Note: Descriptions are shown in the official language in which they were submitted.
CA 02389514 2002-04-30
WO 01/34561 PCT/EP00/10581
The present invention relates to processes for preparing phenylhydrazines
which
are useful intermediates in the preparation of indoles which are useful as
therapeutic agents.
Several indole derivatives are currently on the market or under development as
pharmaceuticals. For example, sumatriptan (Glaxo Wellcome) 3-[2-(dimethyl-
amino)ethyl]-I~methyl-1 H-indole-5-methanesulphonamide is currently used in
the treat-
ment of migraine. This compound is described in US 4,816,470 and US 5,737,845.
Other indoles of pharmaceutical interest are: almotriptan (WHO Drug
Information,
Vo1.10, No. 4, [1996]), avitriptan (WHO Drug Information, Vo1.10, No. 4,
[1996]), eletriptan
(WHO Drug Information, Vol.9, No. 4, [1995]), frovatriptan (Br.]. Pharmacol.
[119, Proc.
Suppl., 109P, 1996]), naratriptan, rizatriptan (WHO Drug Information, Vo1.10,
No. 2, [1996])
and zolmitriptan (J. Med. Chem. [38, No.l8, 3566-80, 1995]).
In the preparation of such indoles phenylhydrazines are key intermediates
which
may be cyclised into indoles using the well known Fischer indole synthesis. In
US 4,816,470
the method of preparation of such compounds involves the diazotisation of an
aniline
followed by reduction of the diazonium salt obtained with stannous chloride
dihydrate. We
have found that the use of tin reagents in this reduction presents a number of
problems.
Firstly there are environmental issues relating to the disposal of toxic
wastes containing tin
residues. Secondly, low temperature vessels are required to carry out the
reduction and
thirdly it is often difficult to remove the last traces of tin salts from the
intermediate and from
later stages of the reaction sequence.
Surprisingly a process has been found which uses a cheaper reducing agent
which
causes minimal environmental problems. In addition the process does not
require the use of
low temperature vessels, allows the telescoping of the process, i.e. that is
the combination of
more than one step, and increases the purity of the intermediate obtained and
further
products in the reaction sequence.
CA 02389514 2002-04-30
WO OI/34561 PCT/EP00/10581
2
The present invention provides a process for preparing a compound of formula I
including salts thereof
I
/ NHNH2
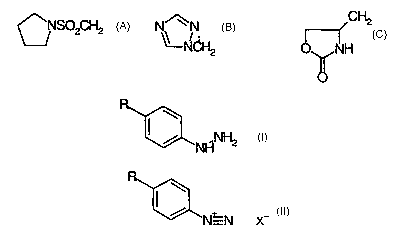
wherein R represents CH2S02NHCH3 , CH2CH2S02Ph, CH2CH2S02NHMe or a group of
structure:
CH2
~NS02CH2 N~ N
NCH2 NH
comprising the reduction of a diazonium salt of formula II
I I
/ NON X-
in which R is as previously defined and X- represents an anion derived from
hydrochloric
acid, sulphuric acid, acetic acid, phosphoric acid, tetrafluoroboric acid or
hydrobromic acid,
with a dithionite salt.
In another aspect the present invention provides a process for preparing a
compound of formula I or a salt thereof
I
/ NHNH2
in which R is as previously defined comprising the steps of
CA 02389514 2002-04-30
WO 01/34561 PCT/EP00/10581
3
a) reacting a compound of formula III or salt thereof
\ III
/ NH2
in which R is as previously defined with a diazotising agent optionally in the
presence of acid
to give a diazonium salt of formula II
I I
/ N~ N x-
in which R and X- are as previously defined and
b) reducing the diazonium salt with a dithionite salt to give the compound of
formula I.
Suitable salts of the compounds of formula I and I I I include acid addition
salts formed
with organic or inorganic acids for example hydrochlorides, hydrobromides,
sulphates,
nitrates, phosphates, formates, mesylates, citrates, benzoates, fumarates,
maleates,
toluene-p-sulphonates and succinates. Preferably the salt is the hydrochloride
or hydro-
bromide salt .
In a further aspect the present invention provides a process in which the
compound
of formula III or salt thereof as previously defined is prepared by reacting a
compound of
formula IV
\
IV
N02
in which R is as previously defined with a reducing agent, optionally in the
presence of an
acid or with an additional salt formation step eg reaction with an acid, where
a salt of the
compound of formula III is required. Preferably the compound of formula III is
used without
isolation.
CA 02389514 2002-04-30
WO 01/34561 PCT/EP00/10581
4
Consequently the present invention provides a process for the preparation of a
compound of formula I which comprises the steps of:
a) reacting a compound of formula IV
IV
N02
in which R is as previously defined with a reducing agent to give a compound
of
formula III
III
/ NH2
in which R is as previously defined and
b) reacting the compound of formula III with a diazotising agent optionally in
the
presence of acid to give a diazonium salt of formula II
\
I I
/ NON X-
in which R and X- are as previously defined and
c) reducing the diazonium salt with a dithionite salt to give the compound of
formula I.
CA 02389514 2002-04-30
WO 01/34561 PCT/EP00/10581
In a further aspect the present invention comprises a process for preparing a
compound of formula V
V
including pharmaceutically acceptable salts thereof in which R is as
previously defined and
R1 is a group of formula a), b), c), d), or e)
CH2CH2NH2 CH2CH2NMe2 NMe
) CH2
CH2CH2CH2~
N
~NMe ~N N
CH I
Me~~ , N
d) e)
comprising performing a Fischer Indole synthesis by reacting a compound of
formula I with
an aldehyde of formula VI in which R1 is as previously defined optionally in
the presence of
an acid catalyst.
R1 Ct~CHO
VI
Optionally a protected form of the aldehyde such as an acetal may be used this
reaction. Optionally an aldehyde or a protected form of the aldehyde of
formula VII may be
used where R2 is a group capable of being transformed into a group R1 upon
cyclisation to a
compound of formula V, for instance when R2 is -CH2CH2C1 to give compounds of
formula V
with R1 is -CH2CH2NH2 using the well known Grandberg version of the Fischer
Indole
synthesis.
R2CI-ECHO
VII
CA 02389514 2002-04-30
WO 01/34561 PCT/EP00/10581
6
Optionally it may be preferred to synthesise one compound of formula V by
reacting
another compound of formula V, for instance, when R1 is CH2CH2NMe2 by
reductive
alkylation of the compound V where R1 is CH2CH2NH2
Optionally it may be preferred to isolate the intermediate hydrazone VIII
R
NHN=CHCH2R1
VIII
In which R and R1 are as previously defined prior to performing the
cyclisation to V.
Suitably the diazotising agent is a metal nitrite salt or alkyl nitrite.
Preferably the
diazotising agent is sodium nitrite or butyl nitrite. Most preferably the
diazotising agent is
sodium nitrite.
Suitably the diazotising agent is present in the range of 0.5-3 molar
equivalents with
respect to the compound of formula II. Preferably the diazotising agent is
present in
the range of 0.8-1.5 molar equivalents with respect to the compound of formula
II. More
preferably the diazotising agent is present in the range of 0.9-1.1 molar
equivalents with
respect to the compound of formula II.
Optionally it may be preferred to add a reagent, for example sulphamic acid,
to
destroy excess nitrous acid at the end of this step.
Suitably the acid is hydrochloric acid, sulphuric acid, acetic acid,
phosphoric acid,
tetrafluoroboric acid or hydrobromic acid. Preferably the acid is hydrochloric
acid.
Suitably the acid is present in the range of 1-10 molar equivalents with
respect to the
compound of formula II. Preferably the acid is present in the range of 2-8
molar equivalents
with respect to the compound of formula II. More preferably the acid is
present in the range of
3-6 molar equivalents with respect to the compound of formula II.
Suitably the dithionite salt is a metal dithionite salt. Most preferably the
dithionite salt
is sodium dithionite.
CA 02389514 2002-04-30
WO 01/34561 PCT/EP00/10581
7
Suitably the dithionite is present in the range of 1-5 molar equivalents with
respect
to the compound of formula II and is used as an aqueous solution or suspension
in the
presence of base, preferably sodium hydroxide. Preferably the dithionite is
present in the
range of 2-4. molar equivalents with respect to the compound of formula II and
is used as an
aqueous solution or suspension in the presence of base, preferably sodium
hydroxide.
Preferably isopropanol can be used as a co-solvent to reduce foaming.
Suitably the diazotisation step is carried out at a temperature in the range
of-20° C to
+20°C. Preferably the diazotisation step is carried out at a
temperature in the range of
-10° C to +10° C. More preferably the diazotisation step is
carried out at a temperature in the
range of-5°C to +5°C.
Suitably the reduction of the diazonium salt is carried out at a temperature
in the
range of -50°C to +50°C. Preferably the reduction of the
diazonium salt is carried out at a
temperature in the range of-10° C to +30°C. More preferably the
reduction of the diazonium
salt is carried out at a temperature in the range of -5°C to
+25°C.
Suitably the reducing agent for the nitro compound is hydrogen in the presence
of a
catalyst, sodium dithionite, iron in the presence of acid or lithium aluminium
hydride.
Preferably the reducing agent for the nitro compound is hydrogen in the
presence of a
catalyst, sodium dithionite, or lithium aluminium hydride. Most preferably the
reducing agent
for the nitro compound is hydrogen in the presence of a palladium catalyst.
The hydrogenation/diazonium reaction/reduction can be carried out as a one-pot
reaction.
In a further aspect the present invention provides a process in which a
compound of
formula V
V
in which R is CH2S02NHCH3and R1 is CH2CH2NMe2, is prepared by reacting a
compound
CA 02389514 2002-04-30
WO 01/34561 PCT/EP00/10581
8
of formula V, in which R is CH2S02NHCH3and R1 is CH2CH2NH2, with a reducing
agent and
a formaldehyde equivalent in the presence of a buffer.
In another aspect the present invention provides a process for the preparation
of
sumatriptan or a pharmaceutically acceptable salt thereof comprising the
following steps:
a) reducing a compound of formula
IV
N02
in which R is CH2S02NHCH3 with a reducing agent, optionally in the presence of
an
acid to give a compound of formula III
\ III
NH2
or optionally a salt thereof in which R is CH2S02NHCH3 and
b) reacting the compound of formula III with a diazotising agent optionally in
the
presence of acid to give a compound of formula II
I I
N~ N x-
in which R is CH2S02NHCH3 and X- represents a chloride, bromide, acetate,
hydro-
sulphate or phosphate anion and
c) reducing the compound of formula II with a dithionite salt to give a
compound of formula I
in which R is CH2S02NHCH3 and
CA 02389514 2002-04-30
WO 01/34561 PCT/EP00/10581
9
I
/ NHNH2
d) reacting the compound of formula I in which R is CH2S02NHCH3 with an
aldehyde of
formula VII
R2C1-ECHO
VII
in which R2 represents CICH2CH2-and the aldehyde group is protected as an
acetal
(preferably the dimethyl acetal) in the presence of a buffer optionally in the
presence
of an acid catalyst to give a compound of formula V
V
or a salt thereof in which R is CH2S02NHCH3 and R1 is CH2CH2NH2 and
e) reacting the compound of formula V obtained in d) with a formaldehyde
equivalent
and a reducing agent in the presence of a buffer to give a compound of formula
V in
which R is CH2S02NHCH3 and R1 is CH2CH2NMe2 or a pharmaceutically
acceptable salt thereof.
Preferably none of the intermediate compounds is isolated in this process.
Suitably the reducing agent is a hydride equivalent such as sodium
borohydride,
sodium cyanoborohydride, sodium triacetoxyborohydride and lithium aluminium
hydride.
Preferably the reducing agent is sodium borohydride or sodium
cyanoborohydride. Most
preferably the reducing agent is sodium borohydride.
Suitably the reducing agent is present in the range of 0.25 - 5 molar
equivalents with
respect to the compound of formula V. Preferably the reducing agent is present
in the range
of 0.5 - 4 molar equivalents with respect to the compound of formula V. More
preferably the
CA 02389514 2002-04-30
WO 01/34561 PCT/EP00/10581
reducing agent is present in the range of 0.75 - 3 molar equivalents with
respect to the
compound of formula V.
Suitably the formaldehyde equivalent is formaldehyde, paraformaldehyde or
dimethoxymethane. Preferably the formaldehyde equivalent is formaldehyde or
para-
formaldehyde. Most preferably the formaldehyde equivalent is formaldehyde as
an
aqueous solution.
Suitably the formaldehyde equivalent is present in the range of 0.5 - 18 molar
equivalents with respect to the compound of formula V. Preferably the
formaldehyde
equivalent is present in the range of 1 -10 molar equivalents with respect to
the compound
of formula V. More preferably the formaldehyde equivalent is present in the
range of
1.9 - 5 molar equivalents with respect to the compound of formula V.
Suitably the buffer used keeps the pH of the reaction solution between pH 6
and pH
14. Preferably the buffer keeps the pH of the reaction solution between pH 7
and pH 11.
More preferably the buffer keeps the pH of the reaction solution between pH 8
and pH 10.
Most preferably the buffer is sodium hydrogenphosphate.
Suitably the buffer is present in the range of 0.1 -10 molar equivalents with
respect
to the compound of formula V. Preferably the buffer is present in the range of
0.2 - 5 molar
equivalents with respect to the compound of formula V. Most preferably the
buffer is present
in the range of 0.5 - 3 molar equivalents with respect to the compound of
formula V.
Suitable pharmaceutically acceptable salts of the compound of formula V
(including
sumatriptan) include acid addition salts formed with organic or inorganic
acids for example
hydrochlorides, hydrobromides, sulphates, nitrates, phosphates, formates,
mesylates,
citrates, benzoates, fumarates, maleates and succinates. Other salts may be
useful in the
preparation of the compound of formula I e.g. creatinine sulphate adducts, and
salts with e.g.
toluene-p-suiphonic acid. When the compound of formula V is sumatriptan the
salt is
preferably the succinate salt or the hemisulphate salt .
The invention is illustrated by the following Examples which are given by way
of
example only. The final products of each of these Examples were characterised
by one or
more of the following procedures: high performance liquid chromatography,
elemental
analysis, nuclear magnetic resonance spectroscopy, mass spectroscopy and
infrared
spectroscopy.
CA 02389514 2002-04-30
WO 01/34561 PCT/EP00/10581
11
EXAMPLES
Example 1
a) A stirred mixture of 4-amino-I~methylbenzenemethanesulphonamide (35.6 g),
concentrated hydrochloric acid (83.7 ml) and water (314 ml) was heated at
50°C for
15 minutes and the solution then cooled to-5°C. A solution of sodium
nitrite (12.5 g)
in water (21 ml) was then added dropwise over 10 minutes. The resulting
solution
was stirred for 1 hour and then added over 10 minutes to a stirred suspension
of
sodium dithionite (sodium hydrosulphite) (96.1 g) in water (420 ml) and 46/48%
wlw
sodium hydroxide solution (34 ml) at-5°C to +5°C. The suspension
was stirred for
2.75 hours. 46/48% w/w Sodium hydroxide (22 ml) was added and the mixture was
stirred for 20 minutes at 20°C and then for 40 minutes at 0-5°C.
The mixture was
filtered and the product was washed with water (3 x 110 ml) and dried to give
the free
base of the product.
The free base of the product (31.5 g) and ethanol (315 ml) were stirred and
heated to
reflux. Concentrated hydrochloric acid (12.3 ml) was added followed by ethanol
(155 ml). The mixture was boiled under reflux for 10 minutes and then cooled
to
0°C. The product was collected by filtration, washed with ethanol (155
ml) and dried
to give 4-hydrazine-I~methylbenzenemethanesulphonamide hydrochloride, yield
31.4 g.
b) Absolute ethanol (50 ml) was added to a stirred suspension of 4-hydrazine-
I~
methylbenzenemethanesulphonamide hydrochloride (6.42 g) in water (20m1) and
the mixture was stirred for 10 minutes. 4-Chlorobutanal dimethyl acetal (3.85
g)
was added and washed in with more absolute ethanol (11.7 ml). 2M Hydrochloric
acid (0.22 ml) was added and the solution was stirred at ambient temperature
for 4.5
hours. Sodium hydrogenphosphate (3.01 g) was added and the mixture was stirred
at ambient temperature for 10 minutes and then gradually heated to boiling
under
reflux over 40 minutes. The mixture was stirred and boiled under reflux for a
further
3 hours and then allowed to stand for 16 hours at ambient temperature. The
mixture
was concentrated under reduced pressure (around 60 ml removed) and then
dichloromethane (25 ml) and water (25 ml) were added, followed by potassium
carbonate (0.74 g). At this point the pH was 5. The mixture was filtered and
the
filtrate was separated. The aqueous layer was washed with more dichloromethane
CA 02389514 2002-04-30
WO 01/34561 PCT/EP00/10581
12
(2 x 25 ml) and the combined organic layer and washings were evaporated to
dryness under reduced pressure to give a solid by-product, yield 1.60 g.
The aqueous layer was mixed with dichloromethane (125 ml), absolute ethanol
(60 ml) potassium carbonate (37 g) and water (12 ml). The mixture was stirred
for
35 minutes and then separated. The organic layer was treated with charcoal and
stirred at ambient temperature for 1 hour. The mixture was filtered and the
filtrate
was concentrated to a mass of 20 g under reduced pressure. The mixture was
stirred for 1 hour and isopropyl acetate (62 ml) was added and the suspension
was
stirred for 64 hours. The solid was collected by filtration, washed with more
isopropyl
acetate (10 ml) and dried under vacuum at 50°C to give 3-(2-aminoethyl-
I~
methyl-1 ~indole-5-methanesulphonamide, yield 3.64 g (53.4%). Purity 93.63%.
Comparative Example
4-Hydrazino-I~methylbenzenemethanesulphonamide hydrochloride, which had
been prepared by diazotisation of 4-amino-I~rnethylbenzenemethanesulphon-
amide with sodium nitrite and then reduced with tin chloride, was reacted
according
to the procedure of Example 2. 3-(2-Aminoethyl-I~-methyl-1 ff-indole-5-me-
thanesulphonamide was obtained in a yield of 48.8% and was found to be 92.25%
pure by HPLC.
Example 2
I~-Methyl-4-nitrobenzenemethanesulphonamide (46.0 g, 0.23 mol), 10%
palladium on carbon (4.6 g) and 2M hydrochloric acid (180 ml) in water (200
ml) were stirred
for 1.5 hours under 2.5 atmospheres of hydrogen at 20°C. The reaction
mixture was then
filtered through a celite bed and washed with further water (100 ml). A
portion of this filtrate
(50 ml) was taken and then diluted with concentrated hydrochloric acid (12.7
ml). The
stirred suspension was cooled to below 0° C and a solution of sodium
nitrite (2.2 g, 32 mmol)
in water (4 ml) added dropwise over 20 minutes under an atmosphere of
nitrogen. After
stirring for 15 minutes, the clear solution was transferred via a cannula to a
solution of
sodium dithionite (17.0 g, 98 mmol) and 46/48% w/w sodium hydroxide (5.9 ml)
in water
(75 ml) at -5° C. The mixture was then warmed to room temperature and
stirred for a further
2.5 hours. 46/48% w/w sodium hydroxide (6.5 ml) was then added until the pH of
the solution
was approximately 9. Stirring at room temperature was continued for a further
0.5 hours
CA 02389514 2002-04-30
WO 01/34561 PCT/EP00/10581
13
followed by cooling in an ice bath for 0.5 hours. Filtration of the reaction
mixture gave
4-hydrazine-I~methylbenzenemethanesulphonamide as an off-white solid (2.5 g,
48%)
Example 3
A stirred mixture of 4-amino-N-methylbenzenemethanesulphonamide (50.0 g, 0.25
mmol),
conc. hydrochlorid acid (117 ml) and water (356 ml) was heated at 50°C
for 15 minutes and
the solution then cooled to -5°C. A solution of sodium nitrite (17.5 g)
in water (30 ml) was
then added dropwise over 15 minutes and the resulting solution stirred for a
further
15 minutes. The reaction mixture was then added via a cannula to a stirred
suspension of
sodium dithionite (135 g, 0.66 mol), water (365 ml), 46/48% w/w sodium
hydroxide solution
(23.5 ml) and IPA (40 ml) at -5°C. The temperature was kept around -
5°C during the
40 minute addition. The suspension was then warmed to room temperature and
stirred for
2.5 hours before 46/48% w/w Sodium hydroxide (53.5 ml) was added to give a pH
of 7-8.
Finally, the mixture was stirred for 30 minutes, product filtered and then
washed with water
(123 ml) to give a cream solid.
To form the hydrochloride salt, conc. hydrochloric acid (20 ml) was added to
the free base
suspended in isopropanol (400 ml) at ambient. After stirring for 15 minutes,
the product was
collected by filtration, washed with isopropanol (125 ml) and dried to give 4-
hydrazino-
N-methylbenzenemethanesulphonamide hydrochloride (45.7 g, 73% at 97% purity by
HPLC).
Example 4
N-Methyl-4-nitrobenzenemethanesulphonamide (23.0 g, 0.1 mol), 5% palladium on
carbon
(9.4 g) and conc. hydrochloric acid (11 ml) in water (245 ml) were stirred for
2 hours under
atmospheres of hydrogen at 50°C. The reaction mixture was then filtered
through a celite
bed and washed with further conc. hydrochloric acid (6 ml) in water (34 ml).
The filtrate was
then diluted with conc. hydrochloric acid (30 ml) and the solution cooled to -
5°C to give a
suspension. Sodium nitrite (7.0 g, 0.101 mol) in water (12 ml) was then added
dropwise
over 20 minutes keeping the temperature around -5°C. After stirring for
15 minutes, the
clear solution was transferred via a cannula to a solution of sodium
dithionite (54.0 g,
0.264 mol), 46/48% w/w sodium hydroxide (9.4 ml), water (197 ml) and
isopropanol (20 ml)
at -5°C. The temperature was kept around -5°C during the 40
minutes addition. The mixture
was then warmed to room temperature and stirred for a further 2.5 hours.
46/48% w/w
sodium hydroxide (21.4 ml) was then added until the pH of the solution was
approximately
CA 02389514 2002-04-30
WO 01/34561 PCT/EP00/10581
14
7.8. Stirring at room temperature was continued for a further 0.5 hours
followed by filtration
of the reaction mixture, washing with water (50 ml) and then isopropanol (150
ml) to give
4-hydrazino-N-methylbenzenemethanesulphonamide as an of-white solid. Forming
the hy-
drochloride salt as above, gave an overall yield of 77% at 97.6% purity by
HPLC.
Example 5
3-(2-Aminoethyl)-I~methyl-11~-indole-5-methanesulphonamide (5.0 g, 18.7 mmol),
prepared by the method of Example 1, and sodium hydrogenphosphate (5.0 g, 35.2
mmol)
were heated to 40°C in methanol (50 ml) for 15 minutes and then
recooled to room
temperature. Solutions of 37% aqueous formaldehyde (5 ml) and sodium
borohydride
(0.72 g) in water (5 ml stabilised with one drop of 46/48% w/w sodium
hydroxide) were added
simultaneously over one hour at a temperature between 17 and 21 °C. The
mixture was
stirred at room temperature for a further 0.5 hours, then filtered and the
filter bed washed with
methanol (10 ml). The combined filtrates were then adjusted to pH 6 by
addition of 4M hydro-
chloric acid, concentrated under reduced pressure (to approximately 20 g) and
acidified to
pH 1 with more 4M hydrochloric acid. Ethyl acetate (30 ml) was added and then
potassium
carbonate was added to give a pH about 11 and the product extracted in a
separating funnel.
The aqueous layer was further extracted with ethyl acetate (30 ml) and the
combined organic
layers dried over sodium sulphate, filtered and concentrated to give
sumatriptan free base
(4.7 g, 85%, HPLC showed 87% compound).
This material was of suitable quality for conversion into sumatriptan mono-
succinate
or sumatriptan hemisulphate as described in GB 2,162,522 and EP 490,689
respectively.
Example 6
3-(2-Aminoethyl)-N-methyl-1 H-indole-5-methanesulphonamide hydrochloride (50.0
g,
0.165 mol) and sodium hydrogenphosphate (47.0 g) was heated to 45°C in
methanol
(420 ml). To this mixture sodium methoxide (29 ml) was added and the solution
was cooled
to room temperature. Separate solutions of 37% aqueous formaldehyde (53 ml) in
methanol
(7 ml) and sodium borohydride (10.0 g) in water (49 ml stabilised with two
drops of
46/48% w/w sodium hydroxide) were then added simultaneously over one hour at a
temperature between 17-21 °C. Stirring the mixture at room temperature
for a further 1 hour
was followed by adjustment of the solution to pH 6 by addition of conc. HCI
(42 ml) in water
(83 ml). The suspension was then removed by filtration and the filter bed
washed with
methanol (59 ml). Further water (90 ml) was added to the filtrate and then the
mixture was
CA 02389514 2002-04-30
WO 01/34561 PCT/EP00/10581
concentrated to remove the residual methanol. Adjustment of the aqueous
solution to pH 2
using conc. NCI, addition of ethyl acetate (100 ml) and methanol (10 ml), was
followed by
basification with potassium carbonate (90 g) in water (130 ml). This
precipitated a grey solid
that was collected by filtration, washed with water (2 X 100 ml) and then
washed with ethyl
acetate (100 ml) to give sumatriptan free base (42.4 g, 87%, HPLC showed 98%
compound).
Comaarative Example.
3-(2-Aminoethyl)-I~-methyl-11~-indole-5-methanesulphonamide was reacted in a
similar fashion to example 3 without the inclusion of the sodium
hydrogenphosphate buffer.
The product free base was obtained in 40% yield and in a purity of 60% by
HPLC.