Note: Descriptions are shown in the official language in which they were submitted.
CA 02394706 2002-06-17
WO 01/48840 PCT/US00/35331
GRAIN BOUNDARY MATERIALS AS ELECTRODES FOR LITHIUM
ION CELLS
STATEMENT OF PRIORITY
This application derives priority from a provisional application filed
December 28, 1999, entitled "Grain Boundary Materials as Anodes for Lithium
Ion
Cells" bearing serial no. 60/173364, the contents of which are hereby
incorporated
by reference.
TECHNICAL FIELD
This invention relates to anode compositions useful in lithium ion cells.
BACKGROUND
Two classes of materials have been proposed as anodes for lithium ion
cells. One class includes materials such as graphite and carbon that are
capable of
intercalating lithium. While the intercalation anodes generally exhibit good
cycle
life and coulombic efficiency, their capacity is relatively low. In
particular,
graphite can intercalate lithium to a maximum of 1 lithium atom per six carbon
atoms. This corresponds to a specific capacity of 373 mAh/g of carbon. Because
the density of graphite is 2.2 g/cc, this translates to a volumetric capacity
of 818
mAh/cc. Other types of carbon have higher specific capacity values, but suffer
from one or more disadvantages such as relatively low density, unattractive
voltage
profiles, and large irreversible capacity that limit their utility in
commercial lithium
ion cells.
A second class includes metals that alloy with lithium metal. These alloy-
type anodes generally exhibit higher capacities relative to intercalation-type
anodes. For example, specific capacity associated with the formation of a
lithium-
aluminum alloy is 992 mAh/g. The corresponding value for the formation of a
lithium-tin alloy is 991 mAh/g. One problem with such alloys, however, is that
they can exhibit relatively poor cycle life and coulombic efficiency due to
fragmentation of the alloy particles during the expansion and contraction
associated with compositional changes in the alloy.
CA 02394706 2002-06-17
WO 01/48840 PCT/US00/35331
SUMMARY
The invention provides electrode compositions suitable for use in lithium
ion batteries in which thr electrode compositions have high initial capacities
that
are retained even after rt~,peated cycling. The electrode compositions, and
batteries
incorporating these compositions, are also readily manufactured.
To achieve these objectives, the invention features, in a first aspect, an
electrode composition that includes particles having a single chemical
composition
formed from (a) at least one metal element selected from the group consisting
of
tin, aluminum, silicon, antimony, lead, germanium, magnesium, zinc, cadmium,
bismuth, and indium; (b) at least one metal element selected from the group
consisting of manganese, molybdenum, niobium, tungsten, tantalum, iron,
copper,
titanium, vanadium, chromium, nickel, cobalt, zirconium, tantalum, scandium,
yttrium, ruthenium, platinum, and rhenium; and, optionally, (c) carbon. The
particles have a microstructure characterized by a plurality of
electrochemically
inactive, nanometer-sized crystalline grains separated by electrochemically
active
non-crystalline regions.
As used herein, a "particle" is a component of a powder. Each particle is
made up of many crystalline "grains." A crystalline grain is a region of the
particle
from which diffraction occurs coherently (i.e., the crystal axes have fixed
directions within the grain). The crystalline grains are separated by non-
crystalline
regions. These regions are characterized by a lower degree of order compared
to
the crystalline grains.
A "single chemical composition" means that when the sample is analyzed
by transmission electron microscopy, the types of atoms that are detected are
the
same, on a nanometer scale range, regardless of where the electron beam is
placed
within the sample.
An "electrochemically active" material is a material that reacts with lithium
under conditions typically encountered during charging and discharging in a
lithium battery.
An "electrochemically inactive" material is a material that does not react
with lithium under conditions typically encountered during charging and
discharging in a lithium battery.
-2-
CA 02394706 2002-06-17
WO 01/48840 PCT/US00/35331
Examples of useful particles include those characterized by the chemical
composition SnMn~C and SnFe3C. These materials have electrochemically
inactive crystalline grains, yet form useful electrode materials owing to the
presence of electrochemically active tin atoms in the non-crystalline regions
separating the crystalline grains. Preferably, the particles have a size
ranging from
about 2 microns to about 30 microns (measured by scanning electron
microscopy).
The crystalline grains preferably are no greater than about 20 nanometers
where
this figure refers to the length of the longest dimension of the grain. The
non-
crystalline regions preferably from at least about 10% by volume of the
particle,
calculated from transmission electron microscopy data assuming spherical
grains.
The details of one or more embodiments of the invention are set forth in the
accompanying drawings and the description below. Other features, objects, and
advantages of the invention will be apparent from the description and
drawings,
and from the claims.
DESCRIPTION OF DRAWINGS
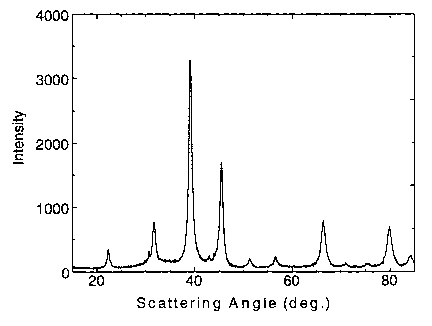
FIG. 1 is an x-ray diffraction profile for a SnMn~C sample prepared by ball
milling for 20 hours. All observed diffraction peaks are from SnMn~C.
FIG. 2 is an x-ray diffraction profile for a SnFe~C sample prepared by ball
milling for 20 hours. All observed diffraction peaks are from SnFe~C.
FIG. 3 illustrates the cycling performance, in terms of voltage versus
capacity and capacity versus cycle number, for two Li/SnMn~C cells.
FIG. 4 illustrates the cycling performance, in terms of differential capacity
versus..voltage, for a Li/SnMn~C cell.
FIG. 5 is a series of x-ray diffraction profiles for a Li/SnMn~C cell obtained
during discharge.
FIG. 6 is a series of Mossbauer spectroscopy scans for a Li/SnMn~C cell
obtained during discharge.
FIG. 7 illustrates the variation of the Mossbauer center shift of the minority
component of a Li/SnMn~C cell during charge and discharge.
FIG. 8 is a series of x-ray diffraction profiles for both an unheated SnMn~C
sample and for samples heated to 400°C, 500°C, and 600°C.
-3-
CA 02394706 2002-06-17
WO 01/48840 PCT/US00/35331
FIG 9 illustrates the cycling performance, in terms of voltage versus
capacity and capacity versus cycle number, for cells constructed using the
samples
described in FIG. 8.
FIG 10 includes a series of x-ray diffraction profiles for both an unheated
SnFe3C sample and for samples heated to 100°C, 200°C, and
300°C, and further
illustrates the cycling performance, in terms of voltage versus capacity, for
cells
constructed using these materials.
FIG. 11 includes a series of x-ray diffraction profiles for both an unheated
SnFe~C sample and for samples heated to 400°C, 500°C, and
600°C, and further
illustrates the cycling performance, in terms of voltage versus capacity, for
cells
constructed using these materials.
FIGS. 12 and 13 are transmission electron micrographs of a SnMn3C
sample.
DETAILED DESCRIPTION
The electrode compositions are in the form of powders made up of
particles. The particles have the chemical composition and microstructure
described in the Summary of the Invention, above. The powders may be prepared
directly using techniques such as ball-milling. Alternatively, the powders may
be
prepared in the form of thin films using techniques such as sputtering,
chemical
vapor deposition, vacuum deposition, vacuum evaporation, melt spinning, splat
cooling, spray atomization, and the like, and then pulverized to form powders.
The electrode compositions are particularly useful as anodes for lithium ion
batteries. To prepare a battery, the electrode powder is combined with a
binder
(e.g., a polyvinylidene fluoride binder) and solvent to form a slurry which is
then
coated onto a backing using conventional coating techniques and dried to form
the
anode. The anode is then combined with an electrolyte and a cathode (the
counterelectrode). The electrolyte may be a solid or liquid electrolyte.
Examples
of solid electrolytes include polymeric electrolytes such as polyethylene
oxide,
polytetrafluoroethylene, fluorine-containing copolymers, and combinations
thereof. Examples of liquid electrolytes include ethylene carbonate, diethyl
carbonate, propylene carbonate, and combinations thereof. The electrolyte is
-4-
CA 02394706 2002-06-17
WO 01/48840 PCT/LTS00/35331
provided with a lithium electrolyte salt. Examples of suitable salts include
LiPF6,
LiBF4, and LiC104.
Examples of suitable cathode compositions for liquid electrolyte-containing
batteries include LiCoO~, LiCoo.~NiO~, and Li,.o7Mn~,9304. Examples of
suitable
cathode compositions for solid electrolyte-containing batteries include LiV30g
and
LiV205.
The invention will now be described further by way of the following
examples.
EXAMPLES
Ball Milling Procedure
A Spex 8000 high-impact mixer mill was used to violently shake sealed,
hardened steel vials for periods up to about 40 hours. In an argon-filled
glove box,
the desired amounts of elemental powders or intermetallic phases were added to
the vial, along with several hardened steel balls measuring 12.7 mm in
diameter.
The vial was then sealed and transferred to the mill where it was shaken
violently.
The milling time was selected to be sufficient to reach milling equilibrium.
In
general, milling times were on the order of about 16 hours.
Cycling Behavior
Electrodes were prepared by coating slurries of the powders onto a copper
foil and then evaporating the carrier solvent. In a typical preparation, about
82%
by weight powder (prepared by ball milling), 10% by weight Super S carbon
black
(MMM carbon, Belgium), and 8% by weight polyvinylidene fluoride (Atochem)
were thoroughly mixed with N-methyl pyrrolidinone by stirring in a sealed
bottle
to make a slurry; the polyvinylidene fluoride was pre-dissolved in the N-
methyl
pyrrolidinone prior to addition of the powder and carbon black. The slurry was
spread in a thin layer (about 150 micrometers thick) on the copper foil with a
doctor-blade spreader. The sample was then placed in a muffle oven maintained
at
105°C to evaporate the N-methyl pyrrolidinone over a 3 hour period.
Circular electrodes measuring 1 cm in diameter were cut from the dried
film using an electrode punch. The electrodes were weighed, after which the
-5-
CA 02394706 2002-06-17
WO 01/48840 PCT/US00/35331
weight of the copper was subtracted and the active mass of the electrode
calculated
(i.e., the total weight of the electrode multiplied by the fraction of the
electrode
made of the active elect,~ode powder). The circular electrodes were then heat-
sealed in polyethylene bag ~ until further use.
The electrodes were, used to prepare coin cells for testing. All cell
construction and sealing was done in an argon-filled glove box. A lithium foil
having a thickness of 125 micrometers functioned as the anode and reference
electrode. The cell featured 2325 hardware, equipped with a spacer plate (304
stainless steel), and a disc spring (mild steel). The disc spring was selected
so that
a pressure of about 15 bar would be applied to each of the cell electrodes
when the
cell was crimped closed. The separator was a Celgard #2502 microporous
polypropylene film (Hoechst-Celanese) that had been wetted with a 1 M solution
of LiPFb dissolved in a 30:70 volume mixture of ethylene carbonate and diethyl
carbonate (Mitsubishi Chemical).
After construction, the cells were removed from the glove box and cycle
tested using a MACCOR constant current cycler. Cycling conditions were
typically set at a constant current of 37 mA/g of active material. Cutoff
voltages of
0.0 V and 1.3 V were used.
X-Ray Diffraction
Powder x-ray diffraction patterns were collected using a Siemens D5000
diffractometer equipped with a copper target x-ray tube and a diffracted beam
monochromator. Data was collected between scattering angles of 10 degrees and
80 degrees unless otherwise noted.
To examine the electrode materials during cycling, in-situ x-ray diffraction
experiments were performed. Cells for in-situ x-ray diffraction were assembled
as
described above in the case of the cycling experiment with the following
differences. The coin cell can was provided with a circular hole measuring 18
mm
in diameter. A 21 mm diameter beryllium window (thickness = 250 micrometers)
was affixed to the inside of the hole using a pressure sensitive adhesive
(Roscobond from Rosco of Port Chester, NY). The electrode material was coated
directly onto the window before it was attached to the can.
-6-
CA 02394706 2002-06-17
WO 01/48840 PCT/US00/35331
The cell was mounted in a Siemens D5000 diffractometer and slowly
discharged and charged while x-ray diffraction scans were taken continuously.
Typically, a complete scan took 2-5 hours and the discharge and charge time
took
40-60 hours, giving approximately 10-30 "snapshots" of the crystal structure
of the
electrode as a function of its state of charge. The voltage of the cell was
continuously monitored during cycling.
Mossbauer Spectroscopy
In-situ ' l9mSn Mossbauer spectroscopy was used to study the local
environment of tin atoms during reaction with lithium. The advantage of
Mossbauer spectroscopy is that it can distinguish between tin atoms within the
non-crystalline regions and tin atoms within the crystalline grains.
Room temperature Mossbauer measurements were made with a Wissel
System II constant acceleration spectrometer operating at a frequency of 23 Hz
and
a krypton/CO~ x-ray proportional counter (Reuter-Stokes Inc.). The detector
employed a Pd filter. Data were collected using an Ortec ACE multi-channel
scaling board. The Ca"9°'SnO~ source had an intrinsic line width of
0.78 mm/s
(F'WHM), and the velocity scale was calibrated using a mixed sample of tin and
BaSnO~. Elevated temperature measurements were made using a small heater
placed around the sample without blocking the gamma rays.
Powder samples were prepared as follows. Powders were manually ground
and sieved (-325 mesh). Typically, 150 mg of powder was uniformly distributed
over a 30 mm piece of Scotch Brand adhesive tape (3M Co., St. Paul, MN), and
was kept in place by another piece of tape on top. Total measurement times
ranged
between 3 and 24 hours.
The cell used for in-situ Mossbauer measurements was similar to the cell
used for in-situ x-ray spectroscopy except that it was designed for maximum
transmission of gamma rays. As such, all steel parts were removed (including
the
spacer and spring), and a second hole (diameter = 13 mm) was cut in the cell
top.
A second piece of beryllium (diameter = 15 mm, thickness = 1 mm) was placed
over the hold and held in place by Roscobond pressure sensitive adhesive. A
thin
bead of Torr Seal (high vacuum grade available from Varian) was applied
CA 02394706 2002-06-17
WO 01/48840 PCT/US00/35331
following cell assembly at the interface between the cell bottom and beryllium
piece, and at the interface between the cell top and beryllium piece.
Electrodes,
prepared as described above, were coated directly onto the beryllium.
The cell was held in place approximately 10 cm from the detector and 1 cm
from the source. Charging and discharging currents were controlled by a
Keithley
220 programmable current source interfaced to a computer equipped with a
general
purpose interface bus. Voltages were measured using a Keithley 196 digital
voltmeter. Spectra were obtained continuously while the cell was discharged
and
subsequently charged. The total experiment time was approximately 180 hours,
during which about 60 three-hour Mossbauer spectra were recorded. The spectra
were fitted with one or more Lorentzian-shaped peaks. The center shift, area,
and
half-width of the fitted peaks were monitored.
Transmission Electron Microscopy
Samples were prepared for transmission electron microscopy by dispersing
the powder in methanol and sonicating the dispersion for one minute. Next, one
drop of the sonicated dispersion was placed on a standard 3 mm transmission
electron microscopy grid (carbon/formvar thin film supported on a copper mesh
grid). Excess solution was wicked away with a wedge of filter paper and the
remaining sample was allowed to dry for 10 minutes before inserting it into
the
microscope.
Transmission electron microscopy and electron diffraction analysis were
performed on a Hitachi H9000 instrument operating at 300 kV. Energy dispersive
x-ray spectroscopy was performed on the same instrument using a Noran Voyager
X-Ray Spectroscopy System.
Specific samples were prepared and tested as follows.
Example 1
An intermetallic compound, SnMn~C, was prepared by adding
stoichiometric ratios of 0.800 g tin powder (Aldrich Chemical), 1.111 g
manganese
powder (Aldrich Chemical), and 0.081 g graphite powder (mesocarbon microbeads
from Osaka Gas Ltd. that had been heated to 2650°C), along with two
12.7 mm
_g_
CA 02394706 2002-06-17
WO 01/48840 PCT/US00/35331
diameter hardened steel balls, to a hardened steel vial in an argon-filled
glove box.
The vial was placed in the Spex 8000 mixer and subjected to maximum milling
intensity for 20 hours following the general procedure described above.
The x-ray diffraction pattern of the milled sample is shown in Fig. 1. It
agrees with the literature pattern for SnMn3C except that the Bragg peaks are
broad
(width = about 1 degree), indicating the presence of nanometer-sized grains.
Using
the Scherer formula, L = 0.9~,,/(BcosO), where L is the grain size, ~, is the
x-ray
wavelength ( 1.54178A), B is the full width at half maximum of a particular x-
ray
peak in radians, and 8 is the Bragg angle of the peak, the grain size is
calculated to
be about 8 nanometers. The particle size of the sample was in the range of 2-
50
micrometers, determined by scanning electron microscopy, demonstrating that
each particle was made up of many grains.
An electrochemical cell was constructed as described above and its cycling
behavior tested. Fig. 3a shows the voltage-capacity for the cell. The cell
exhibited
a reversible capacity of about 130 mAh/g.
Fig. 3b shows the capacity versus cycle number for the cell depicted in Fig.
3a, and for an identical cell. Both show no loss in capacity over 100 cycles.
One
of the cells was slowed to 18.5 mA/g at cycle 120, and to 9 mA/g at cycle 160.
At
the lowest current, a capacity of 150 mAh/g was observed. This corresponds to
a
volumetric capacity of about 1200 mAh/g (calculated based upon a density value
of 7.9 g/cc for SnMn~C.
Fig. 4 shows the differential capacity versus voltage at several cycle
numbers for the cell that was slowed. The differential capacity shows a stable
pattern over the first 150 cycles, characteristic of nanometer-sized tin
grains in a
matrix. No sharp peaks in differential capacity develop, indicating that there
is no
aggregation of tin into large regions and that the tin atoms are active. If
all the tin
atoms were active, and each could react with 4.4 Li/Sn, then the specific
capacity
of SnMn~C would be about 400 mAh/g. The observed value of 150 mAh/g
corresponds to about 1.5 Li/Sn.
In-situ x-ray diffraction measurements were made using a specific current
of 2.2 mA/g. X-ray scans of 3 hours duration were taken successively. Figs.
5(a)-
(d) show the x-ray diffraction pattern from the electrode during discharge;
Fig. 5(e)
-9-
CA 02394706 2002-06-17
WO 01/48840 PCT/US00/35331
shows voltage versus capacity (bottom axis) and versus scan number (top axis)
for
the sample. Each diffra:;tion pattern represents the sum of five adjacent x-
ray
scans to improve the signal to noise ratio. The x-ray data demonstrate that
even
though approximately 2 Li.~Sn have reacted with the electrode (calculated
coulombmetrically based on the current, electrode mass, and time of current
flow),
there is no change in the position or intensity of the main Bragg peaks
attributed to
SnMn3C at 32, 39, and 40°. On the other hand, the broad "hump"
near 22°
intensifies as the discharge process proceeds.
The fact that the Bragg peaks do not change is evidence that the
nanocrystalline grains do not react with lithium at all. Accordingly, the only
materials available to react with lithium are the tin atoms located in non-
crystalline
regions separating the grains. The intensification of the "hump" near
22° may be
the result of small amounts (e.g., on the order of a few atoms) of Li4Sn in
the non-
crystalline regions.
In-situ Mossbauer spectroscopy measurements were made using a
discharge current of 2.2mA/g following the procedure described above. Spectra
of
3 hours duration were collected continuously. Figs. 6(a), (b), and (c) show
the
first, twentieth, and fortieth scans. Fig. 6(d) shows voltage versus capacity
(bottom
axis) and versus scan number (top axis) for the sample. The first spectrum
(Fig.
6(a)) was fitted with a major component with a center shift near 1.7 mm/s and
a
minor component with a center shift near 2.5 mm/s. A third component with a
center shift near 0.0 mm/s was also included, but it was not needed in order
to
obtain a good fit. Because x-ray diffraction data showed that the nanometer-
sized
crystalline grains did not react with lithium, the center shift and half-width
of the
major component were kept fixed while fitting the spectra taken as the
discharge
proceeded.
Figs. 6(b) and (c) show that the minor component shifts to smaller velocity
as lithium reacts with the sample. The Mossbauer spectra demonstrate that the
average center shift changes from about 2.5 to about 1.8 as lithium reacts
with tin.
Accordingly, the shift of the minor component is consistent with the reaction
of
lithium with tin.
-10-
CA 02394706 2002-06-17
WO 01/48840 PCT/US00/35331
Fig. 7 shows the variation of the center shift of the minor component as a
function of scan number taken during discharge and charge. The current used
during charge was 3.3 mA/g. The change in the center shift is reversible. This
is
evidence for the reversible reaction of lithium with tin atoms located within
the
non-crystalline regions of the sample.
Figs. 12 and 13 are transmission electron micrographs taken of the sample
at both high (400,000X) and low (20,000X) magnification. The micrographs show
the presence of two types of particles. The first type ranges in size from 10
nm to
over 10 microns. These particles are composed of crystalline grains having a
size
in the 8 nanometer range. The grains are separated from each other by non-
crystalline regions that are significantly less ordered than the crystalline
grains.
The scanned area exhibited a single diffraction pattern. The second type of
particle
is a single crystal roughly on the order of 10-30 nanometers by 100-300
nanometers with a large aspect ratio (somewhere between 10:1 and 20:1 ).
Example 2
Three additional samples of SnMn~C were prepared following the
procedure of Example 1. The samples were heat-treated at 400°C,
500°C, and
600°C, respectively, under vacuum for 3 hours. The x-ray diffraction
spectra for
the three samples, as well as the sample from Example 1 prepared without heat-
treating, are shown in Fig. 8. As shown in Fig. 8, the widths of the Bragg
peaks of
the SnMn~C phase narrow as the temperature increases, consistent with a growth
of
the size of the nanometer-sized crystalline grains and a reduction in the
number of
atoms in the non-crystalline regions. Fig. 8 also shows evidence of some minor
impurities, representing Fe-C phases, formed during heating as a result of
iron
contamination during milling.
Fig. 9 shows the voltage versus capacity and capacity versus cycle number
results for cells made from these samples. The cells containing heat-treated
material show much smaller capacity compared to the cell containing unheat-
treated material, of which about 15 mAh/g originates from the Super S carbon
black used to prepare the electrode composition. These results are further
evidence
that heat treatment induces grain growth, thereby decreasing the size of the
non-
-11-
CA 02394706 2002-06-17
WO 01/48840 PCT/US00/35331
crystalline regions and reducing the reversible capacity of the materials. .
The
reduction in capacity, in turn, is related to a decrease in the number of tin
atoms in
the non-crystalline regions available for reaction with lithium.
Example 3
The procedure of Example 1 was followed except that 0.823 g tin powder,
1.160 g iron powder (Aldrich Chemical Co.), and 0.084 g graphite powder were
used to prepare a material having the formula SnFe~C. The x-ray diffraction
pattern of the material is shown in Fig. 2. It agrees with the literature
pattern for
SnFe~C except that the Bragg peaks are broad, indicating the presence of
nanometer-sized grains. The particle size of the sample was in the range of 2-
50
micrometers, determined using scanning electron microscopy, demonstrating that
each particle was made up of many grains.
Example 4
Six additional samples of SnFe~C were prepared following the procedure of
Example 2. The samples were heat-treated at 100°C, 200°C,
300°C, 400°C, 500°C,
and 600°C, respectively, under vacuum for 3 hours. The x-ray
diffraction spectra
for these six samples, as well as the sample from Example 2 prepared without
heat-
treating, are shown in Fig. 10. As shown in Fig. 10, the widths of the Bragg
peaks
of the SnFe3C phase narrow as the temperature increases, consistent with a
growth
of the size of the crystalline grains and a reduction in the number of atoms
in the
non-crystalline regions.
Fig. 11 shows the voltage versus capacity and capacity versus cycle number
results for cells made from these samples. The cells containing heat-treated
material show much smaller capacity compared to the cell containing unheat-
treated material, of which about 15 mAh/g originates from the Super S carbon
black used to prepare the electrode composition. These results are further
evidence
that heat treatment induces grain growth, thereby decreasing the width of the
non-
crystalline regions and reducing the reversible capacity of the materials. The
reduction in capacity, in turn, is related to a decrease in the number of tin
atoms in
the non-crystalline regions available for reaction with lithium.
-12-
CA 02394706 2002-06-17
.~"~. , . ~ . ;jy.'y I !1ltt'i
w.i)vuvv).77»1
'3121-02-2002~~e Properties Company «~ :; >':~~j. ;f~~ Q'~ ' ~ ~ ~
r~'~'US003533'
2214 PCT r "' "~'-_ .'~.: :,-'.'~E.. -.
fur tce z . . r _
a ..,;, ~EP3
' ' S : :yin: .::,~~x~<~i
~ a .. ....t.e.. ..s _..,~~,.e~
less, it will be understood that various modificatio n c without
departing from the spirit o the nvention. Accordingly, other
w s.
-13-
_- A-MENDED-SHEET _ _ _ ____ _ -____ _: _ .