Note: Descriptions are shown in the official language in which they were submitted.
CA 02387563 2002-04-12
WO 01/28714 PCT/US00/28563
CATALYTIC POWDER AND ELECTRODE MADE THEREWITH
The present invention is directed to electrocatalytic electrodes. More
particularly, the
present invention is directed to cathodes useful in electrolysis cells such as
a chlor-alkali cell.
Chlorine and caustic soda arc typically produced by electrolysis of aqueous
solutions
of sodium chloride, a process commonly referred to as a chlor-alkali process.
The most widely used chlor-alkali processes employ either diaphragm or
membrane
type cells. In a diaphragm cell, an alkali metal halide brine solution is fed
into an anolyte
compartment where halide ions are oxidized to produce halogen gas. Alkali
metal ions
migrate into a catholyte compartment through a hydraulically-permeable
microporous
Zo diaphragm disposed between the anolyte compartment and the catholyte
compartment.
Hydrogen gas and aqueous alkali metal hydroxide solutions are produced at the
cathode. Due
to the hydraulically-permeable diaphragm, brine may flow into the catholyte
compartment
and mix with the alkali metal hydroxide solution.
A membrane cell functions similarly to a diaphragm cell, except that the
diaphragm is
is replaced by an hydraulically-impermeable, cation-selective membrane which
selectively
permits passage of hydrated alkali metal ions to the catholyte compartment. A
membrane cell
produces aqueous alkali metal hydroxide solution essentially uncontaminated
with brine.
Electrodes are usually prepared by providing an electrocatalytic coating on a
conducting substrate. Useful catalytic coatings include, for example, the
platinum group
2 o metals, such as ruthenium, rhodium, osmium, iridium, palladium and
platinum. Useful
conducting substrates include, for example, nickel, iron, and steel.
Production of chlorine gas at the anode and the concurrent production of the
hydroxide ion and evolution of hydrogen gas at the cathode almost always
require a cell
voltage higher than the thermodynamic energy for the following reaction.
2 s 2 NaCI + 2 HZO ~ Cl~ + H~+2NaOH
-1-
CA 02387563 2002-04-12
WO 01/28714 I'CT/US00/28563
The extra energy, that is, overvoltage, is provided to overcome, among various
other
para~ncters, the electrolyte resistance and the overpotential related to the
chlorine gas
evolution at the anode and the overpotential related to hydrogen gas evolution
and hydroxide
ion formation at the cathode.
Various methods have been proposed to decrease the overpotential requirements
of
the electrodes by altering surface characteristics. The term "ovcrvoltage" is
used herein to
refer to the excess voltage required for an electrolytic cell, while the term
"overpotential" is
used herein to refer to the excess voltage required for an individual
electrode within the
electrolytic cell.
1o The overpotential for an electrode is a function of its chemical
characteristics and
current density. Current density is defined as the current applied per unit of
actual surface
area on an electrode. Techniques which increase the actual surface area of an
electrode, such
as acid etching or sandblasting the surface of the electrode, result in a
corresponding decrease
of the current density for a given amount of applied current and also decrease
overpotential
15 requirements.
Efforts to reduce overpotential requirements include, for example, those
described in
U.S. Patent No. 4,66,8370 and U.S. Patent No. 4,798,662, which disclose
electrodes useful as
cathodes in an electrolytic cell. These are prepared by coating an
electrically conducting
substrate such as nickel with a catalytic coating comprising one or more
platinum group
2 o metals from a solution comprising a platinum group metal salt. Both of
these Patents disclose
electrodes designed to reduce the operating voltage of an electrolytic cell by
reducing the
overpotential requirements of the electrodes. In addition, U.S. Patent. No.
5,035,789, U.S.
Patent. No. 5,227,030, and U.S. Patent. No. 5,066,380 disclose cathode
coatings which
exhibit low hydrogen overpotentials.
2 s A desirable characteristic of a cathode coating is high porosity with
large internal
surface areas. Large internal surface areas result in lower effective current
density and,
accordingly, lower overpotentials. Another result of a porous electrode is
higher resistance to
impurity poisoning. Rough outer surfaces of a typical porous electrode render
difficult the
electrodeposition of metal ions as impurities and the large internal
electroactive surface areas
3 o are not easily accessible to the impurity ions present in the electrolyte
because of long
Pathways for diffusion. Such characteristic is described in U.S. Patent No.
5,645,930
-2-
CA 02387563 2002-04-12
WO 01/28714 PCT/US00/28563
Metal plating is often used to form a reinforcement layer on the electrode.
For
example, U.S. Patent. No. 4,061,802 and U.S. Patent. No. 4,704,401 describe
using palladium
chloride to activate plastic or metal substrates prior to nickel plating by
clcctroless deposition.
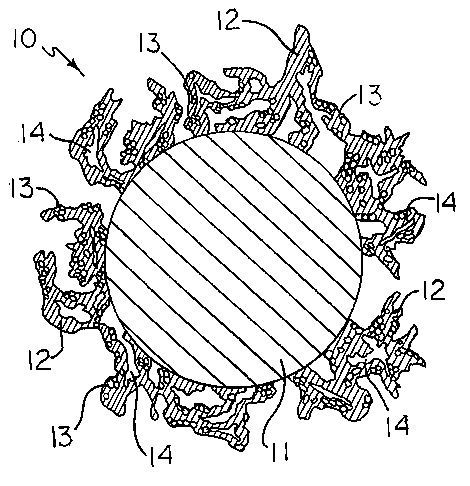
Figure 1 is a magnified representation of a cross section of a catalytic
powder particle
of the present invention.
Figure 2 is a magnified representation of a cross section of a portion of an
electrode of
the present invention.
In one aspect, the present invention is a catalytic powder comprising a
plurality of
support metal particles comprising a transition metal or an alloy thereof; and
a coating
to surrounding the support metal particles, the coating either comprising an
electrocatalytic
metal coating or comprising a coating with a metal continuous phase in
admixture with a
particulate material.
In a second aspect, the present invention is an electrode comprising a
conductive
metal substrate; and a first layer comprising a matrix with a catalytic powder
dispersed
15 therethrough, the matrix comprising a platinum group metal oxide or a
mixture of a platinum
group metal oxide and a valve metal oxide, the catalytic powder comprising
support metal
particles covered either with an electrocatalytic metal coating, or with a
coating comprising
an electrocatalytic metal in admixture with a particulate material.
In a third aspect, the present invention is a process for making an electrode
comprising
2o the steps of forming a catalytic powder; mixing the catalytic powder with a
dispensing
medium to form a mixture; applying the mixture to a conductive metal substrate
to form a
covered substrate; and baking the covered substrate in the presence of oxygen;
and optionally
reinforcing the coating adhesion and strength with an alloy coating process.
The present invention is advantageous because a porous coating mixture is
first
2 s applied to a powder rather than being applied directly to a metal
substrate, thereby creating a
larger internal surface area relative to the prior art. Large internal surface
areas result in
lower effective current density and, accordingly, lower overpotentials.
Therefore, because the
surface area is enhanced using the present invention, the overpotential
required for electrodes
-3-
CA 02387563 2002-04-12
WO 01/28714 PCT/US00/28563
made according to the present invention is also reduced relative to electrodes
of the prior art
cited above.
Figure 1 illustrates a magnified view of a catalytic powder particle 10 of the
present
invention. As shown, the catalytic powder particle 10 comprises a support
metal particle 1 1
s surrounded by a porous coating comprising a continuous phase 12 with a
particulate material
13 dispersed therethrough.
Preferably, the support metal particle 1 1 is a transition metal or alloy
thereof.
Preferred transition metals include nickel, cobalt, iron, steel, stainless
steel or copper.
Preferred transition metal alloys include nickel, cobalt, or copper, alloyed
with phosphorous,
1 o boron or sulfur.
Preferably, the support metal particles, before the porous coating is applied
thereto,
have an average diameter of at least 0.2 microns, more preferably at least
about 1 micron,
even more preferably at least 2 microns, and yet even more preferably at least
3 microns.
Preferably, the metal particles have an average diameter of up to 20.0
microns, more
15 preferably up to 10.0 microns and even more preferably up to 6.0 microns.
The support metal particle 11 is coated with either an electrocatalytic metal
or with a
porous coating comprising an electrocatalytic metal continuous phase 12 in
admixture with a
particulate material 13. Because the coating on the support metal particle is
porous and has a
dendritic nature, the resulting catalytic powder particle 10 has a large
internal surface area
2 o with pores 14 throughout.
Preferably, the electrocatalytic metal continuous phase 12 is ruthenium,
iridium,
osmium, platinum, palladium, rhodium, rhenium, or an alloy of any one or more
of these.
In one embodiment, the continuous phase 12 has a particulate material 13
dispersed
therethrough. Preferably, particulate material 13 comprises the metal oxides
of ruthenium,
2s iridium, osmium, platinum, palladium, rhodium, rhenium, technetium,
molybdenum,
chromium, niobium, tungsten, tantalum, manganese or lead, with the oxides of
ruthenium,
iridium osmium, platinum, palladium and rhodium being more preferred..
To make the catalytic powder, a plurality of support metal particles is
covered with a
porous coating comprising an electrocatalytic metal either alone or in
admixture with a
-4-
CA 02387563 2002-04-12
WO 01/28714 PCT/US00/2A563
particulate material which comprises either a metal or metal oxide. Generally,
the first step in
making the catalytic powder is to prepare a deposition solution comprising at
least a
palladium promoter and an organic or inorganic acid.
It is known from U.S. Patent. No. 5,066,380 that the presence of palladium
metal ions
s in the deposition solution, in addition to the metal ions of the
electrocatalytic metal precursor
compound, promotes deposition of the electrocatalytic metal onto the metal
particles.
Example of suitable palladium metal compounds are palladium halides and
palladium nitrate.
The concentration of the palladium metal ions in the porous coating solution
should be
sufficient to promote improved electrocatalyst loading on the metal particles.
The palladium
so precursor compounds when present are, generally, included in an amount
sufficient to yield a
palladium metal ion concentration in the coating solution of at least 0.001
percent by weight
based on the weight of the solution. The palladium metal ion concentration
suitably can be
0.001 percent to 5 percent; preferably from 0.005 percent to 2 percent and,
most preferably,
from 0.01 percent to 0.05 percent, by weight of the coating solution. A weight
percent of less
15 than 0.001 percent is generally insufficient to promote deposition of the
electrocatalytic
metal. A weight percentage greater than percent 5 results in the deposition of
an excessive
amount of electrocatalytic metal primary phase of the coating on the
substrate.
The pH of the deposition solution may be adjusted by inclusion of organic
acids or
inorganic acids therein. Examples of suitable inorganic acids are hydrobromic
acid,
2 o hydrochloric acid, sulfuric acid, perchloric acid, and phosphoric acid.
Examples of organic
acids are acetic acid, oxalic acid, and formic acid. Hydrobromic acid and
hydrochloric acid
are preferred. The pH range for the deposition solution is, generally, 0 pH to
2.8 pH.
Precipitation of hydrous platinum group metal oxide results at higher pHs. A
low pH can
encourage competing side reactions such as the dissolution of the substrate.
2 s At least one electrocatalytic metal compound soluble in water or an
aqueous acid is
added to the deposition solution. A suitable electrocatalytic metal is,
generally, one that is
more noble than the metal employed for the metal particles, that is, the
electrocatalytic metal
precursor compound has a Gibbs free energy greater than the Gibbs free energy
of the metal
compound from dissolution of the metal particles, such that non-electrolytic
reductive
3 o deposition occurs on the metal particles. Preferably, such
electrocatalytic metal is a platinum
group metal. More details non-electrolytic reductive deposition can be found
in U.S. Patent
-5-
CA 02387563 2002-04-12
WO 01/28714 PCT/US00/28563
5,(,45,930.
The electrocatalytic metal precursor compound can be present in the deposition
solution in amounts sufficient to deposit an effective amount of the metal on
the metal
particles. The concentration of electrocatalytic metal ions in the deposition
solution, in terns
s of weight percent, is, generally, from 0.01 percent to 5 percent,
preferably, from 0.1 percent
to 3 percent and, most preferably, from 0.2 percent to 1 percent by weight of
solution. An
electrocatalytic metal ion concentration of greater than 5 percent is not
desired, because an
unnecessarily large amount of platinum group metal is used to prepare the
coating solution.
An electrocatalytic metal ion concentration of less than 0.01 percent is not
desired, because
Zo undesirably long contact times are required.
The optional particulate material is suspended in the deposition solution at a
concentration of from 0.002 to 2 percent, preferably, 0.005 to 0.5 percent,
and most
preferably, 0.01 to 0.2 percent.
After the deposition solution comprising the palladium promoter, the acid, and
the
z5 optional particulate material is prepared, it is held at an elevated
temperature and stirred at a
high speed, while a powder comprising support metal particles is added
thereto. After a
period of time, the electrocatalytic metal precursor compound is added, and
the electro-
catalytic metal is formed and deposited on the support metal particles with
simultaneous
partial dissolution of the support metal particles.
2 o The rate at which the electrocatalytic metal deposits to form the porous
coating on the
metal particles is a function of the solution temperature. The temperature,
generally, ranges
from 25°C to 90°C. Low temperatures are not practical, since
uneconomically long times are
required to deposit an effective amount of electrocatalytic metal on the metal
particles.
Temperatures higher than 90°C are operable, but generally result in an
excessive amount of
2 s metal deposition and side reactions. A temperature ranging from between
40°C to 80°C is
preferred, with 45°C to 65°C being most preferred.
Generally the time allowed for contact between the deposition solution and the
metal
particles can vary from one minute to 60 minutes. However, it should be
understood that the
contact time required will vary with deposition solution temperature,
electrocatalytic metal
3 o concentrations, and palladium ion concentration. Contact times of from 5
minutes to 60
-6-
CA 02387563 2002-04-12
WO 01/28714 PCT/US00/28563
minutes are preferred, with from 10 minutes to 40 minutes hcing most
preferred. Generally,
if shorter contact times arc desired, the method described herein may be
repeated a plurality
of times until an effective amount of the platinum group electrocatalytic
metals deposit on the
surface of the metal particles.
s The catalytic powder 10 is advantageously used to form electrodes for
electrolysis
cells. Figure 2 illustrates a magnified view of a portion of an electrode 20
of the present
invention. The electrode 20 comprises a conductive metal substrate 21 and a
first layer, the
first layer comprising a matrix 22 with the above described catalytic powder
10 dispersed
therethrough. The porous dendritic nature of the catalytic powder creates a
porous surface on
z o the electrode, which in turn reduces the overpotential required for
efficient operation of the
electrode and electrolytic cells.
Preferably, the conductive metal substrate 21 is nickel, iron, steel,
stainless steel,
cobalt, copper or silver. The shape of the substrate is not critical and can
be, for example, a
flat sheet, a curved surface, a punched plate, a woven wire screen, or a mesh
sheet.
15 The matrix 22 of the first layer comprises either a platinum group metal
oxide or a
mixture of a platinum group metal oxide and a valve metal oxide. Platinum
group metal
oxides include oxides of ruthenium, iridium, rhodium, osmium, platinum,
palladium or a
mixture of any one or more of these. Valve metal oxides include oxides of
titanium,
zirconium, tantalum, tungsten, niobium, bismuth, or a mixture of any one or
more of these.
2o To make an electrode of the present invention, the above described
catalytic powder is
mixed with a dispensing medium to form a mixture which is applied to the
conductive metal
substrate to form a covered substrate. The covered substrate is then baked in
the presence of
oxygen.
The dispensing medium forms the matrix of the electrode and comprises either a
2 s platinum group metal oxide precursor or a mixture of a platinum group
metal oxide precursor
and a valve metal oxide precursor. Platinum group metal oxide precursors are
those materials
that form platinum group metal oxides upon baking in the presence of oxygen.
Preferred
platinum group metal oxide precursors include platinum group metal halides,
sulfates,
nitrates, nitrites, and phosphates. More preferred are platinum group metal
halides, nitrates
3 o and phosphates, with platinum group metal chlorides being the most
preferred. Valve metal
CA 02387563 2002-04-12
WO 01/28714 PCT/US00/28563
oxide precursors are those materials that form valve metal oxides upon baking
in the presence
of oxygen. Preferably, the valve metal oxide precursor is titanium alkoxidc,
tantalum
alkoxidc, zirconium acetylacctonate, or niobium alkoxide.
Preferably, the dispensing medium further comprises a solvent. Suitable
solvents
include methanol, ethanol, 1-propanol, 2-propanol, butanol, or a mixture of
any of these.
Preferably, the dispensing medium further includes a compound soluble in
alkaline
solutions. Examples of such soluble compounds include aluminum chloride and
zinc
chloride. Such alkaline soluble compounds are useful in generating pores in
the coating after
they are dissolved in an alkaline solution.
Any appropriate method may be used for dispersing the catalytic powder in the
dispensing medium. Examples include mechanical stirring, sonicating, or
combinations
thereof.
The application of the catalytic powder/dispensing medium mixture can be
accomplished using any suitable method. An example is spraying through a
nozzle. The
is spraying forms a platinum group metal loading in the resulting electrode
of, generally, 50
ug/cm' to 2000 ug/cm2 calculated as the metal in the "atomic" form. The amount
of metal in
the electrode is measured by x-ray fluorescence. A preferred loading for both
the elemental
metal and combined oxide is from 400 ug/cm'' to 1500 ug/cm' with a most
preferred loading
of from 500 ug/cm'' to 1000 ug/cm'. Loading less than 50 ug/cm'' are generally
insufficient to
2 o provide a satisfactory reduction of cell overvoltage. Loadings greater
than 2000 ug/cm' do
not significantly reduce the applied overvoltage when compared to lesser
loadings within the
preferred range. It should be understood that the effective amount of
deposition specified
above refers only to loading of the platinum group electrocatalytic metal and
metal oxides in
the electrode and does not include the amount of the palladium metal promoter
which can be
2 s used to provide increased loading or any optional secondary
electrocatalytic metal or the
metal particles.
In a preferred embodiment, the substrate is protected before the mixture is
applied
thereto, by, for example, electroless nickel plating. Such a process is
described in U.S. Patent
No. 4,061,802.
_g_
CA 02387563 2002-04-12
WO 01/28714 PCT/US00/28563
A baking step is used to convert the platinum group metal oxide precursor and
valve
metal oxide precursor to an oxide form. The coated substrate is baked in the
presence of
oxygen at a temperature of preferably at least 350°C more preferably at
least 420°C and even
more preferably at least 450°C. Preferably, the coated substrate is
baked at a temperature of
s not more than 550°C more preferably not more than 500°C even
more preferably not more
than 480°C Preferably the baking step occurs for anywhere from 30 to 90
minutes. It is
important that the coated substrate be baked in the presence of oxygen, be it
air or some other
oxygen-containing substance, so that the platinum group metal oxide precursor
and the valve
metal oxide precursor convert to platinum group metal oxide and valve metal
oxide. The
so result is a two-phase first layer of the electrode, one phase being the
matrix, and the second
phase being the catalytic powder particles dispersed through the matrix.
In a preferred embodiment, the electrode of the present invention further
comprises a
reinforcement layer 23. Such a reinforcement layer 23 preferably comprises a
transition
metal or alloy thereof. More preferably, the reinforcement layer is nickel,
cobalt, copper, or
1s alloys thereof with boron, phosphorous or sulfur.
To make the optional reinforcement layer, a second electroless plating step,
which
consists of plating the coated substrate with a transition metal or a
transition metal alloy.
Such a reinforcement layer helps hold the catalyst powder and matrix together
and also helps
ensure that the first layer adheres to the substrate. More details forming the
reinforcement
20 layer can be found in U.S. Patent No. 5,645,930.
-9-
CA 02387563 2002-04-12
WO 01/28714 PCT/US00/28563
Unless otherwise specified, all parts and percentages arc by weight. The
following
examples are not meant to be limiting.
Examples 1-3: Preparation of Catalytic Powder With Metal Particulate Material
s A porous coating solution was prepared, with PdCI~ as palladium promoter and
0.5 N
HC1 as acid. The solution was heated to a reaction temperature and
continuously stirred.
RuCljxH~O was added as the electrocatalytic platinum group metal compound. The
resulting
solution was held at the reaction temperature and stirred using a COWLES high-
speed
disperser, while 3-micron nickel powder (Aldrich) was added. After stirring
the mixture at
to the elevated temperature for a desired contact time, the resulting Ru-
coated nickel powder
was collected on a filter paper, dried for several hours at 90 °C and
weighed. The amount of
Ru in the powder was determined using X-ray fluorescence. Table I lists the
variables and
the results.
Table I
Example 1 2 3
O.SN HCl solution 500 1420 1577
(grams)
PdCl2 (milligrams) 20.9 59.3 6.32
Reaction temp. (C) 64 61 61.7
RuCl3 added (grams) 2.095 5.95 2.681
Nickel powder (grams)35.2 100 100.4
Reaction time (minutes)15 5 5
Total weight after 28.13 83.7 85.22
drying
(grams)
Percent Ru in powder3.1 3.0 3.14
-10-
CA 02387563 2002-04-12
WO 01/28714 PCT/US00/28563
Examples 4-6: Preparation of Catalytic Powder With Metal/Metal Oxide
Agglomerates As Particulate Material
A porous coating solution was prepared, with PdCI~ as palladium promoter and
0.5 N
HCl as acid. The solution was heated to a reaction temperature and
continuously stirred.
RuO~ was added as the platinum group metal oxide. The resulting solution was
held at the
reaction temperature and stirred using a COWLES high-speed disperser operated
at 3000
rpm, while 3-micron nickel powder (Aldrich) was added. RuCI~xH~O was then
added as the
electrocatalytic platinum group metal compound. After stirring the mixture at
the elevated
to temperature for a desired contact time, the resulting Ru-coated nickel
powder was dried and
weighed. The amount of Ru in the powder was determined using X-ray
fluorescence. Table
II lists the variables and the results.
Table II
Example 4 5 6
0.5N HCl solution 1405 1402 1413
(grams)
PdCl2 (milligrams) 60 60 60
Reaction temp. (C) 51 52.8 50.6
Ru02 (grams) 0.714 0.720 0.714
Nickel powder (grams)38.84 38.63 38.14
RuCl3 (grams) 8.74 8.75 8.57
Reaction time (minutes)50 50 110
Total weight after 16.57 16.70 15.98
drying
(grams)
Percent Ru in powder26.0 25.86 26.5
-11-
CA 02387563 2002-04-12
WO 01/28714 PCT/US00/28563
Examples 7-9 - Preparation of Cathode With a metal Particulate Material
A 5 inch by 6 inch plate was electroless nickel-plated according to procedures
described in U.S. Patent No. 4,061,802. The plate was then sprayed with a
mixture of a
dispensing medium and a Ru-coated nickel powder (Ru=3.1 percent) dispersed
thcrcthrough.
The powder weight percent in the spraying mixture was around 10 percent. The
platinum
group metal oxide precursor in the dispensing medium was RuCI~, the valve
metal oxide
precursor compound in the dispensing medium was titanium isopropoxide. The
solvent in the
dispensing medium was a combination of methanol and 2-propanol, the compound
soluble in
1o alkaline solutions was aluminum chloride or zinc chloride, and the acid
used to adjust pH,
when used, was HC1 gas.
The sprayed sample was allowed to dry at 90°C for 20 minutes and baked
at 490°C for
60 min. X-ray fluorescence of the sample was used to determine loading of the
metal on the
substrate. Table III lists the parameters and results.
Table III
Example 7 8 9
RuC13xH20 2.37 2.37 2.37
(wt. percent)
Ti(isopropoxide)6.69 6.69 6.69
(wt. percent)
Methanol 76.5 76.5 5.00
(wt. percent)
2-propanol 9.73 9.73 81.07
(wt. percent)
Compound soluble3.43 3.43 3.62
in alkaline (AlCl~x6H20) (AIC13x6H20) (Zn(N03)2x6H~0
solution
(wt. ercent) )
HCl gas 1.28 1.28 1.25
(wt. percent)
Metal loading 151 133 169
(l~g~cm2)
-12-
CA 02387563 2002-04-12
WO 01/28714 PCT/US00/28563
Examples 10- I 3 - Preparation of Cathode with a Metal/Metal Oxide Agglomerate
Particulate Material
A Sinch by 6 inch plate was electrolcss nickel-plated according to procedures
described in U.S. Patent 4,061,802. The plate was then sprayed with a mixture
of a
dispensing medium and a Ru/RuO~-coated nickel powder (Ru=25.86 percent)
dispersed
therethrough. The powder weight percent in the spraying mixture is around 10
percent. The
dispensing medium comprises 2.37 weight percent RuCI~xH~O as the platinum
group metal
oxide precursor, 2.87 weight percent titanium isopropoxide as the valve metal
oxide
1o precursor, 8.86 weight percent methanol and 83.80 weight percent 2-propanol
as the solvent,
and 2.10 weight percent AlCl~x6H~0 as the compound soluble in an alkaline
solution.
The sprayed sample was allowed to dry at 90°C for 20 minutes and baked
at 490°C for
60 min. X-ray fluorescence of the sample was used to determine loading of the
metal on the
substrate. Table IV lists the parameters and results.
Table IV
Example 10 11 12 13
Percent Ru in 25.86 25.86 25.86 26.0
catalytic powder
Metal loading 521 555 766 428
(pg/cm2)
Examples 14-16 - Preparation of Electrodes having a second reinforcement layer
The samples from Examples 7-9 above are coated with the second reinforcement
layer
of Ni-P by the following steps:
2 o The plates were dipped in the following mixture of solutions for a period
of five
minutes at ambient temperature: 25cc 0.01 M (NH4)2PdC14 in methanol, SOcc 0.1
M poly(4-
vinylpyridine) in methanol, and 425cc methanol. The coated plates were then
dried in a
horizontal position at 90°C. The dipping and drying steps were
repeated.
-13-
CA 02387563 2002-04-12
WO 01/28714 PCT/US00/28563
Thereafter, the coated plates were placed in a plastic horizontal container
with the
thread of the plate fitted in a tilting in the bottom of the container. The
container was first
filled with an aqueous solution containing 36g/1 of NaH,P02xH,0 at pH=2.95 for
5-10
minutes to reduce Pd(II) to Pd°. The solution was then poured out and
SOOmI of an
s electroless nickel-plating solution was then added to the container and
electroless plating was
conducted for 20 min. The composition of the electroless plating solution is:
NiCl2 x6H20 17.4g/1
Sodium Citrate 30.24g/1
NaH2P02xH20 25.2g/1
1 o NH4Cl 21.26g/1
NH40H add to get pH = 8.8
Weight gains for Example 4 (Example 7), Example 5 (Example 8) and Example 6
(Example 9) were 2.63 mg/cm'', 3.26 mg/cm~', and 2.69 mg/cm', respectively.
To measure the hydrogen potential, the plates were connected to a nickel rod
and
15 placed in a caustic bath at an elevated temperature. A platinum plate
welded to a nickel rod
was used as the anode. Current densities of 0.46 amps per square inch (ASI),
1.0 ASI, and/or
1.09 ASI were applied to the cathode sample and the anode from a rectifier.
The potential of
the cathode was measured with the aid of a LUGGIN probe with a Hg/Hg0
reference
electrode. The parameters and results were listed in Table V.
-14-
CA 02387563 2002-04-12
WO 01/28714 PCT/US00/2A563
Table V
Example 14 15 1 G
Percent causticI I .75 11.75 32
in
bath
Temperature 70 70 90
of
caustic bath
(C)
Voltage at 0.46-0.960 -0.962 -1.007
ASI
Voltage at 1.0 -- -0.979 --
ASI
Voltage at 1.09-- -- -1.025
ASI
Examples 17-20 - Preparation of Electrodes having a Second Reinforcement Layer
The samples from Examples 10-13 above were coated with the second
reinforcement
s layer of Ni-P by the following steps:
Initiation was conducted at 0.8-0.9 amperes at ambient temperature for 2-3
minutes.
The plate was then placed in an electroless plating solution for 20-30
minutes. The
composition of the electroless plating solution is:
NiCl2 x6H20 17.4 g/1
Zo Sodium Citrate 30.24g/1
NaH2P02xH20 25.2g/1
NH4C1 21.26g/1
NH40H add to get pH = 8.8
Weight gains for Example 10 (Example 17), Example 11 (Example 18), Example 12
is (Example 19) and Example 13 (Example 20) are 0.550g, 0.578g., 0.683g, and
0.489g,
respectively.
Examples 20-23 - Hydrogen Potential Measurements
To measure the hydrogen potential for the plates prepared in Examples 17-20,
the
plates were connected to a nickel rod and placed in an 11.75 percent caustic
bath at 70°C. A
-15-
CA 02387563 2002-04-12
WO 01/28714 PCT/US00/28563
platinum plate welded to a nickel rod was used as the anode. A current density
of 0.46 ASI
was applied to the cathode plate and the anode from a rectifier. The potential
of the cathode
was measured with the aid of a LUGGIN probe versus a Hg/Hg0 reference
electrode. The
hydrogen potential measurements for Example 17 (Example 20), Example 18
(Example 21 ),
Example 19 (Example 22), and Example 20 (Example 23) are -0.956 volts, -0.960
volts, -
0.949 volts and -0.956 volts, respectively.
-16-