Note: Descriptions are shown in the official language in which they were submitted.
WO 01/21211 CA 02382834 2002-03-19 PCT/US00/25602
1
PROCESS FOR LESSENING POLYMORPHIC CONVERSION OF A DRUG
FIELD OF THE INVENTION
This invention pertains to a novel composition and process for
s maintaining a drug in a preferred therapeutic form. More particularly, the
present invention pertains to a composition, method of administration and
process for substantially lessening the conversion of a drug from one
polymorphic form to a different polymorphic form.
BACKGROUND OF THE INVENTION
In the practice of pharmacy and medicine, it is most important to both
know and to maintain a drug in its active form for good therapy. Many drugs,
for example, can exhibit more than one morphological form, generally known
as polymorphs. Polymorphs, or polymorphic forms, as used for the purpose
~s of this invention, denote the possible existence of a drug in one or more
crystalline and/or amorphous forms. As generally applied to drugs, it refers
to
the different structures of the same drug. More importantly, drugs that can
exhibit different solubilities and/or release rates as associated with this
scientific phenomenon can exhibit different polymorphic forms. With many
2o drugs, for which release rate from a dosage form in which dissolution is a
rate-limiting factor, different solubilities of drug in the gastrointestinal
environment is a rate-limiting step in drug absorption. Thus, different ,
solubilities associated with various polymorphic forms can exhibit an increase
or a decrease in the drug's therapeutic effect. This can lead to uncertainty
in
Zs therapy, in the absence of knowing the drug is administered to the patient
in
the desired polymorphic form.
The above presentation dictates of the critical need for a composition
and process for maintaining a drug in its desired polymorphic form. It is
thus,
a purpose of this invention to provide a composition and process for
so maintaining a drug in the desired polymorphic form and methods of
administering a dose of drug to a patient for good therapy.
WO 01/21211 CA 02382834 2002-03-19 PCT/11500/25602
2
SUMMARY OF INVENTION
In one aspect, the invention comprises a therapeutic composition
comprising a polymorphic drug in a preselected morphological form and a
stabilizing pharmaceutically-acceptable excipient maintaining greater than
90% of the drug in the desired morphological form prior to and during delivery
of the drug to the environment of use. The excipient may a polymer, and the
pre-selected morphological form may be amorphorous or crystalline. The
composition may be formed as a granule and may optionally comprise a
binder and/or a solubility regulating agent. The solubility regulating agent
may
control the pH of the composition. The composition may be formed as a
granule by a wet or dry granulation process. The composition may be formed
as an aggregate of granules, such as in a tablet by compression or in a
capsule containing the granules.
In another aspect, the invention comprises a method for maintaining a
15 pre-selected morphological form of a polymorphic drug in a therapeutic
composition which comprises granulating the drug in its pre-selected
morphological form with a pharmaceutically-acceptable excipient. The
excipient may be a polymer, and the polymer may be selected from the group
consisting of poly(olefin), poly(vinyl), poly(carbohyate) and poly(peptide)
2o polymers. The method may optionally include granulation with a binder
and/or a solubility regulating agent that may also control the pH of the
granule. Granulation may occur as a wet granulation process or a dry
granulation process. The composition may be formed as an aggregate of
granules, such as in a tablet by compression or in a capsule containing the
2s granules.
In yet another aspect, the invention comprises a method of
administering a drug to an environment of use comprising a patient, wherein
the method comprises administering the drug from a dosage form comprising
the drug and a pharmaceutically acceptable excipient that substantially keeps
3o the drug in a pre-selected morphological form and delivers the drug at a
controlled rate that correlates with the dissolution rate of the drug when in
the
environment of use. The method may comprise administering the dose of
WO 01/21211 CA 02382834 2002-03-19 PCT/US00/25602
3
drug from a dosage form comprising granules of the drug and a
pharmaceutically acceptable polymer that substantially maintains the drug in a
pre-selected morphological form, the dosage for optionally comprising one or
more binders or solubility regulating agents. The dose of drug may be formed
an aggregate of granules in tablet or capsule form.
DESCRIPTION OF THE DRAWINGS
Figure 1 is a graphical representation of the cumulative amount of drug
dissolved and released as a function of time from a representative
composition of the invention;
Figure 2 is a graphical representation of the cumulative amount of drug
released as a function of time from a representative composition of the
invention prepared in accordance with Example 5;
Figure 3 is a graphical representation of the cumulative amount of drug
~s dissolved as a function of time from a representative composition of the
invention prepared in accordance with Example 5;
Figure 4 is a graphical representation of the cumulative amount of drug
released as a function of time from a representative composition of the
invention prepared in accordance with Example 6;
zo Figure 5 is a graphical representation of the cumulative amount of drug
dissolved as a function of time from a representative composition of the
invention prepared in accordance with Example 6; and
Figure 6 is a graphical representation of the cumulative amount of drug
released as a function of time from a representative composition of the
25 invention prepared in accordance with Example 7.
DESCRIPTION OF THE INVENTION
The present invention has unexpectedly discovered the sciences of
pharmacy and medicine and their accompanying therapy can be advanced by
so providing a composition and process for substantially lessening the
polymorphic conversion of a drug.
WO 01/21211 CA 02382834 2002-03-19 PCT/US00/25602
4
The therapeutic compositions of the invention comprise a polymorphic
drug in a preselected morphological form and a pharmaceutically-acceptable
stabilizing excipient maintaining greater than 90% of the drug in the desired
morphological form prior to and during delivery of the drug to the environment
of use. The excipient may a polymer, and the pre-selected morphological
form may be amorphorous or crystalline. The composition may be formed as
a granule and may optionally comprise a binder and/or a solubility regulating
agent. The solubility regulating agent may control the pH of the composition.
The composition may be formed as a granule by a wet or dry granulation
process. The composition may be formed as an aggregate of granules, such
as in the form of a tablet by compression using conventional techniques or as
a capsule in which the granules are contained.
The process generally comprises granulating the drug with a polymer
and with optional excipients to provide a formulation that substantially
lessens
conversion of one morphological form of a polymorphic drug to another
morphological form. The term lessening as used herein includes inhibiting
and it denotes substantially keeping greater than 90% of the drug in the
polymorphic form in the dosage form. Often greater than 95% of the drug will
be maintained in a pre-selected polymorphic form.
2o As used herein, a "polymorphic drug" means a drug that may exist in
one or more crystalline and/or one or more amorphous forms. For example,
and without limitation, a polymorphic drug may have multiple crystalline
forms;
or it may have a simple crystalline form and an amorphous form; or it may
have multiple crystalline forms and an amorphous form; and so on.
2s A "polymorphic form," "polymorph" or "morphological form," as used
with respect to a drug means a single form of the drug selected from the
multitude of polymorphic forms in which the drug may exist.
The process comprises a granulation technique. Granulation is a
process of size enlargement. In a granulation process, small particles are
3o gathered into larger aggregates in which the original particles can still
be
identified. Granulation can be divided into a dry method, wherein no liquid is
used for the aggregation, or into a wet method wherein a liquid is used for
WO 01/21211 CA 02382834 2002-03-19 PCT/US00/25602
granule agglomeration of powder particles followed by a drying process.
In the wet granulation technique, for example, the drug and other
ingredients comprising composition are blended using a solvent, such as an
organic solvent, or a cosolvent such an organic-aqueous solvent like ethyl
s alcohol-water, 98:2 V:V (volume:volume) as the granulation fluid. Other
granulating fluid, such as denatured alcohol 100% can be used for this
purpose. The ingredients forming the drug composition are individually
passed through a mesh screen, such as a U.S. Sieve Series screen, and then
thoroughly blended in a mixer. Other ingredients comprising the drug
,o composition are dissolved in a portion of the granulating fluid, such as
the
solvent or cosolvent described above. Then, the latter prepared wet blend is
produced, which wet mass is next forced through a mesh screen onto oven
trays. The blend is dried for 18 to 24 hours at 30° - 50°C. The
dry granules
are then sized with a mesh screen. Next, a lubricant is screened and added
~s to the dry screened granule blend. The granulation is placed into a blender
and blended for up to 15 minutes. Additional compositions are made by the
same granulation techniques, consisting in suspending and tumbling the
granule-composition in a current of air with a coating that forms a membrane
that surrounds the drug granulation. Drug granulations used for the present
2o invention comprise roller compactions and slugging, and granulation by
extrusion and globulation can be used additionally for producing granules or
pellets for controlled release dosage forms.
In using dry granulation, in one manufacture, powder particles are
aggregated under high pressure and aggregate because of bonding forces
25 established by the direct contact between solid surfaces. The high pressure
serves to improve the contact area between the surtace and thus the overall
bonding strength. A binding agent can be added to the powder mix.
Polymeric binders form bridges between the particles and contribute thereby
to the strength of the composition. Dry granulation does not utilize heat or
so moisture, and therefore has application where heat-or moisture-sensitive
powders are processed alternatively, a wet granulation process may be used.
The advantages of using wet or dry granulation to maintain a drug in a chosen
WO 01/21211 CA 02382834 2002-03-19 PCT/US00/25602
6
polymorphic form additionally include improving the flow properties and hence
the mass uniformity of a dose of drug, to lessen the incidence of segregation
of the drug and other ingredients in a granulation, to improve the
manufacturing characteristics of a granulation, to enhance the solubility of a
s polymorphic form of a poorly water soluble drug by granulating the drug with
a
polymer, and to keep certain drugs in an amorphous form to enhance the
solubility and the bioavailability compared to the crystalline form of the
drug.
The amount of a polymer used for homogenously blending with a drug
to provide the granule dose of drug is 10 ng to 100 mg. The granulation
,o processes used by the invention produce granules with a size distribution
in
the range of 0.1 mm to 3.0 mm. Techniques for granulation are reported in
Encyclopedia of Pharmaceutical Technoloay, Vol. 7, pp 121-160, (1960),
published by Marcel Dekker, Inc; Pharmaceutical Sciences, Remington, 17tn
Ed, pp 1610-1615, (1985), published by Mack Publishing Co., air suspension
15 procedures are described in U.S. Pat. No. 2,799,241; J Amer Pharm Assoc ,
Vol 48, pp 451-454, (1979); and ibid., Vol. 49, pp 82-84 (1960). Other
standard manufacturing procedures are described in Modern Plastic
Encyclopedia, Vol. 46, pp 62-70 (1969); and Pharmaceutical Sciences,
Remington, 14tn Ed., pp 1626-1678 (1970), published by Mack Publishing Co.,
2o granulation techniques are described in ibid., pp1655-1660 (1970). The
granular compositions of the invention may be formed into aggregates of
granules, such as tablets formed by compression of the granules or capsules
containing the granules. Such forms are convenient for administering the
drug in its perselected polymorphic form, which will be throughout the tablet
or
2s capsule, as the case may be.
Representative of drugs that possess polymorphic forms comprise a
member selected from the group consisting of acetamide, acetaminophen,
amitriptyline, amobarbitol, amiperone, amcinonide, apronalide, acemetacin,
amisometradine, betamethasone acetate, hupicomide, buspirone,
3o bentiromide, biotiizolam, benoxaprofen, bupranolol hydrochloride,
butoxycaine
hydrochloride, butyrophenone, bolandiol dipropionate, benzocaine picrate,
cephalexin, chlordiazepoxide hydrochloride, carazolal, chlorpropamide,
WO 01/21211 CA 02382834 2002-03-19 PCT/LTS00/25602
7
codeine, clomipramine hydrochloride, clominorex, dimethoxanate
hydrochloride, diphenidol, dobutamine hydrochloride, erythromycin, enitabas,
ethinyl estradiol, etafedrine hydrochloride, flurbiprofen, fenbufen,
famotidine,
flupirtine maleate, griseofulvin, heptolamide, ibuprofen, indomethacin,
s indalpine, imipramine, levobunolol hydrochloride, mefenamic acid,
meprobomate, methisazone, methylprednisolone, methyltestosterone,
metahexamide, moclobemide, moperone, medrogestone; nifedipine, nystatin,
naftifine hydrochloride, noxiptiline hydrochloride, oxamiquine, piracetam,
piretanide, paxamate, propentofylline, piroxicam, propranolol hydrochloride,
penothiazine, phensuximide, protionamide, piribedil, pentobarbital,
phenylpropylmethylamine, phenytoin, resorantel, suloctidil, spironolactone,
sulfameter, sulfabenzamide, sulfapyridine, triclabendozole, terconazole,
tolbutamide hydrochloride, testosterone cypronate, theophylline, tolbulamide,
and leukotriene-antagonist exemplified by acitaganolast, iralukast,
montelukast, pranlukast, verlukast, zafirlukast, and zileton. The dose of
polymorphic drug blended with a polymer and/or other granule forming
ingredients is 10 ng (nanogram) to 40 mg (milligram) per granule.
Polymorphic drugs are known in Pharmaceutical Manufacturing, pp 35-42,
Feb. (1986); Drug Dev. Ind. Pharm., Vol. 13 (15) pp 2749-2769, (1987);
2o Pharmacy International, pp 233-237, Sept. (1986); Pharmaceutical
Manufacturing, pp 27-30, Jan. (1985); J Pharm Sci, Vol. 88(1 ), pp 103-108,
(1999); Sci Pharm, Vol. 62, pp 307-316, (1994); Sci Pharm, Vol. 58, pp 37-53,
(1990); Sci Pharm, Vol. 48, pp 55-67, (1990); Sci Pharm, Vol. 2, pp 81-96,
(1989); J Anal Chem, Vol. 338, pp 752-758, (1989); Sci Pharm, Vol. 55, pp
z5 13-25, (1987); Sci Pharm., Vol. 55, No. 1, pp 27-39, (1987); Sci Pharm Vol.
54, No. 2, pp 61-69, (1986); J Anal Chem., Vol. 322, No. 2, pp 164-169,
(1985); Mikrochim Acta, Vol. 2, pp 205-217, (1984); Mikrochim Acta, Vol. 2,
No. 1-2, pp 103-119, ( 1984); Arch Pharm, Vol. 311, No. 9, pp 757-761,
(1978); Sci Pharm, Vol. 46 (No. 1 ), pp 62-67, (1978), and Arch Pharm,:Vol.
so 307, No. 5, pp 377-384, (1974).
Representative of pharmaceutically-acceptable polymers for
granulating a drug to maintain the drug substantially in a dispensable form
WO 01/21211 CA 02382834 2002-03-19 PCT/US00/25602
8
comprise a pharmaceutically acceptable polymer selected from the group
consisting of a poly(olefin), poly(vinyl), poly(carbohydrate), poly(peptide),
poly(addition), poly(condensation), and poly(erodible) polymer. The term
poly(addition) refers to polymers prepared by addition polymerization
s processes, and the term poly(condensation) refers to polymers prepared by
condensation polymerization. The term poly(erodible) refers to polymers that
erode in the environment of use, namely the gastrointestinal tract. The
pharmaceutically acceptable polymers are selected from the group consisting
of poly(alkylene oxide) possessing a 1 x,000 to 5,250,000 weight average
,o molecular weight, exemplified by polyethylene oxide) of 100,000 molecular
weight, polyethylene oxide) of 200,000 molecular weight, polyethylene
oxide) of 300,000 molecular weight, polyethylene oxide) of 400,000
molecular weight, polypropylene oxide) of 600,000 molecular weight,
copoly(ethylene-propylene oxide) of 1,250,000 molecular weight;
carboxyalkylcellulose, comprising alkali carboxyalkylcellulose including
sodium carboxymethylcellulose, potassium carboxymethylcellulose, and
calcium carboxyethylcellulose, wherein the carboxyalkylcellulose possess a
molecular weight of 10,000 to 2,750,000; poly(hydroxyalkyl methacrylate) of
5,000 to 5,000,000 molecular weight; poly(vinylpyrrolidone) of 10,000 to
20 360,000 molecular weight; polysaccharides such as agar, acacia, karaya,
tragacanth, algin, and guar of 5,000 to 750,000 molecular weight;
poly(glucan); poly(amine); and poly(amino acid). The amount of polymer in
the polymorphic granulation is 10 ng to 100 mg. The polymers are known in
U.S. Pat. Nos. 3,865,108; 4,002,173; 4,207,893; 4,327,725; and 4,844,984;
25 and in Handbook of Common Polymers, by Scott and Roff, published by
Cleveland Ruther Company, Cleveland, Ohio.
The drug-polymer granule provided by the invention to substantially
maintain, wherein substantially maintain denotes ninety percent or higher to
100% of the drug, in a preselected polymorphic form optimally comprises 10
so ng to 20 mg of a binder. The pharmaceutically acceptable binders used for
the purpose of this invention comprise a member selected from the group
consisting of a 2,500 to 3,000,000 viscosity-average molecular weight
WO 01/21211 CA 02382834 2002-03-19 PCT/US00/25602
9
poly(vinylpyrrolidone) polymer and copolymer thereof, such as a copolymer of
poly(vinylpyrrolidone) with vinyl acetate. copolymer of polyvinylpyrrolidone
with vinyl alcohol, copolymer of polyvinylpyrrolidone with vinyl chloride,
copolymer of polyvinylpyrrolidone with vinyl fluoride, copolymer of
s polyvinylpyrrolidone with vinyl butyrate, copolymer of polyvinylpyrrolidone
with
vinyl laurate, and a copolymer of polyvinylpyrrolidone with vinyl stearate.
The
binder can be selected from a hydroxypropylalkylcellulose of 9,200 to 225,000
number-average molecular weight wherein alkyl is one to seven carbon
atoms, as selected from the group consisting of hydroxypropylmethylcellulose,
,o hydroxypropylethylcellulose, hydroxypropylbutylcellulose, and
hydroxypropylpentylcellulose. Additional binders that can be used for the
purpose of this invention comprise a member selected from the group
consisting of acacia, alginic acid, acrylic acid cross-linked with
allylsucrose,
acrylic acid cross-linked with allyl ether of pentaerythritol, dextrin,
gelatin, guar
15 gum, liquid glucose, maltodextrin, pregelatinized starch, sodium alginate,
starch, and zein. The binder may be present in the granule on the order of 10
ng to 200 mg per granule. The binders impart cohesive qualities to the
manufacture. Binders are known in Handbook of Pharmaceutical Excipients,
Second Edition, by Wade and Welter, (1994), published by the American
2o Pharmaceutical Association, Washington, D.C.
The polymorphic-drug granule may comprise a solubility regulating
agent for increasing the concentration of drug and for and concomitantly
regulating the pH of drug in a dispensed granule dose. The regulating agent
regulates the pH environment of the manufactured product. For poorly
2s soluble drug in aqueous fluids that need an increase in solubility, an
increased
dose often can be delivered without the drug conversion to a different form.
The solubility regulating agents useful for this invention comprise a member
selected from the group consisting of acidic and basic groups represented by
tromethamine also known as Iris(hydroxymethyl)-aminomethane;
3o diethanolamine; glycineamide; triethanolamine; N-[tris-
(hydroxymethyl)methyl]
glycine; sodium acetate; sodium lactate; sodium glycocholate; sodium
propionate; sodum butyrate; sodium glycocholate; glycocholate sodium
WO 01/21211 CA 02382834 2002-03-19 pCT/L1S00/25602
phosphate; potassium phosphate monobasic; potassium biphthalate; boric
acid; sodium borate; and sodium phosphate; acidic groups such as glycine,
leucine, methionine, serine, and other acids to regulate a basic compound
wherein the acid group is represented by adipic acid, succine acid, citric
acid,
s tartaric acid, malefic acid, and malic acid. The amount of regulator when
present in a granule composition is 10 ng to 200 mg per granule. The
regulating agents are disclosed in Handbook of Pharmaceutical Excipients,
Second Edition, edited by Wade and Waller, (1994), published by American
Pharmaceutical Association, Washington, D.C.
The drug-polymer polymorphic composition can be manufactured by a
wet granulation technique, for example, the drug and the preselected polymer
and/or additional ingredients comprising the drug-polymer polymorphic
composition are blended using a solvent, such as ethyl alcohol-water 98:2 V:V
(volume:volume) as the granulation fluid. Other fluids, such as denatured
,s alcohol 100% can be used for this purpose. The drug and other ingredients
optionally are passed through a mesh screen, such as the U.S. Sieve Series
Screen, and then blended thoroughly. Other ingredients are dissolved in a
portion of the fluid, such as the cosolvent described above. Then, the latter-
prepared wet blend is added slowly to the drug-polymer blend with mixing
2o continually. The fluid is added until a wet blend is produced, which wet
mass
is forced through a mesh screen onto oven trays. The polymorphic drug-
polymer blend is dried for 15 to 24 hours at 20° to 50°C. The
dry polymorphic-
drug blend are sized then with a mesh screen and formulated into a dosage
form.
2s The polymorphic-drug polymer formulation can be provided by a dry-
granulation method. This method can be used when the ingredients possess
inherent binding or cohesive properties, slugging may be used to form
granules of polymorphic-drug polymer formulation. This method is known in
the formulation art as precompression, or the double-compression method.
so This method comprises the conventional steps including weighing, mixing,
slugging, dry screening, and dosage form formulation. Prior art procedures
for effecting these methods are disclosed in Pharmaceutical Sciences, by
WO Ul/21211 CA 02382834 2002-03-19 PCT/US00/25602
11
Remington, 17t" ed., pp 1610-1615, (1985), published by Mack Publishing
Co., Easton, PA.
Exemplary solvents suitable for manufacturing the polymorphic-drug
polymer complex and dosage form provided by this invention comprise
inorganic and organic solvents. The solvents include a member selected from
the group consisting of aqueous, alcohol, ketone, ester, ether, aliphatic
hydrocarbon, halogenated, cycoaliphatic, aromatic, heterocyclic solvents, and
mixtures thereof. Representative solvents comprise acetone, methanol,
ethanol, isopropyl alcohol, butyl alcohol, methyl acetate, ethyl acetate,
isopropyl acetate, n-butyl ketone, methyl isobutyl ketone, n-hexane, m-
heptane, ethylene glycol monoethyl ether, ethylene glycol monoethyl acetate,
methylene dichloride, ethylene dichloride, propylene dichloride, carbon
tetrachloride, chloroform, nitroethane, nitropropane, tetrachloroethane, ethyl
ether, isopropyl ether, cyclohexane, cyclooctane, benzene, toluene, naptha,
tetrahydrofuran, diglyme, aqueous and nonaqueous cosolvents, such as,
acetone and water, acetone and methanol, acetone and ethyl alcohol,
methylene dichloride and methanol, ethylene dichloride and methanol,
trisolvents such as acetone-methanol and water, and, acetone-water and
isopropyl alcohol.
2o The compositions of the invention may be formed into aggregates of
granules, such as in the form of tablets or filled capsules prepared by
conventional manufacturing process. Such dosage forms are convenient for
administration of the drug which is distributed throughout the tablet or
capsule
in its pre-selected polymorphic form.
EXAMPLES PROVIDED BY THE INVENTION
The following examples are illustrative of the present invention. The
examples should not be considered as limiting the scope of the invention in
any way, as these examples and equivalents thereof, will become apparent to
so those versed in the art in light of the present disclosure, the drawings
and the
accompanying claims.
WO 01/21211 CA 02382834 2002-03-19 PCT/US00/25602
12
EXAMPLE 1
A dosage form is prepared for maintaining a drug in an amorphous
polymorphic form as follows: first, a compositional binder solution is
prepared
by adding 7650 g of purified water into a solution vessel. Next, 1350 g of
s polyvinylpyrrolidone possessing a viscosity-average molecular weight of
40,000 is added slowly to the vessel, and the solution mixed gently for about
40 minutes to produce a homogenous solution.
Next, a drug composition is prepared as follows: first, 2010 g of
tromethamine, (2-amino-2-hydroxymethyl-1, 3-propanediol) is passed through
a 20 mesh screen, U.S. Sieve Series. Then, 60 g of colloidal silicon dioxide
is
passed through a 40 mesh screen. Then, 2400 g of sodium
carboxymethylcellulose possessing a viscosity, measured by a Brookfield
viscometer, at 25°C, of a 2% concentration between 25-45 cps, a degree
of
substitution of 0.70 - 0.80 mol, about 35,000 molecular weight, is placed into
a
15 plastic bag. Next, 2280 g of polyvinylpyrrolidone of 40,000 viscosity-
average
molecular weight is passed through a 40 mesh screen and added to the same
plastic bag. Then, the screened colloidal silicon dioxide is added to the same
plastic bag and the bag tumbled for 1 minute to obtain a blend of sodium
carboxymethylcellulose, polyvinylpyrrolidone and colloidal silicon dioxide.
2o Next, a fluid bed granulator bowl is heated to 28°C. Then, 4320 g of
the leukotriene-receptor antagonist, amorphous zafirlukast is added to the
bowl. Next, the 2010 g of tromethamine is added to the granulator bowl,
followed by the triblend comprising the sodium carboxymethylcellulose, the
polyvinylpyrrolidone and the colloidal silicon dioxide. Then, 6000 g of the
2s binder solution is sprayed into the bowl at a rate of 80-125 ml/min. Then,
3000 g of purified water is added to the granulation bowl. During spraying the
air flow is maintained at 50 slpm (standard liters per minute). Also, during
the
granulation process, the binder solution is sprayed for 40 seconds, followed
by shaking for 15 seconds. Next, the granulation is dried to obtain a moisture
so content of 5.0-7.5%. The granulation is passed through an 8 mesh screen
into a laboratory mill. Then, 30 g of magnesium stearate is passed through a
40 mesh screen, added to the blend and blended for 2 minutes. The drug
WD 01/21211 CA 02382834 2002-03-19 PCT/ITS00/25602
13
composition is pressed into dosage form tablets, comprising 10 mg, 20 mg, 40
mg, or 80 mg of the leukotriene-receptor antagonist zafirlukast indicated for
the prophylaxis and chronic treatment of asthma in adult and children
patients.
EXAMPLE 2
A composition for use in a sustained-release dosage form is provided
by first providing a composition possessing expandable kinetics. The
composition is prepared as follows: first, a binder solution is prepared by
,o adding 9660 g of hydroxypropylmethylcellulose possessing a number-average
molecular weight of 13,000 (5 cps) into a mixing vessel containing 133,400 g
of purified water. Next, 6900 g of hydroxypropylcellulose possessing a
molecular weight of 80,000 is added to the mixing vessel. This mixture is
stirred until the ingredients dissolve in the water and to obtain a homogenous
,s solution.
Next, granules for forming an expandable osmotic composition are
prepared as follows: first, 36,000 g of osmagent sodium chloride are milled in
a grinder and passed through a 21 mesh screen. Also, 600 g of red ferric
oxide colorant is milled and passed through a 21 mesh screen. Then, a
2o granulator is heated to 40°C and 68,400 g of sodium
carboxymethylcellulose
possessing a viscosity at 25°C in a 1 % concentration of 3,000 to 4,000
cps, a
degree of substitution of 0.8 to 0.9 mol, and a molecular weight of 300,000 is
placed into the bowl of a granulator. Then, the 36,000 g of milled sodium
chloride is added to the granulator followed by 600 g of the colorant red
ferric
25 oxide, accompanied by granulation. Next, 30,400 g of the binder solution is
sprayed onto the powder bed at a rate of 1100 g/minute, during a processing
temperature of 40°C, with constant shaking to dislodge powders from
adhering from the granulator. At the end of the process, the moisture content
is adjusted to 6.5 to 8.5%. Next, the granulation is screened and placed into
a
so blender, followed by the addition of 586 g of colloidal silicon dioxide
screened
through a 30 mesh screen. The mixture is blended at 7 rpm for four minutes,
WO 01/21211 CA 02382834 2002-03-19 PCT/LTS00/25602
14
to yield an expandable composition for use in a controlled release dosage
form that delivers a drug at a controlled rate of release up to thirty hours.
EXAMPLE 3
s A pair of compositions, expressed as a bilayer core for use in a
controlled-sustained release dosage form for oral administration is
manufactured by compressing into layered arrangement the leukotriene-
receptor antagonist zafirlukast and the expandable osmotic composition. The
drug layer comprising the leukotriene-receptor antagonist zafirlukast
composition and the expandable-push layer disclosed above comprising the
expandable composition are compressed in a bilayer tablet press fitted with a
9.53 mm round cavity and concave punches and dies. The drug composition
is filled into the first hopper attached to the bilayer tablet press, and the
expandable composition is filled into the second hopper attached to the
~s bilayer tablet press. The press automatically dispenses 242 mg of the drug
composition into a die cavity which is tamped under a force of 80 Ibs,
(pounds). Next, 155 mg of the expandable composition is added to the die
cavity and both the drug layer and the expandable layer are compressed
under a force of 1800 Ibs. This process produces a thickness of the bilayer
zo core of 5.33 mm and an average hardness of 10 kp (kilopons). The bilayer
core produced by this manufacture comprises a drug layer comprising 80 mg
of amorphous zafirlukast, 47.4 mg of sodium carboxymethylcellulose, 62 mg
of polyvinylpyrrolidone, 39.6 mg of tromethamine, 11 mg of water, 1.3 mg of
colloidal silicon dioxide and 0.6 mg of magnesium stearate; the expandable
layer comprises 88.4 mg of sodium carboxymethylcellulose, 46.5 mg of
sodium chloride, 7.8 mg of hydroxypropylcellulose, 10.8 mg of
hydroxypropylmethylcellulose, 0.8 mg of ferric oxide, and 0.8 mg of colloidal
silicon dioxide. The bilayer can be administered as a bilayer dosage form to a
patient in need of leukotriene-antagonist therapy, and/or it can be enveloped
3o with a semipermeable wall and administered as a drug delivery device. The
bilayer comprises the drug-polymeric composition drug with the maintained in
its original pre-selected polymorphic form.
WO 01/21211 CA 02382834 2002-03-19 PCT/US00/25602
EXAMPLE 4
A dosage form provided by the invention is prepared as follows: first,
the bilayer core, described immediately above, is coated with a wall
comprising a semipermeable composition as follows: a closed, mixing vessel
s is used to manufacture a mixing solution. The mixing vessel is purged with
nitrogen. Then, 47,600 g of acetone is charged to the mixing vessel, and the
vessel heated to 25°C to 30°C. Next, 1 g of Poloxamer°
188, a
polyoxyethylene-polyoxypropylene glycol copolymer of the formula
HO(C2H40)a(C3Hs0)b(C2H402)aH wherein a equals 80 and b equals 27 having a
average molecular weight between 7680 and 9510 is slowly added with
stirring to the mixing vessel. Next, the Poloxamer and the acetone are mixed
for 10 to 15 minutes. Then, 1979.1 g of cellulose acetate comprising an
acetyl content of 39.8% is added to the mixing vessel. Then, the ingredients
are mixed for 2 hours to produce a clear solution.
15 Next, the bilayer cores are coated in a 24-inch perforated pan coater.
The coating pan is heated to an exhaust temperature of 40°C to
45°C. Then,
11,000 g of the bilayer compressed cores are placed into the pan coater.
Then, the pan is rotated at 13 rotations per minute. Next, the wall-forming
coating solution is sprayed onto the rotating cores at a rate of 110 ml/min
from
2 spray guns. During the coating process, the air volume in the coater is
maintained between 350 and 370 cfm, cubic feet per minute. The coating
process is stopped when the desired amount of semipermeable wall-forming
composition is sprayed onto the cores.
Next, a 30 mil (0.76 mm) orifice is drilled through the semipermeable
2s wall on the drug side of the just manufactured dosage forms. Then, the
residual acetone is removed by drying at 45°C and at 45% relative
humidity in
an oven for 68 hours. At the end of the drying cycle, the humidity is turned
off
and the dosage forms are dried at 45°C, for an additional 4 hours, to
yield an
osmotic dosage form.
W~ 01/21211 CA 02382834 2002-03-19 PCT/US00/25602
16
EXAMPLE 5
The dosage form provided by the above examples is analyzed in this
example. The dissolution of the drug zafirlukast indicates the drug entered
into solution upon its delivery from a dosage form provided by this invention
s as measured by the following procedure. First, an aqueous sodium dodecyl
sulfate, 1 % (w/v) (weight/volume) solution is used as the dissolution media.
A
dosage form prepared by this invention is placed into the dissolution media
and the drug released by the dosage form into the dissolution media is
sampled at a constant time interval over the time period of dissolution. The
,o filtered samples are assayed by a reversed high pressure liquid
chromatography with detection by UV at 224 nm. The concentration of the
samples are measured against a standard curve containing at least five
standard points. The dissolution test indicates the zafirlukast remains in its
amorphous state in the dissolution media. Procedures for dissolution testing
are reported in The United States Pharmacopoeia, The National Formulary,
pg. 1791 to 1796, (1995); Pharmaceutical Sciences, by Remington, 17th. Ed.,
pg. 653 to 666 (1985); and USP XXII, Dissolution Paddle Analysis, pg. 1578-
1579 ( 1990).
The release rate of drug, zafirlukast, from a dosage form manufactured
zo by this invention is ascertained by the following procedure. The procedure
comprises placing the dosage form in a solution, usually water, and taking
aliquots of the release rate solution, followed by their injection into a
chromatographic system to quantify the amount of drug released during
specified test intervals. The drug, for example, zafirlukast, is resolved on a
25 column and detected by UV absorption at 224 nm. Quantitation is performed
by linear regression analysis of peak areas from a standard curve containing
at least five standard points.
The release rate procedure comprises attaching a dosage form to a
plastic rod with the orifice exposed to the drug receiving solution. Then,
so attaching the rod to a release rate arm, with the arm affixed to an up/down
reciprocating shaker. which operates at an amplitude of about 3 cm and 2
seconds per cycle. Then, continuously immersing the dosage form in 50 ml
WO 01/21211 CA 02382834 2002-03-19 PCT/US00/25602
17
test tubes containing 30 ml of H20, equilibrated in a constant temperature
water bath at 37°C ~ 0.5°C. Next, at the end of each interval,
transfer the
dosage form to the next row of new test tubes containing water. After the
release pattern is complete, remove the tubes and allow the tubes to cool to
s room temperature, followed by filling the calibrated tubes to the 50 ml mark
with acetone. The samples are mixed immediately, transferred to sample
vials, followed by chromatography analysis. The dosage form prepared by
the example comprises a drug layer comprising 80 mg of micronized,
amorphous zafirlukast, 47.4 mg of sodium carboxymethylcellulose, 62 mg of
polyvinylpyrrolidone, 39.6 mg of tromethamine, 11 mg of water, 1.3 mg of
colloidal silicon dioxide, and 0.6 mg of magnesium stearate; a push layer
comprising 88.4 mg of sodium carboxymethylcellulose, 46.5 mg of sodium
chloride, 7.8 mg of hydroxypropylcellulose, 10.8 mg of hydroxypropyl-
methylcellulose, 0.8 mg of red ferric oxide, and 0.8 mg of colloidal silicon
15 dioxide; a wall comprising 26 mg of cellulose acetate comprising 39.8
acetyl
content and 5 mg of surtactant Poloxamer 188; and, a 0.76 mm orifice.
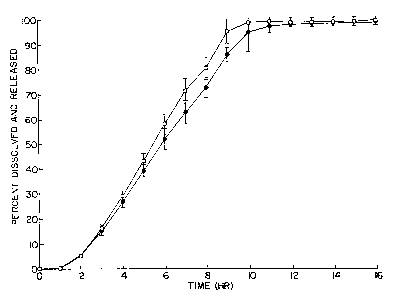
Accompanying drawing Figure 1 illustrates the average cumulative drug
dissolved depicted by clear boxes and the average cumulative drug released,
depicted by dark diamonds, over an extended period of 16 hours. The graph
2o illustrates zafirlukast is delivered by the dosage form without conversion
to a
crystalline monohydrate form, and the release rate and the dissolution rate
are substantially the same, with dual scientific analysis confirming the
disclosed and claimed invention unexpectedly lessens the incidence of the
starting polymorph converting to a different polymorph. That is, the release
Zs rate and dissolution rate are substantially the same, indicting that the
conversion of the amorphous polymorph to the crystalline polymorph is
inhibited as seen by the solubility of the drug form. Figure 2 depicts the
cumulative percent of zafirlukast released versus time for the dosage form;
and Figure 3 depicts the cumulative percent dissolved for zafirlukast versus
so time.
WO ~l/21211 CA 02382834 2002-03-19 PCT/US00/25602
18
EXAMPLE 6
The above procedures are followed with all manufacturing conditions
as set-forth, except for the present manufacture the semipermeable wall
comprises 85% cellulose acetate with an acetyl content of 39.8% and 15%
s Poloxamer° 188, a ethylene oxide-propylene oxide-ethylene oxide
triblock
copolymer available as Pluronic F68 from BASF Corporation, Mt. Olive, NJ. to
provide a dosage form comprising the bilayer drug and push layers with a
mean release rate of 10.51 mg/hr. AccomNanying Figure 4 depicts the
cumulative release rate percent over a time period of 20 hours from the
dosage form. Accompanying Figure 5 depicts the cumulative percent of the
drug dissolved versus time on release from a dosage form.
EXAMPLE 7
A dosage form designed and shaped like an osmotic dosage form to
~s deliver the crystalline form of zafirlukast is manufactured as follows:
first, a
drug layer comprising zafirlukast is made by passing 16.75 g of tromethamine
through a 20 mesh screen. Next, 33 g of crystalline zafirlukast is added to a
500 ml beaker, then 20 g of polyvinylpyrrolidone with an average molecular
weight of 40,000 is added to the beaker. Next, 30 g of
2o carboxymethylcellulose sodium, having an average molecular weight of
35,000 is added to the beaker. Next, all the ingredients are mixed with a
spatula and 60 ml of denatured anhydrous ethanol is added to the beaker with
mixing to change the consistency of the dry powder ingredients to granules.
The granulation then are placed in a hood overnight to dry. The dried
25 granules are passed through a 20 mesh screen to obtain a consistency in
granule size. Next, the granulation is transferred to a glass jar, 0.25 g of
magnesium stearate is added thereto, and the granulation mixed on rollers to
produce a zarfirlukast drug composition for processing into a drug-
composition layer.
so An expandable-push layer is prepared by following the expandable-
push procedure described above.
WO 01/21211 CA 02382834 2002-03-19 PCT/US00/25602
19
Next, a tablet press was used to compress the two layers comprising
the drug and expandable layers into a bilayered arrangement to form a tablet.
First, 260 mg of the drug layer provided immediately above is added to a
10.32 mm die cavity and lightly tamped to yield the drug layer. Next, 140 mg
s of the expandable-push layer is placed in the same cavity and the two layers
compressed under 1 ton of pressure to form a bilayer tablet. Next, the
bilayered tablet is surrounded with a wall comprising a semipermeable
polymeric composition by following the procedures set-forth above. The wall-
forming composition comprises 100% cellulose acetate possessing an acetyl
content of 32.0%. The wall-forming composition is dissolved in a mixture of
acetone and water to provide a cosolvent ratio of 88:12 (v:v), with a solid
composition of 5%.
The dosage form provided by this example exhibited an average
release rate of 12 mg/hr for 16 hours. The percent cumulative zafirlukast
15 (crystalline) released for this osmotic dosage form is depicted in drawing
Figure 6. The dosage form comprised a 0.76 mm drug-releasing orifice.
EXAMPLE 8
A dosage form tablet designed and shaped for oral administration to a
zo patient wherein the drug maintains its polymorphic form is manufactured as
follows: first 16.75 g of tromethamine is passed through a 20 mesh screen.
Then, 33 g of a drug selected from the group consisting of an analgesic, anti-
inflammatory, antihypertensive, antibiotic, anesthetic, antidiabetic,
antimicrobial, antifungal, antiepileptic, antihistaminic, anticonvulsant,
2s antiparkinson, antimalarial, antiparasitic, antiarthritic, cold-cough,
cardioprotective, corticoid, central nervous system acting drugs,
contraceptive, cardiovascular, diuretic, depressant, decongestant, dietary
supplement, electrolyte, hypnotic, hormonal, hypoglycemic, gastrointestinal
drugs, gonadotropins, leukotriene-receptor antagonist, minerals, muscle
so relaxant, muscle contractant, neoplastic modifiers, proteins, peptide,
psychic
energizers, sedative, sympathomimetic, steroid, tranduilizer, vasodilator, and
vitamin, 20 g of polyvinylpyrrolidone to the beaker, possessing a average
WO ~l/21211 CA 02382834 2002-03-19 PCT/US00/25602
molecular weight of 40,000. Then, 5.2 g of osmagent sodium chloride is
added to the beaker with mixing to produce a homogenous mass. Next, 30 g
of osmagel polyethylene oxide) possessing a 100,000 molecular weight is
added to the beaker and all the ingredients are mixed in the presence of
s anhydrous ethanol, to form granules. The granules are dried at 25°C
for 12
hours and then passed through a 20 mesh screen. Next, the granules are
transferred to a glass jar, and, 0.25 g of magnesium stearate is added
thereto,
and the granules mixed to produce a therapeutic composition comprising the
selected polymorphic drug.
Next, 260 mg of the therapeutic composition is added to a tablet press
and compressed under a pressure of 1-1/2 tons to a tablet core. Then, the
tablet core is enveloped with a wall composition comprising 90% cellulose
acetate having an acetyl content of 32% and 10% polyethylene glycol having
a molecular weight of 3,350 dissolved in a solvent. The solvent comprises
~s acetone and water, 88:12, wt:wt, to effect a solid composition of the
solution
of 5%. The coating temperature is 35°C to apply the semipermeable wall
around the drug compositional core. Next, a 50 mil (1.27 mm) passageway is
drilled through the semipermeable wall and the residual solvent is removed by
drying at 45°C and 45% relative humidity in an oven for 48 hours. At
the end
20 of the drying, the humidity is turned off, and the dosage form dried at
45°C for
an additional 4 hours to provide an elementary osmotic dosage form for orally
administering the drug in its unchanged polymorphic form to a patient in need
of therapy.
zs EXAMPLE 9
A therapeutic composition comprising a pharmaceutically acceptable
polymer for maintaining the polymorphic state of a drug is selected from the
generic group consisting of a poly(olefin), poly(vinyl), poly(carbohydrate),
poly(addition), poly(condensation), poly(erodible), poly(hydrophilic),
hydrogel,
so osmagel, and osmopolymer, as represented by a species selected from the
group consisting of a poly(aklylene oxide), polyethylene oxide),
polypropylene oxide), carboxyalkylcellulose, sodium carboxymethylcellulose,
WO ~l/21211 CA 02382834 2002-03-19 PCT/US00/25602
21
potassium carboxymethylcellulose, calcium carboxyethylcellulose, agar,
carrageenan, amylpectin, and starch graft copolymer; and a drug selected
from from the group consisting of acitazanolast, iralukast, montelukast,
pranlukast, verlukast, zafirlukast, and zileuton is prepared as follows:
first,
s 2,580 g of polyethylene oxide having a weight-average molecular weight of
200,000 is passed through a 40 mesh screen. Then, 1,290 g of the screened
polyethylene oxide) is placed into the bowl of a mixer. Then, 2,400 g of a
drug listed above is placed in the mixer over the polyethylene oxide). Next,
300 g of polyvinylpyrrolidone of 40,000 viscosity-average molecular weight is
passed through a 40-mesh screen and added to the mixer. The remaining
1,290 g of polyethylene oxide then is added to the bowl. Next, 300 g of
sorbitol and 360 g of tromethamine (2-amino-2-hydroxymethyl-1, 3-
propanediol) is passed through a 40 mesh screen and added to the mixer.
The addition of the dry ingredients is performed with the drug located between
15 the two layers of polyethylene oxide). The granulation process is initiated
by
the gradual addition of 3,200 g of ethyl alcohol with continuous mixing to the
mixer. Mixing is continued over a period of 5 to 10 minutes to effect a
consistency to change the dry powder to granules. The wet granulation is
dried at 40°C for 16 hours and then passed through a fluid air mill
with a 7-
2o mesh screen for size reduction. Next, the size-reduced granules are placed
into a blender. Then, 60 g of magnesium stearate that is passed through a
60-mesh screen is added to the granulation, and all the ingredients mixed for
4 minutes. This composition provides a drug, polyethylene oxide),
polyvinylpyrrolidone, tromethamine, sorbitol, and magnesium stearate, useful
2s for the therapy.
Next, a composition possessing expandable kinetics is prepared as
follows: first, a binder solution is prepared by adding 300 g of polyvinyl-
pyrrolidone of 40,000 average-molecular weight to a mixer containing 2,700 g
of water. Then, the mixture is stirred until the polyvinylpyrrolidone
dissolves in
so the water and forms a clear binder solution.
Next, the granules for forming an expandable, osmotic composition are
prepared as follows: first, 7,370 g of polyethylene oxide) having an average-
WO 01/21211 CA 02382834 2002-03-19 PCT/US00/25602
22
molecular weight of 7,000,000 is placed into the bowl of a fluid bed
granulator.
Then, 200 g of polyvinylpyrrolidone possessing an average-molecular weight
of 40,000 is added to the granulator. Next, 2,000 g of sodium chloride and
100 g of red ferric oxide, which is milled using a 20-mesh screen are added to
s the granulator. The powder ingredients are fluidized for 3 minutes to
produce
a uniform mixing of the powders. Next, the binder solution is sprayed onto the
powders at a solution spray rate of 50 g/min. During the spraying process the
process air flow is maintained at 500 cfm and the temperature maintained at
24°C. During the spraying operation the solution is sprayed for 30
seconds,
,o followed by a shaking time of 10 seconds. At the end of the spraying
operation, the granules are dried in the granulator for an additional 10 to 15
minutes to obtain a dry granulation. The granules are passed through a fluid
air mill with a 7-mesh screen for size reduction. The size reduced granules
then are placed into a blender. Then, 25 g of magnesium stearate, previously
screened through a 40-mesh screen, and 5 g of powdered butylated
hydroxytoluene, previously screened through a 60-mesh screen, are added to
the granules and mixed together to provide an osmotically expandable
composition.
Next, a bilayered core is manufactured by compressing in layered
2o arrangement the drug composition and the osmotic, expandable composition
described above as follows: first, 750 mg of the drug composition is added
into the cavity of a 5/16-in. (8-mm) diameter, and then 300 mg of the osmotic
expandable composition is placed into the die and the two compositions
compressed into layered arrangement with 1 ton (2,000 Ib.) of pressure.
2s Next, a wall forming composition comprising 90% cellulose acetate
having an acetyl content of 32% and 10% polyethylene glycol having a
molecular weight of 3,350 is dissolved in a solvent. The solvent comprises
acetone and water, 88:12, wt:wt, to effect a solid composition of the solution
of 5%. Then, the bilayer cores are placed into a 12-inch (30-cm) coating pan
so and the coating solution is sprayed onto the bilayer cores at a spray rate
of 25
g/min. The coating temperature is 35°C to apply 140 mg of the
semipermeable wall around and in contact with the bilayer core.
WO 01/21211 CA 02382834 2002-03-19 PCT/US00/25602
23
Next, a 50-mil (1.27 mm) passageway is drilled through the
semipermeable wall into the drug side of the dosage form. The residual
solvent is removed by drying at 45°C and 45% relative humidity in an
oven for
48 hours. At the end of the drying, the humidity is turned off and the dosage
s forms are dried at 45°C for an additional 4 hours, to provide an
osmotic
dosage form for orally administering a controlled, sustained delivery of the
polymorphic drug over thirty-hours of therapy.
PROCESSES AND METHODS OF USING THE INVENTION
,o The mode and the manner of the invention comprises applying the
invention in a plurality of embodiments. The applications comprise; (1 ) a
composition and process for maintaining a therapeutic drug in a known,
preselected polymorphic state selected from the group consisting of
amorphous and crystalline by blending the therapeutic drug with a
pharmaceutically-acceptable excipient that keeps the therapeutic drug in the
desired state; (2) a composition and process for maintaining a drug in a
polymorphic form by blending the drug with a polymer, wherein the polymer
member selected from the group consisting of poly(vinyl), poly(olefin),
poly(addition), poly(condensation), poly(cellulose), and poly(erodible) that
2o provides the chemokinetics for maintaining the drug in the polymorphic
form;
(3) a process for lessening the conversion of a polymorphic drug to a
different
polymorphic form, wherein the process comprises blending the drug with a
gastrointestinal administrable polymer that lessens the incidence of
conversion of the polymorphic drug to a different polymorphic form when
2s administered from a dosage form and in a gastrointestinal tract; (4) a
composition and process for keeping a drug in a first-therapeutic polymorphic
form by blending the drug with a pharmaceutically-acceptable polymer that
retards the conversion of the drug to a second polymorphic form and
simultaneously provides granules comprising the drug in the pre-selected
3o polymorphic form and the polymer; (5) a method of administering a drug to
an
environment of use comprising a patient, wherein the method comprises
administering the drug from a dosage form comprising the drug and a
WO 01/21211 CA 02382834 2002-03-19 PCT/US00/25602
24
pharmaceutically acceptable-polymer that substantially keeps the drug in its
original pre-selected polymorphic form and delivers the drug at a controlled
rate that correlates with the dissolution rate of the drug when in the
environment of use; and (6) a method of administering a dose of drug to a
s patient, wherein the method comprises administering the dose of drug from a
dosage form comprising granules of the drug and a pharmaceutically-
acceptable polymer that substantially keeps the drug in its preferred
pre-selected therapeutic polymorphic form for essentially providing a complete
dose of the drug to the patient.
And, while the above examples, figures and disclosure are set forth for
illustrating the mode and manner of the invention, various modifications and
embodiments can be made by those skilled in the pharmacy, medicine, and
the drug delivery and, in the light of the invention, without departing from
the
spirit of the invention.