Note: Descriptions are shown in the official language in which they were submitted.
WO 01/32624 CA 02389300 2002-04-29 pCT/US00/30328
4-PHENYL-SUBSTITUTED TETRAHYDROISOQUINOLINES AND USE THEREOF TO BLOCK REUPTAKE
OF NOREPINEPHRINE, DOPAMINE AND SEROTONIN
Field of the Invention
The present invention relates to compounds, compositions, methods for the
treatment of various
neurological and psychological disorders, and the use of the compounds in
combination therapy. In
particular, the present invention relates to such compounds, compositions and
methods wherein the
compounds are novel 4-phenyl substituted tetrahydroisoquinolines derivatives.
Background of the Invention
Serotonin, dopamine and norepinephrine are known to be important chemical
messengers
participating in the transmission of nerve impulses in the brain. These
messengers are liberated at
specific sites on pre-synaptic cells and received, to complete transmission of
the impulse, at specific sites
on post-synaptic cells. Their effect is then terminated by metabolism or by
uptake into the pre-synaptic
cells. Drugs capable of blocking the pre-synaptosomal uptake of either of
these chemical messengers in
the brain, are useful in alleviating disorders associated with decreased
levels of these chemical
messengers. For example, duloxetine and fluoxetine which are known serotonin
reuptake inhibitors have
been found to be useful in the treatment of depression, obesity and obsessive-
compulsive disease (Wong,
et al., U.S. Patent No. 5,532,244). Also, Moldt, et al., U.S. Patent No.
5,444,070, discloses the use of
dopamine reuptake inhibitors in the treatment of depression, Parkinsonism,
drug addiction and/or abuse,
cocaine and/or amphetamine addiction and/or abuse. Freedman, et al., U.S.
Patent No. 6,136,803 also
discloses synaptic norepinephrine or serotonin uptake inhibitors which are
useful in treating depression
in a patient. Furthermore, Norden, U.S. Patent No. 5,789,449 discloses the use
of serotonin re-uptake
inhibitors in treating psychiatric symptoms consisting of anger, rejection
sensitivity, and lack of mental
or physical energy. Also, Foster, et al., U.S. Patent No. 4,902,710, discloses
the use of serotonin and
norepinephrine uptake inhibitors in suppressing the desire of humans to smoke
or consume alcohol.
Thus, there continues to remain a need to develop novel compounds which block
reuptake of
norephinephrine, dopamine or serotonin.
Compounds which inhibit the reuptake of serotonin or norephinephrine, have
also been used in
combination therapy. For example, Glatt, et al., U.S. Patent no. 6,121,261
discloses the use of selective
serotonin reuptake Inhibitors or norephinephrine uptake inhibitiors, in
combination with neurokinin-1
receptor antagonist for treating attention deficit disorder in a patient.
Also, Hohenwarter, U.S. Patent No. 4,843,071 discloses the use of a
norepinephrine re-uptake
inhibitor and a norepinephrine precursor in the treatment of obesity, dnig
abuse, or narcolepsy in a
patient. Furthermore, Wong, et al., U.S. Patent No. 5,532,244, discloses the
use of serotonin reuptake
inhibitors in combination with a serotonin 1A receptor antagonist, to increase
the availability of
serotonin. norepinephrine and dopamine in the brain.
-1-
WO 01/32624 CA 02389300 2002-04-29 pCT/US00/30328
The treatment of a variety of neurological and psychiatric disorders is
characterized by a number
of side effects believed to be due to the compounds' inability to selectvely
block certain neurochemicals,
and not others. ADHD, for example, is a disease affecting 3-6°Io of
school age children, and is also
recognized in percentage of adults. Aside from hampering performance at
school, and at work, ADHD is
a significant risk factor for the subsequent development of anxiety disorders,
depression, conduct
disorder and drug abuse. Since current treatment regimes require
psychostimulants, and since a
substantial number of patients (30%) are resistant to stimulants or cannot
tolerate their side effects, there
is a need for a new drug or class of drugs which treats ADHD and does not have
resistance or side effect
problems. In addition, methylphenidate, the current drug of choice for the
treatment of ADHD, induces
a number of side effects; these include anorexia, insomnia and jittery
feelings, tics, as well as increased
blood pressure and heart rate secondary to the activation of the sympathetic
nervous system. However,
Methylphenidate also has a high selectivity for the dopamine transporter
protein over the norepinephrine
transporter protein (DAT/NET Ki ratio of 0.1 ), which can lead to addiction
liability and requires multiple
doses per day for optimal efficacy. Thus, there continues to remain a need to
develop novel compounds
which block reuptake of norephinephrine, dopamine, and serotonin with
particular selectivity ratios.
U.S. Patent No. 3,947,456, discloses tetrahydroisoquinolines which are said to
have utility as
anti-depressants. U.S. Patent No. 3,666,763, describes the use of phenyl
tetrahydroisoquinoline
derivatives as antidepressants and antihypotensives. Canadian Patent
Application No. 2,015,114,
discloses the use of phenyl tetrahydroisoquinoline derivatives as
antidepressants; moreover, described
therein are apparently nonselective as to norepinephrine, serotonin and
dopamine uptake. UK Patent
Application No. 2,271,566 , discloses the use of phenyl tetrahydroisoquinoline
derivatives as anti-HIV
agents. PCT International Application No. W098/40358 discloses the use of
phenyl
tetrahydroisoquinoline derivatives to be useful in the treatment of disorders
of glucose metabolic
pathways. W097/36876 discloses the use of phenyl tetrahydroisoquinoline
derivatives as anticancer
agents. W097/23458 also describes 4 phenyl-substituted tetrahydroisoquinolines
as NMDA receptor
ligands useful for conditions associated with neuronal loss. Phenyl-
substituted tetrahydroisoquinolines
are also described in Mondeshka et al Il Farmaco, 1994,49 pp. 475-481.
Nomofensine~ which is a 4 phenyl-substituted tetrahydroisoquinoline derivative
is known to
inhibit the neuronal uptake of dopamine and other catecholamines and has shown
clinical efficacy for
ADHD. However, long term administration of Nomofensine~ results in fatal
immune hemolytic
anemia. Thus, there continues to remain a need to develop novel compounds
which treat ADHD but do
not have the serious side effects associated with Nomifensine0 or the
currently prescribed
psychostimulants.
The present invention discloses novel aryl and heteroaryl substituted
tetrahydroisoquinoline
derivatives compounds which block reuptake of norephinephrine. dopamine. or
serotonin, and are usefu:
_z_
WO 01/32624 CA 02389300 2002-04-29 PCT/US00/30328
as alternatives to methylphenidate, and known psychostimulants, in the
treatment of ADHD and other
neurological and psychiatric disorders.
The present inventors have discovered that the claimed compounds which block
reuptake of
norephinephrine, dopamine, and serotonin with particular selectivity ratios,
e.g., being more selective for
the norepinephrine transporter (NET) protein than dopamine transporter (DAT)
protein or serotonin
transporter (SERT) protein (lower Ki for NET than for DAT and SERT). It is
postulated that the
compounds would therefore be effective as an ADHD treatment with reduced
addictive liability profiles.
In particular, some of the compounds of this invention are surprisingly and
particularly selective for
NET over the SERT protein, thus also affording compounds without the known
side effect profiles of the
selective serotonin reuptake inhibitor (SSRI) class of compounds.
Summary of the Invention
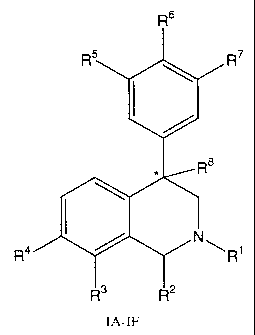
This invention is directed to a compound of formulae (IA-F)
Rs
R~
R4 .Ri
IA-IF
wherein:
the carbon atom designated * is in the R or S configuration;
R' is C,-C~ alkyl, Ci Cfi alkenyl, Cz Cfi alkynyl, C,-Cfi cycloalkyl or C~ C~
cycloalkylalkyl, each of which
is optionally substituted with 1 to 3 substituents independently selected at
each occurrence thereof from
C,-C, alkyl, halogen, aryl, -CN, -ORy and -NRyR"';
R~ is H, C,-Cfi alkyl, CZ C~ alkenyl, C,-C~ alkynyl, C;-Cfi cycloalkyl, C4 C,
cycloalkylalkyl or C~-Cfi
haloalkyl;
R' is H, halogen, -OR", -S(O)~R'-, -S(O)~NR"R'-, -CN, -C(O)R'~, -C(O)NR"R'z,
C,-C~ alkyl, C2 C
alkenyl, CZ Cfi alkynyl, C~ Cfi cycloalkyl, C4 C~ cycloalkylalkyl, -O(phenyl)
or -O(benzyl), wherein each
of -O(phenyl) and -O(benzyl) is optionally substituted from 1 to 3 times with
a substituent selected
-3-
WO 01/32624 CA 02389300 2002-04-29 PCT/US00/30328
independently at each occurrence thereof from halogen, cyano, C~-C, alkyl, C,-
C~ haloalkyl, or C,-Ca
alkoxy, or wherein R' is a C~-C~ alkyl, CZ C~ alkenyl, Cz C~ alkynyl, Ci C~
cycloalkyl or CQ C
cycloalkylalkyl group, then said group is optionally substituted with from 1
to 3 substituents selected
independently at each occurrence thereof from C,-C3 alkyl, halogen, aryl, -CN,
-ORy and -NRyR" ;
provided that for compounds of formula IA, R; is C,-Cfi alkyl, C2-C~ alkenyl,
CZ C~ alkynyl, C,-C
cycloalkyl or CQ C~ cycloalkylalkyl, each of which is optionally substituted
with from 1 to 3 substituents
selected independently at each occurrence thereof from C,-C; alkyl, halogen,
aryl, -CN, -ORy and -
NRyR"'~
provided that for compounds of formula IB, R' is -O(phenyl), -O(benzyl), -
OC(O)R" or -S(O)~R'~, each
of -O(phenyl) and -O(benzyl) is optionally substituted from 1 to 3 times with
a substituent selected
independently at each occurrence thereof from halogen, cyano, C,-Ca alkyl, C,-
C~ haloalkyl, or C,-Ca
alkoxy;
R° is H, halogen, -OR", -S(O)~R'-, -S(O)NR"R'-, -CN, -C(O)R'-, -
C(O)NR"R'-, - NR"R'2, C,-C~ alkyl,
CZ Cfi alkenyl, C~ C~ alkynyl, C.-C~, cycloalkyl, C~ C, cycloalkylalkyl, -
O(phenyl) or -O(benzyl), wherein
each of -O(phenyl) and -O(benzyl) is optionally substituted from 1 to 3 times
with a substituent selected
independently at each occurrence thereof from halogen, cyano, C~-C~ alkyl, C~-
C~ haloalkyl, or C~-Ca
alkoxy and wherein Ra is a C,-C~ alkyl, C,-Cfi alkenyl, C~-C~ alkynyl, C;-Cfi
cycloalkyl or C~-C7
cycloalkylalkyl group, then said group is optionally substituted with from 1
to 3 substituents selected
independently at each occurrence thereof from C~-C, alkyl, halogen, aryl, -CN,
-OR'' and -NRyR"';
provided that for compounds of formula IC, R~ is C,-C~, alkyl, C:-C~, alkenyl,
C=-C~; alkynyl, C,-C,,
cycloalkyl, or C,-C, cycloalkylalkyl, each of which is is optionally
substituted with from 1 to 3
substituents selected independently at each occurrence thereof from C~-C,
alkyl, halogen, aryl, -CN, -OR"
and -NRvR"', or R5 and R'' or R~ and R' may be -O-C(R"),-O-;
provided that for compounds of formula ID, R~ is -O(phenyl), -O(benzyl), -
OC(O)R", -NR"R'~ or -
S(O)~R'~, each of -O(phenyl) and -O(benzyl) is optionally substituted from 1
to 3 times with a substituent
selected independently at each occurrence thereof from halogen, cyano, C,-C~
alkyl, C~-C; haloalkyl, or
C,-C, alkoxy;
Rs, R'' and R' in compounds of each of the formulae IA, IB, IC, ID, IE and IF
are each independently H.
halogen, -OR", -S(O),R'-, -CN, -C(O)R'-, -NR"R'~, -C(O)NR"R'-, -NR"C(O)R'~, -
NR"C(O),R'~, -
NR"C(O)NR'zR", C,-Cfi alkyl, C=-C~ alkenyl, C~-C~ alkynyl, C~ Ch cycloalkyl or
C~ C~ cycloalkylalkyl,
wherein each of R5, R'' and R' is a C,-Cfi alkyl, C_-C~, alkenyl, C,-C~,
alkynyl, Cz-Ch cycloalkyl or Ca-C
cycloalkylalkyl group, then said group is optionally substituted with from 1
to 3 substituents selected
independently at each occurrence thereof from C~-C, alkyl, halogen, aryl, -CN,
-OR" and -NRyR'°, or R'
and Rfi or R" and R' may be -O-C(R'-),-O-;
provided that for compounds of formula IE at least one of R~ or R' is fluoro,
chloro, or meth~'1;
CA 02389300 2002-04-29
WO 01/32624 PCT/US00/30328
or RS and Rfi are each independently -O-C(R'Z)= O- in compounds of the
formulae IE, but only where R' is
fluoro, chloro or methyl;
or R' and R~ can independently also be -O-C(R'-),-O- in compounds of the
formulae IE, but only where
R5 is fluoro, chloro or methyl;
RR is H, halogen, or OR", provided that for compounds of formula IF, R" is
halogen;
Ry and R"' are each independently H, C,-C~ alkyl, C,-C~ haloalkyl, C,-C~
alkoxyalkyl, C,-Cfi cycloalkyl,
C4 C~ cycloalkylalkyl, -C(O)R", phenyl or benzyl, where phenyl or benzyl is
optionally substituted from
I to 3 times with a substituent selected independently at each occurrence
thereof from halogen, cyano,
C,-C, alkyl, C,-C~ haloalkyl, or C,-C, alkoxy;
or Ry and R"~ are taken together with the nitrogen to which they are attached
to form piperidine,
pyrrolidine, piperazine, N-methylpiperazine, morpholine, or thiomorpholine;
R" is H, C,-Ca alkyl, C,-C~ haloalkyl, C,-C~ alkoxyalkyl, C,-Cfi cycloalkyl,
C~-C~ cycloalkylalkyl, -
C(O)R", phenyl or benzyl, where R" is a C~-C~ alkyl, phenyl or benzyl group,
then said group is
optionally substituted from 1 to 3 times with a substituent selected
independently at each occurrence
thereof from halogen, cyano, C,-C; alkyl, C,-C~ haloalkyl, or C,-C~ alkoxy;
R'~ is H, amino, C,-C, alkyl, (C,-C, alkyl)amino, C,-C~ haloalkyl, C,-C~
alkoxyalkyl, Cz C~ cycloalkyl, C
C~ cycloalkylalkyl, phenyl or benzyl, where phenyl or benzyl is optionally
substituted from 1 to 3 times
with a substituent selected independently from halogen, cyano, C,-Ca alkyl, C,-
C, haloalkyl and C,-C
alkoxy;
or R" and R'' are taken together with the nitrogen to which they are attached
to form piperidine,
pyrrolidine, piperazine, N-methylpiperazine, morpholine, or thiomorpholine;
provided that only one of R'' and R'° or R'' and R'° are taken
together with the nitrogen to which they are
attached to form piperidine, pyrrolidine, piperazine, N-methylpiperazine,
morpholine, or thiomorpholine;
R" is C,-C; alkyl, C,-C~ haloalkyl or phenyl;
n is 0, 1, or 2, and;
aryl is phenyl which is optionally substituted 1-3 times with halogen, cyano,
C~-C, alkyl, C~-C~ haloalkyl
and C,-C, alkoxy, or
an oxide thereof, a pharmaceutically acceptable salt thereof, a solvate
thereof, or prodrug thereof.
DETAILED DESCRIPTION OF THE INVENTION
As used above, and throughout the description of the invention, the following
terms, unless
otherwise indicated, shall be understood to have the following meanings:-
The term "Alkyl" means an aliphatic hydrocarbon group which may be straight or
branched
having about 1 to about 6 carbon atoms in the chain. Branched means that one
or more lower alkyl
-5-
WO 01/32624 CA 02389300 2002-04-29 pCT~JS00/30328
groups such as methyl, ethyl or propyl are attached to a linear alkyl chain.
Exemplary alkyl groups
include methyl, ethyl, n-propyl, i-propyl, n-butyl, t-butyl, n-pentyl, and 3-
pentyl.
The term "Alkenyl" means an aliphatic hydrocarbon group containing a carbon-
carbon double
bond and which may be straight or branched having about 2 to about 6 carbon
atoms in the chain.
Preferred alkenyl groups have 2 to about 4 carbon atoms in the chain. Branched
means that one or more
lower alkyl groups such as methyl, ethyl or propyl are attached to a linear
alkenyl chain. Exemplary
alkenyl groups include ethenyl, propenyl, n-butenyl, and i-butenyl.
The term "Alkynyl" means an aliphatic hydrocarbon group containing a carbon-
carbon triple
bond and which may be straight or branched having about 2 to about 6 carbon
atoms in the chain.
Preferred alkynyl groups have 2 to about 4 carbon atoms in the chain. Branched
means that one or more
lower alkyl groups such as methyl, ethyl or propyl are attached to a linear
alkynyl chain. Exemplary
alkynyl groups include ethynyl, propynyl, n-butynyl, 2-butynyl, 3-
methylbutynyl, and n-pentynyl.
The term "Aryl" means an aromatic monocyclic or multicyclic ring system of 6
to about 14
carbon atoms, preferably of 6 to about 10 carbon atoms. Representative aryl
groups include phenyl and
naphthyl.
The term "Heteroaryl" means an aromatic monocyclic or multicyclic ring system
of about 5 to
about 14 ring atoms, preferably about 5 to about 10 ring atoms, in which one
or more of the atoms in the
ring system is/are elements) other than carbon, for example, nitrogen, oxygen
or sulfur. Preferred
heteroaryls contain about 5 to 6 ring atoms. The prefix aza, oxa or thia
before heteroaryl means that at
least a nitrogen, oxygen or sulfur atom, respectively, is present as a ring
atom. A nitrogen atom of a
heteroaryl is optionally oxidized to the corresponding N-oxide. Representative
heteroaryls include
pyrazinyl; furanyl; thienyl; pyridyl; pyrimidinyl; isoxazolyl; isothiazolyl;
oxazolyl; thiazolyl; pyrazolyl;
furazanyl; pyrrolyl; pyrazolyl; triazolyl; 1,2,4-thiadiazolyl; pyrazinyl;
pyridazinyl; quinoxalinyl;
phthalazinyl; 1(2H)-phthalazinonyl; imidazo[1,2-a]pyridine; imidazo[2,1-
b]thiazolyl; benzofurazanyl;
indolyl; azaindolyl; benzimidazolyl; benzothienyl; quinolinyl; imidazolyl;
thienopyridyl; quinazolinyl;
thienopyrimidyl; pyrrolopyridyl; imidazopyridyl; isoquinolinyl;
benzoazaindolyl; azabenzimidazolyl,
1,2,4-triazinyl; benzothiazolyl and the like.
The term "Alkoxy" means an alkyl-O- group wherein the alkyl group is as herein
described.
Exemplary alkoxy groups include methoxy, ethoxy, n-propoxy, i-propoxy. n-
butoxy and heptoxy.
The term "Compounds of the invention", and equivalent expressions, are meant
to embrace
compounds of general formulae (IA-F) as hereinbefore described, which
expression includes the
prodrugs, the pharmaceutically acceptable salts, and the solvates, e.g.
hydrates, where the context so
permits. Similarly, reference to intermediates, whether or not they themselves
are claimed, is meant to
embrace their salts, and solvates, where the context so permits. For the sake
of clarity, particular
_g_
WO 01/32624 CA 02389300 2002-04-29 PCT/US00/30328
instances when the context so permits are sometimes indicated in the text, but
these instances are purely
illustrative and it is not intended to exclude other instances when the
context so permits.
The term "Cycloalkyl" means a non-aromatic mono- or multicyclic ring system of
about 3 to
about 7 carbon atoms, preferably of about 5 to about 7 carbon atoms. Exemplary
monocyclic cycloalkyl
include cyclopentyl, cyclohexyl, cycloheptyl, and the like.
The term "Cycloalkylalkyl" means an cycloalkyl-alkyl- group in which the
cycloalkyl and alkyl
are as defined herein. Exemplary cycloalkylalkyl groups include
cyclopropylmethyl and
cyclopentylmethyl.
The term "Halo" or "halogen" means fluoro, chloro, bromo, or iodo.
The term "Haloalkyl" means both branched and straight-chain alkyl substituted
with 1 or more
halogen, wherein the alkyl group is as herein described.
The term "Haloalkoxy" means a C,~, alkoxy group substituted by at least one
halogen atom,
wherein the alkoxy group is as herein described.
The term "Substituted" or "substitution" of an atom means that one or more
hydrogen on the
designated atom is replaced with a selection from the indicated group,
provided that the designated
atom's normal valency is not exceeded. "Unsubstituted" atoms bear all of the
hydrogen atoms dictated
by their valency. When a substituent is keto (i.e., =O), then 2 hydrogens on
the atom are replaced.
Combinations of substituents and/or variables are permissible only if such
combinations result in stable
compounds; by "stable compound" or "stable structure" is meant a compound that
is sufficiently robust
to survive isolation to a useful degree of purity from a reaction mixture, and
formulation into an
efficacious therapeutic agent.
The term "Pharmaceutically acceptable salts" means the relatively non-toxic,
inorganic and
organic acid addition salts, and base addition salts, of compounds of the
present invention. These salts
can be prepared in situ during the final isolation and purification of the
compounds. In particular, acid
addition salts can be prepared by separately reacting the purified compound in
its free base form with a
suitable organic or inorganic acid and isolating the salt thus formed.
Exemplary acid addition salts
include the hydrobromide, hydrochloride, sulfate, bisulfate, phosphate,
nitrate, acetate, oxalate, valerate,
oleate, palmitate, stearate, laurate, borate, benzoate, lactate, phosphate,
tosylate, citrate, maleate,
fumarate, succinate, tartrate, naphthylate, mesylate, glucoheptonate,
lactiobionate, sulphamates,
malonates, salicylates, propionates, methylene-bis-b-hydroxynaphthoates,
gentisates, isethionates,
di-p-toluoyltartrates, methane-sulphonates, ethanesulphonates,
benzenesulphonates,
p-toluenesulphonates, cyclohexylsulphamates and quinateslaurylsulphonate
salts, and the like. (See, for
example S. M. Berge, et al., "Pharmaceutical Salts," J. Pharm. Sci., 66: p.1-
19 (1977) and Remington's
Pharmaceutical Sciences, 17th ed., Mack Publishing Company, Easton, PA, 1985,
p. 1418, which are
incorporated herein by reference.) Base addition salts can also be prepared by
separately reacting the
-7-
WO 01/32624 CA 02389300 2002-04-29 PCT/US00/30328
purified compound in its acid form with a suitable organic or inorganic base
and isolating the salt thus
formed. Base addition salts include pharmaceutically acceptable metal and
amine salts. Suitable metal
salts include the sodium, potassium, calcium, barium, zinc, magnesium, and
aluminum salts. The
sodium and potassium salts are preferred. Suitable inorganic base addition
salts are prepared from metal
bases which include sodium hydride, sodium hydroxide, potassium hydroxide,
calcium hydroxide,
aluminium hydroxide, lithium hydroxide, magnesium hydroxide, zinc hydroxide.
Suitable amine base
addition salts are prepared from amines which have sufficient basicity to form
a stable salt, and
preferably include those amines which are frequently used in medicinal
chemistry because of their low
toxicity and acceptability for medical use. ammonia, ethylenediamine, N-methyl-
glucamine, lysine,
arginine, ornithine, choline, N,N'-dibenzylethylenediamine, chloroprocaine,
diethanolamine, procaine,
N-benzylphenethylamine, diethylamine, piperazine, tris(hydroxymethyl)-
aminomethane,
tetramethylammonium hydroxide, triethylamine, dibenzylamine, ephenamine,
dehydroabietylamine, N-
ethylpiperidine, benzylamine, tetramethylammonium, tetraethylammonium,
methylamine,
dimethylamine, trimethylamine, ethylamine, basic amino acids, e.g., lysine and
arginine, and
dicyclohexylamine, and the like.
The term "Pharmaceutically acceptable prodrugs" as used herein means those
prodrugs of the
compounds useful according to the present invention which are, within the
scope of sound medical
judgment, suitable for use in contact with the tissues of humans and lower
animals with undue toxicity,
irritation, allergic response, and the like, commensurate with a reasonable
benefitlrisk ratio, and effective
for their intended use, as well as the zwitterionic forms, where possible, of
the compounds of the
invention. The term "prodrug" means compounds that are rapidly transformed in
vivo to yield the parent
compound of the above formula, for example by hydrolysis in blood. Functional
groups which may be
rapidly transformed, by metabolic cleavage, in vivo form a class of groups
reactive with the carboxyl
group of the compounds of this invention. They include, but are not limited to
such groups as alkanoyl
(such as acetyl, propionyl, butyryl, and the like), unsubstituted and
substituted amyl (such as benzoyl
and substituted benzoyl), alkoxycarbonyl (such as ethoxycarbonyl),
trialkylsilyl (such as trimethyl- and
triethysilyl), monoesters formed with dicarboxylic acids (such as succinyl),
and the like. Because of the
ease with which the metabolically cleavable groups of the compounds useful
according to this invention
are cleaved in vivo, the compounds bearing such groups act as pro-drugs. The
compounds bearing the
metabolically cleavable groups have the advantage that they may exhibit
improved bioavailability as a
result of enhanced solubility and/or rate of absorption conferred upon the
parent compound by virtue of
the presence of the metabolically cleavable group. A thorough discussion of
prodrugs is provided in the
following: Design of Prodrugs, H. Bundgaard, ed., Elsevier, 1985; Methods in
Enzymology, K. Widder
et al, Ed., Academic Press, 42, p.309-396, 1985; A Textbook of Drug Design and
Development,
Krogsgaard-Larsen and H. Bundgaard, ed., Chapter 5; "Design and Applications
of Prodrugs" p.1 13-191,
_g_
WO 01/32624 CA 02389300 2002-04-29 PCT/US00/30328
1991; Advanced Drug Delivery Reviews, H. Bundgard, 8, p.1-38, 1992; Journal of
Pharmaceutical
Sciences, 77, p. 285, 1988; Chem. Pharm. Bull., N. Nakeya et al, 32, p. 692,
1984; Pro-drugs as Novel
Delivery Systems, T. Higuchi and V. Stella, Vol. 14 of the A.C.S. Symposium
Series, and Bioreversible
Carriers in Drug Design, Edward B. Roche, ed., American Pharmaceutical
Association and Pergamon
Press, 1987, which are incorporated herein by reference. Examples of prodrugs
include, but are not
limited to, acetate, formate and benzoate derivatives of alcohol and amine
functional groups in the
compounds of the invention.
The term "Therapeutically effective amounts" is meant to describe an amount of
compound of
the present invention effective in increasing the levels of serotonin,
norepinephrine or dopamine at the
synapse and thus producing the desired therapeutic effect. Such amounts
generally vary according to a
number of factors well within the purview of ordinarily skilled artisans given
the description provided
herein to determine and account for. These include, without limitation: the
particular subject, as well as
its age, weight, height, general physical condition and medical history; the
particular compound used, as
well as the carrier in which it is formulated and the route of administration
selected for it; and, the nature
and severity of the condition being treated.
The term "Pharmaceutical composition" means a composition comprising a
compound of
formulae (IA-F) and at least one component selected from the group comprising
pharmaceutically
acceptable carriers, diluents, adjuvants, excipients, or vehicles, such as
preserving agents, fillers,
disintegrating agents, wetting agents, emulsifying agents, suspending agents,
sweetening agents,
flavoring agents, perfuming agents, antibacterial agents, antifungal agents,
lubricating agents and
dispensing agents, depending on the nature of the mode of administration and
dosage forms. Examples
of suspending agents include ethoxylated isostearyl alcohols, polyoxyethylene
sorbitol and sorbitan
esters, microcrystalline cellulose, aluminum metahydroxide, bentonite, agar-
agar and tragacanth, or
mixtures of these substances. Prevention of the action of microorganisms can
be ensured by various
antibacterial and antifungal agents, for example, parabens, chlorobutanol,
phenol, sorbic acid, and the
like. It may also be desirable to include isotonic agents, for example sugars,
sodium chloride and the
like. Prolonged absorption of the injectable pharmaceutical form can be
brought about by the use of
agents delaying absorption, for example, aluminum monosterate and gelatin.
Examples of suitable
carriers, diluents, solvents or vehicles include water, ethanol, polyols,
suitable mixtures thereof,
vegetable oils (such as olive oil) and injectable organic esters such as ethyl
oleate. Examples of
excipients include lactose, milk sugar, sodium citrate, calcium carbonate,
dicalcium phosphate
phosphate. Examples of disintegrating agents include starch, alginic acids and
certain complex silicates.
Examples of lubricants include magnesium stearate, sodium lauryl sulphate,
talc, as well as high
molecular weight polyethylene glycols.
_g_
WO 01/32624 CA 02389300 2002-04-29 PCT~S00/30328
The term "Pharmaceutically acceptable" means it is, within the scope of sound
medical
judgement, suitable for use in contact with the cells of humans and lower
animals without undue toxicity,
irritation, allergic response and the like, and are commensurate with a
reasonable benefit/risk ratio.
The term "Pharmaceutically acceptable dosage forms" means dosage forms of the
compound of
the invention, and includes, for example, tablets, dragees, powders, elixirs,
syrups, liquid preparations,
including suspensions, sprays, inhalants tablets, lozenges, emulsions,
solutions, granules, capsules and
suppositories, as well as liquid preparations for injections, including
liposome preparations. Techniques
and formulations generally may be found in Remington's Pharmaceutical
Sciences, Mack Publishing
Co., Easton, PA, latest edition.
PREFERRED EMBODIMENTS
Another embodiment of the invention is a compound of formulae IA-IF
wherein:
the carbon atom designated * is in the R or S configuration.
Another embodiment of the invention is a compound of formulae IA, IB, IC, ID,
IE and IF,
wherein:
R' is C,-Cfi alkyl, C_-C~ alkenyl, C,-Cfi alkynyl, C;-Cn cycloalkyl or C~ C~
cycloalkylalkyl, each of
which is optionally substituted with from 1 to 3 substituents selected
independently at each occurrence
thereof from C,-C, alkyl, halogen, aryl, -CN, -OR" and -NRyR"'.
Another embodiment of the invention is a compound of formulae IA, IB, IC, ID,
IE and IF,
wherein:
R-is H, C,-Cfi alkyl, C=-Cfi alkenyl, C=-C, alkynyl, C,-C,, cycloalkyl, C,-C,
cycloalkylalkyl or C,-
C~ haloalkyl.
Another embodiment of the invention is a compound of formulae IA, wherein:
R' as C,-C~ alkyl, C= Cfi alkenyl, C; C~, alkynyl, C~-C~ cycloalkyl or C,-C,
cycloalkylalkyl, each of
which is optionally substituted with from 1 to 3 substituents selected
independently at each occurrence
thereof from C,-C. alkyl, halogen, aryl, -CN, -OR" and -NRyR"'
Another embodiment of the invention is a compound of formulae IB, wherein:
R' as -O(phenyl), -O(benzyl), -OC(O)R" or -S(O)"R'~, each of -O(phenyl) and -
O(benzyl)
optionally substituted with 1 to 3 substituents selected independently at each
occurrence thereof from
halogen, cyano, C,-C, alkyl, C,-C, haloalkyl or C,-C~ alkoxy.
Another embodiment of the invention is a compound of formulae IC, ID, IE and
IF, wherein:
R' is H, halogen, -OR", -S(O)"R'~, -S(O)NR"R'-, -CN, -C(O)R'~, -C(O)NR"R'~, -
O(phenyl),
O(benzyl), -OC(O)R'' or -S(O)~R'-, C,-C~ alkyl, C,-C~ alkenyl, C,-Cfi alkynyl,
C,-Cfi cycloalkyl and C~ C
cycloalkylalkyl, wherein each of C,-C" alkyl, C,-C~ alkenyl, C~-C~, alkynyl,
C,-C~ cycloalkyl and C;-C~
-10-
WO 01/32624 CA 02389300 2002-04-29 PCT/US00/30328
cycloalkylalkyl is optionally substituted with from 1 to 3 substituents
selected independently at each
occurrence thereof from C,-C, alkyl, halogen, aryl, -CN, -ORy and -NRyR"', and
wherein R' is a -
O(phenyl) or -O(benzyl) group, then said group is optionally substituted with
1 to 3 substituents selected
independently at each occurrence thereof from halogen, cyano, C,-C4 alkyl, C,-
C~ haloalkyl or C,-C4
alkoxy.
Another embodiment of the invention is a compound of formula IC, wherein:
R° is C,-C~ alkyl, Cz C~ alkenyl, C; C~ alkynyl, C,-C~ cycloalkyl, or
C4 C~ cycloalkylalkyl, each of
which is optionally substituted with from 1 to 3 substituents selected
independently at each occurrence
thereof from C,-C; alkyl, halogen, aryl, -CN, -OR' and -NRyR"'.
Another embodiment of the invention is a compound of formula ID, wherein:
R° is -O(phenyl), -O(benzyl), -OC(O)R", -NR"R'~ or -S(O)~R'-, and said -
O(phenyl) or -
O(benzyl) is optionally substituted with 1 to 3 substituents selected
independently at each occurrence
thereof from halogen, cyano, C,-C, alkyl, C,-C~ haloalkyl and C,-C, alkoxy.
Another embodiment of the invention is a compound of formula IA, IB, IE and
IF, wherein:
R° is H, halogen, -OR", -S(O)"R'~, -S(O)NR"R'-, -CN, -O(phenyl), -
O(benzyl), -OC(O)R",
C(O)R'~, -C(O)NR"R'~, - NR"R'~, C,-C~ alkyl, C,-Cfi alkenyl, C_ C~ alkynyl, C~
Cfi cycloalkyl and C~-C
cycloalkylalkyl, wherein R' is a C,-C~ alkyl, C=-C~ alkenyl, C,-Cfi alkynyl,
C,-C'; cycloalkyl or C~ C
cycloalkylalkyl group, then said group is optionally substituted with 1 to 3
substituents selected
independently at each occurrence thereof from C,-C, alkyl, halogen, aryl, -CN,
-OR" and -NRvR"', and
wherein R' a -(O)phenyl or -(O)benzyl group, then said group is optionally
substituted from 1 to 3 times
with a substituent selected independently at each occurrence thereof from
halogen, cyano, C,-C; alkyl,
C,-C, haloalkyl, and C,-C~ alkoxy.
Another embodiment of the invention is a compound of formulae IA, IB, IC, ID
and IF, wherein:
R5, R° and R' are each independently H, halogen, -OR", -S(O),~R'-, -CN,
-C(O)R'-, -NR"R'~,
C(O)NR"R'~, -NR"C(O)R'~, -NR"C(O),R'~, -NR"C(O)NR'2R", C,-C~ alkyl, C_-Cfi
alkenyl, C,-C', alkynyl,
C,-Cfi cycloalkyl or C,-C~ cycloalkylalkyl, wherein each Rs, R° and R'
is independently a C,-C~ alkyl, C:-
C~ alkenyl, C~ C~ alkynyl, C~-C~ cycloalkyl or C~ C~ cycloalkylalkyl group,
then said group is optionally
substituted from 1 to 3 times with substituents selected independently at each
occurrence thereof from
C,-C, alkyl, halogen, aryl, -CN, -OR" and -NRyR"~, or R' and R'' or R°
and R' may be -O-C(R''),-O-.
Another embodiment of the invention is a compound of formula IE, wherein:
when R5 is fluoro, chloro, or methyl; then R' and R" are each independently H,
halogen, -OR",
S(O)~R'~, -CN, -C(O)R'~, -NR"R'-, -C(O)NR"R'-, -NR"C(O)R'-, -NR"C(O)=R'', -
NR"C(O)NR'ZR", C,-C"
alkyl, C~ C~ alkenyl, C,-C~ alkynyl, C,-Cfi cycloalkyl or C~ C~
cycloalkylalkyl, wherein each of R' and R''
are a C,-C~ alkyl, C_-C~ alkenyl, C= C~ alkynyl, C,-C~ cycloalkyl orC~ C,
cycloalkylalkyl group, said
group is optionally substituted with from 1 to 3 substituents selected
independently at each occurrence
-11-
WO 01/32624 CA 02389300 2002-04-29 PCT/US00/30328
thereof from C,-C~ alkyl, halogen, aryl, -CN, -OR9 and -NR9R"', provided that
R' is not fluoro, chloro, or
methyl
Another embodiment of the invention is a compound of formula IE, wherein:
R' is fluoro, chloro or methyl, then R5 and R6 together can also be -O-C(R'Z)2
O-.
Another embodiment of the invention is a compound of formula IE, wherein:
R5 is fluoro, chloro or methyl, then R' and R~ together can also be -O-C(R'2)z
O-.
Another embodiment of the invention is a compound of formulae IA-IE, wherein:
R" is H, halogen, or OR".
Another embodiment of the invention is a compound of formula IF, wherein
RR is halogen.
Another embodiment of the invention is a compound of formulae IA-F, wherein:
Ry and R"' are each independently H, C,-C~ alkyl, C,-C4 haloalkyl, C,-C4
alkoxyalkyl, C~ Cfi
cycloalkyl, C4 C~ cycloalkylalkyl, -C(O)R", phenyl or benzyl, where said
phenyl or benzyl is optionally
substituted from 1 to 3 times with a substituent selected independently at
each occurrence thereof from
halogen, cyano, C,-C, alkyl, C,-C~ haloalkyl, or C,-CQ alkoxy; or
Ry and R'° are taken together with the nitrogen to which they are
attached to form piperidine,
pyrrolidine, piperazine, N-methylpiperazine, morpholine, or thiomorpholine
rings.
Another embodiment of the invention is a compound of formulae IA-F, wherein:
R" is H, C,-C; alkyl, C,-Ca haloalkyl, C,-C~ alkoxyalkyl, C,-C~ cycloalkyl, CQ
C~ cycloalkylalkyl,
-C(O)R", phenyl or benzyl, where said phenyl or benzyl is optionally
substituted from 1 to 3 times with
a substituent selected independently at each occurrence thereof from halogen,
cyano, C,-Ca alkyl, C,-C,
haloalkyl, or C,-Ca alkoxy.
Another embodiment of the invention is a compound of formulae IA-F, wherein:
R'' is H, C,-C~ alkyl, C,-C~ haloalkyl, C,-C~ alkoxyalkyl, C~-C~ cycloalkyl,
C~ C~ cycloalkylalkyl,
phenyl or benzyl, where said phenyl or benzyl is optionally substituted from 1
to 3 times with a
substituent selected independently at each occurrence thereof from halogen,
cyano, C,-C~ alkyl, C~-C,
haloalkyl and C,-C~ alkoxy; or
R" and R'~ are taken together with the nitrogen to which they are attached to
form piperidine,
pyrrolidine, piperazine, N-methylpiperazine, morpholine or thiomorpholine
rings.
Another embodiment of the invention is a compound of formulae IA-F, wherein:
R" is C,-C~ alkyl, C,-C~ haloalkyl or phenyl; and n is 0, 1, or 2.
Another embodiment of the invention is a compound of formulae IA-F, wherein:
substituents R'-R" are as set forth in the following table:
-12-
WO ~l/32624 CA 02389300 2002-04-29 pCT~JS00/30328
Table A
IA IB IC ID IE IF
R' C,-C6
alkyl,
CZ C~
alkenyl,
Cz C6
alkynyl,
C~ C~
cycloalkyl
or C4
C
cycloalkylalkyl,
each
of which
is optionally
substituted
with
from
1 to
3
substituents
selected
independently
at each
occurrence
thereof
from
C,-C~
alkyl,
halogen,
aryl,
-CN,
-OR9
and -NR9R'
R2 H, C,-C~
alkyl,
CZ C~
alkenyl,
C2 C~
alkynyl,
Cz-C~
cycloalkyl,
C4 C
cycloalkylalkyl
or C,-C~
haloalkyl
R' C,-C~ -O(phenyl),H, halogen,
alkyl, -OR'',
-S(O)~R'2,-S(O)NR"R'2,
-CN,
-
C2 C~ -O(benzyl),C(O)R'',
-C(O)NR"R'2,
C,-C~
alkyl,
Cz C~
alkenyl,
C
alkenyl, -OC(O)R",C~ alkynyl,
CZ C~ C~
cycloalkyl,
C4 C~
cycloalkylalkyl,
-
C~ alkynyl,-S(O)~R'',O(phenyl),
- -O(benzyl)
and -OC(O)R",
wherein
C,-Cfi
C~-C~ wherein,alkyl,
- C; C~
alkenyl,
CZ C~
alkynyl,
Cz Cfi
cycloalkyl
cycloalkylO(phenyl)and C,-C~
cycloalkylalkyl
are optionally
substituted
or CQ and - with I
C~ to 3
substituents
selected
independently
at each
cycloalkyl-O(benzyl)occurrence
thereof
from
C,-Cz
alkyl,
halogen,
aryl,
-
alkyl, are CN, -ORy
each and -NRyR"'
and wherein
-(O)phenyl
and -
of which optionally(O)benzyl
is are optionally
substituted
as described
for
optionallysubstitutedthese
groups
in R'
of IB
substituted1 to
3 times
as set with
forth cyano,
above halogen,
for
the groupsC,-C,
alkyl,
in R~ C,-C
of IC-
IF haloalkyl,
Or C,-Ca
alkoxy
-13-
WO 01/32624 CA 02389300 2002-04-29 PCT/iJS00/30328
Table A (continued)
IA IB IC ID IE IF
R4 H, halogen, C,-C~ -O(phenyl),H, halogen,
-OR", alkyl, -OR",
- -
S(O)~R'Z, CZ C6 -O(benzyl),S(O)~R'z,
-S(O)NR"R'z, -S(O)NR"R'Z,
- -
CN, -C(O)R'Z, alkenyl,-OC(O)R",CN, -C(O)R'~,
- CZ -
C(O)NR"R'2, C~ alkynyl,-NR"R'' C(O)NR"R'~,
- NR"R'', or - NR"R'2,
C,-C~ Ci C~ -S(O)~R'z,C,-Ch
alkyl, - alkyl,
Cz C~ C2-C
alkenyl, cycloalkyl,O(phenyl)alkenyl,
Cz C~ C_ Ch
alkynyl, alkynyl,
C; C~ or CQ and - Cz Cfi
cycloalkyl, C~ cycloalkyl,
C4 C~ C~ C
cycloalkylalkyl, cycloalkyl-O(benzyl)cycloalkylalkyl,
wherein wherein
C,-Cfi alkyl, optionallyC,-C~
alkyl, each alkyl,
C_ C~ C~ C
alkenyl, optionallysubstitutedalkenyl,
C2-C~ Cz C~
alkynyl, alkynyl,
C,-C~ substituted1 to 3 C,-Cfi
cycloalkyl times cycloalkyl
and CQ and C
C~ cycloalkylalkyl as for with cyano,C~ cycloalkylalkyl
R; in
optionally IA, IB, halogen, optionally
substituted IE substituted
with from and IF C,-C, with from
1 to alkyl, 1 to
3 3
substituents C,-C~ substituents
selected selected
independently haloalkyl,independently
at each at each
occurrence or C,-C~ occurrence
thereof thereof
from from
C,-C, alkoxy C,-C,
alkyl, alkyl,
halogen, halogen,
aryl, aryl,
-CN, -ORy -CN, -OR"
and -NRyR"' and -NRyR"'
R' H, halogen, at least see R5,
-OR", one R
-S(O),~R'-,
-CN,
-C(O)R'~,
-NR"R'~,
-C(O)NR"R'z, of Rs and R'
-NR"C(O)R'2, or R' for
-NR"C(O),R'~,
-
Rfi NR"C(O)NR'2R", is F, IA, IB,
C,-Cfi Cl, or IC
alkyl,
C=-C~
alkenyl,
C,-C~
alkynyl, Me; the and ID
Ci C,,
cycloalkyl
or Cy
C~ cycloalkylalkyl,
R' wherein other
each of RS
of C,-C~
alkyl,
C,-C~
alkenyl,
C,-Cfi
alkynyl, or R'
Cz-Cfi and
cycloalkyl
and Ca-C~
cycloalkylalkyl
is
optionally R are
substituted any
with
from
1 to
3 substituents
selected of the
independently
at each
occurrence
thereof
from C,-C, groups
alkyl,
halogen,
aryl,
-CN,
-OR'
and -
,
NRyR"', described
or
R' and
Rfi or
R'' and
R' may
be -O-C(R'-)~-
for RS-'
in
IA-ID.
R5,
R'' (or
R'',
R') are
-O-
C(R~_)~_O-
only where
R' (or
R5)
is F,
C1, or
Me
R~ H, halogen, halogen
-OR"
Preferred embodiments of this invention are compounds of fomullae IA-IF,
wherein:
R' is C,-C, alkyl;
R- is H, C,-C~ alkyl or C,-Cfi haloalkyl.
-14-
WO 01/32624 CA 02389300 2002-04-29 PCT/US00/30328
Preferred embodiments of this invention are compounds of formulae IA, IC, ID,
IE and IF,
wherein:
R~ is C,-C~ alkyl, C~ C~ cycloalkyl or C~ C~ cycloalkylalkyl, each of these
groups being
optionally substituted with from 1 to 3 substituents selected independently at
each occurrence
thereof from C,-C, alkyl, halogen, aryl, -CN, -ORy and -NR9R'°.
Preferred embodiments of this invention are compounds of formula IB, wherein:
R' is -O(phenyl) or -O(benzyl), is optionally substituted from 1 to 3 times
with a substituent
selected independently at each occurrence thereof from halogen, cyano, C,-C4
alkyl, C,-CQ haloalkyl, or
C,-C4 alkoxy.
Preferred embodiments of this invention are compounds of formulae IC, ID, IE
and IF:
wherein R' is -O(phenyl) or -O(benzyl), and is optionally substituted from 1
to 3 times with a
substituent selected independently at each occurrence thereof from halogen,
cyano, C,-CQ alkyl, C,-CQ
haloalkyl, and C,-C, alkoxy.
Preferred embodiments of this invention are compounds of formulae IC-IF,
wherein:
R'isH.
Preferred embodiments of this invention are compounds of formulae IA, IB, IC,
IE and IF,
wherein:
R4 is C,-C, alkyl, C~-C~ cycloalkyl or CQ C~ cycloalkylalkyl, each of these
groups being optionally
substituted with from 1 to 3 substituents selected independently at each
occurrence thereof from C,-C,
alkyl, halogen, aryl, -CN, -OR~ and -NRyR"'.
Preferred embodiments of this invention are compounds of formulae IA, IB, IE
and IF, wherein:
R° is H.
Preferred embodiments of this invention are compounds of formulae IA, IB, IE
and IF, wherein:
R' is -NR"R'-, -O(phenyl) or -O(benzyl), each of these aryl groups being is
optionally substituted
from 1 to 3 times with a substituent selected independently at each occurrence
thereof from
halogen, cyano, C,-C, alkyl, C,-C~ haloalkyl, or C,-C~ alkoxy.
Preferred embodiments of this invention are compounds of formulae IE and IF,
wherein:
R' and R' are both halogen.
Preferred embodiments of this invention are compounds of formulae IA, IB, IC,
ID and IF,
wherein:
Rs, Rfi and R' are each H, halogen, -OR~~, -NR"R'~, C,-C~ alkyl or C,-C~ alkyl
optionally substituted with
from 1 to 3 substituents selected independently at each occurrence thereof
from C,-C; alkyl, halogen,
aryl, -CN, -OR' and -NRyR"~.
Preferred embodiments of this invention are compounds of formulae IA, IB, IC,
ID, IE and IF,
wherein:
-15-
WO 01/32624 CA 02389300 2002-04-29 PCT/US00/30328
R5 is fluoro, chloro or methyl;
one of R6 or R' is H; and the other of R~ or R' which is not H is halogen, -
OR", -NR"R'2, C,-C
alkyl or C,-C6 alkyl each of which is optionally substituted with from 1 to 3
substituents selected
independently at each occurrence thereof from C,-C, alkyl, halogen, aryl, -CN,
-ORy and -
NRvR'°.
Preferred embodiments of this invention are compounds of formulae IA, IB, IC,
ID and IE,
wherein:
R" is H or halogen.
Preferred embodiments of this invention are compounds of formula IF, wherein:
RR is halogen.
Preferred embodiments of this invention are compounds of formulae IA, IB, IC,
ID, IE and IF,
wherein:
the substituents R'-R~ are as set forth in the following table B:
-16-
WO 01/32624 CA 02389300 2002-04-29 pCT~S00/30328
Table B
IA IB IC ID IE IF
R1 C,-C; alkyl
R2 H, C,-C4
alkyl or
C,-C~ haloalkyl
R3 C,-C6 alkyl,-O(phenyl)H; or,
C; alternatively,
C,-C~
alkyl,
C~-Ch
cycloalkyl
or C4-
C~ cycloalkylor - C~ cycloalkyl-alkyl,
or each
optionally
substituted,
or -
CQ C~ O(benzyl),O(phenyl)
or -O(benzyl),
each
optionally
substituted
cycloalkyl-alkyl,each
each optionallyoptionally
substituted substituted
R4 H; or, alternatively, C,-C; -O(phenyl)H; or,
C,-CQ alkyl, alkyl, alternatively,
C,-C
C; C~ cycloalkyl C~-C~ or - alkyl,
or C4 C~ C,-Cfi
cycloalkyl
or
cycloalkyl-alkyl, cycloalkylO(benzyl),C~ C~
each cycloalkyl-alkyl,
optionally or C4 each each optionally
substituted, C~ substituted,
-
NR11R12; cycloalkyl-optionallyNR11R12;
or, -(O)phenyl or, -(O)phenyl
or -
O(benzyl), alkyl, substitutedor -O(benzyl),
each optianally each each
substituted optionally optionally
substituted
substituted
R5 H, halogen, F> Cl, See RS
-OR 11, Me for
-NR 11 R
12> C 1
_C6 alkyl
or C 1-C6
alkyl
optionally IA-ID
substituted
R6 R~ H, halogen, one is See
-OR 1 l H and 6, R~
, -NR 11
R 12 C 1
_C6 alkyl
or C 1-C6
alkyl
R
optionally the otherfor IA-ID
substituted is
halogen,
-
OR11,
_
NR11R12>
C1-C6
alkyl
or C1-C6
alkyl
optionally
substituted
R8 H, halogen, halogen
-OR"
-17-
WO 01/32624 CA 02389300 2002-04-29 pCT/iJS00/30328
More preferred embodiments of this invention are compounds wherein:
R' is C,-C, alkyl;
R~ is H or C,-C, alkyl;
R' is H, C,-C, alkyl, -O(phenyl) or optionally substituted -O(phenyl), more
preferably halogen;
R° is H, C,-C~ alkyl, -O(phenyl) or optionally substituted -O(phenyl),
more preferably halogen;
RS is F, Cl or Me, more preferably -OR", wherein R" is C,-C, alkyl;
R~ is H or more preferably Cl, F, C,-C~ alkyl, halo-substituted C,-C, alkyl,
or -OR",
R" is C,-C, alkyl or -NR"R'-;
R' is H or more preferably Cl, F, C,-Cz alkyl or -OR", wherein R'~ is C,-Cz
alkyl.
A further more preferred embodiments of this invention are compounds wherein:
R' is CHz;
R~ is H or CH,;
R' is H, CH;, or -O(phenyl) or -O-CH~-(phenyl), each of said -O(phenyl) or -O-
CHI (phenyl) is
optionally substituted from 1 to 3 times with a substituent selected
independently at each occurrence
thereof from halogen, cyano, C,-C~ alkyl, C,-C~ haloalkyl, or C,-C, alkoxy;
R' is H, F, CH,, CH_CH~, CH_CH,CH,, CH=CH(CH,)CH,, -O(phenyl) or -O-CH_-
phenyl, where
each of said -O(phenyl) or -O-CH_-(phenyl) is optionally substituted from 1 to
3 times with a substituent
selected independently at each occurrence thereof from halogen, cyano, C~-Ca
alkyl, C~-C~ haloalkyl, or
C,-C~ alkoxy;
R' is H, CH,, OCH" F or C1;
R° is H, CH" -OCH,, F, Cl or CF,;
R' is H, F, Cl, CH. or OCH,; and
R~ is halogen.
_18_
WO 01/32624 CA 02389300 2002-04-29 PCT/~JS00/30328
A further more preferred embodiments of this invention are compounds of
formulae IA-IF,
wherein:
R'-R8 are as follows:
Table C
R1 R2 R3 R4 RS R6 R7 R8
Me H H Me H H H H
Me H H Me H OMe H H
Me H H Me H F H H
Me H H Me F H H H
Me H H Me F F H H
Me H H Me Me F H H
Me H H Me Cl F H H
Me H H Me Cl H H H
Me H H Me H Me H H
Me H H Me F Me H H
Me H H Me H C1 H H
Me H H Me F Cl H H
Me H H Me Cl Cl H H
Me H H Et H H H H
Me H H Et F F H H
Me H H F H OMe H H
Me H H F F OMe H H
Me H H F F Me H H
Me H H F F CI H H
Me H H F F F H H
Me H H F CI H H H
Me H H CN H H H H
Me H H CF3 H H H H
Me Me H Me H H H H
Me Me H H H C1 H H
Me Me H H F F H H
Me H Me Me H H H H
-19-
WO 01/32624 CA 02389300 2002-04-29 PCT/US00/30328
Table C, (continued)
R1 R2 R3 R4 RS R6 R7 R8
Me H F Me H H H H
Me H Me F H H H H
Me H OMe Me H H H H
Me H OH Me H H H H
Me H H OCF3 H H H H
Me H H OMe F F H H
Me H H OMe Me F H H
Me H H OMe F Me H H
Me H H OMe Me H H H
Me H H O(Ph) H H H H
Me H H O(4-OMePh) H H H H
Me H H O(CH2Ph) H H H H
Me H H OH Me H H H
Me H H OH F Me H H
Me H H OH Me F H H
Me H H OH F F H H
Me H H H CN H H H
Me H Me H H H H H
Me H Me H H F H H
Me H Me H F F H H
Me H Me H F H F H
Me H Me H F H H H
Me H Me H Me F H H
Me H Me H CI F H H
Me H Me H Cl Cl H H
Me H Me H CI H H H
Me H Me H H Cl H H
Me H Me H F C1 H H
Me H Me H H OMe H H
Me H Me H H CN H H
Me H Me H H CF3 H H
Me H Me H H Me H H
Me H CH,NHMe H H H H H
Me H CH20H H H H H H
Me H SO~NH2 H H H H H
Me H SO~NHMe H H H H H
Me H OMe H H Me H H
Me H OMe H F H F H
Me H OMe H Cl H H H
Me H OMe H CI Cl H H
Me H OMe H F C1 H H
Me H OMe H CI F H H
Me H H H F H F H
Me H H H F H CI H
Me H H Me F H F H
-20-
W~ 01/32624 CA 02389300 2002-04-29 pCT/US00/30328
Table C (continued)
R1 R2 R3 R4 RS R6 R7 R8
Me H H Me F H C1 H
Me H H H F F F H
Me H H H F H H H
Me H H H F Me H H
Me H H H Me F H H
Me H H H F F H H
Me H H H Cl H H H
Me H H H F Cl H H
Me H H H Cl F H H
Me H H H CN H H H
Me H H H H NHCOMe H H
Me H H H H Cl H F
Me Me H Me F H F H
Me H H Me F F F H
Et H H Me H F H H
Me H H Me H F H OH
Me H F CH,Me H H H H
Me H H CH~NHzH H H H
Me H H CH,NHMe H H H
H
~
Me H OH CN H H H
H
Me H H CH20H H H H H
Et H H H H H H H
That is, the specifically preferred compounds are:
2,7-dimethyl-4-phenyl-1,2,3,4-tetrahydroisoquinoline;
4-(4-methoxy)phenyl-2,7-dimethyl-1,2,3,4-tetrahydroisoquinoline;
2,7-dimethyl-4-(4-fluoro)phenyl-1,2,3,4-tetrahydroisoquinoline;
2,7-dimethyl-4-(3-fluoro)phenyl-1,2,3,4-tetrahydroisoquinoline;
4-(3,4-difluoro)phenyl-2,7-dimethyl-1,2,3,4-tetrahydroisoquinoline;
2,7-dimethyl-4-(4-fluoro-3-methyl)phenyl-1,2,3,4-tetrahydroisoquinoline;
4-(3-chloro-4-fluoro)phenyl-2,7-dimethyl-1,2,3,4-tetrahydroisoquinoline;
4-(3-chloro)phenyl-2,7-dimethyl-1,2,3,4-tetrahydroisoquinoline;
2,7-dimethyl-4-(4-methyl)phenyl-1,2,3,4-tetrahydroisoquinoline;
2,7-dimethyl-4-(3-fluoro-4-methyl)phenyl-1,2,3,4-tetrahydroisoquinoline;
4-(4-chloro)phenyl-2,7-dimethyl-1,2,3,4-tetrahydroisoquinoline;
4-(4-chloro-3-fluoro)phenyl-2,7-dimethyl-1,2,3,4-tetrahydroisoquinoline;
4-(3,4-dichloro)phenyl-2,7-dimethyl-1,2,3,4-tetrahydroisoquinoline;
7-ethyl-2-methyl-4-phenyl-1,2,3,4-tetrahydroisoquinoline;
4-(3,4-difluoro)phenyl-7-ethyl-2-methyl-1,2,3,4-tetrahydroisoquinoline;
-21-
WO 01/32624 CA 02389300 2002-04-29 PCT/LTS00/30328
7-fluoro-4-(4-methoxy)phenyl-2-methyl-1,2,3,4-tetrahydroisoquinoline;
7-fluoro-4-(3-fluoro-4-methoxy)phenyl-2-methyl-1,2,3,4-tetrahydroisoquinoline;
7-fluoro-4-(3-fluoro-4-methyl)phenyl-2-methyl-1,2,3,4-tetrahydroisoquinoline;
7-fluoro-4-(4-chloro-3-fluoro)phenyl-2-methyl-1,2,3,4-tetrahydroisoquinoline;
4-(3,4-difluoro)phenyl-7-fluoro-2-methyl-1,2,3,4-tetrahydroisoquinoline;
4-(3-chloro)phenyl-7-fluoro-2-methyl-1,2,3,4-tetrahydroisoquinoline;
7-cyano-2-methyl-4-phenyl-1,2,3,4-tetrahydroisoquinoline;
2-methyl-4-phenyl-7-trifluoromethyl-1,2,3,4-tetrahydroisoquinoline;
4-phenyl-1,2,7-trimethyl-1,2,3,4-tetrahydroisoquinoline;
4-(4-chloro)phenyl-1,2-dimethyl-1,2,3,4-tetrahydroisoquinoline;
4-(3,4-difluoro)phenyl-1,2-dimethyl-1,2,3,4-tetrahydroisoquinoline;
4-phenyl-2,7,8-trifluoromethyl-1,2,3,4-tetrahydroisoquinoline;
2,7-dimethyl-8-fluoro-4-phenyl-1,2,3,4-tetrahydroisoquinoline;
2,8-dimethyl-7-fluoro-4-phenyl-1,2,3,4-tetrahydroisoquinoline;
2,7-dimethyl-8-methoxy-4-phenyl-1,2,3,4-tetrahydroisoquinoline;
2,7-dimethyl-8-hydroxy-4-phenyl-1,2,3,4-tetrahydroisoquinoline;
2-methyl-4-phenyl-7-trifluoromethoxy-1,2,3,4-tetrahydroi soquinoline;
4-(3,4-difluoro)phenyl-7-methoxy-2-methyl-1,2,3,4-tetrahydroisoquinoline;
4-(4-fluoro-3-methyl)phenyl-7-methoxy-2-methyl-1,2,3,4-tetrahydroisoquinoline;
4-(3-fluoro-4-methyl)phenyl-7-methoxy-2-methyl-1,2,3,4-tetrahydroisoquinoline;
7-methoxy-4-(3-methyl)phenyl-2-methyl-1,2,3,4-tetrahydroisoquinoline;
2-methyl-7-phenoxy-4-phenyl-1,2,3,4-tetrahydroisoquinoline;
7-(4-methoxy)phenoxy-2-methy I-4-pheny I-1,2,3,4-tetrahydroisoqui nol ine;
7-benzyloxy-2-methyl-4-phenyl-1,2,3,4-tetrahydroisoquinoline;
7-hydroxy-2-methyl-4-(3-methyl)phenyl-1,2,3,4-tetrahydroisoquinoline;
4-(3-fluoro-4-methyl)phenyl-7-hydroxy-2-methyl-1,2,3,4-tetrahydroisoquinoline;
4-(4-fluoro-3-methyl)phenyl-7-hydroxy-2-methyl-1,2,3,4-tetrahydroisoquinoline;
4-(3,4-difluoro)phenyl-7-hydroxy-2-methyl- I ,2,3,4-tetrahydroisoquinol ine;
4-(3-cyano)phenyl-2-methyl-1,2,3,4-tetrahydroisoquinoline;
2,8-dimethyl-4-phenyl-1,2,3,4-tetrahydroisoquinoline;
2,8-dimethyl-4-(4-fluoro)phenyl-1,2,3,4-tetrahydroisoquinoline;
4-(3,4-difluoro)phenyl-2,8-dimethyl- I ,2,3,4-tetrahydroisoquinol ine;
4-(3,5-dif7uoro)phenyl-2,8-dimethyl-1,2,3,4-tetrahydroisoquinoline;
2,8-dimethyl-4-(3-fluoro)phenyl-1,2,3,4-tetrahydroisoquinoline;
2,8-dimethyl-4-(4-fluoro-3-methyl)phenyl-1,2.3,4-tetrahydroisoquinoline;
-22-
WO 01/32624 CA 02389300 2002-04-29 pCT/US00/30328
4-(3-chloro-4-fluoro)phenyl-2,8-dimethyl-1,2,3,4-tetrahydroisoquinoline;
4-(3,4-dichloro)phenyl-2,8-dimethyl-1,2,3,4-tetrahydroisoquinoline;
4-(3-chloro)phenyl-2,8-dimethyl-1,2,3,4-tetrahydroisoquinoline;
4-(4-chloro)phenyl-2, 8-dimethyl-1,2,3,4-tetrahydroisoquinoline;
4-(4-chloro-3-fluoro)phenyl-2,8-dimethyl-1,2,3,4-tetrahydroisoquinoline;
2,8-dimethyl-4-(4-methoxy)phenyl-1,2,3,4-tetrahydroisoquinoline;
4-(4-cyano)phenyl-2,8-dimethyl- I ,2,3,4-tetrahydroisoquinoline;
2,8-dimethyl-4-(4-trifluoromethyl)phenyl-1,2,3,4-tetrahydroisoquinoline;
2,8-dimethyl-4-(4-methyl)phenyl-1,2,3,4-tetrahydroisoquinoline;
2-methyl-8-(N-methylamino)methyl-4-phenyl-1,2,3,4-tetrahydroisoquinoline;
8-(hydroxy)methyl-2-methyl-4-phenyl-1,2,3,4-tetrahydroi soquinoline;
2-methyl-4-phenyl-8-sulfonamide-1,2,3,4-tetrahydroisoquinoline;
2-methyl-8-(N-methyl)sulfonamide-4-phenyl-1,2,3,4-tetrahydroisoquinoline;
8-methoxy-2-methyl-4-(4-methyl)phenyl-1,2,3,4-tetrahydroisoquinoline;
4-(3,5-difluoro)phenyl-8-methoxy-2-methyl-1,2,3,4-tetrahydroisoquinoline;
4-(3-chloro)phenyl-8-methoxy-2-methyl-1,2,3,4-tetrahydroisoquinoline;
4-(3,4-dichloro)phenyl-8-methoxy-2-methyl-1,2,3,4-tetrahydroisoquinoline;
4-(4-chloro-3-fluoro)phenyl-8-methoxy-2-methyl-1,2,3,4-tetrahydroisoquinoline;
4-(3-chloro-4-fluoro)phenyl-8-methoxy-2-methyl-1,2,3,4-tetrahydroisoquinoline;
4-(3,5-difluoro)phenyl-2-methyl-1,2,3,4-tetrahydroisoquinoline;
4-(3-chloro-5-fluoro)phenyl-2-methyl-1,2,3,4-tetrahydroisoquinoline;
4-(3,5-difluoro)phenyl-2,7-dimethyl-1,2,3,4-tetrahydroisoquinoline;
4-(3-chloro-5-fluoro)phenyl-2,7-dimethyl-1,2,3,4-tetrahydroisoquinoline;
2-methyl-4-(3,4,5-trifluoro)phenyl-1,2,3,4-tetrahydroisoquinoline;
4-(3-fluoro)phenyl-2-methyl-1,2,3,4-tetrahydroisoquinoline;
4-(3-fluoro-4-methyl)phenyl-2-methyl-1,2,3,4-tetrahydroisoquinoline;
4-(4-fluoro-3-methyl)phenyl-2-methyl-1,2,3,4-tetrahydroisoquinoline;
4-(3,4-difluoro)phenyl-2-methyl-1,2,3,4-tetrahydroisoquinoline;
4-(3-chloro)phenyl-2-methyl-1,2,3,4-tetrahydroisoquinoline;
4-(4-chloro-3-fluoro)phenyl-2-methyl-1,2,3,4-tetrahydroisoquinoline;
4-(3-chloro-4-fluoro)phenyl-2-methyl-1,2,3,4-tetrahydroisoquinoline;
4-(3-cyano)phenyl-2-methyl-1,2,3,4-tetrahydroisoquinoline;
4-(4-acetanilide)-2-methyl-1,2,3,4-tetrahydroisoquinoline;
4-(4-chloro)phenyl-4-fluoro-2-methyl-1,2,3,4-tetrahydroisoquinoline;
(3,5-difluoro)-4-phenyl-1,2,7-trimethyl-1,2,3.4-tetrahydroisoquinoline;
-23-
WO 01/32624 _ CA 02389300 2002-04-29 PCT/US00/30328
(8-fluoro-2-methyl-4-phenyl-1,2,3,4-tetrahydro-7-isoquinolinyl)-N-
methylmethanamine;
(2-methyl-4-phenyl-7-isoquinolinyl)-N-methylmethanamine;
N-methyl(2-methyl-4-phenyl-7-isoquinolinyl)-N-methylmethanamine;
8-hydroxy-2-methyl-4-phenyl-1,2,3,4-tetrahydro-7-isoquinolinecarbonitrile;
(2-methyl-4-phenyl-1,2,3,4-tetrahydro-7-isoquinolinyl)methanol; and
2-ethyl-4-phenyl-1,2,3,4-tetrahydroisoquinoline; or
an oxide thereof, a pharmaceutically acceptable salt thereof, a solvate
thereof, or prodrug thereof.
Further more preferred compound of this invention include those (+)
enantiomers of compounds
of formulae IA-IF, selected from table D:
TABLE D
R3
R2 \ Ra
H O
*-_
OH
R1 ~ N~ OH
O
D
Ex. R R2 R3 R4 Chiral Technologies%IPA Peak Mp ~oC)
1 C olumn exanes Order
H in
I H H Me F Chiralcel~ OD 10 1st 190.0-190.5
2 OM H F F Chiralpak~ AD 10 2nd 160.0-163.5
a
3 Me H F F Chiralpak~ AD 2.5 2nd 136.0-138.0
4 H H Cl F Chiralcel~ OD 10 1st 171.0-172.0
5 H H F F Chiralcel~ OD 10 1st 138.0-139.0
6 Me F H F Chiral ak~AD 10 2nd 174.0-175.0
7 Me H F H Chiral ak~ AD 10 2nd 144.5-146.0
8 Me H H F Chiralpak~ AD ~ 10 2nd ~ 172.0-173.5
Another preferred aspect of the invention is a mixture of compounds of
formulae (IA-F)
wherein the compound of formulae (IA-F) is radiolabeled, i.e., wherein one or
more of the atoms
described are replaced by a radioactive isotope of that atom (e.g., C replaced
by "C and H replaced by
-24-
W~ 01/32624 CA 02389300 2002-04-29
PCT/US00/30328
;H). Such compounds have a variety of potential uses, e.g., as standards and
reagents in determining the
ability of a potential pharmaceutical to bind to neurotransmitter proteins.
Another aspect of the invention is a therapeutically effective amount of the
compound of
formulae (IA-F) and a pharmaceutically acceptable carrier.
Another aspect of the invention is a method of treating a disorder which is
created by or is
dependent upon decreased availability of serotonin, norepinephrine or
dopamine, which comprises
administering to a patient in need of such treatment a therapeutically
effective amount of a compound of
formulae (IA-F), or a pharmaceutically acceptable salt thereof.
Another aspect of the invention is a method of treating a disorder which is
created by or is
dependent upon decreased availability of serotonin, norepinephrine or
dopamine, which comprises
administering to a patient in need of such treatment a therapeutically
effective amount of a compound of
formulae (IA-F), or a pharmaceutically acceptable salt thereof and a
therapeutically effective amount of
a serotonin 1A receptor antagonist, or pharmaceutically acceptable salt
thereof.
Another aspect of the invention is a method of treating a disorder which is
created by or is
dependent upon decreased availability of serotonin, norepinephrine or
dopamine, which comprises
administering to a patient in need of such treatment a therapeutically
effective amount of a compound of
formulae (IA-F), or a pharmaceutically acceptable salt thereof and a
therapeutically effective amount of
a compound selected from the group consisting of WAY 100135 and spiperone, or
pharmaceutically
acceptable salt thereof.
WAY 100135 (N-(t-butyl)-3-[a-(2-methoxyphenyl)piperazin-1-yl]-2-
phenylpropanamide) is
disclosed in Abou-Gharbia et al., U.S. Pat. No. 4,988,814,as having an
affinity for the 5-HT~~ receptor.
Also, Cliffe et al., J. Med. Chem. 36, 1509-10 ( 1993) showed that the
compound is a 5-HT,~ antagonist.
Spiperone (8-[4-(4-fluorophenyl)-4-oxobutyl]-1-phenyl-1,3,8-
triazaspiro[4,5]decan-4- one) is a well-
known compound, and is diclosed in U.S. Pat. Nos. 3,155,669 and 3, I 55,670.
The activity of Spiperone
as a 5-HT~~ antagonist is shown in Middlemiss et al., Neurosci. and Biobehav.
Rev. 16, 75-82 (1992).
Another aspect of the invention is a method of treating a disorder which is
created by or is
dependent upon decreased availability of serotonin, norepinephrine or
dopamine, which comprises
administering to a patient in need of such treatment a therapeutically
effective amount of a compound of
formulae (IA-F) , or a pharmaceutically acceptable salt thereof and a
therapeutically effective amount of
a selective neurokinin-1 receptor antagonist, or pharmaceutically acceptable
salt thereof.
Neurokinin-1 receptor antagonists of use in combination a compound of formulae
(IA-F) in the
present invention, are fully described, for example, in U.S. Pat. Nos.
5,373,003, 5,387,595, 5,459,270,
5,494,926, 5,162,339, 5,232,929, 5,242,930, 5,496,833, 5,637,699; PCT
International Patent Publication
Nos. WO 90/05525, 90/05729, 94/02461, 94/02595, 94/03429,94/03445, 94/04494,
94/04496, 94/05625,
94/07843. 94/08997. 94/10165, 94/10167, 94/10168, 94/10170, 94/11368,
94/13639, 94/13663,
-25-
WO 01/32624 CA 02389300 2002-04-29 PCT/US00/30328
94/14767,94/15903, 94/19320, 94/19323, 94/20500, 91/09844, 91/18899, 92/01688,
92/06079,
92/12151,92/15585, 92/17449, 92/20661, 92/20676, 92/21677, 92/22569, 93/00330,
93/00331, 93/01159,
93/01165, 93/01169, 93/01170, 93/06099, 93/09116,93/10073, 93/14084, 93/14113,
93/18023, 93/19064,
93/21155, 93/21181, 93/23380, 93/24465, 94/00440, 94/01402, 94/26735,
94/26740, 94/29309,
95/02595, 95/04040, 95/04042, 95/06645, 95/07886, 95/07908, 95/08549,95/11880,
95/14017, 95/15311,
95/16679, 95/17382, 95/18124, 95/18129, 95/19344, 95/20575, 95/21819,
95/22525, 95/23798,
95/26338, 95/28418, 95/30674, 95/30687, 95/33744, 96/05181, 96/05193,
96/05203, 96/06094,
96/07649, 96/10562, 96/16939, 96/18643, 96/20197, 96/21661, 96/29304,96/29317,
96/29326, 96/29328,
96/31214, 96/32385, 96/37489, 97/01553, 97/01554, 97/03066, 97/08144,
97/14671, 97/17362,
97/18206, 97/19084,97/19942, 97/21702, and 97/49710; and in U.K. Patent
Application Nos. 2 266 529,
2 268 931, 2 269 170, 2 269 590, 2 271 774, 2 292 144, 2 293168, 2 293 169,
and 2 302 689; European
Patent Publication Nos. EP 0 360 390, 0517 589, 0 520 555, 0 522 808, 0 528
495, 0 532 456, 0 533 280,
0 536 817, 0 545 478, 0 558 156, 0 577 394, 0 585 913, 0 590 152, 0 599 538,
0610 793, 0 634 402, 0
686 629, 0 693 489, 0 694 535. 0 699 655, 0 394 989, 0 428 434, 0 429 366, 0
430 771, 0 436 334, 0 443
132, 0 482 539, 0 498 069, 0 499 313, 0 512 901, 0 512 902, 0 514 273, 0 514
274, 0 514 275, 0 514 276,
0 515 681, 0 699 674, 0 707 006, 0 708 1 O 1, 0 709 375, 0 709 376, 0 714 891,
0 723 959, 0733 632 and 0
776 893. The preparation of such compounds are fully described in the
aforementioned patents and
publications.
Another aspect of the invention is a method of treating a disorder which is
created by or is
dependent upon decreased availability of serotonin, norepinephrine or
dopamine, which comprises
administering to a patient in need of such treatment a therapeutically
effective amount of a compound of
formulae (IA-F) , or a pharmaceutically acceptable salt thereof and a
therapeutically effective amount of
a norepinephrine precursor, or pharmaceutically acceptable salt thereof.
Another aspect of the invention is a method of treating a disorder which is
created by or is
dependent upon decreased availability of serotonin, norepinephrine or
dopamine, which comprises
administering to a patient in need of such treatment a therapeutically
effective amount of a compound of
formulae (IA-F) , or a pharmaceutically acceptable salt thereof and a
therapeutically effective amount of
a compound selected from L-tyrosine and L-phe.nylalanine, or pharmaceutically
acceptable salt thereof.
Another aspect of the invention is a method of treating a disorder referred to
in the above-
mentioned embodiments, wherein the disorder is selected from the group:
attention deficit disorder,
hyperactivity disorder, anxiety, depression, post-traumatic stress disorder,
supranuclear palsy, eating
disorders, obsessive compulsive disorder, analgesia, nicotine addiction, panic
attacks, Parkinsonism and
phobia, obesity, late luteal phase syndrome or narcolepsy, cocaine addiction,
amphetamine addiction,
and psychiatric symptoms anger such as, rejection sensitivity, and lack of
mental or physical energy.
-26-
WO 01/32624 CA 02389300 2002-04-29 PCT/US00/30328
Another aspect of the invention is a method of inhibiting synaptic
norepinephrine uptake in a
patient in need thereof comprising administering a therapeutically effective
inhibitory amount of a
compound of formulae (IA-F) .
Another aspect of the invention is a method of inhibiting synaptic serotonin
uptake in a patient in
need thereof comprising administering a therapeutically effective inhibitory
amount of a compound of
formulae (IA-F) .
Another aspect of the invention is a method of inhibiting synaptic dopamine
uptake in a patient
in need thereof comprising administering a therapeutically effective
inhibitory amount of a compound of
formulae (IA-F) .
Another aspect of the invention is a therapeutic method described herein
wherein the (+)-
stereoisomer of the compound of formulae (IA-F) is employed.
Another aspect of the invention is a therapeutic method described herein
wherein the (-)-
stereoisomer of the compound of formulae (IA-F) is employed.
Another aspect of the invention is a kit comprising a compound of formulae (IA-
F) and at least
one compound selected from the group consisting of: a serotonin 1 A receptor
antagonist compound, a
selective neurokinin-1 receptor antagonist compound, and a norepinephrine
precursor compound.
Another aspect of the invention is a method of treating depression in a
patient in need thereof
comprising inhibiting synaptic serotonin and norepinephrine uptake by
administering a therapeutically
effective inhibitory amount of a compound of formulae (IA-F) which functions
as both a serotonin and
norepinephrine uptake inhibitor.
Another aspect of the invention is a method of treating depression in a
patient in need thereof
comprising inhibiting synaptic serotonin and dopamine uptake by administering
a therapeutically
effective inhibitory amount of a compound of formulae (IA-F) which functions
as both a serotonin and
dopamine uptake inhibitor.
Another aspect of the invention is a method of treating depression in a
patient in need thereof
comprising inhibiting synaptic dopamine and norepinephrine uptake by
administering a therapeutically
effective inhibitory amount of a compound of formulae (IA-F) which functions
as both a dopamine and
norepinephrine uptake inhibitor.
Another aspect of the invention is a method for inhibiting serotonin uptake in
mammals which
comprises administering to a mammal requiring increased neurotransmission of
serotonin a
pharmaceutically effective amount of a compound of formulae (IA-F) .
Another aspect of the invention is a method for inhibiting dopamine uptake in
patients which
comprises administering to a mammal requiring increased neurotransmission of
dopamine a
pharmaceutically effective amount of a compound of formulae (IA-F) .
-27-
WO 01/32624 CA 02389300 2002-04-29 PCT/US00/30328
Another aspect of the invention is a method for inhibiting norepinephrine
uptake in patients
which comprises administering to a mammal requiring increased
neurotransmission of norepinephrine a
pharmaceutically effective amount of a compound of formulae (IA-F) .
Another aspect of the invention is a method of suppressing the desire of
humans to smoke
comprising administering to a human in need of such suppression an effective
dose, to relieve the desire
to smoke, of a compound of formulae (IA-F) .
Another aspect of the invention is a method of suppressing the desire of
humans to consume
alcohol comprising administering to a human in need of such suppression an
effective dose, to relieve
the desire to consume alcohol, of a compound of formulae (IA-F) .
It is appreciated that certain feactures of the invention, which are, for
clarity, described in the
context of separate embodiments, may also be provided in combination in a
single embodiment.
Conversely, various feactures of the invention which are, for brevity,
described in the context of a single
embodiment, may also be provided seperately or in any suitable subcombination.
Preparation of Compounds of the Invention
Compounds according to the invention, for example, starting materials,
intermediates or
products, are prepared as described herein or by the application or adaptation
of known methods, by
which is meant methods used heretofore or described in the literature.
Compounds useful according to the invention may be prepared by the application
or adaptation
of known methods, by which is meant methods used heretofore or described in
the literature, for example
those described by R. C. Larock in Comprehensive Organic Transformations, VCH
publishers, 1989.
A compound of formulae (IA-F) including a group containing one or more
nitrogen ring atoms.
may be converted to the corresponding compound wherein one or more nitrogen
ring atom of the group
is oxidized to an N-oxide, preferably by reacting with a peracid, for example
peracetic acid in acetic acid
or m-chloroperoxybenzoic acid in an inert solvent such as dichloromethane, at
a temperature from about
room temperature to reflux, preferably at elevated temperature.
In the reactions described hereinafter it may be necessary to protect reactive
functional groups,
for example hydroxy, amino, imino, thio or carboxy groups, where these are
desired in the final product,
to avoid their unwanted participation in the reactions. Conventional
protecting groups may be used in
accordance with standard practice, for examples see T.W. Green and P.G.M.Wuts
in "Protective Groups
in Organic Chemistry" John Wiley and Sons, 1991; J. F. W. McOmie in
"Protective Groups in Organic
Chemistry" Plenum Press, 1973.
Compounds provided herein are synthesized, for example, using the methods
described below
(see Schemes 1-4), together with methods known in the art of synthetic organic
chemistry, or variations
-28-
WO 01/32624 CA 02389300 2002-04-29 PCT/US00/30328
thereon as appreciated by those skilled in the art. Preferred methods include,
but are not limited to, those
methods described below.
Compounds of formulae (IA-F) of this invention are, for example, prepared
according to Scheme
1. Treatment of an optionally substituted acetophenone of formula (II) with
common brominating agents
such as, but not limited to, bromine, NBS, or tetrabutylammonium tribromide
readily affords the desired
bromoacetophenones of formula (III, X=Br). These reactions are optimally
conducted in acetic acid or
methylene chloride with methanol used as a co-solvent for the tribromide
reagent with reaction
temperatures at or below room temperature. Another embodiment of this
methodology would include
compounds of formula (III, X=Cl).
The acetophenones of formula (II) are available from commercial sources or are
conveniently
obtained via several well known methods, including the treatment of the
corresponding benzoic acid
intermediates with two stoichiometric equivalents of methyllithium as
thoroughly described in the
review of Jorgenson, M.J. (Organic Reactions, 1970, I8, pg. 1 ).
Alternatively, one may treat the
corresponding benzaldehydes with an alkyl-Grignard (for example, MeMgBr) or
alkyl-lithium (for
example, MeLi) nucleophile follwed by routine oxidation to the ketone as well
demonstrated by Larock,
R.C. (Comprehensive Organic Transformations, VCH Publishers, New York, 1989,
p. 604).
Treatment of intermediates of formula (III) with intermediates of formula
(R3,R4-Ph)-CH(R2)-
NHRI cleanly generates the alkylation products of formula (V). The alkylation
reactions may be run
under a wide variety of conditions familiar to one skilled in the art of
organic synthesis. Typical solvents
include acetonitrile, toluene, diethyl ether, tetrahydrofuran,
dimethylsulfoxide, dimethylformamide,
methylene chloride, and lower alkyl alcohols including ethanol. The reactions
may be successfully run
at temperatures ranging from 0oC up to the boiling point of the solvent
employed. Reaction progress is
conventionally determined by standard chromatographic and spectroscopic
methods. The alkylation
reaction is optionally run with the addition of a non-nucleophilic organic
base such as, but not limited to,
pyridine, triethylamine and diisopropyl ethylamine.
The R1-substituted N-benzyl amines of formula (R3,R4-Ph)-CH(R2)-NHRI may be
purchased
from commercial sources, or alternatively, obtained from a simple reductive
amination protocol. Thus,
carbonyl containing compounds of Formulae (IV, Scheme 1 ) may be treated with
HEN-R 1 in lower alkyl
alcoholic solvents (preferably methanol) at temperatures at or below room
temperature. The resulting
imine may be reduced most commonly with alkaline earth borohydrides
(preferably sodium borohydride)
to provide the desired amine intermediate.
Reductions of compounds of formula (V) to the benzyl alcohols of formula (VI)
proceeds with
many reducing agents including, as example, sodium borohydride, lithium
borohydride, borane,
diisobutylaluminum hydride. and lithium aluminum hydride. The reductions are
carried out for a period
-29-
WO 01/32624 CA 02389300 2002-04-29 PCT/US00/30328
of time between 1 hour to 3 days at room temperature or elevated temperature
up to the reflux point of
the solvent employed. If borane is used, it may be employed as a complex for
example, but not limited
to, borane-methyl sulfide complex, borane-piperidine complex, borane-
tetrahydrofuran complex. One
skilled in the art will understand the optimal combination of reducing agents
and reaction conditions
needed or may seek guidance from the text of Larock, R.C. (Comprehensive
Organic Transformations,
VCH Publishers, New York, 1989, p. 527).
Compounds of formula (VI) may be cyclized to the target compounds of formulae
IA-IF of this
invention by brief treatment with a strong acid. Suitable acids include, but
are not limited to,
concentrated sulfuric acid, polyphosphoric acid, methanesulfonic acid and
trifluoroacetic acid. The
reactions are run neat or in the optional presence of a co-solvent such as,
for example, methylene
chloride and 1,2-dichloroethane. The cyclizations may be conducted at
temperatures ranging from 0°C
up to the reflux point of the solvent employed. One skilled in the art of
heterocyclic chemistry will
readily understand these conditions or may consult the teachings of Mondeshka,
et al. (1l Farmaco,
1994, 49, 475-480) or Venkov, et al. (Synthesis, 1990, 253-255). Cyclizations
may also be effected by
treatment of compounds of formula (VI) with strong Lewis Acids, such as for
example, aluminum
trichloride typically in halogenated solvents such as methylene chloride. One
skilled in the art will be
familiar with the precedent taught by Kaiser, et al. (J. Med. Chem., 1984, 27,
28-35) and Wyrick, et al.
(J. Med. Chem., 1981, 24, 1013-1015).
Compounds of formulae IA-IF may be obtained in enantiomerically pure (R) and
(S) form by
crystallization with chiral salts as well known to one skilled in the art, or
alternatively, may be isolated
through chiral HPLC employing commercially available chiral columns.
Alternatively, compounds of formulae (V) and (VI) may be arrived at as
described in Scheme 2.
Thus, the haloacetophenones of formula may be treated with simple amines of
formula H2N-R1 under
alkylation conditions as described above (vide supra) to provide compounds of
formulae (VII). A
second alkylation may then be performed utilizing reagents of formula (VIII)
where X represents a
leaving group, such as for example, but not limited to, halogen, mesylate, or
tosylate to afford the
common intermediate of formula (V). Reagents of formula (VIII) are in turn
available from the
appropriately substituted carbonyl compound of formula (IV) via reduction
(vide supra) and activation.
Activation to leaving group X is effected by treatment of the alcohol with
methanesulfonyl
chloride or p-toluenesulfonyl chloride in the presence of a non-nucleophilic
base such as, but not limited
to, 1,5-diazabicyclo[4.3.0)non-5-ene (DBN), pyridine or triethylamine. The
reaction is commonly
performed in halogenated organic solvent, for example, methylene chloride, and
at temperatures from -
78oC up to the boiling point of the solvent employed. Benzylic activation to
Leaving Group X may also
be effected by treatment with halogenating agents such as, but not limited to,
S02C12, C12, PCIS, Br2,
-30-
WO 01/32624 CA 02389300 2002-04-29 pCT/LTS00/30328
CuBr2, NBS, and CBr4. The various conditions necessary to accomplish this
transformation will be
readily apparent to those skilled in the art of organic chemistry and
additional reference on benzylic
activiation may be sought from Larock, R.C. (Comprehensive Organic
Transformations, VCH
Publishers, New York, 1989, p. 313).
The flexibility of the synthesis is further demonstrated by an alternative
sequence of reactions,
wherein (VII) may be reduced (vide supra) and either i) alkylated as above
with (VIII) to afford (VI) or
ii) condensed with (IV) followed by in-situ imine reduction to also afford
(VI). Where R5=R6=R7=H,
and the (methylaminomethyl)benzyl alcohol derivative may be obtained from
commercial sources.
Compounds of formulae IA-IF of this invention may also be prepared according
to Scheme 3.
Treatment of an appropriately substituted 2-iodobenzaldehyde (or a 2-
bromobenzaldehyde) (X) with an
amine H2N-R1 in lower alkyl alcohol solvents followed by reduction of the
resultant imine as described
above in Scheme 1 (vide supra) affords an intermediate (2-I or Br),R2,R3-PhCH2-
NH-RI which, when
treated with an optionally substituted bromoacetophenone (as described for the
synthesis of (V), Scheme
1 ) provides the alkylation product (XI).
Compounds of formula (XI) may be treated with strong bases, such as, but not
limited to lower
alkyl (C 1 _6) lithium bases (preferably t-BuLi or n-BuLi) to afford the
anticipated halogen-metal
exchange followed by intramolecular Barbier cyclization to generate compounds
of formulae (IA-IE,
R8=OH). Inert solvents such as dialkyl ethers (preferably diethyl ether),
cyclic ethers (preferably
tetrahydrofuran ar 1,4-dioxane), etc. are necessary, and reaction temperatures
are kept low (-78°C to -
25oC) to avoid by-products. Alternatively, halogen-metal exchange may also be
effected in the presence
of zerovalent nickel, in which case N,N-dialkylformamides (preferably
dimethylformamide) serve as
ideal solvents. One skilled in the art of organic synthesis will understand
the optimal combination of
conditions and may seek further reference from Kihara, et al. (Tetrahedron,
1992, 48, 67-78), and
Blomberg, et al. (Synthesis, 1977, p. 18-30). Additionally, compounds of
formulae (IA-E, R~=OH) may
be readily alkylated (vide supra) to afford compounds formulae (IA-E, R8=ORI
1). Finally, further
treatment of compounds of formulae (IA-E, R8=OH) with a halogenating reagent
or specifically a
fluorinating reagent such as, but not limited to, diethylaminosulfur
trifluoride (DASTj, readily provides
compounds of formulae (IA-F, R8=F). Further reference may be gained from the
review of Hudlicky
(Organic Reactions. 1985, 35, p. 513-637).
Compounds of formulae IA-F of this invention may also be prepared according to
Scheme 4. 4-
Bromoisoquinolines (XII) may be treated with an aryl boronic acid or aryl
boronic acid ester where Y is
equivalent to B(OH)2 or B(ORa)(ORb) (where Ra and Rb are lower alkyl, ie. CI-
C6, or taken together,
Ra and Rb are lower alkylene, ie. C~-Cl ~) in the presence of a metal catalyst
with or without a base in
-31-
WO 01/32624 CA 02389300 2002-04-29 pCT/US00/30328
an inert solvent to give isoquinoline compounds of formula (XIII). Metal
catalysts include, but are not
limited to, salts or phosphine complexes of Cu, Pd, or Ni (eg. Cu(OAc)2,
PdCl2(PPh3)2, NiCl2(PPh3)2).
Bases may include, but are not limited to, alkaline earth metal carbonates,
alkaline earth metal
bicarbonates, alkaline earth metal hydroxides, alkali metal carbonates, alkali
metal bicarbonates, alkali
metal hydroxides, alkali metal hydrides (preferably sodium hydride), alkali
metal alkoxides (preferably
sodium methoxide or sodium ethoxide), alkaline earth metal hydrides, alkali
metal dialkylamides
(preferably lithium diisopropylamide), alkali metal bis(trialkylsilyl)amides
(preferably sodium
bis(trimethylsilyl)amide), trialkyl amines (preferably diisopropylethylamine
or triethylamine) or
aromatic amines (preferably pyridine). Inert solvents may include, but are not
limited to acetonitrile,
dialkyl ethers (preferably diethyl ether), cyclic ethers (preferably
tetrahydrofuran or 1,4-dioxane), N,N-
dialkylacetamides (preferably dimethylacetamide), N,N-dialkylformamides
(preferably
dimethylformamide), dialkylsulfoxides (preferably dimethylsulfoxide), aromatic
hydrocarbons
(preferably benzene or toluene) or haloaalkanes (preferably methylene
chloride). Prefered reaction
temperatures range from room temperature up to the boiling point of the
solvent employed. The
reactions may be run in conventional glassware or in one of many commercially
available parallel
synthesizer units. Non-commercially available boronic acids or boronic acid
esters may be obtained from
the corresponding optionally substituted aryl halide as described by Gao, et
al. (Tetrahedron, 1994, 50,
979-988).
Compounds of formula (XIII) are converted into the target
tetrahydroisoquinolines of formula
via a two-step procedure employing first amine quaternization with a reagent
RI-LG, where LG
represents a suitable leaving group such as I, Br, O-triflate, O-tosylate, O-
methanesulfonate, etc. The
reactions are optimally conducted in haloaalkanes (preferably methylene
chloride), dialkyl ethers
(preferably diethyl ether), cyclic ethers (preferably tetrahydrofuran or 1,4-
dioxane) or other inert solvent.
The reactions are optimally conducted at or below room temperature and
reaction times vary from 10
minutes to 24 hours. The second step of the sequence involves reduction to the
tetrahydroisoquinolines
of formulae IA-F. Optimally, a mild reducing agent is employed, such as for
example, sodium
cyanoborohydride in the presence of acid catalyst to facilitate the reaction.
Additional guidance for
effectively conducting this chemistry may be located from the works of Miller,
et al. (Synthetic
Communications, 1994, 24, I 187-I 193) and Terashima, et al. (Heterocycles,
1987, 26, 1603-1610).
-32-
WO 01/32624 CA 02389300 2002-04-29 PCT/US00/30328
Scheme 1
Rs Rs
R~ R5 R~ R5
\ \
/ ~ /
O O
Ra ~ / O H2N_Ri
R3 R2
IV
OH O
\ I \ Rs ~ I \ I \ R5
Ra / N,R1 / Rs Ra / N.R~ / Rs
R3 R2 R~ R3 R2 R~
VI V
Rs
R~ R5
Ra / N~Ri
R3 R2
I (R8 = H)
-33-
WO 01/32624 CA 02389300 2002-04-29 PCT/US00/30328
Scheme 2
Ra / O
R3 R2
IV \
R4 I / X
Rs Rs Ra R2
R~ \ R5 H2N-R1 R~ \ R5 VIII
V
~/ ~/
O X O NHR~
III VII
R R5
IV VIII
VI V)
NHR~
IX
Rs
HO
-34-
WO 01/32624 CA 02389300 2002-04-29 pCT~S00/30328
Scheme 3
O
I ~ ~ VII I \ ~ I \ R
Ra / CHO Ra / N~R1 / Rs
R3 R3 R~
X XI
Rs
R~ R5
I\
/ s
F R
I \ R~ \ R5
Ra / N~R1 I /
Rs Ra
I \ 1
Rs / N-R1
~5
11 I (R2= H; R8=OH)
R4 ~1
R'~
I (R2= H)
Scheme 4
Rs
R7 Rs R5 R7 \ R5
I
Br ~ /
\ \ Y I \ \ I
Ra / ~ N ~ Ra /
R3 R2 R3 R2 (R = H)
XII XIII
-35-
WO 01/32624 CA 02389300 2002-04-29 pCT/LTS00/30328
It will be appreciated that compounds useful according to the present
invention may contain
asymmetric centres. These asymmetric centres may independently be in either
the R or S configuration
and such compounds are able to rotate a plane of polarized light in a
polarimeter. If said plane of
polarized light is caused by the compound to rotate in a counterclockwise
direction, the compound is
said to be the (-) stereoisomer of the compound. If said plane of polarized
light is caused by the
compound to rotate in a clockwise direction, the compound is said to be the
(+) stereoisomer of the
compound. It will be apparent to those skilled in the art that certain
compounds useful according to the
invention may also exhibit geometrical isomerism. It is to be understood that
the present invention
includes individual geometrical isomers and stereoisomers and mixtures
thereof, including racemic
mixtures, of compounds of formulae (IA-F) hereinabove. Such isomers can be
separated from their
mixtures, by the application or adaptation of known methods, for example
chromatographic techniques
and recrystallisation techniques, or they are separately prepared from the
appropriate isomers of their
intermediates.
Radiolabelled compounds of the invention are synthesized by a number of means
well known to those of
ordinary skill in the art, e.g., by using starting materials incorporating
therein one or more radioisotopes.
This invention provides compositions containing the compounds described
herein, including, in
particular, pharmaceutical compositions comprising therapeutically effective
amounts of the compounds
and pharmaceutically acceptable carriers.
It is a further object of the invention to provide kits having a plurality of
active ingredients (with
or without carrier) which, together, may be effectively utilized for carrying
out the novel combination
therapies of the invention.
It is another object of the invention to provide a novel pharmaceutical
compositions which is
effective, in and of itself, for utilization in a beneficial combination
therapy because it includes a
plurality of active ingredients which may be utilized in accordance with the
invention.
The invention also provides kits or single packages combining two or more
active ingredients
useful in treating a disorder described herein. A kit may provide (alone or in
combination with a
pharmaceutically acceptable diluent or carrier), the compound of formulae (IA-
F) and the additional
active ingredient (alone or in combination with diluent or carrier) selected
from a serotonin 1A receptor
antagonist, a selective neurokinin-1 receptor antagonist, and a norepinephrine
precursor.
In practice compounds of the present invention may generally be administered
parenterally.
intravenously, subcutaneously intramuscularly, colonically, nasally,
intraperitoneally, rectally or orally.
The products according to the invention may be presented in forms permitting
administration by
the most suitable route and the invention also relates to pharmaceutical
compositions containing at least
one product according to the invention which are suitable for use in human or
veterinary medicine.
These compositions may be prepared according to the customary methods, using
one or more
-36-
W~ 01/32624 CA 02389300 2002-04-29
PCT/US00/30328
pharmaceutically acceptable adjuvants or excipients. The adjuvants comprise,
inter alia, diluents, sterile
aqueous media and the various non-toxic organic solvents. The compositions may
be presented in the
form of tablets, pills, granules, powders, aqueous solutions or suspensions,
injectable solutions, elixirs or
syrups, and can contain one or more agents chosen from the group comprising
sweeteners, flavorings,
colorings, or stabilizers in order to obtain pharmaceutically acceptable
preparations.
The choice of vehicle and the content of active substance in the vehicle are
generally determined
in accordance with the solubility and chemical properties of the product, the
particular mode of
administration and the provisions to be observed in pharmaceutical practice.
For example, excipients
such as lactose, sodium citrate, calcium carbonate, dicalcium phosphate and
disintegrating agents such as
starch, alginic acids and certain complex silicates combined with lubricants
such as magnesium stearate,
sodium lauryl sulfate and talc may be used for preparing tablets. To prepare a
capsule, it is advantageous
to use lactose and high molecular weight polyethylene glycols. When aqueous
suspensions are used they
can contain emulsifying agents or agents which facilitate suspension. Diluents
such as sucrose, ethanol,
polyethylene glycol, propylene glycol, glycerol and chloroform or mixtures
thereof may also be used.
For parenteral administration, emulsions, suspensions or solutions of the
products according to
the invention in vegetable oil, for example sesame-oil, groundnut oil or olive
oil, or aqueous-organic
solutions such as water and propylene glycol, injectable organic esters such
as ethyl oleate, as well as
sterile aqueous solutions of the pharmaceutically acceptable salts, are used.
The solutions of the salts of
the products according to the invention are especially useful for
administration by intramuscular or
subcutaneous injection. The aqueous solutions, also comprising solutions of
the salts in pure distilled
water, may be used for intravenous administration with the proviso that their
pH is suitably adjusted, that
they are judiciously buffered and rendered isotonic with a sufficient quantity
of glucose or sodium
chloride and that they are sterilized by heating, irradiation or
microfiltration.
Suitable compositions containing the compounds of the invention may be
prepared by
conventional means. For example, compounds of the invention may be dissolved
or suspended in a
suitable carrier for use in a nebulizer or a suspension or solution aerosol,
or may be absorbed or adsorbed
onto a suitable solid carrier for use in a dry powder inhaler.
Solid compositions for rectal administration include suppositories formulated
in accordance with
known methods and containing at least one compound of formulae (IA-F) .
The percentage of active ingredient in the compositions of the invention may
be varied, it being
necessary that it should constitute a proportion such that a suitable dosage
shall be obtained. Obviously,
several unit dosage forms may be administered at about the same time. The dose
employed will be
determined by the physician, and depends upon the desired therapeutic effect,
the route of administration
and the duration of the treatment, and the condition of the patient. In the
adult, the doses are generally
from about 0.01 to about 100, preferably about 0.01 to about 10, mg/kg body
weight per day by
-37-
WO 01/32624 CA 02389300 2002-04-29 pCT/US00/30328
inhalation, from about 0.01 to about 100, preferably 0.1 to 70, more
especially 0.5 to 10, mg/kg body
weight per day by oral administration, and from about 0.01 to about 50,
preferably 0.01 to 10, mg/kg
body weight per day by intravenous administration. In each particular case,
the doses will be determined
in accordance with the factors distinctive to the subject to be treated, such
as age, weight, general state of
health and other characteristics which can influence the efficacy of the
medicinal product.
The products according to the invention may be administered as frequently as
necessary in order
to obtain the desired therapeutic effect. Some patients may respond rapidly to
a higher or lower dose and
may find much weaker maintenance doses adequate. For other patients, it may be
necessary to have
long-term treatments at the rate of 1 to 4 doses per day, in accordance with
the physiological
requirements of each particular patient. Generally, the active product may be
administered orally 1 to 4
times per day. It goes without saying that, for other patients, it will be
necessary to prescribe not more
than one or two doses per day.
The present invention provides compounds which inhibit synaptic
norepinephrine, dopamine and
serotonin uptake and are therefore believed to be useful in treating a
disorder which is created by or is
dependent upon decreased availability of serotonin, norepinephrine or
dopamine. Although the
compounds of the formulae (IA-F) inhibit synaptic norepinephrine, dopamine and
serotonin uptake, in
any individual compound these inhibitory effects may be manifested at the same
or vastly different
concentrations or doses. As a result, some compounds of the formulae (IA-F)
are useful in treating such
a disorder at doses at which synaptic norepinephrine uptake may be
substantially inhibited but at which
synaptic serotonin uptake or dopamine uptake is not substantially inhibited,
or visa versa. Also, some
compounds of the formulae (IA-F) are useful in treating such a disorder at
doses at which synaptic
dopamine uptake may be substantially inhibited but at which synaptic
norepinephrine or serotonin
uptake is not substantially inhibited, or visa versa. And, conversely, some
compounds of the formulae
(IA-F) are useful in treating such a disorder at doses at which synaptic
serotonin uptake may be
substantially inhibited but at which synaptic norepinephrine or dopamine
uptake is not substantially
inhibited, or visa versa. Other compounds of formulae (IA-F) are useful in
treating such a disorder at
doses at which synaptic norepinephrine, dopamine and serotonin uptake are
substantially inhibited.
The concentrations or doses at which a test compound inhibits synaptic
norepinephrine,
dopamine and serotonin uptake is readily determined by the use of standard
assay and techniques well
known and appreciated by one of ordinary skill in the art. For example, the
degree of inhibition at a
particular dose in rats can be determined by the method of Dudley, et al., J.
Pharmacol. Exp. Ther. 217,
834-840 ( 1981 ), which is incorporated by reference.
The therapeutically effective inhibitory dose is one that is effective in
substantially inhibiting
synaptic norepinephrine uptake, synaptic dopamine uptake, or synaptic
serotonin uptake or inhibiting the
synaptic uptake of two or more of norepinephrine, dopamine and serotonin
uptake. The therapeutically
-38-
WO 01/32624 CA 02389300 2002-04-29 PCT/US00/30328
effective inhibitory dose can be readily determined by those skilled in the
art by using conventional
range finding techniques and analagous results obtained in the test systems
described above.
Compounds of this invention provide a particularly beneficial therapeutic
index relative to other
compounds available for the treatment of similar disorders. Without intending
to be limited by theory, it
is believed that this is due, at least in part, to some of the compounds'
having higher binding affinities,
e.g. their ability to be selective, for the norepinephrine transporter protein
("NET") over the transporters
for other neurochemicals, e.g., the dopamine transporter protein ("DAT") and
the serotonin transporter
protein ("SERT")
Binding affinities are demonstrated by a number of means well known to
ordinarily skilled
artisans, including, without limitation, those described in the Examples
section hereinbelow. Briefly, for
example, protein-containing extracts from cells, e.g., HEK293E cells,
expressing the transporter proteins
are incubated with radiolabelled ligands for the proteins. The binding of the
radioligands to the proteins
is reversible in the presence of other protein ligands, e.g., the compounds of
this invention; said
reversability, as described below, provides a means of measuring the
compounds' binding affinities for
the proteins (Ki). A higher Ki value for a compound is indicative that the
compound has less binding
affinity for a protein than is so for a compound with a lower Ki; conversely,
lower Ki values are
indicative of greater binding affinities.
Accordingly, the difference in compound selectivity for proteins is indicated
by a lower Ki for
the protein for which the compound is more selective, and a higher Ki for the
protein for which the
compound is less selective. Thus, the higher the ratio in Ki values of a
compound for protein A over
protein B, the greater is the compounds' selectivity for the latter over the
former (the former having a
higher Ki and the latter a lower Ki for that compound). Compounds provided
herein induce fewer side
effects during therapeutic usage because of their selectivity for the
norepinephrine transporter protein, as
indicated by the ratios of their Ki's for binding to NET over those for
binding to other transporter
proteins, e.g., DAT and SERT. Generally, some of the compounds of this
invention have a Ki ratio for
DAT/NET of at least about 2:1; generally also have a SERT/NET ratio of at
least about 20:1.
Moreover, in vivo assessment of the activity of compounds at the NE and DA
transporters is, for
example, by determining their ability to prevent the sedative effects of
tetrabenazine (TBZ) (see, e.g., G.
Stille, Arzn. Forsch 14:534-537, 1964, the contents of which are incorporated
herein by reference).
Randomized and coded doses of test compounds are administered to mice, as is
then a dose of
tetrabenazine. Animals are then evaluated for antagonism of tetrabenazine-
induced exploratory loss and
ptosis at specified time intervals after drug administration. Exploratory
activity is, for example,
evaluated by placing the animal in the center of a circle and then evaluating
the amount of time it takes
for the animal to intersect the circle's perimeter - generally, the longer it
takes for the animal to make this
intersection, the greater is its loss of exploratory activity. Furthermore, an
animal is considered to have
-39-
CA 02389300 2002-04-29
WO 01/32624 PCT/US00/30328
ptosis if its eyelids are at least 50% closed. Greater than 95% of the control
(vehicle-treated) mice are
expected to exhibit exploratory loss and ptosis; compound-related activity is
then calculated as the
percentage of mice failing to respond to the tetrabenazine challenge dose,
with therapeutically more
effective compounds expected to better at reducing loss of exploratory
behavior and ptosis.
Accordingly, this invention provides methods of treating subjects afflicted
with various
neurological and psychiatric disorders by administering to said subjects a
dose of a pharmaceutical
composition provided herein. Said disorders include, without limitation,
attention deficit-hyperactivity
disorder, anxiety, depression, post-traumatic stress disorder, supranuclear
palsy, feeding disorders,
obsessive compulsive disorder, analgesia, smoking cessation, panic attacks,
Parkinson's and phobia. The
compounds provided herein are particularly useful in the treatment of these
and other disorders due, at
least in part, to their ability to selectively bind to the transporter
proteins for certain neurochemicals with
a greater affinity than to the transporter proteins for other neurochemicals.
The compounds of the invention, their methods or preparation and their
biological activity will
appear more clearly from the examination of the following examples which are
presented as an
illustration only and are not to be considered as limiting the invention in
its scope.
-40-
WO 01/32624 CA 02389300 2002-04-29 PCT/US00/30328
Examples
The compounds listed in the following Table 1 were made by the processes
described above.
Specific reaction and processing conditions for the preparation of 2,7-
dimethyl-4-phenyl-1,2,3,4-
tetrahydroisoquinoline (example I), 2,7-dimethyl-4-(3-fluorophenyl)-1,2,3,4-
tetrahydroisoquinoline
(example 4), 2,7-dimethyl-4-(4-fluoro-3-methylphenyl)-1,2,3,4-
tetrahydroisoquinoline (example 6), 2,7-
dimethyl-8-fluoro-4-phenyl-1,2,3,4-tetrahydroisoquinoline (example 28), 4-(4-
chloro-3-fluorophenyl)-2-
methyl-1,2,3,4-tetrahydroisoquinoline (example 70), 4-(3,4-difluorophenyl)-2-
methyl-1,2,3,4-
tetrahydroisoquinoline (example 78) and 4-(3,5-difluorophenyl)-2-methyl-
1,2,3,4-tetrahydroisoquinoline
(example 80) are given following the table.
Rs
R5
s
Ra .Ri
R'' R'
I
TABLE I:
Ex. R1 R2 R3 R4 R5 R6 R7 R8 Mp (C)
1 Me H H Me H H H H 245-250'
2 Me H H Me H OMe H H 186-188"
3 Me H H Me H F H H 151-153"
4 Me H H Me F H H H Oil,
MS'
5 Me H H Me F F H H 235-240a
6 Me H H Me Me F H H Oil,
MS'
7 Me H H Me Cl F H H 243-253a
8 Me H H Me Cl H H H 226-230'
9 Me H H Me H Me H H 257-260"
10 Me H H Me F Me H H 230-23
I
I1 Me H H Me H Cl H H 208-210"
12 Me H H Me F Cl H H 240-249a
13 Me H H Me CI Cl H H 245-246a
14 Me H H Et H H H H I 60-162
15 Me H H Et F F H H 140-141
-41-
WO 01/32624 CA 02389300 2002-04-29 PCT/US00/30328
Table 1, (continued)
Ex R1 R2 R3 R4 R5 R6 R7 R8 Mp (C)
16 Me H H F H OMe H H 100-102'
17 Me H H F F OMe H H 225-230'
18 Me H H F F Me H H 240-241
'
19 Me H H F F Cl H H 225-230'
20 Me H H F F F H H 232-235'
21 Me H H F C1 H H H 255-256'
22 Me H H CN H H H H Oil,
MS'
23 Me H H CF3 H H H H 257-275a
24 Me Me H Me H H H H 87-89F
25 Me Me H H H Cl H H Oil,
MS'
26 Me Me H H F F H H Oil,
MS'
27 Me H Me Me H H H H 108-113''
28 Me H F Me H H H H 215-216
29 Me H Me F H H H H 185-186'
30 Me H OMe Me H H H H 130-131'
31 Me H OH Me H H H H 260-261"
32 Me H H OCF3 H H H H 150-151
33 Me H H OMe F F H H 94-95'
34 Me H H OMe Me F H H 215-217'
35 Me H H OMe F Me H H 165-166
36 Me H H OMe Me H H H 173-177J
37 Me H H O(Ph) H H H H 175-176
38 Me H H O(4-OMePh) H H H H 165-166
39 Me H H O(CH2Ph) H H H H 155-156'
40 Me H H OH Me H H H 254-265
41 Me H H OH F Me H H 186-187h
42 Me H H OH Me F H H 190-191"
43 Me H H OH F F H H 236-237"
44 Me H H H CN H H H Oil,
MS'
45 Me H Me H H H H H Oil,
MS'
46 Me H Me H H F H H 165-166h
47 Me H Me H F F H H 125-127'
48 Me H Me H F H F H 250-252
49 Me H Me H F H H H 125-127"
50 Me H Me H Me F H H Oil,
MS'
51 Me H Me H C1 F H H 243-260
52 Me H Me H Cl Cl H H 246-248''
53 Me H Me H Cl H H H 228-230
54 Me H Me H H Cl H H 200-202r
55 Me H Me H F Cl H H 218-228''
56 Me H Me H H OMe H H 79-81
57 Me H Me H H CN H H Oil,
MS'
58 Me H Me H H CF3 H H 214-216"
59 Me H Me H H Me H H Oil,
MS'
60 Me H CH2NHMe H H H H H 278-282'
-42-
WO 01/32624 cA 02389300 2002-04-29 PCT/US00/30328
Table 1, (continued)
Ex. R1 R2 R3 R4 R5 R6 R7 R8 Mp (C)
61 Me H CH20H H H H H H144-146
62 Me H S02NH2 H H H H H 231-242'
63 Me H S02NHMe H H H H H 258-265a
64 Me H OMe H H Me H H 225-260'
65 Me H OMe H F H F H 165-166"
66 Me H OMe H Cl H H H 147-148"
67 Me H OMe H Cl Cl H H 230-235
68 Me H OMe H F Cl H H 179-183'
69 Me H OMe H Cl F H H 245-252'
70 Me H H H F H F H 230-233F
71 Me H H H F H Cl H 205-207'
72 Me H H Me F H F H 230-231
a
73 Me H H Me F H Cl H 180-200J
74 Me H H H F F F H 227-230'
75 Me H H H F H H H 218-220'
76 Me H H H F Me H H 215-217
77 Me H H H Me F H H 193-195"
78 Me H H H F F H H 200(Sub.)'
79 Me H H H Cl H H H 218-220'
80 Me H H H F Cl H H 230-235J
81 Me H H H Cl F H H Oil,
MS'
82 Me H H H CN H H H Oil,
MS'
83 Me H H H H NHCOMe H H 183-1894
84 Me H H H H CI H F 205-210
85 Me Me H Me F H F H 194-197'
86 Me H H Me F F F H 269-274
87 Et H H Me H F H H Oil-Ms'
88 Me H H Me H F H OH Oil-Ms'
89 Me H F CH~Me H H H H 185-205'
90 Me H H CH,NH,H H H H 176-177
91 Me H H CH,NHMe H H H 160-163"
H
92 Me H OH CN H H H H 234-238'
93 Me H H CH~ H H H 237-240'
OH H
94 Et H H _ H H H 172-174"
H H
Footnotes for Table 1 for Salt Forms of the examples:
a - Mono Hydrochloride
b - Mono Maleate
c - Mono Hydrochloride 0.2 Hydrate
d - Mono Fumarate
a - Free Base - mass spectrum shows molecular ion
f - Mono Hydrochloride ~ 0.25 Hydrate
g - Mono Hydrochloride ~ 0.10 Hydrate
h - Mono Hydrochloride ~ 0.75 Hydrate
i - 1.5 Fumarate ~ 0.25 Hydrate
j - Mono Fumarate ~ 0.5 Diethyl ether
k - Mono Hydrobromide ~ 0.25 Hydrate
1- Mono Hydrochloride ~ 0.33 Hydrate
-43-
WU 01/32624 CA 02389300 2002-04-29 PCT/LTS00/30328
m - Mono Fumarate ~ 0.25 Hydrate
n - Mono Hydrobromide
o - Mono Maleate ~ 0.25 Hydrate
p - Mono Hydrochloride ~ 0.5 Hydrate
q - 0.25 Hydrate
r - Mono Maleate ~ 0.25 Hydrate ~ 0.13 Ethanol
s - Mono Sulfate
t - Di Hydrochloride ~ 0.5 Hydrate
a - Bis Maleate
Example 1
Preparation of 2,7-dimethyl-4-phenyl-1,2,3,4-tetrahydroisoquinoline
Step A: A solution of m-tolualdehyde (500 mg, 4.16 mmol), ~;- -
(methylaminomethyl)benzyl
alcohol (630 mg, 4.16 mmol) and acetic acid (0.5 ml) was stirred in methanol (
16 ml) at 0°C under
nitrogen as sodium cyanoborohydride (784 mg, 12.5 mmol) was added in small
portions. The reaction
mixture was stirred for 5 minutes at 0°C and two days at ambient
temperature. The reaction mixture was
brought to pH 12 with 2N sodium hydroxide, diluted with water, and extracted
with diethyl ether (3X).
The combined organic extracts were washed with brine, dried over anhydrous
magnesium sulfate, and
the solvent removed in vacuo to provide the desired intermediate ( 1.24 g): I
H NMR (300 MHz, CDC13)
8 7.08-7.35 (m, 9H), 4.73-4.77 (m, 1H), 3.71 (d, J=13.0 Hz, 1H), 3.50 (d,
J=13.0 Hz, 1H), 2.46-2.67 (m,
2H), 2.36 (s, 3H), 2.32 (s, 3H); CI MS m/z = 256 [C 17H21 NO+H]+.
Ste~B: The product from Step A (1.24 g, 4.90 mmol) was stirred in methylene
chloride (208
ml) and treated dropwise with concentrated sulfuric acid (98%, 10 ml) over 3
minutes. After stirring for
20 minutes, the reaction was diluted with ice chips and made basic with 25%
aqueous ammonium
hydroxide. The reaction mixture was extracted with methylene chloride (3X) and
the organic extracts
combined, dried over anhydrous sodium sulfate, filtered, and concentrated in
vacuo. Purification by
column chromatography, eluting with hexanes / ethyl acetate (5/I), afforded
the desired
tetrahydroisoquinoline (0.23 g): I H NMR (300 MHz, CDCl3) 8 7.17-7.31 (m, 5H),
6.87-6.89 (m, 2H),
6.75 (d, J=7.8 Hz, 1 H), 4.20-4.26 (m, 1 H), 3.72 (d, J=14.8 Hz, 1 H), 3.57
(d, J= I 4.8 Hz, 1 H), 2.96-3.10
(m, 1H), 2.51-2.58 (m. 1H), 2.42 (s, 3H), 2.29 (s, 3H).
Step C: The product from Step B (0.23 g) was treated with ethereal HCl in
methanol (5 ml) to
afford a precipitate. The solvents and excess HCI were removed in vacuo and
the resultant solid
recrystallized from ethanol / diethyl ether to provide the HC1 salt of the
target (0.21 g) as a white solid:
mp 245-250oC; 1 H NMR (CD30D) 8 6.86-7.40 (m, 7H), 6.74 (d, J=7.8 Hz, 1 H),
4.52-4.64 (m, 3H),
3.72-3.88 (m, 1H), 3.45-3.55 (m, 1H), 3.08 (s, 3H), 2.32 (s, 3H); 13C NMR (75
MHz, CD30D)
130.6, 130.3, 129.1, 127.8, 59.3, 56.8, 44.5, 44.0, 21.1; IR (KBr) 2937, 2474,
1454, 701 cm-I ; CI MS
-44-
WO 01/32624 CA 02389300 2002-04-29 pCT/iJS00/30328
m/z = 238 [C17H19N+H]+. Anal. Calcd. for C17H19N-HCI: C, 74,57; H, 7.36; N,
5.12. Found: C,
74.20; H, 7.34; N, 4.82.
Example 4
Preparation of 2 7-dimethyl-4-(3-fluoro~henyl)-1 2.3 4-tetrahydroisoquinoline
Step A: m-Tolualdehyde ( 1.66 g, 14.0 mmol) was treated with methyl amine (40%
aqueous,
1.39 ml, 18.0 mmol) in methanol (20 ml) at room temperature. The reaction was
stirred 20 minutes and
treated with sodium borohydride (0.26 g, 7.0 mmol) portionwise. The reaction
was stirred 1 hour and
treated with 3'-fluoro-2-bromoacetophenone (3.0 g, 14.0 mmol) followed by
stirring for 45 minutes at
room temperature. The reaction was finally treated with sodium borohydride
(0.52 g, 14.0 mmol)
portionwise and stirring continued overnight. The reaction was diluted with
water ( 100 ml) and
extracted with methylene chloride (3 X 100 ml). The combined organic extracts
were washed with brine
and dried over anhydrous sodium sulfate, followed by filtration and
concentration in vacuo. Purification
by column chromatography on silica gel eluting with hexanes / ethyl acetate
(3/1) provided the amino
alcohol (4.3 g) as a yellow oil; 1H NMR (300 MHz, CDCI3) 8 7.08-7.30 (m, 7H),
4.73 (t, J=6.0 Hz,
1H), 3.60 (ABq, JAB=14.0 Hz, 2H), 2.55 (d, J=8.0 Hz, 2H), 2.36 (s, 3H), 2.31
(s, 3H); CI MS m/z = 274
[C 17H20NF0+H]+.
Step B: The product from Step A (1.0 g, 4.0 mmol) was stirred in methylene
chloride (100 ml)
and treated dropwise with concentrated sulfuric acid (98%, 7.0 ml) over 3
minutes. After stirring for 1
hour, the reaction was diluted with ice chips and and made basic with 25%
aqueous ammonium
hydroxide. The reactions mixture was extracted with methylene chloride (3 X
100 ml) and the organic
extracts combined, dried over anhydrous sodium sulfate, filtered, and
concentrated in vacuo.
Purification by column chromatography, eluting with hexanes / ethyl acetate
(3/ 1 ), afforded the desired
tetrahydroisoquinoline as a yellow oil: 1 H NMR (300 MHz, CDC13) 8 6.89-7.00
(m, 5H), 6.75 (d, J=8.0
Hz, 1 H), 4.21 (t, J=7.0 Hz, 1 H), 3.64 (ABq, JAB=15.0 Hz, 2H), 3.02 (m, 1 H),
2.56 (m, 1 H), 2.41 (s, 3H),
2.29 (s, 3H); CI MS mlz = 256 [C 17H 1 gNF+H]+.
Step C: The product from Step B was subjected to chiral HPLC separation
employing a Chiral
Technologies Chiracel~ AD column (5 cm X 50 cm) eluting with hexanes /
isopropanol (9/1) to afford
the (R), [a] D -16.3 (c=0.498, MeOH) and (S), [a] D +16.3 (c=0.476, MeOH)
enantiomers in order of
elution. The (S)-(+) enantiomer was treated with malefic acid ( 1.0
equilvalent) and the resultant maleate
salt filtered and dried to constant weight. (S)-(+)-2,7-dimethyl-4-(3-
fluorophenyl)-1,2,3,4
tetrahydroisoquinoline, maleate salt: mp 172-173.5oC.
Example 6
Preparation of ~ 7-dimethvl-4-(4-fluoro-3-methylphenyl)-1 ~ 3 4-
tetrah~roisoquinoline
-45-
WO 01/32624 CA 02389300 2002-04-29 PCT/US00/30328
St. ep A: m-Tolualdehyde (4.0 g, 33.0 mmol) was treated with methyl amine (40%
aqueous, 3.36
ml, 43.0 mmol) in methanol (40 ml) at room temperature. The reaction was
stirred 20 minutes and
treated with sodium borohydride (0.64 g, 33.0 mmol) portionwise. The reaction
was stirred 1 hour and
treated with 4'-fluoro-3'-methyl-2-bromoacetophenone (7.69 g, 33.0 mmol)
followed by stirring for 45
minutes at room temperature. The reaction was finally treated with sodium
borohydride ( 1.0 g, 33
mmol) portionwise and stirring continued overnight. The reaction was diluted
with water ( 100 ml) and
extracted with methylene chloride (3 X 100 ml). The combined organic extracts
were washed with brine
and dried over anhydrous sodium sulfate, followed by filtration and
concentration in vacuo. Purification
by column chromatography on silica gel eluting with hexanes / ethyl acetate
(2/1) provided the amino
alcohol (65.3 g) as a yellow oil; CI MS m/z = 286 [C18H22NF0+H]+.
Step B: The product from Step A (0.52 g, 2.0 mmol) was dissolved in methylene
chloride (20
ml) and treated dropwise with concentrated sulfuric acid (98%, 3 ml). The
reaction was stirred overnight
at room temperature, then diluted with ice chips and and made basic with 25%
aqueous ammonium
hydroxide. The reaction mixture was extracted with methylene chloride (3 X 50
ml) and the organic
extracts combined, dried over anhydrous sodium sulfate, filtered, and
concentrated in vacuo.
Purification by column chromatography, eluting with hexanes / ethyl acetate
(3/1) afforded the desired
tetrahydroisoquinoline (0.08 g): IH NMR (300 MHz, CDCI~) 8 6.87-7.00 (m, SH),
6.74 (d, J=8.0 Hz,
1H), 4.17 (t, J=7.0 Hz, 1H), 3.64 (ABq, JAB=15.0 Hz, 2H), 3.01 (m, IH), 2.53
(m, 1H), 2.40 (s, 3H),
2.29 (s, 3H), 2.23 (s, 3H); CI MS m/:. = 270 [C 1 gH2pNF+H]+.
Example 28
Preparation of 2 7-dimethyl-8-fluoro-4-phenyl-1,2,3,4-tetrahydroisoquinoline
Step A: A solution of -(methylaminomethyl)benzyl alcohol (745 mg, 4.9 mmol)
and
triethylamine (0.79 ml, 5.66 mmol) in acetonitrile (45 ml) at OoC under
nitrogen was treated dropwise
with 2-fluoro-3-methylbenzyl bromide ( 1.0 g, 4.9 mmol) as a solution in
acetonitrile (25 ml). The
reaction was stirred at 0°C for 1 hour and at room temperature for 1.5
hours, followed by dilution with
water and extraction with methylene chloride (3X). The combined organic
extracts were dried over
anhydrous magnesium sulfate, filtered, and concentrated in vacuo to provide
the alkylation product (1.35
g): 1 H NMR (CDC13) 8 7.23 (m, SH), 7.08-7.17 (m, 2H), 6.97-7.06 (m, 1 H),
4.71-4.82 (m, 1 H), 3.79
(d, J=13.1 Hz, 1 H), 3.62 (d, J=13.2 Hz, 1 H), 2.33 (s, 3H), 2.29 (s, 3H).
Step B: The product from Step A (0.5 g, 1.8 mmol) was treated with sulfuric
acid (3.7 ml) and
purified by column chromatography as described for Example 1, Step B to afford
the desired product
(0.33 g) as an oil: 1 H NMR (CDCl3) 8 7.06-7.37 (m, SH), 6.88 (t, J=7.8 Hz, 1
H), 6.54 (d, J=7.8 Hz,
-46-
WO 01/32624 CA 02389300 2002-04-29 PCT/US00/30328
1 H), 4.18-4.27 (m, 1 H), 3.86 (d, J=15.6 Hz, 1 H), 2.94-3.04 (m, 1 H), 2.49-
2.59 (m, 1 H), 2.45 (s, 3H),
2.22 (s, 3H).
Ste~C: The product from Step B (0.33 g, 1.3 mmol) was treated with ethereal
HCl as described
in Example 1, Step C to provide the anticipated hydrochloride salt (0.30 g):
mp 215-216oC; IH NMR
(300 MHz, CD30D) 8 7.31-7.44 (m, 2H), 7.21-7.28 (m, 2H), 7.15 (t, J=7.9 Hz,
IH), 6.61 (d, J=8.0 Hz,
1H), 4.67-4.78 (m, 1H), 4.42-4.62 (m, 2H), 3.77-3.88 (m, IH), 3.55 (t, J=12.0
Hz, 1H), 3.1 I (s, 3H), 2.26
(s, 3H); IR (KBr) 3432, 2954, 2376, 1497, 1457, 1216, 1043, 704 cm-1; CI MS
m/z = 256
[C 17H 18NF+H]+. Anal. Calcd. for C 17H 18NF-HCI: C, 69.98; H, 6.56; N, 4.80.
Found: C, 69.64; H,
6.49; N, 4.65.
Example 70
Preparation of 4-(4-chloro-3-fluorophenyl)-2-methyl-1,2,3,4-
tetrahydroisoquinoline
Step A: Methylmagnesium bromide was added dropwise over 5 minutes to a stirred
solution of
4-chloro-3-fluorobenzaldehyde ( 10.86 g, 68.5 mmol) in anhydrous
tetrahydrofuran (100 ml) at -78°C
under nitrogen. After stirring for 15 minutes, the cooling bath was removed,
and the solution allowed to
warm to room temperature. After stirring 3 hours, the solution was poured
slowly with stirring into
saturated ammonium chloride (100 ml), then diluted with water (50 ml) and
extracted with diethyl ether.
The organic extracts were washed with water and saturated sodium chloride,
dried over anhydrous
sodium sulfate, filtered and the solvent removed in vacuo to provide the
benzylic alcohol (I 1.89 g) as a
clear, yellow oil: 1 H NMR (300 MHz, CDC13) S 7.35 (t, J=7.8 Hz, 1 H), 7.18
(dd, J=2.0, 10.0 Hz, 1 H),
7.07 (dd, J=1.7, 8.1 Hz, 1 H), 4.83-4.92 (m, 1 H), 2.01 (d, J=3.6 Hz, 1 H),
1.47 (d, J=6.3 Hz, 3H), CI MS
m/~ = 175 [C8H8C1F0+H]+.
Step B: The product from Step A (9.0 g, 52.0 mmol) in anyhdrous methylene
chloride (60 ml)
under nitrogen was added by cannula to a stirred suspension of pyridinium
chlorochromate ( 16.7 g, 77.0
mmol) and diatomaceous earth ( 15 g) in anhydrous methylene chloride ( 150 ml)
at 0°C under nitrogen.
After stirring for 26 hours, the heterogeneous mixture was diluted with
diethyl ether (300 ml), stirred for
1 hour, and filtered. The filtrate was concentrated in vacuo and the volatile
product purified by column
chromatography on silica gel (60 g) eluting with hexanes / ethyl acetate (9/1)
to provide the desired
acetophenone in quantitative crude yield: 1H NMR (300 MHz, CDC13) 8 7.65-7.75
(m, 2H), 7.51 (t,
J=7.6 Hz, 1H), 2.60 (s, 3H), CI MS m/z = 173 [C8H6C1F0+H]+.
Step C: The product from Step B (52 mmol) was treated with tetrabutylammonium
tribromide
(25.5 g, 52.9 mmol) in methanol / methylene chloride (I/3, 240 ml) under
nitrogen. After stirring 3 days
at room temperature, the solvents were removed in vacuo, and the residue
dissolved in diethyl ether (200
ml), washed with water (4 X 50 ml), dried over anhydrous sodium sulfate,
filtered and concentrated in
-47-
WO 01/32624 CA 02389300 2002-04-29 pCT/LJS00/30328
vacuo. Purification by column chromatography on silica gel ( 120 g) eluting
with hexanes / ethyl acetate
(30/1) afforded the desired ~~-bromoacetophenone (6.23 g) as a crystalline
solid: 1H NMR (300 MHz,
CDC13) 8 7.70-7.81 (m, 2H), 7.55 (t, J=7.7 Hz, 1H), 4.39 (s, 2H); CI MS m/z =
251
[C8H5BrC1F0+H]+.
Step D: Methylamine (40 wt% aqueous, 18.0 mmol) was added to a stirred
solution of
benzaldehyde ( 1.8 g, 17 mmol) in methanol (20 ml) under nitrogen. After
stirring 10 minutes at room
temperature, the solution was cooled to 0°C and treated with sodium
borohydride (0.32 g, 8.5 mmol)
portionwise. The reaction was stirred for 15 minutes, warmed to room
temperature and stirred an
additional 1 hour, whereupon the product from Step C (4.3 g, 17 mmol) was
added. The reaction was
stirred 1 hour, cooled to 0°C and treated again with sodium borohydride
(0.32 g, 8.5 mmol) and allowed
to stir overnight with warming to room temperature. The solution was diluted
with water (100 ml) and
extracted with methylene chloride (3 X 50 ml). The organic extracts were dried
over anhydrous sodium
sulfate, filtered, and concentrated in vacuo to provide the desired product as
a clear yellow oil ( I .77 g):
1 H NMR (300 MHz, CDC13) 8 7.25-7.39 (m, 6H), 7.17 (dd, J=I .8, I 0.0 Hz, 1
H), 7.04 (d, J=8.3 Hz, 1 H),
4.69 (dd, J=5.8, 8.2 Hz, 1 H), 3.74 (d, J=13.0 Hz, 1 H), 3.52 (d, J=13.0 Hz, 1
H), 2.45-2.57 (m, 2H), 2.32
(s, 3H), CI MS m/z = 294 [C 16H 17C1FN0+H]+.
Step E: The product from Step D ( I .77 g, 6.0 mmol) was stirred in
concentrated sulfuric acid
(4.0 ml) and methylene chloride (40 ml) for 15 minutes at room temperature.
The reaction was poured
on ice, made alkaline with concentrated ammonium hydroxide, and extracted with
diethyl ether. The
combined ether extracts were dried over sodium sulfate, filtered and
concentrated in vacuo to afford the
crude product as a cloudy yel low oil ( 1.7 g): 1 H NMR (300 MHz, CDC13) b
7.30 (t, J=7.9 Hz, 1 H),
7.06-7.22 (m, 3H), 6.92-7.03 (m, 2H), 6.85 (d, J=7.4 Hz, 1H), 4.28 (t, J=6.7
Hz, 1H), 3.77 (d, J=15.1 Hz,
1H), 3.70 (d, J=15.1 Hz, 1H), 3.05 (dd, J=5.6, 11.9 Hz, IH), 2.62 (dd, J=8.0,
I 1.5 Hz, 1H), 2.46 (s, 3H).
Step F: The product from Step E ( I .7 g, 6.0 mmol) was treated with ethereal
HCl ( 1.0 M, 12.0
ml, 12.0 mmol) in methanol (20 ml) to afford a precipitate. The solvents and
excess HCl were removed
in vacuo and the resultant solid recrystallized from methanol / diethyl ether
to provide the HCI salt of the
target ( 1.1 g) as a white solid: mp 230-235°C; 1 H NMR (CD30D) b 7.51
(t, J=8.0 Hz, 1 H), 7.26-7.39
(m, 3H), 7. I 8 (dd, J=2.0, 10.2 Hz, 1 H), 7.11 (dd. J=1.8, 8.3 Hz, I H), 6.92
(d, J=7.9 Hz, 1 H), 4.68 (dd,
J=6.3, 11.3 Hz, 1 H), 4.59 (bs, 2H), 3.87 (dd, J=6.2, 12.4 Hz, 1 H), 3.56 (t,
J=I 1.8 Hz, 1 H), 3.08 (s, 3H);
IR (Kbr) 3448, 2928, 2365, 1491, 1060, 747 cm-I ; CI MS n~lz = 276 [C 16H
15NC1F+H]+; Anal. Calcd.
for C16H15NCIF-HCI: C, 61.55; H, 5.17; N, 4.49. Found: C, 61.20; H, 5.07; N,
4.32.
-48-
WO 01/32624 CA 02389300 2002-04-29 pCT~JS00/30328
Step G: The product from Step E was subjected to chiral HPLC separation
employing a Chiral
Technologies Chiracel~ OD column (2 cm X 20 cm) eluting with hexanes /
isopropanol (9/1) to afford
the (S) and (R) enantiomers in order of elution. Each enantiomer was treated
with malefic acid ( 1.0
equilvalent) and the resultant maleate salts filtered and dried to constant
weight. (S)-(+)-4-(4-chloro-3-
fluorophenyl)-2-methyl-1,2,3,4-tetrahydroisoquinoline, maleate salt: mp 171-
172°C; [a] D +16.0
(c=0.200, MeOH).(R)-(-)-4-(4-chloro-3-fluorophenyl)-2-methyl-1,2,3,4-
tetrahydroisoquinoline, maleate
salt: mp 171-172°C; [a] 1j -15.5 (c=0.200, MeOH).
Example 78
Preparation of 4-(3,4-difluorophenXl)-2-meth-1,2,3,4-tetrahydroisoquinoline
Step A: 3,4-Difluoroacetophenone (25.0 g, 160.0 mmol) was treated with acetic
acid (250 ml)
and bromine (8.23 ml, 160.0 mmol, solution in 13 ml acetic acid) at room
temperature under nitrogen.
The reaction was stirred at room temperature for 1 hour and concentrated in
vacuo to remove acetic acid.
The residue was suspended in saturated sodium carbonate and extracted with
methylene chloride several
times. The combined organic extracts were dried over anhydrous sodium sulfate,
filtered and
concentrated in vacuo to afford the desired bromoacetophenone derivative (37.0
g) as a yellow
crystalline solid: 1 H NMR (300 MHz, CDCl3) 8 7.81 (m, 2H), 7.32 (m, 1 H),
4.39 (s, 2H).
Step B: The product from Step A (37.0 g, 158.0 mmol) was dissolved in
methylene chloride
(290 ml) and added dropwise to a solution of N-benzyl-N-methylamine (20.3 ml,
158.0 mmol) and
triethylamine (22.0 ml, 158.0 mmol) in methylene chloride (312 ml). The
addition was carried out over
45 minutes at 0°C, warmed to room temperature and allowed to stir an
additional 4 hours. The reaction
was diluted with water (300 ml) and extracted with methylene chloride. The
combined organic extracts
were dried over anhydrous sodium sulfate, filtered, and concentrated in vacuo.
The product was purifed
by column chromatography on silica gel (600 g) eluting with hexanes / ethyl
acetate (7/3) to afford the
desired alkylation product as a clear, light brown oil (30.2 g): 1 H NMR (300
MHz, CDC13) 8 7.87-7.73
(m, 2H), 7.35-7.15 (m, 6H), 3.68 (s, 2H), 3.64 (s, 2H), 2.34 (s, 3H).
Step C: The product from Step B (15.0 g, 54.0 mmol) was dissolved in methanol
(65 ml), chilled
in an ice bath and treated with sodium borohydride ( I .38 g, 36.0 mmol). The
reaction was stirred at 0°C
for 1 hour and at room temperature for 1 hour, followed by quenching with
water and extraction with
methylene chloride. The combined organic extracts were dried over sodium
sulfate, filtered and
concentrated in vacuo to directly provide the pure benzylic alcohol ( 14.4 g)
as a yellow oil: 1 H NMR
(300 MHz, CDC13) b 7.38-7.00 (m, 8H), 4.67 (t, J=7.0 Hz, 1H), 3.74 and 3.35
(ABq, JAB=13.2 Hz, 2H),
2.50 (d, J=7.0 Hz, 2H), 2.31 (s, 3H). Anal. Calcd. for C 16H 17N 1 O 1 F2: C,
69.30; H, 6.19; N, 5.05.
Found: C, 68.94: H, 6.21; N, 4.94.
-49-
WO 01/32624 CA 02389300 2002-04-29 PCT/L1S00/30328
SteRD: The product from Step C (14.4 g, 52.0 mmol) was stirred in concentrated
sulfuric acid
(27.0 ml) and methylene chloride (333 ml) for 15 minutes at room temperature.
The reaction was poured
on ice, made alkaline with concentrated ammonium hydroxide, and extracted with
diethyl ether. The
combined ether extracts were dried over sodium sulfate, filtered, and
concentrated in vacuo. The product
was purified by column chromatography on silica gel eluting with hexanes /
ethyl acetate (1/1) to
provide the pure tetrahydroisoquinoline (11.4 g): IH NMR (300 MHz, CDCl3) 8
7.29-7.36 (m, 1H),
6.83-7.20 (m, 6H), 4.20 (t, J=6.3 Hz, 1 H), 3.66 (s, 2H), 2.95 (dd, J=5.4,
11.5 Hz, 1 H), 2.58 (dd, J=7.4,
11.3 Hz, 1H), 2.41 (s, 3H).
Step E: The product from Step D (0.8 g, 3.0 mmol) was treated with ethereal
HCl as described
in Example 1, Step F to provide the anticipated hydrochloride salt (0.6 g): mp
200°C (sublimed); 1H
NMR (300 MHz, CD30D) 8 7.24-7.39 (m, 4H), 7.14-7.23 (m, I H), 7.06-7.13 (m, 1
H), 6.92 (d, J=7.8
Hz, 1 H), 4.65 (dd, J=6.1, 11.4 Hz), 4.58 (s, 2H), 3.85 (dd, J=6.2, 12.4 Hz, 1
H), 3.54 (t, J=11.8 Hz, 1 H),
3.07 (s, 3H); IR (KBr) 3448, 2932, 2549, 1512, 1465, 1276, 742 cm-1; CI MS m/z
= 260
[C 16H 15NF2+H]+. Anal. Calcd. for C 16H 15NF2-HCl-0.25 H20: C, 64.00, H,
5.54; N, 4.66. Found:
C, 64.11; H, 5.30; N, 4.62.
Step F: The product from Step D was subjected to chiral HPLC separation
employing a Chiral
Technologies Chiracel~ OD column (2 cm X 20 cm) eluting with hexanes /
isopropanol (9/1 ) to afford
the (S) and (R) enantiomers in order of elution. Each enantiomer was treated
with malefic acid (I.0
equilvalent) and the resultant maleate salts filtered and dried to constant
weight. (S)-(-)-4-(3,4-
difluorophenyl)-2-methyl-1,2,3.4-tetrahydroisoquinoline, maleate salt: mp 138-
139oC; [a] D -2.6
(c=0.366, MeOH). (R)-(+)-4-(3,4-difluorophenyl)-2-methyl-1,2,3,4-
tetrahydroisoquinoline, maleate salt:
138-139oC; [a] D +2.5 (c=0.386, MeOH).
Example 80
Preparation of 4-(3 5-difluorophen~)-2-methyl-1,2,3,4-tetrahydroisoquinoline
St_ ep A: Tetrabutylammonium tribromide (18.6 g, 38.6 mmol) was added to a
stirred solution of
3,5-difluoroacetophenone (6.0 g, 38.6 mmol) in methanol / methylene chloride
(1/3, 180 ml) under
nitrogen. After stirring at room temperature for 72 hours, the solvents were
removed in vacuo. The
residue was dissolved in diethyl ether (200 ml), washed with water (4 X 50
ml), dried over anhydrous
sodium sulfate, filtered and the solvent removed in vacuo to give a mixture of
the a-bromoacetophenone
and the corresponding dimethyl ketal (9.0 g): 1 H NMR (300 MHz, CDCl3) 8 7.50
(dd, J=2.0, 4.0 Hz,
2H), 7.08 (m, l H), 4.39 (s, 2H).
Ste~B: To the product mixture from Step A (3.S g, 14.7 mmol) and N-methyl-N-
benzylamine
(1.8 g, 14.7 mmol) in methylene chloride (15 ml) was added diisopropyl ethyl
amine (3.0 ml, 17 mmol).
-50-
WO ~l/32624 CA 02389300 2002-04-29 pCT/LTS00/30328
The reaction was stirred at room temperature for 5.5 hours, then washed with
water and dried over
anhydrous sodium sulfate. After filtration and concentration in vacuo, the
material was purified by
column chromatography on silica gel (140 g) eluting with hexanes / ethyl
acetate / triethylamine
(9/1/0.1) to provide the desired alkylation product (1.2g) as an orange oil:
IH NMR (300 MHz, CDC13)
8 7.48 (dd, J=2.0, 4.0 Hz, 2H), 7.33 (m, 5H), 7.00 (m, 1H), 3.69 (s, 2H), 3.66
(s, 2H), 2.36 (s, 3H).
St_ e~,C: The product from Step B (1.1 g, 4.0 mmol) was dissolved in methanol,
chilled in an ice
bath and treated with sodium borohydride (0.1 g, 2.7 mmol). The reaction was
stirred at 0°C for 1 hour
and at room temperature for 1 hour, followed by quenching with water and
extraction with methylene
chloride. The combined organic extracts were dried over sodium sulfate,
filtered and concentrated in
vacuo to provide the benzylic alcohol (0.8 g) as an orange oil: IH NMR (300
MHz, CDC13) 8 7.40-7.30
(m, 5H), 6.90-6.82 (m, 1 H), 6.70-6.60 (m, 1 H), 4.70 (m, 1 H), 3.73 (d, J= I
4.0 Hz, 1 H), 3.52 (d, J=14.0
Hz, 1H), 2.55-2.40 (m, 2H), 2.29 (s, 3H).
Step D: The product from Step C (0.4 g, 1.4 mmol) was stirred in concentrated
sulfuric acid ( 1.5
ml) and methylene chloride ( 10 ml) for 15 minutes at room temperature. The
reaction was poured on
ice, made alkaline with concentrated ammonium hydroxide, and extracted with
diethyl ether. The
combined ether extracts were dried over sodium sulfate, filtered and
concentrated in vacuo. Purification
by column chromatography on silica gel (15 g) eluting with hexanes / ethyl
acetate / triethylamine
(9/1/0.1) afforded the target (70 mg): 1H NMR (300 MHz, CDC13) 8 7.40-7.07 (m,
4H), 6.87 (d, J=7.0
Hz, 1 H), 6.77-6.62 (m, 2H), 4.21 (t, J=6.0 Hz, 1 H). 3.66 (d, J=2.0 Hz, 2H),
2.95 (dd, J=5.0, 6.0 Hz, 1 H),
2.61 (dd, J=6.0 Hz, 7.0 Hz, 1 H). 2.41 (s, 3H).
Step E: The product from Step D (70 mg, 0.27 mmol) was treated with ethereal
HCl ( 1.0 M, 0.6
ml, 0.6 mmol) in methanol ( I .4 ml) to afford a precipitate. The solvents and
excess HCl were removed
in vacuo and the resultant solid recrystallized from methanol / diethyl ether
to provide the HC1 salt of the
target (53 mg) as a white solid: mp 230-233°C; 1 H NMR (300 MHz, CD~OD)
8 7.36-7.28 (m, 3H),
6.99-6.90 (m, 4H), 4.67 (dd. J=6.0, 6.0 Hz, 1 H), 4.58 (bs, 1 H), 3.87 (dd,
J=6.0, 6.0 Hz, 1 H), 3.57 (m,
l H), 3.08 (s, 3H); IR (KBr) 2931, 2473, 1625, 1598, 1462, 11 19 cm-1; CI MS
m/z = 260
[C 16H I 5F2N+HJ+; Anal. Calcd. for C 16H 15F2N-HCI-0.1 H20: C, 64.58; H,
5.49; N, 4.71. Found: C,
64.45; H, 5.43; N, 4.49.
Example 85
Preparation of (3,5-difluoro)-4-t~henvl-1.2.7-trimethvl-1,2,3.4-
tetrahvdroisoquinoline
Std A: Nitromethane ( 1.6 mL, 30 mmol) was added dropwise to an ice-cold
solution of
tetrabutylammonium fluoride (7.5 mmol) in dry THF (20 mL). A solution of 3,5-
difluorobenzaldehyde
(2.85 g, 20.1 mmol) in dry THF (5 mL) was added dropwise. Triethvlamine (2.8
mL, 20 mmol) was
-51-
WO 01/32624 CA 02389300 2002-04-29 PCT/IJS00/30328
then added dropwise. A solution of tert-butyldimethylsilyl chloride (4.54 g,
30.1 mmol) in dry THF (15
mL) was added dropwise, causing a white precipitate to form. The reaction was
stirred at 0 °C for 30
min and then was filtered. The solid was washed with ether/hexanes. The
filtrate was washed (2 x) with
water. The organic layer was dried over MgS04, filtered, and concentrated
under reduced pressure
leaving a yellow oil. The yellow oil was purified by column chromatography on
silica gel (300 g)
eluting with 30% EtOAc/hexanes to give compound the product (2.65 g, 65%) as a
colorless oil: 'H
NMR (300 MHz, CDCI;) 8 6.98-6.95 (m, 2H), 6.80 (tt, J = 8.8, 2.3 Hz, I H),
5.49-5.44 (m, 1 H), 4.56-4.53
(m, 2H), 3.00 (d, J = 2.9 Hz, 1H).
Step B: A slurry of the product from Step A (2.35 g, 1 1.6 mmol) and platinum
oxide (0.20 g) in
absolute ethanol (20 mL) was hydrogenated at 40 psig for 4 h. The reaction was
filtered throgh a plug of
Celite, which was washed with additional absolute ethanol. The solvent was
removed in vacuo leaving
the amine product (1.97 g, 98%) as a white solid: mp 54-58 °C; 'H NMR
(300 MHz, CDzOD) b 7.01-
6.98 (m, 2H), 6.87-6.81 (m, 1 H), 4.70 (dd, J = 8.2, 3.8 Hz, 1 H), 2.90 (dd, J
= 13.0, 3.8 Hz, 1 H), 2.76
(dd, J = 13.0, 8.2 Hz, 1 H).
Step C: A solution of 3-methylacetophenone (1.36 g, 10.1 mmol) and the product
from Step B
(1.75 g, 10.1 mmol) in toluene (20 mL) was heated at reflux with azeotropic
removal of water for 4 h
under nitrogen. The toluene was removed in vacuo leaving an orange oil. To an
ice-cold solution of the
orange oil in methanol ( 10 mL), was added NaBH4 (0.44 g, 12 mmol). The
reaction was stirred for 1 h at
0 °C and then slowly allowed to warm to room temperature over 4 h. The
reaction was concentrated
under reduced pressure. The residue was taken up in water and extracted (3 x)
with ether. The
combined organic extracts were dried over Na=SO~, filtered, and concentrated
in vacuo to give the
product as a mixture of diastereomers (3.00 g, > 100%) as a yellow oil: ~H NMR
(300 MHz, CDCI~) b
7.22-7.18 (m, 2H), 7.08-7.06 (m, 2H), 6.91-6.8 I (m, 2H), 6.70-6.64 (m, 1 H),
4.69-4.45 (m, I H), 3.81-
3.67 (m, 1H), 2.83-2.75 (m, 1H), 2.58-2.40 (m, IH), 2.34 (s, 3H), 1.39-1.36
(m, 3H).
Step D: Concentrated H,SO~ ( 12.0 mL) was added to a stirred, ice-cold
solution of the crude
product from Step C (3.00 g, 10.3 mmol) in CH~C12 (105 mL). After stirring 15
min, the mixture was
poured onto ice, made strongly alkaline with excess conc. NHaOH, and extracted
(2 x) with Et,O. The
combined organic extracts were dried over Na,SO;, filtered, and the solvent
was removed in vacuo. The
residue (1.75 g) was purified by column chromatography on silica gel (145 g)
eluting with 10%
EtOAc/hexanes containing 1 % Et,N and then 20% EtOAc/hexanes containing 1 %
Et;N to afford the
product, a mixture of diastereomers, (426 mg, 15%) as a yellow oil: 'H NMR
(300 MHz, CDCIz) 8 7.04-
6.61 (m, 6H), 4.22-3.99 (m, 2H), 3.49-3.29 (m, 1 H), 3.19-2.92 (m, 1 H), 2.34-
2.32 (m, 3H), 1.52-1.47 (m,
3H).
Step E: Formaldehyde (37 wt°~o, 0.70 mL, 9.4 mmol) was added to a
solution of the product
from Step D (426 mg, 1.56 mmol) in methanol ( 16 mL). After 1.5 h, Raney
nickel (0.51 g) was added.
-52-
WO 01/32624 CA 02389300 2002-04-29 PCT/LTS00/30328
and the reaction was hydrogenated at 35 psig for 21 h. The reaction was
filtered through a pad of Celite,
which was washed with methanol. The filtrate was evaporated in vacuo, leaving
a milky liquid, which
was extracted with ether. The ether extract was dried over Na2S04, filtered,
and the solvent was removed
in vacuo. The residue (392 mg) was purified by column chromatography on silica
gel ( 150 g) eluting
with 10% EtOAc/hexanes containing I % Et,N to give the desired compound (82
mg, 18%) as a colorless
oil.: 'H NMR (300 MHz, CDCI,) b 6.97 (s, 1 H), 6.92 (d, J = 7.7 Hz, 1 H), 6.78-
6.61 (m, 4H), 4.11 (t, J =
6.4 Hz, 1 H), 3.65 (q, J = 6.6 Hz, 1H), 3.04-2.86 (m, 2H), 2.45 (s, 3H), 2.32
(s, 3H), 1.45 (d, J = 6.6 Hz,
3H).
Step F : A 1 M HCl solution in ether (1.0 mL, 1.0 mmol) was added dropwise to
a stirred
solution of of the product from Step E (82 mg, 0.28 mmol) in methanol (3 mL).
After 30 min, the
solvents and excess HCl were removed in vacuo, and the residue precipitated
from ether and sonicated
for 30 min. The off-white solid was isolated by filtration and then dried at
room temperature under
vacuum for 24 h to give the product (78 mg, 83%) as an off-white solid: mp 194-
197 °C (with
decomposition); 'H NMR (300 MHz, CD,OD) 8 7.14-7.12 (m, 2H), 7.00-6.81 (m,
4H), 4.65-4.59 (m,
2H), 3.66-3.64 (m, 2H), 3.03 (s, 3H), 2.35 (s, 3H), 1.75 (d, J = 6.5 Hz, 3H);
IR (KBr) 2928, 2480, 1624,
1599, 1464, 1119, 975, 859 cm-'; CI MS m/z = 288 [C~HH,yFzN+H]'; HPLC >99%, t,
= 16.96 min; Anal.
Calcd. for C,nH~~,F,N-HCl-0.25H=O: C, 65.85; H, 6.29; N, 4.27. Found: C,
65.98; H, 6.12; N, 4.16.
Example 89
Preparation of (8-fluoro-2-methyl-4-phenyl-1.2 3,4-tetrahydro-7-isoquinolinvl)-
N-methylmethanamine
Ste~A: Methylamine (15.3 mL, 40% aq. solution, 177 mmol) was added to a
stirred solution of
3-fluorobenzaldehyde (20.0 g, 161 mmol) in MeOH (150 mL) at room temperature.
After stirring for 6
h, the reaction was cooled to 0 °C and then NaBH, (6.10 g, 161 mmol)
was added portionwise. The
cooling bath was removed and the reaction was warmed to room temperature and
stirred for 16.5 h. The
reaction was quenched with H,O, and cautiously acidified with 2 N HCI, and
then extracted (3 x) with
CH~CI=. The aq. phase was then basified using 6 N NaOH and then extracted (4
x) with CH=Cl,. The
latter organic extracts were combined, dried over Na~SO~, filtered, and
concentrated in vacuo to afford
the product (21.51 g. 96%), as a clear oil: ~H NMR (300 MHz, CDCI,) 8 7.32
(td, J = 7.5, 1.7 Hz, 1 H),
7.28-7.19 (m, 1 H), 7.14-6.98 (m, 2H), 3.80 (s, 2H), 2.45 (s, 3H), 1.47 (6r s,
1 H).
Steo B: Triethylamine (8.40 mL, 60.0 mmol) was added to a stirred solution of
the product from
Step A (8.35 g, 60.0 mmol) and phenacyl bromide (11.94 g, 60.0 mmol) in CH,CI~
(200 mL) at room
temperature under N,. After stirring for 18 h, the reaction was quenched with
a mixture 10:1 mixture of
H~O/6 N NaOH (33 mL) and organic layer was dried over Na~SO~, filtered, and
the solvent evaporated in
vacuo, affording crude product ( 17.08 g, theoretical = 15.44 g), as a yellow
oil: 'H NMR (300 MHz,
CDCI~) 8 8.00-7.94 (m, 2H), 7.59-7.52 (m, 1 H), 7.48-7.37 (m, 3H), 7.30-7.21
(m, 1 H), 7.15-7.10 (m,
2H), 3.85 (s. 2H). 3.79 (s, 2H), 2.39 (s, 3H): IR (CH,CI, solution) 3055,
2925, 2850, 1682. 1598, 1490,
-53-
WO 01/32624 CA 02389300 2002-04-29 PCT/US00/30328
1450, 1266, 1225, 738, 703 cm-'; CI MS m/z = 258 [C,~H~~FNO+H]+. This material
was used without
further manipulation.
Step C: Sodium borohydride (4.54 g, 120 mmol) was added portionwise to a
stirred solution of
the product from Step B (17.1 g, 60.0 mmol) in MeOH (150 mL), cooled to 0
°C under N2. After
stirring for 4.5 h at room temperature, the reaction was diluted with HZO (300
mL) and extracted (4 x)
with CHzCI,. The organic extracts were combined, washed with sat. NaCI, dried
over NazS04, filtered,
and the solvent evaporated in vacuo. Chromatography of the residual yellow oil
(15.81 g) using silica
(200 g) and elution with 50% EtOAc/hexanes afforded the product (14.81 g, 95%
over 2 steps), as a
yellow oil: 'H NMR (300 MHz, CDCI,) 8 7.39-7.22 (m, 7H), 7.15-7.01 (m, 2H),
4.75 (dd, J = 8.3, 5.6
Hz, 1 H), 3.79 (d, J = 13.3 Hz, I H), 3.64 (d, J = 13.3 Hz, 1 H), 2.65-2.53
(m, 2H), 2.33 (s, 3H); IR
(CHzCl2 solution) 3062, 2849, 1587, 1491, 1455, 1333, 1266, 1228, 1094, 1062,
1023, 897, 877, 758,
738, 701 cm-'; CI MS m/z = 260 [C,~H,hFNO+H]'.
St-ep D: Cone. sulfuric acid (24 mL) was added dropwise to a stirred solution
of the product
from Step C (14.8 g, 57.1 mmol) in CH=Cl~ (280 mL), cooled to 0 °C,
using an ice-water bath. The
cooling bath was removed after addition was complete and the reaction was
vigorously stirred at room
temperature for 20 min. The reaction was then poured into an ice / water
mixture (400 mL) and the
resultant mixture basified with cone. NH~OH solution to pH - 10. The aq. layer
was extracted (3 x) with
CHzCI,. The organic extracts were combined, washed with a 2:1 mixture of sat.
NaCI/1 N NaOH, dried
over Na,S04, filtered and concentrated in vacuo. Chromatography of the residue
( 13.91 g) on silica (450
g) and elution with 33% EtOAc/hexanes afforded the product ( 12.66 g, 92%), as
a yellow oil: 'H NMR
(300 MHz, CDCI~) 8 7.33-7.15 (m, SH), 7.08-6.98 (m, 1 H), 6.90-6.82 (m, 1 H),
6.66 (d, J = 7.7 Hz, 1 H),
4.30-4.22 (m, 1 H), 3.86 (d, J = 15.6 Hz, 1 H), 3.53 (d, J = 15.6 Hz, 1 H),
3.02 (dd, J = 11.4, 5.6, 1.1 Hz,
1H), 2.57 (dd, J = 11.6, 8.7 Hz, 1H), 2.47 (s, 3H); IR (CH,CI, solution) 2941,
2782, 1583, 1494, 1468,
1457, 1378, 1248, 1139, 1040, 887, 792, 764, 736, 701 cm-~; CI MS m/:, = 242
[C,fiH,~FN+H]'.
Step E: t-Butyl lithium (30 mL, 1.7 M in pentane, 50.5 mmol) was added
dropwise to a stirred
solution of the product from Step D (5.50 g, 22.8 mmol) and TMEDA (7.6 mL,
50.2 mmol) in Et_O ( 120
mL) cooled to -60 °C under N,. After stirring for 45 min, DMF (7.0 mL,
91.2 mmol) was added and the
reaction mixture was stirred at -60 °C for 1.5 h. The reaction was
quenched with MeOH ( 10 mL),
warmed to room temperature, and then diluted with H,O (200 mL) and the aqueous
layer was extracted
(4 x) with CH~C12. The combined CH,CI= extract was dried over Na,SOa, filtered
and concentrated in
vacuo. Chromatography of the residue (9.05 g) on silica (350 g) and elution
with 33% EtOAc/hexanes
afforded the product (1.21 g, 20%), as a brown oil: 'H NMR (300 MHz, CDCI,) ~
10.32 (s, 1H), 7.56 (t,
J = 7.6 Hz, 1 H), 7.34-7.21 (m, 3H), 7.19-7.10 (m, 2H), 6.79 (d, J = 8.2 Hz,
IH), 4.31-4.23 (m, 1H), 3.90
(d, J = 15.8 Hz, 1 H), 3.58 (d, J = 15.8 Hz, 1 H), 3.04 (dd, J = 11.9, 5.6,
1.0 Hz, I H), 2.61 (dd, J = 11.7,
8.3 Hz, IH), 2.49 (s, 3H); CI MS ntl= 270 [C"H~fiFNO+H)'
-54-
WO 01/32624 CA 02389300 2002-04-29 PCT/US00/30328
Step F: Methylamine (0.05 mL, 40% aq. solution, 0.62mmo1) was added to a
stirred solution of
impure aldehyde 147 (0.15 g, 0.57 mmol) in MeOH (3 mL) at room temperature.
After stirring for 6 h,
the reaction was cooled to 0 °C and then NaBH~ (0.022 g, 0.57 mmol) was
added. The cooling bath was
removed and the reaction was warmed to room temperature and stirred for 18 h.
The reaction was
quenched with H~O extracted (4 x) with CH~CI=. The organic extracts were
combined, dried over Na,S04,
filtered, and concentrated in vacuo. Chromatography of the residue (0.18 g)
using silica ( 10 g) and
elution with 88:12:1 CHCIz:MeOH:conc. NH~OH afforded methylamine 147 (0.10 g),
as a brown oil: 'H
NMR (300 MHz, CDCI,) 8 7.32-7.12 (m, 5H), 7.02 (t, J = 7.8 Hz, 1 H), 6.63 (d,
J = 7.9 Hz, 1 H), 4.28-
4.20 (m, 1 H), 3.86 (d, J = 15.6 Hz, 1 H), 3.75 (s, 2H), 3.52 (d, J = 15.6 Hz,
1 H), 3.00 (dd, J = 11.3, 5.6,
0.9 Hz, 1 H), 2.55 (dd, J = 11.5, 8.7 Hz, I H), 2.46 (s, 3H), 2.43 (s, 3H); CI
MS m/z = 285
[C~RH2~FN=+H]'.
Step G: An ethereal HCl solution (1.80 mL, 1 N, 1.80 mmol) was added to a
solution of the
product from Step F (0.10 g, 0.35 mmol) in MeOH (0.5 mL) and Et,O (5 mL) at
room temperature,
resulting in the formation of a off-white solid. The solid was isolated and
then recrystallized from
MeOH/Et,O (3x) and the solid was dried in vacuo (54 °C) to afford the
salt (0.083 g, 66%) as a light
green solid: mp 185-205 °C; 'H NMR (300 MHz, CD;OD) 8 7.50-7.24 (m,
6H), 6.86-6.78 (m, 1 H), 4.80-
4.50 (m, 3H), 4.29 (s, 2H), 3.92-3.83 (m, 1 H), 3.70-3.55 (m, 1 H), 3.15 (s,
3H), 2.76 (s, 3H); IR (KBr)
3422, 2956, 2698, 1635, 1497, 1456, 1218, 1032, 895, 770, 703, 560 cm-'; CI MS
m/z = 285
[C,~H=,FN=+H]'; HPLC 95.5%, t, = 10.96 min; Anal. Calcd. for C,~H_,FN_-
2HC1Ø5H,0: C, 59.02; H,
6.60; N, 7.65. Found: C, 59.13; H, 6.73; N, 7.42.
Example 90
Preparation of (2-methyl-4-phenyl-7-isoquinolinyl)-N-methylmethanamine
Step A: Methylamine (40 wt% aqueous, 2.6 mL, 30 mmol) was added to a stirred
solution of 3-
bromobenzaldehyde (5.44 g, 29.4 mmol) in MeOH (30 mL) under N~. After stirring
1 h, the colorless
solution was cooled to 0 °C and then NaBHa (0.60 g, 16 mmol) was added
portionwise. After stirring 1
h, the cooling bath was removed. After stirring for 90 min, the reaction was
cooled to 0 °C and then
phenacyl bromide (5.90 g, 29.6 mmol) was added portionwise over 30 min. The
reaction was allowed to
warm to room temperature. After stirring for 2 h at room temperature, the
solution was cooled to 0 °C
and then NaBHa ( 1.20 g, 31.7 mmol) was added portionwise over 10 min. The
solution was stirred for
24 h, during which time the temperature rose from 0 ° to 25 °C.
The solution was diluted with H,O (400
mL), extracted (4 x) with ether. The ether extracts were dried over Na,SOa,
filtered, and the solvent
removed in vacuo to give the product (9.21 g, 98%) as a yellow oil: 'H NMR
(300 MHz, CDC1.) b 7.47-
7.21 (m, 9H), 4.77 (dd, J = 10.0, 4.0 Hz, 1 H), 3.71 (d, J = 13.3 Hz, 1 H),
3.51 (d, J = 13.3 Hz, 1H), 2.61-
2.49 (m, 2H), 2.32 (s, 3H).
-55-
WO 01/32624 CA 02389300 2002-04-29 pCT/US00/30328
Ste~B: Conc. HzSO~ (40.0 mL) was added dropwise over 15 min to a stirred
solution of the
product from Step A (9.18 g, 28.7 mmol) in CH~Cl2 (300 mL). After stirring 45
min, the mixture was
poured onto ice, made strongly alkaline with excess conc. NH~OH, extracted (3
x) with Et20. The ether
extracts were dried over Na=S04, filtered, the solvent was removed in vacuo,
and the residue (7.29 g) was
purified by column chromatography on silica gel (300 g) eluting with 10%
EtOAc/hexanes containing
I % Et,N the product (2.05 g, 24%) as an orange oil: 'H NMR (300 MHz, CDCI,) 8
7.32-7.27 (m, 4H),
7.25-7.14 (m, 3H), 6.74 (d, J = 8.3 Hz, 1 H), 4.22-4.17 (m, 1 H), 3.71 (d, J =
15.1 Hz, 1 H), 3.57 (d, J =
15.1 Hz, 1 H), 3.05-2.99 (m, 1 H), 2.54 (dd, J = 11.5, 8.7 Hz, 1 H), 2.42 (s,
3H).
Sten C: A slurry of bromide the product from Step B ( I .15 g, 3.81 mmol),
zinc cyanide (271 mg,
2.31 mmol) , and tetrakis(triphenylphosphine)palladium(0) (266 mg, 0.230 mmol)
in dry DMF (5 mL)
was heated at 83 °C for 24 h. After allowing the reaction to cool to
room temperature, the reaction was
diluted with toluene and washed with 2 N NaOH. The toluene extract was dried
over Na~SO,, filtered,
and concentrated in vacuo. The residue ( 1.20 g) was purified by column
chromatography on silica gel
(95 g) eluting with 20% EtOAc/hexanes containing 1 % Et,N to give the product
(673 mg, 71 %) as a
yellow solid: mp 103-104 °C; 'H NMR (500 MHz, CDCI,) 8 7.38 (s, 1H),
7.34-7.23 (m, 4H), 7.16-7.14
(m, 2H), 6.98 (d, J = 8.0 Hz, 1 H), 4.27 (t, J = 7.0 Hz, I H), 3.75 (d, J =
15.2 Hz, 1 H), 3.61 (d, J = 15.2
Hz, 1 H), 3.07-3.03 (m, 1 H), 2.59 (dd, J = 11.7, 8.4 Hz, 1 H), 2.44 (s, 3H);
CI MS m/z = 249
[CmHmN:+H]'.
St-e~D: A solution of the product from Step C (201 mg, 0.809 mmol) in dry THF
(4 mL) was
added dropwise to an ice-cold slurry of lithium aluminum hydride (61 mg, I .6
mmol) in dry THF (2
mL). The reaction was stirred for 90 min with cooling and then was allowed to
warm to room
temperature. The reaction was stirred for 5 h and then was quenched with EtOAc
and then a saturated
Na,SO~ solution. The reaction was diluted with ether, dried over solid Na_SO,,
filtered, and concentrated
in vacuo. The residue was purified by column chromatography on silica gel (26
g) eluting with l2olc
methanol/chloroform containing 1 % conc. NHaOH to give the product (134 mg,
66%) as a colorless oil:
'H NMR (300 MHz. CDCl.) b 7.31-7.18 (m, 5H), 7.04 (s, 1 H), 7.00 (d, J = 8.0
Hz, 1 H), 6.83 (d, J = 8.0
Hz, 1 H), 4.25 (t, J = 7.0 Hz, 1 H), 3.81 (s, 2H), 3.75 (d, J = 14.9 Hz, 1 H),
3.60 (d, J = 14.9 Hz, 1 H), 3.06-
3.00 (m, 1 H), 2.56 (dd, J = 11.4, 8.7 Hz, 1 H), 2.43 (s, 3H).
Sten E: A slurry of the product from Step D (53 mg. 0.21 mmol) and malefic
acid (25 mg, 0.22
mmol) in absolute EtOH (10 mL) was heated in a 40 °C water bath until
all of the solid had dissolved.
After 1 h, the reaction was concentrated in vacuo. The residue was
recrvstallized from ethanol/ether
producing the bis maleate salt (43 mg, 42%) as a green solid: mp 176-177
°C (with decomposition); 'H
NMR (300 MHz, CD;OD) 8 7.40-7.30 (m, 5H), 7.22 (dd, J = 8.0, l .3 Hz, 2H),
6.97 (d, J = 8.0 Hz, 1 H),
6.24 (s, 4H), 4.58 (dd. J = 11.3, 6.1 Hz, I H), 4.52 (s, 2H), 4.12 (s, 2H),
3.78 (dd, J = 12.3, 6.2 Hz, 1 H),
-56-
WO 01/32624 cA 02389300 2002-04-29 PCT/US00/30328
3.45 (t, J = 11.8 Hz, 1 H), 3.02 (s, 3H); HPLC 95.8%, t~ = 10.81 min; Anal.
Calcd. for C,~Hz~N~-
2(C,H404): C, 61.98; H, 5.82; N, 5.78. Found: C, 61.86; H, 5.82; N, 5.60.
Example 91
Preparation of N-methyl(2-methyl-4-phenyl-7-isoquinolinyl)-N-methylmethanamine
Ste~A: A 1 M HCl solution in ether (3.0 mL, 3.0 mmol) was added dropwise to a
solution of
the product from Step C, Example 90 (82 mg, 0.32 mmol) in methanol (6 mL). The
solvents and excess
HCl were removed in vacuo leaving a green solid. A slurry of this green solid,
potassium carbonate (199
mg, 1.44 mmol), and ethyl chloroformate (0.20 mL, 2.1 mmol) in methanol ( I
mL) and acetone (6 mL)
was heated at 50 °C for 20 h. After allowing the reaction to cool to
room temperature, the reaction was
diluted with brine and extracted (4 x) with EtOAc. The combined organic
extracts were dried over solid
Na~S04, filtered, and concentrated in vacuo leaving the carbamate product (99
mg, 88%) as an orange
oil: 'H NMR (300 MHz, CDCh) 8 7.31-7.14 (m, 5H), 6.98-6.93 (m, 2H), 6.83-6.76
(m, 1 H), 4.30-4.10
(m, SH), 3.77-3.58 (m, 2H), 3.07-3.01 (m, 1 H), 2.61-2.54 (m, 1 H), 2.43 (s,
3H), 1.24 (t, J = 7.1 Hz, 3H);
CI MS ml~ = 325 [C,"H,~N,O=+H]'.
Step B: Lithium aluminum hydride (60 mg, 1.6 mmol) was added in portions to a
solution of the
product from Step A (99 mg, 0.30 mmol) in dry THF (5 mL). The reaction was
heated at reflux for 6 h
and then allowed to cool to room temperature. The reaction was quenched with
EtOAc and then a
saturated Na,SO~ solution. The reaction was diluted with ether, dried over
solid Na:SOa, filtered, and
concentrated in vacuo. The residue (81 mg) was purified by column
chromatography on silica gel (8 g)
eluting with 12% methanol/chloroform containing 1 % cone NH~OH to give
compound the product (49
mg, 61 %) as a colorless oil: 'H NMR (300 MHz, CDCI~) S 7.32-7.17 (m, SH),
7.04 (s, 1 H), 7.00 (d, J =
8.0 Hz, 1 H), 6.82 (d, J = 8.0 Hz, I H), 4.26 (t, J = 7.1 Hz, 1 H), 3.83-3.57
(m, 4H), 3Ø7-3.01 (m, 1 H),
2.54 (dd, J = 1 I .4, 8.9 Hz, 1 H), 2.45 (s, 3H), 2.43 (s, 3H); CI MS m/<. =
267 [C,~H=_N=+H]'.
Stet/ C: A slurry of the product from Step B (20 mg, 0.075 mmol) and malefic
acid (9 mg, 0.08
mmol) in absolute EtOH (5 mL) was heated in a 40 °C water bath until
all of the solid had dissolved.
After 2 h, the reaction was concentrated in vacuo. The residue was
recrystallized from ethanol/ether
producing the bis maleate product (l3 mg, 35%) as a tan solid: mp 160-163
°C (with decomposition); ~H
NMR (300 MHz, CD,OD) 8 7.41-7.31 (m, SH), 7.24-7.21 (m. 2H), 6.99 (d, J = 8.0
Hz, 1 H), 6.24 (s,
4H), 4.57 (dd, J = 10.9, 5.7 Hz, 1 H), 4.50 (s, 2H). 4.18 (s, 2H), 3.76 (dd, J
= 12.3, 6.2 Hz, I H), 3.50-3.38
(m, 1H), 3.00 (s, 3H), 2.72 (s, 3H); HPLC 95.8°~0, t = 11.09 min.
Example 92
Preparation of 8-hydroxy-2-methyl-4-phenXl-1,2,3,4-tetrahydro-7-
isoduinolinecarbonitrile
St-ep A: A solution of N-methyl-2-methoxy amine (8.00 g, 52.9 mmol) and
triethylamine (5.40
g, 53.0 mmol) in dichloromethane ( 100 mL) was cooled in an ice water bath.
The 2-bromoacetophenone
(10.5 g, 53.0 mmol) was added, and the reaction was allowed to warm to room
temperature. The
-57-
WO 01/32624 CA 02389300 2002-04-29 pCT/LTS00/30328
reaction mixture was diluted with water (200 mL) and MTBE (200 mL). Layers
were separated, and the
organic layer was washed with HZO and brine. The organic layer was dried over
MgS04, filtered, and
concentrated to yield a red oil which was chromatographed (SiO:, 20%
EtOAc/hexanes) to yield the
desired amino ketone as ayellow oil (12.6 g, 89%): 'H NMR (300 MHz, CDCI,) 8
7.97 (d, J = 7.4 Hz,
2H), 7.53-7.50 (m, 1H), 7.41 (t, J=7.5 Hz, 2H), 7.32 (d, J = 7.4 Hz, 1H), 7.28-
7.21 (m, 1H), 6.92 (t, J =
7.5 Hz, 1 H), 6.85 (d, J = 8.1 Hz, 1 H), 3.81 (s, 2H), 3.77 (s, 3H), 3.73 (s,
2H), 2.39 (s, 3H).
St_~ B: The product from Step A (12.6 g, 46.8 mmol) was taken up in methanol
(120 mL) and
cooled in an ice-water bath. Sodium borohydride (1.76 g, 46.8 mmol) was added
portionwise. The
reaction was stirred for 1 h at ambient temperature. The reaction mixture was
concentrated to half of the
original volume. Water ( 100 mL) was added, and the mixture was extracted (3
x) with dichloromethane.
The combined organic layers were dried over MgSO~, filtered, and concentrated
to provide the desired
amino alcohol as a light yellow oil (10.0 g, 79%): 'H NMR (300 MHz, CDCI,) 8
7.39-7.21 (m, 6H),
6.94-6.85 (m, 3H), 4.78 (dd, J=4.3, 9.6 Hz, 1H), 3.85 (s, 3H), 3.82 (d, J=12.8
Hz, 1H), 3.47 (d, J=12.8
Hz, 1H), 2.62-2.57 (m, 2H), 2.28 (s, 3H).
St. ep C: Methanesulfonic acid (47.7 mL, 735 mmol) was added at ambient
temperature to a
solution of the product from Step B (4.20 g, 13.7 mmol) in dichloromethane
(250 mL). The reaction
mixture was stirred at room temperature under nitrogen for 24 h. After the
reaction was complete, the
reaction was made basic (pH ~ 11 ) with 2 N NaOH, and extracted (3 x) with
methylene chloride. The
combined organic layers were washed with brine, dried over MgSOa and
concentrated in vacuo. The
residue was purified by chromatography (Si0=, EtOAc/hexanes, 2/3) to give the
desired product as a
yellow oil (5.67 g, 61 %): 'H NMR (300 MHz, CDCI;) 8 7.30-7.15 (m, SH), 7.02
(t, J=8.0 Hz, 1 H), 6.65
(d, J=8.1 Hz, 1 H), 6.47 (d, J = 7.6 Hz, 1 H), 4.25 (t, J=6.8 Hz, I H), 3.82
(s, 3H), 3.81 (d, J=16.2 Hz, 1 H),
3.36 (d, J=16.2 Hz, 1 H), 2.96 (dd, J = 4.1, 15.3 Hz, 1 H), 2.58 (dd, J=8.5, 1
I .4 Hz, 1 H), 2.43 (s, 3H).
Step D: A solution of the product from Step C (5.60 g, 22.1 mmol) in 48%
hydrobromic acid
(60 mL) was refluxed at 100 °C for 3 h. The reaction mixture was
concentrated in vacuo and
recrystallized from ethanol to yield the desired product (4.74 g, 67): 'H NMR
(300 MHz, DMSO-d~) 8
9.92 (s, 1 H), 7.48-7.25 (m, 3H), 7.21 (d, J=7.8 Hz, 1 H), 6.98 (t, J=7.7 Hz,
I H), 6.67 (d, J=7.8 Hz, 1 H),
6.24 (d, J=7.7 Hz, I H), 4.26 (t, J = 6.0 Hz, 1 H), 3.80 (d, J = 15.8 Hz, 1
H), 3.32 (d, J = 15.8 Hz, 1 H), 2.99
(dd, J = 5.2, l 1.3 Hz, 1 H), 2.66 (dd, J = 7. I , I I .4 Hz, I H), 2.39 (s,
3H).
Step E: A mixture of the product from step D (4.70 g, 14.7 mmol) and
hexamethylenetetramine
(2.06 g, 14.7 mmol) in trifluoroacetic acid (50 mL) was heated to 80 °C
for 7 h. The reaction mixture
was concentrated in vacuo then diluted with water (100 mL). The solution was
made basic with solid
Na,CO.. The resulting solution was extracted with ethyl ether (3 x), and the
combined organic layers
were concentrated in vacuo. The residue was purified by chromatography (Si0=,
EtOAc/hexanes, 4/1 ) to
afford the desired product as an off-white solid (2.47 mg, 49%): 'H NMR (500
MHz, CDCI,) b 11.42
-58-
WO 01/32624 CA 02389300 2002-04-29 pCT/[JS00/30328
(bs, 1H), 9.82 (s, 1H), 7.28 (d, J=8.1 Hz, 1H), 7.12-6.90 (m, 3H), 6.54 (d,
J=8.1 Hz, 1H), 4.19 (t, J=6.1
Hz, 1H), 3.72 (d, J=16.1 Hz, 1H), 3.62 (d, J=16.2 Hz, 1H), 2.93 (dd, J=11.9,
6.28 Hz, 1H), 2.60 (dd,
J=11.4, 7.0 Hz, 1H), 2.47 (s, 3H).
Ste~F: The product from Step E (I.00 g, 2.87 mmol) was dissolved in water (20
mL) before
treatment with sodium sulfate (100 mg) and hydroxyl amine sulfonate (0.32 mg
2.87 mmol). Reaction
was stirred for 2 h. Reaction was cooled in an ice-water bath and treated with
CHZCh (20 mL). Sodium
bicarbonate (600 mg) was added and the reaction was allowed to warm to ambient
temperature. The
solids were filtered off and combined with the organic layer. The mixture was
concentrated and
chromatographed (Si0" EtOAc/hexanes, 1/1). Two compounds eluted
simultaneously. The mixture was
treated with ethanol (5 mL) and filtered. The filtrate was concentrated to
yield the desired nitrile as an
off-white powder ( 130 mg, 17%): mp 234-238 °C (decomposed); 'H NMR
(300 MHz, CD,OD) 8 7.31-
7.14 (m, 6H), 6.40 (d, J=8.1 Hz, 1 H), 4.21 (t, J=6.1 Hz, 1 H), 4.12 (bs, 1
H), 3.61-3.50 (m, 2H), 2.72 (dd,
J=5.4, 11.7 Hz, 1H), 2.58 (dd, J=7.1, 11.5 Hz, 1H), 2.38 (s, 3H). IR (KBr)
3427, 3026, 2940, 2207,
1590, 1454 cm-'; ESI MS m/~ = 265 [C,~H,~N.O+H]'; HPLC 96.3%, t = 13.54 min.
Example 93
PreQaration of L-methyl4-phenyl-1 2 3 4-tetrahydro-7-isoquinolinyl)methanol
Step A: A solution of Step C, Example 90 ( 127 mg, 0.511 mmol) in dry toluene
( 13 mL) was
cooled to -16 °C and then 1 M DIBAL-H in toluene ( 1.7 mL, 1.7 mmol)
was added dropwise. The
reaction was stirred for 45 min with cooling and then EtOAc ( 1.1 mL) was
added. The reaction was
allowed to warm to room temperature. The reaction was stirred for 45 min and
then 1 N H,SO, ( 12 mL)
was added. The reaction was heated at reflux for 30 min. After allowing the
reaction to cool to room
temperature, the reaction was diluted with water, made basic with 2 N NaOH,
and extracted (2 x) with
CH=Cl=. The CH,CI. extracts were dried over Na,SO~, filtered, and concentrated
in vacuo to give the
desired product (112 mg, 87%) as a yellow oil: 'H NMR (300 MHz, CDCI,) b 9.95
(s, 1H), 7.62 (s, 1H),
7.59-7.56 (m, 1 H), 7.34-7.16 (m, SH), 7.05 (d, J = 8.0 Hz, 1 H), 4.32 (t, J =
7.1 Hz, I H), 3.84 (d, J = 15.1
Hz, 1H), 3.67 (d, J = I5.1 Hz, 1H), 3.10-3.04 (m, IH), 2.60 (dd, J = I 1.6,
8.6 Hz, 1H), 2.46 (s, 3H).
Step B: To an ice-cold solution of the product from Step A (110 mg, 0.438
mmol) in methanol
(20 mL) was added NaBH, (36 mg, 0.95 mmol). The reaction was slowly allowed to
warm to room
temperature overnight. The reaction was quenched with water and brine and then
was extracted (3 x)
with CH=Cl,. The combined organic extracts were dried over Na,SO~, filtered,
and concentrated under
reduced pressure. The residue ( 106 mg) was purified by column chromatography
on silica gel (31 g)
eluting with EtOAc to give the desired alcohol (44 mg, 40%) as a yellow oil:
'H NMR (300 MHz.
CDCI,) 8 7.32-7.22 (m, 3H). 7.17 (dd, J = 6.6, 1.6 Hz, 2H), 7.03 (d, J = 7.6
Hz, 1 H), 7.02 (s, 1 H), 6.83
(d, J = 7.6 Hz, 1 H), 4.61 (s, 2H), 4.26 (dd, J = 8.6, 6.0 Hz, I H), 3.69 (d,
J = 14.9 Hz, 1 H), 3.55 (d, J =
14.9 Hz, IH), 3.07-3.01 (m, IH). 2.53 (dd..l= I I.S, 9.1 Hz, IH). 2.42 (s,
3H).
-59-
WO 01/32624 CA 02389300 2002-04-29 PCT/i1S00/30328
Ste~C: A 1 M HCl solution in ether (1.0 mL, 1.0 mmol) was added dropwise to a
stirred
solution of theproduct from Step B (44 mg, 0.17 mmol) in MeOH (2 mL). The
solvents and excess HCl
were removed in vacuo, and the residue recrystallized from MeOH-Et=O to give
the salt (32 mg, 62%) as
a green solid: mp 237-240 °C (with decomposition); 'H NMR (300 MHz,
CDzOD) 8 7.42-7.31 (m, 3H),
7.27-7.23 (m, 4H), 6.88 (d, J = 7.2 Hz, 1 H), 4.60 (bs, SH) 3.84 (dd, J = 12.4
, 6.0 Hz, 1 H), 3.65-3.45 (m,
1H), 3.08 (s, 3H); IR (KBr) 3356, 2934, 2596, 1495, 1456, 1428, 1049, 758, 703
cm-'; ESI MS m/z =
254 [C,~H~~NO+H]'; HPLC 94.9%, t = 12.83 min; Anal. Calcd. for C"H,yNO-HCI-
0.33 H,O: C, 69.03;
H, 7.04; N, 4.74. Found: C, 68.89; H, 6.87; N, 4.61.
Example 94
Preparation of 2-ethyl-4-phenyl-1,2,3,4-tetrahydroisoquinoline
Steo A: Ethylene glycol dimethyl ether (20 mL) and 2 N NazCOa ( 12.2 mL) were
sparged with
Nz and charged to a round bottom flask containing 4-bromoisoquinoline (2 g,
9.6 mmol), phenylboronic
acid ( 1.76 g, 14.4 mmol), and Pd(PPh;)~ ( 1.11 g, 0.96 mmol). The entire
solution was sparged with N..
The resulting reaction mixture was heated to reflux under N_ overnight. The
solution was cooled,
quenched with saturated NaHCO~ (230 mL), and extracted five times with ethyl
ether. The combined
organic was dried over Na~SO;, filtered, and the solvent was removed in vacuo
to yield an orange oil.
Column chromatography (1:1 ethyl acetate/hexanes) afforded the pure
isoquinoline as a yellow oil which
crystallized upon refrigeration (2.21 g). 'H NMR (300 MHz, CDCI,) 8 9.29 (s,
1H), 8.52 (s, 1H), 8.04
(d, 1 H, J = 8.4 Hz), 7.91 (d, 1 H, J = 8.1 Hz), 7.66 (m, 2H), 7.46 (m, SH).
Step B: Ethyl triflate (383 mg, 2.15 mmol) was added dropwise to a solution of
the product
from Step A (400 mg, I .95 mmol) in CH,CI, (24 mL) at 0"C under N~. The
solution was stirred for I S
min. at room temperature. The solvent was removed in vacuo to yield the
triflate salt of the isoquinoline
as a white solid (420 mg, 56% yield). The triflate salt (420 mg, 1.09 mmol)
was dissolved in MeOH (16
mL), and NaCNBH, ( 159 mg, 2.53 mmol) was added to the solution. The resulting
reaction mixture was
stirred for 5 min., and a few drops of bromocresol green in MeOH were added.
Methanolic HCI was
added to the solution until a yellow color was observed. The reaction mixture
was stirred at room
temperature for 30 min, while adding methanolic HCl as needed to maintain a
yellow color. The
reaction mixture was quenched with H,O (100 mL) and basified with 5% NaOH
until a blue color was
observed. The resulting solution was extracted four times with ethyl ether.
The combined organic was
washed with brine, dried over MgSOa, filtered, and solvent was removed in
vacuo to yield the
tetrahydroisoquinoline product as a clear oil (140 mg, 30% yield).
Step C: The maleate salt was prepared by adding malefic acid (68 mg, 0.59
mmol) and EtOH (2 mL) to
the product from Step B . After refrigeration and removal of EtOH, a white
solid was obtained (130
mg), mp=172-174°C. Free base: 'H NMR CDCIz 8 7.17 (m, 8H), 6.85 (d, 1H,
J = 7.7 Hz), 4.28 (t, IH, J
-60-
WO 01/32624 CA 02389300 2002-04-29 PCT/US00/30328
= 7.5 Hz), 3.89 (d, 1 H, J = 14.65 Hz), 3.62 (d, 1 H, J = 14.65 Hz), 3.15 (dd,
IH, J = 5.7, 11.7 Hz), 2.57
(m, 2H), 1.16 (t, 3H, J = 7.2 Hz).
Binding Assays
Primary binding assts:
In order to evaluate the relative affinity of the various compounds at the NE,
DA and 5HT
transporters, HEK293E cell lines were developed to express each of the three
human transporters.
cDNAs containing the complete coding regions of each transporter were
amplified by PCR from human
brain libraries. The cDNAs contained in pCRII vectors were sequenced to verify
their identity and then
subcloned into an Epstein-Barr virus based expression plasmid (E. Shen, GM
Cooke, RA Horlick, Gene
156:235-239, 1995). This plasmid containing the coding sequence for one of the
human transporters was
transfected into HEK293E cells. Successful transfection was verified by the
ability of known reuptake
blockers to inhibit the uptake of tritiated NE, DA or SHT.
For binding, cells were homogenized, centrifuged and then resuspended in
incubation buffer
(50mM Tris, 120mM NaCI, 5mM KCI, pH 7.4). Then the appropriate radioligand was
added. For NET
binding, ['H] Nisoxetine (86.0 Ci/mmol, NEN/DuPont) was added to a final
concentration of
approximately 5 nM. For DAT binding, ['H] WIN 35,428 (84.5 Ci/mmol) at 15 nM
was added. For
5HTT binding, [;H] Citolapram (85.0 Ci/mmol) at 1 nM was added. Then various
concentrations (10~-5
to 10~-11 M) of the compound of interest were added to displace the
radioligand. Incubation was carried
out at room temperature for 1 hour in a 96 well plate. Following incubation,
the plates were placed on a
harvester and washed quickly 4 times with (SOmM tris, 0.9% NaCI, pH 7.4) where
the cell membranes
containing the bound radioactive label were trapped on Whatman GF/B filters.
Scintillation cocktail was
added to the filters which were then counted in a Packard TopCount. Binding
affinities of the
compounds of interest were determined by non-linear curve regression using
GraphPad Prism 2.01
software. Non-specific binding was determined by displacement with 10
micromolar mazindol.
TBZ assay:
In order to assess in vivo activity of the compounds at the NE and DA
transporters, their ability
to prevent the sedative effects of tetrabenazine (TBZ) was determined (G.
Stille, Arzn. Forsch 14:534-
537, 1964). Male CFI mice (Charles River Breeding Laboratories) weighing 18-25
gm at the time of
testing, are housed a minimum of06 days under carefully controlled
environmental conditions (22.2 +
I .1 C; 50% average humidity; 12 hr lighting cycle/24 hr). Mice are fasted
overnight ( 16-22 hr) prior to
testing. Mice are placed into clear polycarbonated "shoe" boxes ( 17 cm x 28.5
cm x 12 cm). Randomized
and coded doses of test compounds are administered p.o. A 45 mg/kg dose of
tetrabenazine is
administered i.p. 30 minutes prior to score time. All compounds are
administered in a volume of 0.1
m1/10 gm body weight. Animals are evaluated for antagonism of tetrabenazine
induced exploratory losa
-61-
mg), mp=172-174°C. Free base: '
WO 01/32624 CA 02389300 2002-04-29 PCT/US00/30328
and ptosis at specified time intervals after drug administration. At the
designated time interval, mice are
examined for signs of exploratory activity and ptosis. Exploratory activity is
evaluated by placing the
animal in the center of a 5 inch circle. Fifteen seconds are allowed for the
animal to move and intersect
the perimeter. This is considered antagonism of tetrabenazine and given a
score of 0. Failure to leave the
circle is regarded as exploratory loss and given a score of 4. An animal is
considered to have ptosis if its
eyelids are at least 50% closed and given a score of 4 if completely closed.
No closure is given a score
of 0. Greater than 95% of the control (vehicle-treated) mice are expected to
exhibit exploratory loss and
ptosis. Drug activity is calculated as the percentage of mice failing to
respond to the tetrabenazine
challenge dose.
Statistical evaluation.:
Median effective doses (EDS~s) and 95% confidence limits are determined
numerically by the
methods of Thompson ( 1947) and Litchfield and Wilcoxon ( I 949).
-62-