Note: Descriptions are shown in the official language in which they were submitted.
CA 02389832 2002-04-18
WO 01/32916 PCT/US00/30280
IMPROVED AUTOMATED LPA ASSAY and
METHODS of DETECTING CANCER
This application claims priority to the provisional application Serial No.
60/163,534
filed on November 4, 1999.
Introduction & Background of the Invention
Cancer is a major cause of death in the United States exceeded only by heart
disease. In 1999 an estimated 563,100 Americans will die of cancer. Moreover,
approximately 1,221,800 new cases of cancer are predicted in the US for the
year. The
major solid tumors in the US include those of the lung, breast, colon,
prostate, and
ovaries. Lung cancer is the most common cause of cancer death for both sexes
with
almost 159,000 lung cancer related deaths expected in 1999. Total colorectal
cancer-
related deaths are second only to lung cancer with over 56,000 expected.
Breast cancer
continues to be the most common form of cancer present in females in the US
with an
estimated 176,300 new cases projected to be diagnosed during the year. In
males,
prostate cancer is the most common form of cancer with projections of 179,300
new
cases diagnosed and 37,000 prostate cancer related deaths occurring during
1999.
Ovarian cancer is the leading cause of gynecologic death.
Procedures used for detecting, diagnosing, staging, monitoring,
prognosticating,
preventing or treating, or determining predisposition of diseases or
conditions of these
organs are of critical importance to the outcome of the patients. It is
generally accepted
that detection of a solid tumor at an early stage dramatically reduces disease-
related
mortality. For example, patients diagnosed with localized prostate cancer have
greater
than a 90% five-year relative survival rate compared to a survival rate of 25
to 31 % for
patients diagnosed with distant metastasis. Staging of the cancer is performed
after its
diagnosis is confirmed because it is a strong predictor of patient outcome and
greatly
influences patient treatment. In addition, patients are monitored after
primary therapy to
detect persistent disease and to detect early distant metastasis. New testing
methods,
however, which are more sensitive and specific than current standard
procedures for the
management of cancer patients are clearly needed.
CA 02389832 2002-04-18
WO 01/32916 PCT/US00/30280
2
Women with gynecological cancers are especially in need of an accurate and
early diagnostic, especially those with ovarian cancer. Patients with ovarian
cancer have
the highest mortality rate among women with gynecologic cancers, with an
estimated
14,500 deaths from ovarian cancer in 1998 in the Unites States. More than two
thirds of
patients with ovarian cancer have widespread metastatic disease at initial
diagnosis. The
outlook for women with advanced disease remains poor, with a 5-year survival
rate of no
more than 15%. This dismal outcome is due, at least in part, to the failure to
detect the
disease at stage I, when the long-term survival rate may approach 90%. Methods
for
earlier detection are essential to improve prognosis and overall survival of
patients with
ovarian cancer.
Prior Art
It is generally known that detection of various lysophospholipids, such as
lysophosphatidic acid ("LPA") is indicative of various types of disease,
including
carcinomas and especially ovarian carcinoma. Xu et al, "Lysophosphatidic Acid
as a
Potential Biomarker for Ovarian and Other Gynecological Cancers", JAMA , 1998
Aug.26;280 (8):719-723. Thus, LPA measurement can be used as a diagnostic to
detect
carcinomas and, especially to detect early stage ovarian cancer.
The prior art generally describes a method of detecting LPA as follows. The
lysophospholipid, such as LPA, is incubated with lysophospholipase to produce
glycerol-
3-phosphate (G-3-P). G-3-P is then converted to dihydroxyacetone phosphate and
hydrogen peroxide using G-3-P oxidase in the presence of oxygen and water. In
the
presence of NADH, G-3-P dehydrogenase converts dihydroxyacetone phosphate back
to
G-3-P and oxidizes NADH to NAD. The measurement of hydrogen peroxide
correlates
with LPA levels. Specifically, optical absorbance at SOSnm indicates an
accumulation of
hydrogen peroxide and, thus, the presence of LPA in the test sample.
To achieve the aforementioned, the prior art teaches a mufti-step process in
order
to measure LPA concentrations. First, the prior art procedure for measuring
LPA in
order to detect cancer involves an initial liquid: liquid organic phase
extraction using a
number of reagents in a mufti-step procedure to a biological sample such as
whole blood
is collected from a patient.
CA 02389832 2002-04-18
WO 01/32916 PCT/US00/30280
Because the sample, usually plasma, is presumed to contain materials which
interfere with the assay, the analyte is extracted from the sample and
reconstituted to its
original concentration in a buffer compatible with the subsequent assay. The
plasma
sample is first vortexed with chloroform:methanol to precipitate protein.
After
centirfugation to pellet the protein, more chloroform and tris buffer are
added and the
mixture again vortexed and centrifuged. At this stage, most of the neutral
phospholipids,
including LPC are in the organic layer, which is separated and discarded. The
aqueous
layer is extracted with an additional portion of chloroform to remove any
remaining
neutral phospholipids, and the layers again separated. The aqueous layer is
then
subjected to a third chloroform:methanol extraction, this time with
hydrochloric acid
added to protonate and so neutralize the charge on LPA, resulting in its
transfer to the
organic layer. This time the aqueous layer, containing water soluble salts
including G3P,
is discarded. The organic layer is then mixed with a small amount of an
aqueous tris
buffer containing detergent and calcium chloride, and the solvent evaporated
off, leaving
the residue which is stored at -80° C until reconstitution and assay.
Because of the
numerous pipetting steps and multiple extractions there is great potential for
loss and
accumulation of error in this procedure. This, along with the use of the
toxic, volatile
solvent chloroform, greatly complicate its use in a clinical laboratory.
Following the aforementioned extraction and reconstitution of the sample to
aqueous solution, the prior art dictates that LPA is hydrolysed in a separate
single step to
G3P by exposure to a lysophospholipase for 60 minutes at 37° C. The G3P
is then
treated with a mixture of enzymes which consumes NADH and produces hydrogen
peroxide at a rate dependent on the G3P concentration. One of the enzymes, G3P
oxidase, oxidizes G3P to DHAP with the evolution of hydrogen peroxide. The
other
enzyme, Glycerophosphate degydrogenase, converts the DHAP back to G3P, this
reverse
reaction being driven by the conversion of NADH to NAD. The rate of this
cycling
reaction, i.e. the rate at which hydrogen peroxide is produced and NADH
converted to
NAD, is dependent (all other variables being held constant) on the
concentration of G3P.
According to the prior art the extent of reaction may be determined in either
of two ways.
The concentration of NADH may be monitored either continuously or at the end
of the
incubation, and its decrease determined by measuring the loss of absorbance at
340 nm.
Alternatively, at the end of the incubation, set at 60 min in the prior art,
the amount of
CA 02389832 2002-04-18
WO 01/32916 PCT/US00/30280
hydrogen peroxide generated may be measured by a colorimetric reaction using
perxidase and a colorigenic substrate
The disappearance (oxidation) of NADH is then monitored
spectrophotometrically at OD3ao (i.e. disappearance of OD3ao). Alternatively,
the
production of hydrogen peroxide may be measured, for example colorimetrically
by
fluorometry or chemiluminescence. For a colorimetric assay any of a number of
chromogenic substrates may be used including 4-aminoantipyrine (AAP),
pyrogallol, 2-
(2'-Azinobis (3-ethylbenzthiazoline-sulfonic acid) (ABTS) and 3,3',5,5'-
tetramethylbernzidine) (TMB).
As one can clearly see, this complicated LPA detection process calls for many
separate reagents to be used in many separate steps in a specified
chronological order.
Further, several of the reagents must be mixed together just prior to use. Due
to this
complexity, it is difficult, if not impossible, to put this LPA detection
assay on an
automated format and, the lengthy incubation periods required for each step
make it
15 difficult to use this assay in a clinical format.
Further problems in the prior art involve poor calibrator stability. In the
past,
calibrators were stored at -70° C until time for use. One disadvantage
is that the
requirement of -70° C storage is both difficult and costly. Further,
easy spoilage of the
calibrator due to a lapse in -70° C conditions leads to false patient
read-outs. The
2o present inventors have invented a calibrator which is stable at 4° C
as well as room
temperature. This novel calibrator has calcium completely eliminated from the
calibrator
storage matrix. It was previously believed that calcium was critical in
complexing LPA
away from the sides of storage containers. The present inventors found that
the presence
of calcium actually was responsible for reduced stability or unavailability of
the LPA
25 itself once the storage was at a temperature greater than -70° C.
Other problems in the prior art involve separate hydrolysis and cycling steps,
as
mentioned previously. In the past at least two separate steps - one hydrolysis
step and
one cycling step - were used. The disadvantage was that many different reagent
formulations were used and two separate 1 hour incubations were required.
Specifically,
3o the prior art methodology teaches keeping the various enzymes as separate
reagents and
storing each at a different temperature. Lysophospholipase is stored at -
80° C and kept
as a separate hydrolysis reagent for use in the 1 hour hydrolysis step. The
cycling
CA 02389832 2002-04-18
WO 01/32916 PCT/LTS00/30280
enzymes were broken out into a multitude of reagents. The reason for the
aforementioned steps and mufti-reagent format is that it was previously
thought that a
complete conversion of LPA to G3P, prior to the cycling reaction, was
necessary in order
to obtain an accurate measurement. The inventors have combined hydrolysis and
cycling
compounds into a single reagent and further optimized the assay such that the
reaction
time can be reduced from over 2 hours to 15 minutes or less. Specifically, the
inventors
have combined the lipase, G3P oxidase, G3P dehydrogenase, CaCl2 and Tris into
a
single reagent stable at 4° C. The NADH is stored separately at -
20° C. The inventors
found, quite unexpected, that the generation of G-3-P from LPA did not
interfere with
1o the cycling steps performed by G-3-P oxidase and G-3-P-dehydrogenase and
that
measurements were highly accurate. Short incubation periods are crucial to
automating
an assay as well as providing an effective assay format to clinicians, both of
which this
invention allows.
Yet another problem in the prior art involves severe lysophosphotidyl choline
(LPC) cross-reactivity. In the past, a mandatory lipid extraction step
mentioned above
was used to separate LPA from LPC and/or G3P. Since the novel single step-
hydrolysis
and cycling format does not even detect LPC, the cumbersome extraction step
can be
completely eliminated. With a lipid extraction step eliminated from the assay,
it is
possible to put this assay on an automated format due to the elimination of
the
2o extraction's complexity and difficulty.
As with the assay of the prior art, sample handling remains critical. Blood
sampling must be collected so as to prevent lysis of platelets, centrifuged
sufficiently to
remove platelets as well as erythrocytes. If not tested promptly after
collection/centrifugation, they must be frozen at -20° C to prevent
changes in LPA
concentration. Additionally, if free G3P is present, it may be necessary to
measure this
separately and subtract to get actual LPA concentration.
Still another problem in the prior art involves the lack of stabilized
detection
reagents, i.e. peroxidase and chromophore precursor solutions. In the past,
the
components of this formulation were stored separately and then mixed just
minutes prior
3o to use. Again, this mufti-step format makes automation of this assay
impossible, or
impractical. What the present inventors have discovered is a combined
formulation of
peroxidase and chromophore precursor solutions that are stable at both room
temperature
CA 02389832 2002-04-18
WO 01/32916 PCT/US00/30280
and 37° C. Specifically, the inventors have combined the chromophore
precursors i.e.
phenol, phenol derivatives and phenazones together as one stable reagent.
Further, the
inventors have added sodium azide (an antimicrobial) TritonX-100, and FG-10
anti-foam
to further improved stability and shelf life.
Also, with the aforementioned improved formulation, the inventors found the
chromophore precursor solution also stabilizes added HRPO, further reducing
reagent
number. The improved simplicity and stability enhances the assay considerably
and
makes automation possible and reliable.
Summary of the Invention
The present invention relates to an improved enzymatic diagnostic assay to
detect
carcinoma by measuring various lysophospholipids, including lysophosphatidic
acid
(LPA), in a patient. In a preferred embodiment, this assay measures the human
plasma
level of LPA in an automated format with a minimal number of reagents and with
reduced incubation periods. The present invention comprises several additional
technical
improvements to the current LPA assays disclosed in the prior art as described
below.
The inventors have also shown that the cross-reactivity of LPC is
significantly
reduced in their improved single step assay versus the prior art multi-step
assay. This
2o completely eliminates the need for the extraction step currently used.
The inventors also illustrate that the hydrolysis and cycling enzymes and
related
solutions could be combined and that this single reagent remained stable and
efficacious.
The inventors have shown that the stability of the LPA calibrators could be
significantly improved by eliminating calcium, previously thought to be
essential to the
calibrator's efficacy, from the calibrator matrix.
Further, the inventors have formulated an LPA assay in which unextracted
plasma is actually the sample.
The inventors have further shown an improved formulation of a detection
reagent, which contains both peroxidase and chromophore precursors, that has
3o significantly improved stability compared with the prior art reagents. The
present
invention not only combines these precursors into a single reagent format, but
also
stabilizes these reagents so that they are efficacious at room temperature,
thereby
CA 02389832 2002-04-18
WO 01/32916 PCT/US00/30280
eliminating the need for refrigeration and further lengthening shelf life.
This also allows
simplification of the assay and enables it to be put onto an automated format.
Further, the inventors have actually automated the assay and have incorporated
it
onto an automated machine by incorporation of a fluorophore that enables
conversion of
the assay from an absorbance readout to a fluorescence readout. This
fluorescent read
out has a higher sensitivity than the prior art colorgenic method.
The invention differs from prior art due to improved assay specificity,
improved
formulation stability, the elimination of numerous steps, reduced incubation
time of the
assay and actual assay automation. Also, the present invention differs in that
it
1o eliminates the extraction step and can use plasma as the actual sample.
Detailed Description of the Invention
The lysophospholipase used in this assay can be any lipase that hydrolyzes the
15 fatty acids (ester bonds) from either position 1 or 2 of
lysoglycerophospholipids (i.e. sn-1
or sn-2 positions). Examples include phospholipase B, phospholipase C,
phospholipase
D, lysophospholipase, phospholipase Al, and phospholipase A2, lecithinase B
and
lysolecithinase.
Cycling enzymes used are any enzymes or combination of enzymes used to
2o convert the glycerol -3-phosphate (G3P) intermediate to and from DHP and in
the
process increase the production of hydrogen peroxide, which is, preferably,
the actual
species that is detected. The two enzymes that are preferred are glycerol-3-
phosphate
oxidase which converts G3P to dihydroxyacetone phosphate (hydrogen peroxide is
also
generated in this step) and glycerol-phosphate dehydrogenase, which in the
presence of
25 NADH, converts the dihydroxyacetone phosphate back to G3P. G3P then goes
through
the same cycle generating additional hydrogen peroxide. Other cycling enzymes
which
can be used include serine dehydrogenase, serine deaminase, aldehyde
dehydrogenase,
ethanolamine deaminase, glycerokinase and glycerol dehydrogenase.
The NADH is preferably stabilized, and the methods to stabilize NADH are
3o described in the U. S. Patent 4,704,365, issued November 3, 1987 entitled
"Composition
and Method for Stabilization of Dinucleotides", herein incorporated by
reference. The
formulation described includes propylene glycol (polyhydroxyl alkyl solvent)
at 50 %,
CA 02389832 2002-04-18
WO 01/32916 PCT/US00/30280
boric acid and buffers. Specifically, this patent discloses a reduced
dinucleotide,
preferably nicotinamide adenine dinucleotide (NADH), stabilized in an aqueous
base
liquid containing propylene glycol, boric acid and a buffer capable of
buffering within a
pH range of 8-11. The stabilized liquid contains greater than 50% (v/v) water.
The
remaining volume contains propylene glycol, which has been chemically treated
to
remove oxidants. The accuracy of this stabilizer is dependent on pH and the
amount of
glycerol as well as the sample volume.
The hydrolysis/cycling mixture may also contain compounds which prevent
degradation or production of lysophospholipids. Reagents for inhibiting
production or
to hydrolysis of lysophospholipids include specific PLA2 inhibitors such as
Aristolic Acid
(9-methoxy-6-nitrophenanthro-(3,4-d)-dioxole-5-carboxylic acid, Biomol
Research
Laboratories, Plymouth Meeting, PA); ONO-R-082 (2-(p-Amylcinnamoyl)amino-4-
chlorobenzoic acid, Biomol); OBAA (3-(4-Octadecyl)-benzoylacrylic acid,
Biomol), 4-
Bromophenacyl Bromide (Sigma); Quincrine (6-Chloro-9-(4-diethylamino)-1-
methylbutyl)amino-2-methoxycridine, Mepacrine, Sigma); Manoalide (Biomol) and
HELSS (Haloenol lactone suicide substrate, Biomol); phosphodiesterase
inhibitors such
as IBMX (3-Isobutyl-1-methylxanthine, CalBiochem, La Jolla, CA); Ro-20-1724
(CalBiochem); Zaprinast (CalBiochem) and Pentoxifylline (CalBiochem); general
protease inhibitors such as E-64 (traps-Epoxysuccinyl-L-leucylamido-(4-
2o guanidino)butane, Sigma); leupeptin (Sigma); pepstatin A (Sigma); TPCK (N-
tosyl-L-
phenylalanine chloromethyl ketone, Sigma); PMSF (Phenylmethanesulfonyl
fluoride,
Sigma); benzamidine (Sigma) and 1,10-phenanthroline (Sigma); organic solvents
including chloroform and methanol; detergents such as SDS; proteases that
would
degrade phospholipases such as trypsin (Sigma) and thermostable protease
(Boehringer
Mannheim Biochemicals, Indianapolis, IN); and metal chelators such as EDTA
(Ethylenediaminetetracetic acid, Sigma) and EGTA (Ethylene glycol-bis-(beta-
aminoethyl ether), Sigma). These reagents are characterized by their ability
to preserve
lysophospholipid levels in a sample by either reducing lysophospholipid
production or
degradation.
3o The peroxidase solution contains peroxidase which is a hemoprotein
catalyzing
the oxidation by hydrogen peroxide of a number of substrates such as
ascorbate,
ferrocyanide, cytochrome c and the leuco form of many dyes. In short,
peroxidases are
CA 02389832 2002-04-18
WO 01/32916 PCT/US00/30280
heme-binding enzymes that carry out a variety of biosynthetic and degradative
functions
using hydrogen peroxide as the electron acceptor. The function of the
peroxidase is to
catalyze the reaction of a suitable substrate to a detectable colored oxidized
state species.
Examples for a liquid assay include 3,3',5,5'-tetramethylbenzidine, 5-
aminosalicylic acid
(SAS), o-dianisidine, o-toluidine, o-phyeylenediamine, 2,2'-azinodi-(3-
ethylbenzothiazoline-6-sulfonate) (ABTS) and those for a strip assay include
3,3'-
diaminobenzidine (DAB), 3-amino-90-ethylcarbazole, 4-chloro-1-naphthol, 3,4-
diamihnotoluene, 4,5-dimethyl-1,2-phenylenediamine, 4-chloro-1,2-
phenylenediamine,
4,5-dichloro-1,2-phenylenediamine.
1 o The chromophore precursor solutions are the mixtures of compounds, that
when
oxidized, result in color. Any chromophore that develops a color that
corresponds to the
spectra of the fluorophore partner could be used in this technology. In other
words, the
preferred chromophore must be able to absorb light resulting in an attenuation
of the
fluorescence of the fluorphore used in the assay. The chromophore precursors
act as
electron donors and as they donate electrons, they are oxidized and thereby
generate
color.
Fluorescent compounds are those compounds that when irradiated with UV or
visible light, re-emit some of this light as longer wavelength light. The
fluorescent
compounds in the present invention are used to measure, via the Radiant Energy
2o Attenuation (REA) method, the amount of signal generated. Alternative
fluorescent
compounds that could be used include those that are characterized by
excitation andlor
emission spectra that coincide with the absorption spectra of the generated
chromophores. The generic REA method is better described in US Patent No.
4,495,293,
issued January 22, 1985, entitled "Fluorometric Assay", herein incorporated by
reference. Specifically, this patent provides a method to fluorometrically
determine a
ligand in an assay solution containing the ligand, reagent system and a
fluorescer
wherein the intensity of the fluorescer emitted by the assay solution is
related to the
change in the transmittive properties of the assay solution produced by the
interaction of
the ligand to be determined and a reagent system capable of producing a change
in the
3o transmittive properties of the assay solution in the presence of the
ligand. In addition,
novel reagent compositions are provided which may be utilized to either
spectrophotometrically or fluorometrically determine the concentration of a
ligand in an
CA 02389832 2002-04-18
WO 01/32916 PCT/US00/30280
assay solution. Examples of fluorphores include R-Phycoerythrin, TexasRed,
Oregon
Green, Fluorescein, Rhodamine Red, Tetramethylrhodamine, BODIPY FL, BODIPY
TR, BODIPY TMR, YOYO-1', DAPI, Indo-1, cascade Blue, Fura-2, Amino
methylcoumariln, Carboxy-Snarf, Lucifer Yellow, Dansyl Derivitive.
5 Cations contemplated in this invention are positively charged ions such as
Na+,
Cap, Zn++, etc. They are used in the present invention to activate the
glycerol-3-
phosphate oxidase and any cation which activates glycerol-3-phosphate oxidase
is
suitable. Others include those disclosed in Table 1.
1 o TABLE 1
This Table illustrates the effect of metal ions on 1-a-glycerophosphate
oxidase
activity.
1-a-Glycerophosphate oxidase activity was measured in 1 mM potassium
phosphate buffer, pH 7.0, and at 10 mM DL-a-glycerophosphate in the presence
of the
salts below by the peroxidase-linked system described under "Experimental
Procedures".
Effector L-a-Glycerophosphast Activation
oxidase activity
Unitslml
None 9.7 1
CaCl2
MgCl2
10 mM 74.5 7.7
10 mM 66.2 6.8
ZnCl2
1 mM 60.7 6.3
10 mM 8.3 0.9
MnCL2b
10 mM 73.6 7.6
CoCl2b
10 mM 99.4 10.2
NaCI
KCl
10 mM 12.9 1.3
100 mM 36.8 3.8
10 mM 25.8 2.7
100 mM 56.1 5.8
°The rate in the presence of effector was divided by the rate in the
absence of effector.
bRate in presence of this salt was linear for only a short time.
CA 02389832 2002-04-18
WO 01/32916 PCT/US00/30280
Esders, T. W. and Michrina, C.A. Purification and Properties of L-alpha--
Glycerophospate
Oxidase from Streptococcus faecium ATCC 12755. Journal of Biological Chemistry
254, 2710-2715,
1979.
Chelators contemplated within the scope of this invention are multidentritic
species (e.g. citrate, EDTA, EGTA) that bind to positively charged metal ions.
Chelators may preferably be used to stabilize calibrators or to temporarily
lower the
availability of divalent cations in our system.
Phenol and phenazones are used as electron donors. Phenol as defined is
l0 hydroxybenzene sometimes referred to as carbolic acid. Derivitives would
include
compounds that have substituants on the positions of the benzene other than at
the
phenolic hydroxyl. Examples of phenazones include antipyrenes.
LPA is the compound preferably detected, but other lysohospholipids are also
contemplated within the scope of this invention in order to detect cancer,
including but
15 not limited to LysoPC, lysophosphatidyl serine (LysoPS), lysophosphatidyl
inositol
(LysoPI), lysophosphatidyl ethanolamine (LysoPE) and lysophosphatidyl glycerol
(LysoPG).
Manual test kits useful for detecting LPA in a test sample are also provided
which comprise a container containing the necessary enzymes and other reagents
for
20 conducting the assay described herein. These test kits further comprise
containers with
tools useful for collecting test samples (such as, for example, food, urine,
saliva and
stool). Such tools include lancets and absorbent paper or cloth for collecting
and
stabilizing blood; swabs for collecting and stabilizing saliva; and cups for
collecting and
stabilizing urine or stool samples. Collection materials, such as papers,
cloths, swabs,
25 cups, and the like, may optionally be treated to avoid denaturation or
irreversible
adsorption of the sample. The collection materials also may be treated with or
contain
preservatives, stabilizers or antimicrobial agents to help maintain the
integrity of the
specimens.
The diseases correlated with altered concentrations of these lysophospholipids
3o include conditions associated with platelet activation such as,
inflammatory conditions.
Altered phospholipid metabolism has been reported in many diseases and can
lead to
altered lysophospholipid and phospholipid levels in biological fluids, such as
blood.
These diseases include, but are not limited to, Alzheimer's, diabetes, heart
disease,
CA 02389832 2002-04-18
WO 01/32916 PCT/US00/30280
12
ischemia, liver disease, lung disease, malaria, muscular dystrophy,
Parkinson's, sickle
cell anemia, and various cancers. In these diseases, defective cellular
functions may
contribute to changes in levels of phospholipids. Other diseases include
bleeding
disorders including those associated with abnormal platelet function resulting
in
coagulopathy.
Various automated instruments can be used in conjunction with this assay,
including, but not limited to Abbott IMx, Abbott Alcyon, Abbott AxSym, and the
Toshiba Aeroset.
to Examples
The following examples are presented to demonstrate the methods of the present
invention and to assist one of ordinary skill in using the same. The examples
are not
intended in any way to otherwise limit the scope of the disclosure or the
protection
granted by Letters Patent granted hereon.
15 Example 1
Reagent Combination/Step Reduction
This example illustrates how an LPA assay can be reduced to only two steps and
only two reagents.
Lysophospholipase was combined with the cycling enzymes (i.e. glycerol-3-
2o phosphate oxidase and glycerophosphate dehydrogenase) and NADH to form one
reagent in the following way. One hundred uL's of lysophosphatidic acid
(Atairgin,
Irvine CA) calibrators were added to the wells of a 96 well microtiter plate.
Fifty uL's of
a solution that contained lysophospholipase (Atairgin) at 5 units/mL, glycerol
phosphate
dehydrogenase (Atairgin) at 17 units mL, glycerol-3-phosphate oxidase
(Atairgin) at 134
25 units/mL, NADH, 1.25 mM, calcium chloride, 20 mM, and Tris, 50 mM pH 8.0
were
added to the wells and the contents of the wells were mixed.
Following a 60 minute incubation at 37° C, 50 uL of 50 mM Tris, pH 8.0
and 50
uL of a solution that contained 0.5% 3,5-dichloro-2-hydroxy benzenesulfonic
acid,
0.15% 4 aminoantipyrene, 10 units/mL horse radish peroxidase (HRPO, Atairgin),
luM
3o fluorescein, 50 mM Tris pH 8.0 were added to the wells and the contents of
the wells
were mixed. The absorbances at 490 nm were then read using a microtiter plate
reader.
CA 02389832 2002-04-18
WO 01/32916 PCT/US00/30280
13
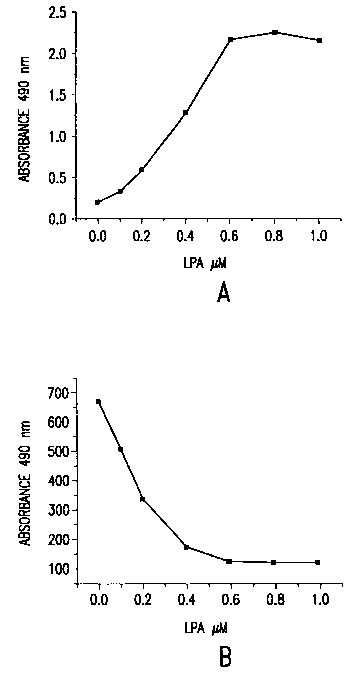
The results shown in Figure 1A and 1B demonstrate that LPA could be detected
by combining the lipase and cycling enzymes. Figure 1 A shows the results as
an
absorbance read while Figure 1 B shows that the results can also be read as
fluoresence
at 520 nm by incorporation of fluorescein in to the chromaphore mixture and
utilizing
the REA ("Radiant Energy Attenuation") method. As one can see, the fluorescent
read-
out has improved sensitivity as compared to the absorbance read-out.
Example 2
Novel Calibrators
1o LPA calibrators were prepared as in the prior art by adding LPA (Sigma) to
2.5%
Triton X-100, SOmM CaCl2, 50 mM Tris, pH 8Ø Novel calibrators were prepared
using
the same solution but only this time the calcium chloride was omitted.
Calibrators were
then stored for approximately 72 hours at both room temperature and 4°
C. After 72
hours fresh calibrators were prepared (+/-) calcium. The fresh and stored
calibrators
were then evaluated in the LPA assay using the microtiter format as follows.
One
hundred uL of sample was added to the wells of a microtiter plate. Fifty uL's
of the
lipase cycling solution that contained 2 units/mL of Lysophospholipase, 80
units/ mL of
glycerol 3-phosphate dehydrogenase, 40 units/mL of glycerol-3- phosphate
oxidase in
20mM calcium chloride, 50 mM Tris. PH 8.0 was added to the sample followed by
50
2o uL of a 1.5 mM solution of nicotinamide adenine dinucleotide reduced
(NADH). The
mixture was mixed and then incubated at 37° C for 20 minutes. Fifty uL
of a color
development solution which contained 0.5% 3,5-dichloro-2-hydroxy
benzenesulfonic
acid, 0.15% 4 aminoantipyrene, l0units/mL horse radish peroxidase(HRPO) in 50
MM
Tris pH 8.0 was then added. Following mixing the absorbances were read at 490
nm.
The results in Figure 2 show that the calibrators prepared without calcium are
more
stable than those prepared in the presence of calcium.
Example 3
Improved Detection Reagent
The REA Chromophore/Fluorophore (C/F) reagent that contained glycine 1.0M;
3,5-dichloro-2-hydroxy benzensulfonic acid, 0.22M; 4 aminoantipyrene, 0.05M;
dimethyl sulfoxide 50%; sodium azide, 0.1 %; Triton X-100, 5.4%; fluorescein ,
4.5
xlOE-6M and FG-10 anti foam (Dow Corning) at 0.01%, pH 7.0 was prepared .
Eight
CA 02389832 2002-04-18
WO 01/32916 PCT/C1S00/30280
14
microliters of an HRPO solution (2.Su/uL) was added to SOOuL's of the C/F-
reagent.
Eight microliters of the HRPO solution was also added to C/F reagent that did
not
contain azide or Triton. Portions of the mixtures were stored at either room
temperature
of 37° C for 24 hours. Fresh solutions were made for comparison to the
ones stored for
24 hours. Fifty microliters of 1/20 dilutions (PBS) of the test solutions were
added to
microtiter plate wells. Fifty microliters of peroxide calibrators were added
to the wells
and the resulting absorbances at 490 nm were read. The results shown in Figure
3
indicate that that HRPO is stable in the C/F reagent (-+/- azide and Triton)
and suggest
that GRPO could be incorporated as a reagent into the C/F reagent formulation.
In other
1o words, this data clearly shows an improved formulation of a detection
reagent, which
contains peroxidase and chromophore precursers, which has significantly
improved
stability compared with the prior art reagent.
Further, this aspect of the invention is critical in achieving an automated
assay
and incorporate it onto an automated machine, such as an Abbott Imx.
Example 4
Automated Assax on Alcyon Analyzer
LPA was detected on the Abbott Alcyon analyzer using the "Dual Reagent End-
Point
Chemistry" method. Briefly, 180u1 of a 0.50 mM solution of NADH (Boehringer
2o Mannheim, Indianapolis, IN) in SOmM Tris, pH 8.0 was added to a reaction
cuvette. Two
measurements of the absorbance at 340nm were recorded at 12 second intervals.
Thirty
microliters of the sample were added to the NADH solution and the reaction
mixture was
mixed. Another absorption measurement at 340nm was made. One hundred eighty
microliters of a solution that contains phospholipase B (Sigma, St.Louis, MO)
at 1 unitlmL,
glycerol phosphate dehydrogenase (Boehringer Mannheim, Indianapolis, IN) at 40
units/mL,
and glycerol phosphate oxidase (Shinko American, NewYork, NY) at 600 units/mL
in
20mM CaCl2 and SOmM Tris, pH 8 were added to the reaction cuvette. As the
reaction
proceeds the absorbance at 340 nm decreases. After an incubation of 12 minutes
48 seconds
at 37° C, a final absorbance measurement was made. The absorbance
difference that occurs
as a result of the reaction was automatically calculated by the Alcyon
analyzer using an input
value of the absorption of the enzyme reagent and the formulas contained in
the Alcyon
operations manual. A typical calibration curve is shown in Figure 4.
CA 02389832 2002-04-18
WO 01/32916 PCT/US00/30280
Example 5
One-Step Assay vs. Multi-Step Assay Comparison
A comparison between the prior art two step (a separate lipase digestion prior
to a
combined lipase/cycling) microtiter format and a one step lipase/ cycling
format was
5 made. The sample size, incubation times and reagent quantities were kept
identical so
that a direct comparison can be properly made.
The following reagents were prepared: Reagent A that contained
lysophospholipase at 5 U/mL, glycerol dehydrogenase at 34 U/mL and glycerol
oxidase
at 134 U/mL in 10 mM calcium chloride, 50 mM Tris pH 8. Reagent B that
contained
to 25 mM NADH in SOmM Tris pH 8. Reagent C, similar to reagent A, but also
containing
12.5 mM NADH. A color development solution, reagent D, that contained 0.5% 3,5-
dichloro-2-hydroxy benzenesulfonic acid, 0.15% 4 aminoantipyrene, l0units/mL
horse
radish peroxidase (HRPO, Atairgin), luM fluorescein in 50 mM Tris pH 8.0 was
prepared immediately before use.
15 Prior Art Assay(Multi Step)
Fifty uL's of Lysophosphatidic acid (LPA, Atairgin, Irvine CA) calibrators
were
added in duplicate to the wells of a 96 well microtiter plate. One hundred
uL's of
reagent A was added to the calibrators. The plates were mixed, covered and
incubated
for 15 minutes at 37° C. At that time 50 ul's of reagent B was added to
the wells. The
2o addition of this reagent initiated the cycling. The plates were mixed then
incubated for
another 15 minutes at 37° C. Fifty uL's of the color development
reagent (D) was added
to all wells. The contents of the wells were mixed and the absorbances at 490
nm were
read. The results are shown in Figure 5.
Novel Assay (One Step)
Fifty uL's of Lysophosphatidic acid (LPA, Atairgin, Irvine CA) calibrators
were
added in duplicate to the wells of a 96 well microtiter plate. One hundred 100
uL's of
reagent C was added. The plates were mixed, covered and incubated for 30
minutes at
37° C. Following this incubation 50 uL's of SOmM Tris pH 8.0 was added
to the wells to
adjust the volume to be the same as the prior art. Fifty uL's of the color
development
3o reagent (D) was added to all wells. The contents of the wells were mixed
and the
absorbances at 490 nm were read. The results shown in Figure 5 demonstrate the
CA 02389832 2002-04-18
WO 01/32916 PCT/LTS00/30280
16
enhanced performance of the single step (Novel Assay) format relative to the
two step
prior art format that utilizes a separate lipase digestion.
Example 6
Novel Color~enic Reagent
A comparison was made between the stabilities of the novel REA
chromophore/fluorophore reagent and horseradish peroxidase (HRPO) mixture and
the
prior art color development reagent and HRPO mixture. The novel REA
chromophore/fluorophore (C/F) reagent contained glycine, 0.1 M; 3,5-dichloro-2-
hydroxy benzensulfonic acid, 0.22M; 4 aminoantipyrene, O.OSM; dimethyl
sulfoxide
50%; sodium azide, 0.1%; Triton X-100, 5.4%; fluorescein , 4.5 xlOE-6M and FG-
10
anti foam(Dow Corning) at 0.01 %, pH 7Ø A modified C/F solution that
contained 0.1 M
Tris, pH 8 instead of glycine was also made.
The prior art color development solution contained 0.5% 3,5-dichloro-2-hydroxy
benzenesulfonic acid, 0.15% 4 aminoantipyrene, in 50 mM Tris pH 8Ø To one mL
of
the C/F and prior art color development solutions 4uL of a HRPO solution (2500
U/mL)
was added. These solutions as well as a solution that contained only HRPO were
incubated at room temperature for three days. The spectra of these were
recorded and
compared with C/F solutions that did not have HRPO added. The results shown in
Figure
6A show that the absorption for the C/F solution (pH 7) + HRPO at S 12 nm
(spectra 1 ) is
slightly lower than that seen for the C/F(pH 8) + HRPO (spectra 2) while the
absorption
for the prior art color development reagent + HRPO (spectra 3) is
significantly higher.
The absorption band near 512 nm is also red shifted for the prior art mixture.
Figure 6B
shows the spectra (5) of the C/F (pH 7) reagent, the spectra (6) of the C/F
(pH 8) reagent
and of HRPO only (spectra 4). After three days the C/F peroxidase mixtures are
still the
same yellow color as they were two hours after addition of the HRPO, while the
prior art
mixture has gone from clear to red. The absorption at 512 nm of these
solutions as a
function of time is shown in Figure 6C. For the prior art color development
reagent
HRPO mixture we observe an initial low absorbance at 512 nm followed by a
steady
increase in absorbance at 512 nm. For the C/F mixtures we see an initial
increase in
absorbance at 512 nm upon addition of HRPO. However this quickly decreases
(after 2
hours) and then remains constant. Although not shown in this experiment the
absorption
CA 02389832 2002-04-18
WO 01/32916 PCT/US00/30280
17
of the prior art color development solution, in the absence of HRPO, at 512 nm
is
insignificant.
These data along with those shown in Example 3 demonstrate that C/F
peroxidase mixture has enhanced stability compared to the prior art color
development
peroxidase mixture.
Example 7
Automated Assay for LPA using IMx Instrument.
The IMx instrument was designed to perform immunoassays in both
to microparticle and fluorescence polarization formats. The fluorescence
polarization
format can be adapted to perform "Radiative Attenuation Assay", which permits
measurements based on optical absorbance. By adding peroxidase and appropriate
dyes
- fluorescein and a colorigenic peroxidase substrate for example - -- to the
mixture of
Example 1 after the cycling reaction has proceeded to a sufficient degree the
concentration of G3P, and therefore, sample LPA can be determined.
Reagents:
Lipase (h~drolvsis)/Cyclin . Enzyme Rea~,ent: lU/mL lysophospholipase,
200U/mL glycerophosphate dehydrogenase, SOOuL glycerol-3-phosphate oxidase,
4UmM
2o calcium chloride, SOmM Tris, SmM sodium benzoate, 20% glycerol pH 8Ø
Chromophore/Fluorophore Reagent: 220mM 3,5 dichloro-2-hydroxy benzene
sulfonic acid, SOmM 4-aminoantipyrene, 100mM glycine, 4.SuM Fluorescein, 0.1%
sodium azide, 5.4% Triton X 100, 50% dimethyl sulfoxide, pH 8.5.
HRPO Mixture: 20U/mL horseradish peroxidase in SOmM tris pH 8Ø
NADH Solution: l.SmM NADH in SOmM tris pH 8Ø
Protocol:
In a preferred embodiment, a plasma sample can be prepared by the following
method. Blood is collected in presence of a stabilizer such as EDTA or
citrate. It is then
3o centrifuged sufficiently to sediment erythrocytes and platelets (15 min at
3000XG) at 4°
C or is filtered to remove these components.
CA 02389832 2002-04-18
WO 01/32916 PCT/US00/30280
18
100uL NADH Solution, 5uL sample and 20uL Lipase/Cycling Enzyme Reagent
are aspirated by the sample probe. 70uL of this is dispensed to the cuvette
and the
remaining NADH Solution in the probe dispensed to waste (This is to prevent
contamination of the cycling mixture by the required line diluent, which
contains
phosphate buffer, which would slow the reaction by complexing the calcium).
The
mixture is incubated for 30 min at 35° C in the instrument, then 40uL
Chromophore/Fluorphore Reagent and 690uL line diluent are added. The mixture
is
incubated for 4 min then the fluorescence intensity is measured and
immediately
thereafter 20uL Chromophore/Fluorphore Reagent, 40uL HRPO mixture and 340uL
line
diluent are added. The mixture is incubated an additional 4 min during which
the color
is formed. Finally the fluorescence intensity is measured again and the data
transferred
to a file for analysis. The ratio of the final fluorescence intensity to the
initial
fluorescence intensity decreases with increasing peroxide-generated color, and
so can be
used with appropriate calibrators to determine the amount of glycerol-3-
phosphate, and
by extension the amount of LPA originally in the sample. When the test is
performed
under identical conditions, but without lysophospholipase in the
Lipase/Cycling Enzyme
Reagent, the LPA does not react and only the free glycerol-3-phosphate is
measured.
For samples collected under conditions in which free glycerol-3-phosphate may
be
present, performing this assay both with and without lysophospholipase can
provide the
2o information needed to determine the actual LPA concentration.
Table 2 shows the results of applying the above protocol to LPA standards
ranging from 0 to SuM. Initial and final fluorescence intensities and their
ratio are
shown along with the LPA concentration of the sample. The first 12 positions
are
duplicates of LPA standards in buffer, then replicates of 4 each of the zero
and 2.0 uM
standards. A 4-parameter log-logit curve fitting algorithm was used with these
results to
generate the curve in Figure 7.
In addition to plasma samples, the standards AS 1 and AS 1 extracted are also
measured. The LPA concentration measured for AS 1 extracted is 0.53 uM,
consistent
with values determined using the microtiter format. The LPA concentration of
the
3o unextracted sample is 2.04 uM, about 4-fold higher than the extracted
sample. LPC
cross-reactivity is ruled out as a cause for reasons shown in Table 3. Most
likely, the
difference results from losses of LPA during the extraction process.
CA 02389832 2002-04-18
WO 01/32916 PCT/LTS00/30280
19
Table 3 shows the application of the method to human plasma. Blood collected
from normal volunteers in EDTA tubes was cooled in an ice bath immediately
after
collection. Within 80 min of collection it was centrifuged 15 min at 3000XG in
a
refrigerated centrifuge at 4° C. The clear supernate was tested by the
above protocol
both with and without lysophospholipase. Standards consisting of 0 to 5uM
glycerol-3-
phosphate in buffer were used for both conditions. The same curve-fitting
algorithm was
used to generate the results. The results in Table 3 illustrate the importance
of obtaining
background measurement in order to accurately measure LPA concentration.
Table 4 shows the effect of sample handling and storage, and demonstrates that
lysophosphatidylcholine, which interferes in the microtiter formatted assay,
does not
interfere in the IMx configured assay if samples are stored at -20° C.
Blood was
collected with EDTA anticoagulant, cooled 10 min in an ice bath 5 min after
drawing,
then centrifuged 15 min at 500XG at 2° C. Of the 12 mL resulting turbid
plasma,
portions were stored at -20° C, 4° C and room temperature =
37° C. The remainder was
centrifuged 30 min at 3100XG at 2° C, and the clear supernate aspirated
from the pellet.
500uL portions of the clear supernate were subjected to the following
treatments, then
aliquoted and stored as above at -20° C, 4° C and room
temperature:
Set B: no treatment
Set C: 2.OuL sample buffer (2.5% Triton X 100 in SOmM tris pH 8.0) added to
SOOuL
2o clear plasma.
Set D: 2.OuL 1.OmM LPA in sample buffer added to SOOuL clear plasma = 4.OuM
LPA.
Set E: lOuL l OmM LPC in sample buffer added to SOOuL clear plasma = 200uM
LPC.
The samples were stored 18 hours at the indicated conditions, then brought to
room
temperature and assayed as above, both with and without lysophospholipase. It
is
immediately apparent that the insufficiently centrifuged turbid plasma
contains a high
background of G3P, most of which is removed by adequate centrifugation. The
LPA
concentration increases with storage at higher temperatures, indicating that
samples
should be stored frozen. Addition of tris/triton buffer makes little
difference at the
concentrations added. The samples spiked with 4uM LPA showed recovery of most
of
3o the spike, but no change with storage conditions. An increase of l.SuM
would be
expected from the results of the unspiked sample. One possibility for its
absence is that
the increase in LPA is compensated by a process which destroys it, such as
some
CA 02389832 2002-04-18
WO 01/32916 PCT/US00/30280
background phosphatase activity. Most interesting here is that spiking the
sample with
200uM LPC increases the measured LPA by only about 0.1 uM when the sample is
stored
at -20° C, and by extension when the sample is fresh. Evidently LPC
interferes much
less with the IMx formatted assay than with the microtiter format of the prior
art,
5 possibly due to decreased exposure to the lysophospholipase. However, when
the
sample is stored at 4° C or room temperature, the LPC spiked sample
shows an
increasing signal for LPA. A possible explanation is the presence of a
phospholipase C
activity in the plasma, which cleaves choline from LPC, leaving LPA.
Table 2
pos uM LPA to I I/lo uM LPA
from
curve
1 0.0 23098 22440 0.972 0.00
2 0.0 23935 22793 0.952 0.05
3 0.5 23386 19709 0.843 0.46
4 0.5 23411 19487 0.832 0.50
5 1.0 24340 16841 0.692 1.05
6 1.0 23672 16614 0.702 1.01
7 2.0 23922 12063 0.504 1.96
8 2.0 24086 11995 0.498 1.99
9 3.0 23623 8391 0.355 2.92
10 3.0 24732 8211 0.332 3.09
11 5.0 24103 3346 0.139 5.01
12 5.0 24495 3439 0.140 5.00
13 0.0 24901 22751 0.914 0.20
14 0.0 24113 22712 0.942 0.09
15 0.0 24218 22476 0.928 0.14
16 0.0 24598 22729 0.924 0.16
17 2.0 24220 11754 0.485 2.07
18 2.0 23981 11759 0.490 2.04
19 2.0 24384 11633 0.477 2.12
20 2.0 24563 11699 0.476 2.12
CA 02389832 2002-04-18
WO 01/32916 PCT/US00/30280
21
TABLE 3
+LYPL uM -LYPL uM uM
to I I/lo from to I I/lo from Sample diff
= LPA
Curve Curve
23084 18338 0.794 0.00 22637 17964 0.794 0.00 0 0.00
23248 16384 0.705 0.47 22959 15958 0.695 0.52 0.5 -0.06
22904 13714 0.599 1.04 22963 14037 0.611 0.99 1 0.04
22598 10212 0.452 2.01 22111 10424 0.471 1.94 2 0.07
22903 7874 0.344 2.96 22675 7827 0.345 3.09 3 -0.14
22200 4103 0.185 5.03 22141 4584 0.207 4.96 5 0.07
22369 13725 0.614 0.95 23106 15106 0.654 0.74 1A 0.21
21873 14281 0.653 0.74 23036 15669 0.680 0.60 2A 0.14
22742 15427 0.678 0.61 22854 16509 0.722 0.38 3A 0.22
22360 15590 0.697 0.51 23303 17214 0.739 0.29 4A 0.21
21973 10687 0.486 1.76 22942 16638 0.725 0.37 5A 1.39
22319 13553 0.607 0.99 22679 14699 0.648 0.78 6A 0.21
21678 12598 0.581 1.14 21831 13697 0.627 0.90 7A 0.25
21533 12097 0.562 1.26 21951 13870 0.632 0.87 8A 0.39
22509 14481 0.643 0.79 20309 15723 0.774 0.11 9A 0.68
21569 14450 0.670 0.65 22029 15817 0.718 0.40 10A 0.25
22584 14812 0.656 0.72 22855 16045 0.702 0.49 11A 0.24
22706 9237 0.407 2.37 21927 16021 0.731 0.33 AS1 2.04
22567 15616 0.692 0.53 22559 18163 0.805 0.00 AS1 0.53
Ext
Table 4
With
LYPL
uM LPA+G3P
turbidclear +buffer+4uM LPA +200uM
LPC
-20 4.69 1.38 1.38 4.95 1.53
4 4.51 1.63 1.51 5.11 2.67
RT 4.82 2.83 2.97 5.06 3.79
Without
LYPL
uM G3P
turbidclear +buffer+4uM LPA +200uM
LPC
-20 4.49 1.01 0.91 1.12 0.99
4 3.67 0.92 0.89 1.14 0.97
RT 3.54 0.96 1.02 1.20 1.01
Difference
uM LPA
turbidclear +buffer+4uM LPA +200uM
LPC
-20 0.21 0.38 0.47 3.83 0.54
4 0.83 0.71 0.62 3.97 1.70
RT 1.28 1.87 1.95 3.85 2.79
CA 02389832 2002-04-18
WO 01/32916 PCT/US00/30280
22
Example 8
Detection of LPA Using a Strip Assay
This assay can also be formatted on a strip. Specifically, whole blood is
collected
in the presence of a stabilizer, such as EDTA or citrate. It is then placed on
a strip,
which wicks the plasma away from the solid components. The plasma, preferably,
passes through a portion of the strip containing calcium and the solid
components are
removed by continued passage through the strip. The lipase and cycling enzymes
are
located downstream on a conjugate pad along with detection reagents or labels.
1 o Example 9
Effect of NADH concentration on rate of HBO production.
50uL of solutions containing 0-I.OuM LPA in SOmM tris pH 8.0 with 2.5%
Triton X 100 and SmM CaCl2 were added to the wells of a microtiter plate. To
this was
added 50uL of a mixture containing SU/mL lysophospholipase, 1 OU/mL
glycerophosphate dehydrogenase, 100U/mL glycerol-3-phosphate oxidase, l OmM
CaCl2
and various concentrations of NADH in SOmM tris pH 8Ø After 20min at
37° C SOuL
of a mixture of l9mM DHBS, 7.5mM 4AAP, l0U/mL HRPO in 50mM tris pH 8.0 was
added to one set of the duplicate samples and the absorbance read at 490nm
(see Fig.
8A). After 60min at 37° C the other set of samples was treated the same
way (see Fig.
8B). Figures 8A-B shows the absorbances for each NADH concentration plotted vs
LPA
concentration. For the shorter reaction time lower concentrations of NADH
result in
more signal for the same LPA concentrations, suggesting an inhibiting effect
of NADH
on the overall reaction. The results shown for the longer reaction time
demonstrate that,
while higher NADH concentrations decrease the rate of H202 production, when
the
NADH is completely consumed no further reaction can take place. These results
point to
the need to limit the NADH concentration in the cycling reaction.
CA 02389832 2002-04-18
WO 01/32916 PCT/US00/30280
23
Example 10
Automated assay for LPA using IMx instrument with combined peroxidase and
color reagent.
This is similar to Example 7 (above) except the peroxidase and color
generating
reagents are combined into a single reagent, simplifying the assay.
Reagents:
Lipase (hydrolysis)/Cycling: 1 U/mL lysophospholipase, 200U/mL
glycerophosphate
dehydrogenase, SOOU/mL glycerol-3-phosphate oxidase, 40mM calcium chloride,
SOmM
to tris, SmM sodium benzoate, 20% glycerol pH 8Ø
Chromophore/Fluorophore/HRPO Mixture: 20U/mL horseradish peroxidase, 220mM
3,5-dichloro-2-hydroxy benzene sulfonic acid, SOmM 4-aminoantipyrene, 100mM
glycine, 4.SuM Fluorescein, 0.1% sodium azide, 5.4% Triton X 100, 50%
15 dimethylsulfoxide, pH 8.5.
NADH solution: I.SmM NADH in SOmM Tris pH 8Ø
Protocol:
20 100uL NADH solution, SuL sample and 20uL Lipase/Cycling Enzyme reagent are
aspirated by the sample probe. 70uL of this is dispensed to the cuvette and
the remaining
NADH reagent in the probe dispensed to waste (This is to prevent contamination
of the
cycling mixture by the required line diluent which contains phosphate buffer
which
would slow the reaction by complexing the calcium). The mixture is incubated
for 15
25 min at 35° C in the instrument, then 40uL
Chromophore/Fluorophore/HRPO reagent and
690uL line diluent are added. The mixture is incubated for 4 min then the
fluorescence
intensity is measured and the data transferred to a file for analysis. The
measured
fluorescence intensity decreases with increasing peroxide-generated color, and
so can be
used with appropriate calibrators to determine the amount of glycerol-3-
phosphate, and
30 by extension the amount of LPA originally in the sample. As with the test
of Example 7
with separate Chromophore/Fluorophore and Peroxidase reagents, the test can be
run
CA 02389832 2002-04-18
WO 01/32916 PCT/US00/30280
24
without lysophospholipase in the Lipase/Cycling Enzyme reagent so as to
determine the
background glycerol-3-phosphate.
Figure 9 shows the results of applying the above protocol to LPA standards
ranging from
0 to lOuM.
Example 11
The effects of pH on the stability of chromogen/peroxidase mixtures has been
studied. Two, one mL solutions of 0.03% 4 aminoantipyrene, 1.0 % 3,5-dichloro-
2-
hydroxy-benzenesulfonic acid were prepared. One contained 35mM Tris at pH 8
while
the other contained 70mM sodium phosphate at pH 7. To each of these solutions
4uL of
horse radish peroxidase (2.5U/uL, HRPO) was added. Control solutions that did
not
contain peroxidase were also prepared. The solutions were allowed to sit
overnight at
room temperature. The absorbance at 512 nm of solutions was then measured. The
results shown in Figure 10 demonstrate that the solution that contained HRPO
prepared
at pH 7 is more stable than the solution that contained HRPO prepared at pH 8.
Example 12
The efficiency of cycling may be increased by covalently linking the cycling
2o enzyme, G3P oxidase and G3P dehydrogenese. Since the product of one is the
substrate
of the other, the linkage of the two would assure availability of the
appropriate enzyme
in the vicinity of its substrate. Covalent linkage of the two may be carried
out by
methods well known in the art.