Note: Descriptions are shown in the official language in which they were submitted.
CA 02386246 2002-04-03
WO 01/25419 PCT/US00/27381
ALTERING GENE EXPRESSION WITH
ssDNA PRODUCED Il~ T~IhO
The present invention relates to alteration of gene expression with a stable
DNA construct, conveniently referred to as a cassette, into which a nucleic
acid
sequence is incorporated for use as a template for subsequent production of
that
sequence in a prokaryotic or eukaryotic host cell, and a system for expression
of that
sequence within eukaryotic host cells without (or with minimal) flanking
sequences.
The construct, or cassette, includes inverted tandem repeats that form the
stem of a
to stem-loop intermediate that functions its vivo to cause expression of the
sequence,
referrAd to as the sequence of interest, as a single stranded DNA (ssDNA)
sequence
that binds to or otherwise interacts with a target gene to alter expression of
that gene.
The expression system of the present invention removes most or all contiguous
plasmid
(or other vector) sequences from the ssDNA either by stem-loop formation with
subsequent termination of a reverse transcription reaction by the stem or by
cleavage
of the stem-loop intermediate. The ssDNA produced by this method is designed
to be
complimentary to and/or to otherwise bind to any endogenous nucleic acid
sequence
target, thereby targeting any desired gene.
Antisense gene therapy has been successfully used in a variety of applications
2o to down regulate gene function. Jain, K.K., Handbook of Gene Therapy, New
York:
Hofgrefe & Huber Publishing ( 1998). To date, however, such therapy has been
characterized by a number of disadvantages and limitations, some serious, that
have
decreased the utility of this type of therapy, including the short half life
of the antisense
molecule, non-specific ef~'ects, uncertainties as to the mode of action of the
antisense
sequence, and potential toxic effects in animal studies. For instance,
antisense
oligonucleotides (ODNs) and their analogs must be administered intravenously,
which
involves problems in cell uptake and distribution (Cossum, P.A., et al.,
Disposition of
the '~C-labeled phosphorothioate oligonucleotide ISIS 2105 after intravenous
administration to rats, 267 J. Pharmacol. Exp. Ther. 1181-1190 (1993), Sands,
H.,
CA 02386246 2002-04-03
WO 01/25419 PCT/US00/27381
et al., Biodistribution and metabolism of internally 3H-labeled
oligonucleotides. II. 3',
5'-blocked oligonucleotides, 47 Mol. Pharmacol. 636-646 (1995)) as well as
toxicity
problems due to high blood concentrations (Henry, S.P., et al., Evaluation of
the
toxicity of ISIS 2302, a phosphorothioate oligonucleotide, in a 4-week study
in CD-1
mice, 7 Antisense Nucleic Acid Drug Dev. 473-481 ( 1997), Henry, S.P., et al.,
Comparison of the toxicity profiles of ISIS 1082 and ISIS 2105,
phosphorothioate
oligonucleotides, following subacute intradermal administration in Sprague-
Dawley
rates, 116 Toxicology 77-88 ( 1997)).
By far the antisense ODN analogs used most in antisense therapies are
to phosphorothioates or methylphosphonates. However, phosphorothioate ODNs
tend to
nonspecifically bind serum and intracellular proteins (Crooke, S.T., et al.,
Pharmocokinetic properties of several novel oligonucleotide analogs in mice,
227 J.
Pharmacol. Exp. Ther. 923-937 (1996), Gao, W.Y., et al., Phosphorothioate
oligonucleotides are inhibitors of human DNA polymerases and RNase H:
implications
for antisense technology, 41 Mol. Pharmacol. 223-229 (1992)), and at higher
concentrations, inhibit RNase H activity (Crooke, S.T., et al., Kinetic
characteristice of
Escherichia coli Rnase H: Cleavage of various ~ntisense oligonucleotide-RNA
duplexes, 312 Biochem. J. 599-608 (1995)). Phosphorothioate ODNs have a lower
Tm (an average of 0.5°C per base pair) for RNA than does DNA (Crooke,
S.T. and B.
LeBleu, Antisense research and application, Boca Raton: CRC Press (1993)).
This
lower Tm requires that phosphorothioate ODNs typically be longer than
phosphodiester DNA oligonucleotides for effective binding. However, an
increase in
the length of the ODN can cause a loss of hybridization specificity (Toulme,
J.J., et al.,
Antisense technology: A practical approach, in C. Lichtenstein and W. Nellen
(Eds.),
New York: IRL Press, pp. 39-74 ( 1997)). In addition, methylphosphonate ODNs
do
not activate RNase H enzyme activity (Maher, L.J, et al., Inhibition of DNA
binding
proteins by oligonucleotide-directed triple helix formation, 245 Science 725-
730
(1989), Miller, P.S., Oligodeoxynucleotides: Antisense inhibitors of gene
expression,
in J.S. Cohen (Ed.), Boca Raton: CRC Press, p. 79 (1989)) and are eliminated
rapidly
3o (Chen, T.L., et al., Disposition and metabolism of oligodeoxynucleoside
- 2 -
CA 02386246 2002-04-03
WO 01/25419 PCT/US00/27381
methylphosphonate following a single i.v. injection in mice., 18 Drug Metab.
Dispos.
815-818 ( 1990)).
Another approach to gene therapy is to administer molecules that have
catalytic
activity against the gene and/or the transcriptional product of the gene.
Ribozymes
comprise only RNA molecules, which can catalyze the cleavage of specific mRNA
sequences, and are thought to be potentially more efficient than antisense
ODNs
because of their catalytic capability (Woolf, T.M., To cleave or not to
cleave:
Ribozvmes and antisense, 5 Antisense Res. Dev. 227-232 ( 1995)). Ribozymes
have
been used as inhibitors of gene expression and viral replication (Jain, supra
( 1998)).
1o Unlike antisense ODNs, ribozymes can be delivered either endogenously, such
as by
using viral vectors, or exogenously. However, ribozymes have limited stability
due to
degradation by RNases irmiao (Jain, supra ( 1998)).
Using in vitro selections, several small single-stranded DNAs have recently
been demonstrated to catalyze the cleavage of RNA (Breaker, R.R., Catalytic
DNA:
15 In training and seeking employment, 17 Nature Biotechnology 422-423 (
1999)),
thereby offering the promise of targeted activity against specific genes. The
patent and
scientific literature describes a number of these short deoxynucleic acid
sequences that
have been shown to have catalytic activity (see, Breaker, R.R. and G.F. Joyce,
1
Chem. Biol. 223-229 (1994); Cuenoud, B. and J.W. Szostak, 375 Nature 611-613
20 (1995); Santoro, S.W. and G.F. Joyce, 94 Proc. Natl. Acad. Sci. USA 4262-
4266
( 1997); Faulhammer and M. Famulok, 269 J. Molec. Bio. 188-203 ( 1997); Carmi,
N,
et al., 95 Proc. Natl. Acad. Sci. USA (1998); Li, Y. and R.R. Breaker, 96
Proc. Natl.
Acad. Sci. USA 2746-2751 (1999) and U.S. Patent Nos. 5,807,718 and 5,910,408),
including the so-called "10-23 DNA enzyme" and other ssDNA sequences that act,
for
25 instance, as copper-dependent DNA ligases and calcium-dependent DNA
kinases.
The catalytic effciency of such sequences has been demonstrated for cleaving
mRNA
targets at 109 rri'/miri' in the presence of divalent magnesium, thereby
oi~ering the
opportunity for targeted destruction of substrate molecules (see, for
instance, R.R.
Breaker, sarpra (1999)). Although the art appears to recognize the potential
for use of
3o this enzymatic activity for therapeutic purposes, so far as is known, no
system is
- 3 -
CA 02386246 2002-04-03
WO 01/25419 PCT/US00/27381
available for producing these target-specific enzymatic nucleic acid sequences
to
produce a therapeutic effect irmivo.
There are, therefore, no systems available that utilize the potential
advantage of
the efficient catalytic activity of these enzymatic nucleic acid sequences for
altering
gene expression. It is, therefore, an object of the present invention to
provide a DNA
construct that directs the synthesis of ssDNA containing a sequence that
specifically
cleaves mRNA targets ifa oioo to alter the expression of the gene producing
that target
mRNA.
Because secondary structure folding may be critical to the catalytic function
of
the enzymatic sequence of the ssDNA, it is another object of the present
invention to
provide methods, and DNA constructs, for producing ssDNA including such a DNA
enzyme sequence of any desired nucleotide sequence within eukaryotic cells
without
undesirable intervening or flanking nucleotide bases so as to preserve the
enzymatic
function of the ssDNA against a target nucleic acid, for use in altering
expression of a
gene including that target nucleic acid.
Another object of the present invention is to provide a method, and a DNA
construct utilized in such methods, for production of ssDNA within eukaryotic
cells
that contains DNA enzyme sequences for overcoming the significant problems
encountered by the use of standard oligonucleotide delivery methods for
therapeutic
2o purposes.
Another object of the present invention is to provide a method, and a DNA
construct, for producing ssDN.A of any nucleotide sequence in vivo that
functions as
(but is not limited to) an inhibitory nucleic acid for, for instance, binding
to mRNA in
an anti-sense fashion to down regulate a gene product or a viral gene product
of
interest or binding to and inhibiting a specific cellular function, for
instance, by binding
to proteins that recognize a nucleic acid sequence.
Another object of the present invention is to provide a method, and a DNA
construct, for producing ssDNA designed to favor binding to duplex (native
DNA) to
form triplex structures that may interfere with normal gene transcription and
3o regulation.
- 4 -
CA 02386246 2002-04-03
WO 01/25419 PCT/LTS00/27381
Another object of the present invention is to produce ssDNA within eukaryotic
cells for the purpose of disrupting one or more cell functions.
Yet another object of the present invention is to provide a method, and a DNA
construct, for producing ssDNA into which secondary structures are designed so
that
the ssDNA oligonucleotides bind to and/or otherwise inhibit or activate
various cellular
functions that rely on the catalytic action of a protein or on nucleic acid
protein
interaction such as transcription, translation, and DNA replication.
Another object of the present invention is to provide a method, and a DNA
construct, for producing ssDNA irmivo for site-directed mutagenesis or gene
1o knockout for therapeutic applications.
Another object of the present invention is to provide a method, and a DNA
construct, for producing ssDNA of precisely defined nucleotide composition
that
favors site-specific insertion into a genome for therapeutic purposes.
Yet another object of the present invention is to provide a method, and a DNA
construct, for producing ssDNA that is complimentary to any endogenous nucleic
acid
target for use in altering expression of a gene including the nucleic acid
sequence
target.
Another object of the present invention is to provide a method, and DNA
expression utilized in such a method, for in vivo production of ssDNA
including a
2o sequence exhibiting catalytic activity against mRNA targets for
transfection into
eukaryotic cells that overcomes the obstacles to delivery of direct
administration of
ssDNA by lipofection, direct cellular uptake, and/or microinjection.
Another object of the present invention is to provide all enzymatic functions
that are necessary to produce ssDNA irmivo containing a sequence with
enzymatic
activity against a target mRNA of choice within a single or dual plasmid
expression
system.
Another object of the present invention is to provide a method, and
pharmacologically acceptable compositions, for delivering an inhibitory
nucleic acid
sequence including a sequence with enzymatic activity to target cells in a
manner which
produces a therapeutic effect.
- 5 -
CA 02386246 2002-04-03
WO 01/25419 PCT/US00/27381
This listing of the objects of the present invention is not intended to be a
list of
all the objects of this invention. There are a vast number of other cellular
functions
that are mediated by the cellular genome which, in the interest of brevity and
practicality, are not mentioned here and which are amenable to regulation by
in vivo
production of ssDNA. For instance, exonucleases digest ssDNA much more
aggressively than double-stranded DNA (dsDNA). Consequently, another object of
the present invention is to provide a ssDNA construct, and a method of
producing that
construct 171 VI7~0, that is not as susceptible to degradation by native
exonucleases in the
cell as dsDNA. It can be seen from this illustration that this list of some of
the objects
to of the present invention is provided for exemplification and is not
intended to limit the
scope of the invention.
These objects are provided by a method of altering expression of an
endogenous nucleic acid target sequence in a target cell comprising the steps
of
introducing a cassette comprised of a sequence of interest flanked by an
inverted
tandem repeat and a 3' primer binding site (PBS) into a target cell and
reverse
transcribing the mRNA transcript of the cassette from the PBS to release a
single
stranded cDNA transcript in the cell. The sequence of interest is comprised of
a
nucleic acid sequence that produces a sequence of nucleic acids that binds to
an
endogenous target nucleic acid sequence when reverse transcribed to alter
expression
2o of the target sequence.
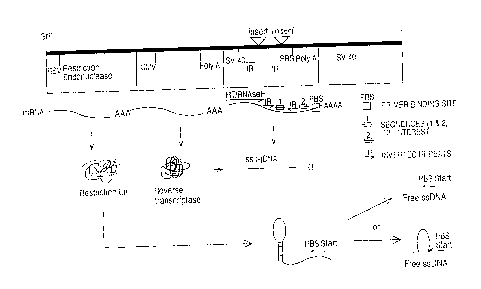
Several embodiments of the invention are illustrated in the figures, in which
Figure 1 is a schematic illustration of a production of ss-cDNA in a host cell
in
accordance with the present invention.
Figure 2 is a schematic illustration of the stem-loop intermediate formed by
the
method illustrated in Fig. 1.
Figure 3 is a schematic illustration of the pssXA plasmid comprising a first
component of a first embodiment of the expression system of the present
invention.
To make pssXA, reverse transcriptase (RT) and MboII genes were subcloned into
the
mammalian expression vector pBK-RSV (Stratagene) and expressed as a single
polypeptide. The RT and MboII domains are separated by a histidine-rich
linker.
- 6 -
CA 02386246 2002-04-03
WO 01/25419 PCT/US00/27381
Figures 4A and 4B are schematic illustrations of the pssXB plasmid comprising
a second component of the first embodiment of the expression system of the
present
invention (Fig. 4A) that includes the sequence of interest and contains ( 1 )
the
MoMuLV reverse transcriptase pomoter region, (2) two NotI sites, one PacI and
one
BamHI, for the subcloning of the DNA sequence of interest, and (3) the tandem
inverted repeats, IR-L and IR-R, and the sequence of the insert region of the
pssXB
plasmid (Fig. 4B).
Figure SA is a schematic illustration of the pssXC plasmid comprising a second
embodiment of the expression system of the present invention and including the
10-23
1o DNA enzyme sequence illustrated schematically in Fig. 5B.
Figures 6A and 6B represent schematic illustrations of the pssXD plasmid
comprising a third embodiment of the expression system of the present
invention Fig.
6A) and an elarged portion of the pssXD plasmid (Fig. 6B).
Figure 7 shows the result of a PCR assay for RT activity in a pssXA
transfected cell lysate. Lanes 1 and 2: A549 cells transiently transfected
with the
pBK-RSV vector; lanes 3 and 4: A549 cells transiently infected with pssXA;
lanes 5
and 6: A549 cells stably transfected with pssXA (E10). Before PCR
amplification,
reverse transcription reaction was carried out for 10 (lane l, 3, and 5) or 30
minutes
(lane 2, 4, and 6), repectively, at 37°C.
2o Figure 8 shows the result of an assay for detection of ssDNA by PCR
analysis.
Total RNA isolated from either E10 cells, transiently transfected with pssXB
vector,
pssXB-I or pssXB-II. Before PCR amplification, total RNA was pre-treated with
either Sl nuclease (lanes 1 and 3) or RNase (lanes 2, 4, and 5) for 30 minutes
at 37°C.
lanes 1 and 2: pssXB-I; lanes 3 and 4: pssXB-II; lane 5: pssXB vector.
Figure 9 shows the results of a dot blot analysis for detection of ssDNA. 1:
E10 cells transfected with pssXB-I; 2. E10 cells transfected with pssXB-II; 3:
E10
cells; 4: A549 cells.
Figure 10 shows a bar graph quantitating a Northern blot of a ssDNA
producing vector constructed in accordance with the present invention
producing an
3o antisense sequence against c-raf kinase. Lanes 1-3: cells harvested 24 hrs
after
CA 02386246 2002-04-03
WO 01/25419 PCT/US00/27381
transfection; lanes 4-6: cells harvested 48 hrs after transfection. Lane I : E
10 cells
transfected with pssXB vector; lanes 2 and 5: E 10 cells transfected with
pssXB-I;
lanes 3 and 6: E 10 cells transfected with pssXB-II.
Figure I 1 shows the results of a dot blot analysis for detection of ssDNA in
A549 cells transfected with control pssXD-I or pssXD-II containing the c-raf
DNA
enzyme sequence. No detectable signal was produced in the presence of S I
nuclease
due to the specific degradation of ssDNA enzyme by S I nuclease.
Figure 12 shows the results of quantitative RT-PCR to determine whether
ssDNA expressed in A549 cells transfected with pssXD-II altered c-raf mRNA
levels.
to Lane 1: control pssXD-I; Lane 2: pssXD-II.
Figure 13 shows the results of a Western blot for suppression of c-raf protein
expression in A549 cells transfected with pssXD-I or pssXD-II. Lane I: pssXD-
II;
Lane 2: control pssXD-I; Lane 3: untransfected cells.
Figure 14 shows the results of a Western blot for genomic DNA cleavage for
15 induction of cell apoptosis by suppression of c-oaf gene expression. Lane
1: pssXD
II; Lane 2: control pssXD-I; Lane 3: untransfected cells.
Figure 15 shows the results of a Western blot for PARP cleavage for induction
of cell apoptosis by suppression of c-raf gene expression. Lane 1: pssXD-II:
Lane 2:
control pssXD-I; Lane 3: untransfected cells.
2o In this description of the present invention, methods and nucleic acid
constructs
are described for producing single-stranded deoxyribonucleic acid (ss-cDNA)
oligonucleotides of virtually any predefined or desired nucleotide base
composition in
vivo in yeast, prokaryotic cells, and/or eukaryotic cells, with or without
flanking
nucleotide sequences, for use in altering the expression of a target gene.
Methods and
25 constructs are described that use biological rather than the irr vitro, or
artificial,
synthesis of ss-cDNA of desired nucleotide base composition. Because
biological, i.e.,
enzymatic reactions, are used in these methods, they are applicable to any in
vivo
system.
In one embodiment, the expression system of the present invention comprises a
3o vector (as used herein, the term "vector'' refers to a plasmid or modified
viral or non-
_ g _
CA 02386246 2002-04-03
WO 01/25419 PCT/LTS00/27381
viral recombinant biological construct used to deliver and manipulate
synthesized
and/or naturally occurring nucleic acid sequences) designed to produce any
sequence
of interest as a ss-cDNA molecule, preferably free of most contiguous vector
sequences, within mammalian cells. The vector system contains all the
necessary
enzymatic functions and signaling instructions for producing ss-cDNA in the
host cell.
The host cell to which the vector of the present invention is delivered
produces an
RNA transcript (Fig. 1 ), driven by an eukaryotic promoter, that is used as a
template
to direct the synthesis of any desired single-stranded DNA sequence (a
"sequence of
interest")
1o In more detail, a first expression system in which the vector comprises two
plasmids that are co-transfected into a suitable host cell, which can be yeast
or any
prokaryotic or eukaryotic cell, to produce the ssDNA sequence of interest in
the cell
for altering gene expression is described herein. A second expression system
is also
described that comprises a single plasmid including the sequence of interest
that is
15 transfected into a suitable host cell for production of the ssDNA sequence
of interest in
the cell for altering gene expression.
The ssDNA produced ifr vivo using the expression systems described herein
may be an inhibitory nucleic acid. Inhibitory nucleic acids may be ssDNA
synthesized
from the mRNA template or the mRNA template itself, which can specifically
bind to a
Zo complementary nucleic acid sequence. By binding to the appropriate target
nucleic
acid sequence, an RNA--RNA, a DNA--DNA, or RNA--DNA duplex or triplex is
formed. More commonly, these nucleic acids are often termed "antisense''
because
they are usually complementary to the sense or coding strand of the gene, but
the
"sense" sequence is also utilized in the cell for therapeutic purposes. The
term
25 "inhibitory nucleic acids" as used herein, therefore, refers to both
"sense" and
"antisense" nucleic acids.
By binding to a target nucleic acid, an inhibitory nucleic acid alters the
function
of the target nucleic acid. This alteration (usually an inhibitory effect)
results from, for
example, blocking DNA transcription, processing or poly(A) addition to mRNA,
DNA
3o replication, translation, or promoting inhibitory mechanisms of the cells
(such as
- 9 -
CA 02386246 2002-04-03
WO 01/25419 PCT/US00/27381
promoting RNA degradation). Inhibitory nucleic acid methods therefore
encompass a
number of different approaches to altering gene expression. These different
types of
inhibitory nucleic acid technologies are described in Helene, C. and J.
Toulme, 1049
Biochim. Biophys. Acta. 99-125 (1990), hereinafter referred to as "Helene and
Toulme," and which is incorporated herein in its entirety by this specific
reference
thereto.
In brief, inhibitory nucleic acid therapy approaches can be classified into (
1 )
those that target DNA sequences, (2) those that target RNA sequences
(including pre-
mRNA and mRNA), (3) those that target proteins (sense strand approaches), and
(4)
1o those that cause cleavage or chemical modification of the target nucleic
acids such as
the ssDNA enzymes, including the so-called "10-23 enzyme" as described herein.
The
first approach contemplates several categories. Nucleic acids are designed to
bmd to
the major groove of the duplex DNA to foam a triple helical or "triplex"
structure.
Alternatively, inhibitory nucleic acids are designed to bind to regions of
single stranded
DNA resulting from the opening of the duplex DNA during replication or
transcription. More commonly, inhibitory nucleic acids are designed to bind to
mRNA
or mRNA precursors. Inhibitory nucleic acids are also used to prevent
maturation of
pre-mRNA. Inhibitory nucleic acids may be designed to interfere with RNA
processing, splicing or translation. In the second approach, the inhibitory
nucleic acids
2o are targeted to mRNA. In this approach, the inhibitory nucleic acids are
designed to
specifically block translation of the encoded protein. Using this second
approach, the
inhibitory nucleic acid is used to selectively suppress certain cellular
functions by
inhibition of translation of mRNA encoding critical proteins. An example of
such an
inhibitory nucleic acid is the sequence that is complementary to regions of c-
myc
mRNA, which inhibits c-myc protein expression in a human promyelocytic
leukemia
cell line, HL60. which overexpresses the c-myc proto-oncogene (Wickstrom E.
L., et
al., 85 Proc. Natl. Acad. Sci. USA 1028-1032 (1988) and Harel-Bellan, A., et
al., 168
Exp. Med. 2309-2318 ( 1988)). As described in Helene and Toulme, inhibitory
nucleic
acids targeting mRNA have been shown to work by several different mechanisms
to
3o inhibit translation of the encoded protein(s).
- 10 -
CA 02386246 2002-04-03
WO 01/25419 PCT/US00/27381
Inhibitory nucleic acids can also utilize the third approach of designing the
"sense" strand of the gene or mRNA to trap or compete for enzymes or binding
proteins involved in mRNA translation as described in Helene and Toulme.
Lastly,
inhibitory nucleic acids are used to induce chemical inactivation or cleavage
of the
target genes or mR~IA. Chemical inactivation occurs, for instance, by
induction of
crosslinks between the inhibitory nucleic acid and the target nucleic acid
within the cell
and by the method contemplated herein, namely, the cleavage of the target
nucleic acid
by the sequence having enzymatic activity that is incorporated into the
cassette of the
present invention.
1o In brief, in a first aspect, the present invention comprises a set of
genetic
elements adapted for delivery into a cell to produce ssDNA ira vitro or in
vivo for
altering gene expression, an expression system comprising the set of genetic
elements,
and one or more stably transfected cells) comprising the set of genetic
elements. The
set of genetic elements is incorporated into an expression system for delivery
into the
~s cell and includes
(A) an RNA dependent DNA polymerase (reverse transcriptase)
gene, and
(B) a cassette including (I) an inverted tandem repeat (IR), (2) one
or more sequences of interest located (a) between the inverted repeat (IR),
(b)
20 3' to the IR, or (c) both between the IR and 3' to the IR and (3) a primer
binding site (PBS) for the reverse transcriptase that is located 3' to the IR
as
shown in Fig. 2.
Although not required, the expression system also preferably includes the
functions and
signaling instructions for transcription of these components in vivo and the
functions
25 and signaling instructions for translation of the reverse transcriptase
(RT) gene.
Additional elements that are optionally included in the set of genetic
elements of the
present invention may include one or more of an RNAse gene, usually associated
with
the RT gene, a restriction endonuclease (RE) gene (for a purpose described
below), a
downstream polyadenylation signal sequence for expression in eukaryotic cells
so that
30 the mRNA produced by the sequence of interest includes a poly(A) tail (see
Fig. 1),
- 11 -
CA 02386246 2002-04-03
WO 01/25419 PCT/US00/27381
and a DNA sequence having enzymatic activity when the linearized ssDNA folds
into
the appropriate secondary configuration. Although the present invention is not
so
limited, in one embodiment of the set of genetic elements, the DNA enzymatic
sequence is located within a sequence of interest, regardless of whether the
sequence
of interest is located between the inverted repeat (IR) or between the 3'
aspect of the
IR and the PBS.
In a first embodiment of the expression system described herein, a vector
system is provided that comprises a two plasmids, and the above-described set
of
genetic elements that is adapted for delivery to the cell to produce ssDNA Ill
vIVO
to includes the RNA-dependent DNA polymerase (reverse transcriptase) gene,
which
additionally contains an RNAse H gene, linked with a histidine-proline linker
to a
restriction endonuclease gene. These genes were constructed and inserted into
a
plasmid vector that contains the necessary transcriptional and translational
control
elements along with polyadenylation tailing sequences. This plasmid is
referred to
15 herein as the "A" plasmid, pssXA, as shown in Fig. 3. A second, "B" plasmid
was
constructed which, in the embodiment described herein, includes the three
above-listed
elements of the cassette, namely, a primer binding sequence (PBS) matched to
the
reverse transcriptase (RT), a sequence of interest (SOI), and an inverted
repeat (IR).
In this second plasmid, exemplified by the plasmid pssXB shown in Fig. 4, the
SOI is
20 located either between the inverted tandem repeats or in a 5' position
(with respect to
the mRNA transcript) to the PBS, the PBS being located at the most 3' aspect
of the
mRNA transcript. In other words, the SOI is lccated ( 1 ) between the IR, (2)
between
the IR and the PBS, and/or (3) both between the IR and between the IR and the
PBS,
and as will be described below, two B plasmids are described herein, one
(pssXB-1)
25 with the SOI between the IR (e.g., NotI sites) and the other (pssXB-II)
with the SOI
between the IR and the PBS (e.g., cloned into the PacIlBamHI sites). Like
plasmid A,
plasmid B also includes a combination of transcriptional control elements.
However,
in another preferred embodiment herein, the B plasmid does not include (or
require)
translational control elements since no protein product is produced from this
construct.
- 12
CA 02386246 2002-04-03
WO 01/25419 PCT/US00/27381
In another embodiment described herein, the expression system of the present
invention comprises a single plasmid vector, shown schematically in Figs. 5
and 6 and
designated as plasmids pssXC and pssXD, respectively, in which the above-
described
set of genetic elements is incorporated. The components of the B plasmid
described
s above, e.g., the PBS, SOI, and IR, reside in the untranslated 3' portion of
the RT
polyprotein in this C plasmid. In other words, when the RT-RNAse H component
of
the C plasmid is transcribed under control of an appropriate promoter (in the
embodiments described herein, the RSV promoter was utilized), the resulting
mRNA
transcript contains the coding region for the RT-RNAse H polyprotein and, at
the end
of translation at the stop signals, the additional mRNA transcript contains
(3' to this
translated protein) the elements from the B plasmid with further 3' downstream
signaling events for polyadenylation signals, which remain intact from the RT-
RNAse
H component.
The particular single plasmid expression system described herein does not
Is contain the restriction endonuclease (RE) gene, and therefore does not
digest the stem
of the stem-loop intermediate formed by the inverted repeats. Consequently,
the SOI
(including the DNA enzyme) is inserted into either the C or D plasmids only in
a 3'
position to the IR and unwanted vector sequences are removed by premature
truncation flf the ss-cDNA product as the transcript encounters the relatively
stable
2o stem of the stem-loop intermediate and is unable to continue transcribing
ss-cDNA
from the mRNA transcript. More specifically, as will be made apparent in the
following description, each SOI was inserted only within the PacIlBanrHI
restriction
sites of the pssXC and pssXD plasmids.
As will also be apparent from the following description of the B, C, and D
2s plasmids, the plasmids include cloning sites for insertion of the SOI. Both
NotI sites
(located between the IR) and PacIlBamHI (3' to the IR, e.g., between the IR
and the
PBS) sites are provided in the preferred embodiment of the B plasmid described
herein.
The C and D plasmids described herein include only the PacIlBamHI sites for
this
purpose. However, those skilled in the art who have the benefit of this
disclosure will
30 recognize that these particular cloning sites were chosen for the
particular systems
- 13 -
CA 02386246 2002-04-03
WO 01/25419 PCT/US00/27381
described herein and that other cloning sites may be equally useful for this
same
purpose. The A plasmid comprising the two plasmid vector system described
herein
was not intended to include the SOI, but those skilled in the art will also
recognize
that, if a two plasmid vector system is to be used, the elements of the set of
genetic
elements of the present invention, and particularly the SOI, may be inserted
into either
plasmid as may be convenient.
The nucleic acid sequence that is referred to herein as a cassette provides
the
template for synthesis of ss-cDNA in target cells. It is this element that
includes the
SOI, IR, and PBS. As is the case for the other elements of the set of genetic
elements
to of the present invention, this genetic element is preferably regulated by
an appropriate
wide spectrum or tissue-specific promoter/enhancer, such as the CMV promoter,
or
combination of promoters/enhancers, located upstream of the genetic element.
Also as
is the case for the other genetic elements, ~he promoter/enhancer can either
be
constitutive or inducible promoter. Those skilled in the art who have the
benefit of this
15 disclosure will recognize that a number of other eukaryotic promoters may
be used to
advantage to control expression of the SOI including SV-40, RSV (non-cell type
specific) or tissue specific glial fibulary acidic protein (GFAP).
The primer binding site (PBS) for initiation of priming for cDNA synthesis is
located between the 3' IR and the polyadenylation signal. The PBS is a
sequence that
2o is complementary to a transfer RNA (tRNA) which is resident within the
eukaryotic
target cell. In the case of the mouse Maloney reverse transcriptase (MoMULV
RT)
described herein as being utilized in conjunction with the present invention,
the PBS
takes advantage of the proline tRNA. The PBS utilized in connection with the
presently preferred embodiment of the invention that is described herein was
taken
25 from the actual 18 nucleotide sequence region of mouse Moloney virus.
Shinnick,
T.M., et al., Nucleotide sequence of Moloney murine leukemia virus, 293 Nature
543-
548 ( 1981 ). In the case of the RT gene from human immunodeficiency virus
that was
also tested as noted below, the PBS used was taken from the nucleotide
sequence of
HIV. Y. Li, et al., 66 J. Virology 6587-6600 (1992). In short, any PBS that is
30 matched to a particular RT is utilized for this purpose. The PBS is
exclusively
- 14 -
CA 02386246 2002-04-03
WO 01/25419 PCT/US00/27381
recognized by a primer tRNA that is endogenous to the target cells. Each tRNA
has
the ability to recognize a unique sequence (i.e., codon) on the mRNA
transcript coding
for an amino acid, and has the ability to covalently link to a specific amino
acid (i.e.,
the tRNA becomes "charged" when bound to a specific amino acid). However, a
primer tRNA, when bound to the mRNA transcript PBS and not covalently linked
with
an amino acid (i.e., "uncharged"), may be used to initiate ssDNA synthesis by
the RT.
For example, the MoMUL..V RT used in the examples described herein recognizes
and
uses an uncharged lysine tRNA that in turn recognizes and binds to its unique
sequence
in the PBS. Thus, each PBS incorporated into the expression system of the
present
1o invention must contain the unique sequence recognized by the primer tRNA,
and the
primer tRNA must be a primer tRNA that is recognized by the particular RT
utilized.
Other retroviral RT,iRNAse H genes may be used to advantage in connection
with the present invention, it being preferred that the RT/RNase H gene be an
RT/RNase H gene that is regulated by an appropriate upstream eukaryotic
promoter/enhancer such as the CMV or RSV promoter for expression in human
cells.
RNA-dependent DNA polymerase/RT genes suitable for use in connection with the
present invention include those from retroviruses, strains of hepatitis B,
hepatitis C,
bacterial retron elements, and retrons isolated from various yeast and
bacterial species.
As found in nature, these RNA-dependent DNA polymerases usually have an
2o associated RNase H component enzyme within the same coding transcript.
However,
the present invention does not require the naturally-occurring RNase H gene
for a
particular RT. In other words, those skilled in the art will recognize from
this
disclosure that various combinations of RT and RNase H genes can be spliced
together
for use in connection with the present invention to fulfill this function and
that
modifications and/or hybrid versions of these two enzyme systems are available
and/or
known to those skilled in the art which will function in the intended manner.
Those
skilled in the art will also recognize that the target cell may itself have
su~cient
endogenous RNase H to fulfill this function. Similarly, those skilled in the
art will
recognize that the target cell may itself have sufficient endogenous RT
activity from,
3o for instance, prior retrovir al infection, to fulfill this function.
- IS -
CA 02386246 2002-04-03
WO 01/25419 PCT/US00/27381
The RT/RNase H gene also preferably includes a downstream polyadenylation
signal sequence so that the mRNA produced from the RTlRNase H gene includes a
3'
poly(A) tail for mRNA stability. As known to those skilled in the art,
multiple poly(A)
tails are available and are routinely used for proc!uction of expressed
eukaryotic genes.
Those skilled in the art will also recognize that a number of tissue-specific
or
wide spectrum promoters/enhancers, or combinations of promoters/enhancers
other
than those listed above may also be used to advantage to regulate the RT/RNAse
H
gene, the RE gene (if utilized), and the sequence of interest. Although a list
of all
available promoters/enhancers is not needed to exemplify the invention, as
noted
to above, the promoters/enhancers may be constitutive or inducible and may
include the
CMV or RSV (non-cell type specific) or GFAP (tissue specific)
promoters/enhancers
listed here and many other viral or mammalian promoters. Representative
promoters/enhancers that are appropriate for use in connection with the
cassette of the
present invention may include, but are not limited to, HSVtk (S.L. McKnight,
et al.,
217 Science 316 (1982)), human 13-globulin promoter (R. Breathnach, et al., 50
Ann.
Rev. of Biochem. 349 ( 1981 )), I~-actin (T. Kawamoto, et al., 8 Mol. Cell
Biol. 267
(1988)), rat growth hormone (P.R. Larsen, et al., 83 Proc. Natl. Acad. Sci.
U.S.A.
8283 ( 1986)), MMTV (A.L. Huang, et al., 27 Cell 245 ( 1981 )), adenovirus 5
E2 (M.J.
Imperiale, et al., 4 Mol. Cell. Biol. 875 (1984)), SV40 (P. Angel, et al., 49
Cell 729
( 1987)), a-2-macroglobulin (D. Kunz, et al., 17 Nucl. Acids Res. 1121 (
1989)), MHC
class I gene H-2kb (M. A. Blanar, et al., 8 EMBO J. 1139 (1989)), and thyroid
stimulating hormone (V.K. Chatterjee, et al., 86 Proc. Natl. Acad. Sci. U.S.A.
9114
( 1989)).
The RT produced in the cell synthesizes a complementary DNA (cDNA) using
as the template the genetic element including the SOI described below. The
RNase H
activity of the RT degrades the mRNA template component of the RNA/cDNA hybrid
to produce a ss-cDNA ira >>ivo.
The gene encoding the RE (used in the two plasmid expression system and not
a required component of that system) may be any of several genes which encode
for
3o REs, and preferably those that are controlled by one or more constitutive
or inducible
- 16 -
CA 02386246 2002-04-03
WO 01/25419 PCT/US00/27381
wide spectrum and/or tissue-specific promoters/enhancers such as those listed
above.
The particular REs tested were MboII and FokI, but those skilled in the art
who have
the benefit of this disclosure will recognize that any RE (type I, II, IIS, or
III) site may
be included in the IR. These enzymes "clip," or digest, the stem of the stem-
loop
intermediate described below to linearize the SOI as single-stranded DNA.
Although expression of the RE gene may be regulated by an appropriate
constitutive or inducible promoter/enhancer located upstream from the
restriction
endonuclease gene such as the CMV or RSV promoter for expression in human
cells,
in plasmid pssXA, the RE gene (MboII) is linked to the RT-RNAse H polypeptide.
1o The RE gene also preferably includes a downstream polyadenylation signal
sequence
so that the mRNA transcript from the RE gene will have a 3' poly(A) tail.
The cassette of the present invention also comprises an inverted tandem repeat
(IR). After digestion of the mRNA from the mRNA-cDNA heteroduplex by RNAse H
and the release of the ss-cDNA, the IR causes the ss-cDNA to fold back upon
itself to
15 form the stem of a stem-loop structure, the stem structure being comprised
of double
stranded, anti-parallel DNA, in the manner described in U.S. Patent No.
6,054,299 and
as shown in Fig. 2, after the cassette is transcribed in the cell and after
the RT/RNase
H produced by transcription of the genes produces the ss-cDNA sequence of
interest
from the mRNA transcript in the cell. One or more RE sites) which is cut by
the RE
2o produced from the RE gene (in the case of those plasmids that include an RE
gene)
may be designed into the double stranded portion, i.e., the IR, that forms the
stem of
the stem-loop intermediate. The ss-cDNA which is produced is transcribed with
the
encoded 5' and 3' regions flanking the stem (made up of the IR) and a loop
containing
the SOI. The stem is then cut (also termed digested or cleaved) by any of the
many
25 RE enzymes that recognize the cut site designed into the stem (note that
the
endonuclease recognition site may be designed into the stem even though the RE
gene
is not included in the vector system of the present invention) to release the
ss-cDNA
loop (see Fig. 1 ). The loop portion of the ss-cDNA, which does not form any
apparent
duplex DNA, is immune to RE activity since REs recognize only double stranded
DNA
3o as a target substrate.
- I7
CA 02386246 2002-04-03
WO 01/25419 PCT/US00/27381
As noted above, those skilled in the art will recognize that the RE sites)
need
not be designed into the IR which forms the stem of the stem-loop intermediate
if it is
desired to produce ssDNA from an SOI located between the PBS and the IR, with
transcription of the cassette terminating at the stem formed by the IR.
Another option
is to design the IR to contain eukaryotic, prokaryotic, or viral protein DNA
binding
sites, which can act to competitively titer out selected cellular proteins.
Combinations
of restriction sites or other sequence specific elements may be included in
the IR
depending on the base pair composition chosen for the IR such that linear or
precisely
cut stem-loop intermediate forms of ssDNA are produced. It is generally
preferred to
to use synthetically constructed sequence specific elements in the IR since it
is unlikely
that a naturally occurring inverted repeat would have the properly aligned
restriction
sites.
As noted above, the cassette which comprises one of the elements of the set of
genetic elements of the present invention may also include a DNA sequence with
catalytic activity. Because of the inclusion of the so-called "DNA enzyme" in
the
cassette (and in the embodiment described herein, the DNA enzyme is located
within
the sequence of interest), the present invention is used to particular
advantage when
the sequence of interest serves as the template for synthesis of an inhibitory
nucleic
acid that is an antisense sequence. For that reason, the examples set out
herein
2o describe production of an antisense SOI as set out in Fig. 5B including a
sequence
having enzymatic activity against mRNA including a c-raf cleaving enzyme
designed
specifically to bind to the 3' untranslated region of the c-raf mRNA, which is
targeted
by antisense ISIS 5132 (Monia, B.P., et al., 2 Nature Medicine 668-675 (1996),
hereby incorporated into the present specification in its entirety by this
specific
reference). The two 9 by target specific binding arms were flanked by the 15
by
catalytic domain (Santoro, S.W. and G.F. Joyce, Mechanism and utility of an
RNA-
cleaving DNA enzyme, 37 Biochemistry 13330-13342 (1998), also incorporated
into
the present specification in its entirety by this specific reference).
Compatible
restriction sites were added to the DNA enzyme oligonucleotides so that they
could be
- 18 -
CA 02386246 2002-04-03
WO 01/25419 PCT/US00/27381
inserted into either NotI sites or PacI and BamHI, and the resulting plasmids
were
designated as pssXB-I and pss-XB-II, respectively.
Those skilled in the art will recognize that the present invention is not
limited
just to antisense sequences, that the antisense sequence need not necessarily
contain a
nucleic acid sequence having catalytic activity, and that the inhibitory
nucleic acid
sequence could also be any of the other types of inhibitory nucleic acid
sequences
described above. The above-described SOI was chosen for demonstration of the
present invention because the c-raf kinase in A549 lung carcinoma cells system
has
been well characterized (Monia, et al., sr~pra ( 1996)). Raf protein is a
serine/threonin
1o protein kinase shown to act as a direct downstream effector of ras protein
within the
MAP kinase signaling pathway with downstream activiatior_ of MEK1/1VIEK2 and
subsequent activiation of ERK1 and ERK2 (Daum, G., et al., The ins and outs of
raf
kinases, 19 Trends Biol. Sci. 474-480 ( 1994)). A number of solid tumors and
leukemias have been demonstrated to harbor either mutations in ras or have
15 upregulations in MAP kinase signal pathways. Signal transduction pathways
such as c-
raf related tumors have been attractive targets for oncological therapies and
the
phosphorothioate ODN ISIS 5132, noted above, has been demonstrated to be a
potent
antisense inhibitor (Monia, et al., supra ( 1996)). Further, ISIS 5132 has
been shown
to induce apotosis (Lau, Q.C., et al., 16 Oncogene 1899-1902 (1998), also
2o incorporated into the present specification in its entirety by this
specific reference) and
appears to represent a potential effective treatment against such tumors. This
antisense
ODN has recently entered Phase I clinical trials (O'Dwyer, P.J., et al., C-raf
1
depletion and tumor responses in patients treated with the c-raf 1 antisense
oligonucleotide ISIS 5132 (CGP 69846A), 5 Clinical Cancer Res. 3977-3982 (
1999)),
25 and may prove to be useful in treating c-raf related tumors. Other SOIs
that have been
cloned into plasmids for expression using the expression system of the present
invention include the partial sequence of the 23'd codon of h-ras antisense
binding
sequence with the 10-23 DNA enzyme sequence ( Santoro and Joyce, supra ( i
997))
inserted between the 5' and 3' complimentary sequences, the partial sequence
of
30 pleiotropin antisense binding sequence with the 10-23 DNA enzyme sequence
inserted
- 19 -
CA 02386246 2002-04-03
WO 01/25419 PCT/US00/27381
between the 5' and 3' complimentary sequences, and the partial sequence of tat
antisense binding region of the SIV sequence with the 10-23 DNA enzyme
sequence
inserted between the 5' and 3' complimentary sequences. Although each of these
sequences included the DNA enzyme sequence, those skilled in the art will
recognize
from this disclosure that the DNA enzyme sequence need not be included with
these,
or any other, SOIs.
The nucleic acid sequence having enzymatic activity utilized in the method of
altering gene expression described herein is the 10-23 DNA enzyme (Santoro and
Joyce, supra ( 1997)). The enzymatic sequence is inserted into the cassette in
either or
to both of the two locations, e.g., (a) between the IR and inside the SOI (at
the NotI site)
or (b) inside the second SOI that is located 3' to the IR and 5' to the PBS
(at the
PacIlBamHI sites). Either way, the resulting aptamer is specific for the
target of the
SOI and is therefore used to target other DNA sequences, mRNA sequences, and
any
other suitable substrate, to inhibit or change DNA or mRNA splicing
mechanisms, or
even to directly alter the cellular genome in a specific manner.
Those skilled in the art will recognize from this disclosure that any DNA
sequence having enzymatic activity will function for the intended purpose when
inserted into the cassette of the present invention. A number of nucleic acid
sequences
with enzymatic activity have been reported in the literature, including:
2o sequences having RNAse activity such us the so-called "10-23" and "8-
17 enzymes" (Santoro, S.W. and G.F. Joyce, sr~pra (1997)) and other metal
dependent RNAses (Breaker, R.R. and G.F. Joyce, 1 Biol. Chem. 223-229
(1994) and Breaker, R.R. and G.F. Joyce, 2 Biol. Chem. 655-660 (1995)) and
histidine-dependent RNAse (Roth, A. and R.R. Breaker, 95 Proc. Natl Acad.
Sci. USA 6027-6031 ( 1998));
sequences having DNAse activity such as copper-dependent DNAse
(Carmi, N., et al., 3 Chem. Biol. 1039-1046 (1996), Carmi, et al., supra
(1997); Sen, D. and C.R. Geyer, 2 Curr. Opin. Chem. Biol. 680-687 (1998))
and the DNAses which required divalent metal ions as cofactors or hydrolyzed
- 20 -
CA 02386246 2002-04-03
WO 01/25419 PCT/US00/27381
the substrate independently of divalent metal ions reported in Faulhammer, D.
.
and M. Famulok (269 J. Molec. Bio. 18-203 ( 1997));
sequences with DNA ligase activity such as copper-dependent DNAse
(Breaker, R.R., 97 Chem. Rev. 371-390 (1997)) and zinc-dependent E47 ligase
(Cuenoud, B. and J.W. Szostak, 375 Nature 611-613 (1995));
sequences with DNA kinase acitivity such as calcium-dependent DNA
kinase (Li, Y. and R.R. Breaker, 96 Proc. Natl. Acad. Sci. USA 2746-2751
( 1999)); and
sequences with RNA kinase acitivity such as calcium-dependent DNA
l0 kinase (Li, Y., supra ( 1999)).
Generally, it is those DNA sequences having enzymatic activity that are
derived from
physiological conditions that are preferred for use in connection with the
cassette of
the present invention.
When the elements comprising the set of genetic elements of the present
1, invention are incorporated into a vector for expression in a target cell,
it is preferred
that the vector contain other specialized genetic elements to facilitate the
identification
of cells that carry the vector and cassette and/or to increase the level of
expression of
the set of genetic elements comprising the cassette. The specialized genetic
elements
include selectable marker genes so that the vector can be transformed and
amplified in
2o a prokaryotic system. For example, the most commonly used selectable
markers are
genes that confer to the bacteria (e.g., E. coli) resistance to antibiotics
such as
ampicillin, chloramphenicol, kanamycin (neomycin), or tetracycline. It is also
preferred
that the vector contain specialized genetic elements for subsequent
transfection,
identification and expression in eukaryotic systems. For expression in
eukaryotic cells,
25 multiple selection strategies (e.g., Chinese Hamster Ovarian: CHO) may be
used that
confer to the cell resistance to an antibiotic or other drug or alter the
phenotype of the
cell such as morphological changes, loss of contact inhibition, or increased
growth
rate. Selectable markers used in eukaryotic systems include, but are not
limited to,
resistance markers for Zeocin, resistance to 6418, resistance to
aminoglycoside
3o antibiotics, or phenotypic selection markers such ~3-gal or green
fluorescence protein.
- 21 -
CA 02386246 2002-04-03
WO 01/25419 PCT/US00/27381
Incorporation of these components into the plasmids comprising the expression
system of the present invention allows two convenient methods for removing
predetermined vector sequences after the production of ssDNA. In the first
method,
the cassette is reverse transcribed from the PBS and the SOI between the IR
comprises
the loop portion of the ssDNA stem-loop intermediate that is produced when the
nucleotides comprising the IR pair up to form the stem of the stem-loop
vector, the
stem comprising an RE site, and after digestion with the appropriate RE, the
loop is
released as linearized, single-stranded cDNA without (and/or with minimal)
flanking
sequences. In the second method, the cassette is reverse transcribed from the
PBS and
1o an SOI included in the cassette 3' to the IR is likewise transcribed, but
reverse
transcription is terminated at the stem of the stem-loop structure formed by
pairing of
the nucleotides of the IR. Either way, the resulting ssDNA is produced without
(and/or with minimal) flanking sequences. If it is desired to produce ssDNA
utilizing
the second method, the cassette is designed with an IR that forms a stem that
is more
stable than the stem needs to be if the ssDNA is produced by digestion of the
stem in
accordance with the first aspect of the present invention (for instance, by
designing the
IR so as not to include an RE site). By designing the cassette with an IR that
forms a
stem that is easily denatured in accordance with the first aspect of the
invention,
reverse transcription proceeds right on through the second SOI (if it is even
designed
Zo into the cassette) to the SOI located between the IR. This ''premature
termination'' of
the reverse transcriptase cDNA transcript at the 3' aspect of the stem
structure
therefore provides a second method for limiting the intervening vector
sequences
contained with an in viro-produced ss-cDNA. A stem that is intermediate in
stability
allows production of both the first and second SOIs.
It will also be evident to those skilled in the art from this description that
the
intact stem-loop ss-cDNA structure can function similarly in many applications
as the
linearized ss-cDNA form. Consequently, the cassette is also used to advantage
without the restriction endonuclease gene and associated regulatory elements
and/or
with a sequence of interest which lacks the corresponding restriction
endonuclease site.
- 22 -
CA 02386246 2002-04-03
WO 01/25419 PCT/US00/27381
It will also be evident to those skilled in the art from this description of
the
preferred embodiments of the present invention that a cassette can be made
which
encodes a ss-cDNA that has a "trimmed" stem-loop structure. The RE sites
encoded
in the IR flanking the SOI are designed such that the stem portion (after
duplex
formation) is digested with the corresponding RE so as to cut the dsDNA
comprising
the stem in a way that removes a portion of the stem and the associated
flanking
sequences, yet leaves su~cient duplex DNA that the transcript retains the
above-
described stem-loop structure. Such a ss-cDNA structure may be more resistant
to
intracellular nucleases by retaining the "ends" of a ssDNA in double stranded
form.
to It will also be evident to those skilled in the art from this description
of the
preferred embodiments of the invention that the stem (duplex DNA) can be
designed
to contain a predetermined sequence (or sequences), i.e., aptamers, that are
recognized
and bound by specific DNA-binding proteins. Among other uses, such stem
structure
is used in the cell as a competitor to titer out a selected proteins) that
regulates
15 specific gene function. For example, a ss-cDNA stem-loop is produced in
accordance
with the present invention in a cell that contains a binding site for a
selected positive
transcription factor such as adenovirus E 1 a. Adenovirus E 1 a, like other
oncogenes,
modulates expression of several adenoviral and cellular genes by affecting the
activity
of cell-encoded transcription factors, resulting in the changing of normal
cells to
2o transformed cells. Jones, et al., 2 Genes Dev. 267-281 (1988). The duplex
stem of
the stem-loop intermediate produced in accordance with the present invention
is
therefore designed to function to "bind up" this transcription factor,
preventing the
protein from binding a promoter, and thus inhibiting expression of the
particular
deleterious gene. To those skilled in the art, it will be clear that the
duplex stem
25 structure may optionally contain multiple binding sites, for example, sites
that are
recognized by various transcription factors that actively regulate expression
of
particular gene. For example., adenovirus E 1 a has been found to repress
transcription
of the collagenase gene via the phorbol ester-responsive element, a promoter
element
responsible for the induction of transcription by 12-O-tetradecanolyphorbol 13-
acetate
30 (TPA), by a number of other mitogens, and by the ras, mos, src, and trk
oncogenes.
- 23 -
CA 02386246 2002-04-03
WO 01/25419 PCT/LTS00/27381
The mechanism involves inhibition of the function of the transcription factor
family
AP-1. Offringa, et al., 62 Cell 527-538 (1990). Any desired nucleotide
sequence can
be inserted into the genetic element that encodes the "loop" portion of the
stem-loop
intermediate to carry out a desired inhibitory function, e.g., antisense
binding, down
s regulation of a gene, and so on as herein described.
In another aspect which will be recognized by those skilled in the art, the
present invention is used to construct complex secondary ssDNA structures that
confer
biologic reactions on the cDNA transcript based on conformational secondary
structure folding. Such secondary structure can be engineered to serve any of
several
1o functions. For instance, the sequence of interest may include (but is not
limited to) a
sequence that is incorporated into the loop portion of the single-stranded
cDNA
transcript to form so-called "clover leaf' or "crucible"-like structures such
as those
found in the long terminal repeats of adeno-associated virus or in
retrotransposons.
Under correct circumstances, such structure is integrated in site-specific
manner into
15 the host genome.
Because the cassette of the present invention is adaptable for incorporation
into
multiple commercially available delivery vectors for mammalian and human
therapeutic
purposes, multiple delivery routes are feasible depending upon the vector
chosen for a
particular target cell. For example, viral vectors are frequently used for
transforming
2o the patient's cells and introducing DNA into the genome. In an indirect
method, viral
vectors carrying new genetic information are used to infect target cells
removed from
the body and the infected cells are then re-implanted (i.e., ex vivo). Direct
irt vivo gene
transfer into postnatal animals has been reported for formulations of DNA
encapsulated in liposomes and DNA entrapped in proteoliposomes containing
viral
25 envelope receptor proteins. Nicolau, et al., 80 Proc. Natl. Acad Sci USA
1068-1072
(1983); Kaneda, et al., 243 Science 375-378 (1989); Mannino, et al., 6
Biotechniques
682-690 (1988). Positive results have also been described with calcium
phosphate co-
precipitated DNA. Benvenisty and Reshef, 83 Proc. Natl. Acad. Sci. USA 9551-
9555
(1986). Other systems that are used to advantage to administer the expression
system
3o including the set of genetic elements of the present invention include
intravenous,
- 24 -
CA 02386246 2002-04-03
WO 01/25419 PCT/US00/27381
intramuscular, and subcutaneous injection, as well as direct intra-tumoral and
intra-
cavitary injection. The cassette, when inserted into the expression system of
choice is
also advantageously administered through topical, transmucosal, rectal, oral,
or
inhalation-type methods of delivery.
The cassette of the present invention is advantageously employed to deliver
anti-sense, triplex, or any other inhibitory nucleic acid or single-stranded
nucleotide
sequence of interest, using known digestion and ligation techniques to splice
the
particular sequence of interest into the cassette (between inverted tandem
repeats or
between PBS and inverted tandem repeats). Those skilled in the art who have
the
to benefit of this disclosure will also recognize that the above-described
signals used for
expression within eukaryotic cells may be modified in ways known in the art
depending
upon the particular sequence of interest. For instance, a likely modification
is to
change the promoter so as to confer advantageous expression characteristics on
the
cassette in the system in which it is desired to express the sequence of
interest. There
are so many possible promoters and other signals, and they are so dependent on
the
particular target cell for which the sequence of interest has been selected,
that it is
impossible to list all the potential enhancers, induc:ble and constitutive
promoter
systems, and/or poly(A) tailing systems which may be preferred for a
particular target
cell and sequence of interest.
2o In one particularly preferred embodiment, the present invention takes the
form
of a kit comprised of a plasmid having the above-described RNA-dependent DNA
polymerase and RE genes cloned therein as well as a multiple cloning site
(MCS) into
which the user of the kit inserts a particular SOI. The cloning site into
which the SOI
is inserted is located between the above-described IR. The resulting plasmid
is then
purified from the cell culture in which it is maintained, lyophilized or
otherwise
preserved for packaging and shipping to the user. The kit preferably also
includes the
RE(s) for the MCS into which the SOI is to be cloned, the ligases and other
enzymes,
along with suitable buffers, for ligating the SOI into the plasmid, and a map
of the
plasmid.
- 25 -
CA 02386246 2002-04-03
WO 01/25419 PCT/US00/27381
In the specific embodiments described herein, the SOI(s) is/are delivered to a
host cell either by co-transfection of the cells with two plasmids, designated
A and B,
each plasmid being designed and constructed to include the components listed
above,
or by a single C or D plasmid. In the two plasmid system, the B plasmid
encodes the
cassette including the SOI, either nested within flanking sequences that
include the IR
or between the IR and the PBS that provides the post-transcriptional
processing
signals that mediate the conversion of the mRNA into ssDNA. Activities
required for
processing the primary gene product of the B plasmid into ssDNA, with the
removal of
vector sequences and processing signals. specifically the RT/RNAse H, and RE
(if
1o utilized), are expressed from the A plasmid. The single-stranded DNA
sequence that is
released by interaction of the transcriptional products of these components in
vivo is
free to bind intracellular targets such as mRNA species and DNA promoters in
antisense and triplex strategies.
As noted above, as described herein, the B plasmid includes cloning sites
(NotI
sites were utilized in the B plasmid described herein) between which any DNA
SOI is
placed (as noted above, in the examples described herein, the SOI is an
antisense
sequence to c-raf kinase including the 10-23 enzyme sequence, but as described
above,
other sequences that have been produced iu vivo using the expression system
described
herein include a "stuffer," or test, sequence, telomeric repeats, h-ras, a
region encoding
2o the angiogenic growth factor pleiotrophin, and the region encoding tat
(from SIV)).
Flanking the cloning sites are signals directing the processing of the primary
mRNA
transcript, produced from a promoter (a CMV promoter was utilized in the B
plasmid
described herein), into the desired single-stranded inhibitory nucleic acid.
After
cloning the desired SOI into the B plasmid, the A and B plasmids are co-
transfected
into a cell line of choice for constitutive expression of ssDNA. Similarly, in
the single
plasmid expression system described herein, the SOI is cloned into that
plasmid and
transfected into the cell line for further processing. Regardless of the
distribution of
the elements of the above-described set of genetic elements between two (or
even
more) plasmids, or if the elements are all contained in a single plasmid, this
processing
- 26 -
CA 02386246 2002-04-03
WO 01/25419 PCT/US00/27381
proceeds in three steps following transcription of the single-stranded DNA
region (i.e.,
SOI, IR, and PBS):
( 1 ) reverse transcription of the plasmid RNA transcript by RT, which in
the embodiments described herein is an RT expressed by the A, C, or D
plasmid (in the embodiment described herein, the RT is MoMuLV RT),
proceeding from the primer binding site lying 3' to the SOI (the SOI
optionally
including the sequence with enzymatic activity), IR, and PBS as shown in Fig.
(2) RNAse H digestion of the resulting heteroduplex, either by RNAse
l0 H activity of the RT polyprotein or by endogenous RNAse H activity, to
release the single-stranded DNA precursor from its RNA complement; and
(3) Removal of flanking sequences by either digestion of the stem of a
stem-loop intermediate formed upon Watson-Crick base pairing of the bases
comprising the IR or by premature termination of the cDNA transcript by
15 formation of the stem-loop secondary structure by the self complementary
IR.
Those skilled in the art will recognize from this disclosure that the
particular cloning
sites flanking the SOI, the particular RT, RE (if utilized), promoter, PBS,
and all the
other elements of the set of genetic elements of the present invention are
chosen
depending upon the particular SOI and/or system in which the ssDNA is to be
2o expressed.
EXAMPLE S
Except where otherwise indicated, star_dard techniques as described by
Seabrook, et al. ( 1989) (J. Seabrook, et al., Molecular Cloning: A Laboratory
Manual
(2nd Ed.), Cold Spring Harbor Press (1989), hereinafter referred to as
"Maniatis, et al.
25 (1989)") and Ausubel, et al. (1987) (F.M. Ausubel, et al., Current
Protocols in
Molecular Biology, New York: John Wiley & Sons ( 1987)), both of which are
hereby
incorporated in their entirety by this specific reference thereto, were
utilized in the
examples set out below. It should be understood that other methods of
production of
ssDNA, both by natural processes and by designed artificial methods using
different
30 enzyme products or systems, may also be utilized in connection with the
method of the
27 -
CA 02386246 2002-04-03
WO 01/25419 PCT/US00/27381
present invention and that the examples set oat herein are set out for
purposes of
exemplification and are not intended to limit the scope of this disclosure or
the
invention described herein.
The plasmid pcDNA3.IZeo+ was purchased from Invitrogen Corp. (Carlsbad,
CA) and plasmid pBK-RSV from Statagene (La Jolla, CA). Oligodeoxynucleotides
(ODN) were synthesized by Midland Certified Reagent Co. (Midland, TX).
Polymerase chain reactions (PCR) were carried out using Taq DNA polymerase
purchased from Boehringer Mannheim Corp. (Indianapolis, IN) in a Robo-gradient
thermal cycler (Stratagene (La Jolla, CA)). Restriction endonucleases and T4
DNA
to ligase were obtained from Boehringer Mannheim Corp. (Indianapolis, IN). The
ODNs
used are listed in the attached Sequence Listing.
All ODNs were allowed to hybridize in 1 u1 (5 pg/pl in water) in separate
tubes
which were incubated at 70°C for ~ min and allowed to hybridize for 15
min at room
temperature. Standard restriction endonuclease digests were carried out (EcoRI
used
m as a negative control) with 10 units of enzyme in a total reaction volume of
15 u1 and
appropriate reaction buffers. DNA fragments were resolved in and isolated from
agarose gels. The selection of positive clones on ampicillin plates was
performed after
transformation into competent XL1-Blue MRF cells (Stratagene) as described by
Maniatis, et al. ( 1989). After positive clones were selected, plasmid DNA was
isolated
2o using the above-described Quiagen plasmid isolation kit.
Construction of plasmids. The construction of four expression plasmids is
described. The first plasmid, pssXB (Fig. 3), was derived from pcDNA3.IZeo(+)
(Invitrogen Corp.) and contains the genetic element which encodes the ss-cDNA
sequence of interest used herein. pcDNA3.IZeo(+) was digested with restriction
25 endonucleases HindIII and NotI at positions 911 and 978, respectively. The
double-
stranded linker region having compatible Hina'~II and NotI ends which is
formed by
annealing the synthetic, single stranded oligodeoxynucleotides ODN-5'-
N/M(link)2-
H/N and ODN-3'-N/M(link)2-H/N was ligated under standard conditions into the
HindIIIlNotI double-digested pcDNA3.IZeo(+) transformed into SureII cells
30 (Stratagene, Inc.). The ODNs were allowed to hybridize in 1 p1 (5 pg/p.l in
water) in
- 28 -
CA 02386246 2002-04-03
WO 01/25419 PCT/US00/27381
Ependorf tubes that were incubated at 70°C for 5 minutes and allowed to
hybridize for
15 minutes at room temperature. Appropriate clones were selected and sequenced
to
assure proper insertion of the linker region. The resulting plasmid was termed
pssXB.
pssXB is shown in Fig. 4A and is the plasmid into which the sequence of
interest (Fig.
4B) is cloned. For cloning sequences of interesu between the inverted tandem
repeats,
the two NotI sites at positions 935 and 978, respectively (see Fig. 4A), were
used.
These two sites are contained within the inverted tandem repeats. For
inserting
sequences of interest between the inverted tandem repeats and the primer
binding site,
two convenient restriction endonuclease sites, PacI and BamHI, at positions
1004 and
l0 1021, respectively, were used.
The second plasmid is also a component of the two plasmid vector system
described herein, pssXA (Fig. 3). This ''A" plasmid contains the Mo-MuLV-RT
(Shinnick, T.M., et al., 293 Nature 543-548 (1981)) and restriction
endonuclease
genes and was derived from pBK-RSV (Stratagene), also using XL-1 Blue MRF' as
the host cell. A mouse cell line expressing Moloney murine leukemia virus was
obtained from the American Type Culture Collection (#CRL-1858). Viral RNA was
isolated from cells in accordance with the method described in Chomczymski, P.
and
N. Sacchi ( 162 Anal. Biochem. 156-159 ( 1987)) using Trizol reagent
(GibcoBRL) and
reverse transcribed using primer 3'-RT-HindIII (S'-
2o CTTGTGCACAAGCTTTGCAGGTCT-3'). The transcript was then PCR amplified
using the TaqPlus long polymerase system (Stratagene) for 35 cycles:
94°C 1 min,
67°C, 1 min. and 72°C, 2.5 min. Primers used for the PCR
reaction were 5'-RT-SacI
(5'-GGGATCAGGAGCTCAGATCATGGGAC-CAATGG-3') and 3'-RT-HifadIII,
same as used for reverse transcription. These primers include compatible SacI
and
HindIII sites, respectively. The 2.4kb product obtained included the sequence
of the
Mo-MuLV between positions 2546 and 4908. The mature viral RT peptide is
encoded
by the sequence between positions 2337 and 4349 (Petropoulos, C.J., Retroviral
taxonomy, protein structure, sequences and genetic maps, in J.M. Coffin, et
al. (Eds.),
Retroviruses, New York: Cold Spring Harbor Laboratory Press, pp. 757-805
(1997)),
3o but the peptide truncated at the amino terminus retains full activity
(Tanese, N. and
- 29 -
CA 02386246 2002-04-03
WO 01/25419 PCT/US00/27381
S.P. Gofl; 85 Proc. Natl. Acad. Sci. U.S.A. 1777-1781 (1988)). The peptide
encoded
by this construct includes part of the integrase gene, which follows the RT in
the
MoMuLV polyprotein (Petropoulos, supra ( 1997)).
The bacterium Moraxella bovis, which encodes the restriction endonuclease
MboII (Bocklage, H., et al., 19 Nucleic Acids Res. 1007-1013 ( 1991 )), was
obtained
from the American Type Culture Collection (ATCC#10900). Genomic DNA was
isolated from M. bovis using the Stratagene DNA extraction kit following the
manufacturer's instructions and used as the template DNA in the PCR. Using two
primers, 5'-MboII-HindIII (S'-CAATTAAGGAAAGCTTTGAAAAATTATGTC-3')
l0 and 3'-MboII-XmaI (5'-TAATGGCCCGGGCATAGTCGGGTAGGG-3'), the MboII
gene was PCR amplified from genomic DNA for 30 cycles: 94°C, 30 sec.,
58°C, 1
min., 72°C, 1 min. These primers were designed to include a HindIII and
an XmaI
site, respectively. The 1.2 kb product, copying the M. bovis genome between
positions 888 and 2206, contains the coding region for the MboII enzyme.
The pBK-RSV vector was digested with XmaI and NheI. The NheI end was
converted to a SacI end using linker formed by two annealed oligonucleotides,
5'-Nhe
Sac-link (5'-CTAGCGGCAAGCGTAGCT-3') and 3'-Nhe-Sac-link (5'
ACGCTTGCCG-3'). The RT and MboII amplimers were ligated through the HindIII
site and the construct was subsequently ligated between the SacI and XmaI
sites of
2o pBK-RSV to give pBK-RSV-RT/MboII.
To insert a flexible linker between the RT and MboII domains of the
polyprotein, a fragment of pBK-RSV-RT/MboII plasmid lying between the AseI and
BgIII sites, which encodes the 5' end of the MboII gene and part of the
integrase gene,
was excised and replaced with an insert containing a 6-His-linker and 5' MboII
DNA
fragment deleted by the double digestion. The insert was obtained by mutually-
primed
DNA synthesis from two templates, Rep(+) (S'-
ATACTATTAATTTTGGCAAATCATAGCGGTTATGC-
'TGACTCAGGTGAATGCCGCGATAATTTTCAGATTGCAATCTTTCATCAATG
AATTTCAGTGATGAATTGCCAAGATTGATGTTGC-3') and Rep(-) (5'-
3o GACGAGATC-
- 30 -
CA 02386246 2002-04-03
WO 01/25419 PCT/US00/27381
TCCTCCAGGAATTCTCGAGAATTCGGATCCCCCGCTCCCCACCACCACCAC
CACCACCCTGCCCCGCGGATGAAAAATTATGTGAGCAACATCAATCTTGGC
-3'), that have complementary sequences of 17 bases at the 3'-ends. These two
oligonucleotides were annealed and extended with the modified T7 DNA
polymerise
(USB) and the double-stranded oligonucleotide was then digested with AseI and
BgIII
and inserted into the pBK-RSV vector to give pssXA (Fig. 3).
In a first embodiment of a single plasmid expression vector system constructed
in accordance with the present invention, the pc3.IDNAZeo(+)-derived "B"
plasmid
and the pBK-RSV-derived "A" plasmid were fused such that resulting plasmid
to encoded all of the elements of the set of genetic elements of the present
invention,
including the ss-cDNA-encoding sequence of interest, the tandem inverted
repeat, the
Mo-MuLV-RT gene, and the restriction endonuclease (MboII) gene. To produce the
C plasmid, plasmid pssDNA-Express-A was digested with SacI XmaI to remove the
MboII gene. A linker region comprised of oligonucleotides 5'-(link)2-Hind/Xba
(5'-
CCGGATCTAGACCGCAAG-CTTCACCGC-3') and 3'-(link)2-Hind/Xba (5'-
GGTGAAGCTTGCGGTCTAGAT-3'), which were allowed to anneal at 70°C
for 15
minutes and slowly cooled to room temperature, was ligated into the plasmid
after
digestion under standard conditions. Positive clones were harvested and
sequenced to
verify linker placement and this plasmid was then digested with Xba and
HindIII. The
2o plasmid pssDNA-Express-B was then digested with HindIII and Xba and the
corresponding 300 base pair DNA fragement containing the previously described
inverted tandem repeats, multiple cloning site, and PBS was cloned into the
digested
plasmid to give pssXC (Fig. 5A). Standard ligation reactions were performed
and
transformed into Sure II cells (Stratagene, Inc.). Transformed positive
colonies were
harvested and positive clones were identified by restriction analysis.
The sequences of interest were cloned into the multiple cloning site of pssXC
by using the BamHI and PacI sites in the multiple cloning site (Fig. 5B). Four
different sequences of interest were synthesized for these constructs as
described
above, and similar procedures were utilized for inserting each of the four
sequences of
3o interest. Each construct was prepared by allowing the paired
oligonucleotides to
- 31 -
CA 02386246 2002-04-03
WO 01/25419 PCTXS00/27381
anneal at 70°C for 1 S minutes and cooling to room temperature,
followed by ligation
into the plasmid under standard conditions. After transformation into SureII
cells,
appropriate colonies were selected with verification by sequencing for the
individual
inserts.
A second plasmid was constructed for use in a single-plasmid expression
system, pssXD, by combining the two plasmids, pssXA and pssXB in the following
manner. pssXA, which contains the Mo-MuLV reverse transcriptase (RT), was
digested with XmaI and BgIII and the resulting XmaI-BgIII fragment was
replaced
with a double-stranded DNA adaptor formed by annealing two oligos, XmaI-BgIII-
to Stop 1 (5'-CCGGATCTAGACCGCAAGCTTCATTTAAA-3') and XmaI-BgIII-Stop
2 (GATCTTTAAATGAAGCTTGCGGTCTCGAT-3'). This adaptor contains a
protein translation stop codon and subcloning sites, XbaI and HindIII. The
resulting
plasmid was designated as pssXD (Fig. 6A). XbaI-HindIII fragments were cleaved
from both pssXB and pssXB-II and then cloned into pssXD between XbaI and
15 HindIII. These DNA fragments contain: 1 ) RT primer binding site (PBS); 2)
stem-
loop structure; and 3) random control sequence (pssXB) or c-raf DNA enzyme
sequence (pssXB-II). The resulting plasmids were designated as pssXD-I and
pssXD-
II, respectively. A RSV promoter regulates gene expression of all elements
necessary
for single-stranded DNA expression and all elements are transcribed as a
single mRNA
2o molecule. Endogenous tRNAp'° binds to the PBS on the 3' end of the
transcript, and is
used as the primer for single-stranded DNA synthesis (Marquet, et al., 77
Biochimie
113-124 (1995)). After reverse transcription of the single-stranded DNA by RT,
the
ssDNA is released when the template mRNA is degraded either by endogenous
RNase
H or the RNase H activity of the RT (Tanase and Goff, 85 Proc. Natl. Acad.
Sci.
25 U.S.A. 1777-1781 (1988)).
Tissue culture studies. Stable and transient transfections were carried out by
using DOTAP liposomal transfection reagent (Boehringer Mannhiem Corp.,
Indianapolis, IN) using the manufacturer's accompanying instructions. All
plasmid
constructs were transfected into A549 lung carcinoma cell line (AT CC CCL-185)
and
3o HeLa cell lines maintained in Dulbecco's Modified Eagles Medium (DMEM)
- 32 -
CA 02386246 2002-04-03
WO 01/25419 PCT/US00/27381
supplemented with 10% Fetal Bovine Serum (FCS) (GibcoBRL, Gaithersburg, MD).
Assays for ssDNA were performed by PCR and by dot-blot analyses 24-48 hours
after
transfection. ssDNA was isolated from cells transfected 48-72-hr earlier. The
ss-
cDNA, which co-localizes with total RNA (Mitrochnitchenko, O., et al.,
Production of
single-stranded DNA in mammalian cells by use of a bacter7al retron, 269 J.
Biol.
Chem. 2380-2383 ( 1994)), was earned out using Trizol reagent (Gibco Life
Technologies, Gaithersburg, MD). Assays for specific ss-cDNA species were
carried
out by both PCR based assays for internal fragment and by denatured single
stranded
gel electrophoresis with subsequent nylon blotting and probing with an
internal biotin
labeled probe.
In more detail, reverse transcriptase activity was assayed using the RT-PCR
assay developed by Silver, J., et al. (An RT-PCR assay for the enzyme activity
of
reverse transcriptase capable of detecting single vir-ions, 21 Nucleic Acids
Res. 3593-
3594 (1993)), with modifications as set out below. pssXA transfected cells
were lysed
with lysis buffer (1% TritonT"r, 1 mM MgCl2, 100 mM NaCI, 10 mM TRIS-HCI, pH
8.0 and 2 nM DTT). After centrifugation at 18,OOOg for 30 min., the
supernatant was
collected and frozen at -80°C until use. Brome mosaic virus (BMV) RNA,
used as a
template, was reverse transcribed by incubation with the lysate, which would
contain
RT activity, for 1C or 30 min. at 37°C. Using primers 5'-
2o CGTGGTTGACACGCAGACCTCTTAC-3' and 5'-TCAACACTGTA-
CGGCACCCGCATTC-3', the product of the reverse transcription was then PCR
amplified for 40 cycles: 94°C, 20 sec., 56°C, 20 sec., and
72°C, 20 sec. RT-PCR
products were analysed by 1.5% agarose gel as shown in Fig. 6.
This RT-PCR assay relies upon RT activity in the cell lysates of transfected
cells to produce a cDNA transcript of the BMV RNA substrate. The replication
cycle
of this virus does not involve a DNA intermediate, eliminating the possibility
that an
amplification product could be produced without prior reverse transcription.
RT
activity was determined in the lysates of A549 cells trar~sfected with the
pssXA plasmid
(lanes 3 and 4) and the E 10 clone, which showed relatively high expression
(lanes 5
3o and 6). RT activity was also determined from A549 cells transiently
transfected with
- 33 -
CA 02386246 2002-04-03
WO 01/25419 PCT/US00/27381
control pBK-RSV plasmid (lanes 1 and 2). For transient transfection, lysates
were
prepared 48 hours after transfection. Results show that cell lysates from both
transient
and stable transfected (E 10) cells support the production of a band of
expected size,
150 by (lanes 3-6), whereas control lysates showed none (lanes 1 and 2).
To detect ssDNA expressed in mammalian cells by the pssXB-I and pssXB-II
plasmids when co-transfected with pssXA into A549 cells (E10), a PCR reaction
was
earned out using T7 primer and c-raf DNA enzyme specific primer 5'-
CTAGCTACAACGAGACATGC-3'. Total RNA fraction was used as template and
pre-treated with either S1 nuclease or RNAse A for 30 min. at 37°C or
left untreated.
to The pre-treated RNA samples were then PCR amplified for 30 cycles:
94°C, 45 sec.,
55°C, 45 sec., and 72°C, 30 sec. PCR products were analyzed by
8% acylamide gel as
shown in Fig. 7 (lanes 1 and 3, S 1 nuclease; lanes 2, 4, and 5, RNAse). A
band of the
expected size was produced from both treated total RNA preparations (lanes 2
and 4)
and untreated preparations (data not shown). Control preparations treated with
S 1
nuclease, a highly specific, ssDNA endonuclease, resulted in no amplified
products
(lanes 1 and 3).
The existence of c-raf DNA enzymes was further confirmed by dot-blot
detection of ssDNA, using the North2South Chemiluminescent Nucleic Acid
Hybridization and Detection Kit (Pierce) following the manufacturer's
instructions.
2o Two ~g of total RNA, isolated from cells transfected with either
pssXA/pssXB-I or
pssXA/pssXB-II, or pssXA or untransfected cells, was used. The sequence of c-
raf
specific, biotin-labeled probe is 5'-
GGCCGCACTAATGCATGTCTCGTTGTAGCTA-GCCCAGGCGGGAAGTGC-3'.
As shown in Fig. 8, a biotin-labeld c-raf specific oligo probe can only detect
signal in
the RNA preparations isolated from E10 cells transfected with pssXB-I or pssXB-
II
but not untransfected E 10 cells or A549 cells.
To determine whether single-stranded c-raf DNA enzyme expressed with the
pssXA/pssXB vector system of the present invention in mammalian cells altered
c-raf
mRNA expression, northern blot analysis was performed. The E 10 cell line was
transiently transfected with either pssXB-I or pssXB-II. At 24 and 48 hrs,
cells were
- 34 -
CA 02386246 2002-04-03
WO 01/25419 PCT/US00/27381
harvested for total RNA preparation. Fifteen pg of total RNA was separated on
denatured agarose gel for Northern blot analysis. After overnight transfer,
membrane
was fixed and probed with both 32P-labeled c-raf DNA fragment and
glyceraldehyde-3-
phosphate dehydrogenase (G3PDH), a housekeeping gene. Using random-primed
labeling kit from Boehringer Mannheim, c-raf probe was prepared from an
IMAGETM
cDNA clone (>D 645539, Research Genetics), which includes a coding region of c-
raf
kinase gene from position 571 to 2028. G3PDH was also 32P-labeled and used for
normalization of the RNA blot. The membrane was washed with 2xSSC, 0.1% SDS
for 15 min. and O.IxSSC for 5 min. The blot was then exposed to X-ray film or
1o quantitated by Molecular Dynamics PhophoImagerTM. The quantitation result
of a
representative experiment by phosphor-imaging is shown in graphical form in
Fig. 9.
Compared to controls transfected with pssXB containing unrelated sequences,
pssXB-
II reduces c-raf mRNA level to 81% in 24 hrs and 66% in 48 hrs. pssXB-I had a
similar effect, reducing c-raf mRNA level by 35% after 48 hrs incubation. It
was also
observed that there was significantly more cell death (approximately by a
third) in the
cells transfected with pssXA/pssXB vector expressing c-raf DNA enzyme compared
to
the control. Only remaining adherent cells were harvested, and not those that
began to
"float," so the degree of mRNA reduction may be greater than the 34-36%
reduction
measured.
2o The single plasmid vector system pssXC, was transfected into HeLa cell
lines.
Assays for ssDNA were performed by PCR and by dot-blot analyses 24-48 hours
after
transfection as described above. Reverse transcriptase activity was assayed
using the
RT-PCR assay developed by Silver, et al. (supra (1993)) also as described
above.
Individual colony isolates of stably substituted HeLa cell lines (A12 and B12)
were
additionally assayed for RT activity. The ss-cDNA was isolated from cells
transfected
48-72-hr earlier. The ss-cDNA, which co-localizes with RNA, was carried out
using
Trizol reagent (Gibco Life Technologies, Gaithersburg, MD). Assays for
specific ss
cDNA species were carried out by both PCR based assays for internal fragment
and by
denatured single stranded gel electrophoresis with subsequent nylon blotting
and
3o probing with an internal biotin-labeled probe.
- 35 -
CA 02386246 2002-04-03
WO 01/25419 PCT/US00/27381
This experiment showed that human tissue culture cells (HeLa cell line),
transfected with plasmids designed to synthesize a processed ss-cDNA, produced
ss-
cDNA of the predicted size. As described in the above-incorporated application
Serial
No. 09/397,782, the ssDNA sequence of interest produced in vivo from pssXC is
produced from either the position between the inverted repeats after digestion
of the
stem of the stem-loop intermediate or from the position between the inverted
repeats
and the primer binding site by premature termination of the reverse
transcriptase
cDNA transcript at the 3' aspect of the stem structure.
Using the total RNA fraction, the expression of intracellular single-stranded
1o c-raf DNA enzyme was determined by a simple dot-blot analysis. The biotin-
labeled c
raf specific oligonucleotide probe used was synthesized by Intergrated DNA
Technologies (Coralville, IA), and was used to detect signals in the RNA
samples
isolated from A549 cells either transfected with control pssXD-I or pssXD-II
containing the c-raf DNA enzyme sequence. Two p.g of total RNA were pretreated
with RNase A to rule out any possible non-specific hybridization to RNA, and
in the
presence and absence of S1 nuclease for 30 min at 37°C. Subsequently,
samples were
loaded onto a Hybond-N+ membrane (Amersham Pharmacia Biotec, Piscataway, NJ),
and fixed by UV exposure for 3 min. Hybridization and signal detection were
performed using the North2South Chemiluminescent Nucleic Acid Hybridization
and
2o Detection Kit (Pierce, Rockford, IL). Fig. 11 shows that only cells
transfected with
pssXD-II displayed a positive signal and that in the presence of S 1 nuclease,
no
detectable signal was observed due to the specific degradation of ssDNA enzyme
by
S 1 nuclease.
To determine whether single-stranded DNA enzyme expressed in A549 cells
altered c-raf mRNA levels, quantitative RT-PCR was conducted. c-raf mRNA was
quantitated by RT-PCR as described by Li, et al. (7 Gene Therapy 321-328
(2000))
with some modification. Briefly, one ~g of total RNA was reverse transcribed
using
the Reverse Transcription System (Promega Corp., Madison, WI). A fraction of
the
resulting cDNA was used as a template for PCR amplification. Forty cycles of
PCR
3o were conducted (95°C, 30 sec, 50 °C, 30 sec, and 72
°C, 60 sec) using specific primers.
- 36 -
CA 02386246 2002-04-03
WO 01/25419 PCT/US00/27381
The specific primer sequences used were as follows: 1 ) c-raf primers: 5'-
TCAGAGAAGCTCTGCTAAG-3' and 5'-CAATGCACTGGACACCTTA-3'; 2)
actin primers: 5'-ACCTTCTACAATGAGCTGCG-3' and 5'-
GCTTGCTGATCCACATCTGC-3'. Actin was used as housekeeping gene control.
s Total RNA, isolated from cells transfected with either control pssXD-I or
pssXD-II
containing c-raf DNA enzyme sequence, was reverse transcripted and PCR
amplified
using a pair of c-raf specific primers. PCR amplification of actin mRNA was
used as a
control to normalize loading quantity among different samples. As shown in
Fig. 12, a
significant reduction (approximately 70-80%) of c-raf mRNA was detected in the
cells
1o transfected with pssXD-II (Lane 2) compared to that of control (Lane 1 ).
The levels of c-raf protein in A549 cells transfected with either pssXD-I or
pssXD-II were assessed by Western Blot analysis. 30 ~g of cell extracts were
subjected to electrophoresis on a 12% sodium dodecyl sulfate-polyacrylamide
gel
(SDS-PAGE). Proteins were electrotransferred using a Mini Trans-Blot
15 Electrophoretic Transfer Cell according to the manufacture's instructions
(BioRad
Laboratories, Hercules, CA) to a Hybond ECL membrane (Amersham Pharmacia
Biotec, Piscataway, NJ). The membrane was subsequently blocked in a buffer
containing 25 mM Tris-HCI, pH 7.5, 500 mM NaCI, 0.05% Tween-20, and 5% non-
fat milk and then incubated with primary and HRP-conjugated secondary
antibodies for
20 45 min each. The polyclonal antibodies (anti-rafl ) against c-raf and
monoclonal
antibodies (Ab-1 ) against actin were purchased from Calbiochem-NovaBiochem
Corp.
(San Diego, CA). Monoclonal antibodies (IgGl, C-2-10) against poly-ADP ribose
polymerase (PARP) were purchased either from Clontech Laboratories, Inc. (Palo
Alto, CA). Proteins were visualized using SuperSignal West Pico
Chemiluminescent
25 Substrate Kit (Pierce, Rockford, IL). As shown in Fig. 13, the level of c-
raf protein in
control pssXD-I transfected cells (Lane 2) was similar to that of
untransfected cells
(Lane 3). However, cells transfected with pssXD-II expressing c-raf DNA enzyme
(Lane I) had lower protein levels (approximately 20-30%) of c-raf compared to
the
controls.
- 37 -
CA 02386246 2002-04-03
WO 01/25419 PCT/US00/27381
To determine whether the expression of c-raf DNA enzyme could induce
A549 cell apoptosis, two standard apoptosis assays, genomic DNA cleavage and
PARP cleavage, were performed. Genomic DNA cleavage was determined using a
LM-PCR Ladder Assay Kit (Clontech Laboratories, Inc., Palo Alto, CA) according
to
the manufacturer's instructions. Briefly, 0.5 ~g of genomic DNA was ligated to
adaptors, supplied by Clontech Laboratories, Inc. with T4 DNA ligase overnight
at
15°C. A fraction of adaptor-ligated DNA was used as template in LM-PCR.
Twenty-
five cycles of PCR (95°C, 1 min and 72 ° C, 3 min) with an
extension of 15 min at 72°C
were conducted. Genomic DNA, isolated from cells transiently transfected with
either
1o pssXD-I (control) or pssXD-II (DNA enzyme), were ligated to specific
adaptors.
Subsequently, LM-PCR was carried out using a c-raf primer and a specific
primer. As
shown in Fig. 14, there was a significant increase in fragmented genomic DNA
in cells
transfected with pssXD-II (Lane 1 ) compared to cells t: ansfected with
control plasmid,
pssXD-I (Lane 2), or cells left transfected (Lane 3). These results suggest
that the
increase in fragmented genomic DNA is a result of DNA cleavage caused by
suppression of c-raf gene expression that was altered by the presence of the c-
raf
DNA enzyme.
Another apoptosis assay, the PARP cleavage assay, was conducted using
Western Blot analysis. Compared to the controls (Lanes 2-3), cells transfected
with
2o pssXD-II (Lane 1) had decreased amounts of fizll-length PARP (Fig. 15),
again
indicating the induction of cell apoptosis by suppression of c-raf gene
expression.
Similar amounts of protein were loaded per lane as determined by the presence
of actin
(Lanes 1-3).
The experiments described above demonstrate a method of production of
ssDNA irr vitro and itr vivo by multiple stepwise reactions using eukaryotic
RT
reactions and various cDNA priming reactions that successfi~lly decreased the
expression of c-raf kinase irmivo. Those skilled in the art will recognize
that the
present invention is not limited to this specific embodiment. It will be
recognized, for
- 38 -
CA 02386246 2002-04-03
WO 01/25419 PCT/US00/27381
instance, that many nucleic acid sequences may be utilized depending upon the
specific
target and/or mode of inhibitory action of the SOI. Similarly, the SOI may be
located
in either or both of the two positions, e.g., between the IR and/or between
the PBS
and the 3' aspect of the IR. Likewise, the SOI may or may not include a DNA
enzyme
sequence depending upon the particular target and/or mode of action of the SOI
and/or
the DNA enzyme sequence. Those skilled in the art who have the benefit of this
disclosure will recognize that any desired therapeutic effect is produced by
this method
by transfecting the appropriate SOI into a eukaryotic cell using the vector
system of
the present invention. By way of example, and not limitation, the following
inhibitory
to nucleic acid sequences are known in the art and may be utilized as the SOI
to alter
gene expression in accordance with the present invention:
Sequences that act as antisense oligonucleotides to one or more RNA
molecules encoding one of the several dopamine receptors for therapy of
Parkinson's disease. The antisense oligonucleotides bind specifically to
expression-controlling sequences of such RNA molecules, thereby selectively
controlling expression of one or more dopamine receptor subtypes, and
alleviating the pathological conditions related to their expression;
Sequences that inhibit expression of KSHV virion protein 26, including
sequences that act as antisense and/or triplex oligonucleotides for treatement
of Karposi's syndrome as described in U.S. Patent No. 5,856,903.
Oligonucleotides for control of the expression of IL-8 and/or IL-8
receptor to control growth, metastasis and/or angiogenesis in tumors as
described in U.S. Patent No. 5,856,903;
Oligonucleotides having a sequence of nucleotide bases specifically
hybridizable with a selected sequence of a cytomegalovirus DNA or RNA,
specifically, sequences targeting cytomegalovirus DIvTA or RNA coding for the
IEI, IE2, or DNA polymerase proteins. It is preferred that such
oligonucleotides have between about 5 and about 50 nucleic acid base units as
described in U.S. Patent No. 5,442,049.
- 39 -
CA 02386246 2002-04-03
WO 01/25419 PCT/US00/27381
Oligonucleotides specifically hybridizable with RNA or DNA deriving
from a gene corresponding to one of the open reading frames ULS, ULB, UL9,
UL20, UL27, UL29, UL30, UL42, UL52 and IE175 of herpes simplex virus
type I comprising nucleotide units su~cient in identity and number to effect
such specific hybridization. It is preferred that the oligonucleotides be
specifically hybridizable with a translation initiation site, coding region or
5'
untranslated region. The oligonucleotides are designed to be specifically
hybridizable with DNA, or preferably, RNA from one of the species herpes
simplex virus type 1 (HSV-1), herpes simplex virus type (HSV-2),
to cytomegalovirus, human herpes virus 6, Epstein Ban virus (EBV) or varicella
zoster virus (VZV). Such oligonucleoticies are conveniently and desirably
presented as a pharmaceutical composition in a pharmaceutically acceptable
carrier as described in U.S. Patent No. 5,514,577. Persons skilled in the art
will recognize that the particular open reading frames described for herpes
simplex virus type I find counterparts in the other viruses named. Thus each
of
herpes simplex virus type 2, cytomegalovirus, human herpes virus type 6,
Epstein Barr virus and varicella zoster virus are believed to have many
analogous open reading frames which code for proteins having similar
functions. Accordingly, the present invention is directed to antisense
oligonucleotide therapy in which the oligonucleotides are directed to any of
the
foregoing viruses, or indeed to any similar viruses which may become known
hereafter, which have one or more of such analogous open reading frames. For
convenience in connection with the present invention, all such viruses are
denominated as herpes viruses.
Antisense oligonucleotides to proto-oncogenes, and in particular to the
c-myb gene, for use as antineoplastic and immunosuppressive agents as
described in U.S. Patent No. 5,098,890.
Antisense oligonucleotides against ICAM-1 gene expression in
interleukin-1 beta-stimulated cells for use as anti-inflammatory agents with
3o activity towards a variety of inflammatory diseases or diseases with an
- 40 -
CA 02386246 2002-04-03
WO 01/25419 PCT/US00/27381
inflammatory component such as asthma, rheumatoid arthritis, allograft
rejections, inflammatory bowel disease, various dermatological conditions, and
psoriasis. In addition, inhibitors of ICAM-l, VCAM-1, and SLAM-1 may be
effective in the treatment of colds due to rhinovirus infection, AIDS,
Kaposi's
sarcoma and some cancers and their metastasis as described in U. S. Patent No.
5,843,738. Similarly, International Application No. PCT/LJS90/02357
discloses DNA sequences encoding endothelial adhesion molecules (ELAMs),
including SLAM-1 and VCAM-1 and VCAM-lb. The oligonucleotides
designated ISIS 1570 and ISIS 2302 are specifically contemplated as being
1o used as the sequence of interest in the method of the present invention for
decreasing the metastatic potential of target cells.
Protein-binding oligonucleotides (aptamers) that specifically bind target
molecules such as proteins, and particularly thrombin, in the host cell as
described in U.S. Patent No. 5,840,867. These non-oligonucleotide target
molecules bind nucleic acids (Blackwell, T.K., et al., 250 Science 1104-1110
(1990); Blackwell, T.K., et al., 250 Science 1149-1152 (1990); Turek, C. and
L. Gold, 249 Science 505-510 (1990); Joyce, G.F., 82 Gene 83-87 (1989)),
specifically controlling the biological activity of the protein.
Although described with reference to the figures and specific examples set out
2o herein, those skilled in the art will recognize that certain changes can be
made to the
specific elements set out herein without changing the manner in which those
elements
function to achieve their intended respective results. For instance, the
cassette
described herein is described as comprising three genetic elements, a sequence
of
interest, a primer binding sequence, and a tandem inverted repeat, and when
transfected into a target cell with a reverse transcriptase gene under control
of a
suitable promoter, produces the inhibitory nucleic acid sequence described
herein.
However, those skilled in the art will recognize that, for instance, the mouse
Moloney
leukemia virus reverse transcriptase gene described for use as the reverse
transcriptase
gene of the cassette can be replaced with other reverse transcriptase genes
(the reverse
3o transcriptase gene from human immunodeficiency virus was one such gene
which was
- 41 -
CA 02386246 2002-04-03
WO 01/25419 PCT/US00/27381
noted above) and that promoters other than she CMV promoter described herein
may
be used to advantage. As noted above, the stem-loop intermediate that is
formed may
or may not include a restriction endonuclease site and its susceptibility to
denaturation
is manipulated to advantage depending upon the particular sequence of interest
that is
intended to be produced from that intermediate. All such changes, and others
that will
be made clear to those skilled in the art by this description modifications
which do not
depart from the spirit of the present invention, are intended to fall within
the scope of
the following claims.
- 42 -
CA 02386246 2002-04-03
WO 01/25419 PCT/US00/27381
Table #1
Oligodeoxynucleotides (ODN's)
Name: 5'-N/M(link)2-H/N
5'AGCTTGGTCGGCGGCCTTGAAGAGCGGCCGCACTCACGATAGAGTGGGAGATGGGCGCGAGAAAGTGCGGCC
GCTCTTCAAGGCCGCCGACCTTAATTAAGTCAGCGGGGGATCCTTTTTGGGGGCTCGTCCGGGATCGGGAGACC
~I CCT-3'
Name: 3'- N/M(link)2-H/N
S'GGCCAGGGGTCTCCCGATCCCGGACGAGCCCCCAAA.AAGGATCCCCCGCTGACTTAATTAAGGTCGGCGGCCT
TGAAGAGCGGCCGCACTTTCTCGCGCCCATCTCCCACTCTATCGTGAGTGCGGCCGCTCTTCAAGGCCGCCGACC
A-3'
Name: 5'- polyNM-gaglink-(Pleio)-DNAse-1023-B/P
5'-GAT GTA AG TCG TTG TAG CTA GCC TCC CCT G -3'
Name: 3'-polyNM-gaglink-(Pleio)-DNAse-1023-B/P
5'-GAT CCA GGG GA GGC TAG CTA CAA CGA CTT ACA TCA T -3'
Name: 5'-polyNM-gagIink-(liras)-DNAse-1023-B/P
5'-GGTGGG CGCCTCGTTGTAGCTAGCCTCGGTGTGGG-3'
Name: 3'- polyNM-gaglink-(liras)-DNAse-1023-B/P
5'-GATCCCCACACCGAGGCTAGCTACAACGAGGCGCCCACCAT-3'
Name: 5'-polyNM-gagiink~rafK)-DNAse-1023-B/P
5'-AATGCATGTCTCGTTGTAGCTAGCCCAGGCGGGA-3
Name: 3'- polyNM-gaslink-(rafIC)-DNAse-1023-B/P
5'-GATCTCCCGCCTGGGCTAGCTACAACGAGACATGCATTAT-3'
Name: 5'-polyN/M-gaglink-(tat-SIV)-DNAse-1023-B/P
5'-AGATGGAGACTCGTTGTAGCTAGCCCCCTTGAGGGCAGATTGGCGCCCGAACAGGGACTTGAAGGA-3'
Name: 3'- polyN/M-gaglink-(tat-SI V)-DNAse-1023-B/P
'~ 5'-
GATCTCCTTCAAGTC'CCTC3TTC'CC'.GCGCCAATC'.TGCCC',TCnAC~(.C;C;C;('TnCICTAC'.AAC:C:
A(7TC'.TCCATCTAT-
Name: 5'-(LINK)2-Hind/Xba
5'-CCG GAT CTA GAC CGC AAG CTT CAC CGC -3'
Name: 3'-(LILIK)2-FGnd/Xba
5'-GGT GAA GCT TGC GGT CTA GAT -3'
- 43 -
CA 02386246 2002-04-03
WO 01/25419 PCT/US00/27381
ODN-PMMV(+) 5'-CTAGGTCGGCGGCCGCGAAGATTGGTGCGCACACACACAACGCGCA
129 bases(#23)CCAATCTTCGCGGCCGCCGACCCGTCAGCGGGGGTCTTTCATTTGGGGG
CTCGTCCGGGATCGGGAGACCCCTGCCCAGGGCC-3'
ODN-PMMV(-) 5' -CTGGGCAGGGGTCTCCCGATCCCGGACGAGCCCCCAAATGAAAGAC
I2lbases(#24)CCCCGCTGACGGGTCGGCGGCCGCGAAGATTGGTGCGCGTTGTGTGTGT
GCGCACCAATCTTCGCGGCCGCCGAC-3'
ODN-Test(+) 5'-GGCCGGAAGATTGGGGCGCCAAAGAGTAACTCTCAAAGGCACGCGC
57 bases (#38)CCCAATCTTCC-3'
ODN-Test(-) 5'-GGCCGGAAGATTGGGGCGCGTGCCTTTGAGAGTTACTCTTTGGCGC
57 bases (#39)CCCAATCTTCC-3'
ODN-Telo(+) 5'-GGCCGGAAGATTGGGGCGTTAGGGTTAGGGTTAGGGTTAGGGTTAG
92 bases(#40)GGTTAGGGTTAGGGTTAGGGTTAGGGTTAGGGCGCCCCAATCTTCC-3'
ODN-Telo (-) 5' -GGCCGGAAGATTGGGGCGCCCTAACCCTAACCCTA2~CCCTAACCCT
92 bases(#41)AACCCTAACCCTAACCCTAACCCTAACCCTAACGCCCCAATCTTCC-3'
ODN-XB(+) 5'-GGCCTTGAAGAGCGGCCGCACTAACACCACCACAGTGCGGCCGCTC
51 bases TTCAA-3'
ODN-XB(-) 5'-GGCCTTGAAGAGCGGCCGCACTGTGGTGGTGTTAGTGCGGCCGCTC
51 bases TTCAA-3'
ODN-RT (+). 5' -GGGATCAGGAGCTCAGATCATGGGACCAATGG-3'
32 bases (#13)
ODN-RT(-) 5'-CTTGTGCACAAGCTTTGCAGGTCT-3'
24 bases (#12).
ODN N>S (+) 5' -CTAGCGGCAAGCGTAGCT-3'
18 bases (#25)
ODN-N>S (-) 5' -ACGCTTGCCG-3'
base's
(#26)
ODN-Mbo (+) 5' -CAATTAAGGAAAGCTTTGAAAAATTATGTC-3'
30 bases (#16)
ODN-Mbo (-) 5' -TAATGGCCCGGGCATAGTCGGGTAGGG-3'
27 bases (#33)
ODN-HisPro 5' -AGCTGGATCCCCCGCTCCCCACCACCACCACCACCCTGCCCCT-3'
(+)
43 bases (#36)
ODN-HisPro 5' -AGCAGGGGCAGGGTGGTGGTGGTGGTGGGGAGCGGGGGATCC-3'
(-)
42 bases (#37)
ODN-Rep(+) 5'-ATATCTATTAATTTTGGCAAATCATAGCGGTTATGCTGACTCAGGT
121bases GAATGCCGCGATAATTTTCAGATTGCAATCTTTCATCAATGAATTTCAG
TGATGAATTGCCAAGATTGATGTTGC-3'
ODN-Rep(-) 5'-GACGAGATCTCCTCCAGGAATTCTCGAGAATTCGGATCCCCCGCTC
lllbases CCCACCACCACCACCACCACCCTGCCCCGCGGATGAAAAATTATGTGAG
CAACATCAATCTTGGC-3'
- 44 -
CA 02386246 2002-04-03
WO 01/25419 PCT/US00/27381
1. Name: 3'-RT/Mol-Hind III (24-mer)
Sequence: 5'-CTT GTG CAC AAG CTT TGC AGG TCT-3'
2. Name: 5'-RT/Mol-Sac I (32-mer)
Sequence: 5'-GGG ATC AGG AGC TCA GAT CAT GGG ACC AAT GG-3'
3. Name: 5'-Mbo II-Hind III (30-mer)
Sequence: 5'-CAA TTA AGG AAA GCT TTG AAA. AAT TAT GTC-3'
4. Name: S'-RT-Not-Mbo-Link (129-mer)
Sequence: S'- CTA GGT CGG CGG CCG CGA AGA TTG GTG CGC ACA CAC ACA ACG
CGC ACC AAT CTT CGC GGC CGC CGA CCC GTC AGC GGG GGT ~TT TCA TTT
GGG GGC TCG TCC GGG ATC GGG AGA CCC CTG CCC AGG GCC
5. Name:3'-RT-Not-Mbo-Link (121-mer)
Sequence: 5'-CT GGG CAG GGG TCT CCC GAT CCC GGA CGA GCC CCC AAA TGA
AAG ACC CCC GCT GAC GGG TCG GCG GCC GCG AAG ATT GGT GCG CGT TGT
GTG TGT GCG CAC CAA TCT TCG CGG CCG CCG AC-3'
6. Name: S' Nhe-Sac-Link (18-mer)
Sequence: S'-CTA GCG GCA AGC GTA GCT-3' .
7. Name:3'-Nhe-Sac-Link (10-mer)
Sequence: S'-ACG CTT GCC G-3'
8. Name: 3'-Mbo II-Xba I (2?-mer)
Sequence: S'-TAA TGG CCC GGG CAT AGT CGG GTA GGG -3'
9. . Name: 5'-Hind-link-Histag (43-mer)
Sequence: S'-A GCT GGA TCC CCC GCT CCC CAC CAC CAC CAC CAC CCT GCC CCT-
3'
10. Name:3'-Hind-link-I~stag (42-mer)
Sequence: 5'-AGC AGG GGC AGG GTG GTG GTG GTG GTG GGG AGC GGG GGA TCC-
~.ic
- 45 -
CA 02386246 2002-04-03
WO 01/25419 PCT/US00/27381
11. Name: S' Not-link-testl (57-mer)
Sequence: 5'-G GCC GGA AGA TTG GGG CGC CAA AGA GTA ACT CTC AAA GGC ACG
CGC CCC AAT CTT CC-3'
12.. Name: 3' Not-link-testl (57-mer)
Sequence: S'-GGC CGG AAG ATT GGG GCG CGT GCC TTT GAG AGT TAC TCT TTG
GCG CCC CAA TCT TCC-3'
13. Name: 5' Not-Mbo-link-telo (92-mer)
Sequence: S'-GGC CGG AAG ATT GGG GCG TTA GGG TTA GGG TTA ~ TTA ~
TTA GGG TTA GGG TTA GGG TTA GGG TTA GGG TTA GGG CGC CCC AAT CTT CC-
3'
14. Name: 3'-Not-Mbo-link-telo (92 mer)
Sequence: S'-GGC CGG AAG ATT GGG GCG CCC TAA CCC TAA CCC TAA CCC TAA
CCC TAA CCC TAA CCC TAA CCC TAA CCC TAA CCC TAA CGC CCC AAT CTT CC-3'
15. 5'-SL-linker-Fok1-RT (111-mer)
Sequence:5'-CTA GTC GGA ~C GGC OGC T~ ACA ACA ACA CAC AAC ACA GOG GCC GCA
TCC GAT CAG CGG GGG TCT T'rC ATT TGG GGG C'1~ GTC CGG ATC GGG AGA CSC CIG CCC
AGC GCC-3'
16. 3'-SIr-linker-Fok1-RT (103-mer)
Sequence:5'-CTG GGC AGG GGT CIC COG ATC CGG ACG AGC CCC CAA AZG AAA GAC OOC
CGC TGA TCG GAT GCG GCC GCT GTG TTG TTr GTr GTr GTG CAG CGG CCG CAT CCG A-3'
17. Name: XmaI-BglII-Stop 1
Sequence: 5'-CCGGATCTAGACCGCAAGCTTCATTTAAA-3'
18. Name: XmaI-BrlII-Stop 2
Sequence: 5'-GATCTTTAAATGAAGCTTGCGGTCTCGAT-3'
- 46 -