cellular events, for example,
to
determine the cellular or tissue-specific distribution of (31A polypeptides
under
different points in the cell cycle. A particularly useful application of such
antibodies
is in purifying native or recombinant (31A, for example, using an antibody
affinity
column. The operation of all such immunolo,gical techniques will be known to
those
of skill in the art in light of the present disclosure.
Means for preparing and characterizing antibodies are well known in the art
(see, e.g., Harlow and Lane, 1988). More specific examples of monoclonal
antibody
preparation are give in the examples below.
As is well known in the art, a given composition may vary in its
immunogenicity. It is often necessary therefore to boost the host immune
system, as
may be achieved by coupling a peptide or polypeptide immunogen to a carrier.
Exemplary and preferred carriers are keyhole limpet hemocyanin (KLH) and
bovine
serum albumin (BSA). Other albumins such as ovalbumin, mouse serum albumin or
rabbit serum albumin can also be used as carriers. Means for conjugating a
polypeptide to a carrier protein are well known in the art and include
glutaraldehyde,
m-maleimidobencoyl-N-hydroxysuccinimide ester, carbodiimide and bis-biazotized
benzidine.
As also is well known in the art, the immunogenicity of a particular
immunogen composition can be enhanced by the use of non-specific stimulators
of
the immune response, known as adjuvants. Exemplary and preferred adjuvants
include complete Freund's adjuvant (a non-specific stimulator of the immune
response
containing killed Mycobacterium tuberculosis), incomplete Freund's adjuvants
and
aluminum hydroxide adjuvant.
The amount of immunogen composition used in the production of polyclonal
antibodies varies upon the nature of the immunogen as well as the animal used
for
immunization. A variety of routes can be used to administer the immunogen
WO 01/23571 cA o23amn 2002-02-0~ pCT~S00/27119
-48-
(subcutaneous, intramuscular, intradermal, intravenous and intraperitoneal).
The
production of polyclonal antibodies may be monitored by sampling blood of the
immunized animal at various points following immunization. A second, booster,
injection may also be given. The process of boosting and titering is repeated
until a
suitable titer is achieved. When a desired level of immunogenicity is
obtained, the
immunized animal can be bled and the serum isolated and stored, and/or the
animal
can be used to generate mAbs.
MAbs may be readily prepared through use of well-known techniques, such as
those exemplified in U.S. Patent 4,196,265. Typically, this technique involves
immunizing a suitable animal with a selected immunogen composition, e.g., a
purified
or partially purified (31A protein, polypeptide or peptide or cell expressing
high levels
of (31A The immunizing composition is administered in a manner effective to
stimulate antibody producing cells. Rodents such as mice and rats are
preferred
animals, however, the use of rabbit, sheep, or frog cells is also possible.
The use of
rats may provide certain advantages (Goding, 1986), but mice are preferred,
with the
BALB/c mouse being most preferred as this is most routinely used and generally
gives a higher percentage of stable fusions.
Following immunization, somatic cells with the potential for producing
antibodies, specifically B-lymphocytes (B-cells), are selected for use in the
mAb
generating protocol. These cells may be obtained from biopsied spleens,
tonsils or
lymph nodes, or from a peripheral blood sample. Spleen cells and peripheral
blood
cells are preferred, the former because they are a rich source of antibody-
producing
cells that are in the dividing plasmablast stage, and the latter because
peripheral blood
is easily accessible. Often, a panel of animals will have been immunized and
the
spleen of animal with the highest antibody titer will be removed and the
spleen
lymphocytes obtained by homogenizing the spleen with a syringe. Typically, a
spleen
from an immunized mouse contains approximately 5 x 10' to 2 x 1 Og
lymphocytes.
The antibody-producing B lymphocytes from the immunized animal are then
fused with cells of an immortal myeloma cell, generally one of the same
species as the
animal that was immunized. Myeloma cell lines suited for use in
hybridoma-producing fusion procedures preferably are non-antibody-producing,
have
high fusion efficiency, and enzyme deficiencies that render them incapable of
CA 02381771 2002-02-07
WO 01/23571 PCT/US00/27119
-49-
growing in certain selective media which support the growth of only the
desired fused
cells (hybridomas).
Any one of a number of myeloma cells may be used, as are known to those of
skill in the art (Goding, 1986; Campbell, 1984). For example, where the
immunized
animal is a mouse, one may use P3-X63/Ag8, P3-X63-Ag8.653, NS1/l.Ag 4 1,
Sp210-Agl4, FO, NSO/LJ, MPC-11, MPC11-X45-GTG 1.7 and S194/SXXO Bul; for
rats, one may use R210.RCY3, Y3-Ag 1.2.3, IR983F and 4B210; and U-266,
GM1500-GRG2, LICR-LON-HMy2 and UC729-6 are all useful in connection with
cell fusions.
Methods for generating hybrids of antibody-producing spleen or lymph node
cells and myeloma cells usually comprise mixing somatic cells with myeloma
cells in
a 2:1 ratio, though the ratio may vary from about 20:1 to about 1:1,
respectively, in
the presence of an agent or agents (chemical or electrical) that promote the
fusion of
cell membranes. Fusion methods using Sendai virus have been described (Kohler
and
Milstein, 1975; 1976), and those using polyethylene glycol (PEG), such as 37%
(v/v)
PEG, by Gefter et al., (1977). The use of electrically induced fusion methods
is also
appropriate (Goding, 1986).
Fusion procedures usually produce viable hybrids at low frequencies, around
1 x 10-6 to 1 x 10-8. However, this does not pose a problem, as the viable,
fused
hybrids are differentiated from the parental, unfused cells (particularly the
unfused
myeloma cells that would normally continue to divide indefinitely) by
culturing in a
selective medium. The selective medium is generally one that contains an agent
that
blocks the de novo synthesis of nucleotides in the tissue culture media.
Exemplary
and preferred agents are aminopterin, methotrexate, and azaserine. Aminopterin
and
methotrexate block de novo synthesis of both purines and pyrimidines, whereas
azaserine blocks only purine synthesis. Where aminopterin or methotrexate is
used,
the media is supplemented with hypoxanthine and thymidine as a source of
nucleotides (HAT medium). Where azaserine is used, the media is supplemented
with
hypoxanthine.
The preferred selection medium is HAT. Only cells capable of operating
nucleotide salvage pathways are able to survive in HAT medium. The myeloma
cells
are defective in key enzymes of the salvage pathway, e.g., hypoxanthine
phosphoribosyl transferase (HPRT), and they cannot survive. The B-cells can
operate
WO 01/23571 cA o23amn 2002-02-0~ pCT~S00/27119
-5 0-
this pathway, but they have a limited life span in culture and generally die
within
about two weeks. Therefore, the only cells that can survive in the selective
media are
those hybrids formed from myeloma and B-cells.
This culturing provides a population of hybridomas from which specific
hybridomas are selected. Typically, selection of hybridomas is performed by
culturing the cells by single-clone dilution in microtiter plates, followed by
testing the
individual clonal supernatants (after about two to three weeks) for the
desired
reactivity. The assay should be sensitive, simple and rapid, such as
radioimmunoassays, enzyme immunoassays, cytotoxicity assays, plaque assays,
dot
immunobinding assays, and the like.
The selected hybridomas would then be serially diluted and cloned into
individual antibody-producing cell lines, which clones can then be propagated
indefinitely to provide mAbs. The cell lines may be exploited for mAb
production in
two basic ways. A sample ofthe hybridoma can be injected (often into the
peritoneal
cavity) into a histocompatible animal of the type that was used to provide the
somatic
and myeloma cells for the original fusion. The injected animal develops tumors
secreting the specific monoclonal antibody produced by the fused cell hybrid.
The
body fluids of the animal, such as serum or ascites fluid, can then be tapped
to provide
mAbs in high concentration. The individual cell lines could also be cultured
in vitro,
where the mAbs are naturally secreted into the culture medium from which they
can
be readily obtained in high concentrations. mAbs produced by either means may
be
further purified, if desired, using filtration, centrifugation and various
chromatographic methods such as HPLC or affinity chromatography.
8. Methods for Screening Active Compounds
The present invention also contemplates the use of (31A and active fragments,
and nucleic acids coding thereof, in the screening of compounds for sodium
channel
modulator activity. Such activity may be a stimulatory activity in which the
activity
of the sodium channel is increased or an inhibitory activity in which the
activity of the
sodium channel is decreased. In certain instances, the modulator may be active
in
stimulating (31 A activity, overcoming the lack of X31 A or blocking the
effect of a
mutant (31A molecule. These assays may make use of a variety of different
formats
WO 01/23571 cA o23amn 2002-02-0~ PCT/US00/27119
-51-
and may depend on the kind of "activity" for which the screen is being
conducted.
Contemplated functional "read-outs" include binding to a compound, inhibition
of
binding to a substrate, ligand, receptor or other binding partner by a
compound, or a
functional readout such as monitoring sodium current density.
To date there are a number of agents known to alter the activity of sodium
channels. These agents include but are not limited to blockers and activators
of
sodium channels. The present section is directed to identifying additional
modulators
of sodium channel function using the findings of the present invention.
Useful compounds in this regard will not be limited to those mentioned above.
The active compounds may include fragments or parts of naturally-occurring
compounds or may be only found as active combinations of known compounds which
are otherwise inactive. However, prior to testing of such compounds in humans
or
animal models, it may be necessary to test a variety of candidates to
determine which
have potential.
Accordingly, in screening assays to identify useful agents which modulate
sodium channel function, it is proposed that compounds isolated from natural
sources,
such as animals, bacteria, fungi, plant sources, including leaves and bark,
and marine
samples may be assayed as candidates for the presence of potentially useful
pharmaceutical agents. It will be understood that the agents to be screened
could also
be derived or synthesized from chemical compositions or man-made compounds.
In these embodiments, the present invention is directed to a method for
determining the ability of a candidate substance to modulate the activity of
the sodium
channel, the method including generally the steps of
(a) obtaining a cell co-expressing a (31 A channel subunit with a sodium
channel a-subunit;
(b) admixing a candidate substance with the cell; and
(c) determining the ability of the candidate substance to alter the
sodium channel function of the cell.
To identify a candidate substance as being capable of modulating sodium
channel function, one would measure or determine the sodium current density of
the
cell that co-expresses a (31A channel subunit with a sodium channel a-subunit
in the
absence of the added candidate substance. One would then add the candidate
WO 01/23571 cA o23amn 2002-02-0~ PCT/LTS00/27119
-52-
substance to the cell and re-determine the sodium current density in the
presence of
the candidate substance. A candidate substance which produces an alteration in
the
sodium current density relative to the density in its absence is indicative of
a
candidate substance with modulatory capability.
The candidate screening assay is quite simple to set up and perform, and is
related in many ways to the assay discussed above for determining sodium
current
density. Thus, after obtaining an suitable test cell, one will admix an
effective amount
of candidate substance with the cell, under conditions which would allow the
sodium
channel to function.
"Effective amounts", in certain circumstances, are those amounts effective at
reproducibly altering sodium current density in comparison to the normal
levels in the
absence of the candidate substance. Compounds that achieve significant
appropriate
changes in activity will be used. If desired, a battery of compounds may be
screened
in vitro to identify other agents for use in the present invention.
A significant alteration in the sodium channel function, e.g., as measured
using sodium current density, are represented by an increase/decrease in
sodium
current density levels of at least about 30%-40%, and most preferably, by
changes of
at least about 50%, with higher values of course being possible.
Quantitative in vitro testing of the inhibitor is not a requirement of the
invention as it is generally envisioned that the agents will often be selected
on the
basis of their known properties or by structural and/or functional comparison
to those
agents already demonstrated to be effective. Therefore, the effective amounts
will
often be those amounts proposed to be safe for administration to animals in
another
context, for example, as disclosed herein.
A. In vitro Testing of Identified Compounds
In certain in vitro assays, the agents identified may be those which bind to
the
(31A molecule or fragment thereof. The polypeptide or fragment may be either
free in
solution, fixed to a support, expressed in or on the surface of a cell. Either
the
polypeptide or the compound may be labeled, thereby permitting determining of
binding.
In another embodiment, the assay may measure the inhibition of binding of
(31A to a natural or artificial substrate or binding partner. Competitive
binding assays
WO 01/23571 cA o23amn 2002-02-0~ pCT/LTS00/27119
-53-
can be performed in which one of the agents ((31A, binding partner or
compound) is
labeled. Usually, the polypeptide will be the labeled species. One may measure
the
amount of free label versus bound label to determine binding or inhibition of
binding.
Another technique for high throughput screening of compounds is described in
WO 84/03564. Large numbers of small peptide test compounds are synthesized on
a
solid substrate, such as plastic pins or some other surface. The peptide test
compounds are reacted with [31A and washed. Bound polypeptide is detected by
various methods.
Purified (31 A can be coated directly onto plates for use in the
aforementioned
drug screening techniques. However, non-neutralizing antibodies to the
polypeptide
can be used to immobilize the polypeptide to a solid phase. Also, fusion
proteins
containing a reactive region (preferably a terminal region) may be used to
link the
(31 A active region to a solid phase.
Various cell lines containing wild-type or natural or engineered mutations in
(31 A can be used to study various functional attributes of (31 A and how a
candidate
compound affects these attributes. Methods for engineering mutations are
described
elsewhere in this document. In such assays, the compound would be formulated
appropriately, given its biochemical nature, and contacted with a target cell.
Depending on the assay, culture may be required. The cell may then be examined
by
virtue of a number of different physiologic assays. Alternatively, molecular
analysis
may be performed in which the function of (31A, or related pathways, may be
explored. This may involve assays such as those for protein expression, enzyme
function, substrate utilization, mRNA expression (including differential
display of
whole cell or polyA RNA) and others.
B. In Vivo Assays
The present invention also encompasses the use of various animal models.
Here, the identity seen between human and rat (31A provides an excellent
opportunity
to examine the function of (31A in a whole animal system where it is normally
expressed.
Treatment of animals with test compounds will involve the administration of
the compound, in an appropriate form, to the animal. Administration will be by
any
WO 01/23571 cA o23amn 2002-02-0~ PCT/US00/27119
-54-
route the could be utilized for clinical or non-clinical purposes, including
but not
limited to oral, nasal, buccal, rectal, vaginal or topical. Alternatively,
administration
may be by intratracheal instillation, bronchial instillation, intradermal,
subcutaneous,
intramuscular, intraperitoneal or intravenous injection. Specifically
contemplated are
systemic intravenous injection, regional administration via blood or lymph
supply and
intratumoral injection.
C. Rational Drug Design
The goal of rational drug design is to produce structural analogs of
biologically active polypeptides or compounds with which they interact
(agonists,
antagonists, inhibitors, binding partners, etc.). By creating such analogs, it
is possible
to fashion drugs which are more active or stable than the natural molecules,
which
have different susceptibility to alteration or which may affect the function
of various
other molecules. In one approach, one would generate a three-dimensional
structure
for (31A or a fragment thereof. This could be accomplished by x-ray
crystallography,
computer modeling or by a combination of both approaches. An alternative
approach,
"alanine scan," involves the random replacement of residues throughout
molecule
with alanine, and the resulting affect on function determined.
It also is possible to isolate a ~31A specific antibody, selected by a
functional
assay, and then solve its crystal structure. In principle, this approach
yields a
pharmacore upon which subsequent drug design can be based. It is possible to
bypass
protein crystallography altogether by generating anti-idiotypic antibodies to
a
functional, pharmacologically active antibody. As a mirror image of a mirror
image,
the binding site of anti-idiotype would be expected to be an analog of the
original
antigen. The anti-idiotype could then be used to identify and isolate peptides
from
banks of chemically- or biologically-produced peptides. Selected peptides
would then
serve as the pharmacore. Anti-idiotypes may be generated using the methods
described herein for producing antibodies, using an antibody as the antigen.
Thus, one may design drugs which have improved (31A activity or which act
as stimulators, inhibitors, agonists, antagonists or (31A or molecules
affected by (31A
function. By virtue of the availability of cloned (31A sequences, sufficient
amounts of
~31A can be produced to perform crystallographic studies. In addition,
knowledge of
WO 01/23571 cA o23amn 2002-02-0~ PCT/US00/27119
-5 5-
the polypeptide sequences permits computer employed predictions of structure-
function relationships.
D. Known Modulators of Sodium Channel Function
As stated above, it may be that known modulators of sodium channel function
are employed as a starting point to generate new and novel compounds that
would be
useful as Mockers or activators of sodium channel function.
Sodium channel blockers are a well characterized group of pharmaceutical
agents especially in the control of arrhythmia. These drugs include but are
not limited
to quinidine, phenytoin, mexiletine, tocainide, procainamide, disopyramide,
moricizine, propafenone, flecainide, and the like. For a more detailed
description of
the mechanisms of action of such drugs, those of skill in the art are referred
to
Goodman and Gilman's "Pharmaceutical Basis of Therapeutics" Chapter 35, ninth
edition, Eds. Hardman et al., 1996.
In addition, local anesthetics which act by preventing the generation and
conductance of the nerve impulse may also be useful as starting materials for
the
rational drug design discussed herein. These agents block conduction by
decreasing
or preventing the large transient increases in permeability of excitable
membranes to
Na+ that normally is produced by a slight depolarization of the membrane. This
is a
result of a direct interaction of the anesthetic with the voltage gated sodium
channels.
Exemplary anesthetic agents that may be used to block these channels include
but are
not limited to lidocaine, benzocaine, bupivacaine, cocaine, etidocaine,
mepivacaine,
promoxine, prilocaine, procaine, proparacaine, ropivacaine and tetracaine. For
a more
detailed description of the mechanisms of action of such drugs those of skill
in the art
are referred to Goodman and Gilman's "Pharmaceutical Basis of Therapeutics"
Chapter 15, ninth edition, Eds. Hardman et al., 1996. Also useful in sodium
channel
blockade are the toxins based on tetrodotoxin and saxitoxin, two of the most
potent
poisons known. These agents may be used in rational drug design to produce a
therapeutic that is less toxic but still an effective modulator of the sodium
channel.
In addition to the above agents commonly being used in the treatment of
arrhythmia, certain sodium channel Mockers also are known to be effective in
anti-
convulsant therapy. These agents include but are not limited to phenytoin,
carbamazepine, valproate, lamotrigine and topiramate. For a more detailed
review of
WO 01/23571 cA o23amn 2002-02-0~ pCT/US00/27119
-56-
these agents and their anticonvulsant activity, those of skill in the art are
referred to a
recent review by Ragsdale and Avoli (1999). It is envisioned that any of these
specific agents or any agents derived from these agents may be useful in
various
applications of the previous invention, including rational drug design,
combination
therapy and the like.
9. Assays for Determining Sodium Channel Function
In various aspects of the present invention it may be necessary to perform a
variety of assays to determine the activity of a particular voltage gated
sodium
channel. Such assays may measure various properties such as repulsion form
TNR,
cell adhesion assays, immunocytochemical assays and the like. The following
section
describes these assays in some detail. Of course it should be understood that
the
conditions described herein are merely exemplary and those of skill in the art
will be
able to modify or alter the conditions depending on the particular cells lines
and
reagents being used.
A. Long-term repulsion assays
The inventors found that sodium channel a, X31 and (32 subunits, when
expressed alone, are modulated specifically by TN-R to produce a repellent
effect of
the transfected cells away from the TN-R substrate. In investigating this
effect,
repulsion assays were carried out as follows.
Tissue culture 4-well or 24-well dishes are coated with methanol-solubilized
nitrocellulose according to Lagenaur and Lemmon (1987). 2.5 ~l aliquots of TN-
R
(15 nM) and TN-C (15 nM) or GST-fusion domains of TN-R (25 nM) are applied to
the nitrocellulose/poly-DL-ornithine (PO)-coated surfaces of the dishes and
incubated
for 2 h at 37 °C in a humidified atmosphere as described previously
(Xiao et al.,
1996). The dishes are then washed three times with Ca2+- and Mg2+-free Hank's
balanced salt solution (CMF-HBSS). The coating efficiency is determined as
described previously (Xiao et al., 1996). Substrate boundaries are marked in
ink. The
source of nitrocellulose (Schleicher and Schuell, catalog #401188, BA85,
0.45~m) is
important to obtain consistent results in this assay. Parental 1610 or
transfected cells
are plated at a density of 105 cells/ml. After 20 h, the cells are fixed with
2.5%
WO 01/23571 cA o23amn 2002-02-0~ pCT/US00/27119
-57-
glutaraldehyde and stained with Coomassie blue (Sigma Chemical Co., St. Louis,
Mo.) and the number of cells adhering to the extracellular matrix-coated
protein areas
is counted under a microscope.
B. Adhesion Assays
Sodium channels appear to recognize substrates such as TN-R by an initial
adhesion. An exemplary adhesion assay that may be employed is described as
follows. Tissue culture 4-well or 24-well dishes are coated with methanol-
solubilized
nitrocellulose according to Lagenaur and Lemmon (1987) and air-dried under a
sterile
hood. For adhesion assays, 2.5 p1 spots of different TN-R fragments or GST
(each at.a
concentration of 25 ~M) are applied to the nitrocelluose-coated surfaces of
the dishes
and incubated for 2 h at 37 °C in a humidified atmosphere. The dried
spots are
washed with PBS and then flooded with CMF-HBSS containing 2% heat-inactivated
fatty acid free BSA (Sigma) and incubated 2 h to block residual non-specific
protein
binding sites. The dishes are then washed with PBS and cells from the various
cell
lines are plated at a density of 105 cells/ml in 0.5 ml of growth medium
containing
10% BSA. After 20 h of growth (5% CO2, 37°C) cultures are fixed with
CMF-HBSS
containing 2.5% glutaraldehyde. For adhesion blocking assays, a mixture of EGF-
L,
EGF-S, and FN6-8 can be added to the culture medium. After fixation, cultures
are
stained with 0.5% toluidine blue in 2.5% sodium carbonate. Cells adhering to
the
various spots of TN-R fragments can then be photographed and counted.
C. Aggregation assays
Stable clones of transfected S2 cells are induced overnight in the presence of
0.7 mM CuSOa followed by aggregation for 4 h at room temperature on a rotary
shaker. Cell aggregates containing at least 10 cells are then analyzed by
phase
contrast microscopy for homophilic aggregation or by a combination of phase
contrast
and fluorescent microscopy for heterophilic aggregation.
D. Immunocytochemical detection of ankyrin in S2 cells
Immunocytochemistry of Drosophila ankyrin distribution in S2 cells is
performed following aggregation experiments. Cells are fixed with 2%
paraformaldehyde, and permeabilized with 0.5% Triton X-100. Mouse anti-
CA 02381771 2002-02-07
WO 01/23571 PCT/US00/27119
-5 8-
Drosophila ankyrin is used as the primary antibody followed by incubation with
fluorescein isothiocyanate-conjugated secondary antibody. Slides are then
viewed
with a Bio-Rad MRC 600 confocal scanning laser microscope.
E. Co-immunoprecipitation of 1 and 2 subunits or subunits with
neurofascin or ankyrin
Immunoprecipitations can be performed from S2 cells following protein
synthesis induction but without inducing cell aggregation (cells not shaken).
Cells are
then pelleted and solubilized in 1.25% Triton X-100 and the soluble fraction
is
incubated overnight with the appropriate antibody. Protein-A-sepharose is then
added
and the incubation continued for 2 h. Immunoprecipitates are eluted from the
Protein-
A-sepharose with SDS-PAGE sample buffer and separated on 10 % SDS-PAGE gels.
Proteins are then transferred to nitrocellulose and probed with the
appropriate second
antibody.
10. Diagnosing Disorders Involving ~ilA
The present inventors have determined that alterations in (31A may be
associated with epileptic and fibrillar seizures. Therefore, (31A and the
corresponding
gene may be employed as a diagnostic or prognostic indicator of such seizures
in
infants. More specifically, point mutations, deletions, insertions or
regulatory
perturbations relating to (31A may be the cause of these seizures. If it can
be predicted
that an individual is predisposed to such seizures, then a prophylactic course
of
treatments can be designed.
A. Genetic Diagnosis
One embodiment of the instant invention comprises a method for detecting
variation in the expression of (31A. This may comprise determining that level
of (31A
or determining specific alterations in the expressed product. Obviously, this
sort of
assay has importance in the diagnosis of related disease.
The biological sample can be any tissue or fluid. Various embodiments
include cells of the skin, muscle, fascia, brain, prostate, breast,
endometrium, lung,
head & neck, pancreas, small intestine, blood cells, liver, testes, ovaries,
colon, skin,
WO 01/23571 cA o23amn 2002-02-0~ PCT/US00/27119
-59-
stomach, esophagus, spleen, lymph node, bone marrow or kidney. Other
embodiments include fluid samples such as peripheral blood, lymph fluid,
ascites,
serous fluid, pleural effusion, sputum, cerebrospinal fluid, lacrimal fluid,
stool or
urine.
Nucleic acid to be used in such an analysis is isolated from cells contained
in
the biological sample, according to standard methodologies (Sambrook et al.,
1989).
The nucleic acid may be genomic DNA or fractionated or whole cell RNA. Where
RNA is used, it may be desired to convert the RNA to a complementary DNA. In
one
embodiment, the RNA is whole cell RNA; in another, it is poly-A RNA. Normally,
the nucleic acid is amplified.
Depending on the format, the specific nucleic acid of interest is identified
in
the sample directly using amplification or with a second, known nucleic acid
following amplification. Next, the identified product is detected. In certain
applications, the detection may be performed by visual means (e.g., ethidium
bromide
staining of a gel). Alternatively, the detection may involve indirect
identification of
the product via chemiluminescence, radioactive scintigraphy of radiolabel or
fluorescent label or even via a system using electrical or thermal impulse
signals
(Affymax Technology; Bellus, 1994).
Following detection, one may compare the results seen in a given patient with
a statistically significant reference group of normal patients and patients
that have
(31A-related pathologies. In this way, it is possible to correlate the amount
or kind of
(31 A detected with various clinical states.
Various types of defects may be associated with (31A. Thus, "alterations"
should be read as including deletions, insertions, point mutations and
duplications.
Point mutations result in stop codons, frameshift mutations or amino acid
substitutions. Somatic mutations are those occurnng in non-germ line tissues.
Germ
line tissue mutations can occur in any tissue and are inherited. Mutations in
and
outside the coding region also may affect the amount of (31A produced, both by
altering the transcription of the gene or in destabilizing or otherwise
altering the
processing of either the transcript (mRNA) or protein, or altering the ratio
of
expression of one subunit in comparison to another.
WO 01/23571 cA o23amn 2002-02-0~ PCT/US00/27119
-60-
It is contemplated that mutations in the (31 A gene may be identified in
accordance with the present invention. A variety of different assays are
contemplated
in this regard, including but not limited to, fluorescent in situ
hybridization (FISH),
direct DNA sequencing, PFGE analysis, Southern or Northern blotting, single-
stranded conformation analysis (SSCA), RNAse protection assay, allele-specific
oligonucleotide (ASO), dot blot analysis, denaturing gradient gel
electrophoresis,
RFLP and PCRTM-SSCP.
a. Primers and Probes
The term primer, as defined herein, is meant to encompass any nucleic acid
that is capable of priming the synthesis of a nascent nucleic acid in a
template-
dependent process. Typically, primers are oligonucleotides from ten to twenty
base
pairs in length, but longer sequences can be employed. Primers may be provided
in
double-stranded or single-stranded form, although the single-stranded form is
preferred. Probes are defined differently, although they may act as primers.
Probes,
while perhaps capable of priming, are designed to binding to the target DNA or
RNA
and need not be used in an amplification process.
In preferred embodiments, the probes or primers are labeled with radioactive
species (32P, laC, 3sS, 3H, or other label), with a fluorophore (rhodamine,
fluorescein)
or a chemillumiscent (luciferase).
b. Template Dependent Amplification Methods
A number of template dependent processes are available to amplify the marker
sequences present in a given template sample. One of the best known
amplification
methods is the polymerise chain reaction (referred to as PCRTM) which is
described in
detail in U.S. Patent Nos. 4,683,195, 4,683,202 and 4,800,159, and in Innis et
al.,
1990.
Briefly, in PCRTM, two primer sequences are prepared that are complementary
to regions on opposite complementary strands of the marker sequence. An excess
of
deoxynucleoside triphosphates are added to a reaction mixture along with a DNA
polymerise, e.g., Taq polymerise. If the marker sequence is present in a
sample, the
primers will bind to the marker and the polymerise will cause the primers to
be
WO 01/23571 cA o23amn 2002-02-0~ PCT/US00/27119
-61-
extended along the marker sequence by adding on nucleotides. By raising and
lowering the temperature of the reaction mixture, the extended primers will
dissociate
from the marker to form reaction products, excess primers will bind to the
marker and
to the reaction products and the process is repeated.
A reverse transcriptase PCRTM amplification procedure may be performed in
order to quantify the amount of mRNA amplified. Methods of reverse
transcribing
RNA into cDNA are well known and described in Sambrook et al., 1989.
Alternative
methods for reverse transcription utilize thermostable, RNA-dependent DNA
polymerases. These methods are described in WO 90/07641 filed December 21,
1990. Polymerase chain reaction methodologies are well known in the art.
Another method for amplification is the ligase chain reaction ("LCR"),
disclosed in EPO No. 320 308. In LCR, two complementary probe pairs are
prepared,
and in the presence of the target sequence, each pair will bind to opposite
complementary strands of the target such that they abut. In the presence of a
ligase,
the two probe pairs will link to form a single unit. By temperature cycling,
as in
PCRTM, bound ligated units dissociate from the target and then serve as
"target
sequences" for ligation of excess probe pairs. U.S. Patent 4,883,750 describes
a
method similar to LCR for binding probe pairs to a target sequence.
Q/3 Replicase, described in PCT Application No. PCT/L1S87/00880, may also
be used as still another amplification method in the present invention. In
this method,
a replicative sequence of RNA that has a region complementary to that of a
target is
added to a sample in the presence of an RNA polymerase. The polymerase will
copy
the replicative sequence that can then be detected.
An isothermal amplification method, in which restriction endonucleases and
ligases are used to achieve the amplification of target molecules that contain
nucleotide 5'-[alpha-thio]-triphosphates in one strand of a restriction site
may also be
useful in the amplification of nucleic acids in the present invention, Walker
et al.,
(1992).
Strand Displacement Amplification (SDA) is another method of carrying out
isothermal amplification of nucleic acids which involves multiple rounds of
strand
displacement and synthesis, i.e., nick translation. A similar method, called
Repair
Chain Reaction (RCR), involves annealing several probes throughout a region
WO 01/23571 cA o23amn 2002-02-0~ PCT/US00/27119
-62-
targeted for amplification, followed by a repair reaction in which only two of
the four
bases are present. The other two bases can be added as biotinylated
derivatives for
easy detection. A similar approach is used in SDA. Target specific sequences
can
also be detected using a cyclic probe reaction (CPR). In CPR, a probe having
3' and
5' sequences of non-specific DNA and a middle sequence of specific RNA is
hybridized to DNA that is present in a sample. Upon hybridization, the
reaction is
treated with RNase H, and the products of the probe identified as distinctive
products
that are released after digestion. The original template is annealed to
another cycling
probe and the reaction is repeated.
Still another amplification methods described in GB Application No. 2 202
328, and in PCT Application No. PCT/US89/01025, may be used in accordance with
the present invention. In the former application, "modified" primers are used
in a
PCRTM-like, template- and enzyme-dependent synthesis. The primers may be
modified by labeling with a capture moiety (e.g., biotin) and/or a detector
moiety
(e.g., enzyme). In the latter application, an excess of labeled probes are
added to a
sample. In the presence of the target sequence, the probe binds and is cleaved
catalytically. After cleavage, the target sequence is released intact to be
bound by
excess probe. Cleavage of the labeled probe signals the presence of the target
sequence.
Other nucleic acid amplification procedures include transcription-based
amplification systems (TAS), including nucleic acid sequence based
amplification
(NASBA) and 3SR (Kwoh et al., 1989; Gingeras et al., PCT Application WO
88/10315). In NASBA, the nucleic acids can be prepared for amplification by
standard phenol/chloroform extraction, heat denaturation of a clinical sample,
treatment with lysis buffer and minispin columns for isolation of DNA and RNA
or
guanidinium chloride extraction of RNA. These amplification techniques involve
annealing a primer which has target specific sequences. Following
polymerization,
DNA/RNA hybrids are digested with RNase H while double stranded DNA~molecules
are heat denatured again. In either case the single stranded DNA is made fully
double
stranded by addition of second target specific primer, followed by
polymerization.
The double-stranded DNA molecules are then multiply transcribed by an RNA
polymerise such as T7 or SP6. In an isothermal cyclic reaction, the RNA's are
reverse transcribed into single stranded DNA, which is then converted to
double
WO 01/23571 cA o23amn 2002-02-0~ pCT/US00/27119
-63-
stranded DNA, and then transcribed once again with an RNA polymerise such as
T7
or SP6. The resulting products, whether truncated or complete, indicate target
specific sequences.
Davey et al., EPO No. 329 822 disclose a nucleic acid amplification process
involving cyclically synthesizing single-stranded RNA ("ssRNA"), ssDNA, and
double-stranded DNA (dsDNA), which may be used in accordance with the present
invention. The ssRNA is a template for a first primer oligonucleotide, which
is
elongated by reverse transcriptase (RNA-dependent DNA polymerise). The RNA is
then removed from the resulting DNA:RNA duplex by the action of ribonuclease H
(RNase H, an RNase specific for RNA in duplex with either DNA or RNA). The
resultant ssDNA is a template for a second primer, which also includes the
sequences
of an RNA polymerise promoter (exemplified by T7 RNA polymerise) S' to its
homology to the template. This primer is then extended by DNA polymerise
(exemplified by the large "Klenow" fragment of E. coli DNA polymerise I),
resulting
in a double-stranded DNA ("dsDNA") molecule, having a sequence identical to
that
of the original RNA between the primers and having additionally, at one end, a
promoter sequence. This promoter sequence can be used by the appropriate RNA
polymerise to make many RNA copies of the DNA. These copies can then re-enter
the cycle leading to very swift amplification. With proper choice of enzymes,
this
amplification can be done isothermally without addition of enzymes at each
cycle.
Because of the cyclical nature of this process, the starting sequence can be
chosen to
be in the form of either DNA or RNA.
Miller et al., PCT Application WO 89/06700, disclose a nucleic acid sequence
amplification scheme based on the hybridization of a promoter/primer sequence
to a
target single-stranded DNA ("ssDNA") followed by transcription of many RNA
copies of the sequence. This scheme is not cyclic, i.e., new templates are not
produced from the resultant RNA transcripts. Other amplification methods
include
"RACE" and "one-sided PCRTM" (Frohman, M.A., In: PCRTM PROTOCOLS: A
GUIDE TO METHODS AND APPLICATIONS, Academic Press, N.Y., 1990; Ohara
et al., 1989.
Methods based on ligation of two (or more) oligonucleotides in the presence
of nucleic acid having the sequence of the resulting "di-oligonucleotide",
thereby
WO 01/23571 cA o23ai~~i 2002-02-0~ PCT/US00/27119
-64-
amplifying the di-oligonucleotide, may also be used in the amplification step
of the
present invention (Wu et al., 1989).
c. Southern/Northern Blotting
Blotting techniques are well known to those of skill in the art. Southern
blotting involves the use of DNA as a target, whereas Northern blotting
involves the
use of RNA as a target. Each provide different types of information, although
cDNA
blotting is analogous, in many aspects, to blotting of RNA species.
Briefly, a probe is used to target a DNA or RNA species that has been
immobilized on a suitable matrix, often a nitrocellulose filter. The different
species
should be spatially separated to facilitate analysis. This often is
accomplished by gel
electrophoresis of nucleic acid species followed by "blotting" on to the
filter.
Subsequently, the blotted target is incubated with a probe (usually labeled)
under conditions that promote denaturation and rehybridization. Because the
probe is
designed to base pair with the target, the probe will bind a portion of the
target
sequence under renaturing conditions. Unbound probe is then removed, and
detection
is accomplished as described above.
d. Separation Methods
It normally is desirable, at one stage or another, to separate the
amplification
product from the template and the excess primer for the purpose of determining
whether specific amplification has occurred. In one embodiment, amplification
products are separated by agarose, agarose-acrylamide or polyacrylamide gel
electrophoresis using standard methods. See Sambrook et al. (1989).
Alternatively, chromatographic techniques may be employed to effect
separation. There are many kinds of chromatography which may be used in the
present invention: adsorption, partition, ion-exchange and molecular sieve,
and many
specialized techniques for using them, including column, paper, thin-layer and
gas
chromatography.
e. Detection Methods
Products may be visualized in order to confirm amplification of the marker
sequences. One typical visualization method involves staining of a gel with
ethidium
WO 01/23571 cA o23amn 2002-02-0~ PCT/US00/27119
-65-
bromide and visualization under UV light. Alternatively, if the amplification
products
are integrally labeled with radio- or fluorometrically-labeled nucleotides,
the
amplification products can then be exposed to x-ray film or visualized under
the
appropriate stimulating spectra, following separation.
In one embodiment, visualization is achieved indirectly. Following separation
of amplification products, a labeled nucleic acid probe is brought into
contact with the
amplified marker sequence. The probe preferably is conjugated to a chromophore
but
may be radiolabeled. In another embodiment, the probe is conjugated to a
binding
partner, such as an antibody or biotin, and the other member of the binding
pair
carnes a detectable moiety.
In one embodiment, detection is by a labeled probe. The techniques involved
are well known to those of skill in the art and can be found in many standard
books on
molecular protocols. See Sambrook et al. (1989). For example, chromophore or
radiolabel probes or primers identify the target during or following
amplification.
One example of the foregoing is described in U.S. Patent No. 5,279,721,
which discloses an apparatus and method for the automated electrophoresis and
transfer of nucleic acids. The apparatus permits electrophoresis and blotting
without
external manipulation of the gel and is ideally suited to carrying out methods
according to the present invention.
In addition, the amplification products described above may be subjected to
sequence analysis to identify specific kinds of variations using standard
sequence
analysis techniques. Within certain methods, exhaustive analysis of genes is
carried
out by sequence analysis using primer sets designed for optimal sequencing
(Pigeon
et al, 1994). The present invention provides methods by which any or all of
these
types of analyses may be used. Using the sequences disclosed herein,
oligonucleotide
primers may be designed to permit the amplification of sequences throughout
the (31A
encoding nucleic acid that may then be analyzed by direct sequencing.
f. Kit Components
All the essential materials and reagents required for detecting and sequencing
(3IA and variants thereof may be assembled together in a kit. This generally
will
comprise preselected primers and probes. Also included may be enzymes suitable
for
amplifying nucleic acids, including various polymerases (RT, Taq, SequenaseTM
etc.),
WO 01/23571 cA o23amn 2002-02-0~ pCT/US00/27119
-66-
deoxynucleotides and buffers to provide the necessary reaction mixture for
amplification. Such kits also generally will comprise, in suitable means,
distinct
containers for each individual reagent and enzyme as well as for each primer
or probe.
g. Design and Theoretical Considerations for Relative
Quantitative RT-PCRTM
Reverse transcription (RT) of RNA to cDNA followed by relative quantitative
PCRTM (RT-PCRTM) can be used to determine the relative concentrations of
specific
mRNA species isolated from patients. By determining that the concentration of
a
specific mRNA species varies, it is shown that the gene encoding the specific
mRNA
species is differentially expressed.
In PCRTM, the number of molecules of the amplified target DNA increase by a
factor approaching two with every cycle of the reaction until some reagent
becomes
limiting. Thereafter, the rate of amplification becomes increasingly
diminished until
there is no increase in the amplified target between cycles. If a graph is
plotted in
which the cycle number is on the X axis and the log of the concentration of
the
amplified target DNA is on the Y axis, a curved line of characteristic shape
is formed
by connecting the plotted points. Beginning with the first cycle, the slope of
the line
is positive and constant. This is said to be the linear portion of the curve.
After a
reagent becomes limiting, the slope of the line begins to decrease and
eventually
becomes zero. At this point the concentration of the amplified target DNA
becomes
asymptotic to some fixed value. This is said to be the plateau portion of the
curve.
The concentration of the target DNA in the linear portion of the PCRTM
amplification is directly proportional to the starting concentration of the
target before
the reaction began. By determining the concentration of the amplified products
of the
target DNA in PCRTM reactions that have completed the same number of cycles
and
are in their linear ranges, it is possible to determine the relative
concentrations of the
specific target sequence in the original DNA mixture. If the DNA mixtures are
cDNAs synthesized from RNAs isolated from different tissues or cells, the
relative
abundances of the specific mRNA from which the target sequence was derived can
be
determined for the respective tissues or cells. This direct proportionality
between the
WO 01/23571 cA o23ai~~i 2002-02-0~ PCT/US00/27119
-67-
concentration of the PCRTM products and the relative mRNA abundances is only
true
in the linear range of the PCRTM reaction.
The final concentration of the target DNA in the plateau portion of the curve
is
determined by the availability of reagents in the reaction mix and is
independent of
the original concentration of target DNA. Therefore, the first condition that
must be
met before the relative abundances of a mRNA species can be determined by RT-
PCRTM for a collection of RNA populations is that the concentrations of the
amplified
PCRTM products must be sampled when the PCRTM reactions are in the linear
portion
of their curves.
The second condition that must be met for an RT-PCRTM experiment to
successfully determine the relative abundances of a particular mRNA species is
that
relative concentrations of the amplifiable cDNAs must be normalized to some
independent standard. The goal of an RT-PCRTM experiment is to determine the
abundance of a particular mRNA species relative to the average abundance of
all
mRNA species in the sample. In the experiments described below, mRNAs for 13-
actin, asparagine synthetase and lipocortin II were used as external and
internal
standards to which the relative abundance of other mRNAs are compared.
Most protocols for competitive PCRTM utilize internal PCRTM standards that
are approximately as abundant as the target. These strategies are effective if
the
products of the PCRTM amplifications are sampled during their linear phases.
If the
products are sampled when the reactions are approaching the plateau phase,
then the
less abundant product becomes relatively over represented. Comparisons of
relative
abundances made for many different RNA samples, such as is the case when
examining RNA samples for differential expression, become distorted in such a
way
as to make differences in relative abundances of RNAs appear less than they
actually
are. This is not a significant problem if the internal standard is much more
abundant
than the target. If the internal standard is more abundant than the target,
then direct
linear comparisons can be made between RNA samples.
The above discussion describes theoretical considerations for an RT-PCRTM
assay for clinically derived materials. The problems inherent in clinical
samples are
that they are of variable quantity (making normalization problematic), and
that they
are of variable quality (necessitating the co-amplification of a reliable
internal control,
WO 01/23571 cA o23ai~~i 2002-02-0~ pCT~S00/27119
-68-
preferably of larger size than the target). Both of these problems are
overcome if the
RT-PCRTM is performed as a relative quantitative RT-PCRTM with an internal
standard
in which the internal standard is an amplifiable cDNA fragment that is larger
than the
target cDNA fragment and in which the abundance of the mRNA encoding the
internal standard is roughly S-100 fold higher than the mRNA encoding the
target.
This assay measures relative abundance, not absolute abundance of the
respective
mRNA species.
Other studies may be performed using a more conventional relative
quantitative RT-PCRTM assay with an external standard protocol. These assays
sample the PCRTM products in the linear portion of their amplification curves.
The
number of PCRTM cycles that are optimal for sampling must be empirically
determined for each target cDNA fragment. In addition, the reverse
transcriptase
products of each RNA population isolated from the various tissue samples must
be
carefully normalized for equal concentrations of amplifiable cDNAs. This
consideration is very important since the assay measures absolute mRNA
abundance.
Absolute mRNA abundance can be used as a measure of differential gene
expression
only in normalized samples. While empirical determination of the linear range
of the
amplification curve and normalization of cDNA preparations are tedious and
time
consuming processes, the resulting RT-PCRTM assays can be superior to those
derived
from the relative quantitative RT-PCRTM assay with an internal standard.
One reason for this advantage is that without the internal
standard/competitor,
all of the reagents can be converted into a single PCRTM product in the linear
range of
the amplification curve, thus increasing the sensitivity of the assay. Another
reason is
that with only one PCRTM product, display of the product on an electrophoretic
gel or
another display method becomes less complex, has less background and is easier
to
interpret.
h. Chip Technologies
Specifically contemplated by the present inventors are chip-based DNA
technologies such as those described by Hacia et al. ( 1996) and Shoemaker et
al.
(1996). Briefly, these techniques involve quantitative methods for analyzing
large
numbers of genes rapidly and accurately. By tagging genes with
oligonucleotides or
WO 01/23571 cA o23amn 2002-02-0~ PCT/US00/27119
-69-
using fixed probe arrays, one can employ chip technology to segregate target
molecules as high density arrays and screen these molecules on the basis of --
hybridization. See also Pease et al. (1994); Fodor et al. (1991).
B. Immunodiagnosis
Antibodies of the present invention can be used in characterizing the (31A
content of healthy and diseased tissues, through techniques such as ELISAs and
Western blotting. This may provide a screen for the presence or absence of a
disorder
or as a predictor of future dysfunction.
The use of antibodies of the present invention, in an ELISA assay is
contemplated. For example, anti-(31A antibodies are immobilized onto a
selected
surface, preferably a surface exhibiting a protein affinity such as the wells
of a
polystyrene microtiter plate. After washing to remove incompletely adsorbed
material, it is desirable to bind or coat the assay plate wells with a non-
specific protein
that is known to be antigenically neutral with regard to the test antisera
such as bovine
serum albumin (BSA), casein or solutions of powdered milk. This allows for
blocking of non-specific adsorption sites on the immobilizing surface and thus
reduces the background caused by non-specific binding of antigen onto the
surface.
After binding of antibody to the well, coating with a non-reactive material to
reduce background, and washing to remove unbound material, the immobilizing
surface is contacted with the sample to be tested in a manner conducive to
immune
complex (antigen/antibody) formation.
Following formation of specific immunocomplexes between the test sample
and the bound antibody, and subsequent washing, the occurrence and even amount
of
immunocomplex formation may be determined by subjecting same to a second
antibody having specificity for ~31A that differs the first antibody.
Appropriate
conditions preferably include diluting the sample with diluents such as BSA,
bovine
gamma globulin (BGG) and phosphate buffered saline (PBS)/Tween°. These
added
agents also tend to assist in the reduction of nonspecific background. The
layered
antisera is then allowed to incubate for from about 2 to about 4 hr, at
temperatures
preferably on the order of about 25° to about 27°C. Following
incubation, the
antisera-contacted surface is washed so as to remove non-immunocomplexed
WO 01/23571 cA o23amn 2002-02-0~ PCT/US00/27119
-70-
material. A preferred washing procedure includes washing with a solution such
as
PBS/Tween~, or borate buffer.
To provide a detecting means, the second antibody will preferably have an
associated enzyme that will generate a color development upon incubating with
an
appropriate chromogenic substrate. Thus, for example, one will desire to
contact and
incubate the second antibody-bound surface with a urease or peroxidase-
conjugated
anti-human IgG for a period of time and under conditions which favor the
development of immunocomplex formation (e.g., incubation for 2 hr at room
temperature in a PBS-containing solution such as PBS/Tween').
After incubation with the second enzyme-tagged antibody, and subsequent to
washing to remove unbound material, the amount of label is quantified by
incubation
with a chromogenic substrate such as urea and bromocresol purple or 2,2'-azino-
di-(3-
ethyl-benzthiazoline)-6-sulfonic acid (ABTS) and HZOZ, in the case of
peroxidase as
the enzyme label. Quantitation is then achieved by measuring the degree of
color
generation, e.g., using a visible spectrum spectrophotometer.
The preceding format may be altered by first binding the sample to the assay
plate. Then, primary antibody is incubated with the assay plate, followed by
detecting
of bound primary antibody using a labeled second antibody with specificity for
the
primary antibody.
The antibody compositions of the present invention will find great use in
immunoblot or Western blot analysis. The antibodies may be used as high-
affinity
primary reagents for the identification of proteins immobilized onto a solid
support
matrix, such as nitrocellulose, nylon or combinations thereof. In conjunction
with
immunoprecipitation, followed by gel electrophoresis, these may be used as a
single
step reagent for use in detecting antigens against which secondary reagents
used in the
detection of the antigen cause an adverse background. Immunologically-based
detection methods for use in conjunction with Western blotting include
enzymatically-, radiolabel-, or fluorescently-tagged secondary antibodies
against the
toxin moiety are considered to be of particular use in this regard.
WO 01/23571 cA o23amn 2002-02-0~ PCT/US00/27119
-71-
11. Methods for Treating (31A Related Disorders
The present invention also involves, in another embodiment, the treatment of
disorders such as epilepsy and fibrillar seizures. The types of disorders that
may be
treated, according to the present invention, are limited only by the
involvement of
(31 A. By involvement, it is not even a requirement that (31 A be mutated or
abnormal -
the overexpression of this protein may actually overcome dysfunction of sodium
channel function within the cell. Thus, it is contemplated that a wide variety
of
disorders may be treated using (31A based therapy, including therapies with
the
modulators identified in the section above, immunotherapeutics, protein
therapy,
gene-based therapies and combination therapies.
A. Immunotherapies
Immunotherapeutics, generally, rely on the use of immune effector cells and
molecules to target and destroy aberrant cells. The immune effector may be,
for
example, an antibody specific for some marker on the surface of a tumor cell.
The
antibody alone may serve as an effector of therapy or it may recruit other
cells to
actually effect cell killing. The antibody also may be conjugated to a drug or
toxin
(chemotherapeutic, radionuclide, ricin A chain, cholera toxin, pertussis
toxin, etc.)
and serve merely as a targeting agent. Alternatively, the effector may be a
lymphocyte carrying a surface molecule that interacts, either directly or
indirectly,
with a tumor cell target. Various effector cells include cytotoxic T cells and
NK cells.
Immunotherapy could be used as part of a combined therapy, in conjunction
with (31 A-targeted gene therapy. The general approach for combined therapy is
discussed below. Generally, the tumor cell must bear some marker that is
amenable
to targeting, i.e., is not present on the majority of other cells.
B. Protein Therapy
Another therapy approach is the provision, to a subject, of (31A polypeptide,
active fragments, synthetic peptides, mimetics or other analogs thereof. The
protein
may be produced by recombinant expression means or, if small enough, generated
by
an automated peptide synthesizer. Formulations would be selected based on the
route
WO 01/23571 cA o23amn 2002-02-0~ PCT/L1S00/27119
-72-
of administration and purpose including, but not limited to, liposomal
formulations
and classic pharmaceutical preparations.
C. Genetic Based Therapies
One of the therapeutic embodiments contemplated by the present inventors is
the intervention, at the molecular level, in the events involved in the
malfunction of
voltage gated sodium channels. Specifically, the present inventors intend to
provide,
to a cell having an asubunit forming the sodium channel pore, an expression
construct
capable of providing (31 A to that cell. The lengthy discussion above of
expression
vectors and the genetic elements are also useful in this embodiment.
Particularly
preferred expression vectors are viral vectors such as adenovirus, adeno-
associated
virus, herpesvirus, vaccinia virus and retrovirus. Also preferred is
liposomally-
encapsulated expression vector.
Those of skill in the art are well aware of how to apply gene delivery to in
vivo
and ex vivo situations. For viral vectors, one generally will prepare a viral
vector
stock. Depending on the kind of virus and the titer attainable, one will
deliver 1 x 104,
1x105,1x106,1x10',1x10g,1x109,1x101°,1x1011or1x101zinfectious
particles to the patient. Similar figures may be extrapolated for liposomal or
other
non-viral formulations by comparing relative uptake efficiencies. Formulation
as a
pharmaceutically acceptable composition is discussed below.
Various routes are contemplated for various tissue types. The section below
on routes contains an extensive list of possible routes. For practically any
tissue,
systemic delivery is contemplated. In addition regional delivery also may be
contemplated
D. Combination Therapy
It is highly desirable to modulate the function of sodium channels in a number
of
clinical and therapeutic environments such as anethesia (Duch et al., 1998;
French et al.,
1998), neuronal disorders (Ureniak and Obrenovitch, 1998), cardiac arrhythmia
(Campbell and Williams, 1998; Capuchi et al., 1998), epilespy (Ragsdale and
Avoli,
1998), and anticonvulsant therapy (Natsch et al., 1997). Given that the
present invention
identifies a sodium channel (3 subunit that can be used to modulate the
activity of the
sodium channel, it is contemplated that protein and gene-based therapies
centered on this
WO 01/23571 cA o23amn 2002-02-0~ pCT/LTS00/27119
-73-
discovery may be combined with other known modulators of sodium channel
function to
achieve a desirable change in sodium channel function in these settings.
To achieve such useful modulation of the sodium channel activity using the
methods and compositions of the present invention, one would generally contact
a
"target" cell with a (31A expression construct, protein, or other (31A based
therapeutic
identified herein and at least one other agent. These compositions would be
provided
in a combined amount effective to alter the voltage gated sodium channel
function of
the cell. This process may involve contacting the cells with the (31 A based
therapeutic and the agents) or factors) at the same time. This may be achieved
by
contacting the cell with a single composition or pharmacological formulation
that
includes both agents, or by contacting the cell with two distinct compositions
or
formulations, at the same time, wherein one composition includes the (31A
based
therapeutic and the other includes the agent.
Alternatively, the (31A based treatment may precede or follow the other agent
1 S treatment by intervals ranging from minutes to weeks. In embodiments where
the
other agent and X31 A based composition are applied separately to the cell,
one would
generally ensure that a significant period of time did not expire between the
time of
each delivery, such that the agent and (31A based composition would still be
able to
exert an advantageously combined effect on the cell. In such instances, it is
contemplated that one would contact the cell with both modalities within about
12-24
hours of each other and, more preferably, within about 6-12 hours of each
other, with
a delay time of only about 12 hours being most preferred. In some situations,
it may
be desirable to extend the time period for treatment significantly, however,
where
several days (2, 3, 4, 5, 6 or 7) to several weeks (1, 2, 3, 4, 5, 6, 7 or 8)
lapse between
the respective administrations.
It also is conceivable that more than one administration of either (31A or the
other agent will be desired. Various combinations may be employed, where (31A
is
"A" and the other agent is "B", as exemplified below:
AB/A B/AB BB/A A/AB B/A/A ABB BBBlA BB/AB
WO 01/23571 cA o23amn 2002-02-0~ PCT/US00/27119
-74-
A/ABB AB/AB ABB/A BB/A/A B/AB/A B/A/AB BBBlA
A/A/AB B/A/A/A AB/A/A A/AB/A ABBB B/ABB BB/AB
S Other combinations are contemplated. Again, to achieve a therapeutic effect,
both agents are delivered to a cell in a combined amount effective to alter
the sodium
current density in said cell.
E. Formulations and Routes for Administration to Patients
Where clinical applications are contemplated, it will be necessary to prepare
pharmaceutical compositions - expression vectors, virus stocks, proteins,
antibodies
and drugs - in a form appropriate for the intended application. Generally,
this will
entail preparing compositions that are essentially free of pyrogens, as well
as other
impurities that could be harmful to humans or animals.
One will generally desire to employ appropriate salts and buffers to render
delivery vectors stable and allow for uptake by target cells. Buffers also
will be
employed when recombinant cells are introduced into a patient. Aqueous
compositions of the present invention comprise an effective amount of the
vector to
cells, dissolved or dispersed in a pharmaceutically acceptable Garner or
aqueous
medium. Such compositions also are referred to as inocula. The phrase
"pharmaceutically or pharmacologically acceptable" refer to molecular entities
and
compositions that do not produce adverse, allergic, or other untoward
reactions when
administered to an animal or a human. As used herein, "pharmaceutically
acceptable
carrier" includes any and all solvents, dispersion media, coatings,
antibacterial and
antifungal agents, isotonic and absorption delaying agents and the like. The
use of
such media and agents for pharmaceutically active substances is well know in
the art.
Except insofar as any conventional media or agent is incompatible with the
vectors or
cells of the present invention, its use in therapeutic compositions is
contemplated.
Supplementary active ingredients also can be incorporated into the
compositions.
The active compositions of the present invention may include classic
pharmaceutical preparations. Administration of these compositions according to
the
present invention will be via any common route so long as the target tissue is
available via that route. This includes oral, nasal, buccal, rectal, vaginal
or topical.
WO 01/23571 cA o23amn 2002-02-0~ PCT/L1S00/27119
-75-
Alternatively, administration may be by orthotopic, intradermal, subcutaneous,
intramuscular, intraperitoneal or intravenous injection. Such compositions
would
normally be administered as pharmaceutically acceptable compositions,
described
supra.
The active compounds may also be administered parenterally or
intraperitoneally. Solutions of the active compounds as free base or
pharmacologically acceptable salts can be prepared in water suitably mixed
with a
surfactant, such as hydroxypropylcellulose. Dispersions can also be prepared
in
glycerol, liquid polyethylene glycols, and mixtures thereof and in oils. Under
ordinary conditions of storage and use, these preparations contain a
preservative to
prevent the growth of microorganisms.
The pharmaceutical forms suitable for injectable use include sterile aqueous
solutions or dispersions and sterile powders for the extemporaneous
preparation of
sterile injectable solutions or dispersions. In all cases the form must be
sterile and
must be fluid to the extent that easy syringability exists. It must be stable
under the
conditions of manufacture and storage and must be preserved against the
contaminating action of microorganisms, such as bacteria and fungi. The
carrier can
be a solvent or dispersion medium containing, for example, water, ethanol,
polyol (for
example, glycerol, propylene glycol, and liquid polyethylene glycol, and the
like),
suitable mixtures thereof, and vegetable oils. The proper fluidity can be
maintained,
for example, by the use of a coating, such as lecithin, by the maintenance of
the
required particle size in the case of dispersion and by the use of
surfactants. The
prevention of the action of microorganisms can be brought about by various
antibacterial an antifungal agents, for example, parabens, chlorobutanol,
phenol,
sorbic acid, thimerosal, and the like. In many cases, it will be preferable to
include
isotonic agents, for example, sugars or sodium chloride. Prolonged absorption
of the
injectable compositions can be brought about by the use in the compositions of
agents
delaying absorption, for example, aluminum monostearate and gelatin.
Sterile injectable solutions are prepared by incorporating the active
compounds in the required amount in the appropriate solvent with various of
the other
ingredients enumerated above, as required, followed by filtered sterilization.
Generally, dispersions are prepared by incorporating the various sterilized
active
ingredients into a sterile vehicle which contains the basic dispersion medium
and the
WO 01/23571 cA o23amn 2002-02-0~ PCT/US00/27119
-76-
required other ingredients from those enumerated above. In the case of sterile
powders for the preparation of sterile injectable solutions, the preferred
methods of
preparation are vacuum-drying and freeze-drying techniques which yield a
powder of
the active ingredient plus any additional desired ingredient from a previously
sterile-
filtered solution thereof.
As used herein, "pharmaceutically acceptable carrier" includes any and all
solvents, dispersion media, coatings, antibacterial and antifungal agents,
isotonic and
absorption delaying agents and the like. The use of such media and agents for
pharmaceutical active substances is well known in the art. Except insofar as
any
conventional media or agent is incompatible with the active ingredient, its
use in the
therapeutic compositions is contemplated. Supplementary active ingredients can
also
be incorporated into the compositions.
For oral administration the polypeptides of the present invention may be
incorporated with excipients and used in the form of non-ingestible
mouthwashes and
dentifrices. A mouthwash may be prepared incorporating the active ingredient
in the
required amount in an appropriate solvent, such as a sodium borate solution
(Dobell's
Solution). Alternatively, the active ingredient may be incorporated into an
antiseptic
wash containing sodium borate, glycerin and potassium bicarbonate. The active
ingredient may also be dispersed in dentifrices, including: gels, pastes,
powders and
slurries. The active ingredient may be added in a therapeutically effective
amount to
a paste dentifrice that may include water, binders, abrasives, flavoring
agents,
foaming agents, and humectants.
The compositions of the present invention may be formulated in a neutral or
salt form. Pharmaceutically-acceptable salts include the acid addition salts
(formed
with the free amino groups of the protein) and which are formed with inorganic
acids
such as, for example, hydrochloric or phosphoric acids, or such organic acids
as
acetic, oxalic, tartaric, mandelic, and the like. Salts formed with the free
carboxyl
groups can also be derived from inorganic bases such as, for example, sodium,
potassium, ammonium, calcium, or ferric hydroxides, and such organic bases as
isopropylamine, trimethylamine, histidine, procaine and the like.
Upon formulation, solutions will be administered in a manner compatible with
the dosage formulation and in such amount as is therapeutically effective. The
formulations are easily administered in a variety of dosage forms such as
injectable
WO 01/23571 cA o23amn 2002-02-0~ pCT/US00/27119
_77_
solutions, drug release capsules and the like. For parenteral administration
in an
aqueous solution, for example, the solution should be suitably buffered if
necessary
and the liquid diluent first rendered isotonic with sufficient saline or
glucose. These
particular aqueous solutions are especially suitable for intravenous,
intramuscular,
subcutaneous and intraperitoneal administration. In this connection, sterile
aqueous
media which can be employed will be known to those of skill in the art in
light of the
present disclosure. For example, one dosage could be dissolved in 1 ml of
isotonic
NaCI solution and either added to 1000 ml of hypodermoclysis fluid or injected
at the
proposed site of infusion, (see for example, "Remington's Pharmaceutical
Sciences"
15th Edition, pages 1035-1038 and 1570-1580). Some variation in dosage will
necessarily occur depending on the condition of the subject being treated. The
person
responsible for administration will, in any event, determine the appropriate
dose for
the individual subject. Moreover, for human administration, preparations
should meet
sterility, pyrogenicity, general safety and purity standards as required by
FDA Office
of Biologic Standards.
12. Transgenic Animals/Knockout Animals
In one embodiment of the invention, transgenic animals are produced which
contain a functional transgene encoding a functional ~31A polypeptide or
variants
thereof. Transgenic animals expressing (31A transgenes, recombinant cell lines
derived from such animals and transgenic embryos may be useful in methods for
screening for and identifying agents that induce or repress function of (31A.
Transgenic animals of the present invention also can be used as models for
studying
malfunctions of voltage gated sodium channels.
In one embodiment of the invention, a (31A transgene is introduced into a non-
human host to produce a transgenic animal expressing a human or murine (31A
gene.
The transgenic animal is produced by the integration of the transgene into the
genome
in a manner that permits the expression of the transgene. Methods for
producing
transgenic animals are generally described by Wagner and Hoppe (U.S. Patent
No.
4,873,191) and Brinster et al. 1985) and in "Manipulating the Mouse Embryo; A
Laboratory Manual" 2nd edition (eds., Hogan, Beddington, Costantimi and Long,
Cold Spring Harbor Laboratory Press, 1994).
WO 01/23571 cA o23ai~~i 2002-02-0~ PCT/US00/27119
_78_
It may be desirable to replace the endogenous (31A by homologous
recombination between the transgene and the endogenous gene; or the endogenous
gene may be eliminated by deletion as in the preparation of "knock-out"
animals.
Typically, a ~31A gene flanked by genomic sequences is transferred by
microinjection
into a fertilized egg. The microinjected eggs are implanted into a host
female, and the
progeny are screened for the expression of the transgene. Transgenic animals
may be
produced from the fertilized eggs from a number of animals including, but not
limited
to reptiles, amphibians, birds, mammals, and fish. Within a particularly
preferred
embodiment, transgenic mice are generated which overexpress (31 A or express a
mutant form of the polypeptide. Alternatively, the absence of a (31A in "knock-
out"
mice permits the study of the effects that loss of (31A protein has on a cell
in vivo.
Knock-out mice also provide a model for the development of X31 A-related
disorders.
As noted above, transgenic animals and cell lines derived from such animals
may find use in certain testing experiments. In this regard, transgenic
animals and
1 S cell lines capable of expressing wild-type or mutant (31 A may be exposed
to test
substances. These test substances can be screened for the ability to enhance
wild-type
(31A expression and or function or impair the expression or function of mutant
(31A.
13. Examples
The following example is included to demonstrate a preferred embodiment of
the invention. It should be appreciated by those of skill in the art that the
techniques
disclosed in the example which follows represent techniques discovered by the
inventor to function well in the practice of the invention, and thus can be
considered
to constitute a preferred mode for its practice. However, those of skill in
the art
should, in light of the present disclosure, appreciate that many changes can
be made in
the specific embodiment disclosed and still obtain a like or similar result
without
departing from the spirit and scope of the invention.
EXAMPLE 1
Materials and Methods
The present example describes certain methods and materials that were
employed in generating the results presented in the present invention. These
are
WO 01/23571 cA o23amn 2002-02-0~ pCT/US00/27119
-79-
exemplary methods and those of skill in the art will understand that
alternative
methods may be employed in order to confirm the results obtained herein.
Library screening
A cDNA probe encoding nucleotides 345-911 of p(31.C1Aa (Isom et al., 1992)
was labeled with digoxigenin following the manufacturer's instructions
(Boehringher
Mannheim Indianapolis, IN) and used to screen a 7~ Express rat adrenal cDNA
library
prepared by Stratagene (LaJolla, CA) as previously described (Isom et al.,
1995a).
pBK plasmids containing cDNA inserts which hybridized strongly to the probe
were
rescued from the ~, phage according to the manufacturer's instructions,
confirmed by
Southern blot analysis and sequenced using ThermoSequenase (Amersham Pharmacia
Biotech, Piscataway, NJ). The nucleotide sequence of (31A has been deposited
in Gen
Bank under accession number AF182949.
RT-PCR from rat adrenal RNA
To confirm independently that the (31A transcript identified by library
screening was expressed by rat adrenal gland, a region of (31 A from the amino
terminus was amplified past the region in which the amino acid sequence
changed
from identity to non-identity to (31, or the putative splice site, by reverse-
transcriptase
polymerase chain reaction (RT-PCR) using adult rat adrenal gland total RNA as
template and (31A3 (5'-GAAGATGAGCGCTTTGAGG-3'(SEQ ID N0:3), primer
sequence common to (31 and (31A) and (31A5 (5'-GAGAGACACAGCAAGC (SEQ
ID N0:4), primer sequence unique to (31A) as oligonucleotide forward and
reverse
primers, respectively. Rat adrenal gland cDNA was synthesized from total RNA
using Superscript II (Gibco/BRL, Gaithersburg, MD) according to the
manufacturer's
instructions in a total volume of 20 ~l. 2.3 ~g of total rat adrenal RNA
(purified using
Trizol reagent, Gibco/BRL) was used in the reaction. The PCR conditions were
as
follows: 1 p1 of cDNA, 0.5 ~M of each primer, 200 ~M of each dNTP (Boehringer
Mannheim), 5 ~l of Mg2+-free l OX PCR buffer (Perkin Elmer-Roche Molecular
Systems, Branchburg, NJ), and 1.5 mM MgCl2 were mixed in a total volume of 50
~1.
Following a hot start at 94°C, 0.25 ~1 of AmpliTaq DNA polymerase
(Perkin Elmer)
WO 01/23571 cA o23ai~~i 2002-02-0~ PCT/US00/27119
-80-
were added to the reaction tube and the amplification cycle was started. The
cycling
parameters were: 40 cyles of 45 sec. at 94°C, 20 sec. at 60°C, 1
min. 30 sec. at 72°C.
This was followed by 10 min. at 72°C and then 4°C until the
tubes were removed
from the thermocyler (GeneAmp 2400, Perkin Elmer). Analysis of the PCR
products
on a 1 % agarose gel revealed a 750 by band. The band was excised form the
gel,
subcloned into pCR2.1 (Invitrogen, Carlsbad, CA), and analyzed using
ThermoSequenase (Amersham Pharmacia Biotech). The sequence obtained from this
PCR clone was identical to that obtained from the original (31A clone plaque-
purified
from the adrenal cDNA library.
Rat ail gene:
Intron 3 of the rat [31 gene (Makita et al., 1994a) was amplified by PCR using
rat genomic DNA as the template, oligonucleotides which encode [31 coding
sequence
flanking intron 3, VVDK (SEQ ~ N0:5) (5'-
AGATCCACCTGGAGGTGGTGGACAAGG-3' (SEQ ~ N0:6)) and ANRD (SEQ
~ N0:7) (5'-ACACGATGGATGCCATATCTCTGTTGG-3'(SEQ ID N0:8)) as
forward and reverse primers, respectively, and the Expand Long Template PCR
System (Boehringer Mannheim). All oligonucleotide primers were synthesized by
Gibco/BRL. The amplification conditions were as follows: 300 ng of rat genomic
DNA (Clontech Laboratories, Inc., Palo Alto, CA), 250 ng of each primer, 1mM
each
dNTP (Boehringer Mannheim), and 5 q1 of Expand Buffer 3 were mixed in a total
reaction volume of 25 ~1. Following a hot start at 95°C, 0.5 ~1 of
Expand DNA
polymerase were added and the amplification cycle was started. 40 cycles of
the
following regimen were performed: 94°C for 10 sec., 58°C for 30
sec., 68°C for 4
min. plus 20 sec. added to each successive cycle. The samples were then held
at 4°C
until removal from the thermocycler (GeneAmp 2400, Perkin Elmer). The 5 kb PCR
product was gel-purified and sequenced directly using oligonucleotide VVDK
(SEQ
B7 NO: 5) as the sequencing primer.
WO 01/23571 cA o23amn 2002-02-0~ PCT/L1S00/27119
-81-
RNAse protection analysis:
A plasmid containing a RNAse protection probe template (pRPA-1) was
constructed corresponding to nucleotides 364-533 in the (31A sequence.
Briefly, a
169-nucleotide Alu I/Acc I fragment was excised from pBK. (31 A and ligated
into the
SmaI and Acc I sites of pBluescript (Stratagene). The resulting plasmid was
then
sequenced using ThermoSequenase (Amersham). To synthesize labeled cRNA, a 10
~g aliquot of pRPA-1 was linearized with Xho I, ethanol-precipitated,
resuspended in
RNAse-free water, and labeled with digoxigenin using the T3 MAXIscript kit
(Ambion, Austin, TX) according to the manufacturer's instructions. Following a
2
hour incubation at 37°C, the reaction was incubated at 95°C for
2 minutes, chilled on
ice, and then treated with RNAse-free DNAse (2 units) for 15 minutes at
37°C.
EDTA (final concentration 30 mM) was added to stop the reaction. Free
nucleotides
were removed by ethanol precipitation with 0.5 M ammonium acetate and the
final
pellet was resuspended in 20 ~l of RNAse-free water. The probe (RPA-1 ) was
quantitated by comparison of serial dilutions of the labeled probe with serial
dilutions
of control digoxigenin-labeled RNA supplied by Boehringer Mannheim following
the
manufacturer's instructions.
RNAse protection experiments were performed using the HybSpeed RPA kit
from Ambion. Briefly, 20 ~g of rat embryonic day 18 brain RNA were mixed with
1
~l of digoxigenin-labeled RPA-1 probe and 30 ~g of yeast tRNA in 0.5 M
ammonium
acetate plus 2.5 volumes of ethanol. The reaction tubes were left at -
20°C for 15
minutes and the RNA was precipitated by centrifugation in a microfuge at top
speed.
The RNA was resuspended in 10 ~1 of HybSpeed hybridization buffer that had
been
preheated to 95°C, vortexed vigorously, and the tubes were placed at
95°C for 3
minutes. The samples were then hybridized for 10 minutes at 68°C, and
digested with
a mixture of RNAse A and T1 (10 U/ml of RNAse A and 400 U/ml RNAse T1) for 30
minutes at 37°C. 150 ~l of Hybspeed Inactivation/Precipitation mix were
added to
each reaction, and the RNA was precipitated and resuspended in 10 p1 of Gel
Loading
Buffer 1. The reactions were electrophoresed on a 1.5 mm thick 6% acrylamide
TBE
denaturing gel containing 7M urea in the Mini-Protean gel format (BioRad,
Hercules,
CA), transferred to nylon (Boehringer Mannheim), and UV crosslinked using a
Stratalinker (Stratagene, LaJolla, CA). Hybridization of the digoxigenin-
labeled
WO 01/23571 cA o23ai~~i 2002-02-0~ PCT/US00/27119
-82-
probe was detected with alkaline phosphatase-conjugated anti-digoxigenin Fab
fragments (1:10,000 dilution) and CSPD chemiluminescent substrate solution
(Boehringer Mannheim) according to the manufacturer's instructions.
Preparation of RNA and Northern blot analysis:
Time-mated pregnant female Sprague-Dawley rats were anesthetized with 60
mg/kg Beuthanasia-D i.p. (Schering-Plough Animal Health Corp., Kenilworth,
N.J.)
and the fetuses were surgically removed. Embryonic day 9 rats were homogenized
in
their entirety in Trizol reagent (Gibco/BRL) to purify total RNA according to
the
manufacturer's instructions. Whole fetal brains were dissected at the
remaining
embryonic time points and total RNA was purified using Trizol reagent. RNA was
also subsequently prepared from the brains and adrenal glands of the adult
female
rats. Postnatal rats at the indicated ages were anesthetized with Beuthanasia-
D, brains
were dissected and total RNA was purified with Trizol reagent. Northern blot
analysis
of 20 ~g of each sample was performed as previously described (Isom et al.,
1995a)
using a digoxigenin-labeled (31A antisense cRNA probe encoding nucleotides 428-
850 or a digoxigenin-labeled antisense cRNA probe specific to the 3'
untranslated
region of X31 (nucleotides 1053-1508 of p(31.C1Aa; Isom et al., 1992).
Construction of (31A expression vector:
A plasmid containing X31 cDNA including an in-frame amino terminal
hemagglutinin (HA) epitope tag was obtained as a generous gift from the
laboratory
of R.A. Maue at Dartmouth University (Shah et al., 1996). This construct has
been
shown to express functional (31 subunits in Xenpous oocytes. The HA-tagged (31
cDNA was recloned into the Eco RI and Not I sites of the mammalian expression
vector pCIneo (Promega. Madison, WI) to create pCL(31-HA. pCL(31-HA was
subsequently digested with Acc I and Not I and agarose gel-purified to remove
the 3'
end of (31. The Acc I restriction endonuclease site is common to (31 and (31A.
pBK.(31A cDNA was digested with Acc I and Not I and gel-purified. The 3' end
of
(31A was then ligated into Acc U Not I-digested pCL(31-HA to create pCL(31A-
HA.
WO 01/23571 cA o23amn 2002-02-0~ PCT/US00/27119
-83-
The junctions were then sequenced to confirm that the segments of (31 and (31A
were
successfully ligated in frame.
Transfection of SNaIIA cells with HA-tagged (31A:
SNaIIA cells were transfected with pCL(31A-HA using DOTAP as previously
described (Isom et al., 1995b). Because SNaIIA cells are resistant to 6418 as
a result
of the original transfection of the aIIA subunit, pCL(31A-HA was cotransfected
with
pSV2*Hyg to confer resistance to the antibiotic hygromycin. Drug selection
with
hygromycin (400 ~g/ml) required approximately 1 week, Clonal cell lines were
selected, analyzed by Northern blot, and expanded as previously described
(Isom et
al., 1995b).
[3H] Saxitoxin binding analysis:
Whole cell saturation binding analysis of SNAIIA and SNaIIA-(31A cells was
performed as previously described (Isom et al., 1995b) over a concentration
range of
0.1 to 10 nM [3H]STX with the addition of 10 ~M unlabeled tetrodotoxin (TTX;
Calbiochem, San Diego, CA) to assess non-specific binding. 3H-Saxitoxin (3H-
STX,
28 Ci/mmol) was obtained from Amersham. Binding data were normalized to
protein
concentration using the BCA Protein Assay kit (Pierce, Rockford, IL).
Saturation
binding data were analyzed by non-linear regression using Prism (GraphPad
Software, LaJolla, CA) to obtain KD and Bn,aX values.
Antibodies:
A multiple antigenic peptide (MAP) with amino acid sequence
RWRDRWKEGDRLVSHRGQ (SEQ ID NO: 9), encoded by nucleotides 160 through
177 of (31A, was synthesized by the Protein and Carbohydrate Structure
Facility at the
University of Michigan. Rabbit polyclonal antibodies were subsequently
generated in
2 separate animals and tested by ELISA against the (31A-MAP to determine the
antibody titer (Research Genetics, Inc., Huntsville, AL). Serum used for the
Western
blots in this study came from animal #86051.
WO 01/23571 cA o23amn 2002-02-0~ PCT/LJS00/27119
-84-
Western blot analysis of (31A protein expression:
Adult female Sprague-Dawley rats were sacrificed by decapitation. Brain,
spinal cord, heart, skeletal muscle, and adrenal gland tissues were
immediately
removed, minced, and briefly stored on ice. SNa(31A-16 cell line cells were
washed
with PBS and scraped into 50 ml conical tubes. Membranes were prepared as
described previously (Isom et al., 1995) and the final pellets were
resuspended in SO
mM Tris pH 8, 10 mM EGTA containing Complete-Mini protease inhibitor tablets
according to the manufacturer's instructions (Boehringer Mannheim). The total
protein in each membrane preparation was quantitated with the BCA Protein
Assay
Kit (Pierce) using BSA as the standard. 250 qg of each membrane preparation
were
separated by SDS-PAGE as previously described (Isom et al., 1995), transferred
to
nitrocellulose (HyBond ECL, Amersham), and stained with Ponceau-S prior to
immunodetection. Western blot analysis was performed as follows: the blot was
washed for 10 minutes in TBS-T (10 mM Tris, pH 7.4, 150 mM NaCI, 0.1% Tween-
20) at room temperature and then blocked for 1 hour in 5% non-fat dry milk in
TBS-
T at room temperature. Primary anti-~31A antibody (1:750 dilution) was applied
in
blocking solution for 30 minutes at room temperature. The blot was then washed
5
times for 15 minutes each in TBS-T. Secondary antibody (horse radish
peroxidase-
conjugated goat anti-rabbit IgG, ICN) diluted to 1:100,000 in blocking
solution was
applied for 30 minutes at room temperature. The blot was then washed 5 times
for 15
minutes each in TBS-T. SuperSignal WestFemto chemiluminescent substrate
solution
(Pierce) was applied according to the manufacturer's instructions, the blot
was placed
between plastic sheet protectors, and exposed to Hyperfilm-ECL (Amersham) for
the
indicated times (typically 10 to 30 seconds) at room temperature.
Immunohistochemical Analysis of ~31A expression:
Rat tissues were routinely fixed in 10% neutral buffered formalin, processed,
embedded in paraffin blocks, sectioned onto slides. The rabbit anti-rat (3A1
antibody
was used at titer of 1:900 for the human tissues and 1:600 for the rat
tissues. Briefly,
slides were incubated (all incubations for 30 min. at room tem) with primary
WO 01/23571 cA o23amn 2002-02-0~ PCT/LTS00/27119
-85-
antibodies then incubated with specific'oiotin conjugated secondary antibodies
(Vector Labs, Burlingham, CA) which were then detected using the ABC-
horseradish
peroxidase system (Vector Labs, Burlingham, CA) followed by 3'-
diaminobenzidine
(Biomeda, Foster City, CA) as the chromogen, stained in Mayer's hematoxylin
and
S coverslipped with Permount (Fisher, Pittsburgh, PA).
Electrophysiological analysis:
Electrophysiological recordings on SNaIIA and SNaIIA(31 A cells were
performed by the patch clamp technique in the whole cell configuration (Hamill
et al.,
1981 ), using an Axopatch 200B patch clamp amplifier and pCLAMP software (Axon
Instruments). Data were filtered at 5 kH and digitally sampled at 50 kH.
Series
resistance was compensated 60-80%. Capacitive transients, elicited by voltage
steps,
were partially canceled using the internal clamp circuitry. Additional
subtraction of
transients and leak currents was obtained using the P/4 procedure (Armstrong
and
Bezanilla 1977). For whole cell recordings, recording pipettes were filled
with 105
mM CsF, 10 mM CsCI, 10 mM NaCI, 10 mM EGTA, 10 mM HEPES, pH 7.4 with
CsOH. Pipette resistances were 1-3 MS2. The bath solution consisted of 130 mM
NaCI, 4 mM KC1, 1.5 mM CaCl2, 1 mM MgCl2, 5 mM Glucose, 10 mM HEPES, pH
7.4 with NaOH. As has been previously described (Isom et al., 1995), the
voltage-
dependence of sodium current activation and inactivation progressively shifted
to
more negative potentials over the first few minutes of experiments with
fluoride-
based intracellular solutions. Thus, all experiments were begun 10 min after
break in,
at which point the shifts in channel gating had stabilized.
For each cell, the voltage-dependence of current activation and steady state
inactivation was examined. Activation was assessed by applying test pulses to
potentials from -50 to +70 mV in 5 mV steps, following a 100 ms prepulse to -
100
mV. Peak current amplitude (Ipeak) was measured at each test potential, and
converted
to conductance (g) according to g = Ipeak/(Vre,,-Vtest), in which Vtest is the
test potential
and Vre" is the current reversal potential, determined by linear extrapolation
of the
straight line portion of the falling phase of the current-voltage
relationship. The
conductance values were normalized with respect to the maximal conductance,
plotted as a function of Vtest, and fit with the Boltzman equation:
1/(1+exp((Vtesr
V~,2)/k), in which V~i2 is the midpoint of the curve and k is a slope factor.
Steady state
WO 01/23571 cA o23amn 2002-02-0~ PCT/US00/27119
-86-
inactivation was examined by applying 100 ms long prepulses to potentials
ranging
from -100 to -10, in 5 mV steps, followed by a test pulse to 0 mV. The peak
amplitude of currents evoked by the test pulses were normalized with respect
to the
largest currents, plotted as a function of prepulse potential and fit with the
Boltzman
S equation.
Spinal Nerve Ligation Surgery:
Spinal nerve ligation (SNL) was performed as described by Kim and Chung
(1992). Briefly, male Sprague-Dawley rats (Harlan, Indianapolis, IN) weighing
approximately 200g were anesthetized with isoflurane. The spinal nerve at the
level
of LS or L6 was exposed through an incision left of the dorsal midline and
tightly
ligated with 6-0 silk. The spinal nerve was visualized without being ligated
in the
sham-operated animals. Naive animals did not undergo surgery. Mechanical
allodynia was assessed according the methods of Chaplan et al. (1994) at
various
times (2 days, 14 days, and 8 weeks) after surgery. SNL animals were included
in the
study if the ipsilateral paw consistently responded with a paw withdrawal
threshold of
less than 4 grams of pressure. The naive animals and sham-operated animals did
not
demonstrate allodynia to the mechanical stimulus, which was greater than 15
grams of
force.
Tissue Preparation of SNL-treated subjects:
At 2 days, 14 days and 8 weeks after surgery, sham-operated and SNL animals
(n=3-5) as well as the naive animals (n=3) were sacrificed by COZ
asphyxiation.
Animals were transcardially perfused with 4% paraformaldehyde, and the dorsal
root
ganglia (DRG) tissue was removed from the ipsilateral and contralateral sides.
These
tissues were processed for paraffin embedding, serially sectioned at S~m,
mounted
onto Superfrost-Plus slides (Fisher, Pittsburgh, PA), and then processed for
immunohistochemistry.
WO 01/23571 cA o23amn 2002-02-0~ PCT/US00/27119
-87-
Quantitation of immunohistochemical staining in SNL-treated subjects:
Tissue sections were processed simultaneously to minimize potential staining
variability. Ipsilateral and contralateral DRG neurons were characterized as
nociceptive if the diameter was < 25 Vim; all others were designated as
sensory
(Waxman, et al., 1999). An average of 30-40 neurons were quantified for
immunoreactivity and scored as follows: 0.0 for no staining, 1.0 for weak
staining,
2.0 for moderate immunoreactivity, and 3.0 for intense immunoreactivity. These
data
were then averaged per animal (n=3-5) per group for the intensity of (31A and
(31
immunolabeling.
The morphologic patterns of the ~31A and (31 immunoreactivity in the
nociceptive and sensory DRG neurons were also characterized as 1 ) homogeneous
for
a diffuse labeling pattern; 2) up nctate for several clumpy, intracellular,
Nissle-like
aggregates of staining; and 3) membrane for peripheral labeling located
predominantly inside the cell, along the cell membrane. Data are presented as
a
percentage of labeling pattern observed per DRG per group.
EXAMPLE 2
Molecular cloning and analysis of (31A
A rat adrenal gland cDNA library prepared in the ,Express vector was
screened with a digoxigenin-labeled cDNA probe encoding nucleotides 345-911 of
p(31.C1Aa (Isom et al., 1992). A clone encoding a protein with a 5' region of
identity
to (31 and a novel 3' region was identified by DNA sequencing. The identity of
this
clone was then confirmed independently by reverse transcriptase polymerase
chain
reaction (RT-PCR) from rat adrenal cDNA using the oligonuceotides (31A3 and
(31A5
followed by DNA sequencing, as described in Experimental Procedures. This
clone,
designated (31A, encoded a novel 253 amino acid protein of 29,055 daltons
(predicted
molecular mass of the mature protein with the signal sequence removed) which
contains a predicted amino terminal region of identity to X31, residues met (-
1) through
lys ( 129), followed by a novel carboxy terminal region (FIG. 1 A and FIG. 1
B).
Hydrophobicity analysis of the novel, carboxy-terminal region revealed an
apparent
66-amino acid extension of the extracellular region of (31 followed by a 19-
amino acid
transmembrane domain and short, 39-amino acid intracellular carboxy-terminus
(FIG.
WO 01/23571 cA o23amn 2002-02-0~ pCT/US00/27119
_88_
1 A and FIG. 1 C). The novel 3' region of (31 A is structurally homologous to
(31 in that
it predicts a transmembrane domain and short intracellular region, yet it
contains little
to no homology at the amino acid level (FIG. 1B). Interestingly, the amino
terminal
region common to (31 and (31A contains the extracellular immunoglobulin fold.
(31A
can thus be characterized as a cell adhesion molecule like (31 and (32.
Analysis of the novel 3' region of (31 A by BLAST-P search of the Swissprot
database revealed a 55-residue region of (31A with 32% identity to an
extracellular
LDL-receptor class A domain of human low-density lipoprotein receptor-related
protein 2 (LRP2), also called megalin or glycoprotein 330 (FIG. 1D; Kounnas et
al.,
1995; Gliemann, 1998; Saito et al., 1994; Korenberg et al., 1994). This region
of
homology in (31A is predicted to be located extracellularly, just proximal to
the
plasma membrane followed by the transmembrane region itself. LRP2 has been
shown to be a cysteine-rich type I membrane protein that forms a multimeric
complex
with receptor-associated protein (RAP). LRP2 binds clusterin with high
affinity and
is localized to clathrin-coated pits, suggesting that it may be an endocytic
receptor.
Interestingly, LRP2 interacts with extracellular matrix components, similar to
sodium
channel (31 and (32 subunits. The BLAST-P search also revealed a 63-residue
region
of (31A with 26% identity to tensin, a protein that has been implicated as the
anchor
for actin filaments at focal adhesions and is thought to act as a link between
the
cytoskeleton and signal transduction proteins (Weigt et al., 1992). The region
of
homology to (31A is located in the insertin domain of tensin. This domain has
been
shown to permit polymerization of actin filaments.
EXAMPLE 3
(31A is encoded by a retained intron in the (31 gene
The genomic organization of the human sodium channel (31 subunit gene has
been reported previously (Makita et al., 1994). The (31 gene contains 5
introns and 6
exons. The (31A cDNA is the result of retention of I3, creating a novel 3'
end. It was
determined that the site of divergence between the (31 and (31A cDNAs was
precisely
located at the boundary between exon 3 andintron 3 of the /31 gene.
Furthermore, a
consensus sequence for exon-intron boundaries in genomic DNA (Mount, 1982) was
WO 01/23571 cA o23amn 2002-02-0~ PCT/US00/27119
-89-
readily identified at this location. Sequencing of a PCR-generated product
from rat
genomic DNA of intron 3 (approximately 5 kb) showed that the sequence of [31 A
beyond the amino acid sequence VVDK was indeed that of intron 3. RNAse
protection experiments were performed using a probe that spanned the exon 3-
intron 3
boundary in the rat sequence; this 169 nucleotide probe was fully protected by
rat
embryonic day 18 brain mRNA. Thus, it is proposed that the novel
extracellular,
transmembrane, and carboxy terminal domains of X31 A are encoded by
alternative
splicing of a retained intron within the X31 gene that includes an in-frame
termination
codon. These data are in agreement with the previously reported observation
that (31
is represented only once in the rat and human genomes (Makita et al., 1994;
Tong et
al., 1993).
EXAMPLE 4
~31A mRNA is expressed in embryonic brain and adult adrenal gland
A comparison ofthe developmental time courses of (31A and (31 mRNA
expression in developing rat brain was determined using specific, non-cross-
hybridizing antisense cRNA probes for (31A and (31, respectively. Time-mated
pregnant female Sprague-Dawley rats were anesthetized with 60 mg/kg
Beuthanasia-
D i.p., and the fetuses were surgically removed. Embryonic day 9 fetuses were
homogenized in their entirety in Trizol reagent to purify total RNA according
to the
manufacturer's instructions. Whole fetal brains were dissected at the
remaining
embryonic time points, and total RNA was purified using Trizol reagent. RNA
was
also prepared from the brains and adrenal glands of the adult female rats.
Postnatal
rats at the indicated ages were anesthetized with Beuthanasia-D, brains were
dissected
and total RNA was purified with Trizol reagent. Northern blot analysis of 20
~g of
each sample was performed as previously described (Isom et al., 1995a) using a
digoxigenin-labeled (31A antisense RNA probe encoding nucleotides 428-850 or a
digoxigenin-labeled antisense RNA probe specific to the 3' untranslated region
of (31
(nucleotides 1053-1508 of p(31.C1Aa; Isom et al., 1992). Interestingly, the
expression
time course of (31A parallels that of the 26 kDa (31-immunoreactive band
described
previously (McHugh-Sutkowski and Catterall, 1990) and complements the
expression
pattern of X31 (Patton et al., 1994). Thus, (31A is expressed early in
development and
WO 01/23571 cA o23amn 2002-02-0~ PCT/US00/27119
-90-
disappears after birth. In contrast, X31 expression is not detectable during
embryonic
brain development and becomes detectable as (31A mRNA expression is decreased.
EXAMPLE 5
Analysis of ~31A protein expression
To determine whether alternative splicing of the (31 gene resulted in
expression of a novel protein, a polyclonal antibody was generated against a
MAP
peptide containing the amino acid sequence RWRDRWKEGDRLVSHRGQ (SEQ >D
N0:9) encoded by the retained portion of intron 3 found in the (31 A cDNA
clone.
Brain, spinal cord, heart, skeletal muscle, and adrenal gland membranes were
prepared as described previously (Isom et al. 1995). Total protein in each
membrane
preparation was quantitated with the BCA Protein Assay Kit. 250 ~g of each
membrane preparation were separated by SDS-PAGE as previously described (Isom
et al., 1995) and transferred to nitrocellulose. Western blot analysis
consisted of the
following steps, all at room temperature: the blot was first washed for 10
minutes in
TBS-T (10 mM Tris, pH 7.4, 150 mM NaCI, 0.1% Tween-20) and then blocked for 1
hour in 5% non-fat dry milk in TBS-T at room temperature. Primary anti-(31A
antibody (1:750 dilution) was applied in blocking solution for 30 minutes.
After
washing the blot 5 times for 15 minutes each in TBS-T, secondary antibody
(horse
radish peroxidase-conjugated goat anti-rabbit IgG, ICN) diluted to 1:100,000
was
applied in blocking solution for 30 minutes. After washing the blot as before,
SuperSignal WestFemto chemiluminescent substrate solution was applied
according
to the manufacturer's instructions, the blot was placed between plastic sheet
protectors, and exposed to Hyperfilm-ECL for 10 seconds at room temperature.
~31A-
immunoreactive bands that migrated at approximately 50 kDa were observed in
heart,
skeletal muscle, and adrenal gland, but were not detected in brain or spinal
cord. An
immunoreactive doublet was observed in adrenal gland. The absence of
immunoreactive (31A protein bands in the CNS tissues brain and spinal cord is
consistent with the Northern blot results.
EXAMPLE 6
Immunocytochemical Analysis of (31A Expression
WO 01/23571 cA o23amn 2002-02-0~ pCT/US00/27119
-91-
Various central and peripheral neuronal populations contained (31A when
analyzed immunocytochemically with anti-[31A primary and horse-radish
peroxidase
conjugated secondary antibodies. These studies demonstrate that [31A was found
in
most but not all Purkinje cells in the cerebellum. In addition, some (31A
positive
neurons in the dentate nucleus of the cerebellum were observed. (31A was
absent
from the granular layer and the molecular layer of the cerebellum. A distinct
population of pyramidal neurons of the cerebral cortex contained (31 A, while
glial cell
populations remained negative. Spinal cord also contained several distinct
populations of [31A containing neurons. The (31A protein was also localized to
a
small population of motor neurons, while [31A-expressing neurons were also
observed
in laminae II-V of the dorsal horn. All neuronal cell types of the dorsal root
ganglia
contained [31 A, while fiber tracks and glial cells did not stain.
In addition, non-neuronal cells were also found to express (31A. These
include endomysium membranes of individual muscle fibers in rat atria, other
areas of
the rat heart such as ventricles, lung alveoli and some bronchus columnar
epithelial
cells expressed (31 A protein.
EXAMPLE 7
Mammalian Cell Expression
To investigate the functional role of [31 A a hemagglutinin (HA) epitope-
tagged version of the (31A cDNA was constructed. An epitope tag was included
for
potential use in the event that the polyclonal antibody production was
unsuccessful.
However, the HA tag experiments was not necessary as this approach was
successful
in raising an anti-(31A antibody. Stably transfected cells lines expressing
[31A were
created in the previously-characterized SNaIIA cell line. SNaIIA cells are a
stable line
expressing type IIA sodium channel a subunits in Chinese hamster lung (CHL)
cells
(Isom et al., 1995). Because SNaIIA cells are 6418-resistant, pcDNA3-[31A was
co-
transfected with pSV2*Hyg in a 10:1 ratio (pcDNA3-(31A: pSV2*Hyg) so that
transfected clones could be selected with the antibiotic hygromycin. A number
of
hygromycin-resistant colonies were analyzed by Northern blot for [31A mRNA
expression. Positive clones were expanded and analyzed further by [3H]-STX
WO 01/23571 cA o23amn 2002-02-0~ PCT/US00/27119
-92-
binding. Western blot analysis of one of these cell lines, SNaIIA(31A-16,
identified
an immunoreactive band of approximately 45 kDa.
[3H]-STX binding analysis revealed a significant increase in the expression
levels of functional sodium channels at the plasma membrane of SNaIIA[31A-16
cells
as compared to the parent line, SNaIIA (Table 2). Non-linear regression
analysis of
saturation binding showed a 4.4-fold increase in BmaX as compared to SNaIIA
with no
significant change in the KD (0.8 nM for SNaIIA vs. 0.9 nM for SNaIIA(31A-16).
WO 01/23571 cA o23amn 2002-02-0~ PCT/US00/27119
-93-
Table 2. 3H-Saxitoxin binding analysis of aIIA- and aIlA/(31A-expressing cell
lines. Whole cell saturation binding analysis of SNAIIA and SNaIIA-(31A cells
was
performed as previously described (Isom et al., 1995b) over a concentration
range of
0.1 to 10 nM [3H]STX with the addition of 10 ~M unlabeled tetrodotoxin to
assess
non-specific binding. 3H-Saxitoxin (3H-STX, 28 Ci/mmol) was obtained from
Amersham. Binding data were normalized to protein concentration using the BCA
Protein Assay kit. Saturation binding data were analyzed by non-linear
regression
using Prism to obtain KD arid BmaX values.
cell line Bmax +/- SEM KD +/- SEM
SNaIIA 7.9 +/- 1.6 fmol/m 0.81 +/- 0.53
nM
SNaIIA- 1A-16 35.1 +/- 8.8 finol/m 0.92 +/- 0.89
nM
WO 01/23571 cA o23amn 2002-02-0~ PCT/US00/27119
-94-
These results are similar to a previous study showing that coexpression of
aIIA and (31 resulted in a 2- to 4-fold increase in the level of [3H]-STX
binding
compared to cells expressing aIIA alone (Isom, et al., 1995). The KD values
obtained
in the present study were very similar to those reported values as well. These
data
suggest that a function common to (31 and [31-A is to increase the level of
sodium
channel expression at the plasma membrane. The inventors suggest that (31 and
(31-A
may stabilize the conformation of channels such that they become more
resistant to
degradation and/or target newly synthesized channels to the plasma membrane
from
intracellular stores. Because (31 and [31A contain a common cell adhesion
molecule
domain, it is proposed that the extracellular Ig loop may be necessary for
this
function.
To determine whether coexpression of (31A subunits affected the functional
properties of type IIA sodium channels in CHL cells, whole cell
electrophysiological
recording was used to compare sodium currents in the parent SNaIIA cell line
and in
three different SNaIIA(31 A cell lines. FIG. 2A shows currents, elicited by
depolarizations to varying test potentials, recorded in a typical SNaIIA cell
and a
typical SNaIIA[31 A cell. As is evident from these traces, coexpression of (31
A did
not dramatically alter the properties of voltage-activated sodium currents.
Nevertheless, currents in [31A-expressing cell lines were subtly different
from currents
in SNaIIA cells. For example, mean steady state inactivation curves for
SNaIIA[31 A
cell lines were shifted to more positive potentials than the mean inactivation
curve for
SNaIIA cells (FIG. 2B and FIG. 2D). Although this difference was quite small,
it was
observed in all three SNaIIA(31A cell lines, and was statistically significant
in two of
the three (31A-containing lines (SNaIIA(31A-7: p = 0.037; SNaIIA(31A-8: p =
0.024).
Thus, one effect of (31A association with aIIA may be a small positive shift
in the
voltage-dependence of steady state inactivation. For activation, mean voltage-
conductance curves for two of the three [31A expressing lines were
statistically
indistinguishable from SNaIIA (FIG. 2B, FIG. 2D); however, for SNaIIA(31A-16,
the
voltage-dependence of activation was shifted to a significantly more negative
membrane potential (p = 0.001). Thus, data from one of the three (31A cell
lines
suggests that (31A may also alter sodium channel activation.
WO 01/23571 cA o23amn 2002-02-0~ PCT/US00/27119
-95-
To determine whether (31A affected the time course of macroscopic sodium
currents, the decaying phase of whole cell currents, elicited over a range of
test
potentials, was fit with single exponential functions (FIG. 2C). For SNaIIA,
the
inactivation time constants determined from these fits were progressively
shorter at
progressively more positive test potentials, approaching a minimum of
approximately
0.5 ms at the most positive test potentials examined. For two of the three
SNaIIA(31A
cell lines, the rate of current inactivation was virtually identical to SNaIIA
(FIG. 2C).
However, for SNaIIA(31A-16, inactivation was faster at all test potentials
examined
(FIG. 2C). Because sodium channel inactivation is coupled to activation
(Aldrich et
al., 1983; Bezanilla, 1977), it is likely that the faster decay rate for
SNaIIA(31-16
currents were, at least in part, secondary to the negative shift in activation
that was
observed in this cell line. In addition, it is also possible that, similar to
(31, ~31A
subunits have direct effects on the rate of sodium channel inactivation.
The most dramatic effect of (31A, detected electrophysiologically, was a large
increase in the amplitudes of macroscopic sodium currents. FIG. 3A shows
amplitudes of currents evoked by depolarization to +10 mV in SNaIIA and
SNaIIA(31A cell lines, converted to current densities to factor out any
differences in
cell surface area. Current densities for SNaIIA cells were 6.9 pA/pF, whereas
current
densities were approximately 2.5 times greater for the three SNaIIA~31A cell
lines
(FIG. 3A). This difference reflected two distinct effects of (31A on sodium
channel
expression. First, (31A greatly increased the proportion of cells with
measurable
whole cell sodium currents. This effect is illustrated in FIG. 3B, which plots
the
number of SNaIIA (black bars) or SNaIIA(31A (white bars) cells with peak
currents
within different amplitude ranges. For SNaIIA, this amplitude-frequency
distribution
was bimodal. In 40% of the cells (16 out of 40) currents were
indistinguishable from
the small inward currents recorded in untransfected CHL cells (i. e. < 100
pA). In the
remaining 60% of the cells, currents, ranged from 500 pA to 5 nA, and thus
were
clearly due to expression of cloned type IIA channels. In contrast, all
SNaIIA(31 A
cells expressed large sodium currents (FIG. 3A). The frequency histogram for
SNaIIA(31A followed a normal distribution with a modal current range of 2-3
nA.
To determine whether the lower mean current density of SNaIIA cells was
solely due to its large proportion of low expressing cells, the mean current
density for
WO 01/23571 cA o23amn 2002-02-0~ PCT/US00/27119
-96-
SNaIIA, excluding these low expressers was recalculated. Eliminating low
expressing cells increased the mean current density for SNaIIA cells from 6.9
pA/pF
to 11.6 pA/pF; however, this value was still significantly smaller than the
mean
current density of cells expressing (31A (p = 0.014). Thus, even when
comparing only
those cells that expressed measurable sodium currents, (31A still increased
the density
of functional sodium channels on the cell surface.
EXAMPLE 8
[31A expression in neuropathic pain
To better understand the relationship it may have in neuropathic pain, (31A
subunit expression and localization was assessed in neural tissues after
spinal nerve
injury. In this example, spinal nerve ligation in rats was used to simulate
injury.
Patterns of (31 and (31A subunit expression levels differed from each other in
the SNL-treated animals. Staining intensity by anti-(31 A subunit antibodies
in the
DRG exhibited a post-surgical time-dependent increase. Labeling intensity in
nociceptive neurons was slightly elevated at two days post-surgery, and
labeling in
both nociceptive and sensory populations was enhanced at two weeks.
Importantly,
and in parallel with the continued expression of allodynic behavior, (31A
labeling
remained elevated at eight weeks post-surgery. Labeling increases were more
prominent in the LS than in the L4 regions, and in the ipsilateral than the
contralateral
DRG neurons.
While (31A levels continued to increase in SNL animals, (31 subunit expression
levels varied. DRG labeling by anti-(31 subunit antibodies was more intense at
the LS
than at the L4 regions in naive animals. Post surgically, the labeling
intensity either
was unchanged or decreased slightly at day 2. At two-weeks post-surgery, a
slight
increase in labeling was observed at the LS and a more pronounced increase at
the L4
level. At the eight week post-surgical time point, labeling in the LS DRG was
further
elevated, whereas the labeling in the L4 DRG was similar to that observed at
the two
week time point. Thus, the (31 subunit exhibits a biphasic post-surgical
expression
pattern.
Immunohistochemical experiments indicated that the subcellular distribution
of the (31 subunit was uniformly diffuse or homogenous, whereas several
distinct
WO 01/23571 cA o23amn 2002-02-0~ PCT/US00/27119
-97
labeling patterns were observed for the X31 A subunit in both nociceptive and
sensory
neurons (Table 3). In naive or sham operated rats, (31A labeling was
predominantly
homogeneous in both nociceptive and sensory neurons of the LS region. At 14
days
after SNL, however. the (31A expression pattern was markedly altered. In
nociceptive
neurons, the dominant labeling was proximal to the membrane, one quarter was
punctate, and less than one quarter of the labeling remained homogeneously
distributed. In sensory neurons in the ipsilateral DRG to the SNL as well as
in both
nociceptive and sensory neurons in the contralateral DRG, similar (31 A
expression
patterns were observed. In these populations, 60-75% of the labeling remained
diffuse, whereas up to 28% of the (31A expression localized proximal to the
membrane in contralateral nociceptive neurons, or was punctate in ipsilateral
and
contralateral sensory neurons.
WO 01/23571 cA o23amn 2002-02-0~ PCT/US00/27119
-98-
Table 3. Relative distribution of the morphologic patterns of ~31A and (31
expression in LS DRG. Immunoperoxidase staining of S ~m sections with
polyclonal antibodies specific for either the X31 or (31A subunit of the
sodium channel.
Labeling patterns were "membrane" if the signal was relegated to the cell
plasma
membrane, "punctate" if the signal was intracellular aggregates, and
"homogenous" if
staining was diffuse. Percentages of labeling patterns are presented as
observed per
DRG per group. Treatment groups: naive, untreated; sham, surgery exposing, but
not
disturbing, the spinal nerve; SNL, spinal nerve ligation surgery-treated
animals.
AntibodySide Neuron Labeling patternsNaiveSham SNL
Class (%) (14d) (14d)
1A IpsilateralNociceptiveMembrane 15 51
Punctate 23 16 26
Homogeneous 77 69 23
Sensory Membrane 11
Punctate 18 11 18
Homogeneous 82 89 71
ContralateralNociceptiveMembrane 28
Punctate 14
Homogeneous 100 100 58
Sensory Membrane 4
Punctate 20
Homogeneous 100 100 76
1 IpsilateralSensory Membrane
Punctate
Homogeneous 100 100 100
NociceptiveMembrane
Punctate
Homogeneous 100 100 100
ContralateralSensory Membrane
Punctate
Homogeneous 100 100 100
NociceptiveMembrane
Punctate
Homogeneous 100 100 100
WO 01/23571 cA o23amn 2002-02-0~ PCT/US00/27119
-99-
The SNL-induced increase in (31A labeling intensity, coupled with the
dramatic post-surgical alteration of the subunit's subcellular distribution,
correlates
with a role for the (31 A subunit in the development and maintenance of
neuropathic
pain observed in this model.
EXAMPLE 9
Production of Mutants
The present Example briefly describes the generation of specific mutants
contemplated by the present inventors. Such mutants will yield specific
information
about the function and properties of particular regions of the ~31A protein.
The
inventors propose that the transmembrane and/or COOH-terminal intracellular
domains of the (3 subunits are responsible for transducing a signal from the
extracellular site of TN-R interaction to cytoskeletal or signaling molecules
inside the
cell. Truncation mutants that eliminate the intracellular COOH-terminal
domains of
(31 and [32 can thus be made and tested in the cell migration assays for
adhesion and
repulsion to determine the role of these domains in transducing signals from
TN-R to
the intracellular environment.
Introduction of COOH-terminal signal peptides that eliminate the
transmembrane domain and add glycophosphatidyinositol (GPI) lipid anchors to
the
~31A or other ~3 subunits will be fused with the extracellular domains of the
(3 subunits.
The inventors also will construct expression vectors that contain point
mutations of amino acids in ~31A that are predicted to be located in the
extracellular Ig
fold. With these constructs it will be possible to determine whether the Ig
fold
contains the site for TN-R interaction. In addition these data will provide
information
as to which residues in the Ig fold are critical for a(31A interaction.
Correlating the
amino acid sequences of (31 A, (31 and X32 with the recently solved crystal
structure of
myelin P o (Shapiro et al., 1996) will allow rational choices regarding which
mutations
might interact with TN-R. These studies will involve the creation of stable
cell lines.
All cell lines, with the exception of those expressing point mutations in the
extracellular regions, described below, will be cloned and characterized for
mRNA
and protein expression.
WO 01/23571 cA o23amn 2002-02-0~ pCT/US00/27119
-100-
Subunit truncation mutants
Truncation mutants of (31A may be constructed, sequenced, and transfected
into Chinese hamster lung 1610 fibroblasts. To construct such mutants, (31 A
cDNA is
amplified by PCR. The 3' reverse primer incorporates a termination codon in
place of
a given residue such that the protein ends. This mutant cDNA is then subcloned
into
an appropraite vector such as pCMVneo (Stratagene), pcDNA3.IZeo (Invitrogen,
Carlsbad, CA) and the like. If the truncation mutants of ~31A and/or other (3
subunits
are expressed at the plasma membrane but are not affected by TN-R in the cell
migration assay, then it can be concluded that the COOH-terminal domains of
these
subunits are critical for transducing intracellular signals between TN-R and
intracellular proteins. If these mutants behave like wild type subunits in the
cell
migration assay, then it can be concluded that the COOH-terminal domains are
not
necessary for these signal transduction events.
Introduction of COOH-terminal signal peptides for GPI linkage
The importance of the transmembrane domains of ~3 subunits can be
determined by replacing them with GPI moieties. A number of CAMS, including
the
TN-R receptor F3/contactin, have been shown to contain a GPI moiety which
anchors
the extracellular portion of the protein directly to the plasma membrane
without a
transmembrane domain. Nascent proteins destined to be GPI-linked have been
shown
to contain both NH z - and COOH-terminal signal peptides (Udenfriend et al.,
1995).
It is thought that the COOH-terminal signal peptide is cleaved and replaced
with GPI
by a putative transamidase enzyme in the endoplasmic reticulum, transamidase
protein + GPI -~ GPI-protein + COOH-terminal signal peptide.
The amino acid residue at which cleavage and GPI linkage occurs is termed
the c~ site. An extensive literature describes the hydrophobicity requirements
for the
COOH-terminal signal peptide, the length of the hydrophilic spacer region, as
well as
the identities of amino acid residues present at the c~, c~ + l, and c~~ + 2
sites (reviewed
by Udenfriend et al., 1995). cDNAs can be constructed which contain a GPI
linkage
in place of their transmembrane domains by creating chimeras of the
extracellular NH
-domain of each (3 subunit with the COOH-terminal signal peptide of the GPI-
WO 01/23571 cA o23amn 2002-02-0~ PCT/US00/27119
-101-
anchored precursor of chicken N-CAM (Cunningham et al., 1987; Furukawa et al.,
1997).
Chimeras
Chimeras can be generated by PCR using the following strategy: Briefly, the
extracellular NH z-terminal regions of (31A or any other (3 subunit is
amplified by
PCR from extracellular epitope-tagged ~31A, (31 or X32 plasmid templates. The
PCR
will incorporate antisense oligonucleotide primers which code for IVSETVIP
(SEQ
ID NO:10) for [31 or PERDTVIP (SEQ ID NO:11 ) for (32 (a fusion protein
between
the 3' end of the (3 subunit extracellular region and the 5' end of the region
of N-
CAM).
The COON-terminal region of N-CAM, will also be amplified by PCR from
chicken brain cDNA. The PCRs incorporate sense oligonucleotide primers that
encode IVSETVIP (SEQ ID NO:10) (for use with (31) or PERDTVIP (SEQ ID
NO:11 ) (for use with X32), creating a fusion protein between the COOH-
terminal
residues of the extracellular region of (31, (32 OR ~31A and the S' end of the
COOH-
terminal region of N-C AM). The PCR products will be purified and mixed
(extracellular (31 + N-CAM in one reaction and extracellular (32 + N-CAM in
another
reaction and extracellular (31A + N-CAM in another reaction) for use in a
second
round of PCR in which the components will anneal at the common IVSETVIP (SEQ
ID NO:10) region for (31 + N-CAM or PERDTVIP (SEQ ID NO:11 ) for ~i2 + N-CAM
and be filled in by the polymerase, thus generating the desired chimeras.
The processed GPI-linked proteins expressed by transfected 1610 fibroblasts
or other suitable cell line will each contain a COON-terminal sequence that
ends with
TVIPA with an attached GPI lipid anchor. To determine if GPI-~31A, GPI-(31 and
GPI-(32 are expressed at the plasma membrane as GPI-anchored proteins,
transfected
cells can be incubated with phosphatidylinositol-specific phospholipase C (PI-
PLC),
an enzyme which specifically cleaves the GPI moiety and releases the protein
into the
cell medium. The cells are then removed by centrifugation and X31, (31A or X32
will be
immunoprecipitated from the supernatant using antibodies specific to the
extracellular
epitope tags and analyzed by Western blot. If GPI-linked (31, (31A or (32 is
indeed
expressed in the plasma membrane of transfected cells, then these lines can be
tested
WO 01/23571 cA o23amn 2002-02-0~ pCT/US00/27119
-102-
in the cell migration assay. If these mutants behave like cells transfected
with full
length (3 constructs, then it is concluded that the (3 subunit transmembrane
domains
are not necessary for transducing TN-R-mediated signaling events and that the
(3 subunits behave similarly to F3/contactin. If these mutants are expressed
but do not
behave like wild type (3 subunits in the cell migration assay, then the
conclusion is
that the transmembrane domains are necessary for TN-R-mediated signal
transduction.
Site-directed mutagenesis of extracellular residues
The crystal structure of the CAM MP o has recently been reported (Smith and
Goldin, 1998). (31, (31A and (32 are structurally homologous to MP o and used
the
alignment of MP o and (31 to make predictions about key residues in a(31
subunit
interactions (Isom et al., 1995b; McCormick et al., 1998). Because (31, (31A,
(32 and
MP o are structurally related CAMs, the alignment of their sequences can be
used to
make some predictions regarding possible interactions of the (3 subunits with
TN-R.
In addition, it has been shown that the third Ig loop of F3/contactin is
responsible for
TN-R binding (Pesheva et al., 1993). This domain of F3/contactin is homologous
to
(32 (Isom et al., 1995b), suggesting that the Ig loop of (32 may be involved
in
interaction with TN-R as well. The region of highest homology between
F3/contactin
and (32, is predicted to be in the hydrophobic core of the Ig loop, based on
the MP o
crystal structure. Mutations in this core region result in (31 subunits which
fail to fold
properly or are unstable (McCormick et al., 1998). Thus, those residues are
not good
candidates for mutagenesis. Instead, other regions may be selected.
Oligonucleotide-directed In Vitro Mutagenesis System Version 2 or Sculptor
kits (Amersham Life Sciences, Arlington Heights, IL) are used to mutate
selected
residues, as described previously (McCormick et al., 1998). Stable cells lines
or
transient transfections may be performed to generate cells to be tested for
the mutant
(3 subunits analysis.
The mutations may be tested for plasma membrane expression by transient
transfection into 1610 fibroblasts as well as immunocytochemistry using
antibodies
recently to (31, X32, and (31A subunits. Mutants that express detectable
protein at the
plasma membrane can then be analyzed further in the cell migration assay.
WO 01/23571 cA o23amn 2002-02-0~ PCT/US00/27119
-103-
All of the compositions and/or methods disclosed and claimed herein can be
made and executed without undue experimentation in light of the present
disclosure.
While the compositions and methods of this invention have been described in
terms of
preferred embodiments, it will be apparent to those of skill in the art that
variations
may be applied to the compositions and/or methods and in the steps or in the
sequence
of steps of the method described herein without departing from the concept,
spirit and
scope of the invention. More specifically, it will be apparent that certain
agents which
are both chemically and physiologically related may be substituted for the
agents
described herein while the same or similar results would be achieved. All such
similar substitutes and modifications apparent to those skilled in the art are
deemed to
be within the spirit, scope and concept of the invention as defined by the
appended
claims.
WO 01/23571 cA o23amn 2002-02-0~ PCT/L1S00/27119
-104-
REFERENCES
Akopian et al., Nature 379, 257-262, 1996.
Aldrich et al., Nature 306(5942):436-41 1983.
Altieri, Nature 306: 436-441, 1994.
Arcone et al., Nucleic Acids Res.,l6(8):3195-207, 1988.
Armstrong and Bezanilla, J. Gen. Physiol. 70, 549-566 1977.
Auld et al., Neuron l, 449-461, 1988.
Baichwal and Sugden, In: Gene Transfer, Kucherlapati R, ed., New York, Plenum
Press,
pp. 117-148, 1986.
Barany and Mernfield "The Peptides, Gross and Meienhofer, eds", Academic
Press,
New York, 1-284, 1979.
Bartlett et al., Proc Natl Acad Sci USA.;93(17):8852-7; 1996.
Bartsch et al., Glia, 9, 57-69, 1993.
Bartsch, Prog. Neurobiol., 49, 145-168, 1996.
Bellus, Sci. Pure. App. Chem, A31(1): 1355-1376, 1994.
Bennett, Jr. et al., FEBSLett. 326, 21-24, 1993.
Bezanilla, J Gen Physiol. 70(5):549-566, 1977.
Brinster et al. Proc Natl Acad Sci U S A. 82(13):4438-42, 1985.
Burgess et al., Nature Genetics 10, 461-465, 1995.
Campbell et al., Br J Clin Pharmacol. Oct;46(4.:307-19, 1998.
Campbell, In: Monoclonal Antibody Technology. Laboratory Techniques in
Biochemistry and Molecular Biology, Vol. 13, Burden & Von Knippenberg
(Eds.), Elseview, Amsterdam, pp. 75-83, 1984.
Cannon et al., Eur. J. Physiol. 423, 155-157, 1993.
Capaldi et al., Biochem. Biophys. Res. Comm., 76:425, 1977.
Capucci, Drugs Aging.13( 1 ):51-70. 1998.
Catterall, Physiological Reviews 72, Suppl. S 15-S48, 1992.
Cech et al., Cell, 27 (3 Pt 2) p487-96, 1981.
Chaplan et al., JNeurosci Meth 53, 55-63, 1994.
Chen and Cannon, Pflugers Arch., 431:186-195, 1995.
Chen and Okayama, Mol. Cell Biol., 7:2745-2752, 1987.
CA 02381771 2002-02-07
WO 01/23571 PCT/US00/27119
-105-
Chiquet-Ehrismann et al., Perspect. Dev. Neurobiol., 2, 3-7, 1994.
Cunningham et al., Science 236, 799-806, 1987.
Davis et al., J. Cell Biol. 135, 1355-1367, 1996.
de la Pene et al., J. Biol. Chem. 267: 25703-25708, 1992.
Dib-Hajj and Waxman, FEBSLett. 377: 485-488, 1995.
Dib-Hajj et al., Proc. Natl Acad. Sci. USA, 95:8963-8968, 1998.
Duch et al., Toxicol Lett. 100-101:255-63, 1998.
Dugandzija-Novakovic et al., J. Neurosci. 15, 492-503, 1995.
EPO No. 320 308.
EPO No. 329 822.
Erickson, Curr. Opin. Cell Biol., 5, 869-876, 1993.
Fodor et al. Science, 251:767-773, 1991.
Forster and Symons, Cell, 49:211-220, 1987.
Fraley et al., Proc. Natl. Acad. Sci. USA., 76:3348-3352, 1979.
French et al., Toxicol Lett. 100-101:247-54, 1998.
Freshner, "Animal Cell Culture: a Practical Approach", Second Edition,
Oxford/New
York, IRL, Press, Oxford University Press, 1992.
Frohman, M.A., In: PCRTM PROTOCOLS.' A GUIDE TO METHODS AND
APPLICATIONS, Academic Press, N.Y., 1990.
Furukawa et al., Biochim. Biophys. Acta 1328, 185-196, 1997.
Fuss et al., J. Cell Biol., 120, 1237-1249, 1993.
Fuss et al., J. Neurosci. Res., 29, 299-307, 1991.
Gee et al., J. Neurosci. 18, 128-137, 1998.
Gefter et al., Somatic Cell Genet., 3: 231-236, 1977.
Gerlach et al., Nature, 328:802-805, 1987.
Gliemann, Biol. Chem. 379: 951-964, 1998.
Goding, In: Monoclonal Antibodies: Principles and Practice, 2d ed. Academic
Press,
Orlando, Fla., pp. 60-61, 65-66, 71-74, 1986.
Goldin et al., Proc. Natl. Acad. Sci. USA 83, 7503-7507, 1986.
Goodman and Gilman's "Pharmaceutical Basis of Therapeutics" Chapter 35, ninth
edition, Eds. Hardman et al., 1996.
Gopal, Mol Cell Biol., 5:1188--1190, 1985.
Graham and Van Der Eb, Virology, 52:456-467, 1973.
WO 01/23571 cA o23amn 2002-02-0~ PCT/LTS00/27119
-106-
Hacia et al. Nature Genetics, 14:441-447, 1996.
Hamill et al., Pflugers Arch., 391(2):85-100, 1981.
Hampson et al., Mol. Cell. Biol. 9: 1604-1610, 1989.
Harland and Weintraub, J. Cell Biol., 101:1094-1099, 1985.
Harlow and Lane, ANTIBODIES: A LABORATORY MANUAL, Cold Spring Harbor
Laboratory, Cold Spring Harbor, N.Y., 1988.
Higashikawa et al., Int. J. Cancer 66: 11-17 1996.
Hortsch et al., Cell Adh. Comm. 5, 61-73, 1998.
Hortsch et al., J. Cell Biol. 142, 251-261, 1998.
Innis et al., PCR Protocols, Academic Press, Inc., San Diego Calif., 1990.
Isom and Catterall, Nature 383, 307-308, 1996.
Isom et al., Cell 83, 433-442, 1995.
Isom et al., J. Biol. Chem. 270, 3306-3312., 1995.
Isom et al., Neuron 12:1183-1194, 1994.
Isom et al., Science 256, 839-842, 1992.
Joe and Angelides Nature 356, 333-335, 1992.
Johnson et al., "Peptide Turn Mimetics" in BIOTECHNOLOGYAND PHARMACY,
Pezzuto et al., Eds., Chapman and Hall, New York 1993.
Joho et al., Mol. Brain Res. 7, 105-113., 1990.
Joyce, Nature, 338:217-244, 1989.
Kaplan et al., Nature 386, 724-728, 1997.
Kayano et al., FEBSLett. 228, 187-194, 1988.
Kim and Cech, Proc. Natl. Acad. Sci. U.S.A., 84:8788-8792, 1987.
Kim and Chung, Pain 50, 355-363, 1992.
Klugbauer et al., EMBO J. 14: 1084-1090, 1995.
Kohler and Milstein, Eur. Jlmmunol., 6:511-519, 1976.
Kohler and Milstein, Nature, 256:495-497, 1975.
Korenberg et al., Genomics, 22:88-93, 1994.
Kounnas et al., J. Biol. Chem. 270:13070-13076, 1995.
KraBe et al., J. Neurosci. 8, 2859-2868., 1988.
Kwoh et al., Proc. Nat. Acad. Sci. USA., 86: 1173, 1989.
Kyte & Doolittle, J. Mol. Biol., 157(1):105-132, 1982.
Lagenaur and Lemmon Proc. Nat. Acad. Sci. U.S.A. 84, 7753-7757, 1987., 1987.
WO 01/23571 cA o23amn 2002-02-0~ PCT/US00/27119
-107-
Lochter et al., Eur. J. Neurosci., 6, 597-606., 1994.
Lochter, and Schachner, J. Neurosci., 13, 3986-4000., 1993.
Macejak and Sarnow, Nature, 353(6339):90-4, 1991.
Makita et al., Genomics 23, 628-634., 1994b.
Makita et al., J. Biol. Chem. 269, 7571-7578., 1994a.
Makita et al., J. Neurosci. 16: 7117-7127, 1996.
Malhotra et al., J. Biol. Chem. 273, 33354-33359.
Manipulating the Mouse Embryo A Laboratory Manual" 2nd edition eds., Hogan,
Beddington, Costantimi and Long, Cold Spring Harbor Laboratory Press, 1994.
McCormick et al., J. Biol. Chem. 273, 3954-3962., 1998.
McHugh-Sutkowski, and Catterall, J. Biol. Chem. 265, 12393-12399., 1990.
Merrifield, Science, 232: 341-347, 1986.
Michel and Westhof, J. Mol. Biol., 216:585-610, 1990.
Mount, Nucleic Acids Res. 10(2):459-72, 1982.
Natsch et al., Drug Saf., 17(4):228-40, 1997.
Nicolas and Rubenstein, Biotechnology,10:493-513, 1988.
Nicolau and Sene, Biochim. Biophys. Acta, 721:185-190, 1982.
Noda et al., Nature 320, 188-192., 1986a.
Noda et al., Nature 322, 826-828., 1986b.
Oh and Waxman, Proc. Nat. Acad. Sci. 91: 9985-9989, 1994.
Ohara et al., Proc. Nat'l Acad. Sci. USA 1989.
Oliviero et al., EMBO J., 6(7):1905-12, 1987.
Patton et al., J. Biol. Chem. 269, 17649-17655., 1994.
Pease et al., Proc. Natl. Acad. Sci. USA., 91:5022-5026, 1994.
Pelletier and Sonenberg, Nature. 28;334(6180):320-5, 1988.
Pesheva et al., J. Cell Biol., 109, 1765-1778, 1989.
Pesheva et al., Neuron, 10, 69-82, 1993.
Pignon et al, Hum. Mutat., 3: 126-132, 1994.
Poli and Cortese, Proc Natl Acad Sci USA.;86(21):8202-6, 1989.
Potter et al., Proc. Nat. Acad. Sci. USA., 81:7161-7165, 1984.
Prowse and Baumann, Mol Cell Biol., 8(1):42-51, 1988.
Qu et al., J. Biol. Chem. 270: 25696-25701, 1995.
Ragsdale and Avoli Brain Res Brain Res Rev. 26(1):16-28, 1999.
WO 01/23571 cA o23amn 2002-02-0~ pCT/US00/27119
-108-
Ragsdale Brain Res Brain Res Rev. 26( 1 ):16-28, 1998.
Reinhold-Hurek and Shub, Nature, 357:173-176, 1992.
Ridgeway, In: Vectors: A survey of molecular cloning vectors and their uses,
Rodriguez
RL, Denhardt DT, ed., Stoneham:Butterworth, pp. 467-492, 1988.
Rippe et al., Mol. Cell Biol., 10:689-695, 1990.
Roden Am J Cardiol.;82(4A):49I-57I. 1998.
Ron, et al., Mol. Cell. Biol., 2887-2895, 1991.
Sadot et al., J. Mol. Biol. 241: 325-331, 1994.
Saito et al., Proc. Natl. Acad. Sci., USA 91: 9725-9729, 1994.
Salzer, Neuron 18, 843-848., 1997.
Sambrook et al., Molecular Cloning: A Laboratory Manual, Cold Spring Harbor
Laboratory Press 1989.
Sangemeswaran et al., J. Biol. Chem. 271: 5953-5956, 1996.
Sarver et al., Science, 247:1222-1225, 1990.
Sashihara et al., Brain Research. Molecular Brain Research. 239-250, 1995.
Scanlon et al., Proc Natl Acad Sci USA., 88:10591-10595, 1991.
Schachner et al., Persp. Devel. Neurobiol., 2, 33-41, 1994.
Schaller et al., J. Neurosci. 15, 3231-42., 1995.
Schreibmayer et al., Pfliigers Arch. 426, 360-362, 1994.
Schwab, Neurochemical Res. 21: 755-761, 1996.
Shah et al., Soc. Neurosci. Abstr. 22 Pt. 1:63, 1996.
Shapiro et al., Neuron. 17, 435-449, 1996.
Shoemaker et al. Nature Genetics 14:450-456, 1996.
Smith et al., J. Neurosci. 18: 6096102, 1998.
Smith, and Goldin J. Neurosci. 18: 811-820, 1998.
Srinivasan et al., Nature 333, 177-180. 1988.
Stewart and Young, Solid Phase Peptide Synthesis, 2d. ed., Pierce Chemical
Co., 1984.
Tabiti et al., Gene 175: 7-13, 1996.
Tam et al., J. Am. Chem. Soc., 105:6442, 1983.
Taylor et al., JNeurosci Res., 35, 347-362. 1993.
Temin, Cell Biophys., 9(1-2):9-16, 1986.
Tong et al., Biochem. Biophys. Res. Commun. 195, 679-685. 1993.
Tur-Kaspa et al., Mol. Cell Biol., 6:716-718, 1986.
WO 01/23571 cA o23ai~~i 2002-02-0~ PCT/US00/27119
-109-
U.S. Patent 4,196,265.
U.S. Patent 4,554,101.
U.S. Patent 4,683,195.
U.S. Patent 4,683,202.
U.S. Patent 4,800,159.
U.S. Patent 4,861,719.
U.S. Patent 4,873,191.
U.S. Patent 4,883,750.
U.5. Patent 5,019,034.
U.S. Patent 5,120,657.
U.5. Patent 5,139,941.
U.S. Patent 5,252,479.
U.S. Patent 5,279,721,.
U.S. Patent 5,328,688.
U.S. Patent 5,354,855.
U.S. Patent 5,474,935.
U.S. Patent 5,507,724.
U.S. Patent 5,585,362.
U.S. Patent 5,612,205.
U.S. Patent 5,620,689.
U.S. Patent 5,622,856.
U.S. Patent 5,631,018.
U.S. Patent 5,658,776.
U.S. Patent 5,661,025.
U.S. Patent 5,661,033.
U.S. Patent 5,670,488.
U.S. Patent 5,686,278.
U.S. Patent 5,693,509.
U.S. Patent 5,707,618.
U.S. Patent 5,714,166.
U.S. Patent 5,749,847.
U.S. Patent 5,770,414.
U.S. Patent 5,773,289.
WO 01/23571 cA o23ai~~i 2002-02-0~ PCT/US00/27119
-110-
U.S. Patent 5,785,987.
U.S. Patent 5,789,213.
U.5. Patent 5,789,390.
U.S. Patent 5,795,581.
U.S. Patent 5,824,544.
U.5. Patent 5,824,547.
U.5. Patent 5,827,703.
U.S. Patent 5,830,725.
U.S. Patent 5,830,727.
U.S. Patent 5,834,441.
U.5. Patent 5,836,905.
U.S. Patent 5,849,571.
U.S. Patent 5,851,521.
U.5. Patent 5,851,818.
U.S. Patent 5,855,910.
U.5. Patent 5,856,152.
U.S. Patent 5,861,314.
U.5. Patent 5,863,541.
U.5. Patent 5,869,326.
U.S. Patent 5,879,934.
U.S. Patent 5,888,502.
Udenfriend and Kodukula, Annu. Rev. Biochem. 64,
563-591. 1995.
Ukomadu et al., Neuron 8, 663-676. 1992.
Urenjak Amino Acids, 14(1-3):151-8, 1998.
Vabnick, and Shrager, J. Neurobiol. 37, 80-96,
1998.
Walker et al., Proc. Nat'l Acad. Sci. U.S.A 89:392-396,
1992.
Wallace et al., Nature Genetics, 19: 366-370,
1998.
Wanner et al., FEBSLett. 336, 535-539, 1993.
Walther and Stein Mol Biotechnol., 6(3):267-86,
1996.
Waxman et al., Proc. Nat'1 Acad. Sci. U.S.A 96,
7635-9, 1999.
Weigt et al., J. Mol. Biol. 227: 593-595, 1992.
Westenbroek et al., Neuron 3:695-704, 1989.
Wilson et al., Mol Cell Biol. 1990 Dec;lO(12):6181-91,
1990.
WO 01/23571 cA o23amn 2002-02-0~ pCT/US00/27119
-111-
Wintergerst et al., Eur. J. Neurosci., S, 299-310, 1993.
WO 88/10315.
WO 89/06700.
WO 90/07641.
Wu and Wu, Biochemistry, 27:887-892, 1988.
Wu and Wu, J. Biol. Chem., 262:4429-4432, 1987.
Wu et al., Genomics, 4:560, 1989.
Xiao et al., Eur. J. Neurosci., 8, 766-782, 1996.
Xiao et al., J. Biol. Chem. 272(51): 32092-32101, 1997.
Xiao et al., Neurosci. Res. 49, 698-709. 1997.
Yang et al., Proc. Natl. Acad. Sci USA., 87:9568-9572, 1990.
Zechner et al., Mol Cell Biol. 1988 Jun;B(6):2394-401, 1988.
Zhang and Longo, J. Cell Biol. 128: 415-431, 1995.
Zhou et al., Neuron 7, 775-785, 1991.
Zisch et al., J. Cell Biol. 119, 203-213, 1992.
WO 01/23571 cA o23amn 2002-02-0~ pCT~S00/27119
SEQUENCE LISTING
<110> Isom, Lori L.
Kazen-Gillespie, Kristen
Rogers, Kathryn E.
Regents of the University of Michigan
Ortho-McNeil Pharmaceutical Inc.
<120> Methods and Compositions Relating to Novel Sodium
Channel b1A Subunits
<130> 8642-92
<140> Unknown
<141> 2000-09-29
<150> 60/156,837
<151> 1999-09-30
<160> 12
<170> PatentIn Ver. 2.1
<210> 1
<211> 850
<212> DNA
<213> Rattus sp.
<400> 1
atggggacgc tgctggctct cgtggtgggc gcggtgctgg tatcctcagc ctgggggggc 60
tgcgtggagg tggattctga gaccgaggca gtgtatggga tgaccttcaa aatcctgtgt 120
atctcctgta agcgtcgtag tgagaccacc gccgagacct tcacggagtg gaccttccgc 180
cagaagggca cagaggaatt tgtcaagatc ctacgctatg agaatgaggt gctgcagctg 240
gaggaagatg agcgctttga gggccgtgtg gtgtggaacg gtagtcgggg caccaaggac 300
ctgcaggacc tgtccatctt catcaccaat gtcacctaca accactctgg cgactacgaa 360
tgtcacgtct accgtctcct cttctttgat aattacgagc acaacaccag cgtcgtcaag 420
aagatccacc tggaggtggt ggacaagggt aagtggagcc ttgtcactct ctggcaagcc 480
agatggaggg acagatggaa agaaggggac aggctggtgt cacacagagg ccagctaaca 540
ccccgcagcc atagggggaa ggacacccct tttctggttc tggagacttc agctcttcag 600
cacacaggag gtcagattag gacccccacc ccacccccca caaatggtat gtgcattgga 660
ctgcactcat gctgtgtgac ctctgacggg tgtattccta tctctgagcc ccaagcctgt 720
ccccagggac cagagagaat attctgtatg gcttgctgtg tctctcaagc gggaccccat 780
tggagagacg tggggactta tttgaggcca cagtgggagt agcagaggcc agtggtgttt 840
ccgagtggaa 850
<210> 2
<211> 272
1
CA 02381771 2002-02-07
WO 01/23571 PCT/US00/27119
<212> PRT
<213> Rattus sp.
<400> 2
Met Gly Thr Leu Leu A1a Leu Val Val Gly Ala.Val Leu Val Ser Ser
1 5 10 15
Ala Trp Gly Gly Cys Val Glu Val Asp Ser Glu Thr Glu Ala Val Tyr
20 25 30
Gly Met Thr Phe Lys Ile Leu Cys Ile Ser Cys Lys Arg Arg Ser Glu
35 40 45
Thr Thr Ala Glu Thr Phe Thr Glu Trp Thr Phe Arg Gln Lys Gly Thr
50 55 60
Glu Glu Phe Val Lys Ile Leu Arg Tyr Glu Asn Glu Val Leu Gln Leu
65 70 75 80
Glu Glu Asp Glu Arg Phe Glu Gly Arg Val Val Trp Asn Gly Ser Arg
85 90 95
Gly Thr Lys Asp Leu Gln Asp Leu Ser Ile Phe Ile Thr Asn Val Thr
100 105 110
Tyr Asn His .Ser Gly Asp Glu Cys His Val Tyr Arg Leu Leu Phe Phe
115 120 125
Asp Asn Tyr Glu His Asn Thr Ser Val Val Lys Lys Ile His Leu Glu
130 135 140
Val Val Asp Lys Gly Lys Trp Ser Leu Val Thr Leu Trp Gln Ala Arg
145 150 155 160
Trp Arg Asp Arg Trp Lys Glu Gly Asp Arg Leu Val Ser His Arg Gly
165 170 175
Gln Leu Thr Pro Arg Ser His Arg Gly Lys Asp Thr Pro Phe Leu Val
180 185 190
Leu Glu Thr Ser Ala Leu Gln His Thr Gly Gly Gln Ile Arg Thr Pro
195 200 205
Thr Pro Pro Pro Thr Asn Gly Met Cys Ile Gly Leu His Ser Cys Cys
210 215 220
Val Thr Ser Asp Gly Cys Ile Pro Ile Ser Glu Pro Gln Ala Cys Pro
225 230 235 290
2
WO 01/23571 cA o23ai77i 2002-02-07 pCT~S00/27119
Gln Gly Pro Glu Arg Ile Phe Cys Met Ala Cys Cys Val Ser Gln Ala
245 250 255
Gly Pro His~Trp Arg Asp Val Gly Thr Tyr Leu Arg Pro Gln Trp Glu
260 265 270
<210> 3
<211> 19
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence:RT-PCR primer
common to b1A and b1
<400> 3
gaagatgagc gctttgagg
<210> 4
<211> 16
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence:RT-PCR primer
sequence unique to b1A
<400> 4
gagagacaca gcaagc
16
<210> 5
<211> 4
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence:bl sequence
flanking intron 3
<400> 5
Val Val Asp Lys
3
WO 01/23571 cA o23ai77i 2002-02-07 PCT/LJS00/27119
1
<210> 6
<211> 27
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence:forward PCR
primer b1 sequence flanking intron 3
<400> 6
agatccacct ggaggtggtg gacaagg ' 27
<210> 7
<211> 4
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence:bl sequence
flanking intron 3 in reverse direction
<400> 7
Ala Asn Arg Asp
1
<210> 8
<211> 27
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence:Reverse PCR
primer of b1 sequence flanking intron 3
<400> 8
acacgatgga tgccatatct ctgttgg
27
<210>g
<211>18
<212>PRT
<213>Rattus
sp.
4
CA 02381771 2002-02-07
WO 01/23571 PCT/US00/27119
<400> 9
Arg Trp Arg Asp Arg Trp Lys Glu Gly Asp Arg Leu Val Ser His Arg
1 5 10 15
Gly Gln
<210> 10
<211> 8
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Fusion Peptide
to add to b1
<400> 10
Ile Val Ser Glu Thr Val Ile Pro
1 5
<210> 11
<211> 8
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Fusion peptide
sequence for b2
<400> 11
Pro Glu Arg Asp Thr Val Ile Pro
1 5
<210> 12
<211> 218
<212> PRT
<213> Rattus sp.
<400> 12
Met Gly Thr Leu Leu Ala Leu Val Val Gly Ala Val Leu Val Ser Ser
1 5 10 15
Ala Trp Gly Gly Cys Val Glu Val Asp Ser Glu Thr Glu Ala Val Tyr
20 25 30
WO 01/23571 cA o23amn 2002-02-0~ PCT/US00/27119
Gly Met Thr Phe Lys Ile Leu Cys Ile Ser Cys Lys Arg Arg Ser Glu
35 40 45
Thr Thr Ala Glu Thr Phe Thr Glu Trp Thr Phe Arg Gln Lys Gly Thr
50 55 60
Glu Glu Phe Val Lys Ile Leu Arg Tyr Glu Asn Glu Val Leu Gln Leu
65 70 75 80
Glu Glu Asp~Glu Arg Phe Glu Gly Arg Val Val Trp Asn Gly Ser Arg
85 90 95
Gly Thr Lys Asp Leu Gln Asp Leu Ser Ile Phe Ile Thr Asn Val Thr
100 105 110
Tyr Asn His Ser Gly Asp Tyr Glu Cys His Val Tyr Arg Leu Leu Phe
115 120 125
Phe Asp Asn Tyr Glu His Asn Thr Ser Val Val Lys Lys Ile His Leu
130 135 140
Glu Val Val Asp Lys Ala Asn Arg Asp Met Ala Ser Ile Val Ser Glu
145 150 155
160
Ile Met Met Tyr Val Leu Ile Val Val Leu Thr Ile Trp Leu Val Ala
165 170
175
Glu Met Val Tyr Cys Tyr Lys Lys Ile Ala Ala Ala Thr Glu Ala Ala
180 185
190
Ala Gln Glu Asn Ala Ser Glu Tyr Leu Ala Ile Thr Ser Glu Ser Lys
195 200
205
Glu Asn Cys Thr Gly Val Gln Val Ala Glu
210 215
6