Note: Descriptions are shown in the official language in which they were submitted.
CA 02389680 2002-05-02
WO 01/32204 PCT/US00/30223
NUCLEIC ACID VACCINE COMPOSITIONS HAVING A
MAMMALIAN CD80/CD86 GENE PROMOTER DRIVING ANTIGEN
S EXPRESSION
Field of the Invention
The present invention relates generally to vaccine compositions and
methods of use thereof. More particularly, the invention pertains to
polynucleotides encoding at least one immunizing antigen whose expression is
controlled by a promoter derived from a co-stimulatory molecule. Methods of
immunization using these polynucleotides are also provided. Also provided are
compositions comprising at least one immunizing agent and at least one
cytokine
involved in maturation of antigen-presenting cells. Methods of eliciting an
immune response using these compositions are also described.
Back. round
Vaccines which induce a cell-mediated immune response are emerging as
important strategies in combating parasites, autoimmune disorders, allergic
diseases and cancers. Conventional vaccination strategies generally involve
administration of either "live" or "dead" vaccines. Ertl et al. (1996) J.
Immunol.
156:3579-3582. The so-called live vaccines include attenuated microbes and
recombinant molecules based on a living vector. The dead vaccines include
those
based on killed whole pathogens, and subunit vaccines, e.g., soluble pathogen
subunits or protein subunits. Live vaccines are generally successful in
providing
an effective immune response in immunized subjects; however, such vaccines
can be dangerous in immunocompromised or pregnant subjects, can revert to
pathogenic organisms, or can be contaminated with other pathogens. Hassett et
al. (1996) Trends in Microbiol. 8:307-312. Dead vaccines avoid the safety
CA 02389680 2002-05-02
WO 01/32204 PCT/US00/30223
-2-
problems associated with live vaccines; however such vaccines often fail to
provide an appropriate andlor effective immune response in immunized subjects.
More recently, direct injection of plasmid DNA by intramuscular (Wolff
et al. (1990) Science 247:1465:1468) or intradermal injection with a needle
and
syringe (Raz et al. (1994) PNAS USA 91:9519-9523) has been described.
Another approach referred to as ballistic or particle-mediated DNA delivery
employs a needless particle delivery device to administer DNA-coated
microscopic gold beads directly into the cells of the epidermis. (Yang et al.
(1990) PNAS USA 87:9568-9572). Thus, a number of delivery techniques can be
used to deliver nucleic acids for immunizations, including particle-mediated
techniques which deliver nucleic acid-coated microparticles into target tissue
(see, e.g., co-owned U.S. Patent No. 5,865,796, issued February 2, 1999).
Particle-mediated nucleic acid immunization techniques have been shown to
elicit both humoral and cytotoxic T lymphocyte immune responses following
epidermal delivery of nanogram quantities of DNA. Pertmer et al. (1995)
Vaccine 13:1427-1430. Such particle-mediated delivery techniques have been
compared to other types of nucleic acid inoculation, and found markedly
superior. Fynan et al. (1995) Int. J. Immunopharmacology 17:79-83, Fynan et
al.
(1993) Proc. Natl. Acad. Sci. USA 90:11478-11482, and Raz et al. (1994) Proc.
Natl. Acad. Sci. USA 91:9519-9523.
A novel transdermal drug delivery system that entails the use of a
needleless syringe to deliver solid drug-containing particles in controlled
doses
into and through intact skin has also been described. In particular, commonly
owned U.S. Patent No. 5,630,796 to Bellhouse et al., describes a particle
delivery
device (e.g., a needleless syringe) that delivers pharmaceutical particles
entrained
in a supersonic gas flow. The particle delivery device is used for transdermal
delivery of powdered drug compounds and compositions, for delivery of genetic
material into living cells (e.g., gene therapy) and for the delivery of
biopharmaceuticals to skin, muscle, blood or lymph. The device can also be
used
CA 02389680 2002-05-02
WO 01/32204 PCT/US00/30223
-3-
in conjunction with surgery to deliver drugs and biologics to organ surfaces,
solid
tumors and/or to surgical cavities (e.g., tumor beds or cavities after tumor
resection). Pharmaceutical agents that can be suitably prepared in a
substantially
solid, particulate form can be safely and easily delivered using such a
device.
One particular particle delivery device generally comprises an elongate
tubular nozzle having a rupturable membrane initially closing the passage
through the nozzle and arranged substantially adjacent to the upstream end of
the
nozzle. Particles of a therapeutic agent to be delivered are disposed adjacent
to
the rupturable membrane and are delivered using an energizing means which
applies a gaseous pressure to the upstream side of the membrane sufficient to
burst the membrane and produce a supersonic gas flow (containing the
pharmaceutical particles) through the nozzle for delivery from the downstream
end thereof. The particles can thus be delivered from the needleless syringe
at
delivery velocities of between Mach 1 and Mach 8 which are readily obtainable
upon the bursting of the rupturable membrane.
Another particle delivery device configuration generally includes the
same elements as described above, except that instead of having the
pharmaceutical particles entrained within a supersonic gas flow, the
downstream
end of the nozzle is provided with a bistable diaphragm which is moveable
between a resting "inverted" position (in which the diaphragm presents a
concavity on the downstream face to contain the pharmaceutical particles) and
an
active "evened" position (in which the diaphragm is outwardly convex on the
downstream face as a result of a supersonic shockwave having been applied to
the
upstream face of the diaphragm). In this manner, the pharmaceutical panicles
contained within the concavity of the diaphragm are expelled at a high initial
velocity from the device for transdermal delivery thereof to a targeted skin
or
mucosal surface.
Transdermal delivery using the above-described device configurations is
generally carried out with panicles having an approximate size that generally
CA 02389680 2002-05-02
WO 01/32204 PCT/US00/30223
-4-
ranges between 0.1 and 250 pm. Particles larger than about 250 pm can also be
delivered from the device, with the upper limitation being the point at which
the
size of the particles would cause untoward damage to the skin cells. The
actual
distance which the delivered particles will penetrate depends upon particle
size
(e.g., the nominal particle diameter assuming a roughly spherical particle
geometry), particle density, the initial velocity at which the particle
impacts the
skin surface, and the density and kinematic viscosity of the skin. Target
particle
densities for use in needleless particle injection generally range between
about
0.1 and 25 g/cm3, and injection velocities generally range between about 150
and
3,000 m/sec.
Summary of the Invention
The present invention is directed to a polynucleotide comprising a first
promoter derived from a gene encoding a co-stimulatory molecule and a first
sequence encoding at least one antigen wherein the first sequence is operably
linked to the first promoter. In particular embodiments, the promoter is
derived
from a CD80 (B7-1) gene or a CD86 (B7-2) gene.
In additional embodiments, the polynucleotide further comprises a second
sequence encoding at least one cytokine operably linked to the first promoter
or,
alternatively, to a second promoter. The promoter may be a constituitive
promoter.
In other embodiments, the invention is directed to a core Garner coated
with a polynucleotide as described above, as well as to pharmaceutical
compositions comprising the polynucleotide and a pharmaceutically acceptable
excipient. The pharmaceutical compositions optionally further include a
cytokine.
In still further embodiments, the invention is directed to a vaccine
composition comprising (a) an expression vector comprising a polynucleotide
encoding at least one antigen; and (b) a cytokine selected from the group
CA 02389680 2002-05-02
WO 01/32204 PCT/US00/30223
-5-
consisting of CD40 ligand (CD40L), tumor-necrosis factor-related activation-
induced cytokine (TRANCE) and Flt3 ligand (flt-3L).
In another embodiment, the invention is directed to a vaccine composition
comprising (a) at least one peptide antigen; and (b) an expression vector
comprising a polynucleotide encoding a cytokine selected from the group
consisting of CD40 ligand (CD40L), tumor-necrosis factor-related activation-
induced cytokine (TRANCE) and Flt3 ligand (flt-3L).
In yet another embodiment, the invention is directed to a vaccine
composition comprising: (a) at least one peptide antigen; and (b) a cytokine
selected from the group consisting of CD40 ligand (CD40L), tumor-necrosis
factor-related activation-induced cytokine (TRANCE) and Flt3 ligand (flt-3L).
Methods for eliciting an immune response in a vertebrate subject
comprising administering the vaccines above are also provided.
The various components of the above vaccine compositions may be
1 S coated onto a core carrier and used in methods for eliciting an immune
response
in a vertebrate subject. In this context, the method comprises administering
the
compositions to the subject using a particle-mediated delivery technique.
In another embodiment, the invention is directed to a method for eliciting
an immune response in a vertebrate subject. The method comprises (a) providing
a nucleotide sequence encoding an antigen operably linked to a promoter
derived
from a gene encoding a co-stimulatory molecule, the promoter capable of
directing the expression of the antigen in the subject; and (b) administering
the
nucleotide sequence to the subject in an amount sufficient for the antigen to
be
expressed and elicit an immune response in the subject.
In a further embodiment, the invention is directed to a method for eliciting
an immune response in a vertebrate subject. The method comprises (a) providing
a particle coated with a nucleotide sequence encoding at least one antigen,
the
nucleotide sequence operably linked to a promoter derived from a gene encoding
a co-stimulatory molecule, wherein the promoter is capable of driving
expression
CA 02389680 2002-05-02
WO 01/32204 PCT/US00/30223
-6-
of the antigen-encoding sequence in the subject; and (b) administering the
particle to the subject using a particle-mediated delivery technique, whereby
the
antigen encoded by the nucleotide sequence is expressed in an amount
sufficient
to elicit an immune response.
S In the methods above, the nucleotide sequence may further comprise a
polynucleotide encoding at least one cytokine, such as a cytokine selected
from
the group consisting of CD40L, tumor-necrosis factor-related activation-
induced
cytokine (TRANCE) and Flt3 ligand (flt-3L).
These and other embodiments of the present invention will readily occur
to those of ordinary skill in the art in view of the disclosure herein.
Brief Description of the Drawings
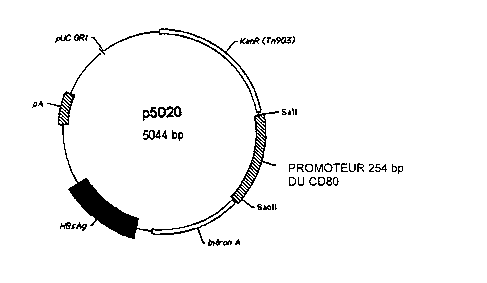
Figure 1 is a schematic representation of the CD80 promoter-driven
expression vector p5020. This plasmid vector was constructed from pWRG7128,
a mammalian expression vector based on a pUC 19 backbone. The CMV
promoter was removed and replaced with a 254 base pair PCR fragment obtained
by amplification of the mouse CD80 promoter, obtained from Life Technologies,
Gibco BRL. The plasmid also contains regulatory elements, and the polyA signal
of bovine growth hormone, operably linked to the full length cDNA encoding the
Hepatitis B surface antigen.
Figure 2 is a schematic representation of the CD80 promoter-driven
expression vector p5021. This plasmid vector was constructed from pWRG7128,
a mammalian expression vector based on a pUC 19 backbone. The CMV
promoter was removed and replaced with a 489 base pair PCR fragment obtained
by amplification of the mouse CD80 promoter, obtained from Life Technologies,
Gibco BRL. The plasmid also contains regulatory elements, and the polyA signal
of bovine growth hormone, operably linked to the full length cDNA encoding the
Hepatitis B surface antigen.
CA 02389680 2002-05-02
WO 01/32204 PCT/US00/30223
_'7_
Figure 3 is a schematic representation of the CD80 promoter-driven
expression vector p5022. This plasmid vector was constructed from pWRG7128,
a mammalian expression vector based on a pUC 19 backbone. The CMV
promoter was removed and replaced with a 3123 base pair PCR fragment
obtained by amplification of the mouse CD80 promoter, obtained from Life
Technologies, Gibco BRL. The plasmid also contains regulatory elements, and .
the polyA signal of bovine growth hormone, operably linked to the full length
cDNA encoding the Hepatitis B surface antigen.
Figure 4 is a schematic representation of the CD80 promoter-driven
expression vector p5023. This plasmid vector was constructed from pWRG7128,
a mammalian expression vector based on a pUC 19 backbone. The CMV
promoter was removed and replaced with a 3357 base pair PCR fragment
obtained by amplification of the mouse CD80 promoter, obtained from Life
Technologies, Gibco BRL. The plasmid also contains regulatory elements, and
the polyA signal of bovine growth hormone, operably linked to the full length
cDNA encoding the Hepatitis B surface antigen.
Figure 5 is a schematic representation of the CD80 promoter-driven
expression vector p5024. This plasmid vector was constructed from pWRG7128,
a mammalian expression vector based on a pUCl9 backbone. The CMV
promoter was removed and replaced with a 578 base pair PCR fragment obtained
by amplification of the human CD80 promoter, obtained from Life Technologies,
Gibco BRL. The plasmid also contains regulatory elements, and the polyA signal
of bovine growth hormone, operably linked to the full length cDNA encoding the
Hepatitis B surface antigen.
Figure 6 is a schematic representation of the CD80 promoter-driven
expression vector p5025. This plasmid vector was constructed from pWRG7128,
a mammalian expression vector based on a pUC 19 backbone. The CMV
promoter was removed and replaced with a 202 base pair PCR fragment obtained
by amplification of the human CD80 promoter, obtained from Life Technologies,
CA 02389680 2002-05-02
WO 01/32204 PCT/US00/30223
_g_
Gibco BRL. The plasmid also contains regulatory elements, and the polyA signal
of bovine growth hormone, operably linked to the full length cDNA encoding the
Hepatitis B surface antigen.
Figure 7 is a schematic representation of the CD80 promoter-driven
expression vector p5026. This plasmid vector was constructed from pWRG7128,
a mammalian expression vector based on a pUC 19 backbone. The CMV
promoter was removed and replaced with a 294 base pair PCR fragment obtained
by amplification of the human CD80 promoter, obtained from Life Technologies,
Gibco BRL. The plasmid also contains regulatory elements, and the polyA signal
of bovine growth hormone, operably linked to the full length cDNA encoding the
Hepatitis B surface antigen.
Figure 8 is a graph depicting cytotoxic T cell (CTL) responses elicited in
mice immunized with plasmids encoding hepatitis B surface antigen (HBsAg)
under the control of CMV promoter or CD80 promoters.
1 S Figure 9 is a histogram depicting anti-hepatitis B core antigen specific
IL-
4 production in splenocytes from mice immunized with plasmids encoding
hepatitis B core/surface antigens and a TRANCE cytokine adjuvant.
Figure 10 is a graph depicting anti-hepatitis B core antigen specific
antibody production in mice immunized with plasmids encoding hepatitis B
core/surface antigens and a TRANCE cytokine adjuvant.
Modes for Carrying Out the Invention
Before describing the present invention in detail, it is to be understood
that this invention is not limited to particular antigens or to antigen-coding
nucleotide sequences. It is also to be understood that different applications
of the
disclosed methods may be tailored to the specific needs in the art. It is also
to be
understood that the terminology used herein is for the purpose of describing
particular embodiments of the invention only, and is not intended to be
limiting.
CA 02389680 2002-05-02
WO 01/32204 PCT/US00/30223
-9-
All publications, patents and patent applications cited herein, whether
supra or infra, are hereby incorporated by reference in their entirety.
It must be noted that, as used in this specification and the appended
claims, the singular forms "a", "an", and "the" include plural referents
unless the
content clearly dictates otherwise. Thus, for example, reference to "an
antigen"
includes a mixture of two or more such agents, reference to "a particle"
includes
reference to mixtures of two or more particles, reference to "a recipient
cell"
includes two or more such cells, and the like.
Definitions
Unless defined otherwise, all technical and scientific terms used herein
have the same.meaning as commonly understood by one of ordinary skill in the
art to which the invention pertains. The following terms are intended to be
defined as indicated below.
The term "vaccine composition" intends any pharmaceutical composition
containing an antigen (e.g., polynucleotide encoding an antigen), which
composition can be used to prevent or treat a disease or condition in a
subject.
The term thus encompasses both subunit vaccines, i.e., vaccine compositions
containing antigens which are separate and discrete from a whole organism with
which the antigen is associated in nature, as well as compositions containing
whole killed, attenuated or inactivated bacteria, viruses, parasites or other
microbes.
The term "transdermal" delivery intends intradermal (e.g., into the dermis
or epidermis), transdermal (e.g., "percutaneous") and transmucosal
administration, i.e., delivery by passage of an agent into or through skin or
mucosal tissue. See, e.g., Transdermal Drug Delivery: Developmental Issues
and Research Initiatives, Hadgraft and Guy (eds.), Marcel Dekker, Inc.,
(1989);
Controlled Drug Delivery: Fundamentals and Applications, Robinson and Lee
(eds.), Marcel Dekker Inc., (1987); and Transdermal Delivery ofDrugs, Vols. 1-
CA 02389680 2002-05-02
WO 01/32204 PCT/US00/30223
-10-
3, Kydonieus and Berner (eds.), CRC Press, (1987). Thus, the term encompasses
delivery of particles from a particle delivery device (e.g., needleless
syringe) as
described in U.S. Patent No. 5,630,796, as well as particle-mediated delivery
of
coated core carriers as described in U.S. Patent No. 5,865,796.
By "core carrier" is meant a carrier particle on which a nucleic acid (e.g.,
DNA) is coated in order to impart a defined particle size as well as a
sufficiently
high density to achieve the momentum required for cell membrane penetration,
such that the DNA can be delivered using particle-mediated delivery
techniques,
for example those described in U.S. Patent No. 5,100,792. Core carriers
typically
include materials such as tungsten, gold, platinum, ferrite, polystyrene and
latex.
See e.g., Particle Bombardment Technology for Gene Transfer, (1994) Yang, N.
ed., Oxford University Press, New York, NY pages 10-11.
By "particle delivery device," or "needleless syringe," is meant an
instrument which delivers a particulate composition transdermally, without a
conventional needle that pierces the skin. Particle delivery devices for use
with
the present invention are discussed throughout this document.
By "antigen" is meant a molecule which contains one or more epitopes
that will stimulate a host's immune system to make a cellular antigen-specific
immune response, or a humoral antibody response. Thus, antigens include
proteins, polypeptides, antigenic protein fragments, oligosaccharides,
polysaccharides, and the like. Furthermore, the antigen can be derived from
any
known virus, bacterium, parasite, plants, protozoans, or fungus, and can be a
whole organism. The term also includes tumor antigens. Similarly, an
oligonucleotide or polynucleotide which expresses an antigen, such as in DNA
immunization applications, is also included in the definition of antigen.
Synthetic antigens are also included, for example, polyepitopes, flanking
epitopes, and other recombinant or synthetically derived antigens (Bergmann et
al. (1993) Eur. J. Immunol. 23:2777-2781; Bergmann et al. (1996) J. Immunol.
157:3242-3249; Suhrbier, A. (1997) Immunol. and Cell Biol. 75:402-408;
CA 02389680 2002-05-02
WO 01/32204 PCT/US00/30223
-11-
Gardner et al. (1998) 12th World AIDS Conference, Geneva, Switzerland, June
28-July 3, 1998).
The term "peptide" is used in it broadest sense to refer to a compound of
two or more subunit amino acids, amino acid analogs, or other peptidomimetics.
The subunits may be linked by peptide bonds or by other bonds, for example
ester, ether, etc. As used herein, the term "amino acid" refers to either
natural
and/or unnatural or synthetic amino acids, including glycine and both the D or
L
optical isomers, and amino acid analogs and peptidomimetics. A peptide of
three
or more amino acids is commonly called an oligopeptide if the peptide chain is
short. If the peptide chain is long, the peptide is typically called a
polypeptide or
a protein
"T cell epitopes" are generally those features of a peptide structure
capable of inducing a T cell response. In this regard, it is accepted in the
art that
T cell epitopes comprise linear peptide determinants that assume extended
conformations within the peptide-binding cleft of MHC molecules, (LTnanue et
al.
(1987) Science 236:551-557). As used herein, a T cell epitope is generally a
peptide having about 8-15, preferably 5-10 or more amino acid residues.
The term "antigen presenting cell" or "APC" as used herein, intends any
cell which presents on its surface an antigen in association with a major
histocompatibility complex molecule, or portion thereof, or, alternatively,
one or
more non-classical MHC molecules, or a portion thereof. Examples of suitable
APCs are discussed in detail below and include, but are not limited to, whole
cells such as Langerhans cells, macrophages, dendritic cells, B cells, hybrid
APCs, and foster antigen presenting cells.
Dendritic cells (DCs) and Langerhans cells are potent antigen-presenting
cells. DCs are minor constituents of various immune organs, for example,
constituting around 1% of epidermal cell suspensions (Schuler et al. (1985) J.
Exp. Med. 161:526; and Romani et al. (1989) J. Invest. Dermatol. 93:600).
Despite their relative scarcity, these cells have been shown to provide all
the
CA 02389680 2002-05-02
WO 01/32204 PCT/US00/30223
-12-
signals required for T cell activation and proliferation. The requisite
signals can
be categorized into two types. The first type, which gives specificity to the
immune response, is mediated through interaction between the T-cell
receptor/CD3 ("TCR/CD3") complex and an antigenic peptide presented by a
major histocompatibility complex ("MHC") class I or II protein on the surface
of
APCs. This interaction is necessary, but not sufficient, for T cell activation
to
occur. In fact, without the second type of signal, the first type of signal
can result
in T cell anergy (e.g., where T-cells are insensitive to additional signals).
The
second type of signal, called a co-stimulatory signal, is neither antigen-
specific
nor MHC-restricted, and can lead to a full proliferation response of T cells
and
induction of T cell effector functions in the presence of the first type of
signal.
Thus, as discussed above, research accumulated over the past several years has
demonstrated convincingly that resting T cells require at least two signals
for
induction of cytokine gene expression and proliferation (Schwartz R.H. (1990)
Science 248:1349-1356; Jerkins M.K. (1992) Immunol. Today 13:69-73). One
signal, the one that confers specificity, can be produced by interaction of
the
TCR/CD3 complex with an appropriate MHC/peptide complex. The second
signal is not antigen specific and is termed the "co-stimulatory" signal.
"Co-stimulatory molecules" act as receptor-ligand pairs expressed on the
surface of antigen presenting cells and T cells. The term encompasses any
single
molecule or combination of molecules which, when acting together with a
peptide/MHC complex bound by a TCR on the surface of a T cell, provides a co-
stimulatory effect which achieves activation of the T cell that binds the
peptide.
Several molecules have been shown to enhance co-stimulatory activity.
These are CD80 (i.e., B7-1), CD86 (i.e., B7-2/B70) (Schwartz R.H. (1992) Cell
71:1065-1068), heat stable antigen (HSA) (Liu Y. et al. (1992) J. Exp. Med.
175:437-445), chondroitin sulfate-modified MHC invariant chain (Naujokas
M.F., et al. (1993) Cell 74:257-268), intracellular adhesion molecule 1 (ICAM-
1)
(Van Seventer, G.A. (1990) J. Immunol. 144:4579-4586). These molecules each
CA 02389680 2002-05-02
WO 01/32204 PCT/US00/30223
-13-
appear to accomplish co-stimulation by interacting with their cognate ligands
on
the T cells. Co-stimulatory molecules mediate signals) which are necessary,
under normal physiological conditions, to achieve full activation of naive T
cells.
One exemplary receptor-ligand pair are the CD80 and CD86 co-stimulatory
molecule on the surface of APCs and their counter-receptors, CD28 and CTLA-4
on T cells (Elks et al. (1996) J. Immunol. 56:2700-2709; Freeman et al. (1993)
Science 262:909-911; Nabavi et al. (1992) Nature 360:266-268). Other
important co-stimulatory molecules are CD40 and CD54. The term thus
encompasses CD80, CD86, or other co-stimulatory molecules) on an antigen-
presenting matrix such as an APC, as well as fragments of the co-stimulatory
molecules) (alone, complexed with another molecule(s), or as part of a fusion
protein) which binds to a cognate ligand and results in activation of the T
cell
when the TCR on the surface of the T cell specifically binds the peptide. Many
of the sequences of the genes encoding co-stimulatory molecules and their
promoter regions are known in the art. Other promoters or fragments thereof
can
also be determined by methods known in the art and described herein.
As used herein the term "adjuvant" refers to any material that enhances
the action of a drug, antigen, polynucleotide, vector or the like. Thus, one
example of an adjuvant is a "cytokine." As used herein, the term "cytokine"
refers to any one of the numerous factors that exert a variety of effects on
cells,
for example, inducing growth, proliferation or maturation. Certain cytokines,
for
example TRANCE, flt-3L, and CD40L, enhance the immunostimulatory capacity
of APCs. Non-limiting examples of cytokines which may be used alone or in
combination in the practice of the present invention include, interleukin-2
(IL-2),
stem cell factor (SCF), interleukin 3 (IL-3), interleukin 6 (IL-6),
interleukin 12
(IL-12), G-CSF, granulocyte macrophage-colony stimulating factor (GM-CSF),
interleukin-1 alpha (IL-la), interleukin-11 (IL-11), MIP-la, leukemia
inhibitory
factor (LIF), c-kit ligand, thrombopoietin (TPO), CD40 ligand (CD40L), tumor
necrosis factor-related activation-induced cytokine (TRANCE) and flt3 ligand
CA 02389680 2002-05-02
WO 01/32204 PCT/US00/30223
-14-
(flt-3L). Cytokines are commercially available from several vendors such as,
for
example, Genzyme (Framingham, MA), Genentech (South San Francisco, CA),
Amgen (Thousand Oaks, CA), R&D Systems and Immunex (Seattle, WA). The
sequence of many of these molecules are also available, for example, from the
GenBank database. It is intended, although not always explicitly stated, that
molecules having similar biological activity as wild-type or purified
cytokines
(e.g., recombinantly produced or mutants thereof) and nucleic acid encoding
these molecules are intended to be used within the spirit and scope of the
invention.
A composition which contains a selected antigen and an adjuvant, or a
vaccine composition which is co-administered with an adjuvant, displays
"enhanced immunogenicity" when it possesses a greater capacity to elicit an
immune response than the immune response elicited by an equivalent amount of
the antigen administered without the adjuvant. Thus, a vaccine composition may
display "enhanced immunogenicity" because the antigen is more strongly
immunogenic or because a lower dose or fewer doses of antigen are necessary to
achieve an immune response in the subject to which the antigen is
administered.
Such enhanced immunogenicity can be determined by administering the adjuvant
composition and antigen controls to animals and comparing antibody titers
and/or
cellular-mediated immunity between the two using standard assays such as
radioimmunoassay, ELISAs, CTL assays, and the like, well known in the art.
The terms "nucleic acid molecule" and "polynucleotide" are used
interchangeably and refer to a polymeric form of nucleotides of any length,
either
deoxyribonucleotides or ribonucleotides, or analogs thereof. Non-limiting
examples of polynucleotides include a gene, a gene fragment, exons, introns,
messenger RNA (mRNA), transfer RNA, ribosomal RNA, ribozymes, cDNA,
recombinant polynucleotides, branched polynucleotides, plasmids, vectors,
isolated DNA of any sequence, isolated RNA of any sequence, nucleic acid
probes, and primers.
CA 02389680 2002-05-02
WO 01/32204 PCT/US00/30223
-15-
A polynucleotide is typically composed of a specific sequence of four
nucleotide bases: adenine (A); cytosine (C); guanine (G); and thymine (T)
(uracil
(L~ is substituted for thymine (T) when the polynucleotide is RNA). Thus, the
term polynucleotide sequence is the alphabetical representation of a
polynucleotide molecule. This alphabetical representation can be input into
databases in a computer having a central processing unit and used for
bioinformatics applications such as functional genomics and homology
searching.
A "gene" as used in the context of the present invention is a sequence of
nucleotides in a genetic nucleic acid (chromosome, plasmid, etc.) with which a
genetic function is associated. A gene is a hereditary unit, for example of an
organism, comprising a polynucleotide sequence (e.g., a DNA sequence for
mammals) that occupies a specific physical location ( a "gene locus" or
"genetic
locus") within the genome of an organism. A gene can encode an expressed
product, such as a polypeptide or a polynucleotide (e.g., tRNA).
Alternatively, a
gene may define a genomic location for a particular event/function, such as
the
binding of proteins and/or nucleic acids (e.g., phage attachment sites),
wherein
the gene does not encode an expressed product. Typically, a gene includes
coding sequences, such as polypeptide encoding sequences, and non-coding
sequences, such as promoter sequences, poly-adenlyation sequences,
transcriptional regulatory sequences (e.g., enhancer sequences). Many
eucaryotic
genes have "exons" (coding sequences) interrupted by "introns" (non-coding
sequences). In certain cases, a gene may share sequences with another genes)
(e.g., overlapping genes).
A "coding sequence" or a sequence which "encodes" a selected
polypeptide, is a nucleic acid molecule which is transcribed (in the case of
DNA)
and translated (in the case of mRNA) into a polypeptide in vivo when placed
under the control of appropriate regulatory sequences (or "control elements").
The boundaries of the coding sequence are determined by a start codon at the
5'
(amino) terminus and a translation stop codon at the 3' (carboxy) terminus. A
CA 02389680 2002-05-02
WO 01/32204 PCT/US00/30223
-16-
coding sequence can include, but is not limited to, cDNA from viral,
procaryotic
or eucaryotic mRNA, genomic DNA sequences from viral or procaryotic DNA,
and even synthetic DNA sequences. A transcription termination sequence may
be located 3' to the coding sequence. Transcription and translation of coding
S sequences are typically regulated by "control elements," including, but not
limited to, transcription promoters, transcription enhancer elements,
transcription
termination signals, polyadenylation sequences (located 3' to the translation
stop
codon), sequences for optimization of initiation of translation (located 5' to
the
coding sequence), and translation termination sequences.
A "promoter" is a nucleotide sequence which initiates transcription of a
polypeptide-encoding polynucleotide. Promoters can include inducible
promoters (where expression of a polynucleotide sequence operably linked to
the
promoter is induced by an analyte, cofactor, regulatory protein, etc.),
repressible
promoters (where expression of a polynucleotide sequence operably linked to
the
promoter is repressed by an analyte, cofactor, regulatory protein, etc.), and
constitutive promoters. In addition, such promoters can also have tissue
specificity, for example, the CD80 promoter is only inducible in certain
immune
cells, and the myoD promoter is only inducible in muscle cells. It is intended
that
the term "promoter" or "control element" includes full-length promoter regions
and functional (e.g., controls transcription or translation) segments of these
regions. A promoter is "derived from" a gene encoding a co-stimulatory
molecule if it has the same or substantially the same basepair sequence as a
region of the promoter region of the co-stimulatory molecule, complements
thereof, or if it displays sequence identity as described below.
A "vector" is capable of transfernng gene sequences to target cells (e.g.,
viral vectors, non-viral vectors, particulate Garners, and liposomes).
Typically,
"vector construct," "expression vector," and "gene transfer vector," mean any
nucleic acid construct capable of directing the expression of a gene of
interest and
CA 02389680 2002-05-02
WO 01/32204 PCT/US00/30223
-17-
which can transfer gene sequences to target cells. Thus, the term includes
cloning and expression vehicles, as well as viral vectors.
An "isolated polynucleotide" molecule is a nucleic acid molecule separate
and discrete from the whole organism with which the molecule is found in
nature;
or a nucleic acid molecule devoid, in whole or part, of sequences normally
associated with it in nature; or a sequence, as it exists in nature, but
having
heterologous sequences (as defined below) in association therewith.
"Operably linked" refers to an arrangement of elements wherein the
components so described are configured so as to perform their usual function.
Thus, a given promoter that is operably linked to a coding sequence (e.g., an
antigen or interest) is capable of effecting the expression of the coding
sequence
when the regulatory proteins and proper enzymes are present. In some
instances,
certain control elements need not be contiguous with the coding sequence, so
long as they function to direct the expression thereof. For example,
intervening
untranslated yet transcribed sequences can be present between the promoter
sequence and the coding sequence and the promoter sequence can still be
considered "operably linked" to the coding sequence.
"Recombinant" as used herein to describe a nucleic acid molecule means
a polynucleotide of genomic, cDNA, semisynthetic, or synthetic origin which,
by
virtue of its origin or manipulation: (1) is not associated with all or a
portion of
the polynucleotide with which it is associated in nature; and/or (2) is linked
to a
polynucleotide other than that to which it is linked in nature. The term "re-
combinant" as used with respect to a protein or polypeptide means a
polypeptide
produced by expression of a recombinant polynucleotide.
Techniques for determining nucleic acid and amino acid "sequence
identity" also are known in the art. Typically, such techniques include
determining the nucleotide sequence of the mRNA for a gene and/or determining
the amino acid sequence encoded thereby, and comparing these sequences to a
second nucleotide or amino acid sequence. In general, "identity" refers to an
CA 02389680 2002-05-02
WO 01/32204 PCT/US00/30223
-18-
exact nucleotide-to-nucleotide or amino acid-to-amino acid correspondence of
two polynucleotides or polypeptide sequences, respectively. Two or more
sequences (polynucleotide or amino acid) can be compared by determining their
"percent identity." The percent identity of two sequences, whether nucleic
acid
or amino acid sequences, is the number of exact matches between two aligned
sequences divided by the length of the shorter sequences and multiplied by
100.
An approximate alignment for nucleic acid sequences is provided by the local
homology algorithm of Smith and Waterman, Advances in Applied Mathematics
2:482-489 (1981). This algorithm can be applied to amino acid sequences by
using the scoring matrix developed by Dayhoff, Atlas of Protein Sequences and
Structure, M.O. Dayhoff ed., 5 suppl. 3:353-358, National Biomedical Research
Foundation, Washington, D.C., USA, and normalized by Gribskov, Nucl. Acids
Res. 14(6):6745-6763 (1986). An exemplary implementation of this algorithm to
determine percent identity of a sequence is provided by the Genetics Computer
Group (Madison, WI) in the "BestFit" utility application. The default
parameters
for this method are described in the Wisconsin Sequence Analysis Package
Program Manual, Version 8 (1995) (available from Genetics Computer Group,
Madison, WI). A preferred method of establishing percent identity in the
context
of the present invention is to use the MPSRCH package of programs copyrighted
by the University of Edinburgh, developed by John F. Collins and Shane S.
Sturrok, and distributed by IntelliGenetics, Inc. (Mountain View, CA). From
this
suite of packages the Smith-Waterman algorithm can be employed where default
parameters are used for the scoring table (for example, gap open penalty of
12,
gap extension penalty of one, and a gap of six). From the data generated the
"Match" value reflects "sequence identity." Other suitable programs for
calculating the percent identity or similarity between sequences are generally
known in the art, for example, another alignment program is BLAST, used with
default parameters. For example, BLASTN and BLASTP can be used using the
following default parameters: genetic code = standard; filter = none; strand =
CA 02389680 2002-05-02
WO 01/32204 PCT/US00/30223
-19-
both; cutoff= 60; expect = 10; Matrix = BLOSLJM62; Descriptions = 50
sequences; sort by = HIGH SCORE; Databases = non-redundant, GenBank +
EMBL + DDBJ + PDB + GenBank CDS translations + Swiss protein + Spupdate
+ PIR. Details of these programs can be found at the following Internet
address:
http://www.ncbi.nlm.gov/cgi-bin/BLAST.
Alternatively, homology can be determined by hybridization of
polynucleotides under conditions which form stable duplexes between
homologous regions, followed by digestion with single-stranded-specific
nuclease(s), and size determination of the digested fragments. Two DNA, or two
polypeptide sequences are "substantially homologous" to each other when the
sequences exhibit at least about 80%-85%, preferably at least about 90%, and
most preferably at least about 95%-98% sequence identity over a defined length
of the molecules, as determined using the methods above. As used herein,
substantially homologous also refers to sequences showing complete identity to
the specified DNA or polypeptide sequence. DNA sequences that are
substantially homologous can be identified in a Southern hybridization
experiment under, for example, stringent conditions, as defined for that
particular
system. For example, stringent hybridization conditions can include 50%
formamide, Sx Denhardt's Solution, Sx SSC, 0.1% SDS and 100 pg/ml denatured
salmon sperm DNA and the washing conditions can include 2x SSC, 0.1% SDS
at 37°C followed by lx SSC, 0.1% SDS at 68°C. Defining
appropriate
hybridization conditions is within the skill of the art. See, e.g., Sambrook
et al.,
supra; DNA Cloning, supra; Nucleic Acid Hybridization, supra.
As used herein, the term "treatment" includes any of following: the
prevention of infection or reinfection; the reduction or elimination of
symptoms;
and the reduction or complete elimination of a pathogen. Treatment may be
effected prophylactically (prior to infection) or therapeutically (following
infection). An "effective amount" is an amount sufficient to effect beneficial
or
desired results. An effective amount can be administered in one or more
CA 02389680 2002-05-02
WO 01/32204 PCT/US00/30223
-20-
administrations, applications of dosages. The term "co-administering" or "co-
administration" refers to administration of at least two substances. Co-
administration can be achieved by administering the substances concurrently or
at
different times. In addition, co-administration includes delivery using one or
more delivery means.
By suitable immune response, it is meant that the methods of the
invention can bring about in an immunized subject an immune response
characterized by the production of B and/or T lymphocytes specific for a viral
antigen, wherein the immune response can protect the subject against
subsequent
infection with homologous or heterologous viral strains, reduce viral burden
and/or shedding during an infection, bring about resolution of infection in a
shorter amount of time relative to a non-immunized subject, or prevent or
reduce
clinical manifestation of disease symptoms.
By "vertebrate subject" is meant any member of the subphylum cordata,
particularly mammals, including, without limitation, humans and other
primates.
The term does not denote a particular age. Thus, both adult and newborn
individuals are intended to be covered.
General Overview of the Invention
Before describing the present invention in detail, it is to be understood
that this invention is not limited to particular formulations or process
parameters
(as such may, of course, vary). It is also to be understood that the
terminology
used herein is for the purpose of describing particular embodiments of the
invention only, and is not intended to be limiting.
DNA-vaccines generally consist of a plasmid that encodes a relevant
antigen for de novo synthesis by cells present in a targeted tissue. Viral .
promoters, e.g., the promoter from Cytomegalovirus (CMV), are generally used
in DNA-vaccine plasmid constructs to drive antigen expression. Delivery of
these DNA-vaccine plasmids, both in "naked" form and attached to particles,
has
CA 02389680 2002-05-02
WO 01/32204 PCT/US00/30223
-21-
been shown to elicit both humoral and cell-mediated immune responses. (See,
e.g., Wang et al. (1993) Proc. Natl. Acad. Sci. USA 90:4156-4160; Tang et al.
(1992) Nature 356:152-154; Fynan, supra).
It is known that in order to provoke a specific CTL (cytotoxic T-cell)
response, an antigen must be presented to T cells. This is accomplished via
antigen presenting cells (APCs), a class of cells which includes dendritic
cells
(DCs), Langerhans cells, monocytes, macrophages, and B cells. DCs were first
described as the morphologically distinct Langerhans cells in the epidermis of
the
skin (as reviewed by Bancheraeau et al. (1998) Nature 392:245-252) and have
since been shown to be the most efficient APC for the activation of naive T
cells.
Lanzavecchia A. (1993) Science 260:937-944 and Bancheraeau et al. (1998),
supra. The antigens encoded by injected DNA-vaccines are processed into
peptides and presented to T-cells by dendritic cells. It has also been shown
that
intraepidermally delivered DNA-vaccines target Langerhans cells. The targeted
Langerhans cells express the DNA-encoded antigen and migrate out of the
epidermis to the draining lymph nodes where they process and present the DNA-
vaccine encoded antigens) to T-lymphocytes. Thus, APCs play an essential role
in the induction of an effective immune response to DNA-vaccines.
APCs exposed to antigens process the antigens into small fragments,
known as epitopes, which are then associated with the major histocompatibility
complex (MHC) Class I for presentation to CD8+ T-lymphocytes and with MHC
Class II for presentation to CD4+ T-lymphocytes. However, certain co-
stimulatory molecules, for example CD80 and CD86 (also known as B7-1 and
B7-2, respectively), are also required for antigen presentation. Thus,
effective
activation of T-lymphocytes requires two signals at the cell surface interface
of
the APC and the target T-cell. It is now known that the first activation
signal is
provided by binding of the T-cell receptor (TCR) to the antigen-MHC complex
and second activation signal is provided by engagement of the CD80/CD86 co-
stimulatory molecules on the APC with the CD28 receptor on the T-lymphocytes.
CA 02389680 2002-05-02
WO 01/32204 PCT/US00/30223
-22-
After maturation, APCs become sensitive to apoptosis, thus limiting their
natural
stimulatory capacity.
The CD80/CD86 co-stimulatory molecules required for successful antigen
presentation are not constitutively expressed by APCs. Rather, upon
activation,
e.g., in response to an infectious pathogen, CD80/CD86 expression on the
surface
of APCs is rapidly up-regulated. Signals for inducing CD80/CD86 expression by
APCs can be provided by cytokines released by epithelial and lymphoid cells in
an inflamed tissue site infected with a pathogen. Furthermore, certain
cytokines
secreted by activated T-lymphocytes, e.g., IFNy, can both induce and maintain
the expression of CD80/CD86 by APCs. To date, two members of the TNF
family, CD40 ligand (CD40L or CD 154), and TNF-related Activation-induced
Cytokine (TRANCE), and a factor known as Flt3 ligand (Flt3L) have been
implicated in APC maturation. see, e.g., Gurunathan et al. (1998) J. Immunol.,
161:4563-4571; Pulendran et al. (1998) J. Exp. Med., 188:2075-2082) and Wong
et al. (1999) J. Immunol 162:2251-2258. TRANCE has also been shown to
prolong the life-span of mature DC. (Josien et al. (1999) J. Immun. 162:2562-
2568; Wong et al. (1997) J. Exp. Med., 186:2075-2080). Co-administration of
CD40L and tumor specific antigens has been shown to result in production of
IgGl antibodies, reflecting a Th2-type immune response (see, Wong et al.
(1999),
supra).
Recently, methods have been described to enhance the T-cell response of
a subject. These methods entail administering nucleotides encoding, under the
same transcriptional regulatory element, an immunizing antigen and full-length
co-stimulatory molecule. (See, e.g., U.S. Patent No. 5,738,852 and
International
Publication WO 97/32987, published September 12, 1997.) It is useful to note
that these studies exemplify and describe expression of an immunizing agent
and
a co-stimulatory molecule under the control of a constitutive promoter, e.g.,
a
CMV promoter.
CA 02389680 2002-05-02
WO 01/32204 PCT/US00/30223
-23-
There remains a need in the art for inducible, APC-targeted DNA
vaccines and methods which effectively enhance maturation and the stimulatory
lifespan of the targeted APCs, for example by targeting specific cells or
being
active only in specific cells. The invention described herein achieves this
goal,
for example by operably linking a polynucleotide encoding an immunizing agent
to a promoter sequence derived from a gene encoding a co-stimulatory molecule,
and/or by co-administering the immunizing agent with one or more cytokines
that
enhance the stimulatory lifespan of APCs.
More particularly, the present invention provides novel polynucleotides
which are particularly useful as vaccines. Typically, the polynucleotides are
carried on vectors, for instance plasmids, which contain suitable regulatory
elements. In one embodiment, the polynucleotides of the present invention
comprise a sequence encoding at least one selected antigen. Expression of the
antigens) is controlled by a transcriptional regulatory element (e.g.,
promoter)
derived from a gene encoding a co-stimulatory molecule, for example CD80 or
CD86. Without being bound by a particular theory, it appears that using a
promoter element derived from a co-stimulatory molecule takes advantage of the
APC's normal up-regulation of these promoters upon activation and during
antigen presentation. Thus, using the polynucleotides described herein
enhances
antigen expression, processing and presentation in APCs as compared to using
polynucleotides driven by, for example, constitutive promoters.
The invention also includes compositions wherein an expression vector
comprising a co-stimulatory molecule promoter and a sequence encoding an
antigen further comprises an adjuvant, for example a cytokine. Preferably, the
cytokine enhances the immune response, for example, by enhancing the
immunostimulatory capacity of the APCs, increasing expression of costimulatory
ligands on the surface of the APCs, stabilizing antigen/MHC complexes and/or
inhibiting apoptosis of the APCs. Methods of co-administering polynucleotides
carrying antigens operably linked to a co-stimulatory molecule promoter along
CA 02389680 2002-05-02
WO 01/32204 PCT/US00/30223
-24-
with adjuvants are also included. The selected adjuvants may be given in the
form of polynucleotides under suitable regulatory control or as polypeptides
(e.g.,
recombinantly produced polypeptides). When administered as nucleotides, the
cytokine-encoding sequence and antigen-encoding sequence may be carried on
the same vector or on different vectors. Thus, the cytokine-encoding sequence
may be under the control of a promoter derived from a co-stimulatory molecule
or, alternatively, a different promoter (e.g., a constitutive promoter).
Furthermore, the cytokine-encoding sequence may be located either 3' or 5' to
the
antigen-encoding sequence.
The invention further includes vaccine compositions comprising antigens
in combination with at least one cytokine that enhances stimulation or
survival of
dendritic cells. As described above, such cytokines (e.g., TRANCE, flt3,
CD40L) may increase expression of co-stimulatory molecules on the surface of
APCs, stabilize the antigen/MHC complex or prevent their apoptosis, thereby
increasing the stimulatory lifespan of APCs. In particular, compositions
comprising polynucleotides encoding at least one antigen and a peptide
cytokine,
particularly a cytokine such as TRANCE, flt-3L or CD40L, are described.
Another composition includes those comprising at least one antigen (e.g.,
peptide) and at least one cytokine (e.g., TRANCE, flt-3L and/or CD40L). Yet
another composition comprises at least one antigen (e.g., peptide) and at
least one
polynucleotide encoding a cytokine (e.g., TRANCE, flt-3L and/or CD40L). It is
to be understood that more than one antigen can be used in combination with
one
or more cytokines.
The polynucleotides of the present invention may be introduced into cells
in vitro or in vivo, for example by transfection or by coating the
polynucleotides
onto particles and administering the coated particles to the cells.
Alternatively,
the polynucleotides and/or peptides may be provided in a particulate (e.g.,
powder) form, discussed more fully below and in the disclosure of
International
CA 02389680 2002-05-02
WO 01/32204 PCT/US00/30223
-25-
Publication Numbers WO 97/48485 and WO 98/10750, which are incorporated
by reference herein.
Thus, the invention includes methods for eliciting an immune response,
preferably a CTL response, in a vertebrate subject by administering a
polynucleotide encoding at least one selected antigen, where the antigen-
encoding sequence is operably linked to a regulatory element of a co-
stimulatory
molecule. Also provided are methods for eliciting an immune response in a
vertebrate subject by co-administering a selected antigen with a cytokine such
as
TRANCE, flt-3L or CD40L.
Antigens
The compositions and methods described herein are useful in eliciting an
immune response against a wide variety of antigens for the treatment and/or
prevention of a number of conditions including, but not limited to, cancer,
allergies, toxicity and infection by pathogens such as viruses, bacteria,
fungi, and
other pathogenic organisms.
Suitable viral antigens for use in the present compositions and methods
include, but are not limited to, those obtained or derived from the hepatitis
family
of viruses, including hepatitis A virus (HAV), hepatitis B virus (HBV),
hepatitis
C virus (HCV), the delta hepatitis virus (HDV), hepatitis E virus (HEV) and
hepatitis G virus (HGV). See, e.g., International Publication Nos. WO
89/04669;
WO 90/11089; and WO 90/14436. The HCV genome encodes several viral
proteins, including E1 and E2. See, e.g., Houghton et al. (1991) Hepatology
14:381-388. Nucleic acid molecules containing sequences encoding these
proteins, as well as antigenic fragments thereof, will find use in the present
methods. Similarly, the coding sequence for the b-antigen from HDV is known
(see, e.g., U.S. Patent No. 5,378,814).
In like manner, a wide variety of proteins from the herpesvirus family can
be used as antigens in the present invention, including proteins derived from
CA 02389680 2002-05-02
WO 01/32204 PCT/US00/30223
-26-
herpes simplex virus (HSV) types 1 and 2, such as HSV-l and HSV-2
glycoproteins gB, gD and gH; antigens from varicella zoster virus (VZV),
Epstein-Barr virus (EBV) and cytomegalovirus (CMV) including CMV gB and
gH; and antigens from other human herpesviruses such as HHV6 and HHV7.
(See, e.g. Chee et al. (1990) Cytomegaloviruses (J.K. McDougall, ed., Springer-
Verlag, pp. 125-169; McGeoch et al. (1988) J. Gen. Virol. 69:1531-1574; U.S.
Patent No. 5,171,568; Baer et al. (1984) Nature 310:207-211; and Davison et
al.
(1986) J. Gen. Virol. 67:1759-1816.)
Human immunodeficiency virus (HIV) antigens, such as gp120 molecules
for a multitude of HIV-1 and HIV-2 isolates, including members of the various
genetic subtypes of HIV, are known and reported (see, e.g., Myers et al., Los
Alamos Database, Los Alamos National Laboratory, Los Alamos, New Mexico
(1992); and Modrow et al. (1987) J. Virol. 61:570-578) and antigen-containing
nucleic acid sequences derived or obtained from any of these isolates will
find
use in the present invention. Furthermore, other immunogenic proteins derived
or obtained from any of the various HIV isolates will find use herein,
including
sequences encoding one or more of the various envelope proteins such as gp 160
and gp4l, gag antigens such as p24gag and pSSgag, as well as proteins derived
from the pol, env, tat, vif, rev, nef, vpr, vpu and LTR regions of HIV.
Antigens derived or obtained from other viruses will also find use herein,
such as without limitation, antigens from members of the families
Picornaviridae
(e.g., polioviruses, rhinoviruses, etc.); Caliciviridae; Togaviridae (e.g.,
rubella
virus, dengue virus, etc.); Flaviviridae; Coronaviridae; Reoviridae (e.g.,
rotavirus,
etc.); Birnaviridae; Rhabodoviridae (e.g., rabies virus, etc.);
Orthomyxoviridae
(e.g., influenza virus types A, B and C, etc.); Filoviridae; Paramyxoviridae
(e.g.,
mumps virus, measles virus, respiratory syncytial virus, parainfluenza virus,
etc.);
Bunyaviridae; Arenaviridae; Retroviradae (e.g., HTLV-I; HTLV-II; HIV-1 (also
known as HTLV-III, LAV, ARV, hTLR, etc.)), including but not limited to
antigens from the isolates HIV,116, HIVSFZ, HIVLAV, HIVLnn HIVE); HIV-lcMZ3s,
CA 02389680 2002-05-02
WO 01/32204 PCT/US00/30223
-27-
HIV-lUSa; HIV-2, among others; simian immunodeficiency virus (SIV);
Papillomavirus, the tick-bourne encephalitis viruses; and the like. See, e.g.
Virology, 3rd Edition (W.K. Joklik ed. 1988); Fundamental Virology, 2nd
Edition (B.N. Fields and D.M. Knipe, eds. 1991), for a description of these
and
other viruses.
In some contexts, it may be preferable that a selected antigen is obtained
or derived from a viral pathogen that typically enters the body via a mucosal
surface and is known to cause or is associated with human disease, such as,
but
not limited to, HIV (AIDS), influenza viruses (Flu), herpes simplex viruses
(genital infection, cold sores, STDs), rotaviruses (diarrhea), parainfluenza
viruses
(respiratory infections), poliovirus (poliomyelitis), respiratory syncytial
virus
(respiratory infections), measles and mumps viruses (measles, mumps), rubella
virus (rubella), and rhinoviruses (common cold).
Suitable bacterial and parasitic antigens can be obtained or derived from
known causative agents responsible for diseases including, but not limited to,
Diptheria, Pertussis, Tetanus, Tuberculosis, Bacterial or Fungal Pneumonia,
Otitis Media, Gonnorhea, Cholera, Typhoid, Meningitis, Mononucleosis, Plague,
Shigellosis or Salmonellosis, Legionaire's Disease, Lyme Disease, Leprosy,
Malaria, Hookworm, Onchocerciasis, Schistosomiasis, Trypamasomialsis,
Lesmaniasis, Giardia, Amoebiasis, Filariasis, Borelia, and Trichinosis. Still
further antigens can be obtained or derived from unconventional pathogens such
as the causative agents of kuru, Creutzfeldt-Jakob disease (CJD), scrapie,
transmissible mink encephalopathy, and chronic wasting diseases, or from
proteinaceous infectious particles such as prions that are associated with mad
cow
disease.
Specific pathogens from which antigens can be derived include M.
tuberculosis, Chlamydia, N. gonorrhoeae, Shigella, Salmonella, Yibrio Cholera,
Treponema pallidua, Pseudomonas, Bordetella pertussis, Brucella, Franciscella
tulorensis, Helicobacter pylori, Leptospria interrogaus, Legionella
pneumophila,
CA 02389680 2002-05-02
WO 01/32204 PCT/US00/30223
-28-
Yersinia pestis, Streptococcus (types A and B), Pneumococcus, Meningococcus,
Hemophilus influenza (type b), Toxoplasma gondic, Complylobacteriosis,
Moraxella catarrhalis, Donovanosis, and Actinomycosis; fungal pathogens
including Candidiasis and Aspergillosis; parasitic pathogens including Taenia,
S Flukes, Roundworms, Amebiasis, Giardiasis, Cryptosporidium, Schistosoma,
Pneumocystis carinii, Trichomoniasis and Trichinosis. Thus, the present
invention can also be used to provide a suitable immune response against
numerous veterinary diseases, such as Foot and Mouth diseases, Coronavirus,
Pasteurella multocida, Helicobacter, Strongylus vulgaris, Actinobacillus
pleuropneumonia, Bovine viral diarrhea virus (BVDV), Klebsiella pneumoniae,
E. coli, Bordetella pertussis, Bordetella parapertussis and brochiseptica.
Typically, a nucleotide sequence corresponding to one or more of the
above-listed antigens) is used in the production of the polynucleotides, as
described below.
Isolation of Genes and Construction of Polynucleotides
The present invention provides polynucleotides encoding at least one
antigen (e.g., antigens derived from and/or expressed by viruses, bacteria,
fungi,
worms, toxins, allergens or cancer cells) operably linked to a non-viral, cell-
or
tissue- specific promoter (e.g., a promoter derived from a regulatory element
which controls transcription of a sequence encoding a co-stimulatory
molecule).
These polynucleotides are useful in eliciting an immune response to the
antigen(s), particularly in activating T-lymphocytes.
Nucleotide sequences selected for use in the present invention can be
derived from known sources, for example, by isolating the same from cells
containing a desired gene or nucleotide sequence using standard techniques.
Similarly, the nucleotide sequences can be generated synthetically using
standard
modes of polynucleotide synthesis that are well known in the art. See, e.g.,
Edge
et al. (1981) Nature 292:756-762; Nambair et al. (1994) Science 223:1299-1301;
CA 02389680 2002-05-02
WO 01/32204 PCT/US00/30223
-29-
Jay et al. (1984) J. Biol. Chem. 259:6311-6317. Generally, synthetic
oligonucleotides can be prepared by either the phosphotriester method as
described by Edge et al., supra, and Duckworth et al. (1981) Nucleic Acids
Res.
9:1691-1706, or the phosphoramidite method as described by Beaucage et al.
(1981) Tet. Letts. 22:1859, and Matteucci et al. (1981) J. Am. Chem. Soc.
103:3185. Synthetic oligonucleotides can also be prepared using commercially
available automated oligonucleotide synthesizers. The nucleotide sequences can
thus be designed with appropriate codons for a particular amino acid sequence.
In general, one will select preferred codons for expression in the intended
host.
The complete sequence is assembled from overlapping oligonucleotides prepared
by standard methods and assembled into a complete coding sequence. See, e.g.,
Edge et al. (supra); Nambair et al. (supra) and Jay et al. (supra).
Another method for obtaining nucleic acid sequences for use herein is by
recombinant means. Thus, a desired nucleotide sequence can be excised from a
plasmid carrying the same using standard restriction enzymes and procedures.
Site specific DNA cleavage is performed by treating with the suitable
restriction
enzyme (or enzymes) under conditions which are generally understood in the
art,
and the particulars of which are specified by manufacturers of commercially
available restriction enzymes. If desired, size separation of the cleaved
fragments
may be performed by polyacrylamide gel or agarose gel electrophoresis using
standard techniques.
Restriction cleaved fragments may be blunt ended by treating with the
large fragment of E. coli DNA polymerase I (Klenow) in the presence of the
four
deoxynucleotide triphosphates (dNTPs) using standard techniques. The Klenow
fragment fills in at 5' single-stranded overhangs but digests protruding 3'
single
strands, even though the four dNTPs are present. If desired, selective repair
can
be performed by supplying only one, or several, selected dNTPs within the
limitations dictated by the nature of the overhang. After Klenow treatment,
the
mixture can be extracted with e.g. phenol/chloroform, and ethanol
precipitated.
CA 02389680 2002-05-02
WO 01/32204 PCT/US00/30223
-30-
Treatment under appropriate conditions with S 1 nuclease or BAL-31 results in
hydrolysis of any single-stranded portion.
Yet another convenient method for isolating specific nucleic acid
molecules is by the polymerise chain reaction (PCR). Mullis et al. (1987)
Methods Enzymol. 155:335-350. This technique uses DNA polymerise, usually a
thermostable DNA polymerise, to replicate a desired region of DNA. The region
of DNA to be replicated is identified by oligonucleotides of specified
sequence
complementary to opposite ends and opposite strands of the desired DNA to
prime the replication reaction. The product of the first round of replication
is
itself a template for subsequent replication, thus repeated successive cycles
of
replication result in geometric amplification of the DNA fragment delimited by
the primer pair used. This method also allows for the facile addition of
nucleotide sequences onto the ends of the DNA product by incorporating these
added sequences onto the oligonucleotide primers (see, e.g., PCR Protocols, A
Guide to Methods and Applications, Innis et al (eds) Harcourt Brace Jovanovich
Publishers, NY (1994)). PCR conditions used for each amplification reaction
are
empirically determined. A number of parameters influence the success of a
reaction. Among them are annealing temperature and time, extension time, Mgz+
and ATP concentration, pH, and the relative concentration of primers,
templates,
and deoxyribonucleotides. One example of suitable PCR conditions is found
below in the Examples.
Once coding sequences for desired proteins have been prepared or
isolated, such sequences can be cloned into any suitable vector or replicon.
Numerous cloning vectors are known to those of skill in the art, and the
selection
of an appropriate cloning vector is a matter of choice. Ligations to other
sequences are performed using standard procedures, known in the art.
As described in detail below, selected nucleotide sequences can be placed
under the control of regulatory sequences such as a promoter, so that the
CA 02389680 2002-05-02
WO 01/32204 PCT/US00/30223
-31-
sequence encoding the desired protein is transcribed into RNA in the host
tissue
transformed by a vector containing this expression construct.
Promoters
The choice of promoter is central to the construction of certain
polynucleotides described herein. Thus, the invention provides for expression
of
a selected antigen driven by a non-viral, preferably mammalian, cell- (or
tissue-)
specific promoter. In a preferred embodiment, the promoter is derived from a
regulatory sequence which controls transcription of a co-stimulatory molecule
for
example, a promoter derived from a CD80 (also known as B7-1), CD86 (also
known as B7-2), CD40 or CD54 gene. Other suitable promoters can be readily
determined using the teachings herein.
Genomic organization, including promoter mapping, of suitable co-
stimulatory factors, such as CD80 and CD86 has been described, for example in
Zhang et al. (1996) Gene 183:1-6; Selvakumar et al. (1993) Immunogenetics
38:292-295 and Fong et al. (1996) J. Immunol. 157:4442-4450. These studies
have shown that the CD80 (B7-1) gene promoter consists of three positively
regulated regions: a distal region from -2597 to -1555 that contains putative
transcription factor binding sites; a proximal region from -130 to -110 that
contains a tandem repeat sequence and a downstream region from +269 to +25
(Zhang et al., supra). Truncations from nucleotide position -906 to -84 have
been
shown to result in increased transcription activity in CD80-expressing Raji
cells
and a regulatory element around -41 has been identified (Fong et al., supra).
Further mapping of the CD80 promoter or other co-stimulatory molecule
promoters can be conducted using methods known in the art in view of these
references and the teachings of this specification.
The present invention also provides methods of eliciting an immune
response using an immunizing agent in combination with at least one cytokine
that enhances the stimulatory lifespan of APCs (e.g., dendritic cells). In
these
CA 02389680 2002-05-02
WO 01/32204 PCT/US00/30223
-32-
embodiments, either the immunizing agent or the cytokine(s) can be delivered
to
the subject as a polynucleotide encoding the polypeptide of interest. These
polynucleotides can include a wide-variety of promoters, including, for
example,
constitutive promoters (e.g., CMV, SV-40, housekeeping gene promoters, and the
like), inducible promoters (e.g., metallothionine, heat-shock, cytochrome,
protein
tyroisne kinase, nitric oxide synthase promoters, and the like), and tissue or
cell-
specific promoters (e.g., CD80/86, muscle creatine kinase, and the like).
In addition to promoters, it may be desirable to add other regulatory
sequences which allow for regulation of the expression of protein sequences
encoded by the delivered nucleotide sequences. Suitable additional regulatory
sequences are known to those of skill in the art, and examples include those
which cause the expression of a coding sequence to be turned on or off in
response to a chemical or physical stimulus, including the presence of a
regula-
tory compound. Other types of regulatory elements may also be present in the
vector, for example, enhancer sequences.
An expression vector is constructed so that the particular coding sequence
is located in the vector with the appropriate regulatory sequences such that
the
positioning and orientation of the coding sequence with respect to the control
sequences allows the coding sequence to be transcribed under the "control" of
the
control sequences (i.e., RNA polymerase, which binds to the DNA molecule at
the control sequences, transcribes the coding sequence). Modification of the
sequences encoding the particular protein of interest may be desirable to
achieve
this end. For example, in some cases it may be necessary to modify the
sequence
so that it is attached to the control sequences with the appropriate
orientation; i.e.,
to maintain the reading frame. The control sequences and other regulatory
sequences may be ligated to the coding sequence prior to insertion into a
vector.
Alternatively, the coding sequence can be cloned directly into an expression
vector which already contains the control sequences and an appropriate
restriction
site.
CA 02389680 2002-05-02
WO 01/32204 PCT/US00/30223
-33-
Generally, nucleic acid molecules used in the subject methods contain
coding regions with suitable control sequences and, optionally, ancillary
nucleotide sequences which encode cytokines or other immune enhancing
polypeptides. The nucleic acid molecules are generally prepared in the form of
vectors which include the necessary elements to direct transcription and
translation in a recipient cell.
Adjuvants
In order to augment an immune response in a subject, the compositions
and methods described herein can further include ancillary substances (e.g.,
adjuvants), such as pharmacological agents, cytokines, or the like. Suitable
adjuvants include any substance that enhances the immune response of the
subject to the polynucleotides of the invention. Non-limiting examples include
cytokines, e.g., Flt3 ligand, CD40L and TRANCE. As detailed above, these
1 S cytokines may enhance the immune response by affecting any number of
pathways, for example, by stabilizing the antigen/MHC complex, by causing
more antigen/MHC complex to be present on the cell surface, by enhancing
maturation of APCs, or by prolonging the life of APCs (e.g., inhibiting
apoptosis). For instance, recent studies suggest that CD40L and Flt3 ligand
can
serve as adjuvants in mouse models (Gurunathan et al., (1998) J. Immunol.
161:4563 and Pulendran et al. (1998), J. Immunol. 188:2075). Wong et al.
(1997)
J. Exp. Med. 186:2075 reports that TRANCE may promote the life-span of
mature dendritic cells. As described herein, these cytokines, delivered as
either
peptides or as polynucleotides encoding functional peptides, are also be
useful in
eliciting immune responses.
Ancillary substances may be administered, for example, as proteins or
other macromolecules at the same time, prior to, or subsequent to,
administration
of the co-stimulatory molecule promoter driven polynucleotides, peptide
antigens
or polynucleotides encoding an antigen of interest. Cytokines can be obtained
CA 02389680 2002-05-02
WO 01/32204 PCT/US00/30223
-34-
from a variety of sources, for example Immunex (Seattle, WA), Genentech
(South San Francisco, CA) and Amgen (Thousand Oaks, CA). Alternatively,
cytokines can be produced using a variety of methods known to those skilled in
the art in view of the teachings of this specification. In particular,
cytokines can
S be isolated directly from native sources, using standard purification
techniques.
Alternatively, the cytokines can be recombinantly produced using expression
systems as described above and purified using known techniques. The cytokines
can also be synthesized, based on known amino acid sequences or amino acid
sequences derived from DNA sequence of a molecule of interest, via chemical
polymer syntheses such as solid phase peptide synthesis. Such methods are
described for example, in J. M. Stewart and J. D. Young, Solid Phase Peptide
Synthesis, 2nd ed, Pierce Chemical Co., Rockford IL (1984) and G. Barany and
R. B. Mernfield, The Peptides: Analysis, Synthesis, Biology, editors E. Gross
and
J. Meinenhofer, vol. 2, Academic Press, New York, (1980), pp.3-254, for solid
phase peptide synthesis techniques.
Alternatively, ancillary nucleic acid sequences coding for peptides known
to stimulate, modify, or modulate a host's immune response (e.g., cytokines),
can
be co-administered as polynucleotides with the above-described antigen-
encoding
polynucleotides or peptide antigens. The gene sequences for a number of these
cytokines are known. (see, e.g., GenBank and other publically available
databases; Wong et al. (1997) .I Biol Chem 272(40):25190-4 (TRANCE); Lyman
et al. (1995) Oncogene 11(6):1165-72 (flt3); Spriggs et al., (1992) J. Exp.
Med.
176:1543-1550 and Armitage et al. (1992) Nature 357:80-82 (CD40L); Spriggs
(1992) Immunol Ser. 56:3-34 (TNF-alpha); Morgan et al. (1976) Science
193:1007-1008 (IL-2); U.S. Patent No. 5,187,077 (LIF); Brankenhoff et al.
(1987) Immunol. 139:4116-4121 (IL-6); etc).
Thus, suitable cytokines can be supplied by administering a
polynucleotide encoding the cytokine (or encoding an active fragment thereof).
These cytokine-encoding nucleotides can be administered either on the same
CA 02389680 2002-05-02
WO 01/32204 PCT/US00/30223
-35-
vector that carries the antigen-encoding sequence, or, alternatively on a
separate
vector. In some cases, it may be desirable to design a polynucleotide in which
both the antigen-encoding sequence and the cytokine-encoding sequence are
under the control of the same promoter, for instance one derived from a co-
y stimulatory molecule. In other cases, it may be desirable to use a cytokine-
encoding sequence under the control of a different promoter, for example a
constitutive promoter.
Administration of Polynucleotides and Adjuvants
The polynucleotides and ancillary substances described herein may be
administered by any suitable method. In a preferred embodiment, described
below, the polynucleotides are administered by coating them onto particles and
then administering the particles to the subject or cells. However, the
polynucleotides may also be delivered using a viral vector as known in the
art, or
by using non-viral systems, as described for example in U.S. Patent No.
5,589,466.
Viral Vectors
A number of viral based systems have been used for gene delivery. For
example, retroviral systems are known and generally employ packaging lines
which have an integrated defective provirus (the "helper") that expresses all
of
the genes of the virus but cannot package its own genome due to a deletion of
the
packaging signal, known as the psi sequence. Thus, the cell line produces
empty
viral shells. Producer lines can be derived from the packaging lines which, in
addition to the helper, contain a viral vector which includes sequences
required in
cis for replication and packaging of the virus, known as the long terminal
repeats
(LTRs). The gene of interest can be inserted in the vector and packaged in the
viral shells synthesized by the retroviral helper. The recombinant virus can
then
be isolated and delivered to a subject. (See, e.g., U.S. Patent No.
5,219,740.)
Representative.retroviral vectors include but are not limited to vectors such
as the
CA 02389680 2002-05-02
WO 01/32204 PCT/US00/30223
-36-
LHL, N2, LNSAL, LSHL and LHL2 vectors described in e.g., U.S. Patent No.
5,219,740, incorporated herein by reference in its entirety, as well as
derivatives
of these vectors, such as the modified N2 vector described herein. Retroviral
vectors can be constructed using techniques well known in the art. See, e.g.,
U.S.
Patent No 5,219,740; Mann et al. (1983) Cell 33:153-159.
Adenovirus based systems have been developed for gene delivery and are
suitable for delivering the polynucleotides described herein. Human
adenoviruses are double-stranded DNA viruses which enter cells by receptor-
mediated endocytosis. These viruses are particularly well suited for gene
transfer
because they are easy to grow and manipulate and they exhibit a broad host
range
in vivo and in vitro. For example, adenoviruses can infect human cells of
hematopoietic, lymphoid and myeloid origin. Furthermore, adenoviruses infect
quiescent as well as replicating target cells. Unlike retroviruses which
integrate
into the host genome, adenoviruses persist extrachromosomally thus minimizing
the risks associated with insertional mutagenesis. The virus is easily
produced at
high titers and is stable so that it can be purified and stored. Even in the
replication-competent form, adenoviruses cause only low level morbidity and
are
not associated with human malignancies. Accordingly, adenovirus vectors have
been developed which make use of these advantages. For a description of
adenovirus vectors and their uses see, e.g., Haj-Ahmad and Graham (1986) J.
Yirol. 57:267-274; Bett et al. (1993) J. Virol. 67:5911-5921; Mittereder et
al.
(1994) Human Gene Therapy 5:717-729; Seth et al. (1994) J. Virol. 68:933-940;
Barr et al. (1994) Gene Therapy 1:51-58; Berkner, K.L. (1988) BioTechniques
6:616-629; Rich et al. (1993) Human Gene Therapy 4:461-476.
Adeno-associated viral vector (AAV) can also be used to administer the
polynucleotides described herein. AAV vectors can be derived from any AAV
serotype, including without limitation, AAV-1, AAV-2, AAV-3, AAV-4, AAV-
5, AAVX7, etc. AAV vectors can have one or more of the AAV wild-type genes
deleted in whole or part, preferably the rep and/or cap genes, but retain one
or
CA 02389680 2002-05-02
WO 01/32204 PCT/US00/30223
-37-
more functional flanking inverted terminal repeat (ITR) sequences. Functional
ITR sequences are necessary for the rescue, replication and packaging of the
AAV virion. Thus, an AAV vector includes at least those sequences required in
cis for replication and packaging (e.g., functional ITRs) of the virus. The
ITR
sequence need not be the wild-type nucleotide sequence, and may be altered,
e.g.,
by the insertion, deletion or substitution of nucleotides, so long as the
sequence
provides for functional rescue, replication and packaging.
AAV expression vectors are constructed using known techniques to at
least provide as operatively linked components in the direction of
transcription,
control elements including a transcriptional initiation region, the DNA of
interest
and a transcriptional termination region. The control elements are selected to
be
functional in a mammalian cell. The resulting construct which contains the
operatively linked components is bounded (5' and 3') with functional AAV ITR
sequences. Suitable AAV constructs can be designed using techniques well
known in the art. See, e.g., U.S. Patent Nos. 5,173,414 and 5,139,941;
International Publication Nos. WO 92/01070 (published 23 January 1992) and
WO 93/03769 (published 4 March 1993); Lebkowski et al. (1988) Molec. Cell.
Biol. 8:3988-3996; Vincent et al. (1990) Vaccines 90 (Cold Spring Harbor
Laboratory Press); Carter, B.J. (1992) Current Opinion in Biotechnology 3:533-
539; Muzyczka, N. (1992) Current Topics in Microbiol. and Immunol. 158:97-
129; Kotin, R.M. (1994) Human Gene Therapy 5:793-801; Shelling and Smith
(1994) Gene Therapy 1:165-169; and Zhou et al. (1994) J. Exp. Med. 179:1867-
1875.
Conventional Pharmaceutical Preparations
Formulation of a preparation comprising the polynucleotides of the
present invention, with or without addition of an adjuvant composition, can be
carried out using standard pharmaceutical formulation chemistries and
methodologies all of which are readily available to the ordinarily skilled
artisan.
For example, compositions containing one or more nucleic acid molecules (e.g.,
CA 02389680 2002-05-02
WO 01/32204 PCT/US00/30223
-3 8-
present in a plasmid or viral vector) can be combined with one or more
pharmaceutically acceptable excipients or vehicles to provide a liquid
preparation.
Auxiliary substances, such as wetting or emulsifying agents, pH buffering
substances and the like, may be present in the excipient or vehicle. These
excipients, vehicles and auxiliary substances are generally pharmaceutical
agents
that do not induce an immune response in the individual receiving the
composition, and which may be administered without undue toxicity.
Pharmaceutically acceptable excipients include, but are not limited to,
liquids
such as water, saline, polyethyleneglycol, hyaluronic acid, glycerol and
ethanol.
Pharmaceutically acceptable salts can also be included therein, for example,
mineral acid salts such as hydrochlorides, hydrobromides, phosphates,
sulfates,
and the like; and the salts of organic acids such as acetates, propionates,
malonates, benzoates, and the like. It is also preferred, although not
required, that
the preparation will contain a pharmaceutically acceptable excipient that
serves as
a stabilizer, particularly for peptide, protein or other like molecules if
they are to
be included in the vaccine composition. Examples of suitable Garners that also
act as stabilizers for peptides include, without limitation, pharmaceutical
grades
of dextrose, sucrose, lactose, trehalose, mannitol, sorbitol, inositol,
dextran, and
the like. Other suitable Garners include, again without limitation, starch,
cellulose, sodium or calcium phosphates, citric acid, tartaric acid, glycine,
high
molecular weight polyethylene glycols (PEGS), and combination thereof. A
thorough discussion of pharmaceutically acceptable excipients, vehicles and
auxiliary substances is available in REMINGTONS PHARMACEUTICAL
SCIENCES (Mack Pub. Co., N.J. 1991), incorporated herein by reference.
Certain facilitators of nucleic acid uptake and/or expression ("transfection
facilitating agents") can also be included in, e.g., non-viral vector
compositions,
for example, facilitators such as bupivacaine, cardiotoxin and sucrose, and
transfection facilitating vehicles such as liposomal or lipid preparations
that are
CA 02389680 2002-05-02
WO 01/32204 PCT/US00/30223
-39-
routinely used to deliver nucleic acid molecules. Anionic and neutral
liposomes
are widely available and well known for delivering nucleic acid molecules
(see,
e.g., Liposomes: A Practical Approach, (1990) RPC New Ed., IRL Press).
Cationic lipid preparations are also well known vehicles for use in delivery
of
nucleic acid molecules. Suitable lipid preparations include DOTMA (N-[1-(2,3-
dioleyloxy)propyl]-N,N,N-trimethylammonium chloride), available under the
tradename LipofectinTM , and DOTAP (1,2-bis(oleyloxy)-3-
(trimethylammonio)propane), see, e.g., Felgner et al. (1987) Proc. Natl. Acad.
Sci. USA 84:7413-7416; Malone et al. (1989) Proc. Natl. Acad. Sci. USA
86:6077-6081; US Patent Nos 5,283,185 and 5,527,928, and International
Publication Nos WO 90/11092, WO 91/15501 and WO 95/26356. These cationic
lipids may preferably be used in association with a neutral lipid, for example
DOPE (dioleyl phosphatidylethanolamine). Still further transfection-
facilitating
compositions that can be added to the above lipid or liposome preparations
include spermine derivatives (see, e.g., International Publication No. WO
93/18759) and membrane-permeabilizing compounds such as GALA,
Gramicidine S and cationic bile salts (see, e.g., International Publication
No. WO
93/19768).
Alternatively, the nucleic acid molecules of the present invention may be
encapsulated, adsorbed to, or associated with, particulate Garners. Suitable
particulate carriers include those derived from polymethyl methacrylate
polymers, as well as PLG microparticles derived from poly(lactides) and
poly(lactide-co-glycolides). See, e.g., Jeffery et al. (1993) Pharm. Res.
10:362-
368. Other particulate systems and polymers can also be used, for example,
polymers such as polylysine, polyarginine, polyornithine, spermine,
spermidine,
as well as conjugates of these molecules.
The formulated vaccine compositions will thus typically include a
polynucleotide (e.g., a plasmid) containing a sequence encoding an antigen of
interest in an amount sufficient to mount an immunological response. An
CA 02389680 2002-05-02
WO 01/32204 PCT/US00/30223
-40-
appropriate effective amount can be readily determined by one of skill in the
art.
Such an amount will fall in a relatively broad range that can be determined
through routine trials. For example, immune responses have been obtained using
as little as 1 ~g of DNA, while in other administrations, up to 2 mg of DNA
has
been used. It is generally expected that an effective dose of polynucleotides
containing the genomic fragments will fall within a range of about 10 ~g to
1000
fig, however, doses above and below this range may also be found effective.
The
compositions may thus contain from about 0.1 % to about 99.9% of the
polynucleotide molecules.
Administration of Conventional Pharmaceutical Preparations
Administration of the above-described pharmaceutical preparations can be
effected in one dose, continuously or intermittently throughout the course of
treatment. Delivery will most typically be via conventional needle and syringe
for the liquid compositions and for liquid suspensions containing particulate
compositions. In addition, various liquid jet injectors are known in the art
and
may be employed to administer the present compositions. Methods of
determining the most effective means and dosages of administration are well
known to those of skill in the art and will vary with the delivery vehicle,
the
composition of the therapy, the target cells, and the subject being treated.
Single
and multiple administrations can be carried out with the dose level and
pattern
being selected by the attending physician. It should be understood that more
than
one antigen sequence can be carried by a polynucleotide vector construct.
Alternatively, separate vectors (e.g., plasmid or viral vectors), each
containing
sequences expressing one or more antigens can also be delivered to a subject
as
described herein.
Furthermore, it is also intended that the polynucleotides delivered by the
methods of the present invention may be combined with other suitable
compositions and therapies. For instance, in order to augment an immune
response in a subject, the compositions and methods described herein can
further
CA 02389680 2002-05-02
WO 01/32204 PCT/US00/30223
-41-
include ancillary substances (e.g., adjuvants), such as pharmacological
agents,
cytokines, or the like. Ancillary substances may be administered, for example,
as
proteins or other macromolecules at the same time, prior to, or subsequent to,
administration of the polynucleotides described herein. The nucleic acid
molecule compositions may also be administered directly to the subject or,
alternatively, delivered ex vivo, to cells derived from the subject, using
methods
known to those skilled in the art.
Coated Particles
In one embodiment, the polynucleotides (e.g., DNA vaccines) and/or
adjuvants are delivered using Garner particles (e.g., core carriers). Particle-
mediated methods for delivering such nucleic acid preparations are known in
the
art. Thus, once prepared and suitably purified, the above-described nucleic
acid
molecules and/or adjuvants can be coated onto carrier particles (e.g., core
Garners) using a variety of techniques known in the art. Carrier particles are
1 S selected from materials which have a suitable density in the range of
particle
sizes typically used for intracellular delivery from an appropriate particle-
mediated delivery device. The optimum Garner particle size will, of course,
depend on the diameter of the target cells. Alternatively, colloidal gold
particles
can be used wherein the coated colloidal gold is administered (e.g., injected)
into
tissue (e.g., skin or muscle) and subsequently taken-up by immune-competent
cells.
For the purposes of the invention, tungsten, gold, platinum and iridium
Garner particles can be used. Tungsten and gold particles are preferred.
Tungsten
particles are readily available in average sizes of 0.5 to 2.0 ,um in
diameter.
Although such particles have optimal density for use in particle acceleration
delivery methods, and allow highly efficient coating with DNA, tungsten may
potentially be toxic to certain cell types. Gold particles or microcrystalline
gold
(e.g., gold powder A1570, available from Engelhard Corp., East Newark, NJ)
will
also find use with the present methods. Gold particles provide uniformity in
size
CA 02389680 2002-05-02
WO 01/32204 PCT/US00/30223
-42-
(available from Alpha Chemicals in particle sizes of 1-3 ,um, or available
from
Degussa, South Plainfield, NJ in a range of particle sizes including 0.95 Vim)
and
reduced toxicity.
A number of methods are known and have been described for coating or
precipitating DNA or RNA onto gold or tungsten particles. Most such methods
generally combine a predetermined amount of gold or tungsten with plasmid
DNA, CaCl2 and spermidine. The resulting solution is vortexed continually
during the coating procedure to ensure uniformity of the reaction mixture.
After
precipitation of the nucleic acid, the coated particles can be transferred to
suitable
membranes and allowed to dry prior to use, coated onto surfaces of a sample
module or cassette, or loaded into a delivery cassette for use in a suitable
particle
delivery instrument.
Peptide adjuvants (e.g., cytokines), can also be coated onto suitable carrier
particles, e.g., gold or tungsten. For example, peptides can be attached to
the
carrier particle by simply mixing the two components in an empirically
determined ratio, by ammonium sulfate precipitation or other solvent
precipitation methods familiar to those skilled in the art, or by chemical
coupling
of the peptide to the Garner particle. The coupling of L-cysteine residues to
gold
has been previously described (Brown et al., Chemical Society Reviews 9:271-
311 (1980)). Other methods include, for example, dissolving the peptide
antigen
in absolute ethanol, water, or an alcohol/water mixture, adding the solution
to a
quantity of carrier particles, and then drying the mixture under a stream of
air or
nitrogen gas while vortexing. Alternatively, the peptide antigens can be dried
onto Garner particles by centrifugation under vacuum. Once dried, the coated
particles can be resuspended in a suitable solvent (e.g., ethyl acetate or
acetone),
and triturated (e.g., by sonication) to provide a substantially uniform
suspension.
CA 02389680 2002-05-02
WO 01/32204 PCT/US00/30223
-43-
Administration of Coated Particles
Following their formation, Garner particles coated with either nucleic acid
preparations, or peptide or protein adjuvant preparations, are delivered to a
subject, for example transdermally, using particle-mediated delivery
techniques.
Various particle delivery devices suitable for particle-mediated delivery
techniques are known in the art, and are all suited for use in the practice of
the
invention. Current device designs employ an explosive, electric or gaseous
discharge to propel the coated core carrier particles toward target cells. The
coated particles can themselves be releasably attached to a movable carrier
sheet,
or removably attached to a surface along which a gas stream passes, lifting
the
particles from the surface and accelerating them toward the target. An example
of a gaseous discharge device is described in U.S. Patent No. 5,204,253. An
explosive-type device is described in U.S. Patent No. 4,945,050. One example
of
an electric discharge-type particle acceleration apparatus is described in
U.S.
Patent No. 5,120,657. Another electric discharge apparatus suitable for use
herein is described in U.S. Patent No. 5,149,655. The disclosure of all of
these
patents is incorporated herein by reference in their entireties.
The coated particles are administered to the subject to be treated in a
manner compatible with the dosage formulation, and in an amount that will be
effective to bring about a desired immune response. The amount of the
composition to be delivered which, in the case of nucleic acid molecules is
generally in the range of from 0.001 to 100.0 pg, more typically 0.01 to 10.0
,ug
of nucleic acid molecule per dose, and in the case of peptide or protein
molecules
is 1 ,ug to 5 mg, more typically 1 to 50 ,ug of peptide, depends on the
subject to
be treated. The exact amount necessary will vary depending on the age and
general condition of the individual being immunized and the particular
nucleotide
sequence or peptide selected, as well as other factors. An appropriate
effective
amount can be readily determined by one of skill in the art upon reading the
instant specification.
CA 02389680 2002-05-02
WO 01/32204 PCT/US00/30223
-44-
Thus, an effective amount of the antigens herein described, or nucleic
acids coding therefor, will be sufficient to bring about a suitable immune
response in an immunized subject, and will fall in a relatively broad range
that
can be determined through routine trials. Preferably, the coated particles are
S delivered to suitable recipient cells in order to bring about an immune
response
(e.g., T-cell activation) in the treated subject.
Particulate Compositions
Alternatively, the antigen of interest (as well as one or more selected
adjuvant) can be formulated as a particulate composition. More particularly,
formulation of particles comprising the antigen and/or adjuvant of interest
can be
carried out using standard pharmaceutical formulation chemistries and
methodologies all of which are readily available to the reasonably skilled
artisan.
For example, one or more antigen and/or adjuvant can be combined with one or
more pharmaceutically acceptable excipient or vehicle to provide an antigen,
1 S adjuvant, or vaccine composition. Auxiliary substances, such as wetting or
emulsifying agents, pH buffering substances, and the like, may be present in
the
excipient or vehicle. These excipients, vehicles and auxiliary substances are
generally pharmaceutical agents that do not themselves induce an immune
response in the individual receiving the composition, and which may be
administered without undue toxicity. Pharmaceutically acceptable excipients
include, but are not limited to, liquids such as water, saline,
polyethyleneglycol,
hyaluronic acid, glycerol and ethanol. Pharmaceutically acceptable salts can
be
included therein, for example, mineral acid salts such as hydrochlorides,
hydrobromides, phosphates, sulfates, and the like; and the salts of organic
acids
such as acetates, propionates, malonates, benzoates, and the like. It is also
preferred, although not required, that an antigen composition will contain a
pharmaceutically acceptable carrier that serves as a stabilizer, particularly
for
peptide, protein or other like antigens. Examples of suitable Garners that
also act
as stabilizers for peptides include, without limitation, pharmaceutical grades
of
CA 02389680 2002-05-02
WO 01/32204 PCT/US00/30223
-45-
dextrose, sucrose, lactose, trehalose, mannitol, sorbitol, inositol, dextran,
and the
like. Other suitable carriers include, again without limitation, starch,
cellulose,
sodium or calcium phosphates, citric acid, tartaric acid, glycine, high
molecular
weight polyethylene glycols (PEGS), and combination thereof. A thorough
discussion of pharmaceutically acceptable excipients, Garners, stabilizers and
other auxiliary substances is available in REMINGTONS PHARMACEUTICAL
SCIENCES (Mack Pub. Co., N.J. 1991), incorporated herein by reference.
The formulated compositions will include an amount of the antigen of
interest which is sufficient to mount an immunological response, as defined
above. An appropriate effective amount can be readily determined by one of
skill
in the art. Such an amount will fall in a relatively broad range, generally
within
the range of about 0.1 pg to 25 mg or more of the antigen of interest, and
specific
suitable amounts can be determined through routine trials. The compositions
may contain from about 0.1 % to about 99.9% of the antigen. If an adjuvant is
included in the composition, or the methods are used to provide a particulate
adjuvant composition, the adjuvant will be present in a suitable amount as
described above. The compositions are then prepared as particles using
standard
techniques, such as by simple evaporation (air drying), vacuum drying, spray
drying, freeze drying (lyophilization), spray-freeze drying, spray coating,
precipitation, supercritical fluid particle formation, and the like. If
desired, the
resultant particles can be densified using the techniques described in
commonly
owned International Publication No. WO 97/48485, incorporated herein by
reference.
These methods can be used to obtain nucleic acid particles having a size
ranging from about 0.1 to about 250 Vim, preferably about 10 to about 150 Vim,
and most preferably about 20 to about 60 pm; and a particle density ranging
from
about 0.1 to about 25 g/cm3, and a bulk density of about 0.5 to about 3.0
g/cm3, or
greater.
CA 02389680 2002-05-02
WO 01/32204 PCT/US00/30223
-46-
Similarly, particles of selected adjuvants having a size ranging from about
0.1 to about 250 pm, preferably about 0.1 to about 150 pm, and most preferably
about 20 to about 60 Vim; a particle density ranging from about 0.1 to about
25
g/cm3, and a bulk density of preferably about 0.5 to about 3.0 g/cm3, and most
S preferably about 0.8 to about 1.5 g/cm3 can be obtained.
Single unit dosages or multidose containers, in which the particles may be
packaged prior to use, can comprise a hermetically sealed container enclosing
a
suitable amount of the particles comprising the antigen of interest and/or the
selected adjuvant (e.g., the vaccine composition). The particulate
compositions
can be packaged as a sterile formulation, and the hermetically sealed
container
can thus be designed to preserve sterility of the formulation until use in the
methods of the invention. If desired, the containers can be adapted for direct
use
in a needleless syringe system, and can take the form of capsules, foil
pouches,
sachets, cassettes, and the like.
The container in which the particles are packaged can further be labeled to
identify the composition and provide relevant dosage information. In addition,
the container can be labeled with a notice in the form prescribed by a
governmental agency, for example the Food and Drug Administration, wherein
the notice indicates approval by the agency under Federal law of the
manufacture,
use or sale of the antigen, adjuvant (or vaccine composition) contained
therein for
human administration.
Administration of Particulate Compositions
Following their formation, the particulate composition (e.g., powder) can
be delivered transdermally to vertebrate tissue using a suitable transdermal
particle delivery technique. Various particle delivery devices suitable for
administering the substance of interest are known in the art, and will find
use in
the practice of the invention. A particularly preferred transdermal particle
delivery system employs a needleless syringe to fire solid particles in
controlled
doses into and through intact skin and tissue. See, e.g., U.S. Patent No.
CA 02389680 2002-05-02
WO 01/32204 PCT/US00/30223
-47-
5,630,796 to Bellhouse et al. which describes a needleless syringe (also known
as
"the PowderJect~ particle delivery device"). Other needleless syringe
configurations are known in the art and are described herein.
The particulate compositions (comprising the antigen of interest and
optionally a selected adjuvant) can then be administered using a transdermal
delivery technique. Preferably, the particulate compositions will be delivered
via
a powder injection method, e.g., delivered from a needleless syringe such as
those
described in commonly owned International Publication Nos. WO 94/24263, WO
96/04947, WO 96/12513, and WO 96/20022, all of which are incorporated herein
by reference. Delivery of particles from such particle delivery devices is
practiced with particles having an approximate size generally ranging from 0.1
to
250 Vim, preferably ranging from about 10-70 Vim. Particles larger than about
250 ~m can also be delivered from the devices, with the upper limitation being
the point at which the size of the particles would cause untoward damage to
the
skin cells. The actual distance which the delivered particles will penetrate a
target surface depends upon particle size (e.g., the nominal particle diameter
assuming a roughly spherical particle geometry), particle density, the initial
velocity at which the particle impacts the surface, and the density and
kinematic
viscosity of the targeted skin tissue. In this regard, optimal particle
densities for
use in needleless injection generally range between about 0.1 and 25 g/cm3,
preferably between about 0.9 and 1.5 g/cm3, and injection velocities generally
range between about 100 and 3,000 m/sec, or greater. With appropriate gas
pressure, particles having an average diameter of 10-70 ~m can be accelerated
through the nozzle at velocities approaching the supersonic speeds of a
driving
gas flow.
If desired, these particle delivery devices (e.g., a needleless syringe) can
be provided in a preloaded condition containing a suitable dosage of the
particles
comprising the antigen of interest and/or the selected adjuvant. The loaded
CA 02389680 2002-05-02
WO 01/32204 PCT/US00/30223
-48-
syringe can be packaged in a hermetically sealed container, which may further
be
labeled as described above.
Compositions containing a therapeutically effective amount of the
powdered molecules described herein can be delivered to any suitable target
S tissue via the above-described particle delivery devices. For example, the
compositions can be delivered to muscle, skin, brain, lung, liver, spleen,
bone
marrow, thymus, heart, lymph, blood, bone cartilage, pancreas, kidney, gall
bladder, stomach, intestine, testis, ovary, uterus, rectum, nervous system,
eye,
gland and connective tissues. For nucleic acid molecules, delivery is
preferably
to, and the molecules expressed in, terminally differentiated cells; however,
the
molecules can also be delivered to non-differentiated, or partially
differentiated
cells such as stem cells of blood and skin fibroblasts.
The powdered compositions are administered to the subject to be treated
in a manner compatible with the dosage formulation, and in an amount that will
be prophylactically and/or therapeutically effective. The amount of the
composition to be delivered, generally in the range of from 0.5 pg/kg to 100
~g/kg of nucleic acid molecule per dose, depends on the subject to be treated.
Doses may be as low as 0.5 pg for 50 kg subject, or approximately 0.01 pg/kg.
Doses for other pharmaceuticals, such as physiological active peptides and
proteins, generally range from about 0.1 p,g to about 20 mg, preferably 10 pg
to
about 3 mg. The exact amount necessary will vary depending on the age and
general condition of the individual to be treated, the severity of the
condition
.being treated, the particular preparation delivered, the site of
administration, as
well as other factors. An appropriate effective amount can be readily
determined
by one of skill in the art.
Thus, a "therapeutically effective amount" of the present particulate
compositions will be sufficient to bring about treatment or prevention of
disease
or condition symptoms, and will fall in a relatively broad range that can be
determined through routine trials.
CA 02389680 2002-05-02
WO 01/32204 PCT/US00/30223
-49-
Vaccination Regimes
As is apparent to those skilled in the art in view of the teachings of this
specification, vaccination with the above-described polynucleotides (DNA
vaccines) can be effected in one dose, continuously or intermittently
throughout
the course of treatment. Methods of determining the most effective means and
dosages of administration are well known to those of skill in the art and will
vary
with the delivery vector, the nature of the composition, the specific therapy
sought, the target cells, and the subject being treated. Single and multiple
administrations can be carried out with the dose level and pattern being
selected
by suitable medical personnel. It should be understood that more than one
antigen can be expressed by the delivered polynucleotide. Alternatively,
separate
vectors, each expressing one or more different antigens under the control of a
co-
stimulatory molecule promoter, can also be delivered to a subject as described
herein.
Furthermore, it is also intended that the immunizing antigen (e.g.,
polynucleotide or peptide) delivered by the methods of the present invention
be
combined with other suitable compositions and therapies. For instance, a T-
cell
response may be enhanced by delivering a polynucleotide or peptide described
herein with one or more additional agents, such as cytokines, can be
administered
prior or subsequent to or simultaneously with (1) the polynucleotides having
co-
stimulatory molecule promoters driving antigen-expression; (2) polynucleotides
encoding at least one antigen or (3) peptide antigens. These additional agents
can
be provided in various forms, for example, as purified molecules, or by
vectors
encoding full-length or functional fragments of the polypeptide. The vectors
may
be distinct from those carrying the sequences encoding the antigen(s), or may
be
carned on the same vector. Whether the cytokine-encoding sequence is on the
same or different vector, it's expression can be driven by the same promoter
that
drives antigen expression or by a different promoter. In this way, APC
CA 02389680 2002-05-02
WO 01/32204 PCT/US00/30223
-50-
maturation cytokines such as CD40 ligand (CD40L or CD 154), tumor-necrosis
factor-related activation-induced cytokine (TRANCE), and Flt3 ligand (flt-3L),
can be provided to enhance the immune response, apparently by increasing
expression of costimulatory ligands on the APC, stabilizing the antigen/MHC
complex or inhibiting apoptosis of the APC.
Below are examples of specific embodiments for carrying out the present
invention. The examples are offered for illustrative purposes only, and are
not
intended to limit the scope of the present invention in any way.
EXAMPLES
Efforts have been made to ensure accuracy with respect to numbers used
(e.g., amounts, temperatures, etc.), but some experimental error and deviation
should, of course, be allowed for.
Example 1: Nucleic Acid Immunization Using CD80 promoter driven
plasmids
In order to assess the specificity and effectiveness of nucleic acid
immunization using DNA vaccine plasmids containing CD80 or CD86
promoters, the following studies were carried out.
A. Plasmid Preparation
The DNA sequence of the mouse and human CD80 gene promoter was
obtained from the GenBank Database. The DNA primers for synthesizing the
mouse CD80 promoter by PCR were obtained from Life Technologies, Gibco
BRL, and had the following sequences:
(1) 5'- ACG CGT CGA CTC TAG AAG GAG ACA TTC AGC TG -3' (SEQ ID
NO:1 )
CA 02389680 2002-05-02
WO 01/32204 PCT/US00/30223
-51-
(2) 5' - ACG CGT CGA CAG CTT TCA TGG CCT AGC TGC TA- 3' (SEQ ID
N0:2)
(3) 5' - ATT CGG CCG CGG TCT AGA GCC AAT GGA GCT TAG G - 3'
(SEQ ID N0:3)
(4) 5' - ATT CGG CCG CGG AGA GTT CTG AAT CAG GGT GT - 3' (SEQ
ID N0:4)
Similarly, DNA primers for synthesizing human CD80 promoter by PCR
were obtained from Life Technologies, Gibco BRL, and had the following
sequences:
(5) 5'- ACG CGT CGA CAG TCT TCC TCA TCC CAC CA -3' (SEQ ID
N0:5)
(6) 5'- ACG CGT CGA CCA TCA CAC AGC AAG GCT AG - 3' (SEQ ID
N0:6)
(7) 5'- ACG CGT CGA CGT TTG TTA GTC CAT GCA CG -3' (SEQ ID
N0:7)
(8) 5'- TCC CCG CGG AGA GAG GCG ACA TTT C- 3' (SEQ ID N0:8)
Fragments of the CD80 promoter (human and mouse) were amplified by
PCR by reacting 200 ~g of human or mouse genomic DNA, 1X TurboTM Pfu
buffer (Stratagene, La Jolla, CA, 20 mM Tris-Cl, pH 8.8, 2 mM MgS04, 10 mM
KCI, 10 mM (NH4)2 S04, 0.1% Triton, 0.1 mg/ml nuclease-free BSA), 20 pm 5'
primer, 20 pm 3' primer, 200 ~m dNTPs, 1U Perfect MatchTM PCR enhancer
(StrataGene, La Jolla, CA) and 2.5 U Pfu TurboTM (Stratagene) for 35 cycles
of:
96°C for 45 seconds; 55 °C for 45 seconds and 72°C for 45
seconds. Following
completion of the 35 cycles, the PCR products were held at 4°C. The PCR
product was purified by QIAquickTM PCR purification kit (Qiagen Corporation),
according to the manufacturer's instructions.
CA 02389680 2002-05-02
WO 01/32204 PCT/US00/30223
-52-
The purified PCR product was double digested with Sal I (New England
BioLabs) and Sac II (New England BioLabs) in buffer #3 (100 mM NaCI, 50 mM
Tris-Cl, 10 mM MgCl2, 1 mM DTT, pH 7.9) at 37°C overnight. The
digested
PCR product was run on a 1 % agarose gel and the correct band was excised from
the gel. The DNA in the gel slice was purified using a GenEluteTM ethidium
bromide spin column (Supelco).
Plasmids were constructed by removing the CMV promoter from plasmid
pWRG7128 (Tacket et al. (1999) Vaccine 17:2826) and replacing it with the
amplified CD80 promoter segments. The plasmid pWRG7128 contains, in
addition to suitable control elements, a sequence encoding the hepatitis B
surface
antigen (HBsAg) which is under the transcriptional control of a
cytomegalovirus
(CMV) promoter, and has been shown to produce HbsAg particles upon
transfection into most cell types. The pWRG7128 plasmid was constructed as
follows. A cloning vector pWRG7077 (Schmaljohn et al. (1997) J. Virol.
1 S 71:9563-9569) was prepared to accept a HBsAg coding sequence by digesting
the
vector to completion with BamHl, followed by a partial digest with Hind3.
After
blunting the 5' overhangs by treatment with Klenow fragemnt and
deoxyribonucleotides, the 4.3 kB vector fragment was isolated. The 1.35 kB
HbsAg insert fragment (containing the untranslated pre-S2 sequence, the 226
amino acid HbsAg coding sequence of the adw strain, and the HBV enhancer
element) was excised from plasmid pAM6 (ATCC, Rockford, MD) by digesting
with BamHl. After blunt-ending by treatment with the Klenow fragment and
deoxyribonucleotides, the fragment was isolated and ligated into the 4.3 kB
vector fragment described above. The resulting recombinants were screened for
proper orientation of the insert and a correct isolate was identified and
designated
as an intermediate plasmid (pWRG7074). In order to remove the start of the
codon of the X protein (present at the 3' end of the pAM6 1.35 kB insert), a
4.86
kB vector fragment was isolated from the pWRG7074 plasmid by digesting with
Bgl2, blunt-ending with the Klenow fragment and deoxyribonucleotides, and then
CA 02389680 2002-05-02
WO 01/32204 PCT/US00/30223
-53-
digesting with BstX 1. Next, a 754 by insert fragment was isolated from the
pWRG7074 construct by digestion with Ncol, treating with mung bean nuclease,
and digesting with BstX l . The resulting vector and insert fragments were
then
ligated together to form the clinical plasmid pWRG7128. The plasmids
constructed are shown in Table 1 and in Figures 1-7.
Table 1: Representative Plasmids
Plasmid Name Primers Length of CD80 promoter obtained
(Size) Used (Source)
p5020 (5044 bp) (2) and 254 by (mouse)
(4)
p5021 (5279 bp) (2) and 489 by (mouse)
(3)
p5022 (7913 bp) (1) and 3123 by (mouse)
(4)
p5023 (8147 bp) (1) and 3357 by (mouse)
(3)
p5024 (5368 bp) (5) and 578 by (human)
(8)
p5025 (5084 bp) (6) and 294 by (human
(8)
p5026 (4992 bp) (7) and 202 by (human)
(8)
pWGR7128 was digested with Sal I and Sac II in buffer #3 at 37°C
overnight. The digested vector was run on a 1% agarose gel and the correct
vector band was excised from the gel and purified using a GenEluteTM ethidium
bromide spin column.
To ligate the promoter into the prepared vector, 20 ng of vector and 100
ng of insert promoter were ligand in lx T4 DNA ligase buffer (50 mM Tris-Cl,
10 mM MgClz, 10 mM DTT, 1 mM ATP, 25 pg/ml BSA, pH 7.8) by 6 weiss
units T4 DNA ligand (New England Biolabs) at 15 °C overnight.
For selection of properly ligated constructs, the ligation mixture was
diluted 1:3 in sterile water. Five ~1 of the diluted mixture added to 50 p1
MAX
Efficiency DHSa Competent CellsTM (Gibco-BRL) and incubated on ice for 30
CA 02389680 2002-05-02
WO 01/32204 PCT/US00/30223
-54-
minutes. The transformation mix was heat shocked at 42°C for 45 seconds
and
immediately put back on ice for a 2 minute incubation. One ml of SOC media
was added to the transformed cells and incubated with shaking at 37°C
for 1
hour. After incubation, the cells were centrifuged briefly, and plated onto a
kanamycin-LB plate. The plates were incubated overnight at 37°C.
To confirm successful ligation, single colonies were picked and
inoculated into 3.0 ml of LB/Kan media and cultured, with shaking, at 37
°C
overnight. Plasmids were isolated from the culture, digested with Sal I and
Sac II
and visualized on a 1 % agarose gel with ethidium bromide staining.
B. Antigen Expression
Two mls B 16 cells were seeded in a 6 well plate at 2 x 105 cells/ml. The
cells were cultured in a 37°C incubation with 5% COZ overnight to reach
60-80%
confluency. The cells were transfected as follows. Two ~g of endotoxin free
plasmid Dna in 100 p1 Opti-MEM~1 reduced serum media (Gibco-BRL) and 10
u1 of lipofectin in 90 u1 in Opti-MEM~1 reduced serum media were separately
incubated at room temperature for 45 minutes. The two solutions were then
mixed and incubated for 15 minutes at room temperature. During this
incubation,
the B 16 cells were washed in serum-free medium 3 times. For each
transfection,
0.8 ml of Opti-MEM~1 reduced serum media was added to the mixed solutions
and the complex overlaid onto the B 16 cells. The cells were then incubated at
37°C for 5 hours. Subsequently, 1.0 ml 20% FBS medium was added. The
medium was collected 48 hours later for antigen expression analysis.
Expression of HBsAg in transfected B16 cells is shown in Table 2. The
cells were treated in one of the three following manners: (1) transfection
with a
positive control, i.e., pWRG7128 which expressed antigen (i.e., HBsAg ) in B16
cells; (2) transfection with plasmids p5020 and p5021 as described above and
(3)
not transfected (negative control).
CA 02389680 2002-05-02
WO 01/32204 PCT/US00/30223
-5$-
TABLE 2. In Vitro Expression of HBsAg by Cells Transfected with Various
Plasmids
Cell Culture Groups Mean Level of Antigen Expresion*
Non-Transfected Cells (Negative0.0075
Control)
Cells transfected with pWRG7128>2.0
Cells transfected with p50200.0045
Cells transfected with p50210.0065
* Data are presented as the mean OD values (492.6 wavelength) for supernatants
obtained from two separate cultures and analyzed using HBV surface antigen
kit, Abbott
Laboratories Diagnostic Division (Auszyme Monoclonal, List No. 1980-24).
As shown in Table 2, unlike the positive control plasmid pWRG7128,
expression of the HBV surface antigen was not detected in cultured B 16 cells
transfected with plasmids p5020and p5021. Thus, the CD80 promoter does not
drive expression of antigen in non-antigen-presenting cells.
C. Preparation of coated microparticles
Plasmid DNA was coated onto 1-3 ,um gold particles (Degussa Corp.,
South Plainfield, NJ) using techniques described by Eisenbraun et al. (1993)
DNA
Cell Biol. 12:791-797. Briefly, gold particles were suspended in 50 mM
spermidine and mixed with an equal volume of plasmid in water. This solution
was mixed on a vortex and volume of 1 M CaClz half that of the gold/DNA
mixture was added dropwise. The mixture was incubated for 10 minutes at room
temperature and centrifuged to pellet the particles. The particles were washed
3
times with 100% ethanol and resuspended in ethanol containing 0.05-0.5%
polyvinyl pyrrilodine (PVP). The DNA-coated gold particles were then loaded
into Tefzel~ tubing as described in U.S. Patent No. 5,584,807 to McCabe, and
the tubing was cut into 1.27 cm lengths to serve as cartridges in a
PowderJect~
CA 02389680 2002-05-02
WO 01/32204 PCT/US00/30223
-S 6-
XR-1 particle delivery device (PowderJect Vaccines, Inc. Madison, WI). The
helium-pulse XR-1 particle delivery device has been previously described (see,
e.g., U.S. Patent Nos. 5,584,807 and 5,865,796). In the vaccinations, each
1.27
cm cartridge contained 0.5 mg gold particles coated with 2 pg of plasmid DNA.
D. Antibody Response
Based on the positive results seen in the above-described in vitro
transfection study, a vaccination trial was initiated using in vivo particle-
mediated
delivery methods. Animal subjects receiving nucleic acid immunizations in the
present study included: (1) a first experimental group of 4 mice that were
vaccinated by particle-mediated delivery to the epidermis with pWGR7128
positive control; and (2) a second experimental group of 6 mice that were
vaccinated by particle-mediated delivery to the epidermis with the CD80-
promoter driven HBsAg plasmids p5020 or p5021. Blood samples were taken
from each animal prior to immunization (naive mice).
Mice were immunized (primed) with plasmids p5020 and p5021 or with
the control pWGR7128 by particle-mediated delivery of the plasmid coated onto
gold-particles (0.5 mg of gold/shot coated with 2 pg DNA/mg gold). For
immunization, mice were shaved and the particles delivered to the abdomen skin
using an XR-1 device with research barrel operated at 500 psi of helium. Two
shots were given to each mouse per immunization. Four weeks later the animals
were boosted, following the same immunization protocol used for the prime
immunization.
Serum was collected from the animals prior to immunization, at 2 and 4
weeks after prime, and at 2 weeks after boost. Serum antibody levels were
analyzed using the AUSAB EIA kit, Abbott Laboratories Diagnostics Division,
according to the directions supplied by the manufacturer. No antibody
reactivity
against hepatitis B virus (HBV) surface antigen was detected in the sera
samples
taken from animals immunized with plasmids p5020 and p5021; however, at 2
CA 02389680 2002-05-02
WO 01/32204 PCT/US00/30223
-$ 7-
weeks post boost the serum antibody titers were >3000 m1U/ml in mice
immunized with pWGR7128 (Table 3).
TABLE 3. HBsAg Specific Serum Antibody Titers in Mice
Immunized with CD80 Plasmids
Treatment Group Mean Level of Antibody Titer*
Naive Mice (Negative Control)223.3
Mice Immunized with pWRG7128>3000.0
Mice Immunized with p5020 17.9
Mice Immunized with p5021 32.1
* Data are presented as the mean HBV surface antigen antibody titer (m1U/ml)
for
designated groups of mice. Serum antibody titers were determined using the
assay kit
from Abbott Laboratories Diagnostic Division (AUSAB EIA, List 9006-24).
Thus, CD80 promoter driven plasmids do not cause a rise in mean
antibody titers in serum.
E. Cell Mediated immune Response
Based on the positive results seen in the above-described analysis of
serum antibody titers, a study of cytotoxic T-cell activity in the immunized
mice
was conducted. Cytotoxic T-cell (CTL) activity was analyzed with splenocytes
obtained from immunized mice that were sacrificed at 2 weeks post boost. As
shown in Figure 8, CTL responses elicited by immunization with plasmids p5020
and p5021 were similar to those elicited by plasmid pWRG7128. These
responses were much greater than that seen in splenocytes collected from naive
mice.
As a result of the above-described studies, it can be seen that nucleic acid
immunization provides a T-cell specific immune response where antigen
expression is driven by a promoter derived from a co-stimulatory molecule.
CA 02389680 2002-05-02
WO 01/32204 PCT/US00/30223
-5 8-
Moreover, the T-cell response is comparable to that seen using the positive
control.
Example 2: SIV-Immune Response
S In order to determine if an expression vector encoding HBsAg driven by a
human CD80 promoter (hCD80-HBsAg) expresses in monkey dendritic cells, the
following studies are conducted. Monkey dendritic cells (DCs) are isolated
from
PBMC, essentially as described in van der Meide et al. (1995) J. Med.
Primatol.
24:271-281. The isolated DCs are than transfected, essentially as described in
Example 1 for B16 cells with plasmids encoding HBsAg driven by a human
CD80 promoter. A non-APC line, for example monkey COS cells are similarly
transfected. Expression of HBsAg in the supernatant and/or in cells is
conducted
by immunohistochemical staining.
In view of the cell-specific expression, studies are conducted to determine
the extent of HBsAg expression in APCs. Plasmid hCD80-HBsAg is delivered
into the epidermis of monkey skin using a PowderJect~ XR particle delivery
device. Various additional epidermal sites are also studied. Gold is used as a
negative control while CMV-HBsAg is used as a positive control. GM-CSF is
administered to test for dendritic cell recruitment. A biopsy is performed on
the
sites of administration after 24 or 48 hours. The tissue is sectioned and
evaluated
for HBsAg expression in dendritic cells by immunohistochemical staining using
specific antibodies for HBsAg and dendritic cell surface markers. Expression
of
HBsAg in dendritic and Langerhans cells is evaluated.
A hCD80-SIV expression vector is constructed, for example, by replacing
the sequence encoding HBsAg with a suitable SIV-encoding sequence.
Monkeys are immunized with the hCD80-SIV construct and compared to
monkeys immunized with a CMV-SIV plasmid (positive control). Delivery of
the plasmids is performed using the particle delivery device. Antigen
expression,
antibody response and CTL activation is evaluated, essentially as described
above
CA 02389680 2002-05-02
WO 01/32204 PCT/US00/30223
-S 9-
in Example 1. The monkeys are then challenged with a pathogenic SIV and
monitored for clinical manifestations of SIV.
Example 3: Use of a Cytokine Adjuvant
In order to assess the ability of a nucleotide encoding a TNF related
activation induced kinase (TRANCE) to enhance an immune response against a
coadministered antigen sequence, the following study was carned out.
A. Plasmid Preparation
A cDNA coding sequence for murine TRANCE was derived from the
mRNA sequence (GenBank No. AF013170) and cloned into the insertion site of a
pFLAG-CMV2 expression vector (Sigma, catalog number E4026) to provide an
expression construct containing the TRANCE coding sequence under
transcriptional control of the CMV2 promoter. The plasmid construct was
termed pTRANCE.
A plasmid containing sequences encoding the hepatitis B core antigen
(HBcAg) and hepatitis B surface antigen (HBsAg) was constructed as follows.
HBcAg and HBsAg coding sequences were both obtained from the HBV clone
pAM6 (ATCC Accession No. 45020). To generate the HBsAg coding region, the
pAM6 construct was cut with NcoI and treated with mung bean nuclease to
remove the start codon of the X-antigen. The resultant DNA was then cut with
BamHI and treated with T4 DNA polymerise to blunt-end the DNA and create an
HBsAg expression cassette. The HBsAg expression cassette is present in the 1.2
kB fragment. The plasmid construct pPJV7077 (Schmaljohn et al. (1997) J.
Virol. 71:9563-9569) which contains the full-length human CMV (Towne strain)
immediate early promoter (with enhancer) was cut with HindIII and BgIII, and
then treated with T4 DNA polymerise and calf alkaline phosphatase to create
blunt-ended DNA, and the HBsAg expression cassette was ligated into the
plasmid to yield the pWRG7128 construct.
CA 02389680 2002-05-02
WO 01/32204 PCT/US00/30223
-60-
To generate the HBcAg coding region, the pAM6 construct was cut to
create an HBcAg expression cassette, after which the HBcAg sequence was
truncated by site directed mutagenesis to remove the C-terminal arginine-rich
region from the core antigen particle (which deletion does not interfere with
particle formation). The truncated HBcAg sequence was then cloned into a
plasmid construct containing the human elongation factor promoter ("hELF",
Mizushima et al. (1990) Nucl. Acids Res. 18:5322) to provide a HBcAg vector
construct.
Expression cassettes containing: (a) the CMV promoter/enhancer, the
Intron A- 5' untranslated region, and the human tissue plasminogen activator
(hTPA) signal peptide ("CMV-IA-TPA"); or (b) the bovine growth hormone
polyA sequence (bGHpA) were each obtained from the JW4303 vector construct
(gift of Dr. Harriet Robinson, University of Massachusetts) and inserted into
a
plasmid backbone. The resultant construct was cut with NheI, filled with
polymerise and then cut with BamHI to generate a vector fragment containing
the
pUC 19 origin of replication, the ampicillin resistance gene and the bGHpA
sequence. The plasmid backbone was cut a second time with SaII, filled with
polymerise, and cut with BamHI to liberate a vector,fragment containing the
CMV-IA-TPA vector fragment. The two vector fragments were ligated together
to yield a construct termed pWRG7054.
The pWRG7054 construct was cut with NheI, filled with polymerise, and
cut with BamHI to produce a vector fragment. The HBcAg vector construct was
cut with NcoI, filled with polymerise, and cut with BamHI to produce an insert
fragment. The two fragments were then ligated together to yield a construct
termed pWRG7063.
PEL-Bos was cut with EcoRI and dephosphorylated with calf intestinal
phosphatase to produce a vector fragment. The pWRG7063 plasmid was cut with
HindIII, filled with polymerise, and cut with EcoRI to produce an insert
fragment
containing the hTPA signal peptide, the HBcAg antigen sequence and the
CA 02389680 2002-05-02
WO 01/32204 PCT/US00/30223
-61-
bGHpA region. These two fragments were ligated together to provide a construct
termed pWRG7145.
The pWRG7128 construct was cut with EcoRI and dephosphorylated with
calf intestinal phosphatase to produce a vector fragment containing the HbsAg
coding region under transcriptional control of the hCMV promoter. The
pWRG7145 construct was cut with MfeI and EcoRI to produce an insert fragment
comprised of the hELF promoter/intron, the hTPA signal peptide sequence, the
HBcAG antigen sequence and the bGHpA region. These fragments were then
ligated together to provide the pPJV7193 plasmid construct containing the
HBcAg and HBsAg coding sequences.
B. Vaccine Preparation and Immunization
A panel of DNA vaccine compositions was assembled using various
combinations of the following DNA plasmids: the pPJV7193 construct (encoding
the hepatitis B surface antigen and hepatitis B core antigen); the pPJV7046
construct (an empty DNA plasmid); and the pTRANCE construct (encoding the
TRANCE cytokine). The final DNA concentration in each vaccine composition
was 2.0 pg DNA/mg gold, and the concentration of the antigen construct
(pPJV7193) was kept constant in all compositions, while the concentration of
the
TRANCE construct was varied. The actual concentrations of each constituent
present in the panel of DNA vaccine compositions are reported in Table 4
below.
CA 02389680 2002-05-02
WO 01/32204 PCT/US00/30223
-62-
Table 4. TRANCE DNA Vaccine Compositions
Concentration
(fig DNA/mg
gold)
Ratio (pPJV7193 pPJV7193 pPJV7046 pTRANCE
:
pTRANCE)
(control) 1.0 1.0 0
1:1 1.0 0 1.0
5:1 1.0 0.8 0.2
25:1 1.0 0.96 0.04
125:1 1.0 0.992 0.008
625:1 1.0 0.9984 0.0016
Plasmid DNA (pPJV7193, pPJV7046 and pTR.ANCE) was combined at
the ratios reported in Table 4 above and then the plasmid mixture was coated
onto
1-3 ~m gold particles using the technique described above in Example 1 to
obtain
a final concentration of 2 ~g DNA/mg gold. The DNA-coated gold particles
were then loaded into Tefzel~ tubing as in Example 1 above, and cut into
lengths
to serve as cartridges in the PowderJect~ XR particle delivery device. Each
cartridge contained 0.5 mg gold particles.
Six experimental groups of 4 Balb/c mice each were assembled and
immunized (primed) with the DNA vaccine compositions listed in Table 4 above
by particle-mediated delivery of the plasmid DNA coated onto gold-particles.
For immunization, mice were shaved and the particles delivered to the abdomen
skin in a single shot using a particle delivery device operated at 500 psi of
helium. The mice were sacrificed at two weeks post immunization, and the
spleens were removed for ELISPOT analysis.
CA 02389680 2002-05-02
WO 01/32204 PCT/US00/30223
-63-
C. ELISPOT Analysis
An ELISPOT filter plate was coated with rat anti-mouse IL-4 antibodies
at a concentration of 0.75 ~g/well in a O.1M carbonate buffer. The plate was
incubated overnight at 4°C. After washing two times with phosphate
buffered
saline (PBS), the plate was blocked with 100 ~1 of RPMI medium supplemented
with 10% fetal bovine serum (FBS) for one hour at 37°C. The media was
discarded after blocking. Splenocytes, with the red blood cells lysed and
resuspended at 1 x 10' cells/ml in RPMI (supplemented with 10% FBS, sodium
pyruvate and non-essential amino acids) were added at a concentration of 1 x
106
cells per well or 0.5 x 106 cells per well. 100 p1 of whole hepatits B virus
core
antigen (BioDesign), diluted to 20 pg/ml in RPMI supplemented with 10% FBS
was added to the wells and the plate was incubated at 37°C for 48
hours. After
incubation, the cells and media were discarded and the plate washed two times
with PBS, followed by a deionized water wash to lyse any remaining cells and
then washed two more times with PBS. Detection antibody (biotinylated rat anti-
mouse IL-4) was diluted to 1 pg/ml in PBS, and 50 ~1 was added to each well
and incubated at room temperature for 1 hour. The plate was then washed five
times with PBS, and 50 p1 of strepavidin alkaline phosphatase conjugate
(diluted
1:1000 in PBS) was added to each well. Following a 1 hour incubation at room
temperature, the plate was washed five times with PBS, and 50 p1 of a
chromagenic alkaline phosphatase substrate was added to each well. As soon as
spots emerged, the reaction was stopped by rinsing the plate with tap water.
Spots were counted and the results were graphed as specific spots per million
splenocytes in Figure 9. As can be seen by review of the data depicted in
Figure
9, IL-4 production specific to the HBcAg was increased relative to the control
(non-pTR.ANCE containing composition), and showed a dose-related decrease in
specific spots.
CA 02389680 2002-05-02
WO 01/32204 PCT/US00/30223
-64-
D. Antibody Analysis (ELISA~
The above-described study was repeated with coated DNA particle
cartridges prepared as before, but with the following modifications: the
pPJV7193 construct (encoding the hepatitis B surface antigen and hepatitis B
core antigen) and the pTRANCE construct (encoding the TRANCE cytokine)
were coated onto separate batches of gold particles (the pTRANCE and
pPJV7046 plasmids were combined as above and the combination coated onto
gold particles, but the pTRANCE plasmid was coated onto a separate batch of
gold particles). The two batches of coated gold particles were then mixed
together prior to coating the Tefzel~ tubing. The same concentrations of DNA
as
reported in Table 4 above were used with the one exception that the 625:1
ratio
was not included in this second study. Experimental groups of 4 Balb/c mice
each were immunized as before, and the mice were sacrificed at 2 weeks post
immunization and serum collected for antibody analysis by ELISA.
For the antibody analysis, an ELISA plate was coated with 100 ~l of
whole hepatits B virus core antigen (BioDesign), diluted to 0.1 ~g/ml in PBS
and
incubated at 4°C overnight. The plate was washed one time with PBS with
0.05% Tween 20 (PBS-T), then blocked with 300 ~1 of blocking solution (PBS
plus 5% dried milk) for 1 hour at room temperature. The blocking solution was
discarded and 100 ~l of serially diluted serum was added. The plate was
incubated for 1 hour at room temperature, followed by washing three times with
PBS-T. A conjugated antibody (goat anti-mouse IgG-horse radish peroxidase)
was diluted 1:5000 in PBS plus 2% dried milk. 100 ~l of the conjugated
antibody as added to each well, and the plate was incubated for 1 hour at room
temperature. The plate was then washed five times with PBS-T and 100 ~l of
TMB substrate was added to each well. After 15 minutes, the reaction was
stopped with 100 ~1 of 1N HzS04. Absorbance was read at 450 nm. The
geometric mean of each experimental group was then calculated and graphed
against the dilution in Figure 10. As can be seen by review of the data
depicted
CA 02389680 2002-05-02
WO 01/32204 PCT/US00/30223
-65-
in Figure 10, there was an increased anti-HBcAg antibody response in all
groups
receiving the pTRANCE adjuvant composition except for the composition
containing the 25:1 ratio.
Accordingly, addition of the pTRANCE cytokine encoding plasmid
construct in the hepatitis DNA vaccine compositions enhanced both the cellular
and humoral immune responses to the antigen of interest.
Accordingly, novel compositions for elicting an immune have been
described. Methods of using these compositions have also been described.
Although preferred embodiments of the subject invention have been described in
some detail, it is understood that obvious variations can be made without
departing from the spirit and the scope of the invention as defined by the
appended claims.